Structural Biochemistry/Volume 8
Nucleic Acids are long linear polymers that are called DNA, RNA. these polymers carry genetic information that passed from generations after generations. They are composed of three main parts: a pentose sugar, a phosphate group, and a nitrogenous base. Sugars and Phosphates groups play as structure of the backbone, while bases carries genetic components, which characterized the differences of nucleic acids. There are 2 types of bases: purines and pyrimidines, and these bases determine whether the nucleic acid is DNA or RNA.

Nucleic acids are composed of smaller subunits called nucleotides. A nucleotide is a nucleoside with one or more phosphoryl group by esterlinkage. When it is in the form of RNA the bases are called adenylate, guanylate, cytidylate, and uridylate. In the form of DNA the bases are called deoxyadenylate, deoxyguanylate, deoxycytidylate, and thymidylate. A nucleoside is a monomer, just the bases attached to a sugar without the phosphate groups. In this state the bases in RNA are called adenosine, guanosine, cytidine and uridine. In this state in DNA the bases are called deoxyadenosine, deoxyguanosine, deoxycytidine and thymidine.
| This page or section is an undeveloped draft or outline. You can help to develop the work, or you can ask for assistance in the project room. |
In organic chemistry, a phosphate, or organophosphate, is an ester of phosphoric acid. Organic phosphates are important in biochemistry and biogeochemistry.

The backbone of the DNA strand is made from alternating phosphate and sugar residues. The sugars are joined together by phosphate groups that form phosphodiester bonds between the third and fifth carbon atoms of adjacent sugar rings.
As you noticed in the deoxyribose sugar, it does not contain a hydroxyl group on the 2' carbon. This absence of the hydroxyl group allows greater stability because the absence of hydroxyl group allows the 2' carbon to resist hydrolysis. This is one of the reasons why the hereditary material is stored in the DNA and not RNA. However, the net negative charge of the phosphate group must be stabilized by metal ions, such as magnesium or manganese.
Phosphodiester bond
[edit | edit source]In the molecular bonding of the deoxyribonucleotide (DNA) and ribonucleotide(RNA), phosphodiester bond is a strong covalent bond between a phosphate group and two 5-carbon ring. The phosphate group contains a negative charge as it bonds to a 3' carbon in one ring and a 5' carbon in another ring.

The phosphodiester is formed when a single phosphate or two phosphates break away and catalyze the reaction by DNA polymerase. dATP would dissociate one phosphates in order to form a phosphodiester bond with a deoxyribose sugar from a nucleotide during the process of DNA elongation.
(DNA)n + dATP <------> (DNA) n+1 + Ppi
Phosphodiesterase is an enzyme that breaks a cyclic nucleotide phosphate due to incorrect hydrolysis of phosphodiester bonds. Phosphodiesterase will be an important clinical significance in repairing DNA sequences.
Carbohydrates
[edit | edit source]Carbohydrates are comprised of monosaccharide units which create sugars ranging from simplest of sugars such as glucose (chemical formula: C6H12O6) to the more complex polysaccharides such as starch. Single nucleotide monomeric units consist of one sugar molecule connected to 1) a heterocyclic nitrogen containing organic base, and 2) a Phosphate group that connects the sugar component of different nucleotides together. The organic base is usually attached to Carbon 1' of the sugar, while the Phosphate group is connected to Carbon 5' of the sugar. When strung together, the phosphate of the neighboring nucleotide attaches to Carbon 3' of the sugar.
Monosaccharides consist of aldehyde or ketone groups with hydroxyl groups as substituents. Sugars that contain an aldehyde group are called aldoses, and the sugars that contain a ketone group are called ketoses.
Sugars that are non-super imposable mirror images of each other are called enantiomers. Sugars that are stereoisomers but mirror images of each other are called diastereoisomers. If sugars that are stereoisomers but differ in configuration at a single chiral center are called epimers.
Sugars can be open-chain form or ring form. To form a six-membered hemiacetal ring, the carbon in the aldehyde group (C-1) attaches to the oxygen atom in the C-5 hydroxyl group. The six membered cyclic hemiacetal is called pyranose because it is similar to the structure of a pyran. To form a five-membered ring, the C-2 of ketone group attacks the oxygen atom of the hydroxyl group on C-6. The five membered cyclic hemiacetal is called furanose because it is similar to the structure of a furan. When a furanose or pyranose ring is formed, a new stereocenter is formed, and this new chiral carbon is called the anomeric carbon. This carbon can have one of two configurations, it is either in the S conformation (the hydroxyl group is pointing up), and it is referred to as the alpha carbon, or it is in the R conformation (the hydroxyl group is pointing down) and it is referred to as the B configuration. These two conformations are diastereomers, not enantiomers, and the α and β forms are called anomers.
A reducing sugar is one that can react because they have a relatively reactive hemiacetal group at C-1 position. Examples include: glucose, fructose, lactose, and maltose. The anomeric carbon in all of these molecules is free to react.
A non-reducing sugar is one that does not react, such as sucrose. The acetal group at the C-1 position makes the sugar non-reactive. Their structures are modified, so that they do not have free aldehyde or ketone groups to react. In sucrose, neither of the monosaccharides in the disaccharide can easily change into an aldehyde or ketone, making it nonreactive, this non-reducing. The glycosidic bond in the disaccharide hinders the molecule from being reactive. The anomeric carbon is not free to react. In order to determine whether or not a sugar is reducing, a Fehling's or Tollen's test is performed. In the Fehling's test a brick red precipitate is the positive result, and in the Tollen's test a silver mirror is the positive result.
In contrast, when a sugar is oxidized, the aldehyde or ketone carbonyl becomes a carboxyl group.
It is called an O-glycosic bond if the anomeric carbon is attached to an oxygen atom of a hydroxyl group. It is called an N-glycosidic bond if the anomeric bond is attached to a nitrogen atom of a amine group.
Glycosidic bonds are also what form the bridges between monosaccharides. If monosaccharides are joined by O-glycosidic bonds, they are called oligosaccharides.
The difference in having an -OH group attached to Carbon 2' of the sugar is the difference between DNA and RNA. In RNA, the carbon 2' contains an -OH group, whereas in the carbon 2' of DNA, there is just a hydrogen attached. The sugar in RNA, or "ribonucleic acid" is "ribose" while the sugar for DNA or "DEOXIribonucleic acid" is "deoxiribose." DEOXI- is used to represent the lack of oxygen from the -OH group on Carbon 2' of ribose.
| ||
||
Importance of sugar in glycoproteins

Sugar attached proteins called glycoproteins is another important component of the cell. Sugar components are oriented toward the watery cell exterior of glycoproteins. These sugar components serve as an identifier like cellular address labels. When signaling molecules pass through bodily fluids they encounter certain patterns of sugars, which either gives them access or dismissal. Therefore, the glyoproteins act as a regulator or gatekeeper in cells. In addition they help direct the formation of organs and tissue by forming correct cells together. Sugar coatings also help cells move through blood vessels by providing traction by latching on cell surface receptors.
References
[edit | edit source]Davis, Alison. "The Chemistry of Health." 'NIGMS August 2006: 36-42. http://publications.nigms.nih.gov/chemhealth/coh.pdf
| This page or section is an undeveloped draft or outline. You can help to develop the work, or you can ask for assistance in the project room. |
Structural Biochemistry/Nucleic Acid/Sugars/Deoxyribose sugar
| This page or section is an undeveloped draft or outline. You can help to develop the work, or you can ask for assistance in the project room. |
Ribose primarily occurs as D-ribose. It is an aldopentose, a monosaccharide containing five carbon atoms that has an aldehyde

functional group at one end. Typically, this species exists in the cyclic form. Ribose composes the backbone for RNA and relates to deoxyribose, as found in DNA, by removal of the hydroxy group on the 2' Carbon.
Ribose is less resistant to hydrolysis and will cause tension in RNA due to the negative charge of the phosphodiester bridge and the hydroxyl group on the 2' Carbon. The hydroxyl group has the capability to attack the phosphodiesr bond that typically links it to another ribose, thereby forming a cyclic form of the sugar. An example of this is cyclic Adenosine Monophosphate (cAMP).
Roles of D-ribose in the body
[edit | edit source]Aside of being the backbone for RNA and DNA, D-ribose is also important in the creation of ATP that all cells require to stay alive. It is currently used in medicinal practice to increase muscle energy and improve exercise performance. People that experiences Fibromyalglia and chronic fatigue syndrome that took a supplement of D-ribose improved their conditions dramatically. D-ribose supplements improved their conditions because it helps the patients produce more ATP in the body, because their body cannot produce a sufficient amount of ATP needed.
D-ribose has an important role in improving heart function for patients that suffer symptoms of congestive heart failure (CHF). Ischaemia, which is sudden decrease of blood supply, reduces myocardial ATP level. The addition of D-ribose will replenish the ATP level because it shortens the time it takes to create and restore ATP levels. Therefore the patient will be able to last longer during exercising before experiencing left chest pain, because the body is getting adequate amount of myocardial ATP. It also aided in regulating blood circulation in the heart by normalizing and readjusting blood flow through the left ventricle and atrium to accommodate the sudden change in blood supply. As a result patients suffering from CHF has an improved quality of life after taking D-ribose supplements because they are able to do more physical activity and return to a near normal lifestyle.
D-Ribose supplement is also important to athletes as well because it quickly replenishes ATP levels in muscle to help increase stamina and aid in strength building. D-ribose shorten the time it takes to create ATP because it directly enter the pentose phosphate pathway to create ribose-5-phosphate without having to go through the glucose-6-phospohate dehydrogenase and 6-phosphogluconate dehydrogenase, both of which require rate-limiting enzymes to form. The rate-limiting enzyme will slow down the creation of ATP, therefore by bypassing those pathways ATP will be produced at a higher rate. Hence, it restores ATP that was loss during exercise faster.
Summary of the roles:
1. Provide a backbone for DNA and RNA
2. Restores ATP in the body
3. Improve muscle stamina
4. Regulate blood circulation in the heart.
Natural sources of D-Ribose
[edit | edit source]D-ribose is a molecule that is naturally produced by the human body and is not found in food sources. However riboflavin, a component of d-ribose that helps aid in the production of d-ribose, is found in a plethora of food. Riboflavin, also known as vitamin B2 is found in found in eggs, milk products, nuts, vegetable, beef, and other proteins. However, these should be kept in areas where it is dimly lit because light can damage riboflavin.
Riboflavin
[edit | edit source]Aside from helping form d-ribose, riboflavin also helps fight off free radicals that can be damaging to cell. Hence it is also a form of antioxidant for the body. Free radicals can damage cells and increase aging and contribute to health conditions, such as heart disease and cancer, therefore riboflavin aids in the reduction of free radicals found in one’s body. Another function of riboflavin is that it helps produce red blood cell and convert B6 vitamin into a form the body can use. Another function of riboflavin is that it helps skin develop properly.
Summary of roles:
1. Helps form ribose that is then converted to d-ribose
2. Acts as an antioxidants
3. Helps produce red blood cells.
4. Convert B6 vitamin into a form the body can use.
5. Helps develop skin properly.
References
[edit | edit source]1. http://eurjhf.oxfordjournals.org/content/5/5/615.long
2. http://www.super-smart.eu/en--Sports-Endurance--D-Ribose--0477
3. http://www.livestrong.com/article/492628-natural-sources-of-d-ribose/
4. http://www.umm.edu/altmed/articles/vitamin-b2-000334.htm
5. http://www.webmd.com/vitamins-supplements/ingredientmono-957-RIBOFLAVIN%20(VITAMIN%20B2).aspx?activeIngredientId=957&activeIngredientName=RIBOFLAVIN%20(VITAMIN%20B2)
Overview
[edit | edit source]
A DNA nucleotide is composed of 3 main units: a 5-carbon monosaccharide (deoxyribose), a phosphate group, and a nitrogenous base. While the monosaccharide and phosphate group alternate in sequence and form the backbone of the DNA double helix, the nitrogenous bases may differ in every adjoining nucleotide. The four nitrogenous bases present in DNA are adenine (A), guanine (G), cytosine (C) and thymine (T). In RNA, the only differing nitrogenous base is uracil (U) (which replaces thymine in DNA and differs thymine only by the missing methyl group at carbon 5 of the pyrimidine ring). Of the nitrogenous bases, adenine and guanine are purines, which are aromatic compounds attached to an imidazole group, while cytosine and thymine and uracil compose a set of pyrimidines, which are one ring-aromatic compounds. Nitrogenous bases, being hydrophobic, tend to face inwards of the double helix, pointing away from the surrounding aqueous environment. If the phosphate backbones were faced inside of the double helix, then there will be too many charges clustered together such that the double helix would be an unlikely product. Bonds between linking nitrogenous bases of two DNA strands are Hydrogen bonds with 3 H-bonds connecting cytosine and guanine and 2 H-bonds connecting adenine and thymine, while the bonds between the stacking of DNA are kept in close contact via van der waals interactions. The aromaticity of the nitrogenous bases accounts for the DNA absorbance peak at 260nm.
== What is a Purine? ==
| This book's factual accuracy is disputed. |

The name was invented by the German chemist Emil Fischer in 1884. A purine is a nucleotide (a nucleoside + phosphate group) that is amine based and planar, aromatic, and heterocyclic. The structure of purine is that of a cyclohexane(pyrimidine group) and cyclopentane(imidazole group) attached to one another; the Nitrogen atoms are at positions 1,3,7,9. Adenine(A) and Guanine(G) are examples of purines which are involved in the construction of the backbone of the DNA and RNA. They are also a part of the structures for Adenosine disphosphate (ADP), triphosphate(ATP), and other enzymes. Purines form bonds with pentoses exclusively through the 9th Nitrogen atom.

6-amino and 2-amino-6-oxy purine
[edit | edit source]One derivative form of purine, adenine (A), is also commonly known as 6-amino purine. The 6-amino purine molecule contains an amine group attached to the carbon atom at position 6 double bonded to the nitrogen atom at position 1 and single-bonded the carbon atom at position 5. Another derivative form of purine, guanine (G), is also known as 2-amino-6-oxy purine. The 2-amino-6-oxy purine contains an amine group attached to the carbon atom at position 2 double bonded to the nitrogen atom on position 3 and single-bonded to the nitrogen atom on position 1. Guanine also has a carbonyl group at position 6 hence the 6-oxy.


Purine content in foods
[edit | edit source]Food is responsible for approximately 30% of uric acid in the blood. Regular diets could affect the level of uric acid. Some food will increase the blood acidity even if the content in purine is low.
Lowest level of Purine: 0–50 mg
[edit | edit source]tea, coffee, soda, nuts, dairy products, vegetables, cereal, fruits, preserve foods, sweets
Moderate level of purine: 50–150 mg
[edit | edit source]spinach, avocado, beef, turkey, lamb, oyster, fish, peanuts, sausages, ducks, chickens
High level of purine: 150–1000 mg
[edit | edit source]kidney, liver, heart, caviar, scallops, lobster, sardines, Thai fish sauce
Risks
[edit | edit source]A diet high in purines can lead to gout, a form of arthritis with symptoms of severe pain, redness, and swelling. Uric acid is a product formed from the breakdown of purines. Uric acid builds up in one's joints, causing the inflammation and resultant pain.
2 Types of Purine Disorders of Nucleotide Synthesis
[edit | edit source]Adenylosuccinase deficiency
[edit | edit source]This causes retardation or heart attacks due to high level of succinyladenosine in urine. Currently, there is no treatment.
Phosphoribosylpyrophosphate synthetase superactivity
[edit | edit source]A recessive disorder which causes too much production of purines, which results in gout or other developmental effects. Treatments could include low purines in daily diet.
Functions
[edit | edit source]Purines are biochemically significant in a myriad of biomolecules besides DNA and RNA, such as ATP, GTP, cyclic AMP, NADH, and coenzyme A. Although purine has not been found naturally in nature, it can be produced through organic synthesis. Purines can also be used as neurotransmitters, acting upon purinergic receptors (i.e., adenosine activates adenosine receptors)
Metabolism
[edit | edit source]Many organisms utilize metabolic pathways in order to synthesize and break down purines. Biologically, purines are synthesized as nucleosides, which are bases attached to ribose.
Laboratory Synthesis
[edit | edit source]Purines can be created artificially, too, and not just through vivo synthesis in purine metabolism. When formamide is heated in an open vessel at 170°C for 28 hours, purine is obtained.
Procedure
[edit | edit source]1. Obtain a sample of formamide
2. Heat in an open vessel with a condenser for 28 hours in an oil bath at 170-190°C
3. Remove excess formamide through vacuum distillation
4. Reflux the residue with methanol
5. Filter the methanol solvent and remove by vacuum distillation
Adenine
[edit | edit source]
Structure & Function
[edit | edit source]Adenine(A) is one of the four bases that make up nucleic acids. It is a purine base that complementarily binds to Thymine (T) in DNA and Uracil (U) in RNA. This bond is formed by two hydrogen bonds, which help stabilize the nucleic acid structures. Different structures of adenine mainly result from tautomerization of adenine, which allows the molecule to be available in isomeric forms in chemical equilibrium. The molecular formula of adenine is C5H5N5 .
An adenine molecule bound to a deoxyribose, a sugar, is known as deoxyadenosine. An adenine bound to ribose, also a sugar, is known as adenosine, a key component in Adenosine Triphosphate. When adenosine attaches to three phosphate groups, a nucleotide, adenosine triphosphate (ATP) is formed. Adenosine triphosphate is an important source of energy that is used in many cellular mechanisms, primarily in the transfer of energy in chemical reactions. The phosphate of ATP can detach, resulting in a release of energy.
In addition to ATP, adenosine also plays a key role in other organic molecules nicotinamide adenine dinucleotide (NAD) and flavin adenine dinucleotide (FAD), both molecules of which are involved in metabolism. Also, adenine can be found in tea, vitamin B12, and several other coenzymes.
Formation and other forms of Adenine
[edit | edit source]In the human body, adenine is synthesized in the liver. Biological systems tend to preserve energy, so usually adenine is achieved through the diet, the body degrading nucleic acid chains to obtain individual bases and reconstructing them through mitosis. The vitamin folic acid is important for adenine synthesis.
Adenine forms adenosine, a nucleoside, when attached to ribose, and deoxyadenosine when attached todeoxyribose; it forms adenosine triphosphate (ATP), a nucleotide, when three phosphate groups are added to adenosine. Adenosine triphosphate is used in cellular metabolism as one of the basic methods of transferring chemical energy between reactions.
In older literature, adenine was sometimes called Vitamin B4. However it is no longer considered a true vitamin (see Vitamin B). Some think that, at the origin of life on Earth, the first adenine was formed by the polymerizing of 5 hydrogen cyanide (HCN) molecules.
Biosynthesis
[edit | edit source]Adenine is one of the byproducts of the Purine metabolism, where inosine monophosphate (IMP) is synthesized with a pre-existing ribose through a complex process involving atoms from the amino acids glycine, glutamine, and aspartic acid, in addition to the formate ions transferred from coenzyme tetrahydrofolate.
]== Tautomerization ==
Tautomers are isomers related by changing the positions of attachment of a single hydrogen and a single double bond, in a three-atom system, such as the keto- and enol tautomers of a ketone. Like, keto-enol tautomers, Adenine, as well as Cytosine, Guanine, Tyrosine, and Uracil may go through tautomerization, interchanging from the amino to the imino functionality by intermolecular proton transfer.
 File:Http://i.imgur.com/l0lfQ.gif
Uracil
File:Http://i.imgur.com/yTNRT.gif
Cystein
File:Http://i.imgur.com/1hwJR.gif
Guanine
File:Http://i.imgur.com/WGmXH.gif
Thymine
File:Http://i.imgur.com/l0lfQ.gif
Uracil
File:Http://i.imgur.com/yTNRT.gif
Cystein
File:Http://i.imgur.com/1hwJR.gif
Guanine
File:Http://i.imgur.com/WGmXH.gif
Thymine
Reference
[edit | edit source]http://www.highbeam.com/doc/1G1-233827562.html http://en.wikibooks.org/wiki/Structural_Biochemistry/Nucleic_Acid/Sugars/Deoxyribose_Sugar http://en.wikibooks.org/wiki/Structural_Biochemistry/Nucleic_Acid/Sugars/Ribose http://en.wikibooks.org/wiki/Structural_Biochemistry/Nucleic_Acid/Phosphate
Guanine
[edit | edit source]
Guanine is among the five nucleobases that is found in DNA and RNA. The formula of guanine is C5H5N5O, and is a planar and bicyclic molecule. Guanine has two forms, keto and enol forms. The keto form is the major form. Guanine, like adenine, is a derivative of purine and binds to cytosine through 3 hydrogen bonds. The amino group in the cytosine is the hydrogen donor and the C2 carbonyl and the N3 amine are the hydrogen-bond acceptors. In Guanine, the group at C6 acts as the hydrogen accepter, and the group at N1 and the amino group at C2 act as the hydrogen donors. The related nucleoside containing guanine and ribose is called guanosine and guanine bound to deoxyribose sugar is called deoxyguanosine.
Guanine is capable of being hydrolyzed by strong acids to form ammonia, carbon monoxide, carbon dioxide, and glycine. Guanine oxidizes more readily than adenine, another purine-derivative nitrogenous base in nucleic acids. Guanine has a high melting point of 350°C due to the intermolecular hydrogen bonds between the oxo and amino groups in the crystal of the molecule. Also because of this intermolecular bonding, guanine is relatively insoluble in water as well as in weak acids and bases.
DNA base pair bonding
[edit | edit source]
From the image on the left, it can be seen that Guanine and Cytosine bond together through noncovalent hydrogen bonding at three distinct sites. Since Cytosin to Guanine has 3 H-bonds and Adenine to Thymine has 2 H-bonds, a higher CG content leads to higher melting point when compare with AT content. An interesting note is that Watson and Crick first hypothesized that Guanine and Cytosine bonded together through hydrogen bonding at two distinct sites. [1]
Tautomerization
[edit | edit source]Guanine may go through tautomerization, interchanging from the keto to the enol functionality by intermolecular proton transfer.

Miscellaneous
[edit | edit source]Guanine is also the name of the white amorphous substance found in fish scales. It serves as an additive to various products such as shampoos, metallic paints, and simulated pearls and plastics providing a pearly iridescent effect. Also, it adds a shimmering luster to eye shadow and nail polish. This pearly luster is produced by the crystalline form of guanine which are rhombic platelets composed of multiple transparent layers that have a high index of refraction that partially reflects and transmits light from layer to layer. To provide this effect, it can be applied by spraying, painting, or dipping.
References
[edit | edit source]- ↑ Crick, Francis H. (1953). "Molecular Structure of Nucleic Acids". Nature. 171: pp. 737-738.
{{cite journal}}:|pages=has extra text (help); Unknown parameter|month=ignored (help)
Berg, Jeremy M. John L. Tymoczko. Lubert Stryer. Biochemistry Sixth Edition. New York: W.H. Freeman and Company, 2007.
| This page or section is an undeveloped draft or outline. You can help to develop the work, or you can ask for assistance in the project room. |
Purine is a heterocyclic aromatic organic compound. Purine consists of a pyrimidine ring fused to an imidazole ring. Purines and pyrimidines make up of two groups of nitrogenous bases. The name was invented by the German chemist Emil Fischer in 1884. Below are the DNA bases.
Hypoxanthine
[edit | edit source]Hypoxanthine (6-Hydroxypurine) is a naturally occurring purine derivative and deaminated form of adenine. It is an intermediate in the purine catabolism reaction and is occasionally found as a constituent in the anticodon of tRNA as the nucleosidic base inosine. It is also utilized as a nitrogen source in bacteria and parasite cultures for energy metabolism and nucleic acid synthesis.

Reactions
[edit | edit source]Hypoxanthine exists as an intermediate in the biodegradation of AMP (adenosine monophosphate). It is first converted to xanthine with xanthine oxidase before it is excreted as urate.

A deleterious reaction that can occur is a spontaneous deamination of adenine to form hypoxanthine. This is a mutagenic process because the result is a pairing of hypoxanthine with cytosine rather than thymine, due to hypoxanthine’s guanine-like form. This could lead to an error in DNA transcription and replication.

References
[edit | edit source]Berg, et al. Biochemistry, 6th Ed. 2007.
Xanthine
[edit | edit source]Xanthine is a purine base that's an antecedent of uric acid and is generally found in muscle tissue, blood, urine and some plants. It is a water insoluble toxic yellowish white powder and acids that's soluble in caustic soda; it sublimes when heated. It is involved in purine degradation and is converted from hypoxanthine and converted to uric acid by xanthine oxidase. Some of its derivatives are widely known as mild stimulants, which include caffeine, a sleep-inhibiting methylated xanthine found in coffee, and theobromine, a bitter alkaloid found in cacao.

Diseases
[edit | edit source]There is a genetic disease of xanthine metabolism, xanthinuria, due to deficiency of an enzyme, xanthine oxidase. Xanthinuria is a rare genetic disorder where individuals are unable to convert xanthine into uric acid because of the lack of enzyme xanthine oxidase resulting in an accumulation of xanthine. Symptoms include renal failure and kidney stones. There is currently no treatment available to cure this disease.
Clinical Use
[edit | edit source]Xanthine derivatives are collectively known as xanthines, which are a group of alkaloids used as stimulants and bronchodilators. As a result of widespread side effects, many of these derivatives have been treated as second-rate asthma treatment medication.
References
[edit | edit source]Berg, et al. Biochemistry, 6th Ed. 2007.
Theobromine
[edit | edit source] Theobromine (xantheose) is a xanthine derivative and bitter alkaloid commonly found in cacao plants. Its name is derived from the name of the genus of the cacao tree. It doesn’t contain bromine, as its name might indicate. It shares a similar structure to that of another well-known purine and xanthine derivative known as caffeine, except it contains one more methyl group. It was first discovered in the cacao plant in 1841, isolated in 1878, and synthesized from xanthine by Hermann Emil Fischer shortly thereafter. In its pure form, it is a water-insoluble, crystalline white powder that has a milder effect than caffeine. Since dark chocolate has higher concentrations of theobromine than milk chocolate, its beneficial effects are better attained from the less diluted dark chocolate.
Theobromine (xantheose) is a xanthine derivative and bitter alkaloid commonly found in cacao plants. Its name is derived from the name of the genus of the cacao tree. It doesn’t contain bromine, as its name might indicate. It shares a similar structure to that of another well-known purine and xanthine derivative known as caffeine, except it contains one more methyl group. It was first discovered in the cacao plant in 1841, isolated in 1878, and synthesized from xanthine by Hermann Emil Fischer shortly thereafter. In its pure form, it is a water-insoluble, crystalline white powder that has a milder effect than caffeine. Since dark chocolate has higher concentrations of theobromine than milk chocolate, its beneficial effects are better attained from the less diluted dark chocolate.
Therapeutic uses
[edit | edit source]Theobromine is known as a diuretic, which promotes the removal of excess fluids accumulated in the body from edema, or the flushing of excess salts through the increase production of urine.
It is also widely used as a vasodilator, which widens blood vessels and improves blood flow. This, in turn, helps reduce blood pressure, although it is reputed that flavanols have a bigger role in promoting that effect.
A 2004 patent on the future use of theobromine for cancer prevention was granted due to recent research that revealed anti-carcinogenic activity.
Effects
[edit | edit source]Humans
[edit | edit source]Theobromine has a weaker effect on the human central nervous system than caffeine because of its weaker inhibition effects on cyclic nucleotide phosphodiesterases and its antagonism of adenosine receptors. As for its effect on the heart, theobromine stimulates it to a much greater degree than caffeine. It is cited as being involved in contributing to chocolate’s role as an aphrodisiac.
Since theobromine is a myocardial stimulator, it increases the heartbeat. As stated above it also dilates blood vessels and reduces blood pressure by enlarging the vessels. It is possible that theobromine might be able to treat cardiac failure since it has properties which allowing draining. Ingesting too much theobromine could lead to some adverse effects. Since it is a diuretic, it will increase the amount of urine produced in the person. It could also possible cause nausea, restlessness, sleeplessness, and anxiety.
Poisoning
[edit | edit source]A helpful hint in responsible pet-keeping is to not feed dogs or cats cacao containing products. This is because they metabolize theobromine much more slowly than humans. Complications that arise from doing such an action is succumbing your pet to theobromine poisoning, which causes digestive issues, dehydration, excitability, and a slow heart rate. Larger quantities of theobromine can result in epileptic-like seizures and even death.
What is a Pyrimidine?
[edit | edit source]A pyrimidine is a 6-membered heterocyclic organic compound made up of 4 carbon atoms and 2 nitrogen atoms at positions 1 and 3.[1] It is one of three isomers of diazine, the other two being pyridazine (1,2-diazine), and pyrazine (1,4-diazine).[2] Pyrimidines are aromatic and planar. The nucleobases Cytosine(C), Uracil(U), and Thymine(T) are all examples of pyrimidines; each with different chemical groups. Pyrimidines can attach to a phosphate sugar group such as a ribonucleotide(which have a hydroxy group positioned axially at carbon-2) or deoxyribonucleotide(which have a hydrogen atom at C-2) through a glycosidic linkage at the 1st Nitrogen to form a nucleotide, the monomeric building block of nucleic acids (DNA and RNA).

Correct mistake:
2. It needs carbonyl phosphate synthetase, which is located in the cytoplasm.
Pyrimidine Biosynthesis
[edit | edit source]1. Unlike in purine, the ring is synthesized first then conjugated after.
2. It needs carbamoyl phosphate synthetase, which is located in the cytoplasm.
3. It also needs an enzyme in order for the reaction to work, but the enzyme should be controlled in 2 steps:
- controlled level at where the reaction occurs & transcriptions must be reduced
- the pyrimidine nucleotides which produces the feedback inhibition level also must be controlled
4. The ring then closes.
5. The C-C bond is formed when the ring oxidizes.



Chemical Properties
[edit | edit source]Pyrimidine has similar properties to that of pyridines. One similarity is that as the number of nitrogen atoms in the ring increase, the ring pi electrons become less energetic and, as a result, electrophilic aromatic substitution gets more difficult while nucleophilic aromatic substitution gets easier. One example is the displacement of the amino group in 2-aminopyrimidine by chlorine and its reverse reaction. Reduction in resonance stabilization of pyrimidines leads to the addition and ring cleavage reactions, and not substitutions. An example of this is in the Dimroth arrangement. Pyrimidines are less basic than pyridines and the N-alkylation and N-oxidation are more difficult in pyrimidines as well.
Cytosine
[edit | edit source]Cytosine is part of the pyrimidine family, and it is one of the 5 nucleotide bases found in both DNA and RNA. The molecular formula of cytosine is C4H5N3O. Cytosine consists of a heterocyclic aromatic ring, an amine group at C4, and a keto group at C2. Cytosine binds with ribose to form the nucleoside cytidine and with deoxyribose to form deoxycytidine.


The molecule is of planar geometry and cytosine forms 3 hydrogen bonds with Guanine in the DNA double helix. The nucleoside of cytosine is cytidine in RNA, which consists of cytosine and ribose. In DNA, it is called deoxycytidine, which consists of cytosine and deoxyribose. The nucleotide of cytosine in DNA is deoxycytidylate which consists of a cytosine, ribose and phosphate.
History
[edit | edit source]In 1894, Cytosine was discovered by the hydrolysis of the calf thymus tissue. The first structure for cytosine was published in 1903 and the structure was validated when it was synthesized that same year.(The Columbia Encyclopedia)
Chemical Activity
[edit | edit source]From the image on the left, it can be seen that Guanine and Cytosine bond together through noncovalent hydrogen bonding at three distinct sites. An interesting note is that Watson and Crick first hypothesized that Guanine and Cytosine bonded together through hydrogen bonding at two distinct sites. [3]
Cytosine is found in DNA and RNA or as a part of a nucleotide. When the nucleoside cytidine binds with three phosphate groups, it forms cytidine triphosphate (CTP). This molecule can act as a co-factor to enzymes and it aids in transferring a phosphate to convert adenosine diphosphate (ADP) to adenosine triphosphate (ATP) to prepare the ATP to be used in chemical reaction.
In DNA and RNA, cytosine binds with guanine through 3 hydrogen bonds. However, this unit is unstable and can change into uracil. This process is called spontaneous deamination. This can possibly lead to a point mutation if DNA repair enzymes such as uracil glycosylase does not repair it by cleaving uracil in DNA.
Tautomerization
[edit | edit source]Cytosine may go through tautomerization, interchanging from the amino to the imino functionality by intermolecular proton transfer.

References
[edit | edit source]- ↑ https://www.sciencedirect.com/science/article/pii/B9780128144534000194
- ↑ https://digitalcommons.unl.edu/cgi/viewcontent.cgi?article=1016&context=physicsuiterwaal
- ↑ Crick, Francis H. (1953). "Molecular Structure of Nucleic Acids". Nature. 171: pp. 737-738.
{{cite journal}}:|pages=has extra text (help); Unknown parameter|month=ignored (help)
Berg, Jeremy M. John L. Tymoczko. Lubert Stryer. Biochemistry Sixth Edition. New York: W.H. Freeman and Company, 2007.
CYTOSINE. The Columbia Encyclopedia, Sixth Edition
Uracil
[edit | edit source]Uracil is among the five nucleobases: adenine, guanine, cytosine, and thymine,but is only found in RNA. It is a naturally occurring pyrimidine derivative with the molecular formula C4H4N2O2. Uracil is planar and unsaturated and has the ability to absorb light.
Properties
[edit | edit source]Uracil is found in RNA and binds to adenine via 2 hydrogen bonds, but is replaced by thymine in DNA. Methylation of Uracil produces thymine. Uracil can pair with any of the base pairs depending on arrangement. Despite this, it readily pairs with adenine because the methyl group is repelled into a fixed position. In the uracil and adenine bond, uracil is the hydrogen bond acceptor and the adenine is the donor. When attached to a ribose sugar, the compound is called uridine, a nucleoside. Then, phosphate attaches to uridine to form uridine 5'-monophosphate. Nucleotides are formed through a series of phosphoribosyltransferase reactions. This produces substrates, aspartate, carbon dioxide, and ammonia.

Uracil, like other bases, undergoes tautomerization. The keto tautomer is referred to as the lactam structure, while the imidic acid tautomer is referred to as the lactim structure. With the lactam structure being the major form of uracil, both tauotemric forms are present under conditions where pH=7.
Uracil is a weak acid.
Chemical Activity
[edit | edit source]Uracil is capable of undergoing reactions such as oxidation, nitration, and alkylation. It can also react with elemental halogens because of the presence of more than one strongly electron donating group. A useful property of uracil is that in the presence of PhOH/NaOCl, it can be visualized in the blue region of UV light.
As stated above, uracil can partake in synthesis, binding with ribose sugars and phosphates to form very useful molecules like uridine, urindine monophosphate (UMP), urindine diphosphate (UDP), urindine triphosphate (UTP).

History
[edit | edit source]Uracil is a nucleotide that was discovered in the 1900s by the hydrolysis of yeast(Brown 1994). Uracil is an important component in helping enzymes to carry out different reactions and the making of polysaccharides (New World Encyclopedia). Because Uracil helps enzymes carry out different reactions in cells, it is important in the drug industry because it helps with delivering drugs throughout the body. Even though it is useful in helping the delivery of drugs in the body, it can increase the risk of cancer when the body is missing the nutrient folate (The Individualist). Uracil is naturally occurring however, it could also be synthesized in the laboratory by mixing water with cytosine. This reaction will produce two compounds which are uracil and ammonia(Wikipedia).
Tautomerization
[edit | edit source]Uracil may go through tautomerization, interchanging from the keto to the enol functionality by intermolecular proton transfer due to rich electrons ring.

References
[edit | edit source]New World Encyclopedia. Uracil. "http://www.newworldencyclopedia.org./entry/Uracil." 17 November 2008.
Wikipedia. Uracil. "http://en.wikipedia.org/wiki/Uracil." 17 November 2008.
Brown, D.J. Heterocyclic Compounds: Thy Pyrimidines. Vol 52. New York: Interscience, 1994.
The Individualist. Uracil. "http://www.dadamo.com/wiki/wiki.pl/Uracil." 17 November 2008.
Thymine
[edit | edit source]5th carbon, hence the other name of thymine, 5-methyluracil. Uracil takes its place in RNA, which also binds to adenine. Thymine is a single ring planar molecule. Thymine combined with deoxyribose yields deoxythymidine while Thymine with ribose makes thymidine.
Thymine binds with deoxyribose to form the nucleoside deoxythymidine, which is the same thing as thymidine. This compound can be phosphorylated with one, two, or three phosphoric acid groups creating thymidine mono-, di-, or triphosphate, respectively.

Thymine is a part of one of the most common mutations of DNA, which involves two adjacent thymines or cytosines. In the presence of UV light, this may form thymine dimers, causing "kinks" in the DNA molecule, interfering with normal function.
Uses of thymine include cancer treatment where it serves as a target for actions of 5-fluorouracil (5-FU). Substitution of this compound to thymine (in DNA) and uracil (in RNA) allows inhibition of DNA synthesis in actively-dividing cells.
Properties
[edit | edit source]Thymine is a heterocyclic aromatic organic compound as a pyrimidine nucleobase. Heterocyclic compounds are organic compounds (those containingcarbon) that contain a ring structure containing atoms in addition to carbon, such as sulfur, oxygen, or nitrogen, as part of the ring. Aromaticity is a chemical property in which a conjugated ring of unsaturated bonds, lone pairs, or empty orbitals exhibit a stabilization stronger than would be expected by the stabilization of conjugation alone.
As the name implies, thymine may be derived by methylation of uracil at the fifth carbon. In DNA, thymine(T) binds to adenine (A) via two hydrogen bonds to support in stabilizing the nucleic acid structures.
Thymine jointed with deoxyribose creates the nucleoside deoxythymidine, which is identical with the term thymidine. Thymidine can be phosphorylated with one, two, or three phosphoric acid groups, creating TMP, TDP or TTP (thymidine mono- di- or triphosphate) correspondingly.
One of the common mutations of DNA involves two neighboring thymine or cytosine, which in existence of ultraviolet light may form thymine dimers, causing "kinks" in the DNA molecule that constrain normal function.
Thymine could also be a goal for actions of 5-fu in cancer treatment. 5-fu can be a metabolic analog of Thymine (in DNA synthesis) or Uracil (in RNA synthesis). Replacement of this analog inhibits DNA synthesis in actively dividing cells.
Tautaumerization
[edit | edit source]Thymine may go through tautaumerization, interchanging from the keto to the enol functionality by intermolecular proton transfer.

References
[edit | edit source]Al Mahroos, M., et al. “Effect of sunscreen application on UV-induced thymine dimers.” Arch Dermatol 138: 1480-5, 2002. Ribonucleotide reductase (or RNR) is the enzyme responsible for catalyzing the reduction of ribonucleotides to deoxyribonucleotides. These deoxyribonucleotides can then be utilized by the cell in DNA replication. Additionally, because of the role RNR plays in the formation of deoxyribonucleotides, RNRs are responsible for regulating the rate of DNA synthesis within the cell.[1]
Classes of RNR[2]
[edit | edit source]- Class I: Class I RNRs consist two subgroups (Ia, Ib, and Ic) which differ only slightly in primary structure; however, both subgroups are common in that they contain two different dimeric subunits (R1 and R2) and require oxygen in order to form a stable radical. Class Ic RNRs are the most recently discovered, first found in Chlamydia trachomatis. Evidence also suggests its existence in archaea and eubacteria. The sequence of class Ic RNRs shows that residues in the PCET pathway and active site for nucleotide reductase are similar between the three subgroups.[3]
- Class II: Class II RNRs form thiyl radicals with the help of adenosylcobalamin – which fulfills the role of the R2 subunit as a radical generator – and utilize thioredoxin or glutaredoxin as electron donors. Therefore, class II RNRs are made up of only one subunit and present as monomers or dimmers and neither require nor are inhibited by the presence of oxygen.
- Class III: Class III RNRs, like Class I RNRs, are made up of two dimeric protein subunits (NrdG and NrdD); however, unlike in Class I RNRs which require R2 continuously to generate radicals, the small NrdG is only required during the activation of NrdD. The mechanism of Class III RNRs uses formate as an electron donor and generates an oxygen-sensitive glycyl radical, thus rendering the enzymes inactive in the presence of oxygen.
Radical Mechanism of RNR
[edit | edit source]Despite the differences in structure and electron donor, all three classes of RNR proceed via a free radical mechanism.[4] Ultimately RNR catalyzes a reaction which results in the replacement of the 2'-hydroxyl group of the ribose with a hydrogen atom resulting in a deoxyribose moiety.
Metallocofactor Assembly in Class I RNR[5]
[edit | edit source]Although the Class I RNR’s (Ia, Ib, and Ic) have comparable structures and pathways, the metallocofactors necessarily involved in the activity of RNRs to catalyze the conversion of nucleotides to deoxynucleotides differ remarkably. The mechanisms which generate these cofactors, both in vitro and in vivo, and examining how damaged cofactors are repaired show the significance of each subgroup’s dependence on different cofactors. Studies of the pathways and activation of these metallocofactors have helped our understanding of how biology prevents mismetallation from occurring and configures cluster formation in high yields. All three class I RNR share a common catalytic mechanism in which the metal cofactor is involved directly or indirectly in the oxidation of the conserved cysteine in the active site of alpha to thiol radical S•). Class I RNR oxidation occurs by the Y• in Ia and Ib.
- Class IA: Class IA RNR requires a FeIIIFeIII-Y• cofactor. It is localized in β2 at the end of a hydrophobic channel, the supposed access route for O2 cluster assembly. In studies of E. coli, the in vivo process showed that incubation of apo-β2 of E. coli with FeII, O2, and reductant, resulted in self-assembly of the FeIIIFeIII-Y• cofactor. This process likely requires at minimum a single small protein or molecule to deliver FeII to apo-β2 and to deliver the extra reducing equivalent required to reduce O2 to H2O. This is also plausible because Ia RNRN binds MnII more tightly than FeII, thus requiring some type of chaperone protein to ensure proper metallation.
- Class IB: Class IB RNR is active with both FeIIIFeIII-Y• and MnIIIMnIII-Y• cofactors. The enzymes can form active FeIIIFeIII-Y• cofactors in vitro, but only the MnIIIMnIII-Y• cofactor was found to be relevant in vivo. The mechanism of this formation has been proposed to occur via oxidation of a MnIIMnII center by a flavoprotein known as NrdI, an oxidant created by reduction of O2. In E.Coli, studies have found that the manganese cofactor is induced when iron is at premature levels in the cell, pointing to the significance of manganese in this and other organisms. There is also an extent of organism-dependent variation in metal homeo-stasis to be considered which may help explain why some organisms rely on either cofactor more frequently.
- Class IC: Class IC RNR is unique from Class Ia and Ib RNRs due to its proposed bimetallocofactor, MnIVFeIII. The class Ic RNRs store a one-electron oxidizing equivalent in its metal cluster. In vitro self-assembly of Ic is similar to Ia and Ib in that it reacts with O2 and a reductant to form its respective MnIVFeIII cofactor; however, it differs in that it can also react with 2 equivalents of H2 O2 to form the active cofactor. The class Ic RNR has been isolated from its native organism in vivo, complicating its assembly as the two different metals have similar affinities for the protein. In vitro studies in C. trachomatis have shown the necessity of regulating levels of the metals, along with the order of addition.
There exists problems with proper metal loading within the three subunits of Class I RNR. In the class Ia RNR, it requires a FeIIIFeIII-Y• cofactor, but the protein tends to bind MnII more tightly than FeII. In e.coli, correct metallation of NrdB relies on the necessity of free MnII and FeII present, while iron chaperones are also present to overcome the preference to bind MnII. The issue in class Ib RNR is that it may bind to either FeIIIFeIII-Y• and MnIIIMnIII-Y• cofactors, but only the manganese cofactor was found to be relevant in vivo. Ib binding is dependent on the preference of individual organisms and the concentrations of each metal that they possess inherently. The class Ic RNR complicates metallocofactor assembly since it requires two different metals with similar affinities for the same protein. Regulation of both levels of the metal is important in order to prevent mismetallation and its success depends on the presence of both types of metals. In C. trachomatis, the absence of MnII or at a lower than required rate may lead to diiron cluster formation instead. Thus if these levels are not regulation, low activity and improper metallation occurs. In general, if there is trouble regulating the levels of any of the required metals in each class I RNR, this leads to low activity and improper metallation and ultimately DNA synthesis is affected.
Biosynthesis and Repair of Metal Cofactors in Class I RNR[6]
[edit | edit source]Certain general principles and challenges exist when studying the metllocofactor formation with different metals and levels of complexity, as summarized below. Physiological expression conditions are taken into account in studies of metalloenzymes to confirm if the form of protein studied in vitro is the same as its active form in vivo. Class I RNRs can control the concentration of the active metal cofactors through biosynthetic and repeair pathways.
- Cofactors of metal proteins are generated by specific biosynthetic pathways.
- The proteins involved in the biosynthetic pathway are often associated with the operon of the metalloprotein of interest, and certain factors can be analyzed by comparing genomic sequences.
- To facilitate the exchange of ligands and protein factors, metals are transferred in their reduced state.
- There exists a variety of protein factors which include: metal insertase or chaperone to deliver the metal to the active site, specific redox proteins which control the oxidation state of the metal, and GTPases or ATPases which aid in the folding and unfolding processes to allow the metal to be inserted in the active site.
- Due to biological redundancy that affect pathway factors, multiple deletions of genes are required in order to identify phenotypes within a gene deletion experiment.
- A hierarchy of metal delivery to proteins and its regulation is inferred but not completely understood.
- Compartmentalization (e.g. periplasm vs cytosol in prokaryotes) and affinities of proteins to bind certain metals preferentially are two likely factors that contribute to prevent mismetatallion at the cellular level.
- Several proteins have not been isolated from their native source and form heterologous expression systems and leading to mismetallation. Since the optimum level of activity is not fully known, incorrect clusters corresponding to low activity may not be recognized.
- Certain oxidants can cause damage to the metal clusters (e.g. NO and O2) and specific pathways are used in their repair.
- During changes of oxidaion states, protons are typically required for this metal oxidation. Ligands to metal binding can reorganize easily and rearrangement of the carboxylate ligands are critical to the cluster assembly process.
One of the biggest complications is that the metal required for activity is often not the metal that has the highest affinity for binding to a specific protein. The Irving-Williams series (MnII < FeII < CoII < NiII < CuII > ZnII) best describes the relative affinities of proteins for divalent metals, in addition to the dependence on the particular protein coordination environment where the binding takes place. For the latter metals in the series, chaperone proteins exist to aid their movement to the active sites, while intracellularly they are likely to exist as "free" metals at a low concentration. These chaperone proteins also have another function beside delivery, which is to help maintain low levels of free concentration of these metals to prevent mismetallation and binding between other proteins that require MnII and FeII. Compartmentalization can overcome a protein's binding preference, as certain activities occur in different parts of the cell which have and require varying amounts of a metal. In cyanobacteria, it was found that MnII dependent perisplasmic protein must fold in the cytosol where MnII exists freely in a higher amount than ZuII, CuI, and CuII.
Techniques to Study RNR Activity[7]
[edit | edit source]There are several techniques used in the laboratory that are used to monitor the activity of the RNR metallocofactors. This contributes to identifying accurate proposed mechanism, generation, and function of these cofactors in vitro and in vivo by studying their movement.
- Whole-Cell Electron Paramagnetic Resonance: EPR was used in studying FeIIIFeIII-Y• biosynthesis in S. cerevisae. It was found that Y• levels were sufficiently high and detectable at endogenous levels in various growth conditions, meaning that the Y• is not modulated as a function of the cell cycle. A small molecule or protein factor must be needed to rapidly reduce the Y• in cell lysates, indicating the presence of a metallocofactor which was later identified to be iron.
- Mossbauer Spectroscopy: This type of spectroscopy monitors iron movement from oxidized and reduced iron pools into the RNR cofactor. It allows for the detection of all oxidation states of iron simultaneously and is sensitive to the surrounding electronic environments of the iron species present. In order for this technique to be accurate, cells first need to be labelled with the Fe57 isotope.
- ↑ Herrick J, Sclavi B. (2007) Ribonucleotide reductase and the regulation of DNA replication: an old story and an ancient heritage Mol Microbiol. 63:22–34
- ↑ Nordlund P, Reichard P (2006). Ribonucleotide Reductases Annu Rev Biochem, 75:681–706
- ↑ Cotruvo, Joseph, Jr., and Stubbe, JoAnne. (2011). Class I Ribonucleotide Reductases: Metallocofactor Assembly and Repair In Vitro and In Vivo Annual Review of Biochemistry, 80: 733-767
- ↑ Eklund H, Eriksson M, Uhlin U, Nordlund P, Logan D (1997). Ribonucleotide reductase--structural studies of a radical enzyme Biol Chem. 378:821–825
- ↑ Cotruvo, Joseph, Jr., and Stubbe, JoAnne. (2011). Class I Ribonucleotide Reductases: Metallocofactor Assembly and Repair In Vitro and In Vivo Annual Review of Biochemistry, 80: 733-767
- ↑ Cotruvo, Joseph, Jr., and Stubbe, JoAnne. (2011). Class I Ribonucleotide Reductases: Metallocofactor Assembly and Repair In Vitro and In Vivo Annual Review of Biochemistry, 80: 733-767
- ↑ Cotruvo, Joseph, Jr., and Stubbe, JoAnne. (2011). Class I Ribonucleotide Reductases: Metallocofactor Assembly and Repair In Vitro and In Vivo Annual Review of Biochemistry, 80: 733-767
Nucleotides
- Nucleotides consist of a base, sugar, and phosphate group. They are the building blocks of nucleic acids. Nucleotides are essential for the body for many reasons. They are needed for gene replication and transcription into RNA. They are also needed for energy. ATP, the body's form of energy, is a nucleotide with adenine as its base. Guanine nucleotides (GTP) are also a source of energy. Furthermore, derivatives of nucleotides are necessary in various biosynthetic processes. Nucleotides are necessary in signal transduction pathways as ewll.
The Biosynthesis of Nucleotides
There are two kinds of pathways in the biosynthesis of nucleotides: de novo and salvage. The following table contains similarities and differences between the two pathways.
| De Novo | Similarities | Salvage |
| Simpler compounds are used in the synthesis of nucleotides. Numerous small pathways are repeated to assemble different nucleotides. | Both synthesize nucleotides, though they utilize different mechanisms. | Bases are preformed, recovered, and reconnected to a ribose. |
| Synthesizes pyrimidine nucleotides. Bicarbonate, aspartate, and glutamine are used to synthesize the ring of the pyrimidine. The ring then links with ribose phosphate, forming the nucleotide. | Both assemble ribonucleotides, which are then used to synthezise deoxyribonucleotides for DNA. | Synthesizes purine nucleotides. Various precurosrs may be used to form the purine ring, which is then added to ribose and phosphate. |
- Feedback inhibition regulates multiple steps in the biosynthesis of nucleotides. Examples of this include activation and inactivation of aspartate transcarbamoylase in the synthesis of pyrimidines by CTP and ATP respectively, and activation and inactivation of glutamine-PRPP amidotransferase by purine nucelotides.
Reduction of Ribonucleotides to Deoxyribonucleotides
- Ribonucleotide reductase is a catalyst in reducing ribonucleoside diphosphates to deoxyribonucleotides. In this process, electrons flow from NADPH to sulfhydryl groups at ribonucleotide reductase's active sites. The reaction is summarized as follows:
- 1. An electron is transferred from cysteine on R1 to tyrosyl on R2. This creates a cysteine thiyl radical on R1, which is highly reactive on the active site.
- 2.A hydrogen from C3 of the ribose is then abstracted. This creates carbon radical.
- 3. The C3 radical helps release OH- at carbon-2. This departs as H2O after protonation from the second cysteine residue.
- 4. A third cysteine residue then provides a hydride to complete the reduction at C2. This returns the C3 to a radicala nd also generates a disulfide bond.
- 5. The c3 radical reacts with the original hydrogen that the first cysteine had extracted. A deoxyribonucleotide has now been generated and can leave the enzyme ribonucleotide reductase.
So What?
- The biosynthesis and metabolism of nucleotides are important to the body because disruptions in them can result in pathology. If nucleotides are not degraded properly, certain conditions may arise. An example of this is gout. Urates are degraded proteins, and gout is when they are accumulated, generating poor joints and arthritis.
- Similarly, if nucleotides are not synthesize properly, or if not enough are synthesized, conditions will arise as well. An example of this is the Lesch-Nyhan syndrome. Symptoms of this include mental deficiency, self-mutilation, and gout. This disease is due to a lack of an enzyme that is needed to synthesize purine nucleotides through the salvage pathway.
Source: Berg, Jeremy and Stryer, Lubert. Biochemistry: Fifth Edition. United States of America: W.H. Freeman and Company, 2002.
DNA and RNA Backbone
[edit | edit source]In macromolecules, such as DNA and RNA, there are linear polymers built and connected together by monomers. These monomers are known as nucleotides, and they consist of a nitrogenous base, a sugar, and a phosphate group. The chains and bonds between these nucleotides form the backbone of DNA and RNA, and these backbones allow the formation of unique genetic sequences. In DNA and RNA backbones, the monomers are connected by phosphodiester bridges. Specifically, the bridges are formed between the 3'-hydroxyl group of either the ribose sugar in RNA or deoxyribose sugar in DNA, and the 5'-hydroxyl group of the adjacent sugar; essentially called a 3'-5' phosphodiester bond. Chemically, to make this bond, the 3'-hydroxyl group of a sugar undergoes esterification with a phosphate group. That phosphate group then gets attacked by the 3'-hydroxyl group to form the phosphodiester bridge.
Once the phosphodiester bond is established, the backbone needs to be preserved in order to maintain the genetic information of the nucleotide sequence. Thus, no more nucleophilic attacks may occur on the backbone. In order to prevent nucleophilic attacks, the phosphate group on the phosphodiester bond has a negative charge which is used to prevent other nucleophilic species such as hydroxyl groups from attacking. The fact that DNA lacks a hydroxyl group on the 2' carbon means that it is more resistant to nucleophilic attacks, and thus, is the more stable hereditary material than RNA is.

Introduction
[edit | edit source]


What is DNA? DNA is a long chain of linear polymers containing deoxyribose sugars and their covalently bonded bases known as nucleic acids. One of the major functions of the DNA is storage of the genetic information. In DNA a sequence of three bases, which is called a codon, is responsible for the encoding of a single amino acid. The amino acid is added to a growing protein during the process of translation. These nucleic acid polymers encode for the all of the materials an organism needs to live in the form of genes. Genes are small blocks of DNA that tell the cell which proteins it should create. The type of genes that a given cell receives depends entirely on the parent cells. Genes are passed on from generation to generation as a way of ensuring an organism's survival genetically.
DNA stands for deoxyribonucleic Acid. The prefix "deoxy" distinguishes DNA from its close relative RNA (ribonucleic acid). The prefix indicates that, unlike Ribose, Deoxyribose does not contain a hydroxyl group at the 2' carbon replacing it with a single Hydrogen atom. The absence of this Hydroxyl group is fundamental in determining the way in which DNA is able to condense itself within the nucleus of a cell.
DNA is a nucleic acid which is capable of duplicating itself via the enzyme known as DNA polymerase. Each of the four bases on DNA, Adenine (A), Cytosine (C), Guanine (G), and Thymine (T) is bonded covalently to a deoxyribose sugar. The four nucleotide units in DNA are called deoxyadenylate, deoxyguanylate, deoxycytidylate, and thymidylate. The nucleotide includes the nucleoside, a nitrogenous base bonded to a deoxyribose or ribose group. The four nucleosides in DNA are deoxyadenosine, deoxyguanosine, deoxycytidine, and thymide. By the joining one or more phosphate groups to a nucleoside through ester linkages, a nucleotide is formed.
The deoxyribose sugars form the structural backbone for DNA via a phosphodiester bond between the 3' carbon of one nucleotide and the 5' carbon of the next. When DNA is not self-replicating it exists in the cell as a double stranded helical molecule with the strands lined up anti-parallel to each other. That is to say if the orientation of one strand is 3' to 5' the other strand would be oriented 5' to 3'. The bases of each strand bind very specifically, A binds with T and C binds with G no other combination exists at least in DNA. The bases are bound to one another internally via hydrogen bonds with the phosphodiester bond backbone oriented to face outward. It is here that the missing 2' hydroxyl group plays an important role in DNA. It is the absence of this group that allows DNA to form its conventional double helix structure. RNA which does have a hydroxyl group at the 2' carbon is unable to obtain this same helical structure. The modern double helix structure of DNA was first proposed by Watson and Crick, and the functions of DNA were demonstrated in a series of experiments which will be discussed in the next few sections.
Why DNA? It is significant to note the reasons why DNA is the primary method through which all cells pass along genetic information. That is to say why has evolution favored a DNA world over an RNA world given that the two molecules are so similar structurally? These reasons involve chemical stability, energy needed to form and break chemical bonds, and the availability of enzymes to perform this task. The primary reason involves the relative stability of the two molecules. DNA is more chemically stable than RNA because it lacks the hydroxyl group on the 2' carbon. In RNA there are two possible OH groups that the molecule can form a phosphodiester bond between, which means that RNA is not forced into the same rigid structure as its deoxy counterpart. Additionally the deoxyribose sugar in DNA is much less reactive than the ribose sugar in RNA. Simply put C-H groups are significantly less reactive than C-OH (hydroxyl) groups. This difference also explains why RNA is not very stable in alkaline conditions, and DNA is. The base in alkaline condition does the same thing as the -OH group at the C2 position. Furthermore, double-strand DNA has relatively small grooves where damaging enzymes can't attach, making it more difficult for them to 'attack' the DNA. Double-stranded RNA, on the other hand, has much larger grooves, and therefore, it is more subject to being broken down by enzymes. The connection between the strands of double-stranded DNA is tighter than double-stranded RNA. In other words, it's much easier to unzip double-stranded RNA than it is to unzip double-stranded DNA. Overall, the breakdown and reform of RNA can be carried out faster and requires less energy than the breakdown and reform of DNA. It is essential to the organism's survival and well-being that its genetic material is encoded into something that is more stable and resistant to changes. In addition, the sequence of DNA and its physical conformation seems to play a part in DNA's selection as well. Another point that helps elucidate DNA's prevalence as the primary storage of genetic information is the availability of the enzyme that breaks down DNA. The body actively destroys foreign nucleases, which are enzymes that cleave DNA. This is only one of the many ways DNA is protected against damage. The body can actually recognize foreign DNA and destroy it, while leaving its own DNA intact.
Hyperchromic Effect Another unique feature of DNA in its double stranded form is the hyperchromic effect, which describes the decreasing absorbance of UV electromagnetic radiation of double helix strands as compared to the non-helical conformation of the molecule. The hydrogen bonding between complementary DNA strands as a result of sugar stacking in the helical conformation causes the aromatic rings to become increasingly stable and thus absorb less UV radiation. This ultimately decreases the amount of UV absorption by 40%. As the temperature is increased these hydrogen bonds dissolve and the helical structure begins to unwind. In this unwound form the aromatic rings are free to absorb much more UV radiation.
Properties of DNA
[edit | edit source]

1. Consists of 2 strands (anti-parallel and complementary): DNA has two polynucleotide chains that twist around a helical axis in opposite direction.
2. It is made up of deoxyribose sugar, a phosphate backbone on the exterior, and nucleic acid bases in the interior.
3. Bases are perpendicular to the helix axis that separated by 3.4 Angstroms.
4. Strands are held together by hydrogen bonds an other various intermolecular forces that form a double helix. The base pairing involves 2 hydrogen bonds for A - T and 3 hydrogen bonds for C - G -see in images to the right
5. Backbone consists of alternating sugars and phosphates, where phosphodiester linkages form the covalent backbone of the DNA.The direction of DNA goes from 5' phosphate group to 3' hydroxide group.
6. Repeats every 10 bases
7. Weak forces stabilize DNA because of the hydrophobic effects and VanDerWaals.
8. DNA chain is 20 Angstroms wide (2 nm)
9. One nucleotide unit is 3.3 Angstroms long (0.33 nm)
Primary Structure
[edit | edit source]DNA is made of two polynucleotide chains (strands) which run in opposite directions around the common axis. As a result, DNA has a double helical structure. Each polynucleotide chain of DNA consists of monomer units. A monomer unit consists of three main components that are a sugar, a phosphate, and a nitrogenous base. The sugar used in the DNA monomer unit is deoxyribose (it lacks an oxygen atom on the second Carbon in the furanose ring). There are also four possible nitrogen containing bases which can be used in the monomer unit of the DNA. Those bases are adenine (A), guanine (G), cytosine (C), and thymine (T). Adenine and guanine are purine derivatives, while cytosine and thymine are pyrimidine derivatives. Polymeric chain forms as a result of joining nucleosides (the sugar which is covalently bonded to the nitrogen containing base) through the phosphodiester linkage. Polymeric chain is a single strand of the DNA molecule. Two strands run in opposite directions to form double helix. The forces that keep those strands together are hydrogen bond, hydrophobic interactions, van de Waal force, and charge-charge interactions. The H-bonds form between base pairs of the antiparallel strands. The base in the first strand forms an H-bond only with a specific base in the second strand. Those two bases form a base-pair (H-bond interaction that keeps strands together and form double helical structure). The base –pairs are: adenine-thymine (A-T), cytosine-guanine (C-G). Such interaction gives us the hint that nitrogen-containing bases are located inside of the DNA double helical structure, while sugars and phosphates are located outside of the double helical structure. The hydrophobic bases are inside the double helix of DNA. The bases, located inside the double helix, are stacked one on the top of another. Stacking bases interact with each other through the Van der Waals force. Even though the van de Waal forces are week, sumation of those forces can be substantial. The distance between two neighboring bases that are perpendicular to the main axis is 3.4 A˚. DNA structure is repetitive. There are ten bases per turn, so every base has a 36° angle of rotation. The diameter of the double helix is approximately 20 A˚. The hydrophobic effect stabilizes the double helix. The structural variation in DNA is due to the different deoxyribose conformations, rotation about the contiguous bonds in the phosphodeoxyribose backbone, and free rotation about the C-1'- N (glycosyl bond).
The technique of southern blotting is often used to uncover the DNA sequence of a sample. The technique is named after Edwin Southern.
DNA Manipulation Techniques
[edit | edit source]When it comes to exploring genes and genomes, it depends on the technical tools that are used. The five important DNA manipulation techniques are:
1.Restriction Endonucleases - also known as restriction enzymes
The restriction of enzymes split the DNA into specific fragments. By having the DNA split into different pieces, it allows the manipulation of DNA segments.
2. Blotting Technique
To separate and characterize DNA, the Southern blotting technique is used. This technique is similar to the Western blot, except that Southern blotting is used for DNA and not RNA. This technique identifies a specific sequence of DNA by electrophoresis through an agarose gel. The DNA is separated by placing the large fragments on top and the small fragments at the bottom. Next, the DNA is transfer into the nitrocellulose sheet. Then a 32-p labeled DNA probe that is complementary to the sequence, is added to hybridize the fragments. Finally, a autoradiography film is use to view the fragment containing the sequence.
3. DNA Sequencing
By using the DNA sequencing technique, a precise nucleotide sequence of a DNA molecule can be determined. The key to DNA sequencing is the generation of DNA fragments whose length depends on the last base of the sequence. Even though there are different alternative methods, they all perform the same procedure on the four reaction mixtures.
A. Chain termination DNA Sequencing
A primer is always needed. To produce fragments, the addition of 2', 3'-dideoxy analog of a dNTP is added to each of the four mixtures. It will stop the sequence at that N-dideoxy. The types of dNTP that can be use are dATP, TTP, dCTP, dGTP. In the end, new DNA strands are separated to electrophoresis.
B. Fluorescence Detection of Bases
Fluorescent tag is used into each of the four chain-terminating dideoxy nucleotides at different wavelengths. It is an effective method because no radioactive reagents are used and large sequences of bases can be determined. The fragments get separated by having the mixture passed through high voltage. Then, the fragments are detected by their fluorescence, which the base sequence is based on the color sequence.
C. Top-down (Shotgun) Method of Genome Sequencing
The top-down method and the shotgun method are similar, the main difference is that the top-down requires a detailed map of the clones. The Shotgun randomly sequences large clones to match them computationally.
D. Microarrays(Green chips)
Using microarrays is useful when it comes to studying the expression of a large number of genes. The microarray is created by using either oligonucleotides or cDNA. Based on the fluorescent intensity, red or green marks will appear. If it is red, it means no fluorescence is present, known as gene induction. If it is green, fluorescence is expressed, known as gene repression.
4. DNA Synthesis
To synthesize DNA, a solid-phase method is used. The solid-phase synthesis is carried out by the phosphite triester method. In this process only one nucleotide is added in each group. The first step that takes place is the binding of the first nucleotide. Another nucleotide is added and activated and reacts with the 3' -phosphoramididte containing DMT. A deoxyribonucleoside 3' -phosphoramidite with DMT and βCE is attached because it has the ability to synthesize any DNA. It is also a basic nucleotide that is modified and protected. Then, the molecule gets oxidized to oxidized the phosphate group. In the end, the DMT is removed by addition of dichloroacetic acid. Overall, the desired product remains insoluble and it is release at the end.
5. Polymerase Chain Reaction (PCR)
PCR is a technique used that allows to amplify DNA sequence between two nucleotides. If the DNA sequence is known, millions of copies of that sequence can be obtained by using this technique. To carried out PCR, a DNA template, a precursor, and two complementary primers are needed. What makes the PCR unique is that the temperature is constantly changing within the three different stages and that the stages get repeated 25 times. The three stages are:
1. Denaturing - DNA gets denature from a double strand (parent DNA molecule) to two single strands by heating thesolution at 94°C.
2. Annealing - After letting the solution cooled, two synthetic oligonucleotide primers are added at the end of the 3' end of target strand, and at the 3' end of complementary strand. This process is done when the temperature is between 50°C - 60°C.
3. Polymerization - Addition of thermostable DNA polymerase to catalyze 5' to 3' DNA synthesis at 72°C.
Structural Variation
[edit | edit source]Structural Variation occurs due to the different deoxyribose conformations, free rotation about the C-1, and rotation about the closest bond in phosphodeoxyribose backbones.
There are secondary structures when it comes to DNA which are forms A, B, and Z. A Form: 1. Right handed 2. Glycosyl bond conformation is ANTI 3. Needs 11 base pairs per helical turn 4. Size of diameter is about 26 angstroms 5. Sugar pucker conformation is at the C-3' endo.
B Form: 1. Like the A form, the B form is right handed. 2. Glycosyl bond formation is ANTI 3. Needs 10.5 base pairs per helical turn 4. Size of diameter is about 20 angstroms 5. Sugar pucker conformation is at the C-2' endo
Z Form: 1. Unlike the A and B form, the obvious difference is that the Z form is left handed. 2. Glycosyl bond formation consists of two components: pyrimidines and purines. ANTI (for pyrimidines) and SYN (for purines) 3. Needs 12 base pairs per helical turn 4. Size of diameter is about 18 angstroms 5. Sugar pucker conformation is at the C-2' endo (for pyrimidines) and C-3' endo (for purines)
DNA libraries
[edit | edit source]A DNA library is a collection of cloned DNA fragments in a cloning vector that can be searched for a DNA of interest. If the goal is to isolate particular gene sequences, two types of library are useful.
Genomic DNA libraries
[edit | edit source]A genomic DNA library is made from the genomic DNA of an organism. For example, a mouse genomic library could be made by digesting mouse nuclear DNA with a restriction nuclease to produce a large number of different DNA fragments but all with identical cohesive ends. The DNA fragments would then be ligated into linearized plasmid vector molecules or into a suitable virus vector. This library would contain all of the nuclear DNA sequences of the mouse and could be searched for any particular mouse gene of interest. Each clone in the library is called a genomic DNA clone. Not every genomic DNA clone would contain a complete gene since in many cases the restriction enzyme will have cut at least once within the gene. Thus some clones will contain only a part of a gene.
cDNA Library
[edit | edit source]A cDNA library is a library of mRNAs. It is made from introns and exons and a cDNA library is made to be able to isolate the genes/the final version of the gene.
A cDNA library i used to screen for colonies. If looking for a gene, you can screen the colonies, use the collection of plasmids, transform the bacteria, and use a probe. You can also use Southern Hybridization. By using an oligonucleotide that is complementary to the gene you are looking for, and that will eventually tell you which colonies of bacteria will have the DNA that corresponds with the mRNA in the plasmids.
How to make a cDNA library:
1. Isolate mRNA from the cell.
2. Use reverse transcriptase and dNTPss so that from the original mRNA, a DNA copy can be created.
3. RNA is easier to degrade than DNA so put in alkali solution to degrade mRNA.
4. Use DNA polymerase to complete the template.
Ultimately, you end up with double stranded DNA, one of which is identical to the mRNA. After doing this all for mRNA, you can clone it in the plasmids. The collection of plasmids will include all of the mRNA but in the form of DNA.[1]
Flow of Genetic Information
[edit | edit source]- Genetic information storage: genome
- Replication: DNA --> DNA
- Transcription: DNA --> RNA
- Translation: RNA --> Proteins
References
[edit | edit source]- ↑ Viadiu, Hector. "Making a cDNA Library." UCSD. Lecture. November 2012.
Sources
[edit | edit source]http://clem.mscd.edu/~churchcy/BIO3600/bio3600_images/phosphodiester_bonds.htm http://www.science-projects.com/hyperChromic1.htm
Berg , Jeremy . Biochemistry . 7. New York : W.H Freeman and Company , 2012. Print.
| This page or section is an undeveloped draft or outline. You can help to develop the work, or you can ask for assistance in the project room. |
Berg, Jeremy, Tymoczko J., Stryer, L.(2012). Protein Composition and Structure.Biochemistry(7th Edition). W.H. Freeman and Company. ISBN1-4292-2936-5
Hames, David. Hooper, Nigel. Biochemistry. Third edition. New York. Taylor and Francis Groups. 2005.
Overview
[edit | edit source]Deoxyribonucleic acid (DNA) stores information for the synthesis of specific proteins. DNA has deoxyribose as its sugar. DNA consists of a phosphate group, a sugar, and a nitrogenous base. The structure of DNA is a helical, double-stranded macromolecule with bases projecting into the interior of the molecule. These two strands are always complementary in sequence. One strand serves as a template for the formation of the other during DNA replication, a major source of inheritance. This unique feature of DNA provides a mechanism for the continuity of life. The structure of DNA was found by Rosalind Franklin when she used x-ray crystallography to study the genetic material. The x-ray photo she obtained revealed the physical structure of DNA as a helix.
DNA has a double helix structure. The outer edges are formed by alternating deoxyribose sugar molecules and phosphate groups, which make up the sugar-phosphate backbone. The two strands run in opposite directions, one going in a 3' to 5' direction and the other going in a 5' to 3' direction. The nitrogenous bases are positioned inside the helix structure like "rungs on a ladder," due to the hydrophobic effect, and stabilized by hydrogen bonding.
| Nitrogenous base | Nucleoside | Deoxynucleoside |
|---|---|---|
 Adenine |
 Adenosine A |
 Deoxyadenosine dA |
 Guanine |
 Guanosine G |
 Deoxyguanosine dG |
 Thymine |
 5-Methyluridine m5U |
Deoxythymidine dT |
 Uracil |
Uridine U |
 Deoxyuridine dU |
 Cytosine |
Cytidine C |
Deoxycytidine dC |
The two strands run in opposite directions to form the double helix. The strands are held together by hydrogen bonds and hydrophobic interactions. The H-bonds are formed between the base pairs of the anti-parallel strands. The base in the first strand forms a H-bond only with a specific base in the second strand. Those two bases form a base-pair (H-bond interaction that keeps strands together and form double helical structure). The base–pairs in DNA are adenine-thymine (A-T) and cytosine-guanine (C-G). Such interactions provide us an understanding that nitrogen-containing bases are located inside of the DNA double helical structure, while sugars and phosphates are located outside of the double helical structure.
The component consisting of the base and the sugar is known as the nucleoside. DNA contains deoxyadenosine (deoxyribose sugar bonded to adenine), deoxyguanosine (deoxyribose sugar bonded to guanine), deoxycytidine (deoxyribose sugar bonded to cytosine), and deoxythymidine (deoxyribose sugar bonded to thymine). The linkage of the bonds between the base to the sugar is known as the beta-N-Glycosidic linkage. In purines, this occurs between the N-9 and C-1' and in pyrimidines this occurs between the N-1 and C-1'. A nucleoside and a phosphate group make up a nucleotide. The bond between the deoxyribose sugar of the nucleoside and the phosphate group is a 3'-5' phosphodiester linkage.
The bases, located inside the double helix, are stacked. Stacking bases interact with each other through the Van der Waals forces. Although the energy associated with a Van der Waals interaction is relatively small, in a helical structure, a large number of atoms are intertwined in such interactions and the net sum of the energy is quite substantial. The distance between two neighboring bases that are perpendicular to the main axis is 3.4 Å. The DNA structure is repetitive. There are ten bases per turn, that is the structure repeats after 34 Å, so every base has a 36° angle of rotation. The radius of the double helix is approximately 10 Å.
An easy way to differentiate between Nucleosides and Deoxynucleosides is the atoms bonded to C-2 on the sugar unit. If the structure is a deoxynucleoside, then C-2 bears two hydrogens. If it is a nucleoside, then C-2 bears one hydrogen and one hydroxide group, in which the hydroxide group faces south.
Structural variations in DNA can occur if:
1. There are different deoxyribose conformations
2. If there are rotations around the contiguous bonds in the phosphodeoxyribose backbone
3. Free rotation about the C-1'N=glycosyl bond (syn/anti)[1]
Terms and Naming
[edit | edit source]There are two types of nucleic acids, ribonucleic acids (RNA) and deoxyribonucleic acid (DNA). Recall that a nucleoside is a base + sugar. A Nucleotide is composed of a base + sugar + phosphate. The deoxy- prefix in Deoxyribonucleotides is the nomenclature used for DNA. The term ribonucleotides is employed when it is nomenclature for RNA, or in other words, C-2 on the sugar unit has an -OH group (versus deoxy which C-2 has 2 hydrogens). Symbols are used to simplify the names. For example, ATP (precursor of RNA). The "A" in the front signifies that the base is Adenine and the "T" in the middle signifies tri-phosphates. AMP on the other hand, also has an adenine, but the M signifies that the sugar is bound to a single phosphate group. Finally, in dAMP, the "d" signifies that it is a 2'-deoxyribo-, versus simply AMP means it is a ribonucleotide.In short, four nucleotide units of DNA are called deoxyadenylate, deoxyguanylate, deoxycitidylate, and thymidylate.
Early foundation for DNA structures
[edit | edit source]The primary structure of a nucleic acid is its covalent structure and nucleotide sequences. One of most important parts of determining the structure of DNA comes from the work of Erwin Chargaff and his colleagues in the late 1940s. They found that the four nucleotide bases of DNA of different organisms and that the amounts of certain bases are closely related. They concluded the following about the structure of DNA:

1. The base composition of DNA generally varies from one species to another.
2. DNA specimens isolated from different tissues of the same species have the same base composition.
3. The base composition of DNA in a given species does not change over time, nutritional states, or environment.
4. In all cellular DNA, regardless of the species, the number of adenine residues is equal to the number of thymine residue (A=T) and the number of guanine residues is equal to the number of cytosine residues (G=C).
Later in 1953, Rosalind Franklin and Maurice Wilkins used a powerful X-ray diffraction technique called X-ray crystallography to deduce the DNA structure. Photographs produced by the X-ray crystallography method are not actually pictures of molecules, however the spots and smudges produced by X-rays that were diffracted (deflected) as they passed through crystallized DNA. Crystallographers use mathematical equations to translate such patterns of spots into information about the three-dimensional shape of DNA. Franklin and Wilkins found that DNA molecules are helical with two periodicities along their long axis, a primary one of 3.4 A and a secondary one of 34 A.

Watson and Crick later based their model of DNA upon the data they were able to extract from Wilkins and Franklin's X-ray diffraction photo.
http://37days.typepad.com/37days/images/2008/03/02/franklin20dna20photo.jpg
They interpreted the pattern of spots on the X-ray photo to mean that DNA consisted of two chains and was helical in shape. Eventually, Watson and Crick formulated a DNA structure from the diffraction pattern of the x-ray photo and gave to incredible insight that is still accepted today. In this structure, they proposed that two helical DNA chains of opposite direction wound around the same axis to form a right handed double helix. The hydrophilic backbones form by phosphodiester bonds of alternating deoxyribose sugar and phosphate group that are faced outside of the helix, surrounded by aqueous environment. The furanose ring of each deoxyribose sugar is in the C-2’ endo conformation. The purine and pyrimidine bases of both strands are stacked inside the double helix and stabilized by Van Der Waals interactions.
The double-helix has a diameter of 10 Å. Each adjacent base on one strand of the double-helix is 3.4 Å apart. Every 10 base-pairs constitutes a 360° turn in the helix, and the length of the helix is determined by 34 Å per 10 base-pairs.

Orientation
[edit | edit source]DNA molecules are asymmetrical, such property is essential in the processes of DNA replication and transcription. A double-stranded DNA molecule consists of two complementary but disjoint strands that are intertwined into a helix formation through a network of H bonds. Although both the right-handed and left-handed helices are among the allowed conformations, right-handed helices are energetically more favorable due to less steric hindrance between the side chains and the backbone. The direction of DNA is determined by the arrangement of the phosphate and deoxyribose sugar groups along the DNA backbone. One of the DNA ends terminates with the 3'-OH group, whereas the other one terminates with the 5'-phosphate group. All sequences of DNA are usually written from 5' to 3' termini. In a double-helix formation, the complementary DNA strands are oriented in opposite directions. DNA is a rather rigid molecule: at physiological conditions, DNA curves at the length scale of about 50 nm, which is 20 times the diameter of the double helix. More so, the alignment of the bases can indicate the global orientation of a DNA strand. For purine nucleotides (A and G) the most probable angle is approximately 88°, whereas for pyrimidine (C and T) that angle is approximately 105°.

Forces involved in DNA helices
[edit | edit source]The DNA double helix is held together by two main forces: hydrogen bonds between complementary base pairs inside the helix and the Van der Waals base-stacking interaction.


Hydrogen bonds
[edit | edit source]Watson and Crick found that the hydrogen bonded base pairs, G with C, A with T, are those that best fit within the DNA structure. It is important to note that three hydrogen bonds can form between G and C, but only two bonds can be found in A and T pairs. On the other hand, A-T pairs seem to destabilize the double helical structures. This conclusion was made possible by a known fact that in each species the G content is equal to that of C content and the T content is equal to that of A content.
Below is the link to the demo of the Hydrogen bondings between base pairs:
http://chemmac1.usc.edu/java/bases/basepairs.html
The three hydrogen bonds that constitute the linkage of Guanine(G) and Cytosine(C) consequently alters the thermal melting of DNA, which is dependent upon base compositions. With varying base composition the melting point of such molecule will either increase or decrease.
Denaturing and Annealing
Ultraviolet (UV) light can detect whether bases are stacked or unstacked. Stacked bases within the DNA structure facilitate shielding from light, therefore the absorbance of UV light of double helical DNA is much less than single stranded DNA. This characteristic is known as the hypochromic effect, in which less color is emitted from the double helix of DNA molecules.
The melting temperature (Tm) is the temperature in which DNA is half way of the DNA is double stranded and half is single stranded. The Tm depends greatly on base composition. Since G-C base pairs are stronger due to more Hydrogen bonds, DNA with high G-C content will have a higher Tm than that of DNA with greater A-T content.
When heat is applied to a double-stranded DNA, each individual strand will eventually separate (denature) because hydrogen bonds are disrupted between base pairs. Upon separation, the separated strands spontaneously reassociate to form the double helix again. This process is known as annealing.
In biological systems, both denaturing and annealing can occur. Helicases use chemical energy (from ATP) to disrupt the structure of double-stranded nucleic acid molecules. The study of the ability of DNA to reanneal within the laboratory is important in discovering gene structure and expression.
Complex Structures
Complex structures can also be formed from single-stranded DNA. A stem-loop is formed when complementary sequences, within the same strand, pair to form a double helix. Hydrogen bonds between base pairs within the same strand occur. Often, these structures include mismatched bases, resulting in destabilization of the local structure. Such action can be important in higher-order folding, like in tertiary structures.
Hypochromic Effect
[edit | edit source]DNA absorbs very strongly at wavelengths close to UV light (~260 nm). A single stranded DNA will absorb more UV light than that of double-stranded DNA. DNA UV absorption decreases when it forms a double strand, this characteristic is an indication of DNA stability. With the increase in light energy, its structure and therefore its function will still remain intact since there is low disturbance to its structure.
The decreased absorbance observed with the DNA double helix with respect to the native and denatured forms is explained by the fact that the stacking of the nitrogenous bases that takes place with the double helix does not leave them as exposed to radiation and thus they are able to absorb less. The aromaticity of the nitrogenous bases (specifically in the purine and pyrimidine like ring structures) accounts for the absorption peak being at 260nm.
Weak forces
[edit | edit source]Various Weak Forces come together to stabilize the DNA structure.
- Hydrogen bonds, linkage between bases, although weak energy-wise, is able to stabilize the helix because of the large number present in DNA molecule.
- Stacking interactions, or also known as Van der Waals interactions between bases are weak, but the large amounts of these interactions help to stabilize the overall structure of the helix.
- Double helix is stabilized by hydrophobic effects by burying the bases in the interior of the helix increases its stability; having the hydrophobic bases clustered in the interior of the helix keeps it away from the surrounding water, whereas the more polar surfaces, hence hydrophilic heads are exposed and interaction with the exterior water
- Stacked base pairs also attract to one another through Van der Waals forces the energy associated with a single van der Waals interaction has small significant to the overall DNA structure however, the net effect summed over the numerous atom pairs, results in substantial stability.
- Stacking also favors the conformations of rigid five-membered rings of the sugars of backbone.
- Charge-Charge Interactions- refers to the electrostatic (ion-ion) repulsion of the negatively charged phosphate is potentially unstable, however the presence of Mg2+ and cationic proteins with abundant Arginine and Lysine residues that stabilizes the double helix.
Nitrogenous Bases
[edit | edit source]Nitrogenous Bases are the foundational structure of DNA polymers, the structure of DNA polymers vary with the different attached nitrogenous bases.
Nitrogenous Bases can tautomerize between keto and enol forms. The aromaticity of the pyrimidine (Cytosine, Thymine, Uracil (RNA)) and purine (Adenine, Guanine) ring systems and their electron-rich nature of -OH and -NH2 substituents enable them to undergo keto-enol tautomeric shifts. The keto tautomer is called a lactam and the enol tautomer is called lactim. The lactam predominates at pH 7. Keto-enol tautomerization is the interconversion of a keto and enol involving the movement of a proton and the shifting of bonding electrons, hence the isomerism qualifies as tautomerism.

Keto-enol tautomerism is important in DNA structure because high phosphate-transfer potential of phosphenolpyruvate results in the phosphorylated compound to be trapped in the less stable enol form, whereas dephosphorylation results in the keto form. Rare enol tautomers of bases guanine and thymine can lead to mutation because of the altered base-pairing properties.
Base-stacking interactions
[edit | edit source]The two strands of double-stranded DNA are held together by a number of weak interactions such as hydrogen bonds, stacking interactions, and hydrophobic effects. Of these, the stacking interactions between base pairs are the most significant. The strength of base stacking interactions depends on the bases. It is strongest for stacks of G-C base pairs and weakest for stacks of A-T base pairs. The hydrophobic effect stacks the bases on top of one another. The stacked base pairs attract one another through Van der Waals forces, typically from 2 to 4 kJ/mol-1. In addition, base stacking in DNA is favored by the conformations of the somewhat rigid five membered rings of the backbone phosphate-sugars. The base-stacking interactions, which are largely nonspecific with respect to the identity of the stacked base, make the major contribution to the stability of the double helix.
Phosphodiester Bond
[edit | edit source]
Phosphodiester linkages form the covalent backbone of DNA. A phosphodiester bond is the linkage formed between the 3' carbon atom and the 5' carbon of the sugar deoxyribose in DNA.
The phosphate groups in a phosphodiester bond are negatively-charged. The pKa of phosphate groups are near 0, therefore they are negatively-charged at neutral pH (pH=7). This charge-charge repulsion forces the phosphates groups to take opposite positions of the DNA strands and is neutralized by proteins (histones), metal ions such as magnesium, and polyamines.
The tri-phosphate or di-phosphate forms of the nucleotide building are blocks, first have to be broken apart to release the energy require to drive an enzyme-catalyzed reaction for a phosphodiester bond to form and for the nucleotide to join. Once a single phosphate or two phosphates (pyrophosphates) break apart and participate in a catalytic reaction, the phosphodiester bond is formed.
An important role in repairing DNA sequences is due to the hydrolysis of phosphodiester bonds being catalyzed by phoshodiesterases, an enzyme that facilitates the repairs.
One reason that made DNA more stable than RNA is absence of the 2'-OH group in DNA. The presence of OH group on 2'C makes RNA more susceptible for reactions. A nucleophile (base) can pull out the H (when everything is in the correct trajectory) and the phosphate part of the backbone will rearrange and eventually a P-O bond is broken to break the connection site between two sugars.
Secondary Structures of DNA
[edit | edit source]Major and Minor Grooves
Base pairing of complementary nucleotides make up the secondary structure of DNA. A single-stranded DNA may participate in intramolecular base pairing between complementary base pairs and therefore make up secondary structure as well. Base pairing between Adenine (A)-Thymine (T) and Guanine (G)-Cytosine(C)are possible because these base pairs are similar in size. This means there are no "bulges" or "gaps" within the double helix.
Irregular placement of base pairs in a double helix will result in consequences that will render the macromolecule nonfunctional. Therefore if there is something wrong with the structure, signals will be sent and DNA repair will work to fix damage.
As a result of the double helical nature of DNA, the molecule has two asymmetric grooves. One groove is smaller than the other. This asymmetry is a result of the geometrical configuration of the bonds between the phosphate, sugar, and base groups that forces the base groups to attach at 120 degree angles instead of 180 degree. The larger groove is called the major groove, occurs when the backbones are far apart; while the smaller one is called the minor groove, occurs when they are close together.
Since the major and minor grooves expose the edges of the bases, the grooves can be used to tell the base sequence of a specific DNA molecule. The possibility for such recognition is critical, since proteins must be able to recognize specific DNA sequences on which to bind in order for the proper functions of the body and cell to be carried out. As you might expect, the major groove is more information rich than the minor groove, allowing the DNA proteins to interact with the bases. This fact makes the minor groove less ideal for protein binding.

A form
[edit | edit source]These following features represented different characteristics of A-form DNA structure:
1. Most RNA and RNA-DNA duplex in this form
2. Shorter, wider helix than B.
3· Deep, narrow major groove not easily accessible to proteins
4· Wide, shallow minor groove accessible to proteins, but lower information content than major groove.
5· Favored conformation at low water concentrations
6· Base pairs tilted to helix axis and displaced from axis
7· Sugar pucker C3'-endo (in RNA 2'-OH inhibits C2'-endo conformation)
8· Right handed
9· Size is about 26 angstroms
10· Needs 11 base pairs per helical turn
11· Glycosyl bond conformation is Anti
B form
[edit | edit source]The double helical structure of normal DNA takes a right-handed form called the B-helix. It is about 20 angstroms with a C-2' endo sugar pucker conformation. The helix makes one complete turn approximately every 10 base pairs (= 34 A per repeat/3.4 A per base). B-DNA has two principal grooves, a wide major groove and a narrow minor groove. Many proteins interact in the space of the major groove, where they make sequence-specific contacts with the bases. In addition, a few proteins are known to make contacts via the minor groove.

Z form
[edit | edit source]DNA sequences can flip from a B form to a Z form and vice versa. Z form of DNA is a more radical departure from the B structure; the most obvious distinction is the left-handed helical rotation.
The Z form is about 18 angstroms and there are 12 base pairs per helical turn, and the structure appears more slender and elongated. The DNA backbone takes on a zigzag appearance. Certain nucleotide sequences fold into left-handed Z helices much more readily than others. Prominent examples are sequences in whichpyrimidines alternate with purines, especially alternating C and G or 5-methyl-C and G residues. To form the left-handed helix in Z-DNA, the purine residues flip to the syn conformation alternating with pyrimidines in the anti conformation. The major groove is barely apparent in Z-DNA, and the minor groove is narrow and deep. For pyrimidines, the sugar pucker conformation is C-2' endo and for purines, it is a C-3' endo.
Z-DNA formation occurs during transcription of genes, at transcription start sites near promoters of actively transcribed genes. During transcription, the movement of RNA polymerase induces negative supercoiling upstream and positive supercoiling downstream the site of transcription. The negative supercoiling upstream favors Z-DNA formation; a Z-DNA function would be to absorb negative supercoiling. At the end of transcription, topoisomerase relaxes DNA back to B conformation.
Tertiary structure (3 dimensional)
[edit | edit source]The tertiary structure of DNA molecule is made up of the two strands of DNA wind around each other. DNA double helix can be arranged in space, in a tertiary arrangement of strands.
- Linking Number( Lk) in a covalently closed circular DNA, where the two strands cannot be separated will result in a constant number of turns in a given molecule. Lk of DNA is an integral composed of two components:
Normally, DNA has Lk of about 25, meaning it is underwound. However, DNA can also be supercoiled with two "underwindings" which is made up of negative supercoils. This is much like the two "turns- worth" of a single stranded DNA and no supercoils. This kinds of interconversion of helical and superhelical turns in important in gene transcription and regulation.
Quaternary structure and other unusual structure
[edit | edit source]DNA is connected with histones and non-histone proteins to form the chromatin. The negative charge due to the phosphate group in DNA makes it relatively acidic. This negative charge binds to the basic histone groups.
Histone Modification
[edit | edit source]Recent studies provide that actively transcribed regions are characterized by specific modification pattern of histone. The experiments carried on by the dynamics of histone modification shows that there is a significant kinetic distinction between methylation, phosphorylation, and acetylation. This suggest that the roles of these modifications has different roles in gene expression patterns.
Histones are proteins which DNA wraps around and forms a chromatin. The basic unit of a chromatin is a nucleosome which are formed by histone octomer of 2 molecules of H2A, H2B, H3, and H4 along with 147 base pairs of DNA wrapped in a superhelix. The accessibility of DNA is regulated by higher-order chromatin structures that of which can be obtained by the packing of nucleosomes. It is believed that the N-Termini tail of the histone molecules contributes to the chromatin function in that it mediates inter-nucleosomal interactions and are involved in the recruitment of non-histone proteins to the chromatin. The N-termini tail directs interactions to the chromatin binders which is thought to be the driving force of modulate chromatin structure. However, there are other ways modifications can occur such as that observed by the unfolding or assembly of nucleosome and how it is involved in gene regulation. It is hoped that this can provided an explanation of epigenetic inheritance (Box 1) the there phenotypic differences in individual cannot be due to differences in DNA, such as that of monozygotic twins.
Epigenetic inheritance are changes in the gene activity that are not encoded by the DNA sequence. These changes include phosphorylation, methylation, ADP-ribosylation, SUMOylation, and ubiquitylation. These modifications can be considered active or repressive depending on their occurrence in active or silent genes. It is show that methylation can have different outcomes depending on the binders of the histone modifications. Nucleosome positioning are found to have an influence on the DNA sequence and may contribute to epigenetic inheritance.[2]
Structural Variation in DNA
[edit | edit source]The Structural Variation in DNA is most due to:
- 1) Varying deoxyribose conformations (4 total conformations)
- 2) Rotations about the contiguous bonds in the phosphodeoxyribose backbone (between the C1-C3 and C5-C6)
- 3) Free rotation about C1'- N-glycosyl bond (resulting in syn or anti conformation)
Because of steric hindrance, purines bases in nucleotides are restricted to two stable conformations with respect to deoxyribose, called syn and anti. On the other hand, pyrimidines are generally restricted to the anti conformation because of steric interference between the sugar and the carbonyl oxygen at C-2 of the pyrimidine.
Comparison of A, B, and Z form of DNA
[edit | edit source]| A form | B form | Z form | ||
|---|---|---|---|---|
| Helical sense | ||||
| Right handed | Right handed | Left handed | ||
| Diameter | ||||
| 26 A | 20 A | 18 A | ||
| Base pairs per helical turn | ||||
| 11 | 10.5 | 12 | ||
| Helix rise per base pair | ||||
| 2.6 A | 3.4 A | 3.7A | ||
| Base tilt normal to the helix axis | ||||
| 200 | 60 | 70 | ||
| Sugar pucker conformation | ||||
| C-3’ endo | C-2’ endo | C-2’ endo for pyrimidines and C-3’endo for purines | ||
| Glycosyl bond conformation | ||||
| Anti | Anti | Anti for pyrimidine and syn for purines |
References
[edit | edit source]Campbell and Reese's Biology, 7th Edition
Nelson and Cox's Lehninger Principles of Biochemistry, 5th Edition Telomeres (from the Greek telos, "an end") are long stretches of repeating non-coding DNA sequences at the ends of the DNA strand. They protect the ends of DNA and prevent DNA strands from shortening or attaching to other molecules by masking the chromosome. Russian Alexei Olonikov was the first to postulate the problem of chromosomes replicating at the tip.[1] He theorized that in every subsequent replication bits of the DNA would be lost until a critical limit had been reached, thereupon cell division would cease.
Telomerase
[edit | edit source]
Telomerase is an enzyme that creates the Telomeres. Telomerase adds specific repeating sequences ("TTAGGG" in all vertebrates) to the ends of four DNA strands.
[2]
The telomerase enzyme has an RNA template that partially attaches to the shortened end of the DNA strand. New nucleic acids then attach to the template, extending the DNA strand. Once the telomerase leaves, the double stranded DNA is completed with the DNA polymerase. Telomerase was discovered in 1985 by Carol W. Greider and Elizabeth Blackburn. For this discovery, they were awarded the 2009 Nobel Prize in Physiology or Medicine along with Jack W. Szostak.[3]
Szostak and Blackburn first discovered telomeres in ciliates. They chose ciliates because at one stage of their life cycle, they make a million new telomeres. The model created includes a telomere-dedicated DNA polymerase, which adds telomeric repeats onto chromosome ends. Therefore, telomeres are represented as a motif in DNA sequences.
Telomerase's presence in humans is somewhat strange. It is located in the nucleus which is unsurprising because that is where DNA replication takes place. However, Telomerase activity is not present in all cells. It was found to be almost absent in the majority of normal adult tissues, including cardiac and skeletal muscle, lung, liver, and kidney. Because of this curious lack of telomerase activity, a theory arose connecting telomere length to aging and cell senescence. According to this theory, human somatic cells are born with a full number of telomeric repeats, but the telomerase enzyme is not present in some tissues. The cells of those tissues would lose about 50 to 100 nucleotides from each chromosome end each time they underwent replication and division. Eventually, the telomeres would cease to exist and the chromosomes themselves would start losing nucleotides, carrying genetic defects into their next division so that neither daughter cell would be viable. Thus after a certain number of divisions a cell will not have enough nucleotides and die.[4]

The function of Telomerase is to allow for short replacements of Telomeres which are gradually lost during cell division.[5] In normal conditions without Telomerase, a cell would divide until it would hit a critical point known as the Hayflick limit.[6] In the presence of Telomerase, however, the cell has the ability to replace lost DNA and divide without limit. But this continuous growth comes with a consequence as this growth may lead eventually to cancerous cells.
While the details are not fully known, it would seem that that shortened Telomeres play a role in aging due to the erosion of the DNA over time. The questions arises whether or not Telomerase has the ability to greatly extend the lifespan of a human due to its importance in the maintenance of the Telomeres.[7]Dr. Michael Fossel, a professor of clinical medicine at Michigan State University, has expressed his views on Telomerase as a viable treatment for cell senescence.
However, several experiments have raised doubts on the ability of Telomerase as an effective anti-aging treatment. An experiment was done with mice having higher levels of Telomerase and it was discovered that they also had a higher rate of cancer which therefore led to a shorter lifespan. In addition, Telomerase favors tumorogenesis.[8] Telomerase fosters cancer development by allowing uncontrolled cell growth which eventually proliferates into tumors. In fact, Telomerase activity has been observed in approximately 90% of all human tumors which suggests that the uncontrolled growth of a cell as conveyed by Telomerase has a key role in cancer.
In addition to using Telomerase as an anti-aging treatment, Telomerase has potential as a drug target against cancer.[9] Since it is necessary for the immortality of many cancer cell types, it is believed that if a drug is able to deactivate Telomerase activity in a cell, Telomeres would shorten, mutations would happen, cell stability would decrease and cancer would be, in essence, effectively treated. Experimental drugs have been tested in mouse models and some drugs have moved onto clinical testing.
Cancer Biology
[edit | edit source]The significance of studying telomeres can be found in telomerase, which rebuilds the telomere so that the cells can keep dividing. The telomerase, however, eventually shortens the telomere, causing the cell to die. In the case of cancer cells, this enzyme builds telomeres long past the cell's average lifetime. These cells then are called to be "immortaled", since they can divide endlessly. This results in a tumor. Many researchers believe that telomere maintenance activity is characterized in most human cancer cells. Though the mechanism by which such phenomena happen has not been well understood, the discovery may reveal key elements of telomere function. Telomerase, on the other hand, is the natural enzyme used for telomere repair, highly abundant in stem cells, germ cells, hair follicles, and most cancers cells, but its expression is low or in some cases absent in somatic cells. Telomerase functions by adding bases to the ends of the telomeres. Cells with sufficient telomerase activity are considered immortal in the sense that they can divide past the Hayflick limit without entering senescence or apoptosis. For this reason, telomerase is viewed as a potential target for anti-cancer drugs such as telomestatin.
2009 Nobel Prize
[edit | edit source]The Nobel Prize 2009 in Physiology and Medicine was awarded to three scientists who have discovered how the chromosomes can be copied in a complete way during cell divisions and how they are protected against degradation. By showing that the ends of the chromosomes, telomeres, and their enzyme, telomerase, are significant in protecting the chromosomes from degradation, they identified telomerase and explained how the telomeres protect the ends of the chromosomes and built by telomerase. On the other hand, if the telomeres become shortened, cells can duplicate damaged as cancer cells. If telomerase is well maintained, conversely, telomere length is maintained and the cell does not become cancerous. In the case of cancer cells, telomerase allows the cell to divide without any limit. Certain genetic disease are caused by a defective telomerase. This discovery can thus be used to stimulate the development of new therapeutic strategies. Understanding such fundamental mechanism is an important first step toward opening new doors for cures for cancer and other related diseases, as well as anti-aging.
Hayflick Limit
[edit | edit source]The Hayflick limit is the number of times a normal cell may divide until it reaches a critical limit and stops dividing based on the idea that Telomeres reach a critical length.[10] This limit was discovered by Leonard Hayflick in the 1960s who demonstrated that the cells in a normal fetus divided around 40 to 60 times before entering into cell senescence. Due to repeated mitosis, the Telomere shortening occurred which inhibited cell division which is analogous to aging. The discovery of this limit, a pillar of Biology, refuted the early contention by Alexis Carrel who, along with the majority of scientists during that time period, believed cells were "immortal".
Role of Telomere
[edit | edit source]Telomeres account for the lost bits of DNA at the ends of chromosomes during DNA replication. Since DNA polymerase moves along the template strand in the 5'--> 3' direction, some of the 5' end of the template strand will not be replicated. This results in the incomplete ends as shown in the diagram below. However, telomeres are usually very long, ranging from 400 to 600 base pairs in yeast to many kilobases in humans. They are made of six to eight base pair long repeats which are usually rich with guanine bases. With long stretches of telomeres at the ends of DNA strands, the incomplete strands of DNA will still contain the genetic code.


The shortening of telomeres in humans induces cell senescence in humans. This mechanism appears to cause the formation of cancerous cells. Telomere length has been theorized in recent publications to account for the aging in humans. Since cells replicate identically, there must be a reason why cells within a body lose function and viability with time. Telomeres may have some influence over the aging process since every consequent DNA replication results in the shortening of telomeres. Two aspects to this question are: (i) whether telomere length, as measured in specific cell populations in the body, correlates with longevity or disease; and (ii) whether telomere shortening in any cell population causes functional impairment of that cell population. However, some may argue telomeres do not correlate to longevity as mice contain long strands of telomeres, but contrarily live much shorter lives than humans who do not have as long telomeres as do mice. And some may argue that telomere length does correlate to longevity as it determines the number of times that a cell can divide before it dies or reaches senescence.
Recent Publications
[edit | edit source]Recently it has been found that telomerase activity is inversely related to length of the telomeres. In other words, telomere elongation happens more often on short telomeres rather than long ones. The research showed a deficiency in telomerase activity in telomeres greater than 125 base pairs,and there was 2 to 3 times more telomerase activity in telomeres shorter than 125 base pairs. This preferential elongation has been demonstrated in yeast and mice, and now human somatic cells. Kinetic data indicates that elongation in yeast cells in a single event in which elongates the telomeres to a certain length, whereas in human cells the elongation seems to be a gradual process. The researchers showed that telomerase adds a regulated length of telomere in each cell division. The researchers showed that human cells expressed telomerase, however long telomeres were maintained and not elongated where as the cells with shorter telomeres elongated, which goes to show that telomeres can not be infinitely extended.[11]
Another interesting paper was focused on the role of DNA damage response (DDR) proteins in the role of telomere maintenance. The review says that early stage DNA repair proteins have a significant role in telomere maintenance where as late stage proteins usually do not take part in telomere repair. The interplay with these proteins and the proteins that cap the telomeres to protect the telomeres is very important too. Many of stronger DDR proteins inhibit cell replication, because of this fact, it would be harmful to the organism for these proteins to be a part of telomere repair. These protein caps on the telomeres inhibit full DNA damage response which keeps the stronger protein from "repairing" the telomere ends. It still isn't clear why some of the DDR proteins participate in telomere maintenance and others do not, but it is clear that the cellular process in repairing a DNA break and repairing telomeres are two different process, with the former halting cellular division.[12]
References
[edit | edit source]- ↑ "Telomeres, telomerase, and aging: Origin of the theory". Alexey M. OlovnikovE-mail The Corresponding Author. 1999. Retrieved 2009-11-05.
{{cite web}}: External link in|publisher= - ↑ "Repeat Expansion–Detection Analysis of Telomeric Uninterrupted (TTAGGG)n Arrays". [1]. 2007. Retrieved 2009-11-05.
{{cite web}}: External link in|publisher= - ↑ "The Nobel Prize in Physiology or Medicine 2009". [2]. 2009. Retrieved 2009-11-05.
{{cite web}}: External link in|publisher= - ↑ "What are telomeres and telomerase?". [3]. Retrieved 2009-11-05.
{{cite web}}: External link in|publisher= - ↑ "Telomerase: regulation, function and transformation". [4]. Retrieved 2009-11-05.
{{cite web}}: External link in|publisher= - ↑ "Hayflick Limit Theory". [5]. Retrieved 2009-11-05.
{{cite web}}: External link in|publisher= - ↑ "Extension of Life-Span by Introduction of Telomerase into Normal Human Cells". [6]. Retrieved 2009-11-05.
{{cite web}}: External link in|publisher= - ↑ "Anti-Aging Medicine". João Pedro de Magalhães. 2008. Retrieved 2009-11-05.
{{cite web}}: External link in|publisher= - ↑ Foreman, Judy. "Telomerase - a Promising Cancer Drug Stuck in Patent Hell?". myhealthsense.com. Retrieved 2009-11-05.
- ↑ "Cellular Senescence". João Pedro de Magalhães. 2008. Retrieved 2009-11-17.
{{cite web}}: External link in|publisher= - ↑ Britt-Compton, Bethan; Capper, Rebecca; Rowson, Jan; Baird, Duncan M. (2009). FEBS Letters (583): 3076–3080.
{{cite journal}}: Missing or empty|title=(help) - ↑ Lyndall, David (2009). The EMBO Journal (28): 2174–2187.
{{cite journal}}: Missing or empty|title=(help)
DNA does not always take the form of a double helix. It can often be found creating structures considered abnormal when compared to what is commonly considered DNA. Normally, DNA contains a B-form helix. Improper formation of base pairs can greatly affect DNA's structure and flexibility.
Single-stranded nucleic acids can form hairpins. Such formations can affect the transcription terminations in prokaryotes. With regard to double-stranded DNA, they can form something called cruciforms.
Hairpins
[edit | edit source]Hairpin loops are formed by a fold in a single strand of DNA, causing several bases to remain unpaired before the strand loops back upon itself. A hairpin loop is only possible if the strand of DNA contains the complimentary bases in correct sequence to those that appear earlier in the strand. For example; if a DNA strand contained CCGT followed by several bases including ACGG, the strand is capable of creating a hairpin loop by folding back on itself.
Hairpin loops can occur in both DNA and RNA, though in RNA the thymine base is replaced by uracil. The number of bases in the loop itself is variable, though it never exists in the length of three bases, as the steric hindrance makes the configuration too unstable.
Here is an image example of hairpin DNA:
(Image is of a Long-alpha hairpin)

Cruciforms
[edit | edit source]Cruciform DNA structure appears as several hairpin loops, creating a crucifix-like structure composed of DNA.
DNA structure is formed by incomplete exchange of the strands between the double-stranded helices.
Cruciform DNA Eukaryotic cells contain DNA-binding protein that can specifically recognize cruciform DNA. Interactions with ubiquitous protein plays a crucial role for the conformation of cruciform DNA.
An example of a DNA-binding Protein is Crp1p. This DNA-binding protein is found in the yeast Saccharomyces cerevisiae
Image of the formation of Cruciform DNA can be found here.
Triple Helix
[edit | edit source]The triple helix form of DNA is similar to the double helix DNA except that it contains another oligonucleotide that hydrogen bonds to the bases that are already included in the double helix strands of DNA.
Background
The triple-stranded DNA was a very common hypothesis in the 1950s when scientists were having trouble figuring out the true structure of DNA. Watson and Crick, Pauling and Corey all published a triple-helix model proposal. Watson and Crick found problems with the model. The problems were as follows:
- Negatively charged phosphates near the axis will repel each other, leaving the question as to how the three-chain structure would stay together.
- In a triple-helix model (specifically Pauling and Corey's model), some of the van der Waals distances appear to be too small.1
For more information on Triple-stranded DNA see DNA Triple-stranded DNA
An image of the triple helix form can be found here.
Hinged DNA
[edit | edit source]Hinged DNA (H-DNA) is a triple helix structure that exists based on hydrogen bonds between DNA bases. The three strands base pair by Hoogsteen base pairing. Hoogsteeen base pairing is a variation of base-pairing in the nucleic acids such as the A-T pair or the G-C pair. The Hoogsteen base pair applies the "N7 position of purine base and c6 amino group which bind the Waston-Crick face of pyrimidine base." More information on the Hoogsteen base pair can be found here. It is also called H-DNA because of its dependence on hydrogen bonds. The H-DNA can be found in vitro or during recombination and also in DNA repair.
An example of H-DNA can be found here.
G-Quadruplex
[edit | edit source]G-quadruplexes are a family of quadruple-stranded structures formed by a guanine-rich sequences of nucleic acids. Members of this family share a common square arrangement of four guanines centered around a monovalent cation and stabilized by Hoogsteen hydrogen bonding. The guanines may adopt either an anti or syn alignment about the glycosidic bond. The backbone strands of the g-tetrad can also adopt a variety of directionalities: all four strands may be oriented in the same direction, three strands are oriented in one direction while the fourth is in another direction, two adjacent strands can be oriented in one direction while the other two will be oriented in another direction, or each strand will have adjacent anti parallel neighbors. The sequence of amino acid that has the potential to form g-quadruplex is: GxNaGxNbGxNcGx, where x is the number of G residues and Na, Nb, and Nc are loops of different lengths. Furthermore, they can form in DNA, RNA, LNA, and PNA, and either be intramolecular, bimolecular and tetramolecular compounds. Their four stranded motifs create four grooves each with varying widths and depths. Their folding depends on many factors; DNA sequence, presence of ions, temperatures, and presence of various ligands. They are a special area of interest due to their biological implications specifically in telomeres and as contributors to gene regulation.
A shows a G-tetrad, B shows the Anti and Syn conformations of Guanine, C shows the various directionailities of the backbone strands, D shows the different types of loops
Structure determination of G-quadruplex based on crystallography or solution NMR demonstrates significant deviations in conformation and loop geometry suggesting heterogeneity in strand topology and loop conformation of G-quadruplexes. Varying conformations can result in varying stability. Furthermore, studies of the various conformations reveal that the nature of the loop sequence and the formation of interactions between loops and the quadruplex core are important elements in controlling quadruplex topology and stability. For example, in examining the bindinging of quinacridine-based ligand to a G-quadruplex, interactions with the sides of the G-stack do not alter the topology but interaction with the loop sequence ended up altering the conformation of the loops. This hints at the notion that the loop sequences of the quadruplex are what actually moderate the binding affinity and specificity of the whole structure.
The four-stranded structure with four grooves instead of the normal two found in typical DNA structure, provides a variety of surfaces for interactions with ligands. Aromatic compounds of various dimensions showed favorable interactions with the planer surfaces of terminal guanine tetrads. Intercalation between layers of G-tetrads does not occur, however because G-tetrads do not allow for bulky aromatic compounds to insert itself between layers of guanine.
In eukaryotic telomeres, there exists repeats of g-rich sequences that can fold into g tetrads. It has been postulated that this structure plays an important role in cell aging and human diseases such as cancer, then making them targets to anticancer drugs.
References
[edit | edit source]- Problems with triple helix model: <http://www.zampwiki.com/?t=Triple-stranded_DNA>
- H-DNA: <http://molbioandbiotech.wordpress.com/2007/10/08/h-dna/>
- Cruciform DNA: <http://www.ncbi.nlm.nih.gov/pubmed/2922595>
- Sannohe,Yuta, Sugiyama, Hiroshi. "Overview of Formation of G-Quadruplex Structures" Wiley Online Library. 01 Mar. 2010. http://onlinelibrary.wiley.com/doi/10.1002/0471142700.nc1702s40/full 20 Nov. 2010.
- Martin Egli, Pradeep S Pallan. "The many twists and turns of DNA: template, telomere, tool, and target" Current Opinion in Structural Biology. 08 Apr. 2010. http://www.sciencedirect.com/science/article/B6VS6-4YT5D86-1/2/cfe41243ea70339783887f259a11b2f8 20 Nov. 2010
- Lubos Bauer,Peter Javorsky, Katarina Tluckova, and Viktor Viglasky. "Evaluation of human telomeric g-quadruplexes: the influence of overhanging sequences on quadruplex stability and folding" Journal of Nucleic Acid. 10 Jun. 2010. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2925402/?tool=pubmed 20 Nov. 2010
Supercoiling
[edit | edit source]

The structure of DNA does not only exist as secondary structures such as double helices, but it can fold up on itself to form tertiary structures by supercoiling. Supercoiling allows for the compact packing of circular DNA. Circular DNA still exists as a double helix, but is considered a closed molecule because it is connected in a circular form. A superhelix is formed when the double helix is further coiled around an axis and crosses itself. Supercoiling not only allows for a compact form of DNA, but the extent of coiling also affects the DNA’s interactions with other molecules by determining the ability of the double helix to unwind.
Although the supercoiling provides an organized way to tightly compact DNA, the structure is relatively unstable as a result of torsional strain. In order to minimize the energy required to maintain the structure, the number of twists and writhes are minimized. Twists refer to the number of turns the double helix makes around the superhelical axis. Writhes refer to the circular distortion, bending, and overall non-planarity of the DNA strand.
Supercoiling changes the shape of DNA. The benefit of a supercoiled DNA molecule is its compactibility. In comparison to a relaxed DNA molecule of the same length, a supercoiled DNA is more compact. How this is reflected in experimentation is that supercoiled DNA moves faster than relaxed DNA. Therefore, the structural differences can be analyzed in techniques such as electrophoresis and centrifugation.
Supercoiled DNA may hinder and favor the DNA to unwind and thus affect the interaction between DNA and other molecules in cells.
Positive and Negative Supercoilings
[edit | edit source]1. Negative supercoiling is the right-handed coiling of DNA thus winding occurs in the counterclockwise direction. It is also known as the "underwinding" of DNA.
2. Positive supercoiling is the left-handed, coiling of DNA thus winding occurs in the clockwise direction. This process is also known as the "overwinding" of DNA. (CORRECTION FIXED on 10/23/17 - DV, original error had the negative and positive supercoiling definitions reversed. Also provided more basic clarity to supercoiling).
Although the helix is underwound and has low twisting stress, negative supercoil's knot has high twisting stress. Prokaryotes and Eukaryotes usually have negative supercoiled DNA. Negative supercoiling is naturally prevalent because negative supercoiling prepares the molecule for processes that require separation of the DNA strands. For example, negative supercoiling would be advantageous in replication because it is easier to unwind whereas positive supercoiling is more condensed and would make separation difficult.
Topoisomerases unwind helix to do DNA transcription and DNA replication. After the proteins have been made,the DNA template supercoils by the force to make chromatin. RNA polymerase also influence DNA strand to have two different supercoiled directions. The region RNA polymerase has passed forms negative supercoil while the region RNA polymerase that have not passed forms positive supercoil. By these processes, supercoils are generated.
Topoisomerase
[edit | edit source]Topoisomerases are enzymes that are responsible for the introduction and elimination of supercoils. Positive and negative supercoils require two different topoisomerases. This prevents the distortion of DNA by the specificity of the topoisomerases. The two classes of topoisomerases are Type I and Type II. Type I stimulates the relaxation of supercoiled DNA and Type II uses the energy from ATP hydrolysis to add negative supercoils to DNA. Both of these classes of topoisomerases have important roles in DNA transcription, DNA replication, and recombinant DNA.
Topoisomerase form loops (unwinded regions of the double helix) of negative supercoils. If the DNA lacks superhelical tension, there is no unwinding of supercoils.
Type I topoisomerase
[edit | edit source]Type I topoisomerase act by creating transient single-strand breaks in DNA. This is further classified as type IA and type IB.
Type IA topoisomerases
Type IA topoisomerases enzyme is a 695-residue monomer and it relaxes negatively supercoiled DNA. First, Type IA cuts a single stranded DNA and catenates two circles of single stranded DNA. Then it unwinds the supercoiled duplex DNA by one turn. Type IA has a specific strand-passage mechanism which is the denaturation of type IA incubated with single stranded DNA that yields a linear DNA by phospho-Tyr diester linkage.
Type IB topoisomerases
Type IB mediates a controlled rotation mechanism to relax both negative and positive supercoils. Type IB cleaves a single strand of a duplex DNA through the nucleophilic attack of an active site with Tyr on a DNA to yield a 3'-linked phospho-Tyr intermediate with 5'-OH group. Type IB consists of several domains and subdomains. Interestingly, type IA topoisomerases form a covalent intermediate with the 5' end of DNA, while the IB topoisomerases form a covalent intermediate with the 3' end of DNA. Historically, type IB topoisomerases were referred to as Eukaryotic Topo I, but Type IB topoisomerases are present in all three kingdoms of life.
Type II topoisomerase
[edit | edit source]Type II topoisomerase is an enzyme that require ATP hydrolysis to complete a reaction cycle in which two DNA strands are cleaved, duplex DNA is passed through the break and the break is resealed. Type II cuts both strands a DNA double helix, passes another unbroken DNA strand through it, and then reanneals the cut strand. It is also split into two subclasses: type IIA and type IIB topoisomerases, which share similar structure and mechanisms. Examples of type IIA topoisomerases include eukaryotic topo II, E. coli gyrase, and E. coli topo IV. Examples of type IIB topoisomerase include topo VI. Supercoiling requires energy because it is torsionally strained. Thus, through the coupling to ATP hydrolysis it can introduce negative supercoils.
In bacteria, Type II topoisomerase is also known as DNA gyrase. Gyrase is an enzyme that acts similarly to human Type II topoisomerase. Antibiotics act on bacterial enzyme by blocking the binding of ATP to gyrase and thus deactivating the breaking and joining of bacterial DNA chains.
Nucleosomes
[edit | edit source]Nucleosomes allow for the compact packing of linear DNA. Nucleosomes are complexes of DNA and histones, consisting of ~145 base pairs of DNA wrapped around in a left-handed superhelix around a histone octomer, which are a group of small proteins. Histones contain a large amount of positively charged amino acids such as lysine and arginine which allow them to bind to the negatively charged DNA molecule. The histone octamer is composed of two copies each of H2A, H2B, H3, and H4. The two loops of DNA around the histone are attached to the histone also using the H1 histone. Nucleosomes are further arranged in a stacked helical complex. Through the extensive wrapping of DNA around the histones, as well is the helical arrangement of the nucleosomes, the linear DNA is able to be compacted. The structural folding of the nucleosomes eventually forms a chromosome.
Chromatin refers to the structure of DNA and its accompanying histones. Chromatin is composed of repeating units called nucleosomes. The five major histones found in chromatin are H2A, H2B, H3, H4, and H1.
In gene clusters, protein genes of histone are present and these are expressed in S phase. Once it is expressed, it forms histone octamers. With interactions of 146 base pairs of DNA double helix, histone octamer becomes a nucleosome. When histones bind to DNA, it is depended on the amino acid sequence of histone, not the nucleotide sequences of DNA.

Histones and Transcription Regulation
[edit | edit source]Histones always appear to remain attached to the DNA even through transcription. The fact that nucleosomes are able to change shape and position allow for transcription to occur and RNA polymerase to move along the DNA strand. Slight loosening of the binding between the histones and DNA are accomplished by acetylation of the histones, which neutralizes the positively charged residues. Meanwhile, binding is made tighter through methylation to restore the positive charge of the histones. By changing the charge of the histones in this manner, gene transcription can be regulated. Histone Chaperones are proteins that mediate the assembly and disassembly of the chromatin to form correct nucleosomes sequences and aid in stable folding conformations. These proteins function to protect and shield the histones from forming incorrect and unwanted aggregates with DNA because of the high ionic strength that exists between DNA and Histones. DNA is primarily negatively charged molecule and histones are positively charged therefore, there exists a strong affinity for each other. Histone Chaperones, which are positively charged, help to guide histones to form octamers and correctly bind to DNA by shielding and masking the negative charge of DNA. There are different types of histone chaperones, including β- sandwich, α/α earmuff, Β-propeller and β-barrel chaperones. Β-sandwich chaperones are chaperone monomers that form β-sheets with the histones. An example of these types of chaperones is ASF1 or anti-silencing function chaperones involved the overexpression during yeast replication. In addition ASF 1 is the first histone used during assembly of the chromatin. α/α earmuff chaperones are dimers that form α helical conformations of histone/DNA complex. An example would include NAP chaperones which are used to transport histones from the cytoplasm to the nucleus during chromatin assembly. Β-propeller chaperones were the first chaperones to be distinguished using NMR and crystallography techniques. These pentamer chaperones function is the storage of histones. Β-Barrel chaperones are heteroligomers that help facilitate chromatin transcription. In addition, there are irregular or variant histone chaperones that do not fit into any specific structural category. All of these different types of chaperones are involved in different stages of assembly of disassembly of chromatin. The energetics of Chromatin assembly and disassembly are regulated by histone chaperones. Assembly which is an energetically favored process because as the histones bind with DNA it forms a more stable structure causing a decrease in energy. On the other hand, disassembly is an energetically unflavored process needing the use of ATP to break apart the stable histone/ DNA interactions.
Nucleosome Sliding
[edit | edit source]Nucleosome sliding is a frequent result of energy-dependent nucleosome remodelling in vitro.
ATP-Dependent Nucleosome Sliding Mechanism
The paper “Mechanisms of ATP-dependent nucleosome sliding” by Gregory D Bowman, researches how ATPase motors engage and manipulates nucleosomal DNA and discusses possible mechanisms for ATP-dependent sliding of nucleosomes. ATPase motors are shared between chromatin remodelers and collections of different protein machines. The ATPase motor generates torsional strain when it engages with DNA at an internal site on the nucleosome. The torsional strain in the nucleosomal DNA is a result of the ATPase motor acting at SHL2 region. Protection of nucleosomal DNA between SHL2 and the entry/exit site is increased Isw2 ATPase is activated. ATP-dependent crosslinking of the Isw2-subunit Dbp4 to SHL4 promotes hydrolysis-dependent changes. Iswi-type remodelers form template-committed complexes that allow for nucleosomes to slide processively.
Bowman also explains possible variations of the bulge/loop propagation model using ATPase motors. One model suggests that the ATPase motor uses translocase abilities to pull DNA from an entry/exit site in a continuous manner. This pumping allows for a remodeler to create a bulge that would rapidly diffuse to a distant entry/exit site. Another model suggests the histone-DNA contacts are disrupted by a DNA loop that is developed by a remodeler ATPase around the SHL2 region. This disruption pulls DNA for the linker and the ATPase motor would move toward dyad along the DNA loop.
Chromatin Remodeler
[edit | edit source]Chromatin remodelers are mainly involved in DNA packaging and facilitating the transcript elongation process. For example, when a DNA strand coils with nucleosomes for packaging into chromatin, chromatin remodelers arrange the nucleosomes in a regular distance for effective condensation of DNA strands. Furthermore, in some processes where nucleosomes have to be modified, chromatin remodelers may disassemble the nucleosome into histones or even detach the whole nucleosome from the DNA. The processes that require nucleosome modification by the chromatin remodeler include DNA repair, recombination, transcription and replication. The following picture displays an example of how a chromatin remodeler may be used during transcription catalyzed by RNA Polymerase II.

Palindromic Sequencing
[edit | edit source]A palindromic sequence is a sequence made up of nucleic acids within double helix of DNA and/or RNA that is the same when read from 5’ to 3’ on one strand and 5’ to 3' on the other, complementary, strand. It is also known as a palindrome or an inverted-reverse sequence.
The pairing of nucleotides within the DNA double-helix is complementary which consist of Adenine (A) pairing with either Thymine (T) in DNA or Uracil (U) in RNA, while Cytosine (C) pairs with Guanine (G). So if a sequence is palindromic, the nucleotide sequence of one strand would be the same as its reverse complementary strand. An example of a palindromic sequence is 5’-GGATCC-3’, which has a complementary strand, 3’-CCTAGG-5’. This is the sequence where the restriction endonuclease, BamHI, binds to and cleaves at a specific cleavage site. When the complementary strand is read backwards, the sequence is 5’-GGATCC-3’ which is identical to the first one, making it a palindromic sequence.
Another restriction enzyme called EcoR1 recognizes and cleaves the following palindromic sequence:
Palindrome
[edit | edit source]
Relationship between Sequence and Protein Structure
[edit | edit source]There have been many researchers who have studied the relationship between palindromic sequences and protein structures. Studies have shown that the frequent appearances of palindromic sequences, also called palindromic peptides, in protein sequences are not just by chance. Scientists suggest that these sequences are important for protein structure and protein function in different proteins. Some of these protein groups include DNA binding proteins, ion channels and Rhodopsin, metal binding proteins and receptors, and etc. By comparing palindromes with set sequences from the database, scientists can try to find the roles of palindromic sequences.
Another topic within palindromic sequences which is being studied is whether the symmetry of palindromic sequences affects the structure and folds of peptides. One hypothesis is that by reversing the sequence, the resulting folds would be mirror-images of the original fold. The conclusion states that because both the original and reverse proteins have identical amino acid compositions which lead to similar hydrophobic-hydrophilic patterns, the reversing sequence results in the same folds as opposed to the mirror-image folds. Another hypothesis guided by research is that by reversing a sequence, the fold could change or possibly be destroyed. This shows evidence that the similarity in reverse sequencing does not reflect structural similarity, which means that they do not form symmetrical protein structures.
Effect on genomic instability in yeast
[edit | edit source]Palindromic sequences have been tied to different genomic rearrangements in different organisms depending on the length of the repeated sequences. Shorter palindromic sequences (shorter than 30 bp) are very stable while longer sequences are not stable in vivo. These sequences occur in both eukaryotes and prokaryotes. These sequences also increase inter and intrachromosomal recombination between homologous sequences. Hairpin structures can form from palindromic sequences due to base pairing in single-stranded DNA. These structures can be substrates for structure-specific nucleases and repair enzymes which can lead to a double-strand break in the DNA. This then leads to loss of genomic material which can cause meiotic recombination. Studies with a 140-bp long mutated palindromic sequence inserted in yeast have shown to lower postmeiotic segregation and increase rate of gene conversions, while shorter sequences do the opposite. Research also found that during meiosis, double-strand breaks are induced by the long 140-bp palindromic sequence. In the long hairpin structure, the entire stem-loop is not covered and the processing endonuclease is exposed, which makes nicks in the loop. This nick creates a gap which is repaired by the wild-type strand. The induction of double-strand breaks during meiosis is what causes genomic instability.
Likelihood of palindromic sequences in proteins
[edit | edit source]There have not been an abundance of studies focusing on the significance of palindromic sequences in protein, but there have been some which tell us a lot about the relationship between palindromic sequencing and protein function. But by understanding the actual formation of these palindromic sequences and their properties, researchers can tie these sequences to functions. It has been found that decreasing amino acid composition complexity increases the likelihood of a palindromic sequence. The next step relates to the likelihood of palindromic sequences in proteins which can be due to the frequent formation of alpha helices by palindromes.
References
[edit | edit source](c) Acdx, from the Wikipedia Commons <http://commons.wikimedia.org/wiki/File:DNA_palindrome.svg/>
Jankowski, Craig, Dilip K. Nag, and Farooq Nasar. "Long Palindromic Sequences Induce Double-Strand Breaks during Meiosis in Yeast." National Center for Biotechnology Information. U.S. National Library of Medicine, 20 May 2000. Web. 7 Dec. 2012. <http://www.ncbi.nlm.nih.gov/pmc/articles/PMC85638/>.
"Palindromic Sequences." Wikipedia. Wikimedia Foundation, 11 Aug. 2012. Web. 07 Dec. 2012. <http://en.wikipedia.org/wiki/Palindromic_sequences>.
Sheari, Armita, Mehdi Kargar, Ali Katanforoush, Shahriar Arab, Mehdi Sadeghi, Hamid Pezeshk, Changiz Eslahchi, and Sayed-Amir Marashi. "A Tale of Two Symmetrical Tails: Structural and Functional Characteristics of Palindromes in Proteins." National Center for Biotechnology Information. U.S. National Library of Medicine, 11 June 2008. Web. 07 Dec. 2012. <http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2474621/>. When a DNA solution is heated enough, the double-stranded DNA unwinds and the hydrogen bonds that hold the two strands together weaken and finally break. The process of breaking double-stranded DNA into single strands is known as DNA denaturation, or DNA denaturing. The temperature at which the DNA strands are half denatured, meaning half double-stranded, half single-stranded, is called the melting temperature(Tm). The amount of strand separation, or melting, is measured by the absorbance of the DNA solution at 260nm. Nucleic acids absorb light at this wavelength because of the electronic structure in their bases, but when two strands of DNA come together, the close proximity of the bases in the two strands quenches some of this absorbance. When the two strands separate, this quenching disappears and the absorbance rises 30%-40%.This is called Hyperchromicity. The Hypochromic effect is the effect of stacked bases in a double helix absorbing less ultra-violet light.
Applications of DNA denaturation
[edit | edit source]Sequence differences between two different DNA sequences can also be detected by using DNA denaturation. DNA is heated and denatured into single-stranded state, and the mixture is cooled to permit strands to re-hybridize. Hybrid molecules are formed between similar sequences and any differences between those sequences will give a disruption of the base-pairing
What determines the Melting Temperature (Tm)?
[edit | edit source]While the ratio of G (Guanine) to C (Cytosine) and A (Adenine) to T (Thymine) in an organism's DNA is fixed, the GC content (percentage of G +C) can vary considerably from one DNA to another. The percentage of GC content of DNA has a significant effect on its Tm. Because G-C pairs form three hydrogen bonds, while A-T pairs form only two, the higher the percentage of GC content, the higher its Tm. Thus, A double-stranded DNA rich in G and C needs more energy to be broken than one that is rich in A and T, meaning higher melting temperature(Tm). Above the Tm, DNA denatures, and below it, DNA anneals. Annealing is the reverse of denaturation.
One aspect of thermal denaturation is never discussed. The heat supplied to effect such denaturation has no preferred direction and is therefore a scalar quantity. However, unwinding a double helix involves unwinding and this has direction and is therefore a vector. The issue is this: how does a scalar change induce a vector change?
Other methods to denature DNA
[edit | edit source]Heating is not the only way to denature DNA. Organic solvents such as dimethyl sulfoxide and formamide, or high pH, could break the hydrogen bonding between DNA strands too. Low salt concentration could also denature DNA double-strands by removing ions that stabilize the negative charges on the two strands from each other.
The central difficulty with denaturation of the double helix remains. How would two strands, typically consisting of many turns, and often many hundreds of turns, actually effect strand separation after the hydrogen bonds have been severed ?
A further, major difficulty lies in the fact that the application of heat to a suspension of nucleic acids amounts to the application of a scalar quantity because the heat applied in this way has no direction. However, unwinding the strands requires an angular force and this is a vector as it has a preferred direction. It has never been explained how a scalar quantity (heat) can effect a vector change (rotation) in a solution. There is simply not enough technology are intellect in this world to explain it.
A solution to these problems is offered by the side-by-side models in which there is no net winding of strands around each other. The Avery-MacLeod-McCarty Experiment was presented by Oswald Avery, Colin MacLeod, and Maclyn McCarty in 1944. During the 1930s and early 1940s, Avery and MacLeod performed this experiment at Rockefeller Institute for Medical Research, after the departure of MacLeoirulency (measure of deadly potency). This experiment would allow them to determine if rough bacteria could be transformed into smooth bacteria, hence passing along the genetic information causing the transformation. By isolating and purifying this chemical component, they could deduce if it had characteristics of a protein or DNA molecule.
Purpose
[edit | edit source]The purpose behind this experiment was to better understand the chemical component that carries the genetic information and transforms one molecule to the next.
Procedure
[edit | edit source]Bacteria grown in petri dishes can grow spots or colonies inside the dish multiplying under certain conditions. Virulent (deadly) colonies look smooth or like tiny droplets, where as non-deadly bacteria formed rigid, uneven edges, basically rough colonies. While analyzing a certain kind of pneumonia caused by bacteria in mice, they were able to isolate a "variant" (mutant) strain that did not kill the mice. During the experiments, Avery and MacLeod injected a mouse simultaneously with "boiled" or dead smooth bacteria and live rough bacteria. Thereafter a short while they were surprised to see that the mouse died. When they took samples from the dead mice, and cultured the samples in a petri dish, Avery and MacLeod found that what grew inside the culture was in fact the smooth deadly bacteria. This suggested that something from the "dead" bacteria somehow converted the rough bacteria into smooth bacteria. The rough bacteria had been permanently converted or transformed into the smooth dangerous bacteria. They had confirmed that they could not grow smooth bacteria from the boiled culture and cause disease if the dead smooth bacteria were injected alone. This all implied that a chemical component in the smooth bacteria survived and transformed the rough bacteria into smooth. Isolating and purifying that chemical component had shown that is was DNA, NOT proteins that transferred the genetic code from the smooth to the rough.
Simpler Experimental
[edit | edit source]
Here is a simpler demonstration for this experiment by Oswald Avery, Colin MacLeod, and Maclyn McCarty. There are two sets of bacteria – one is smooth (virulent), one is rough (nonvirulent).
1) They first inject deadly encapsulated bacteria into the mouse – the mouse dies at the end.
2) They then inject non-encapsulated, nonvirulent bacteria into the mouse – the mouse lives.
3) Next, they heated the virulent bacteria at a temperature that kills them and injected these bacteria into the mouse – the mouse lives.
4) After that, they then have the denatured fatal bacteria mix into the living non-encapsulated, nonfatal bacteria. The mixture was then injected into the mouse – the mouse dies.
5) Finally, they mix the live, non-encapsulated harmless bacteria with the DNA that was extracted from the heated, lethal bacteria. These “harmless” bacteria injected to the mouse after being mixed – the mouse dies.
From these experiments, Avery and his group showed that nonvirulent bacteria become deadly after mixing with the DNA of the virulent bacteria . Such a demonstration shows that nonvirulent bacteria became virulent because of the genetic information that originally came from the virulent bacteria. The protein from the virulent bacteria was already denatured during Step 3. Thus, it was DNA and not protein that transferred the genetic information to the nonvirulent bacteria.
Griffith Experiment
In 1928, Frederick Griffith performed a DNA experiment using pneumonia bacteria and mice. This experiment provided evidence that some particular chemical within cells is genetic material. The objective of the experiment was to find the material within the cells responsible for the genetic codes.
For the experiment, Griffith used Streptococcus pneumoniae, known as pneumonia. Pneumonia contains two strains - a smooth and a rough strain. The smooth strain causes pneumonia and contains a polysaccharide coating around it. The rough strain does not cause pneumonia and also lacks a polysaccharide coating. For his first experiment, Griffith took the S strain (smooth strain) and injected it into the mice. He found that the mice contracted pneumonia and ended up dying. He then took the R strain (rough strain) and injected it into the mice and found that they did not contract the pneumonia illness and survived the insertion of the strain. Through these first two experiments Griffith concluded that the polysaccharide coating on the bacteria somehow caused the pneumonia illness, so he used heat to kill the bacteria (polysaccharides are prone to heat) of the S strain and injected the dead bacteria into the mice. He found that the mice lived, which indicated that the polysaccharide coating was not what caused the disease, but rather something living inside the cell. Then he hypothesized that the heat used to kill the bacteria denatured a protein within the living cells, which caused the disease. He then injected the mice with a heat killed S strain and a live R strain, which resulted in the mice dying.
Griffith performed a necropsy on the dead mice and isolated the S strain bacteria from the corpses. He concluded that the live R strain bacteria must have absorbed the genetic material from the dead S strain bacteria, which is called transformation, a process where one strain of a bacterium absorbs genetic material from another strain of bacteria and turns into the type of bacterium whose genetic material it absorbed. Since heat denatures proteins, the protein in the bacterial chromosomes was not the genetic material. However, evidence pointed to DNA. This experiment that Griffith performed was a precursor to the Avery experiment. Avery, Macleod and McCarty followed up on the experiment because they wanted a more definitive experiment and answer.
Avery, MacLeod and McCarty used heat to kill the virulent Streptococcus pneumonia bacteria and extracted RNA, DNA, carbohydrates, lipids and proteins - which were considered possible candidates for the carriers of genetic information - from the dead cells. Each molecule was added to a culture of live non-virulent bacteria to determine which was responsible for changing them into virulent bacteria. DNA was the only molecule that turned the non-virulent cells into virulent cells, which they concluded was the genetic material within cells.
References
[edit | edit source]Lockshin, Richard A., The Joy of Science 2007.
History
[edit | edit source]In 1944 Oswald Avery and colleagues did an experiment involving the use of pathogenic bacteria to determine the material that contained genetic information. Their experiment concluded that it was DNA and not proteins that is the hereditary material. Despite the findings, the popular and widely accepted conclusion remained that protein encoded genetic information, accounting for its diversity in function and much greater number compared to DNA. In order to gain more evidence on DNA scientists by the name Alfred Hershey and Martha Chase decided to perform a simple but effective experiment involving bacteriophages.
Bacteriophage
[edit | edit source]In order to understand the experiment that was performed we must examine first the vector used which played a crucial role in the experiment – Bacteriophages. Bacteriophages are types of viruses which infect bacteria such as Escherichia coli. They consist of a protein coat, collar, base plate, tail fiber and most importantly a head which houses the genetic material. They have a unique feature, which makes them the perfect candidate to prove whether DNA or proteins house the genetic information. They have an outer capsule of proteins, which surround an inner core of DNA. Bacteriophages, being viruses, are unable to proliferate on their own accord, as they lack the necessary system to do so. Viruses invade a host cell and inject their genetic material to the host’s own gene and allow the host to replicate the viral gene. Knowing this, Martha and Hershey Chase saw that if they labeled the bacteriophages they would be able to track what genetic information is passed on to the host bacteria - the labeled DNA or the labeled protein coat.
Experimental
[edit | edit source]A bacteriophage was taken and its encased DNA was labeled with radioactive 32P and its protein coat was left nonradioactive. The bacteriophage was exposed to a sample of bacteria. The phage attached to the surface of the bacterial cell and injected the labeled DNA. The sample was then chilled to arrest growth. The sample was then shaken vigorously for several minutes in a Waring Blender. This process separated the phages coat from the surface of the bacteria. The sample was then centrifuged very quickly. The bacterial cells were at the bottom of the tube and the phage particles were in the supernatant. Hershey and Chase discovered that there was no disruption in the reproduction of viral phages inside the bacteria. A new generation of viruses had successfully propagated inside the host cell and these phages exhibited 32P radioactivity in their own DNA.
Another set of Bacteriophage was then examined, this time nnznwith a radioactive protein coat 35S and a nonradioactive DNA. The same procedure was followed and the phage attached to the bacterial wall and was allowed to inject its genetic material. Vigorous shaking of the bacteria caused the radioactive viral sheath to detach from the bacteria. Injection of the viral DNA into the bacteria still occurred and new phages were observed to have been produced. However, analysis of the new phages inside the bacteria showed that it had no radioactive properties; a property which should be present in the new phages, if proteins were in fact the genetic material that is passed on to new progeny. This experiment therefore illustrated that DNA, not protein, is the source of genetic information.
Results
[edit | edit source]The first bacterial cell contained phages with observable radioactivity illustrating that the radioactive property present on the parent phage was passed on to the new phages. The second bacterial cell however showed no hint of 35S, showing that it was removed along with the protein coat and did not enter the bacteria. Hershey and Chase then deduced that the genetic material that is being passed on is DNA and not protein as previously accepted before.
This famous 1952 experiment allowed Hershey and Chase to demonstrate that it was DNA, not protein, that functioned as the T2 phage's genetic material. Viral proteins, labeled with radioactive sulfur, remained in the ouside of the host cell during infection. In contrast, the viral DNA, which was label with radioactive phosphorus, entered the bacterial cell. Concluding that the DNA is in fact the material within cells that contains useful genetic information.
Additional Information
[edit | edit source]An animated video of the Hershey and Chase experiment can be viewed by clicking on this link
http://highered.mcgraw-hill.com/sites/0072437316/student_view0/chapter14/animations.html#
The published papers of Martha Chase and Alfred Hershey can be viewed in this link
http://osulibrary.orst.edu/specialcollections/coll/pauling/dna/papers/hersheychase.html
Sources
[edit | edit source]Berg, Jeremy, John Tyzmozcko, Lubert Stryer. Biochemistry
http://www.emc.maricopa.edu/faculty/farabee/BIOBK/BioBookDNAMOLGEN.html
http://www.accessexcellence.org/RC/VL/GG/hershey.php
http://highered.mcgraw-hill.com/sites/0072437316/student_view0/chapter14/animations.html#
http://en.wikipedia.org/wiki/Hershey-Chase_experiment
Historical information
[edit | edit source]Determination of the DNA structure would not have been possible if it was not for the work of Erwin Chargaff, an Austro-Hungarian biochemist. Originally a scientist who did his first work in lipids and lipoproteins, after reading about an experiment of Oswald Avery which showed that DNA was material encoding the genetic information, he turned his work onto DNA.
Tetranucleotide hypothesis was the mainstream theory on Chargaff’s time which was proposed by Phoebus Levene. The theory suggested “that DNA was made up of equal amounts of four bases – adenine, guanine, cytosine, and thymine – but that it was organized in a way that was too simple to enable it to carry genetic information.” The four bases are held together by hydrogen bonds and they are located inside the DNA helical structure. However, the sugar and phosphate backbone are on the outside of the DNA structure. The two strands are complementary to each other and thus one strand depends on the other. Despite the results of Avery’s experiments that DNA encodes life the scientific community was convinced DNA was relatively too simple to carry genetic information. Chargaff was not satisfied with the tetranucleotide postulation because of the minimal data that supported it.
Experiment
[edit | edit source]Erwin and his colleagues collected several DNA samples throughout their discovery. Using the fairly new technique of paper chromatography, Chargaff and his associates proceeded to separate DNA. The DNA that they collected was subjected to acid. The acid would then hydrolyze the phosphodiester bonds as it would cause a nucleophilic attack on the bond and result in the backbone breaking up. Once the phosphodiester bonds were broken then the individual nucleotides would then be separated and be free to analyze. Ultraviolet spectrophotometry was used to analyze the exact amounts of bases that were present in the DNA sample.
UV spectrophotometry showed that there was not an equal amount of purine bases (Adenine and Guanine) and pyrimidine bases (Cytosine and Thymine). Chargaff and his partners showed that the tetranucleotide hypothesis was in fact wrong in assuming that all four bases were in equal amounts. In other words, the concentration of GC equals to the concentration of AT. However, in RNA, Thymine is replaced with Uracil. What Chargaff noticed however was that although not all were in equal amounts certain bases were equal to each other. The base Guanine was equivalent to the amount of cytosine present; and the same held true for Adenine and Thymine. The ratio of A/T and C/G bases held true for all organisms and for both of the strands that were separated. The noticeable proportionality between one purine base to another pyrimidine base as well as it being true for both strands would be crucial in determining the helical structure of DNA although Chargaff was unable to see it.
The experiment gave two discoveries which is now summarized as Chargaff’s Rule:
1. The number of Adenine bases is equal to the number of Thymine bases, and number of Cytosine bases is equal to Guanine bases.
Ratio of A=T
Ratio of C=G
Ratio of A + T +C +G = 100%
2. The proportion of A:T and C:G holds true for both strands.
For example: in human DNA, the four bases Adenine (A), Thymine (T), Cytosine (C), and Guanine (G) are present in these percentages: A= 30.9% and T= 29.4%; G=19.9% and C=19.8%. The A=T and G=C equalities, displays Chargaff's Rule, which actually remained unexplained until the discovery of the double helix by Watson and Crick.
Sources
[edit | edit source]Berg, Jeremy. Biochemistry. 6th edition. ISBN-13 9780716787242
Campbell,Neil. Biology. Pearson Publishing. Dec 2004
Watson, James. DNA : The Secret of Life. Knopf Publishing Group. Aug 2004.
http://www.answers.com/topic/erwin-chargaff
http://www.cumc.columbia.edu/news/in-vivo/Vol2_Iss10_may26_03/CPMC-history.html
http://www.ncbi.nlm.nih.gov/books/bv.fcgi?rid=genomes.figgrp.5265
Inspirations to the Discovery of DNA Structure
[edit | edit source]James Watson began his research on DNA structure when he was in college. In 1945, during his third year of college, he reads Erwin Schrödinger's What Is Life? and takes away the message: Genes are the key components of living cells, so "we must know how genes act". In 1950 at an international conference in Naples, Maurice Wilkins of King's College, London, shows his clear X-ray pictures of DNA to Watson. Determined to work on DNA structure, Watson moves to Sir Lawrence Bragg's biophysics unit of the Cavendish Laboratory at Cambridge, England, where he meets biophysicist Francis Crick. Many scientists, including Rosalind Franklin, began her research on DNA structure with the help of X-ray diffraction. During the same year, she held a seminar at King's College in London, where Watson was invited. Her X-ray photo revealed the physical structure of DNA as a helix. During the seminar, Watson learned that Franklin's research confirmed that DNA had a helical structure, which consisted of two to four interlaced helical chains. Each helix had a phosphate-sugar backbone, with attached bases (adenine, guanine, thymine, and cytosine). The bases were proved to attached to the inside of the helix, possibly forming links between the helical chains. After Franklin's seminar, Watson decided to build DNA models.
Continuation on the Discovery
[edit | edit source]Nevertheless, the diffraction pictures of these models did not fit that of real DNA. The models that Watson built turned out to be wrong—the bases are on the outside of the helix and the helix is dehydrated—because he misinterpreted Franklin's findings. Watson's and Crick's research on DNA structures was terminated by King's College, and they must continue with their previous researches, which are tobacco mosaic virus (TMV) for Watson and proteins for Crick.
Although banned from researching on the structure of DNA, Watson was able to continue because one of the main components of TMV was nucleic acid and Francis Crick continued with it outside of his research. In 1952, Watson described Alfred Hershey's discovery that the genetic material of viruses is DNA, comparing the DNA in virus heads to "a hat in a hatbox". Watson and Crick had a disastrous meeting with Erwin Chargaff of Columbia University, who had discovered the ratios of the amount of the DNA bases. From John Griffiths, the nephew of Fred Griffiths who contributed to the fact that DNA is a genetic carrier, Crick learned that guanine (G) is attracted to cytosine (C), and adenine (A) to thymine (T), and Crick deduced that the bases must fit together like two interleaved decks of cards—they were stacked on top of one another inside the entwined backbones.
Watson was convinced that DNA must be helical due to Crick's proposed DNA structure and the X-ray diffraction plates. When Franklin showed Crick and Watson the X-ray pictures of DNA, even though the pictures did not show the radial symmetry necessary for helices, they show that the crystals were overlapping.
In autumn of 1952, Watson became friends with Linus Pauling's son, Peter. At that time, Linus Pauling was one of the few men in the scientific community who pondered the importance of DNA structure. From Peter, Watson learned that Linus Pauling published a paper on DNA structure—there are three helically entwined chains with sugar phosphate backbone outside of the coil, and the outdated X-ray pictures "proved" the structure to be true. Such structure is known as alpha helix. Watson immediately knew that Pauling's structure was incorrect because of the previous models that Watson had built. In fact, Pauling's structure left out important details: he had omitted to assign ionization charges on the phosphate groups. When there is no electric charge holding the long thin chains together, the chains would unravel and fall apart; without the charges, the nucleic acid structure was not even an acid.
Watson and Crick knew that Linus Pauling was their main competitor in determining the structure of DNA. Knowing that one of the greatest scientists made several mistakes in deducing DNA structures, Watson and Crick resolved to tackle the DNA structure at Cavendish laboratory. They worked on the DNA model using metal plates and Franklin's pictures of DNA by X-ray crystallography, provided by Maurice Wilkins and Max Perutz. Besides matching the bases, they also determined that the width between the two DNA strands must be less than two nanometers. In order to fit the bases inside the strands, Watson believed that the base pairs that are alike should be put together. However, they were unable to fit the similar bases within a small width. Watson then discovered that the keto-form base pairs joined A-T and C-G, and now the base pairs are able to fit inside the double strands. In five weeks of time, Watson and Crick built a DNA model that is indeed the correct structure of DNA.
In April 25, 1953, Watson and Crick published their article "Molecular structure of Nucleic Acids: A Structure for Deoxyribose Nucleic Acid" in Nature, becoming the first to publish the structure of DNA as a double helix.
Importance of Discovery
[edit | edit source]This discovery shed light on how genetic material could be passed on from generation to generation, and proves the simplicity of the transfer of genetic material. In fact, our present understanding of the storage and utilization of a cell’s genetic information is based on work made possible by this discovery.
DNA Structure – Leading to Function
[edit | edit source]After looking at the X-ray crystallography made by Rosalind Franklin, Watson and Crick were able to deduce that the shape of DNA was a double helix, and by Chargaff’s experiment, were able to deduce that the G pairs with C and A pairs with T. The pairs’ base lengths are equal, and fit exactly between the two chains of phosphates. The bonds between the two phosphate groups are hypothesized by Watson and Crick to be hydrogen bonds, which are easily broken. The discovery of DNA structure thus gave them a very good idea on how DNA might replicate itself, and thus the passing of genetic material.
Within Watson and Crick's article they claim that DNA is a double helical structure and that Pauling's previous attempt to define the structure noting that it did not have the much needed hydrogen bond stabilization and underestimated the van der Waals interactions of base stacking. The helix would of right handed as the two chains run in opposite directions. Bases were linked towards the inside of the helix and the sugar phosphate linkage created the outer backbone. The helix would repeat every 10 residues or 3.4 Angstroms, as they saw in the crystallographic data from Rosalind Franklin. The diameter of the helix was found to be 20 Angstroms and there was a rotation of 36 degrees per base, thus having 10 bases every 360 degrees. The most innovative ideas of Watson and Crick's model was that the two chains were held together by bases of purines and pyrimidines. By hydrogen bonding, a purine must be bonded to a pyrimidine creating a complementary pair. Using experimental data that showed the ratios of adenine and thymine were very close as were guanine and cytosine they stated that adenine bonds to thymine and guanine binds to cytosine. They discovered this based on comparing the ratios of A-T, C-G and A-G, and they found that the first two ratios were the closest to 1 where as the second was varied. This helped them make the conclusion that A bonds with T and C bonds with G only. The pairs’ base lengths are equal, and fit exactly between the two chains of phosphates. The bonds between the two phosphate groups are hypothesized by Watson and Crick to be hydrogen bonds, which are easily broken. The DNA nucleotide must also contain deoxyribose and not ribose because the extra oxygen on ribose would interfere with the structure due to van der waals interactions. The discovery of DNA structure thus gave them a very good idea on how DNA might replicate itself, and thus the passing of genetic material. Also, they found that each of the bases was capable of tautomerizing between the enol and keto forms. Experimentally, it was determined that the keto form predominates at a physiological pH. Thus, they also came up with a method for demonstrating how DNA may denature as pH changes due to conversion from the keto to the enol form.
-
 Two complementary regions of nucleic acid molecules will bind and form a double helical structure held together by base pairs.
Two complementary regions of nucleic acid molecules will bind and form a double helical structure held together by base pairs.

Later Discoveries
[edit | edit source]Watson and Crick’s discovery led to many new investigations, such as the structure of RNA, how DNA contains all the information for protein production, and the Human Genome Project, whereby all the 100,000 human genes are attempted to be mapped.
Although the discovery of the structure of DNA was attributed to Watson and Crick, a keynote player in helping them discover this structure was a scientist by the name of Rosalind Franklin. Rosalind Franklin, along with Francis Wilkins, worked on DNA applying X-ray crystallography to find out its structural properties. X-ray crystallography required the process of exposing a crystal specimen (DNA) to X-rays to determine the locations of the atoms in the “molecules that comprises basic unit of crystal called unit cell”. The task however was not an easy one to attain.
Obtaining a clear diffraction pattern of an object required that the crystal be pure and the x-ray strong enough. However, as Franklin realized, DNA existed in two forms in equilibrium which resulted in a very unclear diffraction pattern. These forms were the A form, which is the dehydrated form of DNA, and the B form, which generated a long and fibrous structure due to humid conditions. Franklin, applying her chemistry background, then proceeded to isolate these two forms using clever laboratory techniques such as “manipulation of the critical hydration of her specimens” [2]. The A and B forms were then separated and subjected to X-ray crystallography obtaining the pictures which would be the basis of Watson and Crick’s helical DNA structure; one of them the famous photo no. 51 of the B DNA form.
Fiber Diffraction
[edit | edit source]Fiber diffraction is a method used to determine the structural information of a molecule by using scattering data from X-rays. Rosalind Franklin used this technique in discovering structural information of DNA. The experiment places a fiber in the trajectory of an X-ray beam at right angle. The diffraction pattern is obtained in the films of a detector placed few centimeters away from the fiber. The fiber diffraction pattern is a two dimension patterns showing the helical symmetry of a molecule rather than a three dimension symmetry if taken by X-ray crystallography. A good fiber diffraction patterns exhibits four quadrant symmetry, the axis aligned to the fiber is called the meridian and the axis perpendicular to the fiber is called equator. Franklin obtained a diffraction pattern using a non-crystalline DNA fiber, and from it she deduced the B-form of DNA.
EXPERIMENT
[edit | edit source]- The diffraction pattern obtained by Franklin and Wilkins showed a X pattern which hinted of a 2 stranded helical form

- They also observed that the patterns was consistent and inferred that the helix’s diameter must also be consistent,

- The helical turn of DNA correlates to the horizontal lines in the picture which measures to 34 Angstroms. They also calculated that the gap between based pairs was 3.4 A as measured on the distance from the center of the X to the ends. Simple math deduced that there are 10 nucleotides per turn
Franklin and Wilkins also showed that the sugar phosphate backbones were found to be in the outside of the helix and not inside as it was previously thought to be. They came to this conclusion because of the A and B forms of DNA. The hydrated and dry forms of DNA showed that water could easily come in and bind to DNA, a fact that could only happen if the feature showed sugar phosphate backbones being on the outside.
SUMMARY OF FINDINGS
[edit | edit source]- DNA’s helical structure was composed of two strands
- establish that DNA’s diameter was similar throughout
- calculated that 1 turn was 34 A, distance between base pairs as 3.4A, and 10 nucleotides per helical turn
- showed that sugar phosphate backbones were located outside of the structure
SOURCES
[edit | edit source]Berg, Jeremy. Biochemistry Textbook
http://www.accessexcellence.org/RC/AB/BC/Rosalind_Franklin.php
http://en.wikipedia.org/wiki/Rosalind_Franklin
http://osulibrary.orst.edu/specialcollections/coll/pauling/dna/pictures/franklin-typeBphoto.html
http://www.vigyanprasar.gov.in/scientists/REFranklin.htm
http://en.wikipedia.org/wiki/Fiber_diffraction
http://www.mpimf-heidelberg.mpg.de/~holmes/fibre/branden.html
DNA Replication is required for all cell division, which allows organisms to grow. In DNA replication, the DNA is first divided into two daughter strands in the genome, which carries the exact genetic information as the original cell. This starting point of the strand being separated is called the "origin". The double strand structure of the DNA aids the mechanism in replicating; these two strands are first separated into two separate strands. The complementary stands of the two separate strands are then recreated by DNA polymerase, an enzyme that specialize in making complementary strands; it will find the correct complementary base for each strand and it will extend from the 5' to 3'. The process by which the original strand is being preserved is called "semiconservative replication".
DNA replication is essential in the life cycles for biological organisms. It is initiated when the double stranded DNA located at the origin of replication is separated or melted. When the double stranded DNA is melted, melted region is propagated and a mature replication fork forms. DNA melting, along with the replication fork formation is coordinated by initiators, helicases, and other cellular factors. Recent advancements in structural biochemistry studies of initiators and replicative helicases have been emphasized in archaeal and eukaryotic cells. The results of these studied have provided new insight to possible mechanisms of the early stages of DNA replication.
Genomic DNA is a common, necessary, and essential process in all living things. Replication can be divided into initiation, elongation, and termination steps.
Initiation of DNA replication
[edit | edit source]During initiation, initiators recognize and then bind the replication origin DNA, converting it to a replication fork. The steps of initiation are made of up of the following steps: initiators assemble around the origin of DNA, and the dsDNA origin is melted. The melting of dsDNA produces a replication fork on each side of the origin to allow bi-directional replication. Before this step can happen, however, there are topological limitations that must be overcome to convert the melted origin to a fork structure. To induce the assembly of initiators at the origin, biochemical methods can be utilized to detect the initial melting of origin dsDNA. In the archaeal and eukaryotic cellular systems, the duration of origin melting is still unsure. However, the origin melting has been shown to be induced by the assembly of LTag. SV40 LTag is capable of inducing origin melting and unwinding, therefore it is considered to be the initiator[check spelling] in the eukaryotic system. It has been used as a model to study origin recognition, assembly, and melting process. To convert from a melted dsDNA origin, an assembly of initiators at an active replication fork expands the melted region and positions the helicase on the fork.
The initiation step is one of 3 steps in DNA replication (along with elongation and termination). In initiation, many replication proteins called initiators convert the DNA into a replication fork. This is accomplished first by the initiator proteins assembling around the DNA which causes melting of the dsDNA (double stranded DNA) origin. The origin melting then starts to produce a replication fork on each side of the melted origin. This produces bi-directional replication. Ring shaped helicases assists in this process. However, the mechanism of how the initiators and helicase melts and unwinds the origin DNA is not well understood due to the lack of high-resolution structures at the intermediate.
In eukaryotic and archaeal cellular system the initiator proteins includes Orc, Cdc6, Cdt1, and MCM (mini-chromosome maintenance) helicase. MCM is one of most important factors in the formation of the unwound fork. MCM forms hexamers that can dimerize into double- hexamers. The helicase for SV40 large T antigen (LTag) is able to recognize the origin DNA and can melt and unwind the DNA into a replication fork without the use of cofactors . SV40 LTag is considered the archetypal initiator/helicase in eukaryotic systems and is a model for studying recognition, assemble and melting.
Crystal structures of LTag hexamer reveals a channel of (13-17Å), which is enough for a ssDNA to go through but not dsDNA (20 Å). It is believed that melted ssDNA is encircled in the central channel for hexameric helicase, even during the assembly at the origin.
LTag also shows a β-hairpins in the central channel that is configured in a planar arrangement. β-hairpins form 2 adjacent planar rings with DR/F loops which contributes to the narrowest part of the channel in the AAA+ domain. It is questioned whether LTag can expand to accommodate dsDNA or is the dsDNA modified by initiator/helicase to fit the narrow channel. However for the latter to occur, LTag must squeeze and crush the dsDNA which disrupts the base pairs and melting of the dsDNA. This models often referred to as the squeeze to open model.
The most widely accepted model for fork unwinding is of the ring-shaped helicase that encircles and migrates down the DNA strand and splitting the dsDNA to ssDNA.
In Prokaryotic cells, bacterial replicases contain a polymerase, polymerase III (Pol III), a β2 factor, and a DnaX complex. They are very processive, and cycle faster during Okazaki fragment synthesis in many ways. DnaA (an origin recognition protein) can start the origin melting into single stranded DNA (ssDNA). The ssDNA is the site for loading hexameric helicase DnaB(which only exist as single-hexamers). One helicase that bacteria has is DnaB6, which can separate two strand at the replication fork. It translocates at the 5'-->3'. The DNA polymerase III holoenzyme (Pol III HE) makes contact at the replication fork and also function as a dimer that appears to have a regulated affinity on the lagging strand in order to recycles between primers during Okazaki fragments synthesis. DnaB uses ATP hydrolysis to go down the strand in order to split the two strand. Primase interacts with the helicase and combines with short RNA primers for Okazaki fragment synthesis. The RNA primers keep extending by the Pol III HE until a signal is received to replace to the next primer at the replication fork. During the process, the gaps between the Okazaki fragments are filled, RNA primers are deleted by DNA polymerase I, and is sealed by DNA ligase. DnaB has its N-terminal end free for docking primases making it easy for the primase to capture the ssDNA emerging from the N-terminal domain during fork unwinding.
Initiating Replication in Archae and Eukaryotes by Melting the Double Stranded DNA
[edit | edit source]Although not much is known about the initiation of replication by the melting of double-stranded DNA, recent studies have shed light on possible mechanisms for this process. Two co-crystal structures from archaea that have both the initiators and the origin DNA have been discovered to show how the initiators recognize the double-stranded origin of DNA. The complexes, Cdc6/Orc-dsDNA show the double stranded DNA deforming and bending, but not melting. Thus, researchers believe that in order to trigger the melting of the double-stranded DNA and to generate higher order complexes at the origin of replication, initiators like MCM mentioned in the above section must be needed.

This image represents an example of the structure of a DNA replication initiator—specifically showing the Cdc21 and Cdc54 (similar to the Cdc6 described above) N-terminal domain. The initiator, Cdc6/ORC 1 (which is not depicted here but can be represented by the picture above) binds to the origin of replication and bends the DNA. Citation: http://www.ebi.ac.uk/ http://upload.wikimedia.org/wikipedia/commons/c/c6/PDB_1ltl_EBI.jpg In eukaryotes, the SV40 Ltag at the origin is able to trigger the melting of the origin of replication and the subsequent unwinding of DNA, making it the initiator-helicase that is used as a model system for examining origin recognition, assembly, and the melting of the double-stranded DNA. The crystal structures of Ltag hexamers that are not bounded to DNA have been shown to have channels that seem to be able to bind to only single stranded DNA, but not double stranded DNA because the channels are usually about 13 to 17 Å (angstroms), while double stranded DNA molecules tend to have a diameter of about 20 Å, making a double stranded DNA molecule unable to fit inside the channel. Generally, studies of DNA translocation have shown that in order for a double stranded DNA to fit inside the channel of an Ltag hexamer, without changing its shape, the channels diameter must be at least 20 Å in diameter. In addition to not being big enough, crystal structures of Ltag hexamers have a planar arrangement of b-hairpins in the middle channel.

Here is an example of a b-hairpin, a component of the LTag hexamer structure. The b-strands in the b-hairpin are antiparallel—meaning that the N-terminus of one b-sheet is aligned with the C-terminus of another b-sheet. In the case of the LTag hexamer, the b-hairpins are on the same plane in the central region of the channel. Citation: http://commons.wikimedia.org/wiki/File:Beta_hairpin.png
Recently, cryo-EM has demonstrated that Ltag hexamer channels can bind double stranded DNA molecules by surrounding the double stranded DNA with two hexamers. Researchers however, still are unsure whether the double stranded DNA changes configuration because of the initiator-helicase or whether the Ltag widens to allow the double-stranded DNA to bind. One model, the squeeze-to-open model, asserts that the Ltag hexamer can fit the origin double stranded DNA into its narrower channel by squeezing the DNA through the channel. As a result, base-pairs are disrupted and the melting of the double stranded DNA origin occurs. This model has been proposed, and is in the process of being confirmed because it appears to be consistent with the known data regarding DNA melting.
The Formation of the Replication Fork by the Squeeze-Pumping Model:
The squeeze-pumping model derives from information that comes from the structure of the Ltag hexamers. The structure includes a narrow channel as mentioned above, an AAA+ motor domain, a side channel where single stranded DNA can exit, and inter-Zn domains. This model is based on the DNA being melted by the squeeze-to-open model described above, where the melted DNA is pumped to the Zn-Domain until it generates the single stranded DNA loop which can then leave the channel and form the replication fork.
Translocation of Single and Partially Hydrolyzed Double Stranded DNA: Researchers have demonstrated that double-hexameric LTag and MCM have the ability to unwind DNA. LTag has been shown to be able to unwind long double stranded DNA that include an internal origin sequence in its double-hexameric form. This differs from the steric exclusion model of fork unwinding—which is the most widely accepted model. This model is based on evidence showing that a ring-shaped helicase surrounds and moves down one of the DNA strands toward the double-stranded DNA fork while exposing the single stranded DNA strand in the process.

The above image represents a ring-shaped hexameric helicase structure that surrounds and moves down the DNA strands (which are not depicted in the photo). Citation: http://www.ebi.ac.uk/Information/termsofuse.html ; http://commons.wikimedia.org/wiki/File:PDB_1g8y_EBI.jpg
Sources:
Curr Opin Struct Biol. 2010 Sep 24. [Epub ahead of print] Origin DNA melting and unwinding in DNA replication. Gai D, Chang YP, Chen XS. Molecular and Computational Biology, University of Southern California, 1050 Childs Way, Los Angeles, CA 90089, USA.
Links between DNA replication and protein synthesis
[edit | edit source]For decades, individual studies were done on DNA replication and protein synthesis. Not many scientists discuss the link between these two critical processes in living organisms. Jonathan Berthon, Ryosuke Fujikane, and Patrick Forterre came together in their article “When DNA replication and protein synthesis come together” to provide a detailed explanation of the connections of these seemingly independent fields of structural biochemistry. They suggest that the unexpected but real connections between DNA replication and protein synthesis are found in the three domains of life, especially in Archaea and Eukarya. They believe that there are mechanisms that couple DNA and protein synthesis. Such mechanisms can be found in the activities of (p)ppGpp – Guanosine polyphosphate derivative – and GTPases or the Obg family.
- Stringent response is a phenomenon that can well link the processes of DNA replication in bacteria’s to the change in amino acid concentration in proteins. As starvation of amino acids occur, a dramatic increase in the intracellular (p)ppGpp concentration is observed that initiates the shut-down of rRNA gene transcription as well as protein synthesis. This process, however, varies among different bodies of bacteria. For instant, inside the system of Bacillus subtilis, amino acid starvation, along with the inhibition of rRNA gene transcription, blocks the elongation step of DNA replication. (p)ppGpp also inhibits the DnaG primase in Bacillus subtilis and could directly affect the Okazaki fragment synthesis in the lagging DNA strand, during the process of self-replication. On the other hand, stringent response in Escherichia coli leads to an instant interference of the DNA replication initiation. Such proofs are important in proving the direct connection between proteins and DNA replication process. The starvation of protein’s amino acid has the potential to stop DNA replication.
- Another source of connection is Obg family. Obg is known for its ability to couple ribosome biogenesis, a critical step in production of proteins as protein synthesis is done inside ribosome through mRNA, with DNA replication. The link between ribosome biogenesis and DNA replication is argued by scientists to start from the proteins that are originally function in the making of ribosome. These proteins participate in the regulation of the stringent response in bacteria as well as in the stabilization of DNA replication forks. A type of Obg, called ObgE is useful in controlling the levels of (p)ppGpp. One important link between DNA replication and protein synthesis found in ObgE is the fact that the depletion of ObgE would cause problems in chromosomes segregation and cell separation. This study is significant in showing that changes in certain proteins within the body would directly affect the pattern of DNA replication and the organism’s genetic processing. For this reason, Obg studies were done to prove the direct role that this type of protein plays in connecting DNA replication and protein synthesis.
- Similarly, a type of protein family called NOG1 – Nucleolar G-protein – also participates in the making of ribosome. Nog1p from this particular family belongs to a complex that contains many other proteins that directly take part in DNA replication such as Orc6p (origin recognition complex), Mcm6p, some subunits of MCM complex, Yph1p, and Rrb1p. A very important statement was made by Kilian that changes in proteins that connect ribosome biogenesis to DNA replication would cause “chromosome instability” and “tumor formation”. He also concludes that there exists a network of proteins that directly link the production of ribosome’s and DNA replication in Eukarya domain.
- All of the above studies and conclusions apply only for Eukarya because there is no clear evidence found for the domain of Archaea. Scientists, however, found that there is a cluster of genes that encode both DNA replication and translation proteins. This cluster includes numerous genes including essential ones such as aIF-2, an excellent source for regulation of DNA replication and protein synthesis. eIF-2 phosphorylation from this cluster is a major component in the mechanism of protein synthesis in eukaryotic cells. Another important component is Nop10 – plays a part in rRNA development. From simply examining these components, a clear conclusion can be drawn that there is, indeed, a close relation in the studies of proteins and DNA replication. One important example is the phenomenon where the two ribosomal proteins L44E and S27E interferes with the DNA replication process under special conditions such as amino acid starvation, previously discussed in the case of stringent response.
- In conclusion, in both Archaea and Eukarya, there are many experimental data that confirm or suggest the close connections between protein synthesis and DNA replication. Stringent response is one example of how starving amino acids would inhibit the process of DNA replication initiation.


The DNA Replication process works in an "assembly line" like fashion. The DNA double helix is ripped apart and a copy of each strand is produced. There are many biological enzymes that take part and must be present for this vital action to occur correctly.
Biological Proteins and Enzymes Required for DNA Replication (in chronological order)
[edit | edit source]Replication Fork
[edit | edit source]When DNA is being replicated, it forms a replication fork that was created during the helicase process that separates the DNA strand. The strands that are separated are called the leading strand and the lagging strand accordingly. The leading strand is synthesized in the 5'-3' direction. It is the new DNA strand, which is being synethized by DNA polymerase. The lagging strand, on the other hand, at the opposite side, which runs from 3' to 5' direction and are synthesized by okazaki fragments. Then primase will build up RNA primers, allowing DNA polymerases to use the 3' OH groups on the RNA primers to act on the DNA running from 5' to 3'. Then these RNA fragments are being substituted with new deoxyribonucleotides and the strand will then be joined together with DNA ligase to complete the chain.
As the DNA unwinds, it will automatically force the DNA to rotate, twisting the structure. This is actually a problem to replicating DNA because it will eventually be physically incapable of replicating when it is over-twisted. To solve this problem, a enzyme called DNA topoisomerases is used. Topoisomerases I will cut the backbone of the DNA to allow the DNA to unwind itself and topoisomerases II will cut the backbones of both strands to allow interconnections with other DNA molecules, eliminating the chances of tangling together.
Helicase
[edit | edit source]Helicases are motor proteins that move along the double-stranded nucleic acids and actively unwind the double helix. The enzyme uses the energy produced by the hydrolysis of ATP to ADP to unwind and separate a strand of DNA. This is done by the breaking of the hydrogen bonds between the annealed nucleotide bases. Helicase opening of the double strand can be categorized into two different cases: active opening and passive opening. In the active opening case, helicase directly destabilizes the double strand nucleic acid (dsNA) to promote the separation of the two strands. In the case of passive opening, the helicase enzyme binds to a single strand nucleic acid (ssNA) that existed due to thermal fluctuation which induces the opening of part of the double strand. It is found that active opening can increase the rate of unwinding of the DNA strand by 7 folds compared to passive opening. The product of this action is two template strands. One is known as the Leading Strand and the other is known as the Lagging Strand.
The leading stand is the single strand of the parental DNA that is synthesized continuously without interruption while the lagging strand of the parental DNA is formed in fragments. These fragments are called the Okazaki fragments. This is important in explaining how both strands of the parental DNA forms in a 5'->3' direction despite the fact that the two strands are antiparallel. The fragmentary synthesis enables the 5'->3' growth while appearing to form in a 3'->5' direction.
Single-Stranded DNA Binding Proteins
[edit | edit source]The Single-Stranded DNA Binding Proteins bind to the DNA templates in a way that ceases the two newly formed strands from reannealing. these proteins keep the strands separated so that both of the strands can serve as templates for replication. This allows the remainder of the replication machinery to get into position and begin making new DNA strands.
DNA Polymerase
[edit | edit source](see DNA Polymerase Section)
RNA Primase
[edit | edit source]The RNA Primase attaches itself to the Lagging Strand in a position adjacent to the Helicase. The RNA Primase's Function in DNA Replication is to lay down RNA Primers in 3' to 5' fashion. These RNA Primers act as starting and ending locations for the DNA Polymerases addition of complementary nucleotides. The nucleotide sequences between RNA Primers are known as Okazaki Fragments. The RNA Primase is only necessary in the Lagging Strand because DNA Polymerase can only add complementary bases in a 5' to 3' direction, and the lagging strand is being unwound in the 3' to 5' direction.
 DNA Replicases from a Bacterial Perspective
DNA Replicases from a Bacterial Perspective
Mitochondrial DNA Replication
[edit | edit source]
Mitochondrial DNA (mtDNA) is maintained apart from nuclear DNA. Because of mtDNA’s small size, it can only boast 37 genes and 13 protein products whereas the haploid nuclear genome encodes over 20,000 genes. However, it can provide a model system for studying nuclear DNA replication. The genome for the circular mtDNA contains approximately 16,600 base pairs in human beings. The encoded genes are also found to be necessary for making ATP by way of oxidative phosphorylation. There seems to be no specific phase for mtDNA to be replicated, meaning the replication can take place over and over during a cell cycle.
The endosymbiont hypothesis is the idea that mitochondria were engulfed to create the first eukaryote. Evidence supporting this hypothesis comes from the existence of mtDNA itself. Because mitochondria were once free-living bacteria, it might be anticipated that the mechanics of mtDNA maintenance would show greater similarity to prokaryotes over eukaryotes.
The mechanism in which mtDNA replicated was discovered in 1972 by electron microscopy. All replicating mtDNA molecules had a single-stranded branch. This further resulted in the leading-strand and lagging-strand synthesis uncoupled in mitochondria, which was different compared to the replication fork for nuclear DNA. The human mtDNA is typically arranged in covalently closed circles that are about one genome in length. In mtDNA replication, there is a strand-displacement replication fork in which leading-strand DNA synthesis occurs in the absence of lagging-strand DNA synthesis. DNA synthesis is carried out by conventional coupled leading- and lagging-strand. Then, delayed lagging-strand DNA synthesis is accompanied by incorporation of RNA on the lagging strand termed RITOLS for RNA incorporation throughout the lagging strand.
The issue of how mammals replicate their mtDNA gave rise to mtDNA replication redux which is an attempt to test the idea that biased segregation of human pathological mtDNA variants was related to replicative advantage, as suggested for yeast mtDNA. 2D agarose gel electrophoresis (2D-AGE) was used to resolve replication intermediates from mitochondria. This was used to define details of the mechanisms of replication for nuclear, plasmid, and viral genomes. It was found that many replication intermediates from crude mitochondrial preparations were sensitive to single-strand nuclease as predicted by SDM, a subset formed arcs indistinguishable from those associated with replicating nuclear and prokaryotic DNA.
However, purer preparations of mitochondria yielded not partially single-stranded DNA but RNA/DNA hybrids. This concluded that the SDM intermediates from earlier studies could be explained by RNA loss during isolation and processing.
In conclusion, there is still controversy about the mechanisms of mtDNA replication. The strand-displacement model of mtDNA replication is where there is a minimum of two primer maturation events for each strand, which applies to the RITOLS replication as well. The identification of Dna2 and Fen1 in mitochondria provides new tools for studying mtDNA replication. By manipulating their expression and studying mutant variants that disrupt mtDNA replication, it might prove to be very informative.
It was found that mutations, deletions, and other problematic arrangements of mtDNA increased in correlation to a mammal's aging. The accumulation of mutated mtDNA in single cells cause respiratory chain deficiency. This causes shorter life spans for the mammals. It can also cause aging phenotypes when there are many mutations such as weight loss and loss of hair.[1]
References
[edit | edit source]Holt, Ian J. "Mitochondrial DNA replication and repair: all a flap."
- Betterton MD, Julicher F, "Opening of nucleic-acid double strands by helicases: active versus passive opening.", Physical Review E. 2005 Jan; 71 (1): 011904.
- Berthon, Jonathan, Ryosuke Fujikane, and Patrick Forterre. “When DNA replication and protein synthesis come together”. Trends in Biochemical Sciences. Vol.34, no.9 (2009): 429-434. Cell Press.
- Dahai Gai, Y Paul Chang and Xiaojiang Chen. "Origin DNA melting and unwinding in DNA Replication." Current Opinion in Structural Biology 2010, 20:1-7. Elsevier.
- Charles S. McHenry. "DNA Replicases from a Bacterial Perspective." Annual Review of Biochemistry Volume 40 2011 July, 403-36.
General information
[edit | edit source]The full process of DNA replication is comprised of the intricate and coordinated interplay of more than 20 proteins. In 1958, Arthur Kornberg and his colleagues separated DNA polymerase from E.Coli. DNA polymerase is the first known of the enzymes whose function is to promote the bond formation of the joining units that make up the DNA backbone. E.Coli has various numbers of DNA polymerases, assigned by Roman numerals, that play important roles in DNA replication and repair.
DNA polymerase is an enzyme. This enzyme synthesizes a new DNA strand from an old DNA template and also works to repair the DNA in order to avoid mutations. DNA polymerase catalyzes the formation of the phosphodiester bond which makes up the backbone of DNA molecules. It uses a magnesium ion in catalytic activity to balance the charge from the phosphate group.
Nucleotides are added to only the 3' end of the new strand; it is impossible for it to start a new chain on its own. Another DNA polymerase function is error correction - the correction of mistakes that were made in the new DNA strand. The entire DNA polymerase family consists of 7 different subgroups: A, B, C, D, X, Y and RT. Eukaryotes have at least 15 different DNA polymerases. However, none of the eukaryotic polymerases can remove primers, and only the elongation polymerases can proofread the sequence.
Although there are different types of DNA polymerases, all have common structural features. Additionally, even though DNA polymerases differ greatly in detail, they have very similar overall shape. There are at least 5 structural classes of DNA polymerase that have been identified. They take the shape of a hand with specific regions referred to as the fingers, the palm, and the thumb. In all classes of DNA polymerase, the thumb and finger wraps the DNA, holding it across the active site of the enzyme, while the palm releases residues that comprise this active site. Moreover, all DNA polymerases use similar strategies in the catalyzation of the reaction.
General Formulation
[edit | edit source]DNA polymerases are the catalysts in the step-by-step addition of deoxyribonucleotide units to a DNA chain. The reaction catalyzed is
(DNA)n + dNTP ↔ (DNA)n+1 + PPi
where dNTP stands for any deoxyribonucleotide and PPi is a pyrophosphate ion.
Requirements
[edit | edit source]1. All four activated precursors are needed for the reaction to occur, the deoxynucleotide 5’-triphosphate dATP, dGTP, dCTP, and dTTP, in addition to Mg2+ ions. Typically, two of the metal ions will take part in the reaction. One will interact with the primer while the other with dNTP. The carboxylate groups of the residues in dNTP bind the two metal ions in place.
2. The new DNA chain is constructed directly on a pre-existing DNA template. DNA polymerases can only work efficiently as a catalyst in the formation of phosphodiester bonds if the base on the incoming nucleotide triphosphate is complementary to that of the template strand. In other words, DNA polymerase is an enzyme that synthesizes a product by interpreting the existing DNA strand as a template and produces the complementary sequence of the template into a new strand.
3. DNA polymerases necessitate the presence of a primer to start synthesis. The reaction catalyzed by DNA polymerases that works to elongate the chain is a nucleophilic attack by the 3’OH terminus of the growing chain on the innermost phosphorus atom of the deoxynucleotide triphosphate. Therefore, a primer strand with a free 3'-OH group must be bound to the template strand from the start. This primer is formed from RNA synthesis. Due to the fact that RNA can form without a primer, it starts the synthesis of DNA. Once the complementary DNA is formed and the synthesis has been initiated, the RNA piece will be removed and then replaced by the proper DNA sequence. A phosphodiester bridge is formed from the reaction and pyrophosphate is released. The ensuing hydrolysis of pyrophosphate that results in the creation of two ions of orthophosphate (Pi) by pyrophosphate assists to drive the polymerization forward. This elongation process of the DNA chain proceeds in the 5’-to- 3’ direction.
4. Many DNA polymerases are able to remove the mismatched nucleotides as a method of mistake correction in DNA. The polymerases possess a distinct nuclease activity that allows them to eliminate incorrect bases through a separate reaction. DNA polymerase will reverse its direction by one base pair and excise the incorrect base to replace it with the proper one and continue with the rest of replication. Due to this 3' to 5' exonuclease activity, DNA replication has a remarkably high dependability. This step process is also called proofreading. However, it is not completely perfect, which is why natural mutations and related diseases can still arise.
Eukaryotic DNA Polymerases
[edit | edit source]DNA Polymerases play a key role in the synthesis of DNA. Without these players, life would cease to exist. These polymerases are multi-subunit complexes that function very uniquely. It requires different components to work together to function efficiently. Polymerases act upon single-stranded strands (specifically to the template), to synthesize a strand that is complementary. In eukaryotic cells, there are 5 families of DNA polymerase. These can encode into different (up to as many as 15) enzymes. Critical for DNA replication are three DNA polymerases: Polymerase α-primase, Polymerase δ, and Polymerase ε. These three polymerases function at the replication fork of the DNA strands. The DNA strands are unwounded by MCM helicase, which is part of a CMG complex (Cdc45-MCM-GINS). It is Polymerase α- primase that initiates replication on the leading and lagging strand. It is here that the RNA primers (about 10 nucleotides) are laid down.
After the initiation, Polymerase δ and ε are brought to the complex and tethered. They function to increase the productivity of the different enzymes. Specifically, Pol δ synthesizes on the lagging strand while the Pol ε synthesizes on the leading strand. The roles of these polymerases were found by genetic experiments. For Pol ε, a mutation was placed on the active site. This increased the rate of enzymes activity, and leave behind a signature in the regions of activity. With the involvement of reporter genes, it proved that the Pol ε did indeed participate in the synthesis of the leading strand. The same genetics were done for Pol δ to prove its activity with the lagging strand.
A consistent correctness is necessary with the implementation of the bases. An incorporation fortunately occurs only every 10,000 replicated base pairs. But when it does occur in the DNA primer strand, it must be moved out from the polymerase and to the exonuclease domain. It is there that it is proofread and allow for continuation of a stable domain. [1]
Polymerase Families
[edit | edit source]Central to life, polymerases have been put under study in search of its structure as well as roles. To date, there have been 7 different families (or domains). There are 5 unique to eukaryotic cells. More families are unique to bacteria and archaea. In these polymerase families, there is a core structure: palm, finger, and thumb domains. From there the families diverge to their specific cellular functions. The 7 families are labeled with letters: A, B, C, D, X, Y, and reverse transcriptase. Family A includes Pol I polymerase, which functions to repair nucleotides. It also includes Okazaki fragments, which takes part in the replication of the lagging strand. Family B includes the eukaryotic polymerase sigma, alpha, as well as epsilon. Family C harbors the Polymerase III, which XXX. Family D includes polymerases that are exclusive to archaea. Family X as well as Y include enzymes that do repairing.
Within the Eukaryotic DNA Polymerase Structures
[edit | edit source]As it was earlier noted that the polymerases are multi-unit entities, it holds true that they are very complex. The structures are comprised of a large catalytic subunit (part of the B family), and then many other smaller subunits. The architecture of the B family polymerases are consistent: a N-terminal domain, 3’-5’ exonuclease domain, palm, finger, and thumb domains; in a ring-like structure. The catalytic subunit of all the eukaryotic polymerases are assumed to be related and come from a common ancestor via gene duplication. But studies do show that the catalytic subunit of the ε is larger than the other two due to additional sequences.
Obtaining structures that are in high resolution is essential for further analysis of polymerases. To date, there has been a lot of progress in formulating the structures of the different subunits that make up the polymerases, but only at low-resolutions. The first structure reported was the cryo-EM structure of the Pol ε. Researchers aim to work towards high resolution structures because it came allow further understanding of the fidelity of DNA synthesis, and the highly regulated genome that is maintained in all of the eukaryotic cells. Furthermore, it would allow design of genetic experiments to explore the interactions of and within the complexes.[2]
References
[edit | edit source][1]
"Molecular Recognition and Catalysis in Translation Termination Complexes" by Bruno P. Klaholz. IGBMC (Institute of Genetics and of Molecular and Cellular Biology), Department of Structural Biology and Genomics, Illkirch, F-67404 France. Trends in Biochemical Sciences, May 2011, Vol 36, No. 5
[2]
"Crystal Structure and Functional Analysis of the Eukaryotes Class" Mol. Cell 14, 233-245. Kong, C. etal (2004)
DNA initiation is the first stage of the DNA replication process. During this stage, the double stranded DNA (dsDNA) is first separated into single strands by breaking up the hydrogen bonds between base pairs. The separation of dsDNA into singled stranded DNA (ssDNA) is known as DNA melting. Proteins that are responsible for breaking up of dsDNA are called initiator proteins. In the next step, proteins called helicase bind to the dsDNA and unwind it to create a replication fork. In Eukaryotic and archaeal cells, melting and unwinding of DNA are mainly accomplished by mini-chromosome maintenance helicase (MGM) along with multiple initiation proteins. However, helicase such as large T antigen (LTag) and E1 which are found in simian virus 40 (SV40) and bovine paillomavirus (BPV) are able to break up and unwind the dsDNA without any additional cofactors (Chen et al.). Because of the great similarity between the viral and eukaryotic and archaeal DNA replication system, LTag and E1 are studied intensively by researchers in hope of gaining a better understanding in replication process.
Mechanism
[edit | edit source]Differences in the arrangement of β-hairpins and mode of ATP binding etc. in the viral proteins can lead to different mechanism of melting and unwinding. For example, LTag of SV40 is believed to use mechanism that follows the double-pump looping model which is described in Chen et al. First, LTag in the shape of a double-hexamer binds to the dsDNA at the replication origin and compress it to break up the hydrogen forces between two strands. Two hexamers ahead of the replication origin then pump the dsDNA into the double-hexamer to create a replication fork that is consisted of ssDNA as loops. Further pumping of the dsDNA will elongate the replication fork to allow fork progression. On the other hand, E1 of BPV uses mechanism that follows closely with the steric exclusion model (Chen et al.). In this model, E helicase exists only as a single hexamer and is separated into two trimers with each binds to one strand of dsDNA at the origin to induce melting. After successfully breaking up the dsDNA, two trimers rejoin to form a hexamer that binds to only one strand of dsDNA and unwinds it to create a replication fork.
Conclusion
[edit | edit source]Although they present plausible mechanisms for DNA melting and formation of replication fork, both models still require support of further evidences. Questions such as how LTag binds to dsDNA or whether the E1 hexamer can separate into two trimers still remain unanswered. More intensive investigation and research are therefore needed.
Reference
[edit | edit source]Chen, Xiaojiang S, Paul Chang and Dahai Gai. "Origin DNA melting and unwinding in DNA replication". Current Opinion in Structural Biology 2010, 20:1-7.
Meselson – Stahl Experiment
[edit | edit source]
Theories of Replication of DNA
[edit | edit source]Conservative
The daughter DNA is composed entirely of new DNA and the parent DNA retains it’s same back-bones and bases.
Semi-conservative
Replication produces two copies of DNA that are made up of 50% DNA from the parent DNA helix, and 50% of new DNA. In this situation, each daughter DNA double-helix contains one strand that is the old DNA (from the parent) and one strand that is new (the complimentary strand resulting from the replication).
Dispersive
This form of replication also produces daughter DNA that is constituted by 50% new DNA, and 50% parent DNA. However, in this case, the new DNA and old DNA are shuffled, and fragments of each are found on both strands on the helices on both copies of DNA following replication.

Schematic of the three theories of replication, by CJHIGGIN
The Experiment
[edit | edit source]Watson and Crick proposed that DNA replicated semi-conservatively, but conservative and dispersive replication were still plausible until the theories could be disproved. In 1957, Matthew Meselson, and Franklin Stahl devised an experiment to determine whether DNA replicated following a conservative, semi-conservative, or dispersive model.
Method
Meselson and Stahl cultured Escherichia Coli in a medium containing a heavy isotope of nitrogen (15N) as the only nitrogen source, as opposed to the more common nitrogen-14 (14N). After several generations, the E. coli contained DNA composed of nucleotide base made of 15N isotope. The (15N) DNA was denser than the common (14N) DNA, and the difference in densities allowed for separation by density gradient equilibrium sedimentation.
To achieve separation of the E. coli DNA by densities, the DNA was mixed with a solution of CsCl and centrifuged. A CsCl density gradient was created as a result of sedimentation and diffusion working against each other. The DNA molecules were found in the area of the CsCl density gradient that was equal to their own density.
The (15N) E. colicells were transferred to a medium that contained only (14N). DNA was isolated from the first generation of cells grown in the (14N) medium, and analyzed by density gradient equilibrium sedimentation. Then DNA from the second generation of E. coli grown in the (14N) medium was extracted and analyzed.
Results
[edit | edit source]The first generation of E. coli grown in the 14N medium contained a single DNA band found halfway in between where the 14N DNA band and the 15N DNA band should have been. This demonstrated the presence of a DNA that was lighter than the DNA from the original population of E. Coli grown in the 15N medium, but still heavier than 14N DNA. Due to the position of this intermediate DNA band in the density gradient, it was apparent that the DNA was a hybrid and contained both 14N and 15N. This automatically eliminated the conservative model of replication, which would have resulted in two distinct bands: one matching the position of the 15N-containing DNA, and one matching the position expected by DNA containing only 14N. Only the dispersive and semi-conservative models fit the situation.
The second generation of E. coli grown in the 14N medium contained two distinct bands. One of the bands was 14N DNA, and the other band was the intermediate (14N/15N) DNA. This result supported the theory of semiconservative replication since dispersive replication would have resulted in a single band of lower density DNA in each consecutive generation.
The figure below illustrates the theoretical outcome of the conservative, dispersive, and semiconservative models along with the experimental outcome obtained by Meselson and Stahl.

Figure: A schematic of the appearance of fractions of DNA samples after centrifuging in a density gradient, by CJHIGGIN
References
[edit | edit source]Berg, Jeremy M., John L. Tymoczko, and Lubert Stryer. "Exploring Genes and Genomes." Biochemistry. New York: W. H. Freeman, 2007. 113-14. Print.
Campbell and Reese's Biology, 7th Edition
Nelson and Cox's Lehninger Principles of Biochemistry, 5th Edition
General Information
[edit | edit source]A knockout mouse is a mouse used by researchers for laboratory experiments aimed at understanding the consequences of inactivation or "knocking out" of a specific gene. In general, the over- and/or under-expression of genes in an organism for experimentation is known as transgenic technology [1]. This process is completed by disrupting or replacing the existing gene with an artificial piece of DNA that is a mutated version of the targeted gene [2]. Due to the disruption, there will be a loss of gene activity, and it will cause changes in the mouse's phenotype. When the mouse's phenotype is affected, the changes in appearance, behavior, and other physical characteristics should be evident in the offspring.
Purpose and Applications
[edit | edit source]As many genes are similar in mice and humans, the extraction or "knocking out" of a particular gene in a mouse can provide evidence to further understand the extent of the function of genes in humans [3]. This usually is manifested by a change in the animal's physical characteristics, behavioral characteristics, or biochemical pathways that regulate the mouse's functions [4]. This laboratory technique has been used in various types of research:
- Cancer Research
- Cystic Fibrosis
- Lung, Heart, Blood, and Parkinson Diseases
- Aging
- Anxiety
- Arthritis
- Diabetes
- Obesity
- Neural Pathway Functions
- Substance Abuse
A specific gene studied from the knockout mouse can also be useful in studying how different recreational drugs affect the animal, which can be used to test therapies for drug abuse in humans [5]. For example, a p53 knockout mouse focuses on a mechanism - p53 - that codes for a protein that inhibits the growth of tumors and stops cell division. By taking out this gene, the mouse is at risk of developing various types of cancer (blood, lung, brain, bone, etc.). This is a useful study because humans with the abnormality in this gene have Li-Fraumeni Syndrome, a rare autosomal dominant hereditary disorder that puts people at a much higher risk of developing cancer.
Limitations and Weaknesses
[edit | edit source]Although knockout mouse technology is an excellent research tool, there are frequent complications that occur when a particular gene is knocked out. For example, the mouse might depend on the gene of interest for other important bodily functions; if it was disrupted, the mouse might die or stop functioning correctly in unexpected ways. In addition, the gene that is knocked out in the mouse may not even produce an observable change in any of the mouse's characteristics. This makes it extremely difficult to correlate the study with that of humans. Gene knockouts in mice embryos may sometimes inhibit the mice from growing into adult mice. This makes the studies limited to the pre-natal stage of the mouse, further distancing the relationship between the gene-knockout in the mouse, and that of humans.
Methods of Preparing Knockout Mice
[edit | edit source]
Knockout mice are created from embryonic stem cell (ES cells) by harvesting them approximately 4 days after fertilization. The reason for using the ES cells so early on is because the swapping of gene sequences can be properly passed on to the rest of the cells during division and develop along with the all the other adult cells. This process is completed in one of two methods:
Gene Targeting
[edit | edit source]In gene targeting, a particular gene is manipulated within the nucleus of the ES cells of the mouse through homologous recombination [6]. To start the homologous recombination, the DNA sequence of the gene that needs to be replaced would need to be known. Next is to make a new DNA sequence that is needed to be inserted into a chromosome. That chromosome is going to take the place of a wild-type allele. The artificial inactive DNA sequence is introduced (this piece is nearly identical in sequence to the knocked out gene). This artificial sequence flanks the DNA sequence in both directions on the chromosome. The cell recognizes the identical stretches of DNA, and "trades" the existing gene with the artificial DNA. Since the artificial DNA is inactive, the function of the existing gene has now been "knocked out" by gene targeting. The new cells will keep growing and dividing with the new gene inside of it.
Example: An embryo from a mice in the blastocyst stage of a species with gray fur is isolated. Then the embryonic stem cells are removed from the blastocyst and put on a tissue culture to be grown. Transfer the homologous recombinant gene and grow them in gancyclvir and neomycin. The cells with the new genes for white fur are then transferred back to the blastocyst. Many of the transformed blastocysts will be implanted into pseudo-pregnant mouse with white fur. The mouse will give birth to some white mice and some with patches of gray, showing the activity of the old gene. The mice with the patches - which means they have both the gray fur and white fur genes - will mate with a white mouse. If the gametes of the gray white mouse were from the recombinant stem cells then it will give birth to all gray mice. All of the cells in the mice are heterozygous for the fur gene. The gray mice will then mate together with the heterozygous mice. Identify which mice has the homozygous recombinant and mate them until both of the alleles are knocked out. The end result is the knock out mouse which is when both of the alleles have been knocked out.[7]
Gene Trapping
[edit | edit source]Gene trapping is done by using a sequence of artificial DNA which holds a "reporter gene" that is made to insert into any gene at random. The artificial DNA prevents RNA splicing in the cell, thus preventing the existing gene from synthesizing its assigned protein and eliminating its function. Now the activity of the artificial "reporter gene" can be observed and studied, to determine the existing gene's normal function in the mouse.
Which method is better?
[edit | edit source]For both of these methods, a DNA vector is used to carry the artificial DNA into the embryonic stem cells of the mice. Once the DNA is injected, the cells are cultured in-vitro, and then injected into mouse embryos. These embryos are given planted into female mice, which then give birth to mice with the knocked out genes.
Both ways have their own advantages. For example, in gene targeting, the target gene is known in the DNA sequence. This method allows researchers to knock out the sequence(s) that they find are interesting. On the other hand, although the specific gene which is knocked out is unknown in gene trapping, it would create different kinds of mice because there is no efficiency or precision in how the "reporter gene" binds; finding the function of specific gene can become cumbersome because of the randomness. The researchers need to spend a lot of time testing the ES cell to identify which gene has been knocked out. Moreover, a certain gene that is not easily chosen may be knocked out in random manner.
Resources
[edit | edit source]References
[edit | edit source]- ↑ "What is transgenic technology". Knockout Mouse and Transgenic Research. Retrieved 2009-11-14.
- ↑ Twyman, Richard. "Knockout Mice". The Human Genome. Retrieved 2009-11-14.
- ↑ "NIH Knockout Mouse Project". National Institute of Health. Retrieved 2009-11-14.
- ↑ "Knockout Mice". National Human Genome Research Institute. Retrieved 2009-11-14.
- ↑ Berg, Jeremy (2006). Biochemistry (6th Ed. ed.). W. H. Freeman. ISBN 0716787245.
{{cite book}}:|edition=has extra text (help) - ↑ Twyman, Richard. "Knockout Mice". The Human Genome. Retrieved 2009-11-14.
- ↑ Campbell, A. Malcom. "Homologous Recombination and Knockout Mouse". Davidson College. Retrieved 2009-11-18.
Transgenic animal are animals that have had foreign genes from another animal introduced into their genome. A foreign gene (such as a hormone or blood protein) is cloned and injected into the nuclei of another animal’s in vitro fertilized egg. Cells are then able to integrate with the transgene, and the foreign gene is expressed, upon which the developing embryo is surgically implanted in a surrogate mother. The result of this process, if the embryo develops, is a transgenic animal housing a particular gene from another species.
Applications of transgenic technology are for example, improving upon livestock, such as higher quality wool in sheep, or increasing the amount of muscle mass of an animal so that it can produce more meat for consumption. Conversely, transgenic animals can also be utilized for medical purposes such as producing human proteins by inserting a desired transgene into the genome of an animal in a manner that causes the target protein to be expressed in the milk of the trangenic animal.
Another example is one that involved mice. Normal mice have the capability to not be infected by the polio virus. They do not have the cell surface molecule that is required as a receptor for polio, unlike humans, who do have this receptor. However, the polio receptor gene can be injected into a mouse, thus developing a transgenic mouse. This allows the mouse to now be successfully infected by the polio virus, and display the similar symptoms that are displayed in humans who are affected by the polio virus.
The most common studies that are currently going on with transgenic animals involve animals, such as the rhesus macaques. These animals contain the human gene of the Huntington’s disease. This allows scientists to research options that can provide a cure to Huntington’s or at least a better treatment option. Other animals, such as mice or those that contain human stem cells, are used to create medicine and treatment options for diabetes, strokes, and blindness.
The human genome project has also been of great help in the role of transgenic animals. With the newfound discovery of the DNA sequence of the human genome, scientists can now study the genes that are involved as drug targets, which can help provide them with the ability to mark the appropriate gene that can aid in providing the cure to any certain disease that they are studying.
The expression of a transgene can also be engineered to take place in plants, such as obtaining the bio-luminescent gene that gives fireflies their glow in the dark ability, and introducing it to a plant.
Transgenic Animals Countless Benefits to Humanity
[edit | edit source]Three of the most widely-used reasons for producing transgenic animals for the benefit of human welfare are agriculture, medical and industrial.
Agricultural Applications
[edit | edit source]Farmers have always wanted to have the best breed for any type of animal and to have the best traits that it can possible have. The normal way of breeding animals can potentially take up a lot of time and is not entirely efficient. With new advances in technology, selected characteristics can be developed in species with a lot less time and more accuracy.
Not only are animals produced more efficiently, the quality of the animals are enhanced as well. Some examples include having cows create milk with lessened milk content and sheep that produced a lot more wool.
Also, with these new qualities in animals they must be protected. Scientists are researching on creating animals that are resistant to particular diseases and to enhance the two reasons stated above.
Medical Applications
[edit | edit source]Animals that have their genes modified to show disease symptoms, may be studied and cure could possibly be contrived in the near future.At Harvard, scientists created a transgenic mouse also known as OncoMouse® or also known as the Harvard mouse which allows it to carry genes that can enhance the development of a variety of cancers that are found in humans.
Xenotransplantation will play a major role in the medical industry in the future. It is the transplantation of living cells, tissues or organs from one species to another. Due to worldwide shortages of organs, advances in gene manipulation of animals can alter their organs to become susceptible to humans. For example, Transgenic pigs may play a major role in the transplant of organs to humans. Because Pig and human organs are closely related, there is a possibility to use pig organs for transplants. However currently, a pig protein inhibits the human body’s immune system acceptance of the organs. If animals such as Pigs can have its protein successfully supplanted by a human protein can be used to meet a major need- transplant organs such as the hearts, liver, or kidneys. It can also be applied to bringing about refined drugs in the pharmacy industry and nutritional supplements. An example is insulin and anti-clotting factors of blood can soon be extracted from milk of transgenic animals such as goats, sheep, cows. This milk being the source of importance is undergoing major research to create a type that will be able to treat diseases such as phenylketonuria or cystic fibrosis.
Human gene therapy is another medical application that is gaining wide acclaim. In essence, it is the transfer of genetic information into patient tissues and organs. As a result, diseased defective copies of genes can be eliminated or their normal functions rescued. Moreover, the procedure can provide new functions to cells. For Example, to combat cancer and other diseases, the insertion of a gene that causes the production of immune system mediator proteins can be introduced. By this therapy, countless genetic disease could have potential cures further down the road. There are two paths to Gene therapy. The first path is direct transfer of genes into the patient. The second path is the use of living cells as vehicles to transport the genes of interest. These two paths both have certain advantages and disadvantages. Direct gene transfer is the most simplistic way of administering the gene of choice. There are two methods to direct gene transfer. The first method is the process in which genes are delivered via liposomes or other biological microparticles into patient’s tissue or bloodstream. The second method of the introduction of genes is using genetically-engineered viruses, such as retroviruses or adenoviruses. However, due to biological safety concerns, viruses must first be altered so they are not infections before introduction. However, due to the simplicity of the direct gene transfer method, there are major weaknesses. For example, it does not allow for the control of where the therapeutic gene will insert. The transferred gene will either randomly insert itself into the patient’s chromosomes or remain unintegrated in the targeted tissue. Moreover, the target tissue may not be easily accessible for direct gene application of the therapeutic gene. The second method of gene therapy is the use of living cells to deliver the therapeutic gene. This method is very complex compared to the direct gene transfer method. There are three major steps to this method. The first step is cells from the patient are isolated and propagated in vitro. The second step is the introduction of the therapeutic gene into these cells using methods similar to the direct gene transfer. The last step is the genetically modified cells are returned to the patient. The advantage of using gene transfer vehicles is, in the laboratory cells can be manipulated more accurately and precisely than in the body. In addition, some of these cells are able to continually propagate under laboratory conditions before reintroduction into the patient. Moreover, some of these cells types have the ability to localize to particular regions of the human body, for example, hematopoietic (blood-forming) stem cells can return to the bone marrow upon reintroduction in to the body. This action can be very useful for applying the therapeutic gene which has regional specificity. However, a major disadvantage is the biological complexity of the living cell’s environment. The isolation of a specific type of cell requires not only extensive information of its biological markers, but also knowledge of the requirements for that cell type to stay alive in vitro and continue to divide. Unfortunately, there are many cells types with unknown information to their specific biological markers. Moreover, many normal human cells cannot be sustained in the lab for long periods of time without amassing deleterious mutations.
Industrial Applications
[edit | edit source]Animals that have transgenes have been produced to for testing on chemical safety as these animals are sensitive to toxic things. Also, these transgenic animals may produce something that can be utilized in biochemical reactions. Microorganisms have been structured to be able to produce enzymes that can make major reactions speed up.
Production of Transgenic Animals
[edit | edit source]The production of transgenic animals is taking the genome, the genetic makeup of the organism and introducing foreign genes into that organism. These insertions of genes are known as transgenes. Most importantly, these foreign genes must be transmitted through the germ line of the organisms. As a result, every cell, including the germ cells, whose function is to transmit genes to the organism’s offspring, contains the same change in genetic material.The predominant method of creating these transgenic animals is the use of DNA Microinjection. However, producing these type of transgenic animals is hardly deemed a success as DNA insertion is arbitrary and success rate very low. The offspring is what's studied for this new transgenic gene. But the ability to produce these type of offspring that is successfully carrying the gene is extremely difficult.
Scientists may produce transgenic animals is three main ways: DNA microinjection, retrovirus-mediated gene transfer and embryonic stem cell mediated gene transfer.
DNA microinjection
[edit | edit source]Technique summary
[edit | edit source]The first animals to be experimented with DNA microinjection was the mouse. DNA Microinjection is the transfer of a desired gene into the pronucleus of the reproductive cell. This cell is first cultured in vitro. Then reaching to a specific stage or threshold of the embryonic phase, it is then transmitted into the recipient female.
Technical Explanation
[edit | edit source]The pipets for this technique must be created especially from glass that are extremely thing and a pipet puller as well as a micro-forge. It must be absolutely flat at the tip or there will be impedance when injecting into the embryo. The specification of the DNA injection pipet should have an internal diameter of about 1 µm or even less. When performing this technique gloves that are covered with talc should be avoided as the power has the potential to clog the pipets and could lead to the failure of the embryos. The embryo that is working with should be put in very low magnification. Using the pipet, with ease suction the embryo into the end and let it stay there. The tip of the pipet is brought to exactly where the pronucleus is and then it is punctured through the cell membrane and into the cytoplasm area. It is often hard to see if the pipet tip has gone through into the pronuclear membrane. The only safe bet in judging if it was transferred successfully is to glance at the pronucleus to see if it swells up and its size in volume amounts to around 1pl. After injection it is then moved to the far end of the dish so that the next one may be done as well. When a bundle of embryos are complete, it is left for incubation and then evaluation for a duration of time. The embryos that are viable will then be transmitted to a female's oviduct and then utilized.
Retrovirus-mediated gene transfer
[edit | edit source]Retroviruses are used as vectors to transfer genetic material in the form of RNA rather than DNA. It is the transfer of genetic material into the host cell, resulting in a chimera, a organism that has various genes aside from its own. These chimeras are inbred for as many as twenty generation until homozygous offspring are formed, carrying two copies of the same transgene in all of its cells. This has been proven successful in 1974, when a virus was used as a vector into embryos of mice. They showed the desired transgene.
Embryonic stem cell mediated gene transfer
[edit | edit source]The technique involves isolation of the totipotent stem cells from embryos(stemcells that can develop into any type of specialized cell). The desired gene is inserted/transfer into the stem cell. These stem cells containing the desired DNA of interest are now incorporated into the host's embryo. Thus resulting in a chimeric animal. A major benefit of this technique is that it may test the transgenes on the molecular level, which essentially saves ample time and using this technique one would not have to wait for living offspring.
Stem Cells (in Further Detail)
[edit | edit source]What exactly are stem cells?
Stems cells are now a hot topic for research because of their seemingly endless potential. They are cells that may develop into numerous different categories of cells in the body during the beginning stages of life and also during the growth stage. Stems cells can also be utilized as an internal repair system, basically dividing incessantly to restock the cells under damage and repair until it reaches back to equilibrium and for the duration of the organism's life span. As stem cells divide, each has the opportunity to choose between sticking as a stem cell or become more specific- one with a specialized function, examples including liver cell, a white blood cell, brain cell, etc.
How are stem cells set apart from other types of cell?
There are two primary properties that are used to do this. The first aspect is that stem cells are initially unspecialized cells and may regenerate through cell division, and even at times after prolong periods of time without activity. Another aspect is that under right, specific physiological conditions can be promoted to turn into either tissue or certain cells of organs and with distinctive abilities. Examples of when stem cells are maximized for their repair function are in organs, bone marrow, gut marrow, where they constantly divide to restore injured cells or ones that have been heavily used.
In the past, researchers mainly worked with two categories of stem cells which were from both humans and animals. The two that were worked on were embryonic stem cells and somatic stem cells, which can also be called adult stem cells. The first embryo cells came from mouse as described above, which occurred around 1981. The human embryonic stem cells were made for reproduction and was made possible through the intense research done with the mouse embryos. Recently, there has been a third category of stem cells known as induced pluripotent stem cells(iPSCs). These cells are unique because they are cells of adults that can be reconditioned by gene modifications to be a stem cell.
Why are stem cells valuable for living organisms?
Typically in blastocysts, which are embryos of only 3–5 days aged, the cells on the inside will turn into the cells for all of the body of that living organism, even specialized cells and organs including the skin, heart, lung, reproductive cells-sperm and egg, and different tissues. Within the tissues of adults including bone marrow and muscle, these stem cells have the ability to replace the cells that are damaged, affected by disease, or simply just used.
Research in the stem cell arena, has continued to add new insights to the development of organisms from cells and the repairing mechanism of affected cells. Stem cells may also be utilized to help select for new drugs to be brought to market and better understand not only cell developments but also the irregularities that induce defects in the infants of organisms.
Special characteristics of stem cells
Stem Cells are very unique and set apart from other cells of the body. All types of stem cells will have 3 defining characteristics- able to divide and replenish themselves for long duration of time, not specialized, able to be turned into numerous different types of cell types. For each of these properties, further depth analysis will be explained below.
The first property discussed was stem cells ability to divide and replenish themselves for a long duration of time. Typically cells of muscle or nerve do not duplicate by themselves, but stem cells have the ability to do this and also done ample times. Stem cells replicated countless times in the laboratory times for months at a time may result in millions of cells. If the cells can go on for a long time and not be specialized just like the parents, these cells are able to perform self-renewal for the longterm. Two sources of profound interest under study about this self-renewal for the longterm is how embryonic stem cells can replicate for an entire year in the laboratory and not differentiate but usually non-embryonic stem cells are not capable of this and which aspects of organisms are the ones that are source of regulation of stem cell replication and this self-renewal.
Finding out how the regulation of stem cells is performed for stem cells normal development may assist in finding out the reasoning for cancer through irregular cell division. This could also lead to more efficient growth embryonic and non-embryonic stem cells performed in the laboratory setting. Having stem cells that continue to stay as unspecialized result from special conditions. These special conditions are set up from signals in the cells that induce the stem cells to replicated and stay as unspecialized.
Stem cells are ones that are not specialized. Since they are not specialized, they are incapable of doing any specialized tasks that could occur in specific tissues or organs. As a result, stem cells cannot work collaboratively with other cells to perform organized tasks such as being a carrier of oxygen molecules throughout the body such as red blood cells. But what is unique about stem cells is their potential to be made into specialized cells such as nerve cells, brain cells, or muscle cells.
Stem cells have the capability to be made into specialized cells. The progression of stem cells that are not specialized being turned into ones that are specialized is known as differentiation. The differentiation process have multiple steps and the progression through these steps increases specialization. Many factors help to control this progression. Signals that both inside and outside of the cell help promote the stem cells through each stage. Outside signals include being in close, touching proximity of nearby cells, chemicals that are given off by other cells, and the presence of specific molecules in the immediate environment. Inside signals are managed by genes present on the DNA that tell it exactly what to do. Understanding the regulation of these stem cells can help to grow cells or even tissues to help in selecting for drugs and cell therapy, which is what makes stem cells so special and a primary source of research.
Different types of stem cells
Embryonic stem cells
These type of cells come from embryos. A major portion of these types of cells come from eggs that are fertilized in vitro or in the laboratory setting and then given to labs so that research may be done on them. The embryos from the human stem cells are usually about 4–5 days aged and are in the blastocysts form, which essentially is a hollow ball of cells. The blastocysts have a total of three structures including the trophoblast, embryoblast or pluriblast, and blastocoel. The trophoblast is a layer that surrounds the blastocoel. The hollow cavity of the blastocyst is the blastocoel and the embryoblast is mass of cells that will turn into defined structures of the fetus.
How are embryonic stem cells identified?
While creating embryonic stem cells, there are various checkpoints to test if the cells have the right properties that allow it to be called embryonic stem cells. This is also known as characterization. There is not a universal test agreed to always be used to mark embryonic stem cells but there are various tests that can be used. The first one that can be used is to grow these stem cells for a number of months. This proves that the cell can do long-term growth and self-renewal. The cells are put under a microscope and observed to see that it is in good condition and still have not differentiated. A second test that is to determine transcription factors that are characteristic of cells that are not differentiated. Specific transcription factors to look for are Nanog and Oct4. Essentially what transcription factors do is aid in turning genes either off or on when needed, which is very integral in cell differentiation and development of embryos. Nanog and Oct4 help to keep the stem cells to be undifferentiated. A third test is to use specific techniques to look for cell surface markers that undifferentiated stem cells will give off. A fourth test is to look at the chromosomes using a microscope and to diagnose if there is damage or if the quantity of chromosomes is different. A 5th test is to see if the cells can be grown again after putting it in the freezer and then allowing it to thaw. The last test which is the 6th one is to test if these human embryonic stem cells are pluripotent. This may be done by permitting the cells to instinctively differentiate in the laboratory, conducting the cells so that it will form a cell that consists of three germ layers, or injecting the cells into a mouse that has an impaired immune system to test for the development of teratoma, a tumor that is benign. The growth of the injected stem cells and its differentiation may be observed since the immune system of the mouse does not reject it. Encompassed in the tumor cells is a combination of differentiated or somewhat differentiated kind of cells, showing that embryonic stem cells have the ability to differentiate into other different types of cells.
How does differentiation of embryonic stem cells occur?
When embryonic stem cells are kept under the right conditions, they can be kept in the unspecialized state. When cells are permitted to aggregate and form what is called embryoid bodies, spontaneously differentiation occurs. These cells are able to form numerous different types of cells. This does show that this sample of embryonic stem cells is good condition, however this method is not efficient in creating certain cell types.

In order to generate cultures of specific types of differentiated cells such as blood cells or brain cells, is done by controlling the differentiation of these embryonic stem cells. Components to modify are the different chemicals the culture medium is made of, the surface of the culture dish, or even the cell themselves by giving them specific genes. After a long time of trial and error there have been some standard protocols established for this directed differentiation to certain cell types to occur. If this directed differentiation of embryonic stem cells is done successfully, they can be used to treat certain diseases which include Parkinson's disease, Duchenne's muscular dystrophy, heart disease, vision loss and traumatic spinal cord injury.
Adult stem cells
Adult stem cells are thought to be undifferentiated type of cells, located with differentiated cells either in a tissues or organs that can revitalize itself and may differentiate to give either some or all of the primary specialized types of cells of an organ or tissue. The main job of adult stem cells in organism are to sustain and restore the tissues where they are located. Unparalleled to embryonic stem cells that are named according to location in which they are found, the stock of some adult stem cells in some tissues that are already mature are still being researched.
As more research is being conducted on adult stem cells, their presence is being found in many additional areas of tissue than ever before. This has opened up the possibility of these adult stem cells to be used as transplants. A widespread use of adult stem cells as transplants are for hematopoietic stem cells from bone marrow, which is blood-forming. It is now evident that stem cells do exist in the heart and the brain. The control of differentiation of these stems cells if done correctly it may be feasible to use them for transplantation therapy treatments.
Adult stem cells were first discovered in bone marrow, which contained two versions: hematopoietic stem cells and bone marrow stromal stem cells, which were discovered second. The stromal cells were small in number but had the ability to make everything including fat, bone, cartilage, and fibrous connective tissue.
Location of adult stem cells and their role?
Adult stem cells are actually located in numerous different organs and tissues which include bone marrow, brain, blood vessels, skin, teeth, heart liver, epithelium part of ovarian, and testis. Within each tissue, stem cells live in a particular area. In a lot of tissues, some stem cells comprise the outside layer of small blood vessel known as pericytes. Stem cells usually do not divid for long durations of time until prompted to for normal maintenance of tissues, after injury, or by disease.
Normally the number of stem cells in each tissue is small and once taken away from the body, their ability to divide becomes limited and duplicating large amounts of stem cells difficult. As a result, researchers are looking for improved ways to grow large quantities of adult stem cells in the laboratory so that specific ones may be created to target and treat diseases and injuries. Uses include to recreate bone from cells located in the bone marrow stroma, making cells that produce insulin to help treat diabetes of type1, and to rejuvenate heart muscles that were greatly impaired after a heart attack event.
Identification of adult stem cells
There are many methods to identifying stem cells. Researchers typically use several methods to identify the adult stem cells. One way it occurs is to tag the cells that are in living tissue with molecular markers and then look to see the produced specialized cell types. Another useful method would be to take the cells from a living organism, tag them in the laboratory and reinsert them into another organism to observe whether or not the cells recreate cells at their original tissue location.
One of the primary things that must be exhibited is that one adult stem cell will be able to produce an entire colony of genetically identical cells that can also create the correct differentiated cell types of that particular tissue. To produce these results experimentally and confirm that the cells are indeed adult stem cells is done through showing that it can create genetically identical cells or that the cells can remake the tissues after inserted into another animal or both of these.
Adult cell differentiation

Normal differentiation
Adult stem cells are free to divide when called and can produce mature cells that have the same shapes, structure, role of that tissue in which it resides. Examples of this will follow. Hematopoietic stem cells will produce any type of blood cells including the b lymphocytes, T lymphocytes, natural killer cells, basophils, monocytes, red blood cells, etc. Mesenchymal stem cells actually produce a whole variety of cell types including bone cells, fat cells, cartilage cells, etc. Neural stem cells of the brain may produce neuron, astroyctyes, and oligodendrocytes. In the lining of digestive tract reside epithelial stem cells and they produce cells including goblet cells, enteroendocrine cells, absorptive cells, etc. The stem cells of the skin reside in the basal layer of the epidermis and produces keratinocytes, that provide the security layer.
Transdifferentiation
Particular adult stem cells can differentiate into other types of cells of other organs or tissues than it's predicted type, such as heart muscle cells differentiating into brain cells. This type of differentiation is better known as transdifferentiation. This occurrence in human beings is still not fully proven. Some possible explanations for this type of differentiation being observed could be the junction of this donor cell with the recipient. Another explanation could be that these injected stem cells give off factors that promote that other organism's own stem cells to initiate the repair mechanisms. When transdifferentiation has been observed, it is only seen in small instances.
Scientists have proved that some adult cells can be remade into different cell types in the laboratory using precise gene alterations. This can prove to be a way to remake cells into the other ones that have been injured or eliminated because of diseases. In diabetes, the cells that produce insulin or beta pancreatic cells can be recreated by reprogramming other cells in the pancreas. These recreated cells were very close i appearance and shape to the actual beta pancreatic cells. These reprogrammed cells when put into mice did improve the regulation of the sugar levels in the blood even though the mice had nonworking pancreatic beta cells.
Adult somatic cells can be reprogrammed to mimic embryonic stem cells through the presence of genes of embryos, and these types of cells are known as induced pluripotent stem cells iPSCs. Through iPSCs cells can be introduced that receptive by the donor and will not be rejected, which is important when recreating new tissue. However, iPSCs are still under study until they can produced to entirely only stick to its designated cell type.
Similarity among stem cells
Both human embryonic and adult stem cells have similarities and its differences in relation to using for regenerative therapy or repairing already damaged tissue and cells. A primary difference between adult stem cells and embryonic is the amount of different abilities that each is capable and the specific kind of differentiated cell types they will turn into. Embryonic stem cells can actually turn into all the different type of cells in the body because of their pluripotent nature. Adult stem cells are very specific and so limited to only differentiating into the type of cells of their original tissue.
A noteworthy difference is that embryonic stem cells can be grown with great ease in the laboratory. Looking within mature tissues, the adult stem cells are limited in number so finding these cells may be difficult. Unlike embryonic stem cells, adult stem cells still do not have a way to be grown in the laboratory. This difference has a great impact as replacing cell mechanisms oftentimes requires an abundance of cells in order to work properly.
Moreover, the tissues created from either embryonic or adult stem cells may be different in probability of rejection rate post-injection or transplantation. Embryonic stem cells have not been researched too heavily yet as testing using cells from hESCs were only just now approved by the FDA(Food and Drug Administration). The adult stem cells and tissues that form as a result are presumed to be less probable to rejection post-transplantation. The success can be attributed to using patient's self cells to be duplicated in the laboratory and then induced to differentiate into a specific cell kind and then re-injected into that same very patient. Utilizing the adult stem cells and the tissue products from the patient's very own cells highly decreases the probably of rejection by the immune system. This proves to be a major benefit since only using immunosuppressive drugs can help fix this problem but then the drugs have side effects that come along as well.
Uses for stem cells

There are many uses for stem cells, especially in research and in clinic. Studying human embryonic stems cells will help give information about development of humans. The principal target is to pinpoint how undifferentiated stem cells become differentiated cells and then later to form organs and tissues. Gene regulation is imperative in this aspect. A lot of the most irregular activity in humans result from aberrant erroneous cell division and differentiation. New research has found that iPS cells show that specific factors are associated with genetic signaling and molecular signaling and introducing these into the cells in a proper manner to command these processes will need a special technique.
Stem cells of humans may be used to select for new drugs. These drugs can be tested to see that it is not damaging using these differentiated cells. A vivid example would be to use cancerous cells to select for drugs that could be anti-tumor. Environment of the drugs should be very similar in order to check if the drugs actually work and this can be done through having a precise command over where the differentiation of stem cells turn into.
Another widespread use of stem cells is to utilize them to create cells and tissues to repair damaged or disease tissue in cell therapy. These regeneration of cells and tissues can aid in treating disease such as Alzheimer's disease, stroke, heart disease, osteoarthritis, and spinal cord injury.
Checklist for successful transplant of stem cells
1) Duplicate in mass amounts and be able to produce enough quantities of tissue
2) Differentiate into wanted type of cells
3) Live to survive in recipient post-transplantation
4) Become integrated into the tissue in the proximity post-transplantation
5) For entire duration of organism's life- be able to correctly function
6) No detrimental effects on recipient
Ethical conflicts with stem cells?
The main concern with stem cells has to do with the human embryonic stem cells, which has created a lot of public interest and conflict. Stem cells that are pluripotent, or may become numerous different types of cells in the human body are created from human embryos that are some days aged. The major debate is of when does life technically commence and if embryos or even fetuses would be considered as such and also who has the power to decide on such an issue.
United States' position on stem cells
The Bush administration in 2001 offered federal funds for research on human embryonic stem cells if certain three criteria were met. However, President Barack Obama issued an Executive Order 13505 known as Removing Barriers to Responsible Scientific Research Involving Human Stem Cells on the 9th of March 2009. This allowed National Institutes of Health or NIH to take a different strategy on doing human stem cell research. Also this Executive Order essentially nullified both the Executive Order 13435 and the presidential statement that occurred on August 9, 2001.
Resources
[edit | edit source]Arlan Richardson. Photo. "Use of Transgenic mice in Aging Research.” 1997 <http://dels.nas.edu/ilar_n/ilarjournal/38_3/38_3Richardsonfig1.jpg>
Gordon, Jon W. Photo. "Transgenic Technology and Laboratory Animal Science." 1997. <http://dels.nas.edu/ilar_n/ilarjournal/38_1/38_1Gordonfig3.jpg>
Gordon, Jon W. Photo. "Transgenic Technology and Laboratory Animal Science." 1997. <http://dels.nas.edu/ilar_n/ilarjournal/38_1/38_1Gordonfig2.jpg>
"2009 Executive Order Disposition Tables: Removing Barriers to Responsible Scientific Research Involving Human Stem Cells." <http://edocket.access.gpo.gov/2009/pdf/E9-5441.pdf>. 11 March 2009. 2 December 2009.
Margawati, Endang Tri. "Transgenic Animals: Their Benefits To Human Welfare." ActionBioscience. Jan 2003. 15 Nov 2009 <http://www.actionbioscience.org/biotech/margawati.html?print>
"Stem Cell Basics." In Stem Cell Information. Bethesda, MD: National Institutes of Health, U.S. Department of Health and Human Services, 2009. <http://stemcells.nih.gov/info/basics/defaultpage>. 3 December 2009.
"Transgenic Animals and Genetic Research."<http://www.cartage.org.lb/en/themes/Sciences/Zoology/AnimalPathology/TransgenicAnimals/TransgenicAnimals.htm>. 16 Nov 2009.
"What are Some Issues in Stem Cell Research."<http://learn.genetics.utah.edu/content/tech/stemcells/scissues/>. 9 November 2009. 3 December 2009.
Winslow, Terese. Photo. stemcells.nih.gov 2001. 2 Dec. 2009. <http://stemcells.nih.gov/staticresources/images/figure1_lg.jpg>
Winslow, Terese. Photo. stemcells.nih.gov 2001. 2 Dec. 2009. <http://stemcells.nih.gov/staticresources/images/figure2_lg.jpg>
Winslow, Terese. Photo. stemcells.nih.gov 2001. 2 Dec. 2009. <http://stemcells.nih.gov/staticresources/images/figure4_lg.jpg>
Zwaka Thomas P. “Use of Genetically Modified Stem Cells in Experimental Gene Therapies.” < http://stemcells.nih.gov/info/2006report/2006Chapter4.htm>
Zwaka Thomas P. Photo. “Use of Genetically Modified Stem Cells in Experimental Gene Therapies.” < http://stemcells.nih.gov/StaticResources/info/scireport/images/4_3.jpg> Transgenic plants are genetically engineered to have genes from other organisms inserted into their genome. Transgenic plants are identified as a class of genetic modified organisms (GMO). The introduced genes do not have to be from the plant kingdom, but can come from animals, viruses, or bacteria as well. The uses of exogenous gene introduction include virus immunity, a replacement for pesticides, the ability to grow in acidic soil, and greater nutritional content.
Making Transgenic Plants
[edit | edit source]Transgenic plants are constructed by inserting genes from other organisms into the host plant's DNA sequence. For this to happen a desired gene must be isolated and cloned. A few changes must be made to the gene so that it can effectively be inserted into the plant. First, a promoter sequence must be added to the gene. The promoter sequence is an on/off switch that controls where and under what cues the gene is expressed. The gene must also sometimes be modified (e.g. The Bt gene for insect resistance has a greater amount of A-T nucleotide pairs than plants, which tend to have more C-T pairs. The A-T nucleotides can be substituted for with C-T pairs in a manner that does not significantly change the amino acid sequence, leading to greater protection of the inserted gene in plant cells.). A terminal sequence must also be added to signal when the end of the gene sequence has been reached. Finally, a selective marker gene must be inserted to identify plant cells which have successfully integrated the transgene.
Agrobacterium System
[edit | edit source]A method that is used to transform plants is the Agrobacterium method and the "Gene Gun" method. The Agrobacterium method uses Agrobacterium tumefaciens, a soil-dwelling bacterium that has the ability to infect plant cells by introducing transfer DNA, or T-DNA of a tumor-inducing (Ti) plasmid (i.e. a DNA sequence that can replicate independently of chromosomal DNA and is often circular) to the host's nuclear DNA. The bacteria is part of the rhizobiaceae family which is responsible for many tumors found in plants. The Ti plasmid contains the T-DNA as well as a series of vir (virulence) genes that direct the infection process. Agrobacterium tumefaciens can be used as a vector for gene transfer into plants. First, a hybrid plasmid that carries only the T-DNA from a Ti plasmid is cut open with a restriction enzyme and a foreign gene is inserted, creating a recombinant plasmid. The recombinant plasmid is then transferred into an Agrobacterium tumefaciens cell that contains a Ti plasmid that has had its T-DNA removed. The Agrobacterium with the engineered plasmid is then used to infect a plant and integrates the T-DNA with the foreign gene into the plant genome. For the Agrobacterium to be used the DNA must be able to penetrate into the plant cells. This is often done with electroporation, where brief high-voltage electrical pulses are administered to naked protoplasts (i.e. plant tissues and DNA). The electrical pulses open the pores in the plasma membrane allowing the DNA to enter the protoplast (which can then be grown into a mature plant by treating it with hormones). In the "Gene Gun" method, gold or tungsten microspheres (about 1 micrometer in diameter) are coated with the DNA or RNA from the specific gene of interest. The microspheres are then accelerated into undifferentiated target cells in a petri dish. Once inside the cells, the gene from the DNA coating the microsphere is released and can be incorporated into the host plant genome. The advantage of this method is that a high percentage of a single copy of T-DNA can used to transform the plant. In addition, they are an abundant of vector system available to carry out this method.
Biolistic Method
[edit | edit source]This method delivers microprojectiles that are coated with DNA by accelerating it into the cell of interest. The microprojectiles are usually made up of tungsten or gold. To carry out the acceleration, an explosion is made with gunpowder under high pressure of helium. Plants that are made using the boilistic method have multiple copies of a gene that is still able to segregate in a Mendelian pattern. This method helps increase the diversity seen in plants. There are some advantages to the biolistic method compared to the Agrobacterium method. The plants that undergo the bombardment of genes in this method are still fertile. Other advantages includes this is the only reliable method to transform the chloroplast and this method does not need any transformation vector.
Importance of Transgenic Plants
[edit | edit source]The new methods developed to transform plants have opened a new field of interest. Transgenic plants are used to solve a lot of problems in the agriculture sector. In addition, transgenic plants can be used in the medical field
Nutrients of Transgenic Plants
[edit | edit source]When people go to the supermarket, they often buy fruits that are not soft or overly ripened. The major problem in the agriculture field with fruits is that the fruits often become soft during processing and transporting because they are being ripened. Using one of the methods for creating transgenic plants, scientists are able to slow down the process of ripening. Three companies have been able to apply this technology to slow down the ripening of tomatoes. And now other companies are hoping to be able to do the same for other fruits such as mangos or papayas. Cereal grains and legume seeds are a big source of protein for many people. However, the cereal grains and legumes seed often lack certain amino acids such lysine in cereal grains and methionine in legume seeds. Many efforts have been put into creating seeds that are higher in nutritional values. Currently, transgenic tobacco and canola seeds have a 33% increase in methionine due to the transgenic technology. In addition, the nutritional values have potatoes have increased by transforming it with AmA1, a gene from amaranth.
Increasing the nutritional values in plants and fruits can address many malnutrition problems and diseases. Vitamin A deficiency is a huge problem in Asia that affects around 124 million children and causes blindness. The main staple in Asia is rice, but rice does not contain any vitamin A. Researches are being performed in hope of developing rice that is rich in vitamin A. Currently, scientist have found the genes that encode for B-carotene (pro-vitamin A) enzymes in the endosperm of transgenic rice seed and they hope to use this information to engineer rice in a way that vitamin A can be produce through the rice.
Uses of Transgenic Crops
[edit | edit source]The use of transgenic plants for pathogen resistance has received the most attention from popular media. The use of GMOs has been a topic of debate since their introduction in the mid-1990s. The two best known cases were virus-immunity in papayas and insect immunity in crops such as corn through a gene from Bacillus thuringiensis (BT). The papaya ringspot virus (PRSV) that severely damages papaya trees was causing a major toll on the papaya industry in Hawaii. Genes for the protein coat of the virus were inserted into papaya tissue by using the gene gun. Some of the papaya cells incorporated the viral genes into their DNA, giving the plant immunity to PRSV. This saved the Hawaiian papaya industry. The introduction and use of BT crops is even more publicized. The BT gene codes for the Cry proteins which are toxic to and that specifically target and kill the larvae of butterflies and moths. By introducing this into plants, crops such as corn, rice, and potatoes were able to exhibit the Cry proteins, and have proved to be very effective at stopping insect pests such as the European corn borer caterpillar. The protein is very selective and does not harm other insects (e.g. beetles, flies, bees, wasps) and is also considered safe for human consumption. The use of the BT endotoxic has greatly reduced the use of pesticides on crops. However, issues concerning immunity of the pests to the BT corn are a problem, and refuge crops that do not contain the toxin are planted to reduce the evolution of the caterpillar immunity to the Cry proteins.
GMOs have also been bred to improve food nutritional quality, to induce a longer shelf-life by delaying senescence, to allow corn to grow in acidic soil, to protect strawberries from cold temperatures, and a variety of other uses.
References
[edit | edit source]Bessin, Ric. Bt-Corn: "What It Is And How It Works". University of Kentucky College of Agriculture. January 2004.
http://www.ca.uky.edu/entomology/entfacts/ef130.asp
Transgenic Crops: An Introduction and Resource Guide. Colorado State University Soil and Crop Sciences. March 2006.
http://cls.casa.colostate.edu/transgeniccrops/index.html
Lipps G (editor). (2008). Plasmids: Current Research and Future Trends. Caister Academic Press. Raven, Peter. "Biology of Plants". W.H. Freeman and Company. New York. 2005. "Harvest of Fear" (Film) - Nova. PBS. 2004
Peña, Leandro. Transgenic Plants: Methods and Protocols. Totowa, NJ: Humana, 2005. Print. Structural Biochemistry/The Hypochromic effect Because DNA contains all of the heredity information and the instruction for protein production, it is crucial that there be very few changes to the DNA. DNA is constantly bombarded by radiation and chemical mutagens that can cause mutation. However the rate of mutation is very low because of the four main type of DNA repairs.
DNA Injury Detection and Signaling
[edit | edit source]The human genome is under constant toxic stress from normal cellular conditions such as free radicals or errors in DNA replication, as well as extrinsic conditions such as UV radiation. To combat these stresses and properly maintain the genome, the DDR pathway, or DNA damage response pathway has evolved. This pathway serves to detect errors or abnormalities, propagate the detection signal, and activate systems to correct the issue. If the damage is irreparable, the cell undergoes apoptosis, or programmed cell death, to avoid passing on the potentially lethal errors in DNA. Cells come across DNA damage constantly, so the DDR pathway is vital to cell survival.
The most lethal form of DNA damage comes from ionizing radiation which causes breaks in the double stand. The repair protein RAD51 quickly collects into foci at sites of DNA damage. It is suggested that damaged induced phosphorylation of the histone variant H2AX indicates the sites of DNA breaks; many other repair proteins also collect at these sites of H2AX accumulation. In mice lacking H2AX, immune system degradation and increased incidence of tumors are found.
The major regulators of cellular response to DNA damage are ATM and ATR kinases (ataxia telangiectasia mutated) through the regulation of phosphorylation of over 700 proteins. This phosphorylation is the initial step in the signaling of DNA damage.
“Structural Dynamics in DNA damage signaling and repair” was an article written by JJ Perry, Elizabeth Cotner-Gohara, Tom Ellenberger, and John A. Tainer. In this article, DNA damage responses are studied in aspects that reveal the role of protein in such pathways. DNA is continually damaged by metabolites and toxicants. Thus, DNA repair and damage response are essential in the function of life. There are three steps in which DNA damage is involved. The damage is first detected, removed, then eventually replaced with the correct DNA sequence. The pathway regenerates a 3’ terminal that will be extended using DNA polymerase with an undamaged strand as the template. The repair is completed with a ligase resealing the DNA backbone. Because this process of repair generates toxic intermediates, strong “genetic selection” is required as the DNA is being restored. Proteins structures are found to be connected to the coordination of steps within the DNA damage response and repair pathways. This is very important because proteins are once again, related in the DNA replication process.
When different methods come together, the dynamics of DNA repair complexes can be studied in great details. Such methods involve X-ray crystallography, NMR, SAXS – small-angle X-ray scattering, DXMS – hydrogen-deuterium exchange mass spectrometry, etc. These methods provide information as small as from the nanoscale to atomic level. For instance, SAXS gives information on the flexibility of macromolecules in solutions. It also provides information on the entire pathways and their interactions in solution. In addition, DXMS shows more on the conformation changes that take place during the repairing process as detailed as the resolution of a single amino acid. Thus, combining different structural biochemistry methods helps scientist in discovering the different coordination’s between DNA repair and damage response system. Current studies found that the “Transition between different enzyme conformations can involve non-native interactions that lower the energy barrier for inter-conversion between different states” (1). This discovery is very important because it describes the connections between the changes in the DNA repair complex (conformation changes) and the biological outcomes occurred through such changes. For instance, as stated, the changes in enzyme conformation cause the lower of activation energy for the conversion between different states during the process of restoring damaged DNA. Another example is that changing the normal protein flexibility and the stability of the repair protein system can cause great genetic diseases. Changes in DNA and ATP binding are found to be related to cancer as well as how the defects in the flexibility and stability of DNA repair framework are related to aging disorders such as Cockayne Syndrome or TTD.
The damage repair is carried out by the multi-domain nucleotide excision repair helicase (NER). This enzyme removes bulky and distorted cut from one strand of the DNA needed to be repaired. This is a very precise process where only the defected strand is removed without affecting the undamaged DNA strand because the undamaged DNA strand serves as the template for the modification and repairing process. The NER proteins are assembled in a way that allows for the verification of the damaged site before the actual removal of the DNA backbone. One example of DNA repairing process is on the performance of Yeast Rad4, a multi-domain protein that binds to the distorted part of the helix being repaired by NER. The binding of the protein is showed to stabilize the distorted DNA structure. Observations show that Rad4 inserts a beta hairpin through the DNA helix to relocate its bases. One surprising discovery was that instead of binding to the damaged DNA strand, Rad4 is bound to the undamaged one. The result was that the helical axis is offset due to the damaged DNA strand, causing a bend in structure that increase the Rad4 DNA interaction surface to the neighboring hairpin regions. This extending interaction creates a more stabilized damaged DNA, though its bases are now exposed to the solvent. This stabilization aids NER as it is repairing the damaged strand.
Another important component in the DNA damage response and repair is BER – base excision repair pathway. The difference between BER and NER is that BER has the ability to detect and remove single nucleotides with the smallest modification such as the addition of one single methyl group. Thus, it is extremely efficient in fixing distorted DNA strands. In BER, the oxidative damage-specific glycosylates OGG1 and MutM are found to interact with 8-oxoG bases. 8-oxoG bases are composed of a hydrogen-bond donor N9 and an accept O8. They interact with OGG1 and provides selective cut of the damaged DNA. This entire complex is known as the pseudo-Michaelis complex. Overall, different mechanisms were observed in the process of DNA damage response and repair from the combination of methods ranging from NMR, X-ray crystallography, to SAXS, etc.
Below is an image of a process of DNA repair where the DNA ligase I is repairing a chromosomal damage.

Role of 9-1-1 in DNA Repair
[edit | edit source]DNA repair consists of the detection of existing damage and the actual healing of this impairment. 9-1-1 is a heterotrimeric protein, consists of three sub-units in which at least one is different than the other two, that wraps around DNA to initiate the recruitment of specific checkpoint proteins and freezes the cell cycle temporarily. More specifically, it causes phosphorylation of Sc-Mec1/Hs-ataxia telangiectasia, where Sc- and Hs- prefixes refer to Saccharomyces cerevisiae (a eukaryotic species) and Homo sapiens respectively, and Rad3. Chk1 and Sc-Rad53/Hs-CHK2 protein kinases are activated resulting in the inhibition of cell cycle phases G1/S intra-S or G2/M. Accumulation of repair genes, fixation of the replication fork, and the decrease in production of cyclins (proteins that progress the cell cycle) also result from this activation. 9-1-1 works with Sc-Cdc28 to selectively accumulate Sc-Ddc2. The presence of Sc-Ddc2/Hs-ATRIP, Sc-Mec1/HS-ATR, and 9-1-1 together activates the checkpoint regardless of the detection of DNA damage.
Mismatch Repair
[edit | edit source]Mismatch repairs corrects any mistakes in nucleotide pairing that escape the proofreading ability of DNA polymerase during replication. Base nucleotides that are incorrectly paired causes deformity in the secondary structure of DNA. The MSH2 and MSH6 dimer binds to the mismatch on the strand. Then, MLH1, an endonuclease, will bind to the MSH and nick the strand. Then exonucleases will degrade the region in between and then allow DNA polymerase delta to place the correct nucleotide and DNA ligase will re-connect the strand. Using this ability, the enzyme cut out the distorted portion of the new DNA strand and then use the old DNA strand as a template to fill in the gap. In E.Coli, the mismatch repair enzyme recognizes the old DNA strand by the presence of methyl groups on certain sequences. In eukaryotic cells, it is unknown how the enzyme is able to distinguish between the old and new DNA strands.
Direct Repair
[edit | edit source]
In direct repair, instead of replacing an entire nucleotide, the wrong nucleotide is structurally changed to the right nucleotide. UV ray from the sun causes pyrimidine dimers by forming covalent bonding between adjacent pyrimidines. Some eukaryotic cells have an enzyme called photolyase. The enzyme breaks the covalent bond between the pyrimidine dimers with the energy from light.
Nucleotide Excision
[edit | edit source]NER Helicase
[edit | edit source]DNA repair is carried out by the nucleotide excision repair (NER) helicase, a protein that is composed of multiple domains. NER assembles around damaged DNA regions (which, because of their error, contain a bulge or lesion that encourages NER to bind) in a stepwise manner, allowing damage to be carefully verified before the actual excision is performed. For example, yeast Rad4 protein (an analogue of mammalian XPC) indirectly detects DNA damage by binding to a nearby undamaged region. The damaged DNA strand is flexible, allowing a stable complex to form which includes Rad23, the protein that actually repairs the damage.
If XPC-Rad4 cannot detect a damaged site, one alternative involves the DDC1-DDC2 dimer. This dimer forms a complex with a damaged DNA region and an ubiquitin ligase. The complex ubiquitinates XPC and DDC2, the latter of which then releases the DNA molecule, passing it on to XPC and the normal NER process.
Nucleotide Excision Repair can be divided into two subcategories: Global Genome Repair and Transcription Coupled Repair.
Global Genome Repair involves the XPC and hHR23B dimer binding to the damages DNA and then Transcription Factor 2H (TFIIH) bind to the complex. Then XPG binds and the DNA is further unwound. The nucleases XPG and XPF cleave the DNA, which essentially removes the damaged DNA. Then DNA polymerase delta fills in the gap with the correct nucleotide and then DNA ligase re-connects the strand.
Transcription coupled repair is when RNA polymerase stalls at the damaged site and then Cockayne Syndrome B protein (CSB) displaces RNA polymerase and recruits TFIIH and XPG. The DNA is unwound before the nucleases XPG and XPF cleave the DNA. Then the damaged section is removed and DNA polymerase delta fills in the gap and ligase re-connects the strand.
Source: Molecular Cell Biology, Lodish et al., 6th edition (2008), pages 145-160
The Base-Excision Repair pathway
[edit | edit source]
Not all damages are large enough to cause the lesions that are detected by NER. The base excision repair (BER) pathway repairs single nucleotide errors, sometimes as slight as the addition of a methyl group. While small, these damages can often be enough to impede DNA replication or produce nonfunctional proteins. Damage detection in the BER pathway is difficult because, in addition to the errors being small, there are a large number of them. Numerous enzymes are used to detect different small errors and initiate the BER pathway.
The first step in base-excision repair is the excision of modified nucleotide. Enzymes called DNA glycosylases, each has its own ability to recognize certain type of modified bases, cleave the bond between the 1'-carbon of the deoxyribose sugar and the base and remove the base. Then enzyme called apurinic or apyrimidinic (AP) endonuclease breaks the phosphodiester bond and another enzyme removes the deoxyribose sugar. DNA polymerase comes and adds the correct nucleotide to a free 3'OH group. Finally, DNA ligase connects the DNA strand by forming phosphodiester bond.
Backbone repair and DNA ligase
[edit | edit source]Damage to the sugar-phosphate backbone of DNA is repaired by DNA ligases. Because the DNA backbone is common to all organisms, these ligases are likewise found in every organism that uses DNA as its genetic material. DNA ligase seals breaks in the backbone by a three-step process. In the first step, several of the enzyme's domains adopt a specific conformation, allowing an active site lysine residue to be adenylated. In the last two steps, the enzyme encircles the broken DNA strand and fuse the two ends together.

Double-Strand Break Repair
[edit | edit source]Breaks in the double strand of DNA are common, but particularly hazardous to the cell due to increased chance of genetic mutation. Major causes of double strand breaks include reactive oxygen from oxidative metabolism, ionizing radiation, and enzyme errors. The strand could be repaired in one of two major ways: homologous-directed repair and the nonhomologous DNA end joining pathway (NHEJ).
Homology-Directed Repair
[edit | edit source]Any diploid organism could use homology-directed repair, even if the diploidy is temporary, as in bacteria. Types of homology-directed repair include homologous recombination, single strand annealing, and breakage-induced replication. In homologous recombination, an identical or nearly identical sequence of DNA is required as a template for repair during the S phase of the cell cycle, which occurs only during and shortly after DNA replication, and before mitosis. Nucleotide sequences are then exchanged between similar strands.
Nonhomologous DNA End Joining Pathway (NHEJ)
[edit | edit source]NHEJ arose as an alternative to homology-directed repair, as template donors are usually not available in nondividing cells. With a remarkably flexible mechanism, NHEJ has a wide diversity of substrates that can be converted into the desired product. Like other DNA repair processes, it requires three main proteins: a nuclease to resect damaged DNA , polymerases to fill in new DNA, and a ligase to the restore the strand. Key components include Ku, DNA-PKcs, Artemis, Pol x polymerases, and the ligase complex consisting of XLF, XRCC4, and DNA ligase IV. Each DNA end could then be modified independently multiple times, and substitutions with other enzymes is permitted due to its flexible nature. The problem of joining heterogenous DNA ends at double-strand breaks was shown to have evolved convergently in prokaryotes and eukaryotes.
References
[edit | edit source]- Huen, M. SY. "Assembly of checkpoint and repair machineries at DNA damage sites." Trends in Biochemical Sciences, Volume 35, Issue 2, 101-108, 28 October 2009
- Perry JJ, Cotner-Gohara E, Ellenberger T, Tainer JA. “Structural dynamics in DNA damage signaling and repair.” Curr. Opin. Struct. Biol. 2010 Jun; 20(3)
- Pierce, Benjamin A., Jung H. Choi, and Mark E. McCallum. Genetics: a Conceptual Approach. New York, NY: W.H. Freeman, 2008. Print.
- Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem. 2010;79:181–211.
- Perry, J. Jefferson P., Elizabeth Cotner-Gohara, Tom Ellenberger, and John A Tainer. “Structural Dynamics in DNA Damage Signaling and Repair”. Current Opinion in Structural Biology. (2010): 283-294. ScienceDirect.
- Eichinger, S. Christian and Stefan Jentsch. "9-1-1: PCNA's specialized cousin." Trends in Biochemical Sciences, Volume 36, Issue 11, 563-568, 04 October 2011.
Introduction
[edit | edit source]Mismatch Repair in mammals is an important mechanism in the overall processes of DNA repair. Mismatch Repair (MMR) works by removing incorrect base pair match-ups in double-stranded DNA and replacing it with the correct base pair. However, MMR has other known functions, including mutagenesis in different in vivo conditions.
Canonical MMR Mechanism
[edit | edit source]Errors in DNA replication pose many problems to both the integrity of the DNA and to the individual. MMR is one way that these errors are fixed, as it is known that deficiency in MMR causes cancerous tumors in animal models.
The basic MMR system relies on the proteins, MutSα, MutLα, EXO1, RFC, PCNA, RPA, polymerase-δ, and DNA ligase I. There are three basic steps of MMR known as licensing, degradation, and resynthesis.
Licensing
[edit | edit source]In licensing, MutSα binds to the mismatch error in the DNA strand, which causes a change in the conformation of MutSα into a sliding clamp. This change is dependent upon an exchange of ADP for ATP. MutLα is recruited to forma ternary complex with MutSα, which then diffuses along the DNA strand until it reaches PCNA.
PCNA, or proliferating cell nuclear antigen, is a protein that can undergo a conformational change to become a ring around DNA. To attach to the DNA, it relies on the function of RFC, or replication factor C. This protein uses ATP hydrolysis to attach the PCNA to the DNA. This attachment is only efficient when there is a "nick" in the DNA, or an apyrimidinic (AP) site. The PCNA can then attach to the 3' end of the nick. While RFC can add PCNA without a nick in the DNA, this is down with extremely low efficiency.
Once it reaches PCNA, the cryptic endonuclease of MutLα is activated and causes additional nicks that are on both sides of the mismatch error on the same strand. This is only necessary for PCNA binding on the 3' side of the mismatch. Nicks are only made on the same strand as the mismatch because PCNA is not symmetric and has distinct sides to it. As such, MutLα can only interact with PCNA on a specific face and the complex will have a certain orientation, which remains constant even when sliding across the DNA. MutLα has endonuclease activity on one of its heterodimer subunit, PMS2, and this will only nick the same strand as the PCNA binding.
The reason that nicks are made close by to the mismatch (which is essential for DNA repair) is because the complex making the nicks, MutSα/MutLα, has the highest number around the mismatch site, correlating with greater PCNA collision frequency. This is especially important in replication, where the PCNA molecules adhere to the DNA for an extended period of time even after replication. Due to RFC, they are loaded at the 3' terminus of an Okazaki fragment of the leading strand. They adhere with a certain orientation, which allows MutSα/MutLα to cleave the nascent DNA strand, even though the gap around the Okazaki fragment has long been linked. As such, the MMR system has the correct directionality due to this nick generation.
Degradation
[edit | edit source]In degradation, EXO1 is loaded at the nicks created by the PCNA-activated MutSα/MutLα complex. This creates a large gap that starts the nick and ends around 150 bases after the mismatch. This gap is single-stranded and on the same side as the mismatch. EXO1 is an exonuclease that can only cut in a 5' to 3' direction.
Resynthesis
[edit | edit source]Resynthesis involves PCNA, polymerase-δ, and DNA ligase I in order to replace the removed bases and, overall, fix the mismatch error.
EXO1 Independent Mechanism
[edit | edit source]Although not proven in humans, EXO1 deficient mice showed less mutations than MSH2 and MLH1 deficient mice, indicating a mismatch repair mechanism that does not require EXO1. Indeed, a 5' nick MMR mechanism could occur without EXO1 through use of polymerase-δ and MutSα, RPA, RFC, and PCNA. When there is a 5' nick from the mismatch error, polymerase-δ can catalyze strand displacement, whereby FEN1 can catalyze the removal of the strand containing the mismatch. DNA ligase I would then seal the nick formed.
Insertion/Deletion Loops and Trinucleotide Repeats
[edit | edit source]Insertion/Deletion loops (IDLs) and trinucleotide repeats (TNRs) interact largely with MMR in both error-preventing and error-propagating ways.
Origin of IDLs
[edit | edit source]IDLs arise due to the activity of polymerase on TNRs. Trinucleotide repeats are large number of repeats of a single tripley of nucleotides. Such repeats have been implicated in diseases such as Fragile X. When polymerase reads these repeats, it slows down. However, helicase does not slow down, and due to being relatively faster, there becomes long strands of single stranded DNA. As such, these strands can bunch up and form an IDL. This would cause polymerase to create shorter than usual DNA strands.
The Error-Preventing Role of MMR
[edit | edit source]When things such as this happen, MMR can work to fix it. If the loops is less than two to three extrahelical nucleotides long, the canonical MMR can fix it. However, if the loop is longer, there is a MutSβ-mediated way for loops to be fixed. However, this happens by some other MMR mechanism, for in the regular process, PCNA would not be able to diffuse past the large loop. Thus, there must be some non EXO1-mediated MMR.
The Error-Propagating Role of MMR
[edit | edit source]In certain cases, MMR may be "hijacked" to cause TNR expansion. In the event that there is a cruciform loop structure, where there are loops in both strands in the same relative position, a cleavage by PCNA attached by RFC onto one of the loops activating MutSβ and MutLα endonucleolytic activity may cause one of the loops to collapse. When polymerase replaces the missing nucleotides, there will be an extension of the trinucleotide repeat. A larger number of repeats has been linked to more severe disease in diseases (such as Huntington's Disease) that are caused by TNRs, and so this is an important field of study.
Antibody Variation and Class-Switching
[edit | edit source]Antibody variation, although due to a variety of reasons, is largely dependent on the role of MMR. After VDJ recombination, a process involving recombination of the variable, diversity, and join regions of the immunoglobulin genes, a variety of IgM antibodies can be made. However, there are further mechnanisms for antibody variability.
The role of MMR in Somatic Hypermutation
[edit | edit source]Somatic hypermutation (SHM) is a process whereby many mutations arise in the variable region of the antibody. It works through activation-induced cytidine deaminase (AID) where C nucleotides are converted to U. This occurs during transcription, because AID works best on single-stranded DNA. When this occurs, there is a mismatch error on the resultant DNA. As such, uracil DNA-glycosylase works to do base-excision repair (BER) and remove the incorrect U nucleotide. However, once this happens there is an apyrimidinic site (AP) remaining. This site can be the target of EXO1 in order for MMR to occur.
In this case, EXO1 cleavage may cause a large swath of DNA to be excised. When polymerase goes to fix it, AP sites and remaining uracil nucleotides may cause incorrect mutations at the sites where AID acts. This would result in changes in the variable site of the antibody and ultimately, different antigen recognition.
The role of MMR in Class-Switch Recombination
[edit | edit source]MMR can also cause the type of antibody to change, such as from IgG to IgM while recognizing the same type of antigen. When there are two AP sites and EXO1 causes the excision of a section of DNA to the other gap, class-switch recombination (CSR) can occur due to a double-strand break.
References
[edit | edit source]- Peña-Diaz, J., & Jiricny, J. (2012). Mammalian mismatch repair: error-free or error-prone? Trends in biochemical sciences, 37(5), 206–14. doi:10.1016/j.tibs.2012.03.001
- Zhao, J. et al. (2009) Mismatch repair and nucleotide excision repair proteins cooperate in the recognition of DNA interstrand crosslinks. Nucleic Acids Res. 37, 4420-4429
- Lopez Castel, A. et al (2010) Repeat instability as the basis for human diseases and as a potential target for therapy. Nat. Rev. Mol. Cell Biol. 11, 165-170.
Introduction
[edit | edit source]DNA strand breaks are often caused by internal and external factors. After the termini of these strands break, they require processing before missing nucleotides can be replaced by DNA polymerase and its strands rejoined by DNA ligases. The enzyme polynucleotide kinase/phosphatase plays an important role in repairing DNA strand breaks by catalyzing the restoration of DNA’s termini. In addition to this, PNKP also helps in other DNA repair pathways through interactions with other DNA repair proteins such as XRCC1 and XRCC4. PNKP is important in maintaining genomic stability of normal tissues, like developing neural cells, and enhancing resistance of cancer cell to genotoxic therapeutic agents.
Polynucleotide kinase/phosphatase
[edit | edit source]When damage is done to cellular DNA, this causes aging, cancer etiology and treatment, and neurological disorders. DNA damage comes in different forms like: base modification and base loss and strand breaks. These damages can be triggered by intracellular agents like primary reactive oxygen species (ROS) and exogenous agents. In order to protect themselves from this damage, cells have evolved a battery of repair pathways. These counter mutational and cytotoxic consequences that occur due to DNA damage. Various mechanisms that cause strand breaks include: cleavage by physical and chemicals means such as ionizing radiation (IR) and ROS, and enzymatic processes. Therefore, strand breaks comes in a wide variety of forms and different strand breaks can be further classified or subdivided based on the nature of their termini. The enzyme PNKP carries 5’-kinase and 3’phosphatase activities that are essential for processing of single and double strand breaks at termini. Research into PNKP has shown that small molecule inhibitors of these enzymes sensitize cells to IR or chemotherapeutic agents. Researchers have also identified that mutations that have lead to changes in PNKP, similar to mutations in other genes that encode other strand break repair proteins, have been connected to a severe autosomal recessive neurological disorder.
Chemistry of strand break termini
[edit | edit source]IR-and free radical-induced breaks
[edit | edit source]Ionizing radiation (IR) causes strand breaks with a variety of end groups at 3’-termini by generating hydroxyl radicals. By generating hydroxyl radicals, reactions at different carbon atoms occur within the deoxyribose group to produce two predominant end groups: phosphate and phosphoglycolate. Phosphoglycolate formation is dependent on the presence of oxygen while 3’-phosphate groups are produced under normoxia and anoxia. On the other end, at the 5’-termini, the major end group is phosphate. In addition to causing strand breaks, ionizing radiation also generates complex lesions. These areas contain two or more damaged bases or strand breaks in close quarters and singly damaged sites. Complex lesions include frank DSBs with a ratio of SSB:DSB determined to be ~25:1. Another factor that causes strand breaks is hydrogen peroxide. Similar to IR-medicated damage, hydrogen peroxide causes far fewer frank DSBs. Bleomycin, a chemotherapeutic agent, additionally produces DSBs at the 3’-phosphoglycolate termini.
Camptothecin-induced breaks
[edit | edit source]The enzyme topoisomerase 1 creates a DNA cut with a 5’-OH terminus and a covalent 3’-phosphate-enzyme intermediate in order to relieve torsional strain. Using topoisomerase, camptothecin prevents resultant strand rejoining, leaving a DNA-enzyme ‘dead-end’ complex. By hydrolyzing this complex with tyrosyl-DNA phosphodiesterase, more cuts with 3’-phosphate and 5’-OH termini are made.
Repair-endonuclease induced breaks
[edit | edit source]Using DNA glycosylases, damaged bases can be removed. The abasic sites are then cleaved by one of two classes of enzymes. One of the enzymes, AP endonuclease, hydrolyses the phosphodiester bond 3’ to the abasic site in order to give 3’-OH and 5’-deoxyribose phosphate termini. By using DNA polymerase β, 5’-deoxyribose phosphate termini can be converted to 5’-phosphate. AP lyase works by cleaving the phosphodiester bond 5’ to the abasic site by a β-elimination reaction to give a β-unsaturated aldehyde attached to 3’-phosphate at one terminus and a 5’-phosphate at the other. Since many DNA glycosylases have this enzyme activity, the pentenal moiety can then be eliminated by an AP endonuclease to give 3’-OH or by an AP lyase to give 3’-phosphate. Enzymes NEIL1 and NEIL2, mammalian DNA glycosylases with β,δ-lyase activity, remove an extensive amount of mutagenic and cytotoxic oxidative pyrimidien lesions and purine-derived formamidopyrimidines.
Molecular architecture of PNKP
[edit | edit source]PNKP is constituted as a multidomain enzyme. It consists of 2 domains: an N-terminal forkhead-associated (FHA) domain and a C-terminal catalytic domain that is composed of fused phosphatase and kinase subdomains. Using a flexible polypeptide segment, the two domains, FHA and catalytic domain are linked together. This flexible polypeptide segment acts to selectively bind acidic casein kinase 2 (CK2)-phosphorylated regions in XRCC1 and XRCC4. XRcc1 and XRCC4 are important scaffolding proteins that repair DNA SSBs and DSBs. Aprataxin and APLF are DNA repair factors that also include FHA domains that likewise bind CK2-phosphorylated XRCC1 and XRCC4. This function could result in coordinated regulation of these proteins leading to binding of the phosphorylated scaffolding factors. PNKP and T4 polynucleotide kinases are similar in their catalytic domain in that they both contain contiguous kinase and phosphatase domains but different in that T4 enzyme lacks a FHA domain and that the kinase subdomain lies N-terminal to the phosphatasesubdomian.
The two catalytic active sites are positioned on the same side of the protein of murine PNKP. Murine and T4 kinase subdomain share resembling structure of a bipartite active site cleft that ahs separate ATP and DNA binding sites. The structure of the ATP binding site includes Walker A (P-loop) and B motifs conserved in various kinases. In addition it also carries aspartic acid that activates the 5’-hydroxly for attack on the ATP γ-phosphate. DNA binding sites between mammalian and phage enzymes are different. While phage PNK DNA binding cleft forms a narrow channel that leads to the conserved catalystic aspartic acid residue that accommodates single-stranded substrates, mammalian enzymes phosphorylates 5’hydroxyl termini within cut, gapped or DSBs with single-stranded 3’ overhanging ends since single-stranded 5’ termini are phosphorylated less efficiently. A broad DNA recognition grove composed of two distinct positively charged surfaces, selectively recognizes larger, double-stranded DNA substrates. By using structural information from small angle X-ray scattering experiments coupled with the effect of amino acid substitutions on surfaces of kinase, researchres found that DNA substrates bind across these surfaces in a defined orientation.
A typical process employed by many phosphatases is the haloacid dehalogenase fold. Mechanisms employed by these enzymes are dependent on Mg2+ while proceeding by a catalytic aspartate and acyl-phosphate intermediate. Mammalian PNKP executes its processes on a multitude of 3’-phoshate ends like those within nick,s gaps, DSBs, and single-stranded termini. Two narrow channels that are surrounded by large positively charged loops make a pathway to the phosphatase active site but aren’t wide enough to take in double-stranded substrates. This shows that either a requirement for remodeling of the phosphatase substrate binding surface or an unwinding of the DNA is needed to accommodate double-stranded substrates.
DNA repairing scaffold proteins, XRCC1 and XRCC4, interacts with PNKP function, mediated by binding of the PNKP FHA domain to phosphorylated motifs on XRCC1 and XRCC4. FHA domains, phospho-peptide binding modules, have a β-sandwich fold where a series of loops jut out from one side of the β-sandwich and provide a peptide binding surface with a marked preference for targets that contain a phospho-threonine residue. Even though XRCC1 and XRCC4 are structurally unrelated, they share similar motifs that are phosphorylated by CK2 and act as the binding sites for the PNKP FHA domain. A significant reduction in the efficiency of SSB repair occurs when a cluster of CK2 phosphorylation sites between residues 515 and 526 in XRCC1 is needed for interaction with PNKP and amino acid substitutions within this certain region. Similarly, a primary CK2 site in XRCC4, THr233, is needed for PNKP binding and for efficient repair of DSBs in vivo. Significant conservation of sequence is show around these sites. Phosphorylation of a conserved serine occurs and structure of the complex with regard to the primary phospho-threonine reveals a dynamic interaction of this residue with ARG35 or ARg44 of PNKP FHA domain. Tyrosine residue is conserved at the -4 position and asparagines residue is conserved at the +3 position. Some reactions aren’t conserved in the complex with XRCC4 in the FHA domain. The +3 position residue is a glutamic acid. Due to the peptides acidic properties and long-range electrostatic interactions between residues, the largely positively charged peptide-binding surface contributes to binding specificity. Threonine phosphorylation in the +4 position also plays a role to binding selectivity through the recruitment of a second PNKP FHA domain.
PNKP and single-strand break repair (SSBR)
[edit | edit source]The multienzyme pathway, SSBR, uses different participants depending on the causative agent. An example would be with IR-induced strand breaks that involves losing at least one nucleotide. The process of damage recognition and correction of the strand that is broken at the termini is carried out by enzymes poly(ADP-ribose)polymerase (PARP), XRCC1, AP endonuclease 1 and PNKP, with other proteins acting as backups to this functionality. By using a short patch pathway that involves DNA polymerase β and DNA ligase III or a long patch pathway that uses DNA polymerase δ and/or ɛ, the FEN1 endonuclease and DNA ligase I, consecutive replacement of nucleotides and strand resealing may occur. When IR occurs, APE1 removes 3′-phosphoglycolates while PNKP hydrolyses 3′-phosphate groups. This occurs when 3′-phosphatase activity of APE1 is much weaker than that of PNKP. Enzyme PNKP also plays a role in confirming that 5′-OH termini are phosphorylated. Due to the fact that phosphatase activity of PNKP is much more active than the kinase activity, when strand breaks with both 3′-phosphate and 5′-OH termini occur, the activity of PNKP is prioritized. Phosphatase activity in PNKP was shown to be important in the rapid repair of hydrogen peroxide-induced SSBs in mammalian cells when a failure of overexpression of phosphatase-defective PNIKP to compensate for Xrcc1defieicney occurred. Correspondingly, another important factor of PNKP phosphate activity involves a small molecule inhibitor that dramatically retards SSBR in irradiated human cells. While it is shown here that important phosphatase activity exists in PNP, the physiological important of the 5′-kinase activity has yet to be determined.
A commonly accepted model for repair of radiation-induced SSBS is when SSB catalyzes the polymerization of chains of ADP-ribose onto acceptor chromatin proteins and itself. BY doing this, SSBR attracts the scaffold protein, XRCC1 and maybe also tightly bound DNA ligase III. The proteins then in turn recruits PNKP or APE1 in order to restore the essential terminal groups for DNA polymerase β so that it can add the missing base and allow DNA ligase III to rejoin the strand. By researching and analyzing protein-protein interactions, it was found that direct interactions between XRCC1 and PNKP exist, as well as with DNA polymerase β and DNA ligase III. This shows that these connected partnerships include tetrameric complex between the four proteins. This formation could form for various models. While there is evidence that shows interactions between XRCC1 and PNKP, evidence also exists that counters the concept that XRCC1 recruits either PNKP or APE1 to the strand break. By using the technique of cross linking proteins to DNA substrates, experiments were conducted to track the temporal association of SSBR proteins in HeLa cell. Through this process of incubation, it was discovered that for substrates with either 3′-phosphoglycolate termini or 3′-phosphate termini, APE1 and PNKP, were recruited to the strand breaks before XRCC1/DNA ligase III. In addition to this discovery, it was found that immunodepletion of APE1 or PNKP diminished the binding of XRCC1 to the following substrates. This indicated that APE1 and PNKP inducted XRCC1 to sites of oxidative damage rather than in reverse. Conversely, PNKP foci were found to be in the nuclei of hydrogen peroxide-treated cells expressing XRCC1, but did not exist in cells lacking XRCC1. This shows that although XRCC1 might not be required in the beginning stages of PNKP or APE1, it expedites the focal accumulation and provocation of these specific enzymes at sites of chromosomal damage
Even though DNA repair protein XRCC1 lacks inherent enzymatic activity, it has the ability to enhance both kinase and phosphatase activities of PNKP. By using florescence measurements to work out the binding mechanism between PNKP and substrates that mimic different strand breaks, the mechanism surrounding XRCC1-induced stimulation was discovered. Even though PNKP bounded tightly to a nicked substrate with a 5′-OH terminus with a Kd value of 0.25 μM, this was only 5- to 6-fold tighter than PNKP binding to the identical duplex bearing a 5′-phosphate. This showed that PNKP stayed bounded to the product of its kinase activity. Results showed that the presence of XRCC1 did not influence the binding of PNKP to the nonphosphorylated substrate. But further results also showed that PNKP interaction with the phosphorylated duplex was abolished thus indicating that XRCC1 did influence the binding and displaced PNKP from the reaction product. By following the evidence of kinetics of product accumulation under limiting enzyme concentration, the result of the addition of XRCC1 increasing PNKP enzymatic turnover was confirmed. Further data has shown that similar kinetic data was observed for PNPK phosphatase activity.
The relationship between PNKP and XRCC1 is further complicated by CK2-mediated phosphorylation of XRCC1. While promoting interaction with other proteins, XRCC1 phosphorylation also works to stabilize the XRCC1-DNA ligase III complex. Observations were found of multiple sites of CK2-mediated XRCC1 phosphorylation involved in vitro, clustered within specific locations. In order to recruit XRCC1 and PNKP to nuclear foci in hydrogen peroxide-treated or γ-irradiated cells, XRCC1 phosphorylation is needed. XRCC1 phosphorylation is also needed to promote more rapid repair of SSBs. If a cell lacked XRCC1 phosphorylation, this would not impact cell survival. But through further research and analysis, it was found that cells without function XRCC1 with triple mutant XRCC1 would fail to fully restore rapid SSBR, showing that there indeed existed an important interaction with PNKP. Repair of the cell could easily be completed by overexpression of PNKP. This shows that XRCC1 plays an important role in increasing PNKP enzyme turnover, especially when the cell contains a limiting concentration of PNKP.
Phosphorylation of XRCC1 by CK2, compared to nonphosphorylated XRCC1, prompts the kinase and phosphatase activities of PNKP that are measured in vitro. In contrast, Stimulation by nonphosphorylated XRCC1 is due to enhanced enzymatic turnover of PNKP. This situation brings up problems since it can be seen that phosphorylated and nonphosphorylated XRCC1 bind PNKP at different site and with different affinities, but both are able to stimulate PNKP by a similar mechanism. Research found that while phosphorylated XRCC1 binds the FHA domain with a Kdvalue of 4 nM, the nonphosphorylated protein binds the catalytic domain of PNKP with a 10-fold weaker affinity. This indicates that a certain possibility of phosphorylation-independent interaction between PNKP and XRCC1 in human cells exists. Researchers found that PNKP co-immunoprecipitated with XRCC1 triple mutant that was expressed in human 293T cells. While 85–90% of the cellular XRCC1 is phosphorylated, this does not indicate that the key cluster of amino acids involved in interaction with the FHA domain is fully phosphorylated. An increase in phosphorylation at the cluster and an approximately 3-fold increase in PNKP copurifying with XRCC1 was due to treatment of cells with hydrogen peroxide. This shows that cells might play a role in enhancing CK2-mediated phosphorylation of XRCC1 and its subsequent interaction with PNKP FHA domain. This enhancement happens directly in response to a confrontation by hydrogen peroxide or radiation to deal with rather high levels of DNA damage in an efficient manner. On the opposite end of the spectrum, unstressed cells are able to cope with comparatively low level of endogenous DNA damage by using a different method. By using nonphosphorylated XRCC1, or XRCC1 with a restricted degree of phosphorylation, it is able to activate PNKP through binding to the catalytic domain.
Cells are sensitive to camptothecin due to PNKP depletion in its cells and Pnk1 deletion in fission yeast. XRCC1 overlooks the repair of these strand breaks by forming a complex with TDP1, DNA ligase III and PNKP. Neurodegenerative disorder, spinocerebellar ataxia with axonal neuropathy-1, is caused by mutation of TDP1. Research shows that SCAN 1 cells have a reduced capacity to repair Camptothecin-induced SSBs and also display slow repair of hydrogen peroxide-induced SSBs. This evidence proffers that TDP1 is important and required to repair lesions generated by oxidative processes, lesions that possibly justify neurodegeneration observed in SCAN1. Evidence for this was shown by experiments for fission yeast in G0, Tdp1 and Pnk1 that act sequentially in order to process the 3′-termini of naturally occurring SSBs .
PNKP and base excision repair (BER)
[edit | edit source]Cellular mechanism BER, base excision repair, is accountable for the repair of most minor base modifications determined by IR, ROS and alkylating agents. First step in the mechanism is to remove the modified base by DNA glycosylases and then cleave the DNA at the newly formed apurinic/apyrimidinic (AO) site using APE1. Another way would be to use glycosylases hydrolyze the AP site with its AP lyase activity. With the discovery of the nei endonuclease endonuclease VIII-like-1 (NEIL1) and NEIL2 mammalian DNA glycosylases, it was indisputable that PNKP was involved in the BER pathway. Nei endonuclease VIII-like-1 (NEIL1) and NEIL2 mammalian DNA glycosylases possess β,δ-AP lyase activity that generates 3′-phosphate termini. Instead of binding directly to PNKP, these glycosylases instead are associated with larger complexes that contain other BER components that include PNKP. The function of these glycosylases are to undertake a variety of base lesions that include: thymine glycol, 5-hydroxyuracil and 8-oxoguanine . In addition to this function, glycosylases can also cleave intact abasic sites that are generated by glycosylases that do not possess AP lyase activity, and the pentenal moiety generated by the β-elimination AP lyases of other DNA glycosylases. Because of this NEIL glycosylases would compete with APE1 thus forming the basis of a different, APE1-independent, BER pathway. Although current research can not indicate to what extent NEIL1- or NEIL2-catalyzed cleavage of abasic sites arises in cells, the cleavage of these sites could possibly explain for the increased sensitivity of PNKP-depleted cells to the alkylating agent methyl methanesulfonate (MMS). This sensitivity to MMS came as a surprise in the experiments due to major lesions inflicted by this agent being N7-methylguanine and N3-methyladenine, with little if at all any direct strand scission . Downregulating aprataxin expression also causes cells to be sensitive to MMS. But since human DNA glycosylase that are responsible for removing these methylated bases do not possess AP lyase activity, the ability to act upon the abasic sites generated by MPG to produce strand breaks with 3′-phosphate termini must fall to NEIL1 or NEIL2.
PNKP and double-strand break repair (DSBR)
[edit | edit source]In the two major double-strand break repair pathways, there is proof for PNKP participating in nonhomologous end joining. But in contrast, due to its failure to influence IR-induced sister chromatic exchange by PNKP deletion, this suggests that PNKP may actually not be involved in homologous recombination. In addition to the other pathways, PNKP plays an additional role as a back-up, XRCC1-dependent, DSB repair pathway. Experiments showed evidence for PNKP participation through using human cell-free extracts. This evidence showed that PNKP kinase activity was required before binding of linearized plasmid substrates bearing 5′-OH termini could happen. XRCC4 and DNA-PK were important in determining how successful phosphorylation was. In parallel to the role of XRCC1 linking PNKP to DNA ligase III, XRCC4 links PNKP to DNA ligase IV. CK2-mediated phosphorylation of XRCC4 Thr233 plays a role in interacting with the PNKP FHA domain and smoothly stimulating XRCC4–DNA ligase IV mediated ligation of a 5′-dephosphorylated plasmid substrate in vitro. In an Xrcc4-deficient cell line, when expression of XRCC4 occurs instead of wild-type XRCC4, the rate of survival is reduced by approximately 30% following irradiation and thus slowing down the rate of DSB repair.
The function role of the XRCC4-PNKP interaction was able to be determined by coming biophysical and biochemical examination. While phosphorylation of XRCC4 advocates a tight affinity for PNKP, nonphosphorylated XRCC4 also have the ability to bind to PNKP. Though in this particular case, binding is to the catalytic domain of PNKP thus weakening the affinity. Similar to the ability of XRCC1 stimulation of PNKP turnover from SSBs, nonphosphorylated XRCC4 has the ability to stimulate pNKP enzymatic turnover from DSBs. Research found that the presence of phosphorylated XRCC4 failed to stimulate PNKP and thus did block PNKP-mediated DNA phosphorylation. But with the additional attendance DNA ligase IV, the complex it forms with phosphorylated XRCC4 has the ability to reverses the inhibition and stimulate PNKP turnover. A ratio of XRCC4:DNA ligase IV:PNKP of ∼7:1:3 was found in the proteins in HeLA cells, with almost half of the XRCC4 vitally phosphorylated at Thr233. This shows that in cells, only a fraction of XRCC4 can be complexed to DNA ligase IV thus indicating a possibility for FHA-independent interaction between XRCC4 and PNKP. Using XRCC4 co-immunoprecipitation with PNKP, the FHA independent interaction between XRCC4 and PNKP was confirmed for expression in cells depleted of endogenous PNKP. PNKP also has an important function of processing DSB 3′-phosphoglycolate termini, especially 3′-overhanging and blunt-ended termini. These termini are produced by IR, bleomycin and enediyne compounds like neocarzinostatin. Even though APE1 has the ability to remove phosphoglycolate groups at SSB termini and recessed DSB termini, with blunt-ended DSB termini it loses its effectiveness and with overhanging termini it is completely ineffective.
Physiological roles and clinical potential of PNKP
[edit | edit source]PNKP is involved in several DNA repair pathways that work to protect cells from endogenous and exogenous genotoxic agents. Neurological disorders with various symptoms occur when disruption of NHEU genes and SSBR/BER genes occur. An example would be microcephaly. Microcephaly occurs in people with mutations in LIG4 that encodes DNA ligase IV. Deletion of Xrcc1 in mice causes seizures. Research has found that PNKP mutations are the cause of a sever neurological autosomal recessive disease that is characterized by microcephaly. Symptoms include intractable seizures and developmental delay. Through analysis of families, mutations were found in both the kinase and phosphatase domains. Through the collection of all the symptoms shown by patients with MCSZ, it shows the involvement of PNKP in multiple DNA repair pathways.
PNKP has also shown to be linked to pathophysiological conditions. It has been observed that elevated expression of PNP in arthrofibrotic tissue shows a role for PNKP in mitigating the effects of ROS generated by macrophages. It has also been observed in another experiment that physiologically and environmentally relevant doses of cadmium and copper are known to elicit neurotoxic and carcinogenic effects, thus inhibiting PNKP.
The concept of DNA repair capacity of tumor cells shows an important point in clinical response to many antineoplastic agents. Thus investigations are underway of inhibitors of several DNA repair enzymes like PNKP. They hold on to the ability to sensitize cells to radiation and chemotherapeutic drugs thus showing an important concept for research. Through this research, a small molecule inhibitor of PNKP phosphatase activity was identified and exhibited to heighten the sensitivity of cells to IR and camptothecin. This is the parent compound of two clinically important topoisomerase I poisons, irinotecan and topotecan, that are frequently used to treat colon and ovarian cancers.
Conclusion
[edit | edit source]PNKP is an important enzyme that is used in cellular processing of strand break termini. PNKP is involved in many DNA repair pathways due to its helpful properties. More research is needed to identify how it is regulated, how it collaborates with other repair enzymes, and physiological role in neurons and other tissues. PNKP is seen as a therapeutic target in treatment of cancer since it is involved in a variety of repair pathways. Therefore, new inhibitory compounds will need to be identified, researched, and optimized for clinical use. Further research should be invested in identifying synthetic lethal partners of PNKP in order to view its potential use as single agents against tumors deficient in proteins.
References
[edit | edit source]Weinfeld, Michael. "Tidying up loose ends: the role of polynucleotide kinase/phosphatase in DNA strand break repair." Trends in Biochemical Sciences 36.5 (2011): 262-71. PubMed. Web. 21 Nov. 2012.
DNA Packaging
[edit | edit source]DNA packaging is an important process in living cells. Without it, a cell is not able to accommodate large amount of DNA that is stored inside. For example, a bacterial cell which ranges from 1 to 2um in length contains amount of DNA that is 400 times as big (Becker et al. 530). Eukaryotic cells face even bigger challenges. A typical human cell has enough “DNA to wrap around the cell more than 15,000 times” (531). Therefore, DNA packaging is crucial because it makes sure that those excessive DNA are able to fit nicely in a cell that is many times smaller.
The DNA in bacterial cells are either circular or linear. To accommodate the size of bacterial cell, supercoiled DNA are folded into loops with each loop resembles shape of bead-like packets containing small basic proteins that is analogous to histone found in Eukaryotes (533).
In eukaryotic cells, DNA packaging is more complicated because they contain amount of DNA that is much larger than that of bacterial cells. More proteins are therefore required for the process with histone being the most important one. This protein is consisted largely of positive amino acids like lysine and arginine which make the overall structure positive. Thus, histone interacts favorably with the negative phosphate groups from DNA. There are five main types of histone, H1, H2A, H2B, H3 and H4 (533). Two of each H2A, H2B, H3 and H4 joins to form an octamer wrapped around by DNA of 146 base pairs like a bead on a string. This bead, consisting of eight histone molecules and 146 DNA base pairs, is known as the nucleosome. Each nucleosome is connected by a DNA linker of 50 base pairs to form a fiber like structure called chromatin. H1 is believed to be found in these DNA linkers. Chromatin fibers can be further compacted to form higher order of structures called heterochromatin or euchromatin depending on the degree of packing. Ultimately, DNA packaging in eukaryotic cells can lead to the formation of chromosome which is only present during cell division or several other situations (533-535). In eukaryotic cells, DNA packaging is not only in the nucleus but is also in mitochondria and chloroplast. The overall shape of their DNA resembles that of bacteria instead that of eukaryotes.
Histone chaperones and the nucleosome assembly processes
[edit | edit source]Histones are proteins that allow DNA to be tightly packaged into units called nucleosomes. The DNA wraps itself around the histones.
Chromatin is made of DNA and proteins (Histones). Chromatin is used to give structure to a chromosome.
Nucleosome consists of the acidic chromatin and the basic histone proteins.
Histone chaperones Histone chaperone guided folding pathways, assists in the folding and unfolding of the DNA around the histone.
Organization The tight coiling of DNA allows easier access to the DNA which makes sequencing faster.
Need for histone chaperones Nucleosomes can be assembled or disassembled and are done in stepwise function. Histone chaperones guide the pathway process, they control and regulate.
Structural forms of histone chaperones Since histone chaperones participate at each step of the nucleosome assembly processes, there are different chaperones needed for each different step.[1]
Reference
[edit | edit source]Becker, Wayne M, et al. The World of the Cell. 7th ed. New York: Pearson/Benjamin Cummings, 2009. Print.
Churchill, Das, Tyler The histone shuffle: histone chaperones in an energetic dance Structural Biochemistry/DNA as nanomaterial Structural Biochemistry/Structural DNA nanotechnology Structural Biochemistry/Holliday junction Structural Biochemistry/Proliferative and Antiproliferative genes Structural Biochemistry/Protein-DNA recognition Structural Biochemistry/Transcription Regulation by mediator Structural Biochemistry/Chromatin and aging
Introduction
[edit | edit source]Saccharomyces cerevisia (Sir2) is an NAD+-dependent histone deacetylase. It's role within the cell is to link chromatin silencing to genomic stability, cellular metabolism, and lifespan regulation. For example, in mice, if there is a deficiency for SIRT6 (family member of Sir2), the mice experience genomic instability, metabolic defects, and degenerative pathologies in terms of aging, everything opposite of the roles of Sir2. With new insights to the previously ambiguous SIRT6, scientists have discovered that SIRT6 is a very substrate-specific histone deacetylase that promotes proper chromatin function in things like telomere stabilization and DNA repair.
Sir2: a chromatin-aging connection
[edit | edit source]Sir2 is the founding member of the family of proteins called sirtuins. These proteins provided the first link between chromatin regulation and aging. Sir2 favors chromatin silencing at sub-telomeric DNA, silent mating-type loci, and rDNA repeats. These effects of Sir2 on chromatin is mediated by having Sir2 catalyzing the deacetylation of lysine residues on the amino terminal ends of histones H3 and H4 and also on the globular core of histone (all by NAD+-dependent histone deacetylase activity). Deacetylation of H4 lysine 16 and H3K56 mediate the silencing effects of Sir2.
Example: In budding yeast, Sir2 regulates replicative lifespan through a couple of chromatin-silencing processes.
First, Sir2 suppresses recombination between rDNA repeats and this prevents
Second, H4K16 acetylation levels increase at telomeres when replicative age increases; Thus, Sir2 protein levels decrease. These chromatin changes create defects in telomere position-dependent transcriptional silencing and trigger replicative senescence.[2]
A few studies have shown that there are aging-related Sir2 functions that might be chromatin-independent, making the relationship between Sir2 and lifespan regulation even more complex.
For example,Sir2 asymmetrically segregates damaged proteins to the yeast mother cell during cell division; this asymmetry can age the mother cell by forming toxic protein aggregates. Also, Sir2 can block lifespan extension in response to nutrient deprivation of mutations in nutrient-sensing pathways.
Mammalian sirtuin proteins: venturing out from chromatin
[edit | edit source]SIRT1 is the most closely related to year Sir2 out of the seven SIR2 family members. However, Sir2 appears to deacetylate histones exclusively while SIRT1 appears to more than 40 substrates. SIRT1 deacetylates many non-histone proteins and impacts on many phsysiologic processes like apoptosis.
SIRT1, SIRT6, and SIRT7 are concentrated in different sub-nuclear patterns; SIRT2 is cytoplasmic; SIRT3, SIRT4, and SIRT5 reside in the mitochondria.
SIRTching for a function through knockout mice
[edit | edit source]SIRT6-deficient mice appear normal when born, but after a couple of weeks, they start to develop degenerative phenotypes like osteoporosis. They also experience metabolic defects - so much that with such low levels of the insulin-like protein IFG-1, these mice die by 1 month.
An orphan enzyme finds its substrates
[edit | edit source]Through experiments, it was found in vitro that SIRT6 promotes mono-ADP-ribosylation, an alternative NAD+-depdendent reaction in sirtuins. Another breakthrough occurred to further understand SIRT6 function through discovery of the enzymatic activity and the first substrate of SIRT6: NAD+-depdendent deacetylation of histone H3 lysine 9. SIRT6 specifically deacetylates H3K9, but lacks activity on a lot of other histone tail residues due to its intense specificity.
Two groups were identified independently as the second substrate for SIRT6: lysine 56 of histone H3 (H3K56Ac).
To the core and beyond: biochemical dissection of SIRT6 function
[edit | edit source]Sirtuin proteins have a conserved central "sirtuin domain" flanked by N- and C- terminal extensions. The sirtuin domain supposedly has an enzymatic core and understanding this domain can show scientists the physiologic regulation of sirtuin proteins.
For SIRT6, a recent study showed that the N- and C- terminal domains regulate SIRT6 function by having the C terminus require proper nuclear localization (but is dispensable for enzymatic activity) and then the N terminus is beneficial for chromatic association and intrinsic catalytic activity.
Why is catalytic activity required for chromatin association in the cell?
- It could be possible that histone deacetylation by SIRT6 might be able to stabilize SIRT6 availability at chromatin or it can promote propagation of SIRT6 molecules along chromatin.
At the ends of chromosomes: SIRT6 regulates telomeric chromatin
[edit | edit source]SIRT6 plays an important role in the chromatin-regulatory context by keeping the integrity of telomeric chromatin stable. Telomeres are specialized DNA-protein structures which protect chromosome ends that are linear from degradation and fusion. SIRT6 plays a huge role at telomeres in humans for a couple of reasons:
- First, telomere structures need to be correct in order to maintain genomic stability; chromosomal instability is apparent in cancer cells.
- Also, telomere length decreases with cellular age. This shows that SIRT6's role at telomeres correlates with aging.
Conclusion
[edit | edit source]With many experiments and discoveries, SIRT6 has been determined as a site-specific histone deacetylase, playing very important roles in keeping up telomere integrity, honing aging-associated gene expression programs, preventing the genome to become unstable, and maintaining metabolic homeostasis.
Not only does SIRT6 function at specific sites in the genome, it plays a role in binding to additional gene promotors. Also, there might be interactions between SIRT6 and other sirtuin proteins.
Lastly, SIRT6 might have an impact on cancer due to the fact that there have been links between SIRT6 and cancer by the SIRT6 chromosomal locus.
References
[edit | edit source]Tennen, Ruth I., and Katrin F. Chua. "Chromatin regulation and genome maintenance by mammalian SIRT6." Trends in Biochemical Sciences 36.1 (2011) 39-46. Academic Search Complete. Web. 05 December. 2012. RNA is also known as ribonucleic acid. It is a part of most living organisms as well as viruses. It contains bases of Adenine, Cytosine, Guanine, and Uracil (instead of Thymine) which all bind to the ribose. RNA can be used to make DNA as well as synthesize proteins. It is the only polymer that can serve as a catalyst to the formation of proteins as well as storing genetic information. The RNA backbone is made of alternating ribose-phosphate groups. RNA can be found usually single stranded in humans, but can appear double stranded in many other organisms, including viruses.
Some viruses have RNA as their primary genetic material. They are known as RNA viruses. These viruses infect cells by first binding to a specific protein or receptor on the surface of the cell. After binding to the cell's surface, the virus injects its genetic material, or RNA, into the cell. The viral RNA, then, associates with the ribosomes of the infected cell. Essentially, a virus seizes control of its host's molecular machinery, uses the host cell's transcriptional abilities to produce viral proteins. The newly-made viral proteins then go on to produce new viruses. Furthermore, viral RNA can form replication complexes where it can copy itself. This newly-replicated RNA then gets packaged into the newly created viruses, which leads the cell to lyse, or break open. Consequently, these released viruses can go on to infect other cells.
RNA is nucleic acid, and its single-stranded, helical structure is constructed by nucleotides of nitrogenous bases, ribose sugar, and phosphate group; the bases are adenine, guanine, cytosine, and uracil, for which, 1’ nitrogen of pyrimidine base and 9’ nitrogen of purines base are bonded to 1’carbon of pentose sugar by glycosidic bond; base pairs of adenine and uracil and of cytosine and guanine are bonded by hydrogen bonds; the ribose is a pentose sugar of carbon numbered from 1’ to 5’ and has a hydroxyl group on the 2’ carbon; the 3’ and 5’ carbons of ribose sugar are bonded to phosphate group by phosphodiester bond; more importantly, the structure is of A-form geometry, which is constructed as of vast and thin major groove and of flat and broad minor groove, the structure can fold on itself to form secondary structure, such as tRNA and rRNA, and the secondary structure that are stabilized by hydrogen bonds, domains of loops, and metal ions, such as Mg 2+, form specific tertiary form.
Double Stranded RNA
[edit | edit source]Double Stranded RNAs, or dsRNA, are RNA's that have a complementary strand, similar to that of DNA. Many viruses are made from dsRNAs that infect a variety of hosts, ranging from animals, humans, fungi, plants, and bacteria. An RNA virus is a virus that contains only RNA as its genetic material, or whose genetic material passes through an RNA intermediate during replication. An example of a RNA virus is Hepatitis B, because even though it has a double-stranded DNA genome, the genome is transcribed into RNA during replication. An interesting fact about RNA viruses is that they have very high mutation rates since they lack DNA polymerases which is responsible for finding and editing mistakes. dsRNA's can also be synthetically produced by the process of in vitro and cloning using PCR to amplify the results. dsRNA's are responsible for the RNAi pathway.
Double strand RNA, dsRNA, is important because it helps regulating genes expression in eukaryotes cells. It triggers different gene silencing known as RNAi-Interfering RNA. Interfering RNA is a dsRNA that gets chopped off into a smaller fragments and binds to mRNA to block the gene expression. It also helps to reduce the production of gene’s encoded protein in order to get just right growth and reduce the self defense.
Structure
[edit | edit source]RNA is usually found in humans as a single stranded linear polymer. The monomeric units (nucleotides) linked together by 3'5' phosphodiester bridges. (A nucleoside is a ribose sugar connected to a base through the 1'C, while a nucleotide is a nucleoside plus a phosphate group connected to the 5'C of the sugar) The secondary structure of RNA is stabilized by Hydrogen bonds, intrastrand pairing of the bases (AU, GC) oftentimes resulting in structures such as hairpin loops. The stability of these loops depend on the number of unpaired bases in the loop, anything more than 10 or less than 5 is not very energetically favorable. There are oftentimes when the structure of RNA is not very stable because of the inability to match up Watson and Crick base pairs in the stem of the hair pin loops. Because it is single stranded, RNA will also fold into more complex structures, there are times when three nucleotides interact together to stabilize the structure. The Mg2+ stabilizes the structure when it is more elaborately structured. In these cases, there are often Hydrogen bond donors or acceptors that aren't already in Watson and Crick base pairs can interact and Hydrogen bond in 'irregular' pairing. Because of the extra hydroxyl group attached to the anomeric Carbon (the 2' Carbon), RNA is not as stable as DNA and will not form double helices as easily, although there have been cases of them found in some viruses. The 2' hydroxyl group on RNA also causes it to self hydrolyze. The hydroxyl group will attack the phosphorous which cleaves the phosophodiester bond on the 5' end. This instability also contributes to DNA being the preferred molecule for genetic storage in humans.
The technique of Northern blotting is often used to uncover the DNA sequence of a sample.
Types
[edit | edit source]There are many different types of RNA, and they carry out different function in the cell.
- mRNA
Messenger RNA
- Transcribes the DNA and is the template for the synthesis of protein. DNA + RNA polymerase makes mRNA.
- tRNA
Transfer RNA
- Brings the activated amino acids from other parts of the cell to the site of translation, or the ribosome. tRNA reads the information in th emRNA and translates that to amino acid. In other words, it translates information from the RNA to proteins.
- rRNA
Ribosomal RNA
- RNA that takes part in translating Messenger RNA into protein, constituent of ribosomes. rRNA is the most common and deals with the activity of the ribosome. rRNA deals with the formation of peptide bonds and is carried by this RNA in the ribosome.
- siRNA
- Small interfering RNA
- Bind to Messenger RNA and help them degrade.
- miRNA
Micro RNA
- Small non-coding RNA that inhibit translation of their complementary mRNA.
- snRNA
- small nuclear RNA
- Responsible for the sorting of proteins by removal of the introns (splicing) from hnRNA as well as maintaining telomeres
- RNAi
- Interference RNA
- inhibition of gene expression by cutting up mRNA.
- Structural insights into RNA interference.
The structures of these different types of RNA will vary depending on what they are supposed to do. The tertiary structure varies by function. Even in the simplest sense, some will be relatively long strands of nucleic acids, such as Messenger RNA up to 1.2 kilobases, while others are relatively short sequences of 21 nucleotides such as miRNA.
References
[edit | edit source]http://science.jrank.org/pages/5892/RNA-Function.html">RNA Function http://en.wikibooks.org/wiki/Structural_Biochemistry/Nucleic_Acid/Nitrogenous_Bases/Purines/Adenine http://en.wikibooks.org/wiki/Structural_Biochemistry/Nucleic_Acid/Nitrogenous_Bases/Purines/Cytosine http://en.wikibooks.org/wiki/Structural_Biochemistry/Nucleic_Acid/Nitrogenous_Bases/Purines/Guanine http://en.wikibooks.org/wiki/Structural_Biochemistry/Nucleic_Acid/Nitrogenous_Bases/Purines/Uracil http://en.wikibooks.org/wiki/Structural_Biochemistry/Nucleic_Acid/Nitrogenous_Bases/Purines/Thymine http://en.wikibooks.org/wiki/Structural_Biochemistry/Nucleic_Acid/DNA http://en.wikibooks.org/wiki/Structural_Biochemistry/Nucleic_Acid/RNA/Transfer_RNA_(tRNA) http://en.wikibooks.org/wiki/Structural_Biochemistry/Nucleic_Acid/RNA/Ribosomal_RNA_(rRNA) http://en.wikibooks.org/wiki/Structural_Biochemistry/Nucleic_Acid/DNA/Replication_Process/DNA_Polymerase http://en.wikibooks.org/wiki/Structural_Biochemistry/Nucleic_Acid/RNA/Interference_RNA http://en.wikibooks.org/wiki/Structural_Biochemistry/Nucleic_Acid/RNA/Messenger_RNA_(mRNA) http://en.wikibooks.org/wiki/Structural_Biochemistry/Nucleic_Acid/RNA/MicroRNA_(miRNA) http://en.wikibooks.org/wiki/Structural_Biochemistry/Nucleic_Acid/RNA/Structural_insights_into_RNA_interference_(RNAi) Viadiu, Hector. "Types of RNA." UCSD. Lecture. November 2012.
| This page or section is an undeveloped draft or outline. You can help to develop the work, or you can ask for assistance in the project room. |
| This page or section is an undeveloped draft or outline. You can help to develop the work, or you can ask for assistance in the project room. |
Messenger ribonucleic acid (mRNA) is the blueprint of protein reproduction. Transcribed from deoxyribonucleic acid (DNA), mRNA transfers genetic information from the cell nucleus into the protein-producing ribosomes located in the cytoplasm. Similar to DNA, the genetic information is encoded in four nucleotides that are arranged in codons, or triplets of nucleotide bases. Each codon corresponds to a specific amino acid, and the sequence of codons ends with a codon that has a stop signal. The protein synthesis process requires transfer RNA (tRNA) and ribosomal RNA (rRNA). mRNA makes up only about 5% of the different types of RNA found in both Prokaryotic and Eukaryotic cells.

Transcription
[edit | edit source]During transcription, an RNA strand is copied by an enzyme, RNA polymerase. RNA is then synthesized in the 5' to 3' direction, as is also done in DNA replication. The template of the two DNA strands is the one in which the RNA is synthesized. RNA polymerase binds to the 3' end and replicates via phosphodiester bonds.
The obvious difference between DNA and mRNA in this stage is in the uracil (U) that is present in RNA instead of thymine (T) in DNA.
The RNA first transcribed from the DNA is known as pre-messenger RNA (pre-mRNA) since the exact copy of the DNA region contains both introns and exons. Messenger RNA contains only exons. Introns are removed via splicing by spliceosomes, which recognize intronic sequences based on a GU beginning, a long pyrimidine chain, and an AG ending. Only exons remains in mRNA mainly because it contains useful genetic information for translation - producing a protein. Introns, however, do not provide useful genetic information.
caps and PolyA tails are added as modification to protect the active ends of mRNA after transcription and before translation.
Pre-mRNA
[edit | edit source]In eukaryotes, the product of transcription of a protein-coding gene is pre-mRNA which requires processing to generate functional mRNA. Several processing reactions occur.
5'processing: capping
[edit | edit source]Very soon after it has been synthesized by RNA polymerase II, the 5' end of the primary RNA transcript, pre-mRNA, is modified by the addition of a 5' cap(a process known as capping). This process involves the addition of 7-methylguanosine(m7G) to the 5'end. To achieve this, the terminal 5' phosphate is first removed by a phosphatase. Guanosyl transferase then catalyzed a reaction whereby the resulting diphosphate 5' end attacks the α phosphorus atom of a GTP molecule to add a G residue in an unusual 5'5' triphosphate link. The G residue is then methylated by a methyl transferase adding a methyl group to the N-7 position of the guanine ring, using S-adenosyl methionine as methyl donor. The ribose of the adjacent nucleotide (nucleotide 2 in the RNA chain) or the riboses of both nucleotides 2 and 3 may also be methylated to give cap 1 or cap 2 structures respectively. In these cases. the methyl groups are added to the 2'-OH groups of the ribose sugars.
The cap protects the 5' end of the primary transcript against attack by ribonucleases that have specificity for 3'5' phosphodiester bonds and so cannot hydrolyze the 5'5' bond in the cap structure. In addition, the cap plays a role in the initiation step of protein synthesis in eukaryotes. Only RNA transcripts from eukaryotic protein-coding genes become capped; prokaryotic mRNA and eukaryotic rRNA and tRNAs are uncapped.
Splicing
[edit | edit source]RNA splicing is a key step in RNA processing because it precisely remove the intron sequences and join the ends of neighboring exons to produce a functional mRNA molecule. The exon-intron boundaries are marked by specific sequences. In most cases, at the 5' boundary between the exon and the intron(the 5' splice site), the intron starts with the sequence GU and at the 3'exon-intron boundary (the 3' splice site) the intron ends with the sequence AG. Each of these two sequences lies within a longer consensus sequence. A polypyrimidine tract (a conserved stretch of about 11 pyrimidines) lies upstream of the AG at the 3' splice site. A key signal sequence is the branchpoint sequence which is located about 20-50 nt upstream of the 3' splice site. In vertebrates this sequence is 5'-CURAY-3' where R=purine and Y=pyrimidine (in yeast this sequence is 5'-UACUAAC-3'). RNA splicing occurs in two steps. In the first step, the 2'-OH of the A residue at the branch site attacks the 3'5' phosphodiester bond at the 5' splice site causing that bond to break and the 5' end of the intron to loop round and form an unusual 2'5' bond with the A residue in the branchpoint sequence. Because this A residue already has 3'5' bonds with its neighbors in the RNA chain, the intron becomes branched at this point to form what is known as a lariat intermediate (named as such since it resembles a cowboy's lasso). The new 3'-OH end of exon 1 now attacks the phosphodiester bond at the 3' splice site causing the two exons to join and release the intron, still as a lariat. In each of the two splicing reacitons, one phophate-ester bond is exchanged for another (i.e. these are two transesterification reactions). Since the number of phosphate-ester bond is unchanged, no ATP is consumed.
3' processing:cleavage and polyadenylation
[edit | edit source]A majority of eukaryotic pre-mRNAs undergo polyadenylation which involves cleavage of the RNA at its 3' end and the addition of about 200A residues to form a poly(A)tail. The cleavage and polyadenylation reactions require the existence of a polyadenylation signal sequence (5'-AAUAAA-3') located near the 3' end of the pre-mRNA followed by a sequence 5'-YA-3' (where Y=a pyrimidine), often 5'-CA-3', in the next 11-20 nt. A GU-rich sequence (or U-rich sequence) is also usually present further downstream. After these sequence elements have been synthesized, two multisubunit proteins called CPSF (cleavage and polyadenylation specificity factor) and CStF (cleavage stimulation factor F) aretransferred from the CTD of RNA polymerase II to the RNA molecule and bind to the sequence elements. A protein complex is formed which includes additional cleavage factors and an enzyme called poly(A) polymerase (PAP). This complex cleaves the RNA between the AAUAAA sequence and the GU-rich sequence. Poly(A) polymerase then adds about 200A residues to the new 3' end of the RNA molecule using ATP as precursor. As it is made, the poly(A) tail protects the 3' end of the final mRNA against ribonuclease digestion and hence stabilizes the mRNA. In addition, it increases the efficiency of translation of the mRNA. However, some mRNAs, notably histone pre-mRNAs, lack a poly(A) tail. Nevertheless, histone pre-mRNA is still subject to 3' processing. It is cleaved near the 3' end by a protein complex that recognizes specific signals, one of which is a stem-loop structure, to generate the 3'end of the mature mRNA molecule.
The primary RNA transcript that continues to be synthesized includes both coding(exon) and noncoding(intron) regions. The latter need to be removed and the exon
Transport
[edit | edit source]In Eukaryotic cells, following synthesis, mRNA typically goes through a series of modifications before being exported to the cytoplasm for translation. These modifications include a 5’ guanine capping and a polyadenylation at the 3' end. This strand of Adenine residues (anywhere from 80-250) is called the Poly-A tail and is needed for the export, protection, translation, and stability of the mRNA. Splicing, the process in which introns are removed and exons are joined, also occurs before exportation.
After all the proper modifications have been carried out, the mature mRNAs are ready to be exported through the nuclear pore into the cytoplasm. Nuclear pores are the channels between the nucleus and cytoplasm, and is a selective barrier that allow macromolecule transportation. Alternate splicing patterns of introns allows the same gene to express in a slightly different way in mRNA creating a different, but similar protein. In order for the mature mRNAs to be carried out, first, the formation of the messenger ribonucleoprotein (mRNPs) export complex with RNA binding proteins and transport factors (carriers) must occur since Mex67-Mtr2 heterodimer, the principal mRNA carrier, binds loosely to bulk mRNA.
Nuclear Transport
[edit | edit source]Nuclear export is a pathway unique to eukaryotic cells because the nuclear and cytoplasmic compartments within the eukaryotic cells enables spatial separation of the two processes, transcription and translation. The separation between the two processes allows for multiple steps in between for further modification and gene expression regulation, which becomes vital for physiological responses to extra- and intracellular signals.
mRNA nuclear export can be simplified to three stages:
- 1) the pre-mRNA is transcribed in the nucleus, the site of mRNA synthesis, processing, and packing into mRNP (messenger ribonucleoprotein) complexes (as briefly described earlier)
- 2) the mRNP molecules are targeted to and translocated through the nuclear pore complexes (NPC) of the nuclear envelope
- 3) the mRNPs are released into the cytoplasm for translation to occur. Each of these stages involves numerous protein factors and other molecules that need to be recruited to carry out processes.
Formation of mRNP in yeasts:
[edit | edit source]- 1) In the nucleus, transcription is mediated through RNA polymerase II. This is followed by modifications like the addition of the 5’ cap, splicing, and 3’ processing. The TREX complex is recruited during these processes and coordinates many of the next steps.
- 2) The 3’ end processing is necessary because it generates the poly-A-tail which is crucial for the mRNA to be exported. This process requires the factors Rna14, Rna15 and Pcf11. Nab 2 is added onto the poly-A-tail mRNA then recruits Yra1 and Sub 2 during this time. When mRNA is in contact with Pcf11, Yra1 is transferred to the TREX subunit Sub2. (Yra1-Pcf11 binding is an important early step). Yra1 is necessary
- 3) The MEx67-Mtr2 heterodimer is drafted.
- 4) mRNPs can now be remodeled by tha DEAD-box helicase Sub2
- 5) Yra1 dissociates itself from mRNP before export, along with the TREX complex.
- 6) mRNP is drawn to the nucleus side of the NPC transport channel, where weak interactions arise with FG nucleoporins (proteins that perforate the nuclear pore). To increase the efficiency of export, several mechanisms exist to concentrate mRNAs at the nucleus side of the NPC. Eg: several actively transcribing genes like GAL1 are concentrated at the NPC.
- 7) mRNP goes through the NPC transport channel, to the cytoplasmic side, where is once again goes through remodeling to prevent from going back into the nucleus.
Cofactors Involved
[edit | edit source]The NPCs itself have very essential proteins that facilitate mRNA nuclear export. Within the NPC, there is a conical, basket-like feature that protrudes into the nucleus called the nuclear basket. It contains proteins like Nup 60 Nup2, and Mlp2. The cytoplasm similarly has proteins that are cofactors to the export process (Nup1259, Dbp5, Gle1). There are several other key proteins and components of mRNA export that will not be discussed, but they the references for this page will provide much more insight on the specific functions of these export factors.
Here is a short summary of the principle export factors for yeast and metazoans:
- Mex67-Mtr2 (yeast) and Nxf1-nxt1 (metazoan): facilitate bulk mRNA export through NPCs
- Yra1 (yeast) and ALY (metazoan): Adaptor linking Mex67-Mtr2 to mRNA molecule
- Sub2 (yeast) and UAP56 (metazoan): DEAD-box helicase involved in assembly of export-competent mRNPs
- Nab2 (yeast): Binds the poly (A) til of mRNA to Mlp1 and regulates length of the 3' poly (A) tail
- Mlp1 (yeast) and (TPR): Nuclear basket protein that binds to Nab2
- TREX (both yeast and metazoan): The complex involved in coordinating and regulating transcription
- TREX-2 (both): directs actively expressing genes to NPCs
- Gle1 and Gdf1 (yeast) and GLE (metazoan): enhances Dbp5 activity
- Nup159 (yeast) and NUP214 (metazoan): cytoplasmic NPC protein that binds to Dbp5
Recruitment
[edit | edit source]Recruiting these factors is an essential step for the trafficking and quality control of the export. Most molecules that need to be transported from the nucleus into the cytoplasm involve karyopherin-mediated receptors, like small mRNA export. Its transport direction is based on the gradient of the GTP-bound state of the small GTPase Ran, making the mRNA export process uncharacteristic of normal protein export such as tRNA. Bulk mRNA is exported using Mex67-Mtr2, a non-karyopherin-mediated receptor, via the Nxfl pathway. The Mex67-Mtr2 molecule is recruited to the mRNP using the TREX component. Furthermore, recent works in vertebres shows that the binding of the Yra1 homologue, ALY, to mRNA is stimulated by the presence of the ATP bound form of the Sub2 homologue UAP56. This binding increases the ATPase activity of UAP56. Moreover, Nxf1 binds mRNA associated ALY, forming a ternary complex, and the RNA-binding affinity of Nxf1 is increased in the presence of ALY. Taken together, the events result in an mRNP with bount export receptors. But it is unclear how many receptors must bind a single mRNA for efficient export to occur.
Bulk mRNA Export Pathway
[edit | edit source]The Nxfl pathway involves a small set of transcripts that are exported via karyopherin Crm1, a protein that also mediates the export of incompletely spliced mRNA from HIV viruses. Therefore, if an mRNA molecule is not properly processed and spliced of its introns, it can be kept in the nucleus to degrade since it is recognized as a viral mRNA molecule. When the mRNP and mRNA are properly processed and have recruited all the necessary receptors and cofactors, it is considered export ready (export competent). The export-competent mRNP is then targeted only to the NPC using its recruited export receptor. The export receptor carries the mRNP to the NPC where it stays and interacts with the NPC proteins to allow recognition. The interactions can be nicely summarized in the figure below.
Bulk Release into Cytoplasm
[edit | edit source]The directionality of the bulk mRNA release is determined by another mechanism since it does depend on the RanGTP gradient for small mRNA export. It is determined by the function of two important export factors, Dbp5 and Gle1. The Dbp5 protein binds to the NPC cytoplasmic face by interacting with the NPC protein Nup214 As the mRNP comes closer to the cytoplasmic side of the NPC, it interacts with Dbp5 and Gle1. The binding and interactions between mRNP and the two proteins causes a conformational change and activates the removal of a set of proteins from the mRNP. It physically and spatially changes the mRNP making it suitable to be exported out of the NPC into the cytoplasm. These removed proteins are recycled and brought back into the nucleus where it goes through another cycle of mRNA export. In addition, as the mRNP enters the cytoplasm, specific cytoplasmic mRNA-binding proteins are incorporated. These specific links to translation further show the inherent connections between steps in gene expression.
Translational Significance
[edit | edit source]Since mRNA export is essential for proper gene expression, this process must be properly conducted. Incorrect steps in this export can lead to errors in transcription, and consequently translation. For example, errors in recruiting export factors can lead to incorrect mRNA production, and if the transcript is not recognized by nuclear surveillance the mRNA may be kept inside the nucleus and degraded by exosomes and various other enzymes. Errors in mRNA export can also be linked to many human diseases and developmental issues. Incorrect mRNA export are connected to perturbations that yield mutations in gene encoding export proteins or mRNA-binding proteins as well as mutations in genes that result in the inhibition of correct export of their own mRNA transcripts. Extreme cases also include the decreased regulation or hijacking of endogenous mRNA export complexes by viruses, which enables specific viral genes to hybridize with the mRNA transcript and be expressed in the organism. But with the vast knowledge of the mRNA export process, these malfunctions can be better understood and more easily preventable, and it may be possible to address many issues of diseases and gain a complete understanding of the way cellular function is generated at the simplest level: molecularly.
Translation
[edit | edit source]In prokaryotes, because the mRNA does not need to be modified or transported, it can be translated by the ribosomes right after transcription.
In eukaryotes, however, mRNA can only be translated after it has been modified and transported to the cytoplasm (the mature mRNAs). mRNA is translated into proteins on the ribosomes located on the endoplasmic reticulum. Translation starts by the ribosomes binding to a site on the 5' side. The ribosome moves along the mRNA until it comes across the start codon AUG. When this binding occurs, the ribosome is joined by an initiator tRNA that carries a formylmethionine (fMet) group that recognizes the start codon. Next, an aminoacyl-tRNA that can base pair with the next codon appears and joins the ribosome complex. Along with the aminoacyl-tRNA is the elongation factor EF-Tu (in bacteria) and a source of energy (usually GTP). The fMet (in bacteria) or Met group covalently bonds to the incoming amino acid of the aminoacyl-tRNA. The initiator tRNA is then released and the ribosome shifts one codon toward the 3' end. A new aminoacyl-tRNA arrives and the amino acid of this aminoacyl-tRNA binds to the previous amino acid. This process continues until the ribosome reaches a stop codon (UAA, UAG, or UGA). The newly bound amino acids are the translated mRNA into a protein. The ribosomal complex containing the tRNA splits back up into its separate parts, re-assembling when new mRNA needs to be translated into protein.
The elongation process "terminates" when a stop codon reaches the A site of the ribosome. Incoming tRNA, which carries the subsequent amino acid, will not be accepted by the ribosome at the A site. The A site will then be specific to a protein called the release factor. The release factor will hydrolyze the bond of the tRNA to the polypeptide in the P site, thus releasing the polypeptide chain. The two ribosomal subunits, release factor, and mRNA then come apart to signify the end of the termination process.
- Stop Codon - A stop codon implies a sequence of three nitrogenase bases in the mRNA that signifies the termination of polypeptide elongation, or translation. The amino acid sequence is then released from the mRNA template to form its final 3D conformation.
| Stop Codon | Sequence |
|---|---|
| RNA | UAA |
| RNA | UAG |
| RNA | UGA |
| DNA | TAA |
| DNA | TGA |
| DNA | TAG |

Editing
[edit | edit source]An mRNA can be changed its nucleotide composition in some instances. This process is called editing. In human, the apolipoprotein mRNA is one of the cases. This editing mRNA takes place in some tissues, but not all of them. In this edition, the mRNA's codon is given an early stop, therefore, it will produce a shorter protein when going to the translation process.
Alteration of mRNA sequence through base modification mRNA editing frequently generates protein diversity. Several proteins have been identified as being similar to C-to-U mRNA editing enzymes based on their structural domains and the occurrence of a catalytic domain characteristic of cytidine deaminases. In light of the hypothesis that these proteins might represent novel mRNA editing systems that could affect proteome diversity, we consider their structure, expression and relevance to biomedically significant processes or pathologies.
Degradating
[edit | edit source]The message transported through mRNA after a certain amount of time will be degraded and be deleted. This process is called degradation. The cell can easily and quickly changed the protein production in case of any changing needs due to the lifetime of the mRNA. The lifetime of different types of mRNA can be different.The life span of mRNA molecules in the cytoplasm is an important key in determining the pattern of protein synthesis within a cell. Prokaryotic mRNA molecules often are degraded by enzymes within a few minutes of their synthesis and this is one reason as to why prokaryotes can vary their patterns of protein synthesis so quickly in response to changes in their environment. Eukaryotic mRNA, on the other hand, typically survives for hours, days, or for some instances, weeks. One example of multicellular mRNA is hemoglobin polypeptides which, in the process of developing red blood cells which are unusually stable, these long-lived mRNAs are translated repeatedly in the cell. Research done on yeasts suggest that a common pathway for mRNA degradation begins with the enzymatic shortening of the poly-A tail which helps trigger the action of enzymes that remove the 5’ cap. This removal of the 5’cap end is crucial as it is regulated by particular nucleotide sequences in the mRNA. Once the cap is removed, nuclease enzymes can then move in and rapidly chew up the mRNA. This process of mRNA degradation relies on deadenylation. The shortening of poly-A tail is initiated by deadenylase and afterward, mRNA is either fully degraded or stored in the case of certain cells.
Another mechanism that blocks expression of specific mRNA molecules known as MicroRNA (miRNA) or miRNAs have also become of interest. They are formed from longer RNA precursors that fold back on themselves, forming a long, double-stranded hairpin structure held together by hydrogen bonds. These small singled stranded RNA molecules can bind to complementary sequences in mRNA molecules and an enzyme, called the Dicer, can then cut the double-stranded RNA molecules into short fragments. One of the two strands is degraded and then the other stand, often the miRNA associates with a large protein complex and which allows the complex to bind to any mRNA molecule with a complementary sequence to either degrade or block translation of mRNAs.
Scientists also observed that gene expression inhibited by RNA molecules was possible. This was observed when they noticed that injecting double stranded RNA molecules into a cell somehow turned off a gene with the same sequence. Scientists called this phenomenon RNA interference or Interference RNA (RNAi). It was later discovered that this interference was due to small interfering RNAs (siRNAs) which are RNAs of similar size and function as miRNAs. Researched showed that the cellular machinery for making siRNAs was the same mechanism for creating miRNAs in the cell. The mechanisms by which these small RNAs function are also the same. Because the cellular RNAi pathway can lead to the destruction of RNA sequences complementary to themselves, it is believed that they originally acted as a natural defense against infection by RNA viruses.
References
[edit | edit source]- David Hames, Nigel Hooper. Biochemistry. Third edition. Taylor and Francis Group. New York,2005.
- Neil A. Campbell, Jan B Reece. Biology Seventh Edition, 2005 Pearson Education, Inc.
Nuclear export of mRNA. Murray Stewart. MRC Laboratory of Molecular Biology, Hills Rd., Cambridge CB2 0QH, UK
- Fabienne Mauxion, Chyi-Ying A. Chen, Bertrand Séraphin, and Ann-Bin Shyu. "BTG/TOB factors impact deadenylases." *http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2787745/?tool=pubmed
- http://en.wikipedia.org/wiki/Release_factor
- http://en.wikipedia.org/wiki/Stop_codon
- http://en.wikipedia.org/wiki/mRNA
- http://en.wikibooks.org/wiki/Structural_Biochemistry/Nucleic_Acid/Transcription
- http://en.wikibooks.org/wiki/Structural_Biochemistry/Nucleic_Acid/RNA/RNA_Polymerase
- http://en.wikibooks.org/wiki/Structural_Biochemistry/Nucleic_Acid/RNA/MicroRNA_(miRNA)
- http://en.wikibooks.org/wiki/Structural_Biochemistry/Nucleic_Acid/RNA/Interference_RNA
- Nuclear export of mRNA. Stewart M. Trends Biochem Sci. Epub 2010 Aug 16. 17 November 2012. <http://www.sciencedirect.com/science/article/pii/S0968000410001337#>
- Nuclear Export of RNA. Manuel S. Rodriguez, Catherine Dargemont, Françoise Stutz. Biology of the Cell. 03 August 2004. 17 November 2012. <http://submit.biolcell.org/boc/096/0639/boc0960639.pdf>
- mRNA nuclear export at a glance. Sean R. Carmody and Susan R.Wente. The Company of Biologists. 2009. 17 November 2012. <http://jcs.biologists.org/content/122/12/1933.full.pdf>
Structural Biochemistry/Nonsense-Mediated mRNA decay
Overview
[edit | edit source]Transfer RNA (tRNA) have a primary, secondary, and tertiary (L-shaped) structure. tRNA bonds to activated amino acids and transfers them to the ribosomes. Once at the ribosome, an initiator tRNA binds the amino acid to the ribosome to start translation. It carries the amino acids and binds to the Messenger RNA (mRNA) to form proteins.
tRNA's structure contains an amino acid attachment-site and a template-recognition site. The template-recognition site is called a anticodon and contains a sequence of three bases that are complementary to the codon on the mRNA. tRNA travels from nucleus to cytoplasm in a cell. Each tRNA can be used repeatably to be transcribed from DNA in nucleus.
There are 61 different anticodon sequences which code for the 20 amino acids. However, most prokayotic cells only have 30-40 different tRNAs and eukaryotes have about 50 different tRNAs. This is the third nucleotide of the codon, also called a wobble base, allows wobble pairing of the anticodon to the codon.

Role in Protein Synthesis
[edit | edit source]In protein synthesis, a tRNA molecule takes a specific activated amino acid to the site. The amino acid is esterified to the 3' or 2' -hydroxyl group of the terminal adenylate of tRNA. This joining of tRNA and an amino acid forms an aminoacyl-tRNA and is catalyzed by a specific enzyme called aminoacyl-tRNA synthetase (aaRS). There are 20 aminoacyl-tRNA synthetase, one for each amino acid. Similarly, there is a specific aaRS for each tRNA. The esterification reaction also called charging of the tRNA is powered by ATP.
The process of protein synthesis starts out when a charged tRNA (a tRNA with an amino acid attached), mRNA, and the small and large ribosomal subunits come together and form the initiation c complex, which consists of a peptidyl binding site (P site) and an aminoacyl binding site (A site). The first tRNA, otherwise known as the initiator RNA, binds to the mRNA start codon, AUG; thus, the first amino acid in the chain is methionine. To add additional amino acids to the polypeptide chain, a second charged tRNA must come in and have its anticodon bind to the next mRNA codon in the vacant A site. The P site and A site are in close proximity, thus allowing a formation of a stable peptide bond by reacting the carboxy terminus of the amino acid in the P site with the amino terminus of the amino acid on the tRNA in the A site. The reaction is catalyzed by peptidyl transferase. The complex moves along the RNA in a process called translocation which causes the tRNA in the P site to be displaced. The tRNA in the A site then moves into the P site so another charged tRNA can move into the A site. This process continues until the stop codon is reached the polypeptide chain is released from the ribosome.
1. amino acid + ATP --> aminoacyl-AMP + PPi 2. aminoacyl-AMP + tRNA --> aminoacyl-tRNA + AMP
tRNA Structure
[edit | edit source]
2. Secondary Structure
[edit | edit source]The secondary structure is formed like cloverleaf structure because of four base-paired stems also called arms. The cloverleaf contains three non-base-paired loops: D, anticodon, and TpsiC loop. The terminal CCA is not base paired. It's duplexed between the 5'segment and 3'segment.
The acceptor stem which is not a loop is the site where the enzyme amino-acyl-tRNA synthase attaches an amino acid. It is located opposite of the anticodon arm which reads the mRNA.
There are different types loops. In D loop, D arm ends. Anticodon arms ends in anticodon loop. In the figure, it shows hydrogen bond present inside the loop structure. The hydrogen bonds stabilized the structure.
3. Tertiary Structure
[edit | edit source]For the tertiary structure, it can be described as a compact of L shape. It is three dimensional. The structure is bonded and stabilized by base pairing and base stacking. Base pairs between nucleotides in the D loop and the TΨC loop. At the end of the L shape is the three base sequence called anti codon.
Anticodon
[edit | edit source]The anticodon region of a transfer RNA is a sequence of three bases. They are complementary to a codon in the messenger RNA. In the translation, the pairing between its anticodon and the messenger codon brings the ribosome. The amino acid is attached at its 3' end. And it will be peptide bond. In prokaryote cells, there are about 35 tRNAs with different anticodons present. In eukaryote cells, there are 50 tRNAs with anticodons present. tRNA with the anticodon CCC is complementary to the anticodon GGG. The anticodon AAA is complementary to the anticodon UUU. Since each type of tRNA has a different one, the anticodon of tRNA is able to identify others well.
tRNA Aminoacylation
[edit | edit source]Aminoacyl-tRNA is an amino acid ester of tRNA. It can be called a charged tRNA. When a polypeptide chain is formed by the anticodon of the tRNA, the reaction is thermodynamically unfavorable. So, aminoacyl-tRNA is used to activate the formation. An amino acid is esterified to the 3'-end of a tRNA containing the corresponding anticodon in amynoaclyation of tRNA molecules. As a result, the aminoacyl-tRNA attaches amino acids to the tRNA. These paring of amino acids and tRNAs define the genetic code. The aminoacyl-tRNA synthestase(AARSs)catalyze the aminoacylation of tRNAs. During transfer the genetic information from the nucleotide sequence of a gene to the amino acid sequence of a protein, this process plays an important role. When errors occur, amynoacyl-tRNA synthetases edit mechanisms structurally. Further, it prevents the error synthesis and releases aminoacylated tRNA that shouldn't be placed.
Aminoacyl tRNA synthetases (AARSs)
[edit | edit source]AARSs is an enzyme that catalyzes the esterification of specific amino acid to a tRNA to form an aminoacyl tRNA. AARSs take a major role in translation during protein synthesis. In recent researches, scientists discovered that AARSs also take role in ex-translation.
Role of AARSs in translation
[edit | edit source]The accuracy of the protein translation depends on the exactness of AARSs' recognition of both the amino acid to be activated and the cognate tRNA molecules. That is a crucial step in the fidelity of the translation. All AARSs carry out the same two-steps reaction:
- Step 1: AARSs binds ATP to the amino acid to induce an aminocyl-adenylate intermediate in which a covalent linkage between the 5'-phosphate in ATP and the carboxyl-end of amino acid.[4][5] Next, the AARSs use the generated energy from ATP hydrolysis to activate the amino acid which results in the formation of aminoacyl-AMP as an energy storage.[6]
- Step 2: The amino acid is transferred to the appropriate tRNA and bind either 2'OH or 3'OH of the 3' adenosine terminal of tRNA covalently. The energy that stored in aminoacyl-AMP is used to transfer the amino acid to the tRNA to form aminoacyl tRNA.
Role of AARSs in ex-translation
[edit | edit source]Modified version of AARSs and natural fragments take role in ex-translational functions as confirmed in recent studies. The interplay of AARSs appears to be at the center of homeostatic mechanisma which controls angiogenesis, inflammation, metabolism, and tumorigenesis.[7] Through some recent experiments, ex-translational functions of AARSs was found to be the interplay between natural extracellular fragments of human TrpRS and TyrRS in angiogenesis. TyrRS was found to have a nuclear localization signal that is controlled by its cognate tRNA (called tRNA-Tyr) so that a decrease in level of tRNA-Tyr will increase the level of nuclear import of the AARSs which will induce effects on many gene regulatory mechanisms. In contrast, an increase in level of tRNA-Tyr will decrease the level of TyrRS. Thus, the subcellular distribution of TyrRS is directly controlled by the demands of protein synthesis and this control is an example of homeostatic mechanism that balances a translational with an ex-translational functions.[8].
However, recent reseaches also showed that the ex-translational function of AARSs is regulated which is a contrast to the discussion above. This regulation is considered an auto-balancing process in which a natural fragment takes control in the activity of its own original protein. Thus, further research is needed to confirm the specific role of AARSs in ex-translational functions.
Binding to Ribosome
[edit | edit source]
tRNA will bind at the A, P and E sites of ribosomes. The A site will bind to aminoacyl-tRNA which was signaled by the codon that is binding to that site. The codon will also signify the next correct amino acid that will be in the peptide chain. But the A site will only work when the P site has an aminoacyl-tRNA attaching to it. The P-site is actually occupied by a chain with a few amino acids called peptidyl-tRNA. It carries synthesized amino acid chains. Lastly, the E site carries the empty tRNA.
Three dimensional image of a tRNA.
Coloring:
ORANGE: CCA tail
PURPLE:Acceptor stem
RED: D arm
BLUE Anticodon arm
BLACK: Anticodon
GREEN: T arm
Diseases Caused by Mitochondrial tRNA Gene Mutation
[edit | edit source]Mitochondria are an organelle in the cell, which contains 22 tRNA. Gene mutation of tRNA will cause serious diseases. There are seven kinds of genes diseases caused by mitochondrial tRNA gene mutation:

1- np5601 G->A and np3243 A->G gene mutations related to MELAS (Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes). Most patients get this disease before 40-year-old with epilepsia and lactic acidosis. Some of them will die during 20~30 age.
2- np8363 G->A, np8356 T->C, and np8344 A->G gene mutations related to MERRF (Myoclonic Epilepsy with Ragged Red Fibers). MERRF affects central nerve system, causing epilepsia, Dementia and epicophosis.

3- np4274 T->C gene mutation related to LIMM (Lethal Infantile Mitochondrial Myopathy). Most patients are newborn, having nerve defect and lactic acidosis, and die in one month.
4- np1644 G->T gene mutation related to subacute necrotizing encephalomyelopathy (SNE). This disease is familial autosomal recessive inheritance, happened to newborn baby.
5- np606 A->G gene mutation related to Rhabdomyolysis. Toxin produced by muscle cells is the main reason that causes Rhabdomyolysis.
6- np4500 G->A gene mutation related to the splenic lymphoma. The splenic lymphoma is a common malignant tumor happened on spleen. Normally, the splenic lymphoma caused by advanced stage lymphoma transfer.
7- np4336 A->G, np15927, and np15928 gene mutations related to Parkinson's Disease and Alzheimer’s Disease. Parkinson's Disease is a degenerative disorder of the central nervous system.
Reference:
1. Inheritance of Mitochondrial Disease: http://wenku.baidu.com/view/fe5a99ea172ded630b1cb69b.html
2. Diseases of Human Mitochondria tRNA: http://wenku.baidu.com/view/95fabd1fa300a6c30c229f93.html
3.http://baike.baidu.com/view/1120527.html
4.http://baike.baidu.com/view/2219175.html
5.http://en.wikibooks.org/wiki/Structural_Biochemistry/Nucleic_Acid/Translation
6. http://en.wikibooks.org/wiki/Structural_Biochemistry/Nucleic_Acid/RNA/Messenger_RNA_(mRNA)
7. http://www.wiley.com/college/boyer/0470003790/structure/tRNA/trna_intro.htm
| This page or section is an undeveloped draft or outline. You can help to develop the work, or you can ask for assistance in the project room. |
Ribosomal RNA, also known as rRNA, is a significant component of the ribosome. rRNA fabricates the polypeptides and provides a mechanism for decoding mRNA into amino acids and interacts with the tRNA during translation. rRNA was once known to be the key structural component of ribosomes, but its actually found to be a catalytic element for protein synthesis. It is the most abundant type of RNA (about 80%) in the cell.

rRNA is comprised of a large and small subunit. Prokaryotic rRNA is 70 svedbergs large. A svedberg is a unit of measurement for the sedimentation coefficient or how fast the molecule sediments when centrifuged. The 70S rRNA contains a large 50S subunit which includes a 23S and 16S subunit and a small 30S subunit which contains a 5S subunit. The 23S, 16S and 5S units are essential during protein synthesis, and the structure and function of the ribosomes. The formation of these RNAs take place by cleaving the primary 30S subunits and processing further by folding the molecule to form internal base pair structures. Experiments involving chemical probing methods have been conducted which have provided a detailed model of the secondary structure of the 16S subunit. The secondary structure was obtained through analyzing and comparing the sequences. Proteins containing the 16S ribosomal rRNA can fold and form the 30S subunit.The conformational change of the 16S ribosomal rRNA plays a crucial role in the assembly of the ribosome. The 5S unit found in the 30S subunit is an important part of the large subunit of most ribosomes found in organisms. rRNA is the most abundant of the three major types of RNAs with a 80% relative amount in E. coli for example, following by tRNA (15%) and finally mRNA (5%). Ribosomal RNA has a mass of 1.2 x 10^3 kd and 3700 number of nucleotides in E. coli.
With the help of x-ray crystallographic technique, scientists are able to reveal the detailed features of secondary structures.
The use of Polymerase Chain Reaction (PCR) has been of great importance in the amplification of rRNA genes. PCR is used to amplify rRNA genes in many organisms, however, it is found that the amplification of rRNA genes via traditional PCR methods cannot be conducted in extremely thermophilic organisms.
rRNA contains two tRNA binding sites, an A site and a P site. At the A site, the rRNA binds to a aminoacyl-tRNA, a tRNA bound to an amino acid. The amino acid is transferred to a peptidyl-tRNA containing the growing peptide chain. After the amino acid is added, the empty tRNA is moved to the P binding site where it is ejected. The mRNA then shifts 3 bases (1 codon) for the next aminoacyl-tRNA to bind to the A binding site.
In prokaryotes, rRNA are formed by cleavage and other modifications of nascent RNA chains. Therefore precursors of transfer and ribosomal RNA are cleaved and chemically modified after transcription (DNA --> RNA) in prokaryotes.
Base Pairing
[edit | edit source]rRNA takes great part of base-pairing between the codon and the anticodon. "Adenine 1493, one of three universally conserved bases in 16S rRNA, forms hydrogen bonds with the bases in both the codon and the anticodon only if the codon and anticodon are correctly paired."
Referrence
[edit | edit source]M. Ogle and V. Ramakrishnan. Annu. Rev. Biochem. 74 (2005):129-177.

Small RNA is a classification of RNA which includes small-interferring RNA (siRNA), micro RNA (miRNA), and piwi-interacting RNA (piRNA). These small RNA play important roles in biological and diseases processes.
siRNA & micro RNA
[edit | edit source]small-interferring RNA (siRNA) is a class of RNA molecules that are around 20-25 nucleotides in length. They are mostly involved with the RNA interference (RNAi) pathway in order to interfere with the expression of a specific gene.
siRNA is a type of double stranded RNA that was found target mRNA cleavage sites and were designed to target transcript silencing through transfection of the siRNA into mammalian cells. This allowed for the development of RNAi-based applications such as a new class of therapeutics.
micro RNA (miRNA) is a class of RNA molecules that are found in eukaryotic cells. They are generally 20-25 nucleotides in length and are also involved in translation repression and gene silencing. They were similar to siRNA and was found to negatively regulate expression of target transcripts.

These two types of RNA were established as guides in governing silencing of target transcripts. This also raised questions of how these small RNAs were produced and it was found that immunoprecipitates in Drosophilia S2 cells processed the dsRNA (double stranded RNA) into the siRNA in vitro. miRNA was found to be derived from a conserved stem-loop precursor. This suggests that a dicing step could be required for miRNA biogenesis. The stem-loop forms part of a several hundred nucleotide long miRNA precursor which is then transcribes into miRNA. The existence of this precursor was found in Drosophia pupae.
In analyzing small RNA pathways in Drosophia, it was found that isolated dicer-1 and dicer-2 mutatnts were responsible for the biogeniss of miRNA and siRNA, respectively. Dicer-1 processed pre-miRNA independent of ATP while Dicer-2 processed dsRNA as ATP dependent. However, in mammalian cells, only one dicer generates both miRNA and siRNA.
siRNA effected silencing as they program RNAi effectors (such as RISC) to target mRNA. RISC is a magnesium dependent endoribonuclease that is affected by miRNA and siRNA to target mRNA cleavage activity.
miRNA has a controversial effector mechanism. This disparity is because there is a lack of a comparable well defined biochemical readout for miRNA induced RISC activity while there is a clear one from siRNA.
Making RISC
[edit | edit source]RISC: the effector complex for small RNAs
It is known that small RNAs aid in the regulation of gene expression. However, small RNAs cannot function individually to catalyze reactions. Instead, they come together and form RNA-induced silencing complexes (RISCs) in order to help with silencing genes and locating RISC targets. In this sense, the assembly of RISC is crucial for the small RNAs to do their job. [9]
Argonaute: the core component of RISC
The Argonaute (Ago) family of proteins is a main component of RISC that is essential to RISC’s function of target recognition and silencing. The Ago family can be divided into the Ago subfamily and Piwi subfamily. These Ago proteins, each with their own characteristics, are in charge of the functions of the small RNAs that they are paired with. SiRNAs and micro RNAs bind to Ago proteins while piRNAs bind to Piwi proteins. In mammals, the four proteins from the Ago subfamily (AGO1, AGO2, AGO3, AGO4) hinder translation in their target mRNAs, with AGO2 having the unique ability within its subfamily to induce RNA interference. In flies, AGO2 also triggers RNA interference in siRNA while AGO1 focuses on miRNA. What is different in flies compared to the case with mammals is that both AGO1 and AGO2 in flies can target cleavage and cause RNA interference. [9]
Two steps in RISC assembly: RISC loading and unwinding
There are two steps involved in RISC assembly. The first step is called RISC-loading, and this is when small RNA duplexes are incorporated into Ago proteins. Prior to this step, the double-stranded siRNAs and miRNAs are converted by RNase III enzymes (Drosha and Dicer) into small RNA duplexes: siRNA duplexes and miRNA-miRNA* duplexes. In the second step, the double-stranded small RNA duplexes are separated into two strands inside the Ago protein. Of the two strands, the strand with a less stable 5’ end is kept, serving as the ‘guide strand’. The other strand, called the ‘passenger strand’, is thrown out in order to produce a functional RISC. This strand selection in which one strand is preferred over the other is referred to as the ‘asymmetric rule’. [9]
Genome Encoded Small RNA
[edit | edit source]The Human Genome Project observed a relatively small number of protein-coding genes relative to genome size. It is believed that only five percent of the genome encodes proteins. miRNA, siRNA, and piRNA are part of the noncoding genome.
miRNA is believed to exist in hundreds of species and are identified through forward genetics by miRNA mutant isolation, bioinformation predictions based on the stem-loop, and direct cloning of small RNA. It is unclear how pre-miRNA is converted and there are studies to indicate that pri-miRNA and pre-miRNA occur separately in the nucleus and cytoplasm. dsRNA is a feature of pri-miRNA and aids in the processing into pre-miRNA. Dicer and Drosha are part of the factors required for the small RNA maturation. They are believed to function with dsRNA binding proteins which aid in the miRNA production.
Endo-siRNA play important roles in regulating genome functions in diverse species. They cleave target mRNA so that RNA-dependent RNA polymerases use the cleaved mRNA as templates to prime synthesis of secondary siRNAs. These are then loaded onto non-slicing agos to contribute to target silencing. This corresponds to the spreading of RNAi in mRNA and linked to silencing of worms.
piRNA are small RNA that also aid in the interference but focus on repetition. piRNA in mammalians are mapped uniquely in the genome and cluster to a small number of around 10 to 83 kb. Findings of the amplication of piRNA led to a ping-pong model in which it switches between Ago3 and Aubergine to create new piRN through each successive round. Different Piwi proteins conduct piRNA functions both cooperatively and independent of one another. piRNA play an important role in germ line development and the maintenance of genomic integrity. They are also involved in silencing but this is still unknown how. However, studies suggest that they regulate DNA methylation.
References
[edit | edit source]- ↑ Churchill, Das, Tyler The histone shuffle: histone chaperones in an energetic dance
- ↑ dsfdf
- ↑ http://www.wiley.com/college/boyer/0470003790/structure/tRNA/trna_intro.htm
- ↑ Desogus, Gianluigi; Flavia Todone; Peter Brick; and Silvia Onesti. "Active Site of Lysyl-tRNA Synthetase: Structural Sudies of the Adenylation Reaction. Biochemistry, 2000 vol 39, 8418-8425.
- ↑ Klug, William, and Michael Cummings. Concepts of Genetics.5th Edition. Upper Saddle River, NJ: Prentice Hall, 1997.
- ↑ Hartweel, Leland; Leroy Hodd; Michael Goldberg; Ann Reynolds; Lee Silver; and Ruth Veres. Genetics: From Genes to Genomes. Boston: Mgraw-Hill, 1999.
- ↑ Guo, Min, and Paul Schimmel. "ScienceDirect.com - Trends in Biochemical Sciences - Homeostatic mechanisms by alternative forms of tRNA synthetases." ScienceDirect.com | Search through over 11 million science, health, medical journal full text articles and books.. N.p., n.d. Web. 7 Dec. 2012. <http://www.sciencedirect.com/science/article/pii/S0968000412001077>.
- ↑ G. Fu et al. tRNA-controlled nuclear import of a human tRNA synthetase J. Biol. Chem., 287 (2012), pp. 9330–9334
- ↑ a b c Kawamata, Tomoko and Tomari, Yukihide. "Making RISC", '[Trends in Biochemical Sciences]', July 2010: 368-375. Retrieved on 21 November 2012.
- ↑ Liu, Qinghua; Paroo, Zain; Biochemical Principles of Small RNA Pathways Annu. Rev. Biochem. 79 (2010): 295-319.
Image from Wikipedia Commons
| This page or section is an undeveloped draft or outline. You can help to develop the work, or you can ask for assistance in the project room. |
Overview
[edit | edit source]MicroRNAs(miRNAs) are short, single-stranded RNAs that are about 21 nucleotides in length. Their function is to regulate gene expression. Like other types of RNA, miRNAs are transcribed from DNA; However, they do not participate in protein translation. miRNAs are non-coding RNAs that bind to complementary mRNA and inhibit their translation. miRNAs and siRNAs both function to interfere with gene expression. However, miRNAs are single-stranded, whereas siRNAs are double-stranded.
miRNAs have been determined to play a crucial role in regulation of DNA damage response. Scientists believe that the transmission of generic information in eukaryotic cells requires accuracy in DNA replication and chromosome as well as the ability to sense and repair spontaneous and induce DNA damage. In order to maintain genomic integrity, cells undergo a DNA damage response, a complex network of signaling pathways. This network is composed of coordinates sensors, tranducers and effectors in cell cycle arrest, apoptosis and DNA repair.[1]
miRNAs have recently been linked to various diseases. Recent researches have shown that there is connection between dysregulation of miRNAs with certain diseases, which leads to the need of further researching in robust regulation of miRNA activity.[2]
miRNAs once were considered to be very stable molecules because miRNAs expression is known to be strictly controlled by the mechanisms acting at the level of transcription and also the processing of miRNA precursors. However, recently, scientists have figured out another mechanism that is important for miRNA homeostasis which is the active degradation of mature miRNAs. Degradation of miRNA takes role in dynamic miRNA expression patterns. Researches showed that miRNAs degradation can have affect on specific sets of miRNAs even though how this specificity comes about still remains unknown.[3]
Formation & Function
[edit | edit source]The main function of miRNAs is to regulate the translation of mRNA. In the nucleus, the miRNAs are first transcribed as primary miRNAs(pri-miRNAs) with caps and a poly-A tail. The pri-miRNAs are then processed into precursor miRNAs(pre-miRNAs) by an enzyme called Drosha. The structure of pre-miRNA is a 70 nucleotide-long stem-loop structure. The pre-miRNAs are then exported into the cytoplasm and split into mature miRNAs by an enzyme called Dicer. These mature miRNAs will integrate into the RNA-induced silencing complex(RISC) and activate the RISC. The activated RISC can then allow miRNAs to bind with the targeted mRNA and silence the gene expression. In animal cells, miRNAs are more commonly base paired with the mRNA and inhibit protein translation. The binding of miRNAs to complementary mRNA can degrade the mRNA and therefore terminate protein translation. Or miRNA can inhibit the reading of the 5'-cap and prevent translation. In plant cells, the miRNAs are more likely to perfectly bind with the target mRNA and promote cleavage. MicroRNA's are formed from the hairpins of long single-srranded RNA's that fold in on themselves. The double-stranded hairpins get cut by enzymes called dicer and results in a single-stranded microRNA. MircroRNA then forms a microRNA-protein complex and can then degrade a targeted mRNA and also block translation of targeted mRNA. In few instances, miRNA have shown signs of promoting translation, especially under starvation conditions. The reason for such activity are not known.
Canonical miRNA Function
[edit | edit source]Developing studies have demonstrated that miRNAs carry on a critical role in interacting with the canonical DNA damage response. The DNA damage response is an active system that includes commencement of transcriptional programs, enhancement of DNA restoration, and apoptosis if damage is severe. Breakages in DNA double-strands are mended by homologous re-fusion and non-homologous end-connecting repair pathways. Other forms of DNA damage are repaired by base excision repair (BER), nucleotide excision repair (NER), and DNA mismatch repair (MMR).
miRNA play a significant part in gene regulation and other cellular functions. Many important genes in the DNA damage response are managed by their corresponding miRNAs. For one, miRNAs monitor DNA damage response by way of target genes. In the process of DNA repair, chromatin remodeling takes place to permit DNA repair proteins access to DNA that are damaged. With more miRNA targets such as ATM, H2AX, and RAD52, DNA responsive genes are under inhibition by miRNAs. It has been revealed that higher expression of a certain miRNA -such as miR-421- will reduce ATM delivery, and downregulate H2AX in particular cellular situations.
DNA damaging agents in various treatments have proven to initiate miRNAs. Occurrences of DNA damage have depicted a correlation with the activation of miRNAs, underlying the significance that miRNAs regulate DNA damage response based on the magnitude of the DNA damage.
Noncanonical miRNA Function
[edit | edit source]Recent study has shown that by miRNA directly targeting the primary transcripts of other miRNA in the nucleus, it can control the biogenesis of the miRNA. A particular example is the miR-709 which negatively regulates the miR-15a/16-1. This particular miR709 is found in the mouse nucleus, and it binds specifically to miR15a/16-1 which are both 19-nucleotide recognition element. It clusters and blocks the processing of primary transcript of miR-15a/16-1 into the precursor. As such it regulates the maturation at a post-transcriptional level, which means post primary transcript but pre precursor. As such, because miR-709 can regulate the miR-15a/16-1, which in turn regulates the cell apoptosis, the miR-709 indirectly regulates the cell apoptosis. This in turn demonstrates that miRNA can affect the expressions of things within a cell because it can regulate the biogenesis of the other miRNA within the cells.
The miRNA can also regulate the long ncRNA. ncRNA are generally longer than 200 nucleotides and are non-protein-coding transcripts. The first experimental evidence that shows long ncRNAs are functional miRNA targets is shown by Hansen. In the experiment, the antisense transcript of the cerebellar degeneration related protein 1 (CDR1), which is a circular ncRNA, has been shown to be near perfect complementary with miR-671, which is in the nucleus. Within the experiment, miR-671 directs the cleavage of the CDR1 antisense transcript in an AGO2-dependent manner. Thus, with the negative regulation of the circular antisense ncRNA, it also decreases the CDR1 sense transcript. The study down shows that the antisense RNA can be destabilized by the miRNA through the AGO2 -mediated cleavage, and the sense mRNA can be stabilized by the circular noncoding antisense RNA.
miRISCs and Its Components
[edit | edit source]miRNA combine with Argonaute proteins and GW182 proteins to form miRNA-induced silencing complexes, or miRISCs. AGO attach to the N-terminus of the GW182 protein, while the miRNA bond to the AGO. The GW182 protein seems to be the more important of the two, as it contains the main silencing region. This was discovered when miRNA induced repression was still effective even after the knockoff of AGO in Drosophila cells.
miRNA Inhibition of Translation
[edit | edit source]miRNA possess several methods of inhibiting translation. Suppression can occur both before and post translation, although the former method seems to be preferred.
Before Initiation
[edit | edit source]- miRISC can suppress translation by interfering with the reading of 5' eIF4F-cap structure. The miRNA prevents the ribosome from reading the cap, thus initiation never starts. On the other hand, mRNA that were able to translate without the cap recognition step were not suppressed by the miRNA.
- miRNA are also able to prevent the creation of a functional ribosomal unit. On normal mRNA, the 40S and 60S ribosomal subunit come together to form the 80S complex, which helps translate the mRNA. miRNA inhibit 60S from joining with the 40S unit, making mRNA translation impossible. Translation is never able to start.
Post Initiation
[edit | edit source]- miRNA blocks the elongation of the new RNA being translated.
- The ribosome is forced to drop-off from the mRNA. The 40S and 60S ribosomal units split up before translation is complete.
- The miRNA induce preteolysis of the newly transcribed polypeptide chain. The chain is broken up by enzymes.
The mechanisms for the three post initiation inhibitors are known.
miRNA stability
[edit | edit source]In contrast to the suggestion in the past that miRNAs are highly stable, recent researches have shown that individual miRNAs in certain environments are subject to accelerated decay, which alters miRNA levels so that affects its activity.[4]
During miRNA biogenesis, miRNAs are transcribed by polymerase II as primary transcripts (pri-miRNAs) and the are matured in a multi-step biogenesis process to produce the mature and functional miRNA form. In one case, the pri-miRNAs are captured by polyaldenylated and are quite long (several kilibases long). Pri-miRNAs possess hairpin structures which includes the mature sequence of miRNA in their stem. In another case, the precursor miRNAs (pre-miRNAs) can be kept in introns of mRNAs or other non-coding RNAs. In either of these two cases, the nuclear RNAse type III enzyme Drosha in a complex with co-factor DiGeorge syndrome critical region 8 homolog (DGCR8), cleaves near the base stem which releases about 70 nucleotides pre-miRNA.[5]
Deadenylation and Decay
[edit | edit source]In deadenylation, the miRNA binds to AGO, GW182, and also poly(A)-binding protein (PABP). The PABP attaches to the GW182 protein, forming a slightly different miRISC. The miRISC removes the 5'-cap from the mRNA, which immediately causes decay of the mRNA. Deadenylation is effective because it rids the cell of excess mRNA, eliminating the chance of accidental translation. The decayed fragments are collected by the P bodies, and reused by the miRNA.
The degradation of miRNAs occurs under the aid of several miRNA-degrading enzymes. Many miRNA-degrading enzymes have been determined including both 3'to 5' and 5' to 3' exoribonucleases. Recent researches have shown that certain RNases were found to take the role in the turnover process of different sets of miRNAs in different organisms. However, the substrate specificity and phylogenetic conservation of individual miRNA turnover enzyme are still in the need of researching.[6]
microRNA-206 and Synapse Repair
[edit | edit source]In a mouse model of ALS: When mice get ALS, production of microRNA-206 is induced/increased. Deficiency of microRNA-206 accelerates the progression of the disease. -MicroRNA-206 is required for regeneration of damaged neuromuscular synapses (the signals between muscle and nerve cells). When synapse is damaged, microRNA-206 turns on repair. Without miRNA-206, synapses cannot be repaired; however, some synapses can grow back. -MicroRNA-206 does this through histone deacetylase and fibroblast growth factor (FGF) signaling pathways. Growth factors are specific signals from other cells that tell the cell to grow. -MicroRNA-206 blocks translation. It then activates histone deacetylation which condenses chromatin, therefore blocks transcription. -MicroRNA-206 slows the progression of ALS by repairing neuromuscular synapses.
MicroRNA genes are found in intergenic regions. These regions have its own miRNA gene promoter and regulatory units. Approximately forty percent of miRNA genes are lie in the introns of the proteins coding, non-proteins coding, and even in the exons. The miRNAs are found in the orientation that are regulated together with its own host gene. Between forty-two to fifty percent of other miRNA genes were shown in a common promoter, which originate from polycistronic units. The polycistronic units have a discrete loops of 3-6 where the mature miRNAs are being processed, but the miRNAs family are not homologous structure function. Hence, the promoters have a few identical motifs to other genes promoters that were transcribed protein coding genes from the RNA polymerase II. Also, in the DNA template does not have the finish during the mature miRNA production, because there is about five percent of human miRNAs shows RNA editing. The site-specific modification of RNA sequences to yield products different from those encoded by their DNA. The yield of the product allows to increases the diversity, the scope of miRNA action implied from the genome alone.
miRNAs and Disease
[edit | edit source]Cancer
[edit | edit source]Recent studies have shown that miRNAs are involved in causing diseases. In the case of cancer, researchers found that miRNAs can inhibit the E2F1 protein that regulates cell proliferation. miRNAs bind to the mRNA first before translating the E2F1 protein. One microRNA, miR-21, was labeled as the first oncomir. It is known to aid in tumor growth and metastasis by targeting natural occurring tumor suppressors in the human body. Tropomyosin 1 (TPM1) is a direct target of miR-21, along with programmed cell death 4(PDCD4) and maspin, all of which are inversely correlated with the expression of miR-21 in the presence of tumors. This shows that miR-21 has the ability to target multiple genes and inhibit multiple metabolic pathways at the same time.
Kidney Fibrosis
[edit | edit source]Renal fibrosis is the excessive accumulation of fibrous tissues (connective tissues), occurring as a reparative process after scarring or trauma to the kidney. This type of nephropathy directly promotes renal dysfunction, which ultimately leads to kidney failure and death. Study has shown that a certain microRNA, miR-21, shows significant elevation in expression during the progression of kidney scarring. Experiments were conducted to validate this specific sequence and its effect in mice.
The abrogation of miR-21 in mice showed no overt abnormalities and no obvious suppression/prevention of tumor growths; however, these mice developed far less interstitial scarring tissue in response to kidney injury. Analysis has detected groups of genes and their subsequent metabolic pathways that were inhibited by miR-21. One of which involves peroxisome proliferator-activated receptor- α(Pparα), which is a lipid metabolism pathway that incorporates the synthesized anti–miR-21 oligonucleotides to inhibit miR-21. Pparα is found to ease the effects of ureteral-obstruction induced kidney fibrosis. miR-21 also regulates the redox metabolic pathway that involves a protein called Mpv171. The repression of Mpv171 in cells enhances kidney damage by reducing the production of oxygen radicals.
These studies demonstrate that miR-21 has a broad spectrum of influences on the microscopic scale and can be a suitable target for antifibrotic and cancer therapies.
Heart Disease
[edit | edit source]Another studies have shown miRNA inhibits the maturation in the murine heart, and plays an essential role during its development. The expression level of the miRNA is been changed in the disease of the human heart; it is the involvement in cardiomyopathies. During the heart disease development, they were several specific miRNAs that were been identified in animal models that were mostly in mice under pathological conditions. Those specific miRNA conditions key factors are important for cardiogenesis, the hypertrophic growth response, and cardiac conductance.
References
[edit | edit source]Fabian, Marc R., Nahum Sonenberg, and Witold Filipowicz. "Regulation of MRNA Translation and Stability by MicroRNAs." Annual Review of Biochemistry (2010): 351-79. Biochem.annualreviews.org. Neil A. Campbell, Jane B. Reece "Biology 8th edition"
External links
[edit | edit source]- http://www.ambion.com/techlib/resources/miRNA/index.html
- http://en.wikipedia.org/wiki/Micro_RNA
- http://www.regulusrx.com/
- Chau, and Tran. "MicroRNA-21 Promotes Fibrosis of the Kidney by Silencing Metabolic Pathways." US National Library of Medicine. N.p., 15 Feb. 2012. Web. 20 Oct. 2012. <http://www.ncbi.nlm.nih.gov/pubmed?term=MicroRNA-21%20Promotes%20Fibrosis%20of%20the%20Kidney%20by%20Silencing%20Metabolic%20Pathways.>.
- Liu, Youhua. "Renal Fibrosis: New Insights into the Pathogenesis and Therapeutics."Nature.com. Nature Publishing Group, 19 Sept. 2005. Web. 20 Oct. 2012. <http://www.nature.com/ki/journal/v69/n2/full/5000054a.html>.
- Chen X, Liang H, Zhang CY, Zen K. "miRNA regulates noncoding RNA: a noncanonical function model" Trends Biochem Sci. 2 Sept. 2012. Web. 25 Oct. 2012 <http://www.ncbi.nlm.nih.gov/pubmed/22963870>.
- Guohui Wan, Rohit Mathur, Xiaoxiao Hu, Xinna Zhang, Xiongbin Lu, miRNA response to DNA damage, Trends in Biochemical Sciences, Volume 36, Issue 9, September 2011. <http://www.sciencedirect.com/science/article/pii/S0968000411000855>.
- ↑ Wan, Guohui, Rohit Mathur, Xiaoxiao Hu, Xinna Zhang, and Xiongbin Lu. "ScienceDirect.com - Trends in Biochemical Sciences - miRNA response to DNA damage." ScienceDirect.com | Search through over 11 million science, health, medical journal full text articles and books.. N.p., n.d. Web. 7 Dec. 2012. <http://www.sciencedirect.com/science/article/pii/S0968000411000855>.
- ↑ Chang, T.C. and Mendell, J.T. (2007) microRNAs in vertebrate physiology and human disease. Annu. Rev. Genomics Hum. Genet. 8, 215–239
- ↑ Großhans, Rüegger . "MicroRNA turnover: when, how, and why. [Trends Biochem Sci. 2012] - PubMed - NCBI." National Center for Biotechnology Information. N.p., n.d. Web. 6 Dec. 2012. <http://www.ncbi.nlm.nih.gov/pubmed/22921610>.
- ↑ Großhans, Rüegger . "MicroRNA turnover: when, how, and why. [Trends Biochem Sci. 2012] - PubMed - NCBI." National Center for Biotechnology Information. N.p., n.d. Web. 6 Dec. 2012. <http://www.ncbi.nlm.nih.gov/pubmed/22921610>.
- ↑ Krol, J. et al. (2010) The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 11, 597–610
- ↑ Großhans, Rüegger . "MicroRNA turnover: when, how, and why. [Trends Biochem Sci. 2012] - PubMed - NCBI." National Center for Biotechnology Information. N.p., n.d. Web. 6 Dec. 2012. <http://www.ncbi.nlm.nih.gov/pubmed/22921610>.
1. Small nuclear RNA (snRNA)
[edit | edit source]Small nuclear RNAs (snRNA) are the small RNA molecules that are found in the nucleus of eukaryotic cells. They are usually 300 nucleotides or smaller and the nucleus contains more than just snRNA. The function of snRNA was discovered before the ribozyme enzyme by a few years. They are transcribed by RNA polymerase II or RNA polymerase III. They are important because they help in the process of pre-mRNA splicing and processing, which is the removal of the introns from hnRNA, and involved in the maintenance of the telomeres, or the ends of chromosomes. 5 snRNAs makes up the spliceosomes which are responsible for removing the introns from nuclear pre-mRNA eukaryotes. The spliceosome interacts with the ends of an RNA intron. It cuts at specific points to release the intron, then immediately joins the two exons that were adjacent to the intron. They are also responsible for mediating catalysis and aligning splice sites. Thomas Cech and Sydney Altman discovered that RNA molecules can serve as catalysts and changed the views of molecular evolution. snRNA are always found with specific proteins which make up the complexes called small nuclear ribonucleoproteins (snRNP), or snurps. The secondary structures are highly conserved in organisms ranging from yeast and human beings. Large groups of snRNA are called small nucleaolar RNA’s (snoRNA’s). snoRNA’s are responsible for cleaving eukaryotic long preRNA. They are important in RNA biogenesis and guide chemical modifications or ribosomal RNA (rRNA) and other RNA genes (tRNA and snRNA). Many snoRNA’s are created by processed introns. The host gene for the snoRNA is a ribosomal protein or translation factor. [43] [44]
2. Small Nucleolar RNA molecule (snoRNA)
[edit | edit source]snoRNAs, or Small Nucleolar RNA, modifies ribosomal RNAs (rRNAs) by mediating the cleavage of long pre-rRNA strands into its functional subunits (18S, 5.8S and 28S molecules). snoRNAs can also add the finishing modifications to rRNA subunits. [45]
3. Micro RNA (miRNA)
[edit | edit source]Micro RNA (miRNA) is a gene regulatory small RNA that is typically 21-23 nucleotides long. It is similar to small interfering RNA (siRNA)in that they bind to complementary mRNA molecules and inhibit their translation, however unlike siRNA which is a double strand RNA, miRNA is a single stranded RNA and it is only partially complementary to mRNA molecules. This class of RNA is non-coding.
Micro RNA has a great range of functions. It is used in cellular growth, development and insulin secretions among other things.
However it has been found that too much miRNA has been found to implicate diseases, such as Fraglie X Mental Retardation, as well as some forms of cancer.
4. Small interfering RNA (siRNA)
[edit | edit source]siRNAs, are known by several different names: small interfering RNA, short interfering RNA, and silencing RNA, were discovered by David Baulcombe’s research group in Norwich, England. They are roughly 20-25 nucleotides in length and are double stranded RNA molecules with overhanging 2 nucleotides on the 3'ends. They are largely responsible for the process of RNA interference (RNAi) pathways, which interferes with the expression of a gene. Other RNAi pathways such as antiviral mechanisms and shaping the chromatic structure of a genome are also mediated by siRNA. The discovery of siRNA’s ability to be synthetically produced, allowed for the induction of RNAi in mammalian cells. This then allows for research in drug development of the biomedical field such as treatment for the cure of Human Immunodeficiency virus (HIV). However using RNAi through the use of siRNA in living animals is difficult, because siRNA responds differently to different types of cells and the effectiveness varies from very well to poor. It is not yet understood why the effectiveness of siRNA in living animals varies so vastly. Artificial siRNA can be made synthetically by a phage enzyme which is called a dicer. It is the dicer enzyme that causes destruction of the double stranded RNA (dsRNA). By transfecting artificial siRNAs, specific transcripts are used to probe gene function. Although this is a useful tool, the high cost of production makes it nearly impossible for most laboratories and researchers to be able to use this method of probing gene functions by transfecting artificial siRNAs. Chemical synthesis, invitro transcription, or RNase 3-dicer digestion of long dsRNA’s (double stranded RNA’, in vivo from plasmids PCR cassettes, or viral vectors CMV or polymerase III transcription unit. SiRNA’s are used for loss of function studies. SiRNA’s are very sequence specific.
5. RNA Interference (RNAi)
[edit | edit source]RNAi was discovered Craig Mello and Andrew Fire in the 1990s. The experimented by antisense RNA experiments. It is the process in which double stranded RNA triggers the degradation of homologous mRNA.
RNA interference occurs when a double strand of RNA is broken down by an enzyme called Dicer. Dicer chops the double stranded RNA into short sequences 20-25 base pairs long. These base pairs then complex with the RISC enzyme and a homologous strand of RNA, which is then catalytically cleaved by RISC.

It is used to degrade mRNA in cells as a defense mechanism against Viral DNA that may have infected the cell and to shut down the effects of specific genes post transcription without having to regulate actual gene expression in the cell's DNA. This can be also used as a gene silencing technique. siRNA is put into a cell by transfection reagents. These reagents increase amount of RNA and DNA that can be absorbed by cultured cells. RNAi is used in the biomedical field to silencing disease causing genes. The RNAi can either be injected into specific cells or using modified viruses to transfect the cells. One common use of RNAi is in the birth control pill which stops sperm from fertilizing the eggs by splicing the gene that encodes protein to allow the sperm to bind to the egg. RNAi is also being used to knock out genes in salamanders in an attempt to discover which genes are responsible for their regenerative capabilities in an attempt to cure diseases previously thought to be incurable, such as Huntington's, Parkinsons, and Alzheimers by attempting to trigger the regeneration of the neurons whose death are responsible for such a disorder.
6. Interference RNA (iRNA)
[edit | edit source]Interference RNA or iRNA is used for gene regulation. It is an antisense RNA (complementary to other RNA, mostly mRNA). It is important for gene regulation and it is being researched currently for collective anti-cancer properties. It has ties to siRNA as siRNA is involved in the RNA interference pathway. RNA interference (RNAi) is a phenomenon of gene silencing at the mRNA level offering a quick and easy way to determine the function of a gene both in vivo and in vitro. [46]
7. RNA is a component of telomerase
[edit | edit source]Telomerase is a ribonucleoprotein (a ribonucleic acid-protein complex). It is an enzyme that maintains the telomeres (ends) of chromosomes during DNA replication. It has been found to be useful in the therapeutical, pharmaceutical, and diagnostic reagents.

8. Non-coding RNA (ncRNA)
[edit | edit source]Non-coding RNA is basically any RNA molecule that is not translated into a protein. Non-coding RNA can be found in many different forms of RNA, such as: ribosomal RNA (rRNA), transfer RNA (tRNA), and small RNAs [microRNA and small interfering RNA (siRNA)]. Non-coding RNA can be small or it can be very large. The small non-coding RNA molecules is also known as sRNA, whereas the large or long non-coding RNA is also known as lncRNA. The non-coding RNA molecule that was transcribed from DNA is often referred to as an RNA gene.
It is significant to note that there exists a growing interest in small, barely detectable non-coding RNA molecules because some of them have been found to play an important role in the regulation of gene expression. These small RNA molecules are known as RNA genes. In the early 1990s, American geneticist Victor Ambros and his colleagues first identified these molecules in the species of worm Caenorhabditis elegans. They were found to be responsible for turning off gene expression during worm development. This novel function was later discovered in other species as well. A decade later, another American geneticist Stephen R. Holbrook of Lawrence Berkeley National Laboratory in California discovered several other potential RNA genes previously undetected via a complex computer program called RNAGENiE. Currently, much research is being conducted over these tiny non-coding RNA molecules. In recent years, biotech and pharmaceutical companies have been looking into the potential of RNA genes as drug targets due to recent interest in RNA genes produced during bacterial infections and their pathogenic effects through the regulation of gene expression of host DNA.
9. Antisense RNA
[edit | edit source]Antisense RNA is an RNA strand that is complementary to the messenger RNA (mRNA) strand that transcribes within the cell. The antisense RNA is a single stranded RNA molecule. The antisense strand is brought into a cell in order to inhibit the translation of the mRNA. It does this through base pairing to the complementary mRNA strand, which obstructs the ability of the mRNA to translate.
Antisense RNA has been previously thought to be useful as a therapeutic technique for disease therapy, however over the past few years only one drug has been synthesized through the use of antisense RNA. It has been found that antisense RNA failed to have an effective design for disease therapy.
10. tmRNA
[edit | edit source]In an RNA, an RNAse can take off the 3’ end of an mRNA so that the mRNA has no stop codon for the ribosome sense and stop translation. Once the entire strand of mRNA is translated, this leads to the ribosome being stuck on the mRNA, with a peptidyl-tRNA in the P site of the ribosome. To fix this there is the tmRNA, which removes ribosomes that are stuck on an mRNA. This tmRNA has characteristics of both a tRNA and a mRNA.
In E. Coli, the tmRNA present is SsrA. The structure of this SsrA is arranged so that at one end there is an alanine attached with a tag sequence, and the SsrA is folded to look like a tRNA. The SsrA will enter the A site of the stuck ribosome and the Alanine on the SsrA will form a peptide bond with the polypeptide that is stuck on the ribosome. The tag sequence on the tmRNA is then translated like a mRNA and added to the amino acids on the stuck polypeptide. The string of about 12 added amino acids are called a proteolysis tag. At the end of the tmRNA, a stop codon will signal the ribosome to stop translation and detach itself as well as the SsrA-tagged peptide. SspB, a helper protein, can then recognize the proteolysis tag on the polypeptide chain and bring it to the protease, ClpXp, to be destroyed. [1]
11. Catalytic RNA
[edit | edit source]Catalytic RNA carry out enzymatic reactions. Catalytic RNAs are usually found near proteins where the catalytic activity is found in the RNA portion, rather than protein. [2]
References
[edit | edit source]http://science.jrank.org/pages/5892/RNA-Function.html
- ↑ Joan L. Slonczewski, John W. Foster. "Microbiology: An Evolving Science."
- ↑ Joan L. Slonczewski, John W. Foster. "Microbiology: An Evolving Science."
RNA polymerase is an enzyme that produces RNA and catalyzes the initiation and elongation of RNA chains from a DNA template. RNA is created using a process known as transcription. The RNA polymerase is a key component to this process. The reaction that this enzyme catalyzes for is: (RNA)n + Ribonucleoside Triphosphate ->/<- (RNA)n+1 +PPi. RNA polymerases are relatively large. The size of RNA polymerase in a typical eukaryotic cell is roughly 500kDa. In bacteria it is roughly 400kDa and in T7 bacteriophage it is roughly 100kDa. Their speed of transcription is about 50 bases per second. A typical mRNA that codes for an average protein takes about 20 seconds in a prokaryotic cell and about 3 minutes in a eukaryotic cell. It is primarily longer in eukaryotes due to the fact that eukaryotic genes contain many segments that contain introns.
Requirements to Function
[edit | edit source]For DNA polymerases to properly carry out their function they must have the following components present for catalysis to occur. 1. A template of DNA. The preferable template is a double stranded DNA. Single stranded DNA may also work as a template but RNA strands or DNA-RNA hybrids may not be used. 2. Activated precursors. The reactions require ribonucleoside triphosphates: ATP (Adenine -ribose-triphosphate), GTP (Guanine-ribose-triphosphate), ATP (Adenosine-triphosphate), and UTP (Uracil-ribose-triphosphate). Nucleotides with three phosphates to the 5’ carbon of the ribose sugar.

3. Divalent metal. Unlike DNA polymerase, a primer is not needed but a divalent metal ion like magnesium ion or manganese ion is effective.
The direction of synthesis is from 5' to 3' and the synthesis is driven by the hydrolysis of pyrophosphate. There have been hybridization experiments that show RNA synthesized by RNA polymerase is complementary to its DNA template.

RNA Biogenesis Pol I, Pol II, and Pol III
[edit | edit source]Gene transcription takes place in the nucleus of eukaryotic cells and transcription is performed by three different multisubunit RNA polymerases, Pol I, Pol II, and Pol III. Still little is known today about the biogenesis of these RNA polymerases: from their origin of synthesis, the cytoplasm, to their arrival in the nucleus for transcription. Only until recently have studies shown that polymerase assembly intermediates, assembly factors and factors required for polymerase nuclear import exist in the cell cytoplasm. Pol II is the most identifiable one so is the basis of most studies on the biogenesis of RNA polymerase.
Structure and Assembly of RNA Polymerase II
[edit | edit source]
RNA Pol II transcribes mRNAs and small non-coding RNAs and contains 12 polypeptide subunits. Each RNA Pol has their own specified role in RNA polymerase. They all have ten identical subunit catalytic cores. The peripheral subunits are what differentiate their structure and function; RNA Pol II has been determined to contain cores that allow it to model the homologous cores in Pol I and Pol III. Pol I and Pol III will bind to opposite sides of Pol II (binding to Rpb1 and Rpb2) and are then divided into three interacting subunits.

The assembly of eukaryotic RNA core was first identified in studies of bacterial RNA polymerase because RNA Pol II core subunits are exactly identical to that of bacteria. Assembly of RNA Pol II is initiated by the formation of the αα dimer which interacts with the β and forms a bound complex intermediate. The active cleft in the RNA Pol II is composed of β subunits which are formed in the final step of assembly, so the polymerase will not be catalytically active until it is complete. RNA Pol II in both bacteria and eukaryotic cells has both exhibited formation in equivalent manner.
Assembly in vitro experiments have also been conducted to determine the origins of RNA Pol II. Using three mutant large subunits, their assembly was followed with the use of pulse chase experiments. Scientists found that Rpb3 and Rpb3 were the first to interact, and the bound complex then interacts with Rpb1. However, because larger mutated subunits were used, final assembly could not be complete without the use of Rpb6, Rpb10, and Rpb12, which are not normally part of final assembly in normal sized RNA Pol II. RNA Nuclear Import
If any RNA subunits are lost during its assembly, there will be an excess of Rpb1 present in the cytoplasm, meaning that the polymerase needs to be fully assembled before it is allowed to enter the nucleus and take place in transcriptase. Pol II nuclear localization factors have been identified to be functional polymerase-interacting proteins in the cell. The accumulation of Rpb1 is caused by the depletion of GPN1 and GPN3. The expression of GPN1 will lead to the depletion of excess Rpb1. GPN1 binding to Pol II can also be directly influence the ability of GTP to bind properly. Homologs of GPN1 also aid in the biogenesis and final assembly of Pol II. GPN1 interacts with the CCT complex, which chaperones many subunits in the formation of Pol II.
Nuclear Import Signal
[edit | edit source]The components of Pol II, the subunits and GPN proteins, are unable to produce a nuclear import signal, therefore, which is why a Pol II cannot enter the nucleus until it is fully assembled, so it can produce a signal. Iwr1 is a factor that interacts with fully assembled Pol II and adapts a nuclear signal onto it. And deletion of Iwr1 leads to a accumulation of all the Pol II subunits, showing that lwr1 is most likely the key to proper final assembly. Iwr1 binds to the active site on Pol II and can “sense” completion by interacting with the Rpb1 and Rpb2 subunits, ensuring that Pol II is fully assembled; this acts as the final checkpoint before entering the nucleus. Because deletion of Iwr1 affects the concentration of subunits in the cytoplasm, a nuclear export signal is used to trigger the recycling of Iwr1. Currently Iwr1 is only know to effect the subunits and factors involving Pol II upon depletion, nothing has been found on how it affects Pol I and Pol II.
Biogenesis for RNA Pol I and Pol III
[edit | edit source]The origins of Pol I and Pol III may depend on the chaperones Hsp90 and R2TP because the client proteins for these two chaperones were discovered to be the subunits of Pol I and Pol III. This makes sense because the deletion of A135, the Pol I subunit, results in Hsp90 binding to Pol I’s larging subunit, A190. Several bleaching experiments have been conducted on Pol I that revealed Pol I is assembled at the promoter sites. It unclear as to what happens to Pol I after transcriptase because it remains as a stable complex and does not dissociate, scientists are trying to determine whether or not Pol I is fundamentally different in other organisms.
Pol III is the least understood polymerase out of the three. A NLS sequence was discovered near the N-terminus of the second larger Pol III subunit, C128, and when this sequence is deleted it leads to the accumulation of C128 in the cytoplasm and other Pol III subunits. However, the other Pol III subunits remained intact and nuclear. This reveals that the core of Pol III is assembled within the cytoplasm and the released subunits bind the core of the nucleus. It appears that Pol III follows the same assembly pathway as that of Pol II, as revealed by native mass spectroscopy.
Due to the fact that all three RNA polymerases have at least ten identical subunits, we can draw the conclusion that all three polymerases can coordinate and simultaneous assembly. The study of a certain subunit in any of three polymerases can be better understood by also studying the other subunits at that stage of biogenesis.
RNA Polymerase Translocates
[edit | edit source]RNA molecules thousands of nucleotides long are synthesized by multi-subunit DNA-dependent RNA polymerases. Nucleotide condensation’s reiterative reaction happens at rates of tens of nucleotides per second. This is consistently linked to the translocation of the enzyme along the DNA template (threading of the DNA and emerging RNA molecule through the enzyme. This reiteration of the nucleotide addition/translocation cycle without separating the DNA from the RNA involves both isomorphic and metamorphic conformational flexibility to such a magnitude that it accommodates the essential molecular motions. [1]
Types of RNA Polymerase
[edit | edit source]Eukaryotes
[edit | edit source]Eukaryotic cells have three types of RNA polymerases. Pol I: This type of RNA polymerase synthesizes RNA for the large subunits of ribosomes. Ribosomes are pretty much the protein making organelle in cells. Pol II: Creates mRNAs. Messenger RNAs provide a template for protein synthesis for ribosomes. It also creates many small nuclear RNAs which help modify RNA after they are formed. Pol III: Creates tRNAs. Transfer RNAs is basically for the small subunit of ribosomes.
These three types of polymerases can be distinguished from one another in lab by the level of inhibition by the alpha-amanitin poison. PolI is completely resistance to this poison. PolII is highly sensitive to this poison. And PolIII is moderately sensitive.
RNA polymerases in eukaryotic cells are composed of several subunits. Majority of them are small and unique to each type of polymerase. However there are two large subunits that are similar among all of the polymerases. This fact highlights that all these polymerases must have evolved from an original polymerase. The two large subunits are the functional core of this enzyme. The other smaller subunits tend to provide the specific functions for each distinct type of polymerase.
Bacteria
[edit | edit source]In bacteria, the RNA polymerase holoenzyme is made up of two parts, a core polymerase and a sigma factor. The core polymerase has the components needed for elongation in transcription, while the sigma factor is only needed for transcriptional initiation. The core polymerase is made up of two α’s, one β, and one β’ unit (α2 β β’), while the sigma factor is only made up of s. In total, there are 5 subunits in RNA polymerase-- alpha (α), beta (β), beta' (β '), sigma (s), and omega (w). However, the function of omega is unknown and is thought to possibly stabilize RNA polymerase.
In bacterial DNA, the promoter sequence is recognized by the sigma unit of the RNA polymerase. Upon recognition of the promoter sequence, the sigma factor will guide the RNA polymerase to the promoter. This sigma factor will then bind the RNA polymerase to the promoter through the α unit of the core polymerase. [2]
Archaea
[edit | edit source]Archaeal RNA polymerases are pretty similar to eukaryotic RNA. Especially similar to RNA Polymerase II. These polymerases may have evolved from stripping down eukaryotic systems. An archea polymerase is used in PCR because it can withstand the high temperature used to split DNA strands.
Structure
[edit | edit source]RNA polymerase have a multitude of structural features that help in the transcription process. A Structure known as the clamp keeps the polymerase anchored to DNA . The flap ensures that the MRNA is retained. The rudder prevents DNA/RNA hybrid from occurring. DNA does not enter the mouth of the polymerase directly. It is usually held sidewise with a sharp bend to its left as it exits the polymerase. mRNA is believed to leave from the back of the polymerase. NTPs enter the active site as the same channel that DNA is pulled through but through a secondary channel .

Similarities and Differences between RNA Polymerase and DNA Polymerase
[edit | edit source]The synthesis of RNA and DNA is similar in many aspects. Both of them follow the synthesis direction of 5'->3'. Another is that the method of elongation is by the 3'OH group at the terminus of the growing chain that makes a nucleophilic attack on the innermost phosphate of the incoming nucleoside triphosphate. Another similarity is that the synthesis is driven by the hydrolysis of pyrophosphate. However the difference between the two is that RNA polymerase does not require a primer unlike DNA polymerase which does. Also although DNA polymerase can actually correct mistakes in the nucleotide transcription, RNA polymerase lacks this ability to excise the mismatches nucleotides.
References
[edit | edit source]Transcription Elongation Complex (TEC)
[edit | edit source]To start transcription, RNA Polymerase (RNAP) must recognize and bind to a promoter sequence. Some factors include assisting the polymerase to an open promoter complex in which the DNA exposes the bases, forming a transcription bubble. Then, RNAP typically undergoes an abortive initiation in which the process synthesizes short strands of RNA transcripts. RNAP returns to the initial promoter site and escapes the region by forming a stable, transcription elongation complex (TEC) which is able to transcribe the whole gene.
Single-molecule Techniques
[edit | edit source]Atomic Force Microscopy (AFM)
[edit | edit source]Atomic force microscopy is a technique used to image the ultrastructural alteration in the TEC such as the change in bend angles of the template DNA induced by RNAP. The TEC is placed on a flat surface then scanned with a AFM cantilever which is a beam anchored at one end. Then, deflections are detected by a laser that reflects the surface. This allows the reconstruction of two-dimensional image of transcriptional complex.

Single Molecule Fluorescence
[edit | edit source]Another technique used to monitor transcription is to fluorescently tag the RNAP itself. This method allows the monitor of promoter search or elongation with minimal perturbation. Specifically, the structural change in TEC can be examined by using the method called Fluorescence resonance energy transfer (FRET). FRET can follow the distance between two nucleotides by measuring the intensity change in fluorescence.
Beads
[edit | edit source]By attaching beads to single RNAP molecules, one can record the position of these beads to determine the change in location or rotational state of the enzyme. Specifically, the beads can be sensitively measured by measuring the light scattered from the bead or the rotational states. One can also apply force on the beads with an OT. OT is a tightly focused beam of infrared laser light that exerts forces on the beads by means of radiation pressure. In addition, force can be applied by means of laminar fluid flow. The end of the DNA template can be attached to a second bead so that fluid flow can exert force on the free bead which place tension on the DNA template.
Transcription Initiation
[edit | edit source]
Promoter Search
[edit | edit source]Transcription requires a binding of the holoenzyme to DNA promoter sequence that is placed throughout an excess of genomic DNA. This is a problem that is common to all sequence-specific DNA-binding proteins. Two independent mechanisms, sliding and intersegment transfer, have been proposed to enhance binding by increasing its efficiency of the search process. Sliding transfer occurs when RNAP associates with nontarget DNA by diffusing in a random “walk” until it reaches the target site. Meanwhile, transegment transfer involves polymerase searching for the promoter by crossing from on position to another, bound simultaneously to both DNA segments.
Open-Complex Formation
[edit | edit source]When locating a promoter site, the RNAP undergoes a structural transition from the closed complex to the open complex (OPC). The RNAP bends and unwinds a segment of DNA with the aid of initiation factors such as "sigma", creating the transcription bubble. "sigma" is dubbed as the “housekeeping” factor that directs RNAP to recognize vast number of promoters in bacteria. For instance, AFM reading of E. Coli promoter revealed that the DNA is bent between 55̊ and 88̊ which is a consistent measurement from the bend angles inferred from gel mobility assays.
Abortive Initiation
[edit | edit source]After forming OPC, RNAP starts the synthesis of RNA oligonucleotide complementary to the DNA template strand. Although RNAP creates highly stable complex during elongation phase, the initially transcribing complex (ITC) is highly unstable causing spontaneous release of short RNA chains and restarting synthesis which is known as “abortive initiation.”
Elongation
[edit | edit source]On-Pathway Elongation
[edit | edit source]During transcription, RNAP translocates along the template DNA synthesizing an mRNA that has thousands of nucleotides in length. When the mRNA reaches 9-11nt in length, RNAP leaves the promoter region and enters the elongation phase. In this step, the TEC complex is very stable and remains tightly bound to both the DNA template and the nascent RNA during nucleotide addition. The major stabilizing factor of the complex is thought to be the base pairing within the RNA:DNA hybrid. The “sliding clamp” model states that the extensive protein-nucleic acid contacts within the polymerase greatly contributes to RNA retention, increasing the overall stability. The “clamp” that consists of narrow protein channels surround the hybrid to prevent any shearing motion between the RNA and the DNA.
Off-Pathway Events
[edit | edit source]The process of on-pathway elongation is frequently interfered by entry into off-pathway states that plays an important role in regulating RNA synthesis. One example of RNA regulation is transcriptional pausing during elongation. The puases can reduce rate of mRNA production, recruit factors for the TEC that modify the subsequent transcription, function as a precursor to termination, or lead to messenger splicing. The long “stabilized” pauses are known to play a regulatory role in formation of RNA hairpins in the transcript which is thought to inactivate RNAP. Series of studies have displayed that pauses lasting 20 seconds or more indicates a rate of base misincorporation during RNA synthesis, suggesting in need for proofreading.
Termination
[edit | edit source]Termination is a tricky step because of the stability of the TEC complex and RNAP must dissociate accurately releasing the mRNA and the DNA template. In prokaryotes, the termination occurs at specific sequence that code for a stable hairpin in the nascent RNA. In general, termination might be caused through allosteric interactions between RNA hairpin and RNAP that trigger the TEC to release the substrates to stop the reaction. Some studies concluded that termination occurs due to an intermediate elongation-incompetent state whereas some studies support that termination occurs rather quickly with no intermediates.
Reference
[edit | edit source]Herbert, Kristina M., William J. Greenleaf, and Steven M. Block. "Single-Molecule Studies of RNA Polymerase: Motoring Along." Annual Review of Biochemistry 77.149-76 (2008): 149-172. Print. RNA-dependent RNA polymerase is an enzyme, which catalyzes the replication of RNA from an RNA template. Usually, the typical RNA polymerase is well known that are catalyzes the transcription of mature RNA from a DNA template.
Virus
[edit | edit source]The most famous RdRP in a virus is the polio virus 3Dpol. The virus is made up of RNA which enters the cell through receptor-mediated endocytosis. The RNA is able to act as a template for complementary RNA synthesis. The complementary strand of the RNA is able to act as a template, in order to produce new viral genomes which are packaged and prepare to lyse from the cell transfer to other cells for more infection. This method of replication there is no DNA; therefore the replication is rapidly.However, the downside is that there is no 'back-up' DNA copy.
There is several eukaryotes that have RdRPs, and the RdRPs are involved in RNA interference; these amplify microRNAs and small temporal RNAs. Also, they produce double-stranded RNA from using the small interfering RNAs as primers. The RdRPs are used in the defense mechanisms, but it can be usurped by RNA viruses for their benefit.
Polio Virus
[edit | edit source]The first interaction for the polio virus is with a host cell; it consists two materials: binding to a specific cell surface protein, and the poliovirus receptor (PVR). The PVR, is a cell surface sialylated glycoprotein, and is a member of the immunoglobulin superfamily (is a loop in the structure of the protein that is a Ig domain). Therefore, PVR has three Ig loops that are on the outside of the cell. The loops begins with the most farthest of the cell surface. In loop 1, the polio virus binds to it receptor, which the receptor molecule binds on the virus particle.
The poliovirus genome is made of positive sense single stranded RNA that encodes a polyprotein of aa's in the range of 2100-2400. Both ends of the genome are modified; in the 5' end is modify by a covalently attached basic protein VPg which consist of 23 aa's, and the 3' end by polyadenylation. In a series of cleavages, viral proteases cleave themselves out and break down the polyprotein into 10 separate gene products involved in replication and packaging.
The viral proteases 2A cleaves the p220 subunit of the cap binding complex; therefore, they make host cell from the mRNA unrecognizable to ribosomes. The 2A protease abrogates most of the host cell's own protein synthesis. Viral mRNA depends on a 5' UTR that contains an internal ribosome entry site; serves as a ribosome docking site to the subunits of ribosomes.
Replication occurs entirely in the cytoplasm. In addition, they serve as a template for protein synthesis, the positive sense strand genome is utilized as a template for the synthesis negative sense strands. On the other hand, the host cells has a lack of necessities to replicate RNA. Poliovirus uses a viral RNA-dependent RNA polymerase to produce RNA molecules of the opposite polarity. Viral protein VPg covalently attached to uridine, which serves as the primer. The first round of replication produces a single antisense molecule. The antisense template is used to produce copies of the original genome, which they are packaged into viral capsids before it gets release.
The virus has been translated to its own RNA, so it produce the necessary proteins, and the virus genome is replicated. However, the virus needs to package the newly synthesized RNA molecules inside capsids, and must need the RNA packaged in order the virus is completed. The capsid proteins self-assemble into an immature capsid that has a structure of which proteins were needed, but the final form of the virus is not finished to cleaved. The mature poliovirus capsid has icosahedral symmetry, and have 60 copies of viral capsid proteins that are VP1, VP2, VP3, and VP4. The viral RNA enters the incomplete capsid and is secured inside when the viral proteases make the final cleavages. Once the genomes have been packaged into mature virions, the virus particles await the cell's lysis in order to be released. As many as 100,000 virions can be released from a single infected cell.
There is a conformational changes in the capsid, because there was a binding in the virus with the receptor. VP4, an internal capsid protein detaches from the capsid. The capsid swells and the poliovirus genome is susceptible to degradation. When VP1 is released, the genome is released onto the cytoplasm of the cell. The viral entry strategy is very inefficient; only 1% of the viruses initiate an infection.
External
[edit | edit source]http://www.brown.edu/Courses/Bio_160/Projects2000/Polio/PoliovirusLifeCycle.htm Discovered in the 1980s, RNA helicases are enzymes that use ATP to bind and remodel RNA and ribonucleoprotein complexes (RNPs). Mostly all helicases work and interact with many other proteins inside a multi-component assembly. While it is it unknown how RNA helicases exactly locate their binding sites on the complexes, experiments show that they most likely either bind to cofactors, which then guides them to the complex, or the helicases themselves find the binding sites according to a complex code of features on the RNAs. RNA helicases also play an important role in eukaryotic RNA metabolism and are found in all kingdoms of life. But little is known about them and how they work in the cell. RNA helicases are similar to DNA helicases and share similar functions.
RNA Helicase Classifications
[edit | edit source]RNA helicases can also be classified into six superfamilies (SFs). SFs 1 and 2 are comprised of helicases that are non-ring forming. All eukaryotic RNA helicases belong to these superfamilies. SFs 3 to 6 are helicases that can form rings and can be found in bacteria and viruses.
SFs 1 and 2 can be broken down into well-defined helicase families. Each family has distinct structural and functional properties. Six of the families have RNA helicases while the rest consist of DNA helicases. Helicases in SF 1 and 2 have a core made of two similar helicase domains and have at least 12 characteristic sequence motifs at positions in the helicase core. Not all helicases in one family will have the same motifs but they have high sequence conservation. In other families, sequence conservation is low. Across superfamilies, sequence conservation is even lower. This suggests differentiation between DNA and RNA helicases was not an evolutionary force in the classification of helicase families.
The helicase core is also surrounded on either side by C- and N-terminal domains. The terminal domains are essential to the helicase’s cellular specificity because they assist specific complexes in recruiting proteins. They accomplish this through their interactions with other proteins or by recognizing specific nucleic acid sections. Unlike the core’s sequence motifs, the C- and N-terminal domains are not conserved between families. Certain families in SF1 and SF2 are also identifiable by their characteristic beta-hairpin in between the VA and VI motifs of the helicase core. The helicase families who show this beta-hairpin are the Ski2-like, DHeAH/RHA and NS3/NPH-II families. Other families, such the Upf1-like and RIG-I-like families, have noticeable inserts between or within the helicase core domains.
NPH-II helicase, found in vaccinia virus, and NS3 helicase from the hepatitis C virus are two RNA helicases that are essential for viral replication, and have had extensive studies. Both of these helicases load on a 3' single strand of RNA and moves toward the 5' end of the strand. These helicases begin to unwind the RNA through bursts and pauses, beginning at the junction of the single and double strand. During pauses, the helicase could be either preparing to continue unwinding, but it could also dissociate from the RNA. As of the present, there is still little known about the fundamental characteristics of the helicases acting on the RNA.
Source: Li PTX, Vieregg J, Tinoco I Jr. How RNA Unfolds and Refolds. Annu Rev Biochem. 2008;77:77-100.
RNA Helicase Mechanisms
[edit | edit source]RNA helicases employ two mechanisms for unwinding: canonical and by local strand separation. Both methods are ATP-dependent because ATP binding is needed not only for the helicase to bind to the duplex but to also keep the two helicase domains together. In both canonical and local strand unwinding, the helicase domains surround the nucleic acid in similar directions and make contact with the RNA’s sugar-phosphate backbone. This allows for complete attachment of the RNA helicase and movement along the RNA by 1nt per ATP consumed. Many translocating helicases can move in bursts of up to 18 nt steps before they perform a rate limiting step, allowing for quick unwinding of the RNA duplex. In local strand unwinding, the bound RNA strand often show bends in its backbone due to the presence of ATP analogs while in canonical unwinding no such bend is exhibited. The bends decrease preference for the duplex structure and most likely represent the RNA conformation of the two strands after the duplex is unwound.
Canonical Duplex Unwinding
[edit | edit source]
When RNA helicases unwind RNA strands canonically, the RNA helicase attaches itself on the single-stranded region of the RNA strand and then translocates along the bound strand. It has defined direction and can either go 3’ to 5’ or 5’ to 3’ as it displaces the complementary strand. Each translocating step has multiple processes, including ATP binding and hydrolysis. ATP binding and hydrolysis drives the process forward. This type of winding requires strands to have single-stranded regions in a defined polarity with respect to the duplex. The RNA helicases families who are known to perform this mechanism are Upf1-like, Ski2-like, RIG-I-like and DEAH/RHA.
Local Strand Separation
[edit | edit source]
Local strand separation occurs when a RNA helicase loads itself directly on a duplex region of the RNA, and uses ATP to separate the strands. Unlike the canonical method, this type of unwinding does not require a single-stranded region with specific orientation nor ATP hydrolysis. ATP binding is sufficient for duplex unwinding to occur, ATP hydrolysis however is needed to successfully detach the helicase from the RNA. Sometimes the enzyme will dissociate before the strands have completely separated because of the strands quickly re-annealing. As the RNA strand gets longer, however, this type of unwinding is unfavorable and inefficient. The DEAD-box family unwinds duplexes this way and can only handle duplexes with 10 to 12 basepairs.
Other Functions
[edit | edit source]RNA helicases also have other functions aside from unwinding duplexes. It can also displace proteins on RNAs. This is called RNP remodeling. RNP remodeling appears to be important in how RNA helicase functions since RNAs are usually attached to a protein in vivo. RNP remodeling, however, is not essential in unwinding but also works for helicases that unwind canonically and for DEAD-box proteins. Some helicases can only remove certain proteins, while others can remove a wider variety of proteins.
RNA helicases have also shown to help in the RNA folding process. An example are the RNA helicases who facilitate and regulate RNA folding in fungal mitochondria as RNA chaperones. They should not be confused with protein chaperones which also help in RNA folding. RNA chaperones guide the RNA through the series of folding steps while continually proofreading. It determines if the substrates formed are correct or incorrect. If correct, the process is continued but if incorrect, the substrate is disregarded and the RNA chaperone opens up a new reaction path for RNA folding. Protein chaperones on the other hand catalyze the steps of the folding pathway and help stabilize the subsequent RNA structure.
Other helicase families show activities in regards to the innate immune system. The RIG-I RNA helicase translocates to a RNA duplex but instead of unwinding it, the helicase acts as a pattern recognition receptor and determines if a viral RNA present in the cytoplasm. It detects the viral RNA based upon the long double stranded RNA’s it creates during viral replication. Only viral RNA's are detected because a majority of RNA’s in eukaryotic cells form only short RNA duplexes, which is ideal for the local strand unwinding process.
DEAH/RHA helicases also help in both the separation of a spliced mRNA from the spliceosome and the ligation of the exon to the mRNA. It separates the spliced mRNA from the spliceosome by attaching to the mRNA and moving from the 3’ to 5’ direction, breaking the RNA-RNA and RNA-protein along the way.
References
[edit | edit source]Jankowsky, Eckhard. "RNA helicases at work: binding and rearranging." Trends in Biochemical Sciences. xx (2010): 1-11. Web.
Riboswitches are recently discovered RNA domains that function as gene expression regulators. It is a portion of the mRNA strand that is able to bind small molecules and alter the gene activity. An mRNA which possesses the riboswitch is able to regulate its own activity depending on whether or not a molecule is attached to it. They are located at the 5' end of untranslated regions of messenger RNA. These functional domains exist in bacteria and have also been engineered in the laboratory[47]. Riboswitches are significant because most believe that proteins are primarily responsible for the complexity, specificity, and efficiency of gene control. Most riboswitches exist in bacterias although some have also been found in plants and fungi[48].
It was first described by Ronald Breaker's lab in 2002 when they utilized in-line probing of Escherichia coli btuB mRNA to show that it could bind a metabolite/substrate and inhibit translation of the strand's product (AdoCbl) -- without proteins[49].
The original meaning of riboswitch was that messenger RNA can sense small molecules of metabolite. While this is still the use today, others have changed the meaning to include other types of RNA, further expanding the meaning. mRNA that contains a riboswitch can regulate its own activity. This opens many doors in the world of biology because it shows that molecules can evolve to be their own masters, or regulating themselves. These RNA were seen to distinguish between very similar molecules or analogs which shows the intricacy of the method. This fact has opened up a world of RNA because it is now known that the capabilities of RNA were much greater than once known. It is interesting because it illustrates how little we humans know about our very own bodies. Riboswitches allow RNA to respond to different concentrations of molecules almost as though the RNA had a mind of its own determining its actions. Due to the expansion of the definition of a riboswitch, there are many different kinds known to mankind today.
As the mantra of structural biochemistry is that structure determines function, it is not a surprise that the structure of the riboswitch allows for such great function. Most RNA do not need to conform to the strict watson and crick model of DNA allowing for many variations in RNA. The great variation in RNA is responsible for riboswitchs abilities. Riboswitches are made of two parts. the aptamer domain and the expression platform. The aptamer domain essentially acts as a receptor that binds to specific ligands. The expression platform is interesting because it can toggle between two different secondary structures when binding to a ligand, creating a plethora of possible structures. In both parts of a riboswitch there is a switching sequence. This switching sequence directs the expression of the genes. [1]
Types of Riboswitches
[edit | edit source]There are several types of riboswitches known, some of which are:
- TPP riboswitch : this riboswitch binds TPP (thiamin pyrophosphate in order to regulate the transport and synthesis of thiamin as well as other metabolites with similar properties.
- Lysine riboswitch : binds to lysine and regulates its biosynthesis, catabolism, and transport.
- Glycine riboswitch : this riboswitch regulates glycine metabolism. This is the only riboswitch known currently to be able to perform cooperative binding.
- FMN riboswitch : this riboswitch binds FMN (flavin mononucleotide) in order to regulate the transport and synthesis of riboflavin.
- Purine riboswitch : binds purines to regulate its transport and metabolism. Different forms of this riboswitch are able to bind either guanine or adenine depending on the pyrimidine in the riboswitch.
- Cobalamin riboswitch : this riboswitch binds adenosylcobalamin, the coenzyme form of B12 vitamin, in order to moderate the synthesis and transport of cobalamin and other similar metabolites.
as well as many others such as SAM riboswitch, PreQ1 riboswitch, SAH riboswitch, glmS riboswitch, and cyclic di-GMP riboswitch.
Structure
[edit | edit source]
Riboswitches consist of two functional components, the conserved aptamer region and the highly variable expression platform. Unlike proteins, only four nucleotides are available to generate the specificity required by the riboswitch to bind[50].
The aptamer domain is usually a single binding site that has a highly conserved primary and secondary RNA structure and forms selective binding pockets for ligands. It essentially acts as a sensor for metabolites within the cell. Since it is located at the 5' end of mRNA, it is usually the first to be transcribed by RNA polymerase.
To improve aptamer-substrate affinity, structural data shows that hydrogen bonds, van der Waals, and other interactions form with the substrate and also adjacent RNA regions. Other aptamers may utilize an induced fit mechanism with deep binding pockets[51].
The expression platform is commonly located downstream from the aptamer.
Function
[edit | edit source]Most riboswitches function within feedback pathways by sensing metabolites and turning "off" the ability to express genes that would produce proteins that would continue the production of that metabolite[52]. The aptamer region tends to recognize ligands that are closely related to the gene products downstream from the riboswitch expression platform.
References
[edit | edit source]- ^ Wang, J., Lee, E., Morales, D., Lim, J., Breaker, R. "Riboswitches that Sense S-adenosylhomocysteine and Activate Genes Involved in Coenzyme Recycling". Molecular Cell 29, 691–702, March 28, 2008.
- ^ Nahvi, A., Sudarsan, N., Ebert, M., Zou, X., Brown, K., Breaker, R., "Genetic Control by a Metabolite Binding mRNA" Chemistry & Biology, Vol. 9, 1043-1049, September, 2002.
- ^ Coppins, R., Hall, K., Groisman, A. "The intricate world of riboswitches" Current Opinion in Microbiology, Volume 10, Issue 2, April 2007, Pages 176-181.
- ^ Breaker, R. "Complex Riboswitches''Science, Vol. 319, 1795-1797, 28 March 2008.
- ↑ Riboswitches, November 14th, 2012.
How RNA Unfolds and Refolds
[edit | edit source]In general, RNA unfolds from the tertiary structure to secondary structure to single stranded RNA and vice-versa is true for how RNA folds. RNA unfolding depends on temperature to denature RNA or sometimes enzymes such as RNA-dependent RNA polymerase (RdRps) or helicases. Moreover, scientists use the techniques called optical tweezers, which is also called laser tweezers, and fluorescence resonance energy transfer, also known as FRET, to study how secondary and tertiary RNA structures unfold and refold. Furthermore, scientists use cation binding to study how ribozymes fold and unfold.
Secondary Structure RNA
[edit | edit source]Unfolding:
Secondary RNA structure can unfold by increasing the temperature or using chemical reagents to denature RNA. Another technique used to study how RNA unfolds is optical tweezers. This technique applies a force that causes RNA to unfold in physiological temperature and buffer solutions (79). For example, the ends of a hairpin RNA have two beads—one that has an optical trap and the other has a micropipette strap. From this, RNA can be pulled and unzipped as the micropipette moves.
Refolding:
RNA refolding occurs in the reverse process of RNA unfolding. When micropipette moves, RNA can be pushed back which makes RNA relaxed and refolds RNA. However, if the relaxation force applied by optical tweezers increases, this can cause RNA to misfold (81).
Misfolding in RNA can be corrected by increasing the force. When force is increased, the RNA will try to refold into an active and functional form.
Tertiary Structure RNA
[edit | edit source]Unfolding
Tertiary RNA structure is relatively weak therefore, by changing the temperature or solutions that are not much different from the physiological state can destabilize RNA interaction.
Refolding
A technique called FRET, fluorescence resonance energy transfer, can be used to understand how RNA folds (78). Scientists label two-dyed nucleotides on RNA strand and through observations of RNA folding, FRET signal allows scientists to measure the distance and motif between those two-dyed labeled nucleotides with respect to time. Furthermore, scientists can also use FRET to understand the changes in RNA conformation when RNA is bound to Mg2+ or ribosomal proteins.
Single-molecule of RNA Enzymes
[edit | edit source]To study the single-molecule of RNA enzymes, scientists use ribozymes and FRET technique. The difference between the study of how ribozyme unfolds and folds and that of the secondary or tertiary RNA structure is that scientists add a series of Mg2+ and they observe the FRET signals in order to tell whether ribozyme is docked (folded) or undocked (unfolded). From this Mg2+ “pulse-chase experiments,” scientists can find the “kinetic fingerprints” of the hairpin ribozymes’ enzymatic states (84). Based on this, scientists were able to figure out that ribozymes participate in chemical reactions such as oxidation or reduction, synthesis of nucleotides, and formation of peptides. Thus, the study of ribozymes reinforces the RNA World hypothesis, which stated that RNA preceded DNA (85).
Effects of Ligand and Protein Binding to RNA
[edit | edit source]Another way that RNA unfolds is through the appearance or the lack of ligands and/or proteins. Specific proteins and/or ligands bind to RNA and cause it to unzip. By using a technique called single-molecule fluorescence, scientists studied ribonucleoproteins (RNP) and its effect on RNA (88). In this technique, scientists can count RNP subunits in bacteriophage through “electron cryomicroscopy and crystallography” (89). Then, when RNA hairpins unfold, RNPs are assembled and proteins bind to RNA causing RNA to change conformation.
There are three commonly used applications of single-molecule fluorescence techniques. The first is simply counting the subunits in an ribonucleoprotein (RNP). The second common technique is annealing two hairpins, requiring the unfolding of both. As of current, the specific protein role is still not entirely clear. The third technique is to use the fact the RNP assembly is sequential. Because RNP assembly is sequential, this is an indication that events that occur early on in protein binding result in conformational changes in the RNA. By labeling a pair of fluorophores at different positions of telomeric RNA scientists have identified the binding of p65 protein can induce conformational change.[1]
In DNA, argininine is the component used to bind and stabilize the molecule. However, in RNA, it is argininamide, and not arginine that stabilizes and binds the TAR hairpin.[2]
Enzymes used to unfold RNA
[edit | edit source]Scientists learned that RNA needs energy input in order to unfold itself however, RNA folding does not require energy because this is a spontaneous reaction. According to the authors in the “How RNA Unfolds and Refolds,” in order to unfold three to four base pairs in RNA, one ATP is used (89). Therefore, enzymes such as helicases or RNA-dependent RNA polymerase help RNA unfold by using chemical energy that is present when nucleoside triphosphates undergo a hydrolysis reaction. For example, helicases take the energy from the ATP hydrolysis reaction to extract the proteins bound on RNA and unfold the double-stranded RNA (90). Therefore, as the concentration of ATP goes up, the faster this step will be. Although RNA-dependent RNA polymerase has not completely explored, scientists believed and expected that it is similar to how helicases work.
Another way that RNA can be unfolded is by binding single stranded RNA to a single-strand specific protein. However, in this situation, the binding must be strong so that it can overcome the forces seen in base pair bonding.[3]
In viral RNA replication, RNA must be single-stranded in order for its sequence to be interpreted during replication and translation. The RNA molecule is first unfolded by an RNA-dependent RNA-polymerase or ribosome. Through the hydrolysis of nucleoside triphosphates, the enzymes can use that energy to be able to unfold the RNA substrates. The RNA needs to have varying sequences.[4]
Work Cited: Li, Pan T.X., Tinoco Jr, Ignacio, and Vieregg, Jeffrey. “How RNA Unfolds and Refolds.” Annual Review of Biochemistry. 2008. 77-100. Print.
Tertiary Structure Folding
[edit | edit source]The tertiary folding refers to the interactions between the distal domains that form the structure needed for the RNA to carry out catalytic and regulatory functions. These interactions are fairly weak and can be easily unfolded using small change in temperature and solutions. Specifically, the FRET technique is utilized to observe the tertiary folding of RNA. This technique is performed by measuring the distance between the two florescence-dyed nucleotides. This enables observation of specific tertiary motifs in real time which consists of distinctive folding of the interacting RNA strands. The FRET technique was crucial in studying the RNA folding by measuring the conformational change during the binding of salt (Mg2+) or ribosomal protein. One prominent motif that was studied extensively is the tetraloop-receptor interaction which is present in many large folded RNAs and has been used to create synthetic RNA “building blocks.”
Using optical tweezers and increasing the force, RNA can be unfolded into four distinct conformations in the following order: kissing complex, two linked hairpins, one hairpin, and single strand. Similarly, when the force is decreased, the single strand can be refolded in the reverse order into the kissing complex. The kissing interaction is defined as the base pairing (complementary sequences) between the two hairpin loops and the hairpin loops is created when two complementary sequences in a single RNA meet and bind.

Salt effect on tertiary structures
[edit | edit source]In tertiary structures folding and stability are highly dependent on ionic conditions, especially of Mg2+. Thus metal ions have a greater effect on tertiary structures than on secondary structures of RNA. Mg2+ slows down the kinetics of breaking tertiary interactions, but only moderately affects the folding rates.
Common motifs that demonstrate salt effects include intron ribozymes, pseudoknots, and loop-loop interactions: In intron ribozymes distinct rips were observed in MgCl2, indicating the unfolding of a structural domain. When there was no Mg2+, no rips were observed.
In pseudoknots compact structures are formed, and have increased stability with bound Mg2+.
In loop-loop interactions force manipulation is used to see how an intramolecular kissing complex changes. This can be seen from the unfolding and refolding of secondary structures. The base pair sequence affects the salt dependence of kissing interactions.[5]
References
[edit | edit source]Work Cited: Li, Pan T.X., Jeffrey Vieregg, and Ignacio Tinoco. "How RNA Unfolds and Refolds." Annual Review of Biochemistry 77.1 (2008): 77-100. Print.
- ↑ Li PTX, Vieregg J, Tinoco I Jr. How RNA Unfolds and Refolds. Annu Rev Biochem. 2008;77:77-100.
- ↑ Li PTX, Vieregg J, Tinoco I Jr. How RNA Unfolds and Refolds. Annu Rev Biochem. 2008;77:77-100.
- ↑ Li PTX, Vieregg J, Tinoco I Jr. How RNA Unfolds and Refolds. Annu Rev Biochem. 2008;77:77-100.
- ↑ Li PTX, Vieregg J, Tinoco I Jr. How RNA Unfolds and Refolds. Annu Rev Biochem. 2008;77:77-100.
- ↑ Li PTX, Vieregg J, Tinoco I Jr. How RNA Unfolds and Refolds. Annu Rev Biochem. 2008;77:77-100.
Structural Biochemistry/Mechanical unfolding of RNA
Introduction
[edit | edit source]Short RNAs play an important role in biochemistry. Short RNAs include transfer RNAs, small nuclear RNAs, micro RNAs, and other ones. In this section, we are gonna talk about how to profile short RNAs using helicos single-molecule sequencing. In order to lengthen the RNAs chain, scientists use the methods of splicing and 3 prime-end processing. And there are many non-protein coding RNAs that can be less than 200nt; they can be called short RNAs.
Profiling short RNAs
[edit | edit source]Materials
[edit | edit source]In RNA isolation, scientists want to purify sRNA from the total RNA or cultured cells. Hence they use certain materials to do this technique. They are mirVana™ miRNA Isolation Kit, miRNeasy Mini Kit, RNA/DNA kit. The RNA/DNA kit can be used to isolate large amounts of sRNA from the total RNA. Then people use TBE-Urea polyacrylamide gel electrophoresis and overnight elution to obtain the sRNA from each kit. In the process of making cDNA, scientists use Escherichia coli PolyA polymerase and 100 mM CTP substrate. Then they apply the process of reverse transcriptase called ThermoScript. Consequently, Phenol, chloroform, and isoamyl alcohol were used with the 5 M Ammonium acetate. And the cDNA synthesis primer and RNAse A are also used. This sequence of this primer was created by the Integrated DNA Technologies and their sequence is TCG CGA GCG GCC GCG GGG GGG GGG GGrG rGrG. In profiling 3 prime-end sRNAs, the reverse transcriptase called SuperScript III, USER enzyme, dTU-V cDNA synthesis primer with the sequence of TTTTUTTUTUTTTUTTTTUTTTUTTV, RNAse H, and RNAse 1f were used. Overall, the two methods use similar materials in the process. They both used 100 and 70% ethanol, 10 mM dNTPs, AMPure ®, Magnetic stand for 1.5-mL tubes beads, and PCR machine. The RNAse inhibitors were also used in the methods. They are ANTI-RNAse inhibitors or RNAseOUT inhibitors. In sequencing cDNA, scientists use 20 U/ mL Terminal Transferase, dATP, 1 mM Biotin-ddATP, 10 mg/mL Bovine serum albumin, Quant-tT™ OliGreen ® ssDNA Reagent, NanoDrop 3300, HeliScope™ Single Molecule Sequencer, and Helicos ® Flow Cells
Methods
[edit | edit source]Before beginning to profile short RNAs, scientists have to understand the goals of the experiment. The desired length sRNA must be separated from the long RNAs. They have to understand that only some of sRNAs have the 3 primed polyA tail of the mRNAs that can be used to convert sRNAs to cDNA. Also, using random hexamers can only be used sometimes to convert the short RNAs to cDNAs since the RNAs are too short for the process of conversion to go smoothly. Some RNAs have modifications at their 3 primed and 5 primed ends that would make it harder to make conversion to cDNA. There are two methods in detecting sRNAs. The first method detects sRNAs with the 3 primed -OH. And the second one detects 3 primed-polyA sRNAs. First, the isolation of sRNAs occur using different kits. If only less than 200nt section of sRNAs is needed, the mirVana kit , miRNeasy, or RNA/DNA kit would be useful. The TBEUrea denaturing polyacrylamide gel-electrophoresis can also be used to isolate sNRA for a specific region.
The general method of profiling sRNAs occurs by tailing RNAs with 3 primed polyC. First, they put RNA into a PCR tube with 30μL. The amount of short RNAs used in this step can be around 5ng to 10ng. Then they incubate the tubes in the PCR machine at 850C for 2 minutes, then put the tubes in the ice for another 2 minutes. To the tube, they add 10 mL of 5× E. coli PolyA polymerase buffer; 5 mL of 25 mM MnCl2, 1 mL of 100 mM CTP, 1 mL of Anti-RNAse or RNAseOUT, and 3 mL of 2 U/ mL E. coli PolyA polymerase. Hence , they mix the solutions and incubate it in PCR machine for 3 hrs at 370C. After that,40μL of water and 10 μL of 5M ammonium acetate were added to the incubated tube. Then they extract the solids twice in the tube with phenol, chloroform, isoamyl acid. Then precipitation of the solution occurs when they add three times the 100% EtOH to the tube at -800C . In the end, they would centrifuge the solution at 40 for 30 mins and wash the precipitation with 70% EtOH, then vaccuum dry the solid. Finally, you have to put the solid in 30.5μL of water.To make cDNA, they first add the 1 mL of 100 mM cDNA synthesis primer to the 30.5μL solution in the previous step. Then they would incubate the solution for 2 minutes at the 700C in the PCR machine. At that temperature, they would add more reagents to the solution like 10 mL of ThermoScript cDNA Synthesis buffer for 5 times ,5 mL of 0.1 M DTT, 2.5 mL of 10 mM dNTPs, and 1 mL of ThermoScript reverse transcriptase. Finally, incubation of the solution is required for 15 mins to inactivate the reverse transcriptase. After the cDNA synthesis process, they purify the cDNA. First, they mix the synthesis cDNA with 1 mL of RNAse A. And the AMPure beads suspension were mixed so that the beads would not be suspended. Then to the mixture of cDNA and RNAse, they added 150 mL of the AMPure beads to incubate the solution at room temperature for 30 minutes. Hence, they collect the beads using the magnetic stand and the solids would be removed from the solution. The solution was washed 2 times with 200 mL of 70% EtOH and the solids would be dried for from 30 to 45 minutes at room temperature. Then they would wash the cDNA two times with 20μL of water. Before beginning to profile short RNAs, scientists have to understand the goals of the experiment. The desired length sRNA must be separated from the long RNAs. They have to understand that only some of sRNAs have the 3 primed polyA tail of the mRNAs that can be used to convert sRNAs to cDNA. Also, using random hexamers can only be used sometimes to convert the short RNAs to cDNAs since the RNAs are too short for the process of conversion to go smoothly. Some RNAs have modifications at their 3 primed and 5 primed ends that would make it harder to make conversion to cDNA.
The second method of profiling sRNAs is to make cDNA by profiling with polyA tails. First, 1 mL of 50 mM dTU-V primer and 1 mL of 10 mM dNTPs are mixed together. Then the solution is incubated for 5 minutes at 650C by using the thermocycler. Then the solution is to be put on the cold aluminum and it was left there for 1 minute. The next step is to add the solution to 2 mL of ten times of SuperScript III reaction buffer, 4 mL of 25 mM MgCl2, 2 mL of 0.1 M DTT, 1 mL of SuperScript III, and 1 mL of RNAseOut. This mixture will be incubated for 50 minutes at 850C. The next step is to remove the dTU-V primer sequences from the mixture obtained previously. In order to carry this reaction, 1 mL USER enzyme is added to the mixture and the solution is incubated again for 15 minutes at 370C. Then d 1 mL of RNAse H and 1 mL of RNAse are added to the solution. And this solution can be used for the next step which is the purification of cDNA
In this reaction, 180μL of AMPure beads is heated up to room temperature. Then the beads will be added to the solution that was mixed and incubated to make the cDNA solution. The beads are then collected by using the magnetic stand and the solids would be collected too. Then the beads are washed with 500 mL of 70% EtOH two times. The solids would then dried up. Finally, to isolate the cDNA from the beads, 20 mL of nuclease-free water is added to the solids. And the liquid will be removed using the pipet until the cDNA solid is obtained.
Sequencing cDNA
[edit | edit source]The 3 primed end of the cDNA will be blocked by polyA residues using terminal transferase (TdT). In order to carry out the process, they need to prepare the 3 primed end of the cDNA. First, they got cDNA to be tailed (<10 ng) in 10.8 mL of water. Then 2 mL of 10× TdT buffer and 2 mL of 2.5 mM CoCl2 are added to the mixture and the mixture is to be incubated for 5 minutes at 950C.4 mL of 50 mM dATP, 0.2 mL of BSA, and 1 mL of TdT are added to the incubated solution. Then the mixture will be incubated in the PCR machine for 60 minutes at 700C. The solution is to be put on ice for 2 minutes. Then 1 mL of 10× TdT buffer, 1 mL of 2.5 mM CoCl2, 0.5 mL of 200 mM Biotin-ddATP, 6.5 mL of water, and 1 mL of TdT are added to the cold solution. Finally, it will be incubated in the PCR machine for 20 minutes at 700C Structural Biochemistry/Salt Effects on mechanical folding

Discovery
[edit | edit source]RNAi was first introduced when plant biologists attempted to introduce genes into a petunia. When they added a gene that attempted to deepen the flowers purple color, the gene actually inhibited it. The resulting flowers had white patches or were completely white.
Soon after this discovery, another group of researchers realized that this same gene-silencing phenomenon was occurring in experiments with C. elegans. These scientists figured out that RNAi is triggered by double-stranded RNA, which is not typically found in healthy cells. Two well known scientists, Andrew Fire and Craig Mello, were awarded the 2006 Nobel Prize in physiology or medicine for this discovery. [53]
Biological Implications
[edit | edit source]RNA interference (RNAi) is a natural mechanism within the cell used to silence the expression of certain genes. Small RNA molecules play essential roles in regulating gene expression by RNA interference. There are three basic characteristics of these pathways:
1) Small RNA biogenesis
2) Formation of RNA-induced silencing complexes.(RISCs)
3) Targeting of complementary mRNAs.
RNA interference is triggered by the enzyme Dicer, which cleaves long double-stranded RNA (dsRNA) producing 20 to 30 nucleotide RNAs whose sequences can base pair with segments of mRNA transcripts. Then the newly generated microRNAs (miRNAs) or small interfering RNAs (siRNAs) will assemble into complexes designed to complementarily fit into target RNA strands that wish to be silenced. The induced silencing complexes are called RNA- induced silencing complexes (RISC) and are constructed into large multiprotein effectors, called RNA-induced silencing complexes (RISCs), which bind to target transcripts and trigger their destruction.
Cognate RNA is then cleaved in the middle region bound to the siRNA strand. This mechanism has been theorized to have a self-defense purpose to protect cells against viral infections or cancerous cells.
RNAi can help to study tissue regeneration. RNAi shuts down individual genes during the tissue regeneration and the scientists can understand what genes in amphibians are involved in regenerating tissue when missing limbs are regrown. By understanding this process, they hope to learn how to regenerate human tissue.
Origin
[edit | edit source]RNAi is proposed to have evolved about a billion years ago, before plants and animals diverged. This is due to the fact that it exists in all living organisms, from plants to animals.
Modern hypotheses state that RNAi evolved as a cellular defense mechanism against invaders such as RNA viruses. When they replicate, RNA viruses temporarily produce a double-stranded form. This double-stranded intermediate would trigger RNAi and inactivate the virus’ genes, preventing an infection.
RNAi may also have evolved to combat the spread of genetic elements called transposons within a cell’s DNA. Transposons can wreak havoc by jumping from spot to spot on a genome, sometimes causing mutations that can lead to cancer or other diseases. Like RNA viruses, transposons can take on a double-stranded RNA form that would trigger RNAi to clamp down on the potentially harmful jumping. [54]
Cellular Mechanism
[edit | edit source]
RNAi is a process in which RNA is used to scilence genes. the main player in this process is the RNA-induced silencing complex (RISC), the complex is activated by short double-stranded RNA molecules. [55]. dsRNA can come from infection by a retro virus or artificially inserted (exogenous), the RNA can also come from within the cell's own genome (endogenous). [56].
dsRNA
[edit | edit source]There are two approaches toward dsRNA depending on its origin, whether it be exogenous (from outside the cell) or endogenous (from inside the cell).
-Exogenous: the foreign dsRNA is detected and bound by an effector protein, the protein initiates the dicer to cut up the dsRNA, the same effector protein helps in transporting the siRNA to RISC.[57]
-Endogenous: the target dsRNA is cut up by the Dicer into single stranded siRNA, which are then transported to an active RISC. When they are incorporated into the RISC the siRNA base pair with their corresponding sequences on mRNA strands, which are then cleaved at those sites. By cleaving the mRNA the synthesis of protein is halted [58].


RISC
[edit | edit source]RISC stands from RNA-induced silencing complex, its active components consist of endonucleases and argonaute proteins. The function of each respectively is to recognize a complimentary sequence on mRNA (complementary to the sequence of the bound siRNA) strands and cleave the mRNA (argonaute proteins). This process is ATP independent and act directly through components of the RISC [59] [60].
How RNA pairs with the argonaute protein, structurally was determined by X-ray crystallography, through x-ray crystallography the active sites were determined which led to accurate information regarding how the RNA binds to the argonaute protein. In the active site, the phosphorylated 5' end of the RNA strand enters and bonds with a cation (i.e. magnesium) and by having an aromatic stacking structure between the 5' nucleotides in the siRNA. It has been inferred that the active site contains the ability to pair the siRNA with its corresponding mRNA. [61].
Research Implications
[edit | edit source]RNAi-based therapies have been proposed as a way to regulate and get rid of several disease causing genes. This path has shown to be the most promising. A good target for this type of therapy would be all forms of cancer. Cancer is often caused by overactive genes and regulating the activity of these could stop the spread of it.
Viral infections are also hypothetical targets for RNAi therapies. Many believe that RNAi actually evolved as a way to combat RNA viruses. Reducing the expression of important viral genes would leave the virus helpless and prone to attack by the immune system. In vitro, studies have already indicated that HIV, polio, HCV and others have been reduced by these therapies.
RNAi is already serving as a way to identify function of certain genes. Prior to this discovery, researchers had often resorted to inserting new genes into an organism to see what the effect would be. More recently however, scientists can merely silence the gene of interest and observe the effects that the target gene has on organism function. It can also shed light upon complex cellular pathways.
RNAi has been a novel and highly important discovery for research. For years, scientists had been intensely studying how proteins regulate gene activity, focusing most of their attention on proteins called transcription factors. Now RNA, through RNAi and related processes, is known as an essential player in the cell’s complex technique of gene regulation. [62]
Research Applications
[edit | edit source]As mentioned in the section above, RNAi can be used to selectively “silence” targeted genes in order to analyze the affects this will incur on the model organism. One area of research focuses on regeneration, the regrowth of lost or damaged body parts. This ability is quite common in nature. For example, tree stumps can grow sprouts that develop into new stems, leaves, and flowers; in lab, a mass of undifferentiated cells can grow into a mature plant; in fact, a section of certain plants composed of fully-differentiated cells can also grow into a mature plant. Animals, too, have regenerative abilities: including invertebrates such as sponges, hydra, planarians, and starfish, as well as vertebrates such as salamanders and amphibians. Humans, on the other hand, have only limited regenerative abilities. Apart from healing wounds, humans can regenerate some of the liver and the tips of fingers and toes [63]. Wouldn’t it be amazing if scientists could find a key to regenerating human tissues? RNAi is currently being used to target specific genes and turn them off in planarians and amphibians in order to analyze the functions of those genes. This way, researchers hope to find out which genes are responsible for regeneration.
Role in Tissue Regeneration
[edit | edit source]RNA interference (RNAi) is a mechanism that organisms use to silence genes when their protein products are no longer needed. The silencing happens when short RNA molecules bind to stretches of mRNA, preventing translation of the mRNA. To focus in on the genes that enable planarians to regenerate, Sánchez Alvarado and his coworkers are using RNA interference (RNAi). RNAi is a natural process that organisms use to silence certain genes. Sánchez Alvarado’s group harnesses RNAi to intentionally interfere with the function of selected genes. The researchers hope that by shutting down genes in a systematic way, they’ll be able to identify which genes are responsible for regeneration. The researchers are hoping that their work in planarians will provide genetic clues to help explain how amphibians regenerate limbs after an injury. Finding the crucial genes and understanding how they allow regeneration in planarians and amphibians could take us closer to potentially promoting regeneration in humans.
Specifically in planarians, which can regrow a whole worm from a small fraction of its body, RNAi’s ability to shut off specific genes has led to the discovery that the location of head and tail formation is controlled by *hedgehog signaling and the Wnt/B-catenin pathway. The Wnt/B-catenin pathway regulates the formation of the anterior-posterior axis. “Silencing” either hedgehog or Wnt/B-catenin with RNAi causes head and tail to grow at wrong ends [64]. In addition, some basic researchers are trying to figure out how stem cells work by planarians. These worms are like stem cells in the sense that they can regenerate. Planarians’ resemblance to stem cells isn’t just coincidence. Scientists have discovered that planarians can perform the amazing act of regeneration due to the presence of specialized stem cells in their bodies. Developmental biologist Alejandro Sánchez Alvarado of the University of Utah School of Medicine in Salt Lake City used the gene-silencing technique RNAi to search for planarian genes that were essential for regeneration. He found 240 genes that caused a physical defect in the worm’s growth and regenerative ability when silenced. Interestingly, 16 percent of these looked very much like genes that had been linked to human disease.
- in addition to regeneration in planarians, hedgehog signaling is vital to brain, intestinal tract, finger, and toe development in mammals.
RNAi and Neurological Diseases
[edit | edit source]RNAi not only protects cells from foreign genes, it is also involved in regulating the cells own genes, including the cell’s own set of noncoding mRNA’s. Thus, improperly functioning RNAi can lead to diseases and inherited disorders, including fragile X syndrome. Fragile X causes mental retardation because of the loss of FMRP, a protein usually synthesized from the FMR1(fragile X mental retardation 1) gene. It was discovered that FMRP is a component of RISC, indicating that the loss of this protein prevents RNAi in neurons from functioning properly, thus causing mental retardation [65]. (More research needs to be done to establish this link.)
On the flip side, RNAi has the potential to treat neurological diseases as well. In a similar fashion to how RNAi eliminates foreign mRNA from viral infections, the high specificity of RNAi can be used to target mutations of normal genes that lead to neurological diseases. This way, RNAi can mediate the effects of detrimental dominant alleles by “knocking out” expression of these mutant genes while leaving normal ones alone. Accordingly, this potential can be expanded to other diseases including those caused by triplet expansion or trinucleotide repeats (Neurodegernative diseases such as Spinobulbar Muscular Atrophy and Hungtington’s in addition to Fragile X). [66]
References
[edit | edit source]- ↑ Macrae I, Zhou K, Li F, Repic A, Brooks A, Cande W, Adams P, Doudna J (2006). "Structural basis for double-stranded RNA processing by dicer". Science. 311 (5758): 195–8. doi:10.1126/science.1121638. PMID 16410517.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - ↑ Hammond S, Bernstein E, Beach D, Hannon G (2000). "An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells". Nature. 404 (6775): 293–6. doi:10.1038/35005107. PMID 10749213.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - ↑ U.S. Department of Health and Human Services. Inside the Cell. September 2005.<http://www.nigms.nih.gov>.
National Institute of General Medical Sciences [67]
Biology Pages [68]
Functional Genomics, Fragile X Syndrome, and RNA Interference [69]
The New Genetics (2006): n. pag. U.S. DEPARTMENT OF HEALTH AND HUMAN SERVICES, National Institutes of Health, National Institute of General Medical Sciences. Web. <http://www.nigms.nih.gov>.
Main Component: Argonaute
[edit | edit source]The main component of RISC is the argonaute (Ago) proteins. These proteins will associate the RNAs. The Ago family can be divided into the Argo subfamily and the Piwi sub family. siRNAs and miRNAs bind to the Argo subfamily and piRNAs bind to the Piwi subfamily. In mammals, each of the four Ago subfamily proteins (AGO1-4) can repress translation but only AGO2 can cleave the RNA and result in RNA interference (RNAi)
Two Steps in RISC Assembly: RISC Loading and Unwinding
[edit | edit source]siRNA and miRNA come from double stranded RNA that has been chopped up by the RNase III enzymes, Drosha and Dicer. The resulting RNA are call RNA duplexes. There are two models of when RNA unwinds when binding to Ago proteins. The ‘helicase model’ propose that the RNAs were separated into single stranded RNA first then incorporated into the Ago proteins. The other model is the ‘duplex-loading model’ which states that the double stranded RNA binds to the Ago proteins then dissociated within the protein. Recent studies show that the ‘duplex-loading model’ may be the model for when RNA unwinds. Therefore, RISC assembly can be divided into 2 steps: small RNA duplex is bound to Ago protein, the double stranded RNA dissociates into two single stranded RNA. RNA duplex bound to Ago protein is called pre-RISC while Ago protein with single stranded RNA is called mature RISC.
Since the double stranded RNA will unwind into two single stranded RNA one of these strands must be discarded. The discarded RNA strand is called the passenger strand and the other strand is called the guide strand. The strand with a less stable 5’ end will serve as the guide strand while the other strand is discarded.
RISC Loading
[edit | edit source]RISC loading machinery
[edit | edit source]Ago proteins need the help of RISC-loading machinery to bind to RNA. RISC-loading machinery is composed of Dicer-2 (DCR-2) and R2D2 for Drosophila Ago2. R2D2 binds to the more stable end of RNA while Dcr-2 binds to the more stable end. Although Dcr-2 can both dice up RNA and load RNA into Ago proteins, studies has shown that the siRNA duplexes must dissociate from Dcr-2 after dicing then rebind to the Dcr-2-R2D2 dimer according to its stability. Human only has one type of Dicer, human Dicer and its partner protein TRBP (TAR binding protein) helps load RNA into AGO2-RISC. However, studies have shown that Dicer is only needed when loading into fly Ago2. It is not needed when loading RNA complexes for other Ago proteins. It seems that there are two pathways of RISC loading, a Dicer dependent pathway and a Dicer independent pathway.
Small RNA sorting
[edit | edit source]siRNA duplexes usually have perfectly complementary sequences so that all the bases are lined up. However, miRNA-miRNA* complexes usually have central mismatches. In flies, the Dcr-R2D2 likes to bind to the perfectly complementary siRNA like complexes but doesn’t like RNA strands with mismatches. On the other hand, Ago1 likes to bind to sequences that has central mismatch around nucleotide 8-11.
Another guild loading in the right orientation is the identity of the nucleotide in the 5’ end of the guild strand. In flies, Ago1 favors U while Ago2 favors C. For plants, the orientation of the strands relies heavily on the identity of the nucleotide as well. Arabidosis AGO1 prefers U, AGO2 and AGO 4 prefer A and AGO 5 prefers C. The MID and PIWI domain of Arabidosis Ago proteins confer recognition of the nucleotide at the 5’ end. However, mammalian Ago proteins only prefer perfectly complementary siRNA like complexes and disfavor RNA with non-central mismatches. However, if the RNA only has central mismatches, Ago protein will incorporate it without any difficulties as well. Also, human Ago protein does not have a preference for the 5’ end nucleotide. Therefore, human Ago proteins do not have a strict small DNA sorting system.
Unwinding
[edit | edit source]Slicer-dependent unwinding
[edit | edit source]A pre-RISC loaded with double stranded RNA is very similar to a mature RISC that is bounded to a target mRNA. Therefore, the passenger RNA is like the target RNA for the guide strand. In slicer-dependent unwinding, the passenger strand is discarded just like how a target mRNA would be discarded. This type of unwinding only occurs in siRNA like complexes that has highly complementary strands.
Slicer-independent unwinding
[edit | edit source]Human AGO1, 3, and 4 does not have any slicer activity, therefore, it cannot use slicer-dependent unwinding. Also, if the RNA strands have mismatches, the slicers would not unwind the two strands. Therefore, another pathway was proposed as the slicer-independent unwinding. In this type of unwinding, the mismatched RNA will actually accelerate the unwinding process and it is essential for this type of unwinding. Therefore, scientists dub this the ‘mirror-image’ process of target recognition. It is basically the opposite of when the guild strand anneals to the target strand.
References
[edit | edit source]Kawamata,T and Tomari,Y. "Making RISC". Trends in Biochemical Sciences.35.7(2010):368-376. DNA and RNA are different from their structure, functions, and stabilities. DNA has four nitrogen bases adenine, thymine, cytosine, and guanine and for RNA instead of thymine, it has uracil. Also, DNA is double-stranded and RNA is single-stranded which is why RNA can leave the nucleus and DNA can't. Another thing is that DNA is missing an oxygen.
Predominant structures
[edit | edit source]DNA is a double-stranded molecule with a long chain of nucleotides while RNA is only single-stranded. In most of its biological roles and has a shorter chain of nucleotides (after transcription and splicing, only exons remain in RNA). DNA exists mainly in a double helix form while RNA will take on many different shapes and sizes such as the 'hair pin formation'. DNA is used to carry an organism's genetic information while RNA takes on many different roles, for instance, RNA can act as an enzyme such as ribozyme. There is one single type of DNA while there are many types of RNA that have different functions such as mRNA (carries DNA message to cytoplasm), tRNA (carries amino acids to mRNA and Ribosomes), rRNA (Ribosomal RNA, workbench for protein synthesis). DNA cannot catalyze its own synthesis while RNA can. This supports the RNA World Hypothesis. The pairing of bases in DNA including A-T(Adenine-Thymine) and G-C(Guanine-Cytosine)is different to that of RNA including A-U(Adenine-Uracil) and G-C(Guanine-Cytosine).
Bases and sugars
[edit | edit source]DNA is a long polymer with deoxyriboses and a phosphate backbone. Having four different nitrogenous bases: adenine, guanine, cytosine and thymine. RNA is a polymer with a ribose and phosphate backbone. Four different nitrogenous bases: adenine, guanine, cytosine, and uracil.
 Structure of ribose in RNA
Structure of ribose in RNA
Functions
[edit | edit source]DNA is a nucleic acid that contains the genetic instructions used in the development and functioning of all known living organisms. It is a medium of long-term storage and transmission of genetic information, while RNA is a nucleic acid polymer that plays an important role in the process of translating genetic information from deoxyribonucleic acid (DNA) into protein products. RNA acts as a messenger between DNA and the protein synthesis complexes known as ribosomes.
Both DNA and RNA start synthesis in the 5'-3' direction. However, no primer is needed for RNA. In addition, only RNA polymerase lacks the ability to detect errors of base pairing.
Stabilities
[edit | edit source]Deoxyribose sugar in DNA is less reactive because of C-H bonds on the second carbon (C2). DNA is stable in alkaline conditions. It has smaller grooves where the damaging enzyme can attach which makes it harder for the enzyme to attack DNA; RNA, on the other hand, has larger grooves which makes it easier to be attacked by enzymes. RNA, ribose sugar is more reactive because of the presence of hydroxyl group on C2. RNA is not stable in alkaline conditions because bases can easily deprotonate the Hydrogen from the -OH on C2. After deprotonation, the negatively charged oxygen may attack the Phosphate at the PO4, kicking off the Oxygen connected to the 5'C of next nucleotide over, resulting in hydrogenation.
Unique features
[edit | edit source]The helix geometry of DNA is of β-Form. DNA is completely protected by the body i.e. the body destroys enzymes that cleave DNA. DNA can be damaged by exposure to ultra-violet rays. The helix geometry of RNA is of α-Form. RNA strands are continually made, broken down and reused. RNA is more resistant to damage by ultra-violet rays.
Comparison chart
[edit | edit source]Here is a chart that shows the differences between DNA and RNA:
| DNA | RNA | |
|---|---|---|
| Structural Name: | Deoxyribonucleic Acid | Ribonucleic Acid |
| Function: | Medium of longterm storage and transmission of genetic information. | Transfer the genetic code needed for the creation of proteins from the nucleus to the ribosome. This process prevents the DNA from having to leave the nucleus, so it stays safe. Without RNA, proteins could never be made. |
| Structure: | Typically a double- stranded molecule with a long chain of nucleotides. | A single-stranded molecule in most of its biological roles and has a shorter chain of nucleotides. |
| Bases/Sugars: | Long polymer with a deoxyribose and phosphate backbone and four different bases: adenine, guanine, cytosine and thymine. | Shorter polymer with a ribose and phosphate backbone and four different bases: adenine, guanine, cytosine, and uracil. |
| Base Pairing: | A-T (Adenine-Thymine), G-C (Guanine-Cytosine) | A-U (Adenine-Uracil), G-C (Guanine-Cytosine) |
| Stability: | Deoxyribose sugar in DNA is less reactive because of C-H bonds. Stable in alkaline conditions. DNA has smaller grooves where the damaging enzyme can attach which makes it harder for the enzyme to attack DNA. | Ribose sugar is more reactive because of C-OH (hydroxyl) bonds. Not stable in alkaline conditions. RNA on the other hand has larger grooves which makes it easier to be attacked by enzymes. |
| Unique Traits: | The helix geometry of DNA is of B-Form. DNA is completely protected by the body i.e. the body destroys enzymes that cleave DNA. DNA can be damaged by exposure to Ultra-violet rays. | The helix geometry of RNA is of A-Form. RNA strands are continually made, broken down and reused. RNA is more resistant to damage by Ultra-violet rays. |
Transcription, also known as RNA synthesis, is a method in which a DNA nucleotide sequence is transcribed into RNA information. In this process, genetic information is simply copied from one molecule to another. In prokaryotic transcription, the mRNA genetic information is made and then translated to make proteins. In prokaryotes, translation and transcription can occur simultaneously in the cytoplasm. In eukaryotic transcription, the genetic material is transcribed in the nucleus. Transcription in eukaryotes is much more complex than in prokaryotes. One reason for this is the presence of histones in eukaryotic DNA. These histones tend to hinder the access of polymerases to the promoter. The process of transcription can be thought of as four sequential steps. The first would be the initiation step, during which the RNA polymerase II (RNAPII) binds to the DNA site in order to form a preinitiation complex with other transcriptional factors. The location of this on the DNA is identified as the "promoter." The second step involves an enzyme called a helicase that unwinds the DNA double helix. After the DNA is unwound, synthesis of RNA can begin based on the DNA template strand. It should be noted that Uracil of RNA is paired with Adenine of DNA. This step is called the elongation step, during which the polymerase leaves the promoter behind through a process called promoter clearance, and transcribes the rest of the DNA strand. The final step of transcription is termination of synthesis. There are different signals that lead to the termination of transcription. This step is also called the termination step, and the RNA polymerase finally releases the DNA.
Promoter Sites
[edit | edit source]RNA transcription from DNA begins with the recognition of promoter sites on the DNA strand by RNA Polymerase. These promoter sites are designated base sequences that mark the beginning of transcription on the long DNA strand. Transcription for RNA from DNA does not simply begin anywhere on the DNA strand. The probability that transcription will begin at a desired location just by chance is very slim, thus requiring sequences that RNA polymerase can recognize and initiate transcription. Promoter sites on the DNA sequence provide these starting points for the synthesis of specific RNA sequences from specific genes on the DNA strand. The first nucleotide to be transcribed is numbered +1. The nucleotide upstream to +1 (adjacent to +1 on the 5' side) will be identified as -1.
Since RNA polymerase, the enzyme that synthesizes RNA from DNA, polymerizes RNA from the 5' to 3' end, the promoter site where it attaches is always upstream, meaning closer to the 5' end of the DNA, from the gene of interest. Oftentimes there are molecules that attach to the promoter site and subsequently recruit the RNA polymerase to attach there and begin transcription; these molecules are called transcription factors.
In bacteria, there are two distinct sequences upstream (5') to the first nucleotide to be transcribed that function as promoter sites and determine where transcription will begin. One of them is located at 10 nucleotides to the 5' end of the first nucleotide to be transcribed (-10 region) and is called the Pribnow box with the consensus sequences of "TATAAT". The other, located further upstream at the -35 region, has a consensus sequence of "TTGACA". Note that most often, the first nucleotide to be transcribed is a purine.
The proteins that guide RNA polymerase to genes are the sigma factors. A sigma factors binds RNA polymerase through the alpha subunit and then helps the core enzyme detect or a recognize a specific DNA sequence, this is called a promoter. A single bacteria species can also make several different sigma factors. They also help core RNA polymerase locate the consensus promoter sequences near the beginning of a gene.
Bacterial DNA template----------------TTGACA(-35)-----------TATAAT/Pribnow(-10)------------Start of RNA (+1)07:15, 21 November 2010 (UTC)07:15, 21 November 2010 (UTC)~~
In eukaryotes, the promoter site exists at the -25 region with a consensus sequences of "TATAAA". This sequences is called the "TATA box" or Hogness box. When the cell wants to transcribe the DNA strand, the "TATA binding protein" (transcription factor) attaches to the TATA box and subsequently helps in getting the RNA polymerase to attach there and begin synthesizing the RNA. In addition to the TATA box, most eukaryotes also have a second promoter site at the -75 region called the CAAT box with a consensus sequence of GGNCAATCT. Finally, RNA transcription in eukaryotes is also stimulated by the presence of enhancer sequences found in distant locations from the +1 region on either the 5' or 3' side.
Eukaryotic DNA template------------CAAT box(-75)/optional----------TATA box(-25)------------Start of RNA(+1)07:15, 21 November 2010 (UTC)07:15, 21 November 2010 (UTC)Anneyoh (talk) 07:15, 21 November 2010 (UTC)
Note: Not all base sequences of promoter sites are identical. They are called consensus sequences because they share common features, however almost all promoter sequences differ from the idealized consensus sequence by one or two bases.
Enzymes that replicate DNA do not rely solely on the sequence of bases when determining binding specificity. The three dimensional structure is also important in determining where replicating proteins will bind. For most DNA-binding proteins, the readout of base pairs through hydrogen bonds or hydrophobic contacts is not sufficient to explain specificity. The shape of the minor groove within a binding site can be “read” by a complementary set of basic side chains of DNA binding molecules, most typically arginines but also lysines, when presented in the correct conformation.
Kinks can contribute to binding specificity by creating conformations that enhance protein-DNA and protein-protein contacts. The DNA-binding site of the catabolite activator protein (CAP) shows large kinks at two steps which cause an overall bending of the DNA of about 90◦ around the protein . The kink at the steps creates a space for an arginine residue to engage in partial stacking interactions with a thymine at that site.
Challenges Associated with the Elongation Step
[edit | edit source]As the RNAPII transcribes along the gene (or chromatin) during the transcript elongation, it has to find a way to deal with nucleosomes. One way of coping with the nucleosome is to disassemble it into separate histones and uncoiled gene before transcribing (as shown in the figure). Then, as the RNAPII transcribes along the uncoiled strand of gene, the separated histones may coil the gene to form a nucleosome back again. Histones are able to disassemble into further subunits, which include H2A/H2B dimer (depicted in red) and H3/H4 dimer (depicted in yellow). This disassembly of nucleosomes into histones is usually assisted by ATP-dependent chromatin remodelers and histone chaperones. Some of the identified ATP-dependent chromatin remodelers include SWI-SNF, ISWI, CHD, AND INO80/SWR. FACT (Facilitates Chromatin Transcription) is an example of Histone chaperone, which also plays a significant role in destabilizing the nucleosomes on a gene in order to facilitate the transcript elongation. Mainly, it functions by removing the H2A/H2B dimer from the nucleosome.
RNAPII also has to be certain on inserting the correct nucleotides. This is achieved by specific structure called trigger loop located under the active site of the RNAPII, where the nucleotides bind. The function of the trigger loop is to align the nucleotide in correct orientation for forming phosphodiester bond with the transcribing strand of gene. Only the right nucleotides are capable of aligning in correct orientation with specific trigger loops, which enable RNAPII to be certain on inserting the correct nucleotides.
The Mechanism of Elongation also plays a significant role in RNAPII fidelity. Transcript elongation is done by Brownian ratchet mechanism, which allows the RNAPII to move back and forth of the gene. By removing the misplaced one and inserting the correct one again, the RNAPII can not only increase the fidelity, but also enhance the rate of insertion of further nucleotides. This removal of a misplaced nucleotide usually requires general factors that encourage transcript cleavage, such as TFIIS.
In the elongation of RNA transcripts, the sigma factor remains associated with the transcribing complex until about nine bases have been joined. [Microbiology]. The original RNA polymerase then continues to move along the template, and synthesized RNA at 45 base pairs per second.
Other Factors Affecting Transcript Elongation
[edit | edit source]Histone modification is positively correlated with transcript elongation. In other words, transcription elongation requires increasing amount of histone modification in order to occur in faster rates. One of the histone modifications include histone acetylation, which is catalyzed by histone acetyltransferases (HATs) and histone deacetylases (HDACs). Histone methylation, which is another type of histone modification, interferes with the transcript elongation in order to regulate the rate of histone acetylation. It is believed that histone modification is associated with the disassembly of histones, and thus enhancing the transcript elongation.
Transcription Repression
Polycomb group proteins (PcG) are necessary for organisms to develop from cells to tissues. PcGs form protein complexes with many units that function as transcriptional repressors controlling thousands to hundreds of thousands of genes during cell differentiation and growth during normal development of the organism. Most multicellular organisms need PcGs for growth and development. Homeotic (HOX) genes are correctly expressed during development because of these same PcGs by regulating cell cycles, cancer, x-body inactivation, fate of cells, stem cell pathways and differentiation, among other developmental matters. As proteins, they also contain enzymatic like function when they target specific genes and thus downregulating their transcription. Their work includes the recruitment of other repressors that work together.
PcG proteins can be best described as two parts: PRC1 and PRC 2. They serve unique purposes. PRC 1 catalyzes the ubiquitylation of histone H2Awhich leads to the repressing of gene transcription by making the chromatin more compacted and less available. PRC 2 serves as the catalyst force for the methylation of histone H3 in order to repress the zeste 12 and development of the ectoderm.
PcG don’t attach similar sets of genes in all cells. The mechanisms and DNA patterns that regulate the binding of PcG proteins to the promoters in the cells have to be specific and complex.
Like previously mentioned, PcG does not work alone- there are other factors and proteins which it recruits to help during gene transcription repression. BCL6 Co-repressor which is also known as BCOR, helps with transcription repression in people who suffer from oculofaciocardiodental (OFCD) disease. The complex of PcG and BCOR targets germ cell genes.
The best known case that shows how signaling pathways help with the expressional methods of PcF is the hedgehog-signaling conserved pathway which is important during embryo development. This sonic hedgehog ligand (SHH) is best known for its work with cancer progression and stem cell maturation. More research is yet to be done.
Source:
Polycomb group protein-mediated repression of transcription Lluı´s Morey1,2 and Kristian Helin1,2 1 Biotech Research and Innovation Centre (BRIC), University of Copenhagen, Ole Maaløes Vej 5, 2200 Copenhagen, Denmark 2 Centre for Epigenetics, Ole Maaløes Vej 5, 2200 Copenhagen, Denmark
DNA Damage and Mutagenesis
[edit | edit source]When DNA is defected by UV, it is hindered from carrying on the transcription with RNAPII. As a result, damaged DNA may be repaired through the mechanism called transcription-coupled nucleotide excision DNA repair (TC-NER). Another way of repairing is polyubiquitylation of RNAPII. Both of these two repairing mechanisms involve the activity of RNAPII and degradation or removal of the damaged part of DNA.
Observations also show some relations between transcript elongation and mutagenesis (or simply mutation among the DNA strands). Although very little is discovered on the specific interactions between transcript elongation and mutagenesis, observations suggest that the increased rate of transcript elongation results in increased level of mutation among DNA strands. This probes a critical relation between transcription level and the fidelity of DNA replication.
Although a highly transcribed region spends a majority of its time being single-stranded, the rate of mutagenesis during DNA replication does not increase, but active transcription can interfere with the precision of DNA polymerase as it adds nucleotides to the template strand. The single-stranded DNA may not be protected by chromatin proteins and nucleosomes, but there is little evidence to argue that transcription is mutagenic to the DNA template strand.
In transcription-associated recombination (TAR), DNA polymerase and RNA polymerase II can produce a hybrid mRNA strand that contains both DNA and RNA nucleotides, these are called R-loops. R-loops lead to genetic instability as the cell has trouble during replication trying to activate the S Phase checkpoints. Mutants with the R-loops usually do not make it past the S phase and are not viable.
RECQL5 Helicase and Genomic Stability
[edit | edit source]The enzyme RECQL5 helicase may also have a role in maintaining genomic stability. RECQL5 is a protein that plays a role in preventing collapse or replication forks, which would lead to DNA damage, and the accumulation of DNA double-strand breaks, which would interfere with future replications and transcriptions if the mutation was in a coding region. Mutations in proteins similar to RECQL5 have lead to an increased rate of cancer.
Gene Traffic
[edit | edit source]Sometimes more than one RNAPII may bind to the same strand of gene for transcript elongation. This promotes gene traffic among the polymerases, which may either cause decrease in the rate of transcript elongation or force the polymerases to move forward in faster rate. However, currently very little is known on this phenomenon such as how and why the traffic causes the way polymerases react to the traffic. Some hypothesize that the main cause is directly related to the frequent collisions among the polymerases resulting from elongating at different rates on the same strand.
tRNA roles in transcription
[edit | edit source]tRNA is an RNA molecule and is thus transcribed from DNA, other than this it has little to do with transcription. The primary role of tRNA lies in translation where it interacts with the mature mRNA to bring the appropriate amino acid which it carries to the growing polypeptide chain. Nucleosides consist of a base linked to a ribose or deoxyribose sugar. A nucleoside can form a glycosidic bond linking to 1+ phophate group. A nucleoside + one phosphate makes a nucleotide.
Nucleic acid is an important macromolecule because it carries the information in a form that can be passed from one generation to the next. These macromolecules consist of a large number of linked nucleotides which makes off a sugar, a phosphate, and a nitrogenous base (either a purine or pyrimidine). Purines- Adenine and Guanine. Pyrimidines- Cytosine, Uracil, and Thymine. Sugars and phosphates are linked through the esterphosphate bond created the common backbone that plays a structural role. On the other hand, the sequence of bases along a nucleic acid chain carries the genetic information.
Two of the most common nucleic acids known are deoxyribonucleic acid (DNA) and ribonucleic acid (RNA). Between the two, they differ by only a hydroxyl group and the bonding between the nucleic acids by which will be discussed further in the deoxyribonucleic and ribonucleic section of the wikibook.
History
[edit | edit source]In 1870, Johann Friedrich Miescher was the first person that isolated the components of DNA. He found a weakly acidic substance of unknown function in the nuclei of human white blood cells and named it "nuclein". In the 1920s, it was discovered that nucleic acids was a major components of chromosomes. Elemental analysis showed the presence of phosphorous, 2-deoxyribose sugar, and four different heterocyclic bases. The two monocyclic bases are classified as pyrimidines, and the other two bicyclic bases are purines. Four nucleic acid
Until the late 1940s and early 1950s, DNA was determined to play a main role in inheritance. The structure of DNA more than ever become a universal interest in scientific world. Erwin Chargaff was a pioneer that tried to construct the composition of DNA. He found that the amount of adenine (A) always equaled the amount of thymine (T), and the amount of guanine (G) always equaled the amount of cytosine (C).
Structure of Nucleic Acid
[edit | edit source]A nucleic acid contains three parts: a phosphate group, a sugar group (deoxyribose or ribose), and a base. The bases are adenine, guanine, cytosine, and thymine (uracil for RNA). When a base is attached to a sugar group it is called a nucleoside. The four nucleosides for DNA are deoxyadenosine, deoxyguanosine, deoxycytidine, and thymidine. The four nucleosides for RNA are adenosine, guanosine, cytidine, and uridine. A nucleotide is when a nucleoside is bound to one or more phosphate groups. The four nucleotide units of DNA are called deoxyadenylate, deoxyguanylate, deoxycytidylate, and thymidylate.
Nucleotides have a distinctive structure composed of three components that held together by covalent bond:a nitrogen-containing base (cytosine, thymine, aconine, guanine, a 5-carbon sugar – ribose or deoxyribose, a phosphate group.

The polymer of nucleotide is nucleic acid. It is built by forming phosphodiester bonds between the 3' carbon of one nucleotide and the 5' carbon of another nucleotide, creating sugar-phosphate backbone.

References
[edit | edit source]Basics of Transcription
[edit | edit source]Traewf Transcription is similar to DNA replication in that DNA is used as a template to make a new nucleotide strand (RNA). The newly synthesized RNA strands are complementary to the DNA template strand.
RNA polymerase uses ribonucleoside triphosphate (rNTP) to synthesize mRNA strands (rATP, rUTP, rCTP, and rGTP) in the 5'->3' direction.
Transcription can be broken down into 3 steps:
1. Initiation. Transcription begins when RNA polymerase binds to a DNA region known as a promoter. Additional transcription factors are required to hold the RNA polymerase to the correct region of the DNA. After RNA polymerase binds to the promoter region, it melts 10-15 nucleotide base pairs around the transcription start site, allowing for rNTPs to bind to the template strand. Initiation ends when the first rNTP is linked to RNA polymerase by a phosphodiester bond. (Unlike DNA replication, no primer is needed)
2. Elongation: RNA polymerase leaves the start site and travels down the template in the 3'->5' direction. The DNA helix opens ahead of RNA polymerase during this process due to helicase.
3. Termination: Rna polymerase releases from the DNA template strand and leaves DNA.
Prokaryotic Transcription: Operons
[edit | edit source]Transcription is very similar in both prokaryotes and eukaryotes in that there is an initiation step, elongation step, and termination step. However, they also have their differences.
Prokaryotes contain operons while eukaryotes do not. Operons are clusters of related genes involved in a similar function and are often found in a contiguous array. Operons are controlled by a single promoter, and as a result, transcription produces 1 mRNA that can be translated into multiple proteins. If there were no operons, there would have to be separate promoters for each gene. Operons help make transcription more efficient; for example:
With Operon:
|Promoter|Gene A|Gene B|Gene C| ---transcription---> mRNA ---translation---> protein A + protein B + protein C
Without Operon:
|Promoter|Gene A| ---transcription---> mRNA A ---translation---> protein A
|Promoter|Gene B| ---transcription---> mRNA B ---translation---> protein B
|Promoter|Gene C| ---transcription---> mRNA C ---translation---> protein C
While an operon provides the advantage of being able to initiate transcription at one point and transcribe many genes, it has its disadvantages as well. One disadvantage is that if the promoter for the operon sequence is mutated, all the genes in the operon cannot be transcribed.
An operon consists of 3 parts:
1. Structural Genes 2. Promoter region 3. Operator region
The structural gene encodes for proteins. All structural genes will turn into a single mRNA that encodes for multiple proteins.
The promoter region is where RNA polymerase binds to DNA to initiate transcription. Not all promoters will have the same sequence. Strong promoters will have a similar nucleotide sequence as a known consensus sequence. Weak promoters will have a different sequence.
The operator region is located next to, and overlaps with the promoter region. It is the site where a repressor can bind. When a represson binds to an operator, RNA polymerase will not be able to bind to the promoter, and as a result transcription will not occur.
Inducible Operons
An inducible operon is an operon where a substance is required to be bound before transcription will occur, it is normally "off" but when a substance binds it is turned "on." The lac operon is an example of an inducible operon. It encodes 3 enzymes involved in the metabolism of lactose. The lac operon has 4 regions:
1. CAP binding site: important to increase the rate of transcription of an operon 2. Promoter: location where RNA polymerase binds 3. Operator: location where a repressor binds 4. Genes (ZYA): Gene Z encodes for Beta-galactosidase, which breaks down lactose Gene Y encodes for galactosidase primase, a transporter protein that allows lactose to get into the cell Gene A encodes for galactosidase transacetylase
Bacteria prefer to metabolize glucose over lactose. Given this fact, we can see three different scenarios:
1. No Lactose, High Glucose
If there is no lactose, the lac operon will not be turned on. A repressor will bind to the operator which overlaps the transcription start site, preventing RNA polymerase from binding at the promoter, and thus preventing transcription of the lac operon. This is important for bacteria to save energy. Because they prefer to metabolize glucose, there is no need to turn on the lac operon in the presence of no lactose and high glucose.
2. High Lactose, High Glucose
Lactose will bind to the repressor, causing a conformational change that forces the repressor to unbind from the operator. As a result, RNA polymerase can bind to the promoter and allows for transcription to occur, however, it is a weak transcription.
3. High Lactose, Low Glucose
Lactose will still bind to the repressor and force it to unbind from the operator (due to the high amount of lactose present). However, the low glucose levels will cause an increase in cyclic AMP. The increase in cAMP will bind to the CAP binding site. This then increases RNA polymerase's binding to the promoter, resulting in high amounts of transcription.
Repressible Operon
A repressible operon is an operon that where transcription is normally "on," but when a substance binds it turns "off." It is the opposite of an inducible operon.
The Trp Operon is an example of a repressible operon, and is involved in the synthesis of the essential amino acid tryptophan. Tryptophan is either made, or obtained from the environment. The Trp Operon is normally on, transcribing the RNA needed to synthesize tryptophan, however, in the presence of tryptophan (obtained from the environment), the operon is turned off in order to save energy.
Prokaryotic Transcription: Transcription Factors (Sigma)
[edit | edit source]The function of a transcription factor is to bring RNA polymerase and the promoter together. It will bind to RNA polymerase and at the same time, associate with the DNA promoter. Transcription factors exist in both prokaryotes and eukaryotes.
An example of a prokaryotic transcription factor is the "Sigma Factor." The sigma factor is involved in locating RNA polymerase to the correct location.
Eukaryotic Transcription: RNA Polymerase
[edit | edit source]There are three different types of RNA Polymerase in eukaryotes:
1. RNA polymerase I: makes rRNA 2. RNA polymerase II: makes mRNA, miRNA, and splicing RNA 3. RNA polymerase III: makes tRNA, rRNA, and splicing RNA
The structures of all three RNA polymerases are very similar and highly conserved. The components include a site for rNTP to enter, and a site for phosphodiester bond formation. RNA polymerase II contains an area known as the carboxy terminal domain (CTD), which is RNA pol II specific. CTD is a string of seven amino acid repeats, which is found to repeat 52 times in vertebrates. It is essential for viability, and RNA polymerase II cannot function without it. Before transcription, CTD is non-phosphorylated, and after the initiation step of transcription, CTD becomes phosphorylated.
Eukaryotic Transcription: Promoter, Proximal Promoters, and Enhancers
[edit | edit source]The promoter tells the cell where to start transcription. Transcription factors identify and bind to promoter regions, and also help to recruit RNA polymerases.
Proximal promoters and enhancers are both sites for transcription factors to bind as well. Proximal promoters are about 200 base pairs upstream of the start site. Enhancers are further away from the start site, and can be found up to 50,000 base pairs up or downstream of the site.
Promoter regions were identified through two different experiments: 5' deletion analysis and linker scanning analysis.
Prokaryotic vs. Eukaryotic Transcription
[edit | edit source]A few differences between Eukaryotic and Prokaryotic transcription include: Eukaryotes have multiple general transcription factors, lack operons, and have a genome packed into chromatin. Prokaryotes on the other hand, have one general transcription factor, have operons, and have a genome located in plasmids.
Looking at and comparing the three steps of transcription between Prokaryotes and Eukaryotes:
1. Initiation: the binding of RNA polymerase to double-stranded DNA.
For prokaryotes: RNA polymerase, in its complexity, contains a core that has to bind to the promoter region of the DNA template. Another subunit, known as sigma, is what makes this possible by finding and binding to the promoter region using lots of weak H-bonds with the base pairs (with the culmination of many weak H-bonding, a strong net force is seen). So, the RNA polymerase simply slides along the DNA, and it either finds a promoter, or dissolves and starts somewhere else. If it finds a promoter, it proceeds to unwind the DNA, which leads to elongation. Prokaryotes have two promotors sites located upstream of the first nucleotide to be transcribed. They are the Pribnow box, which is located 10 nucleotides upstream and has the sequence TATAAT and the -35 region which has the sequence TTGACA.
For Eukaryotes: Eukaryotes are more complicated in their initiation phase. Firstly, the RNA polymerase doesn't randomly scale a DNA for promoter regions. Rather, transcription factors are used to create specific instances in the promoter regions for the RNA polymerase to bind to. In eukaryotic cells, there are also 3 different kinds of RNA polymerase, each that transcribes a different type of RNA. Once a RNA polymerase binds to its respective promoter region (equipped with transcription factors), it creates a transcription initiation complex, which traverses the DNA. Eukaryotes also have two promotor sites, one is called the TATA or Hogness box which is at location -25 and has the sequence TATAAA, and the CAAT box, which is located at -75 and has the sequence GGNCAATCT. Transcription is initiated by stimulation by the enhancer sequence.
2. Elongation: the covalent addition of nucleotides to the 3' end of the growing polynucleotide chain.
For prokaryotes: As the DNA is unwound, its base pairs are now available for binding. The first ribonucleoside triphospate (RNA building blocks) binds to it. The RNA polymerase loses its sigma subunit, leaving only the core. The unwound DNA sites provide sites for the RNA building blocks to H-bond to (in their correct base pair). Also, the triphosphates are used like train links, that covalently form phosphodiester bonds with each new RNA block.
For Eukaryotes: As the complex moves across the DNA, it unzips it and allows for Watson-Crick base-pairing to occur with transient RNA building blocks, linking them together using phosphodiester bonds.
3. Termination: the recognition of the transcription termination sequence and the release of RNA polymerase.
For prokaryotes: Elongation continues until it reaches a stop signal found on the DNA. The RNA polymerase core dissolves, and the DNA rewinds. The termination signal in E. Coli is a base-paired hairpin which is rich in guanine and cytosine. These two nucleotides binds complementary to one another creating a hair-pin turn which is then followed by several uracil nucleotides. That hairpin acts like a knot to the RNA strand that is being made, so the RNA detaches from the DNA template. The polymerase leaves shortly after, and the DNA is rewound. Transcription can also be stopped by a rho protein which causes the mRNA to fall of the template DNA strand.
For Eukaryotes: When the RNA complex reaches a termination signal on the DNA, the RNA polymerase is simply detached from the DNA allowing it to rewind. The resulting RNA is then processed. The newly-transcribed mRNA is future processed by adding a cap to the 5' end and a poly(A) tail to the 3' in a process called polyadenylation. It adds several adenine residues to the 3' end of the mRNA. The cap and poly(A) tail act to stabilize the mRNA molecule and prevent it from degrading.

Chromatin Modification
[edit | edit source]DNA and proteins make up complex chromatin, and is can be found as either enchromatin or heterochromatin. Enchromatin is loosely condensed while heterochromatin is tightly condensed. Chromatin condensation is important because it determines transcription activity: if chromatin is too tightly condensed (heterochromatin), then the transcription factors and RNA polymerase are not able to get in; however, if chromatin is loosely condensed like in enchromatin, then transcription factors and RNA polymerase can more easily access the chromatin and start transcription. This means that genes containing enchromatin are highly likely to be transcribed, whereas heterochromatin genes are less likely to be transcribed.

Chromatin compaction and relaxation is regulated by modifying histone tails. When histone is attached to the acetyl group, due to the neutralization of the positive charge on the histone, the interaction between the negative charge on the DNA and the positive charge on the histone becomes weaker. As a result, RNA polymerase can easily access to the DNA, and thus, this process facilitates the transcriptional activity in vivo. In contrast, when histone is deacetylated, meaning that acetyl group is removed from the histone tail, the chromatin structure becomes more compacted, and accordingly, transcription is repressed.
http://www.broadinstitute.org/chembio/lab_schreiber/anims/animations/smdbHDAC.php
Introduction
[edit | edit source]Reverse transcription is the process in which a double stranded DNA molecules are made from a single stranded RNA. The name of this method is formed by its opposite direction to transcription. It also involves the presence of a reverse transcriptase enzyme, a primer, DNTAs and a RNase inhibitor.
Reverse transcriptase
[edit | edit source]A reverse transcriptase, also known as RNA-dependent DNA polymerase, is a DNA polymerase enzyme that transcribes single-stranded RNA into double-stranded DNA. It also helps in the formation of a double helix DNA once the RNA has been reverse transcribed into a single strand cDNA. Normal transcription involves the synthesis of RNA from DNA; hence, reverse transcription is the reverse of this.
Mechanism for a reverse transcription
[edit | edit source]Step a: The minus strand known as primer to transfer RNA to form the first DNA strand and to interact with the tRNA 3 end in a polymerase binding mode.
Step b: The enzyme cleaves the RNA template by binding to it in a RNase H mode.
Step c: the reverse transcriptase use the PPT sequence as a primer to bind in the polymerase mode for the synthesis of the second DNA strand.
Retroviruses
[edit | edit source]Retroviruses store genetic information on RNA. An example of retroviruses are H.I.V. and A.I.D.S.. Retroviruses flow from RNA to DNA. Viruses are enclosed in protein coats and are not capable of independent growth and therefore cannot live without the host.
Although AIDS is a terrible disease which utilizes reverse transcriptase, mankind owes a considerable debt to it. Reverse transcriptase has seen extensive use in the study of gene expression (via gene chips), and protein synthesis (mRNA---reverse transcriptase--->cDNA-->infuse in recombinant plasmid-->insert into E. coli-->have E. coli synthesize more of the original mRNA-->have that mRNA translated into its respective protein).
Disadvantage
[edit | edit source]Sometimes, the enzyme helping in the method such as reverse transcriptase makes mistakes, leading to the wrong reading of the RNA sequence. It causes in the difference of the single infected cells produced in all viruses. Instead, they form a diversity of molecular differences in their surface coat and enzymes, giving scientists difficulty in inventing the corresponding drug for the disease. As a result, it is difficult to fight HIV with vaccines due to its continual changing in surface molecules.
Invention of drugs for HIV
[edit | edit source]For a while, reverse transcriptase was considered as a great target for many HIV studies. It is discovered that without reverse transcriptase, the segment of mutated DNA can't become incorporated into the host cell, and therefore, can't be reproduced.
As a result, the very first major class of drugs were found to aim on this enzyme to slow down HIV infections called reverse transcriptase inhibitors. They are: AZT, 3TC, d4T, ddc, and ddl that block the recoding of viral RNA into DNA. Yet, the continual change in HIV surfaces molecules limits the effect of these drugs.
Common HIV drugs and How They Work
[edit | edit source]There have been quite a few drug therapies starting from the 1990s till 2006, yet still the research for new enhanced drugs are still occurring. Highly Active Antiretroviral Therapy (HAART) consist of three different HIV treatments; Protease Inhibitors, Non-nucleoside reverse transcriptase inhibitor, and Nucleoside reverse transcriptase inhibitor. (Introduction to HAART video HAART)For the Protease Inhibitor (PI) the main target point of this drug is to inhibit the viral protease, which in turn is responsible for Proteolytic processing of the viral polypeptide. There is also a non-nucleoside reverse transcriptase inhibitor (NNRTI) in combination with two nucleoside reverse transcriptase inhibitors (NRTI & NtRTI). NNRTI is a non-competitive inhibitor, which means that it binds to the reverse transcriptase enzyme by binding at a different site. In results in the change of the binding site shape and retarding of the catalyst ability. Relating this to the viral DNA, the movement of protein domains of our target enzyme are stopped. This means that the DNA synthesis doesn't occur. The nucleoside reverse transcriptase inhibitors (NRTI & NTRI) instead work as competitive substrate inhibitors. Competitive inhibitors occur when the substrate competes with the inhibitor at the active site. Relating it to the reverse transcriptase enzyme, we see in the process the deoxynucleotide of the normal DNA competes with the deoxynucleotide aimed towards growing the viral DNA chain. Thus, there is now a 3'-OH group on the deoxyribose unit. This means that deoxyribonucleotide is unable to form the next 5'-3' phosphodiester bond essential for the elongation of the DNA chain. This is called chain termination. (For a general visual of the process NRTI). [70]
Another HIV/AIDS inhibitor is the Diketoaryl (DKA) Integrase inhibitors. Integrase is the 3rd viral enzyme that has a two step catalyses.
1. 3' Processing: The integrase catalyses the processing of the 3'-ends of the viral cDNA. The processing corresponds to an endonucleotlytic cleave of the 3'-ends of the viral cDNA.
2. Strand Transfer: From 3'-processing, the viral 3'-OH cDNA ends are ligated to the 5'-DNA phosphate of an acceptor DNA, which is the host chromsome
Pre-Integration Complex: This macromolecule molecule is formed during and after the 3'-processing which undergoes nuclear translocation. It carries the 3'-processed viral cDNA ends with viral and cellular proteins to the nucleus before integration occurs.
DKA aims to block the stran-transfer step (step 2). The other inhibitors block the strand transfer step and 3'-processing. This integrase inhibitors are still on trial to aid in the future discovery of more specific Anti-AID drugs.[71]

List of Current Anti-AID Drugs
[edit | edit source]Here are a list of current anti-AID therapies that have been approved by the FDA in order of; FDA approval year, brand name, generic name and manufacturer.
Fusion inhibitors
2003 Fuzeon Enfuvirtide (T-20) Roche Pharmaceuticals & Trimeris
Nucleoside reverse transcriptase inhibitors (NRTIs)
1987 Retrovir Zidovudine (AZT) GlaxoSmithKline
1991 Videx Didanosine (ddI) Bristol-Myers Squibb
1992 Hivid Zalcitabine (ddC) Roche Pharmaceuticals
1994 Zerit Stavudine (d4T) Bristol-Myers Squibb
1995 Epivir Lamivudine (3TC) GlaxoSmithKline
1997 Combivir Lamivudine+ Zidovudine GlaxoSmithKline
1998 Ziagen Abacavir GlaxoSmithKline
2000 Trizivir Abacavir + lamivudine + zidovudine GlaxoSmithKline
2000 Videx EC Didanosine (ddI) Bristol-Myers Squibb
2001 Viread Tenofovir disoproxil Gilead Sciences
2003 Emtriva Emtricitabine (FTC) Gilead Sciences
2004 Epzicom Abacavir+ Lamivudine GlaxoSmithKline
2004 Truvada Emtricitabine+ Tenofovir Gilead Sciences
Non-nucleoside reverse transcriptase inhibitors (NNRTIs)
1996 Viramune Nevirapine Boehringer Ingelheim
1997 Rescriptor Delavirdine (DLV) Pfizer
1998 Sustiva Efavirenz Bristol-Myers Squibb
Protease inhibitors (PIs)
1995 Invirase Saquinavir Roche Pharmaceuticals
1996 Norvir Ritonavir Abbott Laboratories
1996 Crixivan Indinavir (IDV) Merck
1997 Viracept Nelfinavir Pfizer
1997 Fortovase Saquinavir Mesylate Roche Pharmaceuticals
1999 Agenerase Amprenavir GlaxoSmithKline
2000 Kaletra Lopinavir+ Ritonavir Abbott Laboratories
2003 Reyataz Atazanavir Bristol-Myers Squibb
2003 Lexiva Fosamprenavir GlaxoSmithKline
[72]
Telomerase Reverse Transcriptase
[edit | edit source]Understanding the structure of Telomeres is vital for genomic stability. The dysregulation of telomere could lead to Apoptosis(Cell death), and abnormality in the cell proliferation. The enzyme telomerase is essential in the process of maintaining telomere repeats in most eukaryotic cells. This Telomerase consists of a reverse transcriptase enzyme and an RNA strand that controls the synthesis of the G-rich strand of telomere terminal repeats. The telomerase reverse transcriptase contains a particular and variable C- and N- terminal extensions that flank a central domain that is reverse transcriptase like. The telomerase reverse transcriptase has two distinguishable properties which are the stable association with the telomerase RNA and the ability reverse transcribe the RNA segment repeatedly.
In Eukaryotes, telomeres are nucleoproteins located at the end of linear chromosomes. It consists of short sequences in addition to proteins that have interaction with these sequences directly as well as indirectly. One of the telomeres jobs is to protect the chromosome terminal from degradation and other reactions that are inappropriate for the chromosome. It also promotes division of chromosomes during meiosis and mitosis. Incomplete replication of telomeres leads to loss of DNA, which is known as “the end replication problem”. The enzyme Telomerase is very crucial in bacteria cells and is required for the reproduction of cell population. On the other hand, in eukaryotes, the telomerase enzyme is suppressed in normal somatic tissues but highly expressed in the sex tissues such as ovaries and testis. Telomerase is considered a plausible target when it comes to cancer therapy because of its up-regulation in cancer cells. [4]
References
[edit | edit source]1-3.^ Integrase Inhibitors To Treat HIV/AIDS Yves Pommier, Allison A. Johnson and Christophe Marchand. Volume 4. March 2005.
4. 1Bloomfield Center for Research in Aging, Lady Davis Institute for Medical Research, Sir Mortimer B. Davis Jewish General Hospital, and Department of Anatomy and Cell Biology and Department of Medicine, McGill University, Montreal, Quebec, Canada Although it has been a common belief that regulation of transcription takes place via regions adjacent to the coding region of the gene, mostly by promoters and enhancers, and that polymerase acts as machine that quickly “reads the gene”, recent evidence suggests that there is much more to the process than just that.
Transcript elongation is extremely complex and highly regulated and the process is significant as it affects both the organization and the integrity of the genome. This will explore some of the intricacies of transcript elongation by RNA Polymerase II, that has been overlooked for some time.
RNA Polymerase II Transcription Cycle
[edit | edit source]RNA Polymerase II transcription cycle can be classified into several distinct steps:
- RNAPII is recruited to the promoter of a gene where it forms a pre-initiation complex with the general transcription factors
- At this point initiation ensues and the promoter is left behind in the process called “promoter clearance”
- RNAPII enters processive transcript elongation and the gene is fully transcribed
- Then, transcriptional termination occurs, and results in the release and recycling of the RNAPII


Overview of Challenges to RNA Polymerase II
[edit | edit source]Challenges that face RNAPII:
- Polymerase needs to escape the promoter
- The production of pre-mRNA transcript needs to be tightly coupled to RNA biogenesis. This includes RNA capping, splicing, transcript, cleavage, and polyadenylation.
- RNAPII must navigate past nucleosomes and other obstacles (like DNA damage) because transcript elongation occurs in the chromatin
- Transcription is affected by DNA metabolic processes: DNA repair, recombination, and replication.
- Transcript elongation in highly transcribed genes is carried out by several polymerase molecules at the same time, so gene “traffic” must be regulated.

The Brownian Ratchet Mechanism
[edit | edit source]This mechanism is what governs transcript elongation. It involves the forward translocation states being stabilized by the binding and hydrolysis of the correct incoming nucleotide. The problem with this mechanism is that although it moves forward rapidly, it can also move backwards. So even though there are newly formed phosphodiester bonds, the enzyme can backtrack for one or several nucleotides so that the newly formed 3’ terminus comes out of alignment with the active site. Brownian motion can bring RNA back in alignment with the active site. General elongation factors affect multiple equilibria between different enzyme states, which is what helps drive the reaction towards forward translocation.
Transcription Fidelity
[edit | edit source]Transcription fidelity is essential, as the correct insertion of nucleotides into the RNA transcript during elongation is highly important for accuracy of gene expression.
There are a few key structures of the RNA polymerase II that ensures fidelity:
- Trigger loop
- Rpb9 (subunit on RNAPII)
- Transcription factors, especially transcription factors known as TFIIS
- RNA Polymerase II itself
The Trigger Loop
[edit | edit source]The trigger loop is located beneath the active site and is involved in multiple interactions with the incoming nucleotide. The trigger loop plays a key role in fidelity, mismatched nucleotides in the active site do not correctly align with the loop and therefore result in a large reduction of the rate of phosphodiester bond formation. So the trigger loop mediates phosphodiester bond formation in this manner. Trigger loop discriminates against dNTPs and interacts with rNTPs bases and with the 2'OH.
Rpb9
[edit | edit source]Another important structure on the RNAPII is the Rpb9 subunit, which was first discovered by scientists studying yeast stains. Rpb9 delays closure of the trigger loop on the incoming nucleotide, which helps to ensure transcription fidelity by allowing more time for mistakes to be fixed.
RNA Polymerase II and TFIIS
[edit | edit source]RNAPII itself plays a key role in its own transcription fidelity, as this polymerase can move forwards and backwards upon the molecule it is transcribing, and can therefore correct any mistakes through transcript cleavage of those erroneously incorporated nucleotides. Transcript cleavage is greatly enhanced by transcription factors known as TFIIS. Recent studies have also suggested that TFIIS also play a role in the process of fidelity as they are tightly coupled to the function of Rpb9, which leads us to believe that Rpb9 might be involved in RNAPII fidelity before and after nucleotide addition by affecting the function of the trigger loop and by mediating TFIIS function.
Transcript Elongation Through Chromatin
[edit | edit source]Transcript elongation occurs on a chromatin template, chromatin is an extremely repressive template to the process of transcription. Thus, certain mechanisms must be utilized to make it a more friendly place for transcription to occur. This involves temporary displacement and modification/dissasembly of nucleosomes. This can occur through a few different mechanisms, and requires;
- Histone chaperones
- Chromatin remodeling factors
- Histone modifying enzymes
These same factors also aid in the resetting of the chromatin structure after RNAPII has transcribed.

Nucleosome Assembly
[edit | edit source]First, the H3-H4 dimers are acetylated by HAT1-RbAp48 complex. Most of the H3 and H4 proteins are then passed on to Asf1, CAF-1, yRtt106, and HIRA to mediate chromatin assembly. The choice between CAF-1, yRtt106, and HIRA depends on the variant of H3. For example, H3.1 targets CAF-1 while H3.3 targets HIRA. CAF-1 and yRtt106 associate with DNA synthesis and HIRA occurs outside of DNA synthesis. Histone hand-off to different histone chaperones depend on the specific acetylation mark H3 K56Ac. H3 K56Ac’s role in yeast is to drive chromatin assembly during DNA repair and replication. However, since H3 K56Ac is harder to detect in humans, it is only speculated that it will do the same task. With H3 K56Ac there is a higher chance that Asf1 will transfer histones to CAF-1 or Rtt106. The choice of the DNA synthesis-dependent pathway or the synthesis-independent pathway depends on the physical interactions between the chaperones. The final chaperone in the hand-off will place the histones on DNA. There are mechanisms for which location of the DNA to place the histone and also mechanisms for increasing the concentration of the histone. [1]
Nucleosome Dissasembly
[edit | edit source]One of the mechanisms used is nucleosome disassembly in front of elongating RNAPII; certain experiments prove this to be a mechanism. One such experiment measuring histone density at the yeast GAL genes, showed that gene activation causes loss of nucleosomes at the promoter and also within the coding region. This loss of histone density is caused by elongating RNAPII . Genome-wide analysis of nucleosome occupancy in yeast revealed that transcription rate and histone density are inversely related, further proof that nucleosomes are disassembled during transcription. Another mechanism is the displacement of all core histones during transcription. There are different types of dimers however. H2A/H2B dimers are localized on the exterior of the nucleosome, and have fewer protein-DNA contacts. These dimers are rapidly exchanged in response to transcription factors. Conversely, histones H3 and H4 are much less mobile and their turnover rate is quite independent of the H2A/H2B dimer. So, although all core histones are displaced during this process, histones H2A/H2B are more readily moved while H3/H4 are not.
Just as the nucleosome assembly process occurred in a stepwise manner with histone chaperones, the nucleosome disassembly process is a stepwise process in the reverse direction. The most significant difference between nucleosome assembly and dissambly is that nucleosome disassembly requires energy. The energy is needed to break the histone-DNA bond so that histone chaperones can bind to the histone and move it away from the DNA. The factors to take into account before is undergoing disassembly is the post-translational histone modifications and histone chaperone availability. It is about equilibrium. If only H2A-H2B’s equilibrium is leaning towards removal from DNA there would only be histone exchange. However, if H3-H4’s equilibrium is leaning towards removal from DNA there would be nucleosome disassembly. H3 K56Ac in promoter region is a common example in nucleosome disassembly because it shifts H3-H4’s equilibrium towards removal from DNA. The equilibrium is all affected by interactions that include histone-histone chaperone interaction, histone-DNA interaction, or histone-histone interactions.[1]
Histone Chaperones
[edit | edit source]Histone chaperones are histone-binding proteins involved in intracellular histone dynamics, recent evidence revealed that one histone chaperone known as FACT has a huge role in transcription elongation. FACT (facilitates chromatin transcription) is a histone chaperone that facilitates elongation by destabilizing nucleosome structure so that H2A/H2B dimer can be removed during the passage of RNAPII. Spt6 is a H3 and H4 histone chaperone that maintains chromatin structure, promoting restoration of normal chromatin structure in the wake of RNAPII transcript elongation. Histone chaperones in general are involved in both removing and re-depositing histones during transcript elongation.
Histone chaperones as proteins not only pack histone and DNA into the nucleosome structure but also dissembles the structure. There are certain ways histones and DNA fold together in the nucleosome and histone chaperones help oversee every step. H2A, H2B, H3, and H4 are histone proteins. The histone chaperones assist in formation of tetrasome from the heterotetramer of H3-H4 on DNA. The tetrasome in combination with the H2A-H2B dimer forms the nucleosome. Histone chaperones are so important because without their help, positively charged histones would form aggregates with negatively charged DNA. The fact that the histones have a hydrophobic part and a slightly acidic part makes the protein even more attracted to DNA. Histone chaperones, although not sequentially similar, can be structurally grouped as beta-sheet sandwich chaperones, alpha-earmuff chaperones, beta-propeller chaperones, and beta-barrel and half barrel chaperones. [1]
Beta-sheet sandwich chaperones
This structure is characterized by an N-terminal core domain of Saccharomyces cerevisiae Asf1 and the absence of an acidic tail. Histone binding was confirmed by mutagenesis and NMR chemical shift to occur at the hydrophobic and acidic surface of Asf1. The C-terminal tail of H4 can bind to either the yAsf1 beta-sheet or H2A mini beta-sheet. Therefore, yAsfi could access the H3-H4 dimer and form a complex that is structurally conserved. The crystal structure of human ASF1a reveals this complex. Asf1 passes off the H3-H4 dimer to other histone chaperones such as CAF-1 or HIRA. Asf1 regulates transcription, replication and repair. In addition to Asf1, Yaf9 is another chaperone in the beta-sheet sandwich chaperone. Yaf9 plays a role in H2AZ acetylation and deposition into euchromatic promoter regions. It also contains structurally conserved features that could serve as H3-H4 binding sites and regulates transcription. [1]
Alpha-earmuff chaperones
This group is responsible for histone delivery from cytoplasm, binding to H1 and assembling and disassembling nucleosomes. These chaperones are characterized by their use of long alpha-helix for dimerization and linking alpha beta earmuff motifs. Mutagenesis confirms the histone binding site at the central and bottom surfaces of the earmuff domains. Vps75 is one of the chaperones in this group and it binds H3-H4 dimer. It is involved in the acetylation of H3 K56, which in turn aids the packing process. Along with acetylation, Vps75 is also responsible for regulating transcription, repair, and maintaining telomere length. The earmuff domains are closer in Vps75 than NAP1. NAP1 is responsible for transcription, H2AZ exchange, linker histone deposition, and histone delivery.[1]
Beta-propeller chaperones
This group is characterized by acidic patches that are not as distinct as other chaperones. Nucleoplasmin is responsible for regulating nucleolar events and histone storage during such events as oogenesis, sperm chromatin decompaction, and nucleosomal assembly. Nucleoplasmin’s pentamer N-terminus has the ability to self associate into a decamer. The nucleoplasmin core works in conjunction with linker histones and core histones to produce five histone octamers. Along with having a beta-propeller chaperone structure, CAF-1 and RbAp46 has an alpha-helix at the N-terminus. These chaperones are negatively charged at the top and hydrophobic at the bottom. H4 alpha helix binds between the chaperones’ alpha helix and a binding loop in an acidic region. Consequently, the H3-H4 dimer structure is disrupted. To counteract, these chaperones take on the role of associating the H3-H4 dimer with several chromatin modifying complexes such as HAT1, PRC2, NURD, and NURF.[1]
Beta-barrel and half barrel chaperones
This family consists of FACT subunits Spt16, Pob3, Nhp6, SPT16 and SSRP1. FACT is the term used to basically mean that these chaperones facilitates chromatin transcription. Pob3’s structure consisting of a helix-capped beta-barrel helps it bind to the yeast replication protein A (RPA) complex to assist in replication. Spt16’s has linked aminopeptidase and pita-bread domains. Rtt106 binds histones acetylated on H3 K56. Its H3-H4 binding site is located in a loop in the C-terminal domain.[1]
Histone variant chaperone Chz1
This histone chaperone group is characterized as not having a defined sequence or structure. Chz1 is a H2AZ-H2B binding protein. NMR confirms Chz1’s unique structure of alpha-helices that bind mainly on one surface of the H2AZ-H2B dimer with high affinity. Therefore, it dissociates more slowly than it associates. [1]
The oligomeric state of the histone chaperone depends on the stage of the nucleosome assembly. Histone chaperone-histone binding could either be simple or multimeric. The weak individual binding adds up to a high affinity multimeric binding. Although Asf1 and Chz1 are both monomeric, they play different roles in nucleosome assembly. Asf1 prevents H3-H4 tetramer formation. H3 would usually dimerize inside the histone octamer, but Asf1 binds to H3 making it exposed to other histone chaperones. Once Asf1 is released, the H3-H4 tetramer can form with Rtt106 and CAF1 attached. It is after this stage that the tetramer is deposited onto DNA. Chz1 on the other hand binds to an already exposed histone octamer surface of H2AZ-H2B. Therefore, the histones are directly deposited on tetrasomes.[1]
- Alpha beta earmuff chaperones assemble nucleosomes in vitro. NAP1 not only assembles histone octamers but also tetrasomes. NAP1 assembles histone octamers through H2A-H2B or
H2AZ-H2B. NAP1 assembles histone tetrasomes through H3-H4 and DNA. This process is not as favorable when there is H2A-H2B around. Vps75 is different from NAP1 in that it is more specific for H3-H4 and has weaker affinity for other histones. NAP1 binds all the histones that have the common histone fold through one surface. This is also the point in which it differs with FACT because FACT binds multiple surfaces of multiple histones. FACT carries out different functions depending on if it’s in vitro or in vivo. When it’s in vitro, FACT removes the H2A-H2B dimer. However, when it’s in vivo FACT puts the H2A-H2B dimers on DNA during the processes of transcriptional elongation, repairs, and replication.[1]
- There’s only one binding site for H4 in dCAF-1 p55 and hCAF-1 RbAp48 subunits but there’s multiple binding sites for H3. The single binding site for H4 is on the opposite face of the H3-H4
dimer. This allows the CAF-1 subunits to either reach histone dimers bound to ASf1 or histone tetramers that are being deposited onto DNA.
- Np is assembled as a dimer of pentamers. Each face of these histone chaperones could bind an octamer. Np binds H2A-H2B dimers and N1 binds H3-H4 tetramers to form octamers. NASP,
nuclear autoantigenic sperm protein, binds H3/H4 and aids the nucleosomes assembly process. [1]
Histone chaperone-guided folding pathways
Histone chaperones are extremely important in guiding the histone-DNA folding process because without it the histone would form intermediates that act as kinetic traps. Nucleosome assembly is an energetically favorable process and to remain that way the histone chaperones need to prevent these kinetic traps that prevent histones from ever binding on DNA. These kinetic traps are low in energy and it is not energetically favorable for histone to get out of these kinetic traps and bind on DNA when it is more stable in these intermediates. The histone chaperones allow histone-DNA complex to fold in a manner that is most stable and low energy. It is an energetically downhill process to get to DNA kinetic traps along the pathway are lower in energy so without the help of histone chaperones the histone would be stuck in that intermediate and never reach its destination. Histone chaperones work together with ATP-dependent chromatin remodelers. They are useful in times of kinetic traps to get the histones back on the correct pathway. Since kinetic traps are low in energy, chromatin remodelers get these intermediates out by raising its energy. These chromatin remodelers are also useful in breaking histone-DNA bonds. Certain acetylation marks on histones either increase or decrease affinity of the binding and is removed depending on if the nucleosomes needs to undergo assembly or disassembly. [1]
Nucleosome Remodeling
[edit | edit source]ATP-dependent chromatin remodeling complexes (remodelers) use the energy of ATP to modify the structure of chromatin.
There are four main families of remodelers:
- SWI-SNF
- ISWI
- CHD
- INO80/SWR
One SWI-SNF remodeler known as RSC can stimulate RNA polymerase II transcript elongation through a mononucleosome in a simple reconstituted chromatin transcription system. This is enhanced by histone acetylation as it increases the affinity of RSC for the nucleosome. The remodeler Chd1 is associated with chromatin at sites of active transcription, and it also plays a role in transcript elongation as it interacts with elongation factors Paf, DSIF, and FACT.
Histone Modifications
[edit | edit source]Covalent modification of histones is another way of modifying chromatin structure and it that does not involve histone removal and replacement. Instead it involves altering the packaging of chromatin by affecting internucleosomal contacts or changing electrostatic charge. As well as using the covalently attached moieties as a binding surface for elongation associated effector complexes.
The aforementioned can be achieved using these three mechanisms:
- Histone acetylation
- Histone methylation
- Histone ubiquitation
Reference
[edit | edit source]Selth, Luke A. Sigurdsson, Stefan. Svejstrup, Jesper Q. "Transcript Elongation by RNA Polymerase II". Annual Review of Biochemistry 2010. Vol. 79: 271-293. 04/01/2010. DOI: 10.1146/annurev.biochem.78.062807.091425
mRNA processing and transfer surrounds the movement of mRNA from the nucleus to the cytoplasm. After transcription, a process which occurs in the nucleus, mRNA must travel to the cytoplasm, where it can reach the ribosomes. mRNA travels past the nuclear membrane in the form of mRNP (messenger Ribonucleoprotein), a structure in which contains cargo-carrier components. The cargo (mRNA) requires the assistance of carriers (proteins) in order to be transported across the nuclear membrane. This mechanism makes usage of recyclable proteins and imposes directionality.
At the nuclear membrane
[edit | edit source]After the mRNP goes through nuclear processing, the mRNP approaches the NPC (Nuclear Pore Complex). Located at this complex is the TREX-2 complex. The TREX-2 complex is comprised of a Sac3 complex containing Sus1, Cdc31 and Thp1. Together, this TREX-2 complex improves the efficiency in which mRNP enters the NPC transport channel, which occurs by promoting active interactions between the TREX-2 complex and that of approaching mRNPs. Located along the NPC nuclear face, the TREX-2 complex becomes a sort of platform in which the necessary interactions between the TREX-2 complex and the mRNP are favorable. The TREX-2 complex serves as an attractive force, which concentrates export-ready mRNPs and helps promote movement through the NPC.
In addition to the Sac3 complex of the TREX-2 complex, a complex known as the SAGA complex is a part of the TREX-2 complex. The SAGA complex, which is attached to the Sac3 complex, is most known to be the location in which active genes are localized. More specifically, the TREX-2 complex localizes the active genes to pores located on the nuclear basket of the NPC transport channel as well as the SAGA complex.
Disassembly of the mRNP export complex
[edit | edit source]Once the mRNP clears the NPC complex, the mRNP becomes exposed to the cytoplasmic conditions. Located in the cytoplasm is an essential DEAD-box helicase known as Dbp5. Dbp5 is responsible for the remodeling of the mRNP complex, essentially removing the carrier aspects of the carrier-cargo relationship of the mRNP. The remodeling of the mRNP, an act whose specifics are unknown, releases Nab2 from the Poly(A) tail of the mRNA and Mex67-Mtr2. As a result, the introduction of Dbp5 isolates the mRNA from the mRNP. However, Dbp5 is not always in an active conformation. Dbp5 flow freely between the nuclear and cytoplasmic side, but only affect the mRNP in the cytoplasmic side. This occurs because of the presence of Gle1 and IP6. Gle1 and IP6 are located on the cytoplasmic face of the NPC. Without these two compounds, Dbp5 remains in a dormant state. This occurs because Gle1 enhances the ATPase efficiency of Dbp5 as well as its affinity for RNA. IP6 enhances the attraction between Gle1 and Dbp5. When an activated Dbp5 remodels the mRNP into the mRNA and the carrier aspects, the Nab2 and Mex67-Mtr2 are recycled. In other words, they flow freely from the cytoplasmic side to the nuclear side and are reused in the formation of the mRNP. The usage of Dbp5 as a means of remodeling the mRNP controls the directionality of the transport. Once the mRNP is separated into the mRNA and carriers, the mRNA cannot re-enter the nucleus. The mRNA that is isolated is ready for translation.
References
[edit | edit source]Stewart M. "Nuclear export of mRNA". Trends Biochem Sci. 2010 Nov;35(11):609-17. Epub 2010 Aug 16. Review. Accessed 2012 Nov 20.
In genetics, translation is the process by which mRNA is decoded and translated to produce a polypeptide sequence, otherwise known as a protein. This process is preceded by the transcription of DNA to RNA. This method of synthesizing proteins is directed by RNA and accomplished with the help of Ribosome.
In translation, a cell decodes the mRNA's genetic message and assembles the brand new polypeptide chain accordingly. This genetic message is composed of a sequence of codons which comprise the mRNA strand. The translator of this message is the transfer RNA, or tRNA. The main function of tRNA is to transfer free amino acids from the cytoplasm to a ribosome, where it is attached to the growing polypeptide chain. Normally, a cell is well stocked with all 20 amino acids, either by producing them or getting them from the food we eat. The ribosome adds on the amino acids that are brought to it by tRNA molecules to the growing end of the polypeptide chain.
![]()
The Ribosome
[edit | edit source]Ribosomes help coordinate the binding of tRNA anticodons with mRNA codons during translation. Ribosome can be like molecular machine that can decode RNA and use the information to build a polypeptide that contains a precise sequence of amino acid. The decoding process of the ribosome may be compared to a language translation machine that converts one language to another. So, the ribosome can be viewed as translating the language of the mRNA code into sensible protein sequences that conduct the activites of the cell. In eukaryotes, the ribosomal subunits are produced in the nucleolus of the cell. A ribosome is made up of two subunits: the large and small subunits. These subunits are made up of proteins and RNA molecules called ribosomal RNA (rRNA). The rRNA in prokaryotes and eukaryotes differ in many ways. For example, eukaryotic large subunits have a size of 60S, while prokaryotes have a size of 50S, meaning that prokaryotic subunits are smaller. Together, the large 50S and the small 30S subunit can form the 70S ribosome. The 30S subunit agrees to the selection of cognate aminoacyl tRNAs by facilitating base-pairing between mRNA codons and tRNA anticodons, while the active site or the PTC exists in the 50S subunit. The typical E. coli cell has approximately 18,000 ribosomes. The PTC catalyzes both the peptidyl transfer during protein elongation and hydrolysis of the petidyl tRNA during termination. In vitro, the 50S subunit can synthesize peptide bonds rapidly as the entire 70S ribosome, indicating that there is a catalyst. In human health, ribosome can serve as a medicinal target for drugs that work by inhibiting translation within pathogens without affecting the host organism. This is medically important because antibiotics such streptomycin and kanamycin targets the smaller subunits of bacteria while leaving the larger eukaryotic subunits unharmed.
The structure of the ribosome consists of a binding site for mRNA and three binding sites for tRNA. For tRNA, the P site (peptidyl-tRNA site) carries the growing polypeptide chain, while the A site (aminoacyl-tRNA site) holds the tRNA that carries the next amino acid that is to be added to the growing chain. The E site (exit site) is the site where discharged tRNAs leave the ribosome. The ribosome holds these two components close together and positions the new amino acid in a way that allows the addition of new amino acids to the carboxyl end of the growing polypeptide chain. As the chain gets longer, it goes through an exit tunnel in the large ribosomal subunit. Once the chain is completed, it is released to the cytosol of the cell through the exit tunnel. A particularly iconic video of Protein Translation shows this process.
Translation
[edit | edit source]In translation, there are three main steps that describe the protein decoding and synthesis process. These steps, in order, are called initiation, elongation, and termination.
Initiation
[edit | edit source]The first step of translation is called initiation. In this step, mRNA, a tRNA containing the first amino acid of the polypeptide, and two ribosomal subunits come together to start the process. The small subunit then binds to both mRNA and a specific initiator tRNA, which contains the amino acid methionine (MET). Next, the subunit scans along the mRNA strand until it reachs the start codon AUG, which indicates the start of translation process. The start codon also establishes the reading frame for the mRNA strand, with is crucial to synthesizing the protein. A shift in the reading frame results in mistranslation of the mRNA. Then, the tRNA initiator then binds to the start codon via hydrogen bonding.
The complex of consisting of mRNA, initiator tRNA, and the small ribosomal subunit attaches to the large ribosomal subunit, which completes the initiation complex. These components are brought together by the help of proteins called initiation factors which bind to the small ribosomal subunit during initiation. In addition, the cell spends GTP energy to help form the initiation complex. Once the formation of the initiation complex is complete, the initiator tRNA attaches to the P site of the ribosome, and the empty A site is ready for the next aminoacyl tRNA. The polypeptide is always synthesized in one direction, which is from the N-terminus to the C-terminus direction.
In bacteria, initiation of protein synthesis is started by binding of proteins called initiation factors (IFs) to the small 30S unit. Followed by the binding of fMet-tRNA to it. (fmet is the Met with formamyl (HCOO-) added to the N-terminus which is later removed). The three small proteins called initiation factors (IF1, IF2, and IF3) are required for the initiation of protein synthesis in bacteria. IF3 first brings mRNA and the 30S ribosome subunits together which allow the ribosome-binding site to find its complementary site on the 16S rRNA. Then, IF1 binds to and blocks the A site. The IF2 bound to GTP then escorts the initiator N-formylmethionyl-tRNA to the start codon located at the P site. IF3 is released when the initiator tRNA is in place. The 50S subunit then docks to the 30S subunit, GTP is hydrolyzed and IF1 and IF2 are released as well. This complex of Ifs-30S-fmet-tRNA now binds to the mRNA to be translated. To establish the reading frame, the mRNA in bacteria has an 8 nucleotide, purine-rich sequence called the Shine-Delgarno sequence, which is complementary to the rRNA of the small (30S) unit and helps in the initial binding of mRNA to the 30S unit complex. The Shine-Delgarno sequence is very close to the AUG codon, where the start of the translation occurs. The Shine-Delgarno sequence is present in all the bacteria.
The process of initiation of translation is complete when the large (50S) subunit binds to the Ifs-30S-fmet-tRNA complex. The binding of 50S subunit is an energy requiring process. First the energy rich GTP binds to another protein IF2 (initiation factor 2), forming the GDP-IF2 complex. The GDP-IF2 complex participates in the formation the final protein synthesizing 70S complex. In eukaryotes, additional initiation factors (IFs) are involved.
Elongation
[edit | edit source]Elongation begins with the aminoactyl tRNA that is delivered to the A site as a ternary complex with the elongation factor. The anticodon of an incoming tRNA pairs the bases with the complementary bases of the mRNA codon at the A site. The tRNA is attached to the amino acid which the mRNA codon codes for. The code which relates the codon to the amino acid is known as the Genetic Code. During this process, an elongation factor (as well as EF-Tu in bacteria) is necessary. The hydrolysis of molecules of GTP to GDP is required for codon recognition with the release of PPi that allows for the accuracy and efficiency of the process of recognizing codons. Next, a new peptide bond is formed between the new amino acid in the A site and the carboxyl group of the growing polypeptide. This peptide formation is carried on by the help of rRNA molecule of the large subunit. Now, the peptide chain has been elongated by one amino acid. The tRNA carrying the elongated polypeptide chain is then moved to the P site while the empty tRNA in the P site is moved to the E side and exits. The process is repeated for the next incoming tRNA and amino acid: tRNA carries the next amino acid to attach to the A site, base-pairs, need energy from the hydrolysis of GTP to GDP, peptide bond formation to connect new amino acid to polypeptide chain, and appropriate movements occur, empty tRNA moves and exist to go back for another "trip", tRNA carrying elongated polypeptide chain moved to P site where it waits for another coming tRNA to take place. Therefore, one by one, an amino acid is added to the preceding amino acid.
The release factors recognize and bind to the A-site stop codon and activate the hydrolysis and release of the polypeptide from the P-site tRNA. the C terminus of the polypeptide chain attached to the P-stie tRNA that undergoes the attack at the ester carbon by a water molecule; therefore, releasing the newly synthesized polypeptide. The ribosome PTC catalyzes the aminolysis of the ester bond, where the alpha amino group of the A-site aminoacyl tRNA attacks the P-site peptidyl tRNA at the carbonyl group. this happens because the amines react faster with esters to form peptide bonds.
Note: The P site is called as such because it only binds to Peptidyl-tRNA molecule, the tRNA anchoring the growing polypeptide chain. The A Site is called as such because it only binds to incoming Aminoacyl-tRNA molecules, the tRNA that contains the free amino acid.
Termination
[edit | edit source]The elongation process stops when any stop codon, a codon signaling the end of translation, reaches the A site of the ribosome. The A site will not accept any incoming tRNA, leaving it free to bind to the release factor. The release factor will hydrolyze the bond between tRNA and polypeptide in the P site, releasing the polypeptide chain. Subsequently, the two ribosomal subunits, release factor, and mRNA come apart when their jobs are done. The polypeptide is released from the mRNA template and allowed to fold into its final 3D conformation. Also, the ribosome arrives at the end of the coding region, not the end of the RNA. So, the end of the coding region is marked by one of the three stop codons. The formation of the last peptide bond and the subsequent translocation of mRNA leads to ejection of tRNA in the E site and also brings the stop codon into the A site.
In bacteria, they have 2 class I release factors that decode the three stop codons: UAG, UAA, and UGA. The class II release factor starts the dissociation of the class I release factor from the post-termination ribosomal complex after peptidyl tRNA hydrolysis. in eukaryotes and archea, they have one class I release factor that can decode all three stop codons.
| Stop Codon | Sequence |
|---|---|
| RNA (DNA) | U(T)AA |
| RNA (DNA) | U(T)AG |
| RNA (DNA) | U(T)GA |
Eukaryote-specific Ribosomal Features
[edit | edit source]Microscopy studies have shown that eukaryotic 40S and 60S ribosomal subunits are largely analogous to prokaryotic 30S and 50S ribosomal subunits. Indeed, a great many structural "landmarks" are conserved, including a central protuberance, two stalks, and a sarcin-ricin loop (SRL). However, within eukaryotic and archaeal ribosomes, subunits have underwent remodeling within specific regions (the 40S subunit, for example, is divided into the "head", "beak", "platform", "body", "shoulder", "left foot", and "right foot" regions). Remodeling through protein addition has occurred primarily on solvent-exposed faces of both the 40S and 60S eukaryotic subunits. With respect to the 40S subunit, rRNA expansion segments (ESs) have networked with a variety of eukaryote-specific protein components, resulting in structural linkage atop the subunit. Similarly, the solvent-exposed face of the 60S subunit possesses two expanded regions, each containing high concentrations of ESs and eukaryote-specific protein elements.
Tertiary contact within the eukaryote-specific ribosomal proteins is essential in producing stable subunit configurations. Such proteins and their extensions are responsible for interconnection that occurs within both subunits. Within the 40S subunit, the presence of extensions rpS10, rpS12, rpS21, and rpS7 serve to link 11 proteins, creating a "daisy-chain" structure. Likewise, protein-modulated subunit connections are quite prevalent, with eukaryote-specific extensions forming networks of interaction. This produces a number of structural effects, including the presence of β sheets and α helices, which may extend tangentially across the ribosomal subunits to interact with distant regions of the structure.
Differences between prokaryotic translation and eukaryotic translation
[edit | edit source]•In general, ribosomes involved in eukaryotic translation are about 30% larger than their prokaryotic counterparts.
•Relative to prokaryotic ribosomes, eukaryotic ribosomes require a very large number of assembly, maturation, and initiation factors. They are also subjected to a great degree of regulation. While prokaryotic ribosome assembly and translational initiation are influenced of a handful of nonribosomal factors, eukaryotic ribosome development and translational initiation are modulated by approximately 200 maturation factors and a minimum of nine initiation factors, respectively.
•The P Site in prokaryotic translation is directly on AUG at the beginning, but in eukaryotic translation the ribosomal subunit scans the chain until reaches to AUG.
•Prokaryotic translation is initiated by the presence of the Shine-Dalgarno (SD) sequence, a short series of base pairs that identifies and binds to an anti-SD sequence located at the end of a 16S rRNA subunit within the ribosome. On the other hand, eukaryotic translation does not involve Shine-Dalgarno (SD) sequences, relying instead on Poly-A-Binding Protein (PABP) within a "scanning mechanism" of sorts.
•The initiating amino acid in eukaryotic translation is methionine. Conversely, the initiating amino acid in prokaryotic translation is N-Formylmethionine (fMet).
Increasing the Rate of Translation
[edit | edit source]The majority of proteins are synthesized in 20 seconds to a few minutes. This can be sped up through a number of different processes.
•A single mRNA can be bound by multiple ribosomes, with each ribosome synthesizing its own protein.
•Ribosomal subunits can be rapidly recycled by the cell. Once the ribosome has reached a stop codon down the 3' end of the mRNA strand, the subunits can reattach to the beginning to immediately begin synthesis of a new protein.
•Polysomes: polysomes are a cluster of ribosomes bound to a circular mRNA molecule. Two proteins are bound to the 5'-CAP and Poly A tail to make the mRNA circular. EIF4 binds to the 5-CAP, and Poly A Binding Protein I (PABPI) binds to the poly A tail. These two proteins associate together to make the mRNA circular, which contributes towards a more efficient translation. The ribosomes in a polysome can travel down from the 5' end to the 3' end, and only have a short distance jump to recycle back to the beginning of the 5' end of the mRNA strand and repeat translation.
Nonsense Mutations in Translation
[edit | edit source]A nonsense mutation is a point mutation (single base substitution/single nucleotide mutation) in a DNA sequence that introduces a premature stop codon in the sequence. For example, if a codon with the sequence 5' U A C 3', which codes for the amino acid tyrosine had a nonsense mutation where the C was mutated into a G, the new codon would be 5' U A G 3', which codes for a STOP codon, truncating the protein. If a nonsense mutation occurs, the cell can respond with Nonsense Mediated Decay.
Nonsense mediated decay degrades mRNA that have a nonsense mutation. The mRNA will initially undergo one round of translation, where the cell will recognize something is wrong due to the premature stop codon, and degrade the mRNA. The nonsense mutation is identified through the exon junction complexes located along the mRNA.
Exon junction complexes are simply proteins that physically associate where 2 exons come together. During translation, as the ribosome moves down the mRNA strand, exon junction complexes are "bumped" off as they come in contact with the ribosome. If translation is stopped early due to a premature stop codon, the exon junction complexes will remain on the mRNA and not be removed. This signals the cell that something is wrong, and nonsense mediated decay is allowed to degrade the mRNA.
Nonsense mediated decays are not necessarily always beneficial. It does not check to see whether or not the truncated protein is functional, but rather only looks for nonsense mutations along the strand. One such example can be seen in the disease Cystic Fibrosis.
Cystic Fibrosis occurs due to a nonsense mutation in the gene "cystic fibrosis transmembrane conductance regulator," or CFTR, which codes for a chloride channel. People without cystic fibrosis have two working copies of the CFTR gene, and only one is needed to prevent the disease. A CFTR nonsense mutation produces a truncated protein, and the cell performs nonsense mediated decay in response, degrading the CFTR mRNA. The degradation of CFTR mRNA results in very little to no CFTR channels, causing cystic fibrosis. However, scientists have discovered that the truncated protein produced due to the premature stop codon is still functional as a CFTR channel. In this case, a response designed to protect the body ultimately ends up hurting it.
References
[edit | edit source]Slonczewski, Joan L. Microbiology. "An Evolving Science." Second Edition. Klinge, Sebastian; Voigts-Hoffmann, Felix; Leibundgut, Marc; Ban, Nenad. "Atomic structures of the eukaryotic ribosome." Trends in biochemical sciences doi:10.1016/j.tibs.2012.02.007 (volume 37 issue 5 pp.189 - 198) RNA modification must be performed in order to form the various proteins needed for eukaryotes to function. RNA modification generates mature RNA. Through RNA modification, a eukaryotic cell can use fewer variations in base pairs of the genetic code (DNA and RNA) while creating proteins with diverse functions. One of the most common methods employed by eukaryotes is to use spliceosomes to cleave out introns (intervening sequences/ non coding proteins) of the pre-RNA, leaving only exons (expressed sequences/coding proteins). Cutting out the intron segments allows for the possibility of exon to rearrangement. Spicing all exons together is called mature mRNA.
What's the advantage of spitting genes? Exons are segments that coding proteins and give proteins specific functions. This leads to the concept of exon shuffling. Exons shuffling is the rearrangement of the exons in the mRNA. These mRNA will come up with different types of proteins with different functions, binding sites, and catalytic sites. This path could lead to the evolution of new proteins.
Even though the use of exons and introns are quite common in eukaryotes, such practice is rarely performed by prokaryotes. In addition, evolution has shown that DNA sequences of genes encoding proteins were conserved. They showed that introns were once appeared in the prokaryotes' ancestral genes, and were vanished over time. The reason for this might be because such processes are not time efficient. Time efficiency is very important for prokaryotes because they multiply at a very fast rate.
Introns are regions within the primary-transcript where part of the fragments are to be removed. They are named introns for intervening sequences. the regions that are saved are called the exons.
Introns are removed from the primary transcript, precursor mRNA (pre-mRNA) after the poly A tail and the 5' cap have been added. Introns usually begin with Guanine-Uracil, and end with Adenine-Guanine that is preceded by a pyrimidine-rich tract, which signal splicing. Introns are spliced from the pre-mRNA by spliceosomes, which are made from proteins and small RNA molecules. Some introns are self-splicing which means they have the ability to remove themselves from an RNA molecule. One advantage of having genes being split by introns is that alternate splicing patterns allows the formation of proteins with varying functions without requiring new genes for each such protein.
There are four types of introns: Group I introns, Group II Introns, Nuclear pre-mRNA Introns, and Transfer RNA Itrons. Group I introns are found in some rRNA genes and splices itself out of genes. Group I introns fold into a type of secondary structure that has a nine-looped stem that is required in order to be spliced. Group II introns are found in mitochondria and chloroplasts. They are self-splicing as well, but they cut themselves out differently than Group I introns. They too fold into a secondary structure like Group I introns, but their splicing produces a lariat structure. A lariat structure is formed when an introns folds back on itself after an exon is cut from it. Nuclear pre-mRNA introns are found in the nucleus in protein-encoding genes. Their removal requires the presence of snRNAs and several other proteins. Transfer RNA introns are found in tRNA genes and needs enzymes in order to be spliced out of the genes.
The average human gene has 8 introns and some have more than 100. The size range ranges from 50 to 10,000 nucleotides. They are longer than exons.
Evolutionary Differences in Existence of Introns
[edit | edit source]Introns are usually found in the genes of higher eukaryotes such as birds or mammals. Lower eukaryotes such as yeast have fewer introns and prokaryotes rarely if ever have introns. Study of genes that have been highly conserved in evolution suggests that introns were present in most organisms long ago but were lost in organisms such as prokaryotes as an evolutionary measure to allow faster replication. The presence of introns is thought to contribute to the development of new genes through exon shuffling. The advantage of the introns is that exons maintained function but are able to interact in new ways. Without introns, the crossover would most likely result in a loss of function.
Discovery of Introns
[edit | edit source]Introns (intervening sequences), were discovered by Phillip Sharp and Richard Roberts in 1977 discovered that several genes are discontinuous. Electron microscopic studies of mRNA and DNA segments combined showed the presence of introns. If the gene was continuous only one of the strands of DNA would be displaced. However, it was observed that the strands were displaced in some regions but remained as double strands in other regions, thus proving the existence of introns.
References
[edit | edit source]Info. obtained (Berg, Stryer, et al.)
General information
[edit | edit source]Exons are protein-coding segments and one of the two major divisions of DNA that is transcribed into RNA. All genes start with exons which are often interrupted by introns (non-protein coding segments). Exons received their name because they exit the nucleus and allow the DNA sequences to be expressed (prefix ex-comes from "expressed")1. Exons are the actual part that contains codes for particular protein parts. The number of exons in DNA can vary from one species to another. Before the functional mRNA is formed, a splicing complex called spliceosome cleaves exons to bring them together, then removes all introns and connects exons to each other. Exons join together and travel out of the nucleus where they eventually code for proteins.

History
[edit | edit source]Exons were first discovered in 1977 by American molecular biologists and Nobel Prize winners (1993) Richard Roberts and Phillip Sharp. They used electron microscopy to study mRNA and DNA hybrids. In the absence of introns, the entire region that is hybridized to the mRNA would be displaced. In their experiment, they observed regions that were not displaced, creating a loop that is indicative of an intron. Initially it was assumed that the sequence of both DNA and mRNA were identical or continuous. The result of their experiments revealed that DNA had stretches of bases not present in mRNA. Based on that they explained the nature of exons.
Roles of exons
[edit | edit source]With the help of introns, exons can undergo recombination or exon shuffling. Crossovers occur in random, but homologous, positions at a frequency that depends on DNA length. Exon shuffling is a natural process that allows the formation of new functional proteins by creating new arrangements and thus new interactions with minimal risk to the sequence encoding of the functional parts. In the absence of introns, crossovers are likely to disrupt the exon sequence and often create a loss of function. Moreover, exon shuffling can produce new and useful proteins which lead to evolution. It encode different domains of the protein products with the processes of transcription, RNA processing, and translation.

References
[edit | edit source]1 Jerry Bergman, The Functions of Introns: From Junk DNA to Designed DNA[73]
RNA Splicing
[edit | edit source]RNA splicing is a modification of an RNA that takes place during the transcription of the primary transcript to the mRNA. Splicing refers to introns being cut out or removed, and the remaining sequence (called exons) being attached. This modification occurs in the nucleus, before the RNA is moved to the cytoplasm.
Splicing happens in all the domains of life, but types of splicing differ immensely between the major divisions. Eukaryotes splice many protein-coding messenger RNAs and some non-coding RNAs. Prokaryotes, on the other hand, splice rarely, and when they do, it is mostly non-coding RNAs.
Discovery of RNA Splicing
[edit | edit source]RNA splicing was discovered by two scientists Phillp Allen Sharp and Richard J. Roberts and they were awarded the 1993 Nobel Prize in Physiology or Medicine for their achievement. The initial discovery of RNA splicing led to the resolution of an earlier paradox in which scientists had discovered RNA in the nucleus that was unusually long compared to the mRNA found in the cytoplasm of the cell. The strange nuclear RNA had a 5’ end containing a cap structure and a 3’ end that contained a polyadenosine [poly(A)] tract and these were similar structures found in the shorter mRNA found in the cytoplasm. The subsequent discovery of splicing explained how the small mRNA could have the same termini as the longer nuclear RNA. While the termini were the same, the lengths were different because introns had been removed from the middle of the strand. These introns, it was discovered, proved to be a problem for the cell because, for example, a nearly a quarter of all mutations in globin genes responsible for beta-thalassemia came from problems in splicing.
It became apparent through development of reactions that replicated RNA splicing that the splicing is done by a branch-shaped section of a lariat RNA and that such RNAs were integral to splicing. Later it was found that these small snRNAs compiled particles found in spliceosomes. Via an intermediatemade up of lariatRNA and the 5’ exon-RNA, the spliceosome was able to remove the intron.
Spliceosome
[edit | edit source]
In eukaryotes, genes are transcribed to messenger RNAs comprising both introns and exons. For the production of validated mRNAs, the introns are to be trimmed off and the exons attached back together by the spliceosome, the molecular tailor of the cell. The spliceosome has the ability to alter its snipping and stitching process in order to generate variation in mRNAs based on a single coil of pre-mRNA cloth. Alternative splicing is the process by which the spliceosome can develop multiple mRNA isoforms from a single bolt of pre-mRNA. Such alternative splicing has enhanced evolutionary possibilities in complex multicellular organisms without the addition of gene number.
The spliceosome is considered as one of the most complicated macromolecular machine in the eukaryotic cell. It is involved with hundreds of RNA and protein mechanisms, specifically with assembly and disassembling pathways. The main role for a spliceosome in eukaryotes is to develop messenger RNAs. Genes are transcribed as precursors to mRNAs, called pre-mRNA, and then the RNAs are generated by the snipping and stitching of intron and exon components. Introns are regions of the pre-mRNA that are cut by spliceosomes to serve as a source of non-coding RNAs. Exons code for proteins so they are usually wanted. Furthermore, a spliceosome can uniquely snip and stitch in ways that will create different types of mRNA, which as allowed evolution to allow organisms to increase in gene number and complexity. The reason why Spliceosome are considered one of the most complicated macromolecule machines in a cell is because they have the responsibility of properly recognizing and processing a large amount of sequences. For example, spliceosomes end up processing five small RNAs and up to 100 different polypeptides in budding yeast. To make things more complicated, humans even need to use a second splicing apparatus, the minor spliceosome. In studying these complex machinery, many barriers were in the way because of the limitations in vivo. However, novel approaches to researching splicing have developed, such as in vitro assembly and purification of active spliceosomes, microscopic visualizations of single spliceosomes, and more. The advantages of these methods are that they are more specific and allow the wider boundary of studying either hundreds or a single RNA molecule. One of the new methods involves using microarrays.
Using splicing-dependent microarrays allows researches to distinguish which features of the splicing cycle are universal or specific to pre-mRNAs. Groups of developed DNA arrays differentiate between spliced and unspliced RNAs and then are probed with cellular RNA to isolate even further. Analysis of the splicing response allowed the observation of how loss of activity in specific protein directly affected the splicing of individual pre-mRNA. Overall, the microarray has proved its importance of pre-mRNA identity by efficiently isolating the desired-protein.

Spliceosome analysis have often times brought up significant obstacles in gaining understanding of more detailed mechanisms. To overcome such difficulties, laboratory techniques have employed methods like in vitro, which involves observing single pre-mRNA molecules, and active spliceosome purification, which includes well- characterized enzymes and controlled conditions. Although each chemical approach are relatively distinct, each contribute a complementary and synergistic view that heighten the knowledge of splicing machinery.
The spliceosome is a complex macromolecular machine consisting of small nuclear riobonucleoprotein particles (snRNPs): U1, U2, U4, U5, and U6, as well as roughly 100 separate splicing factors. The snRNAs range in length from 107-210 nucleotides; the snRNAs link with proteins to make small ribonucleoprotein particles (snRNPs). The snRNP contains a single snRNA and multiple proteins.
Splicing is carried out in multi-megadalton complexes. This means that the spliceosome is made of several components in an ordered manner. First, U1 snRNP binds to the 5’ splice site (SS). At the same time, branchpoint bridgeing protein (BBP) and Mud 2 binds to the branch site. Then, U2 snRNP will displace the BBP/Mud 2 and bind to the branch site. Next, U4/U6.U5 tri-snRNP will also binds to the complex. Before splicing the RNA U1 and U4 will leave the complex and Prp19 will bind. After the splicing is completed, the spliceosome will undergo conformational change for the ligation process. After the ligation process, the components of the Spliceosome will degrade and be recycled. Therefore, each spliceosome is a single turnover enzyme
From all the new and developed techniques, it is not for certain that the spliceosome cycle in the body is far from simple, but rather an "extraordinary dynamic and flexible machine" The new technologies constantly bring new evidence of the detailed reversible, irreversible, kinetic, and mechanism interactions of the pre-mRNA substrate. There are still many things unknown about the spliceosome and its process. There is still limited structural information, which means many of its functional details are unavailable. Same with unknown kinetic understanding. However, the research of this dynamic machinery is still developing and continuing to discover new methods and information on its purpose.
Pre-mRNA Splicing Process
[edit | edit source]Splicing requires there to be three sequences in the introns. One end of the intron is the 5' splice site and the other end is the 3' splice site. At these sites are short consensus sequences. The sequence that exons are ordered in the mRNA usually correlates with the sequence in the corresponding DNA. The process is aided by spliceosomes, which are small RNA molecules that recognize the beginning of introns (usually GU) and the end (usually AG) and catalyze splicing at these sites. Changing a single nucleotide at these sites may prevent splicing to occur. There are also self-splicing introns. The third sequence important to splicing is located at the branch point. The branch point is where an adenine nucleotide lies from 18 to 40 nucleotides before the 3' splice site. The deletion or mutation of the adenine nucleotide at the branch point would prevent splicing. Splicing occurs in large structures called spliceosomes.
Before splicing takes place, an intron between exon 1 and exon 2. Pre-mRNA splices in two steps. In step one, the pre-mRNA is spliced at the 5' splice site, separating exon 1 from the intron. The 5' end of the intron then attaches to the branch point folding back on itself and forming a structure called a lariat. The folding back occurs by the guanine nucleotide in the 5' consensus sequence bonding with the adenine nucleotide at the branch point through transesterification. In step two a splice is made at the 3' splice site and the 3' end of exon 1 is attached to the 5' end of exon 2. The intron is separated as a lariat and becomes linear when the bond breaks at the branch point and is then degraded by nuclear enzymes. And finally, the mature mRNA consisting of only the exons spliced together are moved to the cytoplasm and translated.
It is important to note that the 5' cap greatly affects pre-mRNA processing and mRNA export and if it were ever to be removed, then it would be known as the first irreversible step in mRNA decay which will affect the entire gene expression.

Pre-mRNA Splicing: Constitutive vs Alternative
[edit | edit source]Constitutive splicing pertains to the way mRNA is spliced in exactly the same way, every time with the splicesome
Alternative splicing allows for different expression of genes through SR proteins, which select alternative sites for splicing, using different exons or expressing them in a different order. By choosing combinations of alternative splice sites, protein isoforms can be created that are structurally and functionally distinct. It is estimated that at least 75% of human genes undergo this mechanism.
Another alternative splicing uses multiple 3' cleavage sites. There are 2 or more potential site for cleavage in a pre-mRNA sequence. However, this may or may not produce different proteins.


Alternative splicing can occur under cellular Stress
[edit | edit source]Because alternative splicing can control gene expression, it is an important mechanism that a cell can use in response to certain stressful environmental and pathological cellular conditions such as heat, cold, UV-light, oxygen, ion balance, infections, inflammation, fever, etc.
Splicing factors can be enhancing (recognizing positive sequence elements) or silencing (recognizing negative sequence elements). These sequence elements can by exonic or intronic which determines whether they are included (exonic) or left out (intronic). The splicing enhancers are typically bound and activated by SR proteins. SR proteins are ‘serine/arginine-rich;’ they are a group of proteins that have been highly conserved throughout evolution that participate in both alternative and constitutive splicing. They are involved in regulating and selecting the splice sites.
Examples:
Alternative or unconventional mRNA splicing can be part of adaptive stress responses in certain cell organelles, such as the endoplasmic reticulum (ER). Moreover, abnormal mRNA splicing could also be related to cell apoptosis. Under stress conditions, unfolded proteins accumulate in the ER and form aggregates. These abnormal agglomerations engage a response process called unfolded protein response (UPR), which is triggered thanks to a few different stress sensors that reside in the ER. One of those sensors is inositol-requiring enzyme 1α (IRE1α), a type I transmembrane protein, which once activated, initiates the abnormal splicing of the mRNA that encodes the transcription factor X-box binding protein 1 (XBP1), leading to the translation of a more stable spliced form of XBP1 (XBP1s). XBP1s translocates to the nucleus, where it controls the upregulation of a subset of UPR-related genes linked to protein folding, quality control, ERAD and ER/Golgi biogenesis. Furthermore, prolonged ER stress leads to the inactivation of IRE1α signaling, which in turn is associated with the attenuation of XBP1 mRNA splicing, process that could sensitize cells to apoptosis.
In the heat-shock protein 47 (HSP47), the selection of the 5’ splice-site in the non-coding region of the pre-mRNA is performed more efficiently. In cold shock, alternative pre-mRNA splicing is induced in neurofibromatosis type 1 (NF1) which brings about a cryptic exon. Stress induced long-term neuronal hypersensitivity is associated with stress-induced alternative splicing of the pre-mRNA of neuronal acetylcholinesterase (ACHE).
Impact of Heat Shock Stress
[edit | edit source]Many types of stress, including heat shock, can immediately block many crucial metabolic processes such as DNA replication until recovery. Heat shock proteins (HSPs) help protect cells from injury and aid cell recovery and after heat shock conditions subside. The blocking of pre-mRNA splicing in heat-shocked proteins is well characterized. HSPs are not affected in their expression, however, because they do not contain any introns.
SRp38 is an SR protein, that when overexpressed, antagonizes the activity of SR protein SF2/ASF (splicing factor 2/alternative splicing factor). SRp38 is unique in that, when phosphorylated, it activates sequence-specific splicing that requires an as of yet unidentified cofactor. This activity stems from SRp38’s entry into a complex with U1 and pre-mRNA which strengthens the interaction of U1 and U2 with pre-mRNA. SRp38 is a strong splicing repressor when dephosphorylated after heat shock; after mild heat shock it is rephosphorylated, accompanying the return of splicing activity.
Nuclear stress bodies (nSBs) are proposed to control splicing activity under stress by bringing a set of splicing factors to the region where they bind to SATIII transcripts. The nSBs are the sites of accumulation of heat-shock factor 1 (HSF1) in human cells, and appear fleetingly after mild heat shock, chemical and hypertonic stresses. They are also the site of accumulation of pre-mRNA splicing factors (SF2/ASF, 9G8, SRp30c for the adenoviral E1A gene). The nSBs are assembled on regions of chromatin that consist of long satellite III (SatIII) DNA. After heat shock, chromatin reorganization occurs along with HSF1 transcription of SatIII RNAs. Recruitment of the SF2/ASF and SRp30c proteins requires the stress-induced SATIII transcripts. Reducing the transcription blocks the SR protein splicing factor recruitment.
Structural Insights into RNA Splicing
[edit | edit source]The study of introns and alternative splicing has lead to the additional classification for introns in both eukaryotes and prokaryotes. Two groups of introns have been discovered. The first group of introns, Group I, were the first self-splicing ribozymes to be discovered. Group II introns were later reported. The Group II intros have highly complex RNA structures and they also possess a unique diverse range of chemical reactivity. The Group II intros possess the capability to catalyze the 2'-5' bond formation, and the ability to retrotranspose onto DNA. Retrotranspositions onto DNA requires the help of intron-encoded proteins. Specific analysis of Group II's secondary structure revealed six structural domains. Domain V is the mos conserved phylogenetically (closely related among various groups of organisms). The lower helix of Domain V possess a catalytic triad that consists of nucleotides that is very similar to that of a spliceosome called U6 spliceosomal RNA. This has led to the belief that Group II introns share acommon ancestor with nuclear introns and the eukaryotic splliceosome. This has led to further meticulous study of Group II introns.
Study of Group II Introns
[edit | edit source]Group II introns have been further classified into three main structural elements based on their RNA secondary sturcture. The three groups are IIA, IIB, and IIC. Analysis by reverse transcriptases have shown that group IIC introns are the most primal and simplistic lineage of the Group II introns. The secondary structure of lineage IIC is much simple than that of IIA and IIB. Because the Group IIC are much smaller and simplistic, they are much more preferred by study of crystallization. Oceanobacillus iheyensis was the first organism to have its Group II intron successfully crystallized and examined via x-ray diffraction. Group IIC is also significantly different from Group IIA and IIB by the fact that the nucleophile used during the first step of the splicing is a water molecule. Group IIA and IIB use a 2'-OH nucleophile from the adenosine in Domain VI. Because Group IIC uses a water molecule the introns released are linear molecules, while Groups IIA and IIB introns will be released as a lariat branched species. The use of the Group IIC intron has further suggest that the active site is composed of the bulge and catalytic triad of Domain V, mentioned above. It has been shown that these regions are influenced by the binding of catalytic metal ions, i.e. magnesium. This meatal ion interaction is very common in protein in order to influence shape, i.e. Fe hemoglobin. The ion helps modulate the eclectrostatic environment at the core of the intron or protein. The metal ion interaction can also make or breack the phosphodiester linkages in the DNA and RNA polymerases.
Possible Splicing Errors
[edit | edit source]Although the rate of splicing errors is very low, it does happen occasionally due to several distinct possibilities. A splicing error will most likely result in a mutation of a splice site and could compound into losing the function of the particular site. An exposure of a premature stop codon or a misplace/misinserted exon or intron could all lead to a mutation. A mutation from variations in the splice location which could cause a wrong amino acid to be interpreted. A misinterpreted amino acid could result in reducing specificity. All mutations could result in wrongly constructed proteins, which can be life threatening, i.e. cickle cell anemia or cystic fombrosis. Fortunately many splicing errors can be safeguarded by Nonsense-mediated mRNA decay.
Nonsense-Mediated Decay
[edit | edit source]Nonsense mediated decay is the cellular mechanism of mRNA that exists to detect incorrectly spliced information, and to prevent the expression of incorrect proteins. After transcription the mRNA will reassemble with ribonucleoprotein. Nonsense mediated decay is initiated by exon junction comples that are cut out from the genetic information during the mRNA processing. Exon junction complexes located past a nonsense codon act as tags for the mRNA ribonucleoprotein, RNP. The RNP is able to recognize disorganizantion and wrong splicing from the pressence of these exons that were supposed to be cut out.The nonsense mediated decay will transport the excon tagged misinformation set out the nucleus and into the cytosol where the misinformation RNA is degraded.
References
[edit | edit source]Cellular stress and RNA splicing. Biamonti G, Caceres JF. Trends Biochem Sci. 2009 Mar;34(3):146-53. Epub 2009 Feb 7.
William Fontaigne De La Tour Dautrieve
Aaron A. Hoskins, Melissa J. Moore, The spliceosome: A Flexible, Reversible Macromolecular Machine, Trends in Biochemical Sciences, Volume 37, Issue 5, May 2012. <http://www.sciencedirect.com/science/article/pii/S0968000412000345>
Sharp, Phillip A. "The Discovery of Split Genes and RNA Splicing." Trends in Biochemical Sciences 30.6 (2005): 279-81.
Woehlbier, U., Hetz, C. Modulating stress responses by the UPRosome: A matter of life and death. Trends in Biochemical Sciences, June 2011, Vol. 36, No. 6.
Small Nuclear Ribonucleoprotein Particles (snRNPs)
[edit | edit source]Small Nuclear RiboNucleoprotein Particles (snRNPs) are the secondary molecules made of small nuclear RNAs (snRNAs) and specific proteins. snRNP molecules make up the larger splicosome molecules. U1 snRNP recognizes the binding site on the 5’ end and the six-nucleotide sequence of the U1 snRNA binds to the splice site on the pre-mRNA. From this, the spliceosome will assemble along the pre-mRNA molecule. The U2 snRNP will bind to the branch site on the intron with its complementary sequence between the U2 snRNA sequence and the pre-mRNA. The U4, U5, and U6 snRNPS then bind with the U1 and U2 complexes and form the necessary spliceosome. The splicing process itself begins with the U5 interaction with the exon sequence on the 5’ splice site. The U6 goes through intramoleculear reorganization after breaking from U4, which allows U2 to base pair and interact with the 5’ end of the intron, taking U1 out of the spliceosome. The U2-U6 complex forms a helix that forms the center of the spliceosome itself. U4 prevents U6 from splicing until the splice sites are correctly aligned. Once alignment has occurred, the transesterification reaction cuts the 5’ exon at the phosphodiester bond and produces a lariat intermediate. Splicing continues with rearrangements with the spliceosome that will then produce the next transesterification reaction on the pre-mRNA. In the rearrangement, the U5 aligns with the 5’ exon so that it is easier to attack the 3’ splice site to produce another spliced product. To finish the splicing process, the U2, U5, and U6 release itself from the lariat intron.
snRNP Biogenesis Cycle
[edit | edit source]The biogenesis of snRNPs begin with the transcription of a monomethyl-guanosine (m7G)capped snRNA-precursor using RNA polymerase II. Following its transcription, the snRNA is exported out of the nucleas to react with Sm proteins, which combine to form the Sm core domain. This then triggers the hypermethylation of them7G-cap, thereby generatingthe trimethylguanosine (TMG)m^(2,2,7)3G-cap. The two-part nuclear localization signal (NLS) consisting of the Sm core domain and the TMG cap causes the relocation of the snRNP back to the nucleus. Before re-entering the nucleus, the snRNP undergoes completion of the biogenesis cycle in subnuclear domains called Cajal bodies. It is still relatively unknown as to which proteins join the snRNP at which stages of the biogenesis cycle. The U6snRNP does not follow the above stated steps and is speculated to carry out its biogenesis within the nucleoplasm.
snRNP Assembly Factors
[edit | edit source]Current work on snRNPs have demonstrated cellular assembly strategies for RNA-protein complexes. snRNPs form in vivo by the synchronized action of a complex assembly line containing assembly chaperones, scaffolding proteins, and catalysts. RNP assembly factors satisfy two functions. One, they augment assembly efficiency by helping the accumulation of higher order building blocks and second, they hinder the collection of Sm proteins and the assembly of snNRP centers that contain wrong RNA, or RNAs that do not entertain an Sm site. Various reports have spoken of the ‘proofreading’ function of the assembly machinery. These new strategies employ affinity to those used by protein complexes and also admit the explanation of common rules on how molecular machines are made in vivo.
References
[edit | edit source]1. Chari, Ashwin, and Utz Fischer. "Cellular Strategies for the Assembly of Molecular Machines." Trends in Biochemical Sciences 35.12 (2010): 676-83. Print. RNA degradation is a very ancient and important process. It is a physiological process of the cell cycle to maintain a balanced RNA concentration. To do this, cells secret an abundance of RNases, or ribonucleases, enzymes that help to break apart RNA. RNases play a significant role in cellular immune system by defending against RNA viruses, eradicating of old and non-viable RNAs, and manipulating of the nucleotides for RNA sequencing and gene expression. Viable RNAs avoid degradation by RNases by protecting themselves with “armors” such as the G-cap and the poly-A-tail. Also there are RNase inhibitors that bind to RNases, prohibiting them from cleaving RNA strands.
- _[74]
Ribonuclease
[edit | edit source]Ribonuclease is a type of enzyme that is capable of cleaving the phosphodiester bond between each unit of nucleic acid that form the RNA backbone. A phosphodiester bond in a single RNA strand is formed by the linkage between the 3’ carbon atom of one ribose sugar molecule and the 5’ carbon atom of another ribose sugar attached to an adjacent nucleoside. These enzymes have overlapping functions as the small nuclear RNAs acting on mRNAs, such that they catalyze RNA degradation by breaking down RNA into shorter partial strands. Ribonuclease can be grouped into two categories: endoribonucleases and exoribonucleases.
File:Nucleic acid.gif a picture of two RNA nucleotides with base adenine and cytosine
Endoribonucleases
[edit | edit source]Endoribonulcases, much like the restriction endonucleases, are able to recognize certain RNA nucleotides within RNA and cleave at a specific site. These enzymes are able to break apart both single or double-stranded RNAs.
Exoribonucleases
[edit | edit source]Exoribonucleases cleave RNA by removing terminal nucleotides from either the 5’ end or the 3’ end of the RNA strand. Enzymes that remove nucleotides from the 5' end are called 5'-3' exoribonucleases, and enzymes that remove nucleotides from the 3' end are called 3'-5' exoribonucleases.
Both endonucleases and exoribonucleases can be further broken down into sub-classes of ribonucleases based on their chemical cleaving mechanisms, such as phosphorolytic and hydrolytic activations. They exist in all kingdoms of life, the bacteria, archaea, and eukaryotes. They are involved in the degradation of many different RNA species, such as messenger RNA, transfer RNA, ribosomal RNA, microRNA, etc.
 The left side shows the hydrolytic activity of RNases in which a water molecule intercepts the 3’ ester bond between the phosphate group and the 5’ –OH group of the adjacent sugar, breaking off one nucleotide. The left side shows the phosphorolytic activity of RNases in which a phosphate group intercepts the 3’ ester bond between the phosphate group and the 5’ –OH group of the adjacent sugar, breaking off a nucleotide with two phosphate groups on it.
The left side shows the hydrolytic activity of RNases in which a water molecule intercepts the 3’ ester bond between the phosphate group and the 5’ –OH group of the adjacent sugar, breaking off one nucleotide. The left side shows the phosphorolytic activity of RNases in which a phosphate group intercepts the 3’ ester bond between the phosphate group and the 5’ –OH group of the adjacent sugar, breaking off a nucleotide with two phosphate groups on it.
The left side shows a strand of mRNA, protected by a G-cap and a poly-A-tail. The Dcp1/2 protein recognizes the G-cap at the 5’ end of the RNA, takes it away, and exoribonuclease Xrn1 comes and starts chewing off the RNA strand from the 5’ end. The right side shows the deadenylase protein recognizing the poly-A-tail and starts chewing it off. Then comes the exosome, another exoribonuclease that degrade RNA from the 3’ to 5’ direction. The middle shows an endoribonuclease binding to a specific sequence within the RNA and cleaving it internally.
Important RNase
[edit | edit source]RNase A
[edit | edit source]The structure of RNase A was first crystalized 50 years ago. It was the first enzyme and the third protein whose amino sequence was determined. It is a single chain protein that contains 4 disulfide bridges. It contains 124 amino residues and 19 out of the 20 amino acids, except for tryptophan. These enzymes were found in the exocrine cells of the bovine pancreas. They are very tough and highly stabilized enzymes, mostly due to their structures and folding patterns. Its molecular formula is C575H907N171O192S12 and the general structure consists of 2 sheets of alpha helices and beta sheets cross-linked by four disulfide bridges. RNase A has basic properties. It specifically attacks at the 3’ phosphate of a pyridine nucleotide. The cleavage involves simply two steps: 1). the 3’,5’-phosphodiester bond is cleaved to generate a 2’,3’-cyclic phosphodiester intermediate and 2). the cyclic phosphodiester is hydrolyzed to a 3’-monophosphate.
Example: pG-pG-pC-pA-pG undergoes RNase A cleavage would result in 2 sequences: pG-pG-pCp and A-pG.
RNase A can be activated by potassium and sodium salt and inhibited by alkylation of His12 or His119 in initiate RNA cleaving. It utilizes both phosphorolytic and hydrolytic activities to cleave a strand of RNA.
RNase P
[edit | edit source]
Sidney Altman discovered and named RNase P while working in the Laboratory of Molecular Biology in Cambridge, England, focusing on the functions of tRNA. He proposed that by altering spatial relationships in tRNA, either by insertion of new nucleotides or deletion of existing nucleotides, would affect or change the function of the tRNA. His experiments with E. coli. have shown that mutated tRNAs could not develop into a full mature tRNA, which in turn could not serve its functions of delivering amino acids during protein synthesis. However, these dysfunctional tRNAs quickly resolved back to wild-type tRNAs. By isolating the DNA to RNA transcript of tRNA, Altman had found that there are extra nucleotides hanging off of the 5’ and 3’ ends of tRNAs. When these tRNAs were introduced to a live medium, an enzyme was observed to cut off the extra nucleotides through the cleave of a phosphodiester bond, exposing the 5’ end of the molecule. This RNase was different than other previous known RNase because of its specificity at the 5’ end of the tRNA. Altman also showed that RNase P-like activities were present in cells extracted from a variety of organisms, including humans. RNase P is unique that it is ribozyme. While it cleave other RNAs, it cleaves itself as well, meaning it self-destructs during reaction. It is a single stranded protein containing 120 amino residues. They are found in many organisms such as archaea, bacteria and eukarya as well as chloroplasts in plants. The make-up of RNase P differ from one organism to another, but their functionalities are the same because of orthogonal properties. RNase P is a crucial component in the production of functional tRNA molecules.
RNase T2
[edit | edit source]
T2 family Ribonucleases are considered to be transferase type RNases and are distinguished from the RNaseA and RNaseT1 protein families based on three features.
- First of all T2 ribonucleases are more evenly distributed and are found in bacteria, plants, protozoans, animals and even viruses, whereas RNase T1 enzymes exist only in bacterial and fungal organisms and RNaseA family enzymes are highly represented in animals.
- Secondly, the optimal pH of activity of many T2 ribonucleases is between 4 and 5. By contrast, RNaseT1 and RNaseA families have optimal pH activity at alkaline (pH 7-8) or weakly acidic (pH ~7).
- Thirdly, T2 ribonucleases do not discriminate their cleavage sites. T2 families generally cleave at all four bases, whereas RNaseA and RNaseT1 families tend to be specific for pyrimidine or guanosine bases respectively.
The biological role for T2 Ribonucleases varies. These endoribonucleases are ubiquitously represented in organisms across kingdoms and have been show to perform a variety of functions in different organisms besides it's ability to hydrolyze RNA. Some examples of biological roles include the scavenging of nucleic acids, the degradation of self-RNA, modulation of a host immune response, and serving as cellular cytotoxins.
Other T2 RNase Properties
[edit | edit source]T2 RNase are transferase-type RNases and catalyze the cleavage of ssRNA (single-stranded RNA) through a 2',3'-cyclic phosphate intermediate. The result of this catalyzed reaction are mono- or oligonucleotides with a 3' phosphate group. Typically, these RNases are secreted from the cell or specific special locations within the cell such as vacuoles, which may prove important to how their activity is modulated within the cell. This family of RNases has a specific structure and mechanism that is well known from x-ray crystallography. Likewise, crystallography has defined specific places such as specific binding sites, called B1 for sites with a 5' end and B2 for site with a 3' end, as well as a core made up of alpha and beta structures. Additionally, the catalysis of T2 RNase starts with one to three histidine residues. It should be noted that alteration or mutation of these residues causes inactivation of the RNase. The two main steps of this catalysis are transphosphorylation and hydrolysis. Further study is being conducted in the following areas for these specific RNases:
- Discovering how RNases from this family can function independent of catalysis
- Mutational analysis to determine the regions necessary for nuclease-independent functions
- How these RNases enter the cell to reveal how proteins cross membranes, while many things cannot
References
[edit | edit source]1. T2 Family ribonucleases: ancient enzymes with diverse roles. Luhtala N, Parker R. Trends Biochem Sci. 2010
2. Raines, Ronald T. "Ribonuclease A." Chem Rev. Wisconsins: University of Wisconsins, 1998. 1045-065. Print.
3. Gopalan, Venkat, Agustin Vioque, and Sidney Altman. "RNase P: Variations and Uses*." RNase P: Variations and Uses. JBC Papers in Press, 10 Dec. 2011. Web. 20 Nov. 2012. <http://www.jbc.org/content/277/9/6759.full>.
4. Cuchillo, C. M.; Nogués, M. V.; Raines, R. T. (2011). "Bovine pancreatic ribonuclease: Fifty years of the first enzymatic reaction mechanism". Biochemistry 50: 7835-7841. PMC 3172371. PMID 21838247.
5. J. Holzmann, P. Frank, E. Löffler, K. Bennett, C. Gerner & W. Rossmanith (2008). "RNase P without RNA: Identification and functional reconstitution of the human mitochondrial tRNA processing enzyme". Cell 135 (3): 462–474. doi:10.1016/j.cell.2008.09.013. PMID 18984158. Telomeres (from the Greek telos, "an end") are long stretches of repeating non-coding DNA sequences at the ends of the DNA strand. They protect the ends of DNA and prevent DNA strands from shortening or attaching to other molecules by masking the chromosome. Russian Alexei Olonikov was the first to postulate the problem of chromosomes replicating at the tip.[1] He theorized that in every subsequent replication bits of the DNA would be lost until a critical limit had been reached, thereupon cell division would cease.
Telomerase
[edit | edit source]Telomerase is an enzyme that creates the Telomeres. Telomerase adds specific repeating sequences ("TTAGGG" in all vertebrates) to the ends of four DNA strands.
[2]
The telomerase enzyme has an RNA template that partially attaches to the shortened end of the DNA strand. New nucleic acids then attach to the template, extending the DNA strand. Once the telomerase leaves, the double stranded DNA is completed with the DNA polymerase. Telomerase was discovered in 1985 by Carol W. Greider and Elizabeth Blackburn. For this discovery, they were awarded the 2009 Nobel Prize in Physiology or Medicine along with Jack W. Szostak.[3]
Szostak and Blackburn first discovered telomeres in ciliates. They chose ciliates because at one stage of their life cycle, they make a million new telomeres. The model created includes a telomere-dedicated DNA polymerase, which adds telomeric repeats onto chromosome ends. Therefore, telomeres are represented as a motif in DNA sequences.
Telomerase's presence in humans is somewhat strange. It is located in the nucleus which is unsurprising because that is where DNA replication takes place. However, Telomerase activity is not present in all cells. It was found to be almost absent in the majority of normal adult tissues, including cardiac and skeletal muscle, lung, liver, and kidney. Because of this curious lack of telomerase activity, a theory arose connecting telomere length to aging and cell senescence. According to this theory, human somatic cells are born with a full number of telomeric repeats, but the telomerase enzyme is not present in some tissues. The cells of those tissues would lose about 50 to 100 nucleotides from each chromosome end each time they underwent replication and division. Eventually, the telomeres would cease to exist and the chromosomes themselves would start losing nucleotides, carrying genetic defects into their next division so that neither daughter cell would be viable. Thus after a certain number of divisions a cell will not have enough nucleotides and die.[4]
The function of Telomerase is to allow for short replacements of Telomeres which are gradually lost during cell division.[5] In normal conditions without Telomerase, a cell would divide until it would hit a critical point known as the Hayflick limit.[6] In the presence of Telomerase, however, the cell has the ability to replace lost DNA and divide without limit. But this continuous growth comes with a consequence as this growth may lead eventually to cancerous cells.
While the details are not fully known, it would seem that that shortened Telomeres play a role in aging due to the erosion of the DNA over time. The questions arises whether or not Telomerase has the ability to greatly extend the lifespan of a human due to its importance in the maintenance of the Telomeres.[7]Dr. Michael Fossel, a professor of clinical medicine at Michigan State University, has expressed his views on Telomerase as a viable treatment for cell senescence.
However, several experiments have raised doubts on the ability of Telomerase as an effective anti-aging treatment. An experiment was done with mice having higher levels of Telomerase and it was discovered that they also had a higher rate of cancer which therefore led to a shorter lifespan. In addition, Telomerase favors tumorogenesis.[8] Telomerase fosters cancer development by allowing uncontrolled cell growth which eventually proliferates into tumors. In fact, Telomerase activity has been observed in approximately 90% of all human tumors which suggests that the uncontrolled growth of a cell as conveyed by Telomerase has a key role in cancer.
In addition to using Telomerase as an anti-aging treatment, Telomerase has potential as a drug target against cancer.[9] Since it is necessary for the immortality of many cancer cell types, it is believed that if a drug is able to deactivate Telomerase activity in a cell, Telomeres would shorten, mutations would happen, cell stability would decrease and cancer would be, in essence, effectively treated. Experimental drugs have been tested in mouse models and some drugs have moved onto clinical testing.
Cancer Biology
[edit | edit source]The significance of studying telomeres can be found in telomerase, which rebuilds the telomere so that the cells can keep dividing. The telomerase, however, eventually shortens the telomere, causing the cell to die. In the case of cancer cells, this enzyme builds telomeres long past the cell's average lifetime. These cells then are called to be "immortaled", since they can divide endlessly. This results in a tumor. Many researchers believe that telomere maintenance activity is characterized in most human cancer cells. Though the mechanism by which such phenomena happen has not been well understood, the discovery may reveal key elements of telomere function. Telomerase, on the other hand, is the natural enzyme used for telomere repair, highly abundant in stem cells, germ cells, hair follicles, and most cancers cells, but its expression is low or in some cases absent in somatic cells. Telomerase functions by adding bases to the ends of the telomeres. Cells with sufficient telomerase activity are considered immortal in the sense that they can divide past the Hayflick limit without entering senescence or apoptosis. For this reason, telomerase is viewed as a potential target for anti-cancer drugs such as telomestatin.
2009 Nobel Prize
[edit | edit source]The Nobel Prize 2009 in Physiology and Medicine was awarded to three scientists who have discovered how the chromosomes can be copied in a complete way during cell divisions and how they are protected against degradation. By showing that the ends of the chromosomes, telomeres, and their enzyme, telomerase, are significant in protecting the chromosomes from degradation, they identified telomerase and explained how the telomeres protect the ends of the chromosomes and built by telomerase. On the other hand, if the telomeres become shortened, cells can duplicate damaged as cancer cells. If telomerase is well maintained, conversely, telomere length is maintained and the cell does not become cancerous. In the case of cancer cells, telomerase allows the cell to divide without any limit. Certain genetic disease are caused by a defective telomerase. This discovery can thus be used to stimulate the development of new therapeutic strategies. Understanding such fundamental mechanism is an important first step toward opening new doors for cures for cancer and other related diseases, as well as anti-aging.
Hayflick Limit
[edit | edit source]The Hayflick limit is the number of times a normal cell may divide until it reaches a critical limit and stops dividing based on the idea that Telomeres reach a critical length.[10] This limit was discovered by Leonard Hayflick in the 1960s who demonstrated that the cells in a normal fetus divided around 40 to 60 times before entering into cell senescence. Due to repeated mitosis, the Telomere shortening occurred which inhibited cell division which is analogous to aging. The discovery of this limit, a pillar of Biology, refuted the early contention by Alexis Carrel who, along with the majority of scientists during that time period, believed cells were "immortal".
Role of Telomere
[edit | edit source]Telomeres account for the lost bits of DNA at the ends of chromosomes during DNA replication. Since DNA polymerase moves along the template strand in the 5'--> 3' direction, some of the 5' end of the template strand will not be replicated. This results in the incomplete ends as shown in the diagram below. However, telomeres are usually very long, ranging from 400 to 600 base pairs in yeast to many kilobases in humans. They are made of six to eight base pair long repeats which are usually rich with guanine bases. With long stretches of telomeres at the ends of DNA strands, the incomplete strands of DNA will still contain the genetic code.
The shortening of telomeres in humans induces cell senescence in humans. This mechanism appears to cause the formation of cancerous cells. Telomere length has been theorized in recent publications to account for the aging in humans. Since cells replicate identically, there must be a reason why cells within a body lose function and viability with time. Telomeres may have some influence over the aging process since every consequent DNA replication results in the shortening of telomeres. Two aspects to this question are: (i) whether telomere length, as measured in specific cell populations in the body, correlates with longevity or disease; and (ii) whether telomere shortening in any cell population causes functional impairment of that cell population. However, some may argue telomeres do not correlate to longevity as mice contain long strands of telomeres, but contrarily live much shorter lives than humans who do not have as long telomeres as do mice. And some may argue that telomere length does correlate to longevity as it determines the number of times that a cell can divide before it dies or reaches senescence.
Recent Publications
[edit | edit source]Recently it has been found that telomerase activity is inversely related to length of the telomeres. In other words, telomere elongation happens more often on short telomeres rather than long ones. The research showed a deficiency in telomerase activity in telomeres greater than 125 base pairs,and there was 2 to 3 times more telomerase activity in telomeres shorter than 125 base pairs. This preferential elongation has been demonstrated in yeast and mice, and now human somatic cells. Kinetic data indicates that elongation in yeast cells in a single event in which elongates the telomeres to a certain length, whereas in human cells the elongation seems to be a gradual process. The researchers showed that telomerase adds a regulated length of telomere in each cell division. The researchers showed that human cells expressed telomerase, however long telomeres were maintained and not elongated where as the cells with shorter telomeres elongated, which goes to show that telomeres can not be infinitely extended.[11]
Another interesting paper was focused on the role of DNA damage response (DDR) proteins in the role of telomere maintenance. The review says that early stage DNA repair proteins have a significant role in telomere maintenance where as late stage proteins usually do not take part in telomere repair. The interplay with these proteins and the proteins that cap the telomeres to protect the telomeres is very important too. Many of stronger DDR proteins inhibit cell replication, because of this fact, it would be harmful to the organism for these proteins to be a part of telomere repair. These protein caps on the telomeres inhibit full DNA damage response which keeps the stronger protein from "repairing" the telomere ends. It still isn't clear why some of the DDR proteins participate in telomere maintenance and others do not, but it is clear that the cellular process in repairing a DNA break and repairing telomeres are two different process, with the former halting cellular division.[12]
References
[edit | edit source]- ↑ "Telomeres, telomerase, and aging: Origin of the theory". Alexey M. OlovnikovE-mail The Corresponding Author. 1999. Retrieved 2009-11-05.
{{cite web}}: External link in|publisher= - ↑ "Repeat Expansion–Detection Analysis of Telomeric Uninterrupted (TTAGGG)n Arrays". [7]. 2007. Retrieved 2009-11-05.
{{cite web}}: External link in|publisher= - ↑ "The Nobel Prize in Physiology or Medicine 2009". [8]. 2009. Retrieved 2009-11-05.
{{cite web}}: External link in|publisher= - ↑ "What are telomeres and telomerase?". [9]. Retrieved 2009-11-05.
{{cite web}}: External link in|publisher= - ↑ "Telomerase: regulation, function and transformation". [10]. Retrieved 2009-11-05.
{{cite web}}: External link in|publisher= - ↑ "Hayflick Limit Theory". [11]. Retrieved 2009-11-05.
{{cite web}}: External link in|publisher= - ↑ "Extension of Life-Span by Introduction of Telomerase into Normal Human Cells". [12]. Retrieved 2009-11-05.
{{cite web}}: External link in|publisher= - ↑ "Anti-Aging Medicine". João Pedro de Magalhães. 2008. Retrieved 2009-11-05.
{{cite web}}: External link in|publisher= - ↑ Foreman, Judy. "Telomerase - a Promising Cancer Drug Stuck in Patent Hell?". myhealthsense.com. Retrieved 2009-11-05.
- ↑ "Cellular Senescence". João Pedro de Magalhães. 2008. Retrieved 2009-11-17.
{{cite web}}: External link in|publisher= - ↑ Britt-Compton, Bethan; Capper, Rebecca; Rowson, Jan; Baird, Duncan M. (2009). FEBS Letters (583): 3076–3080.
{{cite journal}}: Missing or empty|title=(help) - ↑ Lyndall, David (2009). The EMBO Journal (28): 2174–2187.
{{cite journal}}: Missing or empty|title=(help)
RNA plays a variety of roles in gene expression, from messenger, catalysis to regulations. For instance, in E.coli, 80% of RNA is ribosomal RNA, 15% is transfer RNA, and only 5% is Messenger RNA. Messenger RNA is the template for protein synthesis. Since the amount of mRNA is relatively small, affinity chromotography must be performed to purify it.
This technique exploits the structure of mRNA,which is polyadenylated at the 3' end to form a Poly A tail. The Poly(A)n can pair with the complementary base Poly(T)n through the formation of Hydrogen bonds. The Poly(A) region is used to selectively isolate mRNA from the rest of the RNA via affinity chromatography. Only mRNA with Poly A can be bound by Poly T in the column. RNA that lack Poly A tails elute out of the column at high salt concentrations. The mRNA is separated for other RNAs. Then the Poly(A) mRNA can be washed out from column by adding a low salt eluting buffer.
Affinity chromotography is very useful for the purification of RNA and the synthesis of cDNA.

RNA Extraction
[edit | edit source]RNA extraction is a technique of isolating and purifying RNA from in-vivo tissues and samples. There are several ways of extracting RNA. The presence of ribonucleases enzymes within the tissue cells complicates the extraction and purification process by quickly degrading the isolated RNA. Isolated and purified RNA can be used to detect gene expression, biomarkers, drug efficacy, and much more.
Trizol RNA extraction
[edit | edit source]File:RNA extraction.jpg First, homogenize the sample tissue in Trizol solution using a vortex mixer. The speed of mixing is very important because the tissue can only be homogenized when mixing at high speed, however, friction may cause heat, which may accelerate RNA degradation. Add chloroform to the finely grinded mixture to perform a liquid phase separation. For every milliliter of Trizol reagent, add 0.2 mL of chloroform. Cap and shake the mixtures vigorous for 15 seconds and incubate them at room temperature. Centrifuge the mixtures at no more than 14,000 rpm for 15 minutes at 2-80C. After centrifugation, the mixture separates into three distinct layers, the lower red chloroform layer, an interphase layer of remaining tissues and fat, and a clear upper aqueous layer. RNA remains in the aqueous layer exclusively. Transfer the aqueous layer to another container. Wash and precipitate the RNA by using isopropyl alcohol. Add 0.5mL of isopropyl alcohol to every mL of Trizol reagent. Incubate at room temperature for 10 minutes and centrifuge again for 4 minutes in RNA elution plates. Remove all supernatant and wash the RNA again with ethanol. Wash the RNA subsequently with buffers. Make sure to dry off any remaining alcohol because they lower the quality of RNA and promote RNA degradation. The last wash should contain RNase free water to elude out the isolated and purified RNA.
Reference
[edit | edit source]1. Gottshall, Susan L., Saban Tekin, and Peter J. Hansen. "EXTRACTION AND PURIFICATION OF TOTAL RNA USING TRIzol OR TRI REAGENT." (n.d.): n. pag. Web. 15 Nov. 2012. <http://www.animal.ufl.edu/hansen/protocols/RNA_extraction.htm>.
Introduction
[edit | edit source]The short RNAs includes different classes of molecules like small nuclear RNA, micro RNA, and transfer RNA and etc. Many functional RNAs are less characterized as short RNA because short RNAs are found at the promoter and 3' termini of gene sequeces [1] . It also involves in paramutation, is a reciprocal action between two alleles at locus, inducing an intermolecular change in the other allele. The short RNAs can be studied in research field for describing and analyzing the stratigies and process to develop short RNA species with single-molecule sequencing (SMS) and its efficiency in many laboratory research. The Short RNA is derived from the final product of functional precursor RNA species in the cell. One of the examples of the short RNA species in the cell can be found in gene splicing in DNA sequence and 3' end RNA processing for synthesizing grown-long mRNAs [2] . However, many classes related to functional RNAs that are not in protein coding are most likely from precursors which are longer than grown up long mRNAs. To fractionate RNA into species some methods are being used to study the various functions of the short RNAs. For example, Dr. Philipp Kapranov, and his researchers uses the method of Helicos single-molecule sequence to study the details of short RNAs in the cell [3].
RNA Isolation for short RNA
[edit | edit source]To proceed the isolation of RNA, sRNA gets purified from total RNA (cultured cells) with mirVana miRNA Isolation Kit and miRNeasy Mini Kit, which also is known as Qiagen. The RNA Kit is used for large amount of sRNAs (from cultured cell) preparation [4] . Through this method of RNA/DNA kit, sRNA of specific fractions gets purified with Elution and Electrophoresis method of TBE-Urea polyacrylamide gel. Electrophoresis is action of spreadout particles in fuild due to spartially uniform electric field throughout the cell structures.
Difficulties of Analysing short RNAs
[edit | edit source]Studying of the short RNAs is very challenging among scientists and researchers. There are some difficult problems that challenges researchers who are studying about short RNAs: 1) Specific Isolation of sRNA must be successful with desired length range. 2) There's lack of interest about sRNAs with molecular handles ike 3' PolyA Tail of the mRNAs which is used for converting itself into cDNAs. 3) sRNA is sometimes too short for efficient and successful conversion into cDNA with hexamers. 4) Some specific sRNAs have modification at gene sequence of their 5' and 3' ends that interrupt the reading with ssubsequent molecular manipulation; therefore, often few sRNAs cannot be detected by some methods that a lot researchers are using. 5) Some classes of sRNAs have firm 2' structures that blocks itself from being detected by enzymatic methods which are often conducted by researchers under nondenaturing condition. 6) Certain sRNAs such as miRNAs have short lengths that are not capable of being used for efficient mapping to complex genomes; thus, discovering sRNAs and its specific functions are very challenging for researchers to study and conduct experiments related to sRNAs. 7) Many methods depend on using ligation process and PCR amplication that can misrepresent the composition and quantification of the RNA species in the cell structures. Moreover, 8) Fraction of sRNA can be dominated with very highly presence of RNA classes like snRNA, rRNA, and sno RNAs (These will require a lot of complexity for completing characterization of the sRNA population in the cell. The most challenging part of studying sRNAs happens to deal with the physical structure in the cell: short length of sRNA makes researchers difficult to conduct an experiement [5] .
Isolation of sRNA Fraction
[edit | edit source]Using diverse methods, sRNA fraction can be detected and isolated with using either miRNeasy, or DNA/RNA Kit (Qiagen) [6] . If wanted, sRNA Isolation can be conducted in specific size range by using TBE-Urea denaturing polyacrylamide gel electrophoresis as another substitutive method for both researchers and scientists who are studying for sRNAs. One of the method of studying sRNAs deals with Tailing of RNAs with 3' PolyC: RNA is mixed with water in PCR amplication tube and it is used with protocol; then, sRNA gets incubated for 2 minutes at 85 degress Celcius in a PCR Machine and put into ice 2 minutes at least. After its incubation, reagents are added to the solution with E. coli Poly A polymerase buffer and mixed. SRNA gets extracted twice with addition of phenol or chloroform or isoamly for few seconds; then, purification with addition of 100% EtOH (Ethanol) for half an hour with minus 80 degrees Celcius. Centrifuge at 4 degrees of Celcius and leave it for 30 minutes. Then, washed with 70% EtOH and finally resuspend with water [7] .
Preparation of cDNA Sequence
[edit | edit source]The cDNA can be sequence from the 3' end with poly-A Polymerase in the method of terminal transferase (TdT) and be blocked at the other 3' end [8].thus, the cDNA can be bind to the oligo-dT present at the surface of the cell. This method is common for protocol and for poly A protocol. Some amounts of cDNA are analyzed under experiement with use of regular NanoDrop if the expected concentration of cDNA is at certain range between 5-10 ng. Tailing part of cDNA is followed: 1) cRNA is prepared to be tailed in water and depending on its volume, its time spending in the Incubation varies and amount of buffer can be differed. After Incubation, certain micro-measured amount of TdT buffer is added into the cDNA soltuion; The cDNA gets incubated again in the PCR Amplication machine at 37 degrees of Celcius for an hour. Incubated cDNA sample gets heat up to 95 degrees of Celcius for 5 minutes. Later, TdT Buffer is added and incubation is repeated to get final sequence of cDNA and measure the concentraion of tailed cDNA in the sample. [9]
Reference
[edit | edit source]1. http://www.ncbi.nlm.nih.gov/pubmed/22144202
2. http://en.wikipedia.org/wiki/Electrophoresis
Nematodes
[edit | edit source]Nematodes are one of the most diverse phylums of all animals. Half of the 28,000 different species of nematodes are parasitic. Nematodes, a type of roundworm, have tubular digestive systems with openings at both ends. Found in nearly all parts of the world, nematodes have adapted to almost all ecosystems, characterized by varying levels of water salinity, temperature, and altitude. Not only have they adapted, nematodes have evolved to outnumber other animals that coexist in the same ecosystems. For example, nematodes constitute 90% of all life on the seafloor.
Although some nematodes are completely dependent on other types of animals for reproduction, some of the strategies used by nematodes seem rather advanced. For example, the parasitic tetradonematid nematode is hypothesized to induce fruit mimicry in tropical ants. Infected ants develop bright red gasters, tend to be more sluggish, and walk with their gasters in a conspicuous elevated position. These changes likely cause frugivorous birds to confuse the infected ants for berries and eat them. Parasite eggs passed in the bird's feces are subsequently collected by foraging Cephalotes atratus and are fed to their larvae, thus completing the tetradonematid life cycle.
The genomes of nematodes are distinct from other metazoans
[edit | edit source]RNA regulation is an important and pervasive process, made possible by both RNA molecules and RNA-binding proteins. RNA molecules function as regulators and targets in diverse pathways pertinent to the proper decoding of the genome. RNA-binding proteins act as effectors of RNA stability and translation efficiency, guide transcripts to defined locations within the cell, control the fidelity of gene decoding, and function as cofactors to promote the activity of functional and structural RNA molecules.

Caenorhabditis elegans is a model organism
[edit | edit source]The facile genetics of the nematode Caenorhabditis elegans make it a useful observational model organism for the study of RNA regulatory mechanisms. The function of specific genes in this organism can be disrupted in a relatively straightforward manner by RNA interference (RNAi). The use of RNAi allows researchers to determine the function of specific genes, by silencing their functions in certain ways. In another important application, it was discovered that this organism showed behavioral responses to nicotine, including acute response, tolerance, withdrawal, and sensitization.
Nematodes contain an expanded genome
[edit | edit source]A surprising discovery based on the laboratory study of C. elegans is that the genome of nematodes contains an expansion of putative RNA-binding proteins relative to other metazoans. The RNA-binding protein Pumilio has 11 homologs in, while it has only two homologs in humans. The CCCH-type tandem zinc finger (TZF) family, which includes the mammalian protein tristetraprolin (TTP) has 16 homologs in roundworms. Lastly, there exists 27 Argonaute homologs in nematodes.
A homologous trait is any characteristic of organisms that is derived from a common ancestor. Paralogs are homologs present in the same species, and usually differ in function. Paralogs arise from gene duplication. Orthologs are homologs present in different species, and usually are similar in function. Orthologs usually arise from speciation when one species diverges into two separate species. Homology among proteins, DNA, and RNA is often concluded on the basis of sequence similarity. It is more effective to compare amino acid sequences than nucleotide base sequences because there are 20 distinct amino acids and only 4 distinct nucleotide bases.
Forward and reverse genetic experiments have provided data, which highlight the basis for the expansion of nematode homologs. Specifically, the data indicate that the RNA-binding family expansions may play roles in germline development, gametogenesis, and early embryogenesis.
The PUF family of RNA-binding proteins
[edit | edit source]The founding members of the PUF family of RNA-binding proteins are Pumilio and FBF, which together maintain the population of progenitor cells in the distal region of the germline. This group promotes the cellular switch from spermatogenesis to oogenesis at the onset of adulthood. During the transition from mitosis to meiosis when the single-celled state becomes a syncytial region, the meiotic nuclei recellularize. Spermatocytes are formed first during the L4 larval stage and stored in the spermatheca, which are then converted to oocytes.
PUF-8 and PUF-9 are biochemically similar to enzymes, in that one of their RNA-binding properties includes a high level of specificity. For instance, the eight nucleotide (5’-UGURNNAUA-3’) that is recognized by the PUF domains differs by a single nucleotide from the nucleotide NRE (Nanos Response Element). NRE is only a single nucleotide shorter than FBE, yet the FBR is discriminated by the two PUF elements more than 30-fold.
The nematode TZF binding specificity is different than the nematode PUF binding specificity. MEX-5 is a TZF protein that binds with a high affinity but relaxed specificity to any uridine rich sequence. The relaxed specificity means that TZF binds to both uridine rich sequences and polyuridine, while TTP binds more that 80-fold more tightly to AREs than polyuridine.
Conclusion
[edit | edit source]The function of RNA-binding proteins, like all proteins, is dictated by their structure. Novel function of RNA-binding protein families, with a common domain characterized by a new binding specificity, is a based on structural changes. Biochemistry and genetics serves as a basis for the identification of critical sequence elements and structural changes, yet fails to provide a mechanistic explanation of how these elements directly relate to novel function. Evidently, further research is necessary in the area of structural studies in an RNA-binding family.
References
[edit | edit source]1 R. Lehmann and C. Nusslein-Volhard, The maternal gene nanos has a central role in posterior pattern formation of the Drosophila embryo, Development 112 (1991), pp. 679–691. View Record in Scopus | Cited By in Scopus (140)
2 B. Zhang, M. Gallegos, A. Puoti, E. Durkin, S. Fields, J. Kimble and M.P. Wickens, A conserved RNA-binding protein that regulates sexual fates in the C. elegans hermaphrodite germ line, Nature 390 (1997), pp. 477–484. View Record in Scopus | Cited By in Scopus (245)
3 B.C Varnum, Q.F. Ma, T.H. Chi, B. Fletcher and H.R. Herschman, The TIS11 primary response gene is a member of a gene family that encodes proteins with a highly conserved sequence containing an unusual Cys-His repeat, Mol Cell Biol 11 (1991), pp. 1754–1758. View Record in Scopus | Cited By in Scopus (85)
Basis of RNA
[edit | edit source]- -RNA bases: A,G,C,U
- -Base Pairs: A-U, G-C
- -non-canonical pairs: G-U
- -Stability: G-C > A-U > G-U
- -Single Stranded: strand folds upon it to form base pairs; can have a diverse form of secondary structure
Secondary Structure
[edit | edit source]Structure Rules
[edit | edit source]- Base pairing stabilize the structure
- Unpaired sections-loops destabilize the structure
- When a base in one position changes, the base it pairs to must also change to maintain the same structure-covariation
Representations
[edit | edit source]- -Most base pairs are non-crossing base pairs: -any two pairs (i, j) and (i’, j’) -> i < i’ <j’ < j or i’ < i < j < j’
Circular Representation
[edit | edit source]- -Base pairs of a secondary structure represented by a circle
- -Arc drawn for each base pairing in the structure
Combinatorics
[edit | edit source]- -The number of RNA secondary structures for the sequence: (Recurrence Relation)
- S(0)=S(1)=S(2)=1
- S(n+1)=S(n)+ƩS(j-1)S(n-j), (n≥2)
- -There are approximately 1.3 billion RNA structures of length n is 27
Types of regions
[edit | edit source]- 1) Hairpin Loop - 4 or more bases long for each loop
- 2) Bulge Loop - bases on one side cannot form base pairs
- 3) Interior Loop - bases on both sides cannot form base pairs
- 4) Multi Loop (Junctions) – 2 or more double stranded regions converge to form a closed structure
Structure Prediction Methods
[edit | edit source]- 1) Maximize Base Pairs
- -Determine set of maximal base pairs
- -Align bases based on ability to pair up to determine the optical structure
- -Nussinov Algorithm: 4 ways to get the optimal structure between i and j
- -Find strucuter with the most base pairs: A-U and G-C
- 2) Minimize Energy
- -Determine maximum of scores for 4 structures at a particular position
- -Stacks are the dominant stabilizing force
- -Energy minimization algorithm predicts the secondary structure by minimizing the free energy
- -Require estimation of energy terms contributing to secondary structure
- -Dynamic programming approach:
- 1) Initialization
- 2) Recursion
- 3) Traceback
- -Does not require prior sequence alighnment
- -Energy associated with any position is only influenced by local sequence and structure
References
[edit | edit source]- How Do RNA Folding Algorithms Work? S.R. Eddy. Nature Biotechnology, 22:1457-1458, 2004.
- <http://en.wikipedia.org/wiki/Biomolecular_structure>
Making RISC’s
[edit | edit source]MicroRNA's, Piwii-interacting RNA's, and small interfering RNA's are unique in the world of RNA catalysts because they cannot perform any designated functions on their own. In order for small RNA's to function, they must first make RISC's. RISC stands for RNA-induced Silencing Complex. RISC's play an important role in regulating a multitude of biological processes by interfering with gene expression. Analyzing the assembly of these effector complexes can help us gain a better understanding of how small RNA's such as siRNA silence specific sequences. The assembly of RISC's has puzzled the scientific community because the final product contains single stranded RNA, while its precursors contain double-stranded RNA.[10]
Argonaute
[edit | edit source]The central protein of an RISC is the Argonaute, abbreviated Ago. The term Argonaute encompasses a family of proteins that act as catalyst in RISC's. The specific function that the small non-coding RNA will perform is determined, in part, by which Ago protein it is associated with. There are two primary classes of Ago proteins. One class binds to miRNA's and siRNA's while the other primarily binds to PiRNA's. Argonaute proteins share the ability to prevent translation. However, they differ in how they interfere with the production of polypeptides. For example, in humans, the AGO2 protein uses a cleaver to create RNAi. Whereas in flies, the AGO1 protein works with miRNA to regulate gene expression.[11]
The Assembly of RISC’s
[edit | edit source]Although Ago proteins are central to the formation of RISC's, the mere binding of a small noncoding RNA to its complementary protein will not result in a complete RISC. Research has shown that RISC assembly is the result of a highly regulated mechanism. This mechanistic pathway results in the processing of small RNA until the desired RISC is produced. The assembly of an RISC can be broken down into two primary steps, loading and unwinding. In the first step, the noncoding RNA is “loaded” onto its corresponding Ago protein. In the second step, the double stranded small RNA is separated inside of the Ago protein. This is the “unwinding” step which results in a single stranded RISC molecule.[12]
ATP Powers RISC Assembly
[edit | edit source]Researchers have found that ATP is needed to load miRNA onto the Ago protein but it is not required to unwind the complex within the protein. These results have been confirmed for both drosophila and humans. Upon closer examination of Ago protein complexes, it was found that these proteins lack any domains that could be used to bind ATP. Scientists hypothesize that the ATP is consumed by machinery in the process of non-coding RNA loading.[13]
References
[edit | edit source]- ↑ Kapranov, Philip (2012). Profiling of short RNAs using Helicos single-molecule sequencing. Cambridge, MA: Helicos Biosciences & Methods Mol Biol.
{{cite book}}: Unknown parameter|coauthors=ignored (|author=suggested) (help) - ↑ Kapranov, Philip (2012). Profiling of short RNAs using Helicos single-molecule sequencing. Cambridge, MA: Helicos Biosciences & Methods Mol Biol.
{{cite book}}: Unknown parameter|coauthors=ignored (|author=suggested) (help) - ↑ Kapranov, Philip (2012). Profiling of short RNAs using Helicos single-molecule sequencing. Cambridge, MA: Helicos Biosciences & Methods Mol Biol.
{{cite book}}: Unknown parameter|coauthors=ignored (|author=suggested) (help) - ↑ Kapranov, Philip (2012). Profiling of short RNAs using Helicos single-molecule sequencing. Cambridge, MA: Helicos Biosciences & Methods Mol Biol.
{{cite book}}: Unknown parameter|coauthors=ignored (|author=suggested) (help) - ↑ Kapranov, Philip (2012). Profiling of short RNAs using Helicos single-molecule sequencing. Cambridge, MA: Helicos Biosciences & Methods Mol Biol.
{{cite book}}: Unknown parameter|coauthors=ignored (|author=suggested) (help) - ↑ Kapranov, Philip (2012). Profiling of short RNAs using Helicos single-molecule sequencing. Cambridge, MA: Helicos Biosciences & Methods Mol Biol.
{{cite book}}: Unknown parameter|coauthors=ignored (|author=suggested) (help) - ↑ Kapranov, Philip (2012). Profiling of short RNAs using Helicos single-molecule sequencing. Cambridge, MA: Helicos Biosciences & Methods Mol Biol.
{{cite book}}: Unknown parameter|coauthors=ignored (|author=suggested) (help) - ↑ Kapranov, Philip (2012). Profiling of short RNAs using Helicos single-molecule sequencing. Cambridge, MA: Helicos Biosciences & Methods Mol Biol.
{{cite book}}: Unknown parameter|coauthors=ignored (|author=suggested) (help) - ↑ Kapranov, Philip (2012). Profiling of short RNAs using Helicos single-molecule sequencing. Cambridge, MA: Helicos Biosciences & Methods Mol Biol.
{{cite book}}: Unknown parameter|coauthors=ignored (|author=suggested) (help) - ↑ Kawamata, Tomoko, and Yukihide Tomari. "Making RISC." Trends in Biochemical Sciences 35.7 (2010): 368
- ↑ Kawamata, Tomoko, and Yukihide Tomari. "Making RISC." Trends in Biochemical Sciences 35.7 (2010): 368
- ↑ Kawamata, Tomoko, and Yukihide Tomari. "Making RISC." Trends in Biochemical Sciences 35.7 (2010): 368-369
- ↑ Kawamata, Tomoko, and Yukihide Tomari. "Making RISC." Trends in Biochemical Sciences 35.7 (2010): 373-374
Introduction
[edit | edit source]Retroviruses are created by Gag, a single virus-encoded protein. Assembling an infectious particle means there are very diverse interactions (specific and nonspecific) between Gag proteins and RNA. These interactions are vital for the particle construction, packing of viral RNA in the particle, and how the primer is placed for viral DNA synthesis.
Gag proteins: the building blocks of retroviruses
[edit | edit source]Retroviruses are composed of 6 genera (alpha-, beta-, gamma-, delta-, epsilon-, and lenti-retrovirsuses) and also a subfamily called Spumaviridae. Th Gag protein is the building block for retrovirus particles; expression of this protein allows for efficient assembly of virus-like particles in most, if not all, mammalian cells. When viruses are being assembled, Gag proteins interact with lipids in the plasma membrane with RNAs and other Gag molecules. These interactions involve the formation of the particle, selecion of what RNA species will be included in the particle, and the refolding of the packages RNAs.
A nonspecific interaction with RNAs drives virus particle assembly
[edit | edit source]Purified, recombinant Gag proteins are soluble in aqueous solutions so they aren't interested for protein-protein interactions that needs to occur for virus particle formation. However, if a single-stranded nucleic acid is added, it triggers in vitro assembly on the Gag proteins into VLPs.
In vivo, particle assembly is dependent on Gag-nucleic acids (NA) interactions because NAs are about 20-40 nucleotides, so they can support assembly in vitro. The NAs are so short, though, that they can only bind to a few Gag molecules.
The NC domain of Gag is where the site of interaction is at with NAs.When the NC domain binds with NA, the conformation is altered of the capsid domain. This ultimately exposes new interfaces for protein-protein interaction.
In many retroviruses, the NC domain of Gag is most attracted to RNA.However, the matrix domain at the N-terminus is positively charged and is also able to interact with RNAs.
Reference
[edit | edit source]Rein, Alan, et al. "Diverse interactions of retroviral Gag proteins with RNAs" Trends in Biochemical Sciences 36.7 (2011) 373-379. Academic Search Complete. Web. 21 Nov. 2012.
Different techniques are commonly used to determine or alter DNA sequences. The most important ones include:
- DNA sequencing: It provides us a lot of information about gene architecture, the control of gene expression, and protein structure.
- solid state synthesis of nucleic acids: The desired sequences of nucleic acids can be synthesized precisely de novo and used to identify or amplify other nucleic acids.
- restriction enzyme analysis: These Restriction Endonucleases provides an investigator to manipulate DNA segments by cutting them from the recognized sites.
- polymerase chain reaction (PCR) : This is one of the most useful methods to amplify DNA sequence billion fold. This technique requires denaturing of DNA and hybridizing primers complementary to strand forward and reverse and the DNA is synthesized on both strands in the 5' to 3' direction. This technique is used to recognize pathogens and genetic diseases, to determine the source of a hair left at the scene of a crime, and to resurrect genes from fossils.
- Blotting techniques: Southern and Northern Blots are used to separate DNA and RNA, respectively.1
References
[edit | edit source]1. Berg, Jeremy M. 2007. Biochemistry. Sixth Ed. New York: W.H. Freeman. 68-69, 78. 2. Voet, Voet, Pratt (2004). - Fundamentals of Biochemistry In the article, "The interface between transcription and mechanisms maintaining genome integrity," the main interfaces between transcription and other process related to DNA and the relevance to genome integrity in eukaryotes is thoroughly explored. Researchers at Cancer Research UK London Research Institute have studied the process of transcription, translation, and RNA mutagenesis and its effect on maintaining genome integrity. Specifically, eukaryotic RNA polymerase II transcription affects processes like chromatin remodeling and DNA repair. Loss of genome integrity causes changes in gene expression. For example loss in a chromosome region causes a mutation and in turn alters the protein that is created based on the genetic code. The research group has also discovered that the movement of RNA polymerases through the chromatin directly and indirectly affects the integrity of the genomic region that is transcribed.
Obstacles to transcription The factors that affect the maintenance of genome integrity during the transcription process are nucleosomes and DNA damager. RNA polymerase and its co-factors attempt to temporarily move the obstructing nucleosomes away from the transcribing region of the DNA. Second, DNA damage like a DNA double-strand break will not allow the RNA polymerase to continue transcribing since the bulky DNA lesion acts as a large obstacle. Thankfully, simple base damage is not a permanent obstacle as there are developed pathways that attempt to fix the lesion.
The pathway that fix the damage-stalling DNA lesion is called TC-NER, or transcription coupled nucleotide excision machinery. The TC-NER pathway attempts to remove areas of DNA damage in the transcribed region that is first encountered by the RNA polymerase II. If there are lesions further down the transcribed region that the RNA polymerase has not yet reached, another pathway called the GG-NER, or general genome nucleotide excision machinery is used. The TC-NER is dependent on proteins called Cockayne Syndrome (CS) A and CSB, that act as cofactors in this pathway. However, the exact mechanism by which the CS protein help catalyze the removal of DNA lesions through TC-NER is still to be determined.
A third pathway (in addition to TC-NER and GG-NER) used to fix the damage-stalling is RNA Polymerase II polyubiquitylation. The polyubiquitylation process has 3 types. The RNA Polymerase II K63 pathway is independent and does not lead to the degradation of the RNA polymerase. However, in the more direct pathway RNA polymerase II is subject to mono-ubiquitylation and the K46 poly-ubiquitylation. After these two steps, the polymerase is degraded. This degradation of the polymerase itself is typically used as the last result to fixing the damage-stalling DNA with the bulky lesion. Once the polymerase is degraded, the DNA lesion is attempted to be removed through a second trial of TC-NER and GG-NER when another RNA polymerase II encounters the lesion, or by DNA recombination.
Another phenomenon that affects genome integrity occurs during the process of DNA replication. During DNA replication, a replication fork is formed where there is a leading and lagging strand that make up the two sides of the fork. At the center of the fork, DNA polymerase slides along and replicates the DNA template. Pol ε is responsible for the leading strand synthesis and the Pol δ is responsible for lagging strand synthesis. In addition to the polymerases, there are primases, helicases and supplemental enzymes all present at this replication fork. The collision of these individual machineries can evidently cause severe consequences.
The realization that these clashes can occur at the replication fork provide further evidence of the phenomenon of transcription-associated mutagenesis or TAM. Highly transcribed region in the genome tend to have a lower percentage of packaging aids like nucleosome present. This leads to a more open structure and 'single strandedness'. The loss of the chromatin proteins and the resulting structural packaging damage of the DNA strands leave the DNA more susceptible to damage and loss of genome integrity. Overall, the spontaneous mutation rate in a eukaryotic gene is proportional to the transcription level. Interestingly, this means that the movement of RNA polymerase II and the repeating transcription processes of the same segment on the DNA strand of interest leading a higher probability of mutagenesis. An example of such mutation has been determined by the research group. In yeast DNA, there is accumulation of dUTP instead of dTTP during DNA replication as a result of highly transcribing a particular strand. This example leads to the conclusion that replication fork breakdown does occur if there is clashing when transcription is in process as well.
RNA mutagenesis does not contribute significantly to the loss of genome integrity like DNA mutagenesis does. This is because the mRNA that is used to eventually produce proteins are short-lived, especially in comparison to tRNA and mrRNA. Due to the short life of mRNA, it is doubtful of mutant protein is caused by RNA mutagenesis.
In conclusion, during transcription and replication of DNA, a loss of genome integrity may occur as a result to clashes and lesions of the proteins and polymerases in the normal process. The cell uses pathways like TC-NER,GG-NER, and RNA PII ubiquitylation to attempt to remove the lesions. Better computational models for studying these pathways may help understand genome instability thoroughly in the future.
Reference 1. Svejstrup, Jesper Q. The interface between transcription and mechanisms maintaining genome integrity. Clare Hall Laboratories, Cancer Research UK London Research Institute, Blanche Lane, South Mimms, EN6 3LD, UK. Trends in Biochemical Sciences Vol. 35 No. 6
2. Mefford, H.C and Eichler, E.E. (2009). Duplication hotspots, rare genomic disorders, and common disease. Curr, Opin. Genet. Dev. 19, 196-204.
History
[edit | edit source]Restriction enzymes were first discovered by Werner Arber and Hamilton Smith. Daniel Nathans pioneered their use which led to recombinant DNA technology.
Restriction Endonucleases
[edit | edit source]Restriction endonucleases, also known as restriction enzymes, are responsible for the phenomenon in bacteria known as host-controlled restriction modification or phenotypic modification. Restriction/Modification enzyme systems are divided into three categories: Type I, Type II, and Type III. The key distinctions between these systems are that Type II enzymes contain separate restriction and methylation systems, while Type I and Type III enzymes carry both restriction and methylation properties in one enzyme, consisting of two or three heterologous subunits. Typical commercial restriction enzymes used in molecular biology are produced by Type II systems. Type II restriction endonucleases recognize specific palindromic sequences (a sequence that reads the same on both strands except one strand is reversed). The restriction enzyme recognizes a particular sequence of base pairs (about 4-8 bp long) with an axis of rotational symmetry. Once this site of recognition is established, it cleaves the phosphodiester bond in each strand of the double helical DNA. The number and size of the fragments produced depends on the frequency of occurrence of the recognition site in the DNA to be cut. The restriction enzyme cuts the DNA into smaller fragments so that they can be analyzed and manipulated easier. Restriction endonucleases can help with the analysis of chromosome structure, sequencing long DNA molecules, isolating genes, and creating new DNA molecules to be cloned.
Cleavage by a restriction enzyme can generate a number of various ends. Often, these ends have 3'-hydroxyl and 5'-phosphate ends. Some cleavages produce single-stranded overhangs, called cohesive ends or sticky ends, while others generate blunt ends. These ends or cleaved sites can be subsequently annealed and ligated to vector DNA or any kind of DNA having compatible ends. Not all cuts may necessarily be symmetrical as BamHI for example, cuts from the ends of DNA sequence in a non-symmetrical fashion.
It is also possible to visualize restriction fragments by gel electrophoresis. There are three different methods that can be used. The first method is a polyacrylamide gel which can separate fragments up to 1000 base pairs. The next one is an agarose gel which can separate up to 20kb. And finally, the Pulsed-Field Gel Electrophoresis (PFGE) which can separate up to millions of nucleotides based on the stretching and relaxation of DNA as the electric field is turned on and off. Autoradiography or staining by ethidium bromide can then be used to visualize the DNA. The gel electrophoresis is run through an electric field which isolates fragments by size, noting that smaller fragments travel farther within the gel and the larger fragments are closer to the start. Compared to a known size standard, the location of where the restriction enzymes cut are known.
Example
Suppose the following segment of DNA is recognized by the restriction enzyme. The red (*) symbolized the axis of symmetry. One major component of these cleavage sites is the presence of twofold rotational symmetry about this axis. The cleavage site is also highlighted on the diagram. Once this site recognition is established, the phosphodiester bond between the highlighted C-G and G-C will be cleaved by the restriction enzyme on the corresponding strands of the double helix. Note that different restriction enzymes have different cleavage sites. Therefore cleavage of phosphodiester bond will not always be between C-G or G-C.
- 5' C-C-G-C-G-G 3'
- *
- 3' G-G-C-G-C-C 5'
- 5' C-C-G-C-G-G 3'


BamHI as an example
[edit | edit source]BamHI is an example of a restriction enzyme. It cleaves palindromic sequences, 6 bases at a time. For example:
File:BamHI palendromic sequence.jpg
This image shows where the BamHI would cleave the palendromic sequence.
BamHI binds to non-specific DNA and slides down the DNA strands with a dimer enzyme that quickly reads the DNA to find palindromic sequences of 6 bases. If it finds 6 bases that are not palindromic, it will still cut the bases, but will do so poorly. BamHI works best when it finds a palindromic sequence to cut.
File:Catalytic mechanism.jpg This image shows the mechanism that BamHI uses to cleave the phosphodiester bond. In the first step, when reacting with water, the substrate obtains a hydroxide group and gives the phosphate two negative charges. A metal is usually in the transition state because the metal is a positive charge that balances out the two negative charges, and makes the transition state more stable. Magnesium is a good metal to use. Calcium is also sometimes used, but it does not work as well because it hinders the cleaving process. Water is then added to the transition state, which results in the donation of a proton to the leaving group, finally breaking the bond and cleaving the DNA.
Everything in the pre-reactive and post-reactive states, excepting the fact that the phosphodiester bond is now broken, cleaving the DNA, basically remains the same as shown in the following picture.


Restriction Enzyme Control
[edit | edit source]Restriction endonucleases exhibit their high specificity due to two major characteristics. One being that they must not degrade the host DNA that contains the sequence recognized by the restriction enzymes. Second, they must only cleave sites that are specifically recognized. The restriction endonucleases must be able to tell the difference between one specific cleaving sequence and a different sequence. Restriction endonucleases are able to exhibit these properties through the process of methylation. Methylate enzymes present in the organism protect the organisms DNA containing the palindromic sequences from being cleaved by the restrictio endonucleases. Once the methylate enzymes methylate the adenines of the organisms personal bases, the restriction enzymes will not cut at these sites. Every restriction endonuclease results in a specific methylate enzyme in the host cell that will methylate the specific sequence sites in the hosts DNA. This system of self-methylation and restriction enzyme action is known as restriction-modification system. The restriction endonucleases produced by the host organism can only cleave sequences that are not marked with the methylated adenines allowing for restriction enzyme control.
Cleavage Mechanism
As stated above, restriction endonucleases cleave the bond between the oxygen 3' and phosphorous atoms. Restriction endonucleases catalyze the hydrolysis of these phosphodiester bonds in DNA. The mechanism of this reaction is based upon nucleophillic attack of the phosphorous creating a pentacoordinate transition state. This results in a bipyramidal structure.
Type I Type I restriction enzymes were the first to be identified and are characteristic of two different strains of E. coli. The recognition site is asymmetrical and is composed of two portions: one containing 3-4 nucleotides, and another containing 4-5 nucleotides which are separated by a spacer. Several enzyme cofactors, are required for their activity. Type I restriction enzymes possess three subunits called HsdR, HsdM, and HsdS; HsdR is required for restriction, HsdM is necessary for adding methyl groups to host DNA (methyltransferase activity) and HsdS is important for specificity of cut site recognition in addition to its methyltransferase activity.
Type II Typical type II restriction enzymes differ from type I restriction enzymes in several ways. They are composed of only one subunit, their recognition sites are usually undivided and palindromic and 4-8 nucleotides in length, they recognize and cleave DNA at the same site, and they do not use cofactors for their activity (except Mg2+). These are the most commonly available and used restriction enzymes.
Type III Type III restriction enzymes recognize two separate non-palindromic sequences that are inversely oriented. They cut DNA about 20-30 base pairs after the recognition site.
Analyzing Restriction Digests
[edit | edit source]
After a restriction enzyme digest of plasmid DNA was done, the DNA fragments were analysis on an argyrose gel. By examining the pattern of bands obtained on the gel, the size of DNA and vector can be determined. This can be use to confirm if the correct plasmid was isolated or not.
From the Figure above conclusion like following can be made:
Lane 3 and 6 were digested by enzyme twice. Two bands represent two pieces of linear DNA. Bottom band is the size of the gene inserted between the two enzyme sites in the multiple cloning sites and the other is the size of the rest of the plasmid.
Lane 4 and 7 were digested by enzyme once. The band moves slower than other linear DNA indicates that DNA is nicked. This DNA uncoils, but remains circular and usually migrates more slowly than linear DNA of the same size.
Lane 5 and 8 were undigested. Super coiled DNA migrates more rapidly than linear DNA of the same size.

Southern blotting, which is named after its inventor, Edwin Southern, is a common technique used in molecular biology to separate and characterize DNA. It is an effective way to identify a specific DNA pattern by the following procedures. Southern blotting is a technique used to determine the presence of a specific DNA sequence within a mixture via agarose gel electrophoresis.
Southern blotting is a hybridization technique that enables researchers to determine the presence of certain nucleotide sequences in a sample of DNA.
Southern Blotting was the first technique of its kind. Soon after however, additional analogous techniques known as Western Blotting and Northern Blotting (among others), were cleverly named eponymously; to follow closely follow the convention behind the naming of Southern Blotting. These techniques don’t detect the presence of DNA, but rather the presence of Proteins and RNA, respectively.
Protocols
[edit | edit source]DNA is digested with one or more restriction enzymes, and the resulting fragments are separated according to size by electrophoresis through an agarose or acrylamide gel. The DNA is denatured and transferred from the gel to a solid support. The reason for transferring the DNA fragments to a solid support (usually a nitrocellulose plate) is that the DNA is inaccessible to DNA probes while embedded in the gel. The relative positions of the DNA fragments are preserved during their transfer to the filter. The DNA fragments attached to the filter are then exposed to a strand of radioactively-labeled DNA that is complimentary to the DNA strand on the plate that is of interest. Autoradiography is then used to locate the positions of bands complementary to the probe.
Method
[edit | edit source]
1. The DNA sample, if necessary, is digested with an appropriate restriction enzymes, or restriction endonucleases. The digest is then separated by gel electrophoresis, usually on an agarose gel. If a large amount of restriction fragments are present in the sample, the sample may likely appear as faint smears rather than discrete bands.
- Gel Electrophoresis:
By the 1970 the DNA fragments was isolated by using gravity. After that time, the scientists found other way to separate DNA fragment. That is Gel Electrophoresis. Electrophoresis uses electricity to separate different sized molecules.
2. The digest is then denatured to allow for transfer onto a membrane. Since the sample DNA is double-stranded and only single-stranded DNA can be transferred, the sample must be denatured by soaking in an appropriate alkaline solution (e.g. 0.5M NaOH). If the sample is still too large to be transferred (more than 15kb), the sample may be treated with an appropriate acid to depurinate (e.g. remove the purines) the sample and break it down into smaller fragments. The sample is then neutralized before continuing with the procedure.
3. The sample is transferred onto a membrane which is a sheet of special blotting paper for analysis. The transfer to another membrane is performed in order to preserve the position of the DNA fragments once electrophoresis has been performed. A Nitrocellulose membrane is generally used, though some may prefer the use of nylon for a better binding capacity. It may also be noted that nylon is less fragile than nitrocellulose. The membrane used is laid on top of the gel, and usually paper towels are placed on top of the membrane to ensure an even distribution by applying pressure evenly. The transfer is done usually be capillary action, which may take several hours. Alternatively, a vacuum apparatus can be used; this is very similar to capillary action, though transferring via vacuum apparatus may be faster. The transfer can also occur by moving the DNA out of the gel and onto the membrane by electrophoresis, a process called electrotransfer.
4. The sample is then treated with UV light to irreversibly cross-link the sample to the membrane covalently. Alternatively, the sample may be baked at around 80°C for several hours (this should only be done if using nylon membrane since nitrocellulose is highly combustible).
5. The membrane is then probed with labeled, single-stranded DNA (this is the target DNA sequence). This process is also known as hybridization. The labeled DNA binds to the membrane DNA via the binding of complementary strands. The label is generally a 32P probe label, though a bioluminescent probe or biotin/streptavidin may also be used. The reason that hybridization is important is that it allows you to physically see it. It should be noted that prior to hybridization, a prehybridization process should be run to restrict the labeled DNA binding to specific sites, because otherwise the labeled DNA may bind to unwanted or unimportant sites within the sample. To do so, salmon sperm DNA treated with varying concentrations of SSC, SDS, and formamide is used.
- 32P labeled:
Treat the dsDNA fragment that you are using as a probe with a limiting amount of Dnase, which causes double-stranded nicks in DNA. Add 32P, dATP, and other dNTPs to DNA polymerase I, which has 5' to 3' polymerase activity and 5' to 3' exonuclease activity.
6. The results are analyzed. If a 32P labeled probe was used, the sample will be analyzed with an autoradiograph, or the use of an X-ray film to reveal whether or not the sample contained the target DNA sequence. If the target sequence is indeed present, the X-ray film will show blackened bands caused by beta emissions from the 32P radiolabeled probe. Sequences in the sample that are very similar to the target sequence will also be shown. If a bio-luminescent probe was used, the luminescence will be a method of visualization. If biotin/streptavidin was used, the sample is analyzed by colorimetric methods, or by looking at the development of color on the membrane.
Method Summary : First, a mixture of restriction fragments is separated by electrophoresis through an agarose gel, denatured to form a single stranded DNA, and then transferred to a nitrocellulose sheet. The positions of the DNA fragments in the gel are preserved on the nitrocellulose sheet because it is directly blotted onto the sheet from the agarose gel. Next, the fragments are exposed to a 32P-labeled single stranded DNA probe which hybridizes with a restriction fragment having a complementary sequence. After hybridization, an autoradiography reveals the position of the restriction-fragment-probe duplex and ultimately the identity of the DNA fragment.
Examples of Southern Blots
[edit | edit source] Southern Blot done by Nisha Patel, UCSD Pediatrics, Sander Lab
Photo uploaded by: Patrick Phuong, UCSD, Sander Lab
Southern Blot done by Nisha Patel, UCSD Pediatrics, Sander Lab
Photo uploaded by: Patrick Phuong, UCSD, Sander Lab
As mentioned above, Southern blots utilize DNA probes to detect the presence of complementary, target DNA sequences. In this case, the Southern blot was run using mouse DNA from mice which were breed in the mouse colony. Southern blotting is a way to track the genetic make up of each new litter of mice and the genotypes of old mice. In the picture, it can be seen that heterozygous, mutant, and wild-type mice are all distinguished by the types of bands that show up in their segment of the blot. Heterozygous mice have sequences for both the top and bottom bands whereas mutants only have the bottom band. Wild-type mice have only the top band of DNA detected by the probe. On the left side is a DNA standard, which is run to show the progress of different sizes of DNA. The labeling at the top shows the mouse line and female number for the mouse that gave birth to the litter whose DNA is being blotted.
Uses of Southern blot
[edit | edit source]By using this technique, we are able to detecting a single specific restriction fragment in the highly complex mixture of fragments produced by cleavage of the entire human genome with a restriction enzyme. In such a complex mixture, many fragments will have the same or nearly the same length and thus migrate together during electrophoresis.
The ability to determine whether or not a specific sequence of DNA is present in a DNA sample can be applied for several uses. For example, Southern blot can be used to determine the presence and amount of a certain gene within the genome of a certain organism, which also reveals the molecular weight of the specific fragment. Due to the procedures involved in the method, the presence of restriction sites for particular restriction endonucleases can also be determined. It can also be used to compare the degree of similarity between the probe sequence and the sample.
Southern Blotting can also be used to follow the inheritance of selected genes. Mutations within restriction sites change the sizes of restriction fragments and as a result, the positions of bands in the Southern-blotting analysis and autoradiography also change. This change in position can later be compared to normal blot-analyses in order to reveal where the possible change has occurred. The existence of genetic diversity created by these mutations in a population, is termed polymorphism. The detected mutation in turn may have different effects. It may cause disease or it may be closely linked to one that does. Some examples of such diseases include sickle-cell anemia, cystic fibrosis, and Huntington chorea. All of these mutations can be detected by comparing the restriction-fragment-length polymorphisms with normal fragments of DNA.
Common Misunderstandings/Differences
[edit | edit source]As previously mentioned, Southern Blotting gave rise to other analytical methods such as Western and Northern Blotting, corresponding to Protein and RNA detection, respectively. Often time, the names of the techniques are erroneously interchanged, or even more importantly, are assumed to use the same method components [e.g. instrumentation, chemicals, etc.]. It is important to recognize that although the methods are very similar, they differ in that each are technique for a specific macromolecule; and therefore are treated differently.
To give a brief example, consider the basic differences of a Southern Blot to a Northern Blot. While both use autoradiography in the final steps to visualize their fluorescent labels, their labels are actually different. A southern blot uses a 32P-label DNA probe to interact with a specific sequence of DNA, whereas Western Blot uses radiolabeled antibodies that will interact with the specific protein. Other examples include different types of sheets to which the macromolecules are transferred to after electrophoresis [Nitrocellulose For Southern; Polymer For Western], to even the type of gel used for the electrophoresis step itself [Agarose For Southern; SDS For Western]. To avoid confusion, Southern, Northern, and Western Blots can also be known respectively as: DNA, RNA, or Protein Blots.
References
[edit | edit source]Bartlett, Linda. "Technology: Recombinant DNA: Southern Blot." Photo. visualsonline.cancer.gov 1 Jan. 2001. 14 Oct. 2009. <http://visualsonline.cancer.gov/details.cfm?imageid=2016>
Berg, Jeremy M., Lubert Stryer, and John L. Tymoczko. Biochemistry. 6th ed. Boston: W. H. Freeman & Company, 2006.
Introduction
[edit | edit source]The Northern blot, also known as the RNA blot, is one of the blotting techniques used to transfer DNA and RNA onto a carrier for sorting and identification. The Northern blot is similar to the Southern blot except that RNA instead of DNA is the subject of analysis in this technique. First, the RNA from each tissue must be purified so that it can be examined for expression of the gene under investigation. The RNA samples are then loaded onto an agarose gel. The gel is then subjected to an electric current that causes the RNA in each sample to migrate toward the bottom of the gel. Smaller RNAs move faster while larger RNAs migrate more slowly. This separation process is known as electrophoresis. The separated RNA fragments are then blotted onto a special filter paper so that each RNA molecule retains its position relative to all the other molecules. The filter is then exposed to radioactive probes that hybridize to complementary sequences in the blot. The filter is then placed on a film for autoradiography and the film is developed. Lastly, a band should be observed on the autoradiograph if the probe has hybridized to a stretch of RNA on the filter. A band will only be visible in the column containing a tissue where the gene represented by the probe has been expressed.
The Northern blot is useful for the study of gene expression in two ways. First, the position of bands on the blot provides a direct measure of RNA size. Knowing the size of the RNA will provide an estimate for the transcript’s coding capacity and thus the size of the protein it encodes. Second, the Northern blot analysis of RNA samples from many different tissues enables researchers to determine which specific tissue a gene is expressed in along with the relative levels of its expression in all cells where transcription is occurring. The Northern blot is a valuable method used by researchers in determining gene expression patterns. For example, many scientists researching Huntington disease or breast cancer are able to determine the expression patterns of the genes responsible for these diseases using blotting techniques. The three blotting techniques including the Northern blot have proven to be important and positive advancements in science.
Northern Blot is a derivative of the Southern blot, which, like the Southern Blot, utilizes electrophoresis separation analysis and a hybridization probe for detection. For both techniques, the hybridization probe for detection can be made from either DNA or RNA. The main difference between the two techniques is that the northern Blot is performed to study gene expression by analyzing RNA instead of DNA. There are 3 types of RNA: tRNA (transfer RNA - active in assembly of polypeptide chains), rRNA (ribosomal RNA - part of the structure of ribosomes) and mRNA (messenger RNA - the product of DNA transcription and used for translation of a gene into a protein). It is mRNA which is isolated and hybridized in northern blots.The formaldehyde was use in electrophoresis gel as a denaturant because the sodium hydroxide treatment used in the Southern blot procedure would degrade the RNA. The northern Blot was developed at Stanford University in 1977 by James Alwine, David Kemp, and George Stark.
Process
[edit | edit source]The steps of Northern Blotting are as follows:

1) RNA isolation: mRNA is extracted from the cells and purified.
2) Probe generation: The mRNA is loaded onto a gel for electrophoresis. Lane 1 has size standards (a mix of known RNA fragments) Lane 2 has the RNA.
3) Denaturing agarose gel electrophoresis: An electric current is passed through the gel and the RNA moves away from the negative electrode. The distance moved depends on the size of the RNA fragment. Since genes are different sizes the size of the mRNAs varies also. This results in a smear on a gel. Standards are used to quantitate the size. The RNA can be visualized by staining first with a fluorescent dye and then lighting with UV.
4) Transfer to solid support and immobilization: RNA is single-stranded, so it can be transferred out of the gel and onto a membrane without any further treatment. The transfer can be done electrically or by capillary action with a high salt solution.
5) Prehybridization and hybridization with probe: A labelled probe specific for the RNA fragment in question is incubated with the blot. The blot is washed to remove non-specifically bount[check spelling] probe and then a development step allows visualization of the probe that is bound.
6) Washing: The probe is bound specifically to the target mRNA and that there is negligible non-specific binding to other mRNA or the nylon membrane itself.
7) Detection: Hybridization signals are then detected.
8) Stripping and reprobing (optional):
RNA Isolation
[edit | edit source]This part of the Northern Blot is an important step because high quality, intact RNA needs to be obtained. There are several ways the isolation can be performed; however, common attributes include cellular lysis and membrane disruption, inhibition of ribonuclease activity, deporoteinization, and recovery of the intact RNA.
Probe Generation
[edit | edit source]Although the Southern blot is named after Edwin Southern [1], it is counter-intuitive to learn that the Northern blot was developed by James Alwine, David Kemp, and George Stark [2]. The name 'Northern Blot' is merely a play on words. Southern, Northern, and Western blots are known respectively for their analysis of DNA, RNA, and proteins, respectively.
Western Blot and Others
[edit | edit source]Western blots allow investigators to determine the molecular weight of a protein and to measure relative amounts of the protein present in different samples.
Procedure:
a) Proteins are separated by gel electrophoresis, usually with SDS-PAGE to have all the proteins carries negative charge(s).
b) The proteins then transferred to a sheet of special blotting paper called nitrocellulose sheet although other types of paper or membranes can be used. The proteins retain in the same pattern of separation they had previous on the gel.
c) The blot is incubated with a generic protein (e.g. milk proteins) to bind to any remaining sticky places on the nitrocellulose. An antibody is then added to the solution which is able to bind to its specific protein. The antibody has an enzyme (e.g. alkaline phosphatase or horseradish peroxidase) or dye attached to it which cannot be seen at this time.
d) The location of the antibody is revealed by incubating it with a colorless substrate that the attached enzyme converts to a colored product, which can be seen and photographed.

Other similar techniques such as far-western blots and south-western blots allow scientists to examine protein-protein interactions and DNA-protein interactions respectively.
References
[edit | edit source]- ↑ Berg, Jeremy M. (2007). Biochemistry (6th Ed. ed.). W. H. Freeman and Company. ISBN 0-7167-8724-5.
{{cite book}}:|edition=has extra text (help) - ↑ Alwine JC, Kemp DJ, Stark GR (1977). "Method for detection of specific RNAs in agarose gels by transfer to diazobenzyloxymethyl-paper and hybridization with DNA probes". Proc. Natl. Acad. Sci. U.S.A. 74 (12): 5350–4. doi:10.1073/pnas.74.12.5350. PMID 414220.
{{cite journal}}: CS1 maint: multiple names: authors list (link)
3. Ambion. Applied Biosystems. http://www.ambion.com/techlib/basics/northerns/index.html DNA sequencing determines the order of Adenine, Guanine, Cytosine, and Thymine within a DNA molecule. Sequencing of DNA allows for the makeup of a wide variety of genetic information. It is one of the simplest techniques when it comes to DNA manipulation.
History of Genome Sequencing
[edit | edit source]About DNA sequencing: Restriction endonucleases led to recombinant DNA. Bacteria make restriction endonucleases, enzymes that cut DNA at positions determined by specific short base sequences. In nature, restriction endonucleases protect bacteria from the foreign DNA of viruses. In the test tube, purified restriction endonucleases were used to "cut and paste" DNA from two unrelated organisms, generating recombinant DNA. The construction of artificially recombinant DNA, by "gene cloning," ultimately made it possible to transfer genes between the genomes of virtually all types of organisms, using processes derived from natural phenomena of bacterial transformation.
The first successful method of sequencing is known as Sanger Sequencing after its creator, Frederick Sanger. The method was revolutionary and opened up untold avenues of research. However, it was and remains inefficient in terms of time, cost, and materials. Bacteriophage fX174, a virus with only 5,368 base pairs, was the first to be sequenced in 1978, with many more to follow. Then, in 1983, the PCR method was developed, which allows for amplification of specific DNA fragments. Later, in 1986, the Leroy E. Hood's laboratory at the CIT and Smith announced the first semi-automated DNA sequencing machine. Next, the shotgun method was created by Craig Venter in 199. This method allowed for much faster sequencing.
Another method of sequencing is fluorescent detection. In this method, a fluorescent tag is used to label each dideoxy analog a different color. An electrophoresis is then performed, and the separate bands of DNA come through. The bases are labeled different colors, making their identities obvious and their sequences very easy to determine.
Perhaps the largest proposal came in 1990, when the Human Genome Project was proposed and launched in an effort to catalog the entire human genome. Later, a team led and created by Craig Venter sequenced the largest bacteria with a total of 1.8 Mb at TIGR's facilities. Such a feat was possible due to the improvement in computational software that was created to work with the extremely large sets of data the Shotgun method created. Instead of shotgunning very small bits of the genome at a time, they did it all at once with the entire genome. Then the TIRG assembler (name of the software) identified the 24,000 base pairs into one whole genome. This new method of sequencing cut the cost and time required nearly in half. The open-access policy of the Human Genome Project allowed any academic facility to use all the date collected, and helped transform our working knowledge of the genome. However, Celera, Craig Venter's project, initially refused to give away their data until he was pressured into it. The group now remains one of the most influential leaders in the industry. Late in 2003, save for a few gaps, the entire human Genome was announced as complete, heralding the entrance of a new era of science.
Fundamental Method
[edit | edit source]Sequencing DNA can be elucidated by applying chemical or enzymatic methods. The fundamental enzymatic method is based on the ability of a DNA polymerase to extend a primer and hybridized to the single-stranded DNA to be sequenced until a chain-terminating dideoxyribonucleotide triphosphate (ddNTP) is incorporated. ddNTPs are missing a hydroxyl group on carbon 3' to which the next dNTP of the growing DNA chain is added. ddNTP is useful as a chain terminator because without the hydroxyl group on carbon 3', no more nucleotides can be added, and DNA polymerase falls off; the DNA chain is stopped at a labeled G, A, T, or C. The resulting fragments share a common origin, but are terminated at different nucleotides, resulting in DNA chains that are a mixture of lengths. Each fragment is labeled with each dideoxy analog (terminators) being tagged with a different fluorescent color. They are separated by using high resolution gel electrophoresis, which is a method which separates them based on their different sizes; the shorter strands move faster though the gel. Once the strands move through the gel, their identities can be detected by fluorescence measurements (depending on their colors). The following techniques described are used in many different methods and are considered general knowledge.

Fluorescence detection
[edit | edit source]An alternative to autoradiography is "fluorescence detection". Fluorescence proteins used as a tag are found in light and color producing cells of many coelenterates such as corals and jellyfish. After fluorescent tag is attached to each of the dideoxy analog, different chain terminator has different color. Fluorescence is the process by which light is absorbed by a molecule and re-emitted at a longer wavelength (sometime falls into the visible range hence can be seen by human eyes), producing particular color. When it is excited by light, the fluorescence will fluoresce without input of energy such as ATP or any other cofactor.Then through a mixture of terminators, reaction can be performed separately and the mixture of fragments can be separated by gel electrophoresis. The bands of DNA can be detected by their color as they emerge. For example, if all of the Adenine dideoxy analogs have been labeled with a green tag and a fragment 5 bases longer than the primer comes out green, it is known that the base that the fifth base after the primer is an adenine. This method allows us to find the sequence of a polynucleotide that have numbers of bases up to 500. This is a viable and competitive solution because the use of radioactive components is eliminated, and can be automated. Thus more than 1 million bases can be sequenced per day.
Fluorescence detectors are probably the most sensitive among the existing modern HPLC detectors. It is possible to detect even a presence of a single analyte molecule in the flow cell. Typically, fluorescence sensitivity is 10 -1000 times higher than that of the UV detector for strong UV absorbing materials. Fluorescence detectors are very specific and selective among the others optical detectors. This is normally used as an advantage in the measurement of specific fluorescent species in samples. When compounds having specific functional groups are excited by shorter wavelength energy and emit higher wavelength radiation which called fluorescence. Usually, the emission is measured at right angles to the excitation.
Roughly about 15% of all compounds have a natural fluorescence. The presence of conjugated pi-electrons especially in the aromatic components gives the most intense fluorescent activity. Also, aliphatic and alicyclic compounds with carbonyl groups and compounds with highly conjugated double bonds fluoresce, but usually to a lesser degree. Most unsubstituted aromatic hydrocarbons fluoresce with quantum yeld increasing with the number of rings, their degree of condensation and their structural rigidity. Fluorescence intensity depends on both the excitation and emission wavelength, allowing selectively detect some components while suppressing the emission of others. The detection of any component significantly depends on the chosen wavelength and if one component could be detected at 280 ex and 340 em., another could be missed. Most of the modern detectors allow fast switch of the excitation and emission wavelength, which offer the possibility to detect all component in the mixture. For example, in the very important polynuclear aromatic chromatogram, the excitation and emission wavelengths were 280 and 340 nm, respectively, for the first 6 components, and then changed to the respective values of 305 and 430 nm; the latter values represent the best compromise to allow sensitive detection of compounds.
Fluorescence Protein
[edit | edit source]Fluorescene Protein is what we use in Fluorescence detection in order to label DNA. In Fluorescene Protein, the fluorophore is typically a double ring structure formed by three amino acids. It is called a beta barrel because it is formed by 11 strands of beta-pleated sheet, with additional amino sequence closing the top and bottom. They can often attach to other protein without changing other proteins' structure or function. Hence, it is often use in lab to label and detect molecules, cells, and organisms. It is also being used for screening durgs, evaluating viral vectors for human gene therapy, monitoring genetically altered microbes in the environment and biological pest control.
Green Fluorescent Protein (GFP) has existed for more than one hundred and sixty million years in one species of jellyfish, Aequorea victoria. The protein is found in the photoorgans of Aequorea. GFP is not responsible for the glow often seen in pictures of jellyfish - that "fluorescence" is actually due to the reflection of the flash used to photograph the jellies.
Because of the unique β-barrel fold of fluorescent proteins, mutations of residues throughout the entire protein have the potential to significantly change their fluorescent properties. As is highlighted in the poster, the most striking result of such mutations is the wide range of different emission colors that is currently available, which greatly increases the usefulness of these proteins as molecular probes. However, most single mutations have a negative impact on the tight packing of the FP β-barrel and, therefore, result in greater environmental sensitivity and reduced brightness. Although some of these defects can be compensated for by additional mutations, derivative FPs are often less bright and/or more sensitive to the environment compared with the original protein. This phenomenon has been especially evident during the search for truly monomeric versions of the tetrameric red fluorescent protein of the coral Discosoma sp.
Sequencing Methods
[edit | edit source]An abundance of sequencing methods have been developed over time and a number of them are listed below with a brief description.
Pyrosequencing
[edit | edit source]Pyrosequencing was developed in 1996 by Pål Nyrén and Mostafa Ronaghi at the Royal Institute of Technology in Stockholm. The sequencing process is done by enzymatically synthesizing a complementary single strand of DNA. The bases A, T, G and C are sequentially added and removed during the process. As DNA polymerase adds to the single stranded chain, ATP is created by ATP sulfurylase which is used by Luciferase to produce light. The light intensity is then detected and recorded for that base. If a base was added and no light was produced, then the incorrect base was added to further the sequencing. The process is repeated several times until the entire strand of DNA has been fully synthesized and sequenced.
Sanger Sequencing
[edit | edit source]Produced in 1974, at its most basic, the Sanger sequencing method uses of chain terminators to extend a specific site of a DNA template with the use of a primer which is complementary to the template at that site. In this method autoradiography is used and DNA polymerase, the four DNA bases, the primer, and a chain terminating nucleotide are used resulting in DNA fragments of different sizes; their size depending on where that particular nucleotide was used. The fragments are separated depending on their size using polyacrylamide gel electrophorese, or PAGE. An easier method would be the fluorescence dye terminator sequencing where one must label the chain terminators with a fluorescent dye. Each chain terminator would be labeled with a separate colored dye. The PAGE is performed and all of the chain terminators can be identified after a single reaction as opposed to the four reactions needed in the labeled-primer method mention before. However, the Sanger method of DNA sequencing has issues because of the need for lots of time and labor due to the gel preparation, and because it requires lots of samples. Prior to sequencing, the DNA must be denatured into single strands, and a primer must be attached to the template strands, which are created with their 3' ends located next to the DNA fragments desired. They are labeled either through radioactivity (autoradiography) or fluorescence. Then the solution is divided into 4 containers, after which one of four reagents are added, ddAPT, ddTPT, ddCTP, DDGTP, along with DNA polymerase and all four dNTP's. The method works because all the reactions begin from the same nucleotide and end with the desired base. So the new chain will terminate at every position in which the nucleotide can add, and bands of different length are created. After this the DNA is again denatured and sent through electrophoresis, then the contents of the froup vessels are run on a polyacrylmide gel to separate the bands from each other.



Because large fragments are difficult to sequence, it is necessary to work with small fragments. Frederick Sanger designed a method that controlled replication termination. In this remarkably simple technique, a 2',3'-dideoxy analog is used to initiate chain termination. Each reaction tube contains A, G, T, and C dideoxy analogs along with regular radioactively labeled dNTPs. Each reaction set is run in one column of an electrophoresis gel after the controlled replication termination. Since replication terminates after the random incorporation of a dideoxy analog, the shortest fragments, which run the farthest, are the first group of bases in the sequence.
The Future of Analyzing DNA
[edit | edit source]Genome sequencing will greatly advance our understanding of genetic biology, and has vast potential for medical diagnosis and treatment. One obstacle is the cost of genome sequencing (The first human genome mapping was done in 2003 and is estimated to have cost 3 billion dollars). The device of a nanopore may reduce this cost to a couple hundred dollars, making personal genome sequencing to be available to everyone. The idea of threading DNA through a tiny pore (nanopore) was envisioned by David Deamer from the University of California, Santa Cruz in the mid 1990’s.
A nanopore is a minuscule hole that a molecule of DNA can be threaded through and read. Currently, the method for nanopore DNA analysis involves inserting proteins into a membrane of lipids. A DNA molecule can be dragged through the nanopore when an electrical voltage is applied. A major drawback of this method is that the lipid membrane is quite fragile.
Researchers from the Delft University of Technology and Oxford University propose a new method that combines artificial and biological materials to create a nanopore on a chip, which can analyze single DNA molecules. The method involves attaching an individual protein to a larger piece of DNA, then threading it through a premade opening on a silicon nitride membrane.
However, the silicon nitride material is a bit too thick, so more than one nucleotide may enter the pore at the same time. Researchers from Delft, Pennsylvania, and Harvard University are working with graphene (one atom thick sheets of carbon), and have drawn DNA through a nanopore drilled into graphene. Graphene could indeed be the future of genome sequencing since it is strong, thin, and a great electrical conductor.
True Single Molecule Sequencing
[edit | edit source]There is a method of directly sequencing DNA or RNA molecule in a fast and low cost manner which allows multiple sequencing of many single DNA strands. Labeled dNTPs (dATP, dGTP, dCTP, dTTP) tagged with a fluorescent indicator and DNA polymerase, are added to a flow cell which is the DNA template and begins complementing. Single molecule sequencers are available to correct errors during sequencing. Wash steps are applied to remove excess nucleotides. The remaining nucleotides are captured and analyzed.
Sequencing by Hybridization (SBH)
[edit | edit source]This refers to an entire class of sequencing methods. Normally used to find a small change in a known DNA sequence, it is sensitive to even single-base mismatches. In this, a SBH chip of short sequences of nucleotides is inserted into a solution of the desired DNA sequence. Then the probes, or a single DNA fragment with a specific sequence, binds the sequence are found, and used to find the entire sequence. The problem with this method is you must find the smallest number of probes to sequence the largest amount of DNA. This technique was first proposed in 1988 and used in 1991 to reconstruct a 100 base pair DNA sequence. There are approximately two broad steps, the first is when you hybridize the DNA with the microarray, and the second when you combine them and algorithmically reconstruct them from the set of k-mers.
Mass Spectrophometry
[edit | edit source]This method can sequence approximately 100 base pairs at a time, however its resolution yet needs improvement. MALDI and ESI are the ionization methods most used, and its competitive edge comes in that it can be done in hours instead of days unlike other methods. Mass spectrometry sequencing is advantageous because frameshift mutations and heterozygous mutations can be identified. Different from other methods, like Sanger sequencing, fragments of DNA of the same lengths but with different DNA compositions can be determined. MALDI-TOF MS of these DNA sequencing fragments can be performed, and the bases can be very accurately determined.
Direct Visualization of Single DNA Molecules by Atomic Force Microscopy (AFM)
[edit | edit source]This method uses Electron Microscopes to scan across the surface of DNA, and get up to nano-meter resolution of the fragment being studied. This only uses very small quantities of DNA, within a few nanograms. With this method you can analyze different types of DNA fragments such as supercoiled, linear, or relaxed DNA. An advantage is it does not require the use of a staining or radioactive agent, and so do less damage to the fragment being sequenced. It also provides 3-D images as opposed to 2D.
Shotgun Sequencing
[edit | edit source]Created by GNN president J. Craig Venter in 1996 this method relies on isolating random pieces of DNA from their genome and then doing this several times to get redundant copies. The increased DNA fragments then are assembled by their overlapping regions and form a continuous transcript, normally done with computers. Then finally custom primers describe the gaps between these transcripts giving the sequenced genome. This method allows for much faster sequencing and was used for example to find the genome of smallpox. Whole genome shotgun sequencing is done in a few steps. First the DNA is again separated into random fragments, then cloned into an appropriate vector. First you isolate the DNA, then shear it into several pieces by a blender, passage through a narrow gauge syringe, or sonication and normally each fragment is about 2,000 base pairs. This DNA is then loaded onto a gel and compared to marker DNA already loaded. The specified DNA is then recovered and ligated into a cloning vector which amplifies the desired DNA sequences. then primers flanking the sequences are annealed and analyzed by a sequencer. Then all of the sequences, normally 500 base pairs are compared by sophisticated computer algorithms and find the largest possible continuous fragments, then put together the entire genome. The most prevalent criticism for this method is that it is not accurate enough, however in 2000 when Celera sequenced the genome of Drosophila melanogaster successfully.
Bac to Bac Sequencing
[edit | edit source]This type of sequencing was used in the human genome project. It is much slower than the shotgun approach, but has is a more certain way of sequencing. It starts off with first creating a map of the human genome prior to sequencing. The human chromosome is cut into pieces and then the order of these are first figured out before the actual sequencing begins. To start off, the genome is cut into pieces that consist of 150kb long. Then, these 150kb pieces are inserted into a BAC, which is an artificial bacterial chromosome. The pieces make up a BAC library, much like an actual library. Each BAC piece is thus like a book, which can be chosen from the library. The next step is to fingerprint the pieces, which is done by cutting the BAC pieces into more smaller pieces with an enzyme. Finding a common sequence in overlaps will then allow the location of the BAC on each chromosome to be figured out. This allows the researcher to determine their order of the fragments along the chromosome. Then next step is break the BAC into even smaller pieces, about 1.5kb long, and then inserted into an artificial DNA. This DNA is called an M13, and the pieces make up the M13 library, much like the BAC library. Then the M13 library is sequenced, and all the pieces are put together to find the order. This is usually done with a computer, because of the complexity of the sequencing. Compared the shotgun sequencing, BAC sequencing takes much longer and is more useful for genomes that are bigger.
Southern Blotting
[edit | edit source]This method is used to check for the presence of specific DNA in a DNA sample. It uses both agarose gel electrophoresis along with methods to transfer the separated DNA into a filter membrane for probe hybridization. First restriction endonucleases cut the DNA into small fragments, then the fragments are electrophoresed on the agarose gel. Next the DNA can be broken into suitably small pieces and then denatured within an alkaline solution. Next a sheet of nitrocellulose is placed on the gel, applying even pressure. This is then set in a high temperature oven or exposed to ultraviolet radiation in order to utilize the covalent crosslinks between the DNA and the membrane. After this a hybriziation probe labeled is set into the membranes, then washed away and the pattern is found on x-ray film through autoradiography. P-32 ATP is the probe that allows for autoradiography.
Dye-terminator sequencing method
[edit | edit source]Another method of DNA sequencing is the dye-terminator sequencing method. This method can be performed in only one reaction, which is quite advantageous. With this method, each of the four dideoxynucleotide chain terminators is labeled with different fluorescent dyes that are at different wavelengths. The sequencing can then be done using a computer with controlled sequence analyzers. One problem to this method is that there may be unequal peak heights and shapes in the electronic DNA sequence trace chromatogram. A way to bypass this problem, however, is to use new DNA polymerase enzymes and dyes that will limit variability and dye blobs. This sequencing method is very commonly used, especially since it is quicker than and not as costly as other methods.
Methods of Sequencing
[edit | edit source]The Sanger Dideoxy method is used to sequence DNA. This process is a fast and simple one in which it involves the use of DNA polymerase to synthesize a complementary sequence containing fluorescent tags on the four deoxyribonucletide bases. The fragments of DNA strands containing the fluorescent bases are then separated via electrophoresis or chromatography then sent through a detector. Another method to sequence genomic DNA is the Shotgun method.
Edman degradation is used to sequence proteins. Phenyl isothiocyanate reacts with the amino group in the N-terminal amino acid, then acidified to remove it. High pressure liquid chromatography (HPLC) is used to identify the amino acid. The process is repeated for each of the following proteins.
DNA Replication
[edit | edit source]The sequential assembly and reorganization of arrays of proteins in DNA sequencing is necessary because it’s crucial for the coordinated execution of initiation, elongation, and termination processes of DNA replication. The physiological significance of this process indicates that defects in proteins associated with the assembly and monitoring of the replication fork can cause genomic instabilities which can further results in carcinogenesis. It can also lead to a series of diseases known was “chromosome instability syndrome”.
A key characteristic of DNA replication in eukaryotic cells is that it is highly adaptable or plastic. This plasticity and ability to adapt is demonstrated with the fork rate and origin selection processes regulating each other and when inactive origins are activated when forks are stalled. During replication, DNA helicase completes replication and DNA polymerase elongates the DNA chain.
Errors or defects in DNA replication can often result in harmful effects such as genomic instability. This type of error can cause mutations and diseases with abnormal tissue growth such as cancer and can also give rise to groups of diseases known as ‘chromosome instability syndromes’.
References
[edit | edit source]- Hall, A.R.; Scott, A.; Rotem, D.; Mehta, K.K.; Bayley, H.; Dekker, C. (2010). Nature Nanotechnology. 5 (12): 874–7. doi:10.1038/nnano.2010.237. PMC 3137937. PMID 21113160 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3137937.
{{cite journal}}: Missing or empty|title=(help); Unknown parameter|tiitle=ignored (help)CS1 maint: PMC format (link) CS1 maint: multiple names: authors list (link)
- Lippincott-Schwartz, J.; Patterson, G.H. (2003). "Development and use of fluorescent protein markers in living cells". Science. 300 (5616): 87–91. doi:10.1126/science.1082520. PMID 12677058.
{{cite journal}}: CS1 maint: multiple names: authors list (link)
- Masai, H.; Matsumoto, S.; You, Z.; Yoshizawa-Sugata, N.; Oda, M. (2010). "Eukaryotic chromosome DNA replication: Where, when, and how?". Annual Review of Biochemistry. 79: 89–130. doi:10.1146/annurev.biochem.052308.103205. PMID 20373915.
{{cite journal}}: CS1 maint: multiple names: authors list (link)
- Slonczewski, J. (2010). Microbiology: An Evolving Science (2nd ed.). New York: W. W. Norton & Company. p. 1097. ISBN 9780393934472.
- Staff Reporter (1 December 2010). "Physicists use graphene to decode DNA". Medical Daily. IBT Media Inc. Retrieved 28 April 2016.
Scientists now have techniques that allow genes and DNA sequences of interest to be stored, through the use of cDNA and phages. DNA cataloging is a process used for recording and storing specific samples of DNA. The basic process is to take a sample of DNA, ligate it to a plasmid, inserting the plasmid into a bacteria, selecting for bacteria of interest, and storing the bacteria in a large freezer.
Sample of DNA
[edit | edit source]The sample of DNA is usually synthesized by PCR from a single DNA strand that contains within it three things: the gene of interest, a restriction enzyme binding site on each end, and a "test" gene. The "test" gene usually codes for a pigment or a pigment inhibitor so later in the process it will be easy to detect which bacterial have the gene of interest.
Ligation to a Plasmid
[edit | edit source]A Plasmid is a circular piece of DNA naturally found in many bacterial cells. The useful features of plasmids to scientists are that they can be absorbed through the bacterial membrane. Once absorbed, the bacteria will begin to express the genes on the plasmids. To ligate a gene of interest to a plasmid, often an artificially produced plasmid will be used. This plasmid will have some sort of antibiotic resistance (usually to Ampicillin) and a specific restriction enzyme (the same one used in the DNA amplified through PCR). when the plasmids and the DNA of interest are mixed, many of the "sticky ends" of the plasmid and the DNA will connect. DNA ligase and ATP will need to be added to the solution to covalently link the strands to each other.
Unfortunately, sometimes the plasmid can simply reconnect with itself where it was broken, which is why there is a test gene in the DNA sequence of interest. We will know (later) which plasmids have the gene, and which do not.[1]
Insertion of Plasmid to Bacteria
[edit | edit source]Once the plasmid is put back together, it can be inserted into a bacteria. The common method of doing this is through "heat-shock" therapy. (note: the bacteria must have the capability of surviving the "heat-shock" often labeled as having the "heat-shock" genes) Heat-Shocking the bacteria is rapidly heating the bacteria up to a temperature higher than the current temperature of the bacteria. While the mechanism is not clearly understood, the effect is that some of the bacteria will absorb the plasmid into its cytoplasm.
Selecting for Bacteria of Interest
[edit | edit source]Some of the bacteria in the solution will not have taken up the plasmid, even still, some of the bacterial may have take up a plasmid whose "sticky ends" have simply recombined with itself. The way to determine which bacteria we want and which we do not want is to use the test gene and the antibiotic resistance gene on the ideal plasmid. The way to do this is to culture the bacteria on an agar petri dish containing the antibiotic, which will kill any bacteria without some form of the plasmid. Next is to examine which colonies are expressing the "test gene" on the DNA of interest that was ligated to the plasmid. Whichever colonies live on the antibiotic plant and exhibit the "test gene" are taken and cultured separately in their own agar dish.
Storing the DNA
[edit | edit source]The most common method of storage is to use a large freezer which keeps the bacteria of interest frozen at around minus seventy(70) degrees C.
References
[edit | edit source]General Information
[edit | edit source]DNA can be synthesized by automated solid-phase technique, just like the synthesize polypeptides. A monomer is attached to a insoluble column, then activated monomers are added in the order of the sequence interested to form an oligonucleotide. The activated monomers are deoxyribonucleoside 3'-phosphoramidites. The meaning of activated is that the reactive group, hydroxyl group on the fifth carbon (5') is replaced by DMT, dimethoxytrityl. It keeps the deoxyribonucleoside 3'-phospohramidite molecule away from any further reaction on 5'. Thus, it was said to be a protecting group. The 3'-phosphoryl group is protected by a β-cyanoethyl (βCE) group. Since it is the 3'-phosphoryl group that is attached to the insoluble support, the chain is synthesized in 3' to 5'* direction. The synthesize of oligonucleotide consist three basic steps.
- 3' to 5', meaning the 5'hydroxyl group perform a nucleophilic attack on the 3'-phosphoramidite of the incoming monomer. It is different from the regular 5' to 3' synthesize of natural DNA and RNA, where the 3' hydroxyl group attacks the inner most phosphorus atom of 5' triphosphate.
Procedure
[edit | edit source]Step one: The procedure starts with deoxygribonucleoside 3'-phosphoramitide (with DMT that was already added to protect the 5' and a βCE that was added to protect 3'-phosphoryl) react with a growing chain to form a phosphite triester group through coupling. This coupling reaction is driven by the nucleophilic hydroxy group on the 5' carbon of the nucleoside that is to be added attacking the electrophilic phosphorus atom on the existing chain with the -NR2, often diisopropylamine (a relative stable secondary amine), as a leaving group. The reaction is carry out in an anhydrous environment to avoid water hydrolyse the 3'-phosphoramidte causing the reaction go backward unexpectedly. And avoid water from reacting with the 3'-phosphoramidite of the incoming monomers. Once the water attack the 3'-phosphoramidite, it is no longer reactive in receiving the nucleophilic attack from the 5'OH of the growing chain.
Step two: Using Iodine, I2, the phosphite triester group oxidized to a phosphotriester group.
Step three: The DMT group on the 5' side of the activated monomer is removed by adding dichloroacetic acid (DCA, CHCl2COOH) while the rest of the molecule unchanged.
The oligonulceotide is now extended by one monomer unit and is ready to react with another incoming activated monomer. When the oligonucleotide of the desired length is synthesized, the final product can be obtained by adding ammonia (NH3) to remove all the protecting groups and remove the product from the insoluble support. Because no synthesis is perfect, not all of the growing oligonucleotide will react with the added monomers every time. Some oligonucleoides will be shorter than the other, for some nucleotides are missing. Thus, that considered to be impurities as they do not have the exact sequence carries the exact genes we wanted. The final product is the longest ones. The mixture of newly synthesized oligonucleotides can be separated by gel electrophoresis to get the desired product that was interested at the first place.

Removal of Protecting group
[edit | edit source]After all the synthesis steps, the mixture of product is then put into concentrated ammonium hydroxide, NH4OH, for an hour at room temperature. Next, the mixture with ammonium hydroxide is placed into an ice-bath and transferred to a vial that has a screw-cap afterwards. In order to remove the heterocyclic hase protecting groups, the solution in a vial is heated at 55 oC over night. Next day, the solution would be cooled in an ice-bath and evaporated off to dryness.
Analysis of Synthetic DNA
[edit | edit source]Four methods can be used to analyze synthesized DNA.
1. High-pressure (performance) liquid chromatography (HPLC).
High pressure liquid chromatography is a technique use to identify or separate compounds. It includes two basic forms, reverse-phase and ion-exchange. The separation and analysis of HPLC are based on the retention time between the stationary phase(chromatographic packing material) and mobile phase(the compounds needed to identify or separate) in the column.
Ion-exchange HPLC is based on the charges between stationary phase and mobile phase. The compound with same charges as stationary phase has a shorter retention time while it has a longer retention time with opposite charges. It is used for oligonucleotides of 10 to 20 bases of nucleotides in length. Two sets of ion-exchange HPLC is used for analyzing DNA. One condition is dNAPac column with 20mM Tris buffer/0.05% acetonitrile and 1M NaCl at a rate of 1.5 mL/min. Another condition is Resource-Q column with 10mM NaOH/80mM NaBr and 10mM NaOH/1.5M NaBr at a rate of 1.5mL/min
Reverse-phase HPLC is based on the affinity between stationary phase and mobile phase. The stationary phase of reverse-phase HPLC is non-polar, a non-polar compound has a longer retention time than a polar compound.
2. Gel electrophoresis
Gel electrophoresis is a technique use to separate DNA, RNA, or protein molecules. For oligonucleotides, 15~20% cross-linked gels are used with 7M urea. The gel electrophoresis takes over 3~6 hours at 50 to 60oC. To see oligonucleotides, a Molecular Dynamics Phosphorimager is used.
Gel electrophoresis can be used to analyze synthesized DNA. It also uses for the purification of synthesized DNA. A longer synthesized DNA is mainly used for cloning and hybridization. Non-full-length synthesized DNA formed during oilgonucleotide synthesis may interfere the applications, so purification is important. Gel electrophoresis is an efficient technique to separate full-length products from other shorter products.
3. MALDI-TOF analysis
MALDI-TOF is an ionization technique in mass spectrometry. It is used to identify compounds like oligonucleotides by their mass. Oligonucleotide is ionized and given a potential energy in the ion chamber of MALDI instrument. The oligonucleotide is then move to the detector of MALDI instrument by converting potential energy to kinetic energy. E=1/2mv2. Since E(energy) is given and v(velocity) is measured, m(mass) of the nucleotide can be determined and compared to value it should have.
4. Enzymatic degradation of oligomers
The products dissolved in the mixture solution of snake venom phosphodiesterase, alkaline phosphatase, and buffer solution. The reaction takes 4 hours at 37 oC. After that the mixture is heated to denature the enzymes. The mixture is then diluted with water and centrifuged. Finally, the mixture is analyzed by HPLC.
Advantages
[edit | edit source]The advantages of solid support synthesize of oligonucleoside is similar to the one of synthesis of polypeptides. Because the growing nucleotides is attached to a insoluble support, all the soluble impurities can be washed away without loosing any product.
Application of Synthesized Oligonucleotides
[edit | edit source]The ability to synthesize oligonucleotide with the exact sequences have important uses. A synthesized oligonuletide can be used in many DNA manipulation techniques.
1. DNA Sequence
A synthesized oligonucleotide with fluorescent tag can used to locate genes within a very long DNA molecule if the gene sequence in known. Because then we will able to synthesized a new oligonucleotides that is complementary to the gene sequence we want to locate. The labeled oligonucleotide use as probe is very important in exploring new genes. The probe can be used as primer to initiate replication of neighboring DNA by DNA polymerase that have not been map yet. With the fluorescent ability of the probe, we are able to tell where the new gene we are trying to map starts.
2. Polymerase Chain Reaction (PCR)
The polymerase chain reaction is the technique to amplify specific DNA. In order to amplify specific DNA sequences, pair of primers to hybridize with the target DNA sequences, four deoxyribonucleoside triphosphates, and a heat-stable DNA polymerase are needed. The specific primers for hybridizing the target DNA sequences can be made by Oligonucleotide Synthesis Technique.
3. Site-Specific Mutagenesis
The Site-Specific Mutagenesis or Oligonucleotide-Directed Mutagenesis can produce mutant proteins with single amino acid substitutions. The key of this technique is to prepare an oligonucleotide primer that is complementary to the DNA except the region that wants to change amino acid. The mismatch of primers of 1 of 15 bases is tolerable at an appropriate temperature. These specific oligonucleotide primers can be prepared by Oligonucleotide Synthesis Technique.
4. Large-Scale Synthesis
The Large-Scale Synthesis of oligonucleotide is almost same with the procedure above except a column used in the procedure. The Large-Scale Synthesis uses a packed column rather that a loosely packed cartridge.
5. Preparation of DNA microarrays on planar glass surfaces
DNA microarrays or gene chips can be used to analyze the pattern and level of pexpression of all genes in a particular cell or tissue. This technique can be done by placing oligonucleotides on a planar glass surfaces. The probes (oligonucleotides) are synthesized on the silicon chip. Thin silicon rubber capillaries are put on a lass slide and the probes are synthesized. Complementary DNA that is fluorescently labeled is hybridized to the gene chips that are made by oligonucleotides. This hybridized gene chips show the expression level for each gene.
6. Southern Blotting
Southern blotting is a technique to identify DNA molecules. In order to identify a DNA fragment with a specific sequence, a 32P-labeled single-stranded DNA probe, a specific DNA molecule made by oligonucleotide synthesis, is needed. The probe is hybridized with the complementary sequence of DNA sample. and the specific sequence can be visualized by autoradiography.
History
[edit | edit source]In 1984, Kary Mullis invented a method to amplify DNA sequences. In previous attempts, DNA polymerase cannot withstand the temperature change. However, Mullis used polymerase from Thermus aquaticus, a bacteria that lives in hot springs in his method, which allowed PCR to be performed at high temperature conditions.
Overview
[edit | edit source]
Polymerase Chain Reaction (PCR) is a very effective technique of obtaining multiple identical copies of a certain DNA strand (amplifying DNA). PCR can be used for amplifying DNA, mutation DNA, delete DNA, and introduce restriction endonuclease site. PCR is performed by repeating a cycle that consists of several steps. This process is performed after regions of target DNA, or the sequence(s) of DNA which are to be amplified (Target Sequence), have been specified. Target Sequence, as its name implies, is a specific part of DNA that is needed for further study. In short, one cycle of PCR consists of these steps:
- Preparation before repetition of PCR cycle:
- Addition of PCR components to solution containing DNA to be amplified
- This step is only performed before the first PCR cycle. Addition of components after the first cycle is not advised because PCR reaction is temperature sensitive (slight change in temperature can greatly affect the reaction)
- Components to be added are as followed:
- DNA template to be amplify during this process.
- A pair of primers (usually between 10 - 30 nucleotides long) that is complementary to the Flanking Sequence (several bases directly left or right of the target sequence). These two different primers will be referred as primer F (for the forward primer) and primer R (for the reverse primer).
- The four dNTPs (deoxyribonucleotide triphosphates); they are: dATP, dGTP, dCTP, and TTP
- Magnesium Chloride (MgCl2) in order to stabilize the charge of phosphate group and activate the replication process.
- Heat stable DNA Polymerase; heat stability is very important because PCR reaction is performed at various temperatures. This heat stable DNA polymerase is obtained from a thermophilic bacteria Thermus aquaticus, the inhabitant of hot springs. Hence, this DNA polymerase is termed as Taq DNA Polymerase. Other polymerases with different properties can be used depending on the purpose and requirements of the PCR; Some polymerases, such as Pfu Turbo, for example, have higher fidelity rates but are much more sensitive.
- Buffer solution to stabilize the DNA sample.
- To maximize the efficiency of this process, a master mix is made and contained of the primer, MgCl2, buffer, and Taq polymerase.
- These component must be added in excess to ensure that when the two DNA strands are separated in the second step, the chance of the two DNA to re-hybridize is minimized.
- Addition of PCR components to solution containing DNA to be amplified
- PCR cycle (perform about 20-30 cycles):
- Strand Separation
- Strand separation is performed to expose target sequence and flanking sequence to primer. This step is done by heating the solution, causing the hydrogen bonds maintaining the double helix structure to break.
- Parameter:
- Temperature: about 95oC
- Time: 15 to 30 seconds (to separate the DNA to 2 single strands)
- Hybridization of Primers (Also known as the annealing step)
- At this step, the solution is cooled to around 54oC. By cooling down the solution, primers are allowed to hybridize with the flanking sequences via hydrogen bonding.
- It is very possible that the two main strand will re-hybridize because both strands are complementary to each other. Fortunately, as explained earlier, it can be easily avoided by having excess primers in solution. Excess primers are expected to hybridize with the flanking sequence before any two complementary DNA can hybridize.
- DNA Synthesis (aka elongation step)
- Solution heated to 72oC. At this optimal temperature, the Taq polymerase will start elongates both primers.
- There are two types of primer; each is the complementary oligonucleotides of the flanking sequence.
- Elongation happens not unlike the regular polymerization of DNA. The primers are elongated from the 5' to the 3' ends (which is the opposite direction of the actual DNA strand; strand that have target sequence, flanking sequence, and un-amplified sequence).
- Strand Separation
- Note:
- By the end of first cycle, we obtain 2 types of new strand in addition to the original DNA strands. These strands are the elongation of each type of primer. They contain:
- One of the primer (exp: primer A)
- The target sequence
- A complementary of a flanking sequence (the one that is not hybridized to the primer contained in this strand; e.g.: complementary of flanking sequence of primer B)
- The rest of the non-target sequence after this complementary of flanking sequence.
- On the second cycle, some of the primers will hybridize with the new type of sequence obtained on first cycle (exp: on the shorter strand elongated from primer A, primer B will attach to its flanking sequence and start elongates the cycle until it reaches the end of the shorter strand, the flanking sequence of primer A).
- On the second cycle, the new type of strand formed, Short Strand consist of:
- One of the primer (exp: Primer B)
- The target sequence
- The flanking sequence that can hybridize with Primer A
- On the second cycle, besides forming the short strand, the same strands as the one formed on the first cycle can also form.
- On the second to the nth cycle, the short strand is amplified exponentially while the slightly longer strand (the only strand that form on the first cycle) is amplified arithmetically.
- This cycle is repeated n times. At the nth repetition, there will be 2n of desired sequence.
- By the end of first cycle, we obtain 2 types of new strand in addition to the original DNA strands. These strands are the elongation of each type of primer. They contain:
- Note:
Finally, the reaction is usually held at 4 degrees Celsius after completion to stabilize the DNA, which is temperature sensitive.
Additional Steps
[edit | edit source]A final elongation step is performed at 70-74°C for at least 5 minutes after the last PCR cycle has been done. This step will guarantee that any remaining DNA strand is fully extended. In addition, a hold step at 4°C for an indefinite time can be utilized for storage of the DNA product.
Thermal Cyclers
[edit | edit source]

The thermal cycler can perform different steps of PCR process by cooling and heating the tubes constantly. They use of the Peltier effect which allows both heating and cooling of the block holding the PCR tubes simply by reversing the electric current. Thin-walled reaction tubes with no RNAase and DNAase are suitable for PCR. Due to their thickness, they can create thermal conductivity to allow for rapid thermal equilibration. As showed in the next picture, most modern thermal cyclers will have heated lids to prevent condensation at the top of the reaction tube and evaporation in the bottom of each tube.
Results Interpretation
[edit | edit source]To ensure the quality of PCR amplification, a gel-electrophoresis is performed. To prepare for this step, a series of steps need to be done before hands
1. Preparation 1% agarose gel: Weight out 1 gram of purified agarose and dissolve to 100 ml with TAE (Tris-Acetate) buffer solution. (Figure A)

2. Heat up. The solution is heated up in microwave until all of agarose particles dissolved completely.
3. Addition of ethidium bromide. After the solution is cooled down, 5 microliters of ethidium bromide are added to the solution. Ethidium Bromide will attached to DNA molecules and make the strand stand out in UV light background (used as a nucleic acid stain).
4. Polymerization of agarose. Wait for the agarose to completely polymerize after inserting a well plate to load the samples into. This process can take up to 30 minutes.


5. Addition of buffer solution. Fill up the chamber with TAE solution to create a barrier between the outside environment and the DNA sample.
6. Addition of DNA sample. Loading dye should be added to the DNA samples, and an appropriate DNA ladder run in order to determine the approximate molecular weight of the sample in kilobases.

7. Apply electrical current. The DNA must be located so it will near the negative charged node and away from the positive node.


8. Visualize the results using UV light.
Advantages
[edit | edit source]PCR is a popular method in amplifying DNA because it brings various benefit. Some of those benefits are:
- Researcher does not need to elucidate the sequence of the target sequence. They only need to know the sequence of the flanking sequence.
- Target that are much larger than the primer can still be synthesized.
- Primers prepared does not have to be an exact match of the flanking sequence.
- PCR is very specific.
- PCR is very sensitive to the point that it is able to amplify a single DNA molecule.
- The resultant DNA can be processed further more such as mutation, deletion, or cloning process
Mechanism
[edit | edit source]In this example, the following convention is used:
- BOLD = flanking sequence and its complementary sequence
- ITALICS = target sequence and its complementary sequence
- NORMAL = the remaining sequence (the non amplified part of DNA strand)
- BOLD/ITALICS= primers
Sequences formed at various stages of PCR:
- Original DNA molecule:
...CGCG TTG CAGCTACTGACTC CCC CGCG...
...GCGC AAC GTCGATGACTGAG GGG GCGC...
- After separation and attachment of primer (beginning of first cycle):
...CGCG TTG CAGCTACTGACTC CCC CGCG...
- AAC
- CCC
...GCGC AAC GTCGATGACTGAG GGG GCGC...
- After first sequence elongation (end of first cycle):
...CGCG TTG CAGCTACTGACTC CCC CGCG...
- AAC GTCGATGACTGAG GGG GCGC...
...CGCG TTG CAGCTACTGACTC CCC
...GCGC AAC GTCGATGACTGAG GGG GCGC...
- After separation of sequence and attachment of the other primer (beginning of second cycle; the formation of sequence identical to sequence formed in first cycle is not shown):
...CGCG TTG CAGCTACTGACTC CCC CGCG...
- AAC GTCGATGACTGAG GGG GCGC...
- CCC
- AAC GTCGATGACTGAG GGG GCGC...
- AAC
...CGCG TTG CAGCTACTGACTC CCC
...GCGC AAC GTCGATGACTGAG CGG GCGC...
- After sequence elongation (formation of Short Sequence, end of second cycle):
...CGCG TTG CAGCTACTGACTC CCC CGCG...
- AAC GTCGATGACTGAG GGG GCGC...
- TTG CAGCTACTGACTC CCC
- AAC GTCGATGACTGAG GGG
...CGCG TTG CAGCTACTGACTC CCC
...GCGC AAC GTCGATGACTGAG CGG GCGC...
- After separation of short sequence and addition of primers (beginning of third and subsequent cycle; only the short sequence formation is shown):
...CGCG TTG CAGCTACTGACTC CCC CGCG...
- AAC GTCGATGACTGAG GGG GCGC...
- TTG CAGCTACTGACTC CCC
- AAC
- CCC
- AAC GTCGATGACTGAG GGG
...CGCG TTG CAGCTACTGACTC CCC
...GCGC AAC GTCGATGACTGAG CGG GCGC...
- After elongation of short sequence (end of third and subsequent cycle; only the short sequence formation is shown):
...CGCG TTG CAGCTACTGACTC CCC CGCG...
- AAC GTCGATGACTGAG GGG GCGC...
- TTG CAGCTACTGACTC CCC
- AAC GTCGATGACTGAG GGG
- TTG CAGCTACTGACTC CCC
- AAC GTCGATGACTGAG GGG
...CGCG TTG CAGCTACTGACTC CCC
...GCGC AAC GTCGATGACTGAG CGG GCGC...
- Short sequences will be re-separated so that new primers can attach in the subsequent cycle:
- TTG CAGCTACTGACTC CCC
- AAC
- CCC
- AAC GTCGATGACTGAG GGG
- TTG CAGCTACTGACTC CCC
- AAC
- CCC
- AAC GTCGATGACTGAG GGG
- Primers will be elongated to form additional short sequences. These last two bullet points are repeated on subsequent cycle until sufficient amount of target sequence has been synthesized:
- TTG CAGCTACTGACTC CCC
- AAC GTCGATGACTGAG GGG
- TTG CAGCTACTGACTC CCC
- AAC GTCGATGACTGAG GGG
- TTG CAGCTACTGACTC CCC
- AAC GTCGATGACTGAG GGG
- TTG CAGCTACTGACTC CCC
- AAC GTCGATGACTGAG GGG
One More PCR Diagram
[edit | edit source]Practical Applications of PCR
[edit | edit source]Polymerase chain reaction has become an important tool for medical diagnosis. PCR can detect and identify bacteria and viruses that cause infections such as Tuberculosis, Chlamydia, viral meningitis, viral hepatitis, HIV and many others. Once primers are designed for the DNA of a specific organism, using PCR to detect the presence or absence of a pathogen in a patient’s blood or tissue is a simple experiment. Because PCR can easily distinguish among the tiny variations in DNA that each of us posses and that make each of us genetically unique, the method is also leading to new kinds of genetic testing. These tests diagnose not only people with inherited disorders but also people who carry deleterious variations (mutations) that could be passed on to their children. These carriers are usually not themselves affected by the mutant gene but they can lead to a disease in the next generations (e.g. mutations that cause cystic fibrosis). Many of the new genetic tests are the result of the Human Genome Project, the huge international effort to identify and study all human genes. The project is progressing rapidly towards its ultimate goal which is to sequence the entire DNA in typical human cells. Sequencing DNA means to determine the precise order of the four different nucleotides that make up any strand of DNA. DNA sequencing reveals crucial variations in the nucleotides that constitute genes. These mutational changes produce diseases and even death by forcing the genes to produce abnormal proteins or sometimes no protein at all. DNA sequencing involves first isolating and duplicating DNA segment for nucleotide analysis. Thus PCR is an essential tool for the Human Genome Project because it can quickly and easily generate an unlimited amount of any piece of DNA for this kind of study. PCR is a direct way of distinguishing among the confusion of different mutations in a single gene, each of which can lead to a disorder such as Duchenne Muscular Dystrophy. It also helps doctors track the presence or absence of DNA abnormalities characteristic of a particular cancer so that they can start and stop drug treatment and radiation therapy as soon as possible. PCR promises to greatly improve the genetic matching of donors and recipients for bone marrow transplantation. The technique incomparable ability to identify and copy the smallest amount of even old and damaged DNA has proved exceptionally valuable in the law, especially the criminal law. PCR is an indispensable adjunct to forensic DNA typing- commonly called DNA fingerprinting. Traces of DNA found at a crime scene can also be amplified by using PCR, thereby providing sufficient amount of DNA to match with the suspect’s DNA. DNA is extremely stable molecule, especially when protected from air, light, and water. Under these circumstances, large pieces of DNA can be preserved for thousands of years or longer. Thus PCR provides an excellent method for amplifying such ancient DNA molecules, so that they can be subjected for analysis. This technique can also be used in amplifying DNA from microorganisms that have not yet been isolated and cultured. Sequences from these PCR products can be sources of considerable insight into evolutionary relationships between organisms.
Variations of PCR
[edit | edit source]Because of the high adaptability of PCR, many new biochemical techniques based on the original PCR method have been invented. Some of the more commonly seen ones are listed below in alphabetical order.
Allele-specific PCR is a cloning technique based on single-nucleotide polymorphism (SNPs). This requires a DNA sequence that has differences between alleles and uses primers whose 3' end encompass the SNP.
Assembly PCR, more formally known as Polymerase Cycling Assembly (PCA), is a method of synthesizing long strands of DNA oligonucleotides by applying PCR to small oligonucleotides and then hybridizing them together. The entire process consists of two sets of PCR reactions. The first reaction utilizes forward and reverse primers to anneal together oligonucleotide fragments with complementary sequences. The second reaction uses another set of primers to perform a regular PCR reaction, which amplifies the results of the first reaction. Only the larger oligonucleotide strands are amplified, distinguishing them from the other incomplete shorter fragments. Once the entire reaction is performed, gel electrophoresis may be performed to isolate and identify the completed strands.
Asymmetric PCR is a biochemical technique that focuses on the amplification of one strand of DNA over the other. To do so, a normal PCR reaction is performed but with either an excess of primers that react to the target strand or absence of primers that react to the complementary strand. This method is utilized in certain types of forensic sequencing and probing, where having only one of the two DNA strands is preferred.
Colony PCR is a biochemical technique of amplifying DNA in a vector, such as E. Coli bacterial colonies, to identify the target DNA. The first step in the process involves removing a sample of the vector from the growth plate by a sterile toothpick or pipette tip. The sample is transferred to either a PCR master mix or autoclaved water, where a normal PCR reaction is performed to determine whether or not the colony contains the DNA fragment or plasmid of interest.
Digital PCR simultaneously amplifies thousands of samples.
Helicase-dependent amplification uses constant temperature instead of cycling through denaturation, annealing, and elongation cycles. DNA helicase, an enzyme, unwinds DNA and used to replace thermal denaturation.
Hot-start PCR is a form of PCR that focuses on the maximization of product yield. To reach this goal, chemical modifications are used to limit polymerase activity prior to the reaction. This limitation reduces non-specific polymerase amplification. Next, the reaction, itself, is performed at extremely high temperatures using polymerase obtained from thermophilic organisms, such as Achaea bacteria from ocean vents. These elevated temperatures further prevent non-specific polymerase amplification, resulting in a larger yield of the target product.
Intersequence-specific PCR a method for DNA fingerprinting. this amplify regions to produce a unique fingerprint of amplified fragments.
Inverse PCR is a method engineered to bypass the limitations of regular PCR, which requires prior knowledge of the flanking sequences around the target sequence. Inverse PCR is aptly named because it can be performed to determine the flanking sequences of a target strand. However, this method has its own limitation: one internal sequence must be known for it to be performed.
Ligation-mediated PCR is a method used for DNA sequencing, genome walking, DNA footprinting. This uses small DNA linkers ligated to desired DNA and multiple primers annealing to the linkers.
Methylation-specific PCR (MSP) detects methylation of CpG islands in genomic DNA. DNA is treated with sodium bisulfate and is recognized by PCR primers. Two PCRs are used to modify the DNA. One primer recognizes DNA with cytosines to amplify methylated DNA, and the other primer recognizes DNA with uracil/thymine to amplify unmethylated DNA.
Miniprimer PCR allows PCR targeting to smaller primer (smalligos) (consists of 9-10 nucleotides) binding regions and used to amplify conserved DNA sequences.
Multiplex PCR is a biochemical technique that focuses on simultaneous amplification of multiple target DNA products through the addition of multiple primers into a single PCR reaction. Multiplex PCR is a preferred method for DNA testing because it allows for the analysis of deletions, mutations, and polymorphisms in a sample.
Multiplex Ligation-dependent Probe Amplification (MLPA) amplifies multiple targets with only a single pair of primer.
Nested PCR is a form of PCR that maximizes accuracy in the amplification of a target DNA fragment. Nested PCR consists of two separate reactions. The first is a normal PCR reaction where a pair of primers is used to amplify a target fragment. The second reaction adds a second pair of primers called “nested primers” that bind to the products of the first reaction and amplify them. By performing two sets of PCR reactions, this method maximizes accuracy since it is very unlikely that two pairs of primers would both bind to the wrong target fragment.
Overlap-extension PCR allows the construction of DNA sequence with an alteration inserted beyond the limit of the primer length.
PAN-AC uses isothermal conditions for amplification.
Quantitative PCR (Q-PCR) measures the quantity of a PCR product. It measures the starting amount of DNA, cDNA, and RNA. this is commonly used to determine if a dNA sequence is present and the number of its copies in the sample. This uses fluorescent dyes, for example TaqMan, to measure the amount of amplified product.
Reverse Transcription PCR (RT-PCR) is a biochemical technique engineered to make good use of our knowledge of how reverse transcription can form complementary DNA from RNA. RT-PCR is performed in two major steps. In the first step, reverse transcription of a target RNA strand into its complementary DNA is performed using oligo-DT, which substitutes for the role of primers. In the second step, a regular PCR reaction is performed by adding primers specific to the DNA to help amplify the target sequence. RT-PCR is a highly sensitive technique that takes advantage of the smaller size of RNA strands (when compared to their DNA counterparts). RT-PCR is a highly applicable technique. In addition to the aforementioned material, this technique can be used for the mass production of target RNA through the synthesis of complementary DNA, which can be subsequently cloned through a vector.
Touchdown PCR is a PCR technique that focuses on maximizing target product yield. In such a way, it can considered an alternative to the Hot-start PCR method. To achieve its goal, this method performs PCR reactions at high annealing temperatures, which reduces non-specific polymerase amplification. In other words, only the fragments containing the sequences of interest will be amplified by the primers. As the fragments are amplified over and over again, the temperature is gradually lowered until it is certain that only target fragments have remained.
Universal Fast Walking is used for genome walking and genetic fingerprinting. This is a more specific two sided PCR than the normal one sided PCR approach.
Variable Number of Tandem Repeats (VNTR) PCR target areas of the genome that exhibit length variation. The analysis of the genotypes involves sizing of the amplification products by gel electrophoresis. Analyzing smaller VNTR segments (short Tandem Repeats, STRs) is the base for DNA Fingerprinting.
DNA Polymerases
[edit | edit source]Klenow fragment is originally derived from the DNA Polymerase I from E. coli (first enzyme to be used in PCR). This polymerase needs to be replenished every cycle due to its lack of stability at high temperature. This is not commonly used in PCR.
Bacteriophage T4 DNA polymerase was initially used in PCR but is also destroyed by heat and has a higher fidelity replication than the Klenow fragment.
Thermus aquaticus (Taq) is commonly used and is the first thermostable (heat-stable) polymerase used in PCR. This enzyme is isolated from a native source or from its cloned gene expressed in E. coli.
Stoffel fragment is made of a truncated gene for Taq polymerase and expressed in E. coli. This enzyme lacks a 5’-3’ exonuclease activity and is able to amplify targets longer than its native enzyme.
Faststart polymerase is a variant of Taq polymerase that requires a strong heat activation. This should be avoid from non-specific amplification because of its low temperature polymerase activity.
Pfu DNA polymerase is isolated from an archean Pyrococcus furiosus. This has a proofreading activity. Since errors increase as PCR progresses, Pfu is preferred when the products are individually cloned for sequencing or expression.
Vent polymerase is extremely thermostable and is isolated from Thermococcus litoralis.
Tth polymerase is a thermostable polymerase from Thermus thermophilus. This polymerase has a reverse transcriptase activity when in presence of Mn2+ ion that allows PCR amplification from RNA targets.
References
[edit | edit source]· ^ Newton CR, Graham A, Heptinstall LE, Powell SJ, Summers C, Kalsheker N, Smith JC, and Markham AF (1989). "Analysis of any point mutation in DNA. The amplification refractory mutation system (ARMS)". Nucleic Acids Research 17 (7): 2503–2516. doi:10.1093/nar/17.7.2503. PMID 2785681.
· ^ Stemmer WP, Crameri A, Ha KD, Brennan TM, Heyneker HL (1995). "Single-step assembly of a gene and entire plasmid from large numbers of oligodeoxyribonucleotides". Gene 164: 49–53. doi:10.1016/0378-1119(95)00511-4. PMID 7590320.
· ^ Innis MA, Myambo KB, Gelfand DH, Brow MA. (1988). "DNA sequencing with Thermus aquaticus DNA polymerase and direct sequencing of polymerase chain reaction-amplified DNA". Proc Natl Acad Sci USA 85: 9436–4940. doi:10.1073/pnas.85.24.9436. PMID 3200828.
· ^ Pierce KE and Wangh LJ (2007). "Linear-after-the-exponential polymerase chain reaction and allied technologies Real-time detection strategies for rapid, reliable diagnosis from single cells". Methods Mol Med. 132: 65–85. doi:10.1007/978-1-59745-298-4_7. PMID · 17876077.
· ^ Myriam Vincent, Yan Xu and Huimin Kong (2004). "Helicase-dependent isothermal DNA amplification". EMBO reports 5 (8): 795–800. doi:10.1038/sj.embor.7400200.
· ^ Q. Chou, M. Russell, D.E. Birch, J. Raymond and W. Bloch (1992). "Prevention of pre-PCR mis-priming and primer dimerization improves low-copy-number amplifications". Nucleic Acids Research 20: 1717–1723. doi:10.1093/nar/20.7.1717.
· ^ E. Zietkiewicz, A. Rafalski, and D. Labuda (1994). "Genome fingerprinting by simple sequence repeat (SSR)-anchored polymerase chain reaction amplification". Genomics 20 (2): 176–83. doi:10.1006/geno.1994.1151.
· ^ Ochman H, Gerber AS, Hartl DL (1988). "Genetic applications of an inverse polymerase chain reaction". Genetics 120: 621–623. PMID 2852134.
· ^ Mueller PR, Wold B (1988). "In vivo footprinting of a muscle specific enhancer by ligation mediated PCR". Science 246: 780–786. doi:10.1126/science.2814500. PMID 2814500.
· ^ Herman JG, Graff JR, Myöhänen S, Nelkin BD, Baylin SB (1996). "Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands". Proc Natl Acad Sci USA 93 (13): 9821–9826. doi:10.1073/pnas.93.18.9821. PMID 8790415.
· ^ Isenbarger TA, Finney M, Ríos-Velázquez C, Handelsman J, Ruvkun G (2008). "Miniprimer PCR, a new lens for viewing the microbial world". Applied and Environmental Microbiology 74: 840–9. doi:10.1128/AEM.01933-07. PMID 18083877.
· ^ Khan Z, Poetter K, Park DJ (2008). "Enhanced solid phase PCR: mechanisms to increase priming by solid support primers". Analytical Biochemistry 375: 391–393. PMID 18267099.
· ^ Y.G. Liu and R. F. Whittier (1995). "Thermal asymmetric interlaced PCR: automatable amplification and sequencing of insert end fragments from P1 and YAC clones for chromosome walking". Genomics 25 (3): 674–81. doi:10.1016/0888-7543(95)80010-J.
· ^ Don RH, Cox PT, Wainwright BJ, Baker K, Mattick JS (1991). "'Touchdown' PCR to circumvent spurious priming during gene amplification". Nucl Acids Res 19: 4008. doi:10.1093/nar/19.14.4008. PMID 1861999, http://www.pubmedcentral.nih.gov/pagerender.fcgi?artid=328507&pageindex=1.
· ^ David, F.Turlotte, E., (1998). "An Isothermal Amplification Method". C.R.Acad. Sci Paris, Life Science 321 (1): 909–914.
· ^ Fabrice David (September–October 2002). "Utiliser les propriétés topologiques de l’ADN: une nouvelle arme contre les agents pathogènes". Fusion.(in French)
· ^ Myrick KV, Gelbart WM (2002). "Universal Fast Walking for direct and versatile determination of flanking sequence". Gene 284: 125–131. doi:10.1016/S0378-1119(02)00384-0. PMID 11891053.
· ^ Park DJ Electronic Journal of Biotechnology (online). 15 August 2005, vol. 8, no. 2
· ^ Park DJ (2005). "A new 5' terminal murine GAPDH exon identified using 5'RACE LaNe". Molecular Biotechnology 29: 39–46. doi:10.1385/MB:29:1:39. PMID 15668518.
· ^ Park DJ (2004). "3'RACE LaNe: a simple and rapid fully nested PCR method to determine 3'-terminal cDNA sequence". Biotechniques 36: 586–588,590. PMID 15088375. In order to concentrate DNA and remove detergents, salts, and other low molecular weight contaminants from our synthesized DNA, Alcohol precipitation is used. The nucleic acids will not precipitate if there is a monovalent cation to neutralize the negative charges and permit the DNA to aggregate. Na or Cl acetate are most often used in the lab to precipitate DNA because it can also serve as a buffer around PH 5.2. which provide an acidic environment that favors precipitation of DNA. NH4 acetate will be use when there are free deoxynucleotide since they co-precipitate less with NH4 than with Na or Cl. After adding salts, isopropanol or ethanol will be added to reduce solubility which will favor DNA precipitation. Alcohol will form hydrogen bonds with water, in other words, reduces the amount of water for dissolved substance. In addition, the polarizability, or capacity of alcohols to shield opposite charges (keep them away from each other) is less than that of water. Gene expression in organisms is carefully regulated to allow them to adapt the differing conditions and most importantly to prevent wasteful overproduction of unneeded proteins which would make the organism not productive. Organisms regulate expression of their genes for the reasons of developmental changes, cellular specialization and adaptation to the new environment.The expression of genetic information in a given cell or organism is neither random nor fully pre-programmed. The information in an organisms genome must be regulated in an orderly fashion during development and yet must be available to direct the organisms responses to changes in internal and external conditions.
In prokaryotes gene expression is almost entirely controlled at the level of transcription. The expression of specific genes may be actively inhibited or stimulated through the effects of proteins that bind to DNA or RNA. Unlike the prokaryotic control systems the eukaryotic mechanisms must contend with much larger amounts of DNA that is packaged in inaccessible structures. Non expressed DNA is typically highly condensed in a form known as heterochromatin. An extreme example is the complete inactivation of one of the two chromosomes in female mammals known as Barr bodies. Virtually every stage of a proteins existence from transcription to posttranslational modification offers opportunities for regulation. Posttranslational control such as alternative mRNA splicing can yield multiple proteins from a single gene. Additional control of eukaryotic gene expression is effected by variable rates of mRNA degradation and regulation of translation initiation.
There are over 25000 genes in our human genome. Each gene codes for a unique protein. Regulation of the expression of proteins occurs at the level of transcription from DNA in to RNA. The regulation takes place at a specific position on the DNA template, called promoter, this is the place where RNA polymerase looks for and knows where to start the transcription of the gene. Sometimes a group of related genes cluster together to form an operon and copied into mRNA molecule.
For example the genes involved in the transport and breakdown of food is highly regulated genes. The food we eat needs a digestive enzyme in our mouth to breakdown the sugar to facilitate the absorption by our body. The genes which code for these enzymes are not expressed when arabinose is absent, they only expressed when arabinose is present in their environment. When arabinose is present in the environment, bacteria take it up. and the arabinose interacts directly with araC which bound to the DNA. The interaction causes araC to change its shape which in turn promotes the binding of RNA polymerase and the genes are grascribed. the enzymes are produced.They break down arabinose and eventually the arabinose runs out. In the absence of arabinose the araC returns to its original shape and transcription is shut off. This is a good example of gene regulation. Structural Biochemistry/Synthetic biology
Zinc fingers
[edit | edit source]A zinc finger is a self-contained domain that formed through the stabilization of a zinc ion binded to a pair of cysteines and a pair of histidines. In addition an inner hydrophobic attaches to it also. The discovery of the zinc finger revealed a new protein fold but also an important aspect of DNA recognition. In contrast to other DNA binding proteins that usually utilize the two-fold symmetry o the double helix, zinc finger are bonded together in a single file to recognize nucleic acid sequences of differing lengths. This standard design gives numerous combinations for the specific recognition of DNA or RNA. Therefore, it is expected that the zing finger is present in nature in great abundance. Also this explains why it is 3% of the genes in the human genome.
The zinc finger's most suitable role is for engineering proteins to find specific genes and target them. One application of them in 1994 used three finger protein to knock off the expression of oncogene transformed and modified into a mouse cell line. Also, targeting a slipped in zinc finger promoter led to the activation of a reporter gene. Therefore, genes can be switched on or off in a carefully chosen manner through the fusion of zinc finger peptides to repression or activation domains. In addition, it is possible that putting zinc fingers with the other effector domains like from nucleases or integrases, to make chimeric proteins, genomes could be controlled and modified. There are a couple applications of engineered zinc finger proteins, which can have therapeutic importance.
It has taken about ten years of research on the structure of chromatin to finally discover the nucleosome and a general depiction of its basic structure. Also it led to the discovery as the next level for the folding of DNA in the 300-A chromatin fiber. This led to an interest to what was labeled "active chromatin" back then. This is the chromatin that is involved in transcription or that was held in a balanced manner to do so, and finding a tractable system. This can lead to many possibilities of extracting relatively big quantities of material for biochemical and structural studies.
Robert Roeder and Donal Brown who studied the 5S RNA genes of Xenopus laevis, which transcribed by RNA polymerase III was interesting. They found out that the accurate starting stage of transcription needs the binding of a 40kDA protein factor, which is also known as factor A or transcription factor IIIA, which if extracted and purified from oocyte extracts. By utilising a method called deletion mapping, it was found that this factor interacts with a region about 50 nucleotides long inside the gene, which is also known as the internal control region. The first eukaryotic transcription factor that was described was this.
Not very developed oocytes serve as place for 5s RNA molecules to be stored in in the form of 7S ribonucleoprotein particles, which each have a single 40-kDa protein, which was displayed to be identical with TFIIIa. TFIIIA hence, attaches to both the 5S RNA and its cognate DNA. In effect, it was thought that it may regulate the auto regulation the 5S gene transcription. Regardless of whether this autoregulation happens in vivo or not, the dual interaction gave an intriguing structural problem that could be gone about because of the big amounts of protein TFIIIA present in immature Xenopus oocytes.
A grad student named Miller, studied TFIIIA and found out an amazing repeating motif within the protein, which was later called a zinc finger because it had zinc and attached onto the DNA. This repeating structure was found through biochemistry and not through computer sequence analysis.

Preparation and Characterization of TFIIA
[edit | edit source]As Miller did his experiments over and over again for the published protocols for purifying the 7S RNP, he got very low yields, which were associated to dissociation. Brown and Roeder utilized buffers that had dithiothritol because the protein contained high level of cysteine content and EDTA to remove any contamination by metals, which hydrolyze nucleic acids. The gel filtration of the complex in 0.1 mM DTT led to a separate elution of protein and 5S RNA. However when the strong reducing agent sodium borohydride did not alter the complex, it was found out that the protein was not bound together by disulfide bridges and that a metal might have been associated with it. Then when after the particle was incubated with an array of chelating agents, the prevention of the particles breaking apart could occur with only the addition of Zn2+ beforehand and not by other types of metals. When analyzing and the partially purified 7S preparation solution using atomic absorption spectroscopy also showed that a good amount of concentration of Zn.
While these experiments occurred,Zinc was present in the 7S RNP at a ratio of two per particle. This was under the accurate value because their buffers had 0.5M or 1mM DTT, which possesses a high binding constant for Zn. Miller repeated this process with pure and intact particle preparations, without DTT. He concluded that the native 7S RNp has about 7 and 11 Zn ions.

Reference
[edit | edit source]↑ Klug Aaron(2009).The discovery of Zinc Fingers and their Applications. "Annual Review of Biochemistry", p. 3-6. Gene regulation is often affected by variations stemming from the chromatin. A multitude of mechanisms play roles in gene regulation, including “regulation of the core transcription machinery, recruitment of the transcriptional activators or repressors, and altering chromatin structure via post-translational covalent modification of histone proteins.” Yet the most studied and proven mechanism roots from variations of chromatin architecture. Recent research has pointed to a variety of metabolic molecules to be responsible for modulation of genes. Some key metabolic regulators found were: Acetyl-CoA, NAD+, SAM and alpha-KG.
In a landmark study by Kaochar et al., acetylation was found to play a major role in histone modifications, which ultimately leads to gene transcription.[1]
Acetyl-CoA
[edit | edit source]History
[edit | edit source]In 1964, Allfrey et al. found that the acetylation of the histone tails facilitated access to DNA for transcription by neutralizing the positive charge of lysine side chains. This is due to the fact that many transcription factors interact directly with the acetylated lysines. A significant correlation was found between the ability of a protein to acetylate histones and activate transcription.
Mechanism
[edit | edit source]The wide range of post-translational modifications of histone proteins allow chromatins to take on a variety of structures. This, in turn, contributes to the cell’s ability to control gene expression.
References
[edit | edit source]RNA, Enzyme, Metabolites
[edit | edit source]It has been conventionally thought that eukaryotic cells have been able to evolve and adapt to their surroundings without the need for gene regulatory mechanisms. Recent studies have shown that in specific cases, this may not necessarily be true as these cases have shown connections between intermediary metabolism and the regulation of gene expression.[1] Currently, there is evidence to show that there are interactions among the RNA, enzymes, and metabolites in gene regulation.
Enzymes Regulating RNA Expression
[edit | edit source]Hentze and Preiss listed several biological cycles which demonstrate that enzymes bind to RNA, which include the tricarboxylic cycle, glycolysis and pentose cycle, fatty acid metabolism, and pyrimidine synthesis.[1] The examples that Hentze and Priess use are cystolic aconitase, GADPH, and the three enzymes used for the thymidylate synthesis cycle.
Cytosolic Aconitase
[edit | edit source]An example of an enzyme that can affect regulation of RNA expression is cytosolic aconitase. This enzyme acts by using its iron-sulfur group as a catalyst. However, cytosolic aconitase can also act as an RNA-binding protein (labeled IRP1) when the cytosolic aconitase loses its iron sulfur cluster. This is significant in that this protein has two mutually exclusive activities that are regulated by whether the iron-sulfur cluster is present or not.
The presence of iron-sulfur clusters, as mentioned above, affects what type of activity cytosolic aconitase can perform. This is an example of how metabolism and the resulting metabolites can regulate interactions between enzymes and RNA. A cell that is iron-deficient will result in cytosolic aconitase losing its iron-sulfur cluster, transforming it into the IRP1 protein that will bind to iron-responsive RNA elements (IRE); the IRE will then regulate the production of necessary compounds to maintain homeostasis in the cell.[1]
GAPDH
[edit | edit source]GAPDH is an enzyme that has been shown to bind to several different types of RNA including, mRNAs, tRNA,s rRNA, and viral RNAs.[1] Hentze and Priess state that this protein is thought to bind to the 3' side of untranslated regions of lymphokine mRNAs because of their richness in AU.
GAPDH is also involved in the GAIT complex, which is a γ-interferon-activated inhibitor of translation.[1] In this complex, the GAPDH controls mRNA translation under the direction of the γ-interferon.

TS, SHMT, and DHFR
[edit | edit source]TS, SHMT (serine hydroxymethl-transferase and DHFR are enzymes involved in the thymidylate synthesis cycle. They are another example of proteins that are known to bind to RNA. These three enzymes regulate mRNA translation by binding to areas in the 5' untranslated regions.[1]

RNA Regulation of Enzyme Activity
[edit | edit source]Hentze and Priess assert that enzymes' catalytic activity can be controlled by RNA binding when enzymatic and RNA functions are competing.[1] The evidence for this activity is shown though the enzyme IDH (isocitrate dehydrogenase), which is a yeast mitocondrial protein that is NAD+ specific. IDH binds to the 5' end of mitochondrial mRNA to stop their activity. Although there is not conclusive evidence, Hentze and Preiss also suggest that noncoding RNAs could regulate enzymatic activity by directly binding to those proteins.
Metabolites
[edit | edit source]There are still vast amounts of research to be done on metabolites. Hentze and Priess assert that metabolites play the role of balancing RNA and enzymatic activity either directly or allosterically (binding a molecule to the active site of a protein). Changes in metabolism can change the body's metabolite concentrations, which will effect proteins with two different functions. One example would be how the cytosolic aconitase switches between its RNA binding role and its catalytic role. Another example of how metabolites are involved in this REM cycle is how the change in nucleotide levels would affect the function of GAPDH. Factors that disrupt nutrition, redox state, or oxygen tension could change the NAD+/NADH ratio, which will change protein functions.[1]
Systematic Exploration of REM Networks
[edit | edit source]As mentioned above, REM regulation requires research for it to be fully explored. Hentze and Priess suggest two steps to begin the study. The first step would to be to catalogue different types of RNA-binding enzymes using corosslinking techniques. The second step would be to identify the different types of RNA that these enzymes bind with.[1]
Conclusion
[edit | edit source]While the evidence (as well as other observations) points to the existence of this idea of REM gene regulation, it should be noted that there lacks a complete underlying concept. Regardless, the findings show that there exists much more in gene regulation than adaptation. This has significant implications, even for medicine; one may use the interactions among the RNA, enzymes, and metabolites to create drugs that specifically target a specific point that can directly affect gene regulation. For now, the RNA-binding enzymes, proteins, and RNAs, must be identified for further study on the REM phenomenon.
References
[edit | edit source]Structural Biochemistry/Bacterial transformation technique/
Overview
[edit | edit source]The genetic code is the relationship between DNA base sequences and the amino acid sequence in proteins. The genetic code features include:
- amino acids are encoded by three nucleotides
- it is non-overlapping
- it has no punctuation (alternatively, the stop codons could be viewed as a "period" or "full stop".)
- it is described as degenerate or redundant.
There are 20 amino acids but 4 bases so a minimum of three bases are needed to code at least 20 amino acids. The set of three nitrogenous bases that code for an amino acid are known as a codon. There are 64 codons in total, 61 that encode amino acids and 3 that code for chain termination. The first letter of the code codes for a certain class of amino acids (for example aromatic rings, etc.) For this reason, a mistake in the first letter would cause the worst damage to an organism, as it would likely code for a completely different class of amino acid.
Nonoverlapping
[edit | edit source]The genetic code is non-overlapping; for example in a sequence of ABCDEF, ABC would code the first amino acid and DEF the second whereas in an overlapping code ABC could code for the first amino acid and BCD the second. The genetic code has no internal punctuation (like commas and semi-colons) such as having X in between each codon like XABCXDEFX... since it is read sequentially from a starting point (however it could be argued that the so called "stop" codons function as "periods" during translation). Therefore, a deletion or insertion mutation that does not occur in a multiple of three results in a frame shift mutation. The reading frame of the codons are shifted after the mutation and often result in a stop codon shortly afterward. Due to the extreme impact, frame shift mutations are often deleterious.
Degeneracy
[edit | edit source]The genetic code is degenerate in that most amino acids are encoded by more than one codon with the exception of tryptophan and methionine which only has one codon. Codons that specify the same amino acids are called synonyms and these codons usually differ in the last base of the triplet. Degeneracy is significant since it helps reduce the deleterious effects of mutations because point mutations, differing in only one amino acid, do not generally significantly alter the protein if at all. But of course, all genetic information does not solely depend on genetic codes, it is also contributed from regulatory sequences, intergenic segments and chromosomal structural areas, which are not as simple as this chart of genetic code. Another term that is synonymous with degeneracy of the genetic code is redundancy.
Translation process
[edit | edit source]Translation is the synthesis of a protein from an mRNA template. This process involves several key molecules including mRNA, the small and large subunits of the ribosome, tRNA, and the release factor. The process is broken into three stages: initiation, elongation, and termination. Eukaryotic mRNA, the substrate for translation, has a unique 3’ end called the Poly-A Tail. Messenger RNA (mRNA) also contains codons that will encode for specific amino acids; a methylated cap is found at the 5’ end. Translation initiation begins when the small subunit of the ribosome attaches to the cap and moves to the translation initiation site. Transfer RNA (tRNA) is another key molecule. It contains an anti-codon that is complementary to the mRNA codon to which it binds. The first mRNA codon is typically AUG. Attached to the end of the tRNA is the corresponding amino acid; methionine corresponds to the AUG codon. The large subunit of the ribosome now binds to create the peptidyl, or (P) site, and the aminoacyl, or (A) site. The first tRNA occupies the P-site, while the second tRNA enters the (A) site and is complementary to the second mRNA codon. The methionine is then transferred to the (A) site amino acid, the first tRNA exits, the ribosome moves along the mRNA, and the next tRNA enters. These are the basic steps of elongation. As elongation continues, the growing peptide is continually transferred to the (A) site tRNA, the ribosome moves along the mRNA, and new tRNAs enter. When a stop codon is encountered in the (A) site, a release factor enters the (A) site and translation is terminated. When termination is reached, the ribosome dissociates, and the newly formed protein is released.
Table of genetic codes
[edit | edit source]| U | C | A | G | ||
|---|---|---|---|---|---|
| U | |||||
| Phe | Ser | Tyr | Cys | U | |
| Phe | Ser | Tyr | Cys | C | |
| Leu | Ser | Stop | Stop | A | |
| Leu | Ser | Stop | Trp | G | |
| C | |||||
| Leu | Pro | His | Arg | U | |
| Leu | Pro | His | Arg | C | |
| Leu | Pro | Gln | Arg | A | |
| Leu | Pro | Gln | Arg | G | |
| A | |||||
| Ile | Thr | Asn | Ser | U | |
| Ile | Thr | Asn | Ser | C | |
| Ile | Thr | Lys | Arg | A | |
| Met | Thr | Lys | Arg | G | |
| G | |||||
| Val | Ala | Asp | Gly | U | |
| Val | Ala | Asp | Gly | C | |
| Val | Ala | Glu | Gly | A | |
| Val | Ala | Glu | Gly | G |
Universal Code?
[edit | edit source]Is the genetic code universal across all species? Since the genetic base sequence is known for many wild-type and mutant genes, the nucleotide and amino acid change in the genes can be correctly predicted by the genetic code. mRNA can be translated correctly by protein synthesizing methods of many different species. For example wheat-germ extract can correctly translate human hemoglobin mRNA and human bacteria can express recombinant DNA molecules encoding human proteins as insulin and such. Although these findings suggest that the genetic code is universal across species it was proven otherwise when the DNA for the human mitochondrial DNA became known. Human mitochondrial DNA differed in the translation of the genetic code that it read UGA as coding for tryptophan rather than as a stop signal. Also AGA and AGG are read as stop signals instead of arginine and AUA codes for methionine rather than isoleucine. It was also found that mitochondrial DNA of other species also have genetic codes that differ slightly. The mitochondrial DNA can differ from the rest of the cell's DNA because it encodes a distinct set of tRNAs. Also some cellular protein-synthesizing systems, at least 16, deviate from the standard genetic code such as the ciliated protozoa which reads UAA and UAG as codons for amino acids rather than stop signals. UGA is used as their only stop signal. Slight variations in genetic code exist in mitochondria and species that have branched off early in eukaryotic evolution. Most variations of the genetic code are for a simpler code and diminish information in the third base of the triple such as both AUA and AUG being codons for methionine. Therefore the genetic code is almost nearly universal but not quite.
The invariance of the genetic code through evolution is likely a result of selection against deleterious mutations that would arise if a mutation that altered the reading of mRNA changed the amino acid sequence of the proteins created by the organism.
References
[edit | edit source]1. Berg, Jeremy M. 2007. Biochemistry. Sixth Ed. New York: W.H. Freeman. 125-127 Structural Biochemistry/Gene expression
Introduction
[edit | edit source]Flexible chromatin is needed for eukaryotes to constantly tuning their gene expression in transcript levels in order for them to respond to the environment. Such constant rapid changes in gene expressions can be done through post-translational modifications of histone proteins, which controls the structure of chromatin. Recent studies have revealed that several particular metabolites, in fact, may have shown to be the key regulators that link gene expression with cellular metabolism.
Chromatin Architecture and Histone Modifications
[edit | edit source]In eukaryotes, 2 meter of 146bp DNA is condensed and packaged into chromatin structure by wrapping it around an octamer, which contains two copies of each histone proteins H2A, H2B, H3 and H4, in order to fit inside a nucleus and at the same time, allow access to the genetic material for replication, repair and transcription in such compacted form. The amino terminal of every lysine-riched histones are exposed to modifications such as acetylation, methylation, phosphorylation, ubiquitylation, SUMOylation and poly-ADP-eibosylation with the help of modifying enzymes, which uses metabolites such as ATP, NAD+, actyl-coenzyme A and S-adenosylmethionine.

Link Between Chromatin and Metabolism
[edit | edit source]The first study of chromatin-level gene regulation was done by Allfrey, in 1964. Allfrey hypothesized that RNA synthesis is closely related to histone acetylation. It was thought that the positively charged lysine tail of histone and the negatively charged backbone are neutralized through acetylation for transcription activities. There are many transcription factors that has bromodomain, which interacts specifically with lysine after acetylation. In addition, acetylation is also important for binding trans-acting factors and chromatin remodeler. Support of this hypothesis was actually first discovered when studying the “silent” chromatin in Saccharomyces cerevisiae. The silent information regulator, a subset of histone deacetylases that is also known as SIR/sirtuin, was found to be responsible for keeping the amino group of histone H3 and H4 in specific genome region in hypoacetylated form. Other sirtuins, such as yeast sir2, were also found to decetylate lysine residue on histone by NAD+, which is used as substrate. The fact that sirtuins are regulated by NAD+ levels suggests a strong connection between metabolism and gene expression regulation.
Acetyl-CoA and Histone Acetylation
[edit | edit source]Histone acetylation was first discovered and identified by Brownell and Allies in the mid 1990s when studying polypeptide in Tetrahymena Thermophila. This study revealed that there is histone acetyltransferase (HAT) activity within the polypeptide chain, which is similar to yeast Gcn5 transciptional coactivator, thus showing a direct relationship between protein and histone acetylation that affects gene transcription. Besides HAT, lysine acetyltransferase (KAT) activity had also been identified to be responsible for acetylation. Both HAT and KAT were found to use acetyl-coenzyme A (acetyl-CoA), an important metabolite that accounts for many metabolic reactions in a cell, as acetyl donor during the process of acetylation. This shows that acetyl-CoA production may be crucial in acetyltransferase regulation and that the levels of acetyl-coA may affect or limit the modifications of histone. In yeast, enzyme that synthesize acetyl-CoA , acetyl-CoA synthetases Acs1p and Acs2p, are shown to be the key regulators of chromatin and gene expression. Studies have proven that mutation of either enzyme leads to growth defects and loss of H3/H4 acetylation, thus altering gene transcription. Similar cases occur in mammalian acetyl-CoA-producing enzymes such as ACL. Further studies have found that acetyl-CoA is not only important for histone acetylation, but also important for modification of nonhistone acetylation. Many cellular processes such as DNA repair, cell cycle progression, differentiation, replication, and apoptosis are found to be regulated by the acetylation of nonhistone proteins.

SAM and Histone Methylation
[edit | edit source]S-adenosylmethionine (SAM), which is produced by SAM synthetases called methionine adenosyl transferase (MAT), gives off its methyl groups to methyltransferases, HMTs, to carry out DNA, RNA and proteins methylation, as well as nonhistone methylation. As shown in figure “Methylation and demethylation of histone”, SAM can also be converted to S-adenosylhomocystein (SAH), which can act as inhibitor of methyltransferases if its level is high.
Through recent studies, SAM has shown a close connection with gene repression. When there is a single point mutation, the synthesis of SAM is prevented due to the breakdown of MatIIα, thus the transcriptional repression is reversed. Moreover, the demethylation of H3K4 and H3K9 greatly decreases.
α-Ketoglutarate (KG) and Histone Demethylation
[edit | edit source]For demethylation, histone demethylases (HDMs) act to remove the methyl groups from histone. As shown in figure "Methylation and Demethylation of Histone",α-Ketoglutarate (α-KG), which is also known as 2-oxoglutarate), is utilized as a substrate by Jmjv-domain-continaing histone demethylases (HDMs) to demethylate histones, while flavin adenine dinucleotide (FAD) is used as cofactor by lysine-specific histone demethylases (LSD). When mutation occurs in isocitrate dehydrogenase (IDH) genes, enzymes are not able to convert isocitrate to α-KG during the TCA cycle, but instead, a competitive inhibitor of HDM called 2-hydroxyglutarate (HG) are produced, which would lead to transcriptional repression. Furthermore, another α-KG-dependent enzyme called TET2 would also greatly reduce. All these would affect the methylation of DNA and thus alter the gene expression.
Reference
[edit | edit source]Kaochar, Salma and Benjamin P. Tu. “Gatekeepers of chromatin: Small metabolites elicit big changes in gene expression.” Trends in Biochemical Sciences. 37.11 (2012):477-483. <http://www.sciencedirect.com/science/article/pii/S0968000412001120>. In general, there are two ways that mutations in DNA sequences could occur:
Environmental effects
[edit | edit source]Altering Nucleotide bases: File:Environmental agents damage DNA.jpgEnvironmental effects such as Ultraviolet light, radiation, or toxic chemicals could modify nucleotide bases to make them look like other nucleotide bases, resulting in damages of DNA. For example, certain environmental agent will change the structure of Guanine base so that it has the shape like Adenine. Thus, during the DNA replication, that “Guanine base” can no longer bind to Cytosine, instead, because it has a shape of Adenine, it will bind to Thymine.
Breaking the Phosphate backbone: Environmental agents can also break phosphodiester bonds between oxygen and phosphate groups. By breaking the phosphate backbone of DNA within a gene, a mutated form of that gene could form. And this mutated gene might results in a mutated protein that functions differently, and might cause protein misfolding diseases.
However, cells usually attempt to fix the broken dens of DNA by joining free ends to other DNA fragments in the cell. This creates “translocation,” another kind of mutation. If this translocation breakpoint happens inside or near a gene, that gene’s functions may be influenced.
DNA replication mistakes
[edit | edit source]During DNA replication, DNA helicase first separates the DNA double-strand into two single strands. Then, DNA polymerase helps add corresponding nucleotides to both template strands, creating two double-stranded DNA molecules. However, DNA polymerase could make mistakes during this process at the rate of once every 100,000,000 bases. The result is mutations of genes, which could lead to many malfunction of translated protein.
In fact, most of the mistakes can be repaired by a type of protein later in the replication process. This protein will replace incorrectly paired nucleotides with correct ones. So that the number of mutations of DNA is actually lower.
Frameshift mutations
[edit | edit source]Frameshift mutation is caused by insertion or deletion of a number of nucleotides that is not evenly divisible by three from a DNA sequence. It often occurs when the addition or loss of DNA bases disrupts a gene’s reading frame. A reading frame consists of groups of three DNA bases (codons), which each code represents one specific amino acid. The resulting protein is usually nonfunctional because the wrong reading frame of the gene translate a very different protein sequence from the normal reading frame.
For example, mRNA with sequence AUG CAG AUA AAC GCU UAA Normal amino acid sequence reading frame should be: MET GLN ILE ASN ALA STOP However, a wrong reading frame (a deletion of the first base 'A') could give a traslation of mRNA of: UGC AGA UAA ACG CUU AA an abnormal amino acid sequence translate would be: CYS ARG STOP
In the case above, a frameshift mutation causes the reading of all codons after the mutation to code for different amino acids. The stop codon ("UAA")cannot be read, which a stop codon could be created at an earlier or later site. The protein being created above is abnormally short, which contain the wrong amino acid; thus, it is nonfunctional.
Frameshift mutations can result in severe genetic diseases such as Tay-Sachs disease, which is caused by the missing enzyme due to genetic mutation that result in the accumulation of fatty substance (Gangliosides) in the nervous system. However, frameshift mutation can be beneficial. For example, a frameshift mutation was responsible for the creation of nylonase, which is capable to digest certain byproducts of nylon 6 manufacture.
Chromosomal Translocation
[edit | edit source]Another type of DNA mutation that can occur is chromosomal translocation, which is a chromosome abnormality that is caused by the rearrangement of parts of nonhomologous chromosomes. There are instances where the two separated genes are joined together, forming a fusion gene, which is common in cancer. This fusion gene can be detected on a karyotype of affected cells. There are two main types of chromosomal translocation that can occur: reciprocal (non-Robertsonian) and Robertsonian. Translocations can also be balanced, where there is an even exchange of genetic material with no information extra or missing, or unbalanced, where the exchange of chromosomal material is uneven resulting in extra or missing genes. Some diseases that result from translocation include cancer, infertility, and Down Syndrome.
Reciprocal Translocation
[edit | edit source]Reciprocal translocations are usually an exchange of material between nonhomologous chromosomes. These kinds of translocations are, for the most part, harmless because the amount of genetic material exchanged is the same. They can usually be detected through prenatal diagnosis. However, carriers of balanced reciprocal translocations have an increased risk of creating gametes that have unbalanced chromosome translocations that end up leading to miscarriages or even children with abnormalities.
Robertsonian Translocation
[edit | edit source]The Robertsonian translocation is most commonly found in children with Down Syndrome. The parents of children with Down syndrome are carriers of unbalanced gametes which lead to miscarriages and/or abnormal offspring. The case of translocation in children with Down syndrome is called trisomy.

Chromosomal Inversion
[edit | edit source]An inversion is a rearrangement of the chromosomes where a segment of the chromosome is reversed end to end. An inversion occurs when a single chromosome undergoes breakage and rearrangement within itself. There are two types of inversions: paracentric and pericentric. Paracentric inversions do not include the centromere and so both breaks occur in one arm of the chromosome. Pericentric inversions do include the centromere and so there is a break point in each arm.
Inversions usually do not cause any abnormalities in carriers so long as the arrangement is balanced. This means there are no extra or missing genetic information. However, those who are heterozygous for an inversion have an increased production of abnormal chromatids, which leads to lowered fertility due to the production of unbalanced gametes.
Point Mutations
[edit | edit source]A point mutation is a mutation where a single base nucleotide is replaced with another nucleotide of the genetic material, DNA or RNA. The term point mutation often includes insertion and/or deletions of a single base pair. Point mutations can be categorized as one of two types:
Transitions: the replacement of a purine base with another purine or the replacement of a pyrimidine with another pyrimidine
Transversions: the replacement of a purine with a pyrimidine or vice versa
Point mutations can also be categorized functionally:
Nonsense mutations: code for a stop, which can truncate the protein
Missense mutations: code for a different amino acid
Silent mutations: code for the same or a different amino acid but there is no functional change in the protein
An example of a missense mutation is sickle-cell disease. A missense mutation occurs in the beta-hemoglobin gene that converts a GAG codon into a GTG codon, which encodes for the amino acid valine rather than glutamic acid.

Insertion
[edit | edit source]Insertion is the addition of one or more nucleotide base pairs into a DNA sequence. This can happen often when DNA polymerase is slipping in microsatellite regions. Insertions can vary in size, some being only a single nucleotide base pair whereas others can be a section of another chromosome being inserted into the DNA sequence. On the chromosomal level, an insertion refers to the insertion of a larger sequence into a chromosome. This usually occurs due to unequal crossover during meiosis. There are a couple different kinds of insertions that can occur based on how and what is inserted. An N region addition is the addition of non-coded nucleotides during recombination by terminal deoxynucleotidyl transferase. A P nucleotide insertion is the insertion of a palindromic sequence encoded by the ends of the recombining gene segments.

Deletion
[edit | edit source]Deletion is a mutation in which a part of the chromosome or a sequence of DNA is missing. Deletion is the loss of genetic material. Any number of nucleotides can be deleted, ranging from a single base pair to an entire piece of the chromosome. Deletions are usually caused by errors in chromosomal crossover during meiosis. Some of the causes of deletions include losses from translocation, chromosomal crossovers within a chromosomal inversion, unequal crossing over, and breaking without rejoining. Some types of deletions are terminal deletion and interstitial deletion. Terminal deletion is a deletion that occurs near the end of a chromosome. Interstitial deletion is a deletion that occurs from the interior of the chromosome.
Small deletions are less likely to be fatal while large deletions can be more fatal because there are always variations based on what genes are lost. Some of the medium-sized deletions can lead to recognizable human disorders. Deletions are responsible for a variety of genetic disorders, such as male infertility and two thirds of cases of Duchenne muscular dystrophy.

Amplification
[edit | edit source]Amplification is the duplication of a region of DNA that contains a gene and can occur as an error in homologous recombination, a retrotransposition event, duplication of an entire chromosome. This duplication arises from unequal crossing-over that takes place during meiosis between misaligned homologous chromosomes. Amplification does not usually constitute a lasting change in a species' genome, not lasting longer than the initial host organism. Amplification is actually a way for a gene to be overexpressed. It can occur artificially via polymerase chain reaction or it can occur naturally, as was just explained.
Gene amplification is believed to play a major role in evolution and this belief has lasted for over 100 years in the scientific community. The duplication of a gene results in an additional copy that is free from selective pressure. The new copy of the gene is then allowed to mutate without deleterious consequences to the organism. With this freedom from these consequences, the mutation of novel genes can occur which could potentially increase the fitness of the organism or code for a brand new function. The two genes that are present after the gene duplication are paralogs and they usually code for proteins that have similar function and/or structure.
Deamination
[edit | edit source]
Deamination is removing the amino group from the amino acid and converting to ammonia. Since the bases cytosine, adenine and guanine have amino groups on them that can be deaminated, Deamination can cause mutation in DNA. For example, If a cytosine were to be deaminated to form uracil (uracil is an analog of thymine) in the template strand of DNA, then the polymerase would put in an adenine at the corresponding position on the nascent DNA strand instead of a guanine. The hydrolysis reaction (deamination) of cytosine into uracil is spontaneous.
In response to this mutation the cell has a repair process. In this process the cell utilizes the enzyme uracil-DNA glycosidase to recognize these uracils and removes them. This enzyme hydrolyzes the N-glycosidic bond between the deoxyribose ring and the uracil base. Therefore, the uracil base is removed.

Since this site on the DNA duplex is without either purine base or a pyrimidine base is called an AP site (either apurinic or apyrimidinic). Then the enzyme AP endonuclease cut the bond on the 3' side of the phosphodiester bond of the nucleotide. In this phase DNA polymerase I recognizes phosphodiester bond at the 3'end on the next nucleotide unit and cleaves the bond. After the ribose-phosphate unit is removed, DNA polymerase I analyzes the complementary strand and finds that the base that corresponds to the AP site is guanine. Then the enzyme inserts a cytosine unit at the AP site on the broken DNA strand. Finally, DNA ligase seals the inserted cytosine into the damaged strand. Spontaneously deamination of cytosine to form uracil can be repaired by the cell. [1]
Quickchange Method
[edit | edit source]Quickchange is a technique used to generate site-specific mutations with minimal hands-on manipulation. The sites of mutations are incorporated in the two complementary primers and the rest of the plasmid DNA is synthesized with a high fidelity DNA polymerase in a thermal cycler. Therefore the whole process is considered quick.
Although the reaction is done in a thermal cycler, it is not PCR. Since the template is circular, the newly synthesized single stranded DNA will terminate at the beginning of the primer on the same strand. This product will not overlap with the primer on the complementary strand. Therefore the newly made DNA cannot be used as a template for further DNA synthesis. Only the original template DNA can be used as templates. In each cycle, the amount of newly synthesized DNA is equal to the template. This is considered linear amplified, rather than exponential amplification in PCR.
Since the template DNA is isolated from bacteria, it contains methylated nucleotides. That makes it sensitive to methylation dependent nucleases, such as DpnI. For example, after 20 cycles of amplification, 10 ng plasmid will be amplified 20 fold and produce 200 ng new DNA. At this moment, restriction endonuclease DpnI will be used to eliminate the original plasmid DNA. The mixture of DNA is then put into bacteria and each DNA species will be separated in different bacteria cells. To see whether a cell contains the right mutation, a single cell needs to be picked, grown up, and the DNA it contains analyzed. Designing the primers is critical. A minimal annealing temperature of 78oC has to be met. Otherwise, the primer will not be attached to its template and the termination will not be stopped precisely.
Quickchange Protocol
[edit | edit source]In general, the way in which quickchange if performed is in the steps below. This technique is similar to PCR. 1. If oligos are from IDT: Spin oligos down at high speed for 1 minute using a table top centrifuge and resuspend in H2O or TE buffer to a 10x stock (1250 ng/µl) If oligos are from Allele: Dilute oligos in H2O or TE buffer to a 10x stock (1250 ng/µl) 2. Further dilute oligos to 1x (125 ng/µl) in H2O or TE buffer 3. Dilute Template DNA to 20-50 ng/µl in H2O or TE buffer 4. Thaw 10x pfu ultra buffer and dNTP mix to room temperature 5. Set up quick change reaction in a 100µl thin walled PCR Tube: H2O: 40µl 10x PFU Ultra Buffer: 5µl dNTP mix (10mM): 1µl Template DNA (20-50 ng/µl): 1µl Forward Primer (125 ng/µl): 1µl Reverse Primer (125 ng/µl): 1µl PFU Ultra HF (2.5u/µl): 1µl Total: 50µl 6. Mix contents of PCR tube gently and place in the thermocycler. Program the thermocycler according to what is needed for the experiment 7. When the reaction has finished, remove the PCR tube from the thermocycler and add 1µl of Dpn1 directly to the contents of the PCR tube and incubate for 1 hour at 37°C 8. Perform a transformation (see transformation protocol) using 2-3µl of the Dpn1 treated PCR reaction into XL1 Blue competent cells and plate the entire volume onto LB agar plates that contain the antibiotic that corresponds to the template DNA. Incubate overnight at 37°C 9. Check plates for colonies the following day. If there are colonies, use them to inoculate 5 ml overnight cultures and perform mini preps the following day. Send 5µl of the mini prep DNA for sequencing and analyze the results

Reference
[edit | edit source]1. Campbell, Neil A. (2005). Biology. Pearson. ISBN 0-8053-7146-0. {{cite book}}: Check |isbn= value: checksum (help); Text "coauthors+ H.C. Van Ness, M.M. Abbott" ignored (help)
2. http://www.answers.com/topic/frameshift-mutation http://www.gmilburn.ca/2009/04/03/human-evolution-and-frameshift-mutations/ http://www-personal.ksu.edu/~bethmont/mutdes.html#types Transposons, also known as jumping genes, are segments of DNA in a single cell that can move in the genome. The process of transposon movement is called transposition. The effects of transposition are mutations and the lengthening or shortening of the genome. Transposons were once called junk DNA because they were seemingly useless. However, research has shown that these mobile segments of DNA are significant in the development of organisms.
Types of Transposons
[edit | edit source]Transposable elements longer and more complex than insertion sequence, called transposons, also move about in the bacterial genome. In addition to the DNA required for transposition, transposons include extra genes that go along for the ride, such as genes for antibiotic resistance. In some bacterial transposons, the extra genes are sandwiched between two insertion sequence. It is as though two insertion sequence happened to land relatively close together in the genome and now travel together, along with all the DNA between them, as a single transposable element. other bacterial transposons do not contain insertion sequences; these have different inverted repeats at their ends.
Transposons are not unique to bacteria and are important components of eukaryotic genomes as well.
Class I
[edit | edit source]Retrotransposons (Class I transposons) move to different areas of the genome through:
1. Transcribing to RNA
2. Reverse transcribing to DNA (via reverse transcriptase)
3. Inserting into a different area of the genome
Retrotransposons are prevalent in plants: the genome of maize and the genome wheat are composed of roughly 50-80% and 70% retrotransposon respectively. Roughly 40% of the human genome is composed of retrotransposons. The mechanism by which retrotransposons work is similar to that of retroviruses, suggesting that there is a relationship between the two.
The three subtypes of Class I transposons are:
1. Viral: encodes the reverse transcriptase enzyme which is used to reverse transcribe to DNA (step 2 of transposition) Behaves most like retroviruses in that it is characteristic of having long terminal repeats (LTRs).
2. Nonviral: does not encode the reverse transcriptase. Use RNA polymerase III
3. long interspersed elements (LINEs): encodes the reverse transcriptase enzyme. Uses RNA polymerase II
LTRs: Observed in retroviruses, long terminal repeats are segments of DNA that repeat from one hundred to a thousand times.
LINEs: Segment of DNA which codes for reverse transcriptase and also may code for an endonuclease.
Class II
[edit | edit source]DNA transposons (Class II transposons) can move to different areas of the genome through two mechanisms:
Mechanism 1: Transposase enzymes can bind to either a specific site of DNA or anywhere. Transposase cuts the DNA at the site it binds to and produces "sticky ends" and also cuts the transposon and ligates it to the specific site. Then DNA polymerase and DNA ligase run through the site to effectively enclose the transposon to the designated area.
DNA transposons that move through the genome via Mechanism 1 can be duplicated during the cell cycle.
Mechanism 2: The second mechanism by which Class I transposons move through the genome is called replicative transposition. In replicative transposition, the DNA transposon replicates itself into a new area of the genome.
DNA transposons are prone to eventually losing the ability to produce the necessary enzymes transposase or reverse transcriptase. However, other types of transposons will produce these enzymes, prolonging the ability of DNA transposons to move through the genome.
Transposons Are Mutagens
When transposons insert into the genome, they may potentially insert into a region encoding for a gene. This causes mutation because the gene will be inhibited. Insertion into introns, exons, and DNA sequences adjacent to the gene encoding region may also cause dysfunction of the gene. In addition to mutation due to insertion, mutation may occur if a sequence is copied several times such that it interferes in cell mitosis and meiosis. The irreparable gap produced when the transposon is removed may cause mutation as well.
Diseases resulting from mutations of this sort include Hemophilia, porphyria, and Duchenne muscular dystrophy. Transposition may also result in cancer.
References
[edit | edit source]Wikipedia.org: http://en.wikipedia.org/wiki/Transposon ; http://en.wikipedia.org/wiki/Long_interspersed_nucleotide_elements#LINEs
Kimball's Biology Pages http://users.rcn.com/jkimball.ma.ultranet/BiologyPages/T/Transposons.html
Copy Number Variants (CNVs) are a type of genetic mutation that results in an abnormal number genes or regions on a specific chromosome. CNVs contrast with Single Nucleotide Polymorphisms, which result in only a single mutated nucleotide. The development of SNPs outnumber CNVs by three orders of magnitude, their significance and contributions to variations are roughly similar. This is due to the fact that CNVs affect tens or thousands of kilobases, and thus its mutation impacts are large.
Importance of CNVs
[edit | edit source]Since CNVs contribute to a significant amount of differences between human genomes, they have been found to be important in both human disease and drug response. A better understanding of CNVs and how they work may help scientists better understand drug targets as well as human genome evolution.
Mutation Mechanisms
[edit | edit source]CNVs can arise from four different mechanisms: Nonallelic Homologous Recombination, Nonhomologous End-Joining, Fork Stalling and Template Switching, and Retrotransposition.
Nonallelic Homologous Recombination (NAHR), most commonly occurs during meiosis when two homologous chromosome pairs in the genome line up for crossing over. The positions of these chromosomes affect the rate at which a CNV mutation occurs. A slight mismatch of homologous chromosomes may result in the extra copy of a specific gene. Nonhomologous End-Joining (NHEJ) occurs when an abnormal repairing of a break of the DNA strand occurs. The repair results in either a deletion or more commonly a duplication of a segment in the genome. Forkstalling and Template Switching (FoSTeS) is induced by errors during DNA replication processes. These variants vary in both size and complexity. Retrotransposition of a DNA element may also result in an abnormal number of genes on a chromosome.
According to the 1000 Genomes Project, approximately 70.8% of deletions were attributed to NHEJ and 89.6% of small insertions were attributable to retrotransposition activity.
Clinical Significance
[edit | edit source]Most genetic mutations are benign. However, there have been few clinical studies showing that some of these genetic mutations may contribute to the onset of diseases. CNVs have been related to onsets of a prevalent amount of neuropsychiatric diseases, as a good proportion of schizophrenia, autism and bipolar cases are linked to CNVs. CNVs have also been found as an underlying cause elevated cancer cells.
References
[edit | edit source]Malhotra, Dheeraj, and Jonathan Sebat. "CNVs: Harbingers of a Rare Variant Revolution in Psychiatric Genetics." Cell 148.6 (2012): 1223-241. CNVs: Harbingers of a Rare Variant Revolution in Psychiatric Genetics. 16 Mar. 2012. Web. 15 June 2012. <http://www.cell.com/abstract/S0092-8674(12)00277-2>.
Daar AS, Scherer SW, Hegele RA. Implications for copy-number variation in the human genome: a time for questions. Nature Reviews Genetics, 2006, 7:414
Implications of Nucleic Acid Mutations
[edit | edit source]Before exploring genetic diseases, scientists must be familiar with possible types of DNA mutations(which include, but are not limited to frameshift, chromosomal translocation, insertion and deletions Structural Biochemistry/DNA Mutation). Multiple genetic mutations can alter biochemical processes, causing different diseases. Understanding the mechanical pathways have led to tremendous discoveries that introduce potential cures for these diseases. Genetic tests improve patients quality of life: earlier diagnosis, fewer misdiagnoses and early medical intervention.
Details on specific disorders are available to the public from the National Institute of Health, Genetic Disorders: Basics and Research Rare Diseases
Chromosomal Abnormalities
[edit | edit source]Duchenne Muscular Dystrophy
[edit | edit source]Duchenne Muscular Dystrophy is a genetic disease that causes the mutation of the Dystrophin protein, which is a rod- shaped protein found in muscle fibers and cardiac cells that function by connecting the muscle fiber cytoskeleton to extracellular matrix of cell membrane. The mutation is found in the DMD gene and is an x-linked recessive disease, which is passed from mother to son. Two thirds of the cases reported are inherited, however, one third of the cases arise from a new mutation that specific gene. The mutation causes the alteration of the shape and therefore function and a lack of production of the dystrophin protein causing symptoms such as, muscle weakness and heart problems, like, arrhythmias, cardiomyopathy and congestive heart failure. Approximately, 1 in 3,500 boys are affected and most of them do not live past their early twenties. Moreover, although females are not affected, they are at higher risk for developing heart problems in their lifetime.
Angelman Syndrome
[edit | edit source]Angelman Syndrome is a genomic imprinting disorder caused by a deletion or silencing of the genes inherited from the mother on the chromosome 15. This disorder is similar to the Prader- Willii syndromewhich affects the paternally inherited chromosome 15. Most people receive both genetic information from mother and father, however, because of this specific deletion of a segment of the chromosome, the maternal genetic information is silenced. This disorder can also be a result of a single gene mutation on the UBE3A gene. Some symptoms include, seizures, developmental delays, mental retardation, frequent laughter and smiling and jerky movement, such as hand flapping. This is not a degenerative disease; therefore, improvement in motor skills and living can be achieved through support and therapy.
Haemochromatosis Type 1
[edit | edit source]Haemochromatosis type 1 is a genetic disorder that causes the over absorption of dietary iron. Excess iron in the body aggregates in the tissues particularly in the liver, adrenal glands, heart and pancreas causing cirrhosis, diabetes, heart failure, and adrenal dysfunction. This disorder is caused by a mutation in the HFE gene, a gene that monitors and conducts the binding a transferrins which are protein that carrier iron in the blood. Typically these proteins are at low concentration in the bloodstream, however, when low iron levels are exhibited these proteins released to simulate the secretion of iron into the blood. Upon mutation, the intestines constantly signal for the release of these proteins causing an over secretion of iron.
Tay-Sachs Disease
[edit | edit source]Tay sachs is a autosomal genetic disorder characterized by loss of motor control, muscle strength and function, dementia and seizures, due to a defective gene on chromosome 15 caused by a deletion or base insertion. This disease is expressed when both copies inherited from both the mother and father are defective. This defective gene causes a decrease in the production of hexosaminidase A, which is a protein, specifically a lysosomal enzyme that functions in the chemical degradation a gangliosides, causing a build up in nerve cells in the brain. Most children afflicted with this disorder usually die before the age of five. Although the cure for this disorder is unknown, there is research conducted for possible therapies. Some of the therapies included: Enzyme replacement therapy, which would replace the defective enzyme with functioning enzymes similar to treatment of diabetes with insulin injections. In addition, Substrate Reduction Therapy which treats the build up of gangliosides by using other enzymes to catabolize it.
Polycystic Kidney Disease
[edit | edit source]Polycystic kidney disease is an autosomal dominant disease inherited from one parent, occurring in both children and adults. Symptoms included development of cysts in liver, kidneys and pancreas, diverticulitis of the colon and aortic and brain aneurysms. The pathophysiology of this disorder is currently unknown and treatment goal is to minimize symptoms by low salt diets, blood pressure medication and possible kidney transplants.
Trinucleotide Repeats
[edit | edit source]Long arrays of repeating sequences of three nucleotides are particularly prone to errors, which can cause serious diseases. Trinucleotide repeats tend to expand when repeats "loop out" of the double helix, such that one strand of the helix contains additional sequences than the other strand. (See Fig. 9 Bissler,1998
Huntington Disease Huntington disease is an autosomal dominant neurological disorder, due to a mutated gene that expresses the protein huntingtin. This protein is expressed in the brain. Huntingtin contains a repetition of glutamine residues encoded in the array of CAG sequences. People affected by this mutation have the CAG array from 36-82 (or longer) repeats while healthy individual have an array between 6-31 repeats. The array becomes longer as it is passed from one generation to the next, a phenomenon known as anticipation. Anticipation causes the children of an affected parent to have an earlier onset of the disease than the parent.
Bloom Syndrome
[edit | edit source]People with Bloom syndrome causes short physique, infertile males, and can be known to cause many types of cancers. It is an autosomal recessive disease that has a mutation in the locus for a gene that encodes a DNA helicase enzyme. With a mutated form of the BLM locus, people can have a higher risk of cancer if they are homozygous for the mutation. Those who are heterozygous are not as affected, but they have a greater risk of contracting colorectal cancer.
Noonan Syndrome
[edit | edit source]Noonan Syndrome is a genetic disorder caused by mutations in the PTPN11, SOS1, or KRAS genes. Approximately half of the people affected by Noonan syndrome have a PTPN11 mutation, and those with mutations in the other genes have a more severe, atypical form of the syndrome. Mutations in these genes cause certain proteins involved with growth and development to become overactive. However, the absence of a mutation in these genes does not exclude a possible diagnosis because there are still some unidentified genes that contribute to the syndrome under mutations. For this reason, genetic testing mainly provides a basis to examine a patient for all the physical symptoms possible and to know what to expect with regards to their health, and the diagnosis of Noonan syndrome is made primarily by clinical features. A physical examination alone can also be the basis for a diagnosis if the majority of symptoms are present. Most common symptoms include:
▪ Heart defects, especially pulmonary stenosis
▪ Delayed puberty
▪ Down-slanting or wide-set eyes
▪ Low-set or abnormally shaped ears
▪ Sagging eyelids
▪ Short stature
▪ Undescended testicles
▪ Unusual sunken chest shape
▪ Webbed and short-appearing neck
▪ Mild mental retardation in 25% of cases
Noonan syndrome can be inherited from a parent or can occur as a random mutation with no family history involved. Noonan syndrome affects males and females equally, and is seen between approximately 1 in 1000 to 1 in 2500 children born worldwide. It is one of the most prevalent genetic syndromes associated with congenital heart disease, very close in frequency to Down syndrome. The symptoms associated with Noonan syndrome can very widely in severity so a diagnosis is not always made at an early age.
A person with NS has up to a 50% chance of transmitting it to a child. However, there is not always and identified affected parent of children with Noonan syndrome. This shows that the manifestations of the syndrome could possibly be subtle enough as to not be identified in some patients (variable expressivity). This could also mean that a random mutation has occurred to cause the condition. Therefore, Noonan syndrome is heterogeneous, where multiple origins cause the disorder in different individuals, whether by inheritance with one or multiple mutations of different genes, or random mutation of one or all of the genes involved.[2]
Triplo-X Syndrome
[edit | edit source]This syndrome occurs when a female has more than two X chromosomes and are usually tall and thin and few cases of mental retardation have occurred although most have normal intelligence. Females that have this condition do not have many phenotypic differences; some may be infertile, but they are usually able to bear children and menstruate regularly. The more X chromosomes a female, the greater chance they will experience more severe effects such as mental retardation and other physical problems.
Progeria
[edit | edit source]Progeria meaning “prematurely old” in Greek, this rare disease is formally known as Hutchinson-Gilford progeria syndrome, this disease was noticed by Jonathan Hutchinson and Hastings Gilford. When babies are first born they seem healthy, but a couple years later they begin to display an unusual accelerated aging process and will experience hair loss, aged skin, stiff joints, heart disease and osteoporosis. Most cases die when they first reach their teenage years. The mutation for this disease is found on a gene called lamin A (LMNA) on chromosome 1.
Williams Syndrome
[edit | edit source]Williams Syndrome is caused by a deletion in chromosome 7, which includes at least 25 genes. This gives rise to a broad range of effects. This is not inherited, but rather due to a random mutation in chromosome 7 of a sperm or egg cell. Yet, it is considered an autosomal dominant condition, because just one abnormal chromosome is needed to cause the disorder. This developmental disorder is characterized by moderate intellectual disabilities, distinct facial and personality features, visual problems and cardiovascular problems. CLIP2, ELN, GTF2I, GTF2IRD1, and LIMK1 are among the genes that are typically deleted in people with Williams syndrome (Williams Syndrome, 2008). The prevalence of Williams Syndrome in the United States is estimated to be about 1 in 7,500-20,000.
Hermansky-Pudlak Syndrome
[edit | edit source]Hermansky-Pudlak Syndrome is anautosomal recessive genetic disorder that causes albinism or the loss of pigment due to the lack of production a melanin and accumulation of ceroids in lungs and kidneys. There are several different types of this disorder caused by mutations of different genes. Type 1, 3, 4, 5,6 are caused by several gene mutations, type 2 caused by a mutation on the AP3B1 gene and type 7 caused by a mutation in the gene expressing the information for the production of the dysbindin protein used in muscle fibers. Symptoms included easy bruising and bleeding, loss of pigmentation in hair, eyes and skin and colitis.
Neurofibromatosis type 1
[edit | edit source]Neurofibromatosis Type 1 is an autosomal dominant genetic disorder caused by a mutation on the NF1 gene, a gene that codes for the production of many different nerve cells, including schwann cells and oligodentrocytes and a protein called Neurofibromin that functions as a tumor suppressor. This disorder is characterized an over production of nerve cells due to the defective neurofibromin resulting in tumors to form. Some symptoms of this disorder include change in skin pigmentations and development of tumors in skin, nerves and brain. Half the cases are due to an inheritance of dominant allele from one parent and most of these cases developed other mutations on other genes due to unchecked cell growth. The other half is due to a new mutation developed on the NF1 gene.
Cystic Fibrosis
[edit | edit source]Cystic fibrosis is a recessive genetic disorder that is more common in the Caucasian population. It is a type of disorder where a thick, sticky mucus plugs up the airways in and out of the lungs as well as clogs the pathways that lead away from the pancreas and into the intestine. This can cause respiratory infections and digestive problems. This disorder can be life-threatening in some cases, depending on how much of the genetic disorder is expressed. Cystic fibrosis is a gene that has a codominance gene expression. The gene that causes cystic fibrosis can be found on chromosome 7. The gene codes a protein called cystic fibrosis transmembrane conductance regulator and people with cystic fibrosis have a mutated form of this protein. The regular protein functions as a gate into the cell membrane and controls the chloride ions in and out of the cell; people with the mutated protein have a closed channel and the chloride ions eventually build up and forming thick mucus. People who suffer this disease usually do not live past their 30s as their lungs become more susceptible to infection and other lung diseases such as pneumonia. Other medical problems include malnutrition and inefficient nutrient absorption within the digestive system.
Sickle Cell Disease
[edit | edit source]Sickle Cell Disease is a recessive genetic disorder that is caused by an error in the hemoglobin gene. Hemoglobin is the protein that carries oxygen in red blood cells. The incorrect amino acid mutation at one position distorts the normal circular, disc-like shape of red blood cells. Instead, the red blood cells are in an abnormal crescent shape. Their distorted shape often results in blood cells jamming together or break into pieces due to their inability to travel through small blood vessels. Consequently, those with sickle cell disease suffer from unpredictable pain in various organs and joints. The pain can either last for hours to up to several days. The blood vessel blockages prevent red blood cells from transferring oxygen to tissues and organs. Symptoms include abdominal pain, poor eyesight/blindness, ulcers and strokes. The disease is commonly found in those of African and Mediterranean descent. In fact, 1 in every 500 African Americans are affected by sickle cell disease. To increase the blood cell count, folic acid supplements can be taken. Treatment options include blood transfusions, pain medications and Hydroxyurea that can be used to reduce pain episodes. Those with the sickle cell trait only get it from one parent and thus do not suffer from the symptoms. However, those with sickle cell anemia inherit it from both parents and they are the ones who suffer from the sickle cell disease. Some people who are carriers or have sickle cell anemia are immune to the contraction of Malaria which is a disease propagated by a parasite. It is believed that due to a large proportion of sickle cell cases occur in the Africa and India, where malaria outbreaks are prevalent this immunity came to be, however, the link between the two pathogens are still unknown. One theory regarding the pathophysiology of the link could be that the malaria parasite is killed by the misshapen red blood cell due to the shorter life cycle of the cell because of its sickled shape halting in replication of this parasite and propagation of disease.
Turner Syndrome
[edit | edit source]Turner syndrome is a genetic disorder that occurs in about 1 in 5,000 births. It is a genetic abnormality that affects the development in only females. Girls born with Turner syndrome show certain physical attributes in which the levels of severity differ greatly between individuals. Most cases of Turner syndrome are not inherited. It is a genetic abnormality in which the female does have the usual pair of two X chromosomes. It is characterized as a chromosomal abnormality when one normal X chromosome is present in a female's cells and the other sex chromosome is missing or structurally altered. [3]
Phenylketonuria
[edit | edit source]Phenylketonuria (also known as PKU) is a autosomal recessive genetic disorder with a mutation in the gene coding for the enzyme phenylalanine hydroxylase (PAH), making it nonfunctional. PAH metabolizes the amino acid phenylalanine into tyrosine. With absence of PAH activity, phenylketonuria patients have a buildup of Phe in their body. Excess Phe in blood levels lead to decreased levels of other large neutral amino acids in the brain, thus hindering the development of brain and causing severe consequences such as mental retardation.
Early Treatment and Prevention of PKU
[edit | edit source]Approximately 1 in 14,000 individuals are affected by PKU. During pregnancy, chorionic villus sampling can be done to screen the unborn baby for PKU. Furthermore, newborns can be tested for the disorder so that they can be treated early if they are diagnosed. Under laboratory analysis, a blood sample taken from a newborn can be tested and screened for PKU. This genetic disorder can be treated by controlling the levels of Phe in the body. Some treatments include certain restrictive diets. For example, people with PKU cannot eat foods that contain aspartame, an artificial sweetener. They can consume a new sugar-substitute called neotame which is similar to aspartame except that it combines the two amino acids differently. The effect is that it is 30 times as sweet as aspartame, so less is needed and thus less phenylalanine is produced when it is metabolized. People are advised to follow the diet throughout their lives. Moreover, foods such as milk and diet drinks contain large amounts of Phe. One who has PKU would be advised to avoid heavy intake of these foods. Another example would be to take supplements and vitamins to ensure a healthy balance of essential amino acids. Another possibility in treating PKU would be treating one with the enzyme PAL. This enzyme facilitates the disposal of excess Phe. Further clinical trials will determine if PAL is safe for human intake.
Further Treatment of PKU
[edit | edit source]For further controlled and treatment of PKU, children and adults should undergo timely cognitive, neurophysical, and social-emotional testings and evaluations. For infants and children up four-years old, annual evaluations should be taken. Children during their elementary school years should undergo evaluations under a psychologist to evaluate any metabolic disorders twice a year. For individuals in and beyond high school, psychological examinations should be taken to evaluate signs of decreased metabolic control or lack of social acceptance.
References
[edit | edit source]- ↑ Berg, Jeremy M. (2010). Biochemistry (7th Ed. ed.). W. H. Freeman and Company. ISBN0-1-42-922936-5.
{{cite book}}:|edition=has extra text (help) - ↑ http://www.mayoclinic.com/health/noonan-syndrome/DS00857
- ↑ "Turner Syndrome." Genetics Home Reference. N.p., 23 July 2012. Web. 31 July 2012.<http://ghr.nlm.nih.gov/condition/turner-syndrome>
- Berg, J.M, Tymoczko, J.L. and Stryer, L. Biochemistry. 6th ed. New York: W.H. Freeman and Company, 2007. Print.
- Neurofirbromatosis: http://ghr.nlm.nih.gov/condition=neurofibromatosistype1
- General information: http://www.nlm.nih.gov/medlineplus/geneticdisorders.html#cat1
- Sickle Cell Disease
- "Proteins Are The Body's Worker Molecules." The Structures of Life. [Bethesda, MD]: U.S. Dept. of Health and Human Services, Public Health Service, National Institutes of Health, National Institute of General Medical Sciences, 2007. 6. Print.
- Williams Syndrome: http://ghr.nlm.nih.gov/condition=williamssyndrome
- BioMarin Pharmaceutical Inc. "Protecting the Brain: Testing and Treatment Approaches". PKU.com, 2009. Web. 29 Oct. 2011. <http://www.pku.com/Protecting-the-Brain/>
- Meister, K. “Sugar Substitutes and Your Health.” Comprehensive Reviews in Food Science and Food Safety, 2006.
Overview
[edit | edit source]Tay-Sachs is a genetically inherited, autosomal recessive condition that is caused a mutation on chromosome 15; the result is the lack of an essential enzyme within the body. In a normal person, there is the presence of an enzyme called hexosaminidase A (HEX A), which is found in the brain. Its primary function is to metabolize a fatty substance in the brain called GN12 ganglioside, which ironically, is needed for properly brain function when present in small amounts. Individuals with Tay-Sachs do not produce this enzyme because the gene that's responsible for making it is mutated. Consequently, these gangliosides, particularly ganglioside GM2, accumulate in cells, especially targeting the ones in the brain.
The Tay-Sachs gene is the most prevalent among the Ashkenazi Jewish population, where approximately 4% of the population carry this lethal gene.
Mode of Inheritance
[edit | edit source]Tay-Sach is inherited when both parents have mutated copies of the gene and both are passed down to the offspring. Normal chromosomes are usually present in pairs, known as alleles, each with identical genes and functions that could dominate just in case of a mutation on the other chromosome. Individuals who have wild-type alleles on both chromosomes have no chance of passing it out to their children. However, if one of the parents has the one copy of the Tay-Sachs gene, he/she becomes a carrier for the disease, but since he/she possesses a normal copy, the disease will not be expressed. However, if both parents are carries of the disease, children who inherit both mutate copies will express the disease to fullest extent. The figure to the right shows inheritance of Tay-Sachs. Because this disease is inherited through one of the 22 autosomal pairs and is only expressed with two copies of the recessive gene have been inherited, it is known as an autosomal, recessive disease.

Diagnosis/Symptoms
[edit | edit source]Tay-Sachs can be diagnosed using blood tests to determine the levels of HEX A in the blood or conducting an eye exam to determine if the cherry red spot, a landmark of Tay-Sachs, is present. There are three forms of Tay-Sachs: infantile, juvenile, and late-onset.
Infants with Tay-Sachs appear normal from birth until about six months of age. After that point, they begin to a experience development complications. Over time, they lose cognitive function, eventually losing the ability to crawl, turn over, or sit up. Other symptoms may encompass inability to swallow and difficulty breathing. By the age of two, infants begin experiencing seizures, loss of mental and muscle function, and overall becoming unconscious of their environment. Most do not live past the age of four or five.

Children afflicted with Tay-Sachs initially begin with a lack of coordination and muscle weakness. Similar to infantile Tay-Sachs, children slowly begin to lose their ability to walk, eat on their own, and communicate; eventually, affected children begin to suffer from seizures as well. In addition, they are also vulnerable to infection, often contracting pneumonia multiple times. Fortunately, the severity of this form is not as severe as infantile Tay-Sachs.
Late-onset Tay-Sachs is perhaps the hardest form to diagnose properly. Adults with these disease of often report of being misdiagnosed with some other illness because they do not possess the cherry red spot that is present on the retina Like the other two forms, adults experience muscle weakness and lack of coordination. The consequences of late-onset Tay-Sachs are not as severe. Many do experience problems breathing and swallowing, but they can still care for themselves. In addition, individual with the disease do not have shortened life spans like the other two forms.
Currently, there are no cures of Tay-Sachs; there are only ways to manage the symptoms.
References
[edit | edit source]Board, A.D.A.M. Editorial. "Tay-Sachs Disease." PubMed Health. U.S. National Library of Medicine, 17 Nov. 2012. Web. 07 Dec. 2012. <http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0002390/>.
Centre for Genetics Education. "Tay-Sachs Disease." N.p., June 2007. Web. 7 Dec. 2012. <http://www.genetics.edu.au/Information/Genetics-Fact-Sheets/Tay-SachsDiseaseandotherconditionsmorecommonintheAshkenaziJewishCommunityFS35>.
Classic InfantileTay-Sachs - Symptoms, Diagnosis and Management. National Tay-Sachs & Allied Diseases, n.d. Web. 7 Dec. 2012. <http://www.ntsad.org/index.php/tay-sachs/tay-sachs-disease-infantile>.
Classic InfantileTay-Sachs - Symptoms, Diagnosis and Management. National Tay-Sachs & Allied Diseases, n.d. Web. 7 Dec. 2012. <http://www.ntsad.org/index.php/tay-sachs/tay-sachs-disease-juvenile>.
Classic InfantileTay-Sachs - Symptoms, Diagnosis and Management. National Tay-Sachs & Allied Diseases, n.d. Web. 7 Dec. 2012. <http://www.ntsad.org/index.php/tay-sachs/tay-sachs-disease-late-onset>.
Mitochondrial Dysfunction
[edit | edit source]Mitochondrial dysfunctions are developed from mutations in the mitochondrial genome or DNA during replication. The mutation may be genetic or simply a random mutation in the genome of the individual itself. The patient does not need to obtain the genetic mutation from both parents; one parent is enough to cause this harmful disease. These mutations lead to a less effective mitochondria. They also usually are caused through stress related effects by the reactive oxygen species (ROS). A less effective mitochondria in this case is when the mitochondria is not able to make enough energy to support the whole body. This in turn will lead to many diseases caused from all different organelles from the body. In severe cases, not only will diseases develop but death may even occur.
Mitochondrion is the organelle in organisms that makes energy for most of the reactions in a cell. Mitochondria are found in plant and animal cells; they exist in almost all cells because they are needed to change energy from one form to another. Mitochondria make most of the energy needed in the human body; thus, without their proper function, they will not be able to carry out many of the reactions of the cell throughout the body. They have many physiological functions; for example, they change the food that is consumed by people into ATP, which is a form of energy that the cells are able to use to carry out reactions that the cells need to accomplish.
Overview
[edit | edit source]Studies and research has shown that Mitochondrial dysfunction is linked to both aging processes and many other diseases. Scientists claim that in order to maintain the function of the mitochondria, then proteins must be observed and watched at all times by chaperones and proteases.
Chaperones are proteins that assist other proteins in forming a polypeptide bond. Thus, in this case, they are proteins that assist the mitochondrial proteins and by watching them. Whereas, proteases are enzymes that breaks down proteins into smaller components like polypeptides or amino acids.
Introduction
[edit | edit source]As mentioned earlier, without proper maintenance of the mitochondria, then there will be a decrease in the effectiveness of the mitochondria. This will then also affect the aging for one over time because of the increasing amount of mutations and deletions that may have occurred. Which also damages and stops the translation and folding of proteins. Therefore, there must be something that scientists came up with which is called, Protein quality control (QC) to help gain the proper structure of the protein and proteases that breaks down the proteins that have undergone mutations or misfolds.
There are many genomes for each organelle and these genomes code for thriteen proteins of the Electron Transport Chain. In order to generate biogenesis and mitochondrial DNA, then the mitochondrian needs to carry out reactions by fission and fusion.
Fission will increase the amount of mitochondrial numbers inside the cell before the process of biogenesis. In addition, they are also able to segregate the mutated organelles from degradation. On the other hand, fusion is when organelles mix to have equal amounts of mitochondrial parts.
Foundation of Misfolding
[edit | edit source]As discussed earlier, there will be a higher risk that proteins will mis-assemble if there are a surmount number of mutations in the mitochondrial DNA (mtDNA). As a result, to reduce proteins from mis-assembling, there must be something that stops the mitochondria from producing errors in its DNA. There was a study that proved this general theory:
- Scientists tested the experiment on mammalian cells. In the experiment they stopped the production of mitochonodrial DNA replication, this resulted in an increase of misfolded proteins.
- This not only could affect the cells in the mitochondria but may also spread into other parts of the body and lead to diseases elsewhere in the body.
- Results also show that because of the mutations that occur in the cells over a long period of time, the proper replication, translation, and folding of proteins will decrease.
Effects of Mitochondrial disease
[edit | edit source]There are many diseases that can result from the mis-folding of the mitochondrial DNA; many of the diseases are linked to the nerves and the brain. This is because the nerves and brain require a lot of the energy made from mitochondria; a dysfunction in the mitochondria is like cutting off the power source from a powerhouse.
Indications of Mitochondrial Disease
[edit | edit source]There are many types of symptoms that mitochondrial diseases are known to posses:
- Some of which include vomiting, seizures, heart attack, muscle weakness or loss of muscle coordination, dementia, stroke, blindness, deafness, droopy eyelids, exercise intolerance, poor growth, heart disease, liver disease, kidney disease, and much more.
- Without much of the energy that the mitochondria provides, the patient diagnosed with the disease will be weak and tired. This will not allow the patient to eat, walk, write, or any daily routines.
People who are diagnosed with mitochondrial disease are usually young, around the age of about 20; but this does not limit to who the disease may result in. They usually start out with the symptoms of loss of coordination in the muscles and they feel very weak and they also cannot exercise. People with this disease cannot exercise because their muscle coordination have decreased; however, it is not just exercise that they are not capable of doing, they simply cannot do any activity that requires physical work.
Treatment
[edit | edit source]Up till now, recent science has not found a complete cure or treatment exactly fit for mitochondrial diseases. However, there are suggestions and ways to help deal with some of the symptoms of the disease.
- Physical therapy is one of the many ways of helping fight mitochondrial disease; it allows the muscles to stretch out which helps counterattack the loss of muscle coordination.
- Another treatment is to take vitamins, which may provide energy for the patients because they are not able to produce enough energy for themselves because of the dysfunction of the mitochondria.
- Recent data also reveals that decreasing the rate of translation might be able to help treat mitochondrial diseases because it will slow down the rate at which the mutation will translate also. Which in turn will slow down the number of mis-assembled proteins that are accumulated in the compartments.
Connection to Biogenesis and Aging
[edit | edit source]Reactive oxygen species damage the proteins and DNA of the mitochondria while cells are in the process of replication. This superoxide anion is produced in the ETC (electron transport chain) of complex I and III and it can severely damage the protein. Reactive oxygen species are able to damage the cells by either changing the way that proteins fold or simply by just adding in mutations to the DNA.
- Age is a factor that is also affected by mitochondrial dysfunctions; as time passes the damage of DNA accumulated in the genome will decrease an individual's lifespan. This will lead to the decrease in the efficiency of organelles in the body because of the defected proteins that the mitochondria has produced.
- They ran a test which shows that mice that had the mutations caused by mtDNA were aging faster than normal mice; although they grew up normally, they were definitely losing their capabilities because the rest of their internal compartments were deteriorating so quickly from the mutations in the mitochondrial DNA.
Biogenesis
[edit | edit source]Complex I of the electron transport chain has many subunits and within these subunits there are or may be mutations and or defects. Depending on the organism, if an individual has the dysfunction then that means there was a mutation or defect and thus this will ruin the whole process of the electron transport chain. As a consequence, this reveals that just one change in one individual nucleic acid such as deletions will greatly change the results of the whole reaction. So it is necessary that efficient QC machinery is needed to take out the bad parts of a protein in order to allow it to fold properly.
QC Machinery
[edit | edit source]As all know, the mitochondria has four compartments which proteins are made and folded; they are the outer membrane (OM), inner membrane space (IMS), inner membrane (IM) and the matrix. These four compartments work together to create a functional protein and if one part has an error then the whole process is destabilized.
Basically, the QC machinery (quality control machinery) is in control and looks over or examines the proteins that are misfolded in order to find the error that is not allowing the protein to fold properly. So there are chaperones and QC proteases that perform these functions and find the foreign proteins that are covered by the proper proteins.
- A study showed that chaperones in the mitochondria will perform some mechanism in which it will rid or treat the accumulation of misfolded proteins. But if the compartment is full or filled with the maximum amount of defected proteins then this will break down proteins into smaller subunits in which will cause the cell to increase transcription to reproduce the polypeptide. Therefore, this shows that the QC machinery is flexible to change to adapt to any conditions.
Four Compartments of the Mitochondria
[edit | edit source]There are QC proteases spread out throughout the mitochondria that scan and protect each of the four mitochondrial compartments along with chaperones. Again, they are present in the mitochondria to prevent the accumulation of defected proteins from compiling in each of the compartments.
- The outer membrane contains ubiquitin ligase which is in charge of breaking down mutated proteins that are stuck in the outer membrane. Scientists know that this enzyme is able to do such a thing but does not know how.
- The inner membrane space is supposed to keep the protein from folding as it is being transported from the outer membrane to the innermembrane. It contains the protease HtrA2 which up to recent study does not show any connection for the regulation of breaking down a folded protein. For example, there was a research done on mice in which the mice did not contain the protease HtrA2; this caused the mice to die in a moth from Parkinson's disease which strongly suggests that protease links to biogenesis.
- The next compartment, the inner membrane, is responsible for recognizing which proteins are properly folded and improperly folded. This is done by the i-AAA and the m-AAA proteases; they both work hand in hand with one another to protect the mitochondria from defects. What is unique about these proteases are that they have active sites that face the inner membrane space and the matrix.
- The last compartment of the mitochondrial compartments is the matrix; this compartment is very high in protein concentration and it is processed by mtHSP70 and HSP60. Both these chaperones require energy in order to work because they are ATP dependent. In addition to the chaperones, they also have AAA proteases, Lon and ClpXP. They predicted the function of these chaperones from studies with bacteria because bacteria have the same proteins. Thus, it is not confirmed that these are the same actions that the human body also does. However, in bacteria, it is proven that they will pin point the defected protein and once they do find it then they will destroy it or remove it.
Significance
[edit | edit source]Why then is all of this important? Researchers continue to do experiments in this field and research on mitochondrial diseases because they want to find out more about this disease. The goal is to be able help cure or treat as many patients that have this disease as possible. And to do this, they must understand the cause of the disease in order to find ways to hinder or prevent the mutations of mtDNA to continue the research. It is also important because this disease leads to other diseases or at least up to recent studies, it is shown that mitochondrial dysfunction results in a decrease in the body;s energy; therefore, it is linked to any cell reactions that require energy. This means that any diseases or illnesses that need energy are going to be affected by the mitochondrial dysfunction. There is also evidence that mitochondrial diseases are connected to cancer and tumors, which range from a big quantity of many different kinds.
- It is a field to be focused on because there are not any treatments or cures designed specifically for this particular disease yet.
- Another point is that there are many people who are born with this dysfunction and research to find a cure would allow millions of people of all ages and especially infants into a better lifestyle. In addition it is like Giving a brand new life to newborn babies that have not experienced anything in this world yet.
- It will provide hope and joy to those suffering from this disease and make children happier to be able to do simple things like run, walk and eat properly like other kids.
Statistics
[edit | edit source]It is a very important filed to look into because the rate at which people get these diseases are so fast and there is no cure to this disease. Therefore, it is a very essential and crucial topic for researchers to focus on.
- According to the statistics, children will develop mitochondrial disease by the age of ten for every thirty minutes that an infant is born.
- Although less but also significant is that there are one out of two hundred that will soon acquire mitochondrial disease in their life.
This disease is fairly new to the biological field and not many people study it because it is not diagnosed in every person out there. Also because it is not found in every person out in the world, companies will not give budget to research on this topic. This is why it is important for people to especially research on this topic because not very much is known about this subject; therefore, there will be more breakthroughs looking into this field of study. As a result, it will help many of the patients out there suffering from the mitochondrial disease. Finding a cure or better treatment to this dysfunction may lead to cures in other diseases; such as, Alzheimers, Parkinsons, diabetes, hypertension, osteoporosis, and many more diseases.
Research Experiments
[edit | edit source]- There was an experiment in which mouses were used to test this experiment. In the experiment, scientists changed one nucleotide in the sequence of the mitochondrial DNA. This experiment showed results in which the mouse became weak because its muscles were losing coordination. It also showed that the health of the mouse was slowly decreasing and ultimately led to heart disease.
- A drug was used to increase thinking and function for patients with Alzheimer's disease; this is done from stabilizing the mitochondria.
- Another link to mitochondrial disease is that it may result in cancer. This is because the signaling must occur inside a cell between the nucleus and the mitochondria. Therefore, if the mitochondria is not working because of the dysfunction then this will lead to cancer.
- Researchers also found that in patients with diabetes, the mitochondria efficiency is decreased. This shows that the mitochondria causes patients to obtain high sugar levels at some degree.
Reference
[edit | edit source]Baker, Brooke M., and Cole M. Haynes. "Mitochondrial Protein Quality Control during Biogenesis and Aging." (n.d.): n. pag. Rpt. in New York: Sloan-Kettering Institute, Cell Biology Program.
"Mitochondrial Disease Causes, Symptoms, Diagnosis, and Treatment on MedicineNet.com." MedicineNet. Government, n.d. Web. 06 Dec. 2012
"Mitochondrial Disease." United Mitochondrial Disease Foundation, n.d. Web. 6 Dec. 2012.
Overview
[edit | edit source]Duchenne Muscular Dystrophy is a fatal genetic disorder that affects the X-chromosome and is commonly found in men. It is found across all cultures and races. Duchenne occurs in approximately 1 in every 3,500 male births. It is caused by a mutation in the X-chromosome were a gene encodes for dystrophin.
Effects
[edit | edit source]Duchenne Muscular Dystrophy results in a loss of strength. This is due to the lack of dystrophin, which allows for muscle cells to be damaged easily. The heart and lungs are affected the most since muscles become progressively weaker.
Cause
[edit | edit source]Dystrophin acts as a glue, which holds muscles together and maintains the structure of cells. It is also known to carry signals inside and outside of muscle fibers. Since dystrophin is carried on the X-chromosome, and boys only carry one X-chromosome, they are more susceptible to damage. Boys is considered diagnosed with Duchenne when they are unable to produce dystrophin at all. Duchenne is normally passed from parent to child but mainly occurs spontaneously.
References
[edit | edit source]1. "About Duchenne." Muscular Dystrophy. N.p., n.d. Web. 07 Dec. 2012. Structural Biochemistry/Enzymes Used for Cloning/ Enzymes that modify nucleic acids are used to synthesize, degrade, join, and/or remove portions of nucleic acids. Cloning enzymes are enzymes that are important in nucleic acid cloning procedures. The activities of the cloning enzymes consist of ligases, kinases, phosphatases, and RecA Protein.
DNA ligase, for example is used in joining DNA molecules together. Within the cell, the double ends of the strands are attached covalently. While RNA primers replace DNA during DNA replication, DNA ligase seals in the gaps.
1. Ligases are used to join nucleic acid segments, especially when one is cloning a DNA fragment into the vector DNA.
2. Phosphatases remove the 5′-phosphate from nucleic acid strands. This prevents vector downgrading, which would reduce the number of background colonies as well as producing substrate to which a kinase can attach a new radio-labeled phosphate.
3. Kinases add new phosphate groups to nucleic acids. This is usually done in order to label the nucleic acid fragments or the synthetically made oligonucleotide.
4. RecA Protein and AgarACE® Enzyme are used primarily to protect in certain cloning procedures or facilitate the nucleic acid purification. The E. coli RecA Protein is able to facilitate the pairing of homologous DNA sequences. AgarACE® Enzyme is a patented agarose-lysing enzyme produced for the harvest of DNA from agarose gels.

DNA ligase is a special type of ligase that can link two DNA strands that have a break in both complementary strands of DNA together by creating phosphodiester bond. DNA ligase has applications in both DNA repair and DNA In addition. It is used a lot in molecular biology laboratories for genetic recombination experiments. DNA ligases are a useful tool in generating recombinant DNA sequences. For example, DNA fragments are cut with restriction enzymes and then recombined with DNA ligase. DNA ligases are commonly found in E. Coli.
T4 DNA ligase is an enzyme that is encoded by the bacteriophage known as T4. In a reaction where the DNA molecules are being joined together at the 3'-hydroxy and 5'-phosophate termini, the ligase is used as a catalyst. Given that there are no missing nucleotides in the repare reaction, the ligase can also catalyze the covalent joining of two segments to one uniterrupted strand in a DNA duplex. In order to accomplish this catalytic activity, ATP and Mg2+ is required. DNA that lacks the required phosphate residues can still be made to ligate through phosphorylation with T4 polynucleotide kinase. Additionally, an exchange reaction of phosphate between pyrophosphate and ATP can also be catalyzed through use of the ligase.
Characteristics of T4 DNA Ligase from E. coli lysogenic NM989
[edit | edit source]The T4 DNA ligase is a single polypeptide with a molecular weight of 68,000 daltons. In order to obtain the maximum amount of activity from the ligase, a pH of 7.5-8.0 is desired. At pH levels of 6.9 and 8.3, the enzyme exhibits 40% and 65% of its full capabilities, respectively. As previously mentioned, Mg2+ is necessary for T4 DNA ligase to be effective, and the optimal concentration of Mg2+ is 10mM. Sulfhydryl reagents (DTT, 2-mercaptoethanol) is also necessary in order to utilize the enzyme. If there is NaCl present, concentrations over 200 mM will stop all enzymatic reactions from occurring. for intermolecular ligation, especially when the substrate DNA consists of large DNA molecules PEG (concentrations of 1-10%) appears to stimulate the enzymatic activity.
Application
[edit | edit source]T4 DNA ligase is mostly used in the joining of DNA molecules with compatible cohesive termini, or blunt-ended, double-stranded DNA to one another, or to synthetic linkers. The reaction that involves blund-ended DNA is slower than the other reactions, but the rate of ligation can be accelerated by adding 150 to 200 mM of NaCl along with a low concentration of PEG. If the 5' phosophate is absent in the DNA, then phosphorylation is necessary before ligation can be performed. Phosphorlyation is achieved through utilization T4 polynucleotide kinase with ATP. If the DNA fragments being joined together have protruding 5' termini that are not compatible with one another, it is still able to join the two fragments together by partial filling of the recessed 3' termini in controlled reactions using the Klenow fragment of E. Coli DNA polymerase I. T4 RNA Ligase catalyzes the ligation of a 5' phosphoryl-terminated nucleic acid donor to a 3' hydroxyl-terminated nucleic acid acceptor through the formation of a 3'→5' phosphodiester bond, with hydrolysis of ATP to AMP and PPi. Substrates include single-stranded RNA and DNA as well as dinucleoside pyrophosphates. This ligase can be formed from Purified E. coli strain ER2497 which contains the plasmid pRF-E35. This enzyme is used for the labeling the 3’-terminus of RNA with a 5’-[32P]. It functions as the inter- and intramolecular joining of the RNA and DNA molecules. It can also be used to synthesize single stranded oligodeoxyribo-nucleotides as well as incorporating additional amino acids into proteins.
Description
[edit | edit source]The T4 Polynucleotide Kinase (T4 PNK) catalyzes the transfer of the phosphate from ATP to the 5'-OH group of single- and double-stranded DNAs and RNAs as well as oligonucleotides. The reaction can be reversed. The enzyme is also a 3'-phosphatase and a homotetramer. It consists of four identical subunits of 28.9 kDa. The T4 polynucleotide kinase can be used in a buffer for restriction enzymes in techniques such as PCR. It can be found in E. coli with a duplicated gene of the bacteriophage T4. It can also be used for labeling 5’-terminus of nucleic acids which can be used as probes for hybridization, transcript mapping; markers for gel electrophoresis. They can also be used as primers for DNA sequencing and PCR technique as well as an enzyme that removes the 3’-phosphate groups.
Storage
[edit | edit source]T4 Polynucleotide Kinase should be stored in -20°C
Applications
[edit | edit source]The T4 polynucleotide kinase is used for labeling the 5'-termini of nucleic acids, and the labeled products can be used in the following ways: - markers for gel-electrophoresis - primers for DNA sequencing - primers for PCR - probes for hybridization - substrates for DNA and RNA ligases - probes for transcrip mapping 5'-phosphorylation of oligonucleotide linkers and DNA or RNA is required before ligation can be performed. using the 32P-postlabeling assay, DNA modification can be detected.
Miscellaneous
[edit | edit source]The 5'-termini of nucleic acids can be labeled either through the forward or the exchange reaction. When the forward reaction is carried out, the phosphate from [32P or 33P]-ATP is transferred to the 5'-hydroxyl end of DNA or RNA. If the nucleic acid already contains a 5'phosphate, though, that portion is removed either through the use of calf intestine alkaline phosphatase or bacterial alkaline phosphatase. When the exchange reaction is carried out, the nucleic acid 5'-phosphate is transferred to ADP and the radiolabeled phosphate from [32P or 33P]-ATP is then transferred to the nucleic acid that does not contain a 5'-hydroxyl group. The ideal buffer to use for the exchange reaction is Imidazole-HCl buffer, which has a pH of 6.4. PEG is used to improve the rate and efficiency of the kinase reaction and should be added to the exchange reaction. When phosphate and ammonium ions are present, the T4 polynucleotide kinase is inhibited. Prior to phosphorylation reaction, DNA should not be precipitate if ammonium ions are present.

Alkaline phosphatase is the enzyme that catalyzes a dephosphorylation of DNA, RNA, ribo-, and deoxyribonucleoside triphosphates. It can also remove phosphates from nucleotides and proteins. They are most active at a basic pH. This enzyme has become very useful in molecular biology because this allows us to cleave the 5' end of DNA and prevent any further ligation thus keeping the DNA molecules linear until they are prepared for analysis. We can then use radiolabeling to easily track a certain section of a DNA through the various processes of DNA replicaton.
This has its uses in vector recombination and cloning genes. After restrictive enzymes cleave a plasmid we have to make sure that this circular DNA doesn't reconnect and form the original circular DNA. So before we can actually insert our gene of interest to allow for its cloning, alkaline phosphatase can remove any phosphate group that might be susceptible to attack by the other end of the DNA or even by an external nucleophile. It's also very valuable for its high rate of activity even in harsh conditions.
There are several main sources of alkaline phosphatase, these are: bacterial alkaline phosphatase(BAP), calf intestinal alkaline phosphatase(CIP), and shrimp alkaline phosphatase. Not to mention it is also present in human tissues and most concentrated at organs such as the kidneys and liver. BAP is the most active of all these enzymes but also is hard to get rid of after the dephosphorylation reaction. CIP is of great interest and is extracted from bovine intestine. It can easily be denatured and destroyed by protease digestion. It has been shown that CIP can be used as antiflammatory agents in human bodies and have been tested on mice.
Alkaline phosphatase must be monitored in the human body because it if the concentration goes too low or too high, health problems may arise. Elevated levels caused by factors that are still not completely clear can cause liver damage and bone disease. However, it is normal for levels to spike during puberty when bone growth is occurring rapidly. Lowered levels of alkaline phosphatase is characterized by different kinds of anemia and leukemia. Postmenopausal women are also more likely to have higher levels.
ALP is associated with cell membranes and present in many tissues in animals and humans. ALP is connected to the membranes
by glycoproteins. When these are cleaved, phosphate groups are recycled throughout the cells and the amount of ALP levels
increases in the plasma. Glucocorticoids can significantly raise the amounts of ALP present in the body while anticonvulsants secrete less
but still a significant amount. Glucocorticoids generate ALp in the C isomer while anticonvulsants generate ALP in an L isomer. Alkaline phosphatase is essential for the mineral deposition in teeth and bones, Bone can become weak and deficient if alkaline phosphatase
is not present, a disease called hypophosphatasia. ALP is nonspecific and mainly cleaves phosphate esters.
RecA can be found in E. Coli and it is needed in order to repair and maintain DNA. A structural and functional homolog for Rec A can be found in every species and as a result, it is used as a method to exchange genetic information by homologous recombination through sexual reproduction. RecA has multiple functions, all of which are related to DNA repair. Its main function which has been most widely studied is that it facilitates DNA recombination in order to repair DNA double strand breaks. The RecA protein can catalyze one directional branch migration. This function makes it possible to complete recombination. Bacterial strains such as E. Coli that are lacking RecA are useful for cloning procedures.
 Structural Biochemistry/AgarACE ®, Enzyme/
Structural Biochemistry/AgarACE ®, Enzyme/
Cloning Tips
[edit | edit source]
Some general considerations in the process of cloning can enhance the efficiency of cloning. The following are some tips:
1. The DNA insert/PCR product and vector digested by appropriate restriction enzymes should be purified directly, or from an agarose gel. This gets rid of the primers or the restriction fragments that are not of interest.
2. When performing ligation, the insert:vector ratio should be tested for the optimal conditions. Generally, a 1:1 or 3:1 ratio works well.
3. Screening for correct clones containing the vector with the DNA of interest inserted is very important. Common methods such as blue/white screening for B-Galactosidase activity and antibiotic screening are applied.
4. Calf Intestinal Alkaline Phosphatase is a hydrolase enzyme that is responsible for removing phosphate groups from nucleotides, proteins, etc. This enzyme can be used after digestion with restriction enzymes.
5.ATP is required for ligation.
6. Enzymes are used to synthesize, degrade, join or remove portions of nucleic acids in a controlled and generally defined manner.
7. Cloning Enzymes, are those important in nucleic acid cloning procedures. Some cloning enzymes are used as ligases, kinases and phosphatases, and RecA proteins.
8. When using vectors with antibiotic resistance characteristics for selection, after transforming the cells allowing the cells to grow in appropriate growth medium and growth temperatures for several hours increases growth efficiency as well as antibiotic-resistance. This will allow for cells that successfully took up the cloned vectors to be better collected once plated onto antibiotic treated growth plates. The preliminary growth period in solution will stimulate cell activity in producing the antibiotic resistance characteristics.
9. Cross contamination when working with multiple cloned DNA vectors with similar antibiotic selections is a concerning issue. Washing, ethanol-treating, and flaming instruments is an important anti-cross-contamination measure to be taken seriously.
10. Sterile technique. Structural Biochemistry/The Stepwise Process of How DNA is Cloned and Inserted into the Vector/ The immune system destroys foreign invaders to the body. It also destroys some non-foreign cells that are no longer performing their functions, such as cancer cells.
Introduction
[edit | edit source]The immune system is a biological system inside an organism which is supposed to fight against diseases. In vertebrates, the immune system is considered the last line of defense against foreign invaders.It fights by destroying pathogens and tumor cells using mechanisms that constantly adapt to recognize and rid these disease causing cells. This mechanism is needed so that the immune system does not attack itself or the organisms healthy cells. Unfortunately, pathogens can evolve rapidly which would allow it to avoid the immune system and attack the host.
There are many mechanisms that have evolved that allow for the immune system to recognize and even neutralize pathogens. Every living organism even the most basic unicellular organisms, bacteria, contain some enzyme systems that are used to protect from viral infections. Similarly, there are many ancient eukaryotes in which basic immune mechanisms that been evolved and have been passed down to their modern descendants. Some of these evolved mechanisms are the complement system, phagocytosis, and defensins which are antimicrobial peptides. The immune system of vertebrates, like humans, is an elaborate network that consists of many types of tissues, cells, organs, and proteins. Due to this advanced immune system, it can more efficiently recognize specific pathogens over time. This adaptation is known as “acquired immunity” and results in an immunological memory that can be compared to a textbook full of information. This immunological memory, formed from direct encounter to a specific pathogen, results in a well-prepared response to the same specific pathogen the second time around. A vaccination serves this purpose.
However, a disease can result in the immune system when it begins to properly operate. One such disease is called immunodeficiency in which the immune system performs at a much lower level than its suppose to and thus results in infections that are constantly recurring and may be life-threatening. This disease can be a result of either genetic disease or an infection such as AIDS which is caused by HIV. On the other extreme end is the disease known as autoimmune disease in which the immune system is so active that is begins to attack its own tissues as if they were foreign invaders.
The immune system has many layers of defenses, to protect against infections, each with increasing specificity. The most basic defense is physical barriers that prevent pathogens from being able to enter the organism. However, if the pathogen is able to get through these physical barriers, the innate immune system takes over and provides a non-specific immediate response. Second line of the defense is the internal defense in the innate immunity. Internal defenses in innate immunity include phyagocytic cells, antimicrobial proteins, inflammatory response, natural killer cells. If the pathogens are able to get through this second line of defense, then the third layer of defense is the adaptive immune system. In order to improve its ability to recognize the pathogen during an infection, the immune system will adapt its response. After the pathogen has been nullified, the improved response is retained forming an immunological memory allowing the adaptive immune system to attack the same pathogen faster and stronger the next time it is encountered.
Both the adaptive and innate immunity require that the immune system is able to differentiate between which molecules are self and non-self. Self molecules are parts of the organism’s body that are distinct and distinguished from outside foreign substances while non-self molecules are often known as antigens that specific immune receptors bind to.
Organisms protect themselves through various barriers. The first line of defense against infection are surface barriers including membranes and exoskeletons. Other bodily systems also protect the body by naturally repelling foreign agents. Tears, urine, mucus, coughing and sneezing are all examples of ways the body expels foreign agents. Another barrier to fight against infection is the chemical barrier which uses enzymes, also known as antibacterials, to kill pathogens.
Innate Immunity
[edit | edit source]Cellular Barriers
[edit | edit source]Certain cells of the mucous membranes produce mucus, a viscous fluid that enhances defenses by trapping microbes and other particles. Saliva, tears, and mucous secretions that bathe various exposed epithelia provide a washing action that also inhibits colonization by microbes. Pathogens that make their way into the body are subject to detection by leukocytes. These cells recognize microbes using Toll-like receptor. Toll-Like Receptor recognizes fragments of molecules characteristic of a set of pathogens. Similarly, on the inner surface of vesicles formed by endocytosis, is the sensor for double-stranded RNA a form of nucleic acid characteristic of certain viruses.
There are four types of internal defenses in the innate immunity:
Phagocytosis
[edit | edit source]Phagocytosis is the ingestion and digestion of bacteria and other foreign substances. It is triggered by hemocytes that circulate within the hemolymph. Six steps of ingestion and destruction of a microbe by a typical phagocytic cell are shown below.
(1) Pseudopodia surround microbes.
(2) Microbes are engulfed into cell.
(3) Vacuole containing microbes forms inside the cell.
(4) Vacuole and lysosome fuse.
(5) Toxic compounds and lysosomal enzymes destroy microbes.
(6) Microbial debris is released by exocytosis
There are four types of phagocytic leukocytes that play a different role in the innate immune system.
(1) Neutrophils are the most abundant phagocytic cells in the mammalian body. neutrophils represent 50-60% of the total leukocytes that circulate in the bloodstream.Signals from infected tissues attract neutrophils, which then engulf and destroy microbes.
(2) Macrophages provide a more effective phagocytic defense than neutrophiles. Some of macrophages migrate throughout the body, while others reside permanently in various organs and tissues. Macrophages in the spleen, lymph nodes, and other tissues of the lymphatic system are particularly well positioned to combat pathogens. Microbes in the blood become trapped in the spleen, whereas microbes in the interstitial fluid flow into lymph and are trapped in lymph nodes.
(3) Eosinohpis have low phagocytic activity but are important in defending against multicellular invaders such as parsitic worms. Rather than engulfing such parasites, eosinophils position themselves against the parasite's body and then discharge destructive enzymes that damage the invaders.
(4) Dendritic cells populate tissues that are in contact with the environment. They mainly stimulate development of adaptative immunity against microbes they encounter. Dendritic cells are also phagocytes in the tissue but have contact with the external environment and thus exiss mainly in the intestines, stomach, nose, lungs, and skin. They are similar to neuronal dendrites as both have spine-like projections. Dendritic cells link both the bodily tissues and the adaptive and innate immune systems as they present antigens to T-cells.
Antimicrobial Proteins
[edit | edit source]Interferons are proteins that provide innate defense against viral infections. Virus-infected body cells secrete interferons, inducing nearby uninfected cells to produce substances that inhibit viral reproduction. Interferons limit the cell-to-cell spread of viruses in the body, helping control viral infections such as colds and influenza. Some white blood cells secrete a different type of interferon that helps activate macrophages, enhancing their phagocytic ability.
Complement System consists of roughly 30 proteins in blood plasma that function together to fight infections. These proteins circulate in an inactive state and are activated in a cascade of biochemical reactions leading to bursting of invading cells.
Inflammatory Response
[edit | edit source]Inflmmatory response is the pain and swelling that alert you to a splinter under your skin. One important inflammatory signaling molecule is histamine, which is stored in the mast cells. Mast cells, which exist in both mucous membranes and connective tissues, regulate the inflammatory response. They secrete chemical mediators that are often found in the defense against parasites and sometimes also are found in allergic reactions like asthma. Natural killer cells are a type of leukocyte that attack tumor cells and cells that have been virus-infected. Histamine released by mast cells at sites of tissue damage triggers nearby blood vessels to dilate and beomce more permeable.
Steps of major events in a local inflammatory response are shown below.
(1) Activated macrophages and mast cells at the injury site release signaling molecules that act on nearby capillaries.
(2) The capillaries widen and become more permeable, allowing fluid containing antimicrobial peptides to enter the tissue. Signaling molecules released by immune cells attract additional phagocytic cells.
(3) Phagocytic cells digest pathogens and cell debris at the site, and the tissue heals.
Natural Killer Cells
[edit | edit source]Natural Killer (NK) cells help recognize and eliminate certain diseased cells in vertebrates. With the exception of red blood cells, all cells in the body normally have on their surface a protein called class I MHC molecule. Following viral infection or conversion to a cancerous state, cells sometimes stop expressing this protein. The NK cells that patrol the body attach to such stricken cells and release chemicals that lead to a cell death, inhibiting further spread of the virus or cancer.
Adaptive Immunity
[edit | edit source]There are two types of immunity in the adaptive immunity:
Cellular immunity
[edit | edit source]Cellular immunity is also known as cell-mediated immunity (CMI). This type of immunity act as the second arm of the immune responses. There are different functions of immune cells. For example, this type of cell-mediated immunity includes the killing of intracellular pathogens and direct cell killing by cytotoxic T cells, natural killer cells, and killer cells (T-cells work by scanning the surface of a cell for anything that appears foreign.) In CMI, the T cells or the lymphocytes would attach to the surface of other cells, then it would display an antigen and trigger a response. This type of immune response may also include white blood cells (leukocytes).
Humoral immunity
[edit | edit source]This type of immunity guards against infections caused by bacteria and extracellular phases of viral infections. The immunity is mediated by a big, diverse collection of proteins which are related termed antibodies or immunoglobins; in mammals, these are produced by the B cells in bone marrow. B cells play a major role in humoral immunity. B cells like T cells also have surface receptors, which allows them to recognize certain antigen. The variable portion in the B cells accepts specific antigens. Once a B cell recognizes this antigen it has the ability to perform two functions. One is to generate plasma B cells that can reproduce more cells with the specific binding site. The other is to form memory B cells, which acts as an immunity to the antibody whenever it is encountered again.
In the immune system inflammation is often one of the first responses. This can be typically seen through swelling and redness which are a result of increased blood flow to a tissue. The injured or infected cells release cytokines and eicosanoids which results in inflammation. The dilation of the blood vessels and the fever are produced by the prostaglandins while the white blood cells are attracted by leukotrienes both of which are part of the eicosanoids. The common cytokines are interferons which can shut down protein synthesis and interleukins which communicate between white blood cells. Cytokines as well as other chemicals, attract immune cells to where the infection is and remove the pathogens followed by healing of the damaged tissue.


Adaptive Immune System
[edit | edit source]The adaptive immune system allows for a strong immune response and is the basis of the immunological memory as each pathogen the body encounters is now remembered by a specific antigen. The adaptive immune response requires that the body recognizes specific non-self antigens during antigen presentation. This antigen specificity allows the body to generate responses that are meant to fight that specific pathogen and the pathogen-infected cells. The ability to generate the correct responses specific to that pathogen is maintained by memory cells in the body. If a pathogen infects the body more than one time, the specific memory cells will quickly eliminate the pathogen.
The adaptive immune system cells are called lymphocytes which are a special type of leukocyte. Major types of lymphocytes include B cells and T cells derived from bone hematopoietic stem cells found in the bone marrow. While T cells are involved in cell-mediated immune response, B cells are involved in the humoral immune response.
Both T cells and B cells contain receptor molecules that are used to recognize specific targets. T cells can recognize a non-self target like a pathogen only after antigens (small part of the pathogen) has been processed together in combination with a self receptor referred to as a major histomcompatbility complex (MHC) molecule. There includes two major types of T cells which are the helper T cells and the killer T cells. While Killer T cells can only recognize antigens coupled with a Class I MHC molecule, helper T cells only can recognize antigens that are coupled with Class II MHC molecules.
On the other hand, B cell antigen-specific receptors are an antibody molecule that exists on the surface of the B cell and recognize pathogens without needing any antigen processing. Different B cells express a different antibody, thus the complete set of B cell antigen receptors is the representation of all the antibodies that the body can make.
Killer T cells kill cells that are infected with viruses and/or other pathogens or cells that are damaged or dysfunctional. Similar to B cells, the different types of T cells recognize a different antigen. Killer T cells are then activated when their respective T cell receptor (TCR) binds to the specific antigen which is in a MHC Class I complex receptor of another cell. CD8, a co-receptor on the T cell helps recognition of this MHC antigen complex. T cells travel throughout the body searching for cells in which the MHC I receptors contain this antigen. When an activated T cell comes in contacts with these cells, it releases cytotoxins which results in the formation of pores in the target cell’s plasma membrane which allows water, toxins, and ions to enter. The entry of granulysin which is another toxin causes the target cell to undergo apoptosis which is basically the self-destruction of the cell. The T cells that kill hosts cells are extremely important in preventing viruses from replicating. T cell activation is controlled extremely tightly and usually requires a really strong MHC/antigen activation signal provided by helper T cells.
Immunological Memory
[edit | edit source]When B and T cells begin to replicate, some of the offspring that they produce will end up becoming long-lived memory cells. These memory cells will remember all specific pathogens encountered during the animal’s lifetime and can thus call forth a strong response if the pathogen ever invades the body again. This is called “adaptive immune system” since it is a result of an adaptation to an infection with the pathogen during the individual’s lifetime and continues to prepare the immune system for potential future pathogens. Immunological memory can either be in active long-term memory or passive short-term memory.
Passive Memory
[edit | edit source]Newborn infants are particularly vulnerable to infections since they have no prior exposure to pathogens. Thus, the mother protects the infant through several layers of passive protection. During pregnancy, TgG, which is a certain type of antibody, is transported to the baby from the mother through the placenta so even babies have high levels of antibodies that have similar antigen specificities as the mother. Even breast milk contains antibodies that are transferred to the infant’s gut and protect against bacterial infections until the baby is capable of making its own antibodies. Since the fetus isn’t making any memory cells or antibodies, it is called passive immunity. The passive immunity is short-lived, ranging from a couple days to a couple months.
Active Memory and Immunization
[edit | edit source]Following an infection, long-term active memory is acquired by activation of B and T cells. Vaccinations take advantage of this by artificially generating active immunity. During a vaccination, the antigen of a pathogen is introduced into the body and stimulates the immune system to develop a specific immunity against that pathogen without actually causing the disease that the pathogen brings. This deliberate introduction of the pathogen is successful since it exploits the immune system’s natural specificity and its inducibility. Vaccination is an extremely effective manipulation of the immune system that helps fight diseases.
Many bacterial vaccinations are the acellular components of the microorganisms while viral vaccinations are the live attenuated viruses as well as harmless toxin components. Since bacterial vaccines derived from acellular components do not induce a strongly adaptive response, most of the bacterial vaccines are thus provided in addition with adjuvants that activate the antigen-presenting cells that are existent in the innate immune system to maximize the immunogenicity.
Immunodeficiencies
[edit | edit source]Immunodeficiencies occurs in a human when parts of the immune system are inactive. Since a component is inactive, its ability to respond to pathogens is reduced . Common causes of poor immune function are obesity, drugs, and alcohol. The most common cause of immunodeficiency is malnutrition in developing countries. The lack of sufficient proteins often result in impaired complement activity, cell-mediated immunity, cytokine production, and phagocyte function. Deficiency of single nutrients also reduces the immune responses. Also the loss of the thymus either through a genetic mutation of removal through surgery also results in severe immunodeficiency as the animal becomes high susceptible to infection.
Immunodeficiency can also be acquired or inherited. An example of inherited immunodeficiency is the chronic granulomatous disease in which the phagocytes’ ability to destroy pathogens have been reduced. An example of an acquired immunodeficiency is AIDS and some types of cancer.
Autoimmunity
[edit | edit source]Autoimmunity occurs when there is an overactive immune response resulting in autoimmune disorders. In these disorders, the immune system is unable to properly distinguish between itself and non-self and as a result, attacks its own body. Usually, the antibodies and T cells react with self peptides. To prevent autoimmunity, one of the functions of specialized cells, often found in the thymus and bone marrow, is to have young lymphocytes that have self antigens produced throughout the body and to get rid of the cells that recognize self-antigens.
Hypersensitivity
[edit | edit source]Hypersensitivity happens when the immune response damages the body’s own tissues. There are four classes of hypersensitivity (Type I-IV). Type I hypersensitivity is an anaphylactice reaction often associated with allergies. The symptoms have a huge range anywhere from just mild discomfort to death. Type I hypersensitivity is often mediated by IgE which is released from basophils and mast cells. Type II hypersensitivity occurs when the antibodies bind to the antigens on the animal’s own cells marking them for destruction often referred to as antibody-dependent hypersensitivity. Type III hypersensitivity reactions are often triggered by immune complexes that are deposited in various tissues. Delayed type hypersensitivity or Type IV hypersensitivity, involve many autoimmune and infectious diseases and often take two to three days to develop. These are often mediated by macrophages, monocytes, and T cells.
Immunology of Tumors
[edit | edit source]An important role that the immune system serves is to identify and eliminate tumors. The tumor’s transformed cells express antigens that aren’t normally found on normal cells. These antigens appear foreign to the immune system and when near tumors, the immune cells attack the transformed tumor cells. The antigens that are expressed by the tumors come from various sources including papillomavirus which is derived from an oncogenic virus which often results in cervical cancer while other sources are the organism’s own proteins that normally only have low levels in normal cells but reach unusually high levels in tumor cells. An example of this is the enzyme tyrosinase that can transform certain skin cells into tumor cells called melanomas when expressed at really high levels. Another source of tumor antigens are the mutation of proteins that are normally important for survival regulating cell growth into cancer inducing molecules.
The main response that the immune system uses for tumors is to use killer T cells to with the assistance of helper T cells to destroy the abnormal cells. The tumor antigens that are present on MHC class I molecules are really similar to viral antigens. This similarity allows the killer T cells to recognize tumor cells as abnormal. NK cells kill tumor cells as well in similar ways especially if on their surface, there are fewer than normal MHC class I molecules; this is a common trait with tumors. Sometimes there are antibodies that are generated against tumor cells to destroy them.
However, some tumors evade the immune system and end up causing cancer. Since the tumor cells often have only a reduced MHC class I molecule count on their surface, they often avoid detection by the killer T cells. Some of the tumor cells release products that inhibit the immune response as well like when they secrete the cytokine TGF-B which is known to suppress the activity of lymphocytes and macrophages. Also sometimes the immune system doesn’t attack the tumor cells anymore when immunological tolerance is developed against tumor antigens.
Macrophages can promote the growth of tumors and thus tumor cells release cytokines that can attract macrophages that release cytokines and growth factors that end up nurturing the tumors for development. Both the combination of hypoxia in the tumor and the cytokines that are released by the macrophages induce tumor cells that decrease the production of a protein that often blocks metastasis that help the spread of cancer cells.
Pathogen’s Evade the Immune System
[edit | edit source]The pathogen’s success is depends on its ability to evade the host’s immune responses. Thus, pathogens have evolved several methods allowing them to infect a host successfully by evading detection and destruction by the immune system. Bacteria usually overcome the physical barriers by secreting enzymes to digest the barrier like type II secretion system. They also use a type III secretion system that allows them to insert a hallow tube providing a direct route for the proteins to enter the host cell. These proteins often shutdown the defenses of the host.
Some pathogens avoid the innate immune system by hiding within the cells of the host also referred to as intracellular pathogenesis. The pathogen hides inside the host cell where it is protected from direct contact with the complement, antibodies, and immune cells. A lot of pathogens release compounds that misdirect of diminish the host’s immune response. Some bacteria even form biofilms which protects them from the proteins and cells of the immune system. Many successful infections often involve biofilms. Some bacteria create surface proteins that will bind to antibodies making them ineffective such as Streptococcus.
Other pathogens invade the body by changing the non-eseential epitopes on their surface rapidly while keeping the essential epitopes hidden. This is referred to as antigenic variation. HIV rapidly mutates so the proteins that are on its viral envelope which are essential for its entry into the host’s target cell are consistently changing. Since these antigens are changing so much, this is why vaccines have not been invented. Another common strategy that is used is asking the antigens with host molecules thus evading detection by the immune system. With HIV, the envelope covering the viron is created from the host cell’s outmost membrane making it hard for the immune system to identify it as a non-self structure.
Medicine Manipulation
[edit | edit source]The immune response system can be manipulated so that the unwanted responses that occur from allergy and autoimmunity can be suppressed. It can also be manipulated to heighten the protective responses against pathogens that evade the immune system. Autoimmune disorders, inflammation due to excessive tissue damage, and prevention of transplant rejection after donation of an organ transplant are controlled by immunosuppressive drugs. Anti-inflammatory drugs are used to control effects of inflammation, however with undesirable side effects such as osteoporosis. Thus anti-inflammatory drugs are often used with immunosuppressive drugs. Cytotoxic drugs can inhibit the immune system by destroying dividing cells like activated T cells. However, the negative part is that it is indiscriminate killing and other constantly dividing cells are also affected resulting in toxic side effects.
Larger drugs can promote a neutralizing immune response especially if it is repeatedly administered or in large doses. This thus limits its effectiveness based on larger proteins and peptides. Methods have been made to predict the immunogenicity of proteins and peptides which is particular useful when designing therapeutic antibodies. Earlier techniques often relied on the observation that hydrophilic amino acids are often more represented in epitope regions than hydrophobic amino acids.
Mechanism
[edit | edit source]An immune response is triggered by the presence of a foreign macromolecule, often a protein or carbohydrate; these are known as an antigen. For example, immunoglobins exist on the surface of B cells.
An innate immune system response is usually triggered by microbes identified by pattern recognition receptors. These defenses are non-specific and does not have long-lasting immunity against a foreign agent, but it is the most dynamic way a defense system responds in most organisms.The innate immune system functions to recruit immune cells to the infection site by producing cytokines, activate complement cascade to identify pathogen, assist white blood cells in identification, and activation of the adaptive immune system through antigen presentation.
The complement system attacks the surface of pathogens. Containing over 20 proteins, the complement system is the biggest humoral component of the innate immune response. This response is triggered by complement binding to antibodies attached to carbohydrates on the surface of microbes triggering the rapid killing response. The complement proteins are initially bound to microbes and activate their protease activity which activates other complement proteases producing a catalytic cascade that amplifies the inial signal using positive feedback. This results in the production of peptides attracting immune cells and increasing the saccular permeability while marking the surface of the pathogen for destruction.
Another quick response to infection is inflammation. Usually marked by redness and swelling caused by increased blood flow to the tissue, inflammation is produce by eicosanoids and cytokins that are released by attacked cells. Eicosanoids induce fever and dilation of blood vessels while leukotrienes attract white blood cells. Cytokins recruit immune cells to the infection site to promote healing of damaged tissue after removing the pathogens.
The adaptive immune system proves to eliminate and prevent pathogens by recognizing and remembering specific pathogens and creating stronger attacks for each encounter of the pathogen. This adaptive property helps prepare the body for future challenges.
When an antigen binds to this immunoglobin, the B cell engulfs the antibody-antigen complex and degrades it. Following this process, the T cells stimulate the B cells to proliferate and the process is repeated for additional antigens. B cells may live for several days; some B cells live for years and are called memory B cells. These can stimulate a more rapid response to an antigen they have encountered in the past.
Image:newImmuneSystemOverview.gif
Structure
[edit | edit source]Antibody structures form a related and big group of proteins. All immunoglobins contain four subunits at the least: 2 identical heavy chains (weighing 53-75 kD) and 2 identical light chains (weighing approximately 23 kD). The subunits are attached by disulfide bonds as well as non-covalent interactions to form a Y shaped structure which is symmetric. There are five different classes of immunoglobins (IgA, IgD, IgE, IgG, IgM) differing mostly in the type of heavy chain they contain and sometimes in their subunit structure. As a result, different immunoglobins have different functions. For example, IgE bids to allergens and protects against parasitic worms while IgA is found in mucus and prevents colonization. The most common of th immunoglobins by far is IgG.

An immunoglobin consists of homology units which all have the same characteristic fold. This fold, which is in the light chain, consists of a 'sandwich'-like structure composed of three and four stranded anti-parallel beta-sheets that are linked by a disulfide bond. This structure can accommodate an enormous variety of antigens. The light chain recognizes antigens through three loops in its variable domain (which is an area part of the light chain). This domain includes the most amino acid variation among antibodies in the whole immunoglobin; these are called hybervariable sequences.
The forces and bonding involved between an antibody and antigen include van der waals, hydrogen bonding, hydrophobic, and ionic interactions. The two are structurally complementary to each other; therefore, strong bonds are formed. Dissociation constants between an antigen and an antibody range from 10^-4 - 10^-10, which is greater than or equal to the dissociation constant associated with an enzyme and its substrate.
For the most part, immunoglobins are divalent molecules capable of binding to two different antigens at the same time. A foreign organism or substance usually has many antigens on its surface. Thus, a typical immune response consists of a mixture of antibodies with different specificities divalently binding to the antigens. This binding allows the cross-linking of the antigens to form an extended lattice formation, which assists and decreases the time in which it takes to remove the antigen. This also triggers further B cell formation and proliferation. [[Media:Media:Example.ogg]]
Antibody generation
[edit | edit source]An antigen does not influence a B cell to produce new immunoglobin to bind to. Instead, an antigen stimulates the proliferation of a pre-existing B cell antibodies that recognize the antigen. Thus, the immune system has the ability to generate a plethora of different antibodies. Most of these are sufficient for a person to respond through his or her immune system to respond to almost any antigen he or she may encounter. The diversity in antibody sequences arises from genetic changes during B lymphocyte development not only from the number of immunoglobin genes.
The immune system is unique in that it only responds to foreign substances and not to the high and diverse amount of endogenous molecules.Because most macromolecules are virtually antigenic, transferring tissues, organs, or blood samples among individuals and within species presents great challenges and is being researched continuously.
The immune system may lose tolerance to some of its self-antigens, causing an autoimmune disease, which at its worst, could be deadly. Autoimmune diseases include: Addison's disease, Crohn's disease, Multiple sclerosis, Psoriasis, and Graves' disease.
Addison's disease is caused when the adrenal glands do not produce enough steroid hormones known as cortisol. This rare genetic disease may develop in children, adults, and even some species of animals. The treatment involves the replacement of hormones.
Crohn's disease is the autoimmune, inflammatory disease of the intestines. The body's immune system attacks the gastrointestinal tract causing inflammation. Commonly believed to be a primary T cell autoimmune disorder, new studies believe it to be an impaired innate immunity due to impaired cytokine secretion by macrophages causing microbial-induced inflammatory response.
Multiple sclerosis is when the body's immune system attacks the central nervous system leading to demyelination. Affecting the communication between the spinal cord and the brain, nerve cells communicate by sending electrical signals (action potentials) down axons which are wrapped in myelin. Myelin is attacked and is damaged resulting in MS.
Psoriasis is a chronic, mostly hereditary, non-contagious auntoimmune disease affecting the skin and joins as it becomes red and scaly patches appear on the skin. These areas of inflammation and excess skin production have a silvery-white appearance. In this special case, inflammation isn't caused by pathogens as T cells move from the dermis to the epidermis.
Graves' disease is an autoimmune disease affecting the thyroid causing it to enlarge its size twice and become overactive. This also affects the eyes and other systems of the body. The body produces antibodies to the receptor for the thyroid-stimulating hormone (TSH) causing hyperthyroidism as the TSH receptor and antibodies bind. There becomes an abnormally high production of T3 and T4 which causes hyperthyroidism and enlargement of the thyroid gland.

Genome regulation consists of many mechanisms that are utilized in the cell. These mechanisms can either increase or decrease the production of specific gene products: RNA and proteins. Gene regulation is used in biology to describe metabolic pathways that respond to the environment. Any step in gene expression (transcription, translation, RNA processing, and modification of a protein) can be seen as gene regulation.
Gene expression plays an essential role in viruses, prokaryotic and eukaryotic cells because it increases the ability of an organism to adapt to its surroundings and allows the cell to express a particular protein when it is needed. Furthermore, in multicellular organisms gene expression comes from the process of cellular differentiation and morphogenesis, which leads to different types of cells being created to synthesis different proteins.
Any step in gene expression can be modulated to increase or decrease the reaction activity. The following list are the stages where gene expression can be regulated
- Transcription
- Chromatin domains
- Post-transcriptional modification
- RNA Transport
- Translation
- mRNA degradation
The purpose of genomics research has been the sequencing and analysis of the human genome. The human genome is made up of about 3 billion base pairs of DNA, which are distributed among 24 chromosomes. This poses the difficulty of producing a complete sequence. By going through the process of an organized international effort of academic labs and private companies, though, the human genome has now progressed from a draft sequence which was first reported in 2001 to a finished sequence in 2004.
The human genome contains a plethora of information about the different characteristics of humanity, including biochemistry and evolution. Analysis of the human genome will continue to grow and expand in order to better understand humans. One of the first tasks in accomplishing this is to develop an inventory of protein-encoding genes. When the genome-sequencing project first began, about 100,000 genes were estimated to exist. When the completed, but unfinished, genome was first made available, this estimate actually dropped down to about 30,000 to 35,000 genes. It dropped again to 20,000 to 25,000 when the sequence was finished. Because there are a relatively large number of pseudogenes, many of which are formerly functional genes that have mutated and are no longer expressed, the estimate of genes dropped by 75%. More than half of the genomic regions for olfactory receptors, the molecules responsible for the human sense of smell, are pseudogenes. The surprisingly small number of genes still belies the complexity of the human proteome, though. Many genes encode more than one protein through mechanisms such as alternative splicing of mRNA and posttranslational modifications of proteins. Important variations in functional properties are shown in the different proteins encoded by a single gene.
A large amount of DNA is contained in the human genome that does not participate in the coding of proteins. Modern-day biochemists and genetic researchers are attempting to elucidate the roles of this noncoding DNA, as a lot of this DNA shows due to the existence of mobile genetic elements. These elements have inserted themselves throughout the genome over the course of time. Most of these elements, though, have gone through mutations and, therefore, are no longer functional. More than 1 million Alu sequences, each sequence consisting of about 300 bases, are present in the human genome. Alu sequences are examples of SINES, short interspersed elements. Additionally, the human genome includes nearly 1 million LINES, long interspersed elements, which are DNA sequences that are 10 kilobase pairs in length. The roles of these elements as neutral parasites or instruments of genome evolution are still being studied.
The exploration of genes relies on key tools 1. Restriction-enzyme analysis 2. Blotting techniques 3. DNA sequencing 4. Solid-phase synthesis of nucleic acids 5. The Polymerase chain reaction
Comparative Genomics
[edit | edit source]Insight into the human genome can be discovered by comparing it with the genomes of other organisms. For example, the sequencing of the genome of chimpanzees, the closest living relative to humans, is almost complete. Genomic comparisons with other mammals used in biological research, such as mice and rats, have already been completed. The results of these comparisons show that 99% of human genes have counterparts in these rodent genomes. These genes, however, have been greatly reassorted among chromosomes in the estimated 75 million years of evolution since humans and rodents have had a common ancestor. The genomes of other organisms has been determined specifically for the purpose of comparative genomics. When comparing the genomes of various other organisms with human genomes, more than 1000 formerly unrecognized human genes are revealed. Furthermore, comparative genomics is a powerful tool for both interpreting the human genome as well as understand major events in the origin of genera and species.
External Links
[edit | edit source]Electrophoresis is the movement and subsequent separation of ions and charge macromolecules through a medium when an electric potential is applied. It is a powerful tool used in fundamental research and diagnostic settings for analysis of biomolecules. It is commonly used for purity and molecular weight determination for both DNA and protein solutions.
Theory
[edit | edit source]The idea behind DNA gel electrophoresis is to separate the fragments of a DNA sample and compare the results to a positive and negative control to determine if a DNA sample contains a particular gene. The size of the DNA fragments in the sample are determined by the specific primers used to fragment the DNA. The primers used will vary depending on the gene that is in question. For example, a DNA gel searching for the MPI (mannose phosphate isomerase) will use different primers than a Vil-Cre gel. Electrophoresis is a very powerful method to separate proteins and other macromolecules such as DNA and RNA. The speed of migration of a protein, DNA, or RNA molecule in an electric field is contingent on the strength of the electric field, the net charge of the protein, and the frictional coefficient. These turns can be summarized in the equation v = Ez/f, where v is the speed of migration, E is the electric field strength, and f is the frictional coefficient. Electrophoresis experiments are generally carried out in gels because the gel can act as a molecular sieve that amplifies the effects of separation. Small proteins move very quickly through the gel, while large proteins do not move as rapidly, staying near the point of application.
Agarose Gel
[edit | edit source]The structure of an agarose polymer is as follows:

In DNA electrophoresis, agarose gel is the common support material used. Agarose is a linear polymer of D-galactose and 3,6-anhydrogalactose that is isolated from seaweed. It forms a macroporous matrix which allows rapid diffusion of high molecular weight macromolecules without significant restriction by the gel. The concentration of agarose in the gel determines the average pore size. The size of a gel controls the mobility and resolution of components because of the sieving effect of the pores on macromolecular species.
Preparing the sample
[edit | edit source]Typically, the DNA sample is prepared by adding the specific primers to fragment the DNA and a loading dye which allows the DNA bands to show after the image is captured.
Loading the sample
[edit | edit source]After the agarose gel sets, the samples are loaded into the wells. Along with the samples in question, two control samples, a positive and negative control, are loaded as well. The positive sample of DNA will already be confirmed to contain the gene in question. The negative control should not contain any genetic material whatsoever and should therefore appear as a blank slot in the gel.
Additionally, a DNA ladder is loaded into one of the wells. The ladder contains fragments of known sizes and essentially serves as the ruler in determining the size of the sample bands. Various ladders can be used, depending on the sample being tested. Two common ladder sizes are 100 base pairs and 1000 base pairs. Each of these have very different ranges.

Running the gel
[edit | edit source]In order to run the sample DNA through the gel, a running buffer (PBS) is added to the gel box until it submerges the gel. Then, the sample is exposed to an electrical current, allowing the DNA to move through the gel and separating the various fragments in the process. After the voltage is applied, the sieving ability of the gel separates the proteins based on their size, with the smallest proteins moving most rapidly. The distance of mobility of polypeptide chains under these conditions can be measured by looking at the inverse logarithm of their mass. The mobility of the polypeptide chains is linearly proportional to this value.

After the DNA sample runs through a significant portion of the gel, an image of the various bands can be collected. By comparing the sample bands to the control bands, a researcher is able to determine if a DNA sample contains the specific gene being tested for. The strength of separation by electrophoresis can be seen through examining fractions produced during the purification scheme. The fractions that are produced initially will show many different proteins. However, as the separation runs for longer time, the number of bands will shrink, and the intensity of one of the bands should increase. The band with the highest intensity should reveal the protein of interest.

DNA Mircoarrays is a collection of genes that allows the study of gene expression in the biological response to further studies of the genome. Examples of the usage in mircoarrays are pathogen response to genetic materials, mutant development in the genes, and the drug discovery. Mircoarrays can be used to study the gene expression of a certain disease and that analysis and data acquired from this method can be used to develop a complementary drug that can cure or suppress the disease. Although the development of DNA microarrays has facilitated many studies in the biology and chemistry, the analysis of microarrays can be difficult. Because there are so many genes in a cell the analysis of such large gene expression presented in the microarrays can be rather time consuming and difficult to analyze.
History
[edit | edit source]The history of microarrays is rather recent. The first array was created in the mid 1980s. It was first called Macro arrays. It was only used to spot DNA probes, but more of it was used for studies of DNA clones, PCR products and oligonucleotides. They were all used with radioactively labeled targets. After further studies, Micro arrays were developed; it was created by pin spotters. Pin spotters were pin based robotic system that can accurately dispense a certain amount of DNA solution into the spots on a glass slide. By the mid 1990s, the technology of the microarrays was used to mainly investigate gene expressions. Some of the studies involved the gene expression profiles for tissues to study the life cycles of a bacteria. Also the study of cell division and which gene was responsible the different stages of cell division. Lastly, another application of microarrays is drug dosing, which can be effectively used to find the correct amount of drugs to use for a certain disease by examining the disease's gene expressions.
Theory
[edit | edit source]
DNA microarrays can be used to explore thousand of sequence of gene in a single run. The fundamental basis of DNA microarray is based on the process of hybridization. The level of hybridization can be detected by the level of detectable chemical level, which is used to mark the target or the probe sequence in the experiment. DAN micrarrays or gene chip, which is high-density array of oligonucleotides, can be built either with light directed chemical synthesis conducted by the photolitographic microfabrication technique used by the semiconductor industry or by putting very small dots of oligonucleotides or cDNA on a solid support such as microscope slide. The expression of the gene level is revealed by the fluorescence level of the cDNA which is hybridized to the chip, which can be identified by the known location on the chip. The extent of the transcription of a particular gene can be seen by the intensity of the fluorescent spot on the chip. This method can be used to detect the variation in expression level shown by specific genes under different growth conditions
A somewhat simpler picture of gene chip theory begins with the goal. The purpose of this technique is to be able to qualitatively determine the amount of a certain mRNA fragment expressed by a gene. First, the mRNA is extracted from cells. The mRNA can then be turned into cDNA via reverse transcriptase, which is tagged with a fluorescent marker. That fluorescent cDNA is then added to a silicon (or glass) chip studded with lots of DNA fragments (oligonucleotides). The gene chip, with all of its DNA fragments, contains one particular fragment which base-pairs perfectly to your desired cDNA (or hybridizes). The RNA/cDNA that don't match the gene chip are washed off (aww), and the gene chip is seen under a special light that allows the fluorescent cDNA to glow. If your mRNA was present in the cell, it can now be seen on the chip in pretty colors. If not, then that part of the chip is blank. It is easy to see now how gene chips can be used to study gene expression by exploring its mRNA.
An example of how one can use microarray testing in order to analyze RNA is in the case of cancerous skin cells. A microarray test can be done to see what genes in the cancerous skin cells are similar to healthy skin cells and which ones are different. This is done by taking a sample of mRNA from healthy skin cells and a sample of mRNA from cancerous skin cells. Both of the mRNA of these skin cells are converted into cDNA. The healthy cDNA can then be labeled with a green fluorescent marker. The cancerous cDNA can then be labeled with a red fluorescent marker. Then a microarray containing all of the DNA in a skin cell is made. The fluorescent cDNAs are then both put onto the microarray. The specific cDNA sequences then hybridize to the corresponding DNA on the microarray. The excess cDNA is then washed off. Then a laser is used to analyze where the fluorescent cDNA hybridized. A computer takes this information and analyzes it giving several dots of color on a sheet indicating which genes are expressed by the healthy skin cell (which would show up as a green dot) and which are expressed by the cancerous skin cell (which would show up as a red dot). The yellow dots would indicate that the healthy and cancerous skin cell both express that certain gene. A black dot indicates that neither the healthy cell nor the cancerous skin cell expressed that gene. Thus by analyzing this data, one can find out what genes are expressed in cancerous skin cells that are not expressed in healthy ones.
Applications
[edit | edit source]Microarrays can be used for a variety of applications including analysis of genomic DNA, however, mRNA gene expression profiling dominates because of the amount of information we can get about the function of genes in cells and tissues using these two applications.
Gene Expression Profiling
[edit | edit source]
Expression profiling using microarrays identifies genes whose expression depends on a specific biological state. For example, the amount of gene mRNAs can be more abundant during a disease state biological condition than in a normal state. Genes expressed in the disease state can then be identified using microarrays. Drugs can then be made to specifically inhibit these genes. Microarrays can also be tack gene expression during cell development, during which genes are grouped according to their pattern of expression over different phases of development. This then helps to identify a gene of unknown function by associating it to a functional role of a known gene whose expression pattern is similar to the unknown gene.
Pathway analysis can also be useful in identifying coordinated changes in expression affecting many genes at a time, which then helps us know the characterizing behavior of these genes that makes them act together to carry out a specific function. The detection of consistent changes in expression of a group of genes that have related function allows us to know the biological aspects that would otherwise not be known from gene expression analysis.
Reconstruction of functional, regulatory networks from gene expression by reverse engineering techniques can also be used to characterize the relationship among genes starting from their expression values in different conditions.
Another way we can use expression profiles is to use them as a fingerprint of a certain biological state by using the analysis of other hybridization experiments to identify common patterns of gene expression among samples with similar biological characteristics. This is useful in finding correlations between gene expression behavior and a phenotype.
Analysis of Genomic DNA
[edit | edit source]Analysis of genomic DNA is the other dominant microarray application. DNA microarrays can be used to directly measure the concentration of genomic DNA fragments from particular genomic regions. For example, microarrays can be applied in this way to scan changes in the gene copy number associated to cancer. Another way microarrays can be used for genomic DNA analysis is to identify the complement of regulatory DNA sequences that are bound by transcriptional regulators. DNA microarrays can be used for a genome-wide identification of in vivo transcription factor binding sites by chromatin immunopercipitation (ChIP) coupled with array hybridization. The overlapping probes on the array yields complete genomic coverage and allows for the identification of all DNA binding regions for a specific transcription factor. Analysis of genomic DNA using Microarrays can also be applied to characterize the presence of specific genetic sequences that can be used for parallel interrogation of pathogen genomes. This can then help us detect the presence of pathogens that cause infective diseases and we can monitor if they are in biological samples, for example, or food/water. Finally, analysis of genomic DNA using microarrays can be applied to genotyping. Microarrays can be designed for the genome-wide identification of single nucleotide polymorphisms (SNPs). Microarray usage for this has contributed greatly to the human genome project regarding human SNPs data. If one is interested in a particular list of known polymorphisms scattered throughout the genome, then probe sets can be designed just for them and chips can be used as a tool for rapid screening of biological samples. Chips were designed for this reason of detecting mutations in genes of interest to human health.
Data Analysis
[edit | edit source]It can be difficult to interpret the data that comes out on the microarray analyzer because there are many factors that can effect the accuracy of the data. The difficulty about getting clear results on the microarray is due to the fact that there are many different methods for normalizing the data of a microarray chip. Another factor to take into consideration is that the genes can produce false positives or false negatives because there are thousands of tests being run at the same time, which cause these errors. A false positive in this case would be concluding that a certain gene is expressed on the microarray when it really is not. A false negative would be concluding that the gene is not expressed on the microarray when it really is.
The statistical p-value is used in microarrays since there are these factors that effect the accuracy of the data. The p-value is the probability that the microarray results for a specific gene being hybridized to the DNA or not hybridized happened by chance alone. It is not accurate to treat the p-value as a statistic for an individual test when you are running a microarray since thousands of tests are being performed simultaneously. That is why one must adjust the p-value in order to account for this error. The Bonferroni correction is a statistical device that corrects the p-value making it more accurate.
Two additional statistical analysis that are useful for microarrays are
1. SAM (statistical analysis of microarrays) This test helps determine which data acquired from the microarray is useful and which is not. It does this by running a bunch of t-tests and then calculates a value for each gene. This value represents the correlation between the strength of the gene expression and its response variable.
2. ANOVA (analysis of variance) This is a statistical test used for analyzing experiments when two or more variables are present.
Reference
[edit | edit source]DNA Microarrays Current Applications. Emanuele de Rinaldis and Armin Lahm. Horizon bioscience. 2007. Due to modern techniques of DNA analysis, many genomes have been sequenced and analyzed. A famous example is the human genome through the Human Genome Project.
Human Genome Project
[edit | edit source]The human genome project was an international scientific research effort to fully map out the human genome. This project was started by James D. Watson at the US Institute of Health, but research centers worked on the project all over the world; such as France, Germany, Japan, China, the United Kingdom, and India. So far about 92.3% of the genome has been sequenced, but its difficult to determine due to non-coding sequences of DNA or "junk" DNA.
The genome project uncovered some key findings such as the genome of the human race is 99.9% alike.
Homology
[edit | edit source]Sequencing genomes allow scientists to identify homologous proteins and establish evolutionary relationships. Furthermore if a newly discovered protein is homologous to a known protein, through homology scientists can make an educated guess on how the new protein functions.
The Impact of Sequencing on Medicine
[edit | edit source]The ability to quickly sequence the human genome in the future may have significant impacts on medicine. Knowledge about genes and an individual's DNA have already given scientists a way to predict the likelihood of certain diseases among individuals. This also allows one to analyze the chromosomal structure, the effects of evolution upon the genome, and protein structures and functions. In the future, gene therapy, genomic medicine, and preventative treatments may reduce the likelihood of disease and allow manufacturers to tailor drugs to specific individuals.
Sequenced Eukaryotic Genomes
[edit | edit source]Eukaryotes are organisms containing cells that enclose complex organelles within a well-defined cell membrane. The defining characteristic that sets Eukaryotes and Prokaryotes apart is Eukaryotes' nucleus, or nuclear envelope, in which an organism's genetic information is contained.
The first eukaryotic genome to be sequenced is that of Saccharomyces cerevisiae (S. cerevisiae) in 1996, and it is commonly known as brewer's yeast. S. cerevisiae is the most useful type of yeast due to its utility in baking and brewing, so it is the most studied eukaryotic model organisms in molecular and cell biology, similar to E. coli's role in the study of prokayortic organisms. Many proteins that are important to humans are studied by examining their homologs in yeasts. For example, signaling proteins and protein-processing enzymes are all discovered through the help of yeast genome.
Other fully sequenced organisms include: roundworm, fruitfly, pufferfish (first vertebrate to be sequenced after humans), and Arabidopsis thaliana.
The tables from below are taken from Wikipedia's list of sequenced eukaryotic genomes.
Protists
[edit | edit source]Chromista
[edit | edit source]The Chromista are a group of protists that contains the algal phyla Heterokontophyta, Haptophyta and Cryptophyta. Members of this group are mostly studied for evolutionary interest.
| Organism | Type | Relevance | Genome size | Number of genes predicted | Organization | Year of completion |
|---|---|---|---|---|---|---|
| Guillardia theta | Cryptomonad | Model organism | 0.551 Mb (nucleomorph genome only) |
464[1] | Canadian Institute of Advanced Research, Philipps-University Marburg and the University of British Columbia | 2001[1] |
| Thalassiosira pseudonana Strain:CCMP 1335 |
Diatom | 2.5 Mb | 11,242[2] | Joint Genome Institute and the University of Washington | 2004[2] | |
| Phaeodactylum tricornutum Strain: CCAP1055/1 |
Diatom | 27.4 Mb | 10,402 | Joint Genome Institute | 2008 [3] |
Alveolata
[edit | edit source]Alveolata are a group of protists which includes the Ciliophora, Apicomplexa and Dinoflagellata. Members of this group are of particular interest to science as the cause of serious human and livestock diseases.
| Organism | Type | Relevance | Genome size | Number of genes predicted | Organization | Year of completion |
|---|---|---|---|---|---|---|
| Babesia bovis | Parasitic protozoan | Cattle pathogen | 8.2 Mb | 3,671 | 2007[4] | |
| Cryptosporidium hominis Strain:TU502 |
Parasitic protozoan | Human pathogen | 10.4 Mb | 3,994[5] | Virginia Commonwealth University | 2004[5] |
| Cryptosporidium parvum C- or genotype 2 isolate |
Parasitic protozoan | Human pathogen | 16.5 Mb | 3,807[6] | UCSF and University of Minnesota | 2004[6] |
| Paramecium tetraurelia | Ciliate | Model organism | 72 Mb | 39,642[7] | Genoscope | 2006[7] |
| Plasmodium falciparum Clone:3D7 |
Parasitic protozoan | Human pathogen (malaria) | 22.9 Mb | 5,268[8] | Malaria Genome Project Consortium | 2002[8] |
| Plasmodium knowlesi | Parasitic protozoan | Primate pathogen (malaria) | 23.5 Mb | 5,188[9] | 2008[9] | |
| Plasmodium vivax | Parasitic protozoan | Human pathogen (malaria) | 26.8 Mb | 5,433[10] | 2008[10] | |
| Plasmodium yoelii yoelii Strain:17XNL |
Parasitic protozoan | Rodent pathogen (malaria) | 23.1 Mb | 5,878[11] | TIGR and NMRC | 2002[11] |
| Tetrahymena thermophila | Ciliate | Model organism | 104 Mb | 27,000[12] | 2006[12] | |
| Theileria parva Strain:Muguga |
Parasitic protozoan | Cattle pathogen (African east coast fever) | 8.3 Mb | 4,035[13] | TIGR and the International Livestock Research Institute | 2005[13] |
| Theileria annulata Ankara clone C9 |
Parasitic protozoan | Cattle pathogen | 8.3 Mb | 3,792 | Sanger | 2005[14] |
Excavata
[edit | edit source]Excavata is a group of related free living and symbiotic protists; it includes the Metamonada, Loukozoa, Euglenozoa and Percolozoa. They are researched for their role in human disease.
| Organism | Type | Relevance | Genome size | Number of genes predicted | Organization | Year of completion |
|---|---|---|---|---|---|---|
| Leishmania major Strain:Friedlin |
Parasitic protozoan | Human pathogen | 32.8 Mb | 8,272[15] | Sanger Institute | 2005[15] |
| Giardia lamblia | Parasitic protozoan | Human pathogen | 11.7 Mb | 6,470[16] | 2007[16] | |
| Trichomonas vaginalis | Parasitic protozoan | Human pathogen (Trichomoniasis) | 160 Mb | 59,681[17] | TIGR | 2007[17] |
| Trypanosoma brucei Strain:TREU927/4 GUTat10.1 |
Parasitic protozoan | Human pathogen (Sleeping sickness) | 26 Mb | 9,068 [18] | Sanger Institute and TIGR | 2005[18] |
| Trypanosoma cruzi Strain:CL Brener TC3 |
Parasitic protozoan | Human pathogen (Chagas disease) | 34 Mb | 22,570[19] | TIGR, Seattle Biomedical Research Institute and Uppsala University | 2005[19] |
Amoebozoa
[edit | edit source]Amoebozoa are a group of motile amoeboid protists, members of this group move or feed by means of temporary projections, called pseudopods. The best known member of this group is the slime mold which has been studied for centuries; other members include the Archamoebae, Tubulinea and Flabellinea. Some Amoeboza cause disease.
| Organism | Type | Relevance | Genome size | Number of genes predicted | Organization | Year of completion |
|---|---|---|---|---|---|---|
| Dictyostelium discoideum Strain:AX4 |
Slime mold | Model organism | 34 Mb | 12,500[20] | Consortium from University of Cologne, Baylor College of Medicine and the Sanger Centre | 2005[20] |
| Entamoeba histolytica HM1:IMSS |
Parasitic protozoan | Human pathogen (amoebic dysentery) | 23.8 Mb | 9,938[21] | TIGR, Sanger Institute and the London School of Hygiene and Tropical Medicine | 2005[21] |
Plants
[edit | edit source]Higher plants
[edit | edit source]| Organism | Type | Relevance | Genome size | Number of genes predicted | Organization | Year of completion |
|---|---|---|---|---|---|---|
| Arabidopsis thaliana Ecotype:Columbia |
Wild mustard | Model plant | 120 Mb | 25,498[22] | Arabidopsis Genome Initiative[23] | 2000[22] |
| Brassica napus | Rapeseed | Oil plant | 1,100 Mb | Bayer CropScience | 2009[24] | |
| Oryza sativa ssp indica |
Rice | Crop and model organism | 420 Mb | 32-50,000[25] | Beijing Genomics Institute, Zhejiang University and the Chinese Academy of Sciences | 2002[25] |
| Oryza sativa ssp japonica |
Rice | Crop and model organism | 466 Mb | 46,022-55,615[26] | Syngenta and Myriad Genetics | 2002[26] |
| Ostreococcus tauri | Green alga | Simple eukaryote | 12.6 Mb | Laboratoire Arago | 2006[27] | |
| Physcomitrella patens | Bryophyte | Model organism
early diverging land plant |
500 Mb | 39,458[28] | US Department of Energy Office of Science Joint Genome Institute | 2008[28] |
| Populus trichocarpa | Balsam poplar or Black Cottonwood | Carbon sequestration, model tree, commercial use (timber), and comparison to A. thaliana | 550 Mb | 45,555[29] | The International Poplar Genome Consortium | 2006[29] |
| Vitis vinifera | Grapevine PN40024 | Fruit crop | 490 Mb[30] | 30,434[30] | The French-Italian Public Consortium for Grapevine Genome Characterization | 2007[30] |
| Zea mays ssp mays |
Corn (maize) | Fruit crop | 2,800 Mb | 50,000-60,000 | NSF | 2008[31] |
Algae
[edit | edit source]| Organism | Type | Relevance | Genome size | Number of genes predicted | Organization | Year of completion |
|---|---|---|---|---|---|---|
| Cyanidioschyzon merolae Strain:10D |
Red alga | Simple eukaryote | 16.5 Mb | 5,331[32] | University of Tokyo, Rikkyo University, Saitama University and Kumamoto University | 2004[32] |
| Thalassiosira pseudonoana[33] | Heterokont | |||||
| Chlamydomonas reinhardtii[34] | Model organism | 2007[34] | ||||
| Ostreococcus tauri[33] | Chlorophyte |
Fungi
[edit | edit source]| Organism | Type | Relevance | Genome size | Number of genes predicted | Organization | Year of completion |
|---|---|---|---|---|---|---|
| Ashbya gossypii Strain:ATCC 10895 |
Fungus | Plant pathogen | 9.2 Mb | 4,718[35] | SyngentaAG and University of Basel | 2004[35] |
| Aspergillus fumigatus Strain:Af293 |
Fungus | Human pathogen | 29.4 Mb | 9,926[36] | Sanger Institute, University of Manchester, TIGR, Institut Pasteur, Nagasaki University, University of Salamanca and OpGen | 2005[36] |
| Aspergillus nidulans Strain:FGSC A4 |
Fungus | Model organism | 30 Mb | 9,500[37] | 2005[37] | |
| Aspergillus niger Strain:CBS 513.88 |
Fungus | Biotechnology - fermentation | 33.9 Mb | 14,165[38] | 2007[38] | |
| Aspergillus oryzae Strain:RIB40 |
Fungus | Used to ferment soy | 37 Mb | 12,074[39] | National Institute of Technology and Evaluation | 2005[39] |
| Candida glabrata Strain:CBS138 |
Fungus | Human pathogen | 12.3 Mb | 5,283[40] | Génolevures Consortium [41] | 2004[40] |
| Cryptococcus (Filobasidiella) neoformans JEC21 |
Fungus | Human pathogen | 20 Mb | 6,500[42] | TIGR and Stanford University | 2005[42] |
| Debaryomyces hansenii Strain:CBS767 |
Yeast | Cheese ripening | 12.2 Mb | 6,906[40] | Génolevures Consortium | 2004[40] |
| Encephalitozoon cuniculi | Microsporidium | Human pathogen | 2.9 Mb | 1,997[43] | Genoscope and Université Blaise Pascal | 2001[43] |
| Kluyveromyces lactis Strain:CLIB210 |
Yeast | 10-12 Mb | 5,329[40] | Génolevures Consortium | 2004[40] | |
| Magnaporthe grisea | Fungus | Plant pathogen | 37.8 Mb | 11,109[44] | 2005[44] | |
| Neurospora crassa | Fungus | Model eukaryote | 40 Mb | 10,082[37] | Broad Institute, Oregon Health and Science University, University of Kentucky, and the University of Kansas | 2003[37] |
| Saccharomyces cerevisiae Strain:S288C |
Baker's yeast | Model eukaryote | 12.1 Mb | 6,294[45] | International Collaboration for the Yeast Genome Sequencing[46] | 1996[45] |
| Schizosaccharomyces pombe Strain:972h |
Yeast | Model eukaryote | 14 Mb | 4,824[47] | Sanger Institute and Cold Spring Harbor Laboratory | 2002[47] |
| Yarrowia lipolytica Strain:CLIB99 |
Yeast | Industrial uses | 20 Mb | 6,703[40] | Génolevures Consortium | 2004[40] |
Animals
[edit | edit source]Mammals
[edit | edit source]| Organism | Type | Shotgun Coverage | Genome size | Number of genes predicted | Organization | Year of completion |
|---|---|---|---|---|---|---|
| Bos taurus | Cow | 6* | 3.0 Gb[48][49] | 22000[50] | Cattle Genome Sequencing International Consortium | 2009 |
| Canis lupus familiaris | Dog | 7.6* | 2.4 Gb[51] | 19,300[51] | Broad Institute and Agencourt Bioscience | 2005[51] |
| Cavia porcellus | Guinea Pig | 2* | 3.4 Gb | The Genome Sequencing Platform, The Genome Assembly Team[49] | ||
| Dasypus novemcinctus | Nine-banded Armadillo | 2* [52] | 3.0 Gb | Broad Institute[49] | ||
| Echinops telfairi | Hedgehog-Tenrec | 2* [52] | Broad Institute | |||
| Equus caballus | Horse | 6.8* | 2.1 Gb [49] | Broad Institute et al.[49] | 2007 [53] | |
| Erinaceus europaeus | Western European Hedgehog | 2* [52] | Broad Institute | |||
| Felis catus | Cat | 2* | 3 Gb | 20,285 | The Genome Sequencing Platform, The Genome Assembly Team[49] | 2007[54] |
| Homo sapiens | Human | 3.2 Gb [55] | 25,000[55] | Human Genome Project Consortium and Celera Genomics | Draft 2001[56][57] Complete 2006[58] | |
| Loxodonta africana | African Elephant | 2* [52] | 3 Gb | Broad Institute | ||
| Macaca mulatta | Rhesus Macaque | 6* | Macaque Genome Sequencing Consortium[49] | |||
| Microcebus murinus | Gray Mouse Lemur | 2* [52] | The Genome Sequencing Platform, The Genome Assembly Team[49] | |||
| Monodelphis domestica | Gray Short-tailed Opossum | 3.5 Gb | 18 - 20,000 | Broad Institute et al. | 2007[49][59] | |
| Mus musculus Strain: C57BL/6J |
Mouse | 2.5 Gb | 24,174[60] | International Collaboration for the Mouse Genome Sequencing[61] | 2002[60] | |
| Myotis lucifugus | Little Brown Bat | 2* [49] | Broad Institute | |||
| Ochotona princeps | American Pika | 2* [52] | Broad Institute | |||
| Ornithorhynchus anatinus [62] | Platypus | 6* [49] | Washington University | |||
| Oryctolagus cuniculus | Rabbit | 2* [52] | 2.5 Gb | Broad Institute et al. [49] | ||
| Otolemur garnettii | Small-eared Galago, or Bushbaby | 2* [52] | Broad Institute | |||
| Pan troglodytes | Chimpanzee | 6* [49] | 3.1 Gb | Chimpanzee Sequencing and Analysis Consortium | 2005[63] | |
| Pongo pygmaeus | Orangutan | 3.0 Gb | Institute for Molecular Biotechnology [49] | |||
| Rattus norvegicus | Rat | 1.8* or better | 2.8 Gb [49] | 21,166[64] | Rat Genome Sequencing Project Consortium | 2004[64] |
| Sorex araneus | European Shrew | 2* [52] | 3.0 Gb [49] | The Genome Sequencing Platform, The Genome Assembly Team[49] | ||
| Spermophilus tridecemlineatus | Thirteen-lined Ground Squirrel | 2* | The Genome Sequencing Platform, The Genome Assembly Team[49] | |||
| Tupaia belangeri | Northern Tree Shrew | 2* | Broad Institute[49] |
Insects
[edit | edit source]| Organism | Type | Relevance | Genome size | Number of genes predicted | Organization | Year of completion |
|---|---|---|---|---|---|---|
| Anopheles gambiae Strain: PEST |
Mosquito | Vector of malaria | 278 Mb | 13,683[65] | Celera Genomics and Genoscope | 2002[65] |
| Apis mellifera | Honey bee | Model for eusocial behavior | 1800 Mb | 10,157[66] | The Honeybee Genome Sequencing Consortium | 2006[66] |
| Bombyx mori Strain:p50T |
Moth (domestic silk worm) | Silk production | 530 Mb | University of Tokyo and National Institute of Agrobiological Sciences | 2004[67] | |
| Drosophila melanogaster | Fruit fly | Model animal | 165 Mb | 13,600[68] | Celera, UC Berkeley, Baylor College of Medicine, European DGP | 2000[68] |
Nematodes
[edit | edit source]| Organism | Type | Relevance | Genome size | Number of genes predicted | Organization | Year of completion |
|---|---|---|---|---|---|---|
| Caenorhabditis briggsae | Nematode worm | For comparison with C. elegans | 104 Mb | 19,500[69] | Washington University, Sanger Institute and Cold Spring Harbor Laboratory | 2003[69] |
| Caenorhabditis elegans Strain:Bristol N2 |
Nematode worm | Model animal | 100 Mb | 19,000[70] | Washington University and the Sanger Institute | 1998[70] |
| Meloidogyne hapla | Northern root-knot nematode | Vegetable pathogen | 54 Mb | 14,420[71] | 2008[71] | |
| Meloidogyne incognita | Southern root-knot nematode | Plant pathogen | 86 Mb | 19,212[72] | INRA, Genoscope and International M.incognita Genome Consortium[73] | 2008[72] |
| Pristionchus pacificus | Nematode worm | Model invertebrate | 169 Mb | 23,500[74] | Max-Planck Institute for Developmental Biology &
Genome Sequencing Center, Washington University School of Medicine |
2008[74] |
Other animals
[edit | edit source]| Organism | Type | Relevance | Genome size | Number of genes predicted | Organization | Year of completion |
|---|---|---|---|---|---|---|
| Ciona intestinalis | Tunicate | Simple chordate | 116.7 Mb | 16,000[75] | Joint Genome Institute | 2003[75] |
| Ciona savignyi | Tunicate | 174 Mb | Broad Institute | 2007[76] | ||
| Gallus gallus | Chicken | 1000 Mb | 20-23,000[77] | International Chicken Genome Sequencing Consortium | 2004[77] | |
| Strongylocentrotus purpuratus | Sea urchin | Model eukaryote | 814 Mb | 23,300[78] | Sea Urchin Genome Sequencing Consortium | 2006[78] |
| Takifugu rubripes | Puffer fish | Vertebrate with small genome | 390 Mb | 22-29,000[79] | International Fugu Genome Consortium[80] | 2002[81] |
| Tetraodon nigroviridis | Puffer fish | Vertebrate with compact genome | 340 Mb[82] | 22,400[82] | Genoscope and the Broad Institute | 2004[82] |
Sequenced Bacterial Genomes
[edit | edit source]There are some techniques which are improving to be fast and high volume DNA sequencing like fluorescent dideoxynucleotide chain terminators, "shot gun" method etc. The bacterial genome of Haemophilus influenza wa determined in 1995 with a "short gun" method. The genomic DNA is cut randomly into fragments and then the computer programs brings out the whole sequence by matching the overlapping regions between these fragments. The H. influenzae genome consists of 1,830,137 base pairs and encodes approximately 1740 proteins. With these similar approaches, more than 100 bacterial and archaeal species including key model of organisms such as E.coli, Salmonella typhimurium, and Archaeoglobus fulgidus, as well as pathogenic organisms such as Yersina pestis (causing bubonic plague) and Bacillus anthracis (anthrax).1
References
[edit | edit source]1. Berg, Jeremy M. 2007. Biochemistry. Sixth Ed. New York: W.H. Freeman. 68-69, 78. 2. Voet, Voet, Pratt (2004). - Fundamentals of Biochemistry
Identification of Single Nucleotide Polymorphisms (SNPs)
[edit | edit source]Major efforts have been made in recent years to identify the SNPs in the Human Genome. While previously known as base substitutions, the term SNP has become more common, indicating the importance of this frequent type of base substitution in molecular genetics. An SNP refers to an abundant base substitution, which differs from a rare substitution in that the population has a frequency of greater than 1% of the least abundant allele. Due to the high density of SNPs in genomes, several can usually be located within a few hundred base pairs. Thus, they are a rich source of information for population studies, and the association of genetic factors with disease states.
SNPs are used constantly in the field of disease study. For instance, they are used to identify the multiple genes that are involved in such common diseases such as cancer, diabetes, and certain versions of mental illness.
Polymorphisms and Disease
[edit | edit source]Approximately five hundred human methyltransferase polymorphisms have been identified and have been linked to disease. A polymorphic hotspot was identified at approximately 20 angstrom from the active site in four out of seven protein studied in the research performed by Karen Rutherford and Valerie Daggett. Simulations outlining the molecular dynamics of proteins demonstrated a mutual mechanism of destabilization. The common mechanism is the fact that the mutations change the side chain contacts within the polymorphic site that are sent throughout the protein causing a distortion to the active site. One theory is that these hot spots may have evolved to modulate enzymatic activity.
Single nucleotide polymorphisms occur at a frequency of 1 in every 1000 nucleotide bases of the 3 billion base human genome. SNPs can affect mRNA transcription, structure, enzymatic activity, and stability. Catechol O-methyltransferase (COMT) is a protein displayed to have ample inter-individual differences in enzymatic activity. Restriction fragment length polymorphism mapping acknowledged that the alterations in activity are accredited to a valine to methionine substitution. The alternative consequently decreased the levels in vivo. This substitution has been linked with an increased risk of breast cancer and neuropsychiatric diseases.
Thiopurine S-methyltransferase (TMPT) is at fault for the metabolism of cytotoxic thioputine drugs employed to medicate multiple conditions like leukemia and inflammatory bowel disease. In eleven percent of the population there has been more than twenty TPMT polymorphisms. NMR is important to understanding how the polymorphism affects protein structure and dynamics because structure dictates function. Molecular dynamic (MD) studies are of peak interest when understanding the effects of polymorphism because aggregation or stability issues do not bother them. MD allows one to notice how the mutations affect protein structure at an atomic level. MD demonstrates how COMT and TMPT polymorphisms affect structural detail. Research has demonstrated that polymorphisms contain destabilizing effects. TPMT was found to decrease the levels of immunological protein. Researchers now contain much epidemiological data for the wild type and variant COMT and TMPT proteins that make them more likely to be studied through MD simulations. The data known can be applied to molecule development for the stabilization of protein structure and activators for the treatments of diseases.
SNP Study Examples
[edit | edit source]The effect of genetic variability on drug response in conventional breast cancer treatment. [75]
Functional SNPs in the lymphotoxin-alpha gene that are associated with susceptibility to myocardial infarction [76]
Genetic polymorphisms of infectious diseases in case-control studies [77]
Bayesian statistical methods for genetic association studies [78]
Conjuring SNPs to detect associations [79]
References
[edit | edit source]1. Vignal, Alain, Denis Milan, Magali SanCristobal, and Andre Eggen. "A Review on SNP and Other Types of Molecular Markers and Their Use in Animal Genetics." Genetics Selection Evolution 34.3 (2002): 275-305. Print.
2. Rutherford, Karen, and Valerie Daggett. "Polymorphisms and Disease: Hotspots of Inactivation in Methyltransferases." Trends in Biochemical Sciences 35.10 (2010): 531-38. Print.
The Ethics and Politics of Genome Sequencing and Science
[edit | edit source]"As in the most important scientific advances, the HGP carries with it benefits and problems. The benefits are immeasurable, and so are the problems. As DNA techniques provide important information on human genes, many diagnostic tools and therapeutic benefits, we do not know if the same information can also be used to discriminate against such individuals, employees and insurance companies. There is a very real prospect of insurance companies insisting on large-scale genetic screening tests for the presence of genes that conferred susceptibility to common disorders such as diabetes, cardiovascular disease, cancer and various mental disorders."1
As we have seen in the past, controversy regarding stem cells, their origin, and their use has had political and scientific consequences. In 2001, President Bush banned the use of federal funds for later stem cell lines. Although this ban was later lifted, the political and human impact of science cannot be ignored; Science shows great promise with respect to human knowledge, technology, and power, but the gains we make through science carry with them great responsibilities. The freeze of federal funding on new stem cell lines significantly hindered scientific progress in a field that appears to show promise with respect to curing disease and improving the welfare of mankind, but it also serves as a reminder to the fact that science, for better or for worse, is intricately related to politics, and that our future scientific progress partly depends not on just scientists and their research, but on legislators and bureaucrats. Although DNA sequencing and genetic research is still a relatively new field, the implications of such knowledge carry social, political, and personal consequences in the field of medicine yet to be discovered.
In addition to the political relationship with science, technology, and medicine legally and financially, there are ethical considerations to be considered with new technologies. In the future, it may be possible to reliably clone human beings, artificially modify DNA to provide physical and mental benefits, and screen for "imperfections." The range of possible consequences of DNA analysis and manipulation technology is nearly infinite, and although it may be hard to forget the impact of scientific technologies and the power scientists already have today to manipulate DNA, it may be too easy to forget the responsibilities we have, both individually and socially, ethically and morally, with respect to the power we have and how we wield it. Whether there will be new laws passed regulating DNA with regard to how it is stored and used or whether there will be a new regulatory agency to protect against the misuse of DNA (through theft, sequencing, cloning, artificial manipulation, etc.) remains to be seen, partly because the technology is not widespread, and current capabilities are relatively limited by the lack knowledge we have about genes and multiple gene interactions and difficulties we have with respect to mammalian cloning.
In the future, however, this may change, and DNA manipulation on a mammalian scale may become very easy, especially with a greater understanding of how genes interact and what they do. What this will mean politically, economically, and ethically, is still anyone's guess. But as science drives new technologies, particularly with respect to DNA and gene manipulation, these questions will come into the public spotlight, as stem cell research has, and the consequences of such technologies will become more apparent and more profound.
Genome Sequencing and Medicine
[edit | edit source]Sickle Cell Anemia
[edit | edit source]
The name Sickle Cell Anemia comes from the shape that the red blood cells take when affected with the sickle-cell trait. The sickle shape, which is caused by red blood cells being deprived of oxygen, causes the small capillaries to clog up, and impair blood flow throughout the body. This can result in painful swelling of the extremities when the capillaries clog up, and cause a higher risk of bacterial infection or stroke because of the poor blood flow. Also, sickle cells do not circulate throughout the body as long as regularly shaped blood cells do, thus leading to anemia because of a decrease in amount of blood circulating. This mutation, though unfavorable in other ways, is slightly beneficial in that this trait makes the infected persons resistant to malaria.
Sickle Cell Anemia is caused by a variation of one specific amino acid sequence in just one hemoglobin chain. This idea was proposed by Pauling, and proved to be correct later on. Around 7% of the world has some sort of disease of the hemoglobin that is caused by a variation in the amino acid sequence. Then, in 1956, Vernan Ingram proved Pauling correct by showing that the specific reason for the sickle cell trait was indeed the substitution of a single amino acid in hemoglobin, more specifically, of a valine substituting a glutamate in position 6 in the beta chain of hemoglobin. With the advancements in genome sequencing, the exact cause of these diseases can be discovered, thus becoming one step closer to discovering a cure. In this way, genome sequencing can be related to medicine, because it helps to find the direct cause of the disease, thus allowing researchers to look at more specific ways to correct the problems, instead of just the symptoms.
Thalassemia
[edit | edit source]Thalassemia is another disorder caused by a change in the normal genome. In this case, it is caused by a loss of a single hemoglobin chain instead of a substitution like in Sickle Cell Anemia. This disorder causes anemia, pale skin, and fatigue. The loss of the single hemoglobin chain can be found by genome sequencing. Thalassemia is an autosomal recessive blood disorder. Unlike sickle cell anemia, where the globin does not function properly, thalassemia is a condition where normal globulins are not produced in high enough quantities. There are three kinds of thalassemia: alpha, beta, and delta. Each is characterized by the inability to produce the appropriate globin chain. Like sickle cell anemia, however, thalassemia provides carriers some measure of resistance against malaria.
References
[edit | edit source]1. Falcón de Vargas, Aída. "The Human Genome Project and its importance in clinical medicine." doi:10.1016/S0531-5131(01)00570-2 <http://www.sciencedirect.com/science?_ob=ArticleURL&_udi=B7581-47W61Y4-BG&_user=4429&_coverDate=07%2F31%2F2002&_rdoc=1&_fmt=full&_orig=search&_cdi=12913&_sort=d&_docanchor=&view=c&_searchStrId=1094477520&_rerunOrigin=google&_acct=C000059602&_version=1&_urlVersion=0&_userid=4429&md5=a44d78db6508ff39d2670fc0ce486bb1#toc9>.
2. Stryer, Lubert, Berg, Jeremy M., Tymoczko, John L. "Biochemistry" Sixth Edition. W.H. Freeman and Company. 2007. A karyotype a complete set of chromosomes of a particular species. The number and appearance of chromosomes can very dramatically between different organisms. Human beings have 46 chromosomes, 22 pairs of autosomal chromosomes and one pair of sex chromosomes. The latter is what determines whether a developing embryo develops as a physiological male or female, with a male karyotype displaying the diminutive Y chromosome beside its larger X chromosome partner (women have two X chromosomes in their karyotype).
A karyotype is generally an image of a completed and arranged set of chromosomes as viewed through a light microscope. A chromosome set can be attained from nearly any type of tissue.[83] Dividing cells are stained with a special dye, usually the Giemsa stain, and then cell division is halted during metaphase. An image of the dividing cells is taken when the chromosomes are all visible, and the individual chromosomes are cut out of the picture and rearranged on a separate medium based on size. Chromosome pairs are matched up also based on their size, banding pattern, and the position of their centromeres.[84]
Once arranged and ordered, scientists can then study the number and appearance of chromosomes. This can lead to medical diagnoses, as is the case with Down’s Syndrome. Down’s Syndrome is a disease in human caused by an extra copy of chromosome 21 (the syndrome is frequently referred to as Trisomy 21 for this reason). Down’s syndrome is easily identified via a karyotype by the obvious extra chromosome present in the image. Another example of a chromosomal abnormality is Turner syndrome (the presence of only a single X chromosome in women instead of the usual two)[85] Edwards syndrome is trisomy 18, and Patua syndrome is a result of trisomy 13. Both Edwards and Patua syndrome result in death while still in infancy.[86]
Indeed, some people insist on doing karyotypes of their unborn babies early in their pregnancies to test for such disorders, the usual method of obtaining cells in amniocentesis.[87] However, some scientific groups are now suggesting newer and more accurate tests to be performed in genetic tests, such as microarray studies.[88]
How do some people end up with extra chromosomes? In humans, both sperm and eggs have one set of chromosomes, 23 in number. Eggs and sperm duplicate via meiosis, with 4 new gametes being produced in each cycle. If there is a mistake during meiosis, a chromosome pair might fail to properly separate and distribute into each forming cell, and a gamete might be left with two copies of a gene instead of one. When this gamete joins with its respective counterpart during fertilization, the number of chromosomes will be abnormal. If a sperm with two copies of a gene fertilizes and egg with one copy, the resulting embryo will have three copies, and suffer the problems listed above associated with trisomy. A sperm lacking a chromosome might also fertilize a normal egg, resulting in an embryo with only one copy of a gene that would also suffer genetic problems (this is called monosomy)[89]
References
[edit | edit source]- ↑ a b Douglas S, Zauner S, Fraunholz M; et al. (2001). "The highly reduced genome of an enslaved algal nucleus". Nature. 410 (6832): 1091–6. doi:10.1038/35074092. PMID 11323671.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ a b Armbrust EV, Berges JA, Bowler C; et al. (2004). "The genome of the diatom Thalassiosira pseudonana: ecology, evolution, and metabolism". Science (journal). 306 (5693): 79–86. doi:10.1126/science.1101156. PMID 15459382.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ Bowler C, Allen AE, Badger JH; et al. (2008). "The Phaeodactylum genome reveals the evolutionary history of diatom genomes". Nature. 456: 239–244. doi:10.1038/nature07410. PMID 18923393.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ Brayton KA, Lau AOT, Herndon DR; et al. (2007). "Genome Sequence of Babesia bovis and Comparative Analysis of Apicomplexan Hemoprotozoa". PLoS Pathogens. 3 (10): e148. doi:10.1371/journal.ppat.0030148.
{{cite journal}}: Explicit use of et al. in:|author=(help)CS1 maint: multiple names: authors list (link) - ↑ a b Xu P, Widmer G, Wang Y; et al. (2004). "The genome of Cryptosporidium hominis". Nature. 431 (7012): 1107–12. doi:10.1038/nature02977. PMID 15510150.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ a b Abrahamsen MS, Templeton TJ, Enomoto S; et al. (2004). "Complete genome sequence of the apicomplexan, Cryptosporidium parvum". Science (journal). 304 (5669): 441–5. doi:10.1126/science.1094786. PMID 15044751.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ a b Aury JM, Jaillon O, Duret L; et al. (2006). "Global trends of whole-genome duplications revealed by the ciliate Paramecium tetraurelia". Nature. 444 (7116): 171–8. doi:10.1038/nature05230. PMID 17086204.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ a b Gardner MJ, Hall N, Fung E; et al. (2002). "Genome sequence of the human malaria parasite Plasmodium falciparum". Nature. 419 (6906): 498–511. doi:10.1038/nature01097. PMID 12368864.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ a b A. Pain, U. Böhme, A. E. Berry, K. Mungall, R. D. Finn, A. P. Jackson, T. Mourier, J. Mistry, E. M. Pasini, M. A. Aslet, S. Balasubrammaniam, K. Borgwardt, K. Brooks, C. Carret, T. J. Carver, I. Cherevach, T. Chillingworth, T. G. Clark, M. R. Galinski, N. Hall, D. Harper, D. Harris, H. Hauser, A. Ivens, C. S. Janssen, T. Keane, N. Larke, S. Lapp, M. Marti, S. Moule, I. M. Meyer, D. Ormond, N. Peters, M. Sanders, S. Sanders, T. J. Sargeant, M. Simmonds, F. Smith, R. Squares, S. Thurston, A. R. Tivey, D. Walker, B. White, E. Zuiderwijk, C. Churcher, M. A. Quail, A. F. Cowman, C. M. R. Turner, M. A. Rajandream, C. H. M. Kocken, A. W. Thomas, C. I. Newbold, B. G. Barrell & M. Berriman (9 October 2008). "The genome of the simian and human malaria parasite Plasmodium knowlesi". Nature. 455: 799–803. doi:10.1038/nature07306.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - ↑ a b JM Carlton, JH Adams, JC Silva; et al. (9 October 2008). "Comparative genomics of the neglected human malaria parasite Plasmodium vivax". Nature. 455: 757–763. doi:10.1038/nature07327.
{{cite journal}}: Explicit use of et al. in:|author=(help)CS1 maint: multiple names: authors list (link) - ↑ a b Carlton JM, Angiuoli SV, Suh BB; et al. (2002). "Genome sequence and comparative analysis of the model rodent malaria parasite Plasmodium yoelii yoelii". Nature. 419 (6906): 512–9. doi:10.1038/nature01099. PMID 12368865.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ a b Eisen JA, Coyne RS, Wu M; et al. (2006). "Macronuclear genome sequence of the ciliate Tetrahymena thermophila, a model eukaryote". PLoS Biol. 4 (9): e286. doi:10.1371/journal.pbio.0040286. PMC 1557398. PMID 16933976.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ a b Gardner MJ, Bishop R, Shah T; et al. (2005). "Genome sequence of Theileria parva, a bovine pathogen that transforms lymphocytes". Science (journal). 309 (5731): 134–7. doi:10.1126/science.1110439. PMID 15994558.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ Pain A, Renauld H, Berriman M; et al. (2005). "Genome of the host-cell transforming parasite Theileria annulata compared with T. parva". Science (journal). 309 (5731): 131–3. doi:10.1126/science.1110418. PMID 15994557.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ a b Ivens AC, Peacock CS, Worthey EA; et al. (2005). "The genome of the kinetoplastid parasite, Leishmania major". Science (journal). 309 (5733): 436–42. doi:10.1126/science.1112680. PMC 1470643. PMID 16020728.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ a b HG Morrison, AG McArthur, FD Gillin; et al. (2007). "Genomic Minimalism in the Early Diverging Intestinal Parasite Giardia lamblia". Science (journal). 317 (5846): 1921–26.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ a b Carlton JM, Hirt RP, Silva JC; et al. (2007). "Draft genome sequence of the sexually transmitted pathogen Trichomonas vaginalis". Science (journal). 315 (5809): 207–12. doi:10.1126/science.1132894. PMC 2080659. PMID 17218520.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ a b Berriman M, Ghedin E, Hertz-Fowler C; et al. (2005). "The genome of the African trypanosome Trypanosoma brucei". Science (journal). 309 (5733): 416–22. doi:10.1126/science.1112642. PMID 16020726.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ a b El-Sayed NM, Myler PJ, Bartholomeu DC; et al. (2005). "The genome sequence of Trypanosoma cruzi, etiologic agent of Chagas disease". Science (journal). 309 (5733): 409–15. doi:10.1126/science.1112631. PMID 16020725.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ a b Eichinger L, Pachebat JA, Glöckner G; et al. (2005). "The genome of the social amoeba Dictyostelium discoideum". Nature. 435 (7038): 43–57. doi:10.1038/nature03481. PMC 1352341. PMID 15875012.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ a b Loftus B, Anderson I, Davies R; et al. (2005). "The genome of the protist parasite Entamoeba histolytica". Nature. 433 (7028): 865–8. doi:10.1038/nature03291. PMID 15729342.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ a b The Arabidopsis Genome Initiative, (2000). "Analysis of the genome sequence of the flowering plant Arabidopsis thaliana". Nature. 408 (6814): 796–815. doi:10.1038/35048692. PMID 11130711.
{{cite journal}}: Unknown parameter|month=ignored (help)CS1 maint: extra punctuation (link) - ↑ Arabidopsis Genome Initiative
- ↑ http://www.research-in-germany.de/coremedia/generator/dachportal/en/07__News_20and_20Events/VDITZ_20-_20News_26Events/Archiv/2009-10-25_2C_20Full_20oilseed_20rape_20genome_20deciphered,sourcePageId=34814.html
- ↑ a b Goff SA, Ricke D, Lan TH; et al. (2002). "A draft sequence of the rice genome (Oryza sativa L. ssp. japonica)". Science (journal). 296 (5565): 92–100. doi:10.1126/science.1068275. PMID 11935018.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ a b Yu J, Hu S, Wang J; et al. (2002). "A draft sequence of the rice genome (Oryza sativa L. ssp. indica)". Science (journal). 296 (5565): 79–92. doi:10.1126/science.1068037. PMID 11935017.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ Derelle E, Ferraz C, Rombauts S; et al. (2006). "Genome analysis of the smallest free-living eukaryote Ostreococcus tauri unveils many unique features". Proc. Natl. Acad. Sci. U.S.A. 103 (31): 11647–52. doi:10.1073/pnas.0604795103. PMC 1544224. PMID 16868079.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ a b Rensing SA, Lang D, Zimmer AD; et al. (2008). "The Physcomitrella genome reveals evolutionary insights into the conquest of land by plants". Science (journal). 319 (5859): 64–9. doi:10.1126/science.1150646. PMID 18079367.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ a b Tuskan GA, Difazio S, Jansson S; et al. (2006). "The genome of black cottonwood, Populus trichocarpa (Torr. & Gray)". Science (journal). 313 (5793): 1596–604. doi:10.1126/science.1128691. PMID 16973872.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ a b c Jaillon O, Aury JM, Noel B; et al. (2007). "The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla". Nature. 449 (7161): 463–7. doi:10.1038/nature06148. PMID 17721507.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ First Draft Of Corn Genome Completed
- ↑ a b Matsuzaki M, Misumi O, Shin-I T; et al. (2004). "Genome sequence of the ultrasmall unicellular red alga Cyanidioschyzon merolae 10D". Nature. 428 (6983): 653–7. doi:10.1038/nature02398. PMID 15071595.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ a b Walker, Tara (16 Nov 2005). "ALGAL TRANSGENICS IN THE GENOMIC ERA". Journal of Phycology. 41 (6): 1077–1093.
{{cite journal}}: Unknown parameter|coauthors=ignored (|author=suggested) (help) - ↑ a b Merchant; et al. (2007). "The Chlamydomonas genome reveals the evolution of key animal and plant functions". Science. 318: 245–250. doi:10.1126/science.1143609. PMID 17932292.
{{cite journal}}: Explicit use of et al. in:|author=(help) - ↑ a b Dietrich FS, Voegeli S, Brachat S; et al. (2004). "The Ashbya gossypii genome as a tool for mapping the ancient Saccharomyces cerevisiae genome". Science (journal). 304 (5668): 304–7. doi:10.1126/science.1095781. PMID 15001715.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ a b Nierman WC, Pain A, Anderson MJ; et al. (2005). "Genomic sequence of the pathogenic and allergenic filamentous fungus Aspergillus fumigatus". Nature. 438 (7071): 1151–6. doi:10.1038/nature04332. PMID 16372009.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ a b c d Galagan JE, Calvo SE, Cuomo C; et al. (2005). "Sequencing of Aspergillus nidulans and comparative analysis with A. fumigatus and A. oryzae". Nature. 438 (7071): 1105–15. doi:10.1038/nature04341. PMID 16372000.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) Invalid<ref>tag; name "Galagan" defined multiple times with different content - ↑ a b Pel HJ, de Winde JH, Archer DB; et al. (2007). "Genome sequencing and analysis of the versatile cell factory Aspergillus niger CBS 513.88". Nat. Biotechnol. 25 (2): 221–31. doi:10.1038/nbt1282. PMID 17259976.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ a b Machida M, Asai K, Sano M; et al. (2005). "Genome sequencing and analysis of Aspergillus oryzae". Nature. 438 (7071): 1157–61. doi:10.1038/nature04300. PMID 16372010.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ a b c d e f g h Dujon B, Sherman D, Fischer G; et al. (2004). "Genome evolution in yeasts". Nature. 430 (6995): 35–44. doi:10.1038/nature02579. PMID 15229592.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ About Génolevures
- ↑ a b Loftus BJ, Fung E, Roncaglia P; et al. (2005). "The genome of the basidiomycetous yeast and human pathogen Cryptococcus neoformans". Science (journal). 307 (5713): 1321–4. doi:10.1126/science.1103773. PMID 15653466.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ a b Katinka MD, Duprat S, Cornillot E; et al. (2001). "Genome sequence and gene compaction of the eukaryote parasite Encephalitozoon cuniculi". Nature. 414 (6862): 450–3. doi:10.1038/35106579. PMID 11719806.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ a b Dean RA, Talbot NJ, Ebbole DJ; et al. (2005). "The genome sequence of the rice blast fungus Magnaporthe grisea". Nature. 434 (7036): 980–6. doi:10.1038/nature03449. PMID 15846337.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ a b Goffeau A, Barrell BG, Bussey H; et al. (1996). "Life with 6000 genes". Science (journal). 274 (5287): 546, 563–7. PMID 8849441.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ International Collaboration for the Yeast Genome Sequencing
- ↑ a b Wood V, Gwilliam R, Rajandream MA; et al. (2002). "The genome sequence of Schizosaccharomyces pombe". Nature. 415 (6874): 871–80. doi:10.1038/nature724. PMID 11859360.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ The Bovine Genome Sequencing and Analysis Consortium (2009-04-24). "The genome sequence of taurine cattle: a window to ruminant biology and evolution". Science. 324 (5926): 522–528. doi:10.1126/science.1169588. Retrieved 2009-04-24.
- ↑ a b c d e f g h i j k l m n o p q r s PubMed Home
- ↑ http://news.bbc.co.uk/1/hi/sci/tech/8014598.stm Cow genome 'to transform farming'
- ↑ a b c Lindblad-Toh K, Wade CM, Mikkelsen TS; et al. (2005). "Genome sequence, comparative analysis and haplotype structure of the domestic dog". Nature. 438 (7069): 803–19. doi:10.1038/nature04338. PMID 16341006.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ a b c d e f g h i Mammalian Genome Project - Broad
- ↑ Horse Genome Assembled, February 7, 2007 News Release - National Institutes of Health (NIH)
- ↑ Pontius JU, Mullikin JC, Smith DR; et al. (2007). "Initial sequence and comparative analysis of the cat genome". Genome Res. 17 (11): 1675–89. doi:10.1101/gr.6380007. PMC 2045150. PMID 17975172.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ a b Human Genome Sequencing Consortium, International (2004). "Finishing the euchromatic sequence of the human genome". Nature. 431 (7011): 931–45. doi:10.1038/nature03001. PMID 15496913.
{{cite journal}}: Unknown parameter|month=ignored (help) - ↑ McPherson JD, Marra M, Hillier L; et al. (2001). "A physical map of the human genome". Nature. 409 (6822): 934–41. doi:10.1038/35057157. PMID 11237014.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ Venter JC, Adams MD, Myers EW; et al. (2001). "The sequence of the human genome". Science (journal). 291 (5507): 1304–51. doi:10.1126/science.1058040. PMID 11181995.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ Gregory SG, Barlow KF, McLay KE; et al. (2006). "The DNA sequence and biological annotation of human chromosome 1". Nature. 441 (7091): 315–21. doi:10.1038/nature04727. PMID 16710414.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ Mikkelsen TS, Wakefield MJ, Aken B; et al. (2007). "Genome of the marsupial Monodelphis domestica reveals innovation in non-coding sequences". Nature. 447 (7141): 167–77. doi:10.1038/nature05805. PMID 17495919.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ a b Waterston RH, Lindblad-Toh K, Birney E; et al. (2002). "Initial sequencing and comparative analysis of the mouse genome". Nature. 420 (6915): 520–62. doi:10.1038/nature01262. PMID 12466850.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ International Collaboration for the Mouse Genome Sequencing
- ↑ Whole Genome Shotgun sequencing project list
- ↑ Chimpanzee Sequencing and Analysis Consortium. (2005). "Initial sequence of the chimpanzee genome and comparison with the human genome". Nature. 437 (7055): 69–87. doi:10.1038/nature04072. PMID 16136131.
{{cite journal}}: Unknown parameter|month=ignored (help) - ↑ a b Gibbs RA, Weinstock GM, Metzker ML; et al. (2004). "Genome sequence of the Brown Norway rat yields insights into mammalian evolution". Nature. 428 (6982): 493–521. doi:10.1038/nature02426. PMID 15057822.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ a b Holt RA, Subramanian GM, Halpern A; et al. (2002). "The genome sequence of the malaria mosquito Anopheles gambiae". Science (journal). 298 (5591): 129–49. doi:10.1126/science.1076181. PMID 12364791.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link)H - ↑ a b Honeybee Genome Sequencing Consortium. (2006). "Insights into social insects from the genome of the honeybee Apis mellifera". Nature. 443 (7114): 931–49. doi:10.1038/nature05260. PMC 2048586. PMID 17073008.
{{cite journal}}: Unknown parameter|month=ignored (help) - ↑ Mita K, Kasahara M, Sasaki S; et al. (2004). "The genome sequence of silkworm, Bombyx mori". DNA Res. 11 (1): 27–35. doi:10.1093/dnares/11.1.27. PMID 15141943.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ a b Adams MD, Celniker SE, Holt RA; et al. (2000). "The genome sequence of Drosophila melanogaster". Science (journal). 287 (5461): 2185–95. PMID 10731132.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ a b Stein LD, Bao Z, Blasiar D; et al. (2003). "The genome sequence of Caenorhabditis briggsae: a platform for comparative genomics". PLoS Biol. 1 (2): E45. doi:10.1371/journal.pbio.0000045. PMC 261899. PMID 14624247.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ a b C. elegans Sequencing Consortium. (1998). "Genome sequence of the nematode C. elegans: a platform for investigating biology". Science (journal). 282 (5396): 2012–8. PMID 9851916.
{{cite journal}}: Unknown parameter|month=ignored (help) - ↑ a b Charles Opperman, David McK. Bird, Mark Burke, Jonathan Cohn, John Cromer, Steve Diener, Jim Gajan, Steve Graham, T. D. Houfek, Jennifer Schaff, Reenah Schaffer, Elizabeth Scholl, Eric Windham, Bryon R. Sosinski, Valerie M. Williamson, Qingli Liu, Varghese P. Thomas, Dan S. Rokhsar, Therese Mitros (2008). "Sequence and Genetic Map of Meloidogyne hapla: A Compact Nematode Genome for Plant Parasitism". Proceedings of the National Academy of Sciences. 105: 14802. doi:10.1073/pnas.0805946105. PMID 18809916.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - ↑ a b Pierre Abad, Jerome Gouzy, Jean-Marc Aury, Philippe Castagnone-Sereno, Etienne G.J. Danchin, Emeline Deleury, Laetitia Perfus-Barbeoch et al. vol. 26, pp. 909-915. (2008). "Genome sequence of the metazoan plant-parasitic nematode : Meloidogyne incognita". Nature Biotechnology. 26: 909–915. doi:10.1038/nbt.1482. PMID 18660804.
{{cite journal}}: Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ International M.incognita Genome Consortium
- ↑ a b Christoph Dieterich, Sandra W Clifton, Lisa N Schuster, Asif Chinwalla, Kimberly Delehaunty, Iris Dinkelacker, Lucinda Fulton, Robert Fulton, Jennifer Godfrey, Pat Minx, Makedonka Mitreva, Waltraud Roeseler, Huiyu Tian, Hanh Witte, Shiaw-Pyng Yang, Richard K Wilson & Ralf J Sommer (2008). "The Pristionchus pacificus genome provides a unique perspective on nematode lifestyle and parasitism". Nature Genetics. 40: 1193. doi:10.1038/ng.227. PMID 17095691.
{{cite journal}}: Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ a b Dehal P, Satou Y, Campbell RK; et al. (2002). "The draft genome of Ciona intestinalis: insights into chordate and vertebrate origins". Science (journal). 298 (5601): 2157–67. doi:10.1126/science.1080049. PMID 12481130.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ Small KS, Brudno M, Hill MM, Sidow A (2007). "A haplome alignment and reference sequence of the highly polymorphic Ciona savignyi genome". Genome Biol. 8 (3): R41. doi:10.1186/gb-2007-8-3-r41. PMC 1868934. PMID 17374142.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - ↑ a b International Chicken Genome Sequencing Consortium. (2004). "Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution". Nature. 432 (7018): 695–716. doi:10.1038/nature03154. PMID 15592404.
{{cite journal}}: Unknown parameter|month=ignored (help) - ↑ a b Sodergren E, Weinstock GM, Davidson EH; et al. (2006). "The genome of the sea urchin Strongylocentrotus purpuratus". Science (journal). 314 (5801): 941–52. doi:10.1126/science.1133609. PMID 17095691.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ International Fugu Genome Consortium. Forth Genome Assembly
- ↑ International Fugu Genome Consortium
- ↑ Aparicio S, Chapman J, Stupka E; et al. (2002). "Whole-genome shotgun assembly and analysis of the genome of Fugu rubripes". Science (journal). 297 (5585): 1301–10. doi:10.1126/science.1072104. PMID 12142439.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ a b c Jaillon O, Aury JM, Brunet F; et al. (2004). "Genome duplication in the teleost fish Tetraodon nigroviridis reveals the early vertebrate proto-karyotype". Nature. 431 (7011): 946–57. doi:10.1038/nature03025. PMID 15496914.
{{cite journal}}: Explicit use of et al. in:|author=(help); Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ↑ "Karyotyping: MedlinePlus Medical Encyclopedia." National Library of Medicine - National Institutes of Health. Web. 10 Nov. 2011. <http://www.nlm.nih.gov/medlineplus/ency/article/003935.htm>
- ↑ "Karyotyping." HudsonAlpha Institute for Biotechnology. Web. 10 Nov. 2011. <http://www.hudsonalpha.org/education/kits/disorder-detectives/karyotyping>.
- ↑ "Genome.gov | Chromosome Abnormalities Fact Sheet." Genome.gov | National Human Genome Research Institute (NHGRI) - Homepage. Web. 10 Nov. 2011. <http://www.genome.gov/11508982>.
- ↑ "KARYOTYPE ALTERNATIVES." The Woodrow Wilson National Fellowship Foundation. Web. 28 Nov. 2011. <http://www.woodrow.org/teachers/bi/1993/karyoteype.html>
- ↑ "Amniocentesis Test: Risks, Benefits, Accuracy, and More." WebMD - Better Information. Better Health. Web. 10 Nov. 2011. <http://www.webmd.com/baby/guide/amniocentesis>
- ↑ Petrone, Justin. "ACMG Recommends Replacing Karyotyping with Chromosomal Microarrays as 'First-Line' Postnatal Test | BioArray News | Arrays."GenomeWeb. Web. 10 Nov. 2011. <http://www.genomeweb.com/arrays/acmg-recommends-replacing-karyotyping-chromosomal-microarrays-first-line-postnat>
- ↑ "Using Karyotypes to Predict Genetic Disorders." Learn.Genetics™. Web. 28 Nov. 2011. <http://learn.genetics.utah.edu/content/begin/traits/predictdisorder/>.
Overview
[edit | edit source]In many areas of biochemical research, retrieving enough quantities of a substance under study is a challenge. For instance, a maximum of 7 mg of DNA polymerase I can be obtained from a 10 L culture of E. coli grown with the highest dilution of ~10^10 cells * mL^-1, and still, other proteins present yield lower amounts. DNA recombinant techniques, also known as genetic engineering and molecular cloning, allow scientists to manipulate, duplicate and apply DNA for research. Deletions, insertions and substitutions are the most useful changes implemented for the synthesis of new genes. Such techniques usually include the cloning or amplification of sequences for further study. Restriction enzymes and DNA ligase play a vital role in the production of recombinant DNA. Polymerase chain reaction (PCR) is often used on interested sequences to be cut by nucleases and then cloned by phages, BACs or YACs. Using a DNA or RNA probe, specific genes can be cloned from a genomic library. The fundamental idea of DNA recombinant techniques is to input a DNA segment into a replicating DNA molecule, also known as a cloning vector or vehicle, so a DNA segment is replicated with the vector.The usage of various recombinant techniques can lead scientists to new understandings of how DNA affects us and other organisms. This section describes the various techniques involved in genetic engineering, and its application to many experiments carried out in biochemistry.
Cloning Vectors
[edit | edit source]A vector is a DNA molecule that can be used to insert a DNA sequence into a cell. Vectors are used for replication. Usual vectors in laboratories, are plasmids , viruses (Lambda Phage),and artificial chromosomes. Vectors must be capable of being replicated by the cell.
Plasmid-Based Cloning Vectors
[edit | edit source]Plasmids are doubled stranded, and commonly circular. They are predisposed with genetic material, like the replication origin (where DNA replication starts) to independently replicate inside of a bacterial host or yeast. Although plasmids may act as parasites, they are beneficial in ways like antibiotic resistance.
On one hand, some plasmids are present in quantities of one or two in a cell and replicate once per cell division and are known to be under stringent control. On the other hand, most plasmids utilized in genetic engineering are under relaxed control where as little as 10 and as many as 700 copies of plasmids can be present in a cell. In addition, when protein synthesis comes to a halt due to inhibition by an antibiotic, cell division also stops and these plasmids can replicate upwards of 3,000 copies in a cell. The types of plasmids synthesized from genetic engineering are under relaxed control, and contain genes that are resistant to antibiotics, while carrying endonuclease sites. These endonuclease sites facilitate the insertion of the DNA desired to be copied. A multiple cloning site, or a polylinker is a small section of a DNA that has an array of restriction sites not found anywhere else in the plasmid. pUC18 ("plasmid Universal Cloning") is a commonly used vector from E. coli. The following link provides more information and a schematic of pUC18.
In 1973, Herbert Boyer and Stanley Cohen demonstrated the first genetically mixed plasmid in DNA cloning. When a host bacterium is mixed with a plasmid, the conditions for optimum conditions is when divalent cations like calcium and heating to ~42 degrees Celsius is applied. This condition allows cell membranes to become more permeable to DNA and are called transformation competent. Once a plasmid vector is absorbed, it is permanently established in the bacterial host with only ~0.1% efficiency. Plasmid vectors are incapable of being used to duplicate DNA more than a certain size, because the time it takes for plasmid replication is directly proportional to the plasmid size.
Virus-Based Cloning Vectors
[edit | edit source]Another method to clone DNA's that are larger in size is using a cloning vehicle named bacteriophage lambda. The central third of the virus's genome space can be inserted by DNA's of larger sizes because that area is not necessary during phage infection. In vitro methods can help the insertion of the chimeric phage DNA by infecting the host cells through phages. Utilization of phages as cloning vectors has advantages in that the chimeric DNA can be made in large amounts and easily purified. In addition, scientists can make use of lambda phages for longer DNA inserts. The only requirement that is needed to allow the viral tool to insert DNA into the heads of phage is the 16-bp sequence known as the cos site at each end. These ends need to be at least 36-51 kb apart, and a combination of two cos sites in vitro creates a cosmid vector. Cosmids do not have phage genes and therefore can make plasmids. The following link is a cartoon picture of a bacteriophage lambda and its important features labeled.
M13- filamentous bacteriophage is a another cloning vector that is single-stranded and circular DNA in a protein tube. The amount of identical helical protein subunits are dependent on the length of phage DNA being coated, but is is normally ~2700 subunits. A longer unit of phage molecule results when an insertion of foreign DNA in a nonessential region occurs. The phages directly make single-stranded DNA that a certain technique requires, even though M13 cannot maintain DNA inserts larger than 1kb. This [[80]] demonstrates a visualization of what an M13 looks like.
Another method of virus based cloning is baculoviruses, which are an array of large and pathogenic viruses that infect insects but not vertebrates, resulting in an easy way to culture them in lab. Like other viruses, a segment of the double stranded DNA that forms the genome of the viruses is not important for viral replication and can be replaced by foreign DNA upward to 15kb. This link is an electron micrograph of the baculovirus.
YAC and BAC Vectors
[edit | edit source]In order to accommodate DNA segments larger than those carried by cosmids, yeast artificial chromosomes (YACs) and in bacterial artificial chromosomes (BACs). YACs contain all molecular necessities of required for replication for yeast such as the replication origin, autonomously replicating sequence (ARS), a centromere, and [[81]]. BACs replicate in E. Coli, and are found from plasmids that normally replicate long segments of DNA. BAC vectors have the bare minimum sequences necessary for self replication, copy number control, and regulated plasmid separation during cell division. The following link is a YAC diagram and [diagram].
Gene Manipulation
[edit | edit source]Through restriction endonucleases, a sequence-defined fragment can be retrieved when cloning a DNA. Oftentimes, reestriction[check spelling] endonucleases split a double stranded region of DNA at specific sites to result in single-stranded ends that complement one another. In 1972, Janet Mertz and Ron Davis shown that a restriction fragment can be inputted into a split made in a cloning vector by the same restriction enzyme. Complementary endings of two DNAs are covalently spliced with the help of DNA ligase. DNA insert can be extracted from a cloned vector by splitting it with the same restriction enzyme by making a chimeric vector.
Terminal deoxynucleotidyl transferase (TdT), was a technique produced by Dale Kaiser and Paul Berg who determined that if foreign DNA and cloning vector do not have a common restriction site, they can be spliced and this procedure can be carried out. The enzyme adds nucleotides to the 3'-terminal OH group of a DNA chain, and it is the only known DNA polymerase where a template is not required. In addition, the cloning vector is cleaved through an enzyme at a specific site, and the 3' ends are extended with poly(dA) tails. Finally, the homopolymer tails are heated and then cooled down, and any gaps due to differences with the other strand is filled in by DNA polymerase I and DNA ligase joins the two together.
Compared to other techniques gene manipulation takes out the restriction sites that were used to generate the foreign DNA insertion. Therefore, it can be hard to retrieve the insert from the cloned vector. However, this problem can be prevented by which a chemically synthesized linker matches the restriction site of the cloning vector is joined with the two ends of the foreign DNA. The adhesion is through the use of T4 DNA ligase and then split with the restriction enzyme. The schematic of splicing DNA with terminal transferase can be seen.
Cells Transformed Need to be Selected
[edit | edit source]It is difficult in choosing the host that have been transformed by the vector due to the low efficiency in transformation and constriction of chimeric vectors. As a result, in plasmid transformation, a double screen using antibiotics or color producing substrates is used. An example would be pUC18 plasmid that contains lacZ' since lacZ' gene codes for the enzyme beta-galactosidase. That gene initiates the hydrolysis of the bond from O1 of sugar beta-D-galactose to a substituent. Therefore, when grown in 5-bromo-4chloro-3-indolyl-beta-D-galactoside short handed for X-gal and hydrolysis, the colorless substance turns blue. Two different scenarios can occur: When E. coli is transformed by an unmodified pUC18 plasmid form blue colonies, then when E. coli is transformed by a pUC18 plasmid containing a foreign DNA insert in a polylinker, it is colorless due to the insert conflicting with the encoding sequence of lacZ' gene. Addition of an ampicillin allows the exclusion of bacteria that would normally become colorless when X-gal is added. This is due to the fact that bacteria that will have color have resistance intact from amp^R gene. These amp^R genes are known as selectable markers. Some of the genetically engineered lambda phages have restriction sites that border the available central third of a phage genome, and that segment can be replaced through a DNA insert. Lambda phages that are unable to obtain a foreign DNA are too short and therefore are unable to become an infectious phage. Cosmids in particular can support conception of larger DNA inserts due to their loss of DNA during random deletion and are not recovered.
Plasmid Vectors
[edit | edit source]Plasmids are naturally occurring circular pieces of DNA that are found in bacteria. Plasmids can be used as vectors to incorporate and replicate a DNA insert of interest by joining that DNA insert into the plasmid DNA. The vector is prepared to accept a DNA insert by treatment with a restriction enzyme (like EcoRI), which cleaves it at specific sites and leaves complementary single stranded "sticky ends." The DNA fragment to be inserted is treated with the same restriction enzyme so that it has complementary ends to those of the vector. DNA ligase is then used to join the DNA insert into the vector plasmid, resulting in recombination. The recombinant plasmid DNA can be cloned and amplified as the bacteria host colony grows.

Enzymes used in Recombinant DNA Technology
[edit | edit source]1. Type II restriction endonucleases always recognize palindromic sequences and the are able to cleave the DNA strand. In other words, they break the phosphodiester bonds of specific base sequences in the DNA.
2. DNA ligase is the opposite of Type II restriction endonuclease in that it joins two DNA molecules or fragments together.
3. DNA Polymerase is an enzyme that is able to use a template DNA and synthesize a complimentary strand by adding nucleotides to the 3' ends.
4. Reverse transcriptase is an enzyme that creates a DNA from an RNA molecule. Reverse transcriptase does the opposite of RNA polymerase, which takes DNA and makes RNA.
5. Polynucleotide kinase adds a phosphate to the 5' -OH end of a polynucleotide. it is useful in radioactively label DNA or permit ligation.[1]
6. Voet, Donald, Judith G. Voet. Biochemistry 3rd ed. New Jersey: John Wiley & Sons, Inc, 2004. Print.
References
[edit | edit source]- ↑ Viadiu, Hector. "Nucleic Acids." University of California, San Diego. November 2012.
Overview
[edit | edit source]Its linear genome was observed to form a circle via complementary base pairing when entering a host cell. The genome was able to act in such a peculiar manner due to the presence of single stranded areas called cohesive sites (cos). These cos sites are able to base pair, and the nature of these sites are the same as those produced by restriction endonucleases. (Ex: EcoR1 produces "sticky ends"). This circular phage DNA is able to recombine with the host cell DNA via attachment sites. Progeny of the phage with the addition of some bacterial gene take place by the lysogenic pathway. What makes lambda phage extremely advantageous is the fact that it can destroy its host (lytic pathway) or it can become part of its host (lysogenic pathway). Studies on the control of these alternative cycles have been very important for our understanding of the regulation of gene transcription.
Lambda phage is a virus particle consisting of a head made up of double-stranded linear DNA as its genetic material, and a tail. It infects the host cell by injecting its own DNA through the tail at which point the phage will enter the lytic or lysogenic pathway. Large segments of the 48 kilobase pair DNA of the lambda phage are not essential for productive infection and can be replaced by foreign DNA, thus making lambda phage an ideal vector.

History
[edit | edit source]Lambda phage was first isolated by Esther Lederberg in 1950 from Escherichia coli. It has been an intensely studied organism, and has been a useful tool in molecular biology. Lambda phage can be used for cloning of recombinant DNA, the use of its site specific recombinase (int) for the shuffling of cloned DNAs by the 'Gateway' method, and in recombineering. The discovery and study of lambda phage led to an understanding of transduction which provides a major mechanism that can explain modes of evolution.
Bacteriophage Lambda was studied by Allan Campbell in 1962 who studied the phage's integration process. He observed that lambda phage had a unique characteristic what some phages would infect and reproduce in some strains of E. coli while other strains seemed immune. Probing further into why this occurred lead to the discovery that the immune strains contained a dormant copy of the phage, but the dormant copy could be activated. Study the phage provided priceless contributions into virus life cycles, the regulation and expression of genetic material, and the mechanism of integration and excision of genetic material.
Mutant Lambda Phages
[edit | edit source]Mutant phages have been genetically designed to make cloning easier. And especially useful one is called lambda(gt)-(lambda)(beta) which contains only two EcoRI cleavage sites instead of the normal five. After cleavage, the middle segment of this phage DNA can be removed leaving 72% of the normal genome length which is too little to be packaged into a lambda particle. Thus, a suitably long DNA insert between the lambda segments of DNA enables a recombinant DNA molecules to be packaged. These modified viruses enter bacteria much more easily than plasmids.
Genomic Library
[edit | edit source]A genomic library is a large collection of DNA fragments. Such libraries assist in identifying DNA fragments that have a gene of interest. Creating a genomic library utilizes lambda phage. First, total genomic DNA is mechanically sheared to produce a random population of overlapping DNA fragments which are separated by gel electrophoresis. All fragments that are about 15 kilobase pairs long are isolated and attached with synthetic linkers to insert into a lambda phage which replicated themselves and then lyse their bacterial hosts. The resulting lysate contains fragments of DNA houssed[check spelling] in virus particles which constitute a genomic library.
Restriction & Modification Systems
[edit | edit source]Restriction: Phages grown in one bacterial cell fail to grow in other bacterial strains. The degradation of phage lambda DNA was observed in certain bacterial strains. This was later found to be a result of restriction endonucleases which would cut the phage DNA after detecting it as being foreign. Thus, restriction endonucleases was termed because of its ability to restrict foreign DNA.
Modification: Phages are modified such that they can be grown normally in other bacterial strains. An example of modification is the methylation of lambda phage DNA so that it would not be cut by restriction endonuclease.
References
[edit | edit source]Allison, et al. Fundamental Molecular Biology, 2007. A plasmid is an accessory chromosomal DNA that is naturally present in bacteria. Some bacteria cells can have no plasmids or several copies of one. They can replicate independently of the host chromosome. Plasmids are circular and double stranded. They carry few genes and their size ranges from 1 to over 200 kilobase pairs. Some functions of their genes include: providing resistance to antibiotics, producing toxins and the breakdown of natural products. However, plasmids are not limited to bacteria; they are also present in some eukaryotes (e.g., circular, nuclear plasmids in Dictyostelium purpureum).
A plasmid is a circular, double stranded DNA that is usually found in bacteria (however it does occur in both eukarya and prokarya). It replicates on its own (without the help of chromosomal DNA)and are used frequently in recombinant DNA research in order to replicate genes of interest. Some plasmids can be implanted into a bacterial or animal chromosome in which it becomes a part of the cell's genome and then reveals the gene of interest as a phenotype. This is how much research is done today for gene identification.
Plasmids contain three components: an origin of replication, a polylinker to clone the gene of interest (called multiple cloning site where the restriction enzymes cleave), and an antibiotic resistance gene (selectable marker).

Plasmids are usually isolated before they are used in recombinant techniques. Alkaline lysis is the method of choice for isolating circular plasmid DNA. This process is quick and reliable. You first obtain the cell that has the plasmid of interest and lyse it with alkali. This step is then followed by extracting the plasmid. The cell fragments are precipitated by using SDS and potassium acetate. This is spun down, and the pellet (cell/cell fragments) is removed. Next, the plasmid DNA is precipitated from the supernatant with the use of isopropanol. The plasmid is then suspended in buffer. Akaline lysis can give you different amounts of plasmid depending if it's a mini-, midi-, or maxi- prep.
Plasmids can be related to viruses because they can be independent life-forms due to their ability to self-replicate inside their host. Though they may be viewed as independent life-forms, they have a sense of dependency on their host. A plasmid and its host tend to have a symbiotic relationship. Plasmids can give their hosts needed packages of DNA carrying genes that can lead to mutual survival during tough times. Providing its host with such genetic information, plasmid allows the host to survive and at the same time allows itself to continue living in the host for generations.
Plasmids are used as vectors to clone DNA in bacteria. One example of a plasmid used for DNA cloning is called pBR322 Plasmid. The pBR322 plasmid contains a gene that allow the bacteria to be resistant to the antibiotics tetracycline and ampicillin. To use pBR322 plasmid to clone a gene, a restriction endonuclease first cleaves the plasmid at a restriction site. pBR322 plasmid contains three restriction sites: PstI, SalI and ecoRI. The first two restriction sites are located within the gene that codes for ampicillin and tetracycline resistance, respectively. Cleaving at either restriction site will inactivate their respective genes and antibiotic resistance. The target DNA is cleaved with a restriction endonuclease at the same restriction site. The target DNA is then annealed to the plasmid using DNA ligase. After the target DNA is incorporated into the plasmid, the host cell is grown in a environment containing ampicillin or tetracycline, depending on which gene was left active. Many copies of the target DNA is created once the host is able to replicate.
Another plasmid used as a vector to clone DNA is called pUC18 plasmid. This plasmid contains a gene that makes the host cell ampicillin resistant. It also contains a gene that allows it produce beta-galactosidase, which is an enzyme degrades certain sugars. The enzyme produces a blue pigment when exposed to a specific substrate analog. This allows the host to be readily identified. The gene for beta-galactosidase contains a polylinker region that contains several restriction sites. The pUC18 plasmid can be cleaved by several different restriction endonucleases which provide more versatility. When the polylinker sequence is cleaved and the target DNA is introduced and ligased, this inactivates the gene that codes for beta-galactosidase and the enzyme will not be produced. The host cell will not produce a blue pigment when exposed to the substrate analog. This allows the recombinant cells to be readily identified and isolated.
Cloning
[edit | edit source]Cloning is a method of recombining genes in order to take advantage of a bacteria's native ability to recreate plasmids. Engineered plasmids can be used to clone genetic material of up to 10,000 base pairs, the amount of genetic material is limited by the size of the plasmid. Because of the repetition of expressive genes within bacterial plasmids, it is possible to remove repeated genetic materials of the plasmid and replace it with desired traits. Most pre-engineered plasmids procured for laboratory use already contain an antibiotic resistance gene, polylinker site, and an origin of replication. The polylinker site is engineered to allow multiple unique cleaving sites that will allow needed DNA fragmentation. The origin of replication will mimic the genetic material of the bacteria that will be used for cloning.
Once the plasmid is acquired, the polylinker will be cleaved at two sites using specific endonucleases. Afterwards, the wanted DNA will also be cleaved from a different source with a different endonuclease. The cleaved DNA is sometimes amplified with a polymerase chain reaction. The desired DNA trait will be inserted into the now empty polylinker site. This replacement of the polylinker site with desired genetic traits is termed a cassette mutagenesis. The newly created plasmid will be mixed with bacteria, which will then be heat shocked or electric shocked to aide in the ability for the plasmid to act as a vector. After allowing the bacteria to reproduce, the antibiotic for which the engineered plasmid conferred resistance will be delivered. All still living bacteria will have acquired the desired traits of both the inserted DNA and the antibiotic immunity. The new proteins or biochemical structures from the inserted DNA can be gathered through different means.

Gene Mutations Using Plasmids
[edit | edit source]Deletions occur when one or more base pairs are removed from the DNA sequence. A large portion of DNA can be removed from the plasmid by using different restriction endonucleases to cut out a certain segment followed by ligation using DNA ligase to reform a new, smaller plasmid. A single or few base pairs can be removed by using multiple restriction endounucleases that cut near the sticky ends, followed by ligation.
Substitutions are a result of the change of a single amino acid in a protein sequence. This is typically accomplished by changing one (a point mutation) or more base pairs on the genetic code sequence in order to alter the amino acid at a particular site and is known as oligonucleotide-directed mutagenesis. An oligionucleotide is designed such that there is a one base pair difference at a particular site and this one base pair different will encode for a new residue. This oligionucleotide is annealed to the plasmid, which acts as the DNA template, and replication using DNA polymerase results in strands that contain this mutation. One stand of the replicated double helical DNA will be the parent chain and contain the original (wildtype) base sequence while the other chain will contain the new (mutant) strand of DNA that encodes for the new desired protein. By expressing the mutant chain, the desired protein can be harvested.
Insertions occur when an entire segment of DNA is introduced to a plasmid. The segment of DNA is known as a cassette and the technique is termed cassette mutagenesis. Plasmids are cut with restriction enzymes, removing a portion of DNA. Then specifically synthesized or harvested DNA is ligated into that region and the plasmid is expressed and studied.
It is also possible to create entirely new proteins and genes by joining together genes that are otherwise unrelated.
Types of Plasmids
[edit | edit source]Modes of Classification
Plamids are not required by their host cell for survival. They carry genes that provide a selective advantage to their host, such as resistance to naturally made antibiotics carried by other organisms. Antibiotic resistant genes produced by a plasmid will allow the host bacteria to grow in the presence of competing bacteria that produce these antibiotics. One way to classify plasmids is based on their ability to transfer to additional bacteria. Conjugative plasmids retain tra-genes, which carryout the intricate process of conjugation, the transfer of a plasmid to another bacterium. Conversely, non-conjugative plasmids are incapable of commencing conjugation, which consequently can only be transferred via conjugative plasmids. A transitional class of plasmids are considered to be mobilizable, contain only a subset of the genes necessry for a successful transfer. They have the ability to parasitize a conjugative plasmid by transferring at a high frequency exclusively in the presence of the plasmid. Currently, plasmids are used to manipulate DNA and could potentially be used as devices for curing disease.
Figure 1-1: Illustrates the process of bacterial conjugation.
It is possible for various plasmids to coexist in a single cell. A maximum of seven different plasmids have been found to coexist in a single E. coli. It is also possible to find incompatible related plasmids, where only one of the plasmids survive in the cell environment, due to the regulation of important plasmid functions. Hence, plasmids can be designated into groups according to compatibility.
Classification of Plasmids by Function Another approach to classify plasmids is according to their function.
There is a total of five major sub-groups:
Fertility Plasmids (F-Plasmids)- carry the fertility genes (tra-genes) for conjugation, the transfer of genetic information between two cells. F plasmids are also known as episomes because, they integrate into the host chromosome and promote the transfer of chromosomal DNA bacterial cells.
Figure 1-2: Illustrates a fertility plasmid.
Resistance Plasmids (R-Plasmids)- contain genes that encode resistance to antibiotics or poisons. Examples of R-plasmids found in Chapter 5 of "Biochemistry" by Berg include the following:
pBR322 Plasmid pBR322 was one of the first plasmids used for the purpose of cloning. It contains genes for the resistance to tetracycline and ampicillin. Insertion of the DNA at specific restriction sites can inactivate the gene for tetracycline (an effect known as an insertional inactivation) or ampicillin resistance.
 Figure 1-3: Illustrates the pBR322 R-plasmid
Figure 1-3: Illustrates the pBR322 R-plasmid
pUC18 Plasmid pUC18/pUC19 has a greater versatility compared to pBR322. Comparable to pBR322, the pUC18 plasmid has an origin of replication and a selectable marker based on ampicillin resistance. Furthermore, this plasmid also contains a gene for beta-galactosidase, an enzyme that degrades certain sugars. while in the presence of a specific substrate analog, this enzyme produces a blue pigment that can be easily detected. Also, this enzyme has been equipped so that it has a polylinker region where many different restriction enzymes or combinations of enzymes can be used to cleave at different locations. Creating a greater variety in the DNA fragments that can be cloned. Interestingly, the insertion of a DNA fragment inactivates the beta-galactosidase. Thus if the blue pigment is not generated, it would be an indication that the DNA fragment was not inserted properly. pUC18 is similar to pUC19, but the MCS region is reversed.
 Figure 1-4: Illustrates the pUC19 R-plasmid
Figure 1-4: Illustrates the pUC19 R-plasmid
Tumor Inducing Plasmids (Ti-Plasmids "Virulence Plasmids")- contain A. tumefaciens, which carry instructions for bacteria to become a pathogen by switching to the tumor state and synthesize opines, toxins and other virulence factors. The plasmid effectively transfers foreign genes into certain plant cells. Ti-Plasmids can can also be found in Chapter 5 of "Biochemistry" by Berg.
 Figure 1-5: Illustrates the Ti-Plasmid
Figure 1-5: Illustrates the Ti-Plasmid
Degradative Plasmids- (Catabolic Plasmid) a type of plasmid that allows the host bacterium to metabolize normally ddifficult or unusual organic compounds such as pesticides.
Col- Plasmids- contain genes that encode for the antibacterial polypeptides called bacteriocins, a protein that kills other strains of bacteria. The col proteins of E. coli are encoded by proteins such as Col E1.
It is possible for a plasmid to belong to more than one of the above subgroups of plasmids.
Those plasmids that exist as only one or a few copies in a bacterium run the risk of being lost to one of the segragating bacteria during cell division. Those single copy plasmids implement systems which actively attempt to distribute a copy to both daughter cells.
Some plasmids include an addiction system, such as a host killing (hok) system of plasmid R1 in E. coli. Producing both a long lived poison and a short lived antidote. Those daughter cells that maintain a copy of the plasmid survive, while a daughter cell that fails to inherit the plasmid dies or suffers a reduced growth rate because of the loitering poison from the parent cell.
Plasmids also replicate autonomously. Each plasmids contains its own origin sequence for DNA replication, but only a few of the genes needed for replication. Some plasmids come equipped with self-preservation genes, and they are called addiction modules. The addiction modules also force the host cell either keep the plasmid or die.
References for Types of Plasmids
[edit | edit source]Berg, Jeremy M., et al. "Biochemistry". 6th ed. W.H. Freeman and Company, NY, 2007.
Uses, Applications, and Significance
[edit | edit source]Plasmid provides a versatile tool in genetic engineering because of its unique properties as a vector. Plasmids are utilized to create transgenic organisms by introducing new genes into recipient cells. For example, the Ti plasmid from the soil bacterium Agrobacterium tumefaciens is very valuable in plant pathology in developing plants with resistance to diseases such as holcus spot on leaves and crown gall tumors.
Plasmid also carries medical significance because of its role in antibiotic synthesis. Streptomyces coelicolor plasmid can give rise to thousands of antibiotics, as well as that of S. lividans or S. reticuli. In another example, E. coli plasmids are used to clone the gene of penicillin G acylase, the enzyme that turns penicillin G into the antibacterial 6-amino-penicillanic acid.[1] Once again, these cloning processes are carried out with the assistance of type II restriction enzyme to put the gene of interest into the plasmid vector.
In DNA recombinant technology, plasmid-based reporter gene are crucial as they allow observation of organisms in real time. The gene for Green Fluorescent Protein can be integrated into a plasmid of the organism under investigation. The encoded protein is small and does not alter the function of the host protein. This feature of GFP makes it very easy to observe cell dynamics.[2]
These are only a few among many techniques, applications and uses of plasmids developed throughout the years. The future of plasmid engineering looks very promising with many more examples and opportunities to come.
Alkaline Lysis Mini Plasmid Prep
[edit | edit source]In order to purify the plasmid DNA from the bacterial cell, alkaline lysis plasmid prep will need to be done. Three solutions will be prepared as following:
Solution1: 50mM glucose, 25mM Tris-HCL pH8, 10mM EDTA, 50 microgram/ml RNase
The purpose of adding glucose is to increase the solute concentration outside the cell, so once the cells are broken open, the osmotic pressure will draw water out of the cell and carry the plasmid with it. EDTA is a chelator of divalent cations such as Mg2+, Ca2+, and Mn2+. These cations will be bind away and remove from the solution. Mg2+ is the cofactor of DNase, so if it is bind away then DNase will not be able to digest DNA hence DNA will remain intact. RNase A will degrade any bacterial RNA present.
Solution2: 0.2M NaOH, 1%SDS
SDS lyses cells by dissolving the cell membrane. The high pH created by the NaOH will denature the bacterial proteins so they will precipitate out of solution. The extreme pH will denature DNA by separate two strands of the double helix giving single strand of bacterial chromosome and plasmid DNA. However, the small interlocked plasmid strands will stay together because it is bind tightly hence NaOH cannot get in.
Solution3: 3M Kacetate( 29.4g KOAc + 11.5 ml glacial acetic acid/ 100ml), pH 5.5
The acetate buffer will bring the pH of the solution to a more neutral pH. This will allow DNA strands to re-anneal. The chromosomal DNA is too complex and big to re-anneal under these condition hence only the plasmid DNA re-anneal. The K+ ions will bind with SDS (-) forming KDS which will precipitate out from the solution.
References
[edit | edit source]Slonczewski, Joan L. Microbiology. "An Evolving Science." Second Edition.
During the lytic pathway, a lambda bacteriophage containing viral DNA attaches to a bacterial cell. The virus DNA is then inserted into the host cell containing plasmid DNA. The viral DNA from the lambda phage is not introduced into the plasmid but remains in the cell. The viral DNA uses the host cells cell machinery in the process of quickly replicating the viral DNA. The replicated viral DNA is then packaged into virus particles made from viral proteins. Then the virus uses the host cell's machinery and metabolism in order to drive the packaging of the viral DNA into protein coat encasements. After the progeny lambda DNA is replicated and encased, the host bacterium lyses or bursts and releases the numerous copies of the lambda phage to repeat the process on other host cells.

Lamda phage in Cloning
[edit | edit source]A lambda phage is also a vector used for DNA cloning. It is a virus containing double-stranded phage DNA that can is introduced into E. Coli for replication. Mutant lambda phages have been created that makes cloning easier. One example is the lambda-gt-lambda-beta phage. It contains two EcoRI cleaves sites. The middle section of the phage DNA is removed by restriction digestion. The two remaining arms of the DNA cannot be packaged in to lambda virion because it is too small. Instead, a foreign piece of target DNA is joined to the two arms with ligase, which replaces the middle section that was removed. The addition of the foreign DNA allows the phage DNA to be long enough to be packed into a virion. The packaged lambda virion can then be inserted into a host cell and then replaced through the lytic or lysogenic pathway.
Genomic library
[edit | edit source]Genomic Library refers to the complete set of thousands of recombinant-plasmid clones, each carrying copies of a particular segment from the initial genome. A researcher can save a library as such and use as a source of gene interest or for genome mapping. A genomic library made using phage is stored is collections of phage clones Whatever cloning vector, the restrictior enzymes do not respect gne boundaries in cutting up genomic DNA, therefore some gene in a genomic library can be divided into two or more clones.
Lambda phages can also be used to house a genomic library. First, a genomic DNA is fragmented by enzymatic digestion. The needed fragments are about 15 kilobases long, which can be separated by gel electrophoresis. Synthetic linkers are attached to the ends of the fragments. The fragments are then introduced into the mutant phage DNA which are then packaged into virions. The virions are introduced to E. coli hosts which then produce many copies of the phage DNA. The progeny virions then contain a genomic library of a DNA that has been fragmented. The genomic library can be screened to isolate a specific gene of interest.
Complementary DNA (cDNA)
[edit | edit source]Researchers are able to make even more limited kinds of gene library using complementary DNA (cDNA). Once they are able to isolate mRNA from cells, they can actually onatin a mixture of all the mRNA molecules in the cell that have transcribed from a number of different genes. Therefore, the cDNA that is made is a library of a collection of genes. cDNA represents only a part of a cell's genome- the genes that were transcribes in the starting cells.
This kind of genomic library has its advantages, for example if a researcher wants to study the genes responsible for a specialized function of a particular kind of cell, such as the liver or brain cell, the smaller more specific library allows this to be done with greater detail. Also, by making cDNA from cells of the same type of an organism at different times, a researcher can trace changes that occur in gene expression.
Lifecycle Stages
[edit | edit source]There are a total of six stages in the Lytic Lifecycle of the Lytic Bateriophage. The six stages are Adsorption, Penetration, Replication, Maturation, Release and Reinfection.
Stage I: Adsorption
[edit | edit source]In the first stage of the lytic bacteriophage life cycle, the attachment sites on a phage is absorbed on the receptor site of the host cell. The phage usually attach itself to the cell-wall fo the cell, sometimes it can attach to the flagella or pilli of a bacteria as well.
Stage II: Penetration
[edit | edit source]In the second stage, the phage has a specific enzyme that makes a hole in the cell wall and then inserts the phage's genome into the cell's cytoplasm. The alternative way to insert the genome into the cell is to make a hollow tube that goes into the bacterium. This is called "contracting a sheath".
Stage III: Replication
[edit | edit source]In the third stage, after the insertion of the phage's genome into the bacterium cell, the cell consequently shuts down the synthesis of RNA, DNA and protein. At the same time, the phage replicates its own genome by using the cell's metabolic components to make phage enzymes.
Stage IV: Maturation
[edit | edit source]In the maturation stage, the genome slows generates itself around the genome to prepare for the next stage.
Stage V: Release
[edit | edit source]The release stage occurs when the lysozymes from the phage genome breaks down the peptidoglycan. This causes a osmotic lysis of the cell, and the bacteriophage is release to the outside of the cell.
Stage VI: Refection
[edit | edit source]About 50 - 200 phages may be produced from this infected bacterium. Thus after the release of the bacteriophages, they can go and infect other bacterium cells near the surroundings.
A specialized lambda transducing phage, containing the rec A gene had been identified in the early 1960s as an essential for homologous recombination in E.coli cells but the product had never been isolated. Robert Lehman and his team decided to embark on this experiment. The rec A protein had ATPase activity dependent on single stranded DNA meaning that it was dependent on DNA ATPase. Rec A protein can promote renaturation, the opposite of denaturation, of the single strand that is complementary but it can also promote ATP dependent strand exchange between single strands and a homologous DNA dublex. It can also form Holiday junctions, a the key factor in recombination where DNA is switched around to form variety. Lehman, Robert. Wanderings of a DNA Enzymologist: From DNA Polymerase to Viral Latency. Annu. Rev. Biochem. January 16, 2006.
Overview
[edit | edit source]During the lysogenic pathway, a lambda phage containing virus DNA attaches itself onto a host bacterium cell containing a plasmid. The lambda phage releases into the host cell where it is integrated into the host's genome. The phage DNA is then incorporated into the host chromosome and is replicated along with the rest of the host's DNA and remains inactive. This can continue for several generations until environmental factors activate the expression of the dormant phage DNA. The progeny DNA is then released from the host genome and is enclosed into virus packages. The bacterium then lyses and releases the progeny virus particles also called virions.
According to an article [citation needed] posted in the September 2008 issue of Biophysical Journal,a new study reveals that these bacteria infecting cells can make collective decisions on whether they should kill host cells immediately after infection or enter a present, but not visible state to remain in the host cell. The fate of a cell is controlled by the number of infecting diseases in a coordinated fashion. According to Joshua Weitz, a Georgia Tech professor, he states, "In the case of perhaps the most extensively studied bacteriophage, lambda phage, experimental evidence indicates that a single infecting phage leads to host cell death and viral release, whereas if two or more phages infect a host, the outcome is typically latency."
Overview
[edit | edit source]Genomic libraries are a catalog of genes of a particular organism. They are also commonly referred to as gene banks. To create a genomic library, the complete genome of an organism is cleaved into fragments and inserted into a cloning vector. It can also refer to the collection of vector molecules.
How to Create a Genomic Library
[edit | edit source]First, a variety of restriction endonucleases are used to cleave at certain base pairs to create the necessary fragments of the DNA. These restriction endonucleases are chosen in such a way that the cohesive ends are compatible with a cloning vector and that the complete genome is represented by these fragments. This also means that the cloning vector is cleaved with the same restriction endonucleases and the original fragments are combined with the cloning vector using a ligase. This mixture is then inserted into a bacterial cell to produce a library. Each cell will have a different DNA molecule. Ordering of individual clones is archived by identifying overlapping sequences. This set of overlapping sequences of the catalog is defined as a contig.

Roles of Genomic Library
[edit | edit source]With the complete information in the library for a specific organism, researchers can perform a variety of experiments on the DNA. By doing so, they can determine the actions and interaction of separated genes along the strand. Also, they can compare the genomic library of healthy and unhealthy individuals of the same species to see where differences in genetic coding may be led to unexpected mutations.
In physical reality, a genomic library for humans is a collection of bacteria, the most common one is E. coli. The usable sequence of DNA from the human genome gets transferred and carried through the E. coli. The DNA is prepared by digesting it with a restriction enzyme. Then the target segments are inserted into the bacteria by using lambda phage. This creates a basic unamplified library, where the bacteria have been allowed to multiply and create additional copies of each section of the DNA.

Overview
[edit | edit source]A genomic library is a collection of all the genes of an organism. After creation of a genomic library, a particular gene usually needs to screened from the genomic library just created. A method to do this is to carry out a colony hybridization experiment.
Colony Hybridization Experiment
[edit | edit source]This technique is used to screen for a plasmid based genomic library. Bacteriophage plaques are screened. First, bacteria is transformed with a plasmid by using a bacteriophage to infect the colony. These are plated on a petri dish and incubated so that phage plaques are allowed to form. A plague with an identical phage will develop on the petri dish wherever the phage is located and infected by the bacterium. Next, replication of the original plate is made by placing a nitrocellulose sheet on top of the original plate. This allows the plaques to be transferred in a certain array. Both infected bacteria and phage DNA will be release from the lysed cells to attach to the nitrocellulose sheet to form spots. This is then treated with 2M NaOH to lyse the phages and separate the strands of DNA. When the DNA is denatured by the 2M NaOH treatment, the DNA strand is more accessible to be hybridize with the 32P probe. One must next neutralize the dish and then allowed to dry to immobilize the DNA. This is then mixed with a 32P probe and allowed to anneal to target any DNA sequence present on the nitrocellulose sheet. This is then subject to autoradiography by placing it on an x-ray film. The spots show where the probe has hybridized with the DNA and this is used to locate the genomic clone from the plaques on the original petri dish. Oligonucleotide mutagenesis is a process in which a specific DNA sequence is incorporated into a vector (such as a plasmid or lambda phage) to introduce a new function into that molecule. Mutagenesis allows the insight into how proteins fold, catalyze reactions, act as substrates and process information. Cloned gene proteins can be used medically in a number of ways. For instance, insulin and interferon can be obtained from production of bacteria. DNA probes may be manufactured for discovery of genetic diseases, cancer, and bacterial infections. Two main forms are used to produce these modified proteins, Site directed mutagenesis and oligonucleotide-directed mutagenesis. The later is used if restriction sites are absent.
Mutagenesis is the process of changing or creating genetic information. This process can occur naturally or made by using different methods. There are more approaches involving mutagenesis methods. One method is a method of site-directed mutagenesis using mismatched oligonucleotides. Second method is cassette mutagenesis by annealing complementary oligonucleotides. Third method is PCR by generating a mutant fragment starting from a double-stranded DNA template using mismatched oligonucleotides. Mutagenesis is also used by experimenters to analyze DNA. Site mediated mutagenesis mutates one sequence of amino acids to determine that specific sequences overall purpose once it stops working. This is an example of experimenters using Mutagenesis.
Site-Directed Mutagenesis
[edit | edit source]In site-directed mutagenesis, individual amino acids in the primary structure of a protein are replaced by changing the DNA sequence of a cloned gene. The process involves removal of a DNA segment and replacement of that segment with a chemically synthesized segment identical to the original but with the desired change. The steps are as follows: A recombinant plasmid containing the gene of interest is treated with a restriction endonuclease to cleave the sequence of interest. The synthetic DNA fragment with the specific base pair change is inserted into the plasmid. This is done using a DNA ligase. The plasmid then contains the gene with the desired nucleotide base pair change.
Site directed mutagenesis is also useful for testing the validity of an enzymatic mechanism.
Oligonucleotide-Directed Mutagenesis
[edit | edit source]This form of mutagenesis creates a specific DNA sequence change. This method makes point mutations, differing in a single nucleotide. In this process, a DNA strand is synthesized with the specific nucleotide base pair change and annealed to the copy of the gene in a plasmid. Increasing the temperature allows for the mismatched base pair to be substituted in order for this reaction to occur. This annealed strand acts as a primer for the complementary strand to be synthesized in the plasmid. Oligonucleotide-directed mutagenesis uses DNA polymerase, dNTPs and DNA ligase. The result are two types of progeny: one with the original sequence and another with the new sequence.
Cassette Mutagenesis
[edit | edit source]
Cassette Mutagenesis is the replacement a region of DNA with new sequences. Any length and sequence can be replaced by this method. Therefore, more variations of proteins can be made. By taking advantage of this, new mutant sequences can be made. The Cassette Mutagenesis uses the original gene containing restriction sites. This site allows cleavage of the gene and produce the region where the new DNA is inserted. After appropriate restriction sites are identified in a vector, the vector is cleaved at the two sites by using a endonucleotide. The new sequence is inserted and ligated in the region produced. The new sequence allows a variety of studies on protein structures or nucleic acid sequences that have not been explored before.
Polymerase Chain Reaction
[edit | edit source]Polymerase chain reaction is used for recombination of sequences and mutagenesis. This method is very useful because it is a fast reaction and it is not limited by any restriction sites. In the PCR, oligonucleotide primers are incorporated into the ends of the product DNA. The 5' ends of these primers can contain any desired sequence. two PCR products are added by PCR. They are mixed, denatured, and reannealed. There are two different sequences produced. One is 5' overlapping strands which is not productive. 3' overllapping strand is produced and reactive. The polymerase extends the 3'strand. In this process, the 3' strand acts as primers. The whole process are repeated by producing synthetic sequences. This overlapping by joining two DNA fragments together can produce mutatation in PCR fragment.
Reference
[edit | edit source]M.J. McPHERSON, Directed Mutagenesis
The Modern Approach to Gene Design
[edit | edit source]A typical gene design cycle starts with defining a desired target protein. From there the scientist, using a gene composer computer program, inputs information such as functional characterization data, sequence alignments or knowledge of related 3D structural information from various sources that pertain to the desired protein. Secondary structural information is annotated and amino acids in the active domains (i.e. - participate in ligand binding sites, are water exposed, or form crystal contacts) are labeled, and multiple amino acid sequence variants are generated. The next step is back-translating designed amino acid sequences into nucleic acid sequences. The program will generate optimized nucleic acid sequences based on defined criteria input by the user, such as codon optimization for highly expressed proteins, suppressing strong mRNA structure forming elements and avoiding undesired restriction sites. Finally, sequences for complete genes are output. The adoption of this new method of protein design allows heterologous expression of proteins to be tested more quickly and minimizes failure rates.
Benefits/Uses of Gene Design
[edit | edit source]Gene/Protein design is currently being utilized for a number of important applications including:
1. Immunotoxins- chimeric protein made from a gene for an antibody and a gene for a toxin, which can be used to kill cells that are recognized by the antibody
2. Synthetic vaccines- recombining DNA to create noninfectious coat proteins of viruses
3. Completely new genes and proteins with functions not found in nature
4. New enzymes
5. New drugs
6. Disease research for Malaria, Anthrax, HIV, Alzheimer's and Various Cancers
Sources
[edit | edit source]http://www.biosciencetechnology.com
http://boinc.bakerlab.org/rosetta/rah_medical_relevance.com
What is the Human Genome Project
[edit | edit source]The human genome project is a 13 year long project run by the US Department of Energy and the National Institutes of Health. The task of sequencing the estimated 3 billion DNA base pairs was intimidating and it took an international effort to complete, the UK’s Wellcome Trust was a partner in the effort and many other countries such as Germany, France, China and Japan also made great contributions. The goals of the project included: sequencing the 3 billion human DNA base pairs, identifying the estimated 20,000-25,000 genes found in human DNA, to store all this information in an accessible databases and improve the tools for data analysis.
The DNA of a set of model organisms was also sequenced and studied to provide comparative information so that scientists could understand how the human genome functioned. The primary reason for this project was to use the sequenced genome to understand and eventually treat the ~4000 genetic diseases that afflict humans, as well as the many multi-factorial diseases in which genetic predisposition plays an important role. Currently the human genome project research and the technologies developed are being used in: molecular medicine, energy sources/environmental applications, risk assessment, Bioarchaeology, anthropology, evolution, human migration, DNA forensics (for identification purposes), agriculture, livestock breeding, bioprocessing and many other fields.
The Human Genome Project (HGP) was a world wide collaboration initiated to discover and create a database for the entire human genome. A genome is the entire DNA of an organism (including its genes). DNA is made up of sequences of four different bases Adenine, Guanine, Thymine and Cytosine. Knowing the specific sequence of these backbone bases allows us to compare and determine certain diseases. For example sickle-cell anemia is caused from the alteration of a single base from A to T. The identification of these differences makes it possible to research and seek medical advances on diseases and DNA disorders. To further understand the importance and significance of the sequence of the human genome studies were carried out on non human species such as the fruit fly, mice, and e-coli.
Creating a means to store the information obtained by the Human Genome Project was a challenge in itself. The human genome contains approximately 3 billion different sequences which alone would take up about 3 gigabytes of memory additional memory would be required for the ongoing advances on these sequences. Bioinformatics Morey Parang, Richard Mural and Mark Adams were the main contributors to designing a means to store this massive amount of information.
Other goals of the HGP include: identifying all 20 to 25 thousand genes in the human DNA, improve tools for data analysis, transfer technologies to private sectors, and address the ethical, legal, and social (ELSI) that might come from the HGP.
The three billion dollar project was supposed to take 15 years to complete, as a combined world wide effort, including scientists from China, Japan, UK, Germany, and France. Strangely enough, the project was finished in 13 years rather than the expected 15. Some believe that the reason behind this anomaly is because of a privately funded only around three hundred million dollar project by Celera Genomics. Because an Celera Genomics declared that they were going to finish sequencing the human genome before the three billion dollar effort put forth by the cooperative effort from scientists around the world. They used a slightly more risky approach called the "shotgun sequencing", instead of sequencing the genome in a linear manner, they "shot" the seqeuences into small segments and found where they overlapped. The competition between the two groups helped fuel the project, as larger efforts were put forth from both parties to finish before the other. Thus, both shared equal amounts of work towards the finished project of the sequencing of the entire human genome.
The project started in 1990 and was completed in the year 2003. Although the goals established by this project were completed the information obtained is still being analyzed and researched in order to make advances in life sciences. The HGP has benefited many areas of science such as molecular medicine, energy sources and environmental applications, risk assessments, bioarchaeology, anthropology, evolution, human migration, DNA forensics, and bioprocessing. Although the human genome is now considered "complete", there are still many sections of the genome that haven't been sequenced as of yet. Many gaps still exist, but the sequences are put onto world wide data bases such as BLAST as the small tadbits are sequenced by different groups around the world.
Why the Human Genome Project?
[edit | edit source]The human genome project was developed in order to open the eyes of mankind.
For instance, through sequencing the human genome, scientists can now study genetic diseases in more detail and depth. The human genome project has helped identify genes associated with different genetic conditions such as myotonic dystrophy, fragile X syndrome, Alzheimer's disease, familial breast cancer and more. This will help researchers develop better ways to treat the root of the conditions rather than just the symptoms of the diseases. In traditional medicine, it will also allow earlier identification of diseases and treatment options customized to individuals. Research is also underway to improve gene therapy—in the future, scientists may be able to fix or replace faulty genes. Another area of research involves the variation in individual response to environment. As researchers determine which genes code for sensitivity to environmental pressures, like carcinogens and irritants, they will be able to better predict the risks involved for an individual exposed to a risky environment. One of the most important results of this area of study is an increased understanding on how low-level exposure to radiation effects cancer risc.
This project also helped boost forensics; scientists can now create DNA fingerprints of small areas of DNA regions that vary between individuals, allowing for accurate identification techniques. Fluids, tissue, and hair at crime scenes have greatly increased in utility as a direct result of genetic research. The importance of the Human Genome Project to forensics extends beyond the criminal sphere. The "DNA fingerprints" can also be used match organ donors, establish familial relationships, and identify microorganisms that might be polluting an environment.
Because of the expansion of the general knowledge related to sequencing genomes, smaller projects such as the microbial genome program were developed to sequence the genomes of bacteria. The goal of this is to find ways to produce energy, reduce toxic wastes, and industrial processing through the use of microbes and microbial enzymes. It can be used to analyze in greater depth the influence of even the tiniest forms of life on an ecosystem. Also, sequencing certain microbial genomes allows scientists valuable insight into the ways pathogenic microbes infect the human body. Because of the dependence humans have on the microbial world, the relationship between human world and the microbial world is also worth researching. This will benefit both human health and the environment.
The human genome project has also helped people understand the road humankind took in the process of evolution. It gives scientists a glimpse of history as it helps connect the three kingdoms of life: determine a complete lack of ethnic divisions Archaebacteria, Prokaryotes, and Eukaryotes. Comparative genomics aid scientists in determining what specific segments of human DNA code for by comparing them to the equivalent segments in other organisms.
As for more recent history, DNA studies have been used to on an ethnic level, proving that ethnicities are the product of society, not different DNA. That said, specific markers on the Y chromosome can be used to trace the migrations of a man's paternal line throughout human history. The field of behavioral genetics has also been aided by the completion of the Human Genome Project. For years, scientists recognized evidence that many behaviors have biological foundations. For example, within a species certain behaviors consistently crop up, and such specific behaviors can be passed along to later generations (like the herding instincts of an Australian Shepherd). Further support for this theory includes cross-species parallels in behavior, particularly with closely related species. While traditionally the area of behavioral genetics has been centered around the study of twins and adoptees in an attempt to clarify the nature vs. nurture debate—that is, how much of our behavior is actually coded into our DNA, and how much has resulted from environmental influences. Behavioral genetics is complicated by both the difficulty of quantifying certain abstract concepts (i.e. intelligence) and by the fact that any behaviors are coded by multiple genes and affected by other factors. In addition, any results from research regarding behavioral genetics will likely be hot-button issues and thus will require greater care before drawing conclusions.
Finally, the human genome project has developed many techniques for genetic engineering, resulting in genetically modified plants and animals for better food and energy production. Examples include crops engineered to require less pesticide use or less water. The human genome project has also allowed the development of plants that break down certain types of waste. This has led to many disputes over the right of man to alter "natural" organisms and concern over the long-term effect of genetically modified organisms—particularly those intended for human consumption.
TimeLine of Major Goals Completed
[edit | edit source]Sep-94: 1-cM resolution Genetic map was produced with ~3,000 markers
Dec-94: High-throughput oligonucleotide synthesis technologies developed
Aug-96: Methanococcus jannaschii genome sequenced; confirms existence of third major branch of life on earth.
Sep-96: First sequenced genome, for yeast, was completed
Dec-96: DNA microarrays technologies developed
Oct-98: A physical map with ~52,000 STS’s (i.e. - a sequence tagged site, a short DNA segment that occurs only once in a genome) was completed
Dec-99: First Human Chromosone Completely Sequenced
Nov-02: Fiscal Report showed financially the project was on track; the project was sequencing more than 1,400 fragments per year at only $0.09 per finished base, well below the estimated costs of 500 fragments a year at $0.25 per base
Dec-02: Genomic-scale technologies: scale-up of two-hybrid system for protein-protein interaction was developed
Feb-03: 3.7 million mapped human SNPs (i.e. - Single nucleotide polymorphisms, DNA sequence variations that occur when a single nucleotide in the sequence is altered)
Mar-03: 15,000 full-length human cDNAs (i.e. - DNA molecules that are complementary to specific messenger RNA) were sequenced
Apr-03: 99% of gene-containing part of human sequence was finished to 99.99% accuracy
Apr-03: Finished genome sequences of E. coli, S. cerevisiae, C. elegans, D. melanogaster, plus whole-genome drafts of several other model organisms, including: C. briggsae, D. pseudoobscura, mouse and rat were completed
May-08: Genetic Information Nondiscrimination Act (GINA) becomes a law
Tools Used To Sequence The Genome
[edit | edit source]The DNA sequence of humans if first broken into smaller projects called a cosmid, BAC, PAC, or P1 clone. These projects can be assigned to private labs all across the world. The following phases are the order in which private or government labs sequence these portions or contigs of the main genome.
The Random Phase For many labs this means using the shotgun approach to sequencing DNA and it utilizes DNA restriction enzyme to cut the project DNA into varying sizes of base pair regions.
Gap Closure Phase Connecting the fragments of DNA caused by restriction enzyme is a bottleneck in the process of DNA sequencing and it has been greatly sped up through gold standard computer programs such as phredPhrap. As these programs become better less overlap is needed to find matching strands, but at the same time the higher the overlap of these strands the higher the accuracy.
Ambiguity Resolution Phase Through the use of programs such as confed, the low quality regions of the sequenced DNA can be analyzed for anomalies such as deletions or contaminant reads. This step is mostly a finishing or spell check function that acts to increase the accuracy of the raw sequenced DNA.
Analysis Phase This portion of the sequencing finds known patterns that are common to DNA. Patterns are found by programs such as BLAST, XGRAIL, and REPBASE. The commonalities looked for by XGRAIL include exons, introns, poly-a sites, promoter regions (TATA boxes, etc.), and repetitive bases. REPBASE will find repetitive sequences that are known to exist in families and subfamilies. BLAST has a large community of scientists who input DNA sequences of a wide range of species and allows the DNA in question to find its nearest evolutionary relative.
Viewing the Genome through DNA Maps
[edit | edit source]There are many different ways of mapping the human Genome. One of the most common ways is in units of Centimorgans. Every centimorgan represents a one percent chance that two genes will separate during meiosis. One example is a gene that is inherited with Huntington’s disease 96 percent of the time. The remaining 4 percent of the time it does not travel with Hunington’s disease and thus it has a 4cM from that gene.
Of the maps that are used to view DNA there are two kinds of maps, genetic-linkage and physical maps. Genetic Linkage maps view DNA in reference to another DNA group and how often they are inherited together. These genetic maps include Cytogenetic maps, Restriction Maps, Cosmid maps, and Sequence Maps.
The Cytogenetic map was created by Victr McKusick, and it utilizes chromosomal staining in order to view groups. This method is limited in resolution as the target gene a scientist may be seeking could be in a stain containing ten million base pairs. That is why this method is useful for broad analysis and narrowing a sequence down to certain regions of the chromosome.
The Restriction map was created by Dr. Raymond White, and it utilizes restriction enzymes. This process takes the genome of a family or generation of people and finds the percentage of genes that are close together between these related people. By using restriction enzymes the same specific sequence of DNA is cut from the genomes of the family and can be analyzed. The resolution of this methods is ten times greater than Cytogenetic mapping and can focus in on a genetic marking within one million base pairs.
Cosmid maps are used from the actual overlapping sequence of base pairs derived from shotgun methods of sequencing. This methods takes bases of roughly 40,000 base pair lengths and overlaps them. The resolution of this method is highly accurate and can find a gene within 10,000 – 100,000 base pairs.
The Sequence Map is the actual culmination of all sequencing and lists the entire order of the known genome of all 46 chromosomes. IT consists of over 3 billion base pairs and has 20,000 - 25,000 protein encoding genes.
Ethical Dilemas
[edit | edit source]"If scientists don't play God, who will?" James Watson, former head of the Human Genome Project.
The Human Genome Project raised many ethical concerns, with knowledge of the entire human DNA sequence and genes they code, people could alter their genes, (for a price) leading to possible genetic discrimination and other moral ramifications from “playing God”. While the intentions may be noble, trying to better understand and help treat the many genetic diseases and defects that afflict mankind, many believe genetic manipulation/alteration is a slippery slope. While now most research is targeted at identifying or treating birth defects that are caused by a single gene, such as cystic fibrosis and Tay-Sachs disease, as well as more daunting ventures such as preventing diabetes, heart disease and other big killers, many worry what will come next. Will the mind will be targeted for improvement—preventing alcohol addiction and mental illness, and enhancing visual acuity or intelligence trying to improve the human design? Even genetic testing of fetuses raises questions about the ethical ramifications of ever more accurate genetic screening. Where should the line be drawn in eliminating perceived defects? On a more mundane note, the completion of the Human Genome Project raised concerns about genetic discrimination by employers and health insurance companies based on individual predisposition to current diseases. As of May 2008, GINA (Genetic Information Nondiscrimination Act) protects individuals from such discrimination, and precludes employers from demanding such tests.
Dr. Marvin Frazier, who fields human genome questions as director of the Life Sciences Division of the U.S. Department of Energy's Office of Biological and Environmental Research, says it will take decades for scientists to figure out how to manipulate human intelligence or athletic ability, because of the complexity of the traits (they rely on a lot of genes) and the unknown role the environment plays on these abilities. To achieve desired goals the costs would be tremendous, not only in fiscal capital but human capital as well due to the large amount of risky experimentation that would be involved. "It is my opinion that this would be wrong," he added, "but that will not stop some people from wanting to try. The key question is not whether human (genetic) manipulation will occur, but how and when it will.”
Related Projects
[edit | edit source]GLT: US Department of Energy's Genomics: explores the diversity of microbial and plant genomes in through their DNA sequences in order to understand how living systems operate
Human Microbiome Project: generates data on the human microbiome to study its role in human diseases. Instead of studying individual species individually, this project studies the microbial community harvested from their natural environment.
Genographic Project: a combined project with National Geographic and IBM, the goal of the project is to analyze the roots of human genetics roots over the course of five years.
Genetic Information Nondiscrimination Act (GINA): Major Impacts
[edit | edit source]On May 21, 2008, President George W. Bush made into law the Genetic Information Nondiscrimination Act (GINA). The law prevents U.S. insurance companies as well as employers from pre-screening potential prospects based on the information of their genetic tests. The bill finally passed both chambers of Congress after certain disagreements were worked out, but not until a few months have passed.
GINA was put into effect to prevent prejudiced situations such as when an arbitrary employer uses a future employee’s genetic information to determine performance level, proneness to tardiness, health risk, etc. It is also illegal to request or demand a genetic test under the law.
Without the fear of affecting their jobs or insurance rates, Americans would be more willing to go through genetic testing for diseases. This is encouraging because it could open doors to new medical discoveries and cures. It also allows early detection of health problems, leading to cost effective preventive solutions.
For more information: http://www.ornl.gov/sci/techresources/Human_Genome/publicat/GINAMay2008.pdf
References
[edit | edit source]- http://www.ornl.gov/sci/techresources/Human_Genome/elsi/legislat.shtml
- http://genomics.energy.gov
- Definitions for LLNL's Shotgun Sequencing. Human Genome Project Information. U.S. Department of Energy Office of Science.
- Davis, Joel. Mapping the Code.
- http://www.ornl.gov/sci/techresources/Human_Genome/project/about.shtml
What is gene therapy?
[edit | edit source]Gene therapy is an experimental technique that uses genes to treat or prevent diseases. Genes are specific sequences of bases that encode instructions on how to make proteins. When genes are altered so that the encoded proteins are unable to carry out their normal functions, genetic disorders can result. Gene therapy is used for correcting defective genes responsible for disease development. Researchers may use one of several approaches for correcting faulty genes. Although gene therapy is a promising treatment which helps successfully treat and prevent various diseases including inherited disorders, some types of cancer, and certain viral infections, it is still at experimental stage. Gene therapy is currently only being tested for the treatment of diseases that have no other cures.
Methods
[edit | edit source]There are many methods utilized to try and correct these altered genes:
• A normal gene can be inserted into a nonspecific location within the genome to replace a nonfunctional gene. This is the most common method.
• An abnormal gene could be swapped for a normal gene through homologous recombination.
• The abnormal gene could be repaired through selective reverse mutation, which returns the gene to its normal function.
• The regulation (act of turning the gene on or off) of a particular gene could be altered.
Using the method of inserting a normal gene to replace the disease-causing gene, a vector is used to deliver the new gene to the target cells. The most commonly used vector is a genetically altered virus that can carry normal human DNA. Target cells are then infected with the viral vector as the vector unload the genetic information into the target cell. This restores the target cell to the normal state.
EX VIVO method: cells are genetically-altered outside of the body and then reintroduced.
IN VIVO method: cells are genetically-altered inside the body after a genetically-altered vector containing therapeutic DNA is injected into the body.


Vectors
[edit | edit source]In order to insert a “normal” gene to replace the abnormal/defective one, a carrier molecule called a vector must be used to deliver the new gene to the target cells. The most common vector used is a virus that has been genetically altered to carry normal human DNA. Because viruses have evolved a way of encapsulating and delivering their genes to human cells in a pathogenic manner, they make excellent vectors, provided they are manipulated to remove disease-causing genes and instead insert therapeutic genes. Target cells are infected with the viral vector and the vector unloads its genetic material containing the “normal” gene into the target cell. The main types of viruses used as gene therapy vectors include: Retroviruses, Adenoviruses, Adeno-associated viruses and Herpes simplex viruses. Retroviruses are a unique class of viruses that make double-stranded DNA copies of their RNA genomes which are integrated into the chromosomes of host cells. Adenoviruses is another class of viruses with the double-stranded DNA genome property that causes respiratory, intestinal, and eye inflections. Adeno-association viruses is a class of viruses that use single-stranded DNA to insert their genetic material at a specific sit. Herpes simplex viruses is a class of double-stranded DNA viruses that infect neurons.
There are also several non-viral options for gene delivery, the simplest being direct introduction of therapeutic DNA into target cells. However, this approach is limited in its application because it can be used only with certain tissues and requires large amounts of DNA. Another non-viral approach is the creation of an artificial lipid sphere with an aqueous core, (i.e. - a liposome), which is capable of carrying the DNA and then passing it through the target cell's membrane. Another way to get the DNA inside target cells is by chemically linking it to a molecule that will bind to special cell receptors. Once the molecule is bound to the receptors, the DNA is engulfed by the cell membrane and passed into the interior of the target cell. However, this delivery system tends to be less effective than other options.
Many of the challenges that make non-viral techniques ineffective is the challenge of getting genetic material past the plasma membrane without the help of viruses and avoiding the host's immune response. Methods using liposomes and plasmids are usually coupled with techniques to help facilitate the process of delivering genetic material past the plasma membrane. Electroporation is the use of electric shock from a high-voltage source to induce the formation of pores along the cell membrane to allow genetic material to enter. Sonoporation is similar, but uses acoustic sound waves to create these openings. While these techniques are used to make the delivery of genetic material by liposomes and plasmids more effective, they run the risk of causing cell death by rupturing the membrane.
In the future scientists hope to apply particle bombardment which has been successfully used to introduce genetic material in plants in the form of a gene gun. The concept would involve using small particles of gold along with liposomes/plasmids in "bullets" to punch pores in the cell membrane and deliver the genes to the interior. Although this is commonly used in the laboratory for research, it is a technique limited to be used ex vitro.
Short-comings of Gene Therapy:
[edit | edit source]•Short-lived nature of gene therapy – There are many problems with integrating the new DNA into the genome, the new DNA introduced into target cells must remain functional and the cells must live long and remain stable. However, rapidly dividing nature of many cells prevents gene therapy from achieving any long-term benefits; patients would have to undergo multiple rounds of gene therapy.
•Immune response - Anytime a foreign object is introduced into human tissues, the immune system is designed to attack the invader, making it difficult to deliver the “normal” gene effectively. Furthermore, the immune system's enhanced response to invaders it has seen before makes it difficult for gene therapy to be repeated in patients.
•Problems with viral vectors – While viral vectors are the carrier of choice in most gene therapy studies, they also present a variety of potential problems: toxicity, immune and inflammatory responses, gene control and targeting issues and the possibility that the virus may recover its ability to cause disease while inside the patient.
•Multi-gene disorders- Conditions or disorders that arise from mutations in a single gene are the best/most effectively treated using gene therapy. Unfortunately, many of most common disorders, such as heart disease, high blood pressure, Alzheimer's disease, arthritis, and diabetes, are caused by the combined effects of variations in multiple genes. These disorders would be especially difficult to treat effectively using gene therapy.
However there have been many recent developments/advancements in gene therapy research, making it safer, and more effective, the field of gene therapy holds great potential for the future of medicine.
Sources
[edit | edit source]General Information
[edit | edit source]Artificial chromosomes are synthetic chromosomes consisting of fragments of DNA integrated into a host chromosome. These artificial chromosomes are introduced into host cells to propagate and can be used to transfect other cells, introducing new DNA. Artificial chromosomes are useful in cloning larger fragments of DNA, as plasmids can only contain up to 10,000 base pairs and phages are hard to work with. Artificial chromosomes can contain anywhere from 300,000(BAC) to 1,000,000(YAC) base pairs, effectively reducing the amount of runs needed for a large fragment to be analyzed. Because these chromosomes are more useful in cloning larger fragments of DNA, it is easier and quicker to clone and transform genes. Artificial chromosome vectors also make it easier to store through bacterial cells rather than mammalian cells.
There are two types of artificial chromosomes: Bacterial artificial chromosome (BAC) and yeast artificial chromosomes (YAC). Although yeast artificial chromosomes can contain more base pairs (over one million) than bacterial artificial chromosomes, bacterial artificial chromosomes are more common than yeast artificial chromosomes because they are more stable, making them easier to work with a smaller risk of rearrangement due to the circular shape of a plasmid. Yeast artificial chromosomes also may produce chimeric effects, while bacteria artificial chromosomes will not.
The usage of artificial chromosomes is mainly for studying DNA fragments. This is done by integrating a non-viral/non-bacterial DNA into a bacterial chromosome and having it express the DNA fragment within the host. Once expressed, the host cell undergoes replication and thus the host chromosome containing the integrated DNA fragment will be replicated. The result is a huge colony of bacteria containing the fragmented DNA. In other words the DNA is cloned into millions of copies. The use of artificial chromosomes has revolutionized every aspect of biological studies.
References
[edit | edit source]Bacterial Artificial Chromosomes (BAC)
[edit | edit source]Bacterial Artificial Chromosome also known as BAC is a vector used to clone a target DNA piece and planted in bacterial content. Vectors are carriers that are also made of DNA. The vector acts as a parent host, which can acts like a guide to carry out the gene for cloning. Bacterial artificial chromosomes can be as large as insects. In order to amplify a gene, DNA sequence must be extracted from a desired source using restriction enzymes to cleave the vector and the target DNA and attach into a host bacterium. Plasmids of bacteria usually play a vital role in the cloning process since they are also made of DNA and they can be compatible with experimental gene. Vectors also contain the origin of replication where replication begins at the vector site and proceed with the attached sample.

Common Gene Components
[edit | edit source]oriS, repE•F
- replicates plasmid replication and regulates copy number
parA and parB
- partitions F plasmid DNA into daughter cells during division and ensures stable maintenance of the BAC
A selectable markers
- because some BACs also have lacZ at the cloning site for blue/white selection, this is used to resist antibiotics
T7 and Sp6
- phage promoters for transcription of inserted genes
Contribution to Models of Disease
[edit | edit source]Inherited disease
[edit | edit source]BACs are being greatly used in modeling genetic diseases in order to study their effects in the experimentation on transgenic mice. Because complex genes often have many regulatory sequences upstream of the encoding sequence, including many promoter sequences that will control a gene's express level, BACs have been found very useful in this area of study. When tested with mice, BAC has been able to help with the study of neurological diseases, such as Alzheimer's disease or Down syndrome. BAC has also been recently used to study specific oncogenes, which are associated with various different cancers. BAC is transferred over to these genetic disease models through the use of electroporation/transformation, which is essentially transfection with a compatible virus or micro injection. BACs have also been used to detect genes or large sequences of interest, and then used to map them onto the human chromosome using BAC arrays. Because this process is able to accommodate much larger sequences without the risk of rearrangement, it is the preferred method of these types of genetic studies and also more stable than other types of cloning vectors.
Infection disease
[edit | edit source]BACs can also act as clones of large DNA viruses as well as RNA viruses. These clones are known as "infections clones," as transfection of the BAC construct into host cells is enough to initiate the viral infection in the host. The infectious property that the BACs contain has aided in the study of viruses such as herpesviruses, poxviruses, and coronaviruses. Using genetic approaches to mutate and transform the BAC while it resides in bacteria, molecular studies of these viruses can be performed.
References
[edit | edit source]http://www.scq.ubc.ca/ THE BIG BAD BAC: BACTERIAL ARTIFICIAL CHROMOSOMES
Bacterial Artificial Chromosomes (BAC)
[edit | edit source]Bacterial Artificial Chromosome also known as BAC is a vector used to clone a target DNA piece and planted in bacterial content. Vectors are carriers that are also made of DNA. The vector acts as a parent host, which can acts like a guide to carry out the gene for cloning. Bacterial artificial chromosomes can be as large as insects. In order to amplify a gene, DNA sequence must be extracted from a desired source using restriction enzymes to cleave the vector and the target DNA and attach into a host bacterium. Plasmids of bacteria usually play a vital role in the cloning process since they are also made of DNA and they can be compatible with experimental gene. Vectors also contain the origin of replication where replication begins at the vector site and proceed with the attached sample.
Common Gene Components
[edit | edit source]oriS, repE•F
- replicates plasmid replication and regulates copy number
parA and parB
- partitions F plasmid DNA into daughter cells during division and ensures stable maintenance of the BAC
A selectable markers
- because some BACs also have lacZ at the cloning site for blue/white selection, this is used to resist antibiotics
T7 and Sp6
- phage promoters for transcription of inserted genes
Contribution to Models of Disease
[edit | edit source]Inherited disease
[edit | edit source]BACs are being greatly used in modeling genetic diseases in order to study their effects in the experimentation on transgenic mice. Because complex genes often have many regulatory sequences upstream of the encoding sequence, including many promoter sequences that will control a gene's express level, BACs have been found very useful in this area of study. When tested with mice, BAC has been able to help with the study of neurological diseases, such as Alzheimer's disease or Down syndrome. BAC has also been recently used to study specific oncogenes, which are associated with various different cancers. BAC is transferred over to these genetic disease models through the use of electroporation/transformation, which is essentially transfection with a compatible virus or micro injection. BACs have also been used to detect genes or large sequences of interest, and then used to map them onto the human chromosome using BAC arrays. Because this process is able to accommodate much larger sequences without the risk of rearrangement, it is the preferred method of these types of genetic studies and also more stable than other types of cloning vectors.
Infection disease
[edit | edit source]BACs can also act as clones of large DNA viruses as well as RNA viruses. These clones are known as "infections clones," as transfection of the BAC construct into host cells is enough to initiate the viral infection in the host. The infectious property that the BACs contain has aided in the study of viruses such as herpesviruses, poxviruses, and coronaviruses. Using genetic approaches to mutate and transform the BAC while it resides in bacteria, molecular studies of these viruses can be performed.
References
[edit | edit source]http://www.scq.ubc.ca/ THE BIG BAD BAC: BACTERIAL ARTIFICIAL CHROMOSOMES
Overview
[edit | edit source]Electroporation can be used to insert genes into eukaryotic cells such as plants cells and animal cells, as well as prokaryotic cells such as bacterial cells. In electroporation, cereal monocots and dicots can be introduced to foreign DNA by applying intense electric fields.
Procedure
[edit | edit source]Lets take a plant cell as an example. First, a plant cell will consist of a cell wall and a plasma membrane. Cellulose is used to digest the cell wall, creating a plant cell that is has an exposed membrane. This is known as a protoplast. Second, foreign DNA is added into the cell. High electric fields are used to create a transiently permeable membrane so that large molecules will be able to pass through the membrane. This is caused by transient electric pulses. Thirdly, the cell wall is able to regrow back. In the end, the plant cell is now a viable plant cell with an insert of foreign DNA.
Considerations for Optimization[1]
[edit | edit source]When in vitro electroporation is performed, there are several factors that must be taken into account.
- Waveform - when the electroporation technique was originally developed, exponential pulses were used. Since then, square waves have been found to be more effective and less damaging to cells. The pulse length and amplitude can be modified to fit the needs of the experimenter.
- Cell size - transmembrane potential is described by the equation ΔV(m) = fE(ext)r cosθ, where V(m) is the transmembrane potential, f is a proportionality constant relating the external field to its impact on the cell, E(ext) is the external applied electric field, r is the cell radius, and θ is the angle of the field with respect to the cell poles. This means that the smaller a cell is, the larger the external field must be to achieve the same electroporetic effect. Also, a more homogenous cell sampling will yield better results of electroporation.
- Resealing - membrane resealing after electroporation is temperature-dependent, with lower temperature correlating to slower resealing times. An incubator often increases the quality of the results. Waiting for 15 minutes after electroporation to disturb cells by pipetting betters results by allowing cells to reseal. The use of surfactant poloxamer 118 increases the rate of resealing for better results as well. Extremely strong pulses can result in irreversible permeabilization.
- Metal electrodes - for longer pulses, the release of aluminum metal from the electrodes in disposable cuvettes is a concern; low-conductivity metal is sometimes recommended.
- Metal in pulsing media - Generally, calcium is to be avoided in the pulsing medium because it disrupts the intracellular levels of this ion. However, calcium or magnesium in media are often recommended for DNA transfer.
Importance
[edit | edit source]Electroporation is usually used in molecular biology to introduce a substance into a cell. This can be done with a molecular probe loading the substance into the cell, by a piece of coding DNA, or by a drug that is able to change the functions of a cell. Electroporation is highly efficient when trying to introduce foreign genes into tissue culture cells. An example is mammalian cells. This process is used in the production of knockout mice. Electroporation is beneficial because it can be used to treat tumors, as well as cell-based therapy and gene therapy. This is known as transfection, the process that is used to introduce foreign DNA into eukaryotic cells.
293T Cells
[edit | edit source]293T cells, or Human Embryonic Kidney (HEK) 293 cells, are cells that are derived from human embryonic kidney cells. These cells are very tough and can be cultured easily through tissue culture. They are normally used in transfection for research, since they are very good models for cancer cells, and normal cells. These cells also have a very high transfectability, depending on the technique used. Some techniques yield close to 100% transfection.
They are usually transfected with a gene of interest and it is allowed to grow as a normal cell. Through translation it makes proteins from the gene of interest, which can then be studied. These cells are basically test tubes in which proteins are grown.
Another use of these cells is to produce viral vectors, which can be used to easily inject genes of interest into other cells. However, viruses can be a danger since they are pathogens, so 293T cells provide the ability to delete genes that allow the viruses to replicate.
Introduction
[edit | edit source]Gram stains’ practical functions mainly revolve around medicinal uses. The use of a Gram test identifies the source of an infection. Appropriate treatments can be determined by doctors through a mere Gram test, as it can describe the type of bacteria present in a sample. Not all bacteria fall conclusively into the Gram positive and negative category, they may stain both purple and pink. Some have waxy layers in their cell walls that are not affected by the stain.
Procedure
[edit | edit source]1. Bacterial sample is applied to a glass slide and heated so as to inactivate it and prevent it from infecting the scientist. 2. Gentian violet Iodine is applied to the sample for 60 seconds. In aqueous solutions, the gentian violet dissociates into CV+ and Cl- ions. These ions have high cell wall penetration regardless if the bacteria Gram positive or negative. 3. The CV+ ion is attracted to the negatively charged portions of the cell wall and thus stains those portions purple. 4. The iodine forms a complex with the CV+ ions and forms iodine and crystal ion complexes, in both the inner and outer membranes. 5. Slide is washed under gentle water and the Gram solution (an iodine and iodine potassium mixture) is applied to the sample, this step instigates a reaction with the gentian violet solution added in the previous step. 6. The iodine washes works to identify levels of lipopolysaccharides and peptidoglycans in the bacterial cell walls. 7. At first the sample will turn dark blue, however rinse the resulting solution with ethyl alcohol will allow the color solution to fade from certain samples. In Gram negative samples, the alcohol breaks down the crystal ion complex. On the other hand, the Gram positive samples are dehydrated from the ethanol and the crystal ion complex grows to be too big and thus trapped within the thick peptidoglycan cell walls. 8. The last set of dye, red in color will be applied and the final product can finally be analyzed. 9. Gram positive bacteria produce a purple stain and represent high levels of peptidoglycan, while Gram negative bacteria are pink and are low in peptidoglycan and lipopolysaccharide concentration.
What Gram Stain Results Mean
[edit | edit source]Gram negative bacteria produce endotoxins that are the sources of cholera and typhoid. Moreover, most Gram negative bacteria are antibiotics-resistant. Gram negative bacteria have thinner cell walls that stains pink, they also possess an additional outer lipid membrane that is separated from the periplasmic space. Gram positive bacteria possess thicker mesh like cell walls rich in peptidoglycan.
Heredity and Related Experiments
[edit | edit source]Gregor Mendel’s Experiment
[edit | edit source]Gregor Mendel was a geneticist who discovered the basic principles of heredity through breeding garden peas. He chose to work with peas because of two reason. First, they were readily available in many varieties during his time, and second, he is able to control which plants mated with which. Mendel controlled their reproductive cycle by altering the biological structure of the pea plant. The reproductive system of pea plants consist of carpels and stamens. Stamens consist of pollen, which is used to fertilize the eggs found in the carpel. Since the stamen is placed right next to the carpel, a mature flower is able to self-fertilize. In his experiment, he has two varieties of flowers: purple flowers and white flowers. Mendel wanted to achieve cross-pollination in order to see if the blending model of inheritance were correct. If it were correct, then Mendel would expect to see two two varieties produce a flower that had a pale purple color, which is a mix between purple and white. To achieve cross-pollination, Mendel removed all of the immature stamens of a purple plant before they produced pollen. Then, Mendel dusted pollen from a white plant onto the altered purple flower.
The purple and white flower, which are known as the P generation (parental generation), hybridized purple flowers, which is known as the F1 generation (first initial generation). This result shows that the blending model is incorrect. Mendel allowed the F1 plants to self-pollinate to see if the white trait had somehow been removed by the purple trait. However, he discovered that the F2 generation, the offspring of the F1 generation, produced both purple and white flowers. Mendel kept records of how many of each flower there were, and he found out that the purple flowers outnumbered the white flowers by a 3 to 1 ratio. Mendel theorized that the white trait never disappeared; it was just dominated by the purple trait. He then concluded that being purple was a dominant trait while being white was a recessive trait.
His experiments lead Mendel to formulate what is now known as the Law of Independent Assortment, which states that different traits are spread to the offspring separately. Today however, we know that this only applies to certain traits while other linked traits do not assort independently. Another important formulation from Mendel's experiments is known as the Law of Segregation, which states that alleles separate during gamete formation and join again when the offspring is created. What is amazing about Mendel's experiments and conclusions is that he had no knowledge of DNA or how the genes are actually passed on, yet he was still able to form the basis of the study of genetics with his laws.
Mendelian Inheritance Model
[edit | edit source]Important Vocabulary
[edit | edit source]Character: a specific heritable feature, e.g. hair color
Trait: a variation of a character, i.e. brown hair, or black hair, etc.
Gene: called “heritable factors” by Mendel, a gene is a piece of hereditary genetic information that corresponds to a specific character, and is represented in DNA by a specific nucleotide sequence.
Phenotype: an organism’s traits, as determined by its genes
Genotype: an organism’s genetic makeup that determines its phenotype
Alleles: alternate versions of a single gene, e.g. the different colors of Mendel’s pea-flowers
“Wild Type”: the naturally occurring phenotype in a population (i.e. red eyes and functional wings in Drosophila)
Gregor Mendel’s theory for inheritance was comprised of four principal concepts. He developed his theory by growing pea plants in an abbey garden and performing controlled crosses between these pea plants. His observations during his experiments led to the following points:
1. Differences in inherited characters are caused by different alleles for the gene that represents that character.
2. All organisms get two alleles for each inherited character, one coming from each parent organism.
3. Varying alleles are either dominant (deciding an organism’s phenotype) or recessive (not visible in an organism’s phenotype, but present in its genotype)
4. In meiosis, two alleles are segregated, or separated and are located in different gametes. (The Law of Segregation)
Thomas Morgan’s Early Experiments
[edit | edit source]Thomas Morgan was an embryologist at Columbia University who studied fruit flies (Drosophila melanogaster) due to their simple maintenance and short reproductive cycle. Additionally, fruit fly chromosomes are easily spotted with a light microscope, making the technological demand on experiments relatively low for modern standards. Also, a fruit fly only has four chromosomes in each sex cell, one of which is a sex chromosome. The female fruit fly has two “X” chromosomes, while the male fruit fly has one “X” and one “Y” chromosome. This fact played an important part in Morgan’s early experiments.
One early problem with Morgan’s choice of experimental subject was the fact that variation in fruit fly population is relatively limited; the wild type fruit fly has red eyes and functional wings. Morgan was forced to breed fruit flies for generation after generation (one fruit fly generation equals two weeks) for years before observing a variation in phenotype; this new trait was white eyes. The white-eyed fruit fly was a male. This is where Morgan’s experimentation truly began.
Morgan took a wild type female and crossed (mated) it with the white-eyed (mutant) male. This was considered the parental generation (P). The resulting offspring made up the first filial generation (F1). All of the flies in this generation were red-eyed, and there was an even distribution of male flies to female flies. From this cross, Morgan hypothesized that the white-eyed trait was recessive to the wild type. Morgan, and his students developed a notation for labeling dominant and recessive traits. The wild type allele was annotated with a (+) superscript (i.e. X+), where X is the first letter of the trait being studied (e.g. “w” for white-eyes in this first experiment). Recessive alleles were labeled only by the letter, without the (+) superscript.
Following the first cross, Morgan decided to mate a F1 male with a F1 female. The resultant offspring were 75% red-eyed, and 25% white-eyed, as predicted in Mendel’s archetypal 3:1 offspring phenotype ratio. However, the 25% white-eyed offspring were all males. Fifty percent of the offspring were females with red eyes, 25% were males with red eyes, and 25% were males displaying the recessive phenotype. This led Morgan to conclude that the trait for white eyes is a sex-linked trait (traits that are linked to an organisms gender), and that the allele for eye-color was absent on the Y chromosome, and carried exclusively on the X chromosomes. In this situation, females cannot display the recessive phenotype, but can carry the allele for it. Males that receive the X chromosome containing the recessive allele do not have a second X chromosome carrying the dominant allele to mask the recessive phenotype. This is analogous to color blindness in humans, where women can carry the recessive allele for color-blindness, but only males display the recessive phenotype. If the trait for white eyes was not sex linked, the F2 generation would have been comprised of 75% red-eyed flies (half of which are male, half of which are female) and 25% white-eyed flies (half of which are male, half of which are female). See Figures Below:

A scheme of the crosses and phenotype results of Morgan's early experiment

A scheme of the crosses with genotype and phenotype results of Morgan's early experiment
Conclusion
[edit | edit source]Morgan’s experiment led him to conclude that trait for white-eyes is sex linked. Additionally, Morgan’s experiment verified Mendel’s hypothesis that genes are carried a specific loci on specific chromosomes. Additionally, Morgan observed that sex-linked traits are found in predictable patterns in subsequent generations.
Resources
[edit | edit source]Personal Notes from A.P. Biology, Lectured by Mr. Bradley Martin, CUHS, 2005.
Reese, Campbell, Biology, 7th Ed. 2005.
Berg, et al. Biochemistry, 6th Ed. 2007.
Nelson and Cox, et al. Lehninger's Principles of Biochemistry, 5th ed. 2008. Proteins with new functions can be created through directed changes in DNA. In the classic genetic approach, mutations are generated randomly throughout the genome. Analysis of mutatants reveal which genes are altered and DNA sequencing identifies the exact changes. Now, utilizing recombinant DNA technology, specific mutations can be made in vitro. New genes can be constructed by three changes to the sequence: deletions, insertions, and substitutions.
Deletions
[edit | edit source]Deletions occur when one or multiple base pairs are cleaved from a DNA sequence. A specific deletion can be produced by cleaving a plasmid at two sites with a restriction enzyme which removes a large segment of DNA. A smaller deletion can be made by cutting a plasmid at a one site and the linear DNA can be digested by an exonuclease that removes nucleotides from both strands. T
Deletion is when a base pair is removed from the DNA sequence. This causes the DNA strand to be shorter.
Substitutions
[edit | edit source]Single amino acid substitutions can be produced by oligonuleotide-directed mutagenesis. This mutation can be made if (1) a plasmid containing the gene or cDNA for the protein is obtained and (2) the base sequence around to site to be altered is known. If serine is to be changed to cystine, the code TCT needs to be changed to TGT, a point mutation. The key to this mutation is to prepare an oligonucleotide primer that is complementary to this region except that it contains TGT instead of TCT. The mismatch of one base pair out of 15 does not make a large difference. Replication of these DNA strands with the mutated primers leads to two kinds of plasmids, one with the original sequence and one with the mutated sequence.
A substitution occurs when one base pair is replaced with another. There are three main effects of substitutions in DNA:
1. The substituted base could encode a stop codon. This will stop the DNA from producing the rest of the amino acids and could lead to a nonfunctioning protein that might be harmful to the biological system or damaging to the DNA.
2. The new substituted base could encode a different amino-acid, which will make the DNA produce a different amino-acid than it was supposed to.
3. Silent Mutation- This is when the changed base still codes for the same amino acid. These mutations are rarely harmful to the DNA or biological system.
Insertions
[edit | edit source]Insertions of a DNA sequence can be made using cassette mutagenesis. In this technique, plasmid DNA is cut with a pair of restriction enzymes to remove a short segment which can be replaced by a synthetic double stranded oligonucleotide.
Insertions are the result of extra bases added into the DNA sequence.
Frame Shifts
[edit | edit source]This is caused by insertions and deletions. Deletions will make the DNA sequence shorter, while insertions will extend the DNA sequence’s length. Codons are encoded in groups of 3 bases. A deletion or insertion of bases will shift the whole sequence, and the DNA could potentially encode a whole new set of amino-acids.
Overview
[edit | edit source]Bacterial and viral genes are not the only sequences that can be introduced into a host. Eukaryotic genes can be introduced into bacteria to produce a desired protein product. It is also possible to introduce DNA into higher organisms which has led to gene therapy. Although the manipulation of eukaryotic genes holds many possibilities and advantages, it is also a source of controversy.
Cloning Eukaryotic DNA
[edit | edit source]Most eukaryotic DNA are interspersed with introns and exons that interrupt genes and cannot be expressed by bacteria. This obstacle is overcome by utilizing DNA that is complementary to mRNA (mRNA splices introns). The key to forming complementary DNA is Reverse Transcription which synthesizes a DNA strand complementary to an RNA template. Complementary DNA for all mRNA that a cell contains can be made, inserted into vectors, and then inserted into bacteria (Such a collection is called a cDNA library).
Complementary DNA molecules can be inserted into vectors that favor their efficient expression in hosts, called expression vectors. In order to maximize transcription, the cDNA is inserted into the vector near a promoter. Clones can be screened on the basis of their capacity to direct the synthesis of a foreign protein in bacteria.
Recombinant DNA in Higher Organisms
[edit | edit source]Bacteria lack the necessary enzymes to carry out posttranslational modifications. Thus, many eukaryotic genes can be correctly expressed only in eukaryotic host cells. Recombinant DNA molecules can be introduced into animal cells in several ways. First, foreign DNA molecules precipitated by calcium phosphate are taken up by animal cells. Another method involves microinjecting DNA into cells. A third method utilizes viruses, specifically retroviruses since they usually do not kill their hosts and becomes randomly incorporated into host chromosomal DNA, to introduce new genes into animal cells.
Moloney murine leukemia virus accepts inserts as long as 6 kilobase pairs and some genes introduced by this vector are efficiently expressed. Vaccinia virus is a large DNA containing virus that replicates in the cytoplasm of mammalian cells where it shuts down host cell protein synthesis. Baculovirus infects insect cells which can be easily cultured.
Plant Cell Modifications
[edit | edit source]What is Electroporation? It is the method used to introduce the foreign DNA into plant cells by helping polar molecules to insert into a host cell through the cell membranes. The process is to initiate a large electric fields to make the plasma membranes permeable to the polar molecules that are allowing the molecules to pass through the cell.
1st: the cellulose wall is removed by adding cellulase to produce protoplast.
2nd: applied electric fulses to disturb the hydrophobic membrane and make its permeable to plasmid DNA to enter.
3rd: The cell wall is then allowed to reform.
Electroporation:Tumor-inducing plasmids (Ti plasmids) carry instructions for the switch to the tumor state. Ti plasmids can deliver foreign genes into some plant cells by integrating into itself into the genome of an infected plant cells (T-DNA). Foreign DNA can be introduced into plant cells by electroporation. The cellulose walls are removed which produces protoplasts. Electric pulses are then applied to a suspension of protoplasts and plasmid DNA. The high electric fields make membranes transiently permeable to large molecules, and the plasmid DNA molecules enter the cells. When the cell wall reforms, the plant cells become viable.
Gene Guns: The most effective means of transforming plant cells fires microprojectiles at the target cells.
Genetically modified organisms create plants with beneficial characteristics such as the ability to grow in poor soils, resistance to climatic variation, resistance to pests, and nutritional fortification. However, their uses are highly controversial because of unknown side effects. There are several single molecule DNA sequencing (SMDS) techniques under development, yet only single molecule sequencing by cyclic synthesis is currently advanced to the point that sequence information is produced in a massively parallel way directly from single DNA molecules. This sequencing technology relies on the use of fluorescently labeled nucleotides by DNA polymerase into complementary strands of DNA that are immobilized to a surface. The individual DNA strands are separated by a few microns and can be monitored as independent entities. The fluorescent signal of each incorporated labeled nucleotide is then sequentially detected using fluorescent microscopy. Since each DNA molecule is sequenced separately, there is no need for synchronization between different molecules. Tens of millions of molecules can be sequenced in parallel in single small reaction volume, and thus this method readily produces high through put sequencing at a minimal cost. Currently this technique produces short reading lengths, which make it suitable to re-sequence applications in which a reference sequence is given. A single reference genome can serve as a template for the thousands of genomes produced by the short DNA fragments. This data can be used to find rare mutations and genetic heterogeneity in multiple target environments with great accuracy, high rates and low cost. The ability to extract a massive amount of sequence information will equip cancer research with a powerful tool needed to defeat various genetic diseases.
Single molecule sequencing is a goal that has been pursued for almost two decades as a possible candidate to replace the Sanger method of sequencing. DNA sequencing by cyclic synthesis (SBS) differs from the Sanger method, which relies on length separation of amplified DNA strands that terminate with a particular color according to the last base in the chain. Instead, in SBS the synthesis itself is followed by various methods, which monitor many reactions in parallel and thus accelerate sequencing rate and reduce cost. Out of all the cycle-extension approaches, single molecule sequencing has the highest sequence information density, i.e. the number of sequence reads per unit area.
Current Sanger sequencing methods require a large amount of DNA to be replicated and then each of the sequencing runs is performed on one sequence at the time, a lengthy and expensive route. The alternative that DNA sequencing by cyclic synthesis offers is the sequencing of millions of fragments in parallel, and in the case of SMDS by cyclic synthesis no duplication of the DNA is needed at all. This combination would not only make whole genome sequencing far cheaper, it would also make it a lot faster. This would allow for rapid sequencing of numerous genomes and generate useful statistical comparisons.
The basic scheme of SMDS by cyclic synthesis is as follows:
1) DNA is sheared and cut into short fragments
2) These fragments are elongated by a common DNA tail
3) The DNA fragments are immobilized onto a glass surface that contains primers that match the common DNA tail.
4) All bound fragments are then sequenced in parallel by -
4a) Polymerase extension of one base with a fluorescently labeled nucleotide.
4b) Detection by TIRM of multiple fields of view to record incorporation events on tens of millions of DNA fragments.
4c) Removal of the dye molecule.
4d) Return to 4a with a different nucleotide.
5) The data of each sequence is compared to a known sequence and aligned with it.
6) Data analysis from this alignment reveals the sequence information in the target DNA.
The sequencing of DNA using single molecule fluorescence calls for careful experimental design in order to be able to observe the incorporation of single nucleotides into the DNA template. The goal is to collect the sequence information from each molecule by itself. As multiple fields of view are imaged in order to monitor incorporations on millions of templates simultaneously, techniques that precisely monitor the position of the molecules should be addressed. The sequence information from each molecule should then be aligned to the reference sequence. For long enough sequences, it is possible to align the found sequences to the reference even if there is disagreement or ‘error’. This ‘error’ could come from either a real error in the sequencing, or from the data under analysis – i.e. the mutations, polymorphism or heterogeneity that the re-sequencing reveals. In order to have enough statistics to provide a meaningful picture of the DNA sequence, an over-sampling is required which averages out random error, and reveals the sequence content of the sample. As the amount of strands that are sequenced at the same time is enormous, this is not a strong limitation on the method.
SMDS by cyclic synthesis is a technique that minimizes cost and enhances throughput over current Sanger sequencing methods. The ability to sequence millions of bases in parallel at very high density and high data rates, without the constraint ofsynchronous incorporations, establishes this method as a viable option for massive DNA resequencing applications. Significant reductions in reagent use, combined with minimal sample preparation, contribute to lower the cost and time of the resequencing, as well as virtually eliminating the amplification biases. The microfluidic implementation of this method could reduce, even further, the cost of the reagents and of the device as a whole. Further, the use of Förster Resonant Energy Transfer as a local illumination source in single molecule sequencing by fluorescence is useful for reducing noise and false positive signals from unspecific binding of nucleotides, and is applicable in other situations where a tightly confined excitation light is desirable. The use of cleavable fluorescent markers substantially increases the read lengths in single molecule sequencing as steric interactions between adjacent dyes are eliminated. Further increase in read length is anticipated by optimizing reaction conditions and by choice of the DNA polymerase used.
References
[edit | edit source]Hebert, Benedict and Ido Braslavsky. Single Molecule Fluorescence Microscopy and its Applications to Single Molecule Sequencing by Cyclic Synthesis. http://www.phy.ohiou.edu/~braslavs/articles/Book_Chapter_Hebert_and_Braslavsky.pdf A. Introduction
One of the most significant contributions of Recombinant DNA technology is the ability to manipulate the DNA of microorganisms. Also, referred to as Recombinant DNA (rDNA) technology, this manipulation of DNA involves the insertion of DNA segments from various organisms into a different host (rDNA host). This is often practiced through the use of a vector which allows for the DNA to replicate in the host cell producing a new manipulated microorganism. Thus, the practice of the manipulation of microorganisms has been the focus of hundreds of companies, laboratories, and even governmental agencies. The reason that the manipulation of DNA microorganisms is such a studied topic is the benefits it could provide on a economic level. Possible areas of the use of manipulated microbes include as a organism for degrading environmental contaminants, pesticides, providing protection for agricultural crops, in medicine, etc. The release of genetically engineered microorganisms (GEM) however has been a much disputed topic. The worries are the effects that would result from the addition of genetically altered microorganisms to the environment. The two main concerns are 1. host cells could contain pathogenic genes in the rDNA that was originally unknown which provides threats to all living organisms. 2. the altered organisms might have effects on the ecosystem especially in terms of evolution and providing an organism an advantage of another possibly altering such things as the nutrient cycle, energy flow, and ecosystem. This is the main reason that the addition of GEM to the environment (genetically engineered microorganisms) must be carefully studied and regulated. Research though therefore has been done therefore do identify genetically altered microorganisms in the environment led by three researchers from the University of Tennessee.
B. Requirements for rDNA Environmental Monitoring
In order for a technique method/applied to observe and search for genetically engineered microorganisms in the environment, a series of questions must first be asked to determine if a method is valid and/or a realistic technique.
1. It should be applicable under a wide variety of environmental conditions 2. It should be suitable for technical application in terms of its simplicity. 3. It should be able to detect, identify and enumerate the GEMs 4. It should be sensitive and specific to detect a small population 5. It should be capable of differentiating the specific GEMS from the other organisms in the environment 6. It should be able to discriminate GEMS from other strains of the same species 7. It should be efficient, cost effective, and time-economic.
These methods reflect the rules determined by the three scientists from the University of Tennessee
C. Conventional Methods
I. Selective Plating and Enrichment Techniques
The process of selective plating of microorganisms involves the presence of a selective media. The selective media can be made differential on basis of physical growth conditions (temperature, oxygen concentration) or constituents. In terms of detecting the rDNA of microorganisms, plating only selects for the rDNA host rather than a specific rDNA sequence. Complex media are used to accommodate the complexity of rDNA host which allows for the differentiation between the non recombinant strains. Furthermore, this technique is only considered a preliminary technique because it must be considered presumptive, which requires conformation for an rDNA to be present.
II. Enumeration by the Most-Probable-Number (MPN) Method
In this method, samples are first diluted to extinction and then viable cells are allowed to grow in tubes of appropriate medium. Then, a probability theory is performed to determine the original density of the population. The medium used determines both selection or enrichment and a species growth and activity thus it is another method of differentiation. There are some drawbacks however to this method which includes the necessity to use a large number of dilutions and tubes in order for precision.
III. Epifluorescence Count Technique
The technique involves concentrating the bacteria onto membrane filters, then staining the bacteria, and then counting the bacteria by microscopy. This technique know is not applied as much for GEM because of the fact that it lacks specificity like the other conventional techniques.
Weaknesses of the Conventional Method
1. weakness is that the technique lacks specificity. 2. technique require the growth of target organisms and consequently will underestimate or produce false-negative results if organisms are stressed at a immediate time of sampling. 3. there is no universal medium that allows growth of all potential host organisms within the sample.
D. Developing Methods
I. Immunological Techniques-The technique involves the use of antibodies (either polyclonal and monoclonal antisera) providing a sensitive and specific technique for identifying GEM.
II. Enzyme-Linked Immunosorbent assay (ELISA)- This biochemical technique involves the use of a particular antibody that is used to find a specific antigen, or vice versa in which a particular antigen is used to detect a particular antibody. This process is sensitive and specific, which might reflect its value when locating GEM (genetically engineered microorganisms).
III. Radioactive Markers-This technique involves the conjugation of some radioactive species, which allows for Genetically engineered microorganisms to be marked radioactively. The main drawback though is the cost and high amounts of radioactive use.
IV. Fluorescent Markers- This technique also involves the use of a antibody that is conjugated to a fluorescent dye. The antibody is added to a sample of interest and therefore could be observed by fluorescence microscopy.
V. Use of Plasmid Epidemiology and Restriction Profiles (DNA fingerprinting)-This technique is a method to study specific genetic engineered microorganisms by specifically studying the plasmid of the microorganisms. The plasmids are separated by agarose gel electrophoresis and studied by staining the gel by ethidium bromide and observing by ultraviolet light.
VI. Use of Selectable Genotypic Markers-Two different markers can be used to study GEM
1. chromogenic markers
2. antibody or heavy resistance markers-many of these markers are found in plasmids or transposons which can be used to incorporate into bacterial chromosomes.
The markers could be placed on either the plasmid or the chromosome of the microorganism.
VII. Use of nucleic Acid Sequence Analysis
VIII. Nucleic Acid Hybridization Techniques
IX. DNA: DNA Colony Hybridization
X. Southern Blot Hybridization
XI. Nucleic Acid Hybridization with DNA Extracts
XII. DNA: RNA Hybridization
XIII. Use of Biotinlyated Probes
REFERENCES
Burlage R, Jain R, Sayler G. "METHODS FOR DETERMINING RECOMBINANT DNA IN THE ENVIRONMENT." Critical Reviews in Biotechnology, 8:1-33-84.

PCR, or polymerase chain reaction, is a technique that can be used to replicate DNA by many orders of magnitude or create mutations in DNA. PCR is a valuable technique for biochemists. For example, it allows us to study the effects of genes and segments of DNA by inducing mutations. In addition, because PCR can be used to amplify even a small amount of DNA, it is a useful tool for forensic scientists. PCR amplifies DNA quickly by thermal cycling. With a thermal cycler, temperatures can be changed constantly through many cycles. This allows a thermal cycler to denature double stranded DNA, anneal primers to it, and then activate a DNA polymerase in order to replicate the DNA. By repeating this process multiple times, DNA is replicated exponentially, allowing us to study it further.
The PCR amplification of DNA undergoes three stages:
1. DNA Denaturation: The two DNA strands are separated by heat (94oC).
2. Oligonucleotide Annealing: Two complementary oligonucleotides (primers) are added to each flanking sequence as the heated DNA strands are cooled (50-60oC).
3. DNA Polymerization: Heat-resistant DNA polymerase catalyzes 5'-3' DNA synthesis (70oC).
After amplification by PCR, the gene of interest is cleaved by restriction endonucleases and ligated to the plasmid cloning vector. The recombinant plasmid is subsequently placed in a bacterial host and propagated.
In addition to DNA amplification, PCR can create mutations and deletions in DNA as well as introduce a restriction endonuclease site.
History
[edit | edit source]
1)Denaturing at 96°C.
2)Annealing at 68°C.
3)Elongation at 72°C (P=Polymerase).
The first cycle is complete. The two resulting DNA strands make up the template DNA for the next cycle, thus doubling the amount of DNA duplicated for each new cycle.
1. Denaturation
DNA denaturation means to heat a double stranded DNA and form two separate single strands.
Basically, this process breaks the hydrogen bonds between the double helix bases in order to overcome the energy that keeps the bases so well stacked together. Many denaturation techniques exist and are possible. The most popular and used technique is by simply raising the temperature of the DNA above its melting point, Tm. The base pairs are then unstacked and can be monitored spectrophotometrically. DNA is absorbed very strongly at 260 nm, and as DNA keeps on melting, its absorbance increases because single strands are absorbed at higher wavelengths and then it stays constant once fully separated. This process is called Hypothermic Effect. This whole technique can be reversed and therefore DNA can be renatured at a certain amount of time allowing the estimation of base composition according to the time. The main biological reasons for denaturing DNA is for replication and transcription.
2. Annealing
Separated complementary strands of nucleic acids spontaneously reassociate to form a double helix when the temperature is lowered below Tm. This renaturation process is called annealing. For PCR reactions, the usual annealing time is 30-45 seconds. Increasing this time by few more minutes does not affect the results of the PCR. On the other hand, as the polymerase has some reduced activity between 45 and 65o C (temperature interval for most annealing processes), longer annealing times may increase the likelihood of unspecific amplification products. Annealing temperature is one of the most important parameters that need adjustment in the PCR reaction. Additionally, the flexibility of this parameter allows optimization of the reaction in the presence of variable amounts of other ingredients, especially template DNA. Annealing temperature is important in finding and documenting polymorphisms. Slight mismatches, (even 1 base-pair mutations) in one of sequences bound by the two primers used to amplify a DNA locus, can be detected by slight variations in annealing temperature and/or by multiplex PCR.
3. Polymerization
Polymerization is an endemic process which means energy input is required to achieve it. Also required joining of the sugar-phosphates, in addition to the nucleotides. Finally, it requires an enzyme, known as DNA polymerase. Triphosphate nucleotides make the polymerization process possible. These triphosphate nucleotides float freely within the nucleus of the cell, and each DNA base exists in a triphosphate nucleotide form. The energy that is released by the breaking of the triphosphate bond is what provides the energy for the polymerization of DNA.
PCR was an idea conceived by Kary Mullis in 1983 who, at that time, was an employee at Cetus Corporation in Berkeley, California.[2] According to Mullis, he got the idea of this technique of DNA amplification while driving his car. Cetus Corporation saw great potential in this method and took Mullis and other scientists off their work to solely concentrate on perfecting PCR.
These scientists spent a year working on PCR, but they encountered numerous obstacles.[3] One in particular was the use of DNA polymerase which was originally obtained from E. coli. However, PCR required heating to denature the DNA strands but this heating, in addition, would inactivate the DNA polymerase. Therefore, for each run DNA polymerase had to be added which, in total, took a lot of DNA polymerase to run the entire polymerase chain reaction. However, the discovery of Taq polymerase, which was purified and isolated from the bacterium Thermus aquaticus, led to dramatic improvements in the PCR technique since Taq polymerase is stable under high temperatures thus virtually eliminating the need to add DNA polymerase after each cycle and allowing the technique to be effectively automatized.[4]
In 1984, the results of the first PCR were analyzed using Southern blotting and it was shown that PCR amplified the target sequence. Cloning results also proved that PCR increased this amount while still retaining high accuracy in the final results. The patent for PCR was approved in 1987.[5] In December of 1985, Cetus started to produce commercial PCR machines and afterwards, PCR began to make its way into forensic science. In 1986, it was used by Edward Blake in the court case "Pennsylvania v. Pestinikas" and in 1989, Alec Jeffreys begins to amplify DNA from old cases thus increasing their sensitivity when tested and allowing innocent prisoners to be set free.[6]
For his contributions to DNA-based chemistry and for the development of the PCR technique, Kary Mullis was awarded the 1993 Nobel Prize in Chemistry.[7]
Variations of PCR
[edit | edit source]Quantitative Real Time PCR (qPCR): Quantitative PCR is a technique of PCR in which a DNA sample is simultaneously amplified and quantified. The general procedure is the same as the original PCR, however, in qPCR the sample of amplified DNA is detected in real time as the reaction progresses whereas in regular PCR, the sample is measured at the end of the reaction.[8] Two common methods are used in qPCR which are: (1) a use of fluorescent dyes which bind to any double-stranded piece of DNA (for example, the EvaGreen dye [9] and (2) specific fluorescent labeled probes which are detected only after hybridization with the complementary DNA strand. For both methods, an increase in measured fluorescence is proportional to an increase in DNA sample. This method of PCR is more efficient and precise than regular PCR since detection happens real time and does not require additional time running the sample on a Southern blot. There are many applications for qPCR which include, but are not limited to, the rapid diagnostic of nucleic acid diseases such as cancer and genetic abnormalities.[10]
Reverse Transcription PCR (RT-PCR): Reverse transcription PCR is a technique of PCR to amplify, detect and quantify mRNA.[11] PCR is first modified in the beginning by first converting RNA to a cDNA by reverse transcriptase. This part of the reaction may or may not be carried out in the same tube as PCR. The next step follows PCR by denaturing the double stranded cDNA, allowing primers to bind, and activation of the DNA polymerase to amplify the amount of DNA in the sample.[12] Like PCR, the products of this reaction may be viewed real time, with fluorescent dyes such as SYBER Green, or the products may be assessed post-reaction.[13] The utility of this reaction is to amplify the amount of RNA in a given sample. In addition, since the products of this reaction is the gene template for which proteins are coded from, this sequence maybe inserted into Prokaryotes for protein production, etc.
Touchdown PCR: Touchdown PCR is a modification of the original PCR technique in which the reaction is initially run at the temperature at which the annealing primers melt. In subsequent cycles, the temperature is gradually reduced, by one degrees Celsius or so, until a specific temperature, or touchdown temperature, is reached.[14] The utility of this technique is that it allows early accumulation of specific products which later serve as templates for later PCR amplification which lowers the non-specific PCR products.[15]
Inverse PCR: Inverse PCR is a variation of the original PCR technique in which one internal sequence of the DNA to be amplified is known. One of the limitations of the original PCR is that it requires primers complimentary to the end sequences of the DNA to be amplified. [16] However, the utility of this technique is that if only one site is identified, then PCR may still be carried out. This is achieved by digesting the ends of the DNA by a restriction endonuclease and ligating the two free ends of the DNA with a DNA ligase to produce a circular DNA. Then the known internal sequence is cleaved with another restriction endonuclease thereby generating a strand of DNA where the end sequences are known which allows amplification of the target through PCR.[17]
Helicase-Dependent Amplification (HDA): Helicase-Dependent Amplification is a method similar to PCR. However, HDA is different in that it relies on an enzyme helicase to split apart the DNA, rather than heat up DNA to denature it. Therefore, it is a method of PCR which operates at a constant temperature.[18] So, the methodology is similar, the helicase enzyme separates the double-stranded DNA into single strands to which primers anneal and the DNA amplified. The utility of this technique is that it may be run almost anywhere, such as at a crime scene, as opposed to PCR which is only feasible in a laboratory setting since it requires large, expensive, and bulky machines.[19] However, despite these positive qualities, HDA is not used as much as PCR since most of these types of reactions, whether from a crime scene or from a hospital, are run in labs most of the time. In addition, HDA cannot run as much samples as PCR and HDA is relatively more expensive than PCR.[20]
Nested PCR: Nested PCR is a variation of PCR in which to minimize the results of primers binding to the wrong places in the DNA sequence thereby generating contamination. This reaction is first performed with a pair of primers that bind to the DNA in various places, but of importance is that they bind on the outside of the sequence of interest to be amplified.[21] During the second cycle, a different pair of primers are used to bind to the specific sequence of DNA on the inside of the first two primers and this sequence is amplified. The reasoning behind this technique is that if a random sequence was amplified by the first primers by accident, the probability is very low that an internal sequence between the first primers would be amplified by the second primers thereby effectively minimizing contamination of side reactions.[22]
Uses
[edit | edit source]Detecting Evolutionary Relationships: PCR can be used to amplify a small amount of DNA found in fossils. This amplified DNA can be sequenced. Using the techniques found in bioinformatics, this newly sequenced DNA can be tested for similarity with other ancient organisms as well as modern organisms. These comparisons reveal what modern species the ancient species was a common ancestor of as well as what other ancient species it was closely related to. PCR removes the restriction of bioinformatics only analyzing the DNA of modern organisms and allows distance evolutionary relationships to be more easily identified. [23]
Detecting the Presence of Cancer Cells: If it is uncertain whether a trace of cancer cells exists in a tissue sample, the DNA can be isolated from the sample and purified. PCR can be performed on the purified DNA. The amplified DNA can more easily be tested for the presence of certain mutations (in growth genes) known to cause cancer. It isn't a foolproof way to detect cancer (some cancer cells might not have any of the list of "cancer causing mutations"), but the point is that PCR makes testing for the presence of a mutation possible when it would have been too small of a sample otherwise. [23]

Forensics: A small amount of DNA obtained from the somewhere at the crime scene (i.e. blood on a suspect's shirt) can be amplified using PCR and compared with the victim's and suspect's DNA. A fraction of a drop of blood or a single hair might not contain enough DNA for testing without PCR, but PCR makes testing this small amount of DNA possible. Sequencing the DNA is too laborious, so restriction enzymes are used to create a DNA fingerprint. The DNA sample is subjected to one or more restriction enzymes for a set length of time. The restriction enzymes cut the DNA at their recognition sequences, but not at every recognition sequence (they would if there was more time). If the DNA is different, the enzymes will cut different lengths of DNA and if the DNA is the same, the enzymes will cut the same distribution of lengths in both samples. The DNA fragments from each sample are run through a gel and compared. This can provide strong evidence of the innocence or guilt of a suspect. [23]
References
[edit | edit source]- ↑ Gehl, J. "Electroporation: theory and methods, perspectives for drug delivery, gene therapy, and research." Acta Physion Scand 177 (2002): 437-447.
- ↑ "History of PCR". [14]. Retrieved 2009-11-17.
{{cite web}}: External link in|publisher= - ↑ "History of PCR". [15]. Retrieved 2009-11-17.
{{cite web}}: External link in|publisher= - ↑ "The History of PCR". [16]. Retrieved 2009-11-17.
{{cite web}}: External link in|publisher= - ↑ "Kary Mullis". [17]. Retrieved 2009-11-17.
{{cite web}}: External link in|publisher= - ↑ "World of Forensic Science: Historical Chronology". [18]. Retrieved 2009-11-17.
{{cite web}}: External link in|publisher= - ↑ "The Nobel Prize in Chemistry 1993". [19]. Retrieved 2009-11-17.
{{cite web}}: External link in|publisher= - ↑ "Quantitative Real-Time PCR". [20]. Retrieved 2009-10-18.
{{cite web}}: External link in|publisher= - ↑ "Characterization of EvaGreen Dye and the implication of its physicochemical properties for qPCR applications" (PDF). [21]. Retrieved 2009-10-18.
{{cite web}}: External link in|publisher= - ↑ "ABgene QPCR Overview". [22]. Retrieved 2009-10-18.
{{cite web}}: External link in|publisher= - ↑ "RT-PCR: The Basics". [23]. Retrieved 2009-10-18.
{{cite web}}: External link in|publisher= - ↑ "RT-PCR Methodology". [24]. Retrieved 2009-10-18.
{{cite web}}: External link in|publisher= - ↑ "Quantitative Real-Time Reverse Transcriptase-PCR Analysis of Deformed Wing Virus Infection in Honeybee" (PDF). Retrieved 2009-10-18.
- ↑ "TOUCHDOWN PCR". [25]. Retrieved 2009-10-18.
{{cite web}}: External link in|publisher= - ↑ "Touchdown PCR for increased specificity and sensitivity in PCR amplification". [26]. Retrieved 2009-10-18.
{{cite web}}: External link in|publisher= - ↑ "Inverse PCR". [27]. Retrieved 2009-10-18.
{{cite web}}: External link in|publisher= - ↑ "Inverse PCR". [28]. Retrieved 2009-10-18.
{{cite web}}: External link in|publisher= - ↑ "Helicase Dependent Amplification HDA". [29]. Retrieved 2009-10-18.
{{cite web}}: External link in|publisher= - ↑ "Application of Isothermal Helicase-Dependent Amplification with a Disposable Detection Device in a Simple Sensitive Stool Test for Toxigenic Clostridium difficile". [30]. Retrieved 2009-10-18.
{{cite web}}: External link in|publisher= - ↑ "Helicase-dependent amplification". [31]. Retrieved 2009-10-18.
{{cite web}}: External link in|publisher= - ↑ "Nested PCR". [32] accessdate=2009-10-18.
{{cite web}}: External link in|publisher=|publisher=(help) - ↑ "Nested Primers for PCR". Retrieved 2009-10-18.
- ↑ a b c Biochemistry, Berg.
Structural Biochemistry/Polyermase Chain Reaction/How PCR is Performed/ Structural Biochemistry/Polyermase Chain Reaction/Uses of PCR/ DNA, deoxyribose nucleic acid, holds all of the instructions for the synthesis of amino acids in the human body and its genes are passed on from parent to offspring. In addition to that, DNA, in the future, could also be used in electronic devices to help it work faster or fit into tiny spaces that today’s large machines that no other equipment can fit into. The new DNA-based machines will use less power and less heat than current equipment. DNA’s unique properties – its size, structure and replication abilities – may allow it to reign superior to materials used to run electronic devices today.
DNA Electrical Mini Wires
DNA has a unique shape known as a double helix, in which the nucleotides of one strand of DNA twist together with its complementary strand. When the nitrogenous bases of the two strands come together, they connect to form a ladder. While the inside of the structure is a ladder, the outside are sugar molecules that connect to each other with phosphate groups. The twisting of DNA gives it a ringed structure. This structure is said to have an orderly display of electrons and form what is dubbed as “pi-ways.” Pi-ways are created by the pi-electrons in the orbitals that hover above atoms that have a ringed structure. Electrons can travel through these pi-ways and conduct electricity. The ability of DNA to conduct electricity could be useful in miniature machines.
DNA Mini-Robots
In the future, factories will be filled with miniature robots instead of humans that will perform repetitive tasks. Nadrian Seemen of New York University in New York City designed a tiny robot, too small to even be seen by the microscope, out of DNA synthesized in the laboratory. He started with a synthetic DNA molecule he calls DNA DX, which has a shape that is rigid enough to be the robot's arm and in addition, looped together three different DNA pieces using enzymes. Seeman’s DNA robot contains strands of DNA that retain its properties, such as the natural twists and turns that occur in the molecule. Since the robot is too small to see through a microscope, fluorescent tags and telltale glows are added to each part of the DNA to monitor when the molecules are close apart. If they are close apart, glowing will occur, if not there will not be any glowing.
DNA Biosensor
In the future, DNA biosensors could be constructed to detect errors in the connection of the two complimentary strands of DNA. The DNA biosensor will be a tiny square chip consisting of sensing elements (a probe), a signal generator, and microlasers. The biosensor will glow when two strands match up at every nucleotide position. Ideally, when immersed in a fluid containing DNA it will detect disease-causing viruses and bacteria.
DNA Computers
A prototype for the first DNA computer was created in 1994. Creating a DNA computer makes sense because evolution has shown that DNA has been selected to hold the instructions for life. In addition, there is mathematics involved in how DNA is passed on from generation to generation, thus it is reasonable to create a DNA computer that will solve complicated mathematical problems. A regular computer would use algorithms and find all the possible routes to get to an answer if given a mathematical problem, while DNA computers will use DNA to create strands of the variables given in the problem and link them together to receive the correct answer without having to go through all of the possible ways to solve the problem.
Introduction
[edit | edit source]The causes of aging and ways to prolong life have always been an interest to many people. Through trials and errors, there had been many theories put forth and denied or accepted by people regarding the phenomenon of aging. Nowadays, there is a general agreement that aging is in fact a multi-factorial process that is not created by some genetic program but is influenced by certain genes.
There are now theories that specific mutations could prolong the life span drastically. For example, the experiment of reduced insulin signaling prolongs the life span of worms, flies and mice. The reduced insulin signaling suggests that it could be a conserved longevity pathway that came about evolutionarily.
For human beings, it is found that the mitochondrial dysfunction heavily impacts the multi-factorial aging process. Aging is a result of the increased levels of somatic mtDNA mutations because those mutations undergo clonal expansion. The expansion then forces mosaic respiratory chain deficiency in various tissues like the heart, brain, skeletal muscles,, and gut, thus showing that somatic mtDNA mutations and mosaic respiratory chain dysfunction are somehow implicated in the multi-factorial aging process. It has been shown experimentally that the increased levels of somatic mtDNA mutations could cause premature aging.
mtDNA
[edit | edit source]mtDNA, short for mitochondrial DNA, is strongly associated with human aging because of the damages it acquire over time as well as the results of clonal expansion and mosaic respiratory chain deficiency. The two strands of mtDNA are categorized by their different base compositions as the heavy strand and the light strand.
mtDNA’s replication, transcription, and translation processes are encoded by nuclear genes and then imported into the mitochondria where the mitochondria’s own ribosomes take charge. It is important to note that the replication, transcription, and translation processes of the mtDNA is very different from the same processes of DNA: in fact, the processes take place within the same mitochondrial matrix without separation. Hence, there is coupling involved when transcription and translation takes place.
mtDNA Transcription
[edit | edit source]Transcription of the mtDNA is extremely necessary for the gene expression of mtDNA. It also produces the RNA primers needed to begin the initiation of the mtDNA replication at the origin of the heavy strand (as indicated in the next section: mtDNA Replication). For mtDNA transcription to occur, there are three major factors that are sufficient and necessary:
1. Mitochondrial RNA polymerase, known as POLRMT.
2. Mitochondrial transcription factor B2, known as TFB2M, is a paralog of TFB2m, meaning that they are homologs that are in the same species but have different functions. However, TFB2M holds no role in mtDNA transcription but is essential for the integrity of the subunit in the mitochondrial ribosome.
3. Mitochondrial transcription factor A, known as TFAM, is a protein that is extremely necessary for transcription initiation as without it, there is no transcription process taking place. It is also responsible for packaging mtDNA.
mtDNA Replication
[edit | edit source]There are two models that depict mtDNA replication. The first model that described mtDNA replication for mammals is called the asymmetric replication model because it is based on the studies done by the electron microscopy, biochemical characterizations, and pulse-chase labeling experiments that involved replication intermediates and nucleic acids respectively. The importance of this model lies in the fact that it is dependent on an RNA primer formed by transcription as well as the initiation of the leading-strand replication occurs at the origin of the heavy strand. When the initiation of the leading-strand is two-thirds complete around the mtDNA circle, the initiation of the lagging-strand replication happens as the origin of the light strand is activated.
Another model that recently came about argues that the mtDNA replication occurs with the coupling of the leading-strand and lagging-strand synthesis. It also stressed that the ribonucleotide on the lagging strand plays an important role. Although there are two models that are still debated by scientists nowadays, it is important to note that both models predict an involvement of a limited number of enzymes.
References
[edit | edit source]Larsson, Nils-Goran. “Somatic Mitochondrial DNA Mutations in Mammalian Aging.” 2010. Annual Review of Biochemistry. Adenosine triphosphate (ATP) is a nucleotide that consists of an adenine and a ribose linked to three sequential phosphoryl (PO32-) groups via a phosphoester bond and two phosphoanhydride bonds. ATP is the most abundant nucleotide in the cell and the primary cellular energy currency in all life forms. The primary biological importance of ATP rests in the large amount of free energy released during its hydrolysis. This provides energy for other cellular work, such as biosynthetic reactions, active transport, and cell movement. ATP is used in cellular metabolism in plants. It involved with light to create energy for plant. Besides, ATP is also one of components of DNA.
Definition
[edit | edit source]
ATP also known as adenosine 5'-triphosphate. It is formed from adenosine diphosphate (ADP) and orthosphosphate (Pi). When fuel molecules are oxidized in chemotrophs or when light is trapped by phototrophs. This nucleotide is tremendously important since it is the most commonly used energy currency. The energy is released from the cleave of the triphosphate group is used to power many cellular processes.[1]
Physical and Chemical Properties of ATP
[edit | edit source]ATP is composed of an adenine ring, ribose sugar, and three phosphate groups (triphosphate). The groups of the phosphate group are usually called the alpha (α), beta (β), and gamma (γ) phosphates. It is typically related to a monomer of RNA called adenosine nucleotide. Gamma phosphate group is the primary phosphate group on the ATP molecules that is hydrolyzed when the energy is needed to drive anabolic reactions. Basically gamma phosphate is typically located the farthest from the ribose sugar and has a higher energy of hydrolysis than either that of the alpha and beta phosphate. The bonds that are formed after hydrolysis or the phosphorylation of a residue by ATP are lower in energy than that of the phosphoanhydride bonds of ATP.
ATP is very soluble in water and is a quite stable solution that has a pH of 6.8-7.4, but is rapidly hydrolysed at extreme pH. Thus, ATP is best stored as an anhydrous salt.
Although, ATP is quite stable in solution, it is an unstable molecule in unbuffered water. This is because, once ATP gets in contact with unbuffered water, it hydrolyses to ADP and phosphate due to the strength of the bonds between the phosphate groups in ATP is commonly seen to be less than the strength of the hydrogen bonds (hydration bonds) between its products (ADP + phosphate) and water. Therefore, if ATP and ADP are in chemical equilibrium in water, almost all the ATP will form into ADP because of the reaction that will occur. Gibbs free energy is when a system is far from equilibrium and it is able to do some kind of work. It is seen that typical living cells maintain the ratio of ATP and ADP at a point ten orders of magnitude from equilibrium. However, this may only occur if ADP is thousand fold lower in concentration than that of ATP. This shows that hydrolysis of ATP in cells usually release a large amount of free energy in reaction.
However, even with releasing a large amount of free energy during reaction, any unstable system of potentially reactive molecules could potentially serve as a way of storing free energy. This is only if the cells maintain their concentration far from the equilibrium point of the reaction. However, the idea of both energy-release and entropy-increase always occur during the breakdown of RNA, DNA, and ATP into simpler monomers.
In an ATP molecule, two high-energy phosphate bonds called phsophoanhydride bonds are responsible for high energy content of this molecule. Based on biochemical reaction, these anhydride bonds are often referred to as high-energy bonds. Also the released of hydrolysis of the anhydride bonds can happen in the energy stored ATP.
Binding to Proteins
[edit | edit source]Rossmann fold is a type of protein fold that some proteins and ATP bind together as. This characteristic protein fold is a general nucleotide-binding structural domain that can also bind the coenzyme NAD. Kinase is the most common ATP-binding protein. They share a small number of common folds and there biggest kinase superfamily all share common structural features specialized for ATP binding and phosphate transfer.
ATP also requires the presence of a divalent cation that is almost as magnesium as a metal used. This metal binds to the ATP phosphate groups. This metal ion can also serve as a mechanism for kinase regulation. The presence of magnesium greatly decreases the dissociation constant of ATP from its protein binding partner without even affecting the ability of the enzyme to catalyze its reaction once the ATP has bound.
Intracellular ATP
[edit | edit source]Intracellular ATP hydrolysis is catalyzed by intracellular ATPases. For example, the (Na+-K+)-ATPase located in the plasma membranes of higher eukaryotes drives active transport of Na+ and K+ coupled to ATP hydrolysis, and generates electrochemical gradients across the cell membrane. Another important intracellular ATPase is myosin. The myosin heads form the cross-bridges to thin filaments in intact myofibrils and its ATP-powered movement is responsible for muscle contraction.
Extracellular ATP
[edit | edit source]ATP is also present in extracellular spaces in nanomolar to micromolar concentrations, which are 3-6 orders of magnitude lower than intracellular ATP concentration (1, 2). ATP is released from cells to extracellular spaces by regulated exocytosis or plasma membrane channels (Figure 1). Regulated exocytosis is an important process used to release substances such as hormones or neurotransmitters from the cell and is triggered by an increase in cytoplasmic Ca2+ concentration (2, 3). ATP efflux also occurs through plasma membrane conductance channels, transporters, or constitutive secretory pathways as residual cargo products (2). Extracellular ATP acts as a neurotransmitter and an autocrine/paracrine chemical messenger in non-neural tissues. Its effects are mediated by the P2 purinergic receptors and elicit a variety of physiological responses, such as neurotransmission, regulation of secretion, modulation of immune functions, pain transmission, apoptosis etc.
P2 receptors consist of two major subfamilies, P2X and P2Y. P2X receptors are ligand-gated ion channels and P2Y receptors are G protein-coupled receptors. The concentration of extracellular ATP is regulated by its hydrolysis that is catalyzed by extracellular ATPases. Thus the physiological responses mediated by the purinergic receptors are modulated by extracellular ATPases (Figure 2). For example, Sesti et al. reported that ATP modulates norepinephrine release from cardiac sympathetic nerve endings and this action of ATP is controlled by purinergic receptors in cardiac synaptosomes and modulated by extracellular ATPases (4). Di Virgilio et al. reported that a potent platelet aggregating factor is ADP and its amount is regulated by the activity of extracellular ATPases on endothelial cells (5). However, the precise relationship of multiple P2X and P2Y receptor subtypes to extracellular ATPases remains to be determined.
Exergonic Reaction
[edit | edit source]The role of ATP is an energy-rich molecule because its triphosphate unit contains two phosphoanhydride bonds. Large amounts of free energy is liberated when ATP is hydrolyzed to adenosine diphosphate (ADP) and orthosphosphate (Pi) or when ATP is hydrolyzed to adenosine monophosphate (AMP) and pyrophosphate (PPi). The precise for these reactions depend on ionic strength of the metal such as Mg 2+. The free energy is liberated in hydrolysis of ATP is harnessed to drive reactions that require an input of energy for muscle contraction. The formation of ATP from ADP and Pi is known as the ATP-ADP cycle is the fundamental mode of energy exchange in biological systems. It is intriguing to note that although, all nucleotide triphosphates are energetically equivalent, ATP is the primary cellular energy carrier. Under cellular conditions, the hydrolysis of ATP shifts the equilibrium of a coupled reaction by a factor of 108[2]

Phosphoryl Potential
[edit | edit source]ATP has a particularly efficient phosphoryl-group donor that can best be explained by features of the ATP structure:
Resonance Structures. ADP and Pi have greater resonance stabilization than does ATP. Orthophosphate has multiple resonance forms of similar energy whereas the phosphoryl group of ATP has a smaller number due to its unfavorability of the positively charged oxygen atom that is adjacent to a positively charged phosphorus atom.

Electrostatic Repulsion. At pH 7, triphosphate unit of ATP carries four negative charges which repel one another due to their close proximity. The repulsion between them is reduced when ATP is hydrolyzed.
Stabilization Due to Hydration. More water can bind effectively to ADP and Pi than can bind to the phosphoanhydride part of ATP, stabilizing the ADP and Pi by hydration. [3]
Consumption of ATP
[edit | edit source]The large amounts of energy provided by the hydrolysis of ATP are necessary to overcome the large free energy changes necessary to create the large macromolecular proteins. The cleavagle of the phosphoanhydride bonds in ATP provides the source for free energy to make biological reactions spontaneous (negative free energy). Because the amount of entropy of the universe is continually increasing it is unfavorable for large macromolecules to form without the use of ATP. Because of this, the free energy generated by the ATP is always immediately consumed by nearby endergonic (energy-reguiring) biological reactions. The exergonic reaction of the ATP is only able to proceed if it is coupled to an endergonic reaction, otherwise thermodynamic equilibrium would not be obtained. The consumption of ATP proceeds with the first step of having an enzyme attache an amino acid to the a-phosphate of ATP. This results in the release of a pyrophosphate. This release is called an aminoacyl-adenylate intermediate. The reaction then proceeds to the enzyme catalyzing transfer of an amino acid to one of two -OH locations on the ribose portion of the adenosine residue. ATP is able to release energy into cells because cells maintain a concentration of ATP that is far higher above the equilibrium concentrations. The high concentration of ATP allows it to be the main provider of driving endergonic reactions in cells. This coupling of energy releasing and consuming systems through a common intermediate is vital to energy exchange in living systems. [4] = Lehninger | firs = Albert | authorlink = |Nelson, David L. and Michael M. Cox | title = Lehninger principles of biochemistry, 4th ed | publisher = W.H. Freeman & Co | date = 2007 | location = New York, New York | pages 22–25 | isbn = 0-7167-4339-6 }}</ref>
Importance of Oxidation of Carbon
[edit | edit source]Formation of ATP
[edit | edit source]ATP is a principal immediate donor of free energy in biological systems meaning that it is consumed within a minute of it formation. The carbon in fuel molecules such as glucose and fats are oxidized to CO2 and the energy released is used to regenerate ATP from ADP and Pi. Oxidation in fuel takes place one carbon at a time and the carbon-oxidation energy is used in some cases to create compounds with high phosphoryl-transfer potential and other cases to create ion gradient as well with the end formation of ATP.[5]
Coupling with Carbon Fuels
[edit | edit source]ATP is coupled with oxidation of carbon fuels directly and through the formation of ion gradients. Energy of oxidation is initially trapped as high-phosphoryl-transfer potential compound and then used to form ATP. In ion gradients the electrochemical potential, produced by oxidation of fuel molecules or by photosynthesis, which ultimately powers the synthesis of most ATP in cells. ATP hydrolysis can be used to form ion gradients of different types and functions.
Energy from Food
[edit | edit source]Described by Hans Krebs the three stages in generation of energy from oxidation of foodstuffs:
1. Large molecules in foods are broken down into smaller units in a process known as digestion. Proteins are hydrolyzed to their 20 different amino acids, polysaccharides are hydrolyzed into simple sugars and lastly fats are hydrolyzed to glycerol and fatty acids.
2. Numerous small molecules are degraded to a few simple units that play a central role in metabolism. Sugars, fatty acids, glycerol and several amino acids are converted into the acetyl unit of acetyl CoA. Some ATP is generated but not a substantial amount.
3. ATP is produced from the complete oxidation of acetyl unit of acetyl CoA. Final stage consist of citric acid cycle and oxidative phosphorylation which are the final pathways in oxidation of fuel molecules. Acetyl CoA brings acetyl units into the citric acid, where they are completely oxidized to CO2. Four pairs of electrons are transferred for each acetyl group that is oxidized. Then a proton gradient is generated as electron flows from the reduced forms of these carriers to O2 and the gradient is used to synthesize ATP.[6]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
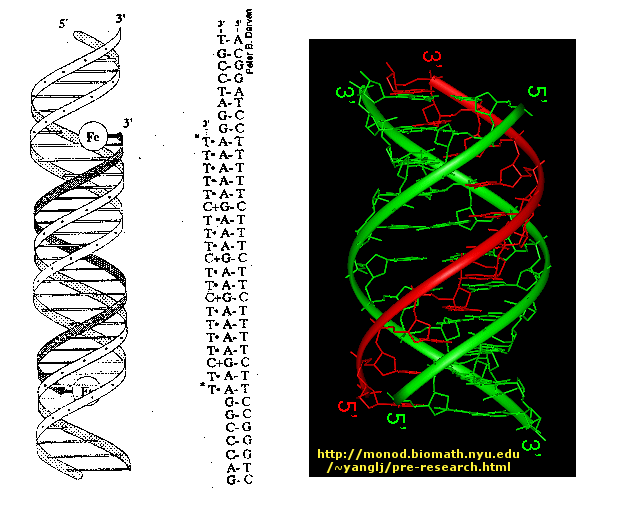{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
![[33]](http://stemcells.nih.gov/StaticResources/info/scireport/images/4_3.jpg){kind=link}
{kind=link}
![[34]](http://dels.nas.edu/ilar_n/ilarjournal/38_3/38_3Richardsonfig1.jpg){kind=link}
{kind=link}
![[35]](http://dels.nas.edu/ilar_n/ilarjournal/38_1/38_1Gordonfig2.jpg){kind=link}
{kind=link}
![[36]](http://dels.nas.edu/ilar_n/ilarjournal/38_1/38_1Gordonfig3.jpg){kind=link}
![[37]](http://stemcells.nih.gov/staticresources/images/figure1_lg.jpg){kind=link}
![[39]](http://stemcells.nih.gov/staticresources/images/figure4_lg.jpg){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
![[80]](https://en.wikipedia.org/wiki/File:Diagram_of_M13_filamentous_phage,_Oct_2012.jpg%7Clink){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
![[84]](https://commons.wikimedia.org/wiki/File:Catabolism.svg){kind=link}
References
[edit | edit source]- ↑ Berg, Jeremy (2007). Biochemistry, 6th Edition. New York, New York: Sara Tenney. pp. 110–111. ISBN 978-0-7167-8724-2.
{{cite book}}: Unknown parameter|coauthors=ignored (|author=suggested) (help) - ↑ Berg, Jeremy (2007). Biochemistry, 6th Edition. New York, New York: Sara Tenney. pp. 413–415. ISBN 978-0-7167-8724-2.
{{cite book}}: Unknown parameter|coauthors=ignored (|author=suggested) (help) - ↑ Berg, Jeremy (2007). Biochemistry, 6th Edition. New York, New York: Sara Tenney. p. 415. ISBN 978-0-7167-8724-2.
{{cite book}}: Unknown parameter|coauthors=ignored (|author=suggested) (help) - ↑ Biochemistry, 6th Edition. New York, New York: Sara Tenney. 2007. p. 110. ISBN 978-0-7167-8724-2.
{{cite book}}:|first=missing|last=(help); Unknown parameter|=ignored (help); Unknown parameter|coauthors=ignored (|author=suggested) (help) - ↑ Berg, Jeremy (2007). Biochemistry, 6th Edition. New York, New York: Sara Tenney. p. 417. ISBN 978-0-7167-8724-2.
{{cite book}}: Unknown parameter|coauthors=ignored (|author=suggested) (help) - ↑ Berg, Jeremy (2007). Biochemistry, 6th Edition. New York, New York: Sara Tenney. pp. 419–420. ISBN 978-0-7167-8724-2.
{{cite book}}: Unknown parameter|coauthors=ignored (|author=suggested) (help)
1. Schwiebert, E. M. ABC transporter-facilitated ATP conductive transport. Am. J. Physiolo., 1999, 276, C1-C8.
2. Lazarowski, E. R., Boucher, R. C., and Harden, K. T. Mechanisms of release of nucleotides and integration of their action as P2X- and P2Y-receptor activating molecules. Mol. Pharmacol., 2003, 64, 785-795.
3. Theander, S., Lew, D. P., and Nüße, O. Granule-specific ATP requirements for Ca2+-induced exocytosis in human neutrophils. Evidence for substantial ATP-independent release. J. Cell Sci., 2002, 115, 2975-2983.
4. Sesti, C., Broekman, M. J., Drosopoulos, J. H., Islam, N., Marcus, A. J., and Levi, R. Ectonucleotidase in cardiac sympathetic nerve endings modulates ATP-mediated feedback of norepinephrine release. J. Pharmacol. and Exp. Ther., 2002, 300, 605-611.
5. http://en.wikipedia.org/wiki/Adenosine_triphosphate
6. Di Virgilio, F., Chiozzi, P., Ferrari, D., Falzoni, S., Sanz, J. M., Morelli, A., Torboli, M., Bolognesi, G., and Baricordi, O. R. Nucleotide receptors: an emerging family of regulatory molecules in blood cells. Blood, 2001, 97, 587-600.
Different Views of Functions of Gene and Gene Products
The concept of function is taken for granted when applying to genes and gene product. People tend to misunderstand, or simplify the concept of function of genes and gene products in terms of different circumstance. Neil S. Greenspan gives us several well organized ways to distinguish the function attribution of gene and gene products in different aspects.
Biochemical function vs. genetic function
Biochemical function: The basis of attribution of biochemical function is merely the behavior of authentic gene product without considering the biological environment. For example, isolating a protein X (which is the “without considering the biological environment”) to detect its particular catalytic activity (which is “the behavior of authentic gene product”). Biochemical function refers to a relatively narrow view of the gene product’s function, as it is determined only by the effect mediated directly by gene product (Greenspan 293).
Genetic function: The basis of attribution of genetic function is the behavior of the whole system (often refer to as cellular or organismal systems) in presence or absence of wild type gene product. For example, when modifying DNA sequence (which is “the presence of absence of wild type gene product”), we could possibly alternate the phenotype of individual cell, or even the whole organism (which is “the behavior of the whole system”). In this example, biological context is required, which is the opposite of biochemical function attribution. Genetic function is a relatively board view of the gene product’s function, as it is determined by looking at the whole system with/without the wild type gene. Genetic function is commonly seen in genetic engineering. For example, when studying behavior of IFN-γ to inflammation, wild-type mice and mice with different concentration of IFN-γ are compared in terms of the level of inflammation. This is a typical example of genetic function attribution, because the experiment needs biological environment, and is comparing the whole systems’ behavior in presence or absence of wild type gene product.
Other senses of function attributed to genes or gene products
Sense1. Function should be the positive contribution to cellular or organismal function. According to this sense, the function attributed to gene or gene products should lead towards survival and reproduction of the associated cell of organism. In another word, this positive function attribution should be parallel to favor evolutionarily adaption.
Sense2. Function should be determined by the net effect of particular gene product on cell or organism. Furthermore, this function attribution is divided into 2 parts: (i) when compare with absence of gene product (ii) when compare with allelic form. This sense is similar with the idea of genetic function. For example, when studying the sickle cell allel at the β-globin locus, we aren’t only look at the heterozygous form, but also the homozygous form, this net effect of combination of sickle cell’s heterozygous form and homozygous form determines the function of gene product encoded by the sickle cell’s allel at the β-globin locus.
Relational nature of molecular function
The classification of function of gene and gene product can be more complicated. Greenspan states that the effects attributable to a given protein or RNA molecule can vary in different cellular contexts. (294) For example, depend on cell type and range of stimuli received by the cell, activation of NF-kB can generate different responses. When people simplified the molecular function, they often make the “fundamental attribution error” when only consider the intrinsic properties without considering cellular environment, because different biological environment might cause completely different effects. This also explain why genetic function is more often used than biochemical function, because it consider the entire systems’ effect rather than the individual gene product’s function when isolated.
The crucial point of molecular function is “the interaction between molecules or between forms of energy and molecules”(Greenspan 295). Therefore, there is a difference between function attributes to isolated gene product and function attributes to gene product in particular context (when interact with other molecules). For example, when modifying gene encoding XR receptor to form a different structure, the hormone or growth factor X no longer works, although the isolated X doesn’t change the structure and the function attributes to isolated X doesn’t change. The function attributes to the whole system (when X interact with XR) change. Therefore, the molecular function also change.
Protein thermodynamics & function
The technique of protein thermodynamics can help us to determine functional contributions of individuals alleles to cellular or oganismal phenotypes. Basically, the technique requires protein-ligand binding, when applying mutagenesis to change ligand recognition. Affinity and free energy will change, from database of properties of different amino acids, protein thermodynamic can assess us to determine the functional or phenotype of interest. The most important advantage of protein thermodynamics is that it allows us to obtain various amino acids at various positions by looking up at the quantitative database.
By using thermodynamic contribution, scientist determined that “thermodynamic contributions of individual amino acids change with single point mutations at other positions in the same polypeptide chain” (Greenspan 295). This idea illustrates that when we change the amino acids of certain gene sequence, although genotype changes, phenotype may not change. Furthermore, Lim and Sauer figured out the in order to keep normal function of certain amino acids, another amino acid is needed in another position. However, this amino acid does not need to be the one in wild-type. For example, in order for methionine at position 36 to function normal, isoleucine needs to occupy position 51, while phenyalanine, which is in wild-type, is not essential. This is a good example to illustrate that there is functional flexibility even if some amino acids are substituted. Protein thermodynamic allows us to determine functional contribution of given structural element by tracking the properties of different amino acids at various positions.
Mathematical functions of gene vs. gene product
Gene and Gene products’ function are often not determined by single factor, thus multiple factors are required. Therefore, the function of gene product can be viewed as a mathematical function of multiple variables. These variables could be temperature, ionic strength, pH and so on.
In terms of the mathematical functions, the function of gene is more complex of the function of gene product, because one gene can generate different gene product by mRNA splicing (which is the abandoned of different introns and recombination of different entrons).
References
[edit | edit source]1. Greespan, N.S. (2011) Opinion-Attributing functions to genes and gene products. Trends in Biochemical Sciences Vol.36, No.6
Presence of 30nm Chromatin fibers, essential?
[edit | edit source]It is essential for eukaryotic genomes to be folded and compacted to fit within the restricted volume of the nucleus. A question arise from this that whether 30nm chromatin fiber must be present or not to constitute the template for active genomic material.
30nm chromatin fiber models
[edit | edit source]The molecular model of the 30nm fiber includes the one-start solenoid, two-start helix zigzag, the cross-linker, and supranucleosome. These models were based on EM of buffer-extracted chromatin fibers and micrococcal nuclease-digested material.
One-start solenoid
[edit | edit source]Solenoid model shows that 10nm fiber coils around a central axis of symmetry with nucleosome which is packed face-to-face and the six or seven nucleosome is packed every helical turn. This model is supported by a molecular tweezers experiment that provides sub-pico Newton force resolution. From the experiment, a conclusion is drawn that the 30nm fiber follows a regular helical structure.
Two-start helix zigzag
[edit | edit source]This model predicts zigzagging pattern of two nucleosomes. Later, the zigzagging of two nucleosomes will turn coil into a helical conformation. This model is demonstrated by the crystal packing of the structure of the tetranucleosome.
Cross-linker
[edit | edit source]It is similar to the two-start helix zigzag model but what is different is the linker DNA crisscrosses back and forth across the helical axis. This model requires folding complexity and precision in linker length and nucleosome spacing.
Supranucleosome model
[edit | edit source]The Supranucleosome model requires clumps of nucleosomes which are separated by linker sequences.
Existence of 30nm fibers in interphase nuclei
[edit | edit source]The 30nm fiber was observed in starfish sperm by EM studies and ESI (electron spectroscopic imaging). It was shown that 10nm fiber was folded and twisted into a fiber and the fiber was about 30nm in width. However, cryo-EM studies demonstrated that the 30nm fibers were not present in the fully condensed chromatin, and it was proved by the absence of a 30nm peak in the power spectrum.
Techniques that prove the existence of 30nm fibers in interphase nuclei
[edit | edit source]EM
[edit | edit source]It relies on heavy metal contrast agent, and thus is hard to visualize chromatin fibers.
ESI
[edit | edit source]It is a high-contrast technique and since it does not rely on the heavy metal contrast agent, but on the only electrons that have interacted with specific elements. Most of the chromatin is observed as 10nm fibers rather than 30nm. This technique proves that majority of genome is compacted into 10nm chromatin fibers.
3C technique
[edit | edit source]This technique utilizes the chromosome conformation capture. Based on this technique, it was also observed that the yeast genome was not compacted into 30nm fiber, but instead as an extended fiber. However, this technique is quite limited to the long-range chromatin interaction from a locus of interest.
FISH(fluorescence in situ hybridization)
[edit | edit source]This technique uses fluorescent probes that bind to only those parts of the chromosomes. This technique lacks high resolution to exclude a highly bent and kinked 10nm fiber from a less bent or kinked 30nm fiber.
Conclusion
[edit | edit source]There are variety of models and techniques to observe the presence of the 30nm fibers but it has been more observed that highly compacted chromatin fiber like 30nm fibers are not necessarily present for any gene regulation such as folding of DNA. Instead, 10 nm chromatin can be condensed enough into compacted domains through frequent bending and making 10nm fibers close to each other. In other words, it does not require to have 30nm fibers but is sufficient to have 10 nm chromatin fibers that is organized in genome to explain the complexities of nuclear organization and gene regulation.
References
[edit | edit source]Fussner E, Ching RW, Bazett-Jones DP. "Living without 30nm chromatin fibers." http://www.ncbi.nlm.nih.gov/pubmed?term=Living%20without%2030nm%20chromatin%20fibers.