macromolecules in living organisms; they are what act out the duties that are encoded in genes. In humans they help our bodies to repair, regulate, and protect themselves. Proteins help in the building and repair of tissues, and in body processes such as water balancing, nutrient transport, and muscle contractions. Many essential enzymes and hormones are proteins. Proteins are basically essential for life. The reason that proteins can carry out such a diverse set of functions is because they are able to bind to other proteins specifically and tightly. Their binding ability can be contributed to their tertiary structure that creates a binding or active site; the chemical properties of the surrounding amino acids' side chains also have a large influence on the binding ability of proteins.
Proteins acting as enzymes are probably their most important function. Enzymes are the biological catalysts that are essential for almost all the biological systems in our bodies to work, they are what catalyze reactions in processes like metabolism, DNA replication, and digestion. Enzymes are extremely specific and will only catalyze certain reactions. The high specificity is related to the structure of the substrate and the enzyme. The enzyme will bind only to an active site only in the substrates which is complementary to its structure, like a key in a lock. Protein-protein interactions regulate this enzymatic activity.
Proteins are also essential for cell signaling and molecular transport systems. Because a protein produced by one cell can bind with a protein from another cell, they provide good cell signal and molecular transport pathways. An example of a protein that acts in this fashion is hemoglobin. Hemoglobin binds iron molecules and transports them from the lungs, through the blood stream, to all the essential organs and tissues. This examples shows how essential proteins are in living systems.
Structure of Hemoglobin
There are also structural proteins such as actin and tubulin that polymerize to form the cytoskeleton of a cell. The essential motor proteins such as myosin, kinesin, and dynein are also structural proteins.
Structural protein are essential for providing structure and rigidity to fluid biological cells and components. Structural proteins are fibrous proteins which provide support for the cells. Structural proteins are usually very large and are made up with up to thousands of amino acids. Insects and spiders use silk fibers to for various tasks such as making their cocoons and webs [1]. Another example of a structural protein can be seeing in Keratin which is the protein of hair, feathers and horns among other things [1]. Actin and collagen are specific examples that fall under this category of proteins. Collagen, recognized as one of the most abundant proteins in mammals, is the main component in connective tissue. Collagen can be found in the tendons, ligament and skin. Collagen can also be found abundant in cornea, cartilage, bone, and blood vessels. Collagen composes about 25-35% of the entire protein content in the human body, which illustrates the importance of structural proteins such as this in the body.
Not only proteins serve an important role as the structural components, it also participates in all of the biological processes. For example, transport, storage, catalytic reactions, immunity, nervous system, growth, and etc. There are four general properties that allow proteins to function in such wide varieties:
Protein is formed with monomers called “amino acids” and it is connected from one end to the other, becoming a linear polymer. The unique sequence of the amino acids causes the chain to fold into three dimensional shapes called “protein,” which the function is also determined by the shape. Because of the endless possible sequences of the amino acids, the folded protein world is also capable of intense diversity and varieties.
Protein carries many kinds of functional groups such as, thiols, alcohols, carboxylic acid, and etc. Because these functional groups are reactive, it gives the protein wide range of reactive properties.
Protein’s ability to interact with other macromolecules also increases the range of functions. Unlike other macromolecules, the ability to interact allows the proteins to develop into complex assembly with other molecules.
The combination of rigidity and the flexibility of the protein is another reason for its usefulness in biological structures. Flexible proteins may work as a spring, hinge, and a lever while ridge proteins can play a role in cytoskeleton structures.
One of the functions of proteins is to bind different molecules together. A ligand is a molecule that is recognized by a protein and is able to bind to the target protein. The site at which the ligand binds to the protein is called the ligand-binding site. The ligand-binding site on the protein is quite flexible, making it easier for the ligand to bind to it. Ligand-binding sites are complementary to the protein to which it binds to. As expected, shape plays a significant role in fitting the ligand to the protein. In addition to that, the charge of the ligand and protein also plays a role.
Similar to the ligand-binding site, an active site is a cavity in the protein surface to which enzymes bind to. The active site is surrounded by amino acids that have the highest affinity to the enzyme that will carry out the reaction. Once again, the shape, charge and polarity of the amino acids affect the binding effects of the enzymes.
There are three models for how an enzymes fits into the active site: the lock-and-key model, the induced fit model, and the transition state model. The lock-and key model assumes that the active site is a perfect fit for the enzyme. This model is a more rigid model that does not allow any modification of the active site or the enzyme. The induced fit model is a derivation of the lock-and-key model which still assumes that an active site is designed specifically for the recognition of one enzyme but both the active site and enzyme are flexible and can slightly modify to create the perfect fit. In the transition state model, the active sites binds to the enzyme in its transition state. This effectively lowers the activation energy needed for the reaction to be carried out.
Note: Above is a phosphate-binding protein
In summary, the properties of proteins that affects the ability of enzymes to bind to it are its flexibility, complementarity, surfaces and non-covalent forces. The flexibility allows an easier fit between binding sites and enzymes. The complementarity and surfaces are important factors that contribute to the specificity of an enzyme to the binding site. It may be assumed that covalent forces are used due to their ability to better bind to the enzyme to its active site. However, the strong binding forces of covalent bonds makes it too difficult for active site to release the enzyme. It must be kept in mind that the enzymes do not bind forever in the active site and as a result non-covalent forces are the best for easy recognition of the substrate and releasing it.
Nature of Binding Sites
1. Generally have a higher than average amount of exposed hydrophobic surface
2. Weak interaction can lead to an easy exchange of partners
3. Displacement of water also drives binding events [2]
The unique structures of repeat proteins grant them their functions. Their surface area to volume ratio is much higher than typical globular proteins. This characteristic makes them very well suited in mediating protein–protein interactions and organizing multiple proteins into functional complexes.
A property about repeat proteins is that individual repeats and the positions relative between those proteins are the same despite in which protein they occur. As repeat proteins bind to ligands, there is little to no conformational change. Scientists have compared different repeat protein structures with and without ligands bound using RMSD, or root mean square deviation. Studying β-catenin which contains 12 armadillo repeats, scientists have also found a Robo complex. This complex helps to develop bilateral symmetry in insects and vertebrates.
Repeat proteins also bind extended ligands. These proteins use multiple repeats to create an extended surface area for interaction with those extended ligands. This efficiently creates tight binding. Usually, a repeat protein interacts with a peptide that is extended or with a secondary structure element from the target protein.
The fact that repeat proteins are extended helps different regions of these proteins interacts with different ligands which bring the two into a functional complex. This multi-protein structure happens many ways. For an Hsp organizing protein or HOP, two discrete sets of TPR or tetratrico peptide repeat modules (one binding to Hsp70 and the other Hsp90) carry chaperones together to form a functional complex.
HEAT Repeat Protein
HEAT repeats are used to make multi-protein complexes in proteins that function differently from average proteins in their nucleocytoplasmatic transport. HEAT repeats in karyopherin form a superhelix and the external convex surface aids in nucleoporin binding while the inner concave face allows for binding with a regulatory protein Ran-GTP. Protein Phosphatase 2A or PP2A is a heterotrimeric protein that has a scaffold subunit to bind to regulatory and catalytic subunits of different HEAT repeat sets. Different versions of the complex exist so different sets of repeats binding within the HEAT domain are independent. An interesting fact is that SV40 small T antigen interferes PP2A’s function by competing with the regulatory subunit which binds to HEAT.
Usually, when there are multiple repeats, a repeat contributes to a binding interface that has the same structural element. There are exceptions to this, though. A helical bundle is formed from the N-terminal capping armadillo repeat when H2 and H3 is packed in the helical BCL9 (β-catenin).Also, a protein like TPR, Fis1, forms complexes with Mdv1 or Caf4 proteins. The N-terminal α-helix of Fis1 takes up the usual hydrophobic groove found on the concave surface on its TPR domain. An α-helix from the target protein comes into a second hydrophobic groove on the concave face. What is atypical, however, is the interaction with Caf4 and a second α-helix from the target protein binding to the convex part of the TPR domain. Finally, a composite surface used for binding the third protein may interact with a repeat protein. Looking at the CSL-Notch-Mastermind complex, we see that Mastermind interacts with Notch1 and CSL simultaneously but neither of these undergo a big conformational change when the complex forms. This means that Mastermind distinguishes the composite surface from both rather than binding to either through allosteric induction.
Repeat proteins do more than just protein-protein interactions. More and more repeat proteins are found to bind to ligands. Instead of specialized folds, the same repeat and fold are binding to many different types of ligands. A well-known example is the toll-like receptor or TLR from mammalian immune systems that bind proteins, lipoproteins/peptides, and nucleic acids. HEAT repeats can also bind in many ways. They are usually found intervening protein-protein interactions but can be found binding nucleic acids.
Protein designers are working on making new repeat proteins because simple and short repeat proteins can be used to bind many ligands using scaffolds. Many sequence alignments and structural characterizations allow for a clear description of structural and functional residues that are important. Two complementary strategies are being used: 1- introducing novel binding specificities onto existing repeat scaffolds and 2 – creating new scaffolds onto which known binding sites are grafted.
[3]
The extended, modular nature of repeat proteins allows for different sections of the protein to be used to bind many different ligands and then bring them together to form functional complexes. An example of this function of repeat proteins is the Hsp organizing protein(HOP), in which two defined sets of TPR modules each bind to Hsp 70 and Hsp 90 to bring them together into a complex.
Typically, when binding involves multiple repeats, each repeat contributes to the binding interface with the same structural element. In a given repeat protein, its binding interface could be formed by only H1 helices, or antiparallel beta strands, etc.
Repeat proteins have become key targets for protein design. Two strategies have been employed in synthesizing new repeat proteins: 1) addition of new binding specificities onto existing repeat scaffolds, and 2) synthesize new scaffolds onto which known binding sites are inserted. For example, Ank repeats have been used extensively in the first strategy presented.In another example, a TPR module has been designed by grafting Hsp90-binding residues onto a synthesized consensus TPR scaffold. What this has done is create a new protein that has greater affinity and specificity for Hsp90 than natural Hsp90 co-chaperones. This has had significant impact in fighting breast cancer. Synthesizing stronger versions of existing repeat proteins is one way in which the second strategy is used.
Myoglobin is a relatively small protein of mass 17.8kDa made up of 153 amino acids in a single polypeptide chain. It was the first protein to have its three-dimensional structure determined by x-ray crystallography by John Kendrew in 1957. Myoglobin is a typical globular protein in that it is a highly folded compact structure with most of the hydrophobic amino acid residues buried in the interior and many of the polar residues on the surface. X-ray crystallography revealed that the single polypeptide chain of myoglobin consist entirely of alpha-helical secondary structure. In fact there are eight alpha-helical secondary structure in myoglobin. Within a hydrophobic cervice formed by the folding of the polypeptide chain is the heme prosthetic group. This nonpolypeptide unit is noncovalently bound to myoglobin and is essential for the biological activity of the protein.
Myoglobin is a small oxygen-binding protein found in muscle cells. Its functions primarily in storing oxygen and facilitating oxygen diffusion in muscle tissue. Myoglobin is a single-chain globular protein that consists of 153 amino acids and a heme group (an iron-containing porphyrin). The globular structure of myoglobin consists mainly of alpha helices linked together by various turns. Myoglobin exists either in an oxygen free-form called deoxymyoglobin or in a oxygen bound form called oxymyoglobin. Whether myoglobin binds to oxygen depends on the presence of the prosthetic group, heme.
When myoglobin is able to bind to oxygen, it serves as the primary oxygen-carrying molecule in muscle tissue. Normally, the iron group in myoglobin has an oxidation state of 2+. However, when oxygen binds to the iron, it gets oxidized to an oxidation state of 3+. This allows the oxygen that is binded to have a negative charge, which stabilizes it. Myoglobin's affinity for oxygen is higher than hemoglobin. And unlike hemoglobin which is found in the red blood cells, myoglobin is found in muscle tissues.
3D structure of myoglobin.
Myoglobin owes its high affinity for oxygen to several factors. First, it has a proximal histidine group that helps it bind oxygen. Once the oxygen has been successfully bound, the structure of myoglobin comes into play. It prevents the reactive oxygen species from escaping by modifying the intrinsic reactivity of the heme group. Specifically, the ferrous ion coordinated with the dioxygen in the heme group can be oxidized to a ferric ion coordinated to superoxide. By keeping the reactivity of the oxygen under control with help from its structure, therefore, myoglobin can bind and hold on to oxygen atoms.
Although it has a much higher affinity for oxygen than its structural analog hemoglobin, myoglobin is a less efficient oxygen carrier for the cell. Because its affinity for oxygen is so high, myoglobin has a difficult time "letting go" of oxygen in the right areas. The cell needs oxygen to be distributed to the appropriate organelles, just as the body needs oxygen to be distributed to the right organ systems. This means that the species that "carries" the oxygen must be capable of releasing it once it reaches its assigned destination. Myoglobin's high affinity for oxygen means that it will be less inclined to release the oxygen once it has been bound; this in turn means that myoglobin will be distributing less oxygen to those areas where it is needed. Thus, hemoglobin is actually a more efficient oxygen carrier for the cell since its affinity for oxygen is lower. A lower affinity means that hemoglobin will have a significantly easier time releasing oxygen in the correct areas of the body. For this reason, the cell relies more upon hemoglobin to distribute oxygen than it does myoglobin; however there are specific areas of the body for which myoglobin is the better oxygen-carrier, such as for muscle cells. More can be read about hemoglobin in the hemoglobin section.
Another consequence of myoglobin's high affinity for oxygen is a higher affinity constant (KA). Since the affinity constant represents the concentration of substrate at which fifty percent of a protein's active sites are saturated, this means that half of myoglobin's active sites will be saturated with oxygen at a much lower concentration than for hemoglobin. More can be read about the affinity constant in its appropriate section.
Haemoglobin, the analog of myoglobin, consists of four poly peptide chains, two identical alpha chains and two identical beta chains. Each of the subunits contains a set of alpha helices in the same arrangement as the alpha helices in myoglobin. This structure that recurs is called a globin fold.
The oxygen-binding properties of proteins can be observed by viewing its oxygen-binding curve. An oxygen binding curve is a “plot of fractional saturation versus the concentration of oxygen”.
Myoglobin is actually used in conjunction with troponin to assist in the diagnosis process of a heart attack. Myoglobin levels appear to rise within two to three hours of a heart attack or other muscle injury. These levels reach their peak within eight to twelve hours, but usually fall back to normal within one day. The reason myoglobin is used as the key marker is because it turns positive far sooner than troponin. A positive reading may or may not signal potential damage of the heart, so it often can be ambiguous. Thus, a positive result is assessed based on troponin testing. However, a negative myoglobin result rules out a heart attack altogether. Another interesting fact regarding myoglobin is that it is highly toxic to the kidneys and if severe muscle injury occurs, blood levels of myoglobin may rise quickly and the kidneys (which function includes: releasing myoglobin in the blood as urine) can be severely damaged due to the increase amount of myoglobin. Another cause of increased myoglobin content is strenuous exercise, in addition to heavy alcohol abuse. In regard to muscle contraction, as fibres contract, they sqeeze the walls of the capillaries thus reducing or even stopping blood flow completely. It is actually during these situations when myoglobin has the ability to release its oxygen. It seems apparent that myoglobin plays the role of a hero. As the muscle relaxes, flow is restored and myoglobin is then recharged using the oxygen supplied by its oxygen-carrier partner, hemoglobin. These actions cause muscle injury and increased myoglobin in blood, which ultimately result in kidney failure.
Myoglobin plays the pivotal role of acting as an oxygen store during times of severely reduced blood oxygen supply. This notion of course is well established. What is also interesting to note is the fact that in terrestrial mammals, myoglobin compensates for the reduced blood flow in the crucial organ of the heart in addition to skeletal muscles during contraction.
The 3D structure of hemoglobin, PDB ID 1hho[1]. Alpha chains in blue, beta chains in tan, and heme with bound oxygen in red
Hemoglobin (Haemoglobin in many varieties of English and often abbreviated to 'Hb') is a tetramer consisting of two dimers that bind to oxygen. Hemoglobin is the oxygen-transporting protein of red blood cells and is a globular protein with a quaternary structure. Hemoglobin consists of four polypeptide subunits; 2 alpha chains and two beta chains. Hemoglobin transports oxygen in the blood from the lungs to the rest of the body. The three-dimensional structure of hemoglobin was solved using X-ray crystallography in 1959 by Max Perutz. The structure of hemoglobin is very similar to the single polypeptide chain in myoglobin despite the fact that their amino acid sequences differ at 83% of the residues. This highlights a relatively common theme in protein structure: that very different primary sequences can specify very similar three-dimensional structures.
There are two states in the hemoglobin, the T state (the tense state) and the R state (the relaxed state). The T state has less of an affinity for oxygen than the R state. In the concerted mode of cooperativity, the hemoglobin must either be in its T state or R state. In the sequential mode of cooperativity, the conformation state of the monomer changes as it binds to oxygen. Actual experimental observation of hemoglobin shows that it is more complex than either of the models and is somewhere in between the two. The conformation of hemoglobin also changes as the oxygen binds to the iron, raising both the iron and the histidine residue bound to it. The oxygen binding changes the position of the iron ion by approximately 0.4 Å. Before oxygenation the iron ion lies slightly outside the plane of the porphyrin upon oxygenation it moves into the plane of the heme.
The oxygen affinity of hemoglobin decreases as the pH decreases. This is useful because, with a high affinity for oxygen in the lungs, hemoglobin can effectively bind to more oxygen. Once it reaches the muscle, where the pH is lower, the lowered affinity for oxygen allows hemoglobin to release its oxygen into the tissues. When carbon dioxide diffuses into red blood cells, its dissociation also causes a decrease in pH.
The affinity of hemoglobin for oxygen is less than its structural analog myoglobin. Interestingly enough, however, this does not affect hemoglobin's usefulness for the body; on the contrary, it allows hemoglobin to be a more efficient oxygen carrier than myoglobin. This is so because hemoglobin can release oxygen more easily than can myoglobin. While it is important for oxygen to be carried to different areas of the body, it is even more important for the oxygen to be released when needed. The higher the affinity of a given protein for oxygen, the harder it will be for that protein to release oxygen when the time comes. Thus, hemoglobin's lower affinity for oxygen serves it well because it allows hemoglobin to release oxygen more easily in the body. Myoglobin, on the other hand, has a significantly higher affinity for oxygen and will, therefore, be much less inclined to release it once it is bound. Thus hemoglobin's lower affinity for oxygen relative to myoglobin allows it to have a higher overall efficiency in binding and then releasing oxygen species. For this reason, the body tends to use hemoglobin more often for oxygen-distributing purposes, although myoglobin is used as well, particularly for carrying oxygen to muscle cells. More can be read on myoglobin in the appropriate section.
Also worth mentioning is the fact that fetal hemoglobin has a noticeably higher affinity for oxygen than does maternal hemoglobin. This is of crucial importance during pregnancy in human females (and presumably in other pregnant mammalian females) because it allows the fetus to obtain much-needed oxygen during development. Basically, the hemoglobin present in the fetus is able to strip oxygen species from the maternal hemoglobin when the mother's blood comes into contact with fetal material. The portion of the mother's blood that does not touch the fetus transfers oxygen as normal to the mother's organ systems.
When oxygen is bound to hemoglobin, the color changes to crimson red. When oxygen is not bound, the color becomes a dark "rustic" shade of red [2] . Hemoglobin's affinity to oxygen increases as more oxygen is bound to it. The disassociation curve represents how hemoglobin is cooperative to oxygen with its sigmoidal shape.
- The left shift shows an increase in oxygen affinity. Hemoglobin has a better chance to hold onto oxygen. This normally occurs with a change in environmental factors such as low temperature, low metabolism rate, and high pH.
- The right shift shows a decrease in affinity. Hemoglobin is more likely to release Oxygen. This is due to high temperature, high metabolism, and low pH.
While Hemoglobin has 4 subunits, Myoglobin has one subunit. It is the enzyme of oxygen storage within the cells (found in skeletal muscle cells). The reason muscles are red is because they contain large amount of myoglobin. Organisms such as diving mammals have very large amounts of myoglobin so that they can go for an extended period of time without breathing.
As mentioned above, hemoglobin exists in two distinct states: the T-state and the R-state. The T-state of hemoglobin is the more "Tense" of the two; this is the deoxy form of hemoglobin (meaning that it lacks an oxygen species) and is also known as "deoxyhemoglobin". The R-state of hemoglobin is more "Relaxed" and is the fully oxygenated form; it is also known as
One of the unique features of hemoglobin is that it exhibits cooperativity. This means that hemoglobin can transmit intramolecular messages to its various functional groups to help it attain a maximum affinity for the ligand of interest, which is oxygen in this case. When a monomer of hemoglobin binds to oxygen, it alerts other nearby hemoglobin monomers to start the binding process as well. This means that, as more and more oxygen is bound by hemoglobin monomers, the affinity of hemoglobin will increase more and more as well. In other words, the affinity of hemoglobin is proportional to the quantity of oxygen bound at a given time. This allows hemoglobin to increase its affinity for oxygen over time, a property that brands it as one of the most flexible proteins in the body. Because it can modify its affinity for oxygen, hemoglobin can exhibit a range of different affinities. As stated before, this makes it quite flexible in terms of how much oxygen it can bind and therefore how much it can release. This is one of the reasons that the body prefers to use hemoglobin, as opposed to myoglobin, for oxygen transport: hemoglobin can modify its own affinity for oxygen to suit the situation at hand, making it capable of handling a wider variety of chemical environments and organ systems while still being able to distribute oxygen effectively.
concerted model for hemoglobinsequential model for hemoglobin
There are two main models of cooperativity for hemoglobin. One of these is the concerted model of cooperativity. This model states that the hemoglobin molecule changes rapidly between its R- and T-states in order to maximize its affinity for oxygen. According to this model, hemoglobin is constantly "flipping" back and forth between states in an attempt to bind as much oxygen as possible. The other model is the sequential model of cooperativity. This model maintains that one strand of hemoglobin starts a sequence of conformational changes in hemoglobin that increase its affinity for oxygen. When one strand of hemoglobin binds oxygen, the hemoglobin rearranges in a manner that favors additional oxygen binding. When the next oxygen is bound, another conformational change occurs to further supplement binding; Thus, hemoglobin can sequentially increase its affinity for oxygen as more and more of its strands bind oxygen.
Experimental data obtained from kinetics experiments with hemoglobin reveals that neither the concerted nor the sequential model of cooperativity is heavily favored. If anything, the data suggests that hemoglobin's behavior represents a hybrid of the two models; thus hemoglobin's cooperativity is somewhere in between the concerted and sequential models.
It is known that hemoglobin undergoes several conformational changes upon binding with oxygen. First of all, as soon as the iron cation within hemoglobin begins to move, the Histidine residues and the alpha-helix of hemoglobin start moving as well to stabilize the changes caused by the movement of iron. Second, the carboxyl terminal end of the alpha-helix usually resides at the interface between the two alpha- and beta-dimers that make up hemoglobin. Finally, the positional changes of the carboxyl terminal end create favorable conditions for transitions between the T- and the R-states of hemoglobin.
The above description makes clear that the concerted and sequential models do not fully explain hemoglobin's behavior, nor the behavior of related classes of proteins. To account for this discrepancy, more complex models have been devised that more accurately reflect the kinetic data gained from experiments with hemoglobin binding.
Oxygen binding to iron in the heme group pulls part of the electron density from the ferrous ion to the oxygen molecule. It is important to leave the myoglobin in the dioxygen form rather than superoxide form when the oxygen is released because the superoxide can be generated by itself to have a new form that gives negative effect on many biological materials, and also the superoxide prevents the iron ion from binding to the oxygen in its ferric state (Metmyoglobin). Superoxide and superoxide-derived oxygen species are so reactive compared to the stable O2 molecule that they could have a destructive effect both within the cell and in its environment. A distal histidine residue in myoglobin regulates the reactivity of the heme group to make it more suitable for oxygen binding. It does this by H-bonding with the oxygen molecule; the additional electron density of the oxygen molecule makes the H-bond unusually strong and therefore even more effective as a stabilizing agent.
An oxygen-binding curve is a plot that shows fractional saturation versus the concentration of oxygen. By definition, fractional saturation indicates the presence of binding sites that have oxygen. Fractional saturation can range from zero (all sites are empty) to one (all sites are filled). The concentration of oxygen is determined by partial pressure.
Hemoglobin's oxygen affinity is relatively weak compared to myoglobin's affinity for oxygen. Hemoglobin's oxygen-binding curve forms in the shape of a sigmoidal curve. This is due to the cooperativity of the hemoglobin. As hemoglobin travels from the lungs to the tissues, the pH value of its surroundings decrease, and the amount of CO2 that it reacts with increases. Both these changes causes the hemoglobin to lose its affinity for oxygen, therefore making it drop the oxygen into the tissues. This causes the sigmoidal curve for hemoglobin in the oxygen-binding curve and proves its cooperativity.
This image shows hemoglobin's oxygen binding affinity compared with myoglobin's affinity and the hypothetical curve that hemoglobin would have to follow if it did not show cooperativity. In this graph, you can see hemoglobin's sigmoidal curve, how it starts out with a little less affinity than myoglobin, but comparable affinity to oxygen in the lungs. As the pressure drops and the myoglobin and hemoglobin move towards the tissues, myoglobin still attains its high affinity for oxygen, while hemoglobin, because of its cooperativity, suddenly loses its affinity, therefore making it the better transporter of oxygen than myoglobin. The gray curve, showing no cooperativity, shows that to have the low affinity for oxygen needed in the tissues, the hemoglobin would have started with a smaller affinity for oxygen, therefore making it less efficient in bringing oxygen in from the lungs.
In red blood cells, the oxygen-binding curve for hemoglobin displays an “S” shaped called a sigmoidal curve. A sigmoidal curve shows that oxygen binding is cooperative; that is, when one site binds oxygen, the probability that the remaining unoccupied sites that will bind to oxygen will increase.
The importance of cooperative behavior is that it allows hemoglobin to be more efficient in transporting oxygen. For example, in the lungs, the hemoglobin is at a saturation level of 98%. However, when hemoglobin is present in the tissues and releases oxygen, the saturation level drops to 32%; thus, 66% of the potential oxygen-binding sites are involved in the transportation of oxygen.
Purified hemoglobin binds much more tightly to the oxygen, making it less useful in oxygen transport. The difference in characteristics is due to the presence of 2,3-Bisphosphoglycerate(2,3-BPG) in human blood, which acts as an allosteric effector. An allosteric effector binds in one site and affects binding in another. 2,3-BPG binds to a pocket in the T-state of hemoglobin and is released as it forms the R-state. The presence of 2,3-BPG means that more oxygen must be bound to the hemoglobin before the transition to the R-form is possible.
Other regulation such as the Bohr effect in hemoglobin can also be depicted via an oxygen-binding curve. By analyzing the oxygen-binding curve, one can observe that there is a proportional relationship between affinity of oxygen and pH level. As the pH level decreases, the affinity of oxygen in hemoglobin also decreases. Thus, as hemoglobin approaches a region of low pH, more oxygen is released. The chemical basis for this Bohr effect is due to the formation of two salt bridges of the quaternary structure. One of the salt bridges is formed by the interaction between Beta Histidine 146 (the carboxylate terminal group) and Alpha Lysine 40. This connection will help to orient the histidine residue to also interact in another salt bridge formation with the negatively charged aspartate 94. The second bridge is form with the aid of an additional proton on the histidine residue.
As carbon dioxide diffuses into red blood cells, it reacts with water inside to form carbonic acid. Carbonic acid dissociated leads to lower pH and stabilizes the T state.
An oxygen-binding curve can also show the effect of carbon dioxide presence in hemoglobin. The regulation effect by carbon dioxide is similar to Bohr effect. A comparison of the effect of the absence and presence of carbon dioxide in hemoglobin revealed that hemoglobin is more efficient at transporting oxygen from tissues to lungs when carbon dioxide is present. The reason for this efficiency is that carbon dioxide also decreases the affinity of hemoglobin for oxygen. The addition of carbon dioxide forces the pH to drop, which then causes the affinity of hemoglobin to oxygen to decrease. This is extremely evident in the tissues, where the carbon dioxide stored in the tissues are released into the blood stream, then undergoes a reaction that releases H+ into the blood stream, increasing acidity and dropping the pH level.
Allosteric regulation is the process by which the behavior of proteins is controlled by other molecules; the molecules that perform this regulation are known as allosteric regulators. This process involves the binding of an allosteric regulator molecule to the protein in question; the result is a distinct effect on the protein's function. Allosteric regulators that increase or supplement a given protein's function are known as allosteric activators. Those that decrease or interrupt a given protein's function are known as allosteric inhibitors.
Hemoglobin, like other proteins, has its share of allosteric regulators. Regulation is highly necessary for a protein as important as hemoglobin, since its affinity for oxygen must be just right for the particular organ system that it is currently dealing with. Thus the main concern for most of hemoglobin's allosteric regulators is tweaking its oxygen affinity to match the situation at hand.
The advantages of cell using allosteric inhibitors are:
- In a typical metabolic pathways, the final product of the pathway acts as an allosteric inhibitor.
- It inhibits the 1st enzyme in the pathway saving the cell from using resources in a metabolic pathway which final product is abundant.
Bisphosphoglycerate, or BPG, is one of many allosteric regulators for hemoglobin. This molecule binds to the central cavity of the deoxyhemoglobin version of hemoglobin (T-state) and stabilizes it. The increased stability of the T-state results in a decreased affinity for oxygen, since normally it is the intense straining of the T-state that drives deoxyhemoglobin to bind to oxygen; once oxygen is bound, the T-state loses its strain and relaxes into the R-state. Thus, by stabilizing the normally tense T-state, BPG makes hemoglobin less likely to bind oxygen in an attempt to release the strain. This mechanism is necessary, because the T state of hemoglobin is so unstable that the equilibrium lies very strongly in favor of the R state and little to no oxygen is released. In other words, pure hemoglobin binds to oxygen very tightly. 2,3-BPG was thus needed to stabilize the T state. Because BPG decreases hemoglobin's affinity for oxygen, it is an allosteric inhibitor of hemoglobin. Without 2, 3-BPG, hemoglobin would be an extremely inefficient transporter of oxygen from the lungs to the tissues, releasing only about 8% of its oxygen content. However, in the presence of 2,3-BPG, more oxygen-binding sites in the hemoglobin tetramer must be filled in order to transition from the T to the R state. Higher concentrations of oxygen must be reached in order for hemoglobin to transition from the lower-affinity T-state to the higher-affinity R state.
The binding of 2,3-BPG has further physiological consequences. Fetal hemoglobin has a higher oxygen-binding affinity than that of maternal hemoglobin. Fetal red blood cells have a higher affinity for oxygen than maternal red blood cells because fetal hemoglobin doesn't bind 2,3-BPG as well as maternal hemoglobin does. The result of this difference in oxygen affinity allows oxygen to be transferred effectively from maternal to fetal red blood cells.
The pH, or proton concentration of a given solution, is another allosteric regulator of hemoglobin. Interestingly enough, pH can act as both an allosteric activator and inhibitor, depending on the direction of pH change. As pH decreases, for example, the affinity of hemoglobin for oxygen decreases as well. This is due to the fact that protons help construct salt bridges in the T-state of hemoglobin. In general, the T-state of hemoglobin is favored by three amino acids that form two salt bridges; one of these salt bridges requires an added proton to form successfully. Thus, the higher the proton concentration (or the lower the pH) in the solution, the easier this salt bridge will form. Better salt bridge formation leads to a better and more stable T-state, and as mentioned before, a more stable T-state means decreased oxygen affinity of hemoglobin. Since higher proton concentration corresponds to lower pH, this means that the lower the pH, the more stable the T-state will be. Finally, the more stable the T-state, the lower the affinity for oxygen will be in hemoglobin molecule; thus pH acts as an allosteric inhibitor of hemoglobin when it is decreasing. Logically, then, the opposite effect would occur when the pH increases. This would signify a lower proton concentration, meaning more difficult salt bridge formation and thus a slower-forming and less stable T-state. A less stable T-state would be much more inclined to bind with oxygen; thus increased pH results in increased oxygen affinity for hemoglobin. The result is that pH acts as an allosteric activator for hemoglobin when it is increasing.
Carbon dioxide, or CO2, is yet another allosteric inhibitor of hemoglobin. There are several reasons for this. The first is that the enzyme carbonic anhydrase can help carbon dioxide react with water to form carbonic acid, which dissociates into bicarbonate and a proton. With enough carbonic anhydrase enzymes present, therefore, carbon dioxide can cause a decrease in the pH of the solution due to all the protons produced from its reaction with water. As mentioned in the previous paragraph, more protons means decreased pH, which in turn means a decreased affinity of hemoglobin for oxygen. Carbon dioxide also neutralizes the positive charge on the amino terminus of hemoglobin (amino groups usually exist in their protonated forms in living systems). This charge neutralization results in production of negatively charged carbamate groups, which form salt bridges that lead to stabilization of the T-state of hemoglobin, which results in a decreased affinity for oxygen. Thus carbon dioxide functions as an effective allosteric inhibitor of hemoglobin.
Bohr effect is a property of hemoglobin which states that at lower pH (more acidic environment), hemoglobin will bind to oxygen with less affinity. Since carbon dioxide is in direct equilibrium with the concentration of protons in the blood, increasing blood carbon dioxide levels leads to a decrease in pH, which ultimately leads to a decrease in affinity for oxygen by hemoglobin.
Physiological role
The Bohr effect facilitates oxygen transport. Hemoglobin binds to oxygen in the lungs and releases it in the tissues predominately to those tissues in most need of oxygen. When a tissue's metabolic rate increases, its carbon dioxide production increases. Carbon dioxide forms bicarbonate through the follow reaction:
CO2 + H2O H2CO3 H+ + HCO3−
This reaction usually progresses very slowly. With the help of the enzyme carbonic anhydrase, the formation of bicarbonate and protons in the red blood cells is accelerated. This causes the pH of the tissue to decrease and promote the dissociation of oxygen from hemoglobin to the tissue, allowing the tissue to obtain enough oxygen to meet its demands. Conversely, in the lungs, oxygen concentration is high. The binding of oxygen causes hemoglobin to release protons, which combine with bicarbonate to drive off carbon dioxide in exhalation. Since these two reactions are closely matched, there is little change in blood pH.
CO2 transport from Tissues to Red Blood CellsCO2 transport from Red Blood Cells to Lungs
BPG binds to hemoglobin and affect oxygen binding:
BPG binds in the central cavity of T-state hemoglobin. The anion groups of BPG are within Hb-bonding and ion-paring distances of the N-terminal amino group of both b subunits. BPG binds to and stabilizes only the T-state hemoglobin. This shifts the T R equilibrium toward the T state, which lowers the O2 binding affinity.
BPG is really important for O2 transport in our body. One example is high altitude adaptation. High altitude will induce a rapid increase in the amount of BPG synthesized in erythrocytes. The increased amount of BPG will shift the oxygen binding curve from sea-level position to a lower affinity position (shift to right). This decreases the amount of O2 binding in the lungs, but, to a greater extent, increases the amount of O2 released at tissues. So hemoglobin can deliver more O2 from lungs to tissues.
Sickle cells can cause hemoglobin cells which transport oxygen to the heart and parts of body change their shapes. It makes the transportation happens not smoothly and cause a disease.
A disease that affects many individuals's hemoglobin functionalities is sickle cell anemia, which cause by substitution of Valine instead of Glutamate at position 6 in amino acid sequence. Symptoms occur when an individual is several months old.
Sickle cell anemia is characterized by decreased breath intake, delayed growth and development, fever, jaundice, rapid heart rate, and many other ailments.
The problem is that hemoglobin in these indivudals are mutated. This mutated form of hemoglobin is called hemoglobin S and is less soluble than regular hemoglobin forms. Examination of the structure of hemoglobin S reveals that a new valine residue lies on the surface of the T-state molecule. As a result of this change deoxyhemoglobin has a hydrophobic patch on its surface. The hydrophobic patch interacts with other hydrophobic patches causing the molecule to aggregate into strands that align into insoluble fibers. Because
this mutated form cannot move freely when they accumulate in the blood stream they end up rupturing or distorting the shape of the red blood cells when delivering oxygen. The red blood cells
end up becoming a sickle or crescent shape. These inflicted cells are much less efficient in deliver oxygen through the body's circulation. They can clog fairly easily in smaller areas of blood flow
causing them to disrupt blood flow. Sickle cell anemia should not be mistaken with hemophilia which is a disease in which an individual's body cannot form blood clots. If gone without proper treatment,
people with this disease usually die from organ failure from ages 20 to 40. Better technology and data on this disease has led to treatments that involve folic acid supplements that activate the production of new
healthy red blood cells. Treatment must be ongoing and is meant to limit the number of pains and emergencies of the disease. Overall immune system also suffers from this disease so often people take antibiotics
and vaccines to prevent themselves from getting sick.
Sickle cell anemia is passed down through families and a child can only receive sickle cell anemia if both parents also have the disease. About 1 in 12 African Americans have this trait. There is a significant correlation between regions with high frequency of sickle cell anemia and regions with high prevalence of malaria. People with the sickle cell trait are resistant to malaria because the parasite that carries the disease needs to live within a red blood cell at some point in its life and is unable to survive in a sickle cell. Therefore, due to natural selection overtime the number of people with sickle cell anemia grew because before there was a cure for malaria the majority of the people who got malaria would die. It is now possible to diagnose sickle
cell anemia during pregnancy. Patients with the disease are encouraged to drink enough fluids, get enough oxygen, responding quickly to infections. Strenuous physical activity should be avoided, smoking should be avoided, and too much
sun exposure should also be avoided. Extreme consequences of sickle cell anemia no doubt includes death but others include blindness, spleen malfunction, tissue death, strokes, and acute chest pains.
Below are pictures comparing healthy red blood cells to blood cells inflicted by sickle cell anemia.
Just as sickle cell anemia is a difference in one amino acid, thalassemia is also an inherited disease where there is a reduction or loss of a hemoglobin chain. This leads to lower levels of hemoglobin and those with the disease experience anemia, fatigue, pale skin as well as spleen and liver malfunctions. Thalassemia branches into two different types: α-thalessemia and β-thalessemia. In α-thalessemia, the α-helix of hemoglobin is in low supply. This makes hemoglobin with high affinity for oxygen and no cooperativity therefore, making the release of oxygen in tissue poor. This is caused by a disruption in 4 alleles on chromosome 16 and is more rare. In β-thalessemia, the β-chain is in low supply. the extra α-helixes will aggregate and precipitate inside red blood cells which can result in anemia. β-thalessemia is caused by disruption on two alleles on chromosome 11.
Carbon Monoxide (CO) is a dangerous gas because it is odorless and colorless. Sources of carbon monoxide include running automobiles and gas-powered appliances. When inhaled, it binds at the same sites as oxygen and can negatively impact the body's ability to absorb oxygen. Carbon monoxide binds to hemoglobin 200 times more tightly than oxygen does. Even at low partial pressures, carbon monoxide will prevent hemoglobin from delivering oxygen to the body. Once carbon monoxide binds to one site of hemoglobin, hemoglobin turns into the R-state which increases oxygen affinity and prevents oxygen dissociation in tissues.
Treatment of carbon monoxide poisoning includes the administration of 100% oxygen at higher partial pressures. Because of the higher pressure, this will displace most of the carbon monoxide from hemoglobin.
Breathing of 100% O2 helps reduce the half-life of COHb, Carboxyhemoglobin, a stable complex of CO and hemoglobin formed in red blood cells with the presence of CO. Measurement of COHb level in red blood cells is used to confirm exposure to CO and assess the severity of poisoning. Elevated level of COHb is determined more than 2% for nonsmokers and more than 9% for smokers.
By replacing oxygen in hemoglobin, CO cuts off the supply of oxygen to tissues and cells, which can result in neurological problems in adults, learning disabilities and developmental issues in children, and miscarriage in women during pregnancy.
CO poisoning symptoms are not obvious, including headache, dizziness, nausea, fatigue and weakness. They can be mistaken as food poisoning, influenza, migraine headache, or substance abuse.
2 main types of CO poisoning: acute, caused by exposure to high level of CO in a short period of time, and chronic or subacute, caused by exposure to low level of CO in a long period of time.
Impact of CO poisoning on body systems
Neurologic: central nervous system depression, causing headache, dizziness in mild cases and coma, seizure in severe cases.
Cardiac: decreased myocardial functions, vasodilatation, and decreased oxygen delivery and utilization by myocardium, causing chest pains, low blood pressure, fast heart rates.
Metabolic: hyperventilation in mild cases, metabolic acidosis in severe cases.
Pulmonary: pulmonary edema occurs in 10-30% of acute cases.
Multiple organ failure: happens at high level of CO poisoning.
Fetal hemoglobin is the main oxygen transport protein. It happens during the last 7 months of development until 8 months later
A fetus obtains its source of oxygen from the mother’s lungs. The oxygen in the mother’s bloodstreams attaches to hemoglobin molecules in the red blood cells and diffuses to the fetal bloodstream at the placenta. By the time the blood reaches the fetus, the pressure is much lower, which is not enough for a normal adult.
During the entire fetal formation period, three different types of hemoglobin are produced, with the succeeding hemoglobin deactivating its predecessor. All three types have the same heme molecules and iron atom, but differ slightly in structure. In the first eight weeks, majority of the hemoglobin is a type called embryonic hemoglobin. The production of Hemoglobin follows by the fetal hemoglobin (Hemoglobin F). It is the predominant form of hemoglobin expressed in the fetus development. The Hemoglobin F is apparent weeks after conception until a few months after birth. Around the thirty-fifth week, the adult hemoglobin (Hemoglobin A) starts production. Eventually, the blood cells will only contain Hemoglobin A, which is the only one produced for the duration of the human life.
Structural differences between the adult hemoglobin and the fetal hemoglobin
From the structural point of view, the adult hemoglobin is composed of 4 heme groups, 2 alpha chains and 2 beta chains. The fetal hemoglobin (also termed haemoglobin F) is also composed of 4 heme groups, 2 alpha chains and 2 gamma chains. The gamma chains are referred to as gamma subunits, which are homologous to the beta chains of the adult hemoglobin. In addition, the fetal hemoglobin and adult hemoglobin are found to be different near the 2,3 BPG binding site. The 2,3 BPG binds less tightly with the deoxy form of fetal hemoglobin as compared to the deoxy form of adult hemoglobin.
Additionally, another form of haemoglobin, termed haemoglobin A2, and comprising of two alpha and two delta globin chains; is produced in small quantities throughout childhood and adulthood. Haemoglobin A2 accounts for around 2-3% of total haemoglobin levels.
There is also a hormone that can induce the increase of red blood cell production. Erythopoietin is a glycoprotein hormone that controls erythropoiesis, or otherwise known as red blood cell production. It is a protein signaling molecule (cytokine) for erythocyte (red blood cell) precursors in the bone marrow. This hormone is produced in the interstitial fibroblasts in the kidney and in the perisinusoidal cells in the liver.
Hemocyanin is a protein found in mollusks that carries oxygen in much the same way as hemoglobin carries oxygen in human blood. Similarly to hemoglobin, a central metal atom binds oxygen differentially, however in hemocyanin, this central metal atom is copper. When the copper is oxidized from its Cu(I) form to its Cu(II) the protein changes color from clear to blue, which is the source of the blue tinge of mollusk hemolymph. The origin of the word hemocyanin (from Latin for heme- blood and cyanin- blue) alludes to this blue tinge. In hemolymph, hemocyanin is present as an extracellular protein that aggregates into large complexes held together by calcium or magnesium ions.[3] The number of monomers and the size of these aggregates can differ between mollusk and arthropod species, but all forms contain the central copper atoms.
The structure and function of the hemocyanin molecule revolves around the two copper atoms embedded at its core. Each copper atom is complexed by three histidine residues that form the distorted pyramidal geometry of each atom. This and the space between the copper atoms facilitates the bonding of the two copper atoms to each dioxygen molecule. In close proximity to the histidine residues are two phenylalanine residues that form a hydrophobic core that protects the active site. Once oxygen is bonded, a geometrical change occurs from trigonal pyramidal to a distorted tetrahedral and it is this change in bonding geometry that explains the change in color that occurs with oxydation of the central copper atoms. Although in both arthropod and mollusk hemocyanin, the binding mechanism and active site are nearly identical, there are various differences in the structure and assembly of subunits.
In arthropods, hemocyanin is made up of monomers of approximately 75 kDa which make up hexamers that aggregate into multiple hexamer groups. Each monomer may take one of several forms, all of which occur in a specific location in the molecule. Arthropod hemocyanin has three regions, the second of which housing the copper atoms and residing within a 4 a- helical set.[4]
Mollusks, however have much larger polymeric subunits on the order of 350-450 kDa. Additionally, the aggregates of subunits are often much larger; for example, cephalopod hemocyanin consists of 5-10 cylindrical aggregates and in other gastropods there can be as many as 160 oxygen accepting units.[5] Despite the differences in quaternary structure between mollusk hemocyanin proteins, the tertiary structure of each subunit is very similar.
Hemocyanin and other proteins that facilitate oxygen transport and aerobic respiration have their evolutionary roots in some of the earliest life forms. Since the atmosphere was mostly anaerobic, oxygen was probably poisonous to many early anaerobic organisms. In an effort to eliminate poisonous oxygen byproducts, early oxidative proteins were evolved that utilized iron or copper to carry out oxidative processes. Over time, the concentrations of oxygen in the atmosphere increased and oxidative proteins began to be used in aerobic systems. Additionally, as body size began to increase (around 700-800 MYA), diffusion would not supply enough oxygen to the entire organism, and iron and copper based molecules began to be diversified. The similarities between hemocyanin structures in mollusks and arthropods suggest a divergence in hemocyanin structure before 750 MYA.[6]
Protein-protein interaction network is bindings of multiple proteins with distinct conformation (3D structure). A node in the network represents a protein and a node that can interact with ten to hundreds of other nodes is considered a hub protein. A hub protein is essential and contains many distinct binding sites to accommodate non-hub proteins.
The problem with understanding a protein-protein network is how one specific hub protein can bind to so many non-hub partners. In certain cases, a change of external environment such as other binding events, partner concentration, pH, ionic strength and temperature can lead to a shift in structural ensemble. But these changes are not capable of accommodating up to hundreds of proteins binding to the same hub.
A new approach in understanding protein-protein interaction is to consider proteins as gene products. Proteins are gene products with different amino acid sequence. A specific set of genes or related genes can have multiple distinct sequences, structures and interactions. Each distinct sequence leads to a distinct structure/conformation. The differences among the conformations may be small, but one gene product can interact with many preferred partners. An example is where a pre-mRNA with four exons and three introns can produce three different mRNAs via exon skipping. This correspond to three gene product with three different protein structure with only minor difference in sequence.
There are several cellular mechanisms that can result in different gene products with many conformations. In alternative splicing, the combinations of exons could result in 38,016 isoforms – different forms of the same protein. All of these isoforms can have different protein-protein interaction due to conformational variability although they are considered the same protein. In cancer, p53 is a tumor suppressor protein encoded by TP53 gene. The isoforms of p53 have many cellular functions. A mutation in TP53 creates multiple gene products of p53, which causes cancer. These p53 variants can regulate hundreds of genes and proteins.
The conclusion is that, although a node in a network is one protein, but the same protein can have multiple gene products with many conformations. Each node of the same protein can be slightly difference in sequence with distinct three-dimensional structure. These differences allows a node to bind with hundreds of partners are different time and perform many essential biological functions.[1]
The network approach helps determine the role of a specific amino acid at a known position in the protein structure. Networks simplify complex system behaviors by splitting the system into a series of links. Links represent the neighboring positions of amino acids in protein molecules. Because proteins are linked in this way and protein structure networks are connected to each other by only a few other amino acid elements, we can determine folding probability. Proteins with denser protein structure networks fold more easily and the folding probability increases as the protein structure becomes more compact.
The network approach can also be applied to the prediction of active centers in proteins. Active centers are protein segments that play key parts in the catalytic reaction of the enzyme function shown by their respective proteins. Scientists have used long-range network topology to create a network skeletons from which they can study only side chains which are essential in the flow of information for the whole protein. Network analysis has showed that active centers occupy a central position in protein structure networks, usually have many neighbors, give unique linkages in their neighborhood, integrate communication for the entire network, do not take part in wasteful actions of ordinary residues, and collect and coordinate most of the energy in the network.[2]
HOT SPOTS – Essential amino acid deposits of protein-binding sites that have a particularly high binding free energy. Can cluster to form densely packed ‘hot regions’.
ACTIVE CENTRE – Protein segment that plays a key part in the catalytic reaction of the enzyme function shown by the respective protein.
BINDING SITE – Amino acid side chains located at the binding interface.
CENTRAL RESIDUES – Contain catalytic residues (active centres) in addition to binding sites and hit spots.
CREATIVE ELEMENTS: Least specialized and best among all network elements to live a separated life away from the rest of the network. This is why they continuously chage their contacts. They must connect to elements that are not directly connected to each other so that they do not create a large cumulative disorder that can lead to permanent change.
↑Chung-Jung Tasi, Buyong Ma and Ruth Nussinov, Center for Cancer Research Nanobiology Program, SAIC-Frederick, Inc., NCI-Frederick, Frederick, MD 21702, USA.
One of the most common functions of enzyme is the ability to catalyze reactions. During a reaction, the reactants must overcome activation energy in order for it to produce the products. The amount of activation energy needed determines how long the reaction takes to proceed. The lower the activation energy makes the faster rate of the reaction. The role of enzymes in catalyzing reactions is to stabilize the intermediate species, which is at the highest point of the activation energy, and thus dropping the activation energy. The enzyme is complementary not to the substrate but its intermediate state. If the enzyme binds to the substrate, it actually increases the activation energy. The equilibrium achieved is the same with or without the catalytic enzyme. However, what is affected is the time and rate in which it is achieved.
Generally, the higher the concentration of the substrate, the easier it is for the enzyme to bind to it. By plotting the amount of product produced as a function of time, the slope is how fast the reaction happens before the amount of substrate is saturated. This value is called the V0. Increasing the substrate concentration will increase the V0. However, there is a certain point in which the substrate concentration is too high and the reaction will not proceed any faster. This point is called the maximum velocity, Vmax. Every enzyme has their unique Vmax value. Another important identity of an enzyme is the Km value, defined as the substrate concentration at half of Vmax. Km is also unique to each enzyme. The turnover rate, the rate at which products are produced is called the Kcat. Dividing Kcat by Km gives the efficiency constant of the enzyme, which tells how fast the reaction is carried out and how likely the enzyme is to find the substrate. For more information, refer to Catalysis.
Phosphoryl-Transfer Reaction: Mechanisms and Catalysis
A key feature of phosphoryl-transfer reactions is that they often have extremely slow nonenzymatic rates and thus require large reaction rate accelerations using catalysts. The reactions that occur at the phosphorous atom of the phosphate ester also form the chemical basis for many of the most important and fundamental processes in living systems because they allow for the inheritance of genetic information through nucleic acids and are also responsible for using energy coupling to drive thermodynamically unfavorable reactions crucial to maintaining cell health and vitality. Phosphoryl transfer reactions also play an important role in metabolic pathways and signal transduction as well.
One possible mechanism for catalysis in these phosphoryl transfer reactions is the hydrolysis of phosphate monoesters. The hydrolysis rate often increases significantly as the pH decreases; this change indicates that the protonated form of the phosphate monoesters react much more quickly than the phosphate monoester dianions. The fact that these reactions are often carried out rapidly, with the help of enzymes, can be attributed to several factors. For example, the activation of the nucleophile can be accomplished in one of three ways; the way that the nucleophile is positioned can affect the nucleophile by increasing or even decreasing it. Another way is to reduce the electrostatic repulsion. One of the most important features of enzymes is their ability to use the binding interactions and positioned groups to carry out catalysis. The ability of enzymes to be able to accomplish this directly combines enzyme specificity with catalysis.
In phosphoryl transfer reactions, it’s also important that the nucleophile is aligned with the phosphorous atom as well as the leaving group for attack at phosphorous. Another factor that can contribute to the catalysis of monoester reactions is the stabilization of the negative charge on the potential leaving group. It has also been found that the transition states for phosphoryl transfer reactions can often be loose, tight, or synchronous depending on whether the compounds are phosphate monoesters, diesters, or triesters. Phosphate monoester usually proceeds through loose transition states, diesters through synchronous transition states, and triesters through tight transition states.
The presence of positively charged functional groups in the enzymes used in phosphoryl transfer reactions can also affect the reaction’s interactions with the oxygen atoms of the transferred phosphoryl group.
http://en.wikibooks.org/wiki/Structural_Biochemistry/Enzyme_Catalytic_Mechanism/Catalysis
Since many enzymes have common names that do not refer to their function or what kind of reaction they catalyze, an enzyme classification system was established. There were six classes of enzymes that were created so that enzymes could easily be named. These classes are Oxidoreductases, Transferases, Hydrolases, Lyases, Isomerases, and Ligases. This is the international classification used for enzymes. Enzymes are normally used for catalyzing the transfer of functional groups, electrons, or atoms. Since this is the case, they are assigned names by the type of reaction they catalyze. The enzymes were numbered 1-6 and from here, they were divided into subdivisions. This allowed for the addition of a four-digit number that would precede EC(Enzyme Commission) and each enzyme could be identified. The reaction that an enzyme catalyzes must be known before it can be classified.[1]
Oxidoreductases catalyze oxidation-reduction reactions where electrons are transferred. These electrons are usually in the form of hydride ions or hydrogen atoms. When a substrate is being oxidized it is the hydrogen donor. The most common name used is a dehydrogenase and sometimes reductase will be used. An oxidase is referred to when the oxygen atom is the acceptor.
Glutathione S-transferase
Transferases catalyze group transfer reactions. The transfer occurs from one molecule that will be the donor to another molecule that will be the acceptor. Most of the time, the donor is a cofactor that is charged with the group about to be transferred. Example: Hexokinase is used in glycolysis.
Hydrolases catalyze reactions that involve hydrolysis. This case usually involves the transfer of functional groups to water. When the hydrolase acts on amide, glycosyl, peptide, ester, or other bonds, they not only catalyze the hydrolytic removal of a group from the substrate but also a transfer of the group to an acceptor compound. These enzymes could also be classified under transferases since hydrolysis can be viewed as a transfer of a functional group to water as an acceptor. However, as the acceptor's reaction with water was discovered very early, it's considered the main function of the enzyme which allows it to fall under this classification. For example Chymotrypsin.
Lyases catalyze reactions where functional groups are added to break double bonds in molecules or the reverse where double bonds are formed by the removal of functional groups. For example, Fructose bisphosphate aldolase used in converting fructose 1,6-bisphosphate to G3P and DHAP by cutting the C-C bond.
Isomerases catalyze reactions that transfer functional groups within a molecule so that isomeric forms are produced. These enzymes allow for structural or geometric changes within a compound. Sometimes the interconversion is carried out by an intramolecular oxidoreduction. In this case, one molecule is both the hydrogen acceptor and donor, so there's no oxidized product. The lack of an oxidized product is the reason this enzyme falls under this classification. The subclasses are created under this category by the type of isomerism. For example phosphoglucose isomerase for converting glucose 6-phosphate to fructose 6-phosphate. Moving chemical group inside the same substrate.
Ligases are used in catalysis where two substrates are ligated and the formation of carbon-carbon, carbon-sulfide, carbon-nitrogen, and carbon-oxygen bonds due to condensation reactions. These reactions are coupled to the cleavage of ATP.
Translocase are enzymes that catalyze the movement of ions or molecules across membranes or their separation within membranes. It is a general term for a protein that assists in moving another molecule, usually across a cell membrane. The reaction is designated as a transfer from “side 1” to “side 2” because the designations “in” and “out”, which had previously been used, can be ambiguous. Translocases are the most common secretion system in Gram-positive bacteria.
EC 4.1 includes lyases that cleave carbon-carbon bonds, such as decarboxylases (EC 4.1.1), aldehyde lyases (EC 4.1.2), oxo acid lyases(EC 4.1.3) and others (EC 4.1.99)
EC 4.2 includes lyases that cleave carbon-oxygen bonds, such as dehydratases
EC 4.3 includes lyases that cleave carbon-nitrogen bonds
EC 4.4 includes lyases that cleave carbon-sulfur bonds
EC 4.5 includes lyases that cleave carbon-halide bonds
Proteases are a protein-digestive enzyme that cleaves protein through hydrolysis, the addition of water to the peptide bond. Although hydrolysis of the peptide bond is thermodynamically favored, it is still a slow reaction without the enzyme. This is due to the fact that the peptide bond is very stable due to its resonance structure forming a partial double bond. The specificity of the peptide bond they hydrolyze is high.
A number of proteolytic enzymes participate in the breakdown of proteins in the digestive systems of mammals and other organisms. An example of a protein-digesting enzyme may be seen in the protease called pepsin.[1] Pepsin is one of two components of gastric juice.[1] Pepsin works by attacking the exposed peptide bonds.[1] Unlike most enzymes which can be denatured when exposed to extreme pH, pepsin works at its optimal performance in a highly acidic environment.[1]
Serine Proteases use serine residue to create a nucleophilic amino acid that cleaves the peptide bond. They are responsible for various functions such as blood clotting, and digestion
One such enzyme, known as Chymotrypsin, cleaves peptide bonds selectively on the carboxyl terminal side of the large hydrophobic amino acids such as tryptophan, tyrosine, phenylalanine, and methionine. Chymotrypsin is a good example of the use of covalent catalysis.
Not all proteases utilize strategies based on activated serine residues. Classes of proteins have been discovered that employ three other approaches to peptide-bond hydrolsis:
In each case, the strategy is to generate a nucleophile that attacks the peptide carbonyl group.
An example of a Cysteine Protease is papain, which is found in the papaya fruit. The catalytic mechanism that this enzyme uses to hydrolyze a peptide bond involves the activation of a cysteine residue by a histidine residue, both present in the active site. The result of this activation is a powerful nucleophile that is able to attack the carbon present in the carbonyl group present next to the peptide bond.
Aspartyl proteases are a type of proteolytic enzymes classified under endonucleases. Aspartyl proteases are known to exist in vertebrates, plants, plant viruses, as well as in retroviruses. Aspartyl proteases is characterized by having a frequent sequence of Asp- Thr- Gly amino acid triad. Most aspartate proteases are found as monomeric enzymes consisting of two domains. Aspartyl proteases are important for the human body in regulating blood pressure, health, and digestion.
An example of Metalloprotease would be Zinc Metalloproteases which include the digestive enzymes carboxypeptidases, various matrix metalloproteases (MMPs) that are secreted by cells, and one lysosomal protease. MMP's have the role of degrading extracellular matrix during tissue remodeling, cell signaling the release of cytokines or growth factors through cleavage of proteins.
Enzymes are extremely useful and effective in many biochemical reactions but only at the right time and place. Enzyme activity is regulated in five different ways:
Allosteric control:Allosteric enzymes contain distinct regulatory sites and multiple functional sites. The protein is significantly controlled when small signal molecules bind to these regulatory sites. Also allosteric enzymes show cooperativity, which means that activity at one functional site will affect the other functional site as well.
Multiple Forms of Enzymes: Isoenzymes or Isozymes are homologous enzymes in an organism that catalyze the same reaction but are a little bit different in their structure, Km and Vmax values, and regulatory properties. Isozymes allow a reaction to be regulated at distinct locations or times.
Reversible Covalent Modification: The catalytic properties of enzymes can be altered by a covalent binding of a modifying group, most commonly to a phosphoryl group. Usually ATP will serve as a donor for these reactions.
Proteolytic Activation: The other regulatory mechanisms mentioned so far can freely change between active and inactive states. However in proteolytic activation, the enzyme is irreversibly converted into from an inactive enzyme to an active one. These enzymes are activated by hydrolysis of a few peptide bonds. Also hydrolysis of an enzyme precursor such as zymogens or proenzymes can also activate the enzyme.
Controlling the Amount of Enzyme Present: Enzyme activity can also be regulated by adjusting the amount of enzymes present. This method of regulation usually takes place during gene transcription.
The first step in the biosynthesis of pyrimidines, the condensation of aspartate and carbamoyl phosphate to form N-carbamoylaspartate and orthophosphate is catalyzed by an allosteric enzyme, aspartate transcarbamoylase or ATCase.
John Gerhart and Arthur Pardee found that ATCase is inhibited by its own final product, the pyrimidine CTP. Therefore as the concentration of CTP increases, the reaction with ATCase slows down. This is a negative feedback loop, or negative inhibition.
As illustrated here the graph of ATCase kinetics is sigmoidal instead of the Michaelis-Menten hyperbolic shape.
ATCase Consists of Separable Catalytic and Regulatory Subunits
ATCase can be separated into regulatory and catalytic substrates by treatment with compounds such as p-hydroxymercuribenzoate. This is evidence that ATCase has distinct regulatory and catalytic sites. John Gerhart and Howard Schachman were the ones to carry out this study. The subunits can then be separate through ion-exchange chromatography or by centrifugation in a sucrose density gradient.
The larger subunit is the Catalytic subunit. This subunit is catalytic as implied by the name but is unresponsive to CTP and does not show sigmoidal kinetics. The other subunit, the regulatory subunit has no catalytic activity but binds to CTP. Therefore ATCase is composed of catalytic and regulatory subunits.
Similar to Hemoglobin, ATCase exists in a T-state and R-state. The T-state is the less active state while the R-state is the active state. CTP inhibits ATCase by binding to the regulatory sites stabilizing the T-state. ATP can also bind to the same sites, but does not stabilize the T-state. Therefore ATP competes with CTP for the sites. ATP is an allosteric activator that binds to the regulatory subunit. ATP as well as CTP are referred to as "heterotropic effects" on a allosteric enzyme such as ATCase. ATP is an allosteric activator of aspartate transcarbamolyase because it stabilizes the R-state of ATCase, effecting neighboring subunits by making it easier for substrate to bind.The increase of the concentration of ATP has two potential explanations. First being, at high concentrations of ATP signals a high concentration of purine and pyrimidine. second, a high concentration of ATP conveys that a source of energy is available for mRNA synthesis and DNA replication follow by the synthesis of pyrimidines needed for these processes.
Enzymes that differ in amino acid sequence but catalyze the same reaction are called isoenzymes. Generally, Isoenzyme have different Km and respond to different regulatory molecules. Different genes encode Isozymes. Isozymes allows specific adjustments to be made to metabolism to accommodate the needs of a tissue or developmental stage.
Different tissues expressing different forms of isozymesIsozymes of lactate dehydrogenase
The activity of an enzyme can be modified by covalently bonding a molecule to it. Most modifications are reversible. The most common covalent modifications are Phosphorylation and dephosphorylation.
Phosphorylation: Almost every metabolic process in eukaryotic cells are regulated by phosphorylation. As much as 30% of eukaryotic proteins are phosphorylated. Phosphoryl groups are usually donated by ATP. The gamma terminal phosphoryl group of ATP is transferred to an amino acid. The amino acid acceptor always has a hydroxyl group in the side chain. Kinases transfers the phosphoryl groups and Protein phosphatases reverses the process. However phosphorylation and dephosphorylation are not reverse reactions of one another. Each reaction is almost irreversible under normal physiological conditions. Phosphorylation will only take place through a specific protein kinase using an ATP and depphosphorylation will only occur through phosphatase.
Protein phosphorylation:
Adds two negative charges
Forms 2 or 3 hydrogen bonds
Phosphorylation is reversible
Kinetics can be adjusted to physiological process
Amplifies sign
ATP coordinates signaling with bioenergetics
Common Covalent Modifications of Protein Activity
Phosphorylation
Donates ATP to glygogen phosphorylase which functions in glucose homeostasis and energy transduction
Acetylation
Donates Acetyl CoA to histones which functions in DNA packing and transcription
Myristoylation
Donates Myristoyl CoA to Src which functions in signal transduction
ADP ribosylation
Donates NAD+ to RNA polymerase which functions in transcription
Farnesylation
Donates Farnesyl Pyrophosphate to Ras which functions in signal transduction
γ-Carboxylation Sulfation
Donates HCO3- and 3'-Phosphoadenosine-5'-phosphosulfate to fibrinogen which functions in blood-clot formation
Ubiquitination
Donates Ubiquitin to cyclin which functions in the control of cell cycle
Many enzymes are inactive until one or a few specific peptide bonds are cleaved. The enzyme exists initially as an inactive precursor called zymogen or proenzyme. Proteolytic cleavage does not need ATP for energy and only occurs once in the life of the enzyme. Some examples of enzymes and biochemical processes that are activated by proteolytic cleavage are:
The digestive system that hydrolyze proteins are initially made as zymogens in the stomach and pancreas
Blood clotting is mediated by a cascade of proteolytic activations
Insulin is derived from the inactive precursor proinsulin
Proteolytic activation of chymotrypsinogen forming an active chymotrypsin is involved in digestive enzymes, blood clotting, protein hormones, and procaspases (a programmed cell death).
Berg, Jeremy M. Tymoczko, John L. Stryer, Lubert. Biochemistry 6th Edition. Copyright 2007, 2002 by W. H. Freeman and Company
Viadiu, Hector. Reversible Covalent Modification. Biochemistry Lecture. Dec. 5, 2012
Protein regulation is essential for biological balance. Too much or too little of any proteinreactivity can cause severe biological damage.
Alzheimer's patients have "plaques" in their brains, which are essentially large chunks of a certain protein believed to contribute to neuronal death. It is also a dementia that gets worse over time. There is no cure for Alzheimer's disease. As you can see in the picture, someone who has Alzheimer's has extreme loss in the different functions of the brain.
Alzheimer's patients have this protein's regulation pathway disturbed, causing it to be overly expressed. Protein regulation is also referred to as Enzyme Regulation. Furthermore, the study of these protein regulation pathways have lead to much growth in the creation of medicinal and pharmaceutical products.
Hunter syndrome is a genetic disease in which mucopolysaccharides do not degrade accurately. As a result, mucopolysaccharides will accumulate inside the body. The main cause of this accumulation is the absence of iduronate sulfatase enzyme. Some of the signs for recognition of the syndrome are big heads and different facial characteristics. The syndrome can be found out with urine test. However, this test is not trusted. The study of fibroblasts that extract from body’s skin is more effective. The gene that contributes to the cause of Hunter syndrome is residing on the X-recessive chromosome. Since males only have one X chromosome, the chance that males inherit this syndrome is higher than that of females.
The problem with Hunter syndrome is how the body is unable to break down mucopolysaccharides, which make up proteoglycan that is part of the extracellular matrix. As a consequence, the adding up of mucopolysaccharides becomes an obstacle for other cells in the body to carry out their jobs. This incident may lead to significant effects that can harm the body. For instance, some early side effects of Hunter syndrome can be the normal sickness such as cold and runny nose.
Hunter syndrome results in a certain effects upon the body. Thus, people who inherit the syndrome tend to have common characteristics. The more extreme consequences of Hunter syndrome include mental retardation, heart problem, joint stiffness, etc. Bone marrow replacement was proven to help expanding the life span of Hunter syndrome patients. Unfortunately, it does not solve the problem of mental retardation. Elaprase, a lysosomal enzyme iduronate sulfatase that can be made by recombinant DNA technique, has demonstrated to be an effective cure for Hunter syndrome through the replace of enzyme. Nevertheless, elaprase is indeed very expensive.
Hurler Syndrome is an inherited disease and is caused by a recessive mutation (both parents would need to have passed down the trait). Historically, it was thought to be caused by the excessive synthesis of 2 mucopolysaccharides: dermatan sulfate and heparan sulfate. Some scientists believed the excessive synthesis of the 2 molecules to be caused by a faulty regulation pathway. Elizabeth Neufeld tested this hypothesis and found it to be false; she found the cause of the disorder to be the inadequate degradation of the 2 sugars. While normal cells leveled off production after a certain point, cells affected by the mutation continued production past normal levels.
Correction of this disease is easily possible in vitro (and works to an extent in vivo.) The addition of healthy cells in the same culture as mutated cells in vitro caused normal levels of dermatan sulfate and heparan sulfate to be created. The normal cells excrete a corrective factor/enzyme in the medium which is taken up by the mutated cells. This enzyme is crucial in the degradation of the 2 mucopolysaccharides; furthermore, very little of it is necessary to fully correct the mutated cells. There are many problems with using α-L-Iduronidase as treatment in vivo; the main issue being that various tissues respond at various degrees to the medication. Most importantly, the central nervous system does not uptake any of the intravenously injected enzyme because of the blood-brain barrier. On top of this issue, a significant number of people with this disease have neurological diseases that need to uptake this enzyme somehow.
α-L-Iduronidase was found to be the key enzyme in restoring proper degradation of dermatan sulfate and heparan sulfate. It was found though that α-L-Iduronidase from some cells was not corrective. Research showed that the carbohydrate mannose 6-phosphate was responsible for proper uptake of the enzyme; a mutation in the structure of it caused the α-L-Iduronidase product to not be taken up properly by cells affected by Hurler Syndrome.
Sanfilippo Syndrome is within a set of neurodegenerative diseases called tauopathies (the most common of which is Alzheimer's Disease). Although there are 4 subtypes of Sanfilippo Syndrome, they are all characterized by reduced degradation of heparan sulfate (see Hurler Syndrome section) due to reduced levels of a lysosomal enzyme.
It was found in a mouse model (MPS IIIB) that there were significantly increased levels of the protein lysozyme; increased levels of this disease were found to cause the creation of hyperphosphorylated tau which is found in the brains of Alzheimer's patients and patients with other tauopathies. Significant research is being done in Alzheimer's disease which may carry over to Sanfilippo Syndrome as well due to their similarities.
1. 4 different subtypes:
Each Sanfilippo subtype is caused by the deficiency of each specific enzyme: heparin N-sulfatase for MPS-III A, N-acetyl-alpha-D-glucosaminidase for MPS-III B, acetyl-CoA: alpha-glucosaminide acetyltransferase for MPS-III C, and N-acetylglucosamine-G-sulfate sulfatase for MPS-III D. Among these four subtypes, Sanfilippo syndrome type A is the most prevalent (60%), followed by B (30%), D (5%), and C (6%). In total, 47% of all cases of mucopolysaccharidosis diagnosed is related with Sanfilippo disease.
2. Mortality/ Morbidity:
Patients with Sanfilippo syndrome tend to develop Central Nervous System degeneration and usually end up at a vegetative state. They usually die before the age of 20 due to cardiopulmonary arrest because of airway obstruction or infection in the pulmonary pathway. Among these 4 subtypes, MPS-III A is the most severe one due to early death in patients (usually during their teenage years).
Sanfilippo syndrome has an equal effect on both males and females as well as on different races since its main cause is the inheritance in an autosomal recessive pattern which has no relations with the sex chromosomes.
3. Diagnosis and History:
In terms of diagnosis, these four subtypes are not indistinguishable clinically; therefore, the only determining factor to identify each specific subtype is the different genes that are responsible for that subtype.
Usually, affected individuals show no symptoms and develop normally during the first two years of their lives. Onset usually takes place between the age of 2 and 6. Developmental delays in infancy may be shown in some of them. Growth might be slowed down at the age of 3 (e.g. short stature). Patients might also become hyperactive and behave aggressively and destructively. Besides disturbing the sleep pattern in patients, this syndrome also interferes severely into the mental development of affected patients – speech impairment, hearing loss, etc. At the same time, patients might show shortened attention spans and find it challenging to concentrate and to be able to perform academic tasks at schools.
By the age of 10, patients’ daily activities and movements are severely limited. They often are in need of wheelchairs to accommodate them and might even have swallowing difficulties and seizures.
Some other physical symptoms might also be shown such as carious teeth, enlarged liver and spleen, diarrhea (which is believed is due to lysosomal glycosaminoglycans (GAG) storage in the neurons of the myenteric plexus3.)
Respiratory compromise can occur and is related to airway obstruction due to anatomical changes, excessive thick secretions and neurologic impairment. Upper respiratory tract infections and sinopulmonary disease are common.
4. Work-up:
In order to diagnose patients with Sanfilippo Syndrome, specific enzymatic assays in cultured skin fibroblasts and in peripheral blood leukocytes are used (e.g. enzymatic cell analysis). One indicating sign for this syndrome is the increase in the level of heparan sulfate in the urinary secretion. Thus a total quantitative or a fractionation test are carried by performing either electrophoresis or chromatography with the purpose of measuring how much Glycosaminoglycans (GAGs) is in the urine. Due to the higher level of GAGs in newborns and infants, age-specific controls and fractionation must be included to accurately quantify the level of GAGs.
Imaging studies can also be used to look for changes in brain structure since the spectrum of skeletal changes can be seen in patients with Sanfilippo syndrome.
5. Treatment:
Currently, there is no available treatment for the real cause of Sanfilippo syndrome. Bone marrow transplantation and Enzyme replacement therapy only work for patients with mucopolysaccharidosis I, II, and VI (not III). However, some promising therapies are making their ways to be FDA-approved.
Interferons are specialized proteins that come in three various classes alpha, beta and gamma. Though each varies slightly in function they are all produced by the immune system and released in the response of a pathogen as an antiviral agent. When released in the body they have the sole function of identifying infection, activating immune cells and increasing antigen production. They are used by cells as a way to warn others of the presences of a viral cell. To do this the cell releases interferons, which then send signals to close cells so they may create a protective defense. Once released the cells within the presence of the viral cell produce two enzymes called protein kinase R (PKR) and RNAse L. At the release of these enzymes RNA is destroyed and protein synthesis is reduced, thus preventing protein synthesis of the viral gene. Interferons also have the ability to increase the activity of the p53 gene and produce numerous other proteins to help combat a viral infection.
Hypoxia-inducible factor (HIF) is the transcription factor responsible for regulating the body’s response to hypoxia. Hypoxia is a state of reduced oxygen in the blood. Without oxygen, the body’s normal pathways and cycles are disrupted, thus resulting in cellular and inevitably human death. HIF translation and degradation are studied because of its link to tumor growth. Tumor hypoxia shows resistance to cancer treatment, such as radiation and chemotherapy and an increased expression for HIF-1 enables tumor growth. Thus the regulation of HIF is a target for advances in cancer research.
HIF is a heterodimer with α and β subunits. Of the three known HIF-α isoforms (HIF-1α, HIF-2α, HIF-3α), HIF-1α is most common.
In a state of hypoxia, HIF-1α responds by promoting anaerobic metabolism through glycolysis. HIF-1α increases the transportation of glucose by upregulating the expression of glucose transporters and glycolytic enzymes. HIF-1α also upregulates pyruvate dehydrogenase kinase 1, which increases the conversion from pyruvate to lactate. This in turn steers activity away from the oxygen-dependent metabolic pathways of the Kreb’s cycle and oxidative phsophorylation by increasing lactic acid production. In addition, HIF-1α also balances the potentially toxic buildup of lactic acid and carbon dioxide by increasing the monocarboxylate transporter 4 and membrane-bound carbonic anhydrase IX which react with the lactic acid and carbon dioxide to prevent the levels from becoming toxic.
HIF-1α is also responsible for activating angiogenesis. Angiogenesis restores the supplies of oxygen and nutrients by forming new blood vessels. Although this may sound like something positive, increased angiogenesis enables tumor growth.
Studies have shown that HIF degradation occurs both in the presence and in the absence of oxygen. Oxygen-dependent degradation (ODD) includes pVHL and SUMOylation. Oxygen-independent degradation includes HAF and RACK1.
The pVHL-HIF-1α degradation pathway
Under aerobic condition, HIF-1α is hydroxylated by prolyl hydroxylases( PHD) at two conserved proline residues located within its oxygen-dependent degradation(ODD) domain. Under hypoxic condition, PHD activity is inhibited by enzyme therefore stabilizing HIF-1α. In addition, hypoxic condition also causes perturbation in the electron transport chain in mitochondria and increases the level of cytoplasmic ROS (reactive- oxygen species) which alters the oxidation state of Fe2+, a cofactor for PHD activity. This effect also inhibits PHD and promotes HIF-1α stabilization. HIF-1α hydroxylation helps pVHL bind to the HIF-1α ODD. Then, pVHL will form the substrate-recognition module of an E3 ubiquitin ligase complex which will direct HIF-1α proteasomal degradation.
In the pVHL (von Hippel Lindau) pathway, the presence of oxygen hydroxylates the HIF-1α. The hydroxylation binds HIF-1α to the pVHL which then forms an E3 ubiquitin ligase complex which essentially tags the HIF-1α for degradation. The SSAT2 regulator also binds and stabilizes the interaction between the HIF-1α and pVHL.
SUMOylation is used to regulate protein properties. This modification in polypeptides have been used to study the amyloid-beta peptide levels which is linked to the human condition, Alzheimer's Disease. Because of sumoylation, it was discovered that familial dilated cardiomyopathy was caused by a decrease in lamin A sumoylation which leads to increased cell death.
SUMOylation leads HIF-1α to bind to the same E3 ligase as in the pVHL pathway but does so under hypoxic conditions. The SUMO protein binds to the HIF-1α, tagging it to attach to the E3 ligase to eventually degrade.
The HAF (Hypoxia-associated factor) is a multi-functional protein. At the C-terminus it has an E3 ligase which binds to the ODD domain of HIF-1α and tags it for degradation. This occurs regardless of the presence or absence of oxygen. At the N-terminus it promotes the translation for some of the HIF-1α targets, thus contradicting the degradation work of the C-terminus. However, HAF selectively activates some of the targets, but not all. This dual functionality provides potential opportunities for therapeutic regulation.
RACK1 binds with HIF-1α and tags it with the E3 ligase for degradation. In contrast to the SSAT2 regulator which promotes the ODD pathway of pVHL, the SSAT1 regulator stabilizes the bond between RACK1 and HIF-1α .Calcium affects this pathway. HIF-1α degradation is inhibited when calcineurin A dephosphorylates RACK1, thus preventing it from binding with HIF-1α.
When in a state of hypoxia, general protein translation is inhibited to in order to decrease the amount of energy consumption. However, the translation of HIF-1α is not disrupted. The exact mechanism and reasoning behind this phenomenon is not completely understood. One proposed pathway is through RNA sequences that do not need a cap-binding complex to form secondary and tertiary structures and to bind directly to the ribosome.
Modern research have disclosed the presence of HIF switch, which are mechanisms that are qualified of directly altering the HIF-α isoform. Examples of HIF switches include Hsp70/CHIP axis, which encourages the particular deterioration of HIF-1α in diabetes-associated hypoxia and hyperglycemia. As a consequence, this gives diabetic complications affiliated with impaired hypoxic response and cell destruction. Histone deacetylase SIRT1, another HIF switch, which has a tendency to deacetylate HIF-2α, and increases HIF-2 activity during hypoxia. Recent evidences have shown that SIRT1 has left traces in regulating HIF-1. HAF, a crucial HIF-α isoform target regulator, specifically attached to and degrades HIF-1α in an oxygen-independent case. However, it also enhances HIF-2α transactivation and constancy. HAF encounters a decrease when exposed to chronic hypoxia, but develops with extended hypoxic exposure. Regardless, the switch from HIF-1α towards HIF-2α is a necessity for cells.
Even though there is much research needed to aid the understanding of HIF-1α to HIF-2α switch, current knowledge holds value in the growth of cancer. Identified HAD is a crucial component has proven to enhance tumor initiation and progression.
Hypoxia refers to the condition of which inadequate oxygen is supplied to tissues and cells. The hypoxic response is facilitated by the hypoxia-inducible transcription factors, HIF-1 and HIF-2. HIF target gene activation is very specific and not indicative of which HIF alpha isoform is active.
Hypoxia and the HIFs in human physiology and disease
The oxygen tension in tissues is a lot lower than the ambient oxygen tensions due to the dramatic decrease in blood oxygen content as oxygen is released in the cells. Low oxygen or hypoxia acts as a stimulus for proper embryogenesis and wound healing and maintains the pluripotency of stem cells. Pathological hypoxia could be a result of high altitude or localized ischemia due to disruption of blood flow to a given area. Solid tumours also have hypoxic regions due to the severe structural abnormality of tumour microvessels. As a response to hypoxia, HIF transcription factors transactivate many genes including those that trigger angiogenesis, anaerobic metabolism and resistance to apoptosis. Structurally, HIFs are heterodimers that consist of one of three major oxygen labile HIF alpha subunits (1alpha, 2alpha, 3alpha) and a constitutive HIF1 beta subunit that combine to form the HIF-1, HIF-2, HIF-3 transcriptional complexes. Majority of the studied has been done on HIF-1alpha and HIF-2alpha. HIF-3alpha has similar basic helix-loop-helix and Per-Amt-SIM (PAS) domains as HIF-1alpha and HIF-2alpha, but it does not have the C-terminal transactivation domain.
HIF-1alpha and HIF-2alpha are non-redundant, and they have distinct target genes and mechanisms of regulation. In some circumstances, HIF-1 drives the initial response to hypoxia, but after long exposure, it is HIF-2 that drives the hypoxic response.
Under aerobic condition, both HIF 1 and 2 alpha are hydroxylated by specific prolyl hydroxylases at two conserved proline residues positions in the oxygen-dependent degradation domain. This reaction requires oxygen, 2-oxoglutarate, ascorbate, and iron (Fe2+) as a factor. Von Hippel-Lindau protein forms the substrate recognition module of an E3 ubiquitin ligase complex that directs HIF – 1 and 2 alpha polyubiquitylation and proteasomal degradation. Under hypoxic conditions, prolyl hydroxylase acitivity is inhibited, von Hippel-Lindau binding is abrogated and HIF-1 and 2 alpha are stabilized. Under normoxic conditions, HIF-1 and 2alpha cannot activate transcription due to oxygen-regulated enzyme, factor inhibiting HIF-1. Asn hydroxylation is also inhibited, which allows the p300/CBP complex to bind to HIF 1 and 2alpha, which results in HIF transactivation.
HIF-1alpha is known to be the master regulator of the hypoxic response and the important node that ensures the survival during hypoxic stress. HIF-2alpha was known as the endothelial PAS domain protein, an endothelium specific HIF-alpha isoform, which was thought to be more specialized that HIF-1alpha. Since HIF-2alpha is expressed in tissues of brain, heart, lung, kidney, liver, pancreas, and intestine, it suggests that it has roles in the hypoxia response. Recent studies show that both HIF-1 and HIF-2 participate in hypoxia-dependent gene regulation through complex and even antagonistic interactions. Post-DNA binding mechanism may be required for transactivation, because studies show that DNA binding does not have to correspond to increased transcriptional activity. Recent research confirms that endogenous HIF-2alpha is the main driver of EPO production. HIF-1 produces genes that encode glycolytic enzymes, enzymes that are involved in pH regulation, enzymes that promote apoptosis. HIF-2 produces genes that are involved in invasion and is proved to regulate enzymes in the glycolytic pathway without HIF-1. Interestingly, HIF-1 and HIF-2 are sometimes able to substitute for the isoform-specific functions of the other, meaning that their ability to activate specific target genes depends on specific context.
Different temporal and functional roles of HIF-1 vs. HIF-2
Multiple mechanisms converge to suggest context-dependent, HIF-alpha isoform-specific activation in response to variations in hypoxic intensities and duration. The balance between HIF-1 and HIF-2 activation allows the coordination regulation of the complex hypoxia-dependent processes that takes place in physiology.
During early embryonic development, the physiological hypoxic environment actives HIFs with the help from other non-hypoxic stimuli such as the renin-angiotensin system, growth factors, and immunogenic cytokines, which all regulates placental development and maturation. Embryonic blood vessels are generated through vasculogenesis, where cells are differentiated into endothelial cells. More blood vessels are made using both sprouting and non-sprouting angiogenesis, which can be remodeled into an adult circulatory system. The differential requirement for HIF-1 and HIF-2 activation during vessel formation and mutation is shown through studies done on mouse. HIF-1alpha knockout mice shows impaired erythropoiesis and finds cephalic vascularization in neural fold formation and the cardiovascular system. With various backgrounds, the HIF-2alpha mice can die either by E12.5 with muscular defects or months after birth due to multi-organ pathology and metabolic abnormalities. Loss of either HIF-1alpha or HIF-2alpha inhibits tumour angiogenesis in adult mouse, which suggests that HIF-1 drives vasculogenesis and early stages of angiogenesis. The formation of complete vasculature requires a smooth transition from the largely HIF-1-dependent transcription, through the period in which both HIF-1 and HIF-2 drive overlapping functions, to the HIF-2 dependent stage of vascular maturation.
Bone can be formed through the mechanisms intramembranous and endochondrial ossification. Intramembranous ossification happens when the flat skull bones are formed and also the mesenchymal cells are differentiated into osteoblasts. Endochondrial ossification happens when other bones are developing; it has a two-staged mechanism. The mesenchymal cells change to chondrocytes, the primary cell type of cartilage, which forms an avascular and highly hypoxic matrix template or growth plate. As a permanent stress, hypoxia influences general chondrocyte metabolism and tissue-specific production of cartilage matrix proteins such as type two collage. The cartilaginous matrix was then replaced by highly vascularized bone tissue through degradation of the matrix and blood vessel invasion. Endochondral ossification requires both the hypertrophic differentiation of chondrocytes and the conversion of avascular cartilage tissue into highly vascularized bone tissue by degradation of the cartilage matrix, and vascular invasion, mainly through the activation of VEGF.
Recent studies show that the HIF pathway is participating in membranous ossification and in both stages of endochondral ossification by binding angiogenesis to osteogenesis and regulating the spatiotemporal onset of angiogenesis in the growth plate. Both HIF-1alpha and HIF-2alpha are expressed in growth plate chondrocytes, HIF-1alpha is expressed in similar levels during all stages of chondrocyte differentiation, with its activity enhanced by hypoxia. HIF-2alpha is independent of oxygen-dependent hydroxylation as its levels increase with chondrocyte differentiation. HIF-1 functions as a survival factor in hypoxic chondrocytes by increasing anaerobic glycolysis and hindering apoptosis. It also promotes autophagy, which could extend the lifespan of chondrocytes. HIF-1 is also crucial in extracellular matrix synthesis, which involves the expression of important components required by proliferating chondrocytes in the proliferating zone. HIF-2 is a potent transactivator of many genes, such as type X collagen. Increased levels of HIF-2alpha have been correlated with the development of osteoarthritis. This suggests that HIF-1 is important in the process of hypoxia-dependent cartilage formation and maintenance. HIF-2 alpha participates in endochondral ossification and cartilage destruction, which may be less hypoxia-dependent. Both HIF-1 and HIF-2 are required for developing skeletal vascularity. HIF-1 is important for early stages during severely hypoxic conditions, whereas HIF-2 is more important for later stages.
Tumour hypoxia promotes tumour regression and resistance to therapy. It promotes the survival of tumour cells by shifting cells towards anaerobic metabolism, neovascularization and resistance to apoptosis. Hypoxia triggers increased genetic instability, invasion, metastasis and de-differentiation, which lead the tumour aggressiveness. Increased levels of tumour HIF-1alpha is associated with poor patient prognosis in multiple tumour types.
(a) This shows the oxygen gradient generated by the lack of oxygen within solid tumours. In (i), vessel occlusion or rapid tumour growth causes acute hypoxia that activates HIF-1alpha and HIF-2alpha. In (ii), only HIF-1alpha is activated to promote acute hypoxia respons, which could lead to angiogenesis or reperfusion or cell dealth shown on (iii). (iv) shows that chronic hypoxia can increase hypoxia-associated factor and HIF-2alpha levels by mediating a switch to HIF-2-dependent transcription that triggers tumour adaptation, proliferation and progression. (b) uses the blue line to show temporal regulation of HIF-1alpha, green line to show HIF-2alpha and red line to show HAF in response to chronic hypoxic exposure. The dashed lines show where the switch from HIF-1alpha to HIF-2alpha occurs.
Tumour HIF-1 provides an immediate response to acute or transient hypoxia due to rapid induction and negative feedback regulation. HIF-2alpha seems to be favoured by chronic hypoxic exposure. The HIF switch is clearly observed during development of RCC, where there is a slow shift from HIF-1alpha to HIF-2alpha expression with increasing tumour grade.
Stem cells have the ability for self-renewal, multilineage differentiation potential, and long-term viability. Embryotic stem cells can be extracted from the inner cell mass of blastocysis. Adult stem cells are found in tissues such as blood, bone marrow and adipose tissue. Both normal and malignant stem cells are situated in specialized areas where factors such as low oxygen play a crucial role in maintaining pluripotency and viability. Tumour cells have been shown to go through de-differentiation under hypoxic conditions. Thus, HIF-1 and HIF-2 both trigger the hypoxia-induced undifferentiated phenotype by activating the Notch pathway and activating the transcription of other stem-cell-specific factors. HIF-1 is the main driver for hypoxia-induced transcription in non-neoplastic embryonic stem cells. It is required for maintenance of the undifferentiated phenotype in GBM stem cells under hypoxic conditions. HIF-2 seems to be nonfunctional for this part. However, it is required for the proliferation of both stem and non-stem GBM cells; it is especially required for the survival of GBM stem cells. Both HIF1 and HIF2 can have hypoxia-independent functions in CSC maintenance. It seems that the HIF2 has similar functions as HIF1; however, it differs from HIF1 in that it has a unique role in stem cell maintenance under physiological oxygen tension, independently of hypoxia.
Studies have been conducted in understanding the complex regulation of HIFs in both physiological and pathophysiological processes. HIF-1 plays an important role in early vascular and bone development. The HIF switch is also seen in solid tumours where HIF-1 triggers the initial response to hypoxia and then HIF-2 triggers the hypoxic response during chronic hypoxia exposure. Thus, it is crucial for cells to switch from HIF-1 to HIF-2 whenever needed.
This research papers delves into the idea of the von Hippel-Lindau protein that is negatively regulated by beta-Catenin and Jade-1 via various mechanisms. One must first understand the proteins to connect the significance of their partnership.
Von Hippel-Lindau is a tumor suppressor protein encoded by the VHL gene. The inactivity of the protein is associated with Von Hippel-Lindau disease. The VHL protein works through E3 ubiquitin ligase activity which targets specific proteins for degradation. However, the inactivity of the protein may cause an excess growth of cells, or a tumor.
Beta-Catenin, or β-catenin, is part of a series of proteins that constitute adherens junctions and maintain epithelial cell layers in organs. The protein is used to regulate the production of VHL through negative feedback. This research paper focuses on Wnt- β-catenin which undergoes signal transduction. Wnts in particular “comprise a conserved family of secreted” (Berndt).
Jade-1 binds to β-catenin to “negatively regulates β-catenin levels in the absence of the Wnts stimulation” (Berndt). The expression of Jade-1 is what primarily determines the reduction of β-catenin. In some cases, the over expression of Jade-1 can reduce the presence of β-catenin by 50-60%. Jade-1 may be used in conjunction with VHL to regulate the production of β-catenin.
Lysozymes, also known as muramidase or N-acetylmuramide glycanhydrolase, are small globular protein enzymes composed of 129 amino acid residues. As one of the first enzymes to be studied, Alexander Fleming had shown them to be produced by phagocytes and epithelial cells (Neufeld).They are part of the glycoside hydrolase family, which are known for damaging the cell walls of bacterial cells by catalyzing hydrolysis of 1,4-beta-linkages. In a peptidoglycan, the 1,4-beta-linkages between N-acetylmuramic acid and N-acetyl-D-glucosamine are hydrolyzed; in chitodextrins, the 1,4-beta-linkages between N-acetyl-D-glucosamine residues are hydrolyzed. Lysozymes can be found in tears, human milk, saliva, and mucus. As such, lysozymes act as part of the body's defense system against bacteria. High concentrations of lysozymes can also be found in egg white. Their ability to break down bacterial cell walls in order to improve protein and nucleic extraction efficiency make lysozymes important proteins in living organisms. In human beings, the LYZ gene is responsible for encoding the lysozyme enzyme.
Laschtschenko first discovered lysozymes in 1909, when he first observed the antibacterial property of hen egg whites. However, it wasn't until about a decade later until scientists used the term. In 1922, Alexander Fleming, who also discovered penicillin, observed the antibacterial effect of treating bacterial cultures with nasal mucus from a patient suffering a cold. As discussed above, he noticed that lysozymes were secreted from such places in the body. It wasn't, however, until 1965 that the three-dimensional structure of lysozyme was determined by David Chilton Phillips. Utilizing X-ray crystallography with a 2 angstrom resolution, a hen egg-white lysozyme model was determined, being the first enzyme structure to be observed using X-ray analysis. The lysozyme was the first enzyme structure to be solved with X-ray diffraction tools. It was also the first enzyme to be fully sequenced and determined to contain all 20 common amino acids. And mechanistically, it was the first enzyme to be well studied and understood.
The best studied lysozymes are from hen egg whites, from which lysozymes are abundant, and bacteriophage T4. This enzyme was the first enzyme to have its structure determined, although crystal structures of other proteins had been determined previously. Lysozymes are often easy to study through X-ray crystallography due in part to their easy ability to be isolated from egg whites and to be crystallized, features used widely in the purification of lysozymes. Lysozymes are a part of the immune system and can protect against E. coli, Salmonella, and also Pseudomonas.
Lysozyme Active Site
Since Fleming's discovery of lysozyme, undoubtedly the most significant contribution to our knowledge concerning this enzyme was the X-ray crystallographic analysis. The X-Ray Crystallography
structure of lysozyme has been determined in the presence of a non-hydrolyzable substrate analog. This analog binds tightly in the enzyme's active site to form the ES complex, but ES cannot be efficiently converted to EP. It would not be possible to determine the X-ray structure in the presence of the true substrate, because it would be cleaved during crystal growth and structure determination.
The active site consists of a crevice or depression that runs across the surface of the enzyme. Look at the many enzymes contacts between the substrate and enzyme active site that enables the ES complex to form. There are 6 subsites within the crevice, each of which is where hydrogen bonding contacts with the enzymes are made. In site D, the conformation of the sugar is distorted in order to make the necessary hydrogen bonding contacts. This distortion raises the energy of the ground state, bringing the substrate closer to the transition state for hydrolysis. [2]
Chicken-type and goose-type lysozyme have a very high anti-bacterial potential mainly against Gram-positive bacteria, this has practical uses in food, pharmaceutical, and medical industries. The lysozyme's antibacterial properties are most highly effective against those Gram-positive bacteria with a cell wall made of a peptidoglycan layer. The properties of the lysozyme have made it possible to be integrated into food packaging materials which can greatly extend the shelf-life of scarcely processed food, protecting it from microbial contamination. It has also been shown to preserve food items such as vegetables, milk, meat, etc. It has been seen to help control the production of lactic acid in the wine-making process. The pharmaceutical industry has seen success with using hen-lysozymes in inflammatory diseases, as well as bacterial and viral diseases. [3]
Neufeld, Elizabeth. "From Serendipity to Therapy",Annu. Rev. Biochem, 2011.
Lesnierowski G., Cegielska-Radziejewska R., 2012. Potential possibilities of production, modification and practical application of lysozyme. Acta Sci. Pol, Technol. Aliment. 11(3), 223-230.
Oct1 and Oct4 are two transcription factors that are key regulators of pluripotency and stem cells and they are the proteins most involved with adult stem cells and cancer. Oct proteins are defined by their ability to interact with a DNA sequence known as the ‘octamer motif’. Oct4 and Oct1 belong to a group of proteins called POU. POU proteins are divided into six classes (POU1 to POU6) based on their DNA binding domains. In this, it can be seen that Oct 4 is an outlier as the DNA sequence it recognizes is different than the other proteins in the POU group. The Oct proteins, in general, can also recognize non octamer sequences due to the conformational flexibility of their linker domain.
Oct4
Oct 4 is the main regulator of pluriotency. Its expression is extremely restricted and limited to the early embroyic development stage. Oct4 is also necessary for ES cell phenotype and for the development of a cell past the blastocyst stage. Because Oct4 is responsible for pluripotency, elevated or reduced expression can lead to excessive changes in the amount of cells. Therefore, the Oct4 protein is highly regulated at multiple levels. While Oct4 is currently used as a min component to generate iPS(induced pluripotent stem) cells there is still a long way to go before iPS can be widely used to generate all types of cells. It has also been found that aggressive human breast carcinomas are devoid of consistent Oct4 expression. Instead, Oct4 paralogs that assume Oct4-like functions are found in these tissues.
Oct1
Oct 1 shares the same functions, targets and regulation of common genes however it cannot substitute for Oct4 in the generation of pluripotency cells. The protein is mainly expressed in adult and embryonic tissues. Because of its function, Oct1 is also heavily regulated on multiple levels. Recent research has pointed to the idea that Oct1 might play a part in regulating its own synthesis. Oct1 deficient cells are extremely sensitive to glucose withdrawal and oxidative stress agents. The metabolic changes in Oct1 deficient cells directly contrast the metabolic changes in tumor cells, implying that Oct1 proteins might have a cancer-protecting effect. Because of this Oct1 is currently one of the main avenues for cancer research. Currently, it has been seen in lab that loss of Oct1 reduces oncogenic transformation in culture and cancer incidences in mouse models.
Kang, J. (2009) Stem cells, stress, metabolism and cancer: a drama in two Octs. Cell Press, 491-499
Lafora Disease, is a fatal autosomal recessive genetic disorder characterized by the presence of inclusion bodies, known as Lafora bodies, within neurons and the cells of the heart, liver, muscle, and skin.[1] Lafora Disease is caused by mutations in the EPM2A (Epilepsy, progressive myoclonus 2A) gene encoding laforin, a dual-specificity phosphatase, or in the EPM2B (Epilepsy, progressive myoclonus 2B) gene encoding malin, an ubiquitin ligase.[2]
Lafora Bodies
Lafora disease is distinguished by the presence of inclusions called "Lafora bodies" within the cytoplasm, the viscous fluidic matrix inside of cells. Lafora bodies are present primarily in neurons, but they have also been found in other organs. Lafora bodies are composed of abnormal glycogen called polyglucosans. These starch-like polyglucosans are insoluble and hence precipitate inside cells (Lafora Bodies shown in Fig. 1). Polyglucosan bodies appear with age; in Lafora disease, their numbers have increased enormously. Lafora bodies have been observed in virtually all organs of patients with the disease. In the brain, their presence appears to be restricted to neurons; they do not seem to present in astrocytes. Their morphology varies from tissue to tissue, but they generally contain a central core and have a peripheral fluffy appearance.[5] Lafora bodies are composed of glucose polymer (polyglucosan) that is chemically but not structurally related to glycogen (Fig.2).
The protein Laforin has two essential roles: First it dephosphorylates glycogen to inhibit excess glycogen phosphorylation and Lafora Body formation, Secondly it brings the protein Malin to the site of glycogen synthesis.The protein Malin can then ubiquinate (give the kiss of death to) PTG (protein targeting to glycogen), GS (glycogen synthase), GDE (glycogen-debranching enzyme), and other proteins to inhibit the formation of Lafora Bodies.[3] Malin, also interacts with laforin and promote the polyubiquitination and degradation of laforin in vitro and in cultured cells.
Since Laforin acts as a phosphatase it removes phosphomonoesters so that glycogen production proceeds normally, without laforin the phosphomonoesters build up and affect glycogen branching and lead to the formation of Lafora Bodies.[4] The Lafora bodies contain more phosphate and its branching is discontinuous compared to glycogen, which make Lafora Bodies insoluble in water. Due to the mutations/defects with the laforin protein, Lafora bodies begin to build up causing Lafora Disease. Also without the laforin protein malin would not be able to locate PTG, GS, and GDE.
Malin functions to maintain laforin associated with soluble glycogen and that its absence causes sequestration of laforin to an insoluble polysaccharide fraction where it is functionally inert.[2]
The laforin-malin complex acts as a controlled 'garbage disposal'[2] to ubiquitinate and degrade proteins involved in glycogen metabolism. Minor defects with these important proteins will lead to Lafora Disease.
Lafora Disease patients with the malin protein defect live 25% long lives than patients with the laforin defect.
Histology of Lafora Bodies
Lafora bodies vary in size from 1 to 30 micron in diameter and one or more Lafora bodies may be present in the cytoplasm. They may be found in nerve cell processes and apparently free in the neuropil(A region between neuronal cell bodies in the gray matter of the brain and spinal cord)[3]. They have a concentric target like lamination, PAS positive, diastases resistant, and Alcian blue positive. The core is more strongly stained than the rim. Lafora bodies are also basophilic, and variably metachromatic (with methyl violet or toluidine blue) inclusion bodies. They are also found in liver, striated muscles, sweat glands. [4]
There are a vast majority of non-common amino acids. There are typically presented as twenty to twenty two common amino acids. The most common amino acids are Glycine, Alanine, Valine, Leucine, Isoleucine, Methionine, Proline, Phenylalanine, Tyrosine, Tryptophan, Serine, Threonine, Asparagine, Glutamine, Cysteine, Lysine, Arginine, Histidine, Asparatate, and Glutamate. There are also two amino acids that aren't typically included in the standard and they are selenocysteine and pyrrolysine.
This amino acid is considered one of the primary amino acids. There is typically considered twenty-two primary amino acids. This one is similar to Cysteine although it does not contain sulfur. Instead the R group on this amino acid is -CH2SeH. The sulfur that is typically present in the amino acid cysteine is now replaced by a selenium atom.
This amino acid is also considered one of the primary amino acids but is relatively not included. It obviously has similarities to lysine in that its R group is lysine but it has an added extra bit to the end of the lysine. The added end ring is considered a pyrroline ring. This is where it gets its name pyrrolysine from by combining the two names together.
There are a number of other amino acids but are not considered primary amino acids. These amino acids are usually the result of changes are the amino acid is joined into a protein. These are called post translational modifications and when this happens it can change the structure of the amino acid.
There are several entire and localized proteins that do not fold into 3-D structures yet are able to function fully. Instead of the usual linear pathway of proteins (sequence-to-structure-to-function), these unfolded protein's functions come from different forms such as structured globules, collapsed disordered ensembles, and extended disordered ensembles). In addition, function can also arise from a disorder-to-structure transition. The understanding of these non 3-D structured proteins can help to diversify the knowledge of proteins and how they function in comparison to the globular 3-D structures.
Since protein folding is directed by the amino acid sequence, the test to determine whether the non-folding proteins were guided by an amino acid sequence was carried out. The development of predictors to test out this hypothesis that the amino acid sequence specified no protein folding showed that the predictor accuracy was much better than expected by chance. This, in turn, revealed that protein non-folding is most likely within the amino acid sequence. The depletion of C, W, Y, F, I, V, and L residues, and the abundance of M, K, R, S, Q, P, and E residues reveals the decrease in residues that form hydrophobic interiors of structured proteins and the increase in residues that form the surface of structured proteins. This decrease and increase in the specific residues shows why the non-folding proteins do not fold into their 3-D structures.
Eukaryotes contain the biggest fraction of disordered proteins while archaea and eubacteria are in similar amounts, but far behind the amount present in eukaryotes. In addition, multicellular eukaryotes have more disordered proteins than mono-cellular eukaryotes.
The partitioning of structured proteins according to their amino acid sequence or function can be very useful because it allows for simple access to a wide variety of proteins and easy grouping of newly discovered ones. However, unstructured proteins and regions are hard to place into distinct groups because of their diversity, lack of a 3-D structure, and variance in their amino acid sequence. An example of this problem can be seen in the short amino acid linker in calmodulin, which forms a helix in the crystallized form but is flexible in solution. The disordered region in calmodulin allows for it to bind to a wide range of target sequence because the side-chains in the methionine-rich hydrophobic areas of the calcium-binding regions are flexible. Another example can be seen in the longer disordered region of PEVK in titin. PEVK can range from about 180 to 2174 residues, depending on the circumstances. The disordered region contains 180 residues in the cardiac muscle isoform while 2174 corresponds to the soleus muscle isoform. Both of these disordered regions help to maintain the appropriate length of muscle fibers. The wide range of function and variability in sequence portrays the difficulty in grouping these disordered proteins together.
Yet, partitioning was still accomplished through grouping the disordered proteins into homogeneous subsets. The disordered regions were randomly grouped into subsets and then different predictors were developed for each separate subset. The assembly of disordered regions were repartitioned into different groups again according to which predictor provided the best results. Then, new predictors would be constructed on the basis of the repartitioned subsets, and the steps would be repeated until there were no more changes with each of the new cycles. From this approach, three types or flavors were found and named V, C, and S. Flavor S contained a large amount of protein-binding regions, flavor V was rich in ribosomal proteins, and flavor C was high in the number of sites of protein modification.
Non-folding proteins and regions have significant duties in biological functions, taking part in signaling and regulatory pathways, through specific protein-protein, protein-nucleic acid, and protein-ligand interactions. Detailed functions of non-folding proteins and regions can be depicted in four categories: 1) molecular recognition, 2) molecular assembly, 3) protein modification, and 4) entropic chain activities. Non-folding proteins and their wide range of partners in interaction allow for the organization of complex protein-protein networks.
The disorder-associated and structure-associated functions in Swiss-Prot, a protein database, were identified recently. There were 310 structure-associated, 238 disorder-associated, and 170 structurally ambiguous, out of 710 functional keywords. This revealed the functional diversity of disordered proteins working in complement with structural proteins. Another test showed that the disordered proteins had more functions than the structural proteins, with the non-folding dealing with the signaling and regulatory processes while the folded proteins were associated with catalysis and transport.
Non-folding proteins and regions usually partook in molecular interactions controlled by localized binding sites such as eukaryotic linear motifs (ELMs), short linear motifs (SLiMs), and molecular recognition features (MoRFs). ELMs and SLiMs were both identified to be short sequence patterns in many proteins that bind to a common target. On the other hand, MoRFs are identified by a pattern in a disorder prediction output. In addition, non-folding regions are also primary loci for alternative splicing.
A summary of some protein functions associated with structural disorder :
The protein San1 functions as an E3 ubiquitin ligase and the role of the disorder consists of recognizing mis-folded substrates. The protein Hsp-33 functions as a redox chaperone and the role of the disorder consists of adhering mis-folded structures. The protein pHD functions as a bacterial antitoxin and the role of the disorder surrounds the allosteric regulation of bacterial toxins. The Sic1 protein functions as a cyclin-dependent kinase inhibitor and the role of the disorder includes "polyelectrostatic" interactions with Cdc4 ubiquitin ligase. The protein WASP functions as a regulator of actin polymerization and the role of the disorder is allosteric regulation. The protein p27 functions as a cyclin-depenedent kinase inhibitor like Sic1, however the role of its disorder is the regulation of targeted degradation. The protein CREB functions as a general transcription co-activator and the role of the disorder consists of interacting through induced folding by a large range of transcription factors. LEA proteins function as stress response proteins in plants and animals and the role of the disorder includes chaperone function in abiotic stress via disorder transfer.
Non-structured proteins have been figured out to be an influence in human diseases since many of the non-structured proteins are either wholly disordered or have a large stretch of disordered sequences. An important malfunction that occurs in the human body because of these disordered proteins is the aggregation of non-folding protein sequences to amyloid fibrils rich in ß-structure, which is associated with the pathogenesis of neurodegenerative diseases such as Alzheimer's, Parkinson's, Huntingtion's, and prion diseases.
Events in alpha synuclein toxicity
Oligomers or protofibrils of the already disordered polypeptides seems to be the pathogenic entities that are involved in diseases such as Alzheimer's. It's been suggested that their mode of action may involve creating pores in the plasma membrane of the affected cells. Techniques have since been used to show that amyloid peptides that are involved in several diseases had similar channels. AFM or atomic force microscopy showed that pore-like structures for amyloid peptides were reorganized into the lipid bilayers. Another example is the family of synucleins which contain three homologous proteins called α-synuclein, ß-synuclein, and γ-synuclein. All of these three proteins contain roughly 130 amino acid residues, which are usually intrinsically disordered proteins. With the α-synuclein, it is typically the aggregation of it into oligomers, protofibrils, and fibrils that makes it closely related to Parkinson's disease, Lewy body dementia, and all kinds of other neurodegenerative diseases which are known as synucleinopathies. However, unlike α-synuclein, ß-synuclein and γ-synuclein have a smaller chance in fibrillating and can also prevent fibril formation in α-synuclein. α-synuclein has shown to be structurally plastic as it can adopt several structurally unrelated conformations. These features are very reliant on the protein environment and on the availability of binding partners. In addition, α-synuclein have also been known to conform to α-helices when associated with phospholipids or micelles.
Structural disorder was also found in multiple other disease-related-proteins such as p53 and the cystic fibrosis transmembrane conductance regulator (CFTR). Within these proteins associated with cancer, neurodegenerative diseases as stated earlier, cardiovascular diseases, and diabetes structural disorder was discovered. Scientists hypothesize that structural disorder allows for the cellular presence of oncogenic protein chimeras. A negative point of structural disorder is apparent in the dosage sensitivity of genes that produce agitation if over-expressed.
Structural disorder is critical for pathogens. Examples consist of virus entry, replication, and budding that have a basis for deregulating the signaling of the host cell and is carried out utilizing differing interactions of viral proteins with key host regulatory proteins.
Studies of these intrinsically disordered proteins ensure a more in depth apprehension of the cause and advancement of multiple disease states. Even better, they assist in improving antidotes against unfavorable conditions.
Intrinsically disordered proteins are different than other solutions to diseases on the market for they do not hold any enzymatic activity. Traditional drugs usually tag the active sites or the ligand-binding pocket of enzymes or receptors. Intrinsically disordered proteins commit in protein-protein interactions that are intervened with by way of small molecules. This partner-targeting method has been advocated for drug development. Current work still needs to improve this method in a cell.
In the past, many researchers were unclear if the structural disorder found in proteins occurred in vivo or in vitro due to the seclusion and large dilution of the protein in the test tube. Multiple studies demonstrate that macromolecular concentrations leading to crowding do not coerce intrinsically disordered proteins to fold completely in the cell. NMR experiments supported the argument of disorder existing in vivo for it was applied to study alpha-synuclein, which is over conveyed in E.coli cells. Also different functional studies prove in vivo for the study of chaperone function affiliated with structural disorder in a live cell. This was an example of indirect evidence.
Dunker, A.K., Silman, I., Uversky, V.N. and Sussman J.L., 2008, Function and structure of inherently disordered proteins, Current Opinion in Structural Biology, p. 756-764.
Lafontaine, Denis L.J. "A ‘garbage Can’ for Ribosomes: How Eukaryotes Degrade Their Ribosomes." Trends in Biochemical Sciences 35.5 (2010): 267-77. Print.
In recent years, scientists have accepted the standard that proteins are able to tolerate most amino acid substitutions but this has been tested and replaced by the concept that the deleterious effects of protein mutations is now the major constraint on protein's ability to change sequences and functions. This article analyzed the different methods for predicting stability effects after a mutation and the different mechanisms that are utilized to compensate for those effects that are destabilizing (and therefore encouraging protein evolability). The most widely accepted idea was that most positions on the protein were able to endure drastic sequence changes while also retaining the protein's configurational stability and function. And although there were exceptions to this view, this hypothesis made the assumption that stability is correlated with activity changes. In 2005, two papers were published that marked the importance of stability effects of mutations to protein evolution and these were then were then studied further to a new link between protein biophysics and it's molecular evolution.
Since mutations can be described as "raw material" for evolution, the selection to continue to sustain the existing structure and function abolishes most protein mutations and this therefore reduces the potential for future adaptions. This concludes the idea that only a small portion of every mutation that occurs will actually be fixed under positive selection to adopt and maintain a new kind of function. Neutral mutations are also termed "neutral drift" which can fix owing to random in small populations. But for the levels of the organisms, the reproduction rates (fitness, W) are not simple and they hardly ever relate with the properties of one type of gene or one type of protein. Because of the effects of redundancy, backup and robustness at a variety of different levels, the effects of mutations are therefore masked. For these reasons, it is safe to conclude that the effects of mutations is a difficulty for evolutionary biologists. But an equation can be utilized to therefore show a simple model of protein fitness. Protein fitness (W) is the fluctuation of an enzyme catalyzed reaction and this is then systematically related to the fitness of the organism in which this particular enzyme functions. So this continual flux is therefore related to (in terms of proportion) to the functional protein's concentration and it's function, f.
Research has shown that the concentration of functional protein is related to the protein stability. The deleterious effects of about ≥80% of mutations have been rooted from their effects on stability and folding. Protein disfunctionalization is then caused when the levels of soluble, functional proteins are reduced and protein disfunctionalization is then caused by destabilizing mutations beyond a certain level. Through measuring experimental different proteins the evidence shows that the probability of a mutation to be deleterious is therefore in the range of 33-40% (with a 36% on average). Therefore, it is clear that as mutations aggregate, protein fitness declines exponentially. This is shown through:
W≈e^(-0.36n)
The following equation then represents how with more mutations, the protein fitness then declines accordingly. The n is the average amount of mutations. Therefore, by the time am average protein accumulates (on average, it is about five mutations), the fitness will then decline <20%. So as the initial stability of a protein can shield some of the destabilizing effects of mutations and it can be concluded that the rate of protein evolution is dictated by the stability of the particular protein and therefore, the rate of protein evolution could thus be related to the obtaining of new functions.
(∆G) is used in various models to describe evolution because it is the definition of stability. Thermodynamic stability is therefore the energy difference between the unfolded and native state of the protein but this thermodynamic stability measurement is only reasonable for small proteins. But this calculation does not represent stability of proteins within cellular environments. Therefore, Kinetic stability is greatly valued because it relates the energy levels of the folding intermediates between the unfolded and native states of the protein and can include the mis-folded forms of the protein. Also, these can potentially lead to aggregation and if not degradation. Experimental data therefore relates the changes of thermodynamic stability of mutations which are available only for a small range of proteins. But recent studies have shown that there are advances in calculating have enabled for the prediction of ∆∆G values of mutations throughout a variety of wider range or proteins. The predictions can be based on methods such as based on sequences or the 3D structure of proteins and the combination of sequences and 3D structure of proteins have been combined as well. This prediction is largely correlated to the effects of mutations on the native state and therefore do not include the effects of the native site mutations. It's been noticed that the effects on folding in vivo overlap greatly with the thermodynamic stability effects. Therefore the predictions of kinetic stability effects would be of great value. So there is a challenge for more accurate predictions of the effects of mutations that can be related to protein levels because they remain in vivo.
Relationship between Stability and Protein Fitness
The relationship between protein fitness and mutation is dictated by the following equation:
This sigmodial relationship shows that more than 99% of folded proteins are given by a stability factor of -3kcal/mol where many proteins exhibit stability factors within the range of several k/mol. However ΔG values lower than -3kcal/mol risk shifting equilibrium away from folded, functional state of proteins. Using the above equation, a stability factor of less than -3kcal/mol indicates a certain amount of misfolded or partially folded proteins, which could lead to the irreversible effects of aggregation and degradation.
Threshold model
Threshold Robustness Model
The above equation also dictates a relationship in which proteins contain a certain limit to the amount of mutations it can handle before decreasing its fitness. [E0] (or protein fitness, as they are both proportional to each other) is fixed as long as ΔG remains above a certain threshold, referred to as ΔGt as shown in the figure “Threshold Robustness Model.” If the threshold were to increase (threshold robustness line in green), there will be a higher tolerance in mutations. Once mutations begin to accumulate, however, protein fitness begins to rapidly decrease. Many mutations leading to monogenic diseases show sigmoidal relationships.
Epistatic effects
The threshold model exemplifies negative epistasis (the increase in the harmful effect of a mutation while other mutations are present). As expected, the first few mutations have no to very little effect on protein fitness because an excess of stability buffers these destabilizing mutations. However, the buildup of more mutations is additive and leads to a reduction in stability which ultimately leads to a fitness decline. Negative epistatic effects dictate that because there is no immediate effect on protein fitness, higher ΔG values are not favored by natural selection.
Environmental Robustness and Phenotypic mutations
Genetic robustness can be explained by the threshold model where proteins maintain a higher tolerance to mutations with an increase in its threshold (ΔGt). Environmental robustness is a theory as to why genetic robustness occurs in proteins. Fluctuations in temperature, salinity, and other environmental factors could have influence the evolution of higher stability. Another factor could be phenotypic mutations. Because these types of mutations occur more frequently than genetic mutations, phenotypic mutations are believed to exert an immediate effect on protein fitness. Therefore the evolution of higher stability thresholds is understood to buffer the effects of phenotypic mutations and other environmental factors.
Gradient Robustness
Another type of robustness to mutations, gradient robustness, is associated with a small initial stability margin, but with a smaller slope so that each mutation causes, on average, a lower loss of stability. In a protein that is not tightly packed, a lower stability change would be expected because there are already few residue contacts so there are not many interactions to be lost. Proteins from RNA viruses do in fact show this type of relationship. These viruses have mutation rates several orders of magnitude higher than most other organisms. Their proteins show lower overall stability and are often loosely packed or partially disordered. Gradient robustness is the notion that proteins with strong well-packed structures exhibit higher stability losses than those whose residues have little contacts.
New Function and Stability
Typically occurring at more buried residues, adaptive new function mutations are more destabilizing than non-adaptive, neutral mutations. If these types of mutations accumulate, protein stability will be below ∆Gt, decreasing [e]o and ultimately decreasing protein fitness. The ability of a protein to acquire mutations that confer new functions is limited by the destabilizing effects of such mutations. The observation that mutations that improved the catalytic efficiency of TEM-1-lactase toward third generation antibiotics were destabilizing suggests that there is a tradeoff between protein stability and the evolution of new functions. On the other hand, compensatory mutations which reestablish protein stability are often seen after a change in function. FoldX predictions of new-function mutations specified that, while destabilizing, new function mutations are not more destabilizing than the average mutation. Observations contradict this prediction; new function mutations were found to be more destabilizing than neutral mutations and often occur more in internal residues. If new function mutations accumulate the protein stability is likely to decrease such that protein fitness decreases, even if the mutations improve function. Stabilized variants of P450 and TEM1 exhibited greater evolvability since they were able to accommodate more new-function mutations without correspondingly lower levels of enzyme.
Stability and Evolutionary Change through Uphill Divergence, Downhill Divergence, and Chaperones
Graphical representation of evolutionary changes with changes in protein stability with different types of mutations. The green area represents protein stability and the red represents instability. Blue arrows represent stabilizing mutations and orange arrows represent new-function mutations (or destabilizing mutations). The area boxed yellow in Figure B represents chaperone buffering.
Uphill Divergence
Compensatory mutations (or global suppressors) restore the stability margin for evolving proteins. Compensatory mutations are also called global suppressors because they can suppress the harmful effects of a wide range of mutations, and they have an important role in the evolutionary dynamics of proteins (they have been observed in both natural and in vitro evolution). Most compensatory mutations are stabilizing, for example, in developing resistance to the antibiotic cefotaxime, TEM-1 showed active site mutations which provided the new resistance followed by the stabilizing compensatory mutation Met182Thr. The rate of evolution is limited by the need for compensatory mutations to restore protein stability to the evolving protein. Still, mutations that change protein function and are destabilizing beyond ∆Gt cannot become fixated, except when buffered by chaperones.
Stabilizing Ancestor/Consensus Mutations and Downhill Divergence
By pairing a compensatory mutation (stabilizing factor) with that of a new function mutation (destabilizing factor), the overall stability of the protein is withheld. However, a large excess of stability could hinder evolvability in that the protein becomes rigid and restricts alternative conformations that could account for new functions. One way of using downhill divergence in protein engineering would be to incorporate compensatory mutations into the library that is selected for the enzyme’s new function; however, this would require the ability to predict stabilizing compensatory mutations. In one neutral drift experiment (multiple rounds of mutating and purifying to maintain the enzyme’s function) which provided a hint to predicting stabilizing compensatory mutations, several different mutations were enriched and five of the mutations showing the highest enrichment increased the stability and acted as compensatory mutations for a range of destabilizing mutations. The enriched mutations had one thing in common: they all changed the sequence of TEM-1 to be closer to its family consensus, and/or its ancestor. If a mutation occurs in a conserved residue, it usually causes a large drop in stability, while stability can be increased by reverting residues that deviate from the consensus amino acid. Ancestral interference, and/or consensus analysis can possibly be used to predict compensatory mutations. These predicted compensatory mutations can then be used to facilitate the engineering of more stable proteins with new functions through downhill divergence.
Chaperones and Protein Evolvability
Chaperones are known to assist in the folding of proteins, but they can also buffer the effects of mutations. Though the extent and the impact on evolutionary rates are unknown, chaperones seem to extend the zone of neutrality, allowing the accumulation of destabilizing mutations. A means of measuring the buffering capacity of the bacterial chaperonin GroEL/ES has been established recently, in which mutation accumulation experiments were performed with overexpression of the GroEL/ES protein. The proteins which accumulated the mutations were then tested for the quantity and type of acquired mutations and the amount of buffering necessary for stability. It was found that under overexpression of GroEL/ES the amount of accumulated neutral mutations was doubled, with increased variability. There were increased levels of mutations in the proteins’ cores, and the mutations had, on average, much higher destabilizing effects than in the absence of GroEL/ES. It has also been shown that overexpression of GroEL/ES can speed up the acquisition of a new enzymatic specificity. One case was observed in which variants of an enzyme selected under overexpression of GroEL/ES had a mutation that largely improved the newly evolving activity but was also largely destabilizing. Variants were selected without GroEL/ES that carried a different mutation which showed lower improvement with no destabilization, and variants selected without overexpression of chaperonins showed no improved function, or even decreased function due to lower enzyme concentrations.
Chaperonins (GroEL) has been known to use ATP in order to help proteins to fold. The process consists of unfolded proteins binding to the GroEL while not block the GroES. Then the ATP will bind to GroEL heptamers which will lead to ATP hydrolysis. ATP hydrolysis consists of releasing 14ADP and GroES. From here, GroEL is then bound to 7ATP and GroES in a pocket which will allow proteins to fold inside. Released proteins from the pockets means they are completely or partially folded while the proteins that are unfolded are sent back to bind to ATP.
Tokuriki, Nobuhiko, and Dan S. Tawfik. "Stability Effects of Mutations and Protein Evolvability." Current Opinion in Structural Biology 19.5 (2009): 596-604. Print.
Protein disorder is still a fuzzy concept for scientists to solve. Nowadays, views about protein are shaped by the structures that have been solved by X-ray crystallography. However, these beautiful structures are not able to tell the dynamic properties and regions that show considerable flexibility of proteins. In fact, only 25% of crystal structures reveal more than 95% of the complete molecular structural of proteins; the others have missing electron density for regions that are usually take on multiple conformations. Additionally, the data from X-ray crystallography are biased toward those crystallizable proteins that fold into a single or a few distinct conformations.
Protein posses a wide range of stability and degree of order/disorder. The continuous spectrum of structural states span from one extreme end of globally intrinsically disordered proteins to another extreme end of well-folded and stable proteins. Due to this wide range of flexibility of the protein, it is difficult to summarize the degree of protein flexibility with a single term; terms such as intrinsically disordered and conditionally disordered are proposed to describe protein structures:
Intrinsically disordered proteins are those proteins that lack a stable structure and show substantial disordered regions when studied as an isolated polypeptide china under physiological conditions in vitro.
Conditionally disorder proteins are those proteins that are intrinsically disordered under some circumstance and gain order under others, such as in the presence of their biding partners. These proteins are majority of intrinsically disorder proteins.
Intrinsic disorder is commonly observed within proteins. Approximate 30% to 50% of eukaryotic proteins contain regions of more than 30 amino acid that do not have a defined secondary structure or unstructured in vitro. Although more sophisticated technology is used to determine the structure the protein, it is still challenge to verify the folding status of the protein, especially within the cell. It is still unclear that whether those proteins that have been experimentally shown to be partially or fully unfolded in vitro are really unstructured in the cell. This is because the fact that molecular crowding and the presence of the appropriate binding partners transfer many disorder proteins to their folded state. Moreover, although intrinsically disordered proteins are sensitive to proteolytic degradation in vitro, they do generally exhibit reduced half-life in vivo, possibly because they are stabilized in cell and decrease the extent of disorder. Therefore, those proteins that are defined as globally intrinsically disordered proteins from in vitro and bioinformatics-based approaches might have gain order in cell.
Various techniques distinguishing intrinsically disorder regions (IDRs) from ordered regions and provide experimental information on protein disorder. Among all different techniques, NMR is unrivaled because it is able to provide detailed residue-by-residue information on the extent of disorder, residual dipolar coupling, and paramagnetic resonance enhancement measurements. In order to determine the information on protein disorder in vivo, new technique called 'in-cell' NMR spectroscopy is developed and used to determine protein structure within living Escherichia coli cells. This 'in-cell' spectroscopy and SUPREX (stability of unpurified proteins from rates of H/D exchange) are used to establish the true in vivo extent of disorder within proteins.
Protein disorder is often misunderstood as the default of the protein. In fact, maximum of only approximate 1 in 1010 random sequences is expected to fold into a defined structure; majority of proteins contains some regions of ordered structure, suggesting that order is selected for during evolution. Since most mutations are destabilizing, protein disorder might be simply a negative consequence of the random mutations occurred during evolution. Therefore, the frequent occurrence of disorder within proteins does not make proteins not functional. Disorder is recognized as providing functional advantages by enhancing binding plasticity, enzymatic catalysis, and allosteric coupling. Thus, disorder might in fact play an important role in molecular recognition and cellular signaling. Additionally, disorder might also increasing conformational entropy and flexibility by decreasing stability. This implies that protein disorder might play a helpful role in in vivo regulation. Lastly, studies have shown that conformational entropy conferred by disordered regions decreases the propensity of proteins to self-aggregate. Base on this fact, scientists hypothesizes that IDRs can prevent unwanted aggregation process within the crowed environment of the cell.
Conditionally disorder proteins are those proteins that can exist in at least two states, one that shows a high degree of flexibility and disorder and a second state that shows a higher degree of order. Many disordered proteins refold when they bind their partners. This is probably because the refolding is guided by thermodynamic principles that dictate that binding will stabilize and strengthen binding interactions. The order-to-disorder-to-order transitions can also occur as part of catalytic cycle of enzymes. The proteins that have only one binding site that engages multiples binding partners exemplify the concept of conditional disorder. Those 'disordered' binding sites are more likely to fold into multiple distinct conformations after binding to different partners than those 'ordered' binding sites. This fact reveals that disordered proteins have multiple distinct conformations upon binding to different partners; thus, disorder is functionally important. There are two model explaining how a partially unfolded surface regains structure by binding to different partners:
Conformational selection hypothesis: molecular recognition mechanism based on the assumption that a small proportion of the intrinsically protein population is in appropriate configuration to interact with specific binding partner. This interaction stabilizes both the proteins and binding partners by shifting the equilibrium towards the binding competent conformation.
Folding upon binding: molecular recognition mechanism based on the assumption that intrinsically disordered regions first bind to binding partner and then subsequently refold.
Disordered proteins may fold into different conformations by binding to different partners.A chameleon-like manner (distinct conformations) is observed when the disordered C terminus of p35 binds to various client. This observation is consistent with its functional role, which is to interact with over 40 different binding partners. However, this chameleon-like manner of intrinsically disordered proteins are rarely observed. This might be because structurally different partners for the same intrinsically disorder proteins and the structure of the protein-binding partner complexes are difficult to identify and determine.
Prototypes of proteins with multiple binding partners
Proteins that engage in multiple mutually exclusive transient interactions are more likely to have a higher degree of disorder. This high degree of disorder is also observed in chaperones, which bind to many different protein folding intermediates to prevent non-specific protein aggregation and facilitate protein folding both in vitro and in vivo. The degree of disorder of chaperones spans from 24% to 100%.
Folding chaperones, such as Hsp70, Hsp60, Hsp90, undergo large conformational rearrangements that are driven ATP-binding and ATP-hydrolysis. The intrinsic disorder enables the function of ATP-dependent chaperones by supporting dynamic conformational rearrangements necessary for client protein maturation. However, it is still unclear about the role of disordered regions in ATP-dependent chaperones.
The unstructured regions provide interdomain flexibility and confer solubility to Hsp90-client complexes.Additionally, several phosphorylation sites might be involved in order-to-disorder transitions during ATP binding and hydrolysis cycle.
ATP-independent chaperones use order-to-disorder transitions to trigger activation and client binding and use disorder-to-order transitions to control client release. The stress conditions that activate these ATP-independent chaperones, including low pH and severe oxidative stress, results in unfolding of proteins, which usually leads to the inactivation. Unlike other proteins, these ATP-independent chaperones is activated by their unfolding due to the stress.
HdeA is an acid-activated conditionally disordered chaperone that protects proteins from aggregation induced by low pH and bacteria from acidic stress. At pH 7, HdeA is well-folded dimer with no chaperone function. While shift to pH 2, HdeA is activated as a chaperone by partially unfolding and menomerizing within 2 seconds. Its nature of being partially disorder at low pH enables it be flexible of interacting with difference substrate that protects proteins from the irreversible damage; thus, it protects the bacteria from the acidic stress . The flexibility of HdeA suggests that HdeA could have the chameleon-like binding property. When the pH goes back to natural, Hdea slowly releases it client protein to minimize the aggregation-sensitive folding intermediates. Thus, the client proteins refolds back to its original structure passively while aggregation is disfavored.
Hsp33 is an oxidative stress-activated intrinsically disordered chaperone that protects proteins from oxidative unfolding. In the inactive state, Hsp33 is a monomeric two-domain protein that contains the tetrahedral, high affinity binding between a single zinc ion and 4 absolutely conserved cysteines in far C terminus. This binding of zinc ion stabilizes the C terminus and a metastable linker region. When Hsp33 is exposed to oxidizing conditions, zinc ion is released and two disulfide bonds are formed; zinc binding domain is destabilized; the linker region is unfolded; the protein is dimerized. The unfolding of the linker region actives the chaperone function of Hsp33. After Hsp33 is activated, it uses its intrinsically disordered linker region to interact with protein folding intermediates that contain significant amount of secondary structure so that a more stabilized conformation of Hsp33 and client protein is adopted. Hsp33 protects client proteins from stress-induced aggregation and shields bacteria from the antimicrobial oxidant bleach. Upon return to non-stress, Hsp33 is refolded, and this refolding of Hsp33 triggers the unfolding of the client proteins; the affinity between Hsp33 and client proteins decreases. Then, client proteins are released to ATP-dependent chaperone foldases, in which the client proteins are bound and refolded back to their native state.
Hsp26, which is a member of small heat shock protein, is a heat-activated conditionally disordered chaperone that protect proteins from aggregation induced by elevated temperature and bacteria from heat stress. At room temperature, Hsp26 is inactivated. Upon the induction of heat shock temperature, Hsp26 undergoes conformational changes by folding its unique thermosensing regions , and its chaperone function is activated. Unlike HdeA and Hsp33, the intrinsically disordered region of Hsp26 does not directly bind the client proteins but does interact with the client proteins.
Some chaperones that are globally disordered in vitro also have anti-aggregation activity.Globally intrinsically disorder chaperones inhibit aggregation by physically shielding and preventing folding intermediates from interacting with other aggregation-sensitive entities. Due to this fact, globally intrinsically disordered chaperones are relatively inefficient.
Caseins shield aggregation-prone surfaces and increases refolding rates through transient hydrophobic interactions. Caseins prevent client proteins from participation by forming soluble micellar complexes with client proteins. Since caseins actively inhibit lysozyme refolding and are unable to prevent activity lose of catalase and alcohol dehydrogenase induced by heat, they are not considered as folding chaperones.
It is unclear whether caseins play a role of chaperon in vivo or not.
It is possible for casein to compensate its inefficiency in the presence of high concentration.
LEA are proteins are highly hydrophilic. When LEA proteins are dehydrated under standard buffer conditions, they are activated as chaperones and adopt α-helical configurations. Similar to caseins, LEA proteins acts via transient hydrophobic interactions to shield aggregation-prone surfaces and increases refolding rates. They protect client proteins from dehydration-mediated and temperature-mediated inaction and aggregation in vitro.
LEA proteins play a role of chaperon and folding protein in vivo.
It is possible for LEA proteins to compensate its inefficiency in the presence of high concentration.
α-synucleins inhibit the aggregation of protein induced by heat. The inhibition of α-synucleins is less efficient than that of small heat shock proteins.
It is unclear whetherα-synucleins play a role of chaperon in vivo or not.
Protein evolution is a key indicator of the progression of proteins through time. These studies have lead scientists to determine the relationships of proteins between species which share similar functions. Similarly proteins which are homologous adapt to perform different functions. Evolution has forced proteins to become more complex and thus lead scientists to question the origins of simpler proteins which preceded modern proteins.
Protein evolution is not an independent process but part of an entire organism. Changes to the proteins are often only occurring on the sequence level leaving the structures and functions rather conserved. This can be used to explain the presence of homology between proteins which share similar structures but have adapted to perform different functions.
The two main approaches to the study of protein evolution are the analysis and comparison of sequences of proteins to prove or disprove evolutionary relationships and the other is the simulation of the evolutionary processes computationally in in vitro studies.
One scenario that was suggested are that the earliest proteins were very small polypeptides with about 10 amino acids and specified by small primitive genes made of RNA. The presence of RNA as the genetic material predated the presence of DNA as the genetic material. The genes coding the proteins probably join together in random sequences while a primitive splicing mechanism pieces together the proteins. Each of the proteins formed would consist of a domain with the characteristic length of amino acids being 100. Further concatenation would lead to multi-domain proteins and thus more complex proteins.
Another scenario which proteins might have evolved starts with small peptides consisting of less than 10 amino acids. These short peptides is said to then form closed loops which consist of 25-30aa and into folds 100-150aa which would lead to multifold proteins. Functionally, the short peptides of less than 10 amino acids do perform any functions, however, the closed loop proteins are functional.
Domains are typically 100-150 a.a. in length. This characteristic lengths are usually present in all proteins. The fold sizes are believed to have appeared during the early stages of the development of DNA genes. DNA being the successor of genetic material from RNA in many organisms was primitive and believed to have existed in circular forms. The optimal size of DNA ring closure is believed to be about 400 base pairs which is determined by DNA's flexibility. The 400 base pairs can be lead to code to approximately 100-150 amino acids seen in a domain. The upper limit of the circularization of DNA thus has a direct impact on the upper limit of a domain of a protein, the two being interconnected. Most proteins can be said to have evolved from these ancient closed loop units.
The PDZ domain is a common domain located in signaling proteins in structures of bacteria, plants, and animals. They are widespread in eukaryotes and eubacteria. Being approximately 90 residues long, they contain critical regions of sequence homology in diverse signaling proteins. Generally, PDZ domains attaches to a small region of the C-terminus of the next consecutive protein. Particularly, these small regions bind to the PDZ domain via beta sheet augmentation. Implicitly, this signifies that the PDZ domain is expanded through the addition of a beta strand from one terminal of the binding partner.
PDZ domains are usually located in the combination with other interaction modules and play a role that is directly specified with receptor tyrosine kinase-mediated signaling. It is also involved with other cellular functions such as protein trafficking, synaptic signal coordination, and cell polarity initiation.
The SRC Homology 3 (SH3) Domain is a relatively small protein that consists of 60 amino acids. The SH3 domain has the tendency to regulate the state activities of adaptor proteins and tyrosine kinases. They also function as a stimulant for substrate specificity of tyrosine kinases that bind at a large distance from the active site.
The SH3 domain is structured in a beta-barrel fold, which is made up of 5-6 beta strands organized in tightly packed anti-parallel beta sheets. The structure of the SH3 domain is a classical fold that is common in eukaryotes and prokaryotes.
WD40 domain is one of the most abundant domains and is one of the most active domains of the eukaryotes. Their functions are deeply involved in cellular processes by playing a crucial role as hubs in cellular networks. WD40 Domain regulates diverse protein-protein interactions, especially those that scaffold. They are present in processes such as signal transduction, cell division, chemotaxis, RNA processing, and cytoskeleton construction.
WD40 domains were first discovered in bovine beta-transducin, a subunit of the trimeric G protein transducin complex. It contains a series of sequence of approximately 44-60 residues with folds into seven-bladed beta-propellers. Each blade is designed in a four-stranded anti-parallel beta-sheet.
WD40 is naturally exploited as seemingly more suitable than other domain candidates because it structurally more compelling. This means that WD40 domains form structures that are highly symmetrical in comparison to other domains that are involved in intracellular processes. The symmetry is of high importance when proteins that lack sequence need to adopt. Additionally, symmetrical folds provide rapid and convenient folding especially for folds that are comprised of discrete and local, non-interlocking units of secondary structures.
Unfortunately, WD40 domains have proven to give difficult management. This is mainly because they are usually subunits of a larger assembly. Moreover, they lack the ability to measure intrinsic activity like catalysis. Regardless, WD40 domains act as scaffolds and clearly characterize one of the most significant domain families for cellular processing.
Sirtuin, a silent information regulator, increases the life span of model organisms. SIR2 has seven family members, SIRT1-SIRT7. SIRT1 is the closest to SIR2. Even though SIRT1 is the closest, the other family members show links to metabolism and aging. SIRT1- SIRT3 and SIRT5- SIRT7 conduct two enzymatic activities in vitro. The two enzymatic activities are NAD+ - dependent protein deacetylase activity and ADP-ribosyltransferase activity. SIRT4's only enzymatic activity is ADP-ribosyltransferase activity. The regulated metabolism and the survival of the cell depend on mammalian sirtuins.
The functions of SIR2, a founding member of the protein family of sirtuins, may provide the link between aging and chromatin regulation. Chromatin is silenced at sub-telomeric DNA, silent mating-type loci and ribosomal DNA by SIR2. SIR2 is effected by its NAD+-dependent histone deacetylase activity. H4 lysine 16 and H3 lysine 56 are the lysine residues where deacetylation happens. This play a key role in SIR2’s silencing effect. The regulation of lifespan in budding yeast is done by SIR2. This is done through two chromatin-silencing activities. The first activity involves suppressing the recombination between repeats of rDNA and thus promoting genomic stability (by preventing senescence-inducing extrachromosomal rDNA circles from being cut out and accumulated). The second activity involves decreasing the Sir2 protein levels by an increase in H4K16 acetylation levels in telomeres. In response to nutrient deprivation or mutation, SIR2 can also block lifespan extension in model organisms.
In yeast cells, SIR2 has the same function as SIRT6 in human cells. It also segregates damaged proteins which leads to the cell aging due to toxic cell aggregates. In response to lack of nutrients and other cell mutations, SIR2 blocks lifespan extension in yeast cells. Furthermore, in model organisms such as Caenorhabditis elegans and Drosophila melanogaster, SIR2 acts to promote longevity through different pathways. Another action promoted by SIR2 is extension of cell lifespan by inducing dietary restriction adaptions.
SIRT1 is one of the seven Sir2p homologues of yeast called sirtuins. SIRT1 along with SIRT6 and SIRT7 are found in the nucleus. SIRT1 requires nicotinamide adenine dinucleotide (NAD+). SIRT1 is the closest homologue to SIR2. Both control replicative senescence. SIRT1 can block oncogene-induced senescence if over expressed. While SIR2 exclusively deacetylates histones, SIRT1 deacetylates more than 40 different substrates. SIRT1 effects the structure of chromatin directly by deacetylating chromatin-regulating enzymes such as TIP6o and SUV39H1. Among other things, SIRT1 also helps regulate many other physiological processes such as apoptosis, metabolism, and stress resistance. SIRT1 is the most studied of the seven SIR2 family members.
For many years after its initial discovery, SIRT6 was thought to not have any deacetylase activity and it wasn't until later that SIRT6 was discovered to be a histone deacetylase that is very substrate-specific. It is in charge of regulating chromatin function, promoting its proper function in telomere and genome stabilization, gene expression, and DNA repair. The function of SIRT6 in humans parallels the function of SIR2 in yeast. Experimentation with SIRT6-deficient mice revealed that these mice are born completely normal but begin to have phenotypic abnormalities at around two weeks. They develop spinal curvature abnormalities, osteoporosis, and other systemic problems that result in death at around one month of age. At the cellular level, lack of this protein results in genomic instability and hypersensitivity to Ionizing Radiation (IR), methylmethanesulfonate (MMS), and hydrogen peroxide (H2O2). Furthermore, problems with base excision repair have been noticed. This revealed the important role SIRT6 plays in maintaining homeostasis, metabolism, and the life span of the organism.
One of the most important roles of SIRT6 in relation to telomeres, is its job in maintaining telomeric chromatin integrity. SIRT6 deacetylates H3K9 and H3K56. In SIRT6 deficient cell, H3K9 and H3K56 are hyperacetylated which leads to stochastic replication-associated telomere sequence loss, accumulation of telomeric DNA damage, and genomic instability with chromosomal end-to-end fusions. With these problems, cell senescence is brought on prematurely in the cell. This discovery has implications in future cancer research because chromosomal instability is corollated with cancer and the healthy function of telomeres plays a large role in maintaining genomic stability in chromosomes.
SIRT6 has been linked to involvement in DNA repair in humans by allowing efficient DNA DSB repair (DNA double-strand break repair). It was discovered that SIRT6 reacts with proteins(DNA-PKcs and Ku70/80) that are involved in the pathways called non-homologous end-joining pathways (NHEJ). SIRT6's association with chromatin increases drastically in response to DSB in order to decrease the levels of H3K9Ac. The SIRT6 structures were found to be useful in flanking chromatins near the breaks and stabilizing the DNA-PKcs required to perform DSB. When SIRT6 is deficient in the cell, DSB in cells is impaired, leading to instability in the cell.
Studies have shown a relationship between SIRT6 and the transcription factor nuclear factor-kappa B (NF-κB)which is in charge of gene expressions related to aging, proliferation,and inflammation. A lack of SIRT6 promotes hyperactivation of this transcription factor leading to over-expression of these genes. This is further seen in experiments on SIRT6-deficient mice where these mice were noticed to have metabolic and degenerative defects. In addition to NF-κB regulation, SIRT6 also plays a role in the transcription factor, HIF1α, which is important in glucose regulation that has been connected with lifespan regulation and even cancer.
Sirtuin and Fatty Acid Oxidation
During fasting, SIRT3 protein expression is increased as well as its levels and enzymatic activity. The phenotype overlap of SIRT3, AceCS2, and Acadl shows that SIRT3 regulate LCAD and AceCS2 acetylation.
Sirtuin and the Electron Transport Chain
Deacetylates Complex I subunits and Succinate Dehydrogenase (Complex II) interacts with SIRT3. Mitochondrial translation is regulated by ATP synthase binding with SIRT3 in proteomic analysis There are less information about the roles of SIRT4 and SIRT5 in the electron transport. A substrate for ATP synthase is created when SIRT4 binds with adenine nucleotide translocator (ANT) which than transports ATP into the cytosol and ADP to the mitochondrial matrix. SIRT5 interacts with cytochrome c. The biological significance of SIRT4 and SIRT5 are unknown
Sirtuin and the Kerb Cycle
Mitochondrial matrix is the location of kerb cycle enzymes. The compartmentalization of the mitochondrial matrix provides the cell to utilize metabolites from carbohydrates, fats, and proteins. Several kerb cycle enzymes interact with SIRT3 including succinate dehydrogenase (SDH) and isocitrate dehydrogenase 2 (ICDH2). With deacetylation and activation of AceCS2 and glutamate dehydrogenase (GDH), SIRT3 influence the kerb cycle indirectly. The carbon entry into the kerb cycle are increased by increasing acetyle-CoA and amino acid utilization. SIRT3 activity may provide the general mechanism of these increases. SIRT4 inhibitates GDH via ADP-ribosylation, and SIRT4 via GDH interacts with the kerb cycle.
A group of proteins called sirtuins can help postpone the death time of certain model organisms (non-human organisms that are studied to better understand biological life). To be more specific, sirtuins are the (Sir)2 (silent information regulator) and its orthologs, which are homologs with the same function from different species.
Seven sirtuins, SIRT1-7, are found in mammals and they change a variety of pathways dealing with metabolism and responding to stress. The sirtuin domain has the devices used to bind a co-substrate involved in metabolism, NAD+. In a controlled environment, all sirtuins perform two important enzymatic processes: NAD+-dependent protein deactylase and ADP-ribosyltransferase. However, SIRT4 cannot recognize specific substrates for acetylation, but it can identify ADP-ribosyltransferase. Because the enzymes rely on NAD+, they can perform their functions with the organism’s excited state and are possibly involved with recognizing metabolism. Furthermore, extensive scientific research on mammals’ sirtuins, specifically SIRT1, has shown that they control metabolic processes and the lifespan of cells. To do these, sirtuins specifically focus on different acetylated protein substrates and are put in separate locations. For example, SIRT1, 6, 7 are located in the nucleus.
Three sirtuins, SIRT3-5, are found in the mitochondria and they help by being an important location for metabolism involving oxidation. Compared to SIRT1, SIRT3-5 are smaller in size. Though intense research has been performed on sirtuins, sirtuins of mitochondria have not been studied to the extent of others such as SIRT1. However, reports and information regarding mass spectrometry have been speculating that SIRT3-5 may play an important role in controlling a wide range of activity in the mitochondria, such as making energy, intracellular signaling, and partaking in apoptosis.
Sirtuins are absolutely dependent on NAD+, meaning the excess of free NAD+ and its biosynthetic and broken down products in the cells are important to how the activity of the enzyme of the sirtuins work. There are essentially two primary ways to the NAD+ biosynthesis in yeast and mammals. One is the a de novo kynurenine pathway, which is formed from tryptophan. The other one is a known as a salvage pathway that is usues nicotinamide that is created from NAD+ by sirtuins in addition to ADP-ribosyl-transferases and polymerases or exogenous nicotinic acid. Two researches Bieganowski and Brenner recently found a special pathway to NAD+ in yeast and humans. It is initiated from nicotinamide riboside, which is provided from the outside. In addition, aonoter significant discovery is that mammalian cells operate differently at a basic level in terms of their pathways compared to bacteria and yeast. In yeast, nicotinamide is deaminated by the enzyme Pnc1p, which transforms to nicotinic acid. Then the nicotinic acid is changed to NaMN by the nicotinic phosphoribosyltransferase. Nicotinamide in mammalian cells, on the other hand, are changed directly to nicotinamide mononucleotide by the Nampt. The level of expression of Nampt in response to a variety of stresses makes the levels of cellular NAD+ higher. In effect this regulated catalytic activity of Sir2. Recent studies have found that changes in the NAD+ metabolites potentially possess tissue-specific effects. Take NAD+ for example. NAD+ makes the level of nuclear neurons higher which prevents axonal degeneration in a SitT1-dependent way. In addition, mammalian de novo biosynthesis is also organized in a different manner compared to plants and prokaryotes.
The relationship between Sir2 proteins, caloric restriction, and aging have been studied in detail. Evidence has shown that sirtuns are associated in encouraging longer life, especially longevity dealing with CR regimens, in a few organisms. There are two key early discoveries that support this in that the discovery that excess amounts of sirtuins encourage longevity in C. elegans. The relationship of yeast mother cell longevity with Sir2 interactions and SIr poteins has been shown that there is a correlation. When the yeast cells divide, they divide in an asymmetric way. The mother cells have only the ability to divide a certain amount of times, which isa bout 20-30 times. Mutants that don't have Sir2 have a decreased life span in the respect that they divide less time. Sir2 mutant mother cells that age prematurely were found to build up extrachromasal rDNA circles, which build up because rDNA combinated is not regulated anymore in the Sir2 mutant.
Due to the large role that sirtuins, particularly SIRT6, play in aiding in genome stability and the regulation of the metabolism, problems with SIRT6 function and availability are thought to be linked to oncogenic transformation and tumorigenesis. For example, certain cancers such as myeloid leukemia have breakages at the SIRT6 chromosomal locus. Furthermore, cancer cells exhibit a change from aerobic respiration to glycolysis that is seen in the Warberg effect which causes cancer cells to switch from oxidative phosphorylation to aerobic glycolysis. Other studies have shown that acetylation of H3K56 is increased in many cancers such as skin, thyroid, breast, liver, and colon cancers.
This is NAD-dependent deacetylase sirtuin-5
SIRT3 and Oral Cancer
It has been studied and reported that SIRT3 has a connection to the beginnings of oral squamous cell carcinoma (OSCC) cancer formation, that is, it inhibits cell growth and induces early cell death. When this sirtuin is produced in excess in breast cancer, it modifies how the protein p53 to prevent cell arrest and deterioration in bladder cancer cells as they age. When compared to human oral keratinocytes, SIRT3 levels were higher than what it should have been and this was evidence for overexpression of SIRT3 in OSCC carcinogenesis. From there, the sirtinol and nicotinamide inhibitors were tested, which resulted in a blockage of cell growth and induced cell death in OSCC cells, which furthers evidence for SIRT3 overexpression in these cells. But people have also studied and reported that SIRT3 does the complete opposite, that is, it helps keep cells alive. It decreases stress on the cells and keeps them away from inducing of cell death and apoptosis. For example, Nampt, which regulates the response to stress and diet, requires SIRT3 to keep cells alive when they are exposed to harmful substances that alter the gene. It also works to keep the heart from failing and protects from other cardiac problems.
Verdin, Eric, et al. Sirtuin regulation of mitochondria: energy production, apoptosis, and signaling. Cell Press. 669-675.
Tennen,Ruth I., and Chua, Katrin F. Chromatin regulation and genome maintenance by mammalian SIRT6
Kwon, Hye-Sook and Ott, Melanie. The Ups and Downs of SIRT1
Anthony A. Sauve,1 Cynthia Wolberger,2 Vern L. Schramm,3 and Jef D. Boeke4(2006). Biochemistry of Sirtuins'. "PubMed", p. 7-9.
Alhazzazi, Turki Y., et al. Sirtuin-3 (SIRT3), a Novel Potential Therapeutic Target for Oral Cancer. (2011). "PubMed".
Collagen Introduction
Collagen, which is the most abundant protein in mammals, is also the main fibrous component of skin, bone, tendon, cartilage, and teeth. Humans' dry weight of skin are made up of over 1/3 collagen. This extracellular protein is a rod-shaped molecule, about 3000 Å long and only 15 Å in diameter. There are at least twenty-eight different types of collagen that are made up of at least 46 different polypeptide chains that have been located in vertebrae and other proteins that contain collagenous domains. The defining characteristic of collagen is that it is a structural proteins that are composed of a right handed bundle of three parallel-left handed polyproline II-type helices. Because of the tight packing of PPII helices within the triple helix, every third residue, which is an amino acid, is Gly (Glycine). This results in a repeating pattern of an XaaYaaGly sequence. Although this pattern occurs in all types of collagen, there is some disruption of this pattern in certain areas located in within the triple helical domain of nonfibrillar collagens. The amino acid that replace the Xaa in the sequence is most likely (2S) –proline (Pro, 28%). The most likely replacement amino acid in the Yaa position is (2s,4R)- 4-hydroxyproline (Hyp, 38%). This means that the ProHypGly sequence is the most common triplet in collagen. Many research has been done on figuring out the structure of the collagen triple helices and how their chemical properties affects collagen's stability. It has been found that stereo electronic effects and preorganization are important factors in determining the stability of collagen. A type of collagen called type I collagen has the structure revealed in detail. Synthesizing artificial collagen fibrils, which are smaller strands of fiber, have now been possible and can now contain properties that natural collagen fibrils have. By continually understanding the mechanical and structural properties of native collagen fibrils, will help research devise and develop ways to create artificial collagenous materials that can be applied to many aspects of our lives such as biomedicine and nanotechnology.
Structure of Collagen
The structure of collagen has been developed intensively throughout history. At first, Astbury and Bell put forth their idea that collagen was made up a single extended polypeptide chain with all their amide bonds in the cis conformation. In 1951, other researches correctly determined the structures for alpha helix and the beta sheet. Pauling and Corey put forth their structure that three polypeptide strands are formed together through hydrogen bonds in a helical conformation. In 1964, Ramachandran and Kartha developed an advanced structure for collagen in that it was a right handed triple helix of three left handed polypeptide 2 helices with all the peptide bonds in the trans conformation and two hydrogen bonds in each triplet. Afterwards, the structure was honed by Rich and Crick to the accepted triple helix structure today, which contains a single interstrand N-H(Gly)...O=C(Xaa) hydrogen bond per triplet and a tenfold helical symmetry with a 28.6 A axial repeat.
Function and diversity
Collagen, which is present in all multicellular organism, is not one protein but a family of structurally related proteins. The different collagen proteins have very diverse functions. The extremely hard structures of bone and teeth contain collagen and a calcium phosphate polymer. In tendons, collagen forms rope-like fibers of high tensile strength, while in the skin collagen forms loosely woven fibers that can expand in all directions. The different types of collagen are characterized by different polypeptide compositions. Each collagen is composed of three polypeptide chains, which may be all identical or may be of two different chains. A single molecule of type I collagen has a molecular mass of 285kDa, a width of 1.5nm and a length of 300nm.
Type
Polypeptide Composition
Distribution
I
[alpha 1(I)]2, alpha 2(I)
Skin,bone,tendon,cornea,blood vessels
II
[alpha 1(II)]3
Cartilage, intervertebral disk
III
[alpha 1(III)]3
Fetal skin,blood vessels
IV
[alpha 1(IV)]2, alpha 2(IV)
Basement membrane
V
[alpha 1(V)]2, alpha 2(V)
Placenta,skin
Overview of Biosynthesis
Collagen polypeptides are synthesized by ribosomes on the rough endoplasmic reticulum (RER). The polypeptide chain then passes through the RER and Golgi apparatus before being secreted. Along the way it is post-translationally modified: Pro and Lys residues are hydroxylated and carbohydrate is added. Before secretion, three polypeptide chains come together to form a triple-helical structure known as procollagen. The procollagen is then secreted into the extracellular spaces of the connective tissue where extensions of the polypeptide chains at both the N and C termini (extension peptides) are removed by peptidases to form troppcollagen. The tropocollagen molecules aggregate and are extensively cross-linked to procuce the mature collagen fiber.
Stability of Triple Helix Structure
Collagen is important for animals as it contains many essential properties such as thermal stability, mechanical strength, and the ability to bond and interact with other molecules. Knowing how these properties are affected require an understanding of the structure and stability of collagen. Replacing amino acids in place of any of the XaaYaaGly positions can affect the structure and stability of collagen in numerous ways.
Glycine Substitutions
Replacing the Glycine position in the XaaYaaGly sequence often cause diseases has it is associated with mutations in the triple helical and non triple-helical domains of a variety of collagens. The damaging mutations to collagen is caused by the substitution of Gly involved in the last hydrogen bods within the triple helix. For example the amino acid replacing the Gly and the location of the substitution can effect the pathology of osteogenesis. Substituting the Gly in proline rich areas of the collagen sequence have less disruption then the areas of proline poor regions. The time delay caused by Glycine substitutions results in an overmodification of the protocollagen chains, which alter the normal state of the triple helix structure and thus contributing the development of osteogenesis.
Higher-Order collagen Structure.
Collagen is made up hieracharcal components from the smaller units of individual TC monomers that self assemble into the macromolecular fibers. In type 1 collagen, monomers make up microfibrils which then make up fibris.
Fibril Structure.
TC monomers of type 1 collagen have a strange feature in that they are unstable at body temperature meaning they prefer to be disordered rather than structured and order. The question is that how can something unstable be a component of something so stable, like the triple helix structure of collagen. The answer to this question is that collagen fibrillogenesis stabilizes the triple helix, meaning when the monomers form together they have a stabilizing effect. This contributes to the strength of the collagen triple helix structure.
Collagen fibrillogenesis occurs through the formation of intermediate-sized fibril segments called microfibrils. There are two essential questions that need to be answered in order to understand the molecular structure of collagen fibrils. The first question is what is the arrangement of the individual TC monomers that make up the microfibril. The second question is then how do those microfibrils make up the collagen fibril. These questions are difficult to answer because individual natural microfibrils cannot be isolated and the big size and insolubility of mature collagen fibrils make it impossible for standard techniques to figure the structure out.
David Hames, Nigel Hooper. Biochemistry. 3rd edition. Taylor & Fancis Group, New York, 2005.
Saad Mohamed (1994) "Low resolution structure and packing investigations of collagen crystalline domains in tendon using Synchrotron Radiation X-rays, Structure factors determination, evaluation of Isomorphous Replacement methods and other modeling." PhD Thesis, Université Joseph Fourier Grenoble 1 Link
Introduction
Serpins are a very large class of proteins that have a wide range of functions, with the most important being inhibition of proteases. The name "serpin" stands for Serine Protease Inhibitors since they replicate the 3D stucture of their respective serine protease and block its structure and pathway. Ultimately, serpines hinder the linkage of amino acids in a polypeptide chain which essentially make up the protein and force it to be inactive.
There are over 1000 serpins known today in humans, plants, bacteria, fungi, and even viruses. Many serpins play significant roles in protein catabolism and the first ones to be studied were antitrypsin and antithrombin, which are types of human plasma proteins. Researchers found that these proteins mediated blood inflammation and coagulation respectively and were crucial in human development. A mishap with either of these two Serpins causes diseases such as thrombosis and emphysema.
Example of serpin Structure
Serpin Structure
Analyzing the structure of serpins has helped figure their function and role in the biological world. Although each serpin slightly varies in conformation to make it distinct from the others, most serpins have a similar ordered structure.
The first serpins that were studied, antitrypsin and antithrombin, showed that all serpins have a distinct fold which allows them to fit inside other proteins and inhibit their functions. Serpins are composed of three β-sheets (referred to as A, B and C), about nine α-helices, and an open region (known as the center loop) which is the site of reaction. The center loop, known as RCL, is the reaction site which initiates inhibition processes and does not always appear on the same area for all serpines. The difference of where the RCL is situated distinguishes each serpine from the next.
Researchers have found around 36 serpin genes in humans which are classified depending on their structure. Naming of the serpin includes the name of the gene, following by the word "SERPIN", followed by a letter that corresponds to the class of serpin and a number for the specific gene in the class.
Serpin Function
The main function of serpins is to inhibit proteases, especially serine protease which gives serpines their name.
Some serpins also perform other functions which are noninhibitory. For example, Ovalbumin (found in egg whites) is a serpine which can store nutrients for the egg, thyroxine-hinding globulin is another serpine which transports hormones to various parts of the body, and Maspin is a serpine which controls gene expression of certain tumors.
Mechanism of Protease Inhibition
Serpins with inhibitory roles hinder functions of other proteins, such as protesaes. The way in which they block activity is by attaching to the other protein in a specific structural orientation to stop them from fully functioning. After they have attached to a target, they oversee structural conformations in their target protein which are usually permanent so that the protein cannot perform any other functions. Although the serpin is generally efficient in performing its job, certain mishaps can lead to mutations or protein misfolding, which would inactivate chains of polymers and confirm long protein chains to be useless.
Serpin Mutations and Diseases
The structure and function of a serpin are easily changed when mutations occur. If protein building is altered, such as a change in the amino acid sequence or a distinction in folding, the identity of the serpin will change. Even a small change in structure will affect the entire function of the serpin and may deem it useless and/or even harmful. Changes in serpin structure leading to genetic disorders or abnormalities are called Serpinopathies. As mentioned earlier, diseases such as emphysema, thrombosis, angiodema, dementia, etc. can result from Serpinopathies.
Two main defects can occur in the body when a Serpin undergoes a mutation. In the first defect, the inactive serpin fails to perform its blocking job and causes the protease to fuel many defects in the body. An example of this defect is emphysema, in which alpha 1-antitrypsin does not perform its duties and the elastase is destructive and incorrectly eliminates useful tissues in the lungs. The second type of defect is when serpins often clump together into bunches and cause harm to the cell by raising the toxicity levels. An example of this defect is clumping of nerve cells in the brain, which results to familial dementia. If many mutations occur, the usual inhibiting functions will not be carried out and the cell can suffer.
Carbohydrates bound to an Asparagine to form a Glycoprotein
Glycoproteins are proteins with oligosaccharide chains (glycans) covalently bound to polypeptide side-chains. The carbohydrate is attached by a process called glycosylation. Proteins that have extending segments in an extracellular manner are often glycosylated. Glycosylation occurs in one of two modes:
N-Glycosylation where the carbohydrate adds onto the amide Nitrogen of an asparagine.
O-Glycosylation where the carbohydrate adds onto the hydroxyl Oxygen of a serine or a threonine.
Note that these glycosylation reactions of a protein will only occur when the protein has the following order: Asn-X-Ser or Asn-X-Thr. X in the sequence can be any protein except proline. Glycoproteins are often important integral membrane proteins, such that they allow cell-cell interactions. Glycoproteins appear in the cytosol as well, but there is a lack of understanding in this subject.
Glycoprotein
The ebola virus operates via an envelope glycoprotein (GP) which is solely responsible for the virus’ ability to infect new cells. The virus’ genome consists of only seven genes; however, one of these genes is responsible for the generation of two proteins via transcriptional editing. The first protein, known as sGP, is primarily produced during the early stages of infection. Its purpose is structural and it does not come in contact with the surface of the cell. The second protein, GP, contains a hydrophobic tail, and thus is present on the surface of the envelope, making it responsible for the infection of new cells. To complicate things further, the immune response is mitigated by the presence of sGP. In fact, immune response demonstrates a preference for sGP over GP, allowing the virus to replicate more quickly. For this reason, the goal is to create an antibody that will target the GP while ignoring sGP.
X-ray crystallography of GP has allowed biochemists to understand its structure. GP contains 676 amino acids broken up into two subsections covalently connected by disulfide bonds. The first subsection is responsible for attachment to the host cell. The second subsection integrates the viral envelope into the host cell membrane. The most promising method for foiling the virus involves the creation of a specific monoclonal antibody that targets GP1 or GP2.
Protein design is the design of new proteins, either from scratch or by making calculated variations of a known structure. There is hope that by designing lattice proteins, or highly simplified computer models of proteins that are used to investigate protein folding and secondary structural modification of real proteins, one can develop better applications in medication and bioengineering.
While the possible amino acid sequences are enormous, only a subset will fold reliably and quickly to a single native state. Protein design involves identifying these sequences through observing their free energy minimum, and the molecular interactions that stabilize proteins. Protein design can be accomplished using computer models, which are able to generate sequences that fold to the desired structure. Using computational methods, a protein with a novel fold—Top7[1], an artificial 93-residue protein—has been designed, as well as sensors for unnatural molecules. This is also referred to inverse folding as a tertiary structure is first specified, and then a sequence is identified which will fold to it.
Other small proteins that have been created include proteins result in chiroselective catalysis[2], ion detection[3], and antiviral behavior[4].
Drug delivery to specific tissues in the human body is a major hurdle that must be overcome for safer, more effective use of therapeutic compounds. Many negative side effects can be avoided if the majority of the bodily tissues are bypassed by the compound on its way through the blood to the target site. One of the best ways to accomplish this is by encapsulating the molecule in a membrane that is more likely to release its contents within specific tissue types. Powerful medicines such as those used in chemotherapy to kill cancer cells need to be prevented from killing healthy cells. Also, gene therapy must be targeted so that the newly introduced genes enter only sick cells. Traditionally, liposomes and viruses were tasked with transportation, but a recent discovery has allowed for the sequencing and synthesis of amphiphilic branched peptides that can form water filled bilayers. These peptide spheres are similar to liposomes in that they self assemble. Otherwise they very different because peptides confer several key advantages over lipids such as increased stability under a variety of physiological conditions, fine tuned specificity, and more favorable interactions with antigens and therefore the immune system. This technique promises to revolutionize drug delivery for cancer patients and those suffering from a multitude of other diseases, including alzheimers which must be treated with care given that the blood brain barrier is a challenge for transport and the brain tissue surrounding the plaques is irreplaceable so collateral damage must be minimized at all costs. In practice, the compound to be delivered is placed in solution to which the self assembling peptide is added, the vesicles form around the compound, and it is ready to be purified and administered. By altering the surface of the peptide, researchers hope to fine tune delivery to specific tissue types throughout the body in order to treat a variety of disorders.
• Gudlur S, Sukthankar P, Gao J, Avila LA, Hiromasa Y, et al. (2012) Peptide Nanovesicles Formed by the Self-Assembly of Branched Amphiphilic Peptides. PLoS ONE 7(9): e45374. doi:10.1371/journal.pone.0045374
1. Yan Hu., Sophia B. Georghiou., Alan J. Kelleher, Raffi V. Aroian, "Bacillus thuringiensis Cry5B Protein Is Highly Efficacious as a Single-Dose Therapy against an Intestinal Roundworm Infection in Mice"
3. Elizabeth C. Wu, Ji-Ho Park, Jennifer Park, Ester Segal, Fre#de#rique Cunin, and Michael J. Sailor, "Oxidation-Triggered Release of Fluorescent Molecules or Drugs from Mesoporous Si Microparticles", ACS Nano, 2008, 2 (11), 2401-2409 • Publication Date (Web): 08 November 2008
4. Michelle Y. Chen and Michael J. Sailor, "Charge-Gated Transport of Proteins in Nanostructured Optical Films of Mesoporous Silica"
5. Jennifer S. Andrew, Emily J. Anglin, Elizabeth C. Wu, Michelle Y. Chen, Lingyun Cheng, William R. Freeman, and Michael J. Sailor, "Sustained Release of a Monoclonal Antibody from Electrochemically Prepared Mesoporous Silicon Oxide"
A beta-propeller is a symmetrical protein that is made with four-stranded antiparallel beta sheets arranged in a circle. The number of antiparallel beta sheets, also known as blades, dictates what function a beta-propeller possesses. Four-, five-, six-, seven-, eight- and ten-bladed beta-propellers and their functionalities are known so far. Beta-propellers are typically shaped like a funnel, where protein-protein interactions can occur on all sides of the funnel, including the central opening. There is a large variety of beta-propellers, all of which are categorized into families that depend on their blade number and amino acid sequence. Some beta-propeller families include: the WD40 family (the largest family of propeller proteins), regulator of chromatin condensation 1 (RCC1) family, kelch family, YWTD family, NHI family, YVTN family, and the Asp-box family.
Function and Structure Association with Number of Blades
Four- and five-bladed propellers: Four- and five- bladed propellers have limited variation in functinonality. Four-bladed propellers act as ligand-binding proteins and are repsonsible for transportation and catalysis. The smallest known form of a four-bladed propeller structure is hemopexin, which is a plasma glycoprotein that has heme-binding abilities. Five-bladed propellers are abundant in nature, but only act as transferases, hydrolases, and sugar-binding proteins.
Six- and seven-bladed propellers: Unlike the four- and five-bladed propellers, six- and seven-bladed propellers have a high variation in functionality. Six-bladed propellers act as ligand-binding proteins, hydrolases, lyases, isomerases, signaling proteins, and structural proteins. Seven-bladed propellers act as ligand-binding proteins, hydrolases, lyases, oxidoreductases, signaling proteins, and structural proteins.
Eight- and ten-bladed propellers: Similar to four- and five-bladed propellers, eight- and ten-bladed propellers also have low variation in functionality. Eight-bladed propellers act as oxidoreductases and structural proteins, and ten-bladed propellers act as signaling-proteins. The only well-known 10-bladed propeller is sortillin, which plays an important role in endocytosis and intracellular sorting. Sortillin is unique in that it is able to regulate ligand- binding by only allowing one ligand to bind at a time to prevent ambiguity in functionality.
Newly-Discovered Beta-Propeller: the Beta-Pinwheel
A newly-discovered beta-propeller called the beta-pinwheel has been found to have a unique property of having strand exchange in the anti-parallel beta-sheets, giving the beta-pinwheel a Velcro-like characteristic which increases its structural stability. Contrary to regular beta-propellers, where beta-propellers are always in closed-ring form, the beta-pinwheel can be in an open-ring form (open barrel form), where the two blade ends of the broken ring can have no interactions with each other. Beta-pinwheels also vary drastically in blade number; they range from three to eight blades. Although beta-pinwheels have a wide range of blade numbers, they all are limited to one function: DNA-binding.
Beta-propellers have been discovered to associate with human diseases when beta-propellers have been mutated. For example, when mutations on the calcium binding pockets of integrin αIIbβ3, a seven-bladed propeller, are present, Glanzmann thrombasthenia (GT), a recessive bleeding disorder, can occur. Kallmann syndrome, a genetic disorder that affects the production of sex hormones, can occur when there are mutations at the protein-protein binding region of WD repeat domain 11 (WDR11) beta-propellers.
Chen, Cammy K.-M.; Chan, Nei-Li; Wang, Andrew H.-J. (2011). "The many blades of the β-propeller proteins: Conserved but versatile". Trends in Biochemical Sciences. 36 (10): 553–61. doi:10.1016/j.tibs.2011.07.004. PMID21924917.
They are the most abundant domain proteins in eukaryotic genomes. As scaffolds, WD40 domain proteins are involved in a variety of cellular process such as signal transduction, cell division, cytoskeleton assembly, chemotaxis and RNA processing. Most distinctive feature of the WD domains is that they mediate diverse interactions between protein and another protein.
WD40 domains have highly symmetrical structure, which supports more rapid and convenient folding. Also, the domains are characterized by the structure of propellers with seven blades. WD40 propellers have three distinctive surfaces, which are top, the bottom, and the circumference, and they take part in various interactions. The top region is the part of the structure where the loops lie, and the loops connect D and A strands of the WD repeats.
Most protein-protein and protein-peptide interactions happen at the specific site of the central channel of the β-propeller. The site is called supersite and this site is and at this site, most of interaction partners bind. Most interactions occur on the top surface of the propeller
Since the core of the central channel is not accessible for interactions and N- or C-terminal of the β-propeller pack against the entry sties of the central channel, majority of interactions happen in WD40 domains. For example, one of the interactions that WD40 domains participate in is the interaction between globular proteins and those involving short peptides/linear motifs.
Peptide-protein interactions
The interaction between WD40 and peptide is important for the assembly of dynamic multi-protein complexes. When the interaction happens, many different peptide motifs bind to different sites of the propeller, but most peptides bind on the top of β propeller close to the central channel. The binding site variability gives WD40 domains the characteristics as ideal platforms for assembling different kinds of proteins and helping the formation of transient complexes.
Transducin;alpha subunit is red, beta is blue, and gamma is yellow
Transducin, which is responsible for the signal transmission from rhodopsin to cyclic guanosine monophosphate phosphodiesterase, consists of three subunits : α-, β-, and γ- subunits α-subunit
It consists of a GTPase domain. A GTPase domain interacts with the α-helical domain , which does not interact with WD40 domain and an N-terminal helix, which contacts the side of the propeller
β-subunit
It forms a stable heterodimer along with γ-subunit
γ-subunit
It consists of two helices. The first helix with an N-terminal helix precedes the β –subunit propeller, and the second helix contacts loops on the bottom of the propeller. γ-subunit controls the expression level of the entire transducin heterotrimer, which plays a major role for normal transducin localization.
Stirnimann CU, Petsalaki E, Russell RB, Müller CW. "WD40 proteins propel cellular networks" Trends Biochem Sci. 2010 Oct;35(10):565-74. Epub 2010 May 5. Review
Xu C, Min J. "Structure and function of WD40 domain proteins." Structural Genomics Consortium, University of Toronto, 101 College St., Toronto, Ontario, Canada
The BUB1 and BUBR1 are the two kinases involved in the checkpoint during mitosis. As seen in the picture, before the two sister chromatids separate in anaphase to create chromatids of two cells, the checkpoint is to ensure that they are separated accurately. This checkpoint is necessary to avoid mutations where cells do not divide which can cause aneuploidy. Aneuploidy results when there is an abnormality in the is number of chromosomes in a human embryo. One of the most well known conditions is Down Syndrome where there is a mutation in the chromosome 21 of a human. The sister chromatids do not separate resulting in a third copy present in the embryo.
The spindle assembly checkpoint is the regulatory checkpoint that allows a dividing cell to continue its division process if everything is going according to planned and the chromosomes align properly along the metaphase plate and attach to microtubules via kinetochores at each of the centromeres. It is essential because if the chromosomes in a cell that is undergoing mitosis do not align and separate correctly, one daughter cell will get two copies of a chromosome, while the other daughter cell completely lacks that same chromosome. This is the cause of many defects in newborns. It is also seen in over 90% of solid tumors. Many different problems arise from incorrect chromosome splitting, depending on the certain chromosomes that does not separate correctly.
Drosophila metaphase chromosomes
There are two kinases which play central roles in the spindle assembly checkpoint that occurs during metaphase in mitosis that we will discuss, even though there are at least fifteen proteins involved in this process altogether. Kinases are enzymatic proteins that transfer phosphate groups from high energy donors, to lower energy acceptors. The two that are highly involved in the checkpoint are BUB1, which stands for budding uninhibited by benzimidazole 1, and BUBR1, which stands for budding uninhibited by benzimidazole-related 1, also called BUB1B. These two proteins, BUB1 and BUBR1, are very similar, due to the fact that they are paralogs, meaning they came from the same ancestor and evolved to slightly different proteins. /but even for paralogs these two share many common attributes and their amino acid composition is basically the same, yet they perform different functions. This is due to the difference in their structures.
In regards to BUB1, it undergoes phosphorylation, the addition of a phosphate group. By doing so, it abolishes the CDC20 gene in order to stop SAC and put a pause to mitosis. This phosphorylation as well as that done by BUBR1 have yielded a few hypothesis and are still in research.
UB1 catalytic activity is of paramount importance because BUB1-mediated CDC20 phosphorylation inhibits APC/C–CDC20 in human cells
PDB 1bub EBI
Both of these enzymes have three main regions in them, an N terminal region and two C regions, one of which is terminal and another that is intermediate. These different regions are involved in different steps, such as spindle checkpoint on one terminus and chromosome congregation on the other. There are several differences in these region in each of the two molecules. The importance of the C region is seen to be insignificant as compared to the N terminal in BUBR1 due to the lack of the C terminus by the Mad3. Mad3, mitotic-arrest deficient, replaces the BUBR1 in in yeast, worms and plants. The Mad3 does not have a C region and since no animals have both BUBR1 and Mad3, the functions of the Mad3 remain the same without the presence of the C region.
BUB1
BUB1 is bound to the kinetochores at it's N-Terminus region. Both BUB1 and BUBR1 contain KEN boxes, which are protein motifs that mediate protein recognition. Two KEN boxes, are located in the N-terminus of BUB1 that are involved in and required for the phosphorylation of CDC20. CDC20 phosphorylation is one of the main tasks of BUB1, which first phosphorylates BUB3 then along with the help of BUB3, it phosphorylates Cdc20. It also plays many other major roles. One of these is the recruitment of BUBR1, Mad1 and Mad2. Upon spindle damage BUB1 is also triggered to phosphorylate Mad1. This kinase is also highly involved in the organization of the centromere.
BUBR1
BubR1 has many roles as well and contains KEN boxes that it depends on to function properly too. It helps connect kinetochores to microtubules and keep the attachment stable. It regulates mitosis by inhibiting anaphase when chromosome segregation is incomplete or incorrect. It also regulates prophase one during meiosis one in eggs.
Effects of Bub1 and BubR1
-Loss of BUB1 can result in aneuploidy, which is the abnormal number of chromosomes resulting from faulty mitosis. Aneuploidy is seen in about 90% of solid tumors.
-Deletion of Bub1 in some species increases the rate of incorrect chromosome segregation while in other species it results in slow growth and loss of chromosomes.
-Many forms of cancer have been related to cell with improper spindle checkpoints
Sources
BUB1 and BUBR1: multifaceted kinases of the cell cycle
Victor M. Bolanos-Garcia, Tom L. Blundell
This image is a derivative work of the following images:
File:Spindle_chromosomes.png licensed with Cc-by-sa-3.0,2.5,2.0,1.0, GFDL
"Spindle checkpoint." Wikipedia. 7 December 2012. 7 December. 2012 <http://en.wikipedia.org/wiki/Spindle_checkpoint#Metaphase_to_Anaphase_Transition>.
APOBEC3G, or A3G for short, is an enzyme that was previously known to fight HIV by inhibiting wild type Viral infectivity factor (Vif). It was the first enzyme that turned cytidine to uridine to be discovered. Some researchers now believe that it acts as a "double agent" by actually facilitating Vif depending on the amounts of DNA deaminase activity. According to the researchers studying this enzyme "The simplest answer might be the little DNA deaminase activity benefits the virus and a high level of activity destroys the virus" (Smith). This enzyme can be antiviral by inhibition of VIF or an HIV facilitator by bringing diversity to the genome of the virus which in turn gives new versions of the virus drug resistance properties.
Protein APOBEC3G PDB 2JYW
APOBEC3G is a deaminase, which removes an amino radical from an amino acid or a compound. A3G deaminase-dependent antiviral activity hinders Vif by changing its genetic code and not allowing reverse transcription to take place.
Glutaredoxins are proteins known to be a part of certain redox reactions by serving as thiol-disulphide oxidoreductases which use thioredoxin (Trx) fold architecture and a Cxx active site. They are able to bind to labile iron-sulphur clusters and carry them to certain acceptor proteins. Glutaredoxins also seem to have a role in the sensing of iron within the cell, and they are also able to serve as scaffold proteins for the de novo synthesis of iron-sulphur clusters.
Glutaredoxins (Grxs) have been known to serve as glutathione (GSH)-dependent electron donors for ribonucleotide reductase in E.coli. They are also able to perform glutathionylation, a protein regulatory or cysteine defense mechanism wherea glutathione is covalently bound as a disulphide on a cysteine residue. They have also been known to serve as catalysts for deglutathionylation.
Grx5P, the glutaredoxin protein found in mitochondrial yeast cells, is known to contribute to the transfer of preassembled Fe-S clusters from a U-type ISC scaffold protein (Isu 1p) to acceptor proteins. Thus, Grx’s are known to participate in the formation of cluster assembly as well as the transfer of these these clusters. Grxs are also known to contribute to the regulation of iron-responsive genes called the iron regulon present in yeast. Stable Fe-S clusters serve as the redox or iron sensor in this case.
TAX1BP1 (Tax1-binding protein 1) plays a role in the negative regulation of NF- kB (transcription factors nuclear factor-kB) and IRF3 (interferon regulatory factor 3) signaling by regulating ubiquitin-editing enzyme A20 in the anti-flammatory and antiviral signaling pathways. This is to regulate the inflammatory and antimicrobial responses that are triggered in the body. TAX1BP1 also serves as a transcriptional coactivator for nuclear receptors and viral transactivators. TAX1BP1 protein is known to be highly conserved across species.
TAX1BP1 is known to form coiled-coil structures as well as two helix-loop-helix regions responsible for homodimerization. The N terminal of TAX1BP1 contains a SKIP carboxyl homology domain (SKICH), although the typical function of SKICH that defines it as a membrane targeting domain has not yet been seen in TAX1BP1’s. At the C terminus there are two zinc finger domains which contain highly conserved ‘PPXY’ motifs that are known to bind to a ‘WW’ domain of proteins. These zinc finger domains are ubiquitin-binding domains that have also been known to allow TAX1BP1 to bind to the motor protein myosin VI which is involved in cellular processes such as endocytosis, secretion, membrane ruffling, and cell motility. TAX1BP1 also contains a 14-3-3 binding motif of unknown function.
The protein 43 kDa TAR DNA binding protein (TDP-43) was first discovered to be involved in neurodegeneration. TPD-43 was discovered to have involvement in amyotrophic lateral sclerosis (ALS) and frontotemporal labor degeneration (FTLD) in 2006, which lead an increase laboratories that studied this protein. This is because unusual TPD-43 aggregation is the main focus of many neuronal diseases that are known as TPD-43 proteinopathies. A new and promising research area had been opened up after TPD-43 was discovered to be involved in neurodegeneration, and findings of other proteins FUS/TLS and C9orf72. These discoveries also opened up the field of RNA binding proteins in neuroscience. But there it is difficult to study TPD-43, because of large amount of process that it is involved in. TPD-43 is a protein that is essential to a cell's life cycle.
TPD-43 is in a family of nuclear factors named hnRNPs. The ability of TPD-43 to bind RNA on specific sequences in a single-stranded behavior. This ability comes from the sixty amino acid residue motifs that are folded in a conserved 3D structure, known as RNA recognition motifs (RRMs). This ability of TPD-43 to bind to RNA is important for RNA processes, especially in alternative splicing. TPD-43 binding to repeats of UG RNA sequences can silence splicing. This can been confirmed in vivo in some labs using in vivo crosslinking and immunoprecipitation-sequencing. Some of these studies have shown that TPD-43 can bind to other conserved sequence motifs, but the function of these binding sites are not well known. UG RNA sequence repeats are common in the human genome and are found mostly in introns and 3' regions that are not translated. TPD-43 also controls neurofilament mRNA stability with the help of other proteins in the ALS pathology.
An extended binding region, TDPBR, of TPD-43 was found in the TPD-43 mRNA 3' UTR. The TDPBR has a few non-UG sequences that are important for autoregulation of TPD-43 mRNA levels. Low levels of nuclear TPD-43 causes the uses of the most efficient polyA1 instead of other choices like polyA2 to polyA4. On the other hand, high levels of nuclear TPD-43 causes less optimal splice sites to be used instead, which leads to rapid degradation of the mRNA. This makes a feedback loop that the cell uses to keep the concentration of TPD-43 leveled. TPD-43 aggregation can occur if the levels are not kept constant. If this happens, TPD-43 aggregates in the cell nucleus and cytoplasm will reduce the amount of free nuclear TPD-43. Then the 3' UTR TDPBR sensor will read protein level drops and increase TPD-43 production. This is bad, because this cycle would lead to cell stress and eventually, death. While high levels of TPD-43 can be bad, low levels and can also have serious effects on expression levels and RNA transcripts.
Another important aspect of TPD-43 is with its ability to bind proteins that help with its RNA processing abilities and aggregation properties. Proteins like hnRNP A1 and A2 are necessary for inhibiting splicing. Another important protein is FUS/TLS, which regulates the expression levels of histone deacetylase 6. All these interactions show that TPD-43 is a very flexible protein in its interactions proteins and RNA. This is important when searching for therapeutic uses of TPD-43 in splicing targets.
It is accepted that the C-terminal is the biggest reason that TPD-43 aggregates. Modifications in TPD-43 such as C-terminal fragments are found in neurons of patients with ALS and FTLD. De novo nuclear cleavage of TPD-43 makes C-terminal fragments, which has been shown to lead to aggregation of TPD-43. Hyperphosphorylation of TPD-43 could be protective to neurons, in contrast to aggregation promotion effects. However, further studies are needed before this can be concluded. TPD-43 aggregation has also been observed when nuclear transport proteins, like karyopherin beta or cellular apoptosis susceptibility proteins, are knocked down.
An external factor that could increase TPD-43 aggregation is their interaction with aggregates of polygluatmine like Ataxin-2, or extracellular kinase (ERK1/2) inhibition. Overexpression of p62 protein and USP14 inhibition go against TPD-43 aggregation. This indicates a possible relationship with autophagosome system in aggregate resolution. Bad autophagosome functioning could contribute to promoting disease growth.
TPD-43 is brought to stress granules after cell stress. TPD-43 controls levels of stress granule factors for formation and support. Some of these factors are GTPase activating protein and TIA-1 binding protein. Long period of cell stress can lead to stress granule aggregates.
Research has shown that TPD-43 can act as biochemical marker in FTLD and ALS. It is important to identify these markers before there is too much neuronal damage from FTLD and ALS. Biomarkers are important when judging the effects of therapeutic techniques. Research has been focused on finding pathological modifications of proteins (including: amyloid beta, tau protein, prion protein, alpha-synuclein, and TPD-43 ) that are important for neurodegeneration. The tissue samples that have been studied for abnormal TPD-43 levels are cerebrospinal fluid, blood plasma, circulating lymphonocytes, and skeletal muscle in people with ALS. These studies can be difficult, especially in cerebrospinal fluid and blood plasma. One method that could be easier is to use TPD-43 solubility tests for proteins from peripheral blood mononuclear cells. Using these tissues makes it easier to compare mRNA expression and polyadenylation levels with normal cells.
Proteins are important macromolecules in living organisms because they are structurally. Therefore, they can take on essential roles in a wide variety of biological processes and functions. Protein structure can be described on several different levels. The primary structure of protein refers to the sequence of amino acids in the polypeptide chains. Different amino acids contain different functional groups. The secondary structure of protein deals with the fact that polypeptide chains fold into a regularly repeating structure, such as an alpha-helix and beta-sheet. The tertiary structure of proteins gives the overall structural arrangement of one single subunit polypeptide chain. The quaternary structure of protein refers to the arrangement and interaction of several subunit polypeptide chains to form a protein molecule. The structural diversity and complexity of proteins enable them to perform a diverse variety of functions. A protein is made up of many amino acids through peptide bonds between a carboxyl group and an amino group of another adjacent amino acid. This makes the protein form long chains. Some proteins are known to function alone; however, there are many proteins that work together to form complexes (for example: ribosomes, lipids, nucleic acids, etc.). This creates functionality of cells, and organisms all together. The main function of proteins depends on the amino acids that make up the protein as well as the way it folds.
Some of these functions are given below:\
Antibody
Antibodies are proteins that participate in the immune response by defending the body against antigens (foreign invaders). Antibodies travel through the blood stream and are utilized by the immune system to identify and defend against bacteria, viruses, and other foreign intruders. Certain antibodies destroy antigens by immobilizing them so that the white blood cells can destroy the antigens. An antibody is made up of 2 heavy and 2 light chains. The two chains are connected by disulfide bonds. There is a variable region and constant region. The variable region is the region where the antigens bind to. Because its variable, different antigens can bind to these regions.
Contractile proteins are the proteins that involved in movement. They include myosin and actin, which participate in muscle contractions and movement. Actin filaments are the major components of the network. Other contractile proteins interact with these filaments in order to create structural rigidity and movement. Contractile proteins's structure and function are striated muscles and well characterized; thus,they contribute a great example of nonmuscle cells. Moreover, the interaction of contractile proteins of various cells may be unique. The study of contractile proteins in cells other than muscle has distinct difficulties. For example, the proteins are presented in a lower concentration than in muscle, and only a few cell types are obtainable for study in quantities comparable to muscle. Also, proteolysis and other detriments may be more severe in nonmuscle cells. Other example can be that the organization of contractile proteins is difficult to define in nonmuscle cells. Nevertheless, the ubiquity of contractile proteins and the importance of their interactions presages increase relevancy for physiology and medicine.
Structural proteins are the proteins that are generally fibrous and stringy. They are the most abundant class of proteins in nature. Their main function is to provide mechanical support. Examples of structural proteins can be keratin, collagen, and elastin. Keratins are found in hair, quills, feathers, horns, and beaks. Collagens and elastin are found in connective tissues such as tendons and ligaments. Collagen is recognized as the most abundant mammalian protein. Structural proteins such as collagen, fibronectin and laminin are utilized in cell culture applications as attachment factors. Sigma offers the most comprehensive collection of structural proteins for extracellular matrix and cytoskeletal research as well as tools for cell culture and material science applications.
Enzymes are the proteins that regulate biochemical processes. They are often called catalysts because they function to lower the activation energy of the reaction and thereby increases the rate of the reaction. Essentially, enzymes are able to do so because they can stabilize the transition state. Lactase and pepsin are examples of enzymes. Lactase is involved in the breakdown of lactose, which are present in milk. Pepsin, on the other hand, helps break down proteins in food. Enzymes are biological catalysts or assistants. Enzymes consist of various types of proteins that work to drive the chemical reaction required for a specific action or nutrient. The chemicals that are transformed with the help of enzymes are called substrates. In the absence of enzymes, these chemicals are called reactants.
Hormonal proteins are the proteins that act as signaling proteins, which help regulate biological activities in the body. Insulin, oxytocin, and somatotropin are examples of hormonal proteins. Insulin is involved in the regulation of glucose metabolism by controlling the blood-sugar concentration. Oxytocin is responsible for stimulating contractions in women during childbirth. Lastly, somatotropin stimulates protein production in muscle cells.
Somatotropin
Storage proteins are the proteins that act as storage for amino acids or specific ligands, such as biologically important metal ions. They include ovalbumin and casein. The former is present in egg whites while the latter is found in milk. Another example is myoglobin, which function as the storage of oxygen for tissues.
Transport proteins are the proteins that are responsible for moving molecules from one place to another. For example, the protein hemoglobin is responsible for the transport of oxygen in the blood. Another example is cytochromes, which acts as electron carrier proteins in the electron transfer chain.
Membrane proteins are the proteins that are found in biological membranes. They can either be peripheral or integral. They may act as biological markers or regulatory channels for ions and molecules.
The protein function of binding is very specific. The ability of binding is dependent on the tertiary structure of the protein, also known as the three-dimensional structure of the protein. The area of the protein that is bound to another molecule, such as a ligand, is called the binding site. The binding site is often a crevice on the surface of the protein. The molecule that binds to the protein changes the chemical conformation of the protein.
•The active site is also the site of inhibition of enzymes
•The active site of an enzyme contains the catalytic and binding sites
•The structure and chemical properties of the active site allow the recognition and binding of the substrate
•Protein functions such as molecular recognition and catalysis depend on complementarity
•Molecular recognition depends on specialized microenvironments that result from protein tertiary structure
•Specialized microenvironments at binding sites contribute to catalysis
An example of binding with a protein is the ligand-binding protein of hemoglobin, which transports oxygen from the lungs to other important organs and tissues within humans.
An enzyme is specific to the substrate it binds to. It is dependent on structure and placement as well as the sequence of amino acids of the substrate. If the sequence is complementary then it ensures the binding of the two components. In the case of proteases, the residues that are not responsible for catalysis may be responsible for the recognition and alignment of the molecule, setting it up so that it can have one of its bonds hydrolyzed by the enzyme.
Proteases in particular tend to recognize the side chains of the amino acid it intends to cleave. Oftentimes it is the carbonyl carbon - amide group that succeeds the recognized side chain.
Note: image is the protease of an HIV-1
It is possible to gauge whether an amino residue plays a role in the enzymatic activity of a protease by inducing site-directed mutagenesis to the amino acid in question. If the amino acid in question does play a role in specificity or catalysis, post-mutation the enzyme will have a decrease in its enzymatic activity.
The concept of complementarity can be understood in terms of the lock and key model of protein binding (as shown in the figure below). Essentially, the surface of the protein involved in binding exhibits a shape that is complementary to the binding ligand. This allows for protein recognition, binding specificity and affinity. Protein binding can be further explained by the induced fit model. In this model, the protein's binding site also exhibits complementarity, but to a lesser degree than the lock and key model. Once, the ligand is already at the binding site, the protein can adjust the shape of its binding site to better fit and bind the ligand. This concept of protein flexibility is explained in more details in the next section.
Binding usually occurs at the surface of the protein. See Nature of Binding Sites (below) for more detailed information. The image below shows an example of how a protein's surface recognizes molecules. This is the binding between a sex hormone and globulin.
Ligand to protein binding typically occurs through non-covalent forces. The weak interactions of non-covalent forces allow for easy exchange between molecules. For instance, hemoglobin traps oxygen in its binding site at the lungs and releases it to the tissues. It is able to perform the task very efficiently because the binding is strong enough to tightly hold on to the oxygen when it is in the oxygen-saturated lungs, yet weak enough to allow for an easy release of oxygen to the oxygen-deficient tissues.
The concept of flexibility can be demonstrated through the idea of induced fit. An enzyme that has a site that originally is not complementary to a substrate may become complementary upon the binding of the substrate. The site of binding changes to a shape that accepts the substrate. This indicates that the enzyme is indeed flexible and the conformation can be changed by the influence of the substrate.
The induced fit model asserts that the binding site of an enzyme is optimized for the transition state of the substrate, not the normal state. This is so that it can easily stabilize the transition state once it is bound to the enzyme, thereby decreasing the activation energy of the reaction and bringing the reaction to equilibrium much quicker .
The binding sites of enzymes has several common features. The active site is a site formed by amino acids that are connected to different parts of the protein. The specific positioning of the amino acids work together to form a three dimensional cleft or crevice. The active site of an enzyme takes up a very small portion of the total volume of the enzyme. The amino acids that do not play a role in enzymatic activity are there to make up for the structure. The many amino acids form a three dimensional structure of the molecule that allows the interaction of the active sites to work with each other and to have reactions with other molecules. Active sites also possess unique micro environments. These unique environments may contain polar or nonpolar residues that each have their own ways for interacting with nearby substrates. In addition, substrates are bound to enzymes by many weak forces that include van der Waals forces, hydrogen bonding, and hydrophobic interactions. Although these forces may be weak individually, the large number of forces acting together contributes to the stability of the binding site of the enzyme-substrate complex. Finally, the specific binding sites of an enzyme depend on the specific arrangement of the molecule. Here is an example where one structure leads to an specific function.
Binding sites for large ligands can be either flat, convex, or concave while binding sites for small ligands tend to be exclusively concave. One characteristic of binding sites is that it contain a significantly greater amount of exposed hydrophobic surfaces than other parts. In addition, it is also important for binding sites to possess the ability to bound to ligands firmly, but not so firmly that it will be hard for the ligand to be released. Therefore, weaker non-covalent interaction is characteristic of binding sites because it allows for an easy exchange. This concept is particularly important for hemoglobin because its major function is to transport oxygen from the lungs to the tissues. In order to efficiently do its job, its binding affinity to oxygen must be sufficiently strong, but at the same time, not too strong otherwise the hemoglobin would not be able to release the bound oxygen once it's in the tissue. Aside from binding affinity, binding specificity is also very important. Specialized and specific micro environments at binding sites are necessary for efficient binding. Lastly, the displacement of water is often typical for binding sites because it is able to promote the binding process.
How Enzyme Catalytic Mechanism/Binding relates to Pharmaceutical field
Pharmaceutical drugs work the same way Enzymes work. They bind to a specific binding site that either inhibits or activates a specific biological action. Caffeine for example is a really known molecule that a lot of people use every day, by drinking coffee or tea, to keep them awake. The reason behind the relationship between Caffeine and alertness and wakefulness is the big structural similarity between the caffeine molecule, adenosine and cyclic adenosine phosphate. The structures similarity allows caffeine molecules to bind to the same binding site of receptors or enzymes that reacts with adenosine derivatives.
Adenosine has a very important role in the regulation of brain activity. The human brain builds up adenosine molecules during the day. When the level of built up adenosine increases in the human brain, adenosine starts binding to its binding sites (receptors) in the human brain which activates mechanisms that lead to drowsiness and sleep.
Since caffeine has the same structural molecule it binds to the same receptors that adenosine binds to, preventing adenosine to bind to that specific receptor in the human brain and delaying the sleeping and drowsiness process.
Pharmaceutical drugs have pretty much the same role. They bind to a protein, enzyme, or a receptor that prevents binding of a specific molecule in the human body to bind to that specific receptor and therefore they inhibit a specific mechanism. A specific drug or molecule can bind to more than one binding site as mentioned earlier in the caffeine case. That means that the same drug can inhibit more than one biological mechanism at the same time by binding to two different binding sites. Binding to more than one binding site can explain the side effects of a specific prescribed drug. Furthermore, pharmaceutical and drug development scientists are currently trying to correlate and create a large network of the current drugs with their side effects, binding sites, structures, and role. The reason there is so much research is done in that field is that scientists strongly believe that discovering drugs that are already there that can cure some other diseases has a very high probability because of the presence of a wide range of drugs. Spending enough money on such projects can save a huge amount of money spent on drugs discovery. Discovering a drug can cost $800 million that is not including the huge amount of time and hassle it takes to test out the drug and then goes in market.
Tamoxifen for instance has thirty six discovered binding sites in the human body. Tamoxifen’s molecule has structural similarity to estrogen but it is missing a part that activates breast cancer cells production. Tamoxifen’s secondary binding sites suggest that the drug can be used to cure some other diseases.
Through bioinformatics, researchers are working hard developing websites and databases that helps ease drug discovery process and makes it more effective and less costly. Promisuous([5]) database is a tool that has a lot of information about drugs, their structures, binding sites (targets) and metabolic pathways that a specific drug goes through. The tool is really helpful and it has all the needed information that were collected through other important websites and databases like PubChem, Protein Data Base, and Uniprot. It a great tool that helps researchers find more information about drugs in just one place faster and eases drug discovery process in the future.
Jeremy Berg, Biochemistry 6ed.
Conformation Selection is when a dynamically fluctuating protein (ligand) binds to a protein and shifts the conformational ensemble towards a stabilized state. You may think that conformation selection and induced fit are the same, but induced fit only concerns about the interaction between a protein and its rigid binding partner. Recent studies and experiments are still trying to find a rigid distinction between the two.
In the late-1990s, X-ray and cryo-electron microscope images, NMR data and kinetics studies were done by scientists to verify the
'lock and key' hypothesis. After years and years of work, models were presented to uncover the complexity of binding scenarios. The Koshland-Nemethy-Filmer (KNF) and the Monod-Wyman-Changeux (MWC) model described the allosteric effect on binding. Allosteric effect is when a ligand binds to a binding site, it induces a conformational change that affect binding ability of the other site. Recent experiments and data show that conformational selection is usually followed by this conformation change or conformation adjustment, thus making the distinction between the induced fit and the original conformation selection models more confusing.
The extended conformational selection model does not only show us that the conformations of the proteins change, but the energy of both binding proteins also changes. As the two proteins approach each other, the electrostatic force and hydrogen bonding change the energy content of the two proteins. These proteins can also undergo different conformational selection and adjustment steps to perform a particular function. These selections and adjustments are step-wise, with one follows another, and dependent, with one step affecting the next or further steps.
There are also a few factors that affect the conformation selection:
Strength of interaction (ionic, hydrogen bond or dipole-dipole)
Concentration of proteins
Size difference (larger proteins often have higher flexibility)
Over years and years of experiments, contributors to conformational changes are identified. The contributors are believed to play a vital role in protein's conformational changes.
Transient Encounter complexes: these complexes are small (small contact area) and are hold by electrostatic forces. However, they cover a relatively large (~15%) surface area at the binding site.
Anchor residues: they are in conformation that are similar to the final conformation after binding. They have a large contact area.
Latch Residues: there residues are presented to stabilize the interaction between proteins.
Protein segments: these segments amass kinetic energy and provide the energy for conformational changes. They are found to exchange up to 65% of their stored kinetic energy during conformational changes, which trigger the induced-fit effect and greatly contribute to the binding.
Ras proteins and protein kinases (guanine and adenine nucleotide triphosphatases) go through big conformational changes when a ligand binds in the course of their functions. Some new computer simulation methods are being combined with experiments to further our knowledge of conformational changes. What scientists refer to as a ‘conformational selection picture’ is surfacing where changes in the relative populations of known conformations can best explain the conformational switching activity of important proteins.
[1]
Over decades, scientists are trying to solve the protein folding problem. In order to thoroughly understand how proteins fold into 3D structure, scientists have to first discover the folding and unfolding of protein. Through experiments, there are a few essential studies that contribute to the biochemical society.
Temperature affects the stability and folding of proteins. At high temperatures, scientists found that most proteins are unstructured. This is because, at high temperature, weak bonding such as ionic interaction and hydrogen bonding is broke apart. This decreases the induced-fit effect of the proteins and increase their conformational selection.
Chaperones aid the assembly of proteins and solve the problem of aggregation (proteins fold into a giant polymorphic aggregate species. A number of chaperones have the ability to unfold proteins and promote the binding of protein complexes.
Peter Csermely, Robin Palotai and Ruth Nussinov. Induced fit, conformational selection and independent dynamic segments: an extended view of binding events. Trends Biochem Sci. 2010 Jun 10.
binding graphMyoglobin(blue) with its ligand heme group(orange).
A ligand is a small molecule that is able to bind to proteins by weak interactions such as ionic bonds, hydrogen bonds, Van der Waals interactions, and hydrophobic effects. In some cases, a ligand also serves as a signal triggering molecule. A ligand can be a substrate inhibitor, activator or a neurotransmitter.
For example, oxygen is the ligand that binds to both hemoglobin and myoglobin.
Binding site: a region of the protein that is complementary to a specific molecule or ion. This site usually exhibits specificity to ligands. The weak interactions of the primary structure of protein, specifically the side chains to the ligand, usually initiates a response. The concentration at which all binding sites are bound to a ligand is termed the point of saturation.
Binding site and the ligand
Induced fit is the concept that an enzyme is a flexible rather than a rigid entity. Interactions between the active site and substrate continually reshape the tertiary structure slightly. Instead of the substrate simply binding to the active site, the enzyme and substrate mold to induce a fit similar to that of a lock and key. This allows the substrate to be in the precise position to enable a catalytic response.
Dissociation constant: Kd is the tendency for a ligand to bind to a binding site. It is measured by the ratio of concentrations of the ligand and enzyme over the concentration of the Enzyme-ligand complex. It is equal to the concentration of the ligand at which the total binding sites are half occupied. Association constant is equal to the reciprocal of the dissociation constant.
Cooperativity:
Allosterism:
Hill equation:
A molecule, atom, or ion that is charged or neutral and of non-bonding pairs of electrons as electron donors or Lewis bases that form bond to a central metal atom or ion to be as complex ion; it is important for control of chemical reactivity of the complex of ligands and metal; monofunctional ligands are complex ions that have one non-bonding pair of electrons, polyfunctional or known as chelates, two or more. Biological ligands are mostly electron-donating groups; important one of biological system is heme that is of nitrogen donating groups and forms chelate structure.
When a ligand binds to the protein, the chemical conformation of the protein changes. The tertiary structure of the protein is altered. The conformation of the protein determines the function of the protein, as structure often denotes a lot about the function. The tendency in which the ligand binds to the protein is known as the term affinity.
The binding affinity depends on the interaction of the binding site with the ligand. When the interaction of the intermolecular forces between the ligand and binding site are high, the affinity is increased. Similarly, when the intermolecular forces between the ligand and binding site are weak, the affinity is low.
When the affinity is high for the ligand binding, the concentration of the ligand does not need to be high in order for the ligand to bind to its maximum potential. Similarly, when the affinity is low, the concentration of the ligand must be large in order for the ligand to bind properly to the binding site.
For example, ligands have an effect on biphosphoglycerate in the T-form of hemoglobin. The ligand binds to the deoxyhemoglobin cavity which decreases the oxygen affinity. Thus, it stabalizes the deoxy form of hemoglobin.
A ligand is a substance that has the ability to bind to and form complexes with other biomolecules in order to perform biological processes. Essentially, it is a molecule that triggers signals and binds to the active site of a protein through intermolecular forces (ionic bonds, hydrogen bonds, Van der Waals forces). The docking (association) is usually a reversible reaction (dissociation). Within biological systems, it is rare to find irreversible covalent bonds between the ligand and its target molecule. The chemical conformation is changed when the ligand bonds to its receptors. For example, the three dimensional shape of the receptor protein is change upon the binding of the ligand. Also, the conformational state of a receptor protein will cause variations in the functional state of a receptor. The strength/tendency of the ligand binding is known as affinity. Different types of ligands include substrates, inhibitors, activators, and neurotransmitters.
Human cells use receptor-mediated endocytosis to take in cholesterol for use in the synthesis of membranes and as a precursor for the synthesis of other steroids. Cholesterol travels in the blood in particles called low-density lipoproteins(LDLs), complexes of lipids and proteins. These particles act as ligands hence they bind to LDL receptors on membranes and enter the cells by endocytosis. In humans with familial hypercholesterolemia, an inherited disease characterized by a very high level of cholesterol in the blood, the LDL receptor proteins are defective or missing so the LDL particles cannot enter cells. Instead, cholesterol accumulates in the blood, where it contributes to early atherosclerosis. Atherosclerosis is the buildup of lipid deposits within the walls of blood vessels, causing of the bulge inwards of vessels and impeding blood flow.
Note: These are the three types of endocytosis that the cell participates in. The third one represents the receptor-ligand binding mentioned for cholesterol in humans.
The binding of a ligand to a protein is greatly affected by the structure of the protein and is often accompanied by conformational changes. As an example, the specificity with which heme binds its various ligands changes when the heme is a component of myoglobin. When carbon monoxide binds to free heme molecules, it binds more than 20,000 times better than oxygen does, but it only binds 200 times better than oxygen when the heme is bound in myoglobin. The difference is most likely due to steric hindrance but there are other factors that have not yet been well-defined that may also affect the interaction of heme with carbon monoxide.
Oxygen is poorly soluble in aqueous solutions and cannot be carried to tissues in sufficient quantity if it is only dissolved in blood serum. The diffusion of oxygen through tissues is also ineffective over distances greater than a couple of millimeters. The evolution of larger, multicelluluar animals, though, depended on the evolution of proteins that could transport and store oxygen, but none of the amino acid side chains in proteins are suited for the reversible binding of oxygen molecules. This function was filled by certain transition metals, among them being iron and copper, that have a strong tendency to bind oxygen. The multicellular organisms make use of the properties of metals, most commonly iron, for oxygen transport. However, iron promotes the formation of highly reactive oxygen species that can damage DNA and other macromolecules. Therefore, the iron used in cells is bound in forms that isolate it or make it less reactive. In order for multicellular organisms to make use of iron, especially when it must be transported over long distances, iron is incorporated into a protein-bound prosthetic group called heme. Iron in the ferrous state binds oxygen reversibly while the ferric state does not bind oxygen. Heme is found in many oxygen-transporting proteins as well as in proteins that participate in oxidation-reduction reactions. When oxygen binds to heme, the electronic properties of the heme iron change, which accounts for the change in color from the dark purple of oxygen-depleted venous blood to the bright red of oxygen-rich arterial blood. Some small molecules, such as carbon monoxide (CO) and nitrogen monoxide (NO), coordinate to heme iron with greater affinity than does oxygen gas. When a molecule of carbon monoxide is bound to heme, oxygen is excluded and this is why carbon monoxide is highly toxic to aerobic organisms. By surrounding and isolating heme, oxygen-binding proteins can regulate the access of CO and other small molecules to the heme iron.
Myoglobin is a relatively simple oxygen-binding protein that is found in almost all mammals, primarily in muscle tissue. It facilitates the oxygen diffusion in muscles. Myoglobin is a single polypeptide consisted of 153 amino acid residues with one molecule of heme. It is typical of the family of proteins called globins, all of which have similar primary and tertiary structures.
The ability of a protein to bind metal ligands is often essential to a protein’s function. Furthermore, recent studies have indicated the existence of a new type of protein-metal interaction in which the metal confers a particular function to the protein. Using the familiar example of hemoglobin and its heme group, this new type of interaction would be analogous to attaching a heme group to a protein not naturally possessing one to allow that protein to transport oxygen. In addition, many metals have the ability to induce a conformational change in the protein that they bind to, thus altering its function once again.
A practical application of this concept is the creation of amyloid mimetics. Because of their strength and mechanical stiffness, amyloids are attractive compounds for the construction of nanomaterials. This can be done by employing peptides with a secondary structure of alpha helices and a primary structure containing amino acids with metal-ligating side chains such as His and Cys. When metals such as Cu(II) or Zinc(II) are introduced, they will bind as ligands at the center of the helix to create the desired nanomaterials. This process can be taken further by creating a coil within a coil. This is usually done using heavy metals such as Pb and As, and results in a diverse array of nanomaterials. Alternatively, peptides can be designed to interact with a metal in a desired way, thus yielding different structures and functions. For example, addition of Cd(II) may induce a conformation in which there are four connections to the metal, whereas addition of Cu(I) may only form two connections with that same peptide.
Another area of interest involving protein-metal interaction; is the for inorganic substrates in the solid state such as metals and carbon. This type of behavior has been observed in the formation of pearls, as well as in bones and tooth enamel in humans. Although a definitive relationship between protein affinity and substrate identity has not yet been established, certain observations pervade this phenomenon. For example, proteins with an affinity for Ag substrates tend to contain proportionally larger amounts of Ser and Pro, whereas proteins with an affinity for C (nanotubes) tend to contain proportionally smaller amounts of Typ and His. Another observed trend is the affinity of unfolded proteins for Au and the affinity of folded proteins for Pt.
Protein-metal interaction is also being studied in green fluorescent protein (GFP). The concept is to create a “metal sensor” by combining the easy visibility of fluorescent compounds with the ability of proteins to bind metal ligands. Through modification of GFP, variations of GFP can be created that are specifically designed to bind a certain number of a certain metal atom, thus the amount of GFP will correspond to the unknown amount of metal ions in the sample solution. Recent studies indicate that the best results are with metals in the 2+ oxidation state, particularly Zn(II), with the exception of Ca(II) and Mg(II).
A binding site is a position on a protein that binds to an incoming molecule that is smaller in size comparatively, called ligand.
In proteins, binding sites are small pockets on the tertiary structure where ligands bind to it using weak forces (non-covalent bonding). Only a few residues actually participate in binding the ligand while the other residues in the protein act as a framework to provide correct conformation and orientation. Most binding sites are concave, but convex and flat shapes are also found.
A ligand-binding site is a place of chemical specificity and affinity on protein that binds or forms chemical bonds with other molecules and ions or protein ligands. The affinity of the binding of a protein and a ligand is a chemically attractive force between the protein and ligand. As such, there can be competition between different ligands for the same binding site of proteins, and the chemical reaction will result in an equilibrium state between bonding and non-bonding ligands. The saturation of the binding site is defined as the total number of binding sites that are occupied by ligands per unit time. High affinity ligands have a high intermolecular force, are able to reside in the binding site longer since it has a low concentration, and cause the receptors to change.
The most common model of enzymatic binding sites is the induced fit model. It differs from the more simple "Lock & Key" school of thought because the induced fit model states that the substrate of an enzyme does not fit perfectly into the binding site. With the "lock & key" model, it was assumed that the substrate is a relatively static model that does not change its conformation and simply binds to the active site perfectly. According to the induced fit model, the binding site of an enzyme is complimentary to the transition state of the substrate in question, not the normal substrate state. The enzyme stabilizes this transition state by having its NH3+ residues stabilize the negative charge of the transition state substrate. This results in a dramatic decrease in the activation energy required to bring forth the intended reaction. The substrate is then converted to its product(s) by having the reaction go to equilibrium quicker.
Proteins are important in our human body. A protein is a chain of amino acids joined by peptide bonds. By understanding protein function, we can solve different problems in our body because proteins are among the main biological components. In order to understand protein functions, there will be four main ideas to study, such as: structure, binding, catalysis, and switching. However, these ideas will be much easier to understand if we know about protein binding. Protein binding has an extremely important role in biochemistry. Protein binding is often reversible and can be stable or unstable depend on the structures and the activities of it. Furthermore, that is also a reason why protein binding can be influence the drug's biological half life in our body. Indeed, many scientists try to perform experiments and research on the binding structure in order to discover diseases and how to destroy it.
Protein binding and the fraction unbound written as the concentration of unbound drug over the total concentration of the drug, which depends on several factors. It is determined by the drug’s affinity for the protein, the concentration of the binding protein, and the concentration of the drug relative to the binding protein.
Protein binding also is very crucial for the hemoglobin and myoglobin. Hemoglobin is involved in the transport of other gases. Also, it carries some of the body's respiratory carbon dioxide, in which CO2 is bound to the globin protein. Hemoglobin exhibits characteristics of both the tertiary and quaternary structures of proteins. Hemoglobin consists mostly of protein, and these proteins, in turn, are composed of sequences of amino acids. Myoglobin is found in the muscle tissues and it was the first protein to have its three-dimensional structure revealed. Even though hemoglobin is found in the blood and myoglobin is found in the muscles, they are have a great connection with each other through functioning and deliver and transport oxygen.
Complementarity
Molecular recognition depends on the tertiary structure of the enzyme which creates unique microenvironments in the active/binding sites. These specialized microenvironments contribute to binding site catalysis.
Flexibility
Tertiary structure allows proteins to adapt to their ligands (induced fit) and is essential for the vast diversity of biochemical functions (degrees of flexibility varies by function). Flexibility is essential for biochemical function.
Surfaces
Binding sites can be concave, convex, or flat. For small ligands – clefts, pockets, or cavities. Catalytic sites are often at domain and subunit interfaces. Catalytic sites often occur at domain and subunit interfaces.
Non-Covalent Forces
Non-covalent forces are also characteristic properties of binding sites. Such characteristics are: higher than average amounts of exposed hydrophobic surface, (small molecules – partly concave and hydrophobic), and displacement of water can drive binding events, and weak interaction can lead to an easy exchange for partners.
Affinity
Binding ability of the enzyme to the substrate (can be graphed as partial pressure increases of the substrate against the affinity increases).
Enzyme inhibitors are molecules or compounds that bind to enzymes and result in a decrease in their activity. An inhibitor can bind to an enzyme and stop a substrate from entering the enzyme's active site and/or prevent the enzyme from catalyzing a chemical reaction. There are two categories of inhibitors.
irreversible inhibitors
reversible inhibitors
Inhibitors can also be present naturally and can be involved in metabolism regulation. For example, negative feedback caused by inhibitors can help maintain homeostasis in a cell. Other cellular enzyme inhibitors include proteins that specifically bind to and inhibit an enzyme target. This is useful in eliminating harmful enzymes such as proteases and nucleases. In addition, many different man made substances are capable of inhibiting enzymes. For example, fluorinated phosphonates (i.e. sarin, VX) bind irreversibly to the enzyme cholinesterase resulting in a buildup of acetylcholine in the body. Moreover, drugs, such as MAOIs (monoamine oxidase inhibitors) bind reversibly to the enzyme monoamine oxidase allowing for the accumulation of monoamine neurotransmitters, which is useful in the treatment of certain medical conditions.
competitive inhibition, the inhibitor and substrate compete for the enzyme (i.e., they can not bind at the same time).[58] Often competitive inhibitors strongly resemble the real substrate of the enzyme. For example, methotrexate is a competitive inhibitor of the enzyme dihydrofolate reductase, which catalyzes the reduction of dihydrofolate to tetrahydrofolate. The similarity between the structures of folic acid and this drug are shown in the figure to the right bottom. Note that binding of the inhibitor need not be to the substrate binding site (as frequently stated), if binding of the inhibitor changes the conformation of the enzyme to prevent substrate binding and vice versa. In competitive inhibition the maximal velocity of the reaction is not changed, but higher substrate concentrations are required to reach a given velocity, increasing the apparent Km.
Irreversible inhibitors covalently bind to an enzyme, cause chemical changes to the active sites of enzymes, and cannot be reversed. A main role of irreversible inhibitors include modifying key amino acid residues needed for enzymatic activity. They often contain reactive functional groups such as aldehydes, alkenes, or phenyl sulphonates. These electrophilic groups are able to react with amino acid side chains to form covalent adducts. The amino acid components are residues containing nucleophilic side chains such as hydroxyl or sulfhydryl groups such as amino acids serine, cysteine, threonine, or tyrosine.
Kinetics of Irreversible Inhibitor
First, irreversible inhibitors form a reversible non-covalent complex with the enzyme (EI or ESI). Then, this complex reacts to produce the covalently modified irreversible comple EI*. The rate at which EI* is formed is called the inactivation rate or kinact. Binding of irreversible inhibitors can be prevented by competition with either substrate or a second, reversible inhibitor since formation of EI may compete with ES.
Example of a reversible inhibitor forming an irreversible product.
In addition, some reversible inhibitors can form irreversible products by binding so tightly to their target enzyme. These tightly-binding inhibitors show kinetics similar to covalent irreversible inhibitors. As shown in the figure, these inhibitors rapidly bind to the enzyme in a low-affinity EI complex and then undergoes a slower rearrangement to a very tightly bound EI* complex. This kinetic behavior is called slow-binding. Slow-binding often involves a conformational change as the enzyme "clams down" around the inhibitor molecule. It changes shape slightly to accommodate the substrate enough to allow the reaction to kinetically proceed forward. Some examples of these slow-binding inhibitors include important drugs such as methotrexate and allopurinol.
Group Specific Reagents
Group specific reagents inactivate an enzyme by reacting with a certain side chain on an amino acid. One example is Iodoacetamide reacting with a cysteine side chain to completely inactivate the enzyme and creating I- and H+ ions along the way. Another example is diisopropylphophofluoridate (DIPF)inactivating enzymes by reacting with a crucial serine side chain in acetylcholinesterase.
Reactive Substrate Analogs (Affinity Labels)
Affinity labels are specific to the binding site on the enzyme. They covalently bind to active-sites in place of the substrate and thus modify a crucial side-chain and inhibit the activity of the enzyme. For example, bromoacetol phosphate binds to triose phosphate isomerase (TPI) at its active site. The covalent bonding inhibits the enzymatic activity since the active side chain (i.e. glutamic acid) is not active.
Mechanism Based Inhibitors (Suicide Inhibitors)
Mechanism based inhibitors affect the enzyme after the initial substrate has already been bound to an enzyme and has undergone a normal catalytic mechanism. The catalysis generally generates an intermediate that covalently inhibits the enzyme.
Reversible inhibitors bind non-covalently to enzymes. Many different types of inhibitions can occur depending on the structure of the enzyme the inhibitors bind to. The non-covalent interactions between the inhibitors and enzymes include hydrogen bonds, hydrophobic interactions, and ionic bonds. Many of these weak bonds combine to produce strong and specific binding. In contrast to substrates and irreversible inhibitors, reversible inhibitors generally do not undergo chemical reactions when bound to the enzyme and can be easily removed by dilution or dialysis.
There are three kinds of reversible inhibitors:
1) competitive
2) mixed
3) uncompetitive/mixed
Competitive inhibitors, as the name suggests, compete with substrates to bind to the enzyme at the same time. The inhibitor has an affinity for the active site of an enzyme where the substrate also binds to. This type of inhibition can be overcome by increasing the concentrations of substrate in order to overcome the inhibitor. Competitive inhibitors are often similar in structure to the substrate. These inhibitors increase the Km.File:Competitive inhibitor.docx
Oxyanion hole stabilizes the tetrahedral intermediate. It is formed by hydrogen bonds linking peptide NH groups to the negatively charged oxygen atom.
Step 3: Instability of the negative charge on the substrate carbonyl oxygen when will leads to collapse of the tetrahedral intermediate, re-formation of a double bond with carbon which breaks the peptide bond between the carbon and amino acid group. The amino leaving group is protonated by His57, facilitating its displacement.
Step 4: The amine component is departed from the enzyme (metabolized by the body) and this completes the first stage (acylation of enzyme). The first product is been made.
Step 5: A water molecule is added.
Step 6: An incoming water molecule is deprotonated by acid-base catalysis, generating a strongly nucleophilic hydroxide ion. Attack of hydroxide on the ester linkage of the acylenzyme generates a second tetrahedral intermediate.
Step 7: collapse of the tetrahedral intermediate form the second product, a carboxylate anion, and displace Ser195.
Step 8: The carboxylic acid is released and the enzyme is reformed to catalyze the next reaction.
Uncompetitive inhibitors bind to the enzyme at the same time as the enzyme's substrate. However, the binding of the inhibitor affects the binding of the substrate, and vice-versa. This type of inhibition cannot be overcome, but can be reduced by increasing the concentrations of substrate. The inhibitor usually follows an allosteric effect where it binds to a different site on the enzyme than the substrate. This binding to an allosteric site changes the conformation of the enzyme so that the affinity of the substrate for the active site is reduced. These inhibitors lower the K mand Vm.File:Uncompetitive inhibition.docx
mixed inhibition the inhibitors can bind to the enzyme at the same time as the enzyme substrate. If the concentrate of substrates is higher than the inhibitor, then this type of inhibition can be reduced.File:Mixed inhibition.docx
Uncompetitive inhibitors are able to bind to both E and ES, but their affinities for these two forms of the enzyme are different. Therefore, these inhibitors increase Km and decrease Vmax because they interfere with substrate binding and hamper catalysis in the ES complex.
Non-competitive inhibitors have identical affinities for E and ES. They do not change Km, but decreases Vmax.
Reaction Rate vs. Substrate
In the Lock and Key Model, first presented by Emil Fisher, the lock represents an enzyme and the key represents a substrate. It is assumed that both the enzyme and substrate have fixed conformations that lead to an easy fit. Because the enzyme and the substrate are at a close distance with weak attraction, the substrate must need a matching shape and fit to join together. At the active sites, the enzyme has a specific geometric shape and orientation that a complementary substrate fits into perfectly. The theory behind the Lock and Key model involves the complementarity between the shapes of the enzyme and the substrate. Their complementary shapes make them fit perfectly into each other like a lock and a key. According to this theory, the enzyme and substrate shape do not influence each other because they are already in a predetermined perfectly complementary shape. As a result, the substrate will be stabilized. This theory was replaced by the induced fit model which takes into account the flexibility of enzymes and the influence the substrate has on the shape of the enzyme in order to form a good fit.
Lock and Key Analogy
The active site is the binding site for catalytic and inhibition reaction of the enzyme and the substrate; structure of active site and its chemical characteristic are of specificity for binding of substrate and enzyme. Three models of enzyme-substrate binding are the lock-and-key model, the induced fit model, and the transition-state model. The lock-and-key model assumes that active site of enzyme is good fit for substrate that does not require change of structure of enzyme after enzyme binds substrate.
Diagrams to show the induced fit hypothesis of enzyme action
Induced fit indicates a continuous change in the conformation and shape of an enzyme in response to substrate binding. This makes the enzyme catalytic which results in the lowering of the activation energy barrier causing an increase in the overall rate of the reaction. In other words, when a substrate binds to an enzyme, it will change the conformation of the enzyme. This forms a transitional intermediate which lowers the activation energy and allows the reactants to proceed towards the product at a faster rate. In the case of macromolecules (e.g. proteins), induced fit shows the changes in the shape of a macromolecule in response to a ligand binding so that the binding site of macromolecule conforms more efficiently to the shape of the ligand. The enzyme will change its shape until it is completely complementary to a substrate to activate the enzyme-substrate complex.
As the Enzyme-substrate complex is formed, free energy is released from the formation of the many weak interactions between the enzyme-substrate complex. The free energy that is released is called binding energy and it is maximized only when the "correct" substrate binds to the corresponding specific enzyme. To maximize the release of free energy, the substrate has to be in its transition state. When this happens, the Enzyme-substrate complex becomes a catalyst, which then makes other activation energies lower.
Of enzyme, the active site is the binding site for catalytic and inhibition reaction of enzyme and substrate; structure of active site and its chemical characteristic are of specificity for binding of substrate and enzyme. Two theories for the ways in which enzyme binds to substrate are lock-and-key model and induced fit model; induced fit is the model such that structure of active site of enzyme can be easily changed after binding of enzyme and substrate.
An illustration of an induced fit interaction between substrates and enzymes
The induced fit model describes the formation of the E-S complex as a result of the interaction between the substrate and a flexible active site. The substrate produces changes in the conformation on the enzyme aligning properly the groups in the enzyme. It allows better binding and catalytic effects.
This model opposes to the lock and key model that explains the formation of the E-S complex as a result of the binding of complementary geometrical rigid structures, as a lock and a key. The concerted model and the sequential model are models used to explain the allosteric changes of conformation of an enzyme from the T structure to the R structure and vice versa. In the concerted model all the subunits that form the allosteric protein change conformation at once, while in the sequential model the change in conformation of one subunit favors the change in conformation of the other subunits.
The Michaelis Menten model is related to the kinetics of enzyme catalyzed reactions, and describes the relationship between the concentration of substrate and enzyme velocity in a reaction assuming that no allosteric effects exist.
Adenylate kinase is a good example of induced fit. This enzyme functions by slightly changing conformation when both the necessary substrate, ATP and NMP are bound. When both ATP and NMP are bound to this kinase a part of this enzyme called the P-loop moves down and forms a lid over the two groups, this in turns helps to hold the two substrate closer together in order to more easily carry out the reaction of transferring a phosphate group from ATP to NMP. This holds the phosphate group of ATP to a closer proximity to NMP, this also holds the two substrate in the proper orientation. This conformational change helps to carry out the reaction more efficiently by placing the substrate in the right position and closer to each other. We see that this enzyme functions through an induced fit, as the substrate bind the conformation of the enzyme slightly changes in order to better interact with the substrate. When Both substrate are bound various conformational changes occurs, this ensures that the reaction only proceeds when both substrate are present and this eliminates any unnecessary transfer of a phosphate group to water if the NMP is not present.
There are 4 types of catalysis mechanisms that occur after the substrate is bound to an enzyme, causing formation of a transition-state complex and the product:
1.Catalysis by Bond Strain: The new arrangement occurs in the binding of the substrate and the enzyme to ultimate bind together in order to form a strained substrate bond. Such binding will rapid the formation of transition-state. However, the final conformation is not allowed for bulky group and substrate atoms.
2.Catalysis by Proximity and Orientation: Enzyme-substrate interactions shows a clear direction to the reactive groups and make them close to one another. The inducing strains are also reactive which play an important role in the catalysis.
3.Cataylsis Involving Acids and Bases: The strain mechanism makes amino acid act as an acid or base to complete the catalysis reaction. Acids are proton donors, and bases are proton acceptors.
4.Covalent Catalysis: Since the substrate is directed to the active site of an enzyme, a covalent bond forms between the substrate and the enzyme. Example: Proteolysis by serine protease is a reaction when proteases have a serine active site that forms a covalent bond between the alkoxyl group of serine and carbonyl carbon of the peptide.
The transition state of any reaction is difficult to study, because it has no visible lifetime. To understand the transition-state Model of enzymatic catalysis, the interaction between the enzyme and this transition in the course of a reaction needs to be understood. An enzyme is complementary to the transition state can be viewed as a requirement for catalysis. The energy "hill" that the transition state sits is what the enzyme must lower if catalysis is to occur. Since if it is too high, the reaction cannot take place. The idea of enzyme-transition state complementarity is shown through a variety of examples.
The structure of the transition state is basically an expanded and spelled out version of an original state where tertiary and secondary structures form concurrently. The transition state model is similar to another model which is known for folding proteins rather than providing a framework model, the "global collapse model". This is a possible common function feature for proteins missing the transition state. An example of the transition state model in action is the folding of barnase. Although barnase has formed complete secondary and tertiary elements in the transition state, a framework process was involved. Unfortunately, the framework could be tampered with by the global collapse model and a unified folding scheme could be presented.
The transition-state model starts with an enzyme which then binds to a substrate. Energy is the required to change the shape of substrate. Once the shape is changed, the substrate is unbound from the enzyme. This ultimately causes a change in the shape of the enzyme. One of the most important aspects of the model is that it increases the amount of free energy.
Without EnzymeEnzyme Not Complementary to Trans StateEnzyme Complementary to Trans StateTrans State
The Transition State Theory is probably the most important rate enhancing mechanism to understand in regards to enzymes. This theory is stating that enzyme binds the transition state of the reaction more tightly than either the substrate or product which is causing the ΔG to be lowered. The weak interactions between the enzyme and substrate are optimized in the transition state.
There is a few important topics to note. If the active site of the "enzyme" is complementary to the substrate, then ΔG is raised. The “enzyme” does not enhance the reaction and stays bound. If the active site is complementary to the transition state, then ΔG is lowered and the enzyme enhances the reaction. ΔG for forming the transition state is favored by the energetics of weak interactions between the enzyme and the transition state.
The enzyme stabilizes and reduces the energy of the transition state structure by forming a non covalent bonds to it . The lowering of energy that results from the binding of the transition state increases the likelihood that the transition state will form and convert to the product. Transition-state stabilization is important to enzyme catalysis. As proof that the active site is most complementary to the transition state structure, chemicals known as transition-state analogs, which resemble the structure of the transition state, have been shown to bind to enzymes with higher affinity than substrates. Due to tight binding, many of these molecules are good inhibitors of enzymes. For example, the antibiotic, penicillin, inhibits the transpeptidase enzyme that catalyzes cross-linking of bacterial cell wall because it resembles the transition state for this reaction. It makes sense that enzymes are more complementary to the transition state than to their substrates. If they were most complementary to the substrate, they might bind them so tightly that the reaction would not be able to proceed.
Nelson, David L.; Cox, Michael M. (2005), Principles of Biochemistry (4th ed.), New York: W. H.
D E Otzen, L S Itzhaki, N F elMasry, S E Jackson, and A R Fersht. "Structure of the transition state for the folding/unfolding of the barley chymotrypsin inhibitor 2 and its implications for mechanisms of protein folding". http://www.pnas.org/content/91/22/10422.short. Last accessed: 30 Nov. 2011.
Cooperativity can be seen in both enzymes and receptors, and describes the trends that occur when these structures contain multiple binding sites. Cooperativity describes the changes that occur when a binding site of one of these structures is activated or deactivated affecting the other binding sites in the same molecule. It can also be described as the increasing or decreasing affinity for binding of the other sites affected by the original binding site.
An example of a sigmoid curve.
Cooperativity can also be noted in large chain molecules that are made of many identical, or near identical, subunits (DNA, proteins, phospholipids), when these molecules go through phase transitions such as melting, unfolding, or unwinding, known as subunit cooperativity. When a substrate binds to the active site of one enzymatic subunit, the other subunits are stimulated and become active.
The activity of an enzyme can be graphed against the concentration of the substrate. For an enzyme that shows a cooperative behavior, the relation between the two shows a sigmoidal curve instead of Michaelis and Menten behavior. The graph shows a rapid increase in speed. This reflects how the binding on one subunit increases the chance that the other subunits will bind to a substrate.
An example of negative cooperativity is the decrease in binding affinity once one of the sites is bound. As ligands bind to the protein, the protein's affinity for the ligand decreases. The relationship between glyceraldehyde-3-phosphate and the enzyme glyceraldehyde-3-phosphate dehydrogenase is a clear example of this process.
Hemoglobin is made up of four subunits. If one subunit binds to oxygen it increases the chance the other three will do the same.
An example of positive cooperativity can be seen when a substrate binds to an enzyme with multiple binding sites and the other binding sites are affected by this change.
This behavior is seen on the binding of oxygen to hemoglobin to form oxyhemoglobin. Hemoglobin is made out of four subunits, two alpha and two beta. They come together to form a tetramer, each subunit having its own active site to bind oxygen to. This active site contains a porphyrin ring structure with an iron atom in the center. When the subunit is not bound to an oxygen the iron is about 0.4 A below the plane of the ring. When the tetramer is in this state, it is considered to be in the T-state or tense state.
The R-state, or relaxed state occurs when hemoglobin has bound to oxygen. Deoxyhemoglobin, or the T-state, has a low affinity for oxygen. When one molecule binds to a single heme, though, the oxygen affinity increases, which allows the following molecules to bind more easily in succession. This occurs when the iron bound to the oxygen is lifted to lie in the same plane as the ring. This forces the histidine residue it is attached to also move, which in turn forces the alpha helix where the histidine is attached, to move. The carboxyl terminal at the end of the helix is located at the interface of the two alpha-beta dimers therefore favoring the R-state transition. Overall the R-state is more stable than T-state but under certain conditions this can change.
The oxygen affinity of the 3-oxyhemoglobin is about 300 times greater than that of its deoxyhemoglobin counterpart. This behavior leads to the affinity curve of hemoglobin to become sigmoidal, not hyperbolic as with the monomeric myoglobin's affinity curve. In the same way, the ability for hemoglobin to lose oxygen is greater as fewer oxygen molecules are bound. This cooperativity can be seen in Hemoglobin when one of the oxygen binds to one of the tetramer's subunits. This will increase the probability that the other three sites will bind to oxygen.
An example of homotropic cooperativity is the effect that the substrate molecule has on its affinity.
An example of heterotropic cooperativity is when a third substance causes a change in the affinity.
The concerted model (symmetry model or MWC model): enzyme subunits are connected in such a way that a conformational change in one subunit is necessarily conferred to all other subunits. Thus all subunits must exist in the same conformation. Example: In hemoglobin, the tetramer changes conformation together (R state) after four oxygen molecules bind to all four monomers. The transition from the T state to the R state occurs in one step.
The Concerted Model, also known as MWC model or symmetry model, of hemoglobin is used to explain the cooperativity in oxygen binding as well as the transitions of proteins made up of identical subunits. It focuses on the two states of the Hemoglobin; the T and R states. The T state of the hemoglobin is more tense as it is in the deoxyhemoglobin form while the R state of the hemoglobin is more relaxed as it is in the oxyhemoglobin form. The T state is constrained due to the subunit-subunit interactions while the R state is more flexible due to the ability of oxygen binding. The binding of oxygen at one site increases the binding affinity in other active sites. Thus in the concerted model of the hemoglobin, it shows that the one oxygen binding to an active site will increase the probability of other oxygen binding to the other active sites. In a concerted model, all oxygen binding sites on Hemoglobin in the T state must be bound before converting to the R state. This is also true in the conversion from the R state to the T state, in which all bounded oxygen must be released before full conversion can take place. At each level of oxygen loading, an equilibrium exists between the T-state and R-state. The equilibrium shifts from strongly favoring the T-state (no oxygen bound) to strongly favoring the R-state (fully loaded with oxygen). Overall, oxygen binding shifts the equilibrium toward the R state. This means that at high oxygen levels, the R form will be prevalent and at lower oxygen levels, the T form will be prevalent. Allosteric effectors of hemoglobin, such as 2,3-BPG, function by shifting the equilibrium towards or away from the T-state, depends on whether it's an inhibitor or a promoter. This model and the sequential model displays the extreme cases of R and T transitions. In a real system, properties from both models are needed to explain the behavior of hemoglobin.
The sequential model: subunits are not connected in such a way that a conformational change in one induces a similar change in the others. All enzyme subunits do not necessitate the same conformation. The sequential model states that molecules of substrate bind through an induced fit. Example: In hemoglobin, the four monomers change conformation (R state) one at a time as oxygen binds to each monomer. This allows hemoglobin to have R state monomers and T state monomers.
The Sequential Model of the hemoglobin explains the cooperativity involved in the binding of oxygen. This model follows the concept that after binding occurs at one site in the active site, the binding affinity in the other sites around the protein will increase as well. Hence, the plot of substrate concentration versus reaction rate is of a sigmoidal shape. Because of this cooperativity, it does not follow Michaelis-Menten Kinetics. The difference between this model and concerted model is that the T states do not have to convert to R states all at one time. In this model, the ligand will change the conformation of the subunit that it is bound to and induce changes in the neighboring subunits. The sequential model does not require the overall state of the molecule to be in only T state or in only R state. Simply, each binding site influences nearby binding sites until all of the binding sites are in the same state. Neither the sequential model or the concerted model fully explains the nature of hemoglobin. Properties from both models appear in a real system.
Cooperativity not only occurs during ligand binding, but occurs any time energetic interactions simplify or complicate the occurrence of something happening that can involve multiple units as compared with single units. An example is the unwinding of DNA. Sections of DNA must first unwind in order for the DNA to carry out its other functions, such as replication, transcription, and recombination. Positive cooperativity among adjacent DNA nucleotides simplifies the process for unwinding a whole group of adjacent nucleotides compared to unwinding the same number of nucleotides spread along the DNA chain. The cooperative unit size is the number of adjacent bases that will unwind as a single unit because of the effects of positive cooperativity. This process applies to other types of chain molecules, too, such as the folding and unfolding of proteins, as well as the melting of phospholipid chains that comprise the cell membrane.
Entropy plays an important role in cooperativity. This can be seen in the example of oxygen binding to hemoglobin, where the first oxygen has four different sites that it can bind to. This shows a relatively higher entropy compared with the binding the last oxygen will have, which has only one site left that will bind. In going from an unbound to a bound state, the first oxygen must overcome a larger entropy change versus the final binding oxygen. This entropy difference is the main reason for the positive cooperativity in binding oxygen to hemoglobin.
When a plot of product formation as a function of substrate concentration produces a sigmoidal curve cooperativity is present. This sigmoidal curve is produced because of the effect of one substrate binding to one active site increasing the activity at the other active sites. The curve increases with a large slope and then levels out to its limit once the substrate saturation is reached.
The Hill equation is an equation describing the amount of ligand bound to the macromolecule, or its saturation. The equation is as follows:
Where:
represents the fraction of binding sites filled
represents the concentration of the ligand
represents the concentration of ligand required for half the binding sites to be occupied
represents the dissociation constant
represents the Hill coefficient, which describes the cooperativity of the reaction.
When:
, the reaction is POSITIVELY cooperative, meaning a ligand binding to a site INCREASES the ligand affinity at other binding sites.
, the reaction is NEGATIVELY cooperative, meaning a ligand binding to a site DECREASES the ligand affinity at other binding sites.
, the reaction is NOT cooperative, meaning a ligand binding to a site DOES NOT alter the ligand affinity at other binding sites.
The Hill Equation was formulated in 1910 by Archibald Hill (1886–1977), pioneer in biophysics.
An integral membrane protein (IMP) is a protein molecule directly attached to a phospholipid bilayer and serves as a structural and functional part of a cells membrane. Structurally, they traverse the hydrophobic phospholipid bilayer and can only be removed by detergents and denaturants that disrupt the hydrophobic interactions.
Currently, in three dimensional structure of only around 160 integral membrane proteins have been visualized through X-ray crystallography and nuclear magnetic resonance because of the difficulty in isolating the proteins and pure crystal growth. Integral membrane proteins can be categorized into two groups: Integral polytopic proteins (Transmembrane proteins) and integral monotopic proteins.
The portions of the protein located in the hydrophobic center of the bilayer are usually arranged into alpha helices so that the polar amino and carboxy groups can interact with each other rather than with the hydrophobic surroundings. The portion that projects out of the bilayer tends to have a large amount of hydrophilic amino acids.
Transmembrane proteins are the most popular IMP and traverse the entire cell membrane. Single pass membrane proteins cross the membrane just once while multi pass membrane proteins cross the membrane several times. Single pass proteins can either have their carboxy end towards the cytosol or their amino end directed at the cytosol.
Integral membrane proteins function as transporters, channels (see Potassium Channel), linkers, receptors, proteins involved in accumulation energy, and proteins responsible for cell adhesion. Examples include insulin receptors, Integrins, Cadherins, NCAMs, and Selectins.
Integral membrane protein movement and distribution
Many proteins are free to move laterally in the plane of the bilayer. One experiment used to show this involved fusing cultured mouse cells with human cells under appropriate conditions to form a hybrid cell known as a heterokaryon. The mouse cells were labeled with mouse protein-specific antibodies to which the green-fluorescing dye fluorescein had been covalently attached, whilst the human cells were labeled with the red-fluorescing dye rhodamine. Upon cell fusion, the mouse and human proteins as seen under the fluorescence microscope were segregated on the two halves of the heterokaryon. After 40 minutes at 37oC, however, the mouse and human protein had completely intermingled. Lowering the temperature to below 15oC inhibited this process, indicating that the proteins are free to diffuse laterally in the membrane and that this movement is slowed as the temperature is lowered. It should be noted, though, that some integral membrane proteins are not free to move laterally in the membrane because they interact with the cytoskeleton inside the cell.
The distribution of proteins in membranes can be revealed by electron microscopy using the freeze-fracture technique. In this technique,a membrane specimen is rapidly frozen to the temperature of liquid nitrogen and then fractured by a sharp blow. The bilayer often splits into monolayers, revealing the interior. The exposed surface is then coated with a film of carbon and shadowed with platinum in order for the surface to be viewed in the electron microscope. The fractured surface of the membrane is revealed to have numerous randomly distributed protuberances that correspond to integral membrane proteins.
The basic mechanism of alpha (α) helical membrane proteins being integrated into the endoplasmic reticulum membrane has been well established. However, scientists seek to find clearer details of these mechanisms as well as their kinetics to understand membrane protein integration as a whole. It is therefore important to use in vivo and in vitro experiments to understand more about membrane protein integration.
The currently proposed mechanism[2] is as follows: Rough endoplasmic reticulum targeting signals, particularly the transmembrane span (TM), are recognized by a signal recognition particle (SRP) in the cytosol. This transmembrane span is attached to the ribosome-nascent polypeptide complex (RNC) which in turn is attached to the SRP; the SRP then forms a stable complex between itself and the endoplasmic reticulum SRP receptor on its membrane. As the SRP dissociates, the RNC attaches to Sec61, a cotranslational translocation channel. Before even passing through this channel, the TM span folds into the α helix orientation. In doing this, there are enough hydrogen bond donors and acceptors for the rest of the protein to adopt the proper α helix folding.
In order to further study the particulars of the aforementioned mechanism, scientists have used in vitro experiments, yet these have problems. One issue is that in vitro experiments do not provide accurate data on the kinetics of membrane protein integration. Obviously, the limiting factor in this case is that in vitro experiments are not in the actual, living cell. In comparison, in vivo studies show that the eukaryotic translation systems synthesize proteins at 5-7 residues per second. In comparison, in vitro experiments are limited to synthesizing only 5-10% of the speed. Another issue is, again, the fact that past experimental data have been received from in vitro environments, unreflective of the actual cells. In this case, the time it took to purify and analyze membrane protein integration intermediates may have resulted in studying the kinetics of what was left after equilibrium of the intermediates, not the actual kinetics of the reactive intermediates.
In vivo experiments have enlightened some details of the mechanism. In one experiment,[2] saccharomyces cerevisiae, or budding yeast, cells were used. While studying the SRP-SR targeting pathway, scientists disrupted the SR and SRP genes. This resulted in a crippled cell growth rate on top of other functional losses, yet the cell adapted to the loss by utilizing an SRP-SP independent pathway. This showed researchers the importance of utilizing in vivo experiments to fully understand how particular mechanisms worked. From this experiment, one could see that the SRP-SR pathway was not the sole way for the RNC to bind to the membrane.
Another in vivo experiment[2] clarified the kinetics of the way RNCs are able to target themselves to the endoplasmic reticulum. In this experiment, the luminal domain of the membrane protein was tagged with a phosphorylation site. Since phosphorylation of the residues could only occur when the kinase was in the cytosol and since the luminal domain was exposed to the cytosol infrequently, scientists were able to calculate the time required for SRP-mediated RNC targeting.
The orientation, or specifically, topology, of TM spans was also studied using in vivo experiments. In normal functioning cells, the topology is determined by the charged residues on the TM span. For this particular cell, there were two topologies: one (type 2) that occurred when there was a net positive charge on the N-terminal and another topology when there was a net positive charge on the C-terminal (type 1).[2] Upon mixing these two topologies in vivo, scientists found that type 2 membrane proteins inserted themselves into the Sec61 complex as the type 1 tomology, but they inverted back to their original type 2 form within 50 seconds.[2] These findings were substantiated by in vitro experiments as well. Scientists also found that this inversion occurred more rapidly when the TM span had positively charged residues in front.
These examples show the importance of in vivo experiments in determining the minute details of the integration of α helical membrane proteins into the endoplasmic reticulum. While it is true that in vitro experiments opened many views into how the mechanism worked, there was a very limited view. In vivo experiments, when combined with in vitro experiments, can provide an unparalleled view of exactly how the mechanism may work as well as how the kinetics of the reactions occur. Furthermore, these two types of experiments, specifically in vivo can open many avenues into other mechanisms and cell functions previously unknown.
Myoglobin was the first protein whose structure was determined. In 1958, Max Perutz and John Kendrew determined the 3D structure of myoglobin by X-ray crystallography. Four years later, they both received the Nobel Prize in chemistry for this innovation.
Myoglobin is a monomeric protein that has 154 amino acids residues. It consists of eight α-helicines connected through the turns with an Oxygen binding site. It has a globular structure. Myoglobin contains a heme (prosthetic) group which is responsible for its main function (carrying of oxygen molecules to muscle tissues). Myoglobin can exist in the oxygen free form, deoxymyoglobin, or in a form in which the oxygen molecule is bound, called oxymyoglobin. Myoglobin is a protein found in muscles that binds oxygen with its heme group like hemoglobin. Heme group consists of protoporphyrin organic component and an iron atom located in its center. The heme group gives muscle and blood their distinctive red color. The organic component consists of four pyrrole rings that are linked by methine bridges. In addition, heme is responsible for the red color of the blood and muscle. Oxidation of the iron atom (Fe2+ -> Fe3+) is mainly responsible for the color of muscle and blood. At the center of protoporphyrin, the iron atom is bonded to nitrogen atoms from four pyrrole rings. The iron atom can form two additional bonds, one on each side of the heme plane. These binding sites are called the fifth and sixth coordination sites. In myoglobin, the fifth coordination site is occupied by the imidazole ring from a histidine residue on the protein. This histidine is referred to as the proximal histidine. The sixth coordination site is available to bind oxygen. The iron atom in deoxymyoglobin lies about four angstroms out of the plane of the protoporphyrin plane because it is too big in that form to fit into the well defined hole.
The normal oxidation state of an iron atom has a positive two charge (ferrous ion) instead of three charge (ferric ion) and it is too large to fit into the plane of protoporphyrin. Thus, a ferrous ion is often 0.4 Å away from the porphyrin plane. However, when iron oxidized from ferrous ion (Fe2+) to ferric ion (Fe3+), because the loss of one extra electron, forces between protons and electrons increases so that the electron cloud will penetrate more towards to the nucleus. As a result, the ferric ion (Fe3+) has a smaller size then ferrous ion (Fe2+) and fits into the protoporphyrin plane when it attaches to an oxygen.
When oxygen leaves the myoglobin, it leaves as dioxygen rather than superoxide. This is because superoxide can be damaging to many biological process, and in the leaving of superdioxide, the iron ion will be in the ferric state which stops biding oxygen.
The distal histidine amino acid from the hemoglobin protein molecule further stabilizes the O2 molecule by hydrogen-bonding interactions.
Myoglobin is a protein molecule that has a similar structure and function to hemoglobin. It is a smaller monomer of polypeptide structure, a globular protein with amino acids and prosthetic heme group binds to proximal histidine group while a distal histidine group interact on the other side of the plane. It binds and stores oxygen without concerning cooperativity. Most importantly, it is the first protein structure to be studied.
Myoglobin follows the Michaelis-Menten Kinetic graph (as seen from the graph above). It follows the Michaelis-Menten kinetics because it is a simple chemical equilibrium.
The binding affinities for oxygen between myoglobin and hemoglobin are important factors for their function. Both myoglobin and hemoglobin binds oxygen well when the concentration of oxygen is really high (E.g. in Lung), however, hemoglobin is more likely to release oxygen in areas of low concentration (E.g. in tissues). Since hemoglobin binds oxygen less tightly than myoglobin in muscle tissues, it can effectively transport oxygen throughout the body and deliver it to the cells. Myoglobin, on the other hand, would not be as efficient in transferring oxygen. It does not show the cooperative binding of oxygen because it would take up oxygen and only release in extreme conditions. Myoglobin has a strong affinity for oxygen that allows it to store oxygen in muscle effectively. This is important when the body is starved for oxygen, such as during anaerobic exercise. During that time, carbon dioxide level in blood streams is extremely high and lactic acid concentration build up in muscles. Both of these factors cause myoglobin (and hemoglobins) to release oxygen, for protecting the body tissues from getting damaged under harsh conditions. If the concentration of myoglobin is high within the muscle cells, the organism is able to utilize the oxygen in its lungs for a much longer period of time.
Myoglobin, an iron-containing protein in muscle, receives oxygen from the red blood cells and transports it to the mitochondria of muscle cells, where the oxygen is used in cellular respiration to produce energy. Each myoglobin molecule has one heme prosthetic group located in the hydrophobic cleft in the protein. The function of myoglobin is notable from Millikan's review (1) in which he put together an accomplished study to establish that myoglobin is formed adaptively in tissues in response to oxygen needs and that myoglobin contributes to the oxygen supply of these tissues. Oxymyoglobin regulates both oxygen supply and utilization by acting as a scavenger of the bioactive molecule nitric oxide. Nitric oxide is generated continuously in the myocyte. Oxymyoglobin reacts with NO to form harmless nitrates, with concomitant formation of ferric myoglobin, which is recycled through the action of the intracellular enzyme metmyoglobin reductase. Flogel (2) conducted a study that showed how the interaction of NO and oxymyoglobin controls cardiac oxygen utilization.
When muscle tissue is damaged, very large concentrations of myoglobin enters the kidneys. When this happens, myoglobin is then considered highly toxic and may contribute to acute renal failure. Muscle injury is commonly associated with the release of myoglobin, and is known to be the cause of heart attacks and many other myoalgia. Studies have shown that acute myocardial infarction can be detected with the help of the monitoring of creatin kinase and troponin by electrocardiogram.
(1) Millikan, G. A. (1939). Muscle hemoglobin. Physiol. Rev. 19,503 -523.
(2) Flogel, U., Merx, M. W., Godecke, A., Decking, U. K. M. and Schrader, J. (2001). Myoglobin: a scavenger of bioactive NO. Proc. Natl. Acad. Sci. USA 98,735 -740
Red blood cells, or erythrocytes are by far the most numerous blood cells. Each red blood cell contains hemoglobin which is the iron-containing protein that transports oxygen from the lungs to other parts of the body. In hemoglobin, each subunit contains a heme group; each heme group contains an iron atom that is able to bind to one oxygen molecules. Since hemoglobin consists of four polypeptide subunits, two alpha chains and two beta chains, and each subunit contains a heme group; each hemoglobin protein can bind up to four oxygen molecules.
The prosthetic group consists of an iron atom in the center of a protoporphyrin which is composed of four pyrrole rings that are linked together by a methene bridge, four methylene groups, two vinyl groups and two propinoic acid side chains. Each pyrrole ring consists of one methyl group. Two of the pyrrole rings have a vinyl group side chain, while the other two rings have a propionate group independently. Heme proteins have some iron-porphyrins such as heme a, heme b, heme c, heme d, heme d1, heme o, etc. They are constituted by tetrapyrrole rings but differ in substituents. For example, heme o contain four methylene groups while heme a contain three methylene groups, the rest structure are similar between two groups. The difference between hemes assigned each of them different functions.
Heme of hemoglobin protein is a prosthetic group of heterocyclic ring of porphyrin of an iron atom; the biological function of the group is for delivering oxygen to body tissues, such that bonding of ligand of gas molecules to the iron atom of the protein group changes the structure of the protein by amino acid group of histidine residue around the heme molecule. A holoenzyme is defined to be an enzyme with its prosthetic group, coenzyme, its cofactor, etc. Therefore an example of a holoenzyme is hemoglobin with its iron-containing heme group.
Heme A is a bimolecular heme that is made up of of macrocyclic ligand called a porphyrin, chelating an iron atom. Heme A differs from Heme B in that it contains a methyl side chain at a ring position that is oxidized to a formyl group and hydroxyethyfarnesyl group. Moreover, the iron tetrapyrrole heme will be attached to a vinyl side and an isoprenoid chain. Heme A is known to be relatively comparable to Heme O since both include farnesyl.
Heme B is present in hemogoblin and myogoblin. Typically, heme B is binded to apoprotein, a protein matrix executed with a single coordination bond between the heme iron and amino-acid side-chain.
The iron contained in heme B is bounded to four nitrogens of the porphyrin and one electron donating atom of the protein, which puts it in a pentacoordinate state. The iron turns into a hexacoordinate when carbon monoxide is bounded.
Heme C differs from heme B in that the two vinyl side from the heme B are substituted with a covalently thioether linkage with the apoprotein. Because of this connection, heme C has difficulty dissociating from holoprotein and cytochrome c.
Heme C functions a crucial role in apoptosis because some molecules of cytoplasmic cytochrome c must contain heme C. As a consequence, this will lead to cell destruction.
Heme D is another form of heme B. Instead, the hydroxylated propionic acid side chain forms a gamma-spirolactone. Heme D reduces oxygen in water of bacteria with a low oxygen tension.
Heme is a porphyrin that is coordinated with Fe(II). One of the most important classes of chelating agents in nature are the porphyrins [1]. A porphyrin molecule can coordinate to a metal using the four nitrogen atoms as electron-pair donors. In the body, the iron in the heme is coordinated to the four nitrogen atoms of the porphyrin and also to a nitrogen atom from a histidine residue, one of the amino-acid residues in hemoglobin) of the hemoglobin proteins. The sixth protein coordination site, around the iron of the heme, is occupied by O2 when the hemoglobin is oxygenated. The heme group is nonplanar when it is not bound to oxygen [2]. The iron is pulled out of the plane of the porphyrin, towards the histidine residue to which it is attached. This nonplanar configuration is characteristic of the deoxygenated heme group, and is often referred to as being "domed shape" [2]. When the Fe heme group binds to an oxygen molecule, the porphyrin ring adopts a planar configuration and hence the Fe lies in the plane of the porphyrin ring [2].
Oxygenated and deoxygenated conformation of Hemoglobin
As may be seen in the figure, the left shows representations of electron-density clouds of the de-oxygenated heme group, depicted in pink, and the attached histidine residue which may be seen in light blue [2]. These regions of electron density repel each other, and the iron atom in the center is drawn out of the plane. The non planar shape of the heme group is represented by the bent line. The right image depicts the electron-density clouds of the oxygenated heme group, shown in pink, the attached histidine residue in light blue, and the attached oxygen molecule which is shown in gray [2]. The oxygenated heme assumes a planar configuration, and the central iron atom occupies a space in the plane of the heme group which is depicted by a straight red line [2].
The shape change in the heme group also has important implications for the rest of the hemoglobin protein. When the iron atom moves into the porphyrin plane upon oxygenation, the histidine residue to which the iron atoms is attached to is drawn closer to the heme group. This movement of the histidine residue then shifts the position of other amino acids that are near the histidine [2]. When the amino acids in a protein are shifted in this manner by the oxygenation of one of the heme groups in the protein, the structure of the interfaces between the four subunits is altered [2]. So when a single heme group in the hemoglobin protein becomes oxygenated, the whole protein changes its shape [2]. In the new shape, it is easier for the other three heme groups to become oxygenated. The binding of one molecule of oxygen to hemoglobin enhances the ability of hemoglobin to bind more oxygen molecules. This property of hemoglobin is known as "cooperative binding" [2].
Association constant is the constant at which the bonding affinity between two different molecules, the substrate and the product, is at stable equilibrium. An example of such a bonding constant occurs in the hapten-antibody interaction.
Dissociation constant is the quantifiable constant in which a compound, molecule, or ion dissociates. A type of dissociation constant is acid dissociation constant. This constant is used to calculate the occurrence of a weak and strong acid dissociation.
The Heme group gives myoglobin and hemoglobin the ability to bind oxygen because of the presence of iron atom. It also contributes to the red color found in muscles and blood. Each heme group contains an iron atom that is able to bind to one oxygen (O2) molecule. Each hemoglobin protein can bind four oxygen molecules. The iron atom, usually in the ferrous oxidation state (Fe2+), lies between four pyrrole rings but slightly bends away from the plane (0.4 Angstrom from the plane). The iron ion has two extra binding sites called the fifth and sixth coordination sites on each side of the protoporphyrin plane. Usually, the fifth coordination binds with proximal histidine where the sixth coordination binds to an oxygen. When oxygen binds to iron, the iron becomes slightly smaller allowing it to move into the plane of the porphyrin ring. A distal histidine binds to oxygen to make sure reactive oxygen is not released. The distal histidine will not allow the release of oxygen when the Iron is in the 3+ state.
The Iron atom is too large in size to fit perfectly inside the porphyrin ring, and sits outside the ring by 0.4 Angstroms. However, upon binding oxygen, the Iron radius shrinks, facilitating a planar alignment with the porphyrin ring. This change causes the proximal histidine bound to the Fe atom to be pulled up and cause a structural change to the alpha helix attached to the histidine residue. This alpha helix's carboxyl terminus interacts with the other alpha-beta dimer, creating a total conformational change in the overall protein. The conformational change facilitates an increased affinity for oxygen, which is shown by a transformation from the T to R state in hemoglobin. The changes that occur in blood upon oxygenation and deoxygenation are visible not only at the microscopic level but also at the macroscopic level. It has been known that blood in the systemic arteries is red-colored while blood in the systemic veins is blue. The blood in the systemic arteries is oxygen-rich, having just traveled from the lungs to the heart and then being pumped throughout the body to deliver its oxygen to the body's cells [2]. The blood in the systemic veins, on the other hand, is oxygen-poor. It has unloaded its oxygen to the body’s cells and must now return to the lungs to replenish the supply of oxygen [2]. Hence, a simple macroscopic observation such as noting the color of the blood, can tell us whether the blood is oxygenated or deoxygenated.
Bilirubin is the product that is formed from further breakdown of heme. As a waste product, bilirubin is secreted into bile and is also what gives urine it's yellow color. Additionally, yellow bruises obtain their color also from bilirubin in a condition called jaundice. Since bilirubin is a breakdown from hemoglobin, it is generated in large quantities as red blood cells undergo turnover. Phagocytes then absorb the dead red blood cells where it is converted into free bilirubin and then released into plasma and eventually absorbed by liver cells. Finally, bacteria in the intestine metabolizes bilirubin where it is eliminated as urine or feces, which is also one of the reasons for its brown color.
As the name suggests, the bilirubin test measures the amount of bilirubin in a blood sample usually taken from a heel stick for a baby or from the vein for an adult. The reason for this test is because it can be used to check for liver function and watch out for early signs of liver disease, diagnose conditions that affect the destruction of red blood cells, or to find out if something blocking
Conditions that may affect the test are caffeine, which can lower bilirubin levels, and fasting, which increases bilirubin levels.
Metal ions play an important role in biochemical processes. Because metal ions are positively charged ions that are stable in multiple oxidation states and can form strong yet conformationally changeable bonds, they become attractive catalytic substances.
Many biochemical reactions depend on the presence of metal ions, which are a part of coordination complexes. Those metal ions function to facilitate or inhibit biochemical reactions in the solution.
Ions, such as Na+, K+, Ca2+, function as charge carriers. Because membrane ion pumps maintain a concentration gradients of those ions from the inside to the outside of the membrane, the movement of the ions would trigger mechanisms. The changes in concentration gradients are signals for nerve and muscle actions. These ions typically are used via intracellular and extracellular concentration differences to create a electric potential gradient. In doing so, ion channels can be controlled via a variety of chemical mechanisms. However, they are not responsible for conformational changes, but instead can cause a cascade of protein activation.
Na+
1. regulation of body fluids, including blood plasma, extracellular fluids in tissues
2. signal transduction in nerves and muscles
3. transmission of heat
4. transportation of nutrients and wastes
K+
1. trigger taste sensations
2. polarization of membrane: muscle contraction, transmission of nerve impulses
3. regulation of body fluids, including cell fluid, plasma
Ca2+
1. trigger for muscle contraction, neurotransmitter release
2. cancer prevention candidate
Metal ions are able to promote reactions by providing appropriate geometry for bond breaking and bond formation. Many coordination sites in the molecules are tetrahedral, octahedral, or square-planar, but metal ions also provide other geometric variation, which enables reactions to take place. Metal ions, transition metals especially, are frequently utilized as a catalyzing group due to their extended octet. They are therefore able to create multiple interactions as well as ionization states that facilitate conformational changes to the protein. Furthermore, they are able to create coordination complexes with ligands, which are considerably weaker bonds than covalent and ionic bonds, but allow for ligand stabilization.
These metal ions participate in catalytic mechanisms in 3 main ways:
1. Binding to substrates to orient them properly for catalytic reaction.
2. Mediation oxidationo-reduction reactions via reversible changes in oxidation state of the central metal ion.
3. Electrostatically stabilizing or shielding negative charges.
Essentially, metal ions serve as a electron sink, much as protonation does. However, metal ions can be in high concentration without an effect on pH. For example, carbonic anhydrase facilitates the creation of carboxylate ion from water and carbon dioxide. It is initiated by the coordination bonding of OH- to Zn2+. This stabilizes the negative charge on the oxygen and facilitates a nucleophilic attack on the carbon atom in carbon dioxide.
Some examples of metal ions used in catalytic reactions include include Fe2+, Fe3+, Cu2+, Mn2+, and Co2+.
Binding to a metal ion makes water molecule more acidic than free water molecule. Coordination to proteins enhances the effect even more, resulting in M-OH species that is able to further react with other biological substances. Mg2+ can activate phosphotransferases and phosphokinase, while Zn2+ and Ca2+ are able to catalyze hydrolysis of phosphates, serving as Lewis acids.
Uncatalyzed hydrolysis rate constant is about ~10-11 s-1, while with enzyme catalyzing the reaction, reaction rate constant increases to k = ~104 s-1.
Carboxypeptidzase A has 307 amino acid residues. One Zn2+ ion is cleft on one side.
- Molecular weight = 34600 g/mol
- Egg-shaped
- Dimension: 50*38A
- Carboxypeptidase catalyzes the hydrolysis of c-terminal amino acid residues of the protein. It is released in pancreatic juice of animals for the digestion of proteins.
Bending with small molecules enables metal ions to adopt unusual angles, bond distances, or specific geometry, and hence increases their reactivity. In hemoglobin and myoglobin, ferric ion is able to bind to oxygen, serving as a oxygen storage and transport. Oxygen molecules that bind to Iron metal ions is accompanied by the partial transfer of an electron from the metal ion to the oxygen. Their structure is best seen as a complex formed between ferric ion and a superoxide anion. Which is important than the heme-group stabilizes this binding or else superoxide is released into the body which could be biologically dangerous. Iron stabilizes this conformation in a strong ionic interaction which prevents this superoxide oxygen anion from leaving out of the blood cell.
Coordination by different ligands changes the reduction-oxidation potentials of a reaction, making it easier or, sometimes, more difficult to take place. The change in redox potential also enables electron transfer.
1. The reaction of O2 -> H2O is catalyzed by Fe2+, where Fe2+ -> Fe3+.
Similarly, N2 is oxidized to flux NH3O while Cu+ is reduced to Cu2+.
2. The reverse reaction mentioned above, H2O -> O2 is catalyzed by the valence charge of Mn.
3. Ribose is reduced to deoxyribose, a reaction catalyzed by Co+, where Co+ is oxidized to Co3+.
The oxygen binding curve for Myoglobin forms an asymptotic shape, which shows a simple graph that rises sharply then levels off as it reaches the maximum saturation. The half-saturation, the point at which half of the myoglobin is binded to oxygen, is reached at 2 torr which is relatively low compared to 26 torr for hemoglobin.
Myoglobin has a strong affinity for oxygen when it is in the lungs, and where the pressure is around 100 torr. When it reaches the tissues, where it's around 20 torr, the affinity for oxygen is still quite high. This makes myoglobin less efficient of an oxygen transporter than hemoglobin, which loses it's affinity for oxygen as the pressure goes down and releases the oxygen into the tissues. Myoglobin's strong affinity for oxygen means that it keeps the oxygen binded to itself instead of releasing it into the tissues.
Neuroglobin and Cytoglobin are two new heme-containing repiratory proteins, which were revealed through the human genome sequence. These proteins are both monomeric, and are structurally similar to myoglobin more than hemoglobin. The expression of neuroglobin is restricted to the brain and is mostly observed in the retina. Its roles include protecting neural tissue from hypoxia and insufficient oxygenation of the blood, through its roles in neuronal oxygen homeostasis. Cytoglobin on the other hand is expressed mainly in fibroblasts and other similar cells throughout the body, and may be involved in collagen synthesis. Spectroscopy data has shown that both proteins have the proximal and sital histidines coordinated to iron in the deoxy form and the distal histidine is displaced on oxygen binding.
Neuroglobin was first identified by an Italian researcher, Thorsten Burmester et al. in 2000, and the 3D structure of the neuroglobin of human body was discovered in 2003. In 2004, the neuroglobin of murine was determined at a further highly resolution.
Neuroglobin, has the symbol of NGB, is an intracellular hemoprotein that involved in cellular oxygen homeostasis. Neuroglobin is a monomer which expressed in the central nervous system, retina, endocrine tissues, and cerebrospinal fluid. Neuroglobin reversibly binds oxygen with affinity which is higher than hemoglobin, and also increase the existence of oxygen in brain tissue and gives the protection under ischemic or hypoxic conditions in order to limiting the brain damages.
Cytoglobin is the protein product that can be found in human body and mammalian.
Just like the Neuroglobin, Cytoglobin is also a globin molecule located in the brain of mostly marine mammals. It has always been considered to be a method to protect the brain under the condition called hypoxia.Cytoglobin is also the transferring reagent of oxygen from arterial blood to the brain.
Cytoglobin is a hexacoordinate hemoglobin. It may scavenge reactive oxygen species such as nitric oxide, and also facilitate diffusion of oxygen through tissues, or serve a protective function during oxidative stress
Hemoglobin is contained in red blood cells, which efficiently carries oxygen from the lungs to the tissues of the body. Hemoglobin also helps in the transportation of carbon dioxide and hydrogen ions back to the lungs.
Hemoglobin or Haemoglobin is able to bind to gaseous nitric oxide (NO) as well as O2. As red blood cells passes through the capillary beds of the lungs, gills (in fish), or other respiratory organs, oxygen is diffused into the erythrocytes and hemoglobin binds O2 and NO. Hemoglobin then unloads its cargo in the capillaries. There O2 is able to diffuse into the body cells. The NO relaxes the walls of the capillaries, allowing them to expand which in effects helps the delivery of O2 to the cells.
Hemoglobin consists of four subunits, each with a cofactor called a heme group that has an iron atom center. The iron is the main component that actually binds to oxygen, thus each hemoglobin molecule is able to carry four molecules of O2. Cooperation among the four subunits of the hemoglobin molecule is necessary for the efficient transportation of O2. The four subunits of hemoglobin actually bind to oxygen cooperatively, the binding of oxygen to one site of the four subunits will increase the likelihood of the remaining sites to bind with oxygen as well.
Hemoglobin is a protein that is used to carry oxygen through the bloodstream from the lungs to the tissues. This is important for survival. Hemoglobin has a lower affinity for oxygen the lower the concentration of oxygen gets. This has great implications for the human body and has helped us adapt very effectively. The lower affinity and lower concentrations means that when we are working out, our bodies are low on oxygen which means hemoglobin has less affinity for oxygen and can more easily drop the oxygen off at human tissues. This gives us greater oxygen in our oxygen dependent state. On the other hand, when oxygen concentration is high, the hemoglobin has a higher affinity for oxygen and therefore does not drop the oxygen where it is not needed. This is a very complex and smart system that has evolved to keep hemoglobin as an important biological molecule for a very long time. On the otherhand, the cousin of hemoglobin, myoglobin is used to store oxygen in muscles. This myoglobin has a slightly higher affinity for oxygen than hemoglobin especially at lower levels. This is because myoglobin has an easier job in that it only needs to store oxygen and release it for the muscles, while hemoglobin also has to transport the oxygen and release it in the correct areas.
Hemoglobin is coded for by DNA just like all the other proteins. Alterations or mutations to hemoglobin causes many blood related diseases such as sickle-cell anemia, where the cell structure is distorted and can no longer carry as much oxygen in the correct way as a normal blood cell. This highlights the underlying ideal in structural biochemistry in that structure determines function. The sickle cell anemia case is extremely interesting because it shows us how and why diseases develop. The gene for sickle cell anemia also provides protection against malaria. Therefore, in countries where malaria presented problems, there was an higher than average amount of individuals carrying the sickle cell anemia gene. The heterozygous state is best because it does not allow sickle cell anemia to develop while still preventing malaria. Whereas, the homozygous states would produce individuals either struck with sickle cell anemia or malaria. This is why in malaria ridden areas, there is a higher than average number of people who are heterozygous for sickle cell anemia which is also why this disease does not die out! The carrier state is actually selected by nature. [3]
Upon O2 binding to an active site of hemoglobin there is a conformational change that results, which helps hemoglobin cooperate. Cooperation refers to the interactions among active sites, in the case of hemoglobin, cooperation allows the binding of oxygen to be increased as one site is filled, the remaining active sites will be more likely to bind to O2 as well.
Hemoglobin Interactions
Once O2 is bound to an active site on the hemoglobin molecule, the iron atom Fe2+) is oxidized to (Fe3+). The interaction that results between iron and oxygen in hemoglobin is a combination of resonance structures, one with (Fe2+)and O2 and another between (Fe3+) and super ion O2.-
The binding of O2 to the iron center results in a conformational change in the histidine residue toward the porphyrin in the structure of the hemoglobin molecule which ultimately results in an increase O2 affinity of hemoglobin. The associated movement of the histidine-containing group will result in a conformational change to the rest of the hemoglobin structure. The COO- group is now interacting with the alpha-beta interface which causes conformational changes of neighboring active sites. These conformational changes will result in an increase of O2 affinity to hemoglobin.
Hemoglobin is an allosteric protein. It's ability to bind to O2 to one of the subunits is affected by its interactions with the other subunits. As mentioned above, the binding of O2 to one hemoglobin subunit induces conformational changes that are relayed to the other subunits, making them more able to bind to O2 by raising their affinity for this molecule. H+, CO2 and 2,3-bisphosphoglycerate are all allosteric effectors as they favor the conformation of deoxyhemoglobin and therefore promote the release of O2. Because these three molecules act at different sites, their effects are additive.
X-ray crystallography revealed that oxyhemoglobin, the form that has four O2 molecules bound, differs markedly in its quaternary structure from deoxyhemoglobin, the form with no O2 bound. In the absence of bound O2, the Fe2+ lies slightly to one side of the porphyrin ring, which itself is slightly curved. As a molecule of O2 binds to the heme prosthetic group it pulls the Fe2+ into the plane of the porphyrin ring, flattening out the ring in the process. Movement of the Fe2+ causes the proximal histidine to move also. This, in turn, shifts the position of helix F and regions of the polypeptide chain at either end of the helix. Thus, movement in the center of the subunit is transmitted to the surfaces, where it causes the ionic interactions holding the four subunits together to be broken and to reform in a different position, thereby altering the quaternary structure,leading to the cooperative binding of O2 to Hb.
The regulation of oxygen binding of hydrogen ions and carbon dioxide is called the Bohr Effect, which was proposed by Christian Bohr, in 1904. The Bohr Effect describes the effect of pH on the oxygen affinity of hemoglobin, the oxygen affinity of hemoglobin decreases as pH decreases from a value of 7.4. As hemoglobin moves into a region of lower pH, its tendency to release oxygen will increase, therefore more oxygen will be released as the environment becomes more acidic.
Protonation occurs in low pH
There is a chemical basis that is responsible for the pH effect. The histidine residue of hemoglobin molecule structure is one factor of the pH effect. At high pH, the side chain of histidine is not protonated and the salt bridge between histidine's terminal carboxylate group and a lysine residue, does not form. However as the pH drops, meaning at low pH levels, the side chain of histidine will become protonated and thus form a salt bridge with Aspartate instead. This electrostatic interaction results in a structural change. The formation of salt bridges stabilizes the hemoglobin structure resulting in a lower O2 affinity of hemoglobin and thus increase the tendency for oxygen to be released.
The effect of 2,3-bisphosphoglycerate (2,3-BPG) in hemoglobin is described as an allosteric effect. 2,3-BPG is an allosteric effector, it binds to a site that is completely remote from the active site for oxygen. The amount of 2,3-BPG in red cells is crucial in determining the oxygen affinity of hemoglobin.
A single 2,3-BPG molecule is bound in the center of the tetramer of a deoxyhemoglobin structure in a central cavity in the T form. Upon the transition of T state to R state, 2,3-BPG is released. Therefore in order for the transition from T to R states to occur, the bonds between hemoglobin and 2,3-BPG needs to be broken. In the presence of 2,3-BPG, oxygen is less tightly bound to hemoglobin. The conformational changes allow a structural stabilization to occur and thus hemoglobin loses oxygen affinity.
Carbon dioxide is able to stimulate oxygen release by two mechanisms:
The presence of carbon dioxide (CO2) in high concentration will DECREASE the affinity of hemoglobin due to a drop in pH with the red blood cell
There are effects of CO2 in hemoglobin through catalysis. A reaction between CO2 and water forms carbonic acid. However, this reaction requires for carbon dioxide to be catalyzed by carbonic anhydrase, an enzyme in red blood cells, which ultimately results in H+ and HCO3-. Once carbonic acid dissociates into these two ions, pH will drop. The drop in pH stabilizes the T state and thus increases the tendency for oxygen release.
CO2 ↔ CO2 + H2O ↔ H2CO3 ↔ HCO3- + H+
A direct chemical interaction between (CO2) and hemoglobin will stimulate the release of O2. (CO2) is able to stable deoxyhemoglobin by reacting with terminal amino groups to form negatively charged carbamate groups. This interaction results in a salt-bridge that stables the T state, which favors the release of O2.
It also explains the transport of carbon dioxides from tissue to lung. CO2 that produced by tissue cells pass through the red blood cell and form H+ and HCO3- as previously mentioned. It allows the exchange of HCO3- for Cl-. Therefore, the concentration of HCO3- increases in the blood capillary and carbon dioxides are carried to lung in this form. When HCO3- reaches lung, the reverse reaction take place and release carbon dioxides in lung.
Several molecules are responsible for substantially lowering hemoglobin's ability to transport oxygen to tissues. The most common is carbon monoxide (CO), which has a binding affinity to hemoglobin 200 times greater than oxygen. Once carbon monoxide binds to the heme group, oxygen affinity is increased, since hemoglobin is a tetrameter that facilitates cooperative ligand binding. However, this prevents oxygen from being released into oxygen-requiring tissue. The CO and hemoglobin complex is known as carboxyhemoglobin. This is known as carbon monoxide poisoning, where CO competitively binds to oxygen and prevents oxygen transport. As such, as small concentration of CO can cause serious harm to an individual. As little as 0.02% of CO concentration can cause headaches, and 0.1% will lead to unconsciousness. Other competitive ligands include cyanide, sulfur monoxide, nitrogen dioxide, and sulfide.
Differences in Embryonic, Fetal and Adult Hemoglobin
Embryonic and fetal hemoglobin differ at the subunit level to that of adult hemoglobin by the subunit interface strengths. Subunit interface in embryonic hemoglobin are much weaker than subunits in fetal hemoglobin which are much weaker than in adult subunit interface. In human red blood cells, hemoglobin can have eight different combinations of dimer formations. Each formation can be present in greater amounts than others or can be present only at distinct times during development. In embryonic development there are three different kinds: ζ_2 γ_2 (Hb Portland-1), ζ_2 ε_2 (Hb Gower-1) and α_2 ε_2 (Hb Gowler-2). Fetal hemoglobin consists of α_2 γ_2(HbF) and adult hemoglobin consists of α_2 β_2 (HbA) as well as trace amounts of α_2 δ_2(HbA2). Although the tertiary structure of all these various hemoglobin is almost identical, their primary structure varies in specific substitutions that accounts for their differing O_2affinity as well as their interactions with allosteric effectors. These amino acid substitutions have an effect on how the subunits fit together and how their interactions take place. Embryonic ζ_2 γ_2 (Hb Portland-1), ζ_2 ε_2 (Hb Gower-1) and α_2 ε_2 (Hb Gowler-2) is found during the first few months of life as well as a fourth, ζ_2 β_2 (Hb Portland-2). ζ_2 β_2 (Hb Portland-2) is a rarely occurring form of hemoglobin. The strengths between the interfaces of the monomer units in each kind of hemoglobin differ significantly and to a greater extent in the deoxy state and become even stronger in the liganded state. The tertramer[check spelling]-dimer dissociation constants differed depending on what subunits they contained. For example, between the two similar embryonic subunits ζ_2 ε_2 (Hb Gower-1) and α_2 ε_2 (Hb Gowler-2) there was a difference in dissociation constants of 13-fold from α to ζ subunits. Therefore by comparing the different types of hemoglobin with one that has one similar subunit, their dissociation constants can give a wide range on information. Little is known about the Hb Portland-2 because it s rare and only occurs in a type of α-thalassemia (genetic defect). Hb Portland-2 differs from other hemoglobins because it dissociates from tetramer to dimers and even more readily from dimers to monomers whereas other types of human hemoglobin will dissociate from from tetramers to dimers rapidly but will not dissociate from dimers to monomers. The dissociation talked about happens at pH 7.5 but will differ significantly once the pH is lowered. In HbA (adult hemoglobin), the dissociation of tetramer to dimer increases with the decrease in pH, for every 1pH unit decrease, there is a 10 fold increase in dissociation. When the pH is changed to 6.3, the dissociation in HbA will be primarily dimers whereas in Hb Portland-2 the dissociation is mainly monomers. With this being true, the formation of tetramers in HbA is favored greater than the formation of tetramers in Hb Portland-2 because ζ_2 β_2 dimer is much weaker than α_2 β_2 dimer. In HbA, the α and β subunits are unstable but in Hb Portland-2 ζ and β subunits are weak interfaces as dimers but are stable as monomers. Embryonic hemoglobins at pH 6.3 are able to dissociate readily but Hb Portland-1 and Gowler-1 dissociate faster than Hb Gowler-2. When monomers of embryonic hemoglobin Hb Gowler-2 and Portland-2 were mixed and allowed to recombine, the stronger tetramer formation was the result. Instead of having α_2 ε_2 (Hb Gowler-2) and ζ_2 β_2 (Hb Portland-2) reform, the formation of α_2 β_2 (HbA) was obtained as well as ζ_2 ε_2 because the αβ interface is the strongest and therefore most favored. And when the ζ_2 γ_2 (Hb Portland-1), α_2 ε_2 (Hb Gower-2) were put through the same process, the interface of Fetal Hemoglobin α_2 γ_2(HbF) was the result because it is by far stronger than embryonic hemoglobins. When the ζ_2 γ_2 (Hb Portland-1), α_2 ε_2 (Hb Gower-2) and ζ_2 β_2 (Hb Portland-2) were mixed, the favored outcome was HbA and HbF was not detectable. All three experiments prove that subunit competition contributes to the rearranging and the notably higher α_2 β_2 formation than any other tetramer due to the stronger interface. The rarity of Hb Portland-2 may be due to the fact that the tetramer-dimer and dimer-monomer interfaces are relatively weaker than any other human hemoglobins. Subunit competition has a lot to do with why some hemoglobins are more likely to form because the formation of hemoglobins with stronger interfaces is favored over the formation of weaker ones. As well as there being formation of stronger ones from the weaker ones.
Hemoglobin is formed by genes that are in charge of the expression of the hemoglobin protein. Failings in these genes can form irregular hemoglobin and anemia, which are conditions termed "hemoglobin disorder". Irregular hemoglobin appears in these three conditions.
1. Structural failure in the hemoglobin molecule. Changes in the gene for one of the two hemoglobin subunit chains, alpha (α) or beta (β), are called mutations. Often, mutations change a single amino acid building block in the subunit. Most commonly the change is innocuous, perturbing neither the structure nor function of the hemoglobin molecule. Occasionally, alteration of a single amino acid dramatically disturbs the behavior of the hemoglobin molecule and produces a disease state. Sickle hemoglobin represents this phenomenon.
2. Reduced production of one of the two sub-units of the hemoglobin molecule. Mutations that form this condition are termed "thalassemias." Equal numbers of hemoglobin alpha and beta chains are essential for normal function. Hemoglobin chain inequity damages and destroys red cells thereby producing anemia. Although there is a death of the affected hemoglobin subunit, with most thalassemias the few subunits created are structurally normal.
3. Irregular relations of otherwise normal sub-units. A single sub-unit of the alpha chain and a single subunit from the β-globin locus combine to create a normal hemoglobin dimer. With severe α-thalassemia, the β-globin subunits start to associate into groups of tetramers due to the scarcity of potential α-chain partners. These tetramers of β-globin subunits are functionally inactive and do not carry oxygen. No similar tetramers of alpha globin subunits form with severe beta-thalassemia. Alpha subunits are quickly destroyed in the absence of a partner from the beta-globin gene cluster.
Berg, Biochemistry (6th Ed) and Campbell Biology (5th Ed)
Johnson RA, Lavesa M, Askari B, Abraham NG, Nasjletti A (February 1995). "A heme oxygenase product, presumably carbon monoxide, mediates a vasodepressor function in rats"
http://sickle.bwh.harvard.edu/hemoglobinopathy.html
↑name="Campbell">Biology, Eight Edition,Pearson, Benjamin Cummings, 2008.
↑ abcdefghijklInvalid <ref> tag; no text was provided for refs named Campbell
↑[ haemoglobin and myoglobin], November 14th, 2012.
Hemoglobin is a protein that is carried by red blood cells. Hemoglobin picks up oxygen in the lungs and delivers oxygen to the tissues to maintain cellular viability. It is composed of four polypetide chains with four prosthetic groups. Two of the units have the same amino acid sequences and are called α-chains, the other two identical amino acid sequences are called β-chains. The α-chains and β-chains combine to form the body of hemoglobin. Usually the α-chains combine with the β-chains rather than α-chains and α-chains while β-chains combine with β-chains. The latter is not expressed before birth. Hemoglobin protein which is found only during fetal development is called gamma (γ). It substitutes for the β-chains. Fetals have γ-chains instead of β-chains because it needs a higher affinity for oxygen, so the fetuses can draw sufficient amount of oxygen from the mother. The fetus accomplishes this task by altering the amino acid sequences of the β-chains. γ-chains altered in the way that it removes the two positive charges on the β-chains in which the 2,3-bisphosphoglycerate (2,3-BPG). This reduces the affinity for 2,3-BPG. Since 2,3-BPG lowers the affinity for oxygen, reduction in that would increase the fetus' ability to bind to an oxygen. The fact that 2,3-BPG helps hemoglobin protein binding oxygen molecule for more oxygen to be delivered to body tissues is known as heterotropic allosteric effect. Bisphosphoglycerate (BPG), pH, and carbon dioxide can have an effect on the hemoglobin. In the cell, when the pH decrease, it lowers the affinity of oxygen in the cell and therefore becomes more efficient oxygen tranporter. When the presence of carbon dioxide increases, it lowers the affinity of oxygen in the cell which is another factor that allows cell to become a better oxygen transporter.
A protein of tetramer, with two α and β units of amino acids or, globin or globular protein and four heme groups that each has an iron atom, and the importance is that Fe2+ is, by which bonding to globin group by proximal histidine and oxygen molecules by bent form of geometry by cooperativeness for the biological function of the protein such that they are circulated through body in red blood cells to be delivered to tissues (to be used for glycolysis and oxidative phosphorylation), and bonding to carbon dioxide for it to be transported to lungs (to be exhaled as waste). Oxyhemoglobin is the form of hemoglobin that the heme group of the protein molecule binds to oxygen; deoxyhemoglobin is that without oxygen.
The iron ion of +2 state of heme group is bonded to four nitrogens in one plane of 4 porphyrin ring and a proximal histidine amino acid of imidazole ring of globin molecule under plane of porphyrin ring. The heme group binds oxygen by bending. This does not increase the oxidation state of the iron ion but it decreases in atomic size. This causes the iron ion to move into the plane of porphyrin ring and brings up the proximal histidine amino acid group. Ultimately, it changes the allosteric conformation of globulin molecule.
As iron binds oxygen, the iron ion changes its position to stay in the same plane of porphyrin ring. This causes the bond distance of iron and oxygen to increase while the bond distance of iron to histidine of imidizole ring decreases. The decrease of iron to histidine of the imidizole ring causes the plane of the ring to move out of tetramer of hemoglobin. Now, the binding sites of the heme groups are opened up for the bonding of oxygen. Overall, the process is classified as positive cooperativeness. The hemoglobin protein's binding affinity to oxygen can be increased by increasing substrate saturation of oxygen molecule. The bonding of the hemoglobin protein and oxygen changes the conformation of the binding site which results in an increase affinity for other oxygen molecules to bind the protein molecule. The indirectly proportional relation for the binding affinity of hemoglobin protein molecules between oxygen and carbon dioxide is known as Bohr effect, for which, the oxygen binding curve of the protein molecule changes place to the right of the graph, as such, the decreased level of carbon dioxide bonding to hemoglobin increases oxygen bonding affinity to the protein molecule.
2,3-BPG binds to hemoglobin in the center of the tetramer to stabilize the T state (E.g. in muscle tissues).
2,3-BPG is also extremely important in the role of stabilizing the T state of hemoglobin. The hemoglobin wants to change into a more favorable R state due to the fact that the T state is quite unstable. Pure hemoglobin (without 2,3-BPG) was tested and found to bind oxygen much more vigorously than hemoglobin in blood. A hemoglobin with 2,3-BPG has a lower affinity for oxygen binding in the tissue which allows it to be a better oxygen transporter than a pure hemoglobin, which does not have 2,3-BPG. When 2,3 BPG is present, it transports about 66% of oxygen while the pure hemoglobin only transports about 8%. The reason is that the 2,3-BPG binds inside of the hemoglobin and somehow stabilizes its T state (the state that has less affinity for oxygen). When enough oxygen has bonded to hemoglobin, a transition occurs from the T state to the R state, which releases the 2,3-BPG. 2.3-BPG stays in the hemoglobin until enough oxygen has come to replace it. This keeps the oxygen in its T state until it is ready to transition to the R state (where its affinity for oxygen increases dramatically). This broadens hemoglobin's oxygen saturation curve.
When the oxygen binds to Hemoglobin, the iron in the Heme group moves from the outside to the inside of the plane. In doing so, the Histidine side group also changes its alpha helix when the iron moves. Thus, this triggers the carbonyl terminal of the alpha helix to change position and that favors transition from T state to R state.
2,3-Bisphosphoglycerate (2,3-BPG) or 2,3-diphosphoglycerate (2,3-DPG) binds to deoxyhemoglobin with larger bonding affinity, such that it makes the T state of hemoglobin protein more stable or increases oxygen affinity of the protein; its biological function is to control bonding between hemoglobin and oxygen molecules for oxygen to be released to body tissues.
This allosteric effector binds to a site on the tetramer that is only present on the Tense(T) form of hemoglobin. The site is in the form of a pocket which is bordered by beta subunits. These positively charged subunits, His143, Lys82, and His2, interact with 2,3-BPG holding it in place. When the equilibrium of the tense form is pushed to the relaxed(R) form, the bonds that hold the 2,3-BPG molecule in place are broken and it is released. The hemoglobin stays in the T form during low concentrations of oxygen, so when 2,3-BPG is present, more of the oxygen binding sites must be filled in order for the transition from T to R form to occur.
The cooperativeness of hemoglobin makes it a much more efficient transporter of oxygen than myoglobin. Although myoglobin and hemoglobin both become highly saturated with oxygen at high concentrations (E.g. in the lungs), hemoglobin is characterized by much weaker binding to oxygen at low concentrations compared to myoglobin. The cooperativeness of tetramers work both ways in hemoglobin. As one oxygen molecule binds to one heme group, the oxygen affinity for the other groups increase. Once an oxygen molecule is released, this stimulates the release of the other oxygen molecules. This makes hemoglobin ideal in transporting and releasing oxygen from lungs to tissues where it is needed.
Cooperativeness occurs as a result of a change in the hemoglobin structure. In the deoxy form (where oxygen is absent), hemoglobin exists in the T (tense)-state. Upon oxygenation, the dimers in hemoglobin shift by 15 degrees and the R (relaxed)-state is adopted. The R-state form has a much higher affinity for oxygen.
For specifics sakes, the cooperativeness experienced in hemoglobin among its four separate monomers occurs as a result of a proximal histidine shift when one monomer binds to an oxygen atom. The heme group of hemoglobin is situated in such a way that it is composed of 4 pyrrole coordinating around an iron ion. In addition, there is a proximal histidine group that is also coordinated the iron group constituting the 5th coordination ligand. In the deoxy form, the iron ion is not completely in the plane of the pyrrole rings, in fact it is about 0.4 angstroms below the plane of the ring. This downward shift is due to the proximal histidine ligand on the bottom of the coordination complex. However, when one of the monomers binds to an oxygen molecule, the iron ion gains a sixth coordination ligand, the oxygen molecule itself, and it pulled up 0.4 angstroms to the plane of the pyrrole rings. This shift upwards also pulls the proximal histidine group up as well. It this movement of the histidine group that contributes to the cooperativeness property of hemoglobin. The proximal histidine is located at the interface of the alpha and beta subunits found in hemoglogin (hemoglobin having two identical alpha units and two identical beta units). When the histidine group moves upwards, it forces a conformational change in that interface, which conforms the next monomer to situate itself in a fashion that increases its affinity to another oxygen molecule. As that monomer binds an oxygen molecule, the whole process happens again. It this cascade of events, the iron shifting up upon binding and the histidine moving up as a result, that describes the cooperativeness that hemoglobin has between its four monomers and the transition it makes from the T state to the R state.
Chemical process by which as active site of enzyme is bonded by substrate, the enzyme can react with substrate with more effect; three forms of which are positive cooperativeness, negative cooperativeness, and non-cooperativeness; for positive cooperativeness, for example, when oxygen binds to hemoglobin, the affinity of the protein for oxygen increases; therefore, binding of oxygen to the protein is more easily done; for negative cooperative, for example, when enzyme binds to ligand, the bonding affinity decreases.
From the oxygen binding curve of the hemoglobin, it is said that hemoglobin follows a sigmoid model because it looks like a "S" shaped curve. The curve also suggested that hemoglobin has a lower oxygen binding affinity. This is due to that fact that hemoglobin binds to 2,3 bisphosphoglycerate inside of the red blood cell.
The sigmoid binding model of the curve indicates that hemoglobin follows a special oxygen binding behavior, known as cooperativeness. The curve shows that binding at one site of the protein will increase the likelihood of other binding at other sites. And also the unloading of oxygen at one site will also facilitate the unloading of oxygen at other sites.
The biological of this sigmoid model of oxygen binding leads to efficient oxygen transport. The unloading of oxygen can be seen in the graph where in the lungs (100 torr) the protein is saturated with oxygen and all of the oxygen binding sites are occupied. However when this is moved to the tissues to release the oxygen, the saturation level drops and the total unloading oxygen level is 66%. This situation is favored because the hemoglobin goes through cooperativeness and it increases the tendency for oxygen binding and unbinding. Unlike myoglobin, which binds to tightly to oxygen for its release.
In the concerted model, T and R states are the only two forms of hemoglobin that exist. T state is the state where hemoglobin has its quaternary structure in the deoxy form, which is also a tense form. The R state is the state where the hemoglobin has its quaternary structure in completely oxygenated form. This state is relaxed, less constrained, and leaves the oxygen binding sites free. An equilibrium exists between these two states that is shifted by the binding of oxygen, which shifts equilibrium towards R-state. This shift (to R-state) increases the affinity of oxygen of its binding sites. All tetramers of the hemoglobin must be in the same state.
In the sequential model, there is no full conversion from the T-state to R-state. The binding of oxygen changes conformation of the subunits, which subsequently induces changes in other subunits to increase their affinity for oxygen. The subunit to which the a ligand binds changes its conformation without interrupting other subunits to have conformational changing.
In the curve of fractional saturation (fraction of possible binding site that include the binded oxygens) vs. the concentration of oxygen measured by its partial pressure in torr, the T-state binding curve is relatively shallow at low concentration of oxygen when all molecules are in the T state because if a molecule is assumed in the R state, the oxygen affinity increases, which means that new oxygen molecules have more chances to bind to the rest of the three unoccupied sites.
The R-state binding curve goes sharply at the beginning but level off when all of the binding sites are occupied by oxygens.
Hemoglobin behavior resembles a mix of these two models. A molecule with only one bound oxygen molecule exists primarily in T-state, but the other subunits have a much higher affinity for oxygen as suggested by the sequential model. Meanwhile, a molecule with three subunits bound exists primarily in the R-state as suggested by the concerted model.
File:Transition.JPG
Le Chatelier's Principle can be seen to play a role in the circulation of CO2 in the body. Within the erythrocyte, by decreasing the concentration of HCO3-, it acts a force in which it requires more CO2 to be in the cell so that it can be converted to HCO3-.
H2O + CO2 <---> H+ + HCO3-
This reaction, which is carried out by carbonic anhydrase, also decreases the pH within the erythrocyte. Consequently this encourages the hemoglobin to take on the T-state as the excess hydrogen in the cell allows for salt bridges to form. These salt bridges then induce the cell to form the T-state more often than the R-state.
An allosteric effector of hemoglobin is a regulation by a molecule that is structurally unrelated to oxygen and binds to a site completely distinct from the oxygen binding site.
2,3-BPG is a highly anionic compound found in hemoglobin, making it an efficient oxygen transporter. It lowers the oxygen affinity of hemoglobin by binding in the center of the tetramer, stabilizing hemoglobin's "T" state. 2,3-BPG, with such high negative charge, interacts with 3 positively charged groups on each beta chain in the two alpha-beta dimer. For the transition of "T" state to "R" state to occur, the 2,3-BPG must be broken. For this to occur, more oxygen-binding sites within the hemoglobin tetramer must be occupied. Therefore, the hemoglobin remains in the lower-affinity T state until a much higher oxygen concentration is reached. In pure hemoglobin with no 2,3-BPG, only 8% of the sites would contribute to oxygen transport. The presence of 2,3-BPG in hemoglobin increases the percentage to 66%.
Oxygen affinity of fetal red blood cells
Fetal hemoglobin has a higher affinity for oxygen than does regular hemoglobin. Regular hemoglobin is made of two alpha/beta dimers, while fetal hemoglobin is made of two alpha/gamma dimers. The gamma subunits have a lower affinity for binding 2,3-BPG. Thus, with less 2,3-BPG, fetal hemoglobin has a higher affinity for oxygen. This is advantageous for the fetus, as oxygen must be carried longer distances (from the mother) than in regular situations.
The Bohr Effect (Hydrogen Ions and Carbon Dioxide)
A hemoglobin traveling from a region of high pH to a region of lower pH has a tendency to release more oxygen. This is because as pH decrease, the oxygen affinity of hemoglobin decreases. The "T" state of the hemoglobin is stabilized by 3 amino acids (alpha2 Lys40, beta1 His146, beta1 Asp94) that form 2 salt bridges. The residue at the C terminus of the His146 forms salt bridge with the lysine residue in the alpha subunit of the other alpha-beta dimer. The salt bridge between the His146 and the Asp94 is formed only when pH drops, protonating the side chains of His146.
Carbon dioxide also stimulates oxygen release in the hemoglobin. Carbonic anhydrase takes carbon dioxide diffused from the tissue into the red blood cell and water to yield carbonic acid (H2CO3), which is a strong acid (pKa 3.5). Once this compound is formed, it dissociates into HCO3- and H+, which increases the acidity of the environment and lowers the pH level. This drop in pH level will again stabilize the T-state of the hemoglobin. In the hemoglobin, there are three key amino acid residues responsible for the bind of oxygen to the active site: lysine (Lys), histidine (His), and aspartate (Asp). The three amino acids are linked by two salt bridges. One of the salt bridge, between histidine and aspartate, does not form until there is an proton added to histidine. Under conditions of low pH, the histidine gets protonated to allow then the formation of the salt bridge and thus, a conformational change that stabilizes the T-state, lower its affinity for oxygen. In addition, carbon dioxide reacts with the amino-terminals of hemoglobin, resulting in the formation of negatively charged carbamate groups which further stabilize the T state by supporting the salt bridge interactions. This is convenient on a physiological sense. Since tissues tend to be low in oxygen and high in carbon dioxide concentration, the low pH environment will lower hemoglobin's affinity for oxygen and cause the red blood cell carriers to release the oxygen at the tissues.
Hemoglobin is an efficient oxygen transporter around the body. How does it release oxygen to the tissue? Hemoglobin releases oxygen where it is a necessity. Examples include working muscles and tissues. When tissue is metabolizing, it releases carbon dioxide and hydrogen ions. Hemoglobin reacts these conditions. These are called the carbon dioxide effect and the pH effect.
Christian Bohr discovered that hemoglobin is found to have a lower oxygen saturation in lower pH. The release of protons signifies a change in pH. The reason is that protons protonate a histidine on the end of one of the beta chains found on the hemoglobin. Consequently, this makes the histidine charged and creates a salt bridge (ion-ion interaction) with aspartate (negatively charged) on the same polypeptide chain. That salt bridge stabilizes the T state of hemoglobin, which favors the release of oxygen.
Carbon dioxide released by cells are mixed with the blood serum to make carbonic acid. Carbonic acid is a relatively strong acid, so it dissociates into bicarbonate and a proton (which can be used above). The carbon dioxide itself, however, can also participate in oxygen release. When the carbon dioxide meets the terminal amino group of hemoglobin's peptides, it can react to form carbamates, which are negatively charged. This reaction also produces an additional acidic proton. These negatively charged groups can also participate in salt bridges that further stabilize the T state of the hemoglobin to further facilitate the release of oxygen. This effect was seen when someone noticed that oxygen saturation was lowest when in an acidic and carbon dioxide-rich environment.
The deficiency or mutations of hemoglobin can be a result of abnormality of structure of protein molecules, which is related to anemia and sickle cell disease - a condition that malformed red blood cells are resulted from structural abnormality of hemoglobin molecules restrict passage of blood vessels for the supply of blood flow to body tissues.
Sickle cell anaemia is a disease caused by one amino acid substitution. In this case, valine was substituted for glutamate in position 6 of the beta chain of hemoglobin. The mutated form is known as Hemoglobin S (HbS). The elimination of the negative charges of glutamate and the substitution of glutamate with hydrophobic valine causes hydrophobic interactions between different hemoglobins. From electron micrographs, studies have shown that hemoglobin in sickle red blood cells form large fibrous aggregates. The fibrous aggregates form across the red blood cells distorting the shape and increasing their potential to clog small capillaries. This detrimental effect leads to a high risk of stoke and bacterial infection from poor blood circulation. Not only does the sufferer attain insufficient amounts of oxygen, but the aggregated hemoglobins make it harder for blood to flow through small vessels which can cause blood clots. It is interesting to note that the areas with high population of people carrying sickle cell traits is correspond to the areas with high prevalence of malaria. Sickle cell traits means that a person have one normal gene and one mutated gene, disease does not take place in these people. The reason is that a person carrying sickle cell traits are resistant to malaria. The malaria causing parasite can not reproduce effectively in people with sickle cell traits. It should be noted however that the sickle cell gene is codominant. This means that in a person carrying one normal gene and one mutated gene, there will be both regular healthy blood cells and mutated blood cells. However, the function of the normal blood cells makes up for the abnormal cells and no symptoms are felt.
Sickle cell anemia is a genetically inherited disease in which the people who suffer from this disease develop abnormally shaped red blood cells - an elongated shape like a sickle instead of the normal spherical shape of hemoglobin - which decrease its affinity to oxygen. Sickle cell anemia is a disease that are passed down from family members in which the red blood cells form an abnormal sickle shape instead of the round (doughnut) shape. A more in depth look at the red blood cells show that the hemoglobin in the affected cells form large fibrous aggregates, resulting from their sickle shape. This shape then clogs capillaries and prevent blood flow, causing poor circulation and leading to higher risks for stroke, organ damage, and bacterial infections. Anemia is caused by the fact that the sickle shaped cells are not retained in circulation for as long as normal blood cells do, and the bone marrow, where the cells are produced, cannot keep up to speed with making new cells. Sickle cell is usually a deadly heritable disease which primarily is associated with those of African descent. The main difference between those affected with sickle cell anemia is that when the hemoglobin is deoxygenated the hemoglobin bends to the "sickle" shape. The oxygenated hemoglobin actually is the same as those oxygenated hemoglobin in normal people. [1]
The hemoglobin mentioned has two parts: the heme and the globin. The heme is contains the iron which is used to transport the oxygen through the blood stream. The globin is a complex protein that keeps the hemoglobin in a liquid like state. [1]
Sickle cells versus regular red blood cells, the colors do not actually vary, but a lighter red is used for the regular cells to indicate the difference. The sickle shape of the cells makes them less effective in transporting oxygen, and increases the likelihood of forming life threatening clots.
-The most well known symptom is anemia-- the delay of healing and growth as well as the affecting the oxygen flow to the rest of the body. This often results in paleness, weakness, and tiredness.
-Other symptoms include jaundice, or yellowing, of the skin, mouth, and eyes caused by the deposits of bilirubin from the excess of dead sickle cells before the liver filters them out.
-Enlargement, scarring, and damage to the spleen if the sickle cells pool there.
-Acute chest syndrome, if the sickle cells occur in the chest, this can lead to lung damage.
-Sickle crisis, caused by sickle cells clustering in blood vessels and constricting the flow of blood. This results in pain, swelling, and possibly death.
The symptoms do not usually occur until after the age of four. Some others symptoms are shortness of breath, rapid heart rate, paleness, fatique, or yellowing of the eyes and skin. [Pubmed Health].
Complications of Sickle cell anemia:
Different parts of the body are effected by sickle cell anemia in different ways. For instance, “hand-foot syndrome” can either effect the hands or the feet or the two simultaneously. For infants, this is often the first sign of sickle cell disease. This occurs when the small vessels in the hands and/or feet are blocked by sickle cells. The result of this process can lead to fever, pain in the bones, and/or swelling on the surface of the hands and/or feet.
Another complication of sickle cell anemia is “Splenic Crisis.” This occurs when the spleen becomes to large due to an overload of sickle cells flowing into it. In this case, the cells clog the spleen and disrupts it usual function, which is to fight infections by filtering out abnormal red blood cells. Without the spleen’s normal function, the overload soon leads to a shrinkage of the spleen. The only way to correct this abnormality is by undergoing blood transfusion. In addition, the spleen is the most vital organ in the body in fighting infections. Without its proper function, infections can occur that would have the ability to kill a person in a matter of days. For young children, pneumonia is the most common infection caused by sickle cell disease.
Acute chest syndrome is very similar to pneumonia and is a life threatening condition caused by sickle cell disease. This occurs when sickle cells flow into the lungs and cause symptoms such as fever and chest pains. When lung damage is extended over a long period of time, pulmonary arterial hypertension is the result. The symptoms include irregular breathing patterns and high blood pressure.
Sickle cell disease can have a major effect on the growth and puberty of children. Basically, children with this disease experience a shortage in red blood cells, which causes their growth rate to decrease relative to a child without the disease. When these children become adults, they are usually thinner and smaller in comparison to the average adult.
Besides, people with sickle cell anemia may have some symptoms of infections as the following: bone infection, gallbladder infection, lung infection, urinary infection, delayed growth and puberty, or even painful joints caused by arthritis. [Pubmed Health].
Two forms of stroke can also occur as a result to sickle cell disease. One form of stroke happens when sickle cells flow into the brain and block blood vessels. The second form of stroke occurs as a result of the first case; bursting blood vessels. If death isn’t the result of this complication, then a person may experience learning disabilities or paralysis.
Vision can also be effected by sickle cell disease. In order to have clear vision, our small blood vessels have to have the ability to transport oxygen into our eyes. If the small blood vessels become blocked by sickle cells, the thin layers of the retina become damaged. Since the retinas are responsible for sending images of what we see to our brain, without this function, blindness is the result.
“Gallstones” is a complication that is the result of the release of hemoglobin from a dead red blood cell. In addition, stones may from in the gallbladder due to the overload of a compound called, bilirubin. When fattening meals are consumed, one may experience discomfort in the right side of the belly, right shoulder, or between the shoulder blades. Nausea, vomiting, sweating, and chills can also occur.
For people between the ages of ten and fifty, ulcers on the legs may form. They are treatable, but reoccurring.
One of the most serious, but rare complications of sickle cell disease is multiple organ failure, when three major organs fail. This will cause serious effects in a person’s mental state, like tiredness and general loss of interest in everyday activities. Fever is also common.
Treatment:
Due to the crisis known as vaso-occlusive, people with sickle cell disease experience painful episodes. There are, however, various types of treatment available. One type of treatment, used for children with sickle cell disease, is folic acid and penicillin. This treatment requires that the patient take 1mg of folic acid every single day for life. As a result of the undeveloped immune system the patient must then take penicillin daily by the age of five.
For children, bone marrow transplants help to suppress this severe disease.
Hydroxyurea is a drug that has the ability to reactivate fetal hemoglobin, which can extend the lifetime for a person living with sickle cell anemia. This drug has also been used in chemotherapy.
Currently, gene therapy is being researched as a possible treatment for these disease. Also, the use of phytochemicals, like nicosan.
Overall, people with sickle cell anemia need to have their treatment continuously and nonstop. In order to get a better treatment, they must seek help from their health care providers and also from clinics that are in charge of sickle cell anemia disease. At the hospital or clinics, patients can receive treatments for sickle cell anemia such as blood transfusion, pain medicines, lots of fluids, and antibiotics to prevent bacterial infections. However, some people might need to do dialysis or kidney transplant, surgery for the eye, or even the wound care for leg ulcers. [Pubmed Health].
By investigating the amino acid sequence of the hemoglobin of affected cells, scientists have shown that there is a single substitution of valine for glutamate in position 6 of the β-chain of hemoglobin that results in this disorder. The mutated hemoglobin is called S-hemoglobin, for sickle cell anemia. When hemoglobin is in its T-state, the additional valine residues bind to a hydrophobic area on other S-hemoglobin molecules, forming a chain of hemoglobin that in turn pulls the red blood cell into its signature sickle shape. This dangerous effect only occurs on hemoglobin in its deoxygenated state because oxygenated hemoglobin has the R conformation, which covers the hydrophobic patch that the extra valine binds to. People who have this disease must have inherited this mutation from both parents as it is a recessive trait. A recessive trait means that the disease will only be expressed if both of the individual's alleles code for that trait. To summarize, sickle cell anemia results from the aggregation of mutated deoxyhemoglobin molecules, where aggregation is caused by interation between Val 6 on a beta chain of one Hb molecule and a hydrophobic patch (formed by Phe 85 and Val 88) on a beta chain of another deoxygenated hemoglobin molecule.
Diagnosis:
Hb levels are usually high for people effected by sickle cell anemia. Hyposplenism may appear on a blood film analysis.
Induction of sickle cells can be promoted by the addition of sodium metabisulfite on a blood film. A “sickle cell solubility test” can also be used to detect the presence of sickle hemoglobin.
Gel electrophoresis can be used to detect abnormalities in hemoglobin.
The distribution of Sickle cell disease in Africa.The distribution of Malaria in Africa.
1/100 of West Africans suffer from sickle cell anemia, and a much larger percentage of the population has the sickle cell trait in comparison to other regions in the world. The mosquito-borne protozoan Plasmodium falciparum, infect erythrocytes, which cause them to adhere to capillary walls and block blood flow to vital organs. The reason of the high probability of West Africans having sickle cell anemia is because of the common disease reign the region, malaria. Individuals who carry the sickle cell anemia gene are found to be more resistant to malaria due to the effect of Plasmodia has on infected erythrocytes. Plasmodia lowers the intracellular pH in erythrocytes by roughly 0.4, which causes hemoglobin to favor deoxyhemoglobin formation. Deoxyhemoglobin has a higher tendency for sickling, which creates a tendency for infected erythrocytes to be removed by the spleen. Furthermore, sickling also mechanistically disrupts erythrocyte adhesion to capillary walls.
As a result, individuals who carry this trait have resistance to malaria. Individuals who are even heterozygous have an adaptive advantage to homozygous individuals who do not carry sickle cell anemia and are unable to seek proper medical treatment. Therefore, the sickle cell trait is extremely common in West Africa, where malaria is also a big factor, contributing to the death toll.
With the sickle cell disorder, Linus Pauling was the first to propose and observe the direct connection between the variation of one single amino acid squence that can result in a sickle shaped red blood cell.Therefore, Sickle Cell studies are important in studying genetically linked diseases. Linus Pauling was able to propose this thought due to his background in studying atoms and his eventual journey through medicine. His diverse academic background gave him a unique viewpoint to which he advanced immunology. Much of his studies were based on normal and abnormal hemoglobin cells. Pauling later went on to write Sickle Cell Anemia, A Molecular Disease, in 1949. In his later years, Pauling used his research on hemoglobin to for social, political and scientific endeavors. [2]
Unlike sickle-cell anemia, which is a disorder that results in the synthesis of an incorrectly functioning globin chain, Thalassemia is a condition in which too few globin strands are synthesized. Thalassemia is generally caused by mutations in regulatory genes. This defect reduces the synthesis rate of one of the necessary globin chains needed to make hemoglobin which leads to the formation of abnormal hemoglobin and causes anemia.
There are three types of Thalassemia (bases on the globin strand affected), α-Thalassemia, β-Thalassemia and ɣ-Thalassemia.
α-Thalassemia: The effects and severity of α-Thalassemia depends on the number of α-globin loci affected, because α-globin chains are encoded by two genes, each containing 2 loci, affects of the disease can be minimal or even non-existent. If only one of the four loci is affected then the person usually doesn’t display any symptoms of the disorder, they are just a carrier. If two loci are affected then the person often has somewhat mild symptoms which mimic those of iron deficiency induced anemia (this condition is generally referred to as the α-Thalassemia trait). However, the effects of the disorder become more evident and severe when three loci are affected because now both genes that encode for the α-globin are defective (this condition is called Hemoglobin H disease), unstable hemoglobin is produced which acts as a much poorer oxygen transporter (because it has a higher than normal affinity for oxygen), which causes anemia and splenomegaly. If all four loci are affected than the fetus can’t survive and will either die in the womb resulting in a miscarriage or will be a stillbirth.
β-Thalassemia: Like α-Thalassemia the severity of β-Thalassemia depends on the nature of the mutation; the mutation can either completely prevent the production of β chains (referred to as βo) or allow limited β chain formation (known as β+). However, unlike α-globin, which is coded for by 4 loci, β-globin is only coded for by one gene with two loci, so the severity of the condition is very different based on whether one or two of the loci are affected. If only one of the loci is mutated then the symptoms are very mild and sometimes even unnoticeable, the only effect is a slight decrease in red blood cell size (this disorder is called β-Thalassemia minor). However, if both alleles are affected the person will experience severe anemia that could even cause death unless they receive periodic blood transfusion (called β-Thalassemia major). There also exists conditions which are intermediates between the two extremes, which often result in anemia but it can be managed with occasional blood transfusions (called Thalassemia intermedia). However, no matter the severity of the disorder (i.e.-the number of loci affected) there is still an excess of α chains produced in relation to the β strands which tend to bind to red blood cells and can cause membrane damage or even form toxic aggregates.
ɣ-Thalassemia: As with the other disorders, ɣ-Thalassemia exist as either a mutation that limits the production of ɣ-strands or completely eliminate the formation altogether. However, unlike the other disorders, ɣ-strands only make up about 2-3% of hemoglobin, so the effects of the condition are not severe even if no ɣ-strands are produced. The biggest risk associated with the disorder is that it counteracts the tested effects of β-Thalassemia (which is an increase in A2 hemoglobin) and can lead to a misdiagnosis. Such a misdiagnosis may lead to extreme sickness and, in some cases, death.
Because alpha chains are coded for by four loci, compared to the two that code for beta chains, excess alpha chains are produced and must be stabilized in order to keep them in solution and prevent them from precipitating out. This is achieved by the production of an alpha hemoglobin stabilizing protein (i.e.-AHSP). This protein binds to the alpha chains in the same manner that the beta chains do and creates a soluble complex. It binds to the alpha hemoglobin as it’s produced and is then displaced by the beta hemoglobin produced which forms a more favorable and stable dimer. This way there is no excess alpha hemoglobin which would accumulate and precipitate out of solution.
AHSP is important not only for dealing with newly synthesized excess α-globin, but also in the assembly of normal Hb tetramers. Studies now show that mice infected with mild α-Thalassemia, a condition which causes a deficit of α-globin and an excess of β-globin, still utilize AHSP. No AHSP was expected to be needed by these mice because there was no risk of excess α-globin accumulating and precipitating out, however, when the mice with no AHSP and α-Thalassemia were compared to both normal mice, and mice with α-Thalassemia and intact AHSP, they were far more anemic, showing AHSP has other roles besides stabilizing excess α-globin.
Besides being an important molecular chaperone in Hb assembly, AHSP may also provide an additional selective advantage to red cells under conditions of oxidative stress induced by drugs, because of its effects on preventing α-globin denaturation and promoting renaturation. AHSP may also be useful to red cells in iron deficiency in which heme availability is limited and apo-α-globin levels are increased and help stabilize red blood cells in the presence of environmental factors that alter Hb’s critical equilibrium.
Berg, Jeremy M. John L. Tymoczko. Lubert Stryer. Biochemistry Sixth Edition. W.H. Freeman and Company. New York, 2007.
The Bohr effect was first discovered by a physiologist Christian Bohr in 1904. This effect explains how hydrogen ions and carbon dioxide affect the affinity of oxygen in Hemoglobin. If pH was lower than it normally was (normal physiological pH is 7.4), then the hemoglobin does not bind oxygen as well. In other words, the lower the pH, the more Hydrogen ions, the higher the carbon dioxide level and the LESS affinity Hemoglobin has for oxygen.
The opposite explains: the higher the pH, the lower the H ion concentration, the lower the carbon dioxide level, and the GREATER affinity hemoglobin has for oxygen. The binding of oxygen to hemoglobin in the lungs is not affected by changing the pH and the oxygen will continue to be loaded normally. This does not prove to be true in tissues however, and a change in the pH results in a lower percent saturation of hemoglobin. More oxygen is delivered to tissues at a lower pH even when the amount of oxygen available remains unchanged.
How can it be determined if a tissue is more active and thus requires more oxygen? One way to determine this is by the amount of oxygen present in a tissue. If a tissue is using more oxygen, then one would expect that the amount of oxygen would be lower. When this is the case, more oxygen is delivered to the tissue.
Another indicator that a tissue has a high metabolic rate, meaning that there is a need for increased oxygen delivery, is the production of Carbon Dioxide. When a tissue is more active, the amount of carbon dioxide produced will be increased.
Carbon dioxide reacts with water as is shown in the following equation:
CO2+ H2O <---------> H+ + HCO-3
This shows that as the amount of carbon dioxide increases, more H+ is formed and the pH will decrease. In other words, the more CO2 present, the more H+ is formed (so the lower the pH; remember pH is inversely related to the H+ concentration by the equation pH = -log[H+])
A lower pH in the blood is suggestive of an increased carbon dioxide concentration which in turn, is suggestive of a more active tissue that requires more oxygen. According to Bohr, the lower pH will cause hemoglobin to deliver more oxygen. If the amount of oxygen and pH should drop together, even more oxygen will be delivered than if only one of the these factors were changed. If the pH of the tissues should rise due to a drop in the carbon dioxide concentration, then less oxygen will be delivered.
The Bohr Effect is dependent upon cooperativity between the hemoglobin tetramer and the Heme group; it is key to note that although myoglobin and hemoglobin are very similar, myoglobin does not exhibit this effect because Myoglobin, a monomer, does not exhibit any cooperative interactions. If the hemoglobin's cooperativity is weak, then the Bohr effect will in turn be low.
This phenomenon explains why Hemoglobin can readily release oxygen in human tissue. The pH of the tissue is much lower than in the human lungs, so the blood will want to release the oxygen creating hemoglobin in its t-state. Once the blood travels back to the lungs, where the pH is higher, the blood will pick up more oxygen for transport. Myoglobin holds onto its oxygen in the tissue because it is not influenced by the Bohr effect. On average, the hemoglobin can release 66% of its oxygen, whereas myoglobin only releases about 7%.
If a person were to increase their physical activity, and take in more oxygen. The transport of oxygen per red blood cell would increase as well because the CO2 levels would rise in the body, leading to a lower pH in the tissues. Another factor that will also affect the binding of oxygen to hemoglobin is temperature, which may be affected due to physical activity among many other factors. A more active tissue will be producing more heat and will be warmer. This increased temperature may lead to changes in hemoglobin's affinity to oxygen in a similar fashion as would be expected from a decrease in pH.
The affinity that hemoglobin has on oxygen is decreased when the pH of the solution is decreased. When the solution is at a lower pH, hemoglobin tends to release more oxygen because it doesn't have as much affinity to keep the oxygen binded to the heme group. The main reason for this is shown by what occurs in deoxyhemoglobin. If the pH is lowered the histidine can be protonated. This triggers salt bridges to form between the now-protonated and positively charged imidazole group on the histidine with the negatively charged carboxylate group on a nearby aspartate. This causes the stabilizing of the deoxyhemoglobin or T state. This causes the T state, which has less affinity for oxygen, to be more prominent which pushes for oxygens to be released from hemoglobin.
The presence of Carbon dioxide gives rise to the release of oxygen from hemoglobin. The first way it does this is that at high concentrations the carbon dioxide reduces the pH. This occurs due to the fact that carbon dioxide reacts with water and forms carbonic acid, and carbonic acid dissociate to release proton H and bicarbonate ion, so it will decrease pH. This reaction is sped up very quickly with an enzyme present in red blood cells, Carbonic anhydrase. Carbonic acid is a relatively strong acid, so it tends to dissociate causing an increase in hydrogen ion presence. This results in a decrease in pH.
The second way it aids in releasing oxygen from hemoglobin is that there is a direct interaction that carbon dioxide has with the hemoglobin itself. What occurs is that carbon dioxide stabilizes the deoxyhemoglobin form by reacting with the terminal amino groups. It basically forms a carbamate group which is negatively charged. These negatively charged groups participate in salt bridges. Due to this the deoxyhemoglobin or T state is stabilized pushing for oxygen to be released from hemoglobin.
Formation of Carbamate group; Due to this reaction occurring deoxyhemoglobin is stabilized therefore releasing oxygen
The affinity constant (also known as the association constant), Ka, is a numerical constant used to describe the bonding affinity of two molecules at equilibrium. In other words, this constant measures the molecular binding strengths between enzymes and substrates - more specifically: protein to protein, protein to ligand, protein to DNA, protein to metal ions, etc.
The binding of molecules can be described as
the corresponding affinity constant would be
where [C] is the complex formed, [S] is the substrate, and [E] is the enzyme, ligand, or ion. Note that the affinity constant Ka is not the acid equilibrium constant Ka. They are completely different constants.
The converse and more widely used of the two constants is the dissociation constant defined as Kd and is the inverse of the affinity constant.
Equilibrium constant used in chemistry, biochemistry, and pharmacology. This constant describes the ability for a large complex to come apart — the larger the constant, the less likely the complex will stay together; the smaller the constant, the more likely the complex will stay together.
In acid and base studies, Ka is used to describe the dissociation of hydrogen from an acid.
Where HA is the acid (most of the time it is used to represent weak acids), H is the hydronium ion (H+), and A is the conjugate base. The equilibrium constant equation for this reaction would be:
In the study of proteins, Kd is used to describe the interactions between either protein to protein, protein to ligand, and protein to DNA. The binding propensity can be described with the formula and the equation below:
where C is the complex, S is the substrate and E is the particular protein or material.
This formula can be described with an equilibrium equation:
The inverse of the dissociation constant is the affinity constant.
Proteins are regulated through a variety of different ways; some are controlled allosterically, some use different forms of enzymes that catalyze the same reaction. There are enzymes that are regulated through reversible, or irreversible covalent modification, other enzymes use proteolytic activation, with zymogens that are inactive until cleaved, and finally, some enzymes are regulated by the control of the amount of enzyme present.
Strategies of Regulation:
- Allosteric Control
- Multiple forms of the same enzyme
- Reversible covalent modification
- Proteolytic activation
- Controlling the amount of enzyme present
Allosteric proteins have two different sites, regulatory and functional. As the name implies, regulatory sites are used by the protein to regulate protein function. This is done by having small signal proteins come and attach themselves to them, and send various signals to control their activity. The functional sites are sites that proteins use to perform its function. Because they contain more than one different site, these proteins often show cooperativity because one active site affects other nearby active sites.
One model of cooperativity is the concerted model, also known as the MWC model. It proposes that an enzyme can only exist in the tense (T) or relaxed (R) state but not both. They either exist completely in T state or completely in R state. When a substrate binds to one monomer of the enzyme, it shifts the equilibrium between the two states. For example, hemoglobin is a tetramer. When oxygen binds to one monomer, the equilibrium shifts from the T state to the R state. The R state favors oxygen binding whereas the T state is stabilized by a different component described later. When an oxygen is bound, the equilibrium of the other monomers also shift from the T state to the R state. As more oxygen is bound to hemoglobin, the affinity for oxygen increases.
The second model of cooperativity is the sequential model. In this model, an enzyme is either in the T or R state. However, in this model, binding of a substrate to an enzyme causes a conformation shift that causes the other monomers in the enzyme to favor binding of the substrate. In hemoglobin, binding of oxygen to a monomer causes the proximal histidine to move closer to the porphyrin ring in the heme group causing one dimer to shift 15 degrees. This conformational change causes the other monomer's affinity for oxygen to increase, thus favoring the T state.
An example of allosteric control is with the enzyme aspartate transcarbamoylase or ATCase. This enzyme catalyzes a reaction that will produce cytidine triphosphate, or CTP. To control the amount of product formed, CTP will inhibit its own formation by inhibiting the catalyst, ATCase, in a process called "feedback inhibition". It does this by making the unbound form, or the T state of the catalyst ATCase more stable. This results in shifting the equilibrium towards the unbound state, and thus lowing the affinity of ATCase to its substrate. This is one way proteins ensure they waste no energy producing excess product.
CTP is not the only NMP molecule that has an effect on ATCase, ATP also has its own effect. Whereas CTP stabilizes the T state ATP stabilizes the R state of ATCase. Stabilizing the R state of ATCase allows the substrate to bind easier and in turn the reaction rate is increased.
ATP as well as CTP are referred to as "heterotropic effects" on a allosteric enzyme such as ATCase. ATP is an allosteric activator of aspartate transcarbamolyase because it stabilizes the R-state of ATCase, effecting neighboring subunits by making it easier for substrate to bind.The increase of the concentration of ATP has two potential explanations. First being, at high concentrations of ATP signals a high concentration of purine and pyrimidine. second, a high concentration of ATP conveys that a source of energy is available for mRNA synthesis and DNA replication follow by the synthesis of pyrimidines needed for these processes.
These can also be called isoenzymes. Isozymes are homologous enzymes that have the same function, but are composed of different amino acid sequences, have slightly different structures, and or respond to different regulatory molecules or kinetic parameters such as kM and Vmax. Isozymes allow for "fine tuning" of the enzymes, resulting in enzymes that have the same function, but work in different environments, or respond to different signals. Isoenzymes may be the result of mutations that are preserved in the genome or a result of convergent evolution.
An example of an isozyme is lactate dehydrogenase, or LDH. This enzyme catalyzes a step in anaerobic glucose metabolism and synthesis. People have two forms of this enzyme, the H and the M, existing in the heart and muscle respectively. These two isozymes differ in their affinity for the substrates and the levels of inhibitors needed to inhibit them. This makes sense because of the highly different environments that exist in the heart and the muscle; the heart is highly aerobic while the muscles are anaerobic. This allows for the same function to be preformed by two "different" enzymes in two different environments.
Since isoenzymes have different structures, they may be separated and identified techniques such as gel electrophoresis.
Enzymatic activity can be modified by attaching a modifying group, such as phosphorous, through a covalent bond. The new group then changes the enzyme's reactivity, size, charge, etc.
A good example of this is phosphorylation, catalyzed by protein kinases. Phosporylation is used in many different cells in many of life's organisms because it has many good attributes making it a good tool.
Of these good attributes include:
~It adds two negative charges to the protein
~Forms 2-3 extra Hydrogen bonds
~Irreversible, due to the amount of energy required to phosphorylate
~has a varying speed that can be changed due to specific needs
~can cause a cascade effect, resulting in an amplified result
Protein kinases are the catalytic enzymes that phosphorylate a protein. They use ATP as the source of phosphoryl groups as well as energy. There are dedicated protein kinases that only phosphorylate a specific protein. They recognize a specific consensus sequence, which usually includes a serine or threonine residue. Other protein kinases are multifunctional protein kinases which can phosphorylate many different proteins.
Protein Kinase A (PKA) is an example of an enzyme that is regulated by reversible covalent modification and allosteric control. The holoenzyme form of PKA forms an inactive R2C2 complex comprised of two regulatory subunits and two catalytic subunits. The two regulatory subunits contain pseudosubstrate sequences that are bound to the active sites of the two catalytic subunits, inhibiting its function. PKA is activated when four cyclic adenosine monophosphate (cAMP) molecules bind to the two regulatory subunits, removing them from the catalytic active sites, thus freeing and activating the catalytic subunits. The protein kinase A is now free to carry on its function and phosphorylate other proteins.
Enzymes can also be controlled by preventing them from functioning until a given time. For example, many enzymes are controlled by hydrolysis of certain bonds, making an inactive enzyme active. The inactive precursors are called zymogens; enzymes such as chymotrypsin, trypsin, and pepsin show this trait.
Take Chymotrypsin for example; the inactive form of it is called chymoprypsinogen which will be cleaved by trypsin to result in pi-Chymostrypin, which will in turn cleave others of its kind and result in the final alpha-chymotrypsin. This will control when and where chymotrypsin cleaves, so it does not cleave in the wrong environments or time.
Chymotrypsinogen is the zymogen (inactive precursor) to the digestive enzyme trypsin. It is synthesized in the pancreas. They are stored in zymogen granules in the acinar cells of the pancreas. When a nerve impulse reaches the pancreas, it stimulates the granules to release chymotrypsinogen into the lumen leading to the small intestine. Chymotrypsin activated when the peptide bond between arginine 15 and isoleucine 16 is cleaved by trypsin. This creates two pi-chymotrypsin peptides. The pi-chymotrypsin then cleaves other pi-chymotrypsin peptides. The final result is the active form alpha-chymotrypsin which is made up of three chains.
Another example of proteolytic activation of enzymes can be seen in blood clotting. When trauma occurs to tissue, it starts a blood-clotting cascade. The activation of one clotting factor triggers the activation of other triggers which creates an amplified effect that allows the body to quickly respond to the injury. When blood vessels rupture, it activates tissue factor (TF). TF then activates thrombin, a protease that cleaves fibrinogen, the zymogen of fibrin. When fibrin is formed, it polymerizes to form clots.
This form of regulation is controlled with transcription, different protein will bind to specific DNA sequences to regulate the transcription of certain segments of DNA. The transcription of a certain enzymatic genes can be adjusted to the changes in a cells environment. This will affect the amount of enzyme present in the system, an thus regulate the catalysis of the reaction.
2,3-bisphosphoglycerate is a three-carbon molecule with two negatively charged (2-) phosphate groups attached to the central carbon, forming a tetrahedral structure. This highly anionic molecule is most commonly found in the red blood cells at about the same concentration as that of hemoglobin (around 2 mM); this is responsible for the great efficiency of oxygen transport that takes place in hemoglobin molecules. 2,3-bisphosphoglycerate, abbreviated as 2,3-BPG and also referred to as 2,3-diphosphoglycerate (2,3-DPG), binds with greater affinity to deoxygenated hemoglobin typically found in tissues than to oxygenated hemoglobin found in the lungs. Pure hemoglobin releases only 8% of oxygen to the tissues, however hemoglobin with 2,3-BPG allows it to release 66% of the oxygen to the tissues. It is for this reason that hemoglobin, and not myoglobin, is more used in transferring oxygen between tissues and the lungs. There are high concentrations of 2,3-BPG found in erythrocytes
2,3-biphosphoglycerate is the product of glucose metabolism. It is formed from 1,3-BPG by the enzyme bisphosphoglycerate mutase. It can form 3-phosphoglycerate from when it is broken down by phosphotase. This synthesis and disassembling takes several steps.
2,3-bisphosphoglycerate is mostly found in human red blood cells, or erythrocytes. It has a less oxygen binding affinity to oxygenated hemoglobin than it does to deoxygenated hemoglobin. It also acts to stabilize the oxygen affinity of the hemoglobin in the tense state, since the oxygen affinity is low. This is due to the position of the 2,3-BPG molecule in the central cavity of the deoxyhemoglobin tetramer, where the 2,3-BPG interacts with the positively charged molecules on each beta chain within the deoxyhemoglobin. As a result, the conformation of the deoxyhemoglobin is altered in such a way that a greater number or concentration of oxygen molecules is needed to bind to the free sites in the deoxyhemoglobin, thereby giving hemoglobin a lower affinity T state until addition of more oxygen. This effect makes it difficult for oxygen to bind to the hemoglobin which allows it be released to areas with low oxygen concentration. This is why hemoglobin is such an effective oxygen carrier. It is able to saturate itself with oxygen at high oxygen level in the lungs and retain the oxygen until it reaches the tissues which has a lower oxygen concentration. However, this does not occur in the relaxed (R) state since the hemoglobin is oxygenated. Thus, 2,3-bisphosphoglycerate helps in the regulation of the oxygen carrying capacity in hemoglobin. The R state conformation of deoxyhemoglobin does not allow for these interactions due to the oxygen bound to the heme group. During the T state to R state transition, the 2,3-bisphophoglycerate is released. It's vital to oxygen transfer, since the T state must be stabilized until the transition point. However, the T state is very unstable resulting for hemoglobin's affinity for oxygen and thus tries to bind to oxygen, disrupting the T state. Without 2,3-BPG, this stabilization cannot occur thanks to its inhibition abilities.
2,3-BPG can also function as an intermediate of phosphoglycerate mutase. The enzyme is used in the Embden-Meyerhof pathway of glycolysis in erythrocytes. The pathway is the anaerobic metabolic pathway that converts glycogen to lactic acid in human muscle.
The effect of 2,3-bisphosphoglycerate is shown between the fetal red cells and the maternal red cells. The maternal red cells are able to bind 2,3-bisphosphoglycerate better than the fetal red cells. Therefore the fetal red cells have a higher oxygen affinity which explains why oxygen flows from oxyhemoglobin to the fetal deoxyhemoglobin. Since the fetal red cells contain a higher oxygen affinity, it allows oxygen to be carried to the placenta. Soon after birth, humans have regular hemoglobin. One of the reasons that fetal cells contain a higher affinity for oxygen is because fetal hemoglobin do not contain a beta subunit, but instead a gamma subunit. Therefore the pocket that binds BPG differs which lowers the affinity for BPG while inducing higher oxygen binding.
Oxygen binding to iron in the heme group pulls part of the electron density from the ferrous ion to the oxygen molecule. It is important to leave the myoglobin in the dioxygen form rather than superoxide form when the oxygen is released because the superoxide can be generated by itself to have a new form that gives negative effect on many biological materials, and also the superoxide prevents the iron ion from binding to the oxygen in its ferric state (Metmyoglobin). Superoxide and superoxide-derived oxygen species are so reactive compared to the stable O2 molecule that they could have a destructive effect both within the cell and in its environment. A distal histidine residue in myoglobin regulates the reactivity of the heme group to make it more suitable for oxygen binding. It does this by H-bonding with the oxygen molecule; the additional electron density of the oxygen molecule makes the H-bond unusually strong and therefore even more effective as a stabilizing agent.
An oxygen-binding curve is a plot that shows fractional saturation versus the concentration of oxygen. By definition, fractional saturation indicates the presence of binding sites that have oxygen. Fractional saturation can range from zero (all sites are empty) to one (all sites are filled). The concentration of oxygen is determined by partial pressure.
Hemoglobin's oxygen affinity is relatively weak compared to myoglobin 's affinity for oxygen. Hemoglobin's oxygen-binding curve forms in the shape of a sigmoidal curve. This is due to the cooperativity of the hemoglobin. As hemoglobin travels from the lungs to the tissues, the pH value of its surroundings decrease, and the amount of CO2 that it reacts with increases. Both these changes causes the hemoglobin to lose its affinity for oxygen, therefore making it drop the oxygen into the tissues. This causes the sigmoidal curve for hemoglobin in the oxygen-binding curve and proves it's cooperativity.
This image shows hemoglobin's oxygen binding affinity compared with myoglobin 's affinity and the hypothetical curve that hemoglobin would have to follow if it did not show cooperativity. In this graph, you can see hemoglobin's sigmoidal curve, how it starts out with a little less affinity than myoglobin, but comparable affinity to oxygen in the lungs. As the pressure drops and the myoglobin and hemoglobin move towards the tissues, myoglobin still maintains its high affinity for oxygen, while hemoglobin, because of its cooperativity, suddenly loses its affinity, therefore making it the better transporter of oxygen than myoglobin . The gray curve, showing no cooperativity, shows that to have the low affinity for oxygen needed in the tissues, the hemoglobin would have started with a smaller affinity for oxygen, therefore making it less efficient in bringing oxygen in from the lungs.
In red blood cells, the oxygen-binding curve for hemoglobin displays an “S” shaped called a sigmoidal curve. A sigmoidal curve shows that oxygen binding is cooperative; that is, when one site binds oxygen, the probability that the remaining unoccupied sites that will bind to oxygen will increase.
The importance of cooperative behavior is that it allows hemoglobin to be more efficient in transporting oxygen. For example, in the lungs, the hemoglobin is at a saturation level of 98%. However, when hemoglobin is present in the tissues and releases oxygen, the saturation level drops to 32%; thus, 66% of the potential oxygen-binding sites are involved in the transportation of oxygen.
Purified hemoglobin binds much more tightly to the oxygen, making it less useful in oxygen transport. The difference in characteristics is due to the presence of 2,3-Bisphosphoglycerate(2,3-BPG) in human blood, which acts as an allosteric effector. An allosteric effector binds in one site and affects binding in another. 2,3-BPG binds to a pocket in the T-state of hemoglobin and is released as it forms the R-state. The presence of 2,3-BPG means that more oxygen must be bound to the hemoglobin before the transition to the R-form is possible.
Other regulation such as the Bohr effect in hemoglobin can also be depicted via an oxygen-binding curve. By analyzing the oxygen-binding curve, one can observe that there is a proportional relationship between affinity of oxygen and pH level. As the pH level decreases, the affinity of oxygen in hemoglobin also decreases. Thus, as hemoglobin approaches a region of low pH, more oxygen is released. The chemical basis for this Bohr effect is due to the formation of two salt bridges of the quaternary structure. One of the salt bridges is formed by the interaction between Beta Histidine 146 (the carboxylate terminal group) and Alpha Lysine 40. This connection will help to orient the histidine residue to also interact in another salt bridge formation with the negatively charged aspartate 94. The second bridge is form with the aid of an additional proton on the histidine residue.
As carbon dioxide diffuses into red blood cells, it reacts with water inside to form carbonic acid, which drops the pH and stabilizes the T state.
An oxygen-binding curve can also show the effect of carbon dioxide presence in hemoglobin. The regulation effect by carbon dioxide is similar to Bohr effect. A comparison of the effect of the absence and presence of carbon dioxide in hemoglobin revealed that hemoglobin is more efficient at transporting oxygen from tissues to lungs when carbon dioxide is present. The reason for this efficiency is that carbon dioxide also decreases the affinity of hemoglobin for oxygen. The addition of carbon dioxide forces the pH to drop, which then causes the affinity of hemoglobin to oxygen to decrease. This is extremely evident in the tissues, where the carbon dioxide stored in the tissues are released into the blood stream, then undergoes a reaction that releases H+ into the blood stream, increasing acidity and dropping the pH level.
Introduction
The globins are a related family of proteins, all of which have similar primary and tertiary structure (amino acid sequence and folding). These proteins all incorporate the globin fold. The haploid human genome contains 1 globin gene for myoglobin, two genes for alpha hemoglobin, and one gene for beta hemoglobin, as well as fetal hemoglobin. Recent examination of the human genome has revealed two additional globins.
Neuroglobin is expressed primarily in the brain and at especially high levels in the tetina. Neuroglobin is a monomer that reversibly binds oxygen with an affinity higher than that of hemoglobin. It also increases oxygen availability to brain tissue and provides protection under hypoxic or ischemic conditions, potentially limiting brain damage. It is of ancient evolutionary origin, and is homologous to nerve globins of invertebrates.
Cytoglobin is a globin molecule located in the brain and most notably utilized in marine mammals. It is thought to be a method of protection under conditions of hypoxia. The predicted function of cytoglobin is the transfer of oxygen from arterial blood to the brain.
Berg, Jeremy M. John L. Tymoczko. Lubert Stryer. Biochemistry Sixth Edition. W.H. Freeman and Company. New York, 2007.
An antibody is a protein that is synthesized by an animal in response to the presence of a foreign substance in our body, called an antigen. They play a great role in the immune system, and are usually found in blood and other bodily fluids. Antibodies are created by white blood cells, or more specifically, B cells. There are five isotypes of antibodies that each play self-defense role to fight off foreign objects in our body. Antibodies are created in response to antigens that include, but are not limited to, foreign proteins, polysaccharides, and nucleic acids. The antibody recognizes a small portion of the antigen called the antigenic determinant or epitope. Each antibody recognizes and binds to a specific antigen in a lock and key type model. Given the sheer amount of antigens present, there are an equally diverse selection of antibodies.
Antibodies are gamma globulin proteins that have sugar groups attached to amino acid chains. They can be classified as glycoproteins. The most basic form is the immunoglobulin monomer, which has only one immunoglobulin unit. Antibodies can also appear in dimeric forms with two immunoglobulin units, tetrameric form with four immunoglobulin units, or even pentameric form with five immunoglobulin units.
Immunoglobulin G, the most common type of antibody, consists of 4 chains. There are 2 light chains and 2 heavy chains. The two heavy chains are bound together by a disulfide bond (S-S), and the two light chains are bound to the heavy chain by disulfide bonds. Together, they roughly form a Y shape.
Basic Structure of Antibody
There are two sites that recognize and bind to antigens located at the top of the Y shaped immunoglobulin.Immunoglobulin G (IgG) all have the same general structure only varying at the antigen binding site. This region is called the variable region (V) and is composed of hypervariable loops. These hypervariable loops give great versatility to the antigen binding site allowing it to bind to multitudes of different antigens. The variable regions (V), which make up the two identical antigen-binding sties, are different in each specific type of antibody, giving these sites specific shapes that fit certain antigenic epitopes. The remainder of the molecule consists of light and heavy chain constant regions (C) where these amino acid sequences vary little form antibody to antibody.
Antibodies obtain their diversity through two processes. The first stage is called somatic or V(D)J, which stand for variable, diverse, and joining regions, recombination. Within each of the three regions are located several sets of genes. During cell maturation, the B cell will splice out the DNA of all but one of the genes from each region and combine the three remaining genes together to form one VDJ segment. This segment, along with a constant region gene, forms the basis for subsequent antibody production. It is estimated that given the number of variants in each of the three regions, approximately 10,000-20,000 unique antibodies are producible.
The second stage of recombination occurs after the B cell is activated by an antigen. When an antigen binds to the B cell, the B cell will begin to reproduce rapidly. During this division process, the variable regions of the gene will undergo rapid mutation called somatic hypermutation. This hypermutation serves to fine tune the antibody binding to the antigen. Cells that have a stronger affinity for the antigen will be given a stronger signal to multiply, leading to a gradual selection of antibodies that bind to the antigen the strongest.
In IgG, the heavy chain has four subunits, CH3, CH2, CH1 (the constant portions) and VH (the variable portion). The light chain has two subunits, CL and VL. The two CH3 units are joined directly, while the CH2 units are separated by oligosaccharides. The CH1 is located past the "hinge" of the heavy chain and is joined to the CL unit by a disulfide bond.
Because antibodies can be produced with thousands of variations, the chance of producing an antibody that cannot fold properly or will otherwise not function properly is high. Folding is therefore a very important step in antibody production, and antibody production is highly dependent on the "quality control" mechanisms of the endoplasmic reticulum (ER). Heavy chains and light chains are synthesized separately and translocated into the ER during the translation process; they begin folding before translation is even complete.
The Ig fold is a highly conserved protein topology, with such a broad presence in nature that it gives the name to the Ig superfamily (IgSF), which consists of proteins that contain an Ig fold. The Ig fold consists of two antiparallel β-sheets, containing 7 to 9 β-strands in total, which form a sandwich-like structure. Typically, the Ig fold is stabilized by internal disulfide bridges that connect two of the β-strands and run perpendicular to the sheets themselves. The two variable regions and one constant region of IgG each contain an Ig fold. An important step in the formation of an Ig fold is proline isomerization. While most peptide bonds have a trans conformation, proline's cyclic structure means that it is only slightly less stable in a cis conformation and can exist that that conformation in nature. A proline residue exists between two of the β-strands in the Ig fold, and this residue's isomerization from trans to cis conformation is often the rate-limiting step in the formation of an Ig fold.
While the final structures are generally very similar, Ig domains can broadly be grouped into three categories based on their folding processes.
In the first category, domains fold autonomously, guided by the internal disulfide bridge, and the final state is a monomer. One major intermediate exists between the unfolded polypeptide and its native folded state. This intermediate persists for a noticeable amount of time because it contains a cis proline residue, while in the final, folded state this residue is trans, and the transformation from cis to trans is relatively slow. The central β-strands in the intermediate are almost completely folded, and small helices link two pairs of the strands, making this intermediate highly stable. The reason for the high stability in the intermediate is because the small helices act as an organizing center in the antibodies and they position the bulky hydrophobic molecules in the center of the protein. These helices are found in the most commonly in the constant domains and not in the variable domains or in immunoglobulin molecules that are prone to misfold, suggesting that these internal linkages are key to the folding of the antibody. Examples of protein domains in this category include the constant region of the light chain (CL) and the second constant region of the heavy chain (CH2) of IgG.
The second category begins folding similarly to the first: it forms a partially-folded monomer, with a trans proline residue, which then isomerizes to a cis residue. At this point, the domain is unable to finish folding until it dimerizes with itself. A representative of this domain is the third constant region of the heavy chain (CH3) of IgG. In fully-formed IgG, the two heavy chains are directly joined at this domain (as opposed to CH2, which are separated by sugars), so its folding by this process makes sense.
The third category is more distinct from the first two. The first constant region of the heavy chain (CH1) of IgG is incapable of folding autonomously, and must associate with already-folded CL, forming a dimeric intermediate. As in the other two categories, the folding domain contains a trans proline residue that must become cis before folding is complete. Unlike the other two processes, this isomerization cannot occur until the CH bonds to the CL. This causes CH1 folding to be the slowest of all IgG domains; it does not happen until the heavy and light chains have already come together.
A commonality in the folding of every antibody is a slow proline isomerization reaction, which is the conversion from the trans proline to a cis proline. This reaction has a very high activation energy (~80 kJ/mol) which makes this a very slow reaction. Because of this fact, the proline isomerization reaction acts as a rate-limiting step in the folding of an antibody. In each of these three categories, there exist very important transition states. The rate-limiting proline isomerization reaction allows for the reactions to proceed slower and for the transition states to be populated since the proline must be in the cis conformation in order to proceed in the folding.
B cells have been observed to go through a series of “quality checks” to in order to regulate the functionality of the antibodies being made. In the pre-B cell state, the folding of all the domains in the heavy chain is tested. After the heavy chain gene is rearranged, the pre-B cells produce IgMs, a short heavy chain with no light chains attached, bound to immunoglobulin heavy chain binding proteins (BiP). The BiP is bound to an unfolded CH1 domain, which is a part of the constant region in the heavy chain. A “surrogate” light chain is then produced from the variable domain of the pre-B cell. If the surrogate light chain induces the unbinding of the BiP with the IgM and the domain CH1 folds appropriately, then the heavy chain proceeds to the plasma membrane. If, however it fails this quality check, then the IgMs act as a substrate for endoplasmic reticulum associated degradation (ERAD). Thus the failure of this quality check leads to the degradation of the heavy chain. Then when the heavy chain is formed and a conventional light chain is formed, the same process as before takes place with the conventional light chains acting as the surrogate lights chains. The conventional light chains must induce the unbinding of the BiP from the CH1 domain and allow for folding in the CH1 domain of the heavy chains in order for the antibody to not undergo ERAD. Once the heavy chain and the light chain have gone through these two quality checks, disulfide bonds are formed between the heavy and light chains and the antibody is then ready for secretion.
Unlike most proteins, which can generally be said to exist in either a specific folded form or the denatured polypeptide form, antibodies are capable of forming some alternate structures under specific conditions. For example, below pH 3, antibodies can exist in a stable form unlike their typical one. While this is unlikely to directly cause any human health issues (a blood pH of 3 would be fatal long before misfolded antibodies caused any problems), it has ramifications for antibody production. Industrial antibody production includes steps that are carried out at low pH, which could affect the final product of the production.
Another, more direct problem that can come from alternative states of antibodies occurs when light chains or truncated heavy chains are secreted from B cells without forming a full antibody. These fragments can clump and become deposited in various organs, inhibited their function. The most common fatal complication from such deposition is light chain amyloidosis, in which monoclonal light chains are produced, secreted, and form deposits in the kidneys. The variable portion of the light chain is more likely to form amyloid deposits, possibly because the need for high variation in its structure make it more able to escape the cell without errors being detected.
Bind to foreign objects and prevent them from attacking normal cells
Can help get rid of pathogens with the aid of macrophages
Can directly damage pathogens by signaling the start of complement pathway, which is another immune response.
An example of antibody function would be in blood types. For instance, people with blood type A produce antibodies that recognize B antigens. If a person with blood type A was transfused with blood type B or blood type AB, the antibodies that recognize the B antigen on these blood cells would cause the person to begin clotting. This phenomenon explains why an individual of type AB blood can receive transfusion of type A or B blood however type A or B individuals cannot receive AB blood. This is the problem that usually occurs when blood donation is low and a match is necessary for medical purposes.
Five Major Classes of Antibodies:
1.) IgM :
a.This is a class of antibody that is produces after the initial exposure to antigen, but afterwards, its concentration in blood start to decrease. Immunoglobulin neutralizes the antigen and is responsible for the agglutination of the antigen. Because of this, it is very effective in complement activation.
b.IgM is a pentamer. It has µ heavy chains and exists as a pentamer in combination with another polypeptide called the J chain, which is responsible for initiating the polymerization to form the pentameric structure. With its large number of antigen-binding sites, each IgM molecule binds very tightly to any pathogen that has multiple copies of the same antigen on its surface. The binding induces the Fc region to activate the complement pathway which eventually causes the death of the pathogen. IgM also activates macrophages to phagocytose pathogens. Not surprisingly given these functions, IgM is the first antibody produced when an animal responds to a new antigen.
2.) IgG:
a.This class of antibody is present in tissue fluids. It is the most abundant class in bloodstream late in the primary immune response and particularly during the secondary immune response. IgG promotes neutralization, agglutination, and opsonization of the antigen, and it is also the only class of antibodies that can cross the placenta. It is secreted into the mother's milk and is taken up from the gut of the newborn animal into the bloodstream, thus it presents passive immunity to the fetus.
b.IgG is a monomer.
3.) IgA:
a.IgA presents the defense of mucous membranes by neutralizing the antigen, and also by agglutination. This antibody is present in bodily secretions such as saliva, tears, breast milk, mucus and in the secretions of the lungs and the intestine. IgA presents in breast milk also presents passive immunity on nursing infants.
b.IgA is a dimer.
4.) IgE:
a.Mast Cells, basophils of Histomine, and other chemicals that cause allergic reactions are released when IgE is triggered. IgE occurs in tissues where having bound the antigen. Some of these in turn activate white blood cells (called eosinophils) to kill various types of parasite. However, the mast cells can also release biologically active amines, including histamine, which cause dilation and increased permeability of blood vessels and lead to the symptoms seen in allergic reactions such as hay fever and asthma.
b.IgE is a monomer
5.) IgD:
a.In the antigen stimulated proliferation and differentiation of B Cells, IgD operates as the antigen receptor. This class of antigen exists on exterior of naïve B cells that have not been exposed to antigens.
b.IgD is a monomer.
Additionally, antibodies have proven to be remarkable because they can block specific protein synthesis within the body, while leaving human cells unharmed. This is due to the differences in ribosome structure found between bacteria and eukaryotic cells. The shapes of ribosomes in bacteria and humans have very specific differences that prove convenient when developing antibiotics meant to target just the bacterial ribosomes in order to halt their protein synthesis. If successful, antibiotics should bind and interfere with protein synthesis initiation complex formation in bacteria, or have some effect on the transcription of the bacteria's messenger RNA. Scientists trying to develop these antibiotics successfully take advantage of these structural differences in ribosomes, attempting to create ways to stop bacterial ribosomes from functioning without affecting the human in which they are located. For instance, mitochondrial cells contain ribosomes resembling those of bacteria, while the eurkaryotic cells hosting those mitochondria have much different ribosome structure. Thus antibiotics can target those bacterial-resembling ribosomes in the mitochondria without harming the eurkaryotic cell itself, which is merely one minor example that proves the remarkable role that structure and specificity can play in drug development.[1]
Antibodies target the functional sites of the virus at hand. A failure of the antibody to target the functional site will usually result in the failure of the antibody to combat the variation of the virus. If the functional site cannot be targeted, the second choice target should be directed at the host molecules (not virus-encoded molecules). The more chemically similar the antibody and the target's natural ligands are, the more effective the antibody will be.
Antibody therapeutics are a valuable form of treating disease due in part to their selectivity for a target protein. However, certain drawbacks exist due to certain diseases having several different mechanisms. Bispecific antibodies (BsAb) create an interesting possible alternative with greater benefits. These antibodies can effective target two different binding sites. Some benefits include limited possibility of escape from therapy, greater tumor targeting, and more efficient cytotoxic selectivity. The main drawback for BsAbs is the difficulty in production. New technology has emerged in antibody production through single-chain variable fragment diabodies, tandem diabodies, two-in-one antibody, and dual variable domain antibodies. Although, these techniques have issues with too frequent dosing or conjugation problems.
CovX-Bodies present a technology based on the aldolase catalytic antibodies. This makes it possible to quickly generate the needed antibodies with proper targeting. A CovX body consists of two covalently bound pharmacophores. They're attached to the lysine at position 93 within the Fab arms of the scaffold antibody. They are created through a process of mixing a branched azetidinone linker with a peptide pharmacophore heterodimer with the aldolase antibody. The peptide is responsible for functional activities and the antibody acts to improve half-life and distribution properties. These antibodies are capable of being created with specific binding affinity, potency and pharmacokinetics. The CovX-Body, CVX-241 is currently taking advantage of angiopoietin-2 (Ang2), and vascular endothelial growth factor (VEGF). This drug has shown impressive phamacokinetics in rodents and nonhuman primates. Xenograft models have shown potent efficacy. CVX-241 is completing phase-1 clinical trials.
↑National Institute of Health, "Inside the Cell", 2005, Pg.10
1. Buchner J, Feige M, Hendershot L. "How antibodies fold." Trends in Biochem. Sci.35 (4): 189-198.
2. Colman, Peter M. "New Antivirals and Drug Resistance", The Walter and Eliza Hall Institute of Medical Research
3. David Hames, Nigel Hooper. Biochemistry.3rd edition. Taylor and Francis Group, New York, 2005.
4. Venkata R. Doppalapudi, et al. "Chemical Generation of Bispecific Antibodies." http://www.pnas.org/content/107/52/22611.full#ref-list-1. Richard A. Lerner. November 9, 2010. CovX. November 30, 2011.
Epitope, or antigenic determinant, is a small, specific portion of an antigen recognized by the immune system such as antibodies. A single antigen usually has several different epitopes. The region on an antibody which recognizes the epitope is called a paratope. Antibodies fit precisely and bind to specific epitopes.
Epitopes exist as tertiary structures of amino acids and are not recognized by antibodies with the same specificity or affinity when denatured by pH or temperature. Immunoglobins which recognize the epitopes will also denature under similar conditions. There is a distinction in the naming of native epitopes and denatured linear epitopes. Epitopes on natural tertiary structures are named cryptotopes and that of linear sequences is called unfoldons. There are two different yet effective ways to study epitope and map their locations. These methods are x-ray crystallography and monoclonal antibodies. The immune response of an animal produces many different types of antibodies that recognize different epitopes with a range of affinity. These various antibodies are called polyclonal antibodies and are found in the serum taken from the blood. A monoclonal antibody is just one of these antibodies out of the many polyclonal antibodies, and it is this monoclonal antibody that is used to define specific epitopes.
Epitope Mapping
The challenges in mapping epitopes and identifying its location becomes challenging because different monoclonal antibodies will recognize different or similar epitopes. An epitope cannot exist without there being a corresponding antibody that recognizes it specifically. This definition makes it harder to specifically identify the range of any given epitope since monoclonal antibodies begin first and polyclonal antibodies all with different affinities and specificity. Some of these antibodies will have different leniency for amino acids and others will overlap in the epitope they recognize. It then becomes troublesome in deciding which polyclonal antibody should correspond to the specific epitope as many recognize the same sequence.
Often the means of the monoclonal antibody, or Mab, influences the definition of the epitope. Because Western Blotting’s SDS-PAGE partially denatures the protein this will affect the recognition of MAb to epitope. Other procedures that utilize native conformations such as liquid phase immunoassays and or frozen tissue samples will simulate in vivo affinities. This creates three artificial categories of Mab to epitope recognition. Monoclonal antibodies that recognize only partially denatured epitopes, those that recognize nativetertiary structure of epitopes, and those that recognize both.
Disulfide bridges that are distinctly visible can be isolated through the precise mapping of Epitope. However, epitopes can be recognized through various different monoclonal antibodies. The specificity and recognition of these features become more pronounced in the tissues. Monoclonal antibodies are rather selective in recognizing Epitope despite the structural subtle and differences that may arise in the conformational change of this protein. Despite the domain not having a concrete protein residue or subunit cavity, the structure can still be detected under x-ray crystallography.
X-ray crystallography is the most precise method in determining native tertiary contact between the epitope and monoclonal antibody. Challenges exist in defining how close the antibody must be to be in “contact” and even if the two are in contact it does not necessarily mean there is binding. Also x-ray crystallogoraphy is an expensive method and requires the antibody to be in crystal form. Methods that circumvent these challenges include NMR which can be used commonly but it sacrifices the precision of x-ray crystallography. NMR is hindered by the size of the antigen. If the antigen is too large then electron-microscopy can be used because it requires that the antigen be sizable.
References
Glen, Morris E. "Choosing a Method for Epitope Mapping." Methods in Molecular Biology. 1996. Epitope Mapping Protocols. 03 Dec. 2008 <http://springerlink.com/content/t5u6u667k23g1725/fulltext.html>.
Antigen is a macromolecule that causes an immune response by lymphocytes. Antigen receptor, a surface protein located on B cells and T cells, binds to antigens and initiates acquired immune responses. The antigen receptors on B cells are called B cell receptors (or membrane immunoglobulins) and the antigen receptors on T cells are called T cell receptors.
Antigens may either be proteins or polysaccharides. In general, an antigen is defined as a substance that binds to specific antibodies, which in the human body are used to find and neutralize any potentially harmful foreign substances in the bloodstream. The specific binding between antigen and antibody is similar to that of the lock-and-key binding model.
In human blood, the different lettering of different blood types is designated by the specific antigen present in the individual's blood cells. While all types contain the oligosaccharide (O) antigen, the A and B blood types are defined by having N-acetylgalactose (A) or galactose (B) monosaccharide. Likewise, the AB blood group has both A and B antigens. Additional antigens are bound to define the positive or negative state of the ABO blood groups. The structures of the enzymes that bind to the antigen are similar and very slightly different, demonstrating antigen specificity.
Besides being used in the indirect ELISA technique to detect the presence of antibody, antigens can also be used to prepare vaccines to establish or to improve immunity to a particular disease.
When pathogens get into the blood and lymph, antigens on the surfaces of the pathogens stimulate lymphocytes to produce specific antibodies which kill the pathogens by lysis, enhanced phagocytosis, clumping the pathogen together, or neutralizing the toxins from pathogens.
In wake of first (primary response) infection involving a particular antigen, the responding naive cells proliferate to produce a colony of cells, most of which transform into plasma cells or effector B cells (which produce the antibodies) to resolve the infection, and the rest persist as the memory cells that can survive for years, or indeed even for a lifetime. This is a complicated set of reactions that needs a latent period for the production of antibodies.
During the invasion of the same type of pathogens, however, previously produced memory cells trigger lymphocytes to produce much a larger amount of antibodies immediately. Unlike antibodies produced in the primary response that can only be maintained for a short period of time, the antibodies produced in the secondary response can be maintained for a longer period, usually for years.
Knowing these facts, vaccines are made to stimulate the production of memory cells to get one ready for the exposure to that kind of pathogens in the future.
Levels of antibodies produced during the primary response and secondary response
There are three types of antigen-based vaccines, namely, purified, recombinant, and synthetic.
Purified antigen vaccines, sometimes called subunit vaccines,composed small fragments of molecules purified directly from the pathogen that generates a "protective" immune response. These molecules can be proteins, polysaccharides or exotoxins (i.e. bacterial proteins either
chemically inactivated or attenuated (derived from mutated
organisms) to prevent toxicity in the host.)
Example: Vaccines against causative agents of meningitis in children
Recombinant antigen vaccines are immunogenic proteins produced by genetic engineering. DNA encoding for an immunogenic protein of a pathogen can be inserted into either bacteria, yeast, viruses which infect mammalian cells, or by transfection of mammalian cells. The cells will then produce the protein endogenously and the protein can
be harvested.
Example: surface protein of Hepatitis B virus (HBsAg)
Synthetic Antigen Vaccines are peptide antigens synthesized by automated machines. Synthetic polynucleotide technology also exists while synthetic polysaccharide technology is still under development. Which sequences to choose requires knowledge of the
conformational structures for B cell epitopes (sequential v.
assembled) and of the anchor residues of MHC for T cell
epitopes. Computer algorithms are available to assist in selection,
but trial-and-error approach is still required. Other aids include the
generation of "protective" monoclonal Abs (B cell epitopes & phage-display libraries) and the peptide-dependent restimulation of T cells from convalescent subjects (T cell epitopes).
Reference: Body Defense Mechanisms for A & H Levels, by Pang King Chee, HKASME
MHC, also known as the Major Histocompatibility Complex is attached to a host cell and it produces a protein that can basically present an antigen fragment to other cells (such as T cells), which will then take an appropriate action depending on what fragment is being presented. The interaction of the antigen fragment with the MHC molecule and T Cell receptor is an event that the acquired immunity (our immune system) uses in order to destroy pathogens that are not meant to be in the body.
The MHC is a large genomic region or gene family that is found in most vertebrates. It is the region that most densely contains the genes of the mammalian genome and is important for the success of the immune system, autoimmunity, and reproduction. Proteins that have been encoded by the MHC are expressed on the surface of cells in all jawed vertebrates and display both self and nonself antigens to a type of white blood cell known as a T cell that has the ability to kill or coordinate the killing of pathogens, infected, or malfunctioning cells.
Different forms of MHC proteins are present in different people.
This variation aids in the prevention of a widespread epidemic, since if all people had the same MHC proteins and a pathogen mutated to avoid being presented by MHC proteins, the entire population would be susceptible to the pathogen.
This variation is also why a "match" is needed for a successful organ transplant. That is, rejections of transplanted organs by the immune system are often caused by too much variation in the MHC genotypes of the organ donor and acceptor. The hereditary nature of MHC structure reveals why immediate family members are the best candidates for an organ donor.
The MHC region is divided into three subgroups: MHC class I, MHC class II, and MHC class III.
MHC class I is responsible for encoding heterodimeric peptide-binding proteins, as well as antigen-processing molecules such as TAP and Tapasin. MHC class I can be found in all nucleated cells. the MHC class I proteins contain an α chain and β2-micro-globulin. They present antigen fragments to cytotoxic T-cells that will bind to the CD8 on cytotoxic T-cells.
MHC class II is responsible for encoding hetrodimeric peptide-binding proteins and proteins that modulate antigen loading onto MHC class II proteins in the lysosomal compartment such as MHC II DM, MHC II DQ, MHC II DR, and MHC II DP. MHC class II can be found on antigen-presenting cells. MHC class II proteins contain α and β chains and they present antigen fragments to T-helper cells by binding to the CD4 receptor on the T-helper cells.
The MHC class III region is responsible for encoding for other immune components, such as complement components (C2, C4, factor B) and some that encode cytokines (TNF-α) and also hsp. Class III has a very different function than do class I and II, but it has a locus between the other two, so they are frequently discussed together.
The MHC proteins act as "signposts" that show fragmented pieces of an antigen on the host cell's surface. These antigens can be either self or nonself. If they are nonself, there are two ways by which the foreign protein can be processed and recognized as being "nonself." The first method is when the phagocytic cells, such as macrophages, neutrophils, and monocytes, degrade foreign particles that are engulfed during a process known as phagocytosis. Degraded particles are then presented on MHC class II molecules. The other method involves the host cell first being infected by a bacterium or virus, or being diagnosed as cancerous, then it may be able to display the antigens on its surface with a Class I MHC molecule. Cancerous cells and cells infected by a virus usually display unusual, nonself antigens on their surface. These nonself antigens, despite which type of MHC molecule they are displayed on, will initiate the specific immunity of the host's body. It is important to remember that cells constantly process endogenous proteins and display them within the context of MHC I. Immune effector cells are then built up to be non-reactive to self peptides within MHC, and are then able to recognize when foreign peptides are being presented during infection or cancer.
The most well known genes in the MHC region are the group that encodes cell surface antigen-presenting proteins. These genes are referred to as human leukocyte antigen genes in humans, though people often abbreviate MHC to mean the HLA gene products. Some of the biomedical literature uses HLA to refer to the HLA protein molecules and uses MHC for the region of the genome that encodes for this molecule, though this is not consistently adhered to. The most intensely studied HLA genes are the nine classical MHC genes: HLA-A, HLA-B, HLA-C. HLA-DPA1, HLA-DPB1, HLA-DQA1, HLA-DQB1, HLA-DRA, and HLA-DRB1. The MHC is divided into three regions: Class I, II, and III. The A, B, and C genes belong to the first class, whereas the six D genes belong to the second class. Besides being scrutinized by immunologists for its pivotal role in the immune system, the MHC has also attracted the attention of many evolutionary biologists, due to the high levels of allelic diversity found within many of its genes. Indeed, much theory has been spent to explaining why this specific region of the genome harbors so much diversity, especially in light of its immunological importance.
The classical MHC molecules have a vital roles in the complex immunological dialogue that must occur between T-cells and other cells of the body. At the time of maturity, MHC molecules are anchored in the cell membrane, where they display short polypeptides to T cells, via the T cell receptors (TCRs). The polypeptides may be "self," meaning they originate from a protein created by the organism itself, or they may be foreign, "nonself," where they originate from bacteria, virus, pollen, etc. The overarching design of the MHC-TCR interaction is that T-cells should ignore self peptides while reacting appropriately to the foreign peptides. The immune system has another and equally important method for identifying an antigen. B-cells with their membrane-bound antibodies, also known as B-cell receptors (BCRs). BCRs of B-cells do not require much outside help in order to bind to antigens while TCRs of T-cells require "presentation" of the antigen, which is the job of the MHC. During the vast majority of the time, MHC are kept busy presenting self-peptides, which the T-cells should appropriately ignore. All MHC molecules receive polypeptides from inside the cells they are part of and display them on the cell's exterior surface for recognition by T-cells.
MHC gene families are found in almost all vertebrates, though the gene composition and genomic arrangement vary widely. Gene duplication is almost certainly responsible for much of the genetic diversity. MHC is littered with many pseudogenes in humans. One of the most striking features of the MHC is the astounding allelic diversity found therein, and especially among the nine classical genes. The most conspicuously diverse loci, HLA-A, HLA-B, and HLA-DRB1, have roughly 250, 500, and 300 known alleles respectively, which is a truly exceptional exemplification of diversity. The MHC gene is the most polymorphic in the genome. Population surveys of the other classical loci routinely find tens to a hundred alleles, which is still relatively high in diversity. Many of these alleles are quite old, too. It is often the case that an allele from a particular HLA gene is more closely related to an allele found in chimpanzees than it is to another human allele from the same gene. The allelic diversity of MHC genes has created fertile grounds for evolutionary biologists. The most important task for theoreticians is to explain the evolutionary forces that have created and maintained such diversity. Most explanations invoke balancing selection, a broad term that identifies any kind of natural selection in which no single allele is absolutely most fit. Frequency-dependent selection and heterozygote advantage are two types of balancing selection that have been suggested to explain MHC allelic diversity. However, recent models have suggested that a high number of alleles is not plausibly achievable through heterozygote advantage alone. Pathogenic co-evolution, a counter-hypothesis, has recently emerged. It theorizes that the most common alleles will be placed under the greatest pathogenic pressure, thus there will always be a tendency for the least common alleles to be positively selected for. This creates a "moving target" for pathogen evolution. As the pathogenic pressure decreases on the previously common alleles, their concentrations in the population will stabilize, and they will usually not go extinct if the population is large enough, and a large number of alleles will remain in the population as a whole. This explains the high degree of MHC polymorphism found in the population, although an individual can have a maximum of 18 MHC I or II alleles.
It has been suggested that MHC plays a role in the selection of potential mates. MHC genes make molecules that enable the immune system to recognize invaders. Generally, the more diverse the MHC genes of the parents, the stronger the immune system of the offspring. It would obviously be beneficial, therefore, to have evolved systems of recognizing individuals with different MHC genes and preferentially selecting them to breed with.
The Class I MHC molecules are found on all nucleated cells in the body (including cells expressing Class II MHC such as antigen presenting cells, dendritic cells, macrophages, and B cells), but are not found on non-nucleated cells such as red blood cells. It is found extending from the plasma membrane of body cells that have been infected with a virus or are otherwise abnormal (for example, cancerous). MHC Class I proteins bind to foreign antigen fragments (peptides) that are derived from intracellular proteins. For example, any cell that can become infected with a form of cancer will synthesize foreign antigens and then present them on the surface of the cell by the MHC I molecule. The T cell receptor of a Cytotoxic T cell that recognizes the MHC I-peptide complex binds to it with the aid of CD8, a protein found on the surface of cytotoxic T cells. This activates the cytotoxic T cell, stimulating its release of perforin, which increases permeability of the target cell membrane, and granzymes, which enter the target cell and initiate apoptosis, killing the target cell and fragmenting both its DNA and that of the virus. The cytotoxic T cell then detaches from the MHC I molecule and proliferates
forming active cytotoxic T cells, which attack other infected body cells expressing the same antigen fragment, and memory cytotoxic T cells which will help produce a faster response to the virus if it attacks again.
Properly functioning MHC I molecules are essential in fighting cancer. That is, the immune cells of a person with defective MHC I molecules would not be able to recognize that cells had become cancerous, so no immune response would take place.
The interaction of T cells with antigen-presenting cells 1 A fragment of foreign protein (antigen) inside the cell associates with the components of an MHC molecule on the endoplasmic reticulum and is transported to the cell surface 2 The combination of MHC molecule and antigen fragment is recognized by a cytotoxic T cell 3 Cytotoxic T cells kill the infected cell.
File:MHC1.gifThe crystal structure of 2DD MHC class I in complex with the HIV-1 derived peptide P18-110.
The Class II MHC molecules are made by antigen presenting cells (dendritic cells, macrophages, and B cells) which help trigger an acquired immune response. Antigen presenting cells ingest foreign antigens via phagocytosis or endocytosis and break it down into peptide fragments that bind to MHC Class II molecules which bring the antigen fragment to the surface of the cell to be recognized by the T cell receptor on the surface of Helper T Cells. The T cell receptor binds to the MHC II-antigen fragment complex with the aid of the CD4, a protein receptor on the surface of helper T cells, and once bound, the helper T cell becomes active and begins proliferating and secreting cytokines.
When Helper T cells proliferate, both memory helper T cells, and to a larger extent, active helper T cells are made. Memory helper T cells contribute to the acquired immune response by quickly proliferating in the case of a secondary "attack" by the antigen producing a faster, stronger response to the antigen. Active helper T cells have a shorter life span than memory helper T cells and help initiate an immune defense against the pathogen by secreting cytokines that stimulate the activation of nearby B cells and Cytotoxic T cells. Once activated, cytotoxic T cells destroy infected cells via apoptosis while the plasma B cells secrete specific antibodies that neutralize and eliminate the pathogen. The cytotoxic T cells and B cells will also proliferate to produce their respective memory cells for use in a second attack by the pathogen.
In the case of HIV, the Helper T cells are the cells that are directly attacked by the virus. They are completely killed off, which means that no more Helper T cells are able to create cytokines and call the other phagocytic cells to reproduce and create an immune response. This causes the death of all Helper T cells, as well as the death of the immune system. Because of this, the person that has HIV/AIDS does not die of the virus, but can die of (i.e.) a common cold, because the immune system has no way to respond to it.
An abzyme is an antibody that expresses catalytic activity [1]. A single molecule of an antibody-enzyme, or abzyme, is capable of catalyzing the destruction of thousands of target molecules [1]. The efficiency of abzyme technology could permit treatments with smaller doses of medicines at lower costs than are possible today. An abzyme is used to lower the activation energy of a reaction allowing for the transition state to be possible and the product to be formed. Abzymes are typically artificially made by having the immune system make antibodies that bind to a molecule that resembles the transition state (Transition State Analogue) of the catalytic process that the researchers want to emulate. Therefore by creating this antibody, now becoming a catalytic antibody allows for this antibody to act as an abzyme reducing the activation energy of the reaction and allowing for the transition state to occur. Abzymes however do occur naturally in the human body.
Abzyme are currently being researched for the possible use against HIV infection. The abzymes could target a specific site on the HIV infected cells that do not mutate and then make the virus inert. This is an on going research project by the University of Texas Medical School.By exploiting the highly specific antigen binding properties of antibodies, experimental strategies have been made to produce antibodies to catalyze that chemical reactions. These abzymes are chosen from monoclonal antibodies which are created by immunizing mice with haptens which mimic the transition states of enzyme-catalyzed reactions. The rate of this reaction is promoted by enzyme catalysts that stabilize the transition state of this reaction, thereby decreasing the activation energy and allowing for more rapid conversions of substrate product [1]. To successfully create abzymes that are complementary in structure to this transition state, mice were immunized with an aminophosphonic acid hapten [1]. The study of catalytic antibodies as a whole has vastly increased current understanding of the mechanisms of enzyme catalysis and represents another step forward in the attempts to create artificially engineered biological enzymes [1].
Myosins are a large super-family of motor proteins that move along actin filaments, while hydrolyzing ATP to forms of mechanical energy that can be used for a variety of functions such as muscle movement and contraction. About 20 classes of myosin have been distinguished on the basis of the sequence of amino acids in their ATP-hydrolyzing motor domains. The different classes of myosin also differ in structure of their tail domains. Tail domains have various functions in different myosin classes, including dimerization and other protein-protein interactions.
Myosin is a common protein found in the muscles which are responsible for making the muscle contract and relax. It is a large, asymmetric molecule, and has one long tail as well as two globular heads. If dissociated, it will dissociate into six polypeptide chains. Two of them are heavy chains which are wrapped around each other to form a double helical structure, and the other four are light chains. One main characteristic of myosin is its ability to bind very specifically with actin. When myosin and actin are combined together, that makes the muscle produce force.
Skeletal muscles are responsible for voluntary movement. Skeletal muscles contain many muscle fibers and these muscle fibers are actually made up of myofibrils, bundles of thick myosin filaments and thin actin filaments. Myofibrils are constructed and lined up in a chain-like formation to create what are called sarcomeres. Sarcomeres contain several regions. One region is called the A-bands and only consist of myosin filaments. The counterpart of A-bands is the I-bands that only contain actin filaments. The ends of each sarcomere are called Z discs. A middle region of each sarcomere called the H-zone only contains myosin.
According to the sliding filament theory by Andrew Huxley and Ralph Niedergerke, muscles contract when Z-discs come closer together thus shortening the sarcomeres. Actin filaments from the I-bands become very short while myosin filaments from the A-bands do not change in length. The actin filaments are actually sliding towards the H-zone and the A-bands thus creating an overlap of myosin and actin filaments. As this overlap occurs, myosin filaments are binding to the actin filaments, allowing myosin to function as the driving motor of filament sliding.
This relative movement between myosin and actin is what results in muscle contraction. The molecular basis for muscle action and contraction is explained in the next section.
This mechanism of contraction is also called "The Sliding Filament Theory."
ATP binding to the myosin head causes and it is in its low-energy conformation
The active site closes and ATP is hydrolyzed to ADP and Pi. This induces a conformational change (cocking of the head) resulting in myosin weakly binding to actin. This forms a cross-bridge.
Pi release results in conformational change that leads to stronger myosin binding, and the power stroke.
ADP dissociation leaves the myosin head tightly bound to actin.
Binding of a new molecule of ATP to myosin head triggers it to let go of actin and the cycle starts all over again.
In the absence of ATP, this state results in muscle rigidity called rigor mortis.
Myosin has groups of protein that divide the motor proteins. The motor proteins are involving actin filaments that hydrolyze ATP. There are 20 different types of Myosin that already distinguished by amino acid sequence.
All 20 types of Myosin have different structure following by tail domain. Because Each classes have characteristic of dimerization, and protein interactions. However, there are known classes in the Myosin. Myosin I, Myosin II, Myosin V and VI.
Myosin I
Among the proteins whose genes have been linked to deafness are several types of myosin. Myosin I appears to cross-link actin filaments to control the tension inside each stereocilium. The ratcheting activity of this myosin motor along the actin filaments may adjust the sensitivity of the hair cells to different sounds. Other types of myosin use their motor
activity to redistribute cellular constituents along the length of the actin filaments.
Myosin II
MyosinII consists of six polypeptide chains: two 220-kD heavy chains and two pairs of different light chain that vary in size between 15 and 22kD, depending on their source. The N-terminal half of each individual heavy chain assumes a globular form that is stretched in one direction. Coming up next is a roughly 100 Angstrom long alpha helix stiffened by the two light chains wrapping around it. This portion of the protein acts as a lever when the muscle contracts. The C-terminal half of the heavy chain takes the form of alpha-helix that ends as a long, fibrous chain. Two of those associate and takes the form a left-handed coiled-coil motif. The overall shape of myosin is a rod 1600 Angstroms long with two globular heads.
Myosin V
MyosinV has a different structure of motor. It has a two headed motor protein which heavy chains diverge. That means actin dependent transport move to axon associated vesicle effect on a melanin. Both microtubule and actin filaments lead to the speculation and affect to the hair color. Myosin V is also a two-headed protein, but it doesn't form a thick filament like Myosin II. Myosin V acts by itself - the domain at the tail-end binds a vesicle that has pigments as its cargo. The lever region of this protein is long enough to have six light chains bound to it, giving it three times greater capacity for those light chains than Myosin II's counterpart lever arm. Under electron microscope (EM) image of Myosin V bound to F-actin filament, it is estimated that the globular heads have thirteen actin subunits between them.
Actin is the most abundant protein found in eukaryotic cells. It is a monomeric unit of microfilaments (actin filaments). The globular actin is often called G-actin. It contains a nucleotide-binding site, which can bind to ATP or ADP. The conformation of actin depends on the ATP or ADP in the nucleotide-binding site.
Actin filament is often called F-actin. It is twisted helical chains of actins, which the actin monomers orient in the same direction of actin filament. It has polarity that contains different ends in its structure. One end is called barbed (+) while the other end is called pointed (-). They are called by the appearance when myosin S1 fragments are bound to it. An actin has a myosin binding site at every 2.7 nm. Actin filament occur as linear filament and also form structural networks, which plays a major role in muscle contraction, cell movement, cell signaling, cytokinesis, and cell division.
The structure of actin monomer was observed to atomic resolution through x-ray crystallography, which scientists have been determined that the structure of actin as filaments. As actin monomers (G-actin) bind together, they form actin filament (F-actin), which has a helical structure. Each monomer has a distance of 27.5pm with a rotation of 166 degrees around the helical axis. Each actin monomer is orientated in the same direction along the helical structure (F-actin), which makes up a polar character of the structure. One end of the helical structure is called the "barbed" end (+), and the other end is called the "pointed" end (-). Actin filaments are self-assembled, which actin monomers come together as very well-structured, polar helical. Since the aggregation of the first two or three monomers to form actin filament is highly unfavorable, specialized protein complexes, such as Arp2/3, would be required to serve as nuclei for actin assembly in cells. Once the first filament nucleus exists, the addition of subunits is more favorable.
A skeletal muscle fiber only moves when it is stimulated. Otherwise at rest, the binding sites are blocked. Actin contains two types of regulatory proteins that modulate the binding site. The first type is tropomyosin, a protein chain that lies along actin and covers the binding sites. Troponin C is attached to tropomyosin and directs the position of tropomyosin on actin. Once a Troponin C binds to calcium, it pulls the tropomyosin to unwrap the binding sites. The exposed binding site allow for myosin to bind to actin. Once myosin binds to actin, it forms a "cross bridge" and is called a rigor complex.
Actin-ATP can polymerize to form actin filament. It is more readily to polymerize than actin-ADP, because actin polymerization occurs when bound ATP in actin is hydrolyzed to ADP.
Actin filament is self-assembled spontaneously, but formins can help the formation. Formin binds to the barbed(+) end of actin filament, and actin monomers can be added on it until a plus-end capping protein binds to the barbed end. This mechanism stabilizes the polymerization and consequently produce a linear unbranched filament due its FH2 domain.
Branched actin networks can form from polymerization with Arp2/3 complexes. Arp2/3 complex is a seven-subunit protein that includes two actin-related proteins, Arp2 and Arp3, and five other smaller proteins. Nucleation of Arp2/3 complex associated with an activator such as WASp can start the actin polymerization. It binds to the side of an actin filament and nucleates to initiate growing of a new Y-shaped filament branch at a 70° angle. In this formation, the structures of two subunits Arp2 and Arp3 are similar to actin. These two subunits are then triggered by the binding of activator to mimic the barbed end of a filament.
Unbranched actin filaments are produced by spire that has WH2 domains each bind to an actin monomer to complete the nucleation. The mechanism associated with spire is totally different from those with formin and Arp2/3 complex. By organizing actin monomers into a prenucleation complex, spire makes template for filmation formation.
Consider the polymerization reaction in detail. Assume an actin filment with n subunits An. This filment can bind an additional actin monomer, A, to form An+1. The given equation is the following:
Kd=[An]*[A]/[An+1]]
Kd is the dissociation constant, which defines the equal concentration of polymers of length n+1 and for the polymers of length n. Therefore, the polymerization reaction will proceed until the monomer concentration is reduced to the value of Kd.
Some major roles of actin include:
(1) Being the structural makeup and support of the cytoskeleton.
(2) Dividing and producing cells in order to enable cells to move spontaneously and actively.
(3) Serving as a supportive framework for myosin proteins during muscle contraction.
(4) Acting as a track for the cargo transport myosins in non-muscle cells.
β- and γ- actin Proteins
The mammalian cytoskeleton proteins β- and γ- actin have amino acid sequences that are extremely similar, yet they both have significantly different functions in the cell. β-actin proteins are partly responsible for the cell mobility by pushing the cell forward, whereas γ-actin proteins promote cell adhesion. The β-actin protein structure changes when arginine is added, while γ-actin structure doesn’t change. Scientists have been trying to distinguish between the two proteins from their form and function. They found that slow translation of γ-actin leads to quick degradation by the proteasome in the cell because both arginylation and ubiquitination are allowed. Ubiquitination is a protein post-translation modification (PTM) process which results in labeling of the proteins so that they are sent to the proteasome to be destroyed. On the other hand, the fast translation of β-actin permits only arginylation and stabilizes the protein. γ-actin is less stable than β-actin due to the occurrences of many codons in its gene that slows down the rate of translation. β-actin proteins use different codons that code for the same amino acids. Lysine is an important amino acid found in both β-actin and γ-actin proteins. Researchers found that slowing the rate of translation experimentally allows for the process of ubiquitination by revealing the Lysine to ubiquitin and leads to the quick degradation of the γ-actin proteins.
The powerstroke of actomyosin is linked through three events which lead to the release of products from ATP hydrolysis (inorganic phosphate and ADP): myosin head binding to actin, structural changes in the head causing strong actomyosin interaction, and the swinging of the lever. The study of ATP hydrolysis-linked enzymatic force generation is difficult to perform because efficient force generation requires the powerstroke to occur while myosin is bound to actin. And this process can only begin when myosin is in a low actin-affinity state, so it is quite rare to observe this occurrence.
Myosin has three different parts, a motor domain, the lever and the tail region. The motor domain is what swings the lever during powerstroke of actomyosin, it has three main parts: the nucleotide pocket, the actin-binding region and the relay region. Three loops: P-loop, Switch 1 and Switch 2 are attached to the nucleotide pocket and face the actin-binding and relay region. Weak interactions with actin is begun in the lower part of the actin-binding region, then when the cleft closes, the upper part of the actin-binding region folds over the actin and produces stronger binding interactions. The relay region interprets the conformation of the now folded actin-binding region and swings the lever from the primed “up” position down, the distance traveled by the lever determines the size of the powerstroke.
Kinetics block the ‘futile’ lever swing in an actin-detached state which leads to an ATP-wasting cycle. The ATP binds to myosin rapidly following a quick conformational equilibrium between down-lever and up-lever states (also known as the recovery step); this is followed by the hydrolysis of ATP. ATP can only be hyrdolized by myosin in the up-lever state. When myosin bings to ADP and P, it results in weaker interactions and the release of the P reduces the complexes stability and is rate-limiting in the absence of actin.; this is contradictory to previously thought rate-limiting step: release of inorganic phosphate. Release of inorganic phosphate is only possible during the down-lever state. In the absence of actin, myosin is mostly in the ADP and Pi bound up-state.
Over the past couple decades; many myosin conformations have been identified via crystallization process which teaches us about the allosteric communication pathways between the actin-binding region and the lever region during powerstroke. Experiments have revealed that energy barriers in myosin enzymatic steps, nucleotide binding, ADP release and conformational changes directly depends on the actions of the lever, meaning that the lever controls energy in the myosin complex during powerstroke.
The actin affinity is determined by the nucleotide content of the active site allosterically. Nucleotide-free and ADP-bound forms of myosin have been found to strongly bind actin, but in complexes where the gamma-phosphate sites are occupied with ATP or ADP-Pi, weak actin affinity is found. This is due to the allosteric coupling between the actin-binding region and the nucleotide pocket which is in the more distant regions of the motor domain. The actin affinity is determined by the conformation of the actin-binding region. The affinity depends primarily on the equilibrium of the switch 1 loop of the nucleotide pocket, which can have an open or closed conformation. The actomyosin powerstroke is initiated by myosin at low actin affinity.
An effective powerstroke stems from the pathway of actin-induced acceleration of the lever swing. The lever swing of ADP-Pi-bound myosin is accelerated by actin by over two orders of magnitude. Therefore actin activation is a crucial part in an effective powerstroke, despite the fact that it starts in a weak actin-affinity, or ADP-Pi, state. The reaction flux is brought into the kinetic pathway involving the lever swing cause by the powerstroke. The reaction flux is then brought towards the actin attachment after the futile lever swing is kinetically blocked. This however is not thermodynamically favorable but this non-equilibrium situation is necessary because this pathway has higher free-energy. This is known a kinetic pathway selection and is used to force a reaction through a more efficient pathway rather than a futile one that would be thermodynamically stable.
Another effective powerstroke pathway also begins with a weak actin attachment to a actomyosin complex. But an opening and closing of the actin-binding region, as opposed to just, is what causes the lever swing. In another method, the powerstroke might begin right after the weak binding of the lower actin-region on the myosin. Both of these alternative reaction pathways will result a reaction flux much like the original one described above. This shows that the reaction flux will also undergo kinetic pathway selection, something that scientists have began studying recently in detail to determine how important it is in physiological function.
Berg, Jeremy; "Biochemistry", W.H. Freeman and Company, New York, 2007, Sixth Ed.
Kashina, Anna, Saha, Sougata, Shabalina, Svetlana A., Zhang, Fangliang. "Differential Arginylation of Actin Isoforms Is Regulated by Coding Sequence- Dependent Degradation." Science 17 September 2010: 1534-1537
Málnási-Csizmadia, A. “Emerging complex pathways of the actomyosin powerstroke” Trends in Biochemical Sciences, Volume 35, Issue 12, 684-690, 31 August 2010
Insulin is a hormone secreted by the pancreas that regulates glucose levels in the blood. Without insulin, cells cannot use the energy from glucose to carry out functions within the body. Insulin was first discovered in 1921 by Frederick Grant Banting and Charles Best from extracted substances from the pancreas of dogs in their laboratory. The material was then used to keep diabetic dogs alive, and then used in 1922 on a 14 year old diabetic boy. The FDA approved insulin in 1939. In 1966 insulin was synthesized by Michael Katsoyannis in his laboratory, which marked the first complete hormone to be successfully synthesized. Synthetic insulin is used as a drug to treat diabetes, and the current forms on the market include insulin from bovine and porcine pancreases, but the most widely used is a form made from recombinant human insulin.
Insulin is made in the pancreas by beta cells. After the body takes in food, these beta cells release insulin, which enables cells in the liver, muscles and fat tissues to take up glucose and either store it as glycogen or allow blood to transfer it to organs in the body for use as an energy source. This process stops the use of fat as a source of energy. When glucose levels are elevated in the blood, insulin is produced at higher rates by the pancreas in order to maintain normal sugar concentrations in the blood. Without insulin, the body cannot process glucose effectively and glucose begins to build up in the blood stream instead of being transported to different cells . In contrast with elevated levels of glucose in the blood, when there is a deficit of glucose available to the body, alpha cells in the pancreas release glucagon, a hormone that causes the liver to convert stored glycogen into usable glucose which is then released into the bloodstream.
Some of the effects of the insulin on the metabolism include:
1. Controlling cell intake of substances like glucose in many organs like muscles and adipose tissues.
2. Controlling amino acid uptake, thus increasing DNA replication and protein synthesis
3. Altering the activity of enzymatic cells
Other Cellular effects of insulin include:
1. Increasing synthesis of glycogen. Glycogen is a type of storage for glucose and is stored in the liver. Levels of blood glucose determine whether glucose is stored as glycogen or is excreted. Low levels of glucose cause the liver to excrete glucose, while higher levels of glucose allows glucose to be stored as glycogen.
2. Increasing the synthesis and esterification of fatty acids. This is caused by the insulin causing fat cells to convert blood lipids to triglycerides. Esterification is caused when the insulin causes the adipose tissue to convert fats from fatty acid esters.
3. Increasing the esterification of fatty
4. Decreasing protein breakdown (proteolysis)
5. Reducing lipolysis
6. Increasing uptake of substances like amino acid and potassium
7. Relaxing wall of arteries of muscles, which vasodilation
8. Increasing secretion of HCl into the stomach
Insulin is a hormone consisting of 2 polypeptide chains. Each chain is composed of a specific sequence of amino acid residues connected by peptide bonds. In humans, chain A has 21 amino acids, and chain B has 30. Post translational modifications result in the connection of these two chains by disulfide bridges. Cysteine residues on A7 and B7, as well as A20 to B19 are covalently connected by disulfide bridges. Chain A also has an internal disulfide bridge connecting A6 to A11. The 3D structure of insulin is composed of 3 helices and the three disulfide bridges. Hydrophobic amino acid residues are clustered on the inside of the molecule while the polar amino acids residues are located on the outer surface. This arrangement of amino acid residues lends stability to the overall molecule. A single molecule of insulin can form a dimer with another insulin molecule, but the most active form is a single unit. The chemical formula for the insulin monomer is: C256H381N65O79S6.
Insulin Synthesis Using Recombinant DNA Technology.
Insulin production takes place in the pancreas, however diabetics lack the capability to produce insulin, so insulin derived by synthetic means is required to maintain normal blood glucose levels. Bovine and porcine insulin is similar to human insulin, however insulin synthesized from these sources can have adverse affects when used to treat diabetic patients due to possible long term effects from the continual injection of a foreign substance into the body. As a result of these possible adverse effects, in 1977, researches at the Genentech corporation developed means to reproduce insulin derived from humans through recombinant DNA technology. The steps involved in cloning human insulin begin with extracting proinsulin mRNA from the pancreas of a human with a functioning pancreas. Next, the enzyme reverse transcriptase is used to synthesize a strand of DNA that is complementary to the proinsulin mRNA. This DNA complement is called cDNA. The cDNA and RNA strands form a double helix hybrid. Next, the RNA is hydrolyzed off by raising the pH, and the DNA strand complementary to the original cDNA strand is formed with the help of an enzyme called terminal transferase. Restriction enzymes can be used to cut the gene and isolate just the sequence that encodes for the insulin protein. Next, circular units of DNA, called plasmids, are extracted from E. coli bacteria cells and cut with the same restriction enzyme that was used to cut the human chromosome. Using the same restriction enzymes creates complementary ends on the plasmid and the insulin gene. Next, the insulin gene is inserted into the plasmid at the proper location and the enzyme DNA ligase is used to form the phosphodiester bonds between the insulin gene and the plasmid. This step essentially “glues” the insulin gene into the E. coli plasmid vector. A certain type of plasmid called an “expression vector” is used in this process, which contains a bacterial promoter that facilitates the formation mRNA. Once the insulin genes are ligated into the vector, the vector is inserted into a bacterial cell. The bacterial cell acts as a host for the translation process of mRNA to protein. These host cells are harvested and allowed to reproduce, which creates a colony of insulin producing bacterial cells. The insulin can then be purified and packaged.
Another possible method to synthesize insulin was recently proposed by a pharmaceutical company SynBioSys in 2006 which used safflower to produce insulin rather than bacteria. Their goal ultimately was to reduce economic costs by exceeding its target and achieving accumulation levels of 1.2 percent of total seed protein. The company claims that this breakthrough in plant-produced insulin have the potential to "fundamentally transform the economics and scale of insulin production." The company announces safflower produced insulin can be up to 60% less expensive than insulin manufactured through bacterial cells.
The clinical testing trials were promising:
SBS-1000 was bioequivalent to most common brand name insulin medications.
SBS-1000 in humans showed no difference in metabolizing bacteria based insulin rather than through safflower.
SBS-1000 was well tolerated at pharmacologically active dosages.
Insulin is released in the body by the Beta cells in the islets of Langerhans. This is done in two phases, which includes a response in a change in blood glucose level and another type of release which is slower and is independent of sugar.
Insulin released by a change in blood glucose level starts when glucose enters glycolysis and the respiratory cycle. During this cycle, ATP is produced by oxidation, and thus the level of ATP produced is representative of the blood glucose level. When the amount of ATP produced gets to a certain point, potassium channels that are activated by ATP close, depolarizing the cell membrane, leading to a change in other voltage activated channels, such as the calcium channels. Due to the depolarization, voltage gated calcium channels open, allowing an influx of calcium ions into the cell. The increased level of calcium in the cell activates phospholipase C. Phospholipase C cleaves the membrane phospholipid phosphatidyl inositol 4,5-bisphosphate, which in turn becomes inositol 1,4,5-triphosphate and diacylglycerol.
The newly formed inositol 1,4,5-triphosphate (also known as IP3) binds to receptors on IP3 gated channels embedded on the membrane of the endoplasmic reticulum. The IP3 gated channels allow an influx of calcium ions in the cell, repolarizing the cell. Insulin, which was synthesized prior to this reaction, is stored in secretory vesicles, and is waiting to be released. The increased levels of calcium due to the binding of IP3 causes the release of the insulin from these vesicles.
The beta cells of the islets of Langerhan regulates the glucose level by this reaction. When the blood glucose level is physiologically normal, the beta cells cease to secrete any more insulin. This is done by the sympathetic nervous system, by the release of the hormon norepinephrine.
When blood glucose level drops, hyperglycemic hormones (glucagon) are released by the alpha cells of the Islets of Langerhans. This causes glucose to be released into the blood from storage within the body, and most mainly comes from the liver. The glucose is stored as glycogen within the liver.
Insulin has two routes of degradation after it has attached to the receptor site on the cell membrane. (1) It may be released into the extracellular environment or (2) it may be degraded by the cell. If insulin is to be degraded by the cell, the insulin-receptor complex is brought into the intracellular area via endocytosis. Subsequently, insulin-degrading enzymes break down the molecule. Insulin is degraded primarily in the liver and the kidneys. The liver is responsible for degrading insulin that is in the bloodstream for the first time; the kidneys are responsible for degrading insulin that is in normal circulation. Natural, endogenously produced insulin is estimated to be degraded within one hour after its initial release into circulation by the pancreatic beta cells. The half life of insulin is approximated to be 4-6 minutes.
Diabetes is a condition which the body either cannot produce insulin or does not respond properly to insulin. There are two types of diabetes. Type 1 diabetes is when insulin is not produced by the body. This is due to an autoimmune condition, in which body attacks the Beta cells of the Islets of Langerhans. This can be treated by insulin injections. Type 2 diabetes is when there is a resistance to insulin, reduction in production of insulin, or both.
Insulin is used to maintain a balance of glucose levels in a body’s bloodstream. After a meal, digestion of the carbohydrates occurs and enters the blood as glucose to provide the body with energy. To maintain a body’s blood sugar level, excess sugar is stored in the liver and is released once the blood sugar level starts to become low. This is where diabetes occurs; when the glucose is unable to enter the cells from the pancreas when the sugar levels are too low. If this disease is left untreated, complications, such as blindness and damage to the kidneys can occur.
1) Intravenous feeding solutions: One of the active ingredients in IV solutions used for feeding the body in hospital patients is insulin. The presence of insulin in the boyd helps improve the adsorption of nutrients and when combined with growth hormones, can help reverse negative protein balance.
2) Intravenous GIK solution: Glucose, insulin and potassium solutions have been used to reduce the mortality rate of acute mycoradial infractions, or otherwise known as heart attacks, along with postoperative cardiac failure. By addition of GIK infusions, it is a quick way to infuse potassium into all the cells of the heart even when circulation has ceased completely to readily restore action potential in cardial muscle to induce contraction.
3) Dialysis shock recovery: A bit insulin can be added to an electrolyte solution that can help patients absorb electrolytes quickly to recover from a dialysis shock.
4) Sports Drinks and Oral Rehydration Solutions: The sugars found in these drinks, such as Gatorade, allow for increased secretion of insulin in the body which speeds up absorption of water and electrolytes into all the cells of the body.
There exists a variety of interactions between proteins and DNA that are necessary for biological processes, in which proteins must recognize specific sections of DNA. These interactions can be categorized by what the proteins use to recognize and interact with in DNA. Proteins use a combination of these interactions in order to achieve specificity in DNA binding.
It was previously thought, based on early low-resolution x-ray structures, that the set of nucleic acids present in the major groove of DNA helices presented a set of bases that correspond with a complementary sequence of amino acids for the sake of recognition specificity. This theory of recognition is referred to as direct readout. Although this mechanism of recognition is common and provides a significant amount of what is in the Protein Data Bank, it has been realized that simple one-to-one correspondence between codes is insufficient in recognizing the specificity of the protein-DNA interactions. In some cases of DNA recognition, interactions of the protein and the DNA strand are less direct, and the interactions are not likely to occur if not for some sort of deformation of the DNA helix conformation. These interactions are defined as indirect readout mechanisms.
The two main categories are base readout and shape readout. Base readout is when the protein recognizes the specific chemical signatures of different nucleic acid bases. Shape readout is the recognition of the shape of DNA sequences.
Base readout can be further categorized into readouts that occur in the major groove versus those that occur in the minor groove. Hydrogen bonding is one mechanism of DNA recognition by proteins; it is a greater source of specificity in the major groove as compared to the minor groove due to the pattern of hydrogen bond donors and acceptors available. In the minor groove, the hydrogen donor/acceptor patterns do not distinguish A:T from T:A and G:C from C:G. Specificity based on hydrogen bonds is based both on the number of donor-acceptor pairs and the unique hydrogen bonding geometry. When A:T bind together they make two hydrogen bonds. When G:C bind together they make three hydrogen bonds. Hydrogen bonding has also been noted to be mediated by water molecules; for example, in the Trp repressor enzyme water molecules are found to bridge hydrogen bonds. However, this type of water-mediated hydrogen bonding mechanism of recognition has only been found in the case of major groove readout, not in cases of minor groove readout.
Displacement of water molecules from the minor groove may also be used as a thermodynamic driving force for the binding of DNA. Hydrophobic effects may also be used in recognizing specific bases, like pyrimidine groups as compared to purine groups. Although hydrogen bonding is effective in recognizing purine bases, like adenine and guanine, contacts with pyrimidines are mainly hydrophobic.
This is an example of a DNA bend with protein 1p78
Shape readout can be divided into global and local shape recognition. Variations of DNA shape are dependent on the chemical interactions of each base pair, which results in a unique conformational signature. Specificity in readouts depends on variations from the usual B-DNA structure, and result in binding less ideal DNA conformations.
Local shape readout is dependent on two main variations: narrow minor groove and DNA kinks. DNA kinks are when the helix's linearity is broken due to base pairs unstacking. This promotes optimal contact between the amino acid and DNA base.
Global shape readout is categorized as when the entire binding site of the DNA is not in the ideal B-DNA structure. Examples of these structures are A-DNA, bent DNA, and Z-DNA. In the A form of DNA, sugar structures that are typically not exposed are due to the expanded minor groove, and thus can contact nonpolar amino acids, such as alanine, leucine, phenylaline, and valine. In Z-DNA, the position of the phosphate groups is recognized. For example, RNA adenosine deaminase recognizes the zig-zag phosphate patterns on the left handed helix.
Fluorescence is the process where light, such as visible light, is absorbed by a molecule and re-emitted at a longer wavelength to generate distinct colors. It is a physical phenomena based on excitation of electrons in an atom or molecule; and emission is at a longer wavelength. Fluorescence allows us to see how things interact in a cell, as well as the localization and pathways taken.
Fluorescent proteins were first discovered through extraction from the jellyfish Aequorea Victoria. They have been instrumental to studies of cellular biology. Fluorescent proteins contain various color variants which emit various colors at different wavelengths thus functioning as valuable probes that can be used for live cell imaging. The proteins can be used as markers in vivo for whole-body imaging and detection of cancer as well as in organelles where protein fusion could be done to monitor intracellular dynamics and aspects of transcription. The important aspect that contributes to the fluorescent properties of the protein is its structure which consists of various amino acids depending on the protein and the local microenvironments. A variety of derivatives of fluorescent proteins have been created for the use of various markers with the most widely used being Green Fluorescent Protein, which was the topic for which Roger Tsien, a scientist from UCSD, received the Nobel Peace Prize.
Understanding the protein structure allows one to understand further protein function. This is the case for the structure of fluorescent proteins. For green fluorescent proteins, it is made up of a beta barrel structure, which consists of a β-Helix and alpha helices and which surrounds the fluorophore. The fluorophore is a part of the molecule responsible for its color. The fluorophore for the helix is formed by three amino acids that form a tripeptide Ser65–Tyr66–Gly67. The cyclicization of the amino acid residues is what forms the imidazolidone ring. However, an important property of the tripeptide is that the amino acid sequence is not the intrinsic property that leads to fluorescence as the same amino acid sequence is found in other proteins that do no have fluorescence. Further oxidation of imidazolidone ring causes the conjugation of the ring with Tyr-66 contributing the maturation of protein in terms of fluorescence. A key component of the fluorophore also is the fact that it is in two states. One state of the fluorophore is the protonated state or the predominated state, which has an excitation maximum of 395 nanometers. The other state of the fluorophore is the unprotonated state, which has an excitation maximum of 475 nanometers. Because of the complexity of the fluorophore for green fluorescent proteins, the molecule can accommodate modification. One feature that is significant is the packing of amino acid residues in the beta barrel is stable resulting in a high fluorescent quantum yield. The tight protein structure contributed by H-bonding also has resistance to pH, temperature, and denaturants such as urea.
These fluorescent proteins are represented by the mutation in one of the amino acid residues of the fluorophore. The tyrosine found in Green fluorescent proteins (GFP) is found to have an mutation, which results in the stabilizing of the dipole movement and the shift in wavelength of the excitation/emission spectra resulting in the yellow fluorescence. Furthermore, the yellow fluorescent proteins was found to have a new threonine residue 203 near the fluorophore. Further modification of the yellow fluorescent protein, can enhance the brightness of the protein, which makes it a Enhanced Yellow Fluorescent Protein (EYFP). Because of the brightness of the fluorescence, the protein is an important tool in multicolor imaging. These proteins are derived from the jellyfish Aequorea Victoria and is modified to create a different emission of fluorescence different from the original Green Fluorescent Protein.
Yellow fluorescence protein TagYFP is a bright yellow fluoresence recommended for protein labeling in protein localization and interaction studies. It may also be used for cell and organelle labeling and for tracking promoter activity, although TurboYFP and Phl-Yellow proteins are preferred. It is a monomeric protein successfully used for fusions and was developed under the bassis of GFP-like protein from the jellyfish Aequorea macrodactyla. TagYFP has a fast maturation phase along with high pH-stability and photstability. It's high pH stability allows it to be more stable that EYFP while its fast maturity allows it to give a brighter fluorescent signal. TagYFP has also been proven to generate stable transfected cell lines.
Blue and Cyan Fluorescent Proteins
Blue and Cyan Fluorescent Proteins result from the modification of the Tyr 66 residue located in the fluorophore. The conversion of the Tyr to histidine leads to the emission of blue fluorescence at a wavelength of 450 nanometers. Another modification is the conversion of Tyr to tryptamine resulting in a different fluorescence at a wavelength of about 480-500 nanometers. Additionally, genetic markers which uses these fluorescent proteins are expressed as TagBFP. One of the advantage of this protein is the ease at which it can be express and detect in a wide range of organisms. Mammalian cells transiently transfected with TagBFP expression vectors give bright fluorescent signals within 10-12 hrs after transfection. No cell toxic effects and visible protein aggregation are observed. TagBFP performance in fusions has been demonstrated in the β -actin and α-tubulin models. It can be used in multicolor labeling applications with green, yellow, red, and far-red fluorescent dyes. A problem though of these modifications is that that they require secondary mutations to increase not only folding efficiency, but also brightness. Fortunately, the genome modification is not life-threatening.
Red Fluorescent Proteins
Various derivatives of Red Fluorescent proteins have been derived in hopes to find a protein that exceeds or equals the fluorescence ability of GFP (green fluorescent protein). One derivative of protein has been from the coral Dicosoma striata also known DSRED. When it is matured, the protein is found to have an emission spectrum of about 583 nanometers. Further modification of DSRED has led to the formation of DSRED2 which have mutations at the peptide terminus preventing formation of protein aggregates and reduce toxic levels. Also, the DSRED2 is found to be more compatible with GFP as well. These proteins are also represented by its tetramer structure.
Astrocytes stained for GFAP, with end-feet ensheathing blood vessels
'Green Fluorescent Proteins'
The green fluorescent protein (GFP) is derived from about 200+ amino acid residues. This protein will exhibit green fluorescence when under to blue light. This GFP is mainly isolated from a marine biological organism: the jellyfish Aequorea victoria. This GFP has three main excitation maximums. An intense one at wavelength 395, a smaller one at 475 nm, and the emission peak which is at 509 nm which is what gives this protein a distinct vibrant green color. Another organism from the sea pansy has another excitation peak around 498 nm. This GFP gene is important for biosensing and reporting locations of gene expression. It can be transformed into other organism's genome through breeding, invitro injection, or through transformation. The GFP gene is introduced into a variety of bacteria, yeast, fungi, and other multicellular organisms. The story of how GFP became a research tool began in 1992, when Martin Chalfie of Columbia University showed that the gene that makes GFP produced a fluorescent protein when it was removed from the jellyfish genome and transferred to the cells of other organisms. Chalfie, a developmental biologist, first put the gene into bacteria and roundworms, creating glowing versions of these animals. Martin Chalfie, Osamu Shimomura, and Roger Y. Tsien were awarded the 2008 Nobel Prize in chemistry on 10 October 2008 for their discovery and development of the green fluorescent protein. Since then, researchers have transferred the GFP gene into many other organisms and even human cells growing in a lab dish.The GFP is unique amongst natural pigments for its ability to autocatalyse its own chromophore, through atmospheric conditions. In this way, a single protein acts as both substrate and enzyme. Other natural pigments require multiple enzymes for their production. Biotechnology has taken advantage of this unique feature of GFP, putting it to use as an in vivo marker of gene expression and protein localisation.
Monomeric Fluorescent Protein Variants
Fluorescent Proteins originally in its natural states exist as dimers, tetramers, and oligomers. Also, the theoretical possibility of fluorescent proteins forming dimers in cellular compartments due to possible high protein concentrations contributing to dimerization. Thus, the use of monomeric fluorescent protein variants has been sought after. One problem however is that the first few monomeric fluorescent proteins had reduced fluorescence capability. Furthermore, the production of the monomers required around 30 amino acid changes to the structure. There are improvements though in the development of these proteins that have increased quantum yields and photostability.
Fluorescent tags are used to label molecules such as proteins, DNA, and antibodies. Fluorescent labeling works with fluorophore reacting with a functional group on the target molecule. Probes are produced from this type of molecular labeling. Western blot assays identify and separate proteins therefore these fluorescent tags are crucial. Size exclusion chromatography removes fluorophore on the target molecules. Fluorescent dyes can be used to specify which organelle of the cell is present in order to distinguish their unique structure and further explore their individual functions. These tags and dyes are important in microscopy and reverse photobleaching because they are less harmful to living cells than quantum dots. Fluorescent dyes have hydrophobic properties hence specific dye columns are used in separations of molecules with dyes.
Example: Western Blot Assay
The purpose of a Western Blot is to locate and determine proteins on the basis of their ability to bind to certain antibodies. A Western Blot analysis allows one to detect a protein of interest from a mixture of a number of proteins. After completing a Western Blot, the information gained from the process is the size of the protein and the expressed amount of protein.
A Western Blot analysis can be done on any protein sample, ranging from cells to tissue, to recombinant proteins synthesized in vitro. A condition the Western Blot is dependent on is the quality of the antibody that is used to probe for the target protein. The antibodies used in a Western Blot must be specific to the protein of interest.
Example: Green Fluorescent Protein (GFP) turns green when exposed to blue light.
on GFP
Hard X-Ray fluorescence microscopy is useful to investigate the trace metal distributions within a whole, unstained, biological tissues.
Trace metal elements are integral to many life forms. Metals help catalyze functions and sometimes even play a structural role within the cell. Take for example zinc finger domains, where Zinc ions help to bind nucleic acids and proteins. Metals are recognized for having influential effects on human health and disease. Therefore, the study of these trace elements can provide important information and reasoning for the functions and pathways of metalloproteins. These studies may even find therapeutic approaches to quantitatively study the intracellular distribution of these trace elements.
These X-ray fluorescence has been used to create tomographs to visualize the structure of a 10-μm cell. Despite the usefulness of using these high penetration X-rays for tomography, there have been limitations that affect the length of time of experiments as well as the accuracy of these images. Due to these limitations in X-Ray fluorescence microscopy, there have been ongoing research to develop better X-ray resolution, detector speed, cryogenic environments, and the pursuance of a auxiliary signals. Thus, there will be many helpful new approaches in X-ray fluorescence tomography in the future.
Hard X-ray fluorescence, also called XRF, microscopy is a useful tool to trace metal distributions in various biological systems. For transition metals such as copper, zinc, and other relevant trace elements, XRF has provided attogram sensitivity at spatial resolutions down to 150 nm. Nowadays, 10-15 elements have been able to be mapped simultaneously. This simultaneous mapping of elements leads to precise elemental colocalisation maps.
Structural visualization has been improved even further with the use of hard X-ray fluorescence, but several technical challenges have presented themselves to lead to two-dimensional and low-definition realizations. Current developments have allowed scientists to overcome some of the most major limitations, and now the scientific world is able to create sub-500-nm resolution XRF tomography. Tomographs with this resolution can provide extreme detail in the realm of elemental specificity. These recent progresses in XRF tomography will definitely be utilized soon. Other developments in progress are cryo-microprobges, which are able to accommodate frozen-hydrated specimens.
The process of X-ray fluorescence is extremely fit for quantifying trace elements. XRF is unique because it does not rely on artificial dyes or flurophores to determine the structure of proteins, rather, X-ray fluorescence can be excited by exposing molecules to particles (electrons and protons) or X-ray beams. With the use of X-ray excitation, the bremsstrahlung background becomes unnecessary, allowing WRF microscopy to show high spatial resolution. Because X-ray penetration enables scientists to have sample thicknesses of tens of microns, XRF microscopy is an ideal method for form tomographic visualization of biological samples.
Due to many technical challenges and the analytical complexities involved in XRF tomography, the method has not found general application. Regardless, recent developments have provided advancements in 3D resolutions down to a few 100 nm for specimen up to 10 µm in size. This means 3D elemental maps of multiple elements can be visualized with high spatial resolution.
There is a bright future ahead for XRF with its recent advancements. Combined with the demand from the biological, environmental, materials, and geological communities, XRF seems very promising.
XRF Tomography:
The term ‘tomography’ is derived from Greek and means ‘slice imaging’, meaning that it is a technique that gets its data from a single slice within its specimen. A 3D reconstruction is made of slices of a specimen from tilted angles. The task of reconstruction can be quite strenuous and time consuming, given that serial sectioning is the direct approach to tomography. This direct method of actually physically sectioning can lead to significant artefacts, which is a defined as a man-made object that taken as a whole. As an alternative to physically sectioning, non-destructive techniques can be utilized because they reduce specimen preparation requirements and are also significantly easier and simpler to perform. In comparison to other methods of tomography, only XRF microtomography is able to map trace elemental distributions in direct relevance to biology.
Full-field tomography:
Recently, X-ray fluorescence tomography has been demonstrating by using full-field imaging and structured detector approaches. However, these novel full-field imaging and structured detector approaches have their pros and cons as well. These nascent technologies have spatial resolution between 2 and 200 μm, an sensitivity levels between 100ppm to percent levels. They also have a wide range of different elemental contrasts, making them ill prepared to be used routinely in studying the cells of biological specimens.
Projection Tomography:
Projection tomography is the technique that makes a tomographic reconstruction algorithm by using projections of the specimen as its input data. There is an energy-dispersive detector that is sensitive to any signal that is produced along the column. 2D data is measured over a range of positions, measurements are made at several angles, and an analytic approach helps to construct a 3D map of the specimen. What distinguishes X-ray fluorescence micrographs from other tomographic methods is its self-absorption effects. This encompasses the re-absorption of the fluorescence by the specimen and the absorption of the incident beam. The self-absorption effects are increased for thick specimens when X-ray micrographs are done using low fluorescent energy.
Self-absorption has significant effects on XRF tomography. To combat this issue, good correction algorithms are required for image clarity they also will expand the specimen size domain, to lower fluorescence energies and maintain accurate data. Absorption maps can be used to estimate the self-absorption at various fluorescence energies. Using the energy-dispersive detector, inelastic and elastic signals were recorded. These inelastic and elastic signals provide access to major light element distributions that are normally hard to attain.
Confocal Tomography:
Confocal tomography is known as a direct-space approach to scanning the XRF tomography when axial resolutions are below 5 µm. In this approach, the signal derives from only a small portion of the illuminated column because the collimator confines the field view of the energy-dispersive direction.
Confocal tomography only gives a direct access to a small region of the specimen. Unfortunately, this can make it quite difficult to target the features of interest within a specimen. Researchers have suggested another approach to dealing with this issue: using projection tomography for a low-resolution overview and then following by using a confocal study of the region of interest.
One of the biggest goals of structural biology is to understand how cells function, and how they sense and process external and internal signals. Genetically encoded fluorescent proteins (FPs) and fluorescent sensors can help researchers visualize how cellular processes work.
Cells, the basic building blocks of all living systems, rely on complex internal processes. These processes take place on the micrometer scale in different membrane compartments and cytoskeletal locations. Fluorescence imaging, employing green fluorescent protein (GFP) from the jellyfish Aequoria victoria and its relatives can help scientists further explore these cellular processes. GFP can be used as an important molecular imaging tool because of its fluorescence, or its capacity to produce a light of a different color other than the illuminating light. GFP and fluorescence can monitor cellular processes over time. Furthermore, GFP is encoded by a single portable DNA sequence that can be easily be fused to a protein of interest and expressed within living cells. Before GFP, researchers relied on fluorescent antibody techniques to examine the proteins and nucleotide sequences, but this could only be done on dead, fixed cells or tissue sections.
As GFP became a more and more popular molecular imaging tool, improvements and advances of GFP also arose. Mutagenesis of GFP caused an increase its brightness ad folding efficiencies and a decrease in its oligomerization. Mutagenesis also created forms of GFP that are photoactivable or photconvertable. It was discovered that GFP is just one member of a bigger family of homologous fluorescent proteins, mainly from marine corals, with different colors resulting from variations in structure and environment. Directed mutagenesis of the FP from these species resulted in a palette of FPs, covering the entire range of visible spectrum. Multiple colors allow for simultaneous imaging of multiple sets of proteins inside cells.
GFP developments ultimately gave rise to different imaging techniques such as fluorescence recovery after photobleaching, fluorescence correlation spectroscopy, FRET, fluorescence cross correlation spectroscopy, total internal reflection microscopy, fluorescence lifetime imaging, and photoactivation localization microscopy (PALM). These imaging techniques allow for in vivo analyses of cell function.
For example FP reporters can be used to monitor the behavior of tagged signaling molecules and their organization as well as detect specific pools of each component in a signaling pathway. This can be done by photobleaching, where an area of the cell is photobleached with a high intensity laser pulse and the movement of unbleached molecules from neighboring areas into the bleached area is recorded by time-lapse microscopy. The kinetic properties of a protein within a cell, such as its movement between compartments, can also be seen when tagged with a genetically encoded FP. In both photobleaching and photoactivation, the overall functions of an FP fusion protein can be determined without disturbing other pathways or cell function.
Use of FPs in fluorescent imaging methods also enables protein-protein interactions to be resolved. This can be done through FRET measurements, which allow for mapping of protein-protein interactions within cells in real-time. In FRET, one reporter is a donor fluorophore, the other reporter is a longer wavelength acceptor fluorophore. The readout is energy transfer from the donor to acceptor. By incorporating GFP variants, these reporters can be attached to different proteins to test for their interaction.
Further processes developed include using probes for monitoring GTP hydrolysis and cell cycle events. One strategy is to use small molecules that can be induced to form dimers. Probes can be used to drive specific biological activities at selected times and places in cells. Another strategy involves optically inducible switches, which employ light to activate signaling molecules. Probes can also react with zinc and nitric oxide indicators, which are inorganic species that drive physiological processes or trigger pathology.
Reporter technologies allow for real-time visualization of biochemical processes in living systems and offer a means to obtain insights into spatial organization and regulation of intracellular signaling networks underlying biological processes.
Molecular imaging approaches have been developed in order to analyze the actions of phosphorylation in live cells. These involve the use of recently introduced fluorescent reporters that allow for high resolution imaging of phosphorylation in the cells. This molecular imaging provides us with understanding and insights of timing and cellular localization of signaling networks. Before the development of these fluorescent imaging techniques, measurements of kinase action was often done through analysis of enzymatic activities ex post facto (meaning retroactive) through techniques of immunoprecipitation or immunocytochemistry. The delayed measurements are not as effective as they have problems of specificity and the inability for reports on the kinase action in real time. In addition, obtainment of information after the kinase action can cause the loss of key information only found in live cells. As such, fluorescent reporters have been developed for the purpose of real time analysis in complex mixtures or living cells.
Biosensors for peptides are often used to measure phosphoylation events with good resolution in vitro with the potetial for live cell imaging. These biosensors follow a basic design of synthetic flurophore into peptides or proteins. These properties change upon phosphorylation which allows for the analysis through a shift in wavelength (increases, decreases, or even both in quantum yield). There are four main types of bio sensors: environmentally sensitive, deep quench, self-reporting, or metal chelation enhanced.
Environmentally sensitive biosensors generally have a phosphospecific amino acid domain that complexes with the phosphoylated peptide. This type of biosensor is often used for Ser/Thr or Tyr phosphorylation. When activated, fluorescence is increased sevenfold.
Deep quench biosensors have a noncovalently attached quencher that shields the fluorescence until phosphorylation. When the molecule is phosphorylated the biosensors obtains a phosphospecific amino acid that separates the quencher from the fluorophore causing an increase in fluorescence. This often has around a 64-fold increases in fluorescence.
Self-reporting biosensors are used to detect tyrosine phosphorylation. For instance, tyrosine can be used to quench a fluorophore through pi-pi stacking interactions. When phosphorylated, the quench is lost and the fluorescence is increased by fivefold.
Lastly, metal chelation-enhanced biosensors use the nonnatural amino acid Sox, chelation-enhanced fluorophore and a series of biosensors for protein kinase activities that respond to Mg2+. These generally have a eightfold increase in fluorescence upon phosphorylation.
These biosensors are generally used in vitro kinase assays and allow for the detection of an initial lag phase. These biosensors also generally monitor activities of several kinases. There is also the possibility of using these biosensors to analyze live cell assays[check spelling] but these are more difficult. While these methods are good for analysis of the protein kinases, they are not as widely adopted as believed as they rely on specilizaed equipment and the instability of peptides and cellular perturbation can occur.
FRET (stands for Forster (fluorescence) resonance energy transfer) is a mechanism to describe energy transfer between proteins.
Most imaging techniques have the difficulty of getting the biosensor into living cells or have limitations that restrict use. FRET-based reporters generally overcome entering the cell as it has the cell manufacture the biosensor itself. These reporters are genetically encoded and can be transferred into cells as DNA. A notable example of these generically encoded biosensors is GFP (Green Flurorescent Protein). The changes in FRET can be seen as changes in emission rations between the fluorophore fluorescence; this is gernally the change in emission between donor and acceptor fluorophores.
FRET imaging is done through photochemical properties of donors using FLIM (Fluorescent lifetime imaging microscopy) which detects shortened fluorescence decay of the donor pair of FRET in the presence of the acceptor. This is advantageous as lifetime measurements are independent of the fluorophore concentration and photobleaching and allow for the distinguishing of actual FRET efficiency and probe concentration.
The FRET pair is selected to generate a maximum dynamic range. A domain is selected on the basis of the phosophoamino acid that is to be detected by the biosensor. The substrate part of the biosensor is what controls the specificity of the reporter. This is generally a short consensus peptide to be specifically recognied and efficiently phosphorylated by the target kinases but inert to other kinases. This method has given rise to a number of successfully genetically encoded biosensors. These have also been modified to include other features such as sequences to target a specific area of the cell or more genertion of the biosensors inside the cell.
These FRET based biosensors provide us with the means to image phosphoyrlation in living cells more efficiently and effectively than before.
Genetically encoded reporters can provide sufficient sensitivity when compared to the fluoresence in peptide-based biosensors and are able to provide more useful information aboue the phosphorylation and regulation of signalling pathways. These reporters can be used to study specific kinases and influence activity. These can also target specific reporters to find correlations between phosphorylation of specific proteins to other features of the cells.
One of the more exciting applications of kinases reporters are through the usage of high throughput chemical screens. These can help pharmaceuticals as the reporters can ensure compounds achieve efficient cell entry and provide kinetic information of protein inhibition. This is a big change as these screens are generally done through in vitro and is unclear is these compounds can target the intracellular kinases. Through the usage of the in-cell kinases reporters, senveral novel PKA inhibitors were able to be identified. This was all done through the analysis of FRET.
Lippincott-Schwartz, Jennifer. "Emerging In Vivo Analyses of Cell Function Using Fluorescence Imaging." Annual Review of Biochemistry. 2011.
The New Genetics (2006): n. pag. U.S. DEPARTMENT OF HEALTH AND HUMAN SERVICES, National Institutes of Health, National Institute of General Medical Sciences. Web. <http://www.nigms.nih.gov>.
G-protein coupled receptors GPCR) is transmembrane receptor protein that stimulates a GTP-binding signal transducer protein(G-protein)which in turn generates an intracellular receptor.
GPCR is large family of receptors that transmembrane protein that is serpentine in shape, crossing the lipid bilayer seven times.
G-protein have two major groups:
1. Small GTP binding protein.
2. Heterotrimeric G protein(directly coupled to receptor and enzyme)
R have alpha, beta and gramma subunit. An alpha subunit contains the GTP/GDP binding site, to identify beta and gramma subunits.
A hormone called epinephrine triggers the communication between inner parts of cells to pass cellular messages. The signal transduction, in detail, is occurred in three distinct steps. First, epinephrine message makes contact with the receptor on the surface of the cell. Second, the connecting transducer, the one with the role of switch, passes the message inward. Lastly, the signal gets amplified and it stimulates the cell to do its work.
Overall, G Proteins simply act as a switch in the process of cell signaling. Put another way, it is the baton in the relay of cell-signaling. However, they must be activated (turned on) only when needed and deactivated (turned off) immediately. If the G protein is left on at all times, illnesses and fatal diseases can and may occur. A G-protein may be left on in the active conformation even in the absence of ligand binding, leading to improper regulation of the proteins. Such fatal diseases that arise from improper regulation of the G-proteins include cholera: a life-threatening condition that occurs when the G-protein controlling water balance is left uncontrollably on. The result of this pathway kept aberrantly on leads to continuous diarrhea which becomes increasingly dangerous to the individual's well-being.
[1][2]
This switch was named G-protein by the 1994 Nobel Prize Winner Alfred G. Gilman from the University of Texas Southwestern Medical Center in Dallas. The name came not from his last name, but from the energy source of these G-proteins: GTP (Guanosine Tri-Phosphate). This discovery sparked considerable activity in further research of this molecule because it gives helpful clues to how cell-signaling works. The impact of this discovery still remains influential, as scientists continue to pay close attention to this molecule today. In 2012, Robert J. Lefkowitz and Brian K. Kobilka were awarded the Nobel Prize in Chemistry for their study of the G-protein coupled receptors, specifically on their studies of its three-dimensional structure.
Lysophosphatidic acid (LPA) is a small, ubiquitous phospholipid that acts as an extracellular signaling molecule by binding to and activating G protein-coupled receptors. There are about five known G protein-coupled receptors (GPCRs): LPA1, LPA2, LPA3, LPA4 and LPA5. LPA has diverse biological roles such as developmental, physiological, and pathophysiological effects.
LPA is a small glycerophospholipid present in low concentrations in all-eukaryotic tissues. It is present in high concentrations in blood plasma. LPA1 was the first high affinity, cognate cell surface receptor identified which then led to the identification of similar receptors LPA2 and LPA3 and divergent receptors LPA4 and LPA5. All five of these receptors are type 1 GPCRs, which differ in tissue distribution and downstream signaling pathways. The effects of LPA at physiological concentrations are mediated by these five receptors. In the brain for example, LPA1, LPA2 and LPA4 are expressed in developing brain and LPA3 is expressed in the postnatal brain. Expression of LPA1 and LPA2 can also be found in neurons.
LPA1 was discovered in the embryonic ventricular zone and shows high gene expression in this region. LPA1 couples and activates three types of G proteins: Gαi/0, Gαq/11, and Gα12/13. The activation of LPA1 prompts many cellular responses such as cell proliferation and survival, cell migration, and cytoskeletal changes. It also plays a role in altered cell-cell contact through serum response element activation as well as Ca2+ mobilization.
LPA2 is a high affinity cognate LPA receptor. It couples to the G proteins Gαi/0, Gαq/11, and Gα12/13. These G proteins carry signals through downstream molecules such as Ras (monomeric GTP-binding protein), mitogen-activated protein activated protein kinase, phosphatidylinositol 3 Kinase, Rac (small GTP-binding protein), phospholipase, diacylglycerol, and Rho, which is comparable to LPA1.
Activation of LPA2 signaling is associated with cell survival and cell migration.
LPA2 promotes cell migration through interfaces with focal adhesion molecule TRIP6 and several PDZ proteins and zinc finger proteins, which work directly with the carboxyl-terminal tail of LPA2. It can also provide inhibitory effects on the epidermal growth factor, which induces migration and invasion of pancreatic cancer cells through the Gα12/13/ Rho pathway. LPA2 signaling has cross-regulation between classical G protein signaling cascades. It also has other signaling pathways that regulate the affinity and specificity of signal transduction.
LPA3 also couples with Gαi/0 and Gαq in order to mediate LPA induced phospholipase C activation, Ca2+ mobilization, adenylyl cyclase inhibition and activation, and mitogenactivated protein kinase activation. LPA3 does not couple with Gα12/13 and therefore does not mediate in neuronal cells. It also has a high affinity for 2-acyl-LPA containing unsaturated fatty acids. LPA3 is not as responsive as LPA1 and LPA2 are to LPA species with saturated acyl chains.
LPA4 is structurally different from classical LPA and is related to P2Y purinergic receptors. It does not respond to nucleotides or nucleosides. LPA4 has a suppressive effect on cell mobility in which deficiency in LPA4 enhances migratory response to LPA on fibroblast and heterologous expressions of LPA4. It also suppresses LPA1 dependent migration of B103 cells (neuroblastoma cells) and LPA induced migration and invasion of colon cancer cells.
Lpa5 is expressed in many parts of the body such as the brain and peripheral nervous system. LPA5 expression can be identified in sensory and motor neurons in the spinal cord and has a functional role in pain processing such as acute and neuropathic pain. LPA5 just like LPA1-4 belong to the rhodopsin-GPCR family and is structurally different from LPA1-3. The stress fiber formation and neurite retraction in LPA5 expressing cells are induced by LPA by coupling to Gα12/13. It increases intracellular calcium levels by activating Gαq. LPA is also responsible for the phosphate production and increased cAMP levels in LPA5 expressing cells. LPA5 is a LPA receptor that can be activated by farnesyl pyrophosphate at high concentrations.
The central nervous system contains high LPA receptor expression. It is seen in various nervous system cell types such as neural progenitors, primary neurons, astrocytes, microglia, oligodendrocytes and Schwann cells. LPA signaling is also involved in developmental processes within the nervous system including cortical development and function, growth and folding of the cerebral cortex, survival, migration and proliferation.
Astrocytes for example, play an important role in neurodevelopmental and neurodegenerative processes and express all LPA receptors. Astrocytes are an abundant glial cell type in the central nervous system (CNS) that regulates biological and pathological processes. LPA signaling regulates morphological changes of astrocytes via the Rho-cAMP pathway and stabilization of stress fibers. LPA signaling is also related to neuronal differentiation, which is a function of astrocytes. LPA-primed astrocytes emit soluble factors to increase neuronal differentiation.
Schwann cells (SCs) are myelinating cells of the peripheral nervous system. They express LPA1 and LPA2 and their activation affect processes dealing with myelination. LPA mediates SC survival and prompts regulation of actin cytoskeleton and cellular adhesion properties
The vascular system involves proliferation, migration, adhesion, differentiation, and assembly of vascular endothelial cells and vascular smooth muscle cells (VSMCs).
LPA induces many responses in endothelial cells such as cell death, proliferation, migration and vasoconstriction. LPA induces cell death though protein nitrosylation (covalently adding a nitric oxide into a thiol group). LPA signaling in cardiovascular systems have been seen in hypotension and hypertension through vasoregulatory actions of LPA.
LPA has many effects in VSMCs. It acts as a phenotypic modulator as atherosclerotic lesions develop. It responds to vascular injury by prompting the dedifferentiations (less specialized cell becomes a more specialized cell) of VSMCs. LPA also prompt the proliferation and migration of VSMCs.
LPA signaling is involved in regulation of biological responses of neural progenitor cells (NPCs) by LPA receptors LPA1, LPA2 and LPA4. NPCs are involved in proliferation, morphogenesis, migration, apoptosis and differentiation, this is known as neurogenesis. The involvement of LPA receptors in NPCs has been revealed by heterologous expression studies that used cell lines in which single or multiple LPA receptors such as LPA1-5 were expressed. Studies that used NPCs, neurospheres, and ex vivo cultures have also showed the control of cell proliferation and differentiation through LPA1.
LPA signaling is suspected to have a relationship with neurological diseases such as schizophrenia and autism. Some studies have compared prenatal fetal or maternal bleeding and other factors to autism and schizophrenia. Since LPA and its metabolic precursors are preset in blood it is probable that the exposure of LPA to the brain can occur through hemorrhage. This can lead to changes in the cerebral cortex related to observation in autism and schizophrenia. Conditions in which blood-brain barrier is compromised or the LPA production is altered can result in abnormal LPA signaling, which can lead to neurological pathologies.
1. Neil A. Campbell, Jane B. Reece "Biology 8th edition"
2. Woong Choi, Ji. D.R. Herr ect. "LPA Receptors: Subtypes and Biological Actions" Annu. Rev. Pharmacol. Toxicol. 2010. 50:157-86
3. Woong Choi, Ji. Jerold Chun. "Lysophospholipids and Their Receptors in the Central Nervous System" Biochimica et Biophysica Acta 2012
G-Protein was first discovered in 1994 by Alfred Gilman and Martin Rodbell who later receive Nobel Prize in Medicine. G-Protein stands for “Guanine-nucleotide binding protein”. Martin Rodbell and Alfred Gilman used genetic and biochemical techniques to identify and purify the G protein. They found that a transducer provided the link between the hormone receptor and the amplifier. They used lymphoma cells that normally can be activated by a receptor to form cyclic AMP. A mutated lymphoma cell was usually found to contain a normal receptor and a normal cyclic AMP-generating enzyme but was yet unable to respond because it lacked the transducer. This was a good system to assay purified G proteins. A G-protein could be isolated from normal brain tissue and inserted in the mutated cell, thereby restoring its function.
Besides, G proteins are a key to the chemical switches. They bind the guanine nucleotides GDP and GTP. Also, they are heterotrimers that are associated with the inner surface of the plasma membrane.
Ras-like G-Protein: molecular switches
Effector: interacts stably with the GTP-bound form
GEF: guanine nucleotide exchange factor
GAP: GTPase Activating protein
The GTPase reaction intrinsic GTPase rates of small G-Proteins are slow in a range of Kcal=10^(-2) to 10^(-3) min^(-1). Then, the reaction performs the Sn2 nucleophilic attack with trigonal bipyramidal transition state. The phosphate hydrolysis reaction is thermodynamically highly favorable but kinetically very slow.
There are mainly two enzymatic strategies for GTP hydrolysis involved:
counteracting negative charge at phosphates with arginine as the catalyst
positing of attacking nucleophile with the catalyst of glutamine.
The non-hydrolysable GTP analogues:
GTP-y-S
GMPPCP
GMPPNP
The GTPase Activating Proteins accelerate intrinsic GTPase by a factor of 10^5 to 10^6. Ras, Rap, Rho, Rab, Ran have completely unrelated GAPs. High affinity binding to the GTP-bound form, low affinity interaction with the GDP-bound form.
The monitors several rounds of GAP catalysed G-Protein hydrolysis. G-Protein as substrate, GAP in catalytic amounts. Vary concentration of G-protein to determine Michaelis-Menten parameters.
2) Single turnover assays:
The analysis of a single cycle of GTP hydrolysis often monitored by fluorescently labeled G-Protein in one cell, excess of GAP in the other cell. It vary concentration of GAP is multiparameter firt allows determination of K1, K2, KD, etc.
The Biochemical features are such binds to adenine and not guanine nucleotides with affinity in the low micromolar range and binds to negatively charged liposome stimulated ATP hydrolysis.
The implications for membrane remodeling factors involved in membrane remodeling/ destabilization: the oligomer formation intro rings around a lipid template; insertion of hydrophobic residues into outer membrane bilayer; interaction of highly curved membrane interaction site perpendicular to curvature of lipid tubule; conformational changes upon ATP hydrolysis.
Vetter and Wittinghofer "The Guanine nucleotide binding switch in three dimensions." Science (2001)
Bos, Rehmann, Wittinghofer "GEFs and GAPs critical elements in the control of G-Proteins." Cell (2007)
A. Wittinghofer, H. Waldmann. "Rad-A molecular switch involved in tumor formation." Angew.Chem.Int.Ed (2000)
Scheffzek,Ahmadian,Kabsch,Wiesmuller,Lautwein,Schmitz& Wittinghofer "The Rass-RasGAP complex: structural basis for GTPase activation and its loss in oncogenic Ras mutants." Science (1997)
Prafcke, McMahon. "The dynamin superfamily: universal membrane tubulation and fission molecules?" Nat Rev Mol cell Biology (2004)
McMahon, Gallop "Membrane curvature and mechanisms of dynamic cell membrane remodelling" Nature (2005)
Ferritin is the cell’s storage compartment for iron. It is a protein that is found in all organisms from plants and animals to bacteria and archaea. Ferritin is present mainly in the cytoplasm of the spleen, liver, and bone marrow in mammals. The amount of ferritin within a cell varies depending on the cell’s function.
Ferritin molecule with 3-fold and 4-fold intersections
A molecule of ferritin protein is comprised of 24-peptide subunits. The subunits are either catalytically active H subunits or the catalytically inactive and specific to mammals, L subunits. The 24-peptide subunits form a hollow spherical shell that encases a core of iron. Channels, or small openings which allows the transportation of certain ions or molecules in and out of the ferritin protein, are formed at the intersections of the subunits.
The intersection of three-peptide subunits forms a three-fold channel. This channel is lined with the polar amino acids, Aspartate and Glutamate. The polarity allows favorable interactions with the iron ion and water, and thus serves as the passageway for iron to enter and leave the ferritin protein. The intersection of four-peptide subunits forms a four-fold channel. It is lined with Leucine amino acids making it a non-polar passageway. Although it is unable to serve as a passageway for the transport of iron ions, it is thought to be the site of electron transport which plays a role in the reduction of Fe(III) to Fe(II) and the vice versa oxidation. The reduction and oxidation of Fe is important because it dictates whether the Fe will be stored in the ferritin protein or release into the cell.
Cross-cut view of ferritin showing the iron lattice core
The protein molecular weight is about 445,000 and up to 4,300 atoms of Fe can be stored in the iron core. The iron is stored as ferrihydrite phosphate, [(Fe(O)(OH)8(FeOPO3H2).xH2PO4] in a crystalline solid lattice. It is known from tracer experiments that all the oxygen atoms in the ferrihydrite are derived from water, rather than O2. The lattice structure prevents the Fe (III) atoms from becoming soluble and exiting the ferritin shell. Reduction of Fe(III) to Fe(II) allows the iron atoms to break away from the lattice in order to be released from the ferritin. A water cage forms around the Fe(II) ion as the positively charged Fe2+ ion attracts the electronegative oxygen in H2O. The polarity of the three-fold channels allows the soluble iron to pass through the channel, thus releases the iron from the ferritin.
Ferritin synthesis decreases when low iron levels are present. Conversely when high iron levels are present, ferritin synthesis increases. The interaction between RNA binding proteins and the iron responsive element (IRE) region of mRNA regulates this process. Ferritin synthesis is inhibited when the two RNA proteins bind to the “stem-loop” structure of the IRE and inhibit mRNA translation. The binding proteins are called iron regulatory proteins (IRP1 and IRP2). IRP1 is regulated by the presence or scarcity of iron. When there is a scarcity of iron, IRP1 has an open configuration and is able to bind to the IRE loop and repress translation. IRP2 is regulated by its degradation in the presence of iron surplus. IRP2 is present when there is no iron, so it is able to inhibit translation. When iron is present in abundance, IRP2 degrades and ferritin synthesis can take place. The ratios of IRP1 and IRP2 are tissue specific. For example. IRP1 is more dominant in the liver, kidney, intestine, and brain tissues whereas IRP2 is more abundant in pituitary and pro-B-lymphocytic cell line.
Other iron proteins, include transferrins and mini-ferritins. Transferrins serve to transport iron as Fe(III) in the blood and other fluids. One of these has iron bound as Fe(III) by two tyrosine phenoxy groups, an aspartic acid carboxyl group, a histidine imidazole, and either HCO3- or CO32-. Mini-ferritins Dps (DNA protection during starvation) proteins are present in bacteria and archaea. They are made with 12 subunits and function oppositely from ferritin. Rather than using dioxygen to concentrate iron like ferritin, mini-ferritin usues iron to detoxify dioxide and peroxide. This process protects the DNA.
Bacteria and fungi also synthesize iron transfer compounds, called siderochromes.The common structures are complex hydroxamates (also called ferrichromes or ferrioxamines) or complex catechols (enterobactin). They have peptide backbones and are very strong chelating agents (Kf ~ 1030 to 1050), allowing the organism to extract iron from surroundings that contain very little iron or are basic enough that iron is present as insoluble hydroxides or oxides. Some of these compounds act as growth factors for bacteria and others act as antibiotics. There are also examples in which the iron is bound by a mixture of phenolic hydroxyl, hydroxamate, amine and alcoholic hydroxyl groups.
Siderochromes are substances which are excreted into the culture medium by micro- organisms in iron-deficient conditions. These compounds characteristically bind Fe3 + very strongly, and a number of them participate in the transport of iron into the cells. Siderochromes are synthesized in large amounts and excreted into the culture medium only when the bacteria
have insufficient iron, and it forms a very stable complex with Fe(II).
Gary L. Miessler, Donald A. Tarr, Inorganic Chemistry, Third Edition, 2004.
The "fuzzy" interactome refers to the concept that the current protein-protein interactome is inaccurate. The research suggests that scientists are satisfied with the protein-protein interactome acquired through insufficient data and inconclusive lab results. Additionally, the study suggests that the current model for defining protein-protein interactions is overly simplified as it neglects several physiological variables.
The main foundation of the "fuzzy" interactome theory is the inability to perfectly replicate all physiological protein-protein interactions in the a lab setting. For example, many experiments do not consider the importance of unstable intermediates, which cannot be isolated. Additionally, results wrongly suggest that protein-protein interactions are extremely simple and can be mapped and identified through simple experimentation. As a result, the current protein-protein interactome tends to reflect the results of technology driven experiments as opposed to hypothesis driven experiments.
This conclusion emphasizes the difference between physiological conditions and what can be replicated in the laboratory.
Protein folding is a highly complex process by which proteins are folded into their biochemically functional three-dimensional forms. The hydrophobic force is an important driving force behind protein folding. The polar side chains are usually directed towards and interact with water, while the hydrophobic core of the folded protein consists of non-polar side chains. Other forces that are favorable for protein folding are the formation of intramolecular hydrogen bonds and van der Waals forces.
Protein folding is entropically unfavorable because it minimizes the dispersal of energy and adds order to the system. However, the summation of the hydrophobic effect, hydrogen bonding, and van der Waals forces is greater in magnitude than the loss of entropy. Protein folding is therefore a spontaneous process because the sign of ΔG (Gibbs free energy) is negative.
For a reaction at constant temperature and pressure, the change ΔG in the Gibbs free energy is:
The sign of ΔG depends on the signs of the changes in enthalpy (ΔH) and entropy (ΔS), as well as on the absolute temperature (T, in kelvin). Notice that ΔG changes from positive to negative (or vice versa) where
T = ΔH/ΔS.
When ΔG is negative, a process or chemical reaction proceeds spontaneously in the forward direction.
When ΔG is positive, the process proceeds spontaneously in reverse.
When ΔG is zero, the process is already in equilibrium, with no net change taking place over time.
A top-view of the GroES/GroEL bacterial chaperone complex model
Intramolecular chaperones are essential for protein folding, but not required for protein function. Considerable evidence shows that chaperones play a critical role in protein folding both in vivo and in vitro. Unlike their molecular counterparts, intramolecular chaperones are encoded in the primary sequence of the protein as an N-terminal or C-terminal sequence extension and are usually termed propeptides or prosequences. Upon mediation of the protein folding, the intramolecular chaperones are removed either by auto-processing in the case of proteases or by an exogenous process in the case of non-protease proteins.
Intramolecular chaperones are classified into two groups on the basis of their roles in protein folding. The type I intramolecular chaperones mediate the folding of proteins into their respective tertiary structures and are mostly produced as the N-terminal sequence extension. The type II intramolecular chaperones mediate the formation of the quaternary or functional structure of proteins, and usually are located at the C-terminus of the protein.
The discovery of the first intramolecular chaperone was based on the studies on subtilisin, an alkaline serine protease from bacillus subtilis. Often, the relation of intramolecular chaperones to the molecular mechanism of protein folding is studied by introducing amino acid substitution mutations in the propeptide region but not in the functional domain of the protein. It was shown that the addition of propeptides in trans allowed for the folding of the subtilisin at a higher efficiency and rate than when folded in cis. It was also shown that if the energy barrier of the transition state in subtilisin was reduced, it was allowed to fold without the intramolecular chaperone, but at a slower rate.
Distinct from the subtilisin protease, the NGF (nerve growth factor) propeptide forms a cysteine knot by virtue of three intramolecular disulfide bonds. Based on the hydrogen-deuterium exchange experiments and the spectroscopic studies, the propeptide acts as a competitive inhibitor for the receptor binding of the mature NGF dimer. It is likely that the quaternary structure may stabilize the tertiary structure.
Competitive inhibition: substrate (S) and inhibitor (I) compete for the active site.
It is suggested that the α-lytic protease folds through a nucleation mechanism, in which the propeptide folds first and acts as a scaffold that stabilizes the C-terminal domain of the mature protease. This allows for the structural arrangement of the two domains to pack into the native structure.
Sometimes the C-peptide has independent physiological functions. For example, the C-peptide of proinsulin both stimulates Na+, K+-ATPase and functions as an intramolecular chaperone for folding of insulin.
Intramolecular chaperones that are involved in the folding of the quaternary structure of proteins are called type II intramolecular chaperones. The E. Coli K1-specific bacteriophages contain tail spikes that exist as homotrimers of endosialidases. These tail spikes are produced with a C-terminal domain (CTD) that is not part of the functional trimer. The fact that the CTD folds independently from the enzymatic domain and forms a hexamer suggests that the CTD is able to associate with each other to initiate the trimerization of endosialidases.
Diagram of a typical tailed bacteriophage structure
There exists both an N-terminal and a C-terminal propeptide in the fibril-forming collagen. The C-terminal propeptide prevents premature fibril formation, while the N-terminal propeptide is important in fibril associate of the collagen triple helix. The propeptide is proteolytically processed in the functional multimer.
Chaperones-protein folding in the cytoplasm of prokaryotic and eukaryotic cells
In vitro, many unfolded proteins attain the native state spontaneously, however within cells such folding efficiency is limited by the conditions. There are two factors expected of the cellular environment to improve propensity of polypeptides misfolding and aggregating.
The Crowding Effect-with high concentration of macromolecules, and close proximity of emerging polypeptide chains developing from polyribosomes. A highly conserved set of proteins chaperones, prevented non-productive protein folding. In the cytoplasm, the Hsp 70 (heat-shockprotein of 70 kDa) and chaperones are the major factors to have efficient protein folding in normal conditions or adverse conditions (heat stress). Chaperonins interact with collapsed folding intermediates so efficient folding advance in the provided environment. For many proteins, both molecular chaperones are need for efficient folding.
It is especially important to decipher the role intramolecular chaperones play in protein folding because human proteins are involved in diseases. These proteins are found to contain the sequence extensions that probably function as intramolecular chaperones. Mutations termed protein-memory mutations in the intramolecular chaperones can cause misfolding of the functional domain, which results in distortion of their function leading to human diseases.
The recent advantages of such technologies as single protein production (SPP) system, in-cell NMR, and cotranslational structural studies will serve as important techniques to further study protein structures and folding in the cell. Furthermore, the mechanisms of intramolecular chaperone-mediated protein folding in vivo can be investigated.
C.B. Anfinsen, Principles that govern the folding of protein chains, Science 181 (1973), pp. 223–230.
H. Ikemura, H. Takagi and M. Inouye, Requirement of pro-sequence for the production of active subtilisin E in Escherichia coli, J Biol Chem 262 (1987), pp. 7859–7864.
J.L. Silen, D. Frank, A. Fujishige, R. Bone and D.A. Agard, Analysis of prepro-alpha-lytic protease expression in Escherichia coli reveals that the pro region is required for activity, J Bacteriol 171 (1989), pp. 1320–1325.
Dean J.Naylor and F.-Ulrich Hartl1, Department of Cellular Biochemistry, Max-Planck-Institut für Biochemie, Am Klopferspitz 18A, Martinsried bei München D-82152, Germany
Determining how a protein will fold has been fairly difficult to predict even though the amino acid sequence is known. Instead of analyzing the structure of the protein and analyzing the mechanism of how a protein folds, understanding the kinetics of folding rates has proven to be a much more efficient way of understanding protein folding. The two-state folding kinetics of proteins is mostly studied, which analyzes the folding progress of a protein from its linear chain form, its primary structure, to its folded state, its tertiary structure. This process is dependent on the cooperative nature of the transition state. The kinetics of protein folding can be illustrated through the funnel energy landscape diagram, which is mathematically explained through the Gibbs free energy equation. This energy landscape diagram can follow the tract of the many pathways a protein can take until it reaches its native, or most stable, folded state. As a protein conforms to its most native state, a free energy barrier ends up controlling the kinetics of the protein folding. To illustrate the folding mechanisms, different Go-model simulations are used, which are coarse-grained topology-based models. However, although Go-model simulations provide the folding mechanism of proteins, they lack the ability to predict the folding rates of proteins based on the kinetic or thermodynamic cooperativity demonstrated by two-state proteins. Because of this reason, studies have been done to understand the cooperative nature of the two-state folding of proteins and the factors that affect the folding rates of proteins.
The folding rates of two-state proteins can be understood through two general properties of the folded conformations. One of the trends is that more structurally complex proteins tend to fold at slower rates in comparison to more simple structural proteins. For example, a tertiary structure containing beta sheet proteins and proteins combined with alpha helices and beta sheets tend to fold slower than proteins that are made up of only alpha helices. The second trend is that larger proteins tend to fold a lot more slowly than smaller proteins. The kinetics of alpha helical proteins and structurally complicated proteins such as globular proteins also differ due to long-range tertiary contacts. The transition states of globular proteins are expected to have a higher transitional energy barrier than alpha helical proteins because more entropic energy is required to make a more structurally complicated protein to fold in a more ordered fashion in comparison to a simpler structural protein. As the chain length of a protein also increases, the free energy barrier exponentially increases as well to reach the transition state of the protein.
In determining the transition state of an in-process folded protein, the native state topology of the protein has to be known in order to predict the structure of the transition state of the protein. Topology refers to the effect of the orientation of objects in space due to deformations of the objects. In the case for proteins, a folded structure might change its orientation in space if the protein is heated up as it would lead to denaturing. To examine this transition state of folded proteins, the formation of the transition state is determined by the free energy barrier that controls the kinetics of the folding reaction. This free energy barrier is the result of the compensation of energy and the loss in entropy due to the new interactions formed in the process of protein folding. The relationship between the kinetics of a folding protein and topology help to explain why the transition state of a protein is dependent upon its native state. This is known as the principle of minimum frustration of energy landscape theory, which can related to the funnel model of folded proteins. The more stable the protein is, the lower the energy it is at, and the energy of the native protein can help give information on how much energy is required for a protein to reach its transition state in the folding process.
The use of Go models helps to give an identification of a protein in its most native state, which is held together by stabilizing interactions between native contacts. These stabilizing interactions are also known as non-additive forces, and these forces play a factor in the kinetics and thermodynamics of protein folding. These non-additive forces can also be thought of as intramolecular interactions that happen spontaneously within the protein such as side-chain ordering and hydrophobic forces. The effect of these non-additive forces have been shown to increase the free energy barrier of the two-state folded protein, and therefore, this makes these Go models more thermodynamically cooperative.
Upon using these Go models, the three-body interactions of the folding rates and what are known as phi values are examined in two-state proteins. The meaning of these phi values gives a relationship between the transition state of a two-state folded protein and its native state. The phi value explains the content of the native structure in its transition state. Therefore, the more native-like the structure of the transition state, the more likely this transition state will conform into its native state in a shorter period of time. In general, phi values improve when the transition state is more like its native state, but the ratio between its transition state and native state is different for each protein that varies in size and its secondary structure.
Many different types of Go models have been developed to better understand the cooperativity of the folding rates of proteins. For example, a Go model has been created in analyzing a small alpha-helical protein also known as a Calpha Go-like model. This model has also been altered by introducing solvent-mediated interactions to the model. The interactions between proteins are instead replaced by a desolvation barrier. Studies have shown that the thermodynamic and kinetic cooperativity of two-state folded proteins increase as the desolvation barrier increases in height. Desolvation is known as the removal of solvent from a material in solution. In general, desolvation has a property whee short-range contact proteins such as those that form alpha-helices have little cooperativity due to desolvation while long-range contacts such as those with a mix of beta sheets and alpha helices are expected to have high cooperativity because long-range contacts require persistence in bringing the proper chains together, and therefore, require a high amount of cooperativity. In conclusion, it is these topological models with nonadditive forces such as hydrophobic forces of proteins that help to better understand the folding rates of certain proteins.
Heat shock proteins, or HSP, are a class of proteins with related functions. Their expression increases when cells are exposed to elevated temperatures or other stress. Heat shock proteins help protect other proteins from heat stress.[5] This response to heat stress can also be seen in heat-stressed animals and microorganisms.[5] Some heat-shock proteins are called chaperone proteins because they function in unstressed cells as temporary scaffolds that help other proteins fold into their functional shapes.[5] The dramatic upregulation, or increase of cellular components, of heat shock proteins plays a key role in heat shock response and is induced primarily by heat shock factor. HSPs can be found in almost all living organisms, ranging from bacteria to humans.
Heat shock proteins are named in accordance to their molecular weight. Some widely-studied HSPs are Hsp60, Hsp70, and Hsp90, whose families consist of heat shock proteins weighing 60, 70, and 90 kilodaltons in size, respectively.
Structure of the ATPase fragment of a 70K heat-shock cognate protein
Heat shock proteins with molecular weight near 70,000, is one of the important part for protein folding to help protect heat stress. It is also a chaperone protein. It was discovered by FM Ritossa in 1960s when a “puffing pattern” –the “heat shock response”-elevated gene transcription was observed.
Structure
N-terminal domain-the one with ATPase activity( 44 kDa) – consists of two lobes, splited by a cleft and adenine nucleotide binds to it.
Substrate binding domain(18 kDa)-- made up of a groove. The groove has an attraction for neutral hydrophobic amino residues. It could also interact with peptides up to seven residues.
C-terminal domain—consists of beta-sandwich damaging and second region full of alpha helical structure for substrate binding domain. The beta-sandwish region is composted of two sheets with 4 anti-parallel beta strands. They form a “pocket” for peptide binds. It open like a lid and peptides bind and release relatively rapidly when Hsp 70 protein is ATP bound. When the lid is closed, and peptides are tightly bind to the substrate HSP 70 proteins is ADP bound.
Reaction cycle with Hsp 70
The ability of Hsp 70 to bind and release hydrophobic amino determines chaperone function, which ATP binding and hydrolysis is depend on this binding and releasing of substrate proteins. First, the N-terminal ATPase domain adjusts the attraction between Hsp 70 and substrate by altering the conformation of the C-terminal region. Once the ATP bind the Hsp 70 open the “lid” and binding and releasing of substrates happens rapidly. When ADP exist, Hsp 70 closed the “lid” and binding and releasing of substrates slow down. Due to ATP hydrolysis to ADP, the interaction is stabilized by converting Hsp 70 to a more active state. It is a repeating cycle when ADP transformed into ATP and followed with the substrate being released.
Activeness of Hsp 70
J-proteins and nucleotide exchange factors (NEFs) both affect the activity of Hsp 70. NEFs stimulate transformation of ADP to ATP, and consequently stimulate the chaperone cycle with Hsp 70. The rate of nucleotides exchange is 10-20 fold faster than the rate of hydrolysis if J-protein is nonexistent. When J-protein exist, hydrolysis is still stimulated but nucleotide release will become limited. Nef with Hsp 70 bond to ADP could excite the reaction of nucleotide exchange rate up to 5000-fold.
Solid ribbon model of the yest Hsp90-dimer in complex with ATP
Heat shock protein 90, or Hsp90, is a molecule chaperone and is a member of the heat shock protein family. Like other heat shock proteins, Hsp90 is upregulated in response to stress and/or elevated temperatures. Hsp90 is found in bacteria and all branches of eukarya. It is one of the most abundant proteins expressed in cells. The functions of Hsp90 includes assisting in protein folding, cell signaling, and tumor repression.
The Structure and Function of the Sections of Hsp90
N-terminal Region: ~25kDa of the N-terminal region of Hsp90 was determined after proteolysis. It was found that there were various sections in the sequence that were homologous to MutL mismatch repair proteins and type II topoisomerases, proteins that alter DNA with the aid of ATP. This evidence pointed researchers in the direction that ADP/ATP had special significance to Hsp90.
The pocket in the N-terminal is capable of binding to adenine nucleotides. Furthermore, the binding site in the N-terminal is especially important for the binding to ATP/ADP, without which, Hsp90 is unable to perform its function properly.
Middle Region (Catalytic loop and binding site to client protein): A tight coil of many α-helices that are small in length connects the N-terminal region to the C-terminal region. A hydrophobic area around Trp 300 and amphiphatic characteristics of the residues between 327-340 promote interactions with client proteins.
An arginine at residue 380 is required for the function of ATPase. The catalytic loop in the middle region is responsible for Hsp90's reaction with ATP/ADP.
C-terminal Region:
This region is responsible for Hsp90 dimerization. A bundle of 4 helices in this region (2 from each protein) is the structure of this region taken in dimerization. This region of Hsp90 diverges most from similar proteins - evidence for this lies in a couple small deletions and a lower sequence similarity.
Although the full structure of a eukaryotic model does not currently exist, with the combination of the structures of the various regions from different model organisms, a good sense of the actual structure can be determined. The largest problem in determining the full structure is the link between the middle region and the n-terminal region because this area is very poorly conserved between organisms (its residues vary widely between various organisms).
The biomedical importance of Hsp90
The clientele of Hsp90 is restricted; unlike many other proteins which require the assistance of molecular chaperones to fold properly. Although not essential in bacteria, Hsp90 plays an important role in eukaryotes in which it maintains both cellular and organismal viability. Furthermore, scientists have discovered that Hsp90 seems to enable cancer cells to survive both hostile environments and chronic genetic instability within the host. Also, many viruses seem to require Hsp90 chaperone machinery to propagate successfully.
The Hsp90 chaperone cycle requires conformational flexibility
Like most chaperones, Hsp90 relies on conformational flexibility for its activity. Although the overall structure of bacterial Hsp90, or HtpG, and eukaryotic Hsp90 proteins are very similar, only the eukaryotic Hsp90 proteins interact with co-chaperones that helps with the stabilization of various Hsp90 conformational states as well as participate in Hsp90-dependent client protein binding, folding, and maturation. Unlike bacterial Hsp90, eukaryotic Hsp90s contain an unstructured flexible region of variable length that links the N-domain with the M-domain, which provides docking sites for client proteins and various co-chaperones. The well preserved N-, M-, and C-domains despite the large conformational rearrangements undergone by the Hsp90 dimer suggests that the conformational flexibility of Hsp90 results from the displacement of the domains with respect to each other.
By using X-ray crystallography and by analogy to other members of the GHKL superfamily of which Hsp90 is a member, scientists have determined that nucleotide binding to and hydrolysis by Hsp90 relays between two stable conformational states: an "open" apo state in which N-domains of nucleotide-free Hsp90 are not dimerized and a "closed" ATP-bound state in which N-domains are dimerized. Furthermore, the use of single particle electron microscopy has proven that bacterial Hsp90 can exist in three distinct conformational states: the "open" apo state that is nucleotide-free, the "closed" state in which both N-domains transiently dimerize in the presence of ATP, and a "compact" state in which the N-domains are no longer dimerized but instead make novel intermolecular contacts with their respective M-domain. These conformational changes are thought to impact client protein and co-chaperone binding and release.
Hsp90 chaperone cycle driven by ATP binding or hydrolysis?
Low Hsp90 ATPase activity makes it difficult to envisage ATP hydrolysis as the driving force behind the Hsp90 chaperone cycle. Researchers Southworth and Agard have brought up evidence that nucleotide binding does not drive conformational change in Hsp90. Instead, their data show that multiple Hsp90 conformations co-exist in a dynamic steady-state equilibrium in the absence of nucleotide and that this equilibrium is only moderately perturbed by nucleotide binding. In consideration of bacterial Hsp90 dynamics, the addition of either AMPPNP (a non-hydrolyzable ATP analog) or ADP can slightly skew the equilibrium to favor the closed or compact state. However, yeast Hsp90, under similar conditions, adopt only two distinct conformations, while human Hsp90 showed no obvious conformational changes in the absence of either nucleotide.
Conservation of the three-step Hsp90 chaperone cycle from bacteria to humans
Although the data collected from bacterial Hsp90 seems to fit the three-state conformational model, the data of both yeast and human Hsp90 seem to not support the model. Yeast and human Hsp90 seem to show different conformational responses to nucleotide. By using a cross-linking technique to trap rarely populated conformational states, Southworth and Agard showed that both yeast and human Hsp90 proteins conform to the three-state model. Southworth and Agard provided evidence that explained recent kinetic studies that suggest a conserved ATPase cycle among Hsp90 proteins from different species and provided support for transient N-domain dimerization in human Hsp90. An important conclusion is that the population occupancy of each conformation at equilibrium is unique for different species. This suggests that evolution have optimized the kinetics of the Hsp90 chaperone cycle to meet the distinct chaperoning requirements of each species.
The CCN family consists of six secreted extracellular matrix associated proteins (CCN1 – CCN6). The CCN family was named as an acronym of the names of the first three CCN proteins discovered: Cyr61 (cysteine rich protein 61), CTGF (Connective Tissue Growth Factor), and NOV (nephoroblastoma overexpressed gene). The CCN family acts as key regulator of the ECM components and as signaling molecules involve in a variety of important biological functions. This includes adhesion and extracellular matrix remodeling, skeletal development and chrondrogenesis, angiogenesis and wound repair, and regulation of cell proliferation.
This picture depicts the four domains that make up the CCN protein structure and the different molecules each domain binds to.
In terms of structure, contains an N-terminal secretory signal peptide, four similar functional domains, the same organization, a common intron/extron pattern, a similar primary structure (a 40 – 60% similarity). All six CCNs have five exons, with the first exon corresponds to a signal sequence and the rest a discrete protein module. The CCN family members feature four functional domains: 1)An insulin-like growth factor bind protein-like module (IGFBP), 2) a von Willebrand factor type C repeat module (VWC), 3) a thrombospondin type-1 repeat module, and 4) a cysteine-knot-containing module (except CCN5). Each domain is separated by linker regions which are susceptible to proteolysis. Proteolysis acts as a regulator of CCN protein activity by cleaving the linker regions resulting in the production of truncated molecules and individual modules. The six members also contain 38 conserved cysteine residues which vary in length and composition throughout the family. These residues are located right after the VWC domain and acts as a hinge between the first and second half of the protein.
IGFBP: The IGFBP family consists of IGFBPs that have a high affinity to and bind to insulin-like growth factors. This allows them to control the transport, localization and metabolic breakdown of the IGFs. IGFBP domains are typically multidomain proteins with distinct cysteine-rich N- and C-terminal domains linked by variable linker region. The N-terminal domains are globular in structure and have 12 conserved cysteine residues while the C-termianl domain has 6 conserved cysteine residues. N- and C –domain work together in concert to contain and bind IGF molecules with high affinity.
CCN family’s IGFBP domain shares strong sequence similarity to the N-terminal domain of traditional IGFBP but lacks the C-terminal domain and thus binds IGF quite poorly. Scientists have created a model of the CCN’s IGFBP domain using a CPH model. To construct the model they used the 80 amino acid sequence residue of the N-terminal domain that both CCN and IGFBP shared 30% sequence identity with. The sequence is L-shaped and divided into two subdomains connected by a short stretch of coil. The first subdomain has a semi-folded with two-stranded antiparallel beta-sheets and two parallel loops. The loops are stabilized by a series of disulphide bonds which form a flat plane with the beta sheets. The second subdomain is a globular domain containing the IGF-binding site and is surrounded by a three-stranded anti-parallel beta-sheet stabilized by disulphide bonds.
VWC:The von Willebrand factor C repeat domain contains a 70-100 amino acid sequence motif which is constantly conserved in most ECM proteins. The VWC domain can be repeated multiple times in a single protein, increasing its activity, but vary in growth factor affinity. This difference among proteins is through to be a means of regulation, accounting for the varying substrate specificity not only in CCN proteins but in others as well. One of the functions of the domain is regulating bone morphogenic proteins, which facilitate bone, cartilage and organ growth, along regulating TFG-beta signaling.
The VWC domain in CCN proteins is made up of two sections. The upper section is made up beta-sheets while the lower section made up of fibronectin-like material held together by disulphide bonds. All the CCN proteins, except CCN6, have the VWC domain’s conserved cysteines. It results in a binding pattern of two disulphide bonds followed by the beta-sheets and then three more disulfide bonds. While the cysteines are conserved in the CCN proteins, their VWC domains vary in electrostatic surfaces. CCN1 and CCN5 have mostly negatively charged surfaces, CCN4 is mostly positively charged, and the rest are a mix of surface charges.
VWC repeats in most proteins functions to regular bone morphogenic proteins (BMP) and TGF-beta signaling. Both BMP and TGF-beta work closely with CCN proteins. BMPs influence bone and cartilage growth, both of which when damaged results in TGF-beta facilitating increased expressions of CCN 1, CCN 2 and CCN 5 with decreased CCN 4 expression.
TSP-1: This domain is made up of a 55 amino acid sequence residues. It has three distinct domain repeats: TSR-1 repeat, three epidermal growth factor-like repeats and seven aspartic acid-rich repeats, all inside a linear structure. The TSR-1 repeat is commonly found inside the human genome as well as in other eukaryotic organisms. It is composed mainly of small three-stranded anti-parallel beta-sheets organized into a right-helical shape and mainly functions as cell attachment sites in signaling and adhesion, inhibition of angiogenesis (blood cell growth), protein-binding sites for various growth factors and other ECM proteins, and glycosaminoglycan-binding sites inside the TSP-1 domain. Similarly to VWC many TSRs bind TGF-beta, however the TSP-1 domain in CCN proteins lack the necessary RFK tripeptide sequence to perform the binding. The TSP-1 domain does however share a 60 amino acid sequence similarity with the TSR superfamilies as well as the conserved cysteine residues, CSxTCG motif, arginine and tryptophan residues at the N-terminal of the domain.
Unlike the original, the TSP domain in the CCN proteins has fewer CWR layers, residues that form hydrogen bonds, along the domain’s three-stranded anti-parallel beta-sheet. Because of this they form fewer hydrogen bonds and only have a one tryptophan and two aginine residues. Three disulfide bonds are all present in the domain and hold the loops together around the beta-sheets. A positively charged domain surface is conserved in all six CCN proteins. Due to the ability of the TSP-1 domain to bind to glycoconjugates and its inhibition of angiogenesis, it may account for CCN proteins’ management of angiogenesis as well as their interactions with the ECM itself. CCN proteins, particularly CCN 1 and CCN 2, interaction with TGF-beta which facilitates their expressions, such in osteoblasts during bone fractures or breaks as well as to mediate angiogenesis. Their interaction with TGF-beta may be coordinated by their TSP-1 domain since original TSP domains also interact with TFG-beta. Mutant and missing TSP domains in CCN proteins are thought to be involved in the formation of colorectal and gastric carcinomas and Wilm’s tumours.
CT: The CT domain contains a cysteine knot motif made up of six conserved cysteine residues. It is thought to mediate many of the CCN protein functions which can be heterodimeric, where both the CT domains of CCN 2 and CCN 3 interact in glutathione S-transferase pull assays, and is involved in heparin binding, one of the components of ECM. All CCN protein CT domains contain a collection of positively charged residues which surround the beta-sheet loops with the heparin-binding site at the N-terminal portion of their domain.One of the main functions of the CCN proteins is regulation and remodeling of the ECM as well as adhesion. Because TSP domains are thought to bind heparin sulphated proteoglycans, scientist think that CT and TSP domains may work in tandem by directing how CCN proteins control and manipulate adhesion processes and ECM composition. For example, in order to induce adhesion in vascular smooth muscle cells, CCN 3 interacts with the integrin cell surface receptors and heparin sulphate proeoglycans.
The CT domain also contains a cysteine knot that is made up of a ring of eight residues linked by two disulphide bonds with a third bond going through the center of the knot. Next to the 3D knot are two two-stranded anti-parallel beta-strands. It is the domain’s cysteine knot motif that scientist think allows it to act as a dimerization molecule similar to those of growth factors. In NGF and TGF-beta, it is the disulphide bond in the center of the knot which helps direct dimerization. While all CCN proteins have similar arrangements, they all slightly differ in electrostatic surfaces. Scientists believed that this different in charge and their diverse sequences, not including their conserved cysteines, may account for the differences in CT domain ligands and binding partners.
Holbourn, Kenneth, K. Ravi Archarya, Bernard Perbal. "The CCN family of proteins: structure-function relationships." Ce Press. 2010. Web.
Prion proteins are those that can be mis-folded and cause harmful neurodegenerative diseases. A commonly known prion disease is one that affects cattle called Bovine Spongiform Encephalopathy, or "Mad Cow Disease". A newer form of this disease that affects humans is the Creutzfeldt-Jakob disease. Prion proteins are in the mammalian genome and are mostly expressed in the brain. In the body there exist two forms: a normal cellular protein and an isoform that is a pathogen. There is no difference in the chemical make-up of the two forms except that their conformations, folding is different. Research into the mechanisms in which a cellular prion protein becomes mis-folded remains unclear. This remains a problem because there is no effective way to combat the infections from such diseases. Unlike cancer or other pathogens that spread through the body by passing genetic material to the host cell and use it to generate more copies of themselves to infect more cells. Prion diseases cannot be targeted like cancer by finding cells that are rapidly multiplying, because the genome of the cell is not being affected only the proteins are. As a consequence the infectious agents can also be much smaller than viruses and bacteria.
The Prion Hypothesis
This idea that proteins can infect other proteins without the use of nucleic acids to transfer genetic material is contested. Part of proving the hypothesis is that the mechanism for the mis-folding is unclear. A search for possible cofactors that might facilitate the changes in conformations of the prion proteins could better explain and prove the hypothesis.
Bacterial proteins are the most powerful human poisons known and belong to two broad categories: lipopolysaccharides (Gram-negative bacteria) and proteins, which are released from bacterial cells. Endotoxins, which are structural components of bacteria, are cell-associated substances that a located in the cell envelope and can be released from growing bacteria or lysed cells as a consequence of effective host defense mechanisms or antibiotics. The extracellular diffusible toxins are referred to as exotoxins and are usually secreted by bacteria during exponential growth. Exotoxins are usually polypeptides that act at tissue sites remote from the original point of bacterial invasion or growth. The location for activity of a particular toxin, like Botulinium, is determined by the site of damage. Enterotoxin, neurotoxin, leukocidin and hemolysin are terms that describe the target site of well-defined protein toxins. Although the tissues affected and the target site may be known, the exact mechanism by which toxins cause death is not clear and is subject to debate.
Botulinum neurotoxins (BoNTs), a family of bacterial proteins produced by the anaerobic bacteria Clotridium botulinum, and the causative agent of botulism, is acknowledged to be the most poisonous protein known. Botulism poisoning is a serious and life-threatening illness in humans and animals. BoNT proteases disable synpathic vesicle exocytosis by cleaving their cytosolic SNARE substrates. There are seven distinct BoNT isoforms (A-G), which show strong amino acid sequence similarity. Human botulism is caused by the BoNT serotypes A, B, E and F. Interestingly, type A is used for various cosmetic and medical procedures, more commonly known as Botox.
BoNTs exert their neurotoxic effect by a multistep mechanism: binding, internalization, membrane translocation, intracellular traffic and proteolytic degradation. The activated mature toxin consists of 3 parts: the N-terminal light chain (~50 kDa), the heavy chain (100 kDa) that encompasses the light chain (HN) and the receptor-binding doman (HC). HC determines the cellular specificity with a protein receptor (SV2 or Syt depending on the isoform) and a ganglioside. HN is a helical bundle that chaperones the light chains across endosomes where it is driven by a transmembrane proton gradient. Then, BoNTs enter the cells via receptor-mediated endocytosis, induces a conformational change and the light chains (LCs) cleave the unique components of the synaptic vesicle docking-fusion complex known as SNARE. As a result, cleavage of SNARE nullifies vesicle fusion and synaptic transmission, which causes the severe paralysis characteristic of botulism.
Tetanus toxin is a very powerful neurotoxin produced by the vegetative cell of Clostridium tetani in conditions that lack oxygen (anaerobic). As the bacterium matures, it developed its characteristic terminal spores which also give them advantage by increasing the bacteria's resistance to heat and most antiseptics. The toxin cause tetanus, a fatal disease that involves unfavorable muscle spasms that can cause respiratory failure and even death. The LD50 of this toxin has been measured to be approximately 1 ng/kg, making it the second most deadliest toxin in the world after the Botulinium neurotoxins.
The mechanism of the toxin is it first travels through the vascular and lymphatic systems of the body, disrupting the neuromuscular junctions and the central nervous system. Tetanus toxin blocks the release of inhibitory gamma-aminobutyric acid (GABA) and glycine by degrading the protein synaptobrevin. This causes the failure of regulating motor reflexes by sensory stimulation, which leads to the muscle depolarizing even with the smallest of action potentials. This continued depolarization causes the antagonist and agonist muscles to contract simultaneously and this generalized contraction causes the symptom known as tetanic spasm.
Diphtheria Toxin is a bacterial exotoxin caused by Corynebacterium Diphtheriae. This toxin exists as a single polypeptide chain, about 60,000 daltons in molecular weight. Outside of the cell, the toxin is produced in its inactive form, later to be activated by trypsin, a proteolytic enzyme, in the presence of thiol. Thiol acts as a reducing agent during this activation process. The toxin consists of two parts: Fragment A and Fragment B. Fragment A, which is responsible for the catalytic activity of the toxin, is masked until it reaches the target cell. The hydrophobic portion of the toxin, named Fragment B, is responsible for interacting with cell membrane receptors on the target cell surface.
Diphtheria toxin may enter a target cell via direct entry or receptor mediated endocytosis. In direct entry, the toxin binds to a target cell surface receptor. This binding induces the formation of a pore on the cell membrane, allowing the catalytic chain of the toxin to enter the target cell’s cytoplasm. During receptor-mediated endocytosis, the toxin is placed in a vesicle, where the pH drops, allowing both Fragment A and Fragment B to unfold. The hydrophobic regions of both chains then enter the vesicle membrane. Next, reduction and proteolytic cleavage of the A chain is released into the cytoplasm, where it regains enzymatic conformation.
Diphtheria toxin utilizes NAD as a substrate, and catalyzes ADP ribosylation, where the ADP-ribose portion of NAD combines with elongation factor-2 (EF-2). This process inactivates protein synthesis (in animal cells), resulting in cell death.
Corynebacerium Diphtheriae consists of two subunits. Subunit A contains an NH3+ group and is responsible for the enzymatic activity during the inhibition of EF-2. This inhibition interferes with the protein synthesis, resulting in cell death. Subunit B contains a carboxylic acid and a Hinge loop, which permits movement of the regulatory domain. Subunit B allows the toxin to bind to the membrane of a target host cell. This subunit possesses a region, known as T (translocation) domain, which is inserted into the target cell’s membrane, thus ensuring the release of Fragment A (catalytic component of the toxin) into the cytoplasm of the host cell.
Ubiquitin is a highly conserved, heat-stable protein found only in eukaryotic cells. It is made up of 76 amino acids and is involved in many cellular processes. It plays a big role in regulating the cell cycle, including DNA repair, embryogenesis, the regulation of transcription, and apoptosis.
The Ub genes exist in two states:
The ubiquitin and ribosomal protein gene that are fused together to make translation products called Ub-ribosomal fusion proteins.
A polyubiquitin molecule: Ub molecules can fused together to make a linear chain of repeated Ub-molecules.
These fusion proteins can be cleaved by protein Ub-C-term hydrolase that can detach an individual UB and ribosomal protein (cleave Ub-ribosomal fusion proteins) or a set of Ub monomers (cleave polyubiquitin molecule).
Ubiquitin’s protein structure is a compact β-grasp fold featuring a flexible C-terminal tail with six residues, and a core with rigid residues as seen in Figure 2a. Despite these rigid core residues, the flexible β1/ β2 loop that contains Leu8 (as seen in Figure 2b) plays a crucial role in allowing the recognition of ubiquitin by ubiquitin-binding proteins. The fact that only three conservative changes are observed from yeast to man indicates the importance of conserving ubiquitin’s structure as preserved by evolutionary pressure to resist change. This is important in facilitating the consistency of recognition of ubiquitin by ubiquitin-binding domains, also known as UBDs.
A hydrophobic surface comprised of the residues Ile44, Leu8, Val70, and His68 (as seen in Figures 2a-c) facilitates the recognition of ubiquitin by other proteins. The different residues built into the ubiquitin structure uniquely contribute to the interactions of ubiquitin with other proteins and to ubiquitin’s many different functions. One example is Ile44 which plays an important role in cell division because it binds proteasomes and most ubiquitin-binding domains. Another example is Ile36 which serves as a mediator for interactions between ubiquitin molecules conformed as chains. This residue is recognized by and specifically interacts with ubiquitin-binding domains (UBDs), deubiquitinating enzymes (DUBs), and HECT (Homologous to E6AP C terminus) E3s which is a type of ubiquitin ligase. Residues can also work together to perform particular functions of ubiquitin. For example, Gln2, Phe4, and Thr12 work together to facilitate cell division in yeast (as shown in Figure 2c). Also, ubiquitin’s TEK-box in higher eukaryotes which features Thr12, Thr14, Glu34, Lys6, and Lys11 plays an important role in mitotic degradation (also shown in Figure 2c). These are only few of the many structural features of ubiquitin and their roles in the many different functions of ubiquitin.
The most essential parts of the ubiquitin structure can be found in the N terminus with its seven lysine residues. These residues serves as chain assembly attachment sites in the process of ubiquitylation, a regulatory mechanism of the cell in which polymeric chains of ubiquitin are used to modify proteins and ultimately decide their fate in the cell. As in Figure 2d, these lysine residues are positioned in the three-dimensional structure in such a way that they face different directions and cover all of ubiquitin’s surfaces. The most dynamic area of ubiquitin’s structure features two particular lysine residues: Lys6 and Lys11. These regions are subject to conformational changes once ubiquitin associates with UBDs or while ubiquitin is in a chain conformation.
Ub primarily exists to regulate protein turnover by regulating degradation of specific proteins. This is a very important process in the cell. By quickly eliminating a particular regulating protein, a turn on of a gene expression can be prevented. Proteins that are to be degraded are tagged by Ub and are then recognized by another protein called 26S proteosome to be degraded. Ub depends on ATP to mark specific proteins to be degraded.
The Ub itself does not degrade proteins but slows down the rate of dissociation between proteasomes and substrate proteins.
There are three types of enzymes that participate in the process: E1- Ub-activating enzymes (turn Ub into reactive state), E2- Ub conjugating enzymes (aid the linking process between Ub and substrate protein), E3- Ub ligases (work together with E2, role mainly on recognizing the substrate protein).
The general reaction pathway starts when Ub is activated by E1 in the present of ATP. After that, E2 and E3 work collaboratively to recognize the substrate protein and conjugate Ub to the substrate. From then the ubiquinated protein is ready for degradation.
These three enzymes can catalyze the binding of a substrate lysine and the C terminus of ubiquitin. This leads to monoubiquitylation. When multiple lysine residues be bound, multimonoubiquitylation will occur. Ubiquitin also can form polymeric chains when the N terminus or one of the lysine residues attached to a substrate. If the chains are elongated by the same residue, this will be called homogeneous ubiquitin chain. If the chains are elongated by the mixed residues, this will be called mixed ubiquitin chain. However, only monoubiquitylation and four homogenous chain types have found that have outcomes in the cell.
Though not fully understood, there are a few theories to help understand what determines a protein will be marked by Ub
N-degron: In 1986, Alexander Varshavsky observed that there is a correlation between the half life of a protein and its N-terminal residue. This suggested that one could predict the lifespan of the protein by its N-terminal amino acid.
Certain amino acid sequences such as PEST signal protein degradation. The PEST sequence is rich in proline, glutamic acid, serine, and threonine. It has been seen that removal of the PEST sequence in the protein increases protein half life.
Mutant proteins revealing degradation signals are more prone to degradation than a normal protein. Usually the signals would be hidden away in the hydrophobic core, but sometimes mutation causes a partial folding that exposes the signal to Ub.
Komander, David and Rape, Michael. “The Ubiquitin Code” Annual Reviews Biochem.
Overview
Cdc48 is a hexamer protein that belongs to a group of enzyme called ATPase. Past research has shown that it helps to control the quality of protein activities such as: endoplasmic reticulum associated protein degradation (ERAD), transcriptional and metabolic regulation, DNA damage response, and many more. New research shows that Cdc48 regulates the degradation of damaged proteins by helping to store them in aggresome when the cell is under stress and later to destroy the damaged proteins.
Structure
Cdc48 is a hexamer protein. It has central core with six protomers attached to it to form a ring. The four domains that make up a protomers an amine terminus, AAA region (D1 and D2), and a carboxyl tail. There are twelve ATPase active sites for ATP hydrolysis reaction. When a hydrolysis reaction occurs, Cdc48 changes its shape by maneuvering the six protomers that allows for the opening and closing of the rings. Furthermore, different temperatures allows for different parts of the domain to be more activity than others. For example, D1domain works at its maximum at the 60˚C while D2 domain works better at 37˚C. In addition, the conformation change is necessary because it affects the function of the protein. If the protein does not work, it can result in a cell death. Furthermore, the carboxyl tail can be phosphorylated and acetylated that will affect the protein’s ATPase activity, localization, and binding ability.
Function
The function of Cdc48 depends heavily on its cofactor. Each cofactor programs that protein to perform a specific function. Meanwhile, the substrate-processing factor will bind to the protein to direct the protein’s path. One of the best known functions of the protein is in endoplasmic reticulum-associated protein degradation. Cdc49 forms a complex with Udf1 and Np14, which then bind to an ERAD ubiquitylated substrate. Consequently, this action causes a chain reaction that leads to the damaged proteins to be degraded in the proteasome. Another role that Cdc48 takes part in is mitochondrial protein degradation. Here, Vms1 replaces Ufd1 to form a complex with Cdc48. When cells are exposed to rapamycin or hydrogen peroxide, this activates the Cdc48 to be moved to the mitochondrial membrane to degrade selected mitochondrial proteins in a process called UPS. New research showed that a protein called E3 ligase Parkin and Cdc48 help to prevent damaged mitochondrial proteins from fusing with healthy ones. Cdc49 also plays a role in cytoplasmic protein degradation. Similar to ERAD, Cdc48 form the same complex. However, its role differs by removing the ubiquitylated enzymes from the glucose-induced degradation deficient (Gid) complex to create loose ends so that the damaged proteins can go into the proteasome to be degraded. In the nucleas protein degradation, Cdc48 complex helps to unfold and disassemble defected proteins with of its cofactors Ubx4 and Ubx5. Ribophagy is another process that depends on Cdc48. Here, mature ribosomes are degraded to make sure that the cell can survive. Cdc49 pairs up with Ubp3 and Bre5 to deubiquitylate the ribosome.
Sources
Buchberger, Alexander, Hilt, Wolfgang, Stolz, Alexandra, Wolf H. Dieter. “Cdc48: a power machine in protein degradation”. Trends in Biochemical Sciences. October 2011.
TGFβ
The transforming growth factor beta is a protein that controls many cellular responses. TGFβ is an important multifunctional cytokine. There are three types of TGFβ: TGFβ1, TGFβ2, TGFβ3. These isoforms have similar functional properties that are non-overlapping and have distinct phenotypes. Furthermore, the forms are expressed by many different cell types and many are capable to respond to TGFβ. The TGFβ isoforms are encoded by separate genes. The protein is produced in its inactive form until integrins activate TGFβ so that it can bind to its receptor to activate the cascades of signals that result in modulation of gene transcription. There have been many different processes that have been experimented in labs to activate TGFβ such as heat, acidic pH, reactive oxygen, and proteases. It is important that TGFβ is produced in its inactive form that makes sure that TGFβ’s potent effects are only apparent during appropriate surroundings and time.
TGFβ Signaling Pathway
The activated TGFβ binds to TGFβRII. Then, TGFβII phosphorylates TGFβRI to activate the cytoplasmic domain’s kinase activity. This create TGFβ-ligated tetrameric receptor complex. The activated cytoplasmic domain phosphorylates Smad2 or Smad3. Then, the phosphorylated Smad2/3 complex binds to Smad4 and migrates to the nucleus. Next, the Smad2/3/4 complex binds to Smad-responsive elements. This can either start gene transcription or repress it. This pathway can be inhibited by Smad7 by competing with Smad2/3 to bind to TGFβ. In addition, other pathways that TGFβ can start are MAPK, Wnt, Notch, and PI3K.
Integrins – Activators of TGFβ
Integrins are large family of cell adhesion and signaling receptors. There are two subunits, α and β, that bind together to form a heterodimeric type 1 transmembrane receptor. There are six types of integrins: αvβ1, αvβ3, αvβ6, αvβ8, α8β1. The αvβ3 and αvβ5 integrins activates TGFβ in fibroblastic cells. Research in mice with defective αvβ3 and αvβ5 show that autoimmune disease scleroderma disease. Increased expression of these integrins resulted in lung fibroblast-to-myofibroblast differentiation that plays a role in pulmonary fibrosis. On the other hand, integrin αvβ6 is usually expressed in the epithelial cells. In research, mice that do not express αvβ6 show signs of mild inflammatory phenotype to the skin and lungs. This shows that the αvβ6 helps with skin and lung cells in controlling immune homeostasis. Integrin αvβ8 is the integrin that is expressed in the most cell types. Mice that do not have αvβ8 do have αvβ8 complex with transforming growth factor beta, that results brain disease. Other problems that these mice would have are cleft palate and chronic pulmonary disease. Thus, the results show that αvβ8 complex with TGFβ is important in controlling neurovascular development. More results that the complex influences homeostasis in the immune system.
TGFβ Activation with Integrins
TGFβ is activated by integrins by protease-independent and dependent mechanisms. In order for TGFβ to bind to its receptors, the protein has to change its shape in order to not be masked by LAP. The latent TGFβ complex binds to ECM by interacting with integrin αvβ3 or αvβ5 receptors . This will cause the TGFβ protein to change its shape. Then, αvβ8 bind to latent TGFβ that will result in the cleavage of LAP and release of active TGFβ to send signals to other cells.
References
Worthington, John J., Joanna E. Klementowicz, and Mark A. Travis. "TGFβ: A Sleeping Giant Awoken by Integrins." Trends in Biochemical Sciences (2010): n. pag. Print.
The AB5 toxins are vital virulence factors for several bacterial pathogens. AB 5 toxins are one of many virulence factors deployed by major bacterial pathogens, which collectively kill over a million people each year. AB5 toxins have recently become of interest for disease pathogenesis, due to the widespread and severe bacterial infections that have resulted from the action of AB5 toxins.
The AB5 toxin is a group of polypeptide chains, also known as a protein complex that pathogenic bacteria secrete in order to assist in overtaking a host. The protein complex is composed of six components; five "B" Subunits (binds to the glycan receptors on the host cell) and one "A" subunit (the toxic subportion that disrupts host functions). The B subunits of the protein complex form a ring that the A subunit is attached to. By doing so, the protein complex is allowed to function; the B subunits attach to the cell while the A subunit employs its toxicity.
This Toxin is important because of its presence in many common or important pathogens including, but not limited to:
-Various forms of Escherichia Coli; Heat-Labile Enterotoxins (Diharrhea)
The study of this toxin is interesting as it provides researchers with valuable insight on how bacteria and cells function. In fact, researchers are investigating ways to incorporate the concept behind this toxin into treatment for different diseases.
In the past two decades, close to 30 AB5 crystal structures have been depicted. These depictions have proven to provide significant structural insights into the biological function and catalytic activity of the holotoxins. Based on sequence homology and catalytic activity, AB5 toxins have been classified into four separate families. Although the toxins share similar structural aspects, they still differ in their host cell surface receptor specificity, catalytic activity and intracellular trafficking.
Millions of people die each year due to bacterial infections. Many of these infections are caused by AB5 toxins that are released by bacteria that humans come into contact on a regular basis.
For example:
E. Coli: People that reside in or are visiting developing countries are at a high risk for exposure to E. Coli and the symptoms it causes.
Vibrio Cholerae: Epidemic cholera outbreaks all over the world that result in the death of anywhere from dozens to thousands.
Shigella Dysenteriae: Similar to E. Coli, dysentery causes gastrointestinal issues that sometimes lead to life-threatening conditions such as systemic sequelae and haemolytic uraemic syndrome. Suprisingly enough, haemolytic uraemic syndrome caused by dysentery has a higher mortality rate in adults then in children.
The A-Subunit of the AB5 Toxin is the part that actually conducts the attack on the target cell. The subunit itself is broken down further into two parts linked through a disulfide bond that forms a polypeptide.
A1: The first part of the A-subunit is the part that actually contains the toxins that disrupts host cell activities
A2: The second part of the A-subunit connects the A1 part with the B-Subunits, linking all of it together and allowing it to function as one cohesive unit.
Furthermore, this subunit is divided into different families according to their catalytic activity and their homology. Each family contains different forms of the same toxin. These different forms share a very similar amino acid composition with the other forms in the same family; toxins from different familys may share some similarities in amino acid composition but the percentage is significantly lower. Some toxins utilize similar methods of attacks even though they derive from different families. For example, both the toxins for Cholera and Whooping cough disrupt the "G-Protein Signal Transduction Pathways" which leads to a failure in the ion transporting system in the cell. Although they result in similar failures, these two toxins ultimately cause different symptoms to be expressed. The toxins in the STX family in which dysentery derives from attacks the cell by disrupting the way the cell synthesizes proteins. It does this by causing an abnormality in the nucleotide sequence in the rRNA of these cells. In doing so, it causes cell death by denying the cell the ability to synthesize proteins.The last family of AB5 toxins is classified as the SubAB toxins due to their subtilase like cytotoxin. This family of AB5 toxins targets Binding immunogloblin protein (BiP) and acts as a protease dismantling BiP. This disrupts the protein folding process in the cell and therefore eventually causes the fatality of the cell.
This subunit is composed of five monomers arranged in a pentameric structure.The B-Subunit is tasked with both identifying the host cell and transporting the toxic A-Subunit to and into the cell. For this reason, the identification of the correct cell is very important for the toxin. The CTX, STX, and SubAB families share a similar structure while the PTX family is composed differently. However, each family responds to and seeks out different glycan cell receptors. They also have a varying amount of binding sites on each monomer of the B-Subunit; varying from one on the SubAB and CTX family to three on each monomer of the STX family. These toxins look to bind to different glycolipids and glycoproteins on the surface of cells and sometimes even use the antigens present on blood as receptors. The toxins that utilize blood as a host cell may recognize certain types of blood cells better than others causing humans with certain blood types to be more susceptible to cholera and e. coli attacks. An interesting case to observe is the inability of humans to synthesize the sialic acid Neu5Gc. Humans still produce Neu5Ac, a similar sialic acid; however, it is believed that humans stopped producing Neu5Gc as a method of avoiding infection due to AB5 toxins.
AB5 toxins as Cellular Tools and Novel Therapeutics
AB5 toxins have been used to specifically manipulate associated signaling pathways in cells. Studying the B-Subunits of AB5 Toxins has helped scientists to understand receptor pathways better by allowing them to view the steps that the toxin takes. AB5 toxin B subunits have been utilized to help counter certain allergens. In essence, by isolating only the non-toxic portion of the AB5 toxin, it is possible to utilize them in ways that may be beneficial. An example of this would be the B-Subunit of the CTX family which has been used in mice to suppress certain allergic reactions. Also, the B-Subunit of the STX family may provide the basis for an anti-tumor vaccine. Both the CTX family and SubAB family may quite possibly have a large effect on inflammatory responses of cells; they have potential as immunomodulatory agents.
In addition to their potential use as immunomodulatory agents, AB5 toxins can be used as therapeutic agents against a range of diseases. AB5 Toxin research is very promising in the fight against cancer. The B-Subunit of these toxins can bind to the glycan sites that many cancer and tumor cells exhibit. The A-Subunit possess the ability to kill the cancer cell by obstructing its protein synthesis ability. One of the issues is how to target only cancer cells since the toxins do exhibit the tendency to target normal cells as well as cancer cells. The Sub-AB shows the most promise as it prefers to bind to glycans that humans cannot synthesize (but uptake through diet).
Plant homeodomain (PHD) zinc fingers are conserved modules found in proteins that modify chromatin and mediate the molecular interactions in gene transcription. PHD zinc fingers were originally discovered to play a role in gene transcription and recognition of lysine-methylated histone H3. Recently, studies have shown that PHD fingers also have a sophisticated histone sequence reading ability that is set by the interplay between various histone modifications. These studies emphasize the functional aspects of PHD fingers as genome readers that can control gene expression through molecular recruitment of multiprotein complexes of chromatin regulators and other transcription factors.
The plant homeodomain consists of approximately 50 - 80 amino acid residues of various sequences containing a certain zinc-binding motif that shows up in many chromatin-associated proteins. The PHD folding pattern consists of two anti parallel β-sheets and a C-terminal α-helix, which is stabilized by two zinc atoms. The PHD fingers read the N-terminal tail of histone H3 (methlyation of H3K4 and to a smaller extent the methylation state of H3R2 and the acetylation of H3K14).
PHD domain
It has been reported that all PHD finger structures bind histone H3 through interactions with the first six N-terminal residues of H3, with the exception of two residues.
The reason that PHD fingers are so fascinating to scientists is that these domains are the smallest in size, which allows them to be versatile epigenome readers. More importantly, the structure-function relationships of the PHD fingers revealed from these recent studies illustrate how functional diversity of a protein module can be achieved by evolutionary changes to the structural features or amino acid residues near ligand binding sites. Also, because of PHD's low sequence conservation and adaptable structural plasticity, it will not be surprsing to see other modifications occur in the PHD recognition domains in the future.
The BPTF-PHD structures reveal the main characteristics of PHD fingers that can read H3K4me3. The binding occurs through an aromatic cage where a trimethyl ammonium group is stabilized by van der Waals and cation-–π interaction, which is similar to the ones observed in chromodomain, MBT, PWWP, and Tudor domains. This aromatic cage is composed of one Trp and three Tyr residues; and it has three faces and a 'lid' that is beyond the tip of H3K4me3. Subsequently determined structures of other fingers in complex with the H3K4me3 peptides show that the cage varies and can contain a combination of two to four aromatic and hydrophobic residues. These residues that participate in aromatic cages tend to appear at similar positions within the sequence. At the most conserved position is the invariable Trp residue that is at the beginning of the β1 strand which is then followed by the aromatic or hydrophobic residue that starts at the β2 position. Generally, the residues that are used to form the aromatic cage exists in parts of the structure that are rigid, such as β-strand or close to the Zn-coordinating Cys residue. At minimum, two aromatic residues that include the invariable Trp at position 1, appears to be necessary for the H3K4me3 binding.
The simplest observed so far aromatic cage is that of jumonji, which is the AT-rich interactive domain 1A (JARID1A), and is composed of only two Trp residues that are at both positions 1 and 2. Together with JARID1A, the aromatic cage of recombination active gene 2 (RAG2) and myeloid/lymphoid or mixed-lineage leukemeia-1 (MLL1) lack the 'lid' residues that are present at position 3 in all other PHD finger aromatic cages. Additionally at Y1581, which is position V, in MLL1 undergoes a conformation change when binding that is not observed within the other PHD fingers. There is a slight variation in H3K4 binding region which is observed in the PHD fingers of yeast homolog of mammalian ING1 (Yng1), transcription initiation factor TFIID subunit 3 (TAF3), pygopus homolog 1(PYGO), inhibitor of group protein (ING4), and other PHD fingers of the ING family that have charged (Asp) or hydrophilic (Ser) residues that are close to the H3K4 residue at position 4 or 5. Of those, only the charged residues at position 5 in PYGO plays a role in the methylated lysine binding by slightly shifting the affinity in favor of the dimethlyated form of K4 (H3K4me2), making the affinities of the free PHD for both H3K4me and H3K4me2 virtually identical, and it shows a slight preference for H3K4me2 over both H3K4me1 and H3k4me3 in the PHD-HD1 complex. Even though the Ser and ASP residues in the other PHDs contribute to H3 binding, they do so through interactions with residues other then the K4.
Mutational studies in the BPTF suggest that the presence of a negatively charged residue in aromatic boxes can alter the binding selectivity of the other PHD fingers. Affinities in the wild-BPTF, which shows a preference for the H3K4me3 over the H3K4me2, change when mutated thus resulting a preference in H3K4me2 over the H3K4me3. Thus the relative affinity of H3K4me3 versus H3K4me2 can be modulated by subtle changes within the sequence of the PHD fingers. The biological impact of these small differences in affinity however, are not clear.
RNA polymerase II accurately transcribes because of the TATA box's directions. TATA box is the core promoter's predominant DNA element. TATA-binding protein is what recognizes the TATA box. TATA-binding protein is found to have strong preferences for the TATA, shown through structural experiments and in vitro binding. Severe DNA bending is also induced by the TATA-binding protein. NC2 and Mot1p regulate the TATA-binding protein turnover at TATA boxes. It is proposed that the TATA-binding protein acts with NC2 and Mot1p to bend TATA and releases the TATA-binding protein at a more rapid pace from promoters of TATA. This occurs in vivo.
RNA polymerase II transcription: TATA, TFIID, and TATA-binding protein discovery
Using Drosophila melanogaster genes, David Hogness and Michael Goldberg discovered that the DNA sequences was found to be rich in A and T. This was also later found in many eukaryotic and viral genes. In eukaryotic genes, the sequences rich in A and T are present on the 5’ start site of mRNA. The TATA box was known as the ‘Goldberg-Hogness’ box, due to its discoverers. The TATA box is really important in the initiation of accurate transcription, which was underscored by mutational analysis. The first steps in assembling pol II pre-initiation complex was marked by the basal transcription factor recognizing the TATA box because of the TATA-binding protein. TATA and TATA-binding protein characterizes an unusual DNA-protein complex.
TATA-promoters is a core promoter, which control viral genes, representing cellular promoter minorities. Five general factors that is required for in vitro transcription has been identified from the strong promoters of TATA box. TATA-binding protein and TFIID were the first to stably bind to the template in directing the assembly of pol II.
Based on the quality of TATA box, TATA-binding protein and TATA sequences can form stable complexes in vitro. This selects the pol II promoters to use in transcription. TATA minor groove binding is mediated by the concave hydrophobic surfaces of TATA-binding surfaces.
The minor groove widens as the beta form goes in and out of the complex
The first T A step and in between the last 2 base pairs is inserted by two pairs of phenylalanine. This creates sharp kinds and bending towards the major groove.
TATA-binding protein is enhanced because of the bending towards major groove.
Reduced bending is due to the sequence TATAAAAG.
A stable complex is yielded by slower isomerization.
TATA-binding protein and TATA dissociates slowly.
In vitro, compared to TATA sequences, TATA-binding protein shows less affinity to non-specific DNA.
In eukaryotic cells, few free TATA-binding proteins are found. TATA-binding protein can also be found in other complexes besides TFIID. Other elements, such as INR, MTE, DCE, and DPE do not contact TATA-binding protein, but they contact TAF of TFIID.
It is proposed that the role of in vivo TATA box for transcription of pol II is to help in dissociating TATA-binding protein of pol II promoters. NC2, ATPases BTAF1, or Mot1p regulators can regulate TATA-binding proteins to release TATA-binding protein quickly from TATA.
Gene specific regulators, basal transcription, and cofactors regulate TATA-binding protein activities. TFIIA is related to NC2 inhibition relief of basal transcription and activated transcription. TFIIA and TFIIB basal factors are blocked by NC2 and TATA-binding protein – TATA complex binding. Non-TATA sequences can be recognized by TATA-binding protein by structural changes due to NC2.
MOT1 is shown to positively and negatively regulate pol II transcription. Yeast genes encode Mot1p and NC2 subunits. Mot1p and TATA-binding protein complexes have a large affinity to DNA while it has a relaxed one for TATA. NC2 and TATA-binding protein complexes do not really prefer TATA sequences. NC2 and TATA-binding protein does not have BTAF1 in human chromatin. TATA-binding protein in pol II transcription is regulated by the collaboration of BTAF1 and NC2.
in vivo Dynamic Regulation of TATA-Binding Protein Mobility
Chromatin structures allow for cells to have TATA-binding protein activity. This activity is regulated by core promoters’ restricted access. Barriers from nucleosome promoters must be defeated. Experiments have shown that TATA binding sites are competed for by nucleosomes and TATA-binding protein. Some observations have shown that nucleosomes have low density in promoters.
Promoters containing TATA have highly regulated transcription and it is usually tissue-specific. In cells, it is very unlikely that TATA is the function that directs TATA-binding protein to PIC assembly. Van Werven et al. demonstrated that the turnover of TATA-binding protein to TATA promoters is largely significant, compared to promoters of non-TATA. This turnover is really dependent on Mo1p, as well as NC2, and SAGA action. Lacking in TATA sequences compared to promoters of pol II, promoters of pol I and pol III have lower TATA-binding protein turnover. These findings indicate that the TATA-binding protein turnover of pol II promoters is contributed by the sequence TATA.
Rapid TATA-Binding Protein Dissociation in vivo, Main Player: TATA
The bending of DNA is really important in distinguishing the functionality of TATA-binding protein between the promoters that contain TATA and the promoters that do not contain TATA. TATA box binding by TATA-binding protein is key in creating the DNA bent conformation. DNA's strained conformation can be released by BTAF1 and NC2, helping the TATA-binding protein dissociation with TATA. In humans, in vitro experiments have shown that NC2 can reduce this bending alone at sequences of TATA box.
It has been proposed that TATA-binding protein cannot be released as quickly as the promoters containing TATA, with the activities of BTAF1 and NC2. SAGA complexes may be involved in the removal of TATA-binding protein from promoters, which can inhibit the TATA-binding protein binding promoter in vitro.
Two Functional Classes, Eukaryotic Core Promoters Dissection
It has been shown that Mot1 and NC2 can repress TATA dependent transcription and activate DPE dependent transcription in insect cells. In yeast, TATA promoters are repressed by Mot1p and NC2. Yeast needs the SAGA complex for promoters containing TATA and recruitment of TATA-binding proteins. Developmentally regulated genes are promoted by promoters that contain TATA to initiate direct transcription at a specific site in mammalian cells. Yeast promoters that do not contain TATA are TFIID dependent.
There are multiple chaperones and co-chaperones in the nucleolus. Chaperones are components of a larger network that promotes protein homeostasis. Studies show that cellular chaperones' network have individual compartments that have specific functions, which then even makes the cellular proteostatsis network more efficient. These unique network chaperones and multitasking proteins are present in the nucleoli, which supports and controls many important biological processes.
In eukaryote cells, many organelles have specific functions. The nucleus directs the largest number of necessary cellular processes. Within the nucleus, there is a nucleolus that is the most prominent compartment, contain a vast number of proteins, so it is therefore associated with many diseases and pathologies. The nucleoli functions due to a of a network or chaperones, co-chaperones, and multitasking proteins all work together to direct activities and adaptions due to stress.
Chaperone proteins helps the folding and unfolding of biological molecules. For example, RNA chaperones control RNA quality and play very important roles in ribosomes biogenesis. Chaperone proteins are also responsible in aging, cancer, protein folding, diabetes, and much much more. Chaperones need networks of co-chaperones and heat shock proteins for processes to work. Thus, their cooperation is organized into dynamic networks that promote effective interaction. Heat shock proteins HSP70s and HSP90s are critical in protein homeostasis to make sure polypeptides are functional and structured, protect cells from proteotoxicity. They bind to proteins. Each chaperone networks are separated by their distinct function.
The network of chaperones and multitasking proteins are in the nucleolus. Scientists have studied the HeLa cell nucleoli which helps them making some interesting conclusions. The abundance of chaperones and co-chaperones in nucleoli is regulated by the physiology of cells, but the quantitative data is unknown. Heat shock protein families were detected in the study by proteomics. Some of these proteins have multiple functions that contribute to many events and biological activities, known as nucleolus multitasking proteins (NoMPs). NoMPs can be in the nucleolus, nucleoplasm, and even the cytoplasm. Research with yeast proteins shows that nucleoli has a chaperone NoMP network committed just to have specific functions, which was tested with an assembly of possible networks that incorporated RNA chaperones, protein kinases, and phosphatases. Nucleolar chaperones NoMP network is made out of a unique set of protein factors, and it is dynamic to modulate nucleolar activities upon cell physiological changes.
There are many roles of chaperone networks. Roles of chaperone/NoMP network are specific to its clients, requiring coordination between co-factors and the designated group of chaperones. There are 4 types of proposed nucleolar networks. One of them is that chaperone networks are composed of multiple abilities that secures coordination of nucleolar processes. Another suggested network is centered around HSP90, its co-chaperones, and NoMps. HSP90 along with CDC37 inputs protein kinases for activation. In the nucleolus, HSP90 is associated with controlling CK2 activity, important for biological function. It's postulated that HSP90-CK2 regulates the activity of RNA polymerase I and ribosomes biogensis. Moreover, since CK2 is required to re-organize the nucleolus filaments due to changes in stress, the network is also responsible for proper nucleolar organization. A 3rd proposal is that chaperone HSPA8 also participates in the organization of the nucleoli by associating with fibrillarin. Protein chaperone networks are involved in a multitude of functions, including regulation of cell cycle, signaling events, and protein turnover.
Nucleolar chaperone / NoMP network promotes survival of the cell, because the processes they control are vital for growth conditions. The networks provide assistance to complete the tasks. Changes in stress or environment or viral infections can change the demand for chaperone or NoMP activities. Because of this, it's implied that nucleoli are at the center of essential cellular activities. NoMP network offer unique opportunities for therapeutic intervention.
HSP90 is a molecular chaperone network which leads to homeostasis through modulating protein-DNA dynamics of components involved in RNA transcription, telomere maintenance, DNA repair, and DNA replication. It displays a protein binding capacity with an affinity for short hydrophobic amino acid motifs. These chaperones usually have short, low affinity interactions with their target protein to avoid interfering with the protein’s activity.
Prokaryotes are known to typically contain one HSP90 gene which isn’t essential, while eukaryotes contain several HSP90 genes that are known to be essential.
HSP90 along with the p23 cochaperone are both proteins that interact with the protein subunit of a human telomerase (hTERT) and contribute to its enzymatic activity. HSP90 proteins have been known to promote DNA binding and nucleotide affinity for telomerase, and so HSP90 aids in maintaining telomere DNA length.
DeZwann, Diane C., and Brian C. Freeman. "Trends Biochem Sci." Trends Biochem Sci. (2010): n. page.
Background
On a microscopic scale, macromolecular structure is seen as a reflection of a specific function. Scientists have concluded that structural patterns found in genes and gene products serve as a key in understanding function. Each macromolecule exhibits certain characteristics, distinct of all other macromolecules. This allows for researchers the ability to interpret molecular function as a product of structure. Albeit this is often mislabeled as a simple and efficient process, the complexity and intricacy required in relating structure to function is immense.
Problem
A major issue facing modern biochemists is the nature in which those structures are interpreted and how they translate into function. Despite common misconceptions this process has been the focus of hundreds of research papers. As a result, structures including genes and gene products have been a focus of study by researchers. In order to decode molecular structure, researchers have employed several techniques, each addressing different attributes. Each technique or method addresses a different attribute. As a result, if a researcher is to observe a certain functional attribution, they must employ the appropriate method.
Interpretation
For example, a certain technique requires a cellular protein to be isolated and prepared in a buffer solution as to allow for the detection of a particular catalytic activity. After the addiction of a substrate, if a certain enzymatic activity is detected by the researcher, that specific activity is then attributed to the cellular protein in question. The formal term attributed to that type of activity would be known as biomedical function. In this particular experiment, the effects observed would be directly attributed to the protein in question. On a different note, if a protein were to be changed by the deletion or modification of its amino acid sequence, major phenotypic changes may be observed in the resulting cellular content. The observed changes may consequently be attributed to the gene and gene product which are associated with protein function. Although minute, such functional changes can have major impacts and are crucial to bodily systems.
The buildup of unfolded proteins in the endoplasmic reticulum (ER) activate sensors like inositol-requiring enzyme-1α (IRE1α). These activated sensors carry out the unfolded protein response (UPR) by creating a protein platform called the UPRosome. The speed and strength of the UPR responses are regulated by a group of modulator and adaptor proteins. Stress to the ER can cause apoptosis (programmed cell death), but some of the apoptotic proteins will interact with IRE1α. This interaction will have two functions: one to regulate apoptosis and one that adapts to stress.[1]
The ER oversees the quality of protein folding; inside the ER lumen, protein chaperones, foldases, and cofactors to make sure that these proteins fold correctly. The proteins that are correctly folded will then be sent to their respective places in the body through vesicular transport, but stress will impair the ER. Under stressful conditions the protein homeostasis will be thrown off balance, which causes a buildup of misfolded proteins called ER stress. The unfolded protein response (UPR)is a set of intracellular signaling pathways that regulate the folding process. The UPR uses UPR transductase to increase the number of proteins that are involved in the regulation such as folding proteins, quality control, and ER-associated degradation (ERAD), which helps restore the protein homeostasis by changing the ER's ability to fold and remove incorrectly folded proteins. If the homeostasis cannot be rebalanced, the UPR triggers apoptosis or programmed cell death to get rid of damaged cells. [2]
Many neurodegenerative diseases like Alzheimer's, Parkinson's and amyotrophic lateral sclerosis (ALS) are caused by abnormally folded proteins, which is a result of severe ER stress. Proteins in these diseases makes unusual aggregations, which lead to the disease. [3]
There are three UPR stress sensors: i) inositol requiring enzyme 1α; ii) protein kinases RNA-activated (PKr)-like ER kinase (PERK); and iii) activating transcription factor 6 (ATF6). They control the expression of certain transcription factors by conveying information to the nucleus of the cell.
Hetz and Wohlbier state that the IRE1α is a type I transmembrane protein that has a RNase domain, a cytosolic kinase domain, an N terminal in the luminal region. These proteins in their activated form will want to create oligomeric complexes in order to trans-autophosphorylate themselves.[4]There are two models that have been suggests to explain how the activation of IRE1α occurs and how it senses stress. The first model asserts that the immunoglobulin-binding protein (BiP) binds to IRE1α to inhibit oligomerization of IRE1α. The buildup of unfolded proteins makes the BiP dissociate with the IRE1α so that BiP can interact with the unfolded proteins. When BiP and IRE1α separate the IRE1α is now free to interact with itself, which causes a spontaneous formation of IRE1α oligomeric complexes. The second model suggests that the unfolded protein induce the creation of these IRE1α complexes by directly interacting with the protein's luminal domain.[5]
PERK is also a type I transmembrane protein that has a cytosolic kinase domain and a N-terminal in the luminal region. It is proposed that the mechanism for PERK is similar to the mechanism for IRE1α. Activated PERK can cause reduce the overproduction of proteins in the ER and upregulate genes that will help restore protein homeostasis during times of ER stress by phosphorylating the eukaryotic translation initiation factor 2α (eIF2α).[6]
Unlike IRE1α and PERK, ATF6 is a type II transmembrane protein. This proteins like to bind to BiP under basic conditions, but the buildup of unfolded proteins will cause them to separate because the BiP wants to bind to the unfolded proteins. When they separate disulfide bond in the ATF6 will be reduced, which caused the a fragment of the protein to be able to go the nucleus of the cell to increase the transciption of ER chaperones and ERAD.
TDP-43 is a binding protein of approximately 43 kildodaltons that has come under the scientific light for its potential neurodegeneration in humans. More specifically, TDP-43 aggregation is at the center of an extensive network of neuronal diseases that are collectively referred to as TDP-43 proteinopathies. In order to understand TDP-43 and its function in neurodegeneration, two biochemical properties were studied—it’s ability to bind RNA and its protein-protein interactions. Establishing direct links between TDP-43 and disease is not easy and may not get any easier in the near future due to the number of processes that can be aberrantly affected by TDP-43 aggregations in neurons and glia, and by nuclear depletion of TDP-43.
TDP-43 Protein
Biochemical basis of the aggregation properties of TDP-43
It is now widely accepted that the C-terminal tail of TDP-43 is responsible for most of its tendency to aggregate, even in the absence of cofactors. The hypothesis is that there is a region that contains an infectious ‘prion domain’ from residues 277-414. This hypothesis has been supported by experimental evidence that in vitro prepared TDP-43 fibrils can be taken up by cultured cells and function to trigger aggregation in the cell. In recent times, experimental evidence has shown that either changes in the protein architecture itself or the surrounding protein environment can affect TDP 43 aggregation. C-terminal fragment expression in TDP-43 proteins in neurons was shown to promote aggregation in a considerable number of cells. Another important note is that TDP-43 aggregates have also been observed following knockdown of nuclear transport proteins such as karyopherin beta or the cellular apoptosis susceptibility proteins (CAS), which results in increased cytoplasmic localization of TDP-43.
TDP-43: Biochemical marker of neurodegenerative diseases
Due to the fact that TDP-43 is extremely prevalent in many areas of the body (muscle, skin, etc.), it is important to determine various thresholds of optimal TDP protein levels. There has been an increase in the effort of identifying biomarkers so that disease onset can be diagnosed in patients before neuronal damage becomes too severe, leaving time for scientists and doctors to attempt therapies that can slow down or even prevent the disease. Biomarkers can also help in evaluating the beneficial and harmful effects of novel therapeutic strategies. Several tissue samples from controls and patients have been analyzed to determine what level of TDP-43 expression might be used as a biomarker for disease diagnosis or prognosis. The tissues have been tested for the presence of abnormal levels of TDP-43. However, unfortunately, there is still limited specificity between patients and controls when it comes to TDP-43 expression levels in CSF (cerebrospinal fluid) & plasma.
Therefore, in addition to determining specific biomarkers for the disease, scientists are beginning to use common molecular biology techniques including western and northern blots, PCR, and SDS-PAGE analyses. The hope is that these techniques will not require an excessive amount of effort and training to be set up efficiently in a clinical setting. Through these techniques scintists can look at several molecular aspects of TDP-43 biochemical properties in order to try and identify abnormal expression and/or function.
TDP-43 causes Neurodegeneration in worms in culture
The biochemical studies performed on TDP-43 suggest that the protein is essential to many aspects of cell cycle (especially RNA metabolism). Any alteration to the functional properties of TDP-43 will greatly enhance the protein’s ability to aggregate and consequently generate lethal effects in the organism. Although environmental effects may alter TDP functionality, studies suggest that aggregation should be a primary feature in all TDP-43 proteinopathies. There are promising indications that TDP-43 is indeed a vital biomarker molecule that detects disease onset or progression.
1. TDP-43: gumming up neurons through protein–protein and protein–RNA interactions
Emanuele Buratti and Francisco E. Baralle
A membrane protein is any protein found in a biological membrane. They participate in various biological processes, such as cell signaling-transduction pathways. The membrane proteins also play a strong role in controlling a wide array of gradients such as chemical, electrical, and mechanical gradients and are responsible for cell structure during key cell events such as division. Due to their many functions in the membrane, they are in high concentration on the surface of the membrane. They may also act as channels that move specific molecules into and out of the membrane. Theses proteins fall into two main categories, depending upon how strongly the protein interacts with the membrane.
The two main categories are listed below:
Integral proteins: (also called intrinsic proteins) These are proteins are characterized by strong interaction with the membrane, which can only be broken by the addition of detergents or some other nonpolar solvent. Essentially, they are permanently bounded to the membrane. They may span across the entire phospholipid bi-layer, or be monotopic.Integral proteins. They have one or more segments that are permanently embedded within the phospholipid bilayer and have their domains on both sides of the membrane. Most integral proteins contain residues with hydrophobic side chains that interact with fatty acyl groups of the membrane phospholipids, thus anchoring the protein to the membrane. Most integral proteins span the entire phospholipid bilayer.It interacts extensively with the hydrocarbon chain of membrane lipid and they can be released by agents that compete for these nonpolar interaction.
Peripheral proteins: (or extrinsic proteins) are proteins that have a much weaker interaction with the membrane than integral proteins. These attachments tend to be much more temporary and can be displaced via treatment with a polar reagent. Peripheral proteins. They are temporarily bound either to the lipid bilayer or to integral proteins by hydrophobic, electrostatic, and other non-covalent interactions. This type of proteins does not interact with the hydrophobic core of the phospholipid bilayer. They are usually bound to membrane by interactions with integral membrane proteins or directly by interactions with lipid polar head groups. This polar interaction can be disrupted by the change in pH.
There is also an alternative method of classification for membrane proteins. It arises from membrane proteins, such as colicin A and alpha-hemolysin. These do not fit to either integral or peripheral classification. In this alternative system of classification, the membrane proteins are divided into integral and amphitropic.
Membrane proteins
Biological membranes have phospholipid bilayer structure which contains a set of proteins which help plasma membrane to carry its distinctive functions. Membrane proteins can be attached to the membrane or associated with the membrane of a cell or an organelle. Membrane proteins can be classified into two groups based on the strength of their association with the membrane:
Some membrane proteins are found bounded to lipid bilayer and generally involved in cell-cell signaling or interactions. Others are embedded within the lipid bilayer of a cell often form channels and pores. Membrane proteins can be attached to both the outside and inside of the cell membrane.
Proteins can be attached to the cell membrane in a variety of ways. One method involves irreversible covalent modification. Both Ras (a GTPase) and Src (protein tyrosine kinase) are known to be modified in this manner. Both of these proteins participate in signal transduction pathways, but upon covalent attachment of a lipid group they become attached to the inner face of the cytoplasmic membrane. When Ras and Src are affixed to the cell membrane they are better able to receive and transmit information being transferred via their respective signal transduction pathways.Membrane proteins can be made of alpha helices or beta strands, or the combination of both alpha helices or beta strands. For example the channel protein called Porin is made up of entirely beta strands, while the enzyme protein called prostaglandin is made entirely of the alpha helices.
Membrane proteins can be alpha - helices or beta - strands. Proteins can span the membrane with alpha helices. Membrane - spanning alpha - helices are the most common structural motif in membrane proteins. An examination of the primary structure reveals that most amino acids in the membrane protein are nonpolar and very few are charged. One of the first alpha - proteins found was the bacteriorhodopsin. It uses light energy to transport protons from inside the cell to outside generating a proton gradient used to form ATP. The seven alpha - helices are closely packed and arranged perpendicular to the plane of the cell and they span 45A in width. Membrane proteins can also be made out of beta strands. Beta Strands form channel proteins. They are less common than alpha - helices. Channel proteins are formed by beta arrangement of beta strands. Each strand is hydrogen bonded to its neighbor in an anti-parallel arrangement, forming a single beta sheet. The beta sheet then curls up to form a hollow cylinder that forms a channel in the membrane. An example is Porin. The outside surface is non-polar and interacts with the hydrocarbon core of the membrane, while the inside channel is hydrophilic and filled with water. The arrangement of polar and non-polar is accomplished by the alternation of hydrophobic and hydrophilic amino acids along with each beta strand.
Many membrane proteins have quaternary structures consisting of multiple subunits. This oligomerization in membrane proteins is beneficial to their functions, stability, genetic efficiency and maybe even optimizing productive output per unit area of the membrane. Cytochrome b6f serves as an example of quarterary structure affecting membrane protein function. This protein consist of two subunits which are connected by a bridge so that electrons can be transferred between them. As for stability, a quaternary protein consisting of 2, 3 or 4 subunits would be 2, 3 or 4 times more stable if a stability improving mutation were to occur on each subunit. It would be more genetically efficient to have all the subunits of a quaternary protein be coded for by 1 gene than to have each of its subunits be coded for by a different gene. In this way, a quaternary protein can be coded for with minimal genetic space. One example of this are the ion channels that span the membrane. the entirety of these quaternary membrane proteins are made from repeating, identical subunits stacked on top of each other. Everyone of these subunits and therefore the iono channel as a whole, is then coded by and translated from 1 single gene. In addition, oligomerization may also contribute to maximizing functional output as it allows membrane proteins to be closely packed in an area of the lipid bilayer without coming into contact with other proteins in energetically unfavorable ways.[1]
Mutations in both Ras and Src have been observed in a number of cancer cells; it is thought that these mutations and the subsequent interruption of the signal transduction pathways predispose a cell to uncontrolled replication. When the presence of a mutation is detected a small protein named ubiquitin is attached to the damaged protein; this modification signals that the marked protein is to be destroyed. It is essential that the protein be destroyed before anaphase so that the damaged DNA is not passed on to other cells. The attachment of ubiquitin to a damaged protein is the first step of apoptosis, which is programmed cell death.
[edit] Integral Proteins
As mentioned earlier, integral proteins, also known as intrinsic proteins, are strongly and permanently bounded to the membrane. One or more parts of these proteins are embedded in the phospholipid bi-layer of the membrane. They exhibit strong interaction with the membrane because their amino acid residues contains hydrophobic side chains that interact with the hydrophobic interior (fatty acyl groups) of the phospholipid bilayer. Because of their strong hydrophobic interaction with the hydrophobic core of the membrane, such proteins can only be dissociated from the membranes using detergents, non-polar solvents, or sometimes denaturing agents. Lastly, it is important to note that integral proteins account for a significant fraction of the proteins encoded in the genome.
There are two basic categories for integral proteins.
These proteins span across the entire membrane. They are the most common among integral proteins.
Transmembrane proteins
They may cross the membrane only once or several times, weaving in and out. The two kinds of transmembrane proteins are alpha-helical and beta-barrels.
The former is the more common of the two and can be found in the inner membrane of bacterial cells or the plasma membrane of eukaryotes. Voltage-gated ion channels, such as potassium and chloride channels, are examples of alpha-helical transmembrane proteins. They are mostly composed of hydrophobic amino acid residues and little hydrophilic residues, such as charged and polar residues. The polar carbonyl oxygen in the backbone doesn’t project outwards the helix, but rather towards the inside, facilitating and strengthening hydrogen bonds within the helix. Van der Waals interactions hold the tertiary and quaternary structures together in the transmembrane region. These interactions allow for flexibility in the structure to accommodate for necessary functions. Two polar residues that are found most frequently in the TM backbone are serine and threonine which can potentially hydrogen bond to the helical backbone. This hydrogen bonding captures polar side chains in a hydrophobic environment, such as a lipid bilayer. The polar side chains in turn hydrogen bond to other helices. Two residues, glycine and proline, known as helix breakers in water make kinks in the helix which play significant roles in functional mechanisms. [2]
Beta-barrels present in the outer membranes of Gram-negative bacteria, cell wall of Gram-positive bacteria, outer membrane of mitochondria and chloroplasts. Porins are examples of a beta-barrel transmembrane protein. They cross cellular membrane and acts as a pore through which molecules can diffuse. Transmembrane proteins can further be categorized into Type I and Type II. In Type I, the N-terminal is positioned on the exterior of the membrane. In Type II, the C-terminal appears on the exterior of the membrane.
Human VDAC
VDAC (voltage-dependent anion channel) is an example of a transmembrane protein found in the mitochondrial outer membrane which provides the pore for substrate diffusion. VDAC is composed of 19 β-strands which make up the β-barrel and a partial α-helix strand totaling 20 strands in the unit. The first and last β-strands of the β-barrel are parallel, while the strands in-between are anti-parallel.[3]
Isoform 1 of VDAC, three high-resolution structures in fact, in detergent micelles and bicelles have been recently published from solution NMR and X-ray crystallography. This helps to solve the membrane topology of VDAC and gives the first eukaryotic β-barrel membrane protein structure. Something different about this integral membrane protein was that it had parallel β-strand pairing and an odd number of strands. The voltage gating mechanism of VDAC and its modulation by NADH are given a structural and functional basis from studies. Since VDAC-1’s de novo structure and six more proteins, the amount of integral membrane protein structures found by solution NMR has doubled in the past two years.[4]
These proteins are permanently bounded to the membrane but only from one side. Many of these proteins are enzymes. Examples include cyclooxygenase and carnitine O-palmitoyltransferase. The former is an enzyme that is involved in the formation of prostanoids. Anti-inflammatory drugs, such as aspirin and ibuprofen, work to relieve symptoms of inflammation and pain by inhibiting this enzyme. The latter is a mitochondria transferase enzyme that participates in the metabolism of palmitoylcarnitine into palmitoyl-CoA.
Peripheral proteins, also known as extrinsic proteins, lack interaction with the hydrophobic interior of the phospholipid bi-layer. Because they lack hydrophobic interaction with the membrane, they can be detached from the membrane much more easily than integral proteins. Dissociation of peripheral proteins can be achieved through treatment with a solution of high pH or high salt concentration. Instead, peripheral proteins attach to the membrane via electrostatic and other non-covalent forces. Typically, they are either attached to the membrane indirectly via interaction with integral proteins, or directly through interaction with the polar heads of the phospholipid (amphitropic). Some peripheral proteins exhibit both types of interaction. These include certain kinases and G proteins. Other examples of peripheral proteins are the regulatory protein subunits of ion channels and transmembrane receptors.
Membrane Protein Functions
Due to the nature of the lipid bilayer, many molecules cannot enter or exit the cell because of size or charge. Membrane proteins function to assist in the transportation of such molecules across the lipid bilayer. Trans-membrane proteins participate in either passive or active transport.
Insertion of ER into lipid bilayer causes newly synthesized integral membrane proteins to be sorted, transferred, and qualitatively maintained. This process is controlled by ubiquitination, a posttranlational redirection of commands which relate to biosynthetic delivery of proteins to the plasma membrane. This process can be followed through the secretory pathways. Ubiquitination can also be used to regulate the deletion of proteins from the plasma membrane through a endocytic pathway. Ubiquitination of integral membrane proteins often is enough to edocytically target the Plasma membrane protein. However, there are still certain functions such as sortin and degradation which fully requires ubiquitin.
This control and change of specific membrane proteins is due to the ubiquitin changing the quality or quantity of the integral membrane protein. As a side effect, defects in this process can also contribute to detrimental diseases such as cystic fibrosis.
Ubiquitin modification can influence cargo trafficking, mechanisms of quality control/maintenance in secretory/endocytic pathway.
Ubiquitin in Membrane Transport and Quality Control in Endoplasmic Reticulum
Ubiquitin transformations do not affect the regulating effects preformed by the ER. However, Ubiquitin activity is initialized during the Endoplasmic Reticulum- associated degradation process, or ERAD, because ubiquitin ligase is needed. This procedure is important because it is responsible for the removal of proteins which are not folded properly. The substrates which are subject to this procedure are relocated to the cytoplasm, waiting to be removed.
The ERAD targets are first ubiquitinated and must negotiate protein Ubx, a ubiquitin-binding protein. This process shows that that ubiquitation of the ERAD substrates provides a signal which is necessary for targeting the protein for degradation. This shows that ubiquitin plays a vital in protein membrane protein transport. It is important to note that ERAD functionalities also provide a key quality assurance aspect. A kink in this procedure could can cause detrimental side effects; this means that the ERAD procedure is monitored carefully and therefore the membrane protein transfers can be assured for quality.
Ubiquitin in protein quality control to regulate Protein Membrane Protein Composition
It is found that ERAD can affect and strengthen communication between ER and Golgi complexes. This can be accomplished by degrading retention factors of the ER. In Cholesterol depleted conditions, cells were ubiquitinated and degraded. This showed that the protein resulted left the ER and was sent into the Golgi for packing.
Conversely, the GAT protein within the Golgi complex contains three surfaces which can bind ubiquitin very well. This causes successful binding of ubiquitin and speed up the transferring of GAT proteins from Golgi to the ER. In addition, the polymerization protein cargo and ubiquitin provides the necessary driving force for localization to the Protein membrane.
Ubiquitin in the turnover of Plasma Membrane proteins
In endocytosis in yeast, ubiquitin is required for almost all processes. It is beneficial that there is a more than sufficient supply of ubiquitin within the yeast. The internalization of protein cargoes that are present in yeast are generally all ubiquitin mediated.
Plasma Membrane protein contains a protective mechanism which are driven by intrinsic factors of the protein. Plasma Membranes also places a limit on the amount of proteins which exhibit error folding. A certain amount over a lifetime span is placed and plasma membrane proteins are there to regulate these levels. This quality check shows that these specific proteins must control integral membrane proteins and the removal of the damaged and misconstructed proteins. Despite the constrained understanding of the chemical process, quality maintenance mechanisms must usually include capabilities such as: the function to refold or fix the damaged protein, and the ability to distinguish healthy and damaged proteins.
As with proteins in the cytoplasm or in aqueous environments, proteolytic processing is key to cellular function in both the cytoplasm and in the lipid bilayer. However, intramembrane proteases present a different challenge to work with than water-soluble proteins. Scientists have been working on methods to decipher the molecular mechanisms of families of intramembrane proteins. Specifically, site-2 intramembrane metalloprotease and serine intramembrane protease rhomboid share common characteristics. The active sites of both families of proteases are entrenched in the membrane. However, to effectively cleave a membrane protein or any other protein, water must be introduced to hydrolyze the peptide bonds. These proteases often recognize a specific sequence of residues and thus cleave proteins at specific sites. To introduce water to the site of cleavage, there is a delivery system to connect the aqueous environment to the site of cleavage.
Molecules are allowed to flow down their concentration gradient. In most cases, this does not require a special protein. However, in facilitated diffusion, molecules that are insoluble in the lipid bilayer or too large to pass through is assisted in crossing the cell membrane through special transport proteins. Examples of facilitated diffusion are amino acids and ions.
Passive transport.
The other types of passive transport, which do not require proteins because the molecules diffuse directly through the cell membrane, are osmosis, diffusion, and filtration.
Facilitated diffusion.
Uniporters are the proteins that move molecules in passive transport. They can either be channel proteins or carrier proteins. Channel proteins open in response to a stimulus and let molecules flow freely through. Carrier proteins bind to a molecule, making it hydrophobic enough to cross the membrane. The following image shows the two kinds of uniporters and how they function.
Energy is expended to transport a molecule up its concentration gradient. There are two types of active transport, primary and secondary. Both involve going against a concentration gradient using ATP, but they differ in how the ATP is used by the protein.
ATP is expended to move a molecule up its concentration gradient. An example of this is the sodium-potassium pump, which pumps both ions against their concentration gradients in order to create a membrane voltage potential.
ATP is not directly coupled to the molecule of interest in secondary active transport. Instead, another molecule is moved up its concentration gradient, which generates an electrochemical gradient. The molecule of interest is then transported down the electrochemical gradient. While this process still consumes ATP to generate that gradient, the energy is not directly used to move the molecule across the membrane, hence it is known as secondary active transport.
Two main types of protein are involved in secondary active transport: antiporters and symporters.
Antiporter
The molecules move in opposite directions. One type of molecule enters the cell while the other exits. An example is the sodium-calcium exchanger, which removes calcium ions from the cell while allowing sodium back in. The sodium is pumped out by the sodium-potassium pump, which generates the concentration gradient required for this to work.
Symporter
The molecules move in the same direction. This usually works by allowing an ion to move down its electrochemical gradient. The other molecule piggy-backs off that movement and goes against its concentration gradient.
Fluidity of Membrane proteins Biological membrane are flexible. This flexibility is attained by the fluidity of the protein. The fluid mosaic model allows lateral movements called the lateral diffusion, and sometimes the transverse diffusion or flip flop can occur, which takes longer time to take place.
Lateral diffusion is the movement of the lipid laterally which is very rapid, unless there is restriction by special interaction.
Lateral Diffusion.
Flip-flop or Transverse diffusion is the condition is when transition of a molecule from one membrane surface to the other occurred. It is a very slow space compared with the lateral diffusion.It happens once in several hours.
Despite the many advances made in the study of membrane proteins, not much is known about the role of the environment in determining membrane protein structure or function because these proteins are easily affected by changes in their environment. The main problem remains in the difficulty of creating an environment that promotes a protein's native functions and structures. However, advances in the study of the influenza virus, more specifically the M2 protein, is giving more insight to this complex challenge.
The M2 protein is a homotetramer with 3 functional domains: the N-terminal, the TM helix, and the C-terminal. Until recent discoveries, drugs were effective in the blocking of the TM helix, which prevented proton conductance functions and thus disabling the virus. However, with recent outbreaks of the H1N1 virus and swine flu, the structure of this protein was scrutinized in 3 different environments, each using a different methodology. The influence of the environment on the proteins can then be seen in the comparison of these 3 results obtained.
The 1st imaging technique, solid-state NMR, concluded that the M2 was stable in a lipid bilayer environment. The drug amantadine was later added giving the protein a 4-fold symmetry structure further indicating more stability in the presence of amantadine.
The 2nd image, crystal structures, not only compared structures at differing pH levels but also showed that membrane proteins can access a range of conformational states.
Finally, the 3rd image made by solution NMR concluded that the membrane protein's amino acids interact to minimize electrostatic potentials and that water, when present, allows for hydrogen bond exchange.
Further screening of this protein is still undergoing, and much has yet to be revealed in the study of the environment's influence on protein structure. However, it is easily seen why this topic remains an important and popular issue. By understanding the environment's influence on membrane proteins, researchers are able to develop drugs to inhibit, for example, the influenza virus. Even mutations such as the H1N1 virus can be disabled as long as researchers have a key understanding of their membrane protein and how they can be manipulated and changed by their environments. The study of the M2 protein will eventually lead to a deeper understanding of other membrane proteins and how they are changed by changes in their environment.
Membrane places a most important role in the human body. It affects strongly in each structure environment. Every times, we talk about membranes, we have to mention protein structures because they related with each other. Proteins also known as amino acids that function in our body. The membrane and amino acid are the main function in the human body to help our body alive. They are supporting each other to form the right structure and sequences in each other to form the right structure and sequences in each part inside the body. Amino acid sequence allows the interpretation of some of the many studies on the chemical and mechanism of the membrane transport protein.
There are different kind of membrane in our body and each of them has different structure and function which also relate to amino acid. For example, integral membrane proteins are present in a heterogeneous environment that poses major obstacle for existing structure methodologies. Each structure could function as different environment and how the bonds are related. It is very difficult to obtain membrane mimetic environments that support the native structures, dynamic and functions of a membrane protein. Membrane protein often necessary to use detergents to mimic the nature lipid bilayer environment. In order to successful understand in which environment they are functioning, we have to know the bonding structure. Bonding is very important in each structure because it connects elements and one or more structures to each other. Nonetheless, it also very important to understand how to break the bond and forming a new bond. That is a reason why it very helpful to know the bond angles and stability of the bonding. Furthermore, by understanding the bonding structure help the scientists study about the differences kind of diseases and medicine to cured all the diseases. Lipid bilayers is a thin membranes. Lipid bilayers have a unique role in characterizing the native structures of membrane proteins and validating structures determined in other membrane mimetic environments.
Indeed, many proteins are membrane proteins which have the function in the cell. The cell need to communicate with the exterior or passing through the cell membrane. Many proteins go to membrane are glycoprotein related. Proteins are very difficult to study because the structures and functions are very complex. However, some proteins function can be predicted.
The study of membrane proteins have been complicated by the difficulty of examining the proteins by X-ray crystallography. Thus far, scientists have been able to examine the detail of their interactions between membrane components and their relative functions by computational simulations of the proteins in the membranes.
The questions of the stability of a membrane protein have eluded scientists. One particularly difficult task relates to studying the reversible transitions between different states. These interactions have been studied thermodynamically and yielded information pertaining to helix-helix interactions and the types of approaches to membrane protein stability. The stability between proteins and lipids have been simulated by methods such as simulating the free energy cost of burying specific amino acid side chains in the bilayer. Atomistic simulations have made these efforts possible including divulging information on complex membrane proteins such as ion channels.
The difficulties in studying the membrane proteins are mainly due to the difficulties of handling of proteins and experimental challenges associated with working with membrane protein. Also, the co-studying of isolated protein molecule and the molecular environment in order to have an appropriate understanding of the system makes it even more difficult to study.
For instance, isolating the protein from remainder components in the biological system is crucial in structural determination. But in order to have any proper thermodynamic analysis, it must include ALL relevant components of system, particularly paying close attention to boundaries where energy is exchanged.
Traditionally while studying membrane proteins, scientists remove the lipids surrounding the membrane proteins in their preparations. Now, scientists recognize the significance of the lipids as important additives for crystallization. Currently scientists have been more successful solving membrane protein structures with the addition of the lipids during analysis. These successes have led to an increasing number of membrane protein structures which bind lipid molecules to become readily visible and possible to classify.
Membrane proteins are assembled into complexes that allow these intricate assemblies to allow complexity that is not possible using single polypeptides. These complex assemblies allow membrane proteins to have many functions involving regulatory mechanisms and chemical reactions. The existence of these membrane protein complexes prevents potential problems such as unwanted interactions, aggregations, or the formation of hazardous intermediates. Furthermore, these complexes are mechanistically invaluable because they follow a process in which parts of the complexes are "pre-fabricated" and replaced in isolation if damaged, meaning that the whole complex does not need to be replaced if only one subunit it damaged. Membrane protein complexes has been analyzed through the use of blue native polyacrylamide gel electrophoresis and split-ubiquitin method.
Membrane protein complexes allow the avoidance of problems such as those listed above (unwanted interactions, aggregations, or the formation of hazardous intermediates) by being assembled in an ordered, even sequential, manner. To understand that the formation of these complexes are ordered, one would need to know what the assembly intermediates are. Thus, the larger the proteins, the more difficult it is to expose the formation order. However, smaller complexes such as cytochrome bo3 of E. Coli complex have allowed scientists to understand that membrane protein complexes follow a linear pathway of assembly. the bo3 complex is made up of four subunits that assemble through two intermediate complexes. It is understood that bo3 assemble linearly because thought it is possible for other intermediates to form leading to the formation of bo3, they are not observed and there is only one assembly pathway indicating that the intermediates follow a sequential, ordered path. Non-linear assembly would be noticeable because there would be several different assembly pathways. Ordered formation is also seen with cell division in divisomes whereby if one protein is missing, all downstream proteins are preventing from interacting properly. Scientists believe that this sort of ordered pathway exists to protect the cell from potentially harmful intermediates.
Chaperones also play a large role in the formation of these complexes. Chaperones act as physical assembly factors that interact with proteins and prevent unproductive interactions from occurring. For instance, chaperones prevent aggregation in the F1 compound of yeast F1F0-ATP synthase. Two chaperones bound to the alpha and beta subunits bind to the hydrophobic interfaces and guide the alpha and beta subunits into a3b3 complex assembly. Research has also shown that the loss of chaperones in some intermediates could be responsible for the activation of a membrane protein complex. It is important that an intermediate remain inactive so that unregulated activity by partially assembled complexes does not occur.
Membrane protein complexes are believed to undergo dynamic exchange as a mechanism for regulating damaged subunits within the complexes. Dynamic exchange allows the assembly of newly imported proteins into complexes to replaced damaged proteins without replacing the entire complex. An example of this is seen with photosystem II chloroplasts whose D1 subunits that become photo-damaged and is replaced as part of its repair mechanism. Dynamic exchange, at first, was only carried out in vitro. Scientists were only able to conclude that dynamic exchange is a possible repair mechanism, but could not conclude that it was what actually occurred in vivo. It was not until the use of fluorescent microscopy that scientists were able to confirm that dynamic microscopy did occur to a degree. Fluorescent microscopy tagged proteins and watched its interactions in vivo. Subunits were seen freely diffusing into and out of complexes. Future research hopes to disclose which proteins are being exchanged and why thee proteins undergo dynamic exchange.[5]
In the article Membrane Protein Structure: Prediction versus Reality, Arne Elofosson and Gunnar von Heijne discussed several current techniques used to predict the insertion and folding of membrane protein; they depicted a realistic and pragmatic view on how those techniques are used and the limitations. They also pointed out unresolved major issues concerning those techniques.
Arne and Gunnar first pointed out alpha-helix bundle and beta-barrel are the two main structures of membrane proteins. While the helix bundle represents about 20% to 25% of all open reading frame, the barrel form represents a few percent of all open reading frame. An open reading frame. ( A reading frame refers to DNA/RNA that can be broken into three letter codon and be transcribed into protein, while an open reading frame refers to a DNA sequence that does not contain a stop codon in its reading frame. ) The similarities between the helix bundle and beta-barrel is that, in order to fit the basic structure of lipid bilayer in membrane, they both contain hydrophobic amino acids in the middle of the protein. The major difference between the two is their secondary structure. The helix bundle is a complex long trans-membrane protein that packs several alpha-helixes; while beta-barrel protein has several beta-sheets rolled up, and it is shorter and less hydrophobic than the alpha helix bundle. Arne and Gunnar points out that the helix bundle form has been paid more attention as they are longer hence easier to be recognized than the beta-barrel.
Arne and Gunnar then depicts how helix-bundle and beta-barrels are synthesized and inserted into lipid bilayer. In the case of helix bundle translation, corresponding ribosome first bind to a translocon, which is a protein in the inner membrane responsible for the translocation of protein across the inner cellular membrane into the periplasm, called SecYEG translocon. Helix bundle is translated and inserted into the inner membrane. Depending on the hydrophobicity of the helix bundle, the interaction among helix bundles varies; either one helix bundle or a couple are synthesized at a time. Beta-barrel, due to the fact that it is less hydrophobic than helix bundle, could not get through the inner membrane just by itself; after its translation in the cytoplasm, it binds to SecB, with the help of SecA ATPase, via SecYEG translocon and transferred to the periplasm. It is inserted into the outer membrane via YaeT hetero-oligomeric outer membrane integration complex. After the membrane proteins are inserted into the lipid bilayer, it is believed that interactions among helix bundles are stronger than that with the lipids, hence the helix dandles are packed together and obtain its conformation. Hydrogen bonding between polar side chains also contributes in the conformation of the protein. Helix bundle and beta-barrel reaches rather stable conformation after inserted into the membrane. Nevertheless, some membrane protein exhibits a higher degree of flexibility, such as those that are in charge of proton or electron transfers.
According to Arne and Gunnar, in the case of helix-bundle membrane protein, the primary structure, hence its amino acid sequence has long been used to distinguish helix-bundle membrane protein from others. Due to the fact that lipid bilayer has hydrophobic character, the helix-bundle that is inserted into such lipid bilayer should consist of residues that are hydrophobic. Two essential amino acids, tryptophan and tyrosine, whose side chain contain aromatic structures contribute the hydrophobicity of helix-bundle membrane protein. The helix-bundle must also be long enough to span through the inner membrane; hence a helix bundle could have an averaged 10 to 20 hydrophobic residues. Loops connect the helixes; depending on whether the loops are facing inside or outside of the cell, the loops contain different amino acid compositions.
Membrane protein has been thought of perpendicularly orientated through the membrane, Arne and Gunnar points out that, membrane protein orientation could be more complex. One of the factors that contribute to the unexpected complexness of membrane protein comes from reentrant loops, as exhibited in the case of glutamate transporter. Concerning beta-barrel, Arne and Gunnar generalized a series of deducted structural principles; for instance, beta-stands have even numbers and tilts about 45 degrees in antiparallel fashion.
Arne and Gunnar surveyed a series of topology and structure prediction schemes in increasing complexness. 2D prediction is the earliest technique; such technique utilized the higher hydrophobicity possessed by trans-membrane protein than loop regions and has been an effective tool. One challenge faced by the 2D prediction is that the topogenic data from signal peptide and trans-membrane helices are similar, so it is hard to distinguish between the two.
In predicting the structure of β-barrel membrane proteins, scientists look for the existence of an N-terminal signal peptide and the protein’s general amino acid composition. Predicting the structure of a β-barrel membrane protein is simpler than that of a helix-bundle because its amino acid sequence is shorter and not as obvious to see.
As mentioned before, membrane protein cannot be simply modeled as all perpendicular through the membrane; reentrant loops are an important feature that elevates the complexity of membrane protein. These reentrant loops, as suggested by Arne and Gunner can be predicted by a recent developed topological technology named as 2.5D prediction. The residues in these reentrant loops, which come in long loops, medium length loops and short loops, usually are smaller than other parts of the protein so they are easily found in between the transmembrane helices. 2.5D membrane protein structure prediction predicts structures based on the type of amino acid sequence that it contains or by predicting how far the residue is from the center of the membrane protein. Characteristics of residues which can be predicted include lipid-exposed (hydrophobic) regions or lipid buried residues and kinks due to proline. Since 2.5D prediction was able to include sub-structures of membrane protein, such as the interfacial helices and reentrant loops, it is helpful in classifying membrane proteins.
3D structure prediction was first attempted via low-resolution experiments such as electron microscopy. Arne and Gunner points out like, 3D prediction of membrane protein, like all other globular proteins that have been tested against 3D predictions, has low accuracy. What has increased the difficulties in 3D prediction of membrane protein is that they sustain their structures in environments different from those of globular protein; also the globular protein that has been successfully predicted are much smaller than the membrane protein of interest.
To date, there are limited 3D models to be tested against, but there have been hypotheses of models to be tested against. One of which is homology modeling, which would potentially result in structures with details at the atomic level and with similar quality as of the models tested against globular proteins.[6]
In Unsolved Mysteries in Membrane Traffic, a paper written by Susanne R. Pfeffer, from the Department of Biochemistry in Stanford University, she explains how there are various hypotheses to how proteins travel and help facilitate transport within the cell but there isn’t a completely proven hypothesis yet. To start off, Soluble N-ethylmaleimide-sensitive factor attachment protein receptor proteins also known as SNARE proteins help facilitate the fusion of vesicles to their target membranes. There are two distinct groups of SNARE proteins. The first is the R- SNARE which is also called the v- SNARE group which is found on the vesicle. The second group of SNARE proteins is the Q- SNARE which is also called the t- SNARE due to the fact that it’s located on the target membrane. The main difference between these two proteins is that the R- SNARE will only be as a single protein on the vesicle whereas the Q- SNARE will form a complex of three Q- SNAREs. Under these two categories lie specific R-SNARE proteins that will pair up with specific Q-Proteins. The method of how these two pair up to facilitate fusion of a Golgi vesicle to the cell surface is still unknown but one can think of it as SNARE proteins being like puzzle pieces because they have certain specificity therefore one SNARE PROTEIN (R-SNARE/v-SNARE) will bind to only a specific SNARE complex (Q-SNAREs/ t-SNAREs). Although scientist still don’t know how the Golgi decides to transport these vesicles R- SNAREs and Q- SNAREs give clues as to what has arrived and what might depart. If there is a concentration of Q- SNAREs at a specific site that can be accounted for by noticing that there was previous fusion activity at that site. Now when we look at R- SNAREs there are two possible answers to why there are at a specific site, one can be because they have recently arrived and fusion just occurred or because a vesicle is about to depart the Golgi membrane. An important thing to note is that less membrane traffic occurs in the trans Golgi than does in the cis Golgi which is proved by the low concentration of SNAREs in trans Golgi rim and a higher in the cis Golgi rim. Therefore, with all these new ideas we must search for concrete answers to better understand how membrane trafficking occurs within cells.[7]
↑Hiller, S., Abramson, J., Mannella, C., Wagner, G., and Zeth, K., "The 3D structures of VDAC represent a native conformation," Trends in Biochemical Sciences, 2010.
↑The role of solution NMR in the structure determinations of VDAC-1 and other membrane proteins. Sebastian Hiller and Gerhard Wagner*
↑Assembly of Membrane Proteins into Complexes by Daniel O. Daleya,at Center for Biomembrane Research, Department of Biochemistry and Biophysics, Stockholm University, SE-106 91 Stockholm, Sweden, 5 June 2009.
↑Membrane Protein Structure: Prediction versus Reality.Annu Elofsson A, von Heijne G. Rev Biochem. 2007.76:125-40
↑Unsolved Mysteries in Membrane Traffic: Annu. Rev. Biochem. 2007. 76:629–45 Pfeffer, Suzanne R. Dept. of Biochemistry, Stanford
Transport of molecules across membrane is the movement of a molecule from inside the membrane to outside or vice versa. There are two factors to determine if a molecule will cross a membrane:
- The permeability of the molecule in a lipid bilayer: Molecules move spontaneously from high concentration to low concentration due to the second law of thermodynamics. However, molecules with high polarity, such as sodium, are not able to freely enter the cell membrane because the charged ion cannot pass through the hydrophobic core of the membrane. This transport of molecule across membrane can be either passive (where movement is driven by a gradient) or active (where movement is against a gradient and requires energy).
- Availability of an energy source: Energy is minimized when all concentrations are equal, so an uneven distribution of molecules is a form of potential energy, which can be used to drive other processes. One process is the transport of molecules from one side of a membrane to the other. The equation that describes the amount of energy required for this process is :
,
where R = 8.315*10 -3 KJ/mol, the molar gas constant, and T = temperature.
When the molecules involved are charged, an electrical potential can build up. The cumulative effect of the electrical potential and the uneven distribution of concentrations gives us a modified free energy equation:
,
where F is Faraday’s constant and Z is electrical charge of transported species and V is potential difference across the membrane.
It requires work to pump a molecule across a membrane against its gradient. Moving ions from low concentration to high concentration leads to decrease in entropy, which requires an input of free energy. Therefore, this type of membrane traffic is called active transport. The transport proteins that move solutes against a concentration gradient are called carrier proteins. On the other hand, channel proteins are involved in passive transport.
Sodium-Potassium Pumps are an example of active transport. It is known that cells contain high concentrations of potassium ions but low concentrations of sodium ions. Therefore, it was deduced that a protein existed on the plasma membrane which actively pumped the two ions against their biological gradients. This protein was discovered in the 1950s by Jens Christian Skou and for his discovery, was awarded the Nobel Prize in 1997. This pump works by binding sodium ions which stimulates phosphorylation by the addition of a phosphate group from ATP. This phosphorylation causes a change in the 3D shape of the protein, making it open up to the extracellular world, and decreases the protein's affinity for sodium ions. In turn, the new shape has a high affinity for potassium ions which bind and force a 3D conformational change while triggering the release of the phosphate group. This causes the protein to open up the intercellular world and the loss of the phosphate causes the pump to have a lower affinity for potassium and a higher affinity for sodium. The cycle repeats.
In this sodium/potassium pump, sodium is transferred out of the plasma membrane and potassium is pumped inside the plasma membrane. Since active transport requires energy, it uses ATP or it couples to molecules moving down the concentration gradient. In the sodium/potassium pump, sodium and phosphate (the phosphate from the breakdown of ATP: ATP → ADP + P) are coupled to the pump, which takes both of them out of the cell and brings the potassium inside. For this process to take place, both the potassium and sodium pumping must occur at the same time because if the ability to pump one of them is lost, then the ability to pump the other ion will be lost as well. The sodium/potassium pump is active transport because there is coupled transport where one molecule's transfer is dependent on the other molecule's transfer. This example of active transport is antiport because molecules are being moved in opposite directions.
cotransport
Cotransport Pumps, or coupled transport, is a type of active transport in which the transport of a specific solute indirectly facilitates the active transport of another solute. The general mechanism is that, through the use of ATP, a specific solute is driven up its concentration gradient, analogous to moving water up a hill. In the second step, the specific solute runs back down its concentration gradient while forcing the other solute up its own concentration gradient, analogous to coupling water running downhill to force the work of another machine.
endocytosisexocytosis
Endocytosis is another type of active transport. In the previous examples, active transport was used on small molecules. In endocytosis, energy is used to take in biological molecules and large particles by the formation of new vesicles. There are multiple types of endocytosis, with the major categories being phagocytosis, pinocytosis and receptor-mediated endocytosis.
Exocytosis is also another type of active transport, utilizing energy to do the opposite of endocytosis. In exocytosis, the vesicle fuses with the plasma membrane thereby releasing all the contents and waste outside of the cell. This type of active transport is mainly used by secretory cells where they secrete insulin or neurotransmitters.
Efflux Pumps
Active Efflux is a type of active transport and is the mechanism largely responsible for the extrusion of drugs such as antibiotics, toxic substances and other xenobiotics. Bacteria efflux pumps are separated into five families.
1. Major facilitator superfamily (MFS)
2. ATP binding cassette (ABC)
3. Small multidrug resistance family (SMR)
4. Resistance-nodulation-cell division superfamily (RND)
5. Multi-antimicrobial extrusion protein (MATE)
These efflux pumps are largely responsible for antibiotic drug resistance due to the presence of the efflux pumps that export toxins out of the cell and inhibit the drug's effects. Gram-negative bacteria have a greater resistance to antiseptics and antibiotics. The RND family of efflux pumps is exclusive to gram-negative bacteria and is very effective in generating resistance against antibiotics.
In the case of E. Coli, two homologues, AcrB and AcrB complex together with the outer membrane protein channel TolC and utilizing the proton-motive force, this complex can effectively export a variety of drugs across the periplasmic place and out of through the outer membrane. This is possible due to AcrB's ability to assume an asymmetric structure in which each subunit exhibits different conformations to attach to the attach to the substrate and move them out of the transporter.
A schematic representation of diffusion.
Passive Transport Molecules have a natural tendency to move and spread out evenly in any available space. This property is called diffusion. An example would be a drop of dye in a section of a beaker separated by a permeable membrane. In this case, the dye, although the molecules move randomly, would have a net movement across the membrane such that the concentration of dye on both sides would be equal. At that point, an equilibrium would be established with the same amount of dye molecules moving across the membrane in both directions. In other words, the natural tendency of any substance is to migrate down its concentration gradient.
The diffusion of a substance across a biological membrane is called passive transport because the cell does not have to expend energy to make it happen. The concentration gradient itself represents potential energy and drives diffusion. However, membranes are selectively permeable and therefore have different effects on the rates of diffusion of various molecules. In the case of water, aquaporins allow water to diffuse very rapidly across the membranes of certain cells. The movement of water across the plasma membrane has important consequences for cells.
Osmosis is a form of passive transport which involves the diffusion of water through the lipid bilayer with the use of aquaporins. However, since this is passive transport with the diffusion of water through the concentration gradient, three scenarios should be established. The first is in an isotonic solution where the concentration of water inside the cell is the same as the concentration outside the cell. In this case, since there is equilibrium, the net movement of water across the permeable membrane is the same. Therefore the cell remains unaffected. The second is in a hypotonic solution where water concentration outside the cell is greater (solute concentration lesser) than the concentration inside the cell. In this scenario, water would diffuse down its concentration gradient and into the cell, thereby causing the cell to burst. The last is in a hypertonic solution where the water concentration is lesser (solute concentration greater) than the concentration inside the cell. In this case, water rushes out of the cell thereby causing the cell to shrink. There are many organisms that live in either hypertonic or hypotonic environments and each have special adaptations for osmoregulation, or control of water flow. For example, Paramecium lives in a hypotonic environment. The protist has adapted by having a plasma membrane which is less permeable to water, thereby limiting the rate at which water enters the Paramecium. In addition, the cell is equipped with a contractile vacuole which literally pumps water out of the cell at the same rate at which it enters the cell.
Facilitated Diffusion
Facilitated Diffusion is another form of passive transport where many polar molecules diffuse with the help of integral proteins in the lipid bilayer. The two main types of this kind of transport are ion channels and gated channels. In the case of ion channels, the protein which spans the lipid bilayer has a channel with polar R-group amino acids which allow small polar molecules to passively diffuse down their concentration gradient and into the cell. In the case of gated channels, the protein binds a molecule which causes an alteration to its 3D shape opening it up to the inside of the cell. Then, the molecule is released into the cell and the protein opens up again to the outside of the cell. In this method, the molecule still diffuses down its concentration gradient, albeit with the help of a carrier protein.
Filtration
Filtration is another type of passive transport, and refers to the movement of water and other molecules across the cell membrane due to hydrostatic pressure generated by the cardiovascular system. Which molecules are permitted to pass through the membrane depends largely on the size of the molecules compared to the membrane pores. For example, the membrane pores of the Bowman's capsule in the kidneys are very small, and thus only albumins (one of the smallest proteins) may be permitted to pass through the membrane. In contrast, the membrane pores of liver cells are very large and allow a variety of molecules to pass through the membrane and be metabolized.
The Second Law of Thermodynamics suggests that particles will naturally diffuse from an area of high concentration to an area of lower concentration. The potential energy or the free energy reserved in a concentration gradient can be mathematically represented. Since free energy is lowest when the distribution of molecules is even, the uneven concentration of particles is an environment with abundant energy. Energy must be added into the system to achieve the unequal distribution of molecules or to form a concentration gradient.
The quantity of energy that must be added can be accounted for by first considering an uncharged solute molecule. The free-energy difference in moving particles from side 1 (with a concentration of c1), to side 2 with a concentration of c2 can be represented by the following equation:
∆G =RT ln(c2/c1) = 2.303RTlog10(c2/c1)
in which R represents the ideal gas constant (8.314 x 10-3) and T is the temperature in units of kelvins.
The pictorial representation of the concentration gradient of an uncharged solute can be analyzed in the diagram below.
For the charged species, a mathematical and pictorial representation can be derived as well. The uneven distribution across the plasma membrane creates stored free energy that needs to be included in the formula because like charges will repel. The electrochemical potential (membrane potential) is the addition of the concentration and electrical factors. The free energy difference is
where ∆V is the potential in volts across the plasma membrane, Z is the electrical charge of the transported species, and F stands for the Faraday constant (96.5 kJ/V. mol).
Note: the charged species across a membrane have the same charge as the transported ion.
∆G is positive if the transport process is active and ∆G negative represents the passive transport.
The membrane potential of a cell is the electrical potential difference between the inside and outside of the cell. The potential is determined by the ion concentration between the inside and the outside. This is maintained by different membrane gradient.
Electrical and Concentration gradient help establish a cell's resting potential
The resting potential of a neuron has a charge of -70 mV, and this is called the electrical gradient. There is a constant exchange of ions between the cell and environment, and ions play a significant role in the resting potential include potassium, sodium, and chloride. The concentration and movement of ions is maintained by a protein pump, that pumps 3 K+ out for every 2 Na+ ion. This pump is called the sodium-potassium pump. There is higher concentration of Na+ outside the cell, and a higher concentration of K+ inside the cell. The mix between the concentration gradient and the electrical gradient cause the Na+ ions to have a tendency to move inside, and the K+ ions to move outside. The Na+ ions have a tendency to move in because of the charge difference, and the K+ have a tendency to move outside because of a concentration difference. When the equilibrium potential is reached, the K+ ions do not have a strong tendency to move out of the cell because of the charge difference. The inside of the cell is -70 mV, and further outflow of K+, despite the concentration gradient, will cause the cell to be even more negative. The equilibrium potential is the point when the electrical gradient and the concentration gradient have stabilized with respect to each other.
The resting potential can be calculated by Goldman’s equation, which is represented by
The influx of K+ and Na+ into a cell affects the charge of a cell positively, while an influx of Cl- affects the charge of a cell negatively. The numerator represents the inside concentration of a cell, while the denominator represents the outside concentration. K+ and Na+ outside correspond Cl- inside, because of the opposite charges. P represents the permeability of the ion. Other ions affect the resting potential, but only these three ions are major contributors.
Biochemistry . 6th ed. New York : W. H. Freeman and Company, 2007. 352-353. Print.
Calcium ATPaseSodium-Potassium ATPase
P-type ATPases are a family of cation transport enzymes present in eukaryotes, archea and bacteria. These alpha helical bundle primary transportser (P-type ATPases) are known for catalysis of self phosphorylation of a key conserved aspartate residue within an ion pump. They generate essential ion gradients that are the basis for a variety of cellular functions like signaling, energy storage and secondary transport. Prominent examples of P-type ATPases are the sodium-potassium pump(Na+,K+-ATPase), the proton pump (H+-ATPase), the proton-potassium pump (H+,K+-ATPase) and the calcium pump (Ca2+-ATPase).
Discovered in 1957 by Nobel Laureate Jens Christian Skou, the first P-type ATPase was the Na+, K+ - ATPase. As of June 2007, 400 unique and confirmed members of the P-type ATPase family have been discovered.
These enzymes have a significant impact on cellular function. Indeed, more than one third of the ATP consumed by a resting animal is used by a single ion pump, the Na+,K+-ATPase. The calcium pump (located in the sarcoplasmic reticulum (SR) membrane) also plays a significant role because it is responsible for pumping calcium from the muscle cell to cause a muscle to relax.
P-type ATPases can be divided into five phylogenetic subfamilies:
Type I — Transition/heavy metal ATPases. These ATPases are involved in transport of metals such as K+, Cu+, Ag+, Cu2+, Zn2+, Cd2+, Pb2+ and Co2+.
Type II — Includes Ca2+-ases, such as SERCA1a. Also includes Na+/K+ and H+/K+ ATPases.
Type IIA and IIB transports Ca2+.
Type IIC contains Na+/K+ and H+/K+ ATPases from animal cells.
Type IID contains few fungal ATPases of unknown function.
Type III — Includes plasma membrane H+-ATPases from plants and fungi and Mg2+-ATPases from three bacterial species
Type IV — Includes ATPases have been shown to be involved in the transport of phospholipids.
Type V — ATPases with unknown specificity. This large group are only found in eukaryotes and are believed to be involved in cation transport in the endoplasmic reticulum.
Structure of the Escherichia coli BtuCD protein, an ABC transporter that mediates vitamin B12 uptake.
ATP-binding cassette transporters, also known as ABC transporters, are transmembrane proteins that utilize the energy of ATP hydrolysis to carry out certain biological processes including translocation of various substrates across membranes and non-transport-related processes such as translation of RNA and DNA repair. They transport a wide variety of substrates across extra- and intracellular membranes, including metabolic products, lipids, and sterols. Medically, ATP-binding cassette (ABC)transporters contribute to the resistance of multi-drug to cytotoxic drugs. Proteins are classified as ABC transporters based on the sequence and organization of their ATP-binding cassette domains.
ATP-binding cassette (ABC) systems are universally distributed among living organisms and function in many different aspects of bacterial physiology. ABC transporters are best known for their role in the import of essential nutrients and the export of toxic molecules, but they can also mediate the transport of many other physiological substrates. In a classical transport reaction, two highly conserved ATP-binding domains or subunits couple the binding/hydrolysis of ATP to the translocation of particular substrates across the membrane, through interactions with membrane-spanning domains of the transporter. Variations on this basic theme involve soluble ABC ATP-binding proteins that couple ATP hydrolysis to nontransport processes, such as DNA repair and gene expression regulation. Insights into the structure, function, and mechanism of action of bacterial ABC proteins are reported, based on phylogenetic comparisons as well as classic biochemical and genetic approaches. The availability of an increasing number of high-resolution structures has provided a valuable framework for interpretation of recent studies, and realistic models have been proposed to explain how these fascinating molecular machines use complex dynamic processes to fulfill their numerous biological functions. These advances are also important for elucidating the mechanism of action of eukaryotic ABC proteins, because functional defects in many of them are responsible for severe human inherited diseases.
ABC transporters utilize the energy of ATP hydrolysis to transport various substrates across cellular membranes. They are divided into three main functional groups. In prokaryotes, importers mediate the intake of nutrients to the cell. Ion, amino acids, peptides, sugars and other molecules that are mostly hydrophilic are the substrates that can be transported. The area of the membrane around the ABC transporter protects hydrophilic substrates from the lipids within the membrane bilayer, providing a pathway across the cell membrane. Eukaryotes do not have any importers. Instead, they have exporters, or effluxers, which are also present in prokaryotes. Exporters function as pumps that remove toxins and drugs from the cell. The third subgroup of ABC proteins do not function as transporters, but are more involved in the process of translation and DNA repair.
Bacterial ABC transporters are necessary in cell viability, virulence, and pathogenicity. For instance, the iron ABC uptake systems are very important effectors of virulence. Siderophores are used by the pathogens to scavenge for iron that is in complex with high-affinity iron-binding proteins called erythrocytes. Erythrocytes are high-affinity iron-complexing molecules that the bacteria secretes and is reabsorbed into iron-siderophore complexes. Bacterial ABC transporters are also essential in cell survival. This is because they function as protein systems that act against any undesirable change that may occur in the cell. One example of this is seen when osmosensing ABC transporters that mediate the uptake of solutes is activated in order to counteract a potentially lethal increase in osmotic strength. In addition to functioning in transport, there are some bacterial ABC proteins that are involved in the regulation of several physiological processes.
Most eukaryotic ABC transporters are effluxers, but there are some that are not directly in the transporting of substrates. For example, in the cystic fibrosis transmembrane regulator (CFTR) and in the sulfonylurea receptor (SUR), ATP hydrolysis is used to regulate the opening and closing of the ion channels carried by the ABC protein itself or other proteins. ABC transporters in humans are involved in quite a few diseases that are a result of polymorphisms in ABC genes rather than the complete loss of function of single ABC proteins. Some of these diseases include Mendelian diseases and complex genetic disorders such as cystic fibrosis, immune deficiencies, Stargardt disease, Tangier disease, Dubin-Johnson syndrome, and many others.
NKA, which stands for Na+,K+ -ATPase, is a sodium pump that uses energy to transport ions across the plasma membrane. The ATP hydrolysis energy is used to transport the sodium and potassium, generating electrochemical gradients. NKA falls under the P-type group, which is a protein complex in the form of αβϒ. The α-subunit is the actual pump that transports the sodium and potassium, and contains the three nucleotide-bindings. It contains M1-M10, which are the ten transmembrane segments. It also contains the three cytoplasmic domains which are: the nucleotide-binding (N-) domain, the phosphorylation (P-) domain, and the actuator (A-) domain. The β-subunit is responsible of transporting the potassium ions and making sure that the α-subunit is inserted correctly in the plasma membrane. The β-subunit contains one membrane-anchoring helix, disulfide bridges, and a extracellular domain. Then, the ϒ-subunit regulates the pumping activity of the sodium pump in the tissue.
The interaction between agrin signaling and the ion sodium-potassium pump is due to the bimodular mechanism. Two homologous sites of agrin are cleaved by neurotrypsin at the synapse. This affects to both of the pumping and the signaling function of the NKA. This causes the C-terminal 22 (C22) to restrain the ion pumping function of NKA. Then, agrin binds to the C-terminal. Due to this, the membrane can affect the activity and insertion since the β-subunit is displace. To know if the α-subunit or the β-subunit will dominate, based it on the binding affinity or the concentration.
ATP-binding cassette transporters constitute a large superfamily of integral membrane proteins that includes both importers and exporters. In recent years, several structures of complete ABC transporters have been determined by X-ray crystallography. These structures suggest a mechanism by which binding and hydrolysis of ATP by the cytoplasmic, nucleotide-binding domains control the conformation of the transmembrane domains and therefore which side of the membrane the translocation pathway is exposed to. A basic, conserved two-state mechanism can explain active transport of both ABC importers and ABC exporters, but various questions remain unresolved. In this article, I will review some of the crystal structures and the mechanistic insight gained from them. Future challenges for a better understanding of the mechanism of ABC transporters will be outlined.
ABC transporters make use of the energy of ATP binding and/or hydrolysis to drive the conformational changes in the transmembrane domain (TMD) and consequently transports molecules. There are two types of ABC transporters, ABC importers and ABC exporters, that have a common mechanism in transporting substrates because they are similar in structure. The mechanism that describes the conformation changes associated with the binding of the substrate is the alternating-access model. In this model, the substrate binding site alternates between outward- and inward-facing conformations. The relative binding affinities of the two conformations for the substrate largely determines the net direction of the transport. For ABC importers, the translocation is directed from the periplasm to the cytoplasm and so the outward-facing conformation will have higher binding affinity for the substrate. Contrastingly, the substrate binding affinity in exporters will be greater in the inward-facing conformation. A model that describes the conformational changes in the nucleotide-binding domain (NBD) as a result of ATP binding and hydrolysis is the ATP-switch model. This model gives two principal conformations of the NBDs: formation of a closed dimer upon binding two ATP molecules and dissociation to an open dimer facilitated by ATP hydrolysis and release of inorganic phosphate and adenosine diphosphate (ADP). The switching between open and closed dimer conformations induces the conformation changes in TMD resulting in substrate translocation.
The general mechanism for the transport cycle of ABC transporters has not fully been clarified but substantial structural and biochemical data has been gathered to support a model in which ATP binding and hydrolysis is coupled to conformation changes in the transporter. The resting state of all ABC transporters has the NBDs in an open dimer configuration with a low affinity for ATP but a high affinity for substrate binding site. This open conformation contains a chamber that is accessible to the interior of the transporter. The transport cycle is initiated by the binding of the substrate to the high-affinity site on the TMDs, which induces the conformational changes in the NBDs and enhances the binding of ATP. Then, two molecules of ATP bind cooperatively to form the closed dimer configuration. The closed NBD dimer then induces a conformational change in the TMDs such that the TMD opens, which forms a chamber with an opening that is opposite that of the initial state. The affinity of the substrate to the TMD is then reduced, which releases the substrate. Following that is the hydrolysis of ATP and the sequential release of inorganic phosphate and ADP. The ADP restores the transporter to its basal configuration. Although this common mechanism has been suggested, the order of substrate binding, nucleotide binding and hydrolysis, and conformational changes as well as the interactions between the domains, is still being debated.
Recent studies have shown that ATP binding, rather than ATP hydrolysis, provides the principal energy input, or "power stroke", needed for transport. This may be because ATP binding triggers NBD dimerization and so the formation of the dimer may represent the "power stroke". Additionally, there are some transporters that have NBDs that do not have the similar abilities in binding and hydrolyzing ATP. Thus, the interface of the NBD dimer consists of two ATP binding pockets, suggesting that there is a concurrent function of the two NBDs in the transport cycle.
The transport mechanism for importers supports the alternating-access model. The importers' resting state is inward-facing, where the NBD dimer interface is held open by the TMDs and facing outward. When the closed, substrate-loaded binding protein attaches towards the periplasmic side of the transmembrane domains, the ATP binds and the NBD dimer closes. The resting state of the transporter then switches into an outward-facing conformation in which the TMDs have reoriented themselves so that they are able to receive the substrate from the binding protein. After the ATP has been hydrolyzed, the NBD dimer opens up and the substrate is released into the cytoplasm. The transporter then reverts back to its resting state upon the release of phosphate and ADP. The only inconsistency in this mechanism is that the conformation in its resting, nucleotide-free state is different from the expected outward-facing conformation. While this may be the case, the key point is that the NBD does not dimerize unless ATP and the binding protein is bound to the transporter.
ABC exporters have a transport mechanism that is consistent with both the alternating-access model and the ATP-switch model. In the apo states of exporters, the conformation is inward-facing while the TMDs and NBDs are relatively far apart to accommodate the amphiphilic or hydrophobic substrates. The binding of the substrate initiates the transport cycle. The binding of the ATP induces NBD dimerization and formation of the ATP sandwich, which then drives the conformational changes in the TMDs. The cavity where the substrate binds is lined with charged and polar residues that are most likely solvated, which creates an energetically unfavorable environment for hydrophobic substrates and an energetically favorable environment for the polar moieties in amphiphilic compounds or sugar groups in LPS. Since they hydrophobic compounds cannot be stable in the chamber environment for a long time, they "flip" into an energetically more favorable position within the outer membrane leaflet. The repacking of the helices will then switch the conformation to an outward-facing state. The hydrolysis of ATP can widen the periplasmic opening, pushing the substrate towards the outer leaflet of the lipid bilayer. The hydrolysis of a second ATP molecule releases phosphate, separating the NBDs, followed by the restoration of the resting state. This will open up the chamber towards the cytoplasm of the cell, readying the exporter for another cycle.
Symporters is an indirect active transport. It couples the thermodynamically unfavorable flow of one species of ion or molecule up a concentration gradient with the favorable flow of a different species down a concentration gradient. In symporters, ions and/or molecules move in the same direction. Lastly, symporters can also be classified under secondary transporters or cotransporters because by definition they are membrane proteins that pump ions or molecules ‘uphill.’
[1]
This symporter uses H+ gradient across the E. coli membrane (outside H+ has higher concentration) generated by the oxidation of fuel molecules to drive the uptake of lactose and other sugars against a concentration gradient.
SGLT1
From the intestinal epithelium, SGLT1 transports sodium ions and glucose across the luminal membrane of the epithelial cells to be absorbed in the blood stream. This is the basis of oral rehydration therapy. Without this symporter, individual sodium channels and glucose uniports wouldn't be able to transfer glucose against the concentration gradient and into the bloodstream.
Na+/K+-ATPase
Na+,K+,2Cl- symporter
Found in the loop of Henle in the renal tubules of the kidney, Na+,K+,2Cl- symporter transports these four molecules. Loop diuretics act on this protein.
The structure of lactose permease has been determined. The structure consists of two halves, each of which comprises six membrane - spanning alpha helices. Some of these helices are somewhat irregular. The two halves are well separated and are joined by a single stretch of polypeptide. In this structure, the sugar lies in a pocket in the center of the protein and is accessible from a path that leads from the interior of the cell. On the basis of these structures, a mechanism for symporter action has been developed (See Figure Below).
1. The cycle begins with the two halves oriented so that the opening to the binding pocket faces the outside of the cell. A proton from outside the cell binds to a residue in the permease.
2. In the protonated form, the permease binds lactose from outside the cell.
3. The structure everts to the form observed in the crystal structure.
4. Lactose is released to the inside of the cell by the permease.
5. A proton is released to the inside of the cell by the permease.
6. The permease everts to complete the cycle.
The site of protonation likely changes in the course of this cyle.
↑Berg, Jeremy (2007). Biochemistry, 6th Edition. New York, New York: Sara Tenney. pp. 360–361. ISBN978-0-7167-8724-2. {{cite book}}: Unknown parameter |coauthors= ignored (|author= suggested) (help)
An antiporter is an integral protein involved in secondary active transport, which couples the energy of a molecule moving down its concentration gradient to another moving up its concentration gradient. The antiporter simultaneously transports two molecules at the same time in the opposite direction.
Antiporter, also called exchanger and counter-transporter, is an integral membrane protein that involves in a secondary active transporter of two or more different substrates (molecules or ions). These substrates are pumped in opposite directions across a membrane. It is a channel that transports different types of substrates across a phospholipid membrane (one into the cell and one out of the cell) either simultaneously or sequentially.
Antiporter
In secondary active transport, species of solute moves along its electrochemical gradient which allows other different species to move against its own electrochemical gradient. There are two main forms of secondary active transport, antiporters and symporters.
Secondary Active Transport
ATP-ADP Translocase
ATP-ADP translocase is one type of antiporter that assist in oxidative phosphorylation, a key component of cellular respiration in eukaryotic cells. This specific transport protein is located in the inner mitochondrial membrane that allows ATP and ADP to be transported across the membrane since ATP and ADP cannot diffuse across the inner mitochondrial membrane by itself. ATP-ADP translocase is in high abundance and it makes up about 15% of the protein content in the inner mitochondrial membrane. The high abundance of ATP-ADP translocase can be attributed to the fact that homo sapiens exchange ATP in very large amounts. In addition, this flow of ATP and ADP are coupled in that ADP can only enter the mitochondrial matrix if ATP exits (vice versa). This means that the translocase catalyzes the couples the entry and exit of ADP and ATP respectively from the matrix. This process is represented by the equation:
ATP-ADP translocase is about 30 kilodaltons in size and has a single nucleotide binding site that has the orientation facing the matrix side and the side of the cytoplasm of the membrane. ATP and ADP can bind to the translocase without the help of Mg2+, and since ATP has an additional negative charge than ADP (see equation above) the ATP transport out of the mitochondrial matrix and ADP transport into the matrix is favored for a mitochondrion with a positive membrane potential that is actively respiring. As soon as ADP binds from the cytoplasm, the transporter favors the configuration that will invert the transporter to release ADP into the matrix. The ensuing binding of ATP from the matrix to the inverted configuration favors the inversion of the translocase back to original configuration in which ATP is released into the cytoplasm (See Figure Below). However, the ATP-ADP exchange is still endothermic and requires about 25% of the energy yielded from electron transfer of respiration to be consumed in order to restore the membrane potential.
Analysis of the primary structure (amino acid sequence) of ATP-ADP translocase revealed that this protein is made up of three sequence repeats of length of 100 amino acids, in which each has two transmembrane segments. The structures of the three sequence repeats have been determined from three dimensional structure of this transporter. The three transmembrane helices form a conical structure that come together to form the binding site of the nucleotide in the center of the cone-like structure.
Uniporters are able to transport a specific species like ion channels in either direction governed only by concentrations of that species on either side of the membrane. Noted as either a channel or carrier protein, uniporter is an integral membrane protein involved in facilitated diffusion.
Uniporter carrier proteins bind to one molecule of solute at a time and transport it with the solute gradient. Uniporter channels open in response to a stimulus and allow the free flow of specific molecules. Since uniporters may not utilize energy other than the solute gradient, they may only transport molecules along with the solute gradient, and not against it. It is good to note that with uniporters the rate of movement is much higher than passive diffusion because the molecule never comes in contact with hydrophobic core of the membrane.
By definition, a uniporter is a channel protein that transfers only a single substrate at a time through the membrane. Multiple uniporters are found in the plasma membrane of most cells, where they allow small molecules such as amino acids, nucleosides, and sugars to enter and exit the cells down their concentration gradients. The movement often known as facilitated diffusion is when a substance moves through a membrane down its concentration gradient, and has the same negative ΔG value regardless of whether a protein transporter is included. This transport is fueled by the potential energy of a concentration gradient, and therefore does not need the assistance of metabolic energy.
Uniporters can be better understood by comparing and contrasting them to enzymes. They are similar to enzymes because they accelerate a reaction that is already thermodynamically favored. On the other hand, they are different from enzymes because the transported substance experience no chemical change when traveling through a membrane.
Uniporters are classified by two categories: channels and carriers. These two categories determine the kind of transmembrane solute transfers by different mechanisms. Channel proteins have hydrophilic pores that expand the lipid bilayer and are used to transport water-soluble solutes from both sides of the membrane at the same time. Carriers also expand the lipid bilayer, but unlike the channel proteins, their substrate binding locations cannot be simultaneously used on both sides of the membrane. Due to this mechanism difference, carriers are able to mediate counter-transports.
Involved in many biological processes, Uniporters use voltage-gated sodium channels in the propagation of nerve impulse across neurons. As the signal is transmitted from one neuron to the next, calcium is transported into the presynaptic neuron by voltage-gated calcium channels. The presynaptic neuron calcium is bound to a ligand-gated calcium channel in the postsynaptic neuron to stimulate an impulse within the neuron.
Potassium leak channels are also regulated by voltage and help restore the resting membrane potential after impulse transmission.
Sound waves in the ear cause stress-regulated channels in the ear to open vestibulocochlear nerve by sending impulses.
GLUT1 – widely distributed glucose transporter
Glucose uniporter shuttles between two conformational states. The net flow is reversed when the concentration of glucose changes.
GLUT4- primary insulin regulated glucose transporter in muscle and adipose tissue
UCP - uncoupler of proton gradient in mitochondria
Ion Channels are porous transmembral proteins which help regulate the voltage gradient across the plasma membrane by allowing ions to diffuse through the pores down their electrochemical gradients. They all regulate the flow of ions across the plasma membrane with several protein subunits assembled in a circular arrangement with the narrowest area in the pore being only a few atoms across in diameter. They selectively allow ions to diffuse either by size or by charge. Other ion channels may act like a gate, allowing ions to pass through by electrical, temperature or other stimulus.
Cyclic-nucleotide channels, also known as CNG channels, are key in the sensory signaling of various types of cells, ranging from neurons to sperm cells. They come in two classes: classical CNG channels and CNGK- and mICNG – type channels. These two classes have helped activation mechanisms evolve overtime.
CNG channels produce voltage responses as they keep track of the intracellular concentration of cNMPs (cyclic nucleotides). Cyclic n[1]ucleotides channels are permeable to Ca2+ ions, and can carry a mixed cation current, therefore making them able to create a chemical symbol.
Due to these characteristics, CNG channels play important roles in the signal pathways of retinal photoreceptors and olfactory receptor neurons. Photoreceptors in the retina respond to light and dark by opening or closing the CNG channels depending on cGMP (cyclic guanosine monophosphate) concentrations.
In the light, cGMP concentrations are low and photoreceptors go through a brief hyperpolarization and close the CNG channels. On the other hand, in the dark, cGMP concentration is high and CNG channels are open. When CNG channels are open, this allows a steady inward current.
In contrast to photoreceptors, olfactory receptor neurons open CNG channels when they are being stimulate, whereas CNG channels are closed at rest.
CNG channels may also play a role in guiding nerve growth cones by utilizing molecules that attract and repulse in the nervous system. Another function CNG channels claim to have is control of chemotactic response of sea urchin sperm. Ca2+ oscillations are produced by a cGMP-signaling pathway, and those oscillations navigate the sperm.
Recent studies of the CNG channel family have found new members within bacteria and sea urchin sperm studies focusing on the function and structure of the new members show that binding a single molecule suffices to open CNG channels. There exist several other differences between the new CNG channels in comparison to the classical CNG channels.
Classical CNG channels are heterotetramers with subunits A and B. In mammals, four different A subunits and two different B units are known to exist in mammals.
There are two classes of CNG channels: low and high ligand sensitivity. Differences in ligand sensitivity are organized by the different interactions between ligand and CNBD. Another way ligand selectivity can be determined is by a specific Asp residue in the CNGA1 channel. Side chains that are negatively cGMP selectivity and reduces the cAMP gating efficacy.
New CNG (cyclic nucleotide-gated ion channels) have been found in sea urchin sperm and bacteria that are uncooperative because they only need a ligand molecule to bind in order to open the gate.
For photoreceptors in the eye, there is a high concentration of cGMP in dark; the light creates hydrolysis at phosphodiesterase. For olfactory receptor neurons, the odorants start the synthesis of cAMP.
CNG polypeptides contain transmembrane domains named S1-S6. S4 which detects voltage in Ca, Na, and K channels has a chain of charge Lys or Arg residues spread out between hydrophobic amino acids. This is unlike the classic CNG channel which does not rely as heavily on voltage. Glutamic acid is a key component of CNGs: in the hairpin turn portion containing pores, the Glu is carried as residue; at the tetramer, a ring of Glu provides an area for organization and movement of Ca+2 ions.
At the C-terminus, the cyclic nucleotide-binding domain or CNBD is attached to S6 by about 80 amino acids which convey the binding of the ligand with the gate of the channel. It is thought that this link supports tetramerization.
At the A and B parts of the CNG, calmodulin binding (CaM) with Ca+2 dictates the channel’s activity. Four of these CaM binds have been found in olfactory neurons, but only those at B and A4 seem to matter due to their N and C-terminus binding. The binding between CaM and Ca+2 has been found to lower sensitivity to ligands, offering a smoother transition to the resting state.
The mlCNG channel from the bacteria Mesorhizobium loti is like classic CNG channels in the sense that it creates tetramers and is made from six transmembrane segments with a C-terminus CNBD. It differs because it contains the GYGD sequence of K channels (Gly-Tyr-Gly-Asp). Also, instead of being about 80 amino acids, the C-link is about 20 amino acids which cause it to not support tetramerization.
CNGK structures are CNG channels which has the sequence of the K channel in its pore region. CNGK’s have a large polypeptide which holds four areas with a similar sequence or ‘repeats’.
Scientists are trying to see how it would be possible to open all four gates at once. They use double electron-electron resonance (DEER) to give the structure and point out where residues and conformation movements are.
CNG channels help us understand the relationship between ligand binding and gates.
[2]
Potassium channels are found basically in most organisms. Potassium channels must be ancient because many channels had originated from them. Generally, there are two types of potassium ion, the six-transmembrane helix voltage gated and the two-transmembrane helix inward rectifier. Usually, potassium channels have the amino acid sequence of TMxTVGYG. In addition, the six-transmembrane helix voltage gated type typically has the unique pattern of lysine and arginine show up at every third or fourth place in the sequence. Scientists had studied potassium in a bacterial protein called KcsA and found that its structure is very similar to that of eukaryotic. Thus, they used KcsA as a model to study about K+ channel.
Potassium channel in a shape of a pore locates in the plasma membrane but not in a random manner. It carries an important role for it contributes in many different processes in the body such as secreting of hormones, regulating vascular system, etc. Potassium channel is also important in plant for guard cells uses K+ channels to control osmotic flow. Potassium channel has two components to it: the filter and the gate. The filter is responsible for choosing the ion, which in this case is potassium, and the gate opens and closes depending on the outside environment such as the effect of voltage or signaled molecules. The structure of the filter is similar to that of hemoglobin in the way that it is a tetramer with four repeating subunits that surround the pore. Also, this structure is known to be asymmetric. Moreover, the subunits are hydrophilic. The flow of potassium ion through the channel happens very quickly. Although the name is potassium channel, it also allows sodium ion to pass through. However, the ratio of potassium to sodium is 10,000 to 1. Another reason why sodium ion wants to get through the channel is because the ions are competing against each other.
Since potassium channel relates to nerve signaling and if the channel does not function normally, this can result in serious damage. What makes potassium channel only permeable to K+ ion is the imitation of oxygen across the channel. Potassium is known for having water around it. The formation of oxygen on the inside wall of the channel facilitates the flow of potassium ion through the channel. Even though much is known about potassium channel, the function of K+ channel in prokaryotic cell is still a mystery.
M2 proton channel resides in the envelope of the virus influenza A. The structure of the M2 proton channel is a homotetramer interconnected by disulfide bonds. is similar to that of potassium channel in which is it also a tetramer, and it consists of four similar subunits. The subunits in this case is the M2 itself. Moreover, the subunits take the shape of helices and are bonded together by disulfide bonds. M2 proton channel functions best when the pH level is low. The M2 protein is composed of three components: the 24 amino acids of the N terminal which face the outside of the cell, the 22 amino acids which are embedded on the membrane and are hydrophobic, and the 52 amino acids of the C terminal which face the cytoplasm. [3]
Superimposition of the X-ray(yellow) and NMR(red) structures of the M2 proton channel. Also depicted are predicted inhibitory hotspots, X-ray in blue and NMR in green.
The 22 amino acid chain forms a trans membrane alpha helix. Since the amino acid Val27 is close to the N terminal the M2 channel is very selective in term of protons. As a result, protons that go through the channel can only pass through in the form of water molecule, and it is not easy for molecules such as hydrated sodium to pass through.
Activation of the proton channel occurs from low pH. The main function of the M2 channel is to allow hydrogen ions from the endosome to enter the virion and lower the pH within the virus. This causes disassociation of the matrix protein surrounding the virus’s genome, which is crucial in exposing the virus’s contents to the host cell’s cytoplasm.[4] The two medicines that were designed to stop the function of M2 channel are amantadine and rimantadine. These two medicines prevent the attack of the virus on the host cell. Nevertheless, because M2 is prone to be affected by mutations, the use of these medications becomes helpless. For instance, if one amino acid in the membrane is changed, the drugs will not be able to block the virus anymore. Two structures of M2 have been discovered: one is mutated and locates in the membrane, the second structure is similar to the first except it is longer, and it has the C-terminal component. Due to the virus’s susceptibility to mutations, substitutions along the trans membrane alpha helix have led to the virus’s immunity to inhibitors. In particular, the Ser31Asn substitution renders the protein highly resistant to amantadine inhibition. This most recently occurred during the 2009 ‘swine flu’ pandemic where 100% of samples tested during the season were resistance to amantadine and its derivative rimantidine. [5]
Scientists began to understand the structure and function of ion channels beginning in the early 1950s when British biophysicists Alan Hodgkin and Andrew Huxley analyzed the properties of currents by ion channels in their 1951 research on action potentials which won them the Nobel Prize. The existence of ion channels were indirectly confirmed by Bernard Katz and Ricardo Miledi in 1971 and later were directly proved by Erwin Neher and Bert Sakmann by using the patch clamp technique. In 2003, the Nobel Prize in Chemistry was awarded to Roderick MacKinnon for x-ray crystallographic studies on ion channel structure and to Peter Agre for similar work on aquaporins.
Ion channels are prominent features of the nervous system which allow for conduction between the synapses. In addition, ion channels facilitate rapid changes in cells and are therefore useful as a target for new drugs. However, the importance of ion channels for nerve cell function result in evolution of toxins secreted by animals and plants which target the central nervous system and result in paralysis. Some examples include tetrodotoxin used by pufferfish for defense and novocaine which block sodium channels. Others include dendrotoxin which is produced by the snake, the Black Mamba, which blocks potassium ion channels. In addition, genetic mutations in DNA which encode for the ion channel protein would cause the same type of reactions.
↑ Helenius, A. (1992) Unpacking the incoming influenza-virus. Cell 69,577–578
↑Crosby, Niall J; Deane, Katherine; Clarke, Carl E (2003). Clarke, Carl E (ed.). "Amantadine in Parkinson's disease". Cochrane Database of Systematic Reviews. doi:10.1002/14651858.CD003468.
To measure what's happening in or on a single, living cell, scientists use a technique called the patch clamp which requires an extremely fine pipet held tightly against the cell membrane. The technique was introduced by Erwin Neher and Bert Sakmann in 1976.
This powerful technique enables the measurement of the ion conductance through a small patch of cell membrane. The stepwise changes in membrane conductance are observed. These changes correspond to the opening and closing of individual ion channels. In this technique, a clean glass pipette with a tip diameter of about 1 micro m is pressed against an intact cell to form a seal. The slight suction from the pipette leads to the formation of a very tight seal. Therefore, the resistance between the inside of the pipette and the bathing solution is many gigaohms. Thus, a gigaohm seal known as a gigaseal ensures that an electric current flowing through the pipette is identical with the current flowing through the membrane covered by the pipette. The gigaseal makes possible high - resolution current measurement while a known voltage is applied across the membrane. A time resolution of microseconds monitors the flow of ions through a single channel and transition between the open and the closed states of a channel.
Patch clamp is a powerful technique in electrophysiology for studying ion channels in cells. This technique is very useful in the study of neutrons, muscle fibers, pancreatic beta cells, etc. Patch clamp can be applied and used to study bacterial ion channels.
In the late 1970s and early 1980s, Erwin Neher and Bert Sakmann developed the patch clamp technique and were awarded the Nobel Prize in Medicine in 1991. Their discovery made it possible to record and measure currents of single ion channels.
The patch - clamp technique for monitoring channel activity is highly versatile. A high - resistance seal (gigaseal) is formed between the pipette and a small patch of plasma membrane. This configuration is called cell attached mode. The breaking of membrane patch by increased suction produces a low - resistance pathway between the pipette and the interior of the cell. The activity of the channels in the entire plasma membrane can be monitored in this whole - cell mode. To prepare a membrane in the excised - patch mode, the pipette is pulled away from the cell. A piece of plasma membrane with its cytoplasmic side now facing the medium is monitored by the patch pipette.
The pipette is pressed against the cell to form a gigaseal by sucking a small area of the membrane to break both the membrane and cytoplasm off the cell. This leaves the patch of membrane attached to the pipette and exposes the interior of the membrane to the external solution. A voltage is applied across the patch, allowing experimenters to see recordings of discrete current steps through single ion channels without disrupting the cell’s interior.
This is an excised patch technique because the patch is excised or removed from the main body of the cell. The internal membrane surface faces the bath solution (the pipette solution). The intracellular space of the membrane is exposed to the bath to allow testing of various intracellular channel modulators.
Fresh glass pipette with a tip diameter of only a few micrometers is pressed on the cell membrane gently to form a gigaseal. Suction is applied to break the membrane and the cytoplasm. The pipette solution starts to mix in to make the cell have a similar ionic environment as the saline filling the pipette uses.
Using the whole-cell mode, the pipette is pulled away from the cell which excises a patch with the extracellular side of the membrane facing the bath solution (the pipette filling solution). The patch is superfused on the extracellular side with the solution. The solution contains agonists (i.e. GABA or glutamate) which activate the receptor channels in the patch.
Once the whole-cell is established, instead of using suction to rupture the patch membrane, an electrode solution that contains antibiotics diffuses into the membrane patch forming small perforations that provides electrical access to the intracellular side of the cell.
This employs a loose seal rather than a tight gigaseal. An advantage of loose seal is that the pipette can be removed from the membrane without rupturing the membrane, the membrane will still remain intact. This allows repeated measurements on the same cell without destroying the membrane’s integrity. A disadvantage is the resistance between the pipette and the membrane is reduced, thus allowing current to leak through the seal.
Transient receptor potential (TRP) ion channels get their name from a Drosophila phototransduction mutant that show a transient instead of a sustained response to bright light. In addition to responses to light, TRPs mediate responses to nerve growth factor, pheromones, olfaction, mechanical, chemical, temperature, pH, osmolarity, vasorelaxation of blood vessels, and metabolic stress. TRP-related ion channel proteins are found in a variety of organisms, tissues, and cell types, including nonexcitable, smooth muscle, and neuronal cells. TRP channels are recruited to diverse signaling pathways, such as gating mechanisms, regulation, and permeability.
Reference:
1. Ajay Dhaka, Veena Viswanath, and Ardem Patapoutian, "TRP Ion channels and Temperature Sensation"
2. Baruch Minke and Boaz Cook, "TRP Channel Proteins and Signal Transduction"
3. Kartik Venkatachalam and Craig Montell, "TRP Channels"
Ligand-gated Ion Channels (LGICs) are a group of transmembrane ion channels that open when a signal molecule (ligand) binds to an extracellular receptor region of the channel protein. This binding changes the structural arrangements of the channel protein, which then causes the channels to open or close in response to the binding of a chemical messenger such as a neurotransmitter. This ligand-gated ion channel, a type of ionotropic receptor, allows specific ions (like Na+, K+, Ca2+, or Cl-) to flow in and out of the membrane. Examples of ligand-gated ion channels include acetylcholine receptors, serotonin receptor, GABAA, and Glutamate receptor.
http://commons.wikimedia.org/wiki/File:Ligand-gated_ion_channel.JPG
3 superfamilies of extraceullarly activated ligand-gated ion channel subunits:
Receptors of the cys-loop superfamily - five protein subunits which form a pentameric arrangement around a central pore
Serotonin (5-HT) receptors are found in the central and peripheral nervous system. They mediate both excitatory and inhibitory neurotransmission and are activated by serotonin. They modulate the release of many neurotransmitters, including epinephrine, acetylcholine, and many hormones. Because of its wide versatility, the serotonin receptors influence many biological processes such as aggression, anxiety, appetite, cognition, learning, memory, mood, nausea, sleep, and thermoregulation. As one can see, this is one very important receptor.
Glycine receptors is the receptor for neurotransmitter glycine. It is one of the most widely distributed inhibitory receptors in the central nervous system and has important roles in mediating inhibitory neurotransmission in the spinal cord and brain stem.
Nicotinic Acetylcholine receptors are cholinergic receptors that form ligand-gated ion channels in the plasma membranes of specific neurons. At neuromuscular junctions, nicotinic acetylcholine receptors form a cation channel that opens when two molecules of acetylcholine bind to the channel's extracellular region. This results in sodium ion influx, which causes depolarization of the muscle cell membrane.
GABAA receptor (GABAAR) is an ionotropic receptor and ligand-gated ion channel. GABAA receptor is the binding site for GABA and several drugs such as muscimol, gaboxadol, and bicuculline. GABAA receptor allows Cl- to pass through its pore which hyperpolarizes the neuron. This inhibits neurotransmission by diminishing the chance of a successful action potential occurring.
ATP-gated channel superfamily
glutamate activated cationic channel superfamily
NMDA receptor is a glutamate receptor that, when activated, triggers the opening of ion channels that creates a flux of calcium into the cell. It is both ligand-gated and voltage-gated, and its activation requires both glutamate and glycine. Calcium influx is thought to control synaptic plasticity and memory function, so NMDA receptor is a topic of interest for those who wish to learn about memory formation.
AMPA receptor is an ionotropic transmembrane receptor for glutamate. It mediates fast synaptic transmission in the central nervous system. Native AMPA receptor channels are impermeable to calcium, a function controlled by the GluR2 subunit in the receptor. The calcium permeability is determined by the post-transcriptional editing of the GluR2 mRNA. AMPA receptor permeabiilty to calcium determine largely the function of the signal transduction behavior.
kainate receptors are ionotropic receptors that respond to glutamate. They are less well understood than AMPA and NMDA receptors. Kainate receptors are involved in excitatory neurotransmission via the activation of postsynaptic receptors, and they also participate in inhibitory neurotransmission by modulating release of GABA through a presynaptic mechanism.
/Gap junctions/Gap junctions The gap junctions are strictly packed into a pattern of hexagonal lattice. With the aid of an electron micrograph, each gap junction can be observed to have a 20 Armstrong central hole, which is the lumen of the membrane channel. The channels are also condensed in localized regions of the cell plasma membrane of contiguous cells, and they are spread out in the gap between neighboring cells. The distance of the gap between the cytoplasm of the two apposed cells is roughly 35 Armstrong.
A typical gap junction is composed of connexin, which is a transmembrane protein that varies in mass from 30 to 42 kd. One connexin possesses four membrane-spanning helices. Connexon (hemichannel) are half channels made up of six connexin molecules that are shaped into an extensive hexagonally lattice. A pair of connexons may connect end to end in the gap between adjacent cells to generate a special channel between neighbor cells in order to communicate. Our genome contains twenty-one different connexins. Different tissues un-codes the various connexins in distinguish ways.
Picture form Wikipedia.
In three significant ways, gap junctions are unique compared to membrane channels:
1. The cell-to-cell channels cross two membranes versus just one membrane.
2. The channels join cytoplasm to cytoplasm, and not to the extracellular gap or the lumen of any organelle.
3. Different cells produced the connexons forming the cell-to-cell channels.
The gap junctions tend to remain open for minutes or seconds after they are made because the channels are quickly formed when to cells are in contact. The junctions will be closed if exposed to acidic conditions or high levels of calcium ion. The gap junction closure by calcium ions and protons is a self-saving mechanism of the cell to protect/seal normal cells from injured or dying cells near by. Membrane potential and hormone- assisted phosphorlyation help maintain the gap junctions.
Again, gap junctions are passageway for cell communication. Small ions and hydrophilic substances can flow through the cell-to-cell channel. The pore size of the channel can be experimentally deduced by microinjecting several fluorescent particle into the observed cells and tracking the fluorescent molecule movement to the adjacent cells. The gap junctions facilitated the passage of inorganic ions, amino acids, sugars, and nucleotides in the insides of cells. Large molecules, such as proteins, polysaccharide, and nucleic acid cannot flow through the gap junctions because of their hefty size. Overall, most polar particles that are less than 1kd may easily pass through the cell junction.
Biological Importance of the Gap Junction
Intercellular nourishment and communication are made possible by the cell-to-cell channels. For instance, the cell-to-cell channel plays a key role in providing nourishment to cells that are further away from the blood vessels; bone and lens cells are among numerous cells that can obtain nourishment via gap junctions rather than directly from the blood vessels. Concerning communication between cells, the channels affect cells in strength tissues, such as the heart muscle, are complemented by the intense rush of ions through the protein channel to signify a quick response to the appropriate trigger or stimuli.
The gap junctions govern some aspects of cellular differentiation and growth. The creation of cell-to-cell channels during labor (delivery of a new born) the uterus no longer shelters the baby but is forced to push the baby out by multiple contractions. This is a classic example of muscle cells acting simultaneously to create contractions because of the formation of gap junctions during delivery.
Since various members of the 21 human connexins are expressed differently in the tissues, a mutation in one of the connexins has major consequences. Take connexin 26 for instance; connexin 26 encodes for vital ear tissues and a mutation in connexin will lead to being deaf (hereditary). This is due to the fact that unsuccessful transportation of secondary messenger molecules (inositol trisphoate) or small ions in between sensory cells for the ears creates deafness.
Reference:
Biochemistry . 6th ed. New York : W. H. Freeman and Company, 2007. 373-374. Print.
Crystallographic structure of the aquaporin 1 (AQP1) channel (Template:PDB).
Water channels play a critical role in controlling the water contents of cells. Their connection to water - the essential substance of life - make them pervasive throughout all kingdoms of life.
Water channels can also be referred to as aquaporins. They do not take part in ion transport at all. Instead, these channels increase the rate at which water flows through membranes. Even though membranes are reasonably permeable to water, these water - specific channels are required because in certain tissues rapid water transport through the membranes is necessary. For example, in the kidneys water must be quickly reabsorbed into the bloodstream after filtration.
Water channels (aquaporin) were discovered by Peter Agre. He noticed a protein present at high levels in red blood cell membranes. The protein was found to be present in large quantities in red blood cells and tissues such as kidneys and corneas. This 24-kd membrane protein was actually a water channel.
Water channels or aquaporins in the cell membrane form tetramers that assist in transporting H2O across the cell membrane. Occasionally, some small solutes such as glycerol may cross over as well. Water channels are impermeable to other charge molecules such as protons. This is key to maintain the electrochemical equilibrium. Water channels (aquaporins) consist six membrane-spanning segments. They are located in the plasma membrane as homotetramers, and each of the segments are made up of 2 hemi-pores that form the aquaporins when folded together. Aquaporin's molecular structure makes sure that ions don't pass between the cells. The channel is made of more than 100,000 atoms, that come together to create a structure that sends water molecules through the channel in a single file line. This is done by orienting the molecules in an electric field that is produced by the channel wall. The positive charged formed in the center of the channel repels any unwanted protons from entering through the channel.
The protein consists of six membrane - spanning alpha helices. There are five interhelical loop regions that form the extracellular and cytoplasmic vestibules. Two of the loops are hydrophobic which contain the highly conserved Asn-Pro-Ala, which overlap the middle of the lipid bilayer of the membrane forming a 3-D 'hourglass' structure where the water flows through. The water molecules pass through in single file at rate of 106 molecules per second. Specifically, positively charged residues toward the center of the channel prevent the transport of protons through aquaporin. Therefore, aquaporin channels will not disrupt protein gradients.
One characteristic that interest researchers about the aquaporins is that the channels only permits the transport of water and other small non-charged solutes across the membrane. All charged species, such as protons, are completely impermeable in the channel. Based on hydrophobicity plots of their amino acids sequences (structure determines function!), each aquaporin monomer contains two hemi-pores, which fold together to form a water channel. The water molecules work their way through the narrow channel in a single file by orienting themselves in the local electrical field established by the atoms of the channel wall. The strictly opposite orientations of the water molecules ensures the lowest electrostatic repulsion between the water and the wall, thereby allowing the water molecules through to the other side. Charged species, on the other hand, are stopped and rejected out of the channel once they reach the positively charged center.
Water permeation through aquaporins is a passive process that relies on the direction of osmotic pressure across the membrane. Although many water channels are always open, a subgroup of aquaporins has evolved a complex mechanism (mainly involves covalent modification phosphorylation and change in pH) through which the channel can be closed in response to harsh conditions of the environment, under which water exchange can be harmful to the organism.
After primary urine exits the glomerulus of the kidney it passes through winding tubes where about 70% of the water is reabsorbed to the blood by the aquaporin 1 protein. Another 10% is absorbed after the tubes by aquaporin 2 protein. In addition to water, solutes such as sodium, potassium and chloride ions are also reabsorbed into the blood. Antidiuretic hormone stimulates the transport of aquaporin 2 proteins and hence increases water resorption from the urine. People deficient in this hormone might have a daily urine output of 10-15 liters!
Macromolecules must move between the nucleus and cytoplasm often in eukaryotic cells because replication and transcription happens in the nucleus and translation happens in the cytoplasm. The nuclear envelope separates the nucleoplasm (that has the genetic material) from the cytoplasm; the RNA molecules that are made in the nucleus must be translated to proteins in the cytoplasm. These macromolecules move through the nuclear envelope by way nuclear pore complexes (NPC).
The nuclear pore complex was discovered in 1950 and has been studied extensively by electron microscopy. Nuclear pore complexes are the openings that macromolecules use to cross from the nucleus to the cytoplasm and vice versa. NPCs are pores in the nuclear envelope and are a part of aqueous transport channels that participate in the transport of macromolecules between the cytoplasm and nucleus.
The nuclear pore complex is made of thirty different proteins known as nucleoporins; there are about 500-1000 proteins in the NPC because the NPC has internal symmetry and each nucleoporin repeats many times. Nucleoporins are made from α- helical regions, β-propellers, and phenylalanine-glycine (FG) repeats.
Electron microscopy revealed that the nuclear pore complex had a donut shaped core and is covered with cytoplasmic filaments and has a nuclear basket. It is unfeasible to use X-Ray crystallography to study the structure of the NPC because of size and flexibility of the complex and because it is very difficult to get a large amount of the pure complex in order to form a crystal.
Structure of the NPC. 1. Nuclear envelope. 2. Outer ring. 3. Spokes. 4. Basket. 5. Filaments.
The function of the nuclear pore complex, as stated above, is to help transport macromolecules back and forth between the nucleus and the cytoplasm. In addition, the nuclear pore complex also serves as a barrier between the cytoplasm and nucleus to prevent harm to genetic material housed in the nucleus. Macromolecules move through the NPC through diffusion channels (up to a size of about 40 kilodaltons), the diffusion barrier is made by spreading nucleoporins that have many FG repeats. FG repeats also serve an important role in the NPC; they act as a docking zone for transport receptors (known as karyopherins) that move molecules between the NPC. For molecules to move through the NPC, they have short localization sequences for import, and nuclear export sequences for export.
The nuclear pore complex has roles outside of nucleocytoplasmic transport between the nucleus and cytoplasm. The NPC is also involved in chromatin organization, gene expression regulation, DNA repair, and many other functions.
Defects in the nuclear pore complex lead to many diseases because it is integral to cellular structure. These diseases include hematological neoplasms, heart arrhythmia, primary biliary cirrhosis, and others.
Cotransporters are protein pumps used to export or import small molecules. It is sometimes equated with symporter, but the term "cotransporter" refers both to symporters and antiporters (though not uniporters). They utilize active transport, meaning that they require some sort of energy to carry out its process. Cotransporters are secondary active transporters, which means they use an electrochemical gradient as a means of energy. It works by binding to two molecules or ions at a time and using the gradient of one solute's concentration to force the other molecule or ion against its gradient.
Primary active transporters, on the other hand, use chemical energy like ATP. The electrochemical gradient for the cotransporter is due to the movement of Na + and H+ ions. This powers the movement of another substance that is pumped either in or out, against the concentration gradient. An example is the movement of glucose. In order for the transportation of glucose to be in the opposite direction of the concentration gradient, sodium ions are needed. Na+ ions are moved across the cell membrane by a transmembrane voltage gradient (movement from an area of positive charge to negative charge), and by a concentration gradient (from an area of high concentration to low concentration). Glucose is then coupled and moved against its concentration gradient. When the Na+ ion, an example of a cotransported ion, moves in the same direction as the coupled substance, it is call symporter; movement in the opposite direction of a substance is called antiporter.'
Transport of glucose in animal cells by symporters
A symporter is an integral membrane protein that is involved in movement of two or more different molecules or ions across a phospholipid membrane such as the plasma membrane in the same direction, and is therefore a type of cotransporter. Cotransporters are used for the transport of glucose in the cell. Glucose is needed in different organs, but there is already an abundance of glucose in these areas. This prevents glucose from entering by passive transport. Active transport is thus used, using a 2Na+/1Glucose symporter. The transporter moves one glucose in for every two sodium ion, and moves them out in the same ratio.
What forces are used to drive the substances in and out of the cells? As mentioned before, the electrochemical gradient and the concentration gradient of the Na+ ion allows this process to happen.
This is done by the free energy change (ΔG) of transportation of Na+ ions. The concentration of Na+ ions is either higher or lower inside the cell than outside. The charge of the cell is also either inside negative or inside positive. The flow of Na+ ions is either to the inside or the outside, depending on which side has more Na+ ions and the charge of each side. Na+, like all particles, will go from an area of low concentration to high concentration. In addition, Na+ will move from an area of positive charge to negative charge, until there is a balance between the concentration of Na+ and a balance between the charges. This is the equilibrium potential, and it can be calculated by the Nernst equation.
There is a free energy change of the cell for the transport of Na+ ions, which will affect the concentration of glucose inside and outside the membrane.
Transport of calcium ions from cardiac muscle cells by antiporter
An antiporter (also called exchanger or counter-transporter) is an integral membrane protein which is involved in secondary active transport of two or more different molecules or ions (i.e. solutes) across a phospholipid membrane such as the plasma membrane in opposite directions.
In secondary active transport, one species of solute moves along its electrochemical gradient, allowing a different species to move against its own electrochemical gradient. This movement is in contrast to primary active transport, in which all solutes are moved against their concentration gradients, fueled by ATP.
Antiporters play a key role in movement of calcium ions out of cardiac muscle cells. For every 3 Na+ ions out (or in) the membrane, 1 Ca2+ ions are pumped oppositely in (or out) the membrane. This is significant for different physiological functions, such as relaxation of cardiac muscles, maintenance of calcium ion concentration in different cellular organelles, control of neurosecretion, and other functions.
The nervous system functions by the almost instantaneous transmission of electrochemical signals. Highly specialized cells called neurons control the means of transmission. They are also the functional unit of the nervous system.
The neuron is an elongated cell with three parts: dendrites, cell body, and an axon.
The typical neurone contains many dendrites which appear to look like thin branches that extend from the long branch of the cell body. The axon is a single long projection that extends from the cell body. It usually ends in a few small branches called axon terminals. Is is also covered by the myelin sheath to promote neural impulses, or to ensure the signals get transferred smoothly and quickly. Neurons are usually connected in chains and networks. They are physically close to each other, yet never actually come in contact with one another. The gap that separates the axon terminals of one neuron from the dendrites of another neuron is called the synse.
When an electrical impulse moves through the neuron, it starts at the dendrites. Basically, dendrites receive messages from other cells. From there, it passes through the cell body along the axon. Impulses always follow the same path from the dendrite to the cell body, and then the axon. When the electrical impulse reaches the synapse at the end of the axon, special chemicals called neurotransmitters are released. The neurotransmitters will carry a signal across the synapse to the dendrites of the next neuron to restart the process in the next cell. They, as chemical messengers, are called agonists because they open cellular locks or receptors to communicate between the outside and inside of the cell (Medicines by Design 10-11), just like a key fitting into lock.
When there is no impulse traveling through a neuron, the cell is said to be at its resting potential. The inside of the cell contains a negative charge in relation to the outside. The cell requires energy to maintain this negative charge. The cell membrane of the neuron contains a protein called Na+/K+ ATPase that uses the energy provided by one molecule of ATP to pump 3 positively charged sodium molecules out of the cell. At the same time, it brings in 2 positively charged K+ ions, creating a system that has a higher concentration of Na+ on the outside and K+ on the inside. A sodium leak channel allows some of the potassium ions to flow out of the cell. Nonetheless, the difference in concentrations creates a net potential difference across the cell membrane of about -70mV, which is the value of the resting potential.
The action potential is the electrochemical impulse that can travel along the neuron. The neuron membrane also contains voltage-gated proteins. These proteins will respond to changes in the membrane potential by opening and allowing certain ions to cross that would normally not be allowed to do so. The neuron has both voltage-gated sodium channels and voltage-gated potassium channels. Each one will open under different circumstances.
The action potential will begin when another neuron sends chemical signals to depolarize, or make less negative, the potential of the cell membrane in one localized area of the cell membrane- usually in dendrites. When the neuron is stimulated so that the cell membrane potential reaches -50mV, the voltage-gated sodium channels in that region will open up. The voltage at which these channels open up is called the threshold potential. (in this case, the threshold potential is -50). When the voltage-gated channels open, the sodium ions outside the cell will follow the concentration gradient and rush into the cell. The flood of sodium ions will cause the cell to depolarize and eventually the membrane potential will increase to +35mV. At this point, the voltage-gated sodium channel will close and the voltage-gated potassium channel will reach their threshold and open up. The positive potassium ions concentrated in the cell will now rush out of the neuron to repolarize the cell membrane to its negative resting potential. The membrane potential will now drop to -90mV and the voltage-gated potassium channels will close. After this occurs, the potassium leak channels will restore the membrane back to its original state with a potential of -70mV. This whole process will take place in about one millisecond.
The vertebrate nervous system can be divided into 2 main parts: the central nervous system (CNS) and the peripheral nervous system (PNS). The central nervous system acts as a central command that receives sensory input from all regions of the body and integrates the information toe create a response. It controls most of the basic functions that are needed for survival, such as breathing, digestion, and consciousness. On the other hand, the peripheral nervous system refers to pathways through which the central nervous system communicates with the rest of the organism. In highly evolved systems, such as the human nervous system, there are 3 types of neural binding blocks: sensory, motor, and interneurons.
Sensory neurons send information to the central nervous system after the organ's sense organs receive a stimulus from the environment. Another name for these neurons is afferent neurons.
Motor neurons carry information away from the central nervous system to an organ or muscle as a response to a stimulus or a voluntary action. Another name for these neurons is efferent neurons.
Interneurons provide connection between sensory neurons and motor neurons.
CENTRAL NERVOUS SYSTEM
The CNS consists of the brain and the spinal chord and plays a role in the communication between sensory receptors, muscles, and glands. The spinal cord is a long cylinder cord that extends along the vertebral column back bone from the head to the lower back.
The brain is made of almost entirely interneurons. The cerebrum is the largest portion of the brain and controls consciousness. It controls all voluntary movement, perception, sensory perception, speech, memory, and creative thought. The cerebellum helps to fine-tune voluntary movement, but does not initiate it. It makes sure that movements are coordinated and balanced. The brainstem is responsible for the control of involuntary functions, such as breathing, cardiovascular regulation, and swallow. It is a portion of the medulla oblongata, which is essential for life and processes a great deal of information. The medulla also helps maintain alertness. The hypothalamus is responsible for maintenance of homeostasis. It regulates temperature, controls hunger, manages water balance and helps to generate emotion.
The spinal cord contains 3 types of neurons: axons, interneurons, and glial cells. Axons of motor neurons extend from the spinal column into the peripheral nervous system. Interneurons link motor and sensory neurons. Glial cells provide physical and metabolic support for neurons. It serves as a link between the body and the brain so that it can regulate simple reflexes
THE PERIPHERAL NERVOUS SYSTEM
It is a sensory system that carries information from the senses into the central nervous system from the body and motor system that branches out from the CN to organs or muscles. The motor system can be divided into 2 parts: somatic system and the autonomic system, however, the two systems work together to ensure a proper internal states to avoid any extreme responses.
The somatic nervous system is responsible for voluntary or conscious movement. Neurons will only target skeletal muscles needed for bodily movement. All of the neurons in the somatic system release acetylcholine, an excitatory neurotransmitter that causes skeletal muscles to contract.
The autonomic nervous system controls tissues other than skeletal muscles. It controls processes that an animal that does not have voluntary control over, such as heart beat, movement of the digestive tract, and contraction of the bladder. It can be subdivided into sympathetic division and parasympathetic division. Sympathetic division works to prepare the body for emergency situations. It increases heart rate, dilates pupils, and increases breathing rate. It also stimulates the medulla of the adrenal glands to release epinephrine and norepinephrine into he bloodstream. Together, it creates the "fight of flight" response. The parasympathetic division is most active when the body is at rest. It slows the heart rate, increases digestion, and slows breathing. it creates the "rest and digest" response.
PARKINSON'S DISEASE
Parkinson’s Disease (PD) is a degenerative disorder affecting the nervous system. This is caused by low levels of the neurotransmitter dopamine produced in the brain. Evident symptoms include trembling or stiffness in body limbs or face, instability in posture, or lack of balance and body coordination. These symptoms can cause labor in simple tasks, such as walking and talking. The severity in the impairment of the motor skills differ between individuals. Some may become critically disabled and others may only suffer from minor motor trembling. This is a chronic disease that usually affects people around the age of 50. There is no cure for this disease, therefore, the symptoms progressively worsens over time.
Wilson Disease
The Wilson Disease is a inherited mutation on chromosome 13. This disorder causes a person's body from removing excess build up of copper. The protein that is in charge of the removal of copper is ceruloplasmin. With the inability for one to be able to remove copper can lead to cirrhosis, which is the damaging of liver cells and hindering the liver function. Evident symptoms of having the Wilson Disease is having extreme body tremors and decrease in speed in body movement. Also, the person's mental capabilities will decrease and emotionally sensitive, especially temperamental. Treatment can include avoid eating any food that contains copper or chelating the excess copper to remove from the body.
The evolution of membrane proteins and the structure of membranes remain unclear despite the advancement of our understanding of molecular membrane mechanisms and the complex topology of the membrane. One model of understanding the origins of the membrane and membrane proteins is that the biological membrane was a co-evolution of three things: lipid bilayers, membrane proteins, and membrane bioenergetics.(Mulkidjanian, Galperin, Koonin)
The goal of this model is to provide an insight into what features would the "LUCA"(last universal common ancestor) have. The premise for this model, relies on the idea that the division between archaea and bacteria was the first among cellular organisms. Using this then one can come to compare these two organism's components and try to deduce common features present in their ancestor.
Possible Molecules for the Formation of the First Membranes
There are two proposed arguments for the nature of the "LUCA" membrane. One is that fatty acids are molecules that are simple and able to from abiogenetically to be used by primordial organisms as their membranes. The other argument is that polyprenyl phosphates are a viable molecule for the first membrane due to the fact that they have the ability to form vesicles in the presence of sodium. This would then be a primitive mechanism for cation transport.
Both F-type and V-type ATPases thought to be present(or some form of these proteins) in the LUCA for the reason that these are common in all of modern cellular life. Both F and V-type ATPases use energy from ATP hydrolysis to transfer cations across membranes. The however do require impermeable membranes to establish ion gradients. It is postulated that sodium gradients were used in primordial membranes for the production of ATP. This is deduced from the observation that F- and V-type ATPases posses the ability to use both proton-motive force(PMF) and/or sodium-motive force(SMF) for the synthesis of ATP. However, only sodium ATPases can translocate protons and sodium but proton ATPases can only translocate protons. This is due to the fact that the binding site for sodium requires six ligands where a proton onl requires one ionisable group. This signifies that proton ATPases were evolved from sodium ATPases and therefore the common ancestor of F- and V-type ATPases had a sodium binding site. Therefore it is deduced that utilization of sodium gradients was a means for the LUCA to synthesize ATP. Along with the common sodium binding site is the structural similarity among both F- and V-ATPases in where amino acid sets are almost identical. However these ATPases' subunits of their central stalk are not close to being homologous but the catalytic hexamer are homologous to hexameric helicases. Hence, the possible ancestor would enable passive transfer of biomaterial across the cell. This with a ATP-driven helicase could potentially give rise to a protein translocase(membrane pore).
One probable theory of proteins inserting into a lipid membrane is using an amphiphilic alpha-helix which dimerizes at the surface of the membrane and the oligomerizes using other alpha-helices and subsequently making a pore. This idea stems from the fact that there is spontaneous insertion of alpha-helices into a lipid-bilayer. The weakness in this theory lies in the fact that alpha-helices would stabilize onto the surface of the membrane by spreading out on the surface and not producing a pore but instead something resembling an F-type ATPase.
Using the understanding the charge inside a cell should be negative, the porous primeval membranes would be sufficient to keep charge separation intact. This could also lead to voltage dependent membrane proteins. The cell through its historically low levels of sodium inside the cell due to a compliance to keep sodium levels similar to the start of life and through a changing environment, where the ocean becomes really sodium concentrated, the cell develops mechanism to make energy from the sodium concentrations. This in essence leads to an adoption of a coupled membrane ion translocation. This mechanism would not arise until ocean salinity would rise and as a consequence would lead to sodium-tight membranes. The development of a proton-tight membrane is more complicated and although there are theories regarding this phenomena nothing is conclusive.
Armen Y. Mulkidjanian, Michael Y. Galperin, and Eugene V. Koonin.(2009). "Co-evolution of primordial membranes and membrane proteins". PMID 19303305
There are three factors that contribute to the function of membrane proteins: structure, dynamics, and environment. These three work in correlation with each other in order to determine the unique function, as seen from the figure below.
Structure
Oligomerization is when a small peptides (i.e. monomer, dimer, trimer) are added to the protein. This affects the structure and therefore changes the function. There are four main explanations for this:
1. Functional Role
Most likely came about from evolution
Ex) Cytochrome b6f dimer forms an electron transfer bridge
Optimizes functional output and eliminates unfavorable protein interactions
Strongly bound lipids and specific site receptors for water molecules gives rise to structural and functional variability. This means that the structure is stabilized and the functionality of the membrane protein is increased.
For example:
In cytochrome b6f, the lipids act as stabilizers or functional restraints
In the ATP/ADP carrier, activity is decreased with the removal of the ligands.
Dynamics
In short, dynamics is important because it regulates how much energy is spent relative to how much work is done.
One model of dynamics in membrane proteins is the Induced Transition Fit Mechanism. In this mechanism, the metabolite binds only at the highest energy states, despite there being many different conformations.
This equation is the total free energy, also known as Gibbs Free Energy. The equation mathematically expresses the sum of all the intermediate(transition) states. The overview of dynamics of membrane protein is depending on which state the metabolite chooses will determine the amount of work that is need, which will determine the protein efficiency and the function.
Environment
Environment is an important factor in understanding the function of the membrane protein. In studying the structure of the membrane protein, it is isolated from the rest of the membrane components. However, in order to get the holistic view of the membrane protein, the environment in which it lives should be examined. The reality of how the protein interacts with the other components of the cell is important in understanding the function. The path in which the protein takes can be altered by the physical constraints set by the environment. Physical properties affect the efficiency of the membrane protein. Several examples that affect the structure and function are: lateral tension, hydrophobic matching, electrostatics, and curvature elastic stress. By changing these properties, the cell can fine-tune it's function.
Sachs, Jonathan N., and Donald M. Engelman. "Introduction to the Membrane Protein Reviews: The Interplay of Structure, Dynamics, and Environment in Membrane Protein Function." Annual Review of Biochemistry 75 (2006): 707-12. Print.
Studying membrane proteins is very difficult. Extracting, purifying, homogenizing or removing them from the membrane could mean serious loss of information and protein unwinding and unfolding. The best way to study protein membranes is to mimic their native environment in the cell, embedded or attached to the cell membrane.
Membrane proteins have proven to be very difficult to crystallize through X-ray crystallography mainly due to the large percentage of non-polar amino acids present within the primary structure. Traditionally, while crystallizing a membrane protein sample, scientists have tried to remove a large amount of the lipids surrounding the membrane proteins during the preparation of the sample. However, now scientists are beginning to recognize the lipids as important additives during the crystallizing process. Either lipid additions or the use of lipid cubic phases have led to an increasing number of solved membrane proteins. With the lipids attached, scientists are able to classify and figure out the different interactions between the proteins and lipids
Traditionally, detergents have been used to extract membrance proteins using PDCs (protein-detergent complexes) for study such as X-Ray Crystallography or NMR. The effectiveness of the specific detergent used is based upon what is the desired protein. A wide variety of detergents form spherical or ellipsoid micelles which poorly imitate the cell membrane to form complexes with the protein. Membrane proteins in this detergent-solubilized state are prone to issues such as irreversible aggregation or the loss of their native structure. Being solubilized in detergent also causes constant and rapid interchanging of the bound detergent molecules with the free molecules in the solution. This causes NMR spectroscopy to be very difficult as it often requires high temperatures over an extended period of time. NMR had been limited to especially hardy proteins. Also, because of the amount of detergent molecules that bind to the protein, the structure appears disordered and is not visible in an electron density map.
This issue with detergents led scientists to believe that membrane proteins were delicate relative to other proteins. When in the cell membrance, however, integral proteins are stabilized by multiple factors including the shielding of the hydrophobic portions of the protein and lateral pressure exerted by the lipid bilayer. These factors are not present in the detergent-solubilized state. Also, the introduction of artifacts from the detergent may appear. That causes the membrane proteins to be more susceptible to their loss of native structure which also results in the deactivation of the protein. The effectiveness of the detergents used is based on the particular protein and the detergent used. A poor detergentwill serve as a poor substitute for the lipid bilayer, causing the protein to degrade. This all interferes with the studies of the proteins. Currently, Dodecyl-B-D-maltopyranoside (DDM) is currently the most effective detergents for maintaining protein stability. This is due to the formation of large micelles by the detergent which better imitate the lipid bilayer.
PDCs are solubilized membrane proteins which exist in a complex with a detergent. This particle exists in solution with free detergent monomers and detergent micelles. There is a constant exchange of bound detergent molecules and the free molecules in the solution. The amount of detergent bound to the protein of interest can range from 30-70% of the mass of the PDC, amounting to hundreds of detergent molecules bound to the protein. These molecules also do no arrange themselves with their hydrophobic chains along the hydrophobic portion of the protein. They, instead, arrange themselves to be normal to the protein and appear disordered in X-ray crystal structures and electron density maps.
Another detergent has been developed to more effectively replicate the membrane bilayer to stabilize the membrane proteins which allows them to be studied more effectively and while they are able to function. Lipopeptide Detergents are amphiphiles with a 25-residue peptide that forms an amphipathic α-helix with two fatty acid groups ranging between 12 and 20 carbons at the 2 and 24 positions. These chains lie along the hydrophobic surface and form a wedge shake. They are able to form small, cylindrical micelles with an interior that more closely resembles the lipid bilayer. This allows the membrane proteins to last longer against aggregation and denaturation. Also, compared to traditional detergents, LPDs are relatively small and keep a rigid exterior surface. The rigidity of the complexes favor both crystallization and NMR. X-Ray Crystallography, for example provided a resolution of 10Å with traditional detergents while LPDs have been able to provide a resolution of 1.20Å due to the highly ordered nature of LPDs. [1]
LPDs, however, are not able to replace PDCs due to their higher cost. Currently, purification of the membrane proteins is still carried out with PDCs to extract the proteins and solubilize them. Once the proteins are extracted, LPDs are added to the solution and the mixture is concentrated by centrifugation in an ultrafiltration concentrator that allows the smaller LPDs to pass through while filtering out the larger PDC complexes. More buffer is added to the solution and the filtration process is repeated for several cycles, the result being the removal of PDCs and the solution of stabilized proteins in LPDs. This method requires the minimal use of LPDs which reduces the cost of membrane protein research.[1]
While LPDs are a relatively new and effective tool for studying the structure of proteins, it is simply another method of studying proteins. It is not the single solution to the various issues in protein studies. It does, however, provide a new avenue to study more integral proteins that have challenged earlier methods. Lipopeptide Detergents.[1]
Importance of Solubilizing Environment on Membrane Proteins[2]
Cell membranes constitute an array of different compositions. The heterogeneous environment effects membrane proteins in their structure and therefore, in their function. This heterogeneous environment makes it difficult for present methods to determine the structure of membrane proteins because the alteration of the membrane environment causes the structure to change. And although membrane proteins are significantly important because of their roles in biology, few of them have had their structures determined.
Protein-membrane interactions are greater than intra-protein interactions for smaller proteins and for proteins with out prosthetic groups. Prosthetic groups help overcome protein-membrane interactions by stabilizing the proteins transmembrane domains. For larger proteins, the intra-protein interaction is significant enough to overcome these interactions. Therefore, special attention must be given to the environment of smaller proteins when deciphering their structure. Since membrane proteins have significant conformational flexibility which may be necessary for their function under native conditions. An example of this would be the structural change that ion channels undergo when opening or closing. Both of these structures contribute to the proteins function and require the membrane to allow this change to occur by overcoming the protein-membrane interactions. There can also be conformational changes that do not contribute to the function of the protein and may well inhibit the protein's function. These changes can be caused by the same protein-membrane interactions. This gives rise to a special concern when unraveling the structure of new proteins.
Membrane Protein
This concern is whether the structure being analyzed is in fact the, or one of the functional structures of the protein. A case such as this is seen in the influenza A virus M2 protein. This protein's structure has been determined from samples in liquid crystalline lipid bilayers, samples crystallized from detergents and samples in detergent micelles. Each of the samples represents different protein structure and it's not clear which contribute to the function of the protein. Understanding which structure is relevant to the function will aid in developing pharmaceuticals that can efficiently disrupt the replication of the virus and prevent flu pandemics mutate and no longer are susceptible to previous drug therapy.
While using detergents to solubilize proteins, it is important to note that differences between lipids and detergents can influence protein structure. One difference is that monomeric lipid concentrations are many orders of magnitude lower than millimolar concentrations of monomeric detergents. In some protein crystal structures, these monomeric detergents have been seen to penetrate aqueous pores of protein domains which can compromise its functionality. Second, detergent micelles are expandable in their hydrophobic sections. This can cause helices, designed to have a hydrophobic mismatch, to induce helical tilt and cause them to be tightly packed. This can also affect protein function. Finally, the micelle surface has a greater degree of curvature when compared to a membrane surface. This change in curvature can also interfere with the surface binding of amphipathic helices. These changes all involve the environment of the membrane protein. These interactions can have significant effect on protein structure and further investigation is needed to uncover all relationships between the two.
↑ abcPrive, G. Lipopeptide Detergents for Membrane Protein Studies, Current Opinion in Structural Biology, Volume 19, Issue 4, Pages 379-385 August 2009
Understanding how membrane proteins function has posed difficulty because they cannot be purified or removed from the membrane without losing vital information. To study them, they must be placed in an environment that mimics conditions they are typically found in and then analyzed through solution NMR spectroscopy. Solution NMR spectroscopy requires certain compounds and assemblies to stabilize membrane proteins as they are analyzed.
Detergent in aqueous solution forms micelle with membrane protein. Red components signify detergent monomer. Gray area is the hydrophobic region of membrane protein. Orange area is hydrophilic region of membrane protein.
Typically, micelle-forming detergents are used to stabilize the proteins when they are in aqueous solution. Detergents are molecules that contain both hydrophobic and hydrophilic regions. Each detergent has a critical micellar concentration (cmc) where if detergent molecules are in concentrations below their cmc, they will exist as monomers; if they are above, they will exist as detergent micelles that are in equilibrium with detergent monomers.
However, detergents are not ideal for studying membrane proteins. Because they are highly dynamic, they can cause the protein to unfold and aggregate. Membrane proteins that contain sections which extend onto the aqueous space are particularly difficult to study with detergents because these regions unfold and become destabilized. The spherical shape of micelles also poses a problem as the membrane the proteins are found in are planar. Detergent molecules can also interfere with experimental conditions used to study membrane proteins and it takes quite a bit of time to find a compatible detergent for the protein to be studied.
Using detergents overall can be time consuming and problematic.
Detergents are essential tools used by scientists for the biochemical and structural study of membrane proteins. The structural study of membrane proteins requires detergents that can effectively mimic lipid bi-layers. Scientists are always struggling between what choice of detergent they want to use. Do scientists want detergents that promote protein stability or detergents that form small micelles?
Schematic representations of membrane protein/amphiphile associations. Green areas represent hydrophobic surfaces, and red areas are hydrophilic. (a) An integral membrane protein embedded in a phospholipid bilayer. (b) Possible mode of detergent binding in a traditional protein–detergent complex (PDC). (c) Representation of a membrane protein solubilized in an LPD. (d) End-on view of a protein/ LPD complex through a plane indicated by the dashed line in panel (c). The number of LPD molecules required to form a ring around the protein depends on the circumference of the protein transmembrane region.[1]
Dodecyl-β-D-maltopyranoside (DDM) is generally considered as one of the best detergents for maintaining protein stability, and the use of this detergent has been influential in the crystallization of landmark proteins.[1] Although DDM is great for maintaining protein stability, its large micelle size is one of the reasons why most membranes proteins crystallized from DDM diffract only low to moderate resolutions.
Lipopeptide Detergents (LPD’s) are a new class of amphiphile (a water loving and fat loving molecule) that consists of a peptide scaffold that supports two alkyl chains, one anchored to each end of an α-helix. [2] The LPD design is an attempt to create an amphiphile that can form small micelles, but create an acyl packing environment that is more comparable to the interior of a bi-layer that of a micelle. Eventually scientists were able to create a Lipopeptide detergent with this exact design. The LPDs were able to self-assemble into small micelles, and are gentle, nondenaturing detergents that preserve the structure of the membrane protein in solution for an extended period of time. [1] The target conformation for an LPD monomer is that of a wedge, with the inside edge consisting of the two hydrophobic acyl chains opposite from a wider surface consisting of the polar face of the helix (Shown in the 2nd picture)
LPDs are highly effective at stabilizing proteins, because of the formation of favorable membrane-like cylindrical acyl packing interactions at the hydrophobic surface of the target protein. (shown in part c and d picture on the left). [1]
LPDs in NMR
Normal detergents used in NMR are generally harsh and aggressive. Conformational exchange in PDCs(protein detergent complex) is a common problem. LPD are useful in NMR because the membrane-like environment achieved from PDC, while the mass stays at a minimum.
Practicality of LPDs
Although LPDs are relatively expensive compared to other detergents, LPD is effective at very low molar mass ratios of 12-20 LPD monomers per protein, very little LPD is required to fully solubilize a target membrane protein. All the added LPD is used in the final sample for crystallization trials or NMR spectroscopy. This makes LPD highly efficient in what it does.[1]
An alternative to detergents are amphipols, or amphiphatic polymers. Developed mainly by Jean-Luc Popot and colleagues, amphipols have “polymeric backbones that are covalently modified with stochastic distribution of hydrophobic and hydrophilic groups”.[1] Amphipols are used as a detergent-free membrane substitute that conserve the membrane protein function.
Another technique is using lipid bilayer systems. Lipid bilayer systems oftentimes preserve the integrity and structure of the membrane protein much better than amphipols and detergents. Three classes of membrane mimics fall under this category: liposomes, bicelles and nanolipoprotein particles (NLPs).
Protein membrane in bicelle assembly in aqueous solutoin. Red components are detergents. Blue components are lipids. Gray area is the hydrophobic area of the membrane protein. Orange area is the hydrophillic area of the membrane protein.
Bicelles are also used to study membrane proteins. Bicelles are binary, water-soluble assemblies of lipids and detergents. Ideally, the lipids will form the central part of the bicelle and the detergents will form the edge of the assembly. The assembly itself is a roughly circular patch of lipid bilayer in aqueous solution.
A way to study membrane proteins in their native environment uses nanolipoprotein particles (NLPs), or nano discs or reconstituted high density lipoprotein particles (rHDLs). NLPs are made of non-covalent assemblies of phospholipids arranged as a discoidal bilayer.
A way to study membrane proteins in their native environment uses nanolipoprotein particles (NLPs), or nano discs or reconstituted high density lipoprotein particles (rHDLs). NLPs are made of "noncovalent assemblies of phospholipids arranged as a discoidal bilayer, surrounded by amphipathic apolipoproteins".[1]
↑ abRaschle, T., Hiller, S., Etzkorn, M., & Wagner, G. (2010). Nonmicellar systems for solution NMR spectroscopy of membrane proteins. Current Opinions in Structural Biology , 471-479.
2. [1] http://www.ncbi.nlm.nih.gov/pubmed/19682888
3. [2] http://www.nature.com/nbt/journal/v21/n2/full/nbt776.html
Electron and x-ray crystallography study of membrane proteins have confirmed the ‘fluid mosaic model’ of the membrane structure, proposed by Singer and Nicholson. The ‘fluid mosaic model’ pictured protein molecules integrated in the fluid lipid bilayer as isolated units or complex. To further understand the function of the membrane protein requires the study of their conformational changes in different physiological settings. Electron crystallography of protein – lipid arrays has played a key role in understanding of how membrane proteins work. In 1975, electron crystallography provided the first view of polypeptide chain of alpha-helices of bacteriorhodopsin traversing the lipid bilayer. Since 1975, electron crystallography has been able to unveil other structures and how polypeptide folds. Electron crystallography is able to study membrane proteins in trapped transition state, and samples frozen rapidly in a defined solution. New methodological advances have also been made allowing the collection of detailed information of small or disordered membrane assemblies.
Electro crystallography is the study of proteins in crystallized form which reveals their 3D structure and amino acid sequence. There are two types of crystals made for studies on protein-lipid arrays (Figure 1). One is in the form of sheets (2D) and the other in the form of tubes (tubular shape). The lattice and symmetry of the two forms define accurately the position and orientation of each molecule. Using the advantage of these two types, the crystals have revealed precise details of individual lipid molecules and protein side chains.
Tubular crystals resemble the natural product of protein crystallizing on the surface of vesicles, but they have not been used extensively for determining exact 3D structure because it requires relatively large proteins (>250 kDa) to obtain accurate 3D shape averaging the isolated proteins.
Traditionally, the Fourier-Bessel methods are used to determine the 3D structure of a protein. The image of the crystal is processed to correct for distortion. The repeat length is divided into short segment and compared to a reference structure to determine the parameters of the 3D alignment. An alternative real-space method is now widely used treating segments as a string of single particles. This method has been applied to tubular protein-lipid crystals (Figure 2), demonstrating its potential to determine their structure at near-atomic resolution.
The principle value in using tubular and sheet crystals is that they probe the structures of membrane proteins in their natural lipid environment; therefore other methods carry the risk of not representing a biological relevant state. To capture protein-in-lipid specimens, a routine step is plunge-freezing of the electron microscope grid into liquid nitrogen-cooled ethane. The low temperature cools the protein specimens very rapidly and trapping them in their transient structure, which has a life-time of a millisecond or longer.
The freeze-trapping technique allowed a structural model of the membrane gating mechanism to be proposed but high quality images of ACh receptor channel in tubular crystals have been difficult to obtain. Recently, the development of an extremely stable helium-cooled top-entry stage has made the entire process of data collection much easier, and should eventually permit the gating mechanism to be described at near-atomic resolution.
Cryo-electron tomography is a recently developed approach to explore membrane protein in their functional state. With this technique, the 3D picture of frozen specimen is built up by combining information from a large number of tilt views. The organization of proteins can be obtained from the tomographic section. The atomic structures obtained from other methods can be docked into the tomographic densities to show proteins in their proper functional context. Recent tomographic study of frozen sections highlights the potential for exploring the architecture of more complex membrane assemblies and organelles.
From electron and x-ray crystallographic studies, the right-handed helix packing is common in membrane protein and more stable in bilayer environment. Helices in membrane also adopt bent configurations, which enable them to pack more tightly against each other. Helices may contain flexible region or hinge point, as in bacteriorhodopsin, to enable a rapid localized conformation change. my name mr. chena ram , my contact no. +918875454158
Many membrane proteins do not in fact undergo rigid quaternary rearrangement typical of other allosteric proteins, but instead work as a result of distinct movements in each domain, which are coupled to one another. Examples are ligand-gated ion channels and ABC transporters (figure 3).
Ion channels in charge of ion flows across membrane share similar principles with soluble enzymes. Ion channels, like an enzyme can incorporate precise stereochemistry changes to bind to specific substrate and catalyze reactions. The potassium channel uses precisely positioned carbonyl group lining its surface to provide a constricting, yet highly conductive pathway for potassium ions across a part of bilayer (figure 4). By contrast, the closed-gate in ACh receptor has a much wider opening but is built of completely hydrophobic residues. This creates an effective permeation barrier to potassium ions, because ions has no opportunity to drop its hydration shell and therefore too large to pass through.
The structural and chemical information about membrane proteins have been acquired from images formed by electron crystallography combined with advances in new techniques such as in cryo-technology. Compared to X-ray diffraction of proteins in detergent, these methods preserve the biology relevance of the structure. The development of tomography and real-space averaging methods are extending the possibilities of obtaining high resolution information from increasingly smaller protein-lipid arrays. Improvements in methods and ultimately the development of better electron detectors are keys to future research on membrane proteins.[1]
↑Yoshinori Fujiyoshi1 and Nigel Unwin2, 1Department of Biophysics, Faculty of Science, Kyoto University, Kitashirakawa, Sakyo-ku, Kyoto 606-8502, Japan. 2MRC Laboratory of Molecular Biology, Hills Road, Cambridge CB2 0QH, UK. Link text, additional text.
Combining cryo-electron tomography and 3D image classification together allow for better opportunities to determine the structure of membrane proteins in undisturbed cells. It relies on averaging a vast array of images of identical copies of a complex to determine its structure.
Tomography is a technique used to assemble a 3D view, called a tomogram, from several images of the structure. First, the cells are frozen very quickly and embedded in plastic. The plastic is then thinly sliced. Using a specialized high-voltage electron microscope, several images of these samples, at different angles (caused by tilting the stage of the microscope) are collected. The assembled images are called the tomogram.
Some protein membranes can be crystallized when they are in the lipid bilayer plane. Electron crystallographic techniques are able to determine structures of these proteins. It combines information from images of thin crystallized proteins whose images are taken at different angles. For proteins high in symmetry that can be purified, single particle cryo-electron microscopic techniques are used to help build 3D structures of these complexes.
Electron tomography is used to determine 3D density of microscopic objects. To minimize damage from electron irradiation, the images captured in this technique are done at cryogenic temperatures. Averaging the 3D images can be used to create density maps of macromolecular complexes. X-Ray Crystallography is also used to determine atomic structures of individual components. Currently, only low resolution images of protein membranes are able to be recorded. Higher resolution images, however, are also possible as this technique gets further developed as there are no significant barriers preventing this.
This technique can also shed light on the conformation of membrane proteins in their native environment. Combining cryo-electron tomography and 3D image classification and averaging also allows for in situ structure determination of non-symmetric viruses.
Though X-Ray Crystallography is advancing and getting better at determining the structure of integral protein membranes, it is still very challenging to determine their structure when they are still in the cell.
A computer assembles these images into the 3D view called the tomogram. A tomogram can help us understand cell function because it allows us to view small organelles like the Golgi. You can even produce a movie of a virtual journey through the cell. Howell’s research of the Golgi for example, shows that there are several pathways for proteins and other molecules to exit the Golgi. The findings are revealing, as earlier studies using different methods had suggested that there was only one road out of this organelle.
Bartesaghi, Alberto and Sriram Subramaniam. “Membrane protein structure determination using cryo-electron tomography and 3D image averaging.” NIH Public Access. Web.
“Inside the Cell.” US Department of Health and Human Services. September 2005.
Proteins embedded within membranes (integral proteins) perform many crucial functions within the cell, and data has shown that membrane proteins function in complexes. Researchers have been working to identify the factors with which these membrane proteins interact with. Only a limited amount of membrane proteins are compatible with the current methods used to study membrane protein function in complexes, and very limited knowledge on this subject is confidently reported. Scientists are still working in attempts to define the composition of membrane protein complexes.
However, even in light of these difficulties, there are a few methods in existence that have helped researchers to make some headway in terms of analyzing certain membrane protein complexes. In particular, the membrane protein complexes of mitochondria, microsomes, bacteria, and chloroplasts have been examined via blue native-polyacrylamide gel electrophoresis (BN-PAGE). Furthermore, the split-ubitquitin method was utilized to analyze yeast membrane protein interactions as well.
In addition to determining the composition of membrane protein complexes, it is also important to understand the vitality of the assembly process in these complexes. Assembly is an ordered process, which is crucial to the overall function of the complex. Larger protein complexes require more intermediates, and it is much more difficult to identify the nature of the assembly intermediates in these cases. In the simpler complexes, scientists have been able to determine the process of assembly order and the intermediates involved. These specific assembly orders play important roles in determining the specific pathway needed to carry out a certain cell function. With a different assembly order, the pathway would possibly lead to a "dead-end" in the systematic process.
A good example of one of these "simpler complexes" that have undergone study is the cytochrome bo3 complex of Escherichia coli. This complex's linear assembly pathway was determined from discovered how the complex is made up of four integral subunit proteins, and assembled by two intermediate complexes. E.coli also proves to be a showing example of the vitality of order in "ordered assembly". For the cell division process within this complex to function normally, 12 proteins are sequentially imported to division site, and they must be ordered in the correct way otherwise correct cell division would not occur, and potentially harmful intermediate complexes can arise from incorrect pathways produced from mistakes in assembly order.
How Chaperones help Membrane proteins assemble. Notice that without the chaperones, the assembly could lead to aggregation, dead-end intermediates, or toxic proteins
A very crucial factor in the function of membrane protein complexes are the chaperones that coordinate their assembly. Such chaperones can interact with the assembled protein complexes and guide them to their ultimate functional goal by preventing unwanted interactions. Unwanted membrane protein complex interactions could then lead to potentially harmful, and non-functional complexes. These unwanted proteins are shown in the figure to the left. Accordingly, without a chaperone, the protein could aggregate, stop at a dead-end intermediate, or become highly toxic. An example of chaperoning is the assembly of F1 in F1F0-ATP synthase. This is a yeast enzyme that is composed of an alpha and beta subunit. The assembly factors Atp12p and Atp11p prevents the assembly from becoming aggregated by binding to the hydrophobic surfaces of the subunits and moving them into an alpha 3 beta 3 complex. The formation of F0 also uses a chaperone called UncI. The UncI chaperones the assembly of the c subunits, which form a homo-oligomeric c-ring. The c subunits would still be capable of assembling with other parts of the F0 machinery in the absence of the UncI chaperone; however without the UncI, it cannot form the c-ring and the complex formed would be an ultimately nonfunction, 'dead-end' intermediate.
Furthermore, chaperones also mediate the process responsible for regulating complex formation. In some cases when there are various proteins and interfaces involved in the assembly process of the complex, it is carried out in a selective manner to which chaperones contribute. Certain interactions may occur at later stages of complex formation, not during intermediate stages as seen previously; chaperones play a role in this regulation. In the study of cbb3-type cytochrome c oxidase in Rhodabacter capsulatas, researchers found that complex assembly occurred through an inactive late assembly intermediate (made up of three structural subunits and two assembly factors). At the end of the process, the complex was only activated when the final structural subunit took the place of the assembly factors previously in place. It is thought that these inactive intermediates may be vital during complex assembly in which incomplete complex assembly could produce harmful consequences, such as in the respiration or photosynthesis complex assembly.
Recent research has also been put into studying dynamic exchange in assembled proteins. This dynamic exchange allows such membrane proteins to easily transfer in and out of the complex they are a part of. Dynamic exchange provides a means of repair: if part of the complex is damaged, instead of rendering the entire complex useless or nonfunctional, that specific protein can be substituted for a functional one. According to this research, membrane proteins are not stable once they are formed. Early research suggested that the proteins in photosystem II in chloroplasts are repaired by exchanges in a new D1 subunit for a photo-damaged D1. Another example of dynamic exchange involves the Translocase of the mitochondrial outer membrane complex. Such experimentation has shown that proteins are carried solely into the mitochondria, and thus they do not pick up any other required subunits needed for complete complex synthesis. Although this brings up a new and exciting fact to protein assembly, it has only been directly proven in vitro or in test tubes and not in vivo. However, three recent research have used fluorescent microscopy on cells containing GFP tagged proteins showing dynamic exchange in vivo.
It has been well established that membrane proteins must be inserted in a specific topology, an orientation in the lipid bilayer, in order to properly function. However, recent evidence from experiments done by Shimon Schuldiner has challenged this conventional thought through studying EmrE, a small homodimeric multidrug transporter from Escherichia coli that transports positively charged aromatic drugs out and takes in two protons.[2]
It should be noted that dimeric membrane proteins can exist in a parallel and/or antiparallel orientation. In the specific case of EmrE, the parallel orientation can be visualized as having the N-terminus and C-terminus on the same side; the antiparallel orientation can be visualized as having the N-terminus on one side and the C-terminus on the other.[2]
While it is well known that proteins such as receptors must be oriented in one way and one way only, this may not necessarily be the case for EmrE. Past experiments on EmrE have indicated that even though EmrE can be found in a parallel topology, only the antiparallel topology can induce normal function. However, each of these experiments may have certain conditions that deserve a counterargument.
Antiparallel Topology: Experiments and Counterarguments
In one experiment, scientists created a 3D artificial model of an antiparallel topology of EmrE. They then used an actual antiparallel 3D model of a native EmrE and compared the two, concluding that both were similar and thus proved that EmrE fits the antiparallel topology.[2] While the evidence is convincing, the fact that the resolution of the structures was very low and so similarities were much easier to “identify.” Furthermore, the crystals used for the antiparallel model came from proteins in detergents that inhibit the protein function. This implies that the way the 3D model was created was by the least energy required for a crystal formation, not the way EmrE naturally exists in.
In another experiment, EmrE was fused to a green fluorescent protein that did not affect the function of EmrE. However, upon manipulation of the positive charges in the protein, parallel mutant EmrE proteins were created. These mutations were shown to be resistant to the green fluorescent protein, leading to the conclusion that this was because of the changed topology. To further support this, the mutant EmrE proteins were coexpressed with the native EmrE and normal function was restored within one generation.[2] A problem with this experiment was that the researchers assumed that the parallel mutants were inactive. In fact, further experiments done by Schuldiner[2] showed that parallel mutant EmrE proteins had continuous growth. In addition, the inactivity of the mutant parallel proteins can be explained by impaired dimerization—if the protein cannot be formed properly due to the mutation, its function will not be normal. This idea is independent of topology.
Two experiments were done to show the ability of EmrE to function properly even with a parallel topology. In one experiment, unique cysteines were created in the termini of the protein. Crosslinkers, or molecules that can form covalent bonds with other similar molecules, were formed within 9 to 11 angstroms apart.[2] Previous studies had established that in antiparallel topology, cysteine residues had to be at least 35 to 40 angstroms apart. To solidify this evidence, repeated experiments on proteins crosslinked in other loops with similar results showed that they were fully functional EmrE proteins.
In another experiment,[2] the C-terminus of one protomer was artificially fused to the N-terminus of another by a very short, hydrophilic linker, ensuring that the terminus of both protomers were on the same side of the membrane (therefore parallel). These artificially created proteins were also shown to be fully functional.
Another study is that parallel homodimers like EmrE and antiparallel heterodimers like EbrAB from Bacillus subtilis both perform identical functions regardless of their topology. In fact, EbrB was observed to be able to form parallel homodimers and function in the same way. This has very strong implications for EmrE. Because EbrAB and EmrE are closely related proteins, EmrE may also have a similar “promiscuity” in that it can be functional in both the parallel and antiparallel topology.
It has been shown that EmrE exists in a parallel topology and can function normally. Furthermore, it can exist in an antiparallel topology and function just as well. A conceivable mechanism[2] into how this can work is that the active sites are conserved in both topologies. This active site is occupied by either an H+ or a substrate, depending on the two glutamate residues that vary the pKa. This in turn, is strong evidence that the topology is determined by the direction of the driving force, mainly the H+ or the substrate.
Although one should note that these evidence came from one protein, the significance of these findings is clear. It should be noted that EmrE is a small membrane protein capable of evolving into much more complex membrane proteins. If a protein can exist in different forms directed by their function, the conventional idea of structure determining function may be challenged. Instead, function may determine the structure.
G protein coupled receptors have seven transmembrane spans(TM), an amino-terminus facing outwards and a carboxy terminus facing the interior of the cell. Evidence and studies have shown that TM5 and TM6 play a more important part in the course of activation. The G protein is inside the cell, and it binds to either GTP or GDP; GTP is the same as ATP except that it has a G (guanine) base nucleotide instead of an A (adenine) nucleotide. When the G protein is bound to GTP, it activates, but when the G protein is bound to GDP, it deactivates; in other words, when there is a signaling molecule the G protein will bind to GTP, but if there is no signaling molecule, the G protein will bind to GDP.
The GPCR is a huge family of proteins that is involved in sight (rod cells have rhodopsin as a GPCR, which bind to a photon of light--a signaling molecule), in smell (olfactory receptors bind to different smells, and there are specific receptors for specific smells), and in taste (sweet, bitter, and umami are GPCR while salty and sour are ion coupled).
Besides the seven transmembrane helices, GPCRs vary a lot in structure. Unfortunately, it is very difficult to actually form spatial models of different GPCRs because the GPCRs have the ability to assume several conformations. Thus, during the course of crystallization, one has to minimize the conformation changes throughout so as to attain reasonable levels of visualization of the crystals. Only a few structures of several GPCRs have been proposed.
The structural differences of GPCRs are responsible for the differences in their specificity towards different G-proteins.
Epinephrin signaling stimulates glycogen and breaks it down. First, epinephrin binds to GPCR, which causes the GPCR to recruit a G protein. This G protein drops its GDP and associates with GTP instead. The G protein then activates production of cAMP (a molecule). As a result, cAMP activates a protein called PKA (protein kinase A). PKA phosphorylates enzymes and activates molecules, which results in the breakdown of glycogen to glucose. PKA can also phosphorylate enzymes to inactivate and convert glucose to glycogen.
In this process, the first messenger is epinephrine while the second messenger is cAMP (the second messenger is a molecule that is produced). Amplification, the hallmark of pathways, is when one epinephrine produces multiple cAMPs to activate PKA.
The pathway can be turned off by doing one of the following: removing the epinephrine, removing cAMP, getting rid of GPCR (this is feasible through endocytosis), hydrolyzing GTP to GDP to deactivate the G protein, or by dephosphorylizing PKA targets to deactivate phosphates by removing them.
Cholera toxin inhibits the switch from GTP to GDP, which does not allow the pathway to be turned off. This increases cAMP production, which then increases ion secretion. This is dangerous because water molecules follow the ions that are secreted out, which can lead to dehydration and eventual death.
Rhodopsin belongs to the class-A receptors of GPCRs and it was through rhodopsin that scientists were able to find the common structure of the seven transmembrane helixes in GPCRs. Rhodopsin has a covalently bound ligand--retinal. Retinal has several different forms; this is the property that can help in structural studies of GPCR, setting it apart from most GPCRs. In the absence of light, retinal exists in 11-cis form. This form is inactive in G protein transducin and rhodopsin containing retinal in this form is called dark rhodopsin. In the presence of light, retinal, through isomerization, changes into its all-trans form. When this happens, through a series of conformational changes, it is activated into metarhodopsin II.
From the study of its structure, ionic locks were discovered in its structure. Ionic lock is basically a collection of hydrogen bonds and ionic interactions that were formed between the residues of rhodopsin. This is responsible for the complete inactivity of dark rhodopsin and its G protein, transducin.
Its three-dimensional structure was proposed in year 2000 but it is still continually being refined.
The structure of β-AR were studied using two methods
GPCR crystals are usually formed by the interactions that take place with its water soluble, exterior parts. Thus, the main strategy is to provide a larger water-soluble surface area for crystals to form on. Therefore, β-AR was made to complex with fragment antigen-binding(Fab). A Fab fragment was prepared from an antigen that could identify TM5 and TM6 β-AR; Fab bound to the receptor, but does not affect β-AR's ability to bind with its ligand. The GPCR-Fab complex was then used for crystallization. The result was partially successful; the interior side formed reasonably well but the exterior side did not.
The other way was to substitute the third intracellular loop(ICL3) between TM5 and TM6 with T4 lysozyme(T4L). T4L was chosen because of the past successes it had with crystallization and its length; it had similar length to ICL3. This will not only increase the water-soluble surface area, but it will also stabilize β-AR. β-AR with T4L has the same affinity for both ligands and inverse ligands. The unaltered β-AR binds 2-3 times more to its ligand than to its inverse ligand. The altered β-AR produced better crystals; Most of it could be visualized, except for the last 71 residues facing the interior of the cell.
In both methods, the inverse ligand, carazolol was used to stabilize its conformation.
Weis, William I; Kobilka, Brian K (2008), "Structural Insights into G-protein-coupled receptor activation", Current Opinion in Structural Biology, doi:10.1016/j.sbi.2008.09.010, PMID18957321{{citation}}: Unknown parameter |acess date= ignored (|access-date= suggested) (help)
Even though there are numerous drugs to kill bacteria, there always remains a small percentage that cannot be killed with the available medicines. Scientists have found intercellular proteins in the cell membranes, called multidrug-resistance (MDR) pumps, which are believed to be self-defense mechanisms. MDRs arise from three different gene families and are widespread in bacteria.
In bacteria,multidrug-resistance pumps confer resistance to chemically unrelated amphipathic toxins. A major challenge in developing efficacious antibiotics is identifying antimicrobial compounds that are not rapidly pumped out of bacterial cells. The plant antimicrobial berberine, the active component of the medicinal plants echinacea and golden seal, is a cation that is readily extruded by bacterial MDRs, thereby rendering it relatively ineffective as a therapeutic agent.
MDR pump
Multidrug efflux pumps have other physiological functions: AcrB of E.Coli, main physiological function is to protect bacteria against bile salts since E.Coli can be found in the intestines. In addition, bile salts have affinity to AcrB transporters
These pumps are found in almost all living organisms, and have a multitude of roles, including moving the body’s natural molecules in and out of cells. In humans, they can be found within the membranes of the brain, liver, kidneys, and digestive tract.[1]
These MDR pumps not only work in bacteria, but unfortunately, also in cancerous cells. The cancerous cells can fend off the chemotherapy drugs by pumping them out of the cell, allowing evasion of cell death.[1]
Kim Lewis of Northeastern University in Boston conducted an experiment to test the hypothesis that the MDR pump is indeed responsible for bacterial evasion of antibiotics. In his experiment, he genetically changed the bacterium Staphylococcus aureus to not have the MDR pump. He then treated this altered bacteria with berberine antibiotic, which is normally very futile against the unaltered bacteria. He saw that the antibiotic actually worked against these bacteria. Lewis also saw that if this usually weak antibiotic was given concurrently with MDR pump inhibitors, unaltered Staphylococcus aureus bacteria would be killed. These results implicate that the missing or deactivated MDR pump prove very critical in the bacteria’s protection and survival.[1]
AcrB
AcrB and its homologues are the major multidrug efflux transporter systems, and it captures some of its substrates from the periplasm in E. coli and other Gram-negative organisms. AcrB forms a complex with AcrA and an outer membrane protein channel, TolC, which harnesses proton-motive force, to export a wide variety of compounds across the periplasmic space to the exterior of the cell.
Although AcrB is a homotrimer, it can undergo structural changes in which each subunit exhibits different conformations that interconvert to move toxic compounds from the initial binding site out of the transporter.[2]
AcrB works with an outer membrane channel TolC and a membrane fusion protein AcrA. This complex removes many types of chemicals that may be toxic to the cell in a reaction that is powered by proton-motive force.
AcrB transports drugs by a three-step functionally rotating mechanism in which drugs undergo an ordered change in binding.[3] The first conformation is the access state. This is where the vestibule is open to the periplasm to allow the substrate to enter into the complex. The second conformation is the binding state. In this state, the binding pocket is expanded and the substrate binds to different locations in the pocket. The third conformation is the extrusion state. The vestibule is close and the exit is opened. The bound drug is pushed out into the top of the funnel allowing the AcrB protomer to shrink and return to the first stage of the mechanism, the access state.
These conformational changes in the trimer are powered by the proton motive force across the membrane through the involvement of three charged residues, Aspartate-407, 408, and Lysine-940, making charge pair in the membrane-embedded region.[3] In the access and binding state, the side chain of Lysine-940 forms a salt bridge with the carboxy-groups of Aspartate-407 and Aspartate-408. This salt bridge is then dissociated in the extrustion state and the Lysine side chain is turned and tilted to form a new polar bond with Threonine-978 where a proton may be attached. To return to the access state, the bound proton is dissociated. These residues,Aspartate-407, 408, and Lysine-940, are essential for exporting drugs and without them the cell would be at a complete loss of drug resistance.
Because this complex has a trimer of AcrB, it expels three drugs consecutively. As the first protomer of AcrB is in the access state, the second protomer is in the binding state, and the third protomer is in the extrustion state. The change in conformation of one protomer affects the conformation of another. This conformation cycle where one protomer affects another can be explained by the principle of cooperativity.
Multidrug resistance came about after the development of drugs because bacteria soon became anti resistant. There are two forms of multidruge resistance occurring. The first way is by genes coding for resistance on R plasmids or transposons. The second way is by efflux pumps which are able to pump out one or more drugs at a time. Bacteria can also become resistant due to mutations that distort the protein so that it is less susceptible to the drug. Sometimes resistance can be transferred by cells passing it to other cells on their plasmids. Other times, the resistance is due to target modification such as a substrate binding onto the protein and altering its shape. For example, Tet(M) or Tet(S) protein bind to ribosomes and change the conformation to prevent the drug from binding to them. Resistance genes also came from microorganisms in the soil because of the evolutionary origin of degradation genes because they use antibiotics as nutrients. An example of when genes are expressed from plasmids are integrons, which contains a gene coding, and is catalytic for the insertion of resistance genes at certain sites on the R plasmids. ISCR delivers resistant genes to integrons to make more resistance genes. Sometimes R plasmids are stable and have a killer element which if the plasmid is loss, then the cell dies. Lastly, multidrug resistance occurs in antibiotic treated patients because when they are sick, they may get into a resistant state without genetic change. Genetic change and mutations is what makes bacteria become resistant to drugs.
Von Heijne and Rees. Current Opinion in Structural Biology ,18:405. August 2008.
"Multidrug efflux transporter, AcrB-the pumping mecahnism." Murakami, Satoshi. Current Opinion in Structural Biology, 18:459-465. August 2008.
A membrane protein is any protein found in a biological membrane. They participate in various biological processes, such as cell signaling-transduction pathways. The membrane proteins also play a strong role in controlling a wide array of gradients such as chemical, electrical, and mechanical gradients and are responsible for cell structure during key cell events such as division. Due to their many functions in the membrane, they are in high concentration on the surface of the membrane. They may also act as channels that move specific molecules into and out of the membrane. Theses proteins fall into two main categories, depending upon how strongly the protein interacts with the membrane.
The two main categories are listed below:
Integral proteins: (also called intrinsic proteins) These are proteins are characterized by strong interaction with the membrane, which can only be broken by the addition of detergents or some other nonpolar solvent. Essentially, they are permanently bounded to the membrane. They may span across the entire phospholipid bi-layer, or be monotopic.Integral proteins. They have one or more segments that are permanently embedded within the phospholipid bilayer and have their domains on both sides of the membrane. Most integral proteins contain residues with hydrophobic side chains that interact with fatty acyl groups of the membrane phospholipids, thus anchoring the protein to the membrane. Most integral proteins span the entire phospholipid bilayer.It interacts extensively with the hydrocarbon chain of membrane lipid and they can be released by agents that compete for these nonpolar interaction.
Peripheral proteins: (or extrinsic proteins) are proteins that have a much weaker interaction with the membrane than integral proteins. These attachments tend to be much more temporary and can be displaced via treatment with a polar reagent. Peripheral proteins. They are temporarily bound either to the lipid bilayer or to integral proteins by hydrophobic, electrostatic, and other non-covalent interactions. This type of proteins does not interact with the hydrophobic core of the phospholipid bilayer. They are usually bound to membrane by interactions with integral membrane proteins or directly by interactions with lipid polar head groups. This polar interaction can be disrupted by the change in pH.
There is also an alternative method of classification for membrane proteins. It arises from membrane proteins, such as colicin A and alpha-hemolysin. These do not fit to either integral or peripheral classification. In this alternative system of classification, the membrane proteins are divided into integral and amphitropic.
Membrane proteins
Biological membranes have phospholipid bilayer structure which contains a set of proteins which help plasma membrane to carry its distinctive functions. Membrane proteins can be attached to the membrane or associated with the membrane of a cell or an organelle. Membrane proteins can be classified into two groups based on the strength of their association with the membrane:
Some membrane proteins are found bounded to lipid bilayer and generally involved in cell-cell signaling or interactions. Others are embedded within the lipid bilayer of a cell often form channels and pores. Membrane proteins can be attached to both the outside and inside of the cell membrane.
Proteins can be attached to the cell membrane in a variety of ways. One method involves irreversible covalent modification. Both Ras (a GTPase) and Src (protein tyrosine kinase) are known to be modified in this manner. Both of these proteins participate in signal transduction pathways, but upon covalent attachment of a lipid group they become attached to the inner face of the cytoplasmic membrane. When Ras and Src are affixed to the cell membrane they are better able to receive and transmit information being transferred via their respective signal transduction pathways.Membrane proteins can be made of alpha helices or beta strands, or the combination of both alpha helices or beta strands. For example the channel protein called Porin is made up of entirely beta strands, while the enzyme protein called prostaglandin is made entirely of the alpha helices.
Membrane proteins can be alpha - helices or beta - strands. Proteins can span the membrane with alpha helices. Membrane - spanning alpha - helices are the most common structural motif in membrane proteins. An examination of the primary structure reveals that most amino acids in the membrane protein are nonpolar and very few are charged. One of the first alpha - proteins found was the bacteriorhodopsin. It uses light energy to transport protons from inside the cell to outside generating a proton gradient used to form ATP. The seven alpha - helices are closely packed and arranged perpendicular to the plane of the cell and they span 45A in width. Membrane proteins can also be made out of beta strands. Beta Strands form channel proteins. They are less common than alpha - helices. Channel proteins are formed by beta arrangement of beta strands. Each strand is hydrogen bonded to its neighbor in an anti-parallel arrangement, forming a single beta sheet. The beta sheet then curls up to form a hollow cylinder that forms a channel in the membrane. An example is Porin. The outside surface is non-polar and interacts with the hydrocarbon core of the membrane, while the inside channel is hydrophilic and filled with water. The arrangement of polar and non-polar is accomplished by the alternation of hydrophobic and hydrophilic amino acids along with each beta strand.
Many membrane proteins have quaternary structures consisting of multiple subunits. This oligomerization in membrane proteins is beneficial to their functions, stability, genetic efficiency and maybe even optimizing productive output per unit area of the membrane. Cytochrome b6f serves as an example of quarterary structure affecting membrane protein function. This protein consist of two subunits which are connected by a bridge so that electrons can be transferred between them. As for stability, a quaternary protein consisting of 2, 3 or 4 subunits would be 2, 3 or 4 times more stable if a stability improving mutation were to occur on each subunit. It would be more genetically efficient to have all the subunits of a quaternary protein be coded for by 1 gene than to have each of its subunits be coded for by a different gene. In this way, a quaternary protein can be coded for with minimal genetic space. One example of this are the ion channels that span the membrane. the entirety of these quaternary membrane proteins are made from repeating, identical subunits stacked on top of each other. Everyone of these subunits and therefore the iono channel as a whole, is then coded by and translated from 1 single gene. In addition, oligomerization may also contribute to maximizing functional output as it allows membrane proteins to be closely packed in an area of the lipid bilayer without coming into contact with other proteins in energetically unfavorable ways.[1]
Mutations in both Ras and Src have been observed in a number of cancer cells; it is thought that these mutations and the subsequent interruption of the signal transduction pathways predispose a cell to uncontrolled replication. When the presence of a mutation is detected a small protein named ubiquitin is attached to the damaged protein; this modification signals that the marked protein is to be destroyed. It is essential that the protein be destroyed before anaphase so that the damaged DNA is not passed on to other cells. The attachment of ubiquitin to a damaged protein is the first step of apoptosis, which is programmed cell death.
[edit] Integral Proteins
As mentioned earlier, integral proteins, also known as intrinsic proteins, are strongly and permanently bounded to the membrane. One or more parts of these proteins are embedded in the phospholipid bi-layer of the membrane. They exhibit strong interaction with the membrane because their amino acid residues contains hydrophobic side chains that interact with the hydrophobic interior (fatty acyl groups) of the phospholipid bilayer. Because of their strong hydrophobic interaction with the hydrophobic core of the membrane, such proteins can only be dissociated from the membranes using detergents, non-polar solvents, or sometimes denaturing agents. Lastly, it is important to note that integral proteins account for a significant fraction of the proteins encoded in the genome.
There are two basic categories for integral proteins.
These proteins span across the entire membrane. They are the most common among integral proteins.
Transmembrane proteins
They may cross the membrane only once or several times, weaving in and out. The two kinds of transmembrane proteins are alpha-helical and beta-barrels.
The former is the more common of the two and can be found in the inner membrane of bacterial cells or the plasma membrane of eukaryotes. Voltage-gated ion channels, such as potassium and chloride channels, are examples of alpha-helical transmembrane proteins. They are mostly composed of hydrophobic amino acid residues and little hydrophilic residues, such as charged and polar residues. The polar carbonyl oxygen in the backbone doesn’t project outwards the helix, but rather towards the inside, facilitating and strengthening hydrogen bonds within the helix. Van der Waals interactions hold the tertiary and quaternary structures together in the transmembrane region. These interactions allow for flexibility in the structure to accommodate for necessary functions. Two polar residues that are found most frequently in the TM backbone are serine and threonine which can potentially hydrogen bond to the helical backbone. This hydrogen bonding captures polar side chains in a hydrophobic environment, such as a lipid bilayer. The polar side chains in turn hydrogen bond to other helices. Two residues, glycine and proline, known as helix breakers in water make kinks in the helix which play significant roles in functional mechanisms. [2]
Beta-barrels present in the outer membranes of Gram-negative bacteria, cell wall of Gram-positive bacteria, outer membrane of mitochondria and chloroplasts. Porins are examples of a beta-barrel transmembrane protein. They cross cellular membrane and acts as a pore through which molecules can diffuse. Transmembrane proteins can further be categorized into Type I and Type II. In Type I, the N-terminal is positioned on the exterior of the membrane. In Type II, the C-terminal appears on the exterior of the membrane.
Human VDAC
VDAC (voltage-dependent anion channel) is an example of a transmembrane protein found in the mitochondrial outer membrane which provides the pore for substrate diffusion. VDAC is composed of 19 β-strands which make up the β-barrel and a partial α-helix strand totaling 20 strands in the unit. The first and last β-strands of the β-barrel are parallel, while the strands in-between are anti-parallel.[3]
Isoform 1 of VDAC, three high-resolution structures in fact, in detergent micelles and bicelles have been recently published from solution NMR and X-ray crystallography. This helps to solve the membrane topology of VDAC and gives the first eukaryotic β-barrel membrane protein structure. Something different about this integral membrane protein was that it had parallel β-strand pairing and an odd number of strands. The voltage gating mechanism of VDAC and its modulation by NADH are given a structural and functional basis from studies. Since VDAC-1’s de novo structure and six more proteins, the amount of integral membrane protein structures found by solution NMR has doubled in the past two years.[4]
These proteins are permanently bounded to the membrane but only from one side. Many of these proteins are enzymes. Examples include cyclooxygenase and carnitine O-palmitoyltransferase. The former is an enzyme that is involved in the formation of prostanoids. Anti-inflammatory drugs, such as aspirin and ibuprofen, work to relieve symptoms of inflammation and pain by inhibiting this enzyme. The latter is a mitochondria transferase enzyme that participates in the metabolism of palmitoylcarnitine into palmitoyl-CoA.
Peripheral proteins, also known as extrinsic proteins, lack interaction with the hydrophobic interior of the phospholipid bi-layer. Because they lack hydrophobic interaction with the membrane, they can be detached from the membrane much more easily than integral proteins. Dissociation of peripheral proteins can be achieved through treatment with a solution of high pH or high salt concentration. Instead, peripheral proteins attach to the membrane via electrostatic and other non-covalent forces. Typically, they are either attached to the membrane indirectly via interaction with integral proteins, or directly through interaction with the polar heads of the phospholipid (amphitropic). Some peripheral proteins exhibit both types of interaction. These include certain kinases and G proteins. Other examples of peripheral proteins are the regulatory protein subunits of ion channels and transmembrane receptors.
Membrane Protein Functions
Due to the nature of the lipid bilayer, many molecules cannot enter or exit the cell because of size or charge. Membrane proteins function to assist in the transportation of such molecules across the lipid bilayer. Trans-membrane proteins participate in either passive or active transport.
Insertion of ER into lipid bilayer causes newly synthesized integral membrane proteins to be sorted, transferred, and qualitatively maintained. This process is controlled by ubiquitination, a posttranlational redirection of commands which relate to biosynthetic delivery of proteins to the plasma membrane. This process can be followed through the secretory pathways. Ubiquitination can also be used to regulate the deletion of proteins from the plasma membrane through a endocytic pathway. Ubiquitination of integral membrane proteins often is enough to edocytically target the Plasma membrane protein. However, there are still certain functions such as sortin and degradation which fully requires ubiquitin.
This control and change of specific membrane proteins is due to the ubiquitin changing the quality or quantity of the integral membrane protein. As a side effect, defects in this process can also contribute to detrimental diseases such as cystic fibrosis.
Ubiquitin modification can influence cargo trafficking, mechanisms of quality control/maintenance in secretory/endocytic pathway.
Ubiquitin in Membrane Transport and Quality Control in Endoplasmic Reticulum
Ubiquitin transformations do not affect the regulating effects preformed by the ER. However, Ubiquitin activity is initialized during the Endoplasmic Reticulum- associated degradation process, or ERAD, because ubiquitin ligase is needed. This procedure is important because it is responsible for the removal of proteins which are not folded properly. The substrates which are subject to this procedure are relocated to the cytoplasm, waiting to be removed.
The ERAD targets are first ubiquitinated and must negotiate protein Ubx, a ubiquitin-binding protein. This process shows that that ubiquitation of the ERAD substrates provides a signal which is necessary for targeting the protein for degradation. This shows that ubiquitin plays a vital in protein membrane protein transport. It is important to note that ERAD functionalities also provide a key quality assurance aspect. A kink in this procedure could can cause detrimental side effects; this means that the ERAD procedure is monitored carefully and therefore the membrane protein transfers can be assured for quality.
Ubiquitin in protein quality control to regulate Protein Membrane Protein Composition
It is found that ERAD can affect and strengthen communication between ER and Golgi complexes. This can be accomplished by degrading retention factors of the ER. In Cholesterol depleted conditions, cells were ubiquitinated and degraded. This showed that the protein resulted left the ER and was sent into the Golgi for packing.
Conversely, the GAT protein within the Golgi complex contains three surfaces which can bind ubiquitin very well. This causes successful binding of ubiquitin and speed up the transferring of GAT proteins from Golgi to the ER. In addition, the polymerization protein cargo and ubiquitin provides the necessary driving force for localization to the Protein membrane.
Ubiquitin in the turnover of Plasma Membrane proteins
In endocytosis in yeast, ubiquitin is required for almost all processes. It is beneficial that there is a more than sufficient supply of ubiquitin within the yeast. The internalization of protein cargoes that are present in yeast are generally all ubiquitin mediated.
Plasma Membrane protein contains a protective mechanism which are driven by intrinsic factors of the protein. Plasma Membranes also places a limit on the amount of proteins which exhibit error folding. A certain amount over a lifetime span is placed and plasma membrane proteins are there to regulate these levels. This quality check shows that these specific proteins must control integral membrane proteins and the removal of the damaged and misconstructed proteins. Despite the constrained understanding of the chemical process, quality maintenance mechanisms must usually include capabilities such as: the function to refold or fix the damaged protein, and the ability to distinguish healthy and damaged proteins.
As with proteins in the cytoplasm or in aqueous environments, proteolytic processing is key to cellular function in both the cytoplasm and in the lipid bilayer. However, intramembrane proteases present a different challenge to work with than water-soluble proteins. Scientists have been working on methods to decipher the molecular mechanisms of families of intramembrane proteins. Specifically, site-2 intramembrane metalloprotease and serine intramembrane protease rhomboid share common characteristics. The active sites of both families of proteases are entrenched in the membrane. However, to effectively cleave a membrane protein or any other protein, water must be introduced to hydrolyze the peptide bonds. These proteases often recognize a specific sequence of residues and thus cleave proteins at specific sites. To introduce water to the site of cleavage, there is a delivery system to connect the aqueous environment to the site of cleavage.
Molecules are allowed to flow down their concentration gradient. In most cases, this does not require a special protein. However, in facilitated diffusion, molecules that are insoluble in the lipid bilayer or too large to pass through is assisted in crossing the cell membrane through special transport proteins. Examples of facilitated diffusion are amino acids and ions.
Passive transport.
The other types of passive transport, which do not require proteins because the molecules diffuse directly through the cell membrane, are osmosis, diffusion, and filtration.
Facilitated diffusion.
Uniporters are the proteins that move molecules in passive transport. They can either be channel proteins or carrier proteins. Channel proteins open in response to a stimulus and let molecules flow freely through. Carrier proteins bind to a molecule, making it hydrophobic enough to cross the membrane. The following image shows the two kinds of uniporters and how they function.
Energy is expended to transport a molecule up its concentration gradient. There are two types of active transport, primary and secondary. Both involve going against a concentration gradient using ATP, but they differ in how the ATP is used by the protein.
ATP is expended to move a molecule up its concentration gradient. An example of this is the sodium-potassium pump, which pumps both ions against their concentration gradients in order to create a membrane voltage potential.
ATP is not directly coupled to the molecule of interest in secondary active transport. Instead, another molecule is moved up its concentration gradient, which generates an electrochemical gradient. The molecule of interest is then transported down the electrochemical gradient. While this process still consumes ATP to generate that gradient, the energy is not directly used to move the molecule across the membrane, hence it is known as secondary active transport.
Two main types of protein are involved in secondary active transport: antiporters and symporters.
Antiporter
The molecules move in opposite directions. One type of molecule enters the cell while the other exits. An example is the sodium-calcium exchanger, which removes calcium ions from the cell while allowing sodium back in. The sodium is pumped out by the sodium-potassium pump, which generates the concentration gradient required for this to work.
Symporter
The molecules move in the same direction. This usually works by allowing an ion to move down its electrochemical gradient. The other molecule piggy-backs off that movement and goes against its concentration gradient.
Fluidity of Membrane proteins Biological membrane are flexible. This flexibility is attained by the fluidity of the protein. The fluid mosaic model allows lateral movements called the lateral diffusion, and sometimes the transverse diffusion or flip flop can occur, which takes longer time to take place.
Lateral diffusion is the movement of the lipid laterally which is very rapid, unless there is restriction by special interaction.
Lateral Diffusion.
Flip-flop or Transverse diffusion is the condition is when transition of a molecule from one membrane surface to the other occurred. It is a very slow space compared with the lateral diffusion.It happens once in several hours.
Despite the many advances made in the study of membrane proteins, not much is known about the role of the environment in determining membrane protein structure or function because these proteins are easily affected by changes in their environment. The main problem remains in the difficulty of creating an environment that promotes a protein's native functions and structures. However, advances in the study of the influenza virus, more specifically the M2 protein, is giving more insight to this complex challenge.
The M2 protein is a homotetramer with 3 functional domains: the N-terminal, the TM helix, and the C-terminal. Until recent discoveries, drugs were effective in the blocking of the TM helix, which prevented proton conductance functions and thus disabling the virus. However, with recent outbreaks of the H1N1 virus and swine flu, the structure of this protein was scrutinized in 3 different environments, each using a different methodology. The influence of the environment on the proteins can then be seen in the comparison of these 3 results obtained.
The 1st imaging technique, solid-state NMR, concluded that the M2 was stable in a lipid bilayer environment. The drug amantadine was later added giving the protein a 4-fold symmetry structure further indicating more stability in the presence of amantadine.
The 2nd image, crystal structures, not only compared structures at differing pH levels but also showed that membrane proteins can access a range of conformational states.
Finally, the 3rd image made by solution NMR concluded that the membrane protein's amino acids interact to minimize electrostatic potentials and that water, when present, allows for hydrogen bond exchange.
Further screening of this protein is still undergoing, and much has yet to be revealed in the study of the environment's influence on protein structure. However, it is easily seen why this topic remains an important and popular issue. By understanding the environment's influence on membrane proteins, researchers are able to develop drugs to inhibit, for example, the influenza virus. Even mutations such as the H1N1 virus can be disabled as long as researchers have a key understanding of their membrane protein and how they can be manipulated and changed by their environments. The study of the M2 protein will eventually lead to a deeper understanding of other membrane proteins and how they are changed by changes in their environment.
Membrane places a most important role in the human body. It affects strongly in each structure environment. Every times, we talk about membranes, we have to mention protein structures because they related with each other. Proteins also known as amino acids that function in our body. The membrane and amino acid are the main function in the human body to help our body alive. They are supporting each other to form the right structure and sequences in each other to form the right structure and sequences in each part inside the body. Amino acid sequence allows the interpretation of some of the many studies on the chemical and mechanism of the membrane transport protein.
There are different kind of membrane in our body and each of them has different structure and function which also relate to amino acid. For example, integral membrane proteins are present in a heterogeneous environment that poses major obstacle for existing structure methodologies. Each structure could function as different environment and how the bonds are related. It is very difficult to obtain membrane mimetic environments that support the native structures, dynamic and functions of a membrane protein. Membrane protein often necessary to use detergents to mimic the nature lipid bilayer environment. In order to successful understand in which environment they are functioning, we have to know the bonding structure. Bonding is very important in each structure because it connects elements and one or more structures to each other. Nonetheless, it also very important to understand how to break the bond and forming a new bond. That is a reason why it very helpful to know the bond angles and stability of the bonding. Furthermore, by understanding the bonding structure help the scientists study about the differences kind of diseases and medicine to cured all the diseases. Lipid bilayers is a thin membranes. Lipid bilayers have a unique role in characterizing the native structures of membrane proteins and validating structures determined in other membrane mimetic environments.
Indeed, many proteins are membrane proteins which have the function in the cell. The cell need to communicate with the exterior or passing through the cell membrane. Many proteins go to membrane are glycoprotein related. Proteins are very difficult to study because the structures and functions are very complex. However, some proteins function can be predicted.
The study of membrane proteins have been complicated by the difficulty of examining the proteins by X-ray crystallography. Thus far, scientists have been able to examine the detail of their interactions between membrane components and their relative functions by computational simulations of the proteins in the membranes.
The questions of the stability of a membrane protein have eluded scientists. One particularly difficult task relates to studying the reversible transitions between different states. These interactions have been studied thermodynamically and yielded information pertaining to helix-helix interactions and the types of approaches to membrane protein stability. The stability between proteins and lipids have been simulated by methods such as simulating the free energy cost of burying specific amino acid side chains in the bilayer. Atomistic simulations have made these efforts possible including divulging information on complex membrane proteins such as ion channels.
The difficulties in studying the membrane proteins are mainly due to the difficulties of handling of proteins and experimental challenges associated with working with membrane protein. Also, the co-studying of isolated protein molecule and the molecular environment in order to have an appropriate understanding of the system makes it even more difficult to study.
For instance, isolating the protein from remainder components in the biological system is crucial in structural determination. But in order to have any proper thermodynamic analysis, it must include ALL relevant components of system, particularly paying close attention to boundaries where energy is exchanged.
Traditionally while studying membrane proteins, scientists remove the lipids surrounding the membrane proteins in their preparations. Now, scientists recognize the significance of the lipids as important additives for crystallization. Currently scientists have been more successful solving membrane protein structures with the addition of the lipids during analysis. These successes have led to an increasing number of membrane protein structures which bind lipid molecules to become readily visible and possible to classify.
Membrane proteins are assembled into complexes that allow these intricate assemblies to allow complexity that is not possible using single polypeptides. These complex assemblies allow membrane proteins to have many functions involving regulatory mechanisms and chemical reactions. The existence of these membrane protein complexes prevents potential problems such as unwanted interactions, aggregations, or the formation of hazardous intermediates. Furthermore, these complexes are mechanistically invaluable because they follow a process in which parts of the complexes are "pre-fabricated" and replaced in isolation if damaged, meaning that the whole complex does not need to be replaced if only one subunit it damaged. Membrane protein complexes has been analyzed through the use of blue native polyacrylamide gel electrophoresis and split-ubiquitin method.
Membrane protein complexes allow the avoidance of problems such as those listed above (unwanted interactions, aggregations, or the formation of hazardous intermediates) by being assembled in an ordered, even sequential, manner. To understand that the formation of these complexes are ordered, one would need to know what the assembly intermediates are. Thus, the larger the proteins, the more difficult it is to expose the formation order. However, smaller complexes such as cytochrome bo3 of E. Coli complex have allowed scientists to understand that membrane protein complexes follow a linear pathway of assembly. the bo3 complex is made up of four subunits that assemble through two intermediate complexes. It is understood that bo3 assemble linearly because thought it is possible for other intermediates to form leading to the formation of bo3, they are not observed and there is only one assembly pathway indicating that the intermediates follow a sequential, ordered path. Non-linear assembly would be noticeable because there would be several different assembly pathways. Ordered formation is also seen with cell division in divisomes whereby if one protein is missing, all downstream proteins are preventing from interacting properly. Scientists believe that this sort of ordered pathway exists to protect the cell from potentially harmful intermediates.
Chaperones also play a large role in the formation of these complexes. Chaperones act as physical assembly factors that interact with proteins and prevent unproductive interactions from occurring. For instance, chaperones prevent aggregation in the F1 compound of yeast F1F0-ATP synthase. Two chaperones bound to the alpha and beta subunits bind to the hydrophobic interfaces and guide the alpha and beta subunits into a3b3 complex assembly. Research has also shown that the loss of chaperones in some intermediates could be responsible for the activation of a membrane protein complex. It is important that an intermediate remain inactive so that unregulated activity by partially assembled complexes does not occur.
Membrane protein complexes are believed to undergo dynamic exchange as a mechanism for regulating damaged subunits within the complexes. Dynamic exchange allows the assembly of newly imported proteins into complexes to replaced damaged proteins without replacing the entire complex. An example of this is seen with photosystem II chloroplasts whose D1 subunits that become photo-damaged and is replaced as part of its repair mechanism. Dynamic exchange, at first, was only carried out in vitro. Scientists were only able to conclude that dynamic exchange is a possible repair mechanism, but could not conclude that it was what actually occurred in vivo. It was not until the use of fluorescent microscopy that scientists were able to confirm that dynamic microscopy did occur to a degree. Fluorescent microscopy tagged proteins and watched its interactions in vivo. Subunits were seen freely diffusing into and out of complexes. Future research hopes to disclose which proteins are being exchanged and why thee proteins undergo dynamic exchange.[5]
In the article Membrane Protein Structure: Prediction versus Reality, Arne Elofosson and Gunnar von Heijne discussed several current techniques used to predict the insertion and folding of membrane protein; they depicted a realistic and pragmatic view on how those techniques are used and the limitations. They also pointed out unresolved major issues concerning those techniques.
Arne and Gunnar first pointed out alpha-helix bundle and beta-barrel are the two main structures of membrane proteins. While the helix bundle represents about 20% to 25% of all open reading frame, the barrel form represents a few percent of all open reading frame. An open reading frame. ( A reading frame refers to DNA/RNA that can be broken into three letter codon and be transcribed into protein, while an open reading frame refers to a DNA sequence that does not contain a stop codon in its reading frame. ) The similarities between the helix bundle and beta-barrel is that, in order to fit the basic structure of lipid bilayer in membrane, they both contain hydrophobic amino acids in the middle of the protein. The major difference between the two is their secondary structure. The helix bundle is a complex long trans-membrane protein that packs several alpha-helixes; while beta-barrel protein has several beta-sheets rolled up, and it is shorter and less hydrophobic than the alpha helix bundle. Arne and Gunnar points out that the helix bundle form has been paid more attention as they are longer hence easier to be recognized than the beta-barrel.
Arne and Gunnar then depicts how helix-bundle and beta-barrels are synthesized and inserted into lipid bilayer. In the case of helix bundle translation, corresponding ribosome first bind to a translocon, which is a protein in the inner membrane responsible for the translocation of protein across the inner cellular membrane into the periplasm, called SecYEG translocon. Helix bundle is translated and inserted into the inner membrane. Depending on the hydrophobicity of the helix bundle, the interaction among helix bundles varies; either one helix bundle or a couple are synthesized at a time. Beta-barrel, due to the fact that it is less hydrophobic than helix bundle, could not get through the inner membrane just by itself; after its translation in the cytoplasm, it binds to SecB, with the help of SecA ATPase, via SecYEG translocon and transferred to the periplasm. It is inserted into the outer membrane via YaeT hetero-oligomeric outer membrane integration complex. After the membrane proteins are inserted into the lipid bilayer, it is believed that interactions among helix bundles are stronger than that with the lipids, hence the helix dandles are packed together and obtain its conformation. Hydrogen bonding between polar side chains also contributes in the conformation of the protein. Helix bundle and beta-barrel reaches rather stable conformation after inserted into the membrane. Nevertheless, some membrane protein exhibits a higher degree of flexibility, such as those that are in charge of proton or electron transfers.
According to Arne and Gunnar, in the case of helix-bundle membrane protein, the primary structure, hence its amino acid sequence has long been used to distinguish helix-bundle membrane protein from others. Due to the fact that lipid bilayer has hydrophobic character, the helix-bundle that is inserted into such lipid bilayer should consist of residues that are hydrophobic. Two essential amino acids, tryptophan and tyrosine, whose side chain contain aromatic structures contribute the hydrophobicity of helix-bundle membrane protein. The helix-bundle must also be long enough to span through the inner membrane; hence a helix bundle could have an averaged 10 to 20 hydrophobic residues. Loops connect the helixes; depending on whether the loops are facing inside or outside of the cell, the loops contain different amino acid compositions.
Membrane protein has been thought of perpendicularly orientated through the membrane, Arne and Gunnar points out that, membrane protein orientation could be more complex. One of the factors that contribute to the unexpected complexness of membrane protein comes from reentrant loops, as exhibited in the case of glutamate transporter. Concerning beta-barrel, Arne and Gunnar generalized a series of deducted structural principles; for instance, beta-stands have even numbers and tilts about 45 degrees in antiparallel fashion.
Arne and Gunnar surveyed a series of topology and structure prediction schemes in increasing complexness. 2D prediction is the earliest technique; such technique utilized the higher hydrophobicity possessed by trans-membrane protein than loop regions and has been an effective tool. One challenge faced by the 2D prediction is that the topogenic data from signal peptide and trans-membrane helices are similar, so it is hard to distinguish between the two.
In predicting the structure of β-barrel membrane proteins, scientists look for the existence of an N-terminal signal peptide and the protein’s general amino acid composition. Predicting the structure of a β-barrel membrane protein is simpler than that of a helix-bundle because its amino acid sequence is shorter and not as obvious to see.
As mentioned before, membrane protein cannot be simply modeled as all perpendicular through the membrane; reentrant loops are an important feature that elevates the complexity of membrane protein. These reentrant loops, as suggested by Arne and Gunner can be predicted by a recent developed topological technology named as 2.5D prediction. The residues in these reentrant loops, which come in long loops, medium length loops and short loops, usually are smaller than other parts of the protein so they are easily found in between the transmembrane helices. 2.5D membrane protein structure prediction predicts structures based on the type of amino acid sequence that it contains or by predicting how far the residue is from the center of the membrane protein. Characteristics of residues which can be predicted include lipid-exposed (hydrophobic) regions or lipid buried residues and kinks due to proline. Since 2.5D prediction was able to include sub-structures of membrane protein, such as the interfacial helices and reentrant loops, it is helpful in classifying membrane proteins.
3D structure prediction was first attempted via low-resolution experiments such as electron microscopy. Arne and Gunner points out like, 3D prediction of membrane protein, like all other globular proteins that have been tested against 3D predictions, has low accuracy. What has increased the difficulties in 3D prediction of membrane protein is that they sustain their structures in environments different from those of globular protein; also the globular protein that has been successfully predicted are much smaller than the membrane protein of interest.
To date, there are limited 3D models to be tested against, but there have been hypotheses of models to be tested against. One of which is homology modeling, which would potentially result in structures with details at the atomic level and with similar quality as of the models tested against globular proteins.[6]
In Unsolved Mysteries in Membrane Traffic, a paper written by Susanne R. Pfeffer, from the Department of Biochemistry in Stanford University, she explains how there are various hypotheses to how proteins travel and help facilitate transport within the cell but there isn’t a completely proven hypothesis yet. To start off, Soluble N-ethylmaleimide-sensitive factor attachment protein receptor proteins also known as SNARE proteins help facilitate the fusion of vesicles to their target membranes. There are two distinct groups of SNARE proteins. The first is the R- SNARE which is also called the v- SNARE group which is found on the vesicle. The second group of SNARE proteins is the Q- SNARE which is also called the t- SNARE due to the fact that it’s located on the target membrane. The main difference between these two proteins is that the R- SNARE will only be as a single protein on the vesicle whereas the Q- SNARE will form a complex of three Q- SNAREs. Under these two categories lie specific R-SNARE proteins that will pair up with specific Q-Proteins. The method of how these two pair up to facilitate fusion of a Golgi vesicle to the cell surface is still unknown but one can think of it as SNARE proteins being like puzzle pieces because they have certain specificity therefore one SNARE PROTEIN (R-SNARE/v-SNARE) will bind to only a specific SNARE complex (Q-SNAREs/ t-SNAREs). Although scientist still don’t know how the Golgi decides to transport these vesicles R- SNAREs and Q- SNAREs give clues as to what has arrived and what might depart. If there is a concentration of Q- SNAREs at a specific site that can be accounted for by noticing that there was previous fusion activity at that site. Now when we look at R- SNAREs there are two possible answers to why there are at a specific site, one can be because they have recently arrived and fusion just occurred or because a vesicle is about to depart the Golgi membrane. An important thing to note is that less membrane traffic occurs in the trans Golgi than does in the cis Golgi which is proved by the low concentration of SNAREs in trans Golgi rim and a higher in the cis Golgi rim. Therefore, with all these new ideas we must search for concrete answers to better understand how membrane trafficking occurs within cells.[7]
↑Hiller, S., Abramson, J., Mannella, C., Wagner, G., and Zeth, K., "The 3D structures of VDAC represent a native conformation," Trends in Biochemical Sciences, 2010.
↑The role of solution NMR in the structure determinations of VDAC-1 and other membrane proteins. Sebastian Hiller and Gerhard Wagner*
↑Assembly of Membrane Proteins into Complexes by Daniel O. Daleya,at Center for Biomembrane Research, Department of Biochemistry and Biophysics, Stockholm University, SE-106 91 Stockholm, Sweden, 5 June 2009.
↑Membrane Protein Structure: Prediction versus Reality.Annu Elofsson A, von Heijne G. Rev Biochem. 2007.76:125-40
↑Unsolved Mysteries in Membrane Traffic: Annu. Rev. Biochem. 2007. 76:629–45 Pfeffer, Suzanne R. Dept. of Biochemistry, Stanford
The study of membrane protein folding has been complicated by several factors. Historically, membrane proteins have been very difficult to crystallize due to their large amounts of hydrophobic residues. Proteins that reside in these biological membranes have different surface properties than water-soluble proteins. The proteins are more so affected by the lateral forces and elastic forces from being within the membrane. Duplicating these conditions in order to observe a membrane protein in vitro has been difficult due to the fact that once the membrane protein has been separated, it either unfolds or no longer stays functional.
Maniuplating the lipid bilayer to observe proteins has been increasingly successful. Studies used on smaller water-soluble proteins are very effective with this method. The combination of kinetic and thermodynamic studies of the small water-soluble proteins have been feasible to observe the helical proteins which contain a lot of alpha helices.
Incorrectly folded proteins are associated with prion-related illnesses such as Creutzfeldt-Jakob disease, bovine spongiform encephalopathy (mad cow disease), amyloid-related illnesses such as Alzheimer's disease and familial amyloid cardiomyopathy or polyneuropathy, as well as intracytoplasmic aggregation diseases such as Huntington's and Parkinson's disease. These age onset degenerative diseases are associated with the aggregation of misfolded proteins into insoluble, extracellular aggregates and/or intracellular inclusions including cross-beta sheet amyloid fibrils. While it is not completely clear whether the aggregates are the cause or merely a reflection of the loss of protein homeostasis, the balance between synthesis, folding, aggregation and protein turnover, the recent European Medicines Agency approval of Tafamidis or Vyndaqel (a kinetic stabilizer of tetrameric transthyretin) for the treatment of the transthyretin amyloid diseases suggests that it is the process of amyloid fibril formation and not the fibrils themselves that causes the degeneration of post-mitotic tissue in human amyloid diseases.[18] Misfolding and excessive degradation instead of folding and function leads to a number of proteopathy diseases such as antitrypsin-associated emphysema, cystic fibrosis and the lysosomal storage diseases, where loss of function is the origin of the disorder. While protein replacement therapy has historically been used to correct the latter disorders, an emerging approach is to use pharmaceutical chaperones to fold mutated proteins to render them functional.
Curved membranes recruit large densities of alkylated proteins independent of affinity, with no requirement for specific recognition motifs. The biological curved membrane is identified the lipid composition and the action of membrane-bending proteins.
The membrane curvature can regulate the localization of proteins with specific recognition motifs. Amphipathic alpha helices are critical membrane curvature sensors with a large range of proteins included with larger affinity for positively curved membranes through the recognition of curve defects in lipid packing. The membrane curvature dependent measurements, mostly made in vitro, are averaged across liposomes of variant diameter. These measurements reduce the accuracy and make calculation of affinity more difficult.
Scientist Dimitrios Stamou and his fellows eliminated the problem of the calculations by using the fluorescence microscopy to determine membrane curvature- selective binding on individual liposomes. Their studies reveal the curved membranes can accommodate at a high density of amphiphilic molecules. Therefore, the sensing of membrane curvature emerges consists the property of lipid membranes without any specific proteins anchored by hydrophobic motif.
The single liposome binding measurements used biotinylated liposomes of varied sizes, which then labeled with a chromosphore and immobilized by streptavidin. The fluorescence intensity was used to measure the individual liposomal diameters. Another chromophore was used to tag the molecule of interest, and the ratio of the two fluorescent signals in order to find the density of labeled molecules to each individual liposome.
The binding of amphipathic alpha-helical peptides to the immobilized liposomes were discovered to plot against the amphipathic alpha-helical diameter. The density of peptide stayed constant for measurements of 200nm and above; the measurement increases sharply below 50nm. The equilibrium constant Kd was discovered with little contribution to curvature-selective binding at concentration below Kd.
A geometrical model in highly-curved membranes with a greater density of lipid packing defects provide sites for amphiphilic molecules to insert themselves. The density of binding increases the area of outer leaflet of a membrane. At higher concentration, the interactions are saturated and the defect density effect will predominate. After all, the peptide density will become proportional to defect density, the results for prptides will then fit with the model.
The model can determine the insertion of any amphiphilic molecules with curvature selective. Scientist uses simple alkyl chains with palmitoyl motif to test the property of model. All alkyl chains are sensors of membrane curvature comparable to amphipathic alpha-helical peptides. The head group of alkyl chains without any influence alter to chain itself. Also, the proteins can be added to the chains without affect the curvature-sensing properties.
Overall, the membrane curvature is a generic mechanism to influence the localization of all amphiphilic molecules. Under the effect of concentration of fusogenic lipids in the membrane, membrane curvature can play an important role for protein trafficking and cell signaling in the study of science.
Hatzakis, N. S., Bhatia, V. K., Larsen, J., Madsen, K. L., Bolinger, P-Y., Kunding, A. H., Castillo, J., Gether, U., Hedegård, P. & Stamou, D. How curved membranes recruit amphipathic helices and protein anchoring motifs. Nature Chemical Biology (13 September 2009). doi:10.1038/nchembio.213
Hydrophobicity scales is a system used by biochemists who study amino acids to relatively define the hydrophobicity of amino acid residues. A hydrophobicity scale is typically within a negative to positive range. Values that are in the negative range are defined as not very hydrophobic, whereas the values in the positive range are defined as somewhat hydrophobic. Hydrophobicity scales also offer great insight as to the thermodynamics of the interactions that take place between lipids and proteins inside the cell membrane.
Hydrophobicity, also known as the hydrophobic effect, is the tendency and readiness of non-polar molecules, such as lipids, to associate themselves together in an aqueous solution while simultaneously excluding water molecules. The hydrophobic effect is created when lipids and other non-polar molecules disrupt the hydrogen bonding network of water and forces it reform itself around the non-polar molecule. The resulting effect is water forming a cage around that particular hydrophobic molecule. The hydrophobic effect plays a crucial role in the regulation of protein folding, as well as formation of lipid bilayers and the insertion of membrane proteins into non-polar lipid environment.
There are many different types of hydrophobicity scale. Below are five different ways to access the hydrophobicity level of a single amino acid residue or a single amino acid mutation of a polypeptide, as discussed by MacCallum in his hydrophobicity scales article. In the Radzicka–Wolfenden experiment, the sidechain of the amino acid arginine is placed into water and cyclohexane solvent. The concentration of the amino acid in each layer is used to calculate the free energy. The problem with this hydrophobicity scale is that cyclohexane, being more nonpolar, does not accurately represent the lipid bilayer of the cell membrane. The MacCallum et al.’s evaluation of the hydrophobicity scale is very similar to that of Radzicka’s. The only difference is that instead of using cyclohexane, MacCallum uses DOPC, which is a more accurate representation of the actual lipid bilayer. The problem is the same in that there is no backbone in this model. A “water defect” is seen because as some of the amino acids pass through the bilayer, its charges attracts and pulls the water with it. Wimley–White uses 1-octanol with water droplets and a pentapeptide Ace-WLxLL instead. They replace a single amino acid each time. This is a more realistic representation of the cell membrane because it takes into consideration the effects of the polypeptide backbone. They also measure the proportion of pentapeptide in each solvent. Moon–Fleming uses the protein OmpLA, which can be in an unfolded state in water or in a folded state when inserted inside the membrance.The equilibrium is shifted by mutating ompLA. Measurement is taken from using fluorescent spectrometry. Hessa et al. uses the protein leader peptidase. They attach an H-segment,a 19-residue-long chain, and 2 glycosylation sites on the H-segment to the leader peptidase. By assaying the glycosylation sites, they can predict if the H-segment have been inserted inside the membrane or not. These five experiments all measured the free energy transfers as the amino acid passes through the membrane. These hydrophobicity scales correlated very well with each other when normalized. Note: It should be kept in mind that these experiments do not fully represent the hydrophobicity inside the biological systems because they do not take into consideration the membrane proteins that are already presented inside the cell membrane, which could have interact with our amino acid.[1] The following image shows the system and the environment for each of the five experiments mentioned.
Even though 20-30% of all proteins are membrane proteins, less than 1% of all structures known in the Protein Data Bank are membrane proteins. Knowing the hydrophobicity scale allows for prediction of the transmembrane protein sequence, as well as allows for a better understanding of the water-protein-lipid interactions.[1]
An example of the components of a typical transmembrane protein.
The thermodynamic and microscopic details for lipid-protein interactions are very important in a number of vital biological factors.
One of such biological factor involves KvAP, which is the first crystal structure of a voltage-gated potassium channel. The structure of KvAP has led to numerous discussions in the biochemistry community about the interactions that take place between arginines, an amino acid, and lipids. This is due to the gating mechanism that the structure suggests in which positively charged arginines were exposed to the interior of the hydrophobic lipid bilayer of the cell membrane.
Another key biological factor in protein-lipid interactions is the action of anti-microbial peptides and cell-penetrating peptides. This is because anti-microbial peptides have specific amino acid sequences that are enriched in cationic and aromatic residues, whereas cell-penetrating peptides are rich in cationic residues.
The last major development is the successful crystallization and determination of the structure of the Sec-translocon system, which has the fundamental task of inserting membrane proteins into the membrane as soon as they are done synthesizing by the ribosome. The complexity of the Sec translocon system has raised questions about the thermodynamics of membrane insertion.
The membrane bilayer is a highly heterogeneous layer with large gradients in density and polarity on a nanometer length scale. The membrane lipid bilayer can be divided into four major regions. The hydrophobicity decreases and the hydrophilicity increases moving along each region. In the first region, which is the center of the bilayer, is very hydrophobic and is significantly disordered with properties similar to decane. In the second region, the lipid tails are more ordered and have a higher density, with features that are very similar to a polymer. In the third region, there is a diverse mixture of functional groups, most of the head group density, as well as water. In the fourth and last region, it is very hydrophilic because it is defined as mostly water that is perturbed by the lipid layer. This layer can be very deep, depending on the cellular conditions.[2]
Different hydrophobicity scales have been developed in the biochemistry community to study the lipid-protein interactions that take place at the membrane protein. Each hydrophobicity scale has been developed independently of one another using different techniques to study these lipid-protein interactions.[1]
Radzicka-Wolfenden small molecule partitioning scale
One of the earliest hydrophobicity scales was developed by Radzicka and Wolfenden to study the folding of globular proteins. This scale was based on the partitioning of small molecule analogs of amino acid sidechains between a polar layer (water), and a lipophilic layer (cyclohexane). This particular scale is relevant for membrane partitioning due to the fact that the center of the membrane has physiochemical properties similar to those of bulk hydrocarbon.[3]
Sidechain analogs of amino acids were added to a biphasic system of water and cyclohexane. After the system reached equilibration, the concentrations of both water and cyclohexane were both measured. This ratio in the concentrations of water and cyclohexane are directly proportional to the free energy of transfer.
Although the Radzicka-Wolfenden hydrophobicity scale is simple, this scale does not accurately reflect real cellular conditions because lipid bilayers do not resemble isotropic solvents and sidechains by themselves ignore important aspects of protein structure.[4]
MacCallum et al. molecular dynamics potential of mean force scale
MacCallum et al.'s molecular dynamics potential of mean force scale focuses on molecular dynamics simulations in order to calculate the distribution of the Radzicka-Wolfenden sidechain analogs (as described above). Instead of using cyclohexane as the lipophilic layer, a more realistic bilayer called 1,2-dioleoylsn-glycero-3-phosphocholine bilayer (DOPC) was utilized instead. Because these are computer simulations, the local environment is able to be known in the molecular level.
One of the most important things about this particular model is the formation of water defects in the bilayer, which are local deformations in the membrane which allow for water to penetrate into the bilayer core and keep polar and charged groups hydrated in the partitioning of polar and charged molecules into the lipid bilayer membrane.[5]
Wimley and White developed a peptide-based system to derive a thermodynamic scale between amino acid side-chain residues and lipids. [6]. The pentapeptide which Wimley and White used was Ace-WLxLL, where x can be any of the 20 naturally occurring amino acids. Water was used as the polar layer and 1-octanol was used as the lipophilic layer, and the partitioning between the two layers was measured. The partitioning between water and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) was also measured via equilibrium dialysis and reverse-phase high-performance liquid chromatography (HPLC).
One of the caveats of this particular scale is that its focus is explicitly interfacial instead of the bilayer core like the other scales. In order to achieve a better understanding of this scale, one will need to dwelve into the microscopic level. However, compared to the other small molecule scales, this scale is more realistic but emphasizes on the interactions of amino acid side-chain residues with either the water-lipid interface or a heterogeneous octanol environment with a hydrophobic environment for neutral amino acids like lycine and a more hydrophilic environment for charged amino acids like arginine. [7][8]
Moon and Fleming developed a hydrophobicity scale that is based on the reversible in vitro equilibrium between the water soluble unfolded state and membrane-inserted folded state of outer membrane phospholipase A (OmpLA) [9]. By making mutations in OmpLA, equilibrium between the folded, membrane-inserted state and the unfolded state in solution can be shifted and subsequently measured by fluorescent spectroscopy.
The three-dimensional structure the outer membrane phospholipase A (OmpLA).
This experiment compares a well-defined folded membrane state with an unfolded state in solution and measures the thermodynamic equilibrium between the two. One of the complications is that the bilayer used, 1,2-dilauroyl-sn-glycero-3-phosphocholine (DLPC), is relatively thin and unstable.
Hessa et al. developed a hydrophobicity scale using a previously developed membrane protein insertion assay using the small membrane protein leader peptidase. They engineered two glycosylation sites and a 19 amino acids long residue, also known as the H segment, which can be inserted into the membrane as a transmembrane helix depending on its hydrophobicity. By assaying the two glycosylation sites, the insertion state of the H segment can be determined [10]. An apparent equilibrium between the inserted and non-inserted state of H segments can be achieved by modulating the sequence.
The insertion of the H segment involves the Sec translocon, which is a cellular machinery that either inserts of secretes a given H segment into the membrane as it gets synthesized by the ribosome. Hessa et al. used this to develop a transmembrane prediction method that relies on a linear combination of single residue results to predict whether or not a helix of an amino acid residue will get inserted into the cellular membrane [11].
Another useful application for this method with the Sec translocon is to test whether a particular sequence from a voltage-gated potassium channel would insert into the membrane, despite having multiple arginine and other polar residues [12].
Despite having used different environments and methods to test for hydrophobicity, each of the five aforementioned derived hydrophobicity scales all exhibit a well-defined correlation with one another [1]. For instance, the Radzicka-Wolfenden and MacCallum scales correlate very well with each other and yield almost identical absolute free energy differences. The Wimley-White scale measures the interactions in the heterogeneous environment of water and 1-octanol, whereas the Moon-Fleming and Hessa et al. scales measure properties that are directly related to membrane protein insertion and stability.
The absolute magnitudes of the Wimley-Hessa-Moon scales and the MacCallum-Radzicka scales differ by a significant amount. Despite this fact, all five of these scales all point to the same result [13].
Although the experiments that have been conducted have had similar results in an attempt to match the reaction of lipids and their membrane proteins, the experiments were overall very simple and not necessarily replicators of a true physiological setting, and therefore do not give much insight to true physiological cellular membranes. The experiments are simply correlated and not necessarily exact, for biological membranes contain a diverse mixture of lipids as opposed to the single component bilayers used. Membrane proteins play up to a 25% role in cellular membranes and these are not included within the experiments. Additionally, charged or polar molecules typically distort the lipid-water interface. Moreover, the scales used in the above experiments have only considered single amino acid residues, while in a true biological setting, multiple amino acid residues are present. The free energy calculations (the free energy being the amount of energy measured from the transfer of each amino acid between a polar, water environment and a lipid bi-layer environment) used to determine the scales in each of the experiments were also very different and this is why a scale factor was used to compare the experiments. Overall, true biological systems have very different settings involved, which have yet to be matched more closely to true settings. Despite the discrepancies within the research of hydrophobicity scales, there are many profound reasons to continue investigating this rather complex and relatively unknown topic, as discussed below.
Researching hydrophobicity and establishing a scale is rather important in creating the ability to discuss the implications of the different scales, as well as in finding a single scale to utilize. The scales help to predicting membrane protein-lipid interactions as well as membrane protein structure, of which little is known. Because many drugs interact strictly with membrane proteins, understanding protein-lipid interactions through measuring hydrophobicity could give great insight to how to cure or treat illnesses through new methods that are not yet known and could be more effective. This would be most applicable in mechanisms of antimicrobial and cell penetrating peptides. Also, hydrophobicity reveals side chain-lipid interactions.
↑ abcdMacCallum, J.L. and Tieleman, D.P. (2011) Hydrophobicity Scales: A Thermodynamic Looking Glass into Lipid–protein Interactions. Trends in Biochemical Sciences. 12, 653-661
↑Engleman, D.M. et al. (1986) Identifying nonpolar transbilayer helices in amino acid sequences of membrane proteins. Annu. Rev. Biophys. Biophys. Chem. 15, 321-353
↑Wolfenden, R. (2007) Experimental measures of amino acid hydrophobicity and the topology of transmembrane and globular proteins. J. Gen. Physiolo. 129, 357-362
↑Radzicka, A. and Wolfenden, R. (1988) Comparing the polarities of the amino-acids - side-chain distribution coefficients between the vapor-phase, cyclohexane, 1-octanol, and neutral aqueous-solution. Biochem. 27, 1664-1670
↑MacCallum, J.L. et al. (2008) Distribution of amino acids ina lipid bilayer from computer simulations. Biophys. J. 94, 3393-3404
↑Wimley, W.C. and White, S.H. (1996) Experimentally determined hydrophobicity scale for proteins at membrane interfaces Nat. Struct. Biol. 3. 842-848
↑Franks, N.P. et al. (1993) Molecular oganization of liquid n-octanol: An X-ray diffraction analysis. J. Pharm. Sci. 82, 466-470.
↑Wimley, W.C. et al. (1996) Solvation energies of amino acid side chains and backbone in a family of host-guest pentapeptides. Biochem. 35, 5109-5124
↑Moon, C.P. and Fleming, K.G. (2011) Side chain hydrophobicity scale derived from transmembrane protein folding into lipid bilayers. Proc. Natl. Acad. Sci. U.S.A. 108, 10174-10177
↑Hessa, T. et al. (2005) Recognition of transmembrane helices by the endoplasmic reticulum translocon. Nature 433, 377-381
↑Hessa, T. et al. (2007) Molecular code for transmembrane-helix recognition by the Sec61 translocon. Nature 450, 1026-1030
↑Hessa, T. et al. (2005) Membrane insertion of a potassium-channel voltage sensor. Science 307, 1427
↑Gumbart, J. et al. (2011) Free-energy cost for translocon-assisted insertion of membrane proteins. Proc. Natl. Acad. Sci. U.S.A. 108, 3596-3601
Membrane fusion is defined as a progress of two separated membranes that would be unified into one continuous membrane through the progress of intermediate transformations. At the beginning of the progress, only contacting monolayers of membranes would merge into one, while the distal membranes would be separated. Hence, the fusion stalk, which is the lipid bridge, connects the two monolayers membranes and result in the first stage of fusion, hemifusion. Eventually, this fusion stalk would allow the unification between the distal monolayer membranes. And such unification would result in the formation of the fusion pore that would merge the separated distal monolayers. As a result, that would be the last step of the unification between monolayers membranes.
Membrane fission occurs when there's a separation of membrane into two separated membranes. This progress takes place through the membrane neck. Hence, fission occurs when there's a self-merger between the inner monolayer of the membrane neck. This step results in the formation of the fission stalk. And the fission progress would end when there's the self-unification between the outer monolayers.
This image illustrates curvature and membrane structures. (a) Membrane structures, like invaginations, are made up of regions with different curvatures. Note that the sign of the curvature depends on the vantage point; hereafter, curvature will be referred to as seen from the cytosol. The top visual indicates the schematic view of a plasma membrane invagination displaying various curvatures. The flat membrane has zero curvature, which is shown in yellow. At the base of the invagination, indicated by the brown color, the curvature is negative. The part of the membrane that reaches further into the cytosol contains a positive curvature. The bottom figure demonstrates a membrane tubule, which is a ubiquitous structure in cells. These structures are maintained by proteins, which are labeld as blue crescents. The curvature is positive along the circumference of the tubule and zero along the tubular axis. (b) Schematic view of the mechanisms by which Bin/amphiphysin/Rvs (BAR) domains can generate curvature. The crescent shaped BAR domain interacts with the bilayer through electrostatic interactions, imposing its intrinsic shape onto the membrane as shown by the top image. This mechanism is known as the ‘scaffolding mechanism’. Alternatively, BAR domains can introduce an amphipathic structure, such as a helix, into one leaflet of the membrane. This ‘wedge’ displaces lipids, which will cause the membrane to bend towards the BAR domain. This mechanism is referred to as the ‘wedging mechanism’ shown in the bottom picture.
Cellular membrane has the ability to execute very important functions like locomotion and receptor turnover. It is necessary for cells to sense changes in the environment and transport signals across the membranes. To perform this, cells need to reconstruct the shape of its membrane with relatively high spatial and temporal accuracy due to the dependency from cellular processes—such as cell division, endocytosis, and cell migration. Therefore, membranes have the power in remodeling the overall shape of the cell. For this to occur, membrane-bending proteins are required and membrane curvature are closely examined. The sign, or sometimes known as the direction, of the curvature is considered to be an arbitrary measure. From a cytosol, plasma membrane invaginations predominantly contain segments with positive curvature. On the contrary, protrusions consist of membranes with a negative curvature. Membrane curvature is critical in the investigation of membrane fusion and fission, which is the analysis of activity of membrane proteins, and the unity of proteins to the location of membrane-cytosol interface. Membrane curvature is not known to be a passive characteristic of the cellular membrane; instead, it has risen to be a regulated state due to its active influences of diverse processes of membrane fusion and fission.
Examples of such membrane proteins include the Bin/amphiphysin/Rvs (BAR) domain, one of the main membrane-bending protein superfamily. The BAR domains lack sequence motifs of characteristic signature in their primary structure making it not entirely recognizable. However, at the structural level, BAR domains are considerably conserved, containing a three helix coiled core which creates a curved homodimers or heterodimers. This gives an overall ‘banana shape’.
The formation of homodimers or heterodimers are still relatively a mystery in the BAR domain. However, researches have shown that a mishap of the dimerization interface contribute malfunction in BAR proteins in terms of remodeling. The nonfunctionality happens due to proper interaction of the BAR domains with the negatively-charged lipid headgroups like phosphinositides, which require the specific positioning of a positively-charged residues in the context of a banana- shaped dimer.
Most BAR proteins contain an extra domain, such as a src-hology 3 (SH3) domain, that permit BAR proteins to communicate with proline-rich domain containing proteins. Therefore, the BAR domain proteins are a type of scaffolding proteins that systemize a selection of proteins intp a “curvature-dependent” form. The three general categories of BAR proteins are classical BAR domain, IMD/Inverse BAR domain, and the FCH domain (F-BAR).
Classical BAR domains are typically found in arfaptin and contain the highest level of intrinsic curvature. It extends to smaller divisions depending on additional emembrane binding domains like an amphipathic N-terminal helix or the phox domain. All classical BAR domains are known to provide and encourage positive membrane curvature.
An example of the F-BAR domain is the Cdc42-interacting protein 4. This time of protein domain signifies the largest and most diverse domains. It is further broken down into six subcategories. Moreover, the intrinsic curvature of this particular protein domain ranges from high to almost planar. Due to this, proteins of this kind are allowed to support a large range of membrane curvatures.
The IMD/Inverse BAR domain contain a negative curvature and symbolize a mechanism for cells to generate extrusions. A similar protein domain to IMD/Inverse BAR domain is PinkBAR domain, which contains no intrinsic curvature. The absence of intrinsic curvature for PinkBAR domains help to form scaffolds on flat membrane surfaces.
Bar proteins function a large role in membrane scaffolding, organelle creation, organismal patterning, and disease. Most of the functions associated with BAR domains narrow down to the intrinsically curved dimers of all BAR domains. The shape of the BAR domain dimers correlate to its curvature, which inherently depicts its ability to bend membranes.
The molecular mechanism of membrane bending for BAR domains arises in the intrinsic curvature of the dimers. The Bar Domain dimers encourage membrane bending by imposing its shape on the membrane substrate, a process known as the ‘scaffolding’ mechanism. Another membrane bending mechanism includes the presence of amphipathic wedges, which can sense membrane curvature, into the bilayer. Such wedges can also encourage formation through the concerted displacement of lipids in the leaflet proximal at its present location. Additionally, substrate selection could potentially cause experimental bias; the composition bilayer and pre-existing curvature for the BAR domain determines the likelihood of recruiting and remodeling for the BAR domain. Some BAR proteins that have amphipathic wedges are known to play a significant role in membrane fission. Furthermore, membrane bending and fission are inversely related.
The property of BAR domains to be involved in bilayers and recruit specific interactive partners requires access to and control of its target. Studies have indicated that BAR proteins show an inclination for membranes with a specified curvatures in vitro, and such membrane preferences correspond to the intrinsic curvature of the BAR domain. Also, amphipathic sequences detect and bind to locations where curvature stress creates packing defects in the region of the lipid headgroup. The amount of defects correlates with the degree of curvature. As a result, the ability of BAR domain proteins to sense the defects is a method to control differential binding to membranes of various curvatures.
Despite the developing progress in gaining an understanding of membrane transport and signaling, studies of how the cells reshape their membranes have provided information on the structural and functional features of the membrane bilayer. However, the molecular mechanisms of which membrane-bending domains process and stabilize membrane curvature and how the events are coordinated remains elusive. For one, the structural complexity of the membrane bilayer is constantly influenced to be susceptible to reconstruct and recruit specific membrane remodelers. Second, the deficiency of standardized methods to investigate membrane remodeling and the tools to computationally examine the complicated lipid mixtures that is generally located in cellular membranes.
Similarity of the membrane fusion and membrane fission
One common feature of the membrane fusion and membrane fission is the creation of the membrane stalk. In fusion, there is the formation of the fusion stalk which is the merging between the monolayer membranes. In fission, there is also the formation of the fission stalk which is the unification of the inner monolayers of the membrane neck. Scientists have done research on the pre-stalk fusion. They believe this progress requires one lipid molecule to be inserted between the two opposing hydrocarbon membranes. As a result, there would be the formation of the lipid bridge between the membranes.
Differences between membrane fusion and membrane fission
One difference is the opposite sequences of shapes of the fission and fusion progress. Membrane fusion results in the unification of the monolayers membranes, while fission results in the separation of the membranes. Hence, fission allows the separated membranes to have a greater curvatures and stronger bent of the membranes. However, fusion results in a smaller curvatures which allows the bending of the membranes to relax. In conclusion, membrane bending would favor membrane fission, while membrane fusion would results in membrane unbending.
The second difference is the self-connectivity between the membrane fusion and membrane fission. Since fission occurs when there's a separation of the continuous membrane, that would result in the limited area for each separated membranes. However, the unification of membranes, fusion, would increase the area over the entire membrane. Therefore, fission is not favored by self-connectivity, while fusion is supported by the forces that result in the merging of the membranes.
Proteins are the forces that generate the merging between the monolayer membranes or the separation of the monolayer membrane. Hence, membrane remodeling is the reconstructing of the lipid bilayer which is determined by the membrane proteins. There are two physical requirements for membrane remodeling. First, the free energy before the beginning of the remodeling must be higher than the energy after the remodeling. This means that there must be a release of energy that would allow for the reconstructing of the membranes. In this case, we use the term "relaxation of the free energy" to summarize the first requirement. Second, the intermediate energy must be low enough to be overcome by the thermal fluctuations.
The progress of membrane remodeling is determined by the proteins. They are proteins that provide the free energy for this spontaneous reaction. Hence, the proteins can change the physical structures of the lipid bilayers and the lipid composition of the monolayers membrane. These physical changes would result in the membrane remodeling.
The free energy used in membrane remodeling is called elastic energy. Elastic energy is created by the three different forms of membranes. They are membrane bending, stretching, and tilting of the lipid chains. First, membrane bending is determined by the curvatures of the membranes surface. The bending of the membrane, curvature, is the shape of the membranes. Mathematically, curvature is defined by two radii R1 and R2 of the arcs plane. We use the equations of inverse radii c1= 1/R1 and c2 = 1/R2 to calculate the sum and product of the curvatures. As a result, those values would determine the shapes of the lipid bilayer. The sum and the product of curvatures are called the total and Gaussian curvatures. And they both require free energy (F) to generate membrane remodeling. The sum of the curvatures has FB, the free energy of the total curvature. This value is based on the membrane shape. And it can change because there are multiple stages from the beginning of the membrane remodeling to the fusion or fission progress. Hence, fission would result in the increase of this free energy, while fission would decrease the free energy FB.
Second, membrane stretching depends on the fusion pore. This fusion pore would allow the increase in the area of the surface membrane when fusions occurs.
Lastly, membrane titling depends on the merging of the lipid hydrocarbon chains when there is the formation of the fusion stalk or fission stalk.
In general, protein-curvature generates fusion by identifying a lipid bilayer area at the top of the membrane. Then the protein would bend the membrane into cylindrical shapes. Proteins choose the area on top of the membrane because there would be "relaxation of elastic energy" when the fusion reaction occurs. Hence, there are two requirements for this fusion reaction to occur. First, the lipid bilayer area must be at the external area where fusion takes place. If it is not at the external area, that would stabilize curvature and results in the non-spontaneous reaction when there is no release of free energy at the end of the reaction. Second, the curvature must be large enough in order generate the fusion reaction. There are two mechanisms that are examples of this fusion reaction.
The first mechanism is called the hydrophobic insertion or wedging mechanism. This reaction requires hydrophobic proteins to be inserted into the lipid bilayer chains. Hence, there would be the process of expanding the polar head of the monolayers membrane. As a result, the proteins would gather and interact with the SNARE complexes.Then Ca2+ inside the hydrophobic proteins would be inserted into the lipid matrix and that would cause the bending of the lipid region. As a result, there would be changes in the shapes of the membranes such that it can be either cylindrical or conical belt that has protein-free end cap at its ends. Lastly, the fusion reaction will take place at these ends of the membranes when they release free energy during bending.
The second mechanism is called the force transmission. Here, SNARE complex has syntaxin and synaptobrevin that are factors that allow the fusion reaction to take place. The SNARE complex will start at the N terminus of the lipid bilayer chain and merge the monolayer in the zipper like fashion. As a result, it would form the stable four-helix bundle which is called the core SNARE complex between the monolayer membranes.
Proteins generate fission through the release of the free energy in the membrane neck. There are two mechanisms that illustrate this fission reaction. The first mechanism is called the scaffolding mechanism. It requires protein complexes to drive the membrane scaffolding. One of the examples of membrane scaffolding is its role in the release of enveloped viruses. As a result, scaffolding would lead to the bending of the membranes into cylindrical shapes. And that would results in the "relaxation of elastic energy". The second mechanism is called the hydrophobic insertion. Like fusion, hydrophobic insertion in fission also requires hydrophobic proteins to be inserted between the lipid matrix. For instance, the protein, BAR domain, would cause the bending of the lipid region. As a result, there would be changes in the shapes of the membranes which then drive the spontaneous reaction in fission
Generating membrane remodeling by membrane tension
Tension inside the lipid bilayer membrane would cause the reaction of the membrane remodeling to occur. This is based on the process of protein scaffolding. And scaffolding would create a lipid bridge between the monolayers and result in the first stage of fusion which is called the hemifusion. Then there would be the formation of the fusion pore which link the distal monolayers and that would complete the remodeling process.
'
Endosomes in the cell have complexes that help sort cellular packages for transport. These are known as ESCRT. They have been found to have a role in pathology, by affecting cytokinesis, virus envelope budding, and vesicular biogenesis.
Removing receptors from the cell membranes and delivering them to lysosomes is needed for controlling signaling in cells. The molecular machinery that sorts all the cellular packages in the endosomes has been greatly researched and progress has been made about its structure and basic function. Target receptors are ubiquitylated and through cell-mediate endocytosis, they are sent to an endosome. Epidermal Growth Factor (EGFR) is a receptor that is made to degrade, but others are recycled back to the membrane or to the endosomal-golgi bodies via tubules. The cellular packages that are assigned to be degraded are placed into vesicles and they bud into the lumen of endosomes, which go to the multivesicular bodies and then fuse further with lysosomes.
The ESCRT complex is composed of 5 subunits, which are regulated by molecules sequentially to the endosomal membranes. However, the ESCRT assembly at the endosomes remains unclear.
ESCRT protein complex is sent to the middle of the cell during cytokinesis by directly interacting with the phosphoprotein CEP55, which is a requirement for membrane fission.
Source:
Divergent pathways lead to ESCRT-IIIcatalyzed
membrane fission
Suman Peel1, Pauline Macheboeuf2, Nicolas Martinelli2 and Winfried Weissenhorn2
1 Department of Biochemistry, School of Medical Sciences, University Walk, University of Bristol, Bristol BS8 1TD, UK
2Unit of Virus Host Cell Interactions (UVHCI) UMI 3265 Universite´ Joseph Fourier-EMBL-CNRS, 6 rue Jules Horowitz 38042 Grenoble,
France
Kozlov,Michael. McMahon, Harvey. Chernomordik, Leonid. Protein-driven membrane stresses in fusion and fission. PubMed. 11/15/12.
Mim, Carsten; Unger, Vinzenz M. 'Membrane curvature and its generation by BAR proteins'. Trends in biochemical sciences doi:10.1016/j.tibs.2012.09.001 (volume 37 issue 12 pp.526 - 533).
Due to many potentially-harmful particles that settle on our airway paths, our bodies have developed innate defense mechanisms that are continually working to protect us from various infections. With the use of the perfluorocarbon/osmium/transmission electron microscopy fixation techniques and with confocal microscopy, the transport of mucus can now be visualized under high resolutions. The airway surface liquid, also identified as ASL, acts as the first line of defense and is lined along the epithelia lining. It is made up of two layers, the perciliary liquid layer, also known as PCL, and the mucus layer. The mucin rich mucus layer lies on top of the PCL, the very watery layer that surrounds the cilia (Matsui). The mucus layer is made up of heavily glycosylated macromolecules, genes MUC5AC and MUC5B. These macromolecules act as a spider web for trapping inhaled particles. Because of the large carbohydrate sequences which comprise these macromolecules, mucins bind to nearly almost every particle so that it can be cleared before it reaches the lungs. The viscoelastic properties of the mucus allows for the conversion of energy from beating cilia into mucus unidirectional transport along the airway surface. The frictional interaction of the PCL and mucus lining also acts as a secondary form of transport. If all else fails, the lungs also have another line of defense, cough clearance. Cough clearance is completely independent of cilia. Just to go over briefly, the efficiency of the cough is related to the height and volume of liquid on the airway surface. the PCL and mucus lining encounter vast amounts of various foreign particles. This can range anything from the fart that the person in front of you let out and the dust you may inhale when playing football to inhaling toxic fumes that may have accidentally slipped out from the lab. To go over some specific irritants, agents such as ammonia, organic vapors, and cigarette smoke induce mucus secretion from both the epithelial secretory cells and the submucosal glands. These cells are stimulated by direct and reflex stimulation, respectively. Prolonged inhalation of SO2 and cigarette smoke can cause goblet cell hyperplasia and enlargement of the submucosal glands. Also, various drugs induce mucus secretion. For example, prostaglandin (PG) stimulates mucus secretion among cats. But when prostaglandin is inhaled by humans, PG stimulates cough receptors.
The mechanism of mucus clearanceis triggered by the effects of pollution or from stress from the body. Now when the mammalian body is stressed from the constant intake of foreign particles, extracellular nucleotides and nucleosides stimulate Cl- transport and inhibit Na+ absorption. This allows for the correct height and volume of the ASL. Under normal circumstances, the epithelial lining along the airways adjust Na+ and Cl− transport so that the PCL liquid levels stay intact. ATP is also released onto the airways, where it is metabolized into adenosine to activate the Cl- secretory channels. All of this is done to balance absorption and secretion for efficient mucus homeostasis. “Mechanotransduction is the process by which physical forces are translated to physiologic responses and is widely important in tissue homeostasis.”Pollution, in this case cigarette smoke, is a direct factor to the disruption of homeostasis among the ASL. When cigarette smoke is inhaled, an obvious effect is lung damage. What most people don’t realize is that it also causes your body to take longer and larger breaths to allow for optimal oxygen inhalation. Recent studies show that when normal human airway epithelial cells were subject to tidal breathing, the height of the ASL doubled, along with an increase of ATP in response to the shear stress.
Secretin is a hormone that controls secretions into the duodenum and water homeostasis in the body. It is first produced in the S cells in the duodenum during the crypts of Lieberkuhn. [1] Secretin works by regulating the pH of duodenal by controlling gastric acid secretion and buffering by utilizing the bicarbonate found in centroacinar cells in the pancreas. [2] It is also noted to be the first hormone to be identified. [3] The secretin protein in the human genome is encoded by the SCT Gene. [4]
During the 1900s, Ernest Starling and William Bayliss were studying how the human nervous system controls how the body digests food. [5] It was already known that the pancreas helped during the digestion by secreting juices to help pass food through the pyloric sphincter into the duodenum. Starling and Bayliss discovered that this process was not controlled by the pancreas but by the nervous system. They did this by first cutting all of the nerves to the pancreases in their model animals and observing how the digestive system responded. Starling and Bayliss determined that the substance that was secreted during digestion by the intestinal lining initiated the pancreas secretion through the bloodstream. They named this this secretion secretin, being the first chemical messenger identified. These types of substances were called hormones, a term Bayliss coined in 1905.
Secretin was synthesized as a 120 amino acid precursor protein also known as prosecretin. Prosecretin contains an n-terminus, secretin, spacer, and a c-terminus.
Secretin is a linear peptide that is made up out of 27 different amino acids with a molecular weight of 3055. It also forms an alpha-helical structure between the 5 and 13th amino acid positions. Some of the amino acid sequences present in secretin have similarities of those amino acids in glyucagon, gastric inhibitory peptide (GIP) and vasoactive intestinal peptide (VIP). Fourteen out of the twenty-seven amino acids in secretin are in the same positions in the protein glucagon, 10 in GIP, and 7 in VIP. [6]
Secretin works by increasing the bicarbonate concentration in the pancreas. The pancreas contains centroacinar cells that have secretin receptors located on their plasma membrane. When secretin binds to the receptors on the membrane, it initiates adenylate cyclase activity and converts cyclic AMP from ATP[1]Cyclic AMP is a second messenger in intracellular signal transduction and increases the concentration of carbonate. It is also known to promote the growth and maintenance of the pancreas.
Secretin increases water and bicarbonate secretion in the duodenal to buffer incoming protons of the acidic chyme[2] In addition, it also enhances the effects of cholecystokinin to produce secretion of digestive enzymes from the gallbladder and pancreas.
It also decreases the blood glucose concentration by increasing insulin being released by the pancreas[2]
In addition, secretin stimulates pepsin secretion from chief cells, which then help break down proteins during digestion. It also stimulates the release of glycagon, somatostatin, and pancreatic polypeptide. [7]
↑ abBayliss W, Starling EH (1902). "The mechanism of pancreatic secretion". J. Physiol. (London). 28: 325–353. {{cite journal}}: Cite has empty unknown parameter: |month= (help)Invalid <ref> tag; name "Bayliss_1902" defined multiple times with different content
↑ abcDeGroot, Leslie Jacob (1989). J. E. McGuigan (ed.). Endocrinology. Philadelphia: Saunders. p. 2748. ISBN0-7216-2888-5. Invalid <ref> tag; name "isbn0-7216-2888-5" defined multiple times with different content
↑Polak JM, Coulling I, Bloom S, Pearse AG (1971). "Immunofluorescent localization of secretin and enteroglucagon in human intestinal mucosa". Scandinavian Journal of Gastroenterology. 6 (8): 739–744. doi:10.3109/00365527109179946. PMID4945081.{{cite journal}}: CS1 maint: multiple names: authors list (link)
The biochemical structure of the M2 proton channel in influenza A was uncovered with the techniques of Nuclear Magnetic Resonance (NMR) for the closed state and X-ray crystallography for the open state. It is believed that the sole inhibitor-binding site shown by the X-ray structure to be directly in the center of the channel is responsible for binding to the drug amantadine, even though NMR revealed the possibility of the exterior binding of four additional inhibitors on the outside of the channel under certain states.
Protein M2 Proton Channel Structure and Importance
The M2 protein is a 97-residue protein that is made up of 23 amino acids; it is a single-pass membrane protein that has all of the N-terminus’s orientated to the outside of the cell. Inside of the cell is a 19 residue transmembrane domain (M2TM) and a 54-residue tail. It is the M2TM, which is the target of the anti-influenza drugs amantadine and rimantadine. The structure of these residues was solved using a combination of X-ray crystallography and NMR techniques; both techniques support a semi-symmetric tetramer proton channel, the section inside the cell membrane coiling in a leftward orientation. The structural determination of this channel differs between these two methods in regards to the bound inhibitor position. Even though both methods suggest similar hot-spots for possible binding sites inside and on the outside of the channel, X-ray crystallography indicates a amantadine binding site in the central pore of the channel whereas NMR indicates four rimantadine binding sites on the surface outside of the channel.
It is important to know the structure of this proton channel, the location of the binding sites and their functionality in order to better assess how the anti-influenza drugs bind to the virus protein and carry out their functions. Most strain of influenza have recently developed a resistance to these two drugs, with a large 97% of people having some of the virus containing a specific Ser31Asn substitution which makes the virus highly resistant to these drugs. Therefore it is vital to further prove the structure of this proton channel’s binding sites in order to fully realize how the drug binds to the channel and how resistance to these drugs has developed. Once these facts are determined, scientists can attempt to uncover other compounds that might inhibit the M2 protein channel as these drugs once did.
New solid state NMR data looking at a closed M2 channel revealed a central binding site with high affinity for amantadine with four outer binding sites with low affinity for the drug. This confirmed earlier X-ray crystallography and NMR data as well as exposing that the main binding site for amantadine is most likely in the central pore of the M2 channel. Although the prominence of a single central inhibition is assumed, it is quite possible that there is a dual inhibition mechanism, so further scientific research is necessary. One additional thought is that the secondary inhibitor sites on the outside of the channel might play a greater role in drug binding for the mutant drug-resistant influenza virus which is now present in 97% of people who are tested.
A number of eukaryotic membrane proteins have a single C-terminal transmembrane domain which casts them to a variety organelles in secretory and endocytic pathways. These tail-anchored (TA) proteins are recently found to be post-translationally inserted into the endoplasmic reticulum by molecular mechanisms. The TA proteins (GET) pathway is found to be conserved from yeast to young man. This GET pathway seems to be complicated and further studies are in need in order to understand more about it. Up to now, the GET pathway is thought to have relation to membrane protein biogeneses.[1]
Membrane protein targeting to the endoplasmic reticulum








 File:Oxygen binding curve with hemoglobin and myoglobin.jpg
File:Oxygen binding curve with hemoglobin and myoglobin.jpg



























![{\displaystyle [E_{0}]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/09236a165b89deb1ca4a526d27507640018c0bf8)
![{\displaystyle W=[E_{0}]f}](https://wikimedia.org/api/rest_v1/media/math/render/svg/17b53325d83fa86960ff3162f74fac3b2583e348)
![{\displaystyle W\propto [E_{0}]=1-{\frac {1}{e^{-{\frac {\Delta G}{RT}}}+1}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/47f2e9431888de8b193fe2ff474103cfea438bfe)





.jpg)




 ]]
BUB1 and BUBR1
]]
BUB1 and BUBR1















 Diagrams to show the induced fit hypothesis of enzyme action
Diagrams to show the induced fit hypothesis of enzyme action







![{\displaystyle \theta ={[L]^{n} \over (K_{A})^{n}+[L]^{n}}={[L]^{n} \over K_{d}+[L]^{n}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/4e5402eb2ec96629d4231c945af8ced8e4d73f9c)

![{\displaystyle [L]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/c1eba88cad4b17de726ed2c779b29eecdf5819a2)


















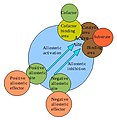 Allosteric regulation of an enzyme
Allosteric regulation of an enzyme












![{\displaystyle \mathrm {[S]+[E]} \rightleftharpoons \mathrm {C} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/c0be4331250336c8a811d6b0af9873da4bcfa245)
![{\displaystyle K_{a}={\frac {[C]}{[S][E]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/f87af491788d8a0a6314c11e4a049df3b82f1934)

![{\displaystyle K_{a}={\frac {[H][A]}{[HA]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/09bea2fe91b0977dbecdb39fab730b6e3de8f1e7)

![{\displaystyle K_{d}={\frac {[S][E]}{[C]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/5df9ea5616b7698325d85de138223acee3758cc3)












 Arp23 side branching model
Arp23 side branching model












.JPG)

































.png)



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
![[1]](https://commons.wikimedia.org/wiki/File:Core_promoter_elements.jpg){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
.jpg){kind=link}
{kind=link}
{kind=link}
{kind=link}









{kind=link}





