Structural Biochemistry/Protein function/Hemoglobin
Hemoglobin
[edit | edit source]
Hemoglobin is contained in red blood cells, which efficiently carries oxygen from the lungs to the tissues of the body. Hemoglobin also helps in the transportation of carbon dioxide and hydrogen ions back to the lungs.
Hemoglobin or Haemoglobin is able to bind to gaseous nitric oxide (NO) as well as O2. As red blood cells passes through the capillary beds of the lungs, gills (in fish), or other respiratory organs, oxygen is diffused into the erythrocytes and hemoglobin binds O2 and NO. Hemoglobin then unloads its cargo in the capillaries. There O2 is able to diffuse into the body cells. The NO relaxes the walls of the capillaries, allowing them to expand which in effects helps the delivery of O2 to the cells.
Hemoglobin consists of four subunits, each with a cofactor called a heme group that has an iron atom center. The iron is the main component that actually binds to oxygen, thus each hemoglobin molecule is able to carry four molecules of O2. Cooperation among the four subunits of the hemoglobin molecule is necessary for the efficient transportation of O2. The four subunits of hemoglobin actually bind to oxygen cooperatively, the binding of oxygen to one site of the four subunits will increase the likelihood of the remaining sites to bind with oxygen as well.
Hemoglobin is a protein that is used to carry oxygen through the bloodstream from the lungs to the tissues. This is important for survival. Hemoglobin has a lower affinity for oxygen the lower the concentration of oxygen gets. This has great implications for the human body and has helped us adapt very effectively. The lower affinity and lower concentrations means that when we are working out, our bodies are low on oxygen which means hemoglobin has less affinity for oxygen and can more easily drop the oxygen off at human tissues. This gives us greater oxygen in our oxygen dependent state. On the other hand, when oxygen concentration is high, the hemoglobin has a higher affinity for oxygen and therefore does not drop the oxygen where it is not needed. This is a very complex and smart system that has evolved to keep hemoglobin as an important biological molecule for a very long time. On the otherhand, the cousin of hemoglobin, myoglobin is used to store oxygen in muscles. This myoglobin has a slightly higher affinity for oxygen than hemoglobin especially at lower levels. This is because myoglobin has an easier job in that it only needs to store oxygen and release it for the muscles, while hemoglobin also has to transport the oxygen and release it in the correct areas.
Hemoglobin is coded for by DNA just like all the other proteins. Alterations or mutations to hemoglobin causes many blood related diseases such as sickle-cell anemia, where the cell structure is distorted and can no longer carry as much oxygen in the correct way as a normal blood cell. This highlights the underlying ideal in structural biochemistry in that structure determines function. The sickle cell anemia case is extremely interesting because it shows us how and why diseases develop. The gene for sickle cell anemia also provides protection against malaria. Therefore, in countries where malaria presented problems, there was an higher than average amount of individuals carrying the sickle cell anemia gene. The heterozygous state is best because it does not allow sickle cell anemia to develop while still preventing malaria. Whereas, the homozygous states would produce individuals either struck with sickle cell anemia or malaria. This is why in malaria ridden areas, there is a higher than average number of people who are heterozygous for sickle cell anemia which is also why this disease does not die out! The carrier state is actually selected by nature. [1]
Conformational Change
[edit | edit source]Upon O2 binding to an active site of hemoglobin there is a conformational change that results, which helps hemoglobin cooperate. Cooperation refers to the interactions among active sites, in the case of hemoglobin, cooperation allows the binding of oxygen to be increased as one site is filled, the remaining active sites will be more likely to bind to O2 as well.

Once O2 is bound to an active site on the hemoglobin molecule, the iron atom Fe2+) is oxidized to (Fe3+). The interaction that results between iron and oxygen in hemoglobin is a combination of resonance structures, one with (Fe2+)and O2 and another between (Fe3+) and super ion O2.-
The binding of O2 to the iron center results in a conformational change in the histidine residue toward the porphyrin in the structure of the hemoglobin molecule which ultimately results in an increase O2 affinity of hemoglobin. The associated movement of the histidine-containing group will result in a conformational change to the rest of the hemoglobin structure. The COO- group is now interacting with the alpha-beta interface which causes conformational changes of neighboring active sites. These conformational changes will result in an increase of O2 affinity to hemoglobin.
Allostery
[edit | edit source]Hemoglobin is an allosteric protein. It's ability to bind to O2 to one of the subunits is affected by its interactions with the other subunits. As mentioned above, the binding of O2 to one hemoglobin subunit induces conformational changes that are relayed to the other subunits, making them more able to bind to O2 by raising their affinity for this molecule. H+, CO2 and 2,3-bisphosphoglycerate are all allosteric effectors as they favor the conformation of deoxyhemoglobin and therefore promote the release of O2. Because these three molecules act at different sites, their effects are additive.
-
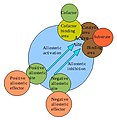 Allosteric regulation of an enzyme
Allosteric regulation of an enzyme

{kind=link}
Mechanism of the allosteric change
[edit | edit source]X-ray crystallography revealed that oxyhemoglobin, the form that has four O2 molecules bound, differs markedly in its quaternary structure from deoxyhemoglobin, the form with no O2 bound. In the absence of bound O2, the Fe2+ lies slightly to one side of the porphyrin ring, which itself is slightly curved. As a molecule of O2 binds to the heme prosthetic group it pulls the Fe2+ into the plane of the porphyrin ring, flattening out the ring in the process. Movement of the Fe2+ causes the proximal histidine to move also. This, in turn, shifts the position of helix F and regions of the polypeptide chain at either end of the helix. Thus, movement in the center of the subunit is transmitted to the surfaces, where it causes the ionic interactions holding the four subunits together to be broken and to reform in a different position, thereby altering the quaternary structure,leading to the cooperative binding of O2 to Hb.
Regulation by pH (Bohr effect)
[edit | edit source]The regulation of oxygen binding of hydrogen ions and carbon dioxide is called the Bohr Effect, which was proposed by Christian Bohr, in 1904. The Bohr Effect describes the effect of pH on the oxygen affinity of hemoglobin, the oxygen affinity of hemoglobin decreases as pH decreases from a value of 7.4. As hemoglobin moves into a region of lower pH, its tendency to release oxygen will increase, therefore more oxygen will be released as the environment becomes more acidic.

There is a chemical basis that is responsible for the pH effect. The histidine residue of hemoglobin molecule structure is one factor of the pH effect. At high pH, the side chain of histidine is not protonated and the salt bridge between histidine's terminal carboxylate group and a lysine residue, does not form. However as the pH drops, meaning at low pH levels, the side chain of histidine will become protonated and thus form a salt bridge with Aspartate instead. This electrostatic interaction results in a structural change. The formation of salt bridges stabilizes the hemoglobin structure resulting in a lower O2 affinity of hemoglobin and thus increase the tendency for oxygen to be released.
Regulation by 2,3-bisphosphoglycerate (2,3-BPG)
[edit | edit source]
The effect of 2,3-bisphosphoglycerate (2,3-BPG) in hemoglobin is described as an allosteric effect. 2,3-BPG is an allosteric effector, it binds to a site that is completely remote from the active site for oxygen. The amount of 2,3-BPG in red cells is crucial in determining the oxygen affinity of hemoglobin.
A single 2,3-BPG molecule is bound in the center of the tetramer of a deoxyhemoglobin structure in a central cavity in the T form. Upon the transition of T state to R state, 2,3-BPG is released. Therefore in order for the transition from T to R states to occur, the bonds between hemoglobin and 2,3-BPG needs to be broken. In the presence of 2,3-BPG, oxygen is less tightly bound to hemoglobin. The conformational changes allow a structural stabilization to occur and thus hemoglobin loses oxygen affinity.
Regulation by CO2
[edit | edit source]Carbon dioxide is able to stimulate oxygen release by two mechanisms:
- The presence of carbon dioxide (CO2) in high concentration will DECREASE the affinity of hemoglobin due to a drop in pH with the red blood cell
There are effects of CO2 in hemoglobin through catalysis. A reaction between CO2 and water forms carbonic acid. However, this reaction requires for carbon dioxide to be catalyzed by carbonic anhydrase, an enzyme in red blood cells, which ultimately results in H+ and HCO3-. Once carbonic acid dissociates into these two ions, pH will drop. The drop in pH stabilizes the T state and thus increases the tendency for oxygen release.
CO2 ↔ CO2 + H2O ↔ H2CO3 ↔ HCO3- + H+
- A direct chemical interaction between (CO2) and hemoglobin will stimulate the release of O2. (CO2) is able to stable deoxyhemoglobin by reacting with terminal amino groups to form negatively charged carbamate groups. This interaction results in a salt-bridge that stables the T state, which favors the release of O2.

It also explains the transport of carbon dioxides from tissue to lung. CO2 that produced by tissue cells pass through the red blood cell and form H+ and HCO3- as previously mentioned. It allows the exchange of HCO3- for Cl-. Therefore, the concentration of HCO3- increases in the blood capillary and carbon dioxides are carried to lung in this form. When HCO3- reaches lung, the reverse reaction take place and release carbon dioxides in lung.
Competitive Inhibitory Ligands
[edit | edit source]Several molecules are responsible for substantially lowering hemoglobin's ability to transport oxygen to tissues. The most common is carbon monoxide (CO), which has a binding affinity to hemoglobin 200 times greater than oxygen. Once carbon monoxide binds to the heme group, oxygen affinity is increased, since hemoglobin is a tetrameter that facilitates cooperative ligand binding. However, this prevents oxygen from being released into oxygen-requiring tissue. The CO and hemoglobin complex is known as carboxyhemoglobin. This is known as carbon monoxide poisoning, where CO competitively binds to oxygen and prevents oxygen transport. As such, as small concentration of CO can cause serious harm to an individual. As little as 0.02% of CO concentration can cause headaches, and 0.1% will lead to unconsciousness. Other competitive ligands include cyanide, sulfur monoxide, nitrogen dioxide, and sulfide.
Differences in Embryonic, Fetal and Adult Hemoglobin
[edit | edit source]Embryonic and fetal hemoglobin differ at the subunit level to that of adult hemoglobin by the subunit interface strengths. Subunit interface in embryonic hemoglobin are much weaker than subunits in fetal hemoglobin which are much weaker than in adult subunit interface. In human red blood cells, hemoglobin can have eight different combinations of dimer formations. Each formation can be present in greater amounts than others or can be present only at distinct times during development. In embryonic development there are three different kinds: ζ_2 γ_2 (Hb Portland-1), ζ_2 ε_2 (Hb Gower-1) and α_2 ε_2 (Hb Gowler-2). Fetal hemoglobin consists of α_2 γ_2(HbF) and adult hemoglobin consists of α_2 β_2 (HbA) as well as trace amounts of α_2 δ_2(HbA2). Although the tertiary structure of all these various hemoglobin is almost identical, their primary structure varies in specific substitutions that accounts for their differing O_2affinity as well as their interactions with allosteric effectors. These amino acid substitutions have an effect on how the subunits fit together and how their interactions take place. Embryonic ζ_2 γ_2 (Hb Portland-1), ζ_2 ε_2 (Hb Gower-1) and α_2 ε_2 (Hb Gowler-2) is found during the first few months of life as well as a fourth, ζ_2 β_2 (Hb Portland-2). ζ_2 β_2 (Hb Portland-2) is a rarely occurring form of hemoglobin. The strengths between the interfaces of the monomer units in each kind of hemoglobin differ significantly and to a greater extent in the deoxy state and become even stronger in the liganded state. The tertramer[check spelling]-dimer dissociation constants differed depending on what subunits they contained. For example, between the two similar embryonic subunits ζ_2 ε_2 (Hb Gower-1) and α_2 ε_2 (Hb Gowler-2) there was a difference in dissociation constants of 13-fold from α to ζ subunits. Therefore by comparing the different types of hemoglobin with one that has one similar subunit, their dissociation constants can give a wide range on information. Little is known about the Hb Portland-2 because it s rare and only occurs in a type of α-thalassemia (genetic defect). Hb Portland-2 differs from other hemoglobins because it dissociates from tetramer to dimers and even more readily from dimers to monomers whereas other types of human hemoglobin will dissociate from from tetramers to dimers rapidly but will not dissociate from dimers to monomers. The dissociation talked about happens at pH 7.5 but will differ significantly once the pH is lowered. In HbA (adult hemoglobin), the dissociation of tetramer to dimer increases with the decrease in pH, for every 1pH unit decrease, there is a 10 fold increase in dissociation. When the pH is changed to 6.3, the dissociation in HbA will be primarily dimers whereas in Hb Portland-2 the dissociation is mainly monomers. With this being true, the formation of tetramers in HbA is favored greater than the formation of tetramers in Hb Portland-2 because ζ_2 β_2 dimer is much weaker than α_2 β_2 dimer. In HbA, the α and β subunits are unstable but in Hb Portland-2 ζ and β subunits are weak interfaces as dimers but are stable as monomers. Embryonic hemoglobins at pH 6.3 are able to dissociate readily but Hb Portland-1 and Gowler-1 dissociate faster than Hb Gowler-2. When monomers of embryonic hemoglobin Hb Gowler-2 and Portland-2 were mixed and allowed to recombine, the stronger tetramer formation was the result. Instead of having α_2 ε_2 (Hb Gowler-2) and ζ_2 β_2 (Hb Portland-2) reform, the formation of α_2 β_2 (HbA) was obtained as well as ζ_2 ε_2 because the αβ interface is the strongest and therefore most favored. And when the ζ_2 γ_2 (Hb Portland-1), α_2 ε_2 (Hb Gower-2) were put through the same process, the interface of Fetal Hemoglobin α_2 γ_2(HbF) was the result because it is by far stronger than embryonic hemoglobins. When the ζ_2 γ_2 (Hb Portland-1), α_2 ε_2 (Hb Gower-2) and ζ_2 β_2 (Hb Portland-2) were mixed, the favored outcome was HbA and HbF was not detectable. All three experiments prove that subunit competition contributes to the rearranging and the notably higher α_2 β_2 formation than any other tetramer due to the stronger interface. The rarity of Hb Portland-2 may be due to the fact that the tetramer-dimer and dimer-monomer interfaces are relatively weaker than any other human hemoglobins. Subunit competition has a lot to do with why some hemoglobins are more likely to form because the formation of hemoglobins with stronger interfaces is favored over the formation of weaker ones. As well as there being formation of stronger ones from the weaker ones.
Roles in disease
[edit | edit source]Hemoglobin is formed by genes that are in charge of the expression of the hemoglobin protein. Failings in these genes can form irregular hemoglobin and anemia, which are conditions termed "hemoglobin disorder". Irregular hemoglobin appears in these three conditions.
1. Structural failure in the hemoglobin molecule. Changes in the gene for one of the two hemoglobin subunit chains, alpha (α) or beta (β), are called mutations. Often, mutations change a single amino acid building block in the subunit. Most commonly the change is innocuous, perturbing neither the structure nor function of the hemoglobin molecule. Occasionally, alteration of a single amino acid dramatically disturbs the behavior of the hemoglobin molecule and produces a disease state. Sickle hemoglobin represents this phenomenon.
2. Reduced production of one of the two sub-units of the hemoglobin molecule. Mutations that form this condition are termed "thalassemias." Equal numbers of hemoglobin alpha and beta chains are essential for normal function. Hemoglobin chain inequity damages and destroys red cells thereby producing anemia. Although there is a death of the affected hemoglobin subunit, with most thalassemias the few subunits created are structurally normal.
3. Irregular relations of otherwise normal sub-units. A single sub-unit of the alpha chain and a single subunit from the β-globin locus combine to create a normal hemoglobin dimer. With severe α-thalassemia, the β-globin subunits start to associate into groups of tetramers due to the scarcity of potential α-chain partners. These tetramers of β-globin subunits are functionally inactive and do not carry oxygen. No similar tetramers of alpha globin subunits form with severe beta-thalassemia. Alpha subunits are quickly destroyed in the absence of a partner from the beta-globin gene cluster.
References
[edit | edit source]Berg, Biochemistry (6th Ed) and Campbell Biology (5th Ed)
Johnson RA, Lavesa M, Askari B, Abraham NG, Nasjletti A (February 1995). "A heme oxygenase product, presumably carbon monoxide, mediates a vasodepressor function in rats"
http://sickle.bwh.harvard.edu/hemoglobinopathy.html
http://www.rockefeller.edu/labheads/chait/pdf/07/07_manning_prot-sci.pdf
- ↑ [ haemoglobin and myoglobin], November 14th, 2012.