Proteins are polymers of multiple monomer units called amino acid, which have many different functional groups. More than 500 amino acids exist in nature, but the proteins in all species, from bacteria to humans, consist mainly of only 20 called the essential amino acids. The 20 major amino acids, along with hundreds of other minor amino acids, sustain our lives. Proteins can have interactions with other proteins and biomolecules to form more complex structures and have either rigid or flexible structures for different functions. Iodinated and brominated tyrosine are also amino acids found in species, but are not included in the 20 major amino acids because of their rarity: iodinated tyrosin is only found in thyroid hormones, and brominated tyrosine is only found in coral. The 20 main amino acids that are found in most but not all proteins are listed below:
Amino acids are molecules which contain both a carboxylic acid and an amine group. In amino acid, the carboxyl group is more acidic than the carboxylic acid. 2-amino acids, also known as alpha-amino acids, are a specific type of amino acid that makes up proteins. These amino acids have many interesting properties which will be discussed in the next sections.
Amino acids play central roles both as building blocks of proteins and as intermediates in metabolism. Proteins are linear polymers formed by linking the a-carboxyl group of one amino acid to the a-amino group of another amino acid. This type of linkage is called a peptide bond or an amide bond. The formation of a dipeptide from two amino acids is accompanied by the loss of a water molecule. The equilibrium of this reaction lies on the side of hydrolysis rather than synthesis under most conditions. Hence, the biosynthesis of peptide bonds requires an input of free energy. Nonetheless, peptide bonds are quite stable kinetically because the rate of hydrolysis is extremely slow; the lifetime of a peptide bond in aqueous solution in the absence of a catalyst approaches 1000 years.
Thus, the 20 amino acids that are found within proteins convey a vast array of chemical versatility. The precise amino acid content, and the sequence of those amino acids, of a specific protein, is determined by the sequence of the bases in the gene that encodes that protein. The chemical properties of the amino acids of proteins determine the biological activity of the protein. Proteins not only catalyze all (or most) of the reactions in living cells, they control virtually all cellular process. In addition, proteins contain within their amino acid sequences the necessary information to determine how that protein will fold into a three dimensional structure, and the stability of the resulting structure. The field of protein folding and stability has been a critically important area of research for years, and remains today one of the great unsolved mysteries. It is, however, being actively investigated, and progress is being made every day.
There are twenty major amino acids which make up proteins. Each of them contains a unique functional group which gives rise to different properties. These properties include size, shape, charge, capacity for hydrogen bonding, hydrophillicity/hydrophobicity(hydrophobic interactions), and chemical reactivity. Amino acids can be broadly hydrophobic and hydrophilic, depending on the chemical properties of the R group side chain. In an aqueous environment, the hydrophobic amino acids are unable to participate in hydrogen bonding. They associate with one another and reside mostly inside the protein. On the other hand, hydrophilic amino acids tend to interact in the aqueous environment due to polarity. These amino acids are normally found on the exterior surface.
An amino acid is in a zwitterionic state when the carboxylic acid group is deprotonated and the amino group is protonated, simultaneously. Zwitterions are dipole ions—meaning that these molecules have two charges, both a positive and a negative charge. The pH of the water solution is a factor determining the state of protonation. Such a state leaves the carboxylic end negatively charged (-COO-) and the adjacent amino end positively charged (-NH3+). The carboxyl group (-COO-) is deprotonated first because the pKa is about 2 and the pKa of the amine group (-NH3+) is about 9. The net charge for the protein in zwitterionic form is zero.
[1]
Molecules which behave in this fashion are called amphoteric. In solid state, the amine functionality deprotonates the carboxylic acid group, giving rise to the zwitterionic, dipolar entity. The charged state of an amino acid in aqueous solution depends largely on the pH. The major form of all amino acids at a pH of 2 to 9 is the zwitterionic form. In strong acid (pH < 2), the predominant form is the fully protonated cationic ammonium with the corresponding protonated form of the carboxylic acid. This species has a net charge of +1. In strongly basic solutions (pH > 9), the predominant form is the fully deprotonated aminocarboxylate anion. This species would have a net charge of -1. These forms interconvert by acid-base equilibria. This leaves a wide pH range wherein the zwitterion would play a large role as a contributing species. The pH at which the extent of protonation equals that of deprotonation is called the isoelectric pH or the isoelectric point (pI). At this pH, the amount of positive charge balances that of negative charge and the concentration of the charge-neutralized zwitterionic form is at its highest. When the side chain of the acid bears an additional acidic or basic function, the pH is either decreased or increased, respectively. Note that at most relevant physiological pH ranges, the zwitterion would be, by far, the species of the most abundance.
Histidine contains an imidazole ring with 2 nitrogen atoms: one is basic and the other is not. The basic nitrogen is involved in the delocalization which is important during enzyme catalysis.
Here is an example of L-amino acids forming zwitterion at neutral pH:
All proteins or polypeptides are a series of linked amino acids. A typical α amino acid consists of a central carbon (which is the alpha carbon in this case) that is attached to an amino group (-NH2), a carboxylic acid (-COOH), a hydrogen atom, and a distinctive R group. The R group, usually referred to as a side chain, determines the properties of each amino acid. Scientists classify amino acids into different categories based on the nature of the side chain. A tetrahedral carbon atom with four distinct groups is called chiral. The ability of a molecule to rotate plane polarized light to the left, L (levorotary) or right, D (dextrorotary) gives it its optical and stereochemical fingerprint. All amino acids within polypeptides are configured in the L form. The L form corresponds to the absolute configuration of S, which is a system used to designate stereochemistry in the field of organic chemistry. Although D-amino acids (designated as R stereoisomers in the field of organic chemistry) exist naturally, they are not found in proteins. Thus far, scientists have not been able to come up with a hypothesis on the preference for the L amino acids in living organisms. It is clear, however, that all of the physiological mechanics downstream of the amino acids are geared towards recognizing and interacting with the specific L conformation. Note: Since the central carbon has four distinct groups attached, all of amino acids are chiral except for glycine, which is achiral. This is due to the fact that the central carbon atom in glycine contains only 3 unique substituents instead of 4 (R sidechain = H).
Within proteins, it is possible to find amino acids which do not correspond to the 20 standard types. Most of these come about by chemical modification of an already incorporated amino acid. For example, a hydroxylated form of proline exists within collagen protein. Also, a selenium analog of cysteine is known to occur in glutathione peroxidase enzymes. Pyrrolysines have also been isolated and characterized. These exceptions to the rule are dictated by and encoded within DNA and RNA and there are many more examples.
Any discussion of amino acids is not complete without mentioning how each amino acid bonds to another. All amino acids bond to one another through a condensation reaction involving the amine group of one amino acid and the carboxylic acid group of another. The enzymatically-catalyzed reaction forms an amide entity: [R1-NH2 + R2-COOH ==> R1-NH-C(=O)-R2 + H2O]. The amide bond has special properties in that it has a resonance form which gives the bond a planar, rigid, double bond character: [R1-N-C(=O)-R2 <==> R1-N+=C(-O-)-R2]. Amino acids can link to each other in small units of only 2 or 3 amino acids called dipeptides and tripeptides, but can also connect in very large chains consisting of hundreds or even thousands of amino acids. Each complete peptide series has an N terminus (amino) and a C terminus (carboxylate). The overall, 4 atom angles involved in the peptide bond system are important to those who study proteins. In particular, the R-[N-C-C(=O)-N]-R group is called a phi torsion angle and the adjacent angle, the psi, φ, torsion angle, involves the R-[C-N-C-C(=O)]-R group. These angles are important to consider and the natural distribution of know peptide angles are summarized on the Ramachandran plot. Peptide bond is formed by condensation reaction and broken by hydrolysis (addition of water).
The network approach helps determine the role of a specific amino acid at a known position in the protein structure. Networks simplify complex system behaviors by splitting the system into a series of links. Links represent the neighboring positions of amino acids in protein molecules. Because proteins are linked in this way and protein structure networks are connected to each other by only a few other amino acid elements, we can determine folding probability. Proteins with denser protein structure networks fold more easily and the folding probability increases as the protein structure becomes more compact.
The network approach can also be applied to the prediction of active centres in proteins. Active centres are protein segments that play key parts in the catalytic reaction of the enzyme function shown by their respective proteins. Scientists have used long-range network topology to create a network skeleton from which they can study only side chains which are essential in the flow of information for the whole protein. Network analysis has showed that active centres occupy a central position in protein structure networks, usually have many neighbors, give unique linkages in their neighborhood, integrate communication for the entire network, do not take part in wasteful actions of ordinary residues, and collect and coordinate most of the energy in the network.
Structure
Alanine, also known as 2-Aminopropanoic Acid, (abbreviated as Ala or A) is an α-amino acid with the chemical formula HOOCCH(NH2)CH3. It has a molar mass of 89.09 g/mol and a density of 1.424 g/cm3. The α-carbon atom of alanine is bound with a methyl group (-CH3), making it one of the simplest α-amino acids with respect to molecular structure and also resulting in alanine being classified as an aliphatic and amino acid. The methyl group of alanine is non-reactive and is thus almost never directly involved in protein function. Alanine is a nonpolar hydrophobic molecule. It is ambivalent, meaning it can be inside or outside of the protein molecule. The α-carbon of alanine is optically active; in proteins, only the L-isomer is found.
Features
Alanine is a non-essential amino acid which means that it can be manufactured by the human body and does not need to be obtained directly through the diet. Alanine is found in a wide variety of foods, but is particularly concentrated in meats. It is a non-essential amino acid that occurs in high levels in its free state in plasma.
Functions
Alanine is the primary amino acids for sugar and acid metabolism. It boosts up the immune system by producing antibodies, and provide energy for muscles tissues, brain, and the central nervous system. It is used in pharmaceutical preparations for injection or infusion. It is also used in dietary supplement and flavor compounds in maillard reaction products. In addition, it is a stimulant of glucagon secretion.
Chemical Synthesis
Alanine can be manufactured in the body from pyruvate and branched chain amino acids such as valine, leucine, and isoleucine. Alanine is most commonly produced by reductive amination of pyruvate. Because transamination reactions are readily reversible and pyruvate pervasive, alanine can be easily formed and thus has close links to metabolic pathways such as glycolysis, gluconeogenesis, and the citric acid cycle. It also arises together with lactate and generates glucose from protein via the alanine cycle. Racemic alanine can be prepared via the condensation of acetaldehyde with ammonium chloride in the presence of potassium cyanide by the Strecker reaction.
Analysis
Alanine can be identified via UV spectrometry, infrared spectroscopy (IR), nuclear magnetic spectroscopy, (NMR), and mass spectroscopy.
Structure
Arginine, 2-Amino-3-carbamoylpropanoic acid, contained of a three-carbon aliphatic straight chain with the end of which is capped by a guanidinium group. Its molar mass is 132.12g/mol. With a pKa of 12.48, the guanidinium group is positively charged in neutral, acidic and even most basic environments. Therefore, arginine has basic chemical properties. Because of the conjugation between the double bond and the nitrogen lone pairs, the positive charge is delocalized and enables the formation of multiple H-bonds.
Features
Arginine is an essential amino acid that plays important role in nitrogen metabolism. It is a chemical precursor to nitric oxide (a blood vessel-widening agent called a vasodilator. Nitric oxide is a powerful neurotransmitter that helps blood vessels relax and also improves circulation. Food that are rich in arginine include red meat, fish, poultry, wheat germ, grains, nuts and seeds, and dairy products.
Functions
Arginine assists in wound healing and help in burn treatment. It is necessary in normal immune system activity by enhancing the production of T-cells. Studied show that arginine may help treat medical conditions that improve with increased vasolidation. Some conditions that are treated with arginine are chest pain, atherosclerosis (clogged arteries), heart disease or failure, erectile dysfunction, intermittent claudication/peripheral vascular disease, and vascular headaches (headache-inducing blood vessel swelling). Arginine also helps with bodybuilding, enhancing sperm production, and preventing tissue wasting in people with critical illnesses. Arginine hydrochloride has high chloride content and has been used to treat metabolic alkalosis.
Biosynthesis
Arginine is synthesized from citrulline with the presence of cytosolic enzymes argininosuccinate synthetase and argininosuccinatelyase. This is energetically costly reaction. Therefore, the synthesis of each molecule of argininosuccinate will be coupling with hydrolysis of adenosine triphosphate (ATP) to adenosine monophosphate (AMP).
Synthesis of arginine in human body occurs principally via the intestinal–renal axis, wherein epithelial cells of the small intestine, which produce citrulline primarily from glutamine and glutamate, then join with the proximal tubule cells of the kidney, which extract citrulline from the circulation and convert it to arginine, which comes back to the circulation.
Arginine and Nitrogen Storage
In order for a cell to grow, it needs nitrogen which can come from ammonia, nitrates, dinitrogen or amino acids. The PII protein is an ancient signaling protein that senses and integrates nitrogen and carbon abundance by binding 2 OG and ATP/ADP. The N-acetyl-L-Glutamate kinase (NAGK) stores nitrogen as arginine which it incorporates into arginine rich copolymers. Since arginine is nitrogen-rich, it is an ideal for nitrogen storage. The osmotic impact of arginine minimizes when arginine is incorporated into proteins. The PII protein binds to NAGK when nitrogen is abundant only in oxygenic phototrophs. But when nitrogen is scarce, 2-oxoglutarate binds to the PII protein with ATP leading to the dissociation of the PII-NAGK complex.
Arginine-insensitive NAGK is a homodimer containing a backbone of 16-stranded Beta sheets in both subunits. However, arginine-sensitive are hexameric and recent studies have shown that these enzymes are ring-like hexameric trimers of dimers. The ring is formed by the link between three E. Coli NAGK-like dimers and the N-terminal alpha-helix. In arginine-sensitive NAGK, the arginine is connected by interlaced N-helices. The helices are needed for making NAGK an arginine-operated switch showing a sigmoidal of the arginine inhibition kinetics. The PII protein is homotrimers having a βαββαβ subunit topology with the alpha helices looking outward and the beta sheet inward. The T-loop is large and flexible loop that contain the phosphorylation and uridylylation sites in cyanobacteria and proteobacteria.
When the protein PII is absent, S. elongates NAGK is inactive having low Vmax and high Km for NAG and requiring a low concentration of argigine for inhibition. However, the enzyme A. thaliana NAGK is highly active having a Km four times lower and a Vmax three times greater for NAG than S. elongates NAGK. When PII binds the S. Elongates NAGK, the Vmax for NAG increases up to four times the original amount and decreases up to ten times the original amount for Km. Km is not affected when it binds to A. thaliana NAGK, but the Vmax for NAG increases by five times the original amount. The original amount is the amount with the protein PII absent.
The S. elongates PII-NAGK complex has one NAGK hexamer that is sandwiched between two PII trimers. Since the PII proteins are not packed tightly on NAGK, PII only interacts with NAGK on the T-loops and B-loops. The A. thaliana PII-NAGK complex has MgATP bounded to the PII protein with all the NAGK active centers containing bound NAG and ADP.
Structure
Asparagine is polar and uncharged derivative of acidic amino acid aspartic acid or aspartate; as a side chain, it has a carboxamide group, which is neutral at physiological pH and can be changed to carboxylic acid by hydrolysis to form aspartate amino acid. The carboxamide group of the amino acid can form hydrogen bonds.
Features
Asparagine is found in abundance in asparagus, and is thus named so. Asparagine is not an essential amino acid, meaning that it is not necessary for humans to ingest it to receive necessary amounts. Asparagine has a high propensity to hydrogen bond, since the amide group can accept two and donate two hydrogen bonds. It is found on the surface as well as buried within proteins. It is a common site for attachment of carbohydrates in glycoproteins. Food sources that contain asparagine is dairy, beef, poultry, and eggs.
Functions
Asparagine, along with glutamate, is an important neurotransmitter. Since Aspartic acid and Asparigine have high concentration in the hippocampus and hypothalamus of the brain, which is important in short-term memory and emotions, the two amino acids serves essential role between the brain and the rest of the body. Asparagine is required by the nervous system to maintain equilibrium and is also required for amino acid transformation from one form to the other which is achieved in the liver.
Synthesis
Synthesis of asparagine requires oxaloacetate, C4H4O5. The double bonded oxygen attached to carbon-2 is replaced by ammonium group from glutamate via a process called transaminase. The newly formed compound, or aspartate, is converted to asparagine by replacing a negatively charged oxygen end with an ammonium group. The asparagine synthesis converts glutamine to glutamate, and ATP into AMP and pyrophosphate.
Analysis
Asparagine can be identified by following methods: UV spectrometry, infrared spectroscopy (IR), nuclear magnetic spectroscopy, (NMR), and mass spectroscopy.
Structure
Aspartic acid (C4H7NO4) is also named as a 2-aminobutanedioic acid. Its molecular weight is 133.1 g/mol.
Also known as aspartate, Aspartic acid is an acidic and polar amino acid that has carboxylic acid group, which loses a proton to be carboxylate group for physiological pH and has a negative charge; the carboxylic acid group of the amino acid has a pKa value of 4.1, which is a little basic than the terminal α-carboxyl group. Its pI is 5.41. Proteins are critical to maintain the pH balance in the body. It is the charged amino acids that are involved in the buffering properties of proteins. Aspartic acid is similar to alanine but with one of the β hydrogens replaced with a carboxylic acid group. This carboxylic acid group is what makes aspartate an acidic amino acid. Aspartate has an α-keto homolog, called oxaloacetate. Aspartate and oxaloacetate are interconvertable by a simple transamination reaction. Oxaloacetate is one of the intermediates of the Krebs cycle. The Krebs cycle is the sequence of reactions by which most living cells generate energy during the process of aerobic respiration.
Features
Aspartic acid is a non-essential amino acid can be obtained from central metabolic systems.
Functions
Aspartic acids are involved in transamination in which oxaloacetate and aspartate is interconvertible. It is also involved in immune system activity by promoting immunoglobulin production and antibody production. Moreover, aspartic acid protects the liver and helps in detoxification of ammonia.
Aspartate, the conjugate base of aspartic acid, also functions as a neurotransmitter. Along with few other amino acids, its primary role is to activate NMDA receptors in brain and; however, its effect is not significant as glutamate's.
Other than its role as an excitatory neurotransmitter, aspartate is proteinogenic amino acids that are used in coding of DNA.
Aspartate plays important roles as acids in enzyme active centers, as well as in maintaining the solubility and ionic character of proteins.
Synthesis
Aspartic acid is synthesized from oxaloacetate via transamination. Aspartic acid can be used as an initial reactant in synthesis of other essential amino acids as well: methionine, threonine, isoleucine, and lysine. Aspartic acid needs to be reduced to its semialdehyde form of HOOCCH(NH2)CH2CHO. Asparagine can be also obtained from aspartic acid via transamidation: aspartic acid + glutamine -> asparagine + glutamic acid
Structure
Cysteine, C3H7NO2S with molecular mass of 121.16 g/mol, is an amino acid that is made of the sulfhydryl or thiol group (-SH), which is more nucleophilic than a hydroxyl group. Its alternate name is 2-amino-3-mercaptopropanoic acid. Two cysteine residues can be oxidized to form stable disulfide bonds. Disulfide bonds can help to give a protein secondary and tertiary structure, e.g. protein folding. The unit of two bonded cysteines is known as cystine. Cysteine is considered to be a hydrophilic amino acid based on the fact that the thiol group interacts well with water. It is also a non-essential amino acid, and can be biosynthesized in human bodies.
Functions
Nucleophilic thiol groups in cysteine can be easily oxidized; thus, cystein is highly reactive with its neutral pKa and has various functions in biology.
Cysteine is capable of inactivation of insulin in bloodstream. Excessive amount of cysteine reduces one of three disulfide bonds in insulin structure. As a result, insulin loses its functionality. Cysteine's capability of inactivation of insulin can be utilized in medicine and pharmaceutic when a patient experiences hypoglycemia attack due to high level of insulin.
Cysteine promotes iron production in iron deficiency anemia. It also assists in lung diseases by increasing production of red blood cells. Cysteine is a key, active site residue in many important proteins. Cysteine is the key residue in glutathione reductases which has protective effects against UV light, radiation, and free radicals. Additionally, glyceraldehyde-3-phosphate dehydrogenase, a key enzyme in glycolysis, uses cysteine in to achieve its most critical functions.
When cysteine is taken as a supplement, it is in the form of N-acetyl-L-cysteine (NAC). The body makes this into cysteine and then into glutathione, a powerful antioxidant. Antioxidants fight free radicals which are harmful compounds in the body that cause damage to the cell membranes and DNA. Researchers believe the free radicals play a role in aging as well as the development of a number of health problems, including heart disease and cancer. NAC can also help prevent side effected caused by drug reactions and toxic chemicals. It also helps break down mucus in the body. NAC also benefits in treating some respiratory conditions, such as bronchitis and COPD. COPD is the acronym for chronic obstructive pulmonary disease.
Doctors often give NAC to people who have taken an overdose of acetaminophen (Tylenol). The NAC helps to prevent or reduce liver and kidney damage. NAC also helps reduce angina. Angina is chest pain or discomfort when the heart muscle does not get enough blood. Taking NAC will open the blood vessels and improve blood flow to the heart. Studies have also shown that NAC may help relieve symptoms of chronic bronchitis, leading to fewer flare ups. Not all studied gave these results. Some studies did not find any reduction in flare ups. Other studies showed that people with COPD who took NAC lowered the number of flare ups about 40% when used with other therapies. Another study shows that people who took NAC two times a day had fewer flu symptoms than those who took placebo. Some research has shown that intravenous NAC may boost levels of glutathione and help prevent and/or treat lung damage cause by ARDS, acute respiratory distress syndrome. Other results did not coincide with these results. For example, giving NAC to people with ARDS helped reduce the severity of their conditions while not reducing the number of overall deaths compared to placebo. Cysteine is important in keratin structure, which is important in hair and nails formation on skin. Wool obtained from sheeps, and other animals is cysteine containing.
Biosynthesis
The precursors of synthesis of cysteine are serine and methionine. Serine has a hydroxide group and methione has a sulfer as their substituents. Methione is initially converted into a homocysteine. With serine, homocysteine becomes cystathione (C7H14N2O4S) with water molecule leaving. Finally, addition of water and departure of ammonia from cystathione result in cysteine and alpha-ketobutyrate as a side-product.
Glutamine, or 2-amino-4-carbamoylbutanoic acid, has a molecular formula of C5H10N2O3 and a molecular mass of 146.16 g/mol. It is a polar and uncharged derivative of acidic amino acid glutamic acid or glutamate; it has a carboxamide group, which is neutral at physiological pH and can be changed to carboxylic acid by hydrolysis to form glutamate amino acid. The carboxamide group of the amino acid can form hydrogen bonds.
Glutamine Final
Synthesis
As previously stated, glutamine is a nonessential amino acid. In the body, glutamine is synthesized from glutamate via the enzyme glutamine synthestase (GS) and through the addition of ATP and ammonia. (See Figure).
The incorporation of ammonia into glutamate is an amidation type reaction and the hydrolysis of ATP to ADP drives the reaction forward. ATP is directly involved in the reaction because it phosphorylates the carboxyl group on the side chain of glutamate and forms an acyl-phosphate intermediate (See Figure: Glutamine Final). The acyl-phosphate intermediate reacts with free ammonia and forms glutamine. Glutamine synthetase (GS) plays a major role because a high-affinity binding-site for ammonia is formed in GS after the formation of the intermediate to prevent hydrolysis of the intermediate. Hydrolysis of the intermediate would not yield glutamine and thus waste a valuable molecule of ATP.
Functions
Glutamine is a non-essential amino acid, which means that it will naturally occur in the human body and does not need to be gathered from exogenous sources. It is one of the most abundant amino acid manufactures in the body. Glutamine circulates in the blood and is able to cross the blood-brain barrier directly.
Glutamine has various functions in biochemistry. Its primary role is protein synthesis, but it also helps to maintain neutral pH in the liver by balancing the acid and base levels.
Like glucose, glutamine is capable of fueling cell bodies. It donates nitrogen to cells via anabolic reactions and provides carbons in the citric acid cycle. It is critical in the gastrointestinal system in that it provides energy to the small intestine. Notably, intestine is the only organ in the body that uses glutamine as a primary energy source. The kidney, activated immune cells, and cancer cells also require glutamine, but not as a primary energy source.
Within a cell, glutamine is essential for cell growth and protein translation. Moreover, it serves as a nitrogen donor and assists in maintaining the gradient across the mitochondrial membrane.
Normal cells require glutamine. On the other hand, cancer cells use glutamine in quantities much higher than normal cells. As discussed in the paper "Glutamine addiction: a new therapeutic target in cancer" by David R. Wise and Craig B. Thompson, cancer cells will sometimes exhibit what is called “glutamine addiction”. In this addiction, cancer cells will uptake glutamine from the body in much larger amounts than is necessary for cellular function. In fact, cancer cells will intake more glutamine than the cell can metabolize. Depriving cancer cells of this excess glutamine causes them to die. Such deprivation is the key to potential glutamine-based cancer therapy. Glutamine consumption can exceed the consumption of any other amino acid in the cell by tenfold. In cancer cells, a metabolic shift occurs so that glutamine replaces glucose as the major source of carbon for the cell.
The body can make enough glutamine for its regular needs, but extreme stress, such as heavy exercise or an injury), will make the body require more glutamine. Most glutamine is stored in muscles followed by the lungs, where much of the glutamine is made. Usually the body can make enough glutamine so it is not necessary to take supplements of glutamine. Certain medical conditions, including injuries, surgery, infections, and prolonged sites, can lower glutamine levels, however. In these cases, taking a glutamine supplement may be helpful.
Glutamine is important for removing excess ammonia, which is a common waste product in the body. Glutamine also helps your immune system function and is need for normal brain function and digestion. Glutamine is important in wound healing and recovery form an illness. When the body is stressed, it releases hormone cortisol into the bloodstream. This high concentration of cortisol will lower the body’s stores of glutamine. Other studies have shown that adding glutamine to enteral nutrition it will help reduce the rate of death in trauma and critically ill people. Clinical studies have found that glutamine supplements strengthen the immune system and reduce infections. Glutamine supplements also help in the recovery of severe burns. Another importance of glutamine is to protect the lining of the gastrointestinal tract known as the mucosa. People who have inflammatory bowel disease (IBD) may not have enough glutamine in their body. Two clinical trials found that taking glutamine supplements did not improve symptoms of Crohn’s disease. People with HIV or AIDs often experience severe weight loss, thus those people take glutamine supplements along with other nutrients including vitamin C and E, beta-carotene, selenium, and N-acetylcysteine to increase weight gain and help the intestines better absorb nutrients. Athletes who train for endurance events may reduce the amount of glutamine in their bodies, thus making them more prone to catch a code after an athletic event. Studies show that taking glutamine supplements resulted in fewer infections.
Glutamine and Cancer
It has been shown that some cancer cells have an addiction to glutamine in that there is an increased rate of glutamine uptake. The increase in glutamine uptake is due to glutamine playing roles other than providing nitrogen for protein (amino acid) and nucleotide biosynthesis.
The first signs of cancer cells relying on an excess of a given compound to produce energy were discovered by Otto Heinrich Warburg. Warburg noticed that the energy produced in most cancer cells was produced through glycolysis of excess glucose, which is in turn converted into lactic acid during lactic acid fermentation. Such a process is in contrast with energy production in normal cells, in which glycolysis still occurs, but is instead followed by oxidation of pyruvate in mitochondria. As such, Warburg concluded that these cancer cells must have devolved into a more primitive form of metabolism as seen in single-celled eukaryotes. Thus this effect of cancer cells up taking excess glucose for their energy needs has been dubbed the "Warburg Effect". Glutamine was later found to mirror this effect in some tumor cells.
Glutamine has been shown to participate in signaling and uptake of essential amino acids. For instance, it is capable of acting as the substrate of the mitochondria to maintain the integrity of the mitochondrion membrane potential. It also plays integral roles in a variety of anaplerotic reactions.
Glutamine donates nitrogen to cancer cells. Like all cells, cancer cells must synthesize nitrogen compounds to produce nucleotides and other amino acids. Glutamine donates the nitrogen that is necessary for the production of these compounds. Glutamine donates its amide group and is converted into glutamic acid. Glutamatic acid transfers its amine group by transaminases to α-ketoacids which is used to generate the nonessential amino acids. This decomposition provides the nitrogen with several amino acids including alanine, serine, aspartate, and proline. Tyrosine is the only nonessential amino acids not produced from either glucose or glutamine.
Glutamine is Needed for the Uptake of Essential Amino Acids in Certain Cancer Cells and as a Molecular Signal
Glutamine is imported through glutamine solute carrier SLC1A5 and quickly exported through the SLC7A5 amino acid transporter in exchange for extracellular essential amino acids. However, when the glutamine importer is impaired, the uptake of essential amino acids is also impaired. Such impairment suggests that glutamine is necessary for essential amino acid uptake. Without essential amino acids, the rapamycin-sensitive (mTORC1) is not activated. mTORC1 plays an essential role in regulation cell growth and protein translation as well as inhibiting macroautophagy. As such, inactivation of mTORC1 inhibits cellular growth and protein translation. Thus, glutamine acts as a signal to mTORC1 and as a resource of essential amino acids in some cancer cells.
Glutamine Provides Anaplerosis in Cancer Cells
Anaplerosis is a term used to describe the replenishing of the carbon pool in the mitochondrion. Oxaloacetic acid (OAA) is one of the substrates in mitochondria that eventually lead to synthesis of many essential biological macromolecules like cholesterol. In glioblastoma cells, glutamine metabolism provides the bulk of the OAA cellular pool. Thus, the increased rate of glutamine metabolism into OAA confirms glutamine as a primary substrate in cancer cells that provides the mitochondria with precursor macromolecules to carry out its metabolic functions.
c-Myc Regulate Glutamine Metabolism in Cancer Cells
The synthesis of purines and pyrimidine uses glutamine as a source of nitrogen in five enzymatic steps. Three out of the five steps are regulated by c-MYC (Myc), a DNA transcription factor. Oncogenic levels of Myc promote increased glutaminolysis at the transcription level and the metabolism of glutamine into lactic acid. The catabolism of glutamine provides cells with carbons for anaplerosis and NADPH production.
Myc is a transcription factor that codes for a protein that binds to DNA. In a cancerous cell, Myc is amplified. Myc uptakes glutamine and converts it to glutamic acid and lactic acid. Myc over expression leads to increased catabolism of glutamine, which leads to a larger amount of carbon in the cell, which allows the cell to produce more NADPH. This over-expression of Myc triggers the metabolic switch from glucose to glutamine as the source of carbon for the cell.
Glutamine-based cancer therapy
Glutamine addiction in some cancer cells is a target for new cancer therapies. Further research is needed to determine a non-toxic dosage; that is, a dosage that does not inhibit glutamine production indiscriminately and does so only in cancerous cells.
Since cancer cells are dependent on glutamine, starving these cells of glutamine will cause them to die. Thus, glutamine has become a target for new cancer treatments. New treatments have attempted to deny cancer cells their source of glutamine by reducing the amount of glutamine in the body. However, as glutamine is essential for many other processes in the body, such as synaptic communication in the brain, removing glutamine from the body is not a feasible treatment and is very dangerous. Other treatment methods have attempted to reduce the ability of the cell to uptake glutamine by targeting Myc and other proteins that are responsible for transporting glutamine into the cell. Other treatments have attempted to reprogram the mitochondria so that it will no longer depend on gluatmine. Another treatment involves targetting mTOR’s glutamine response. These treatments show more promise and less harm than removing all glutamine from the body.
These therapeutic methods target major glutamine activity in cancer cells:
Glutamine uptake and mTOR activation: L-γ-glutamyl-p-nitroanilide (GPNA) inhibits SLC1A5, a target for Myc. Such inhibition suppresses glutamine uptake in the cell. 2-aminobicyclo-(2,2,1)heptanecarbozylic acid (BCH) also inhibits SLC7A5 and blocks mTOC activation, inducing autophagy.
Glutamine-dependent anaplerosis and activity in mitochondria: Studies suggest that carbons derived from glutamine enter the citric acid cycle via transaminase. Therefore, Amino-oxyacetic acid (AOA), a transaminase inhibitor, shows potential as a promising cancer therapeutic. Additionally, the regeneration of mitochondrial NAD+ may prevent the entry of glutamine through the citric acid cycle. Metaformin, a biguanide class drug, inhibits this mechanism.
Structure
The molecular formula of glutamic acid is C5H9NO4. Its molecular mass is 147.13 g/mol. Also known as glutamate, Gluctamic acid is a polar amino acid that has carboxylic acid group, which loses a proton to become carboxylate group for physiological pH and has a negative charge; the carboxylic acid group of the amino acid has a pKa value of 4.3, which is a little basic than the terminal α-carboxyl group and that of aspartic acid. The pKa of glutamic acid is significantly higher than that of aspartic acid due to the inductive effect o the additional methylene group. In some proteins, due to a vitamin K dependent carboxylase, some glutamic acid will be dicarboxylic acids, referred to as γ carboxyglutamic acid, that form tight binding sites for calcium ion. Glutamic acid and α-ketoglutarate, an intermediate in the Krebs cycle, are interconvertible by transamination. Glutamic acid can therefore enter the Krebs cycle for energy metabolism, and be converted by the enzyme glutamine synthetase into glutamine, which is one of the key players in nitrogen metabolism.
Function
Glutamic acid is highly involed in metabolism. In citric acid cycle, transamination of alpha-ketoglutarate with alanine or aspartate each gives off glutamate and pyruvate or oxalatate respectively. Pyruvate and oxalatate formed fram transamination play critical roles in cellular metabolism.
Glutamic acid is a non-essential amino acid. It plays an important role in DNA synthesis. It also assists in wound and ulcer healing. Glutamic acid takes places in the excitatory neurotransmitter and the metabolism of sugars and fats. It aids potassium move through the blood-brain barrier. Glutamic acid is a source of fuel for the brain. It is capable to attach to amine group to form glutamine. The process of forming glutamine will detoxifies ammonia that the body contains.
Glutamic acid can be used in correcting personality disorders and treating childhood behavioral disorders. It also takes places in treating epilepsy, mental retardation, muscular dystrophy, ulcers, and hypoglycemic coma.
Other minor uses include flavor enhancer, GABA precursor, nutrients, and fertilizers for plants
Synthesis
A biosynthesis of glutamic acid involves various schemes. The most common scheme is the conversion of glutamine to glutamic acid by adding water molecules with glutaminase as a helper enzyme. The side product is an ammonia group. Addition of water to a N-Acetylglutamic acid also produce glutamic acid and acetate. Ketoglutaric acid is another common precursor in synthesis of glutamic acid. Addition of NADPH ad ammonia or alpha amino acid produces glutamic acid. Such enzymes involved are glutamate dehydrogenase and transaminase. Other methods include 1-pyrroline-5-carboxylate + NAD+ + HOH and N-formimino-L-glutamate + FH4.
Glutamic acid is easily converted into proline. First, the γ carboxyl group is reduced to the aldehyde, yielding glutamate semialdehyde. The aldehyde then reacts with the α-amino group, eliminating water as it forms the Schiff base. In a second reduction step, the Schiff base is reduced, yielding proline.
Structure
Glycine's molecular formula and mass are C2H5NO2 and 75.07 g/mol. Being the smallest amino acid out of all 20 amino acids, glycine only has a hydrogen atom as its substituent. For this reason, it has the ability to fit into tight spaces of molecules where no other amino acid could possibly fit therefore glycine is evolutionarily conserved. Most proteins contain small amount of glycine, however collagen is one of the exception that contains 35% glycine. Thus, if glycine were cleaved from an amino acid chain composing a whole protein, it would either alter the function of that protein, or denature it entirely. It is also the only achiral amino acid since its R group is simply a H atom. In particular it does not favor the helix formation.
Functions
Glycione is non-essential amino acids meaning the human can manufacture it in their body. It serves an important role in maintaining central nervous and digestive systems. Glycine prevents the breakdown of muscle by increase creatine, which is a compound that helps build muscle mass. Glycine also keeps the skin firm and flexible. Without glycine, the skin can be damage from the UV rays, oxidation and free radical.
Glycine regulates blood sugar levels and helps provide glucose for the body.
Glycine serves as an inhibitory neurotransmitter in the central nervous system, especially in the spinal cord. When glycine binds to receptors, it activates chloride ion channels to open. As chloride ions enter the channels, the membrane becomes hyperpolarized, causing an inhibitory postsynaptic potential (IPSP).
Some disorders that can be treating using glycine is used for treating schizophrenia, stroke, benign prostatic hyperplasia (BPH), and some rare inherited metabolic disorders. It is also used to protect kidneys from the harmful side effects of certain drugs used after organ transplantation as well as the liver from harmful effects of alcohol. Other uses include cancer prevention and memory enhancement.
Some people apply glycine directly to the skin to treat leg ulcers and heal other wounds. The body uses glycine to make proteins. Glycine is also involved in the transmission of chemical signals in the brain, so there is interest in trying it for schizophrenia and improving memory. Some researchers think glycine may have a role in cancer prevention because it seems to interfere with the blood supply needed by certain tumors.
Biosynthesis
Glycine is a derivative form of serine and 3-phosphoglycerate. The conversion of serine requires a specific enzyme called serine hydroxymethyltransferase and co-factor pyridoxal phosphate. The process can be simplied as the following reaction: serine + tetrahydrofoate -> glycine + N5, or N10-methylene tetrahydrofolate + water.
The reaction continues to carry out in the liver. Glycine synthase is used as enzyme in the conversion of N5, or N10-methylene tetrahydrofolate. In this reaction, carbon dioxides, ammonium, NADH, and protons transform the tetrahydrofolate molecule into glycine.
Degradation of glycine has three pathways. The most common pathway is the opposite of the previous reaction: conversion of glycine into a tetrahydrofolate molecule. Another pathway is the conversion of serine into pyruvate and serine dehydratase. The last pathway involves converting glycine to gloxylate by D-amino acid oxidase. This pathway leaves glycoxylate oxidized to oxalate.
Structure
Histidine, C6H9N3O2, is also called 2-amino-3-(1H-imidazol-4-yl)propanoic acid. Its molecular mass is 155.15 g/mol. It is a basic, polar amino acid with an imidazole group, which is an aromatic ring that can be of positive charge and hydrophilic. The imidazole group of the amino acid has a pKa value of 6, which can be either uncharged or positively charged at neutral pH. This amino acid is often present in active sites of enzymes wherein the imidazole group acts as a buffer (proton acceptor or donor) for chemical reactions. Histidine is a precursor of histamine, a compound released by the immune system cells during an allergic reaction.
Features
At a physiological pH of around 7, the Henderson-Hasselbalch equation can be used to give a ratio of deprotonation/protonation of the imidazole side chain (pKa = 6). As it turns out, the histidine side chain is approximately 10% protonated at a neutral pH. That is not a negligible amount and it gives the histidine residue a certain amount of buffering capacity. The basic nitrogen activates imidazole sites as a nucleophile.
Functions
Histidine is found in high concentrations in hemoglobin. As a result, it aids in treatment of anemia and maintaining optimal blood pH. Also, histidine is the precursor of histamine, which is involved in local immune responses.
Histidine is an essential amino acid, which means that the body cannot manufacture it. Histidine plays important roles in stimulating the inflammatory response of skin and mucous membranes. It also stimulates the secretion of the digestive enzymes gastrin and acts as the source and control for histamine levels. Histidine is required for growth and for the repair of tissues, as well as the maintenance of the myelin sheaths that act as protector for nerve cells. Histidine is also required to manufacture both red and white blood cells. With histidine in the body, it helps protect the body from damage caused by radiation and in removing heavy metals from the body. Histidine is also in the stomach. It is helpful in producing gastric juices, and people with a shortage of gastric juices or suffering from indigestion, may also benefit from this nutrient. It is thought that histidine may be beneficial to people suffering from arthritis and nerve deafness. This is not conclusively proven. Histidine is also used for sexual arousal, functioning and enjoyment. Histidinemia is an inborn error of the metabolism of histidine due to a deficiency of the enzyme histidase, where high levels of histidine are found in the blood and urine, and may manifest in speech disorders and mental retardation. There are no reported side effects with histidine, but too high levels of histidine may lead to stress and mental disorders such as anxiety and people with schizophrenia have been found to have high levels of histidine. People suffering from schizophrenia or bipolar (manic) depression should not take a histidine supplement without the approval of their medical professional.
Metabolism
Histidine can be converted into histamine by histidine decarboxylase. The carboxyl group leaves histidine.
Food sources
Food that contain histidine are dairy, meat, poultry, fish, rice, wheat, and rye.
Structure
Isoleucine, HOOCCH(NH2)CH(CH3)CH2CH3, is also known as a 2-amino-3-methylpentanoic acid and has a molar mass of 131.17 g/mol. Isoleucine is a nonpolar, aliphatic or hydrophobic amino acid that has two chiral centers for α-carbon atom and the R group. Isoleucine, because it contains two stereocenters, is a diastereomer. If it weren't for the selectivity of living things for one particular stereoisomer, there would be 4 possible stereoisomers because of the 2 chiral centers. However, only one version persists in living organisms: the 2S, 3S version. The structure stabilizes water-soluble proteins by hydrophobic effect.
Features
Isoleucine cannot be distinguished by MS from leucine because of the simple fact that they have the same molecular weight. Instead, these two residues would usually have to be isolated and characterized by HPLC or TLC against known standards.
Isoleucine is also degraded into succinyl CoA and acetal CoA and consumed by TCA cycle.
Functions
Isoleucine is an essential amino acid, meaning the human body cannot manufacture it. It is needed for the formation of Hemoglobin and to regulate blood sugar and energy levels. Isoleucine serves important roles in muscle strength and endurance and is a source of energy for muscle tissues.
Isoleucine promotes muscle recovery after an intense workout. Isoleucine is necessary for the formation of hemoglobin as well as assisting with regulation of blood sugar levels as well as energy levels. It is also involved in the formation of blood clots.
Symptoms of people with a deficiency of isoleucine may result in headaches, dizziness, fatigue, depression, confusion as well as irritability. Symptoms of deficiency may mimic the symptoms of hypoglycemia. This nutrient has also been found to be deficient in people with mental and physical disorders, but more research is required on this. Consuming higher amounts of isoleucine is not associated with any health risks for most people but those with kidney or liver disease should not consume high intakes of amino acids without medical advise. People who take in higher amounts of isoleucine report elevated urination. People involved with strenuous athletic activity under extreme pressure and high altitude may benefit from supplementation of this nutrient.
Food sources of isoleucine
Food containing isoleucine are almonds, cashews, chicken, eggs, fish, lentils, liver, meat, etc.
Biosynthesis
Synthesis of iseoleucine involves multiple steps. Isoleucine can be derived from pyruvate and ketoglutarate. Catalytic enzymes required are the followings:
Structure
Leucine's molecular formula and mass are C6H13NO2 and 131.17 g/mol respectively. Leucine, also known as a 2-amino-4-methylpentanoic acid, has aliphatic R group. It is one of the three amino acids with branched hydrocarbon side chains (generally buried in folded proteins) and result as a nonpolar or hydrophobic amino acid. The hydrophobic effect counts for stabilization of water-soluble proteins.
Features
Leucine cannot be distinguished by MS from isoleucine for the simple fact that they have the same molecular weight. Instead, these two residues would usually have to be isolated and characterized by HPLC or TLC.
Functions
Leucine has all functions of the amino acid Isoleucine as their similarity in branched hydrocarbon side chain. Leucine facilitates skin healing and bone healing by modulating the release of natural pain-reducers, Enkephalins. It is also a precursor of cholesterol and increases the synthesis of muscle tissues by slowing down their degradation process. Leucine is an essential amino acid. It is essential in promoting growth in infant and regulating nitrogen concentration in adults. Leucine is generally used as a flavor enhancer.
Deficiency and Excess
Deficiency of this particular amino acids can result in Hypoinsulinemia, Depression, Chronic fatigue syndrome, Kwashiorkor (or starvation), etc. Excess of Leucine leads to Ketosis.
Biosynthesis
As an essential amino acid, leucine cannot be synthesized in human bodies, and must be obtained from outside sources. Starting from pyruvic acid, the conversion includes valine, ketovalerate, isopropylmalate, and ketoisocaproate via reduction. Enzymes required are: 1. acetolactate synthase, acetohydroxy acid isomeroreductase, dihydroxyacid dehydratase, isopropylmalate synthase and isomerase, and leucine aminotransferase.
Structure
Lysine is an essential amino acid. This means that is is necessary for human health but the body cannot produce it so you have to get the amino acid from food or supplements. Lysine are the building blocks of protein. Lysine has a positively charged amine group chain. The ε-amino group has a significant high pKa value of about 10.8, which is more basic than the terminal α-amino group. This basic amino group is highly reactive and participates in the reactions at the active center of enzymes. Although the terminal ε-amino group is charged under physiological condition, the hydrocarbon side chain with three methylene group is still hydrophobic.
Features
Lysine is a naturally occurring essential amino acid in human body. It promotes optimal growth of infants and nitrogen equilibrium in adults.
Functions
Lysine can be a treatment of Herpes Simplex and virus-associated Chronic Fatigue Syndrome as it inhibits viral growth. It facilitates the formation of collagen, which is the main component of fascia, bone, ligament, tendons, cartilage and skin. It also helps in absorption of calcium, which is critical in bone growth of infants.
Lysine is important for proper growth, and it plays an essential role in the production of carnitine, a nutrient responsible for converting fatty acids into energy and helping to lower cholesterol. Lysine helps the body absorb calcium, and it plays an important role in the formation of collagen, a substance important for bones and connective tissues including skin, tendon, and cartilage.
Herpes Simplex Virus (HSV)
Consuming lysine on a regular basis may help prevent outbreaks of cold sores and genital herpes. Lysine has antiviral effects by blocking the activity or arginine, which promotes HSV replication. It has been studied that lysine at the beginning of a herpes outbreak did not reduce symptoms. Studies show that lysine with L-arginine makes bone building cells more active and enhances production of collagen. No studies have examined whether lysine helps prevent osteoporosis in humans.
Osteoporosis
Lysine helps the body absorb calcium and thus decreases the amount of calcium that is lost in urine. Calcium is essential for strong bones so some researchers assumed lysine may help prevent bone loss associated with osteoporosis.
Deficiency and Excess
Deficiency of lysine is seen in Herpes, Chronic Fatigue Syndrome, AIDS, Anemia, hair loss, and weight loss, etc. Having excessive lysine can result in high concentration of ammonia in the blood. Most people get enough lysine in their diet, although athletes, vegans who do not eat beans, as well as burn patients may need more. Not enough lysine can cause fatigue, nausea, dizziness, loss of appetite, agitation, bloodshot eyes, slow growth, anemia, and reproductive disorders. For vegans, legumes such as beans, peas, and lentils are the best sources of lysine.
Food Sources
Foods rich in lysine are meat, cheese, fish, nuts, eggs, soybeans, spirulina, and fenugreek seed. Brewer's yeast, beans, and other legumes, and dairy products also contain lysine, Many nuts contain lysine.
Structure
Methionine is one of the two amino acids with side chain containing sulfur. It contains a largely aliphatic side chain that includes a thioether (-S-) group. Unlike Cysteine, the chemical linkage of the sulfur in methionine is thiol ether. This sulfur does not participate in covalent bonding like that of cysteine. The high inclination of the sulfur oxidation in methionine is one of the causes of smoking-induced emphysema in the human lung tissue.
Features
Methionine is a naturally occurring essential amino acid, which plays a critical role in supplying free methyl groups and sulfur in metabolism. It is also one of only two amino acids coded for by a single codon.
Functions
Methionine helps the breakdown of fat and reduces blood cholesterol levels. It is an antioxidant that neutralizes free radicals and removes waste in the liver. Synthesis of DNA and RNA requires the presence of Methionine. It is also a precursor of several critical amino acids, hormones, and neurotransmitters in human body. Its AUG codon also serves as a "start" signal for ribosomal translation of messenger RNA or mRNA; this means that every peptide chain began with an methionine residual at its N-terminal. It may however be removed later on by cleavage.
Deficiency and Excess
Methionine deficiency can be seen in chemical exposure and vegetarians. Severe liver disease can result from having excessive methionine.
Structure
The amino acid phenylalanine is a derivative of alanine wherein a phenyl group takes the place of one of the hydrogens on the CH3 group. Phenylalanine has stronger hydrophobic properties when compared to the other aromatic amino acids, i.e. tyrosine and tryptophan. Tyrosine and tryptophan are less hydrophobic than phenylalanine due to their hydroxyl and indol substituents. Phenylalanine is often found buried in the proteins due to its hydrophobicity. Neighboring phenyl rings (on adjacent amino acids) can stabilize each other by pi stacking.
Features
Individual amino acids as well as peptides are occasionally analyzed by UV light. Phenylalanine, along with the few other aromatic amino acids, fluoresces when UV light is applied. UV light can be a useful technique for verifying the presence of Tyr, Phe, and Trp. It can also quantify those amino acids if a sensitive enough assay is developed.
Functions
Phenylalanine is a precursor of the amino acid tyrosine, which gives rise to neurotransmitters, such as dopamine, norepinephrine and epinephrine. It can be used to manage certain types of depression as a powerful anti-depressant and can also enhance memory, thought, and mood. This amino acid also plays a role in decreasing blood pressure in hypertension. The D form of phenylalanine can be used to reduce pain in arthritis which is a rare instance among amino acids. Phenylalanine is a naturally occurring amino acid that promotes growth in infants and regulates nitrogen concentration in adults.
Deficiency and Excess
Deficiency of Phenylalanine can be seen in depression, AIDS, obesity, Parkinson's Disease, etc. Some people have a autosomal recessive genetic disorder called phenylketonuria, or PKU. This disorder is due to the lack of an enzyme that breaks down phenylalanine amino acids, which leads to a large accumulation of this amino acid, and in large quantities, phenylalanine is toxic, particularly to the brain. This leads to the possibility of mental retardation from this disorder. As a result, babies were blood tested early for signs of PKU, and if they have it then they must follow a strict diet that reduces the amount of natural phenylalanine in the food.
Structure
Proline is one of the twenty DNA-encoded amino acids. It is unique among the 20 protein-forming amino acids because the α-amino group is secondary rather than primary as other amino acid. The distinctive cyclic structure of proline side chain locks its φ backbone dihedral angle at approximately -75°, giving proline an exceptional conformational rigidity compared to other amino acids. Hence, proline loses less conformational Entropy upon folding, which may account for its higher prevalence in the proteins. Proline, strictly speaking, can also be referred to as an imino acid. It greatly influences protein architecture because of its ring structure that makes it more conformationally restricted than the other amino acids.
Functions
Proline behaves as a structural disruptor in the middle of regular secondary structure elements. However, proline is commonly found as the first residue of an alpha helix and in the edge strands of beta sheets. Proline is most commonly found in turns, which may account for the curious fact that proline is usually solvent-exposed although it has a completely aliphatic side chain. Because proline lacks of hydrogen on the amide group, it cannot act as a hydrogen bond donor, only as a hydrogen bond acceptor. Proline is important in healing, cartilage building, and in flexible joints and muscle support. It also helps reduce the sagging, wrinkling, and aging of skin resulting from exposure to the sun. Proline by breaking down protein and helps create healthy cells. It is essential both to skin health, and for the creation of healthy connective tissues and also muscular tissue maintenance.
Deficiency and Excess
Proline deficiency is generally caused by people who perform prolonged exercises. Vitamin C deficiency will also cause proline to be lost in the urine because of collagen breakdown. Generally, people's body with proline deficiency tends to metabolize muscle cells instead of carbohydrates first if glucose levels are low. Proline is needed to maintain proper collagen creation and stabilize muscular tissue as well. The lack of proline could lead to symptoms such as fatigue, weight loss, dehydration, dizziness, and nausea.
Structure
This amino acid's R group is a hydroxyl group attached to a CH2 group. The hydroxyl group is polar giving serine polar/hydrophilic properties. It has a pH of 5.68. pKa = 2.21, 9.15.
Features
Serine is a non-essential amino acid which means it can be synthesized by the human body. For instance, serine can be synthesized from glycine. Serine is a precursor of glycine and cysteine.
Biosynthesis
The biosynthesis of serine begins with the oxidation of 3-phosphoglycerate (an intermediate in glycolosis) to 3-phosphohydroxypyruvate which is then transaminated to 30phosphoserine. This last intermediate is then hydrolyzed to serine.
Function
Serine is a non-essential amino acid which means it can be synthesized by the human body. For instance, serine can be synthesized from glycine. Serine is also a precursor of glycine and cysteine. Serine is found in phospholipids, active sites of trypsin and chymotrypsin. It can synthesize pyrimidines and proteins, cysteine and tryptophan. It is also involved in fat and fatty acid formation, muscle synthesis. Serine can be deaminated by the catalyst serine dehydratase, yielding to pyruvate and ammonium. The deamination of threonine follows a similar process.
Structure
Threonine is a polar, uncharged amino acid. Its side chain contains a secondary alcohol and a methyl group; hence it can be characterized as a hydrophilic amino acid. Threonine incorporates two chiral centers, just like isoleucine. If it weren't for the selectivity of living things for one particular stereoisomer, there would be 4 possible stereoisomers because of the 2 chiral centers. However, only one version persists in living organisms: the 2S, 3R version.
Features
Threonine is an essential amino acid, which means it cannot be synthesized by the human body. Humans must ingest it in the form of threonine-containing foods.
Functions
Threonine aids the formation of elastin and collagen. In the immune system, threonine aids in the formation of antibodies. It also promotes growth and function thymus glands and absorption of nutrients. In addition, threonine is the precursor to isoleucine. Threonine can be deaminated by the catalyst threonine dehydratase, yielding to α-ketobutyrate and ammonium. The deamination of Serine follows a similar process.
Structure
Tryptophan is an amino acid of aromatic group of an indole group bonded to a methylene group as the side chain, which is of two aromatic rings of nitrogen and hydrogen group and is hydrophilic. One of the side chains is 5-membered while the other is 6, and 2 carbons are shared by both aromatic rings.
Features
Individual amino acids as well as peptides are occasionally analyzed by UV light. Tryptophan, along with the few other aromatic amino acids, fluoresces when UV light is applied. UV analysis can be a useful technique for verifying the presence of Tyr, Phe, and Trp. It can also quantify those amino acids if a sensitive enough assay is developed.
Functions
Tryptophan is the precursor for various proteins, serotonin and niacin. It also promotes the formation of peptides and proteins. It is an essential amino acid, meaning it cannot be produced by the human body. It is usually present in peptides, enzymes, and structural proteins.
Deficiency and Excess
Excess tryptophan has been linked with eosinophilia-myalgia syndrome (EMS). A deficiency of tryptophan is known as Pellagra which causes a deficiency of niacin. However, with vitamin supplements, this disease is no long as prominent. Symptoms of the disease include dementia and schizophrenia. Hartnup Disease is a genetic autosomal recessive disease in which a person cannot effectively digest this amino acid in their digestive tract. Although the disease of experiences symptoms similar to those of pellagra, however being slightly less severe. Patients suffering from the disease are generally seen with red rashes that are further aggravated by UV light from the sun. Further mental retardation could occur if not treated correctly with vitamin supplementation.
Tyrosine
Tyrosine is a nonpolar aromatic amino acid that contains a hydroxyl group attached to an aromatic ring. The hydroxyl group is particularly important because these residues are utilized in the phosphorylation of other proteins.
Tyrosine is a non essential amino acid meaning it can be synthesized in the body. It is synthesized using phenylalanine in the body.
Features
Individual amino acids as well as peptides are occasionally analyzed by UV light. Tyrosine, along with the few other aromatic amino acids, fluoresces when UV light is applied. UV light can be a useful technique for verifying the presence of Tyr, Phe, and Trp. It can also quantify those amino acids if a sensitive enough assay is developed.
Functions
Tyrosine plays crucial roles in the human body: It helps deal with stress by becoming an adaptanogen helps minimize effects of the stress syndrome, in drug detoxification such as for cocaine, coffee and nicotine addictions. It reduces withdrawals and abuse. It assists in treating Vitiligo, pigmentation of skin, Phenylketonuria, the condition where phenylalanine is not metabolized. In addition, it is effective for depression treatment.
Tyrosine is also important in the production of epinephrine, norepinephrine, serotonin, dopamine, melanin, and enkephalins, which has pain-relieveing effects in the body. It also affects the function of hormones by regulating thryoid, pituatary and adrenal glands. For example, one need only look at the thyroid hormone thyroxine to see that it is synthesized from tyrosine. Tyrosine is known to dislodge molecules that may be harmful to cells, therefore it has protective qualities.
Deficiency and Excess
Deficiency of tyrosine can result in low blood pressure, depression, and low body temperature. Tyrosine is a major amino acid responsible for skin, hair, and eye pigments. A loss of tyrosine amino acid in the body may lead to failure to form melanin pigments, resulting partial or full albinism. Interestingly enough, Tyrosine is produced mainly from phenylalanine in which a loss of one would lead to the increase of the other amino acid present in the organism's body.
Structure
Valine is an amino acid with an aliphatic, isopropyl side chain and is therefore a hydrophobic amino acid. Valine differs from threonine in that the OH group of threonine is replaced by a CH3 group. This is a nonpolar amino acid. It is an essential amino acid; therefore it cannot be produced by the human body. Being hydrophobic, this amino acid is often found in the interior of proteins.
Features
In animals, valine must be ingested. In plants, it is created by using pyruvic acid, converting it to leucine followed by the reductive amination with glutamate. Valine is found in the following foods: soy flour, fish, cheese, meat and vegetables.
Functions
Valine is essential in muscle growth and development, muscle metabolism, and maintenance of nitrogen balance in the human body. It can be used as an energy source in place of glucose. It can also be used as a treatment for brain damage caused by alcohol.
Deficiency and Excess
Deficiency of valine affects myelin sheets of nerves. Maple Syrup Urine Disease is caused because leucine, valine and isoleucine cannot be metabolized.
The 20 standard amino acids have two acid-base gorups: the alpha-amino and the alpha-carboxyl groups attached to the Cα atom. Those amino acids with an ionizable side-chain (Asp,Glu,Arg,Lys,His,Cys,Tyr) have an additonal acid-base group. At low pH (i.e. high hydrogen ion concentration) both the amino group and the carboxyl group are fully protonated so that the amino acid is in the cationic form H3N+CH2COOH. As the amino acid in solution is titrated with increasing amounts of a strong base (e.g. NaOH), it loses two protons, first from the carboxyl group which has the lower pK value (pK=2.3). The pH at which Gly has no net charge is termed its isoelectric point, pI. The α-carboxyl gorups of all the 20 standard amino aicds have pK values in the range 1.8-2.9, while their α-amino groups have pK values in the range 8.8-10.8. The side-chains of the acidic amino acids Asp and Glu have pK values of 3.9 and 4.1, respectively, whereas those of the basic amino acids Arg and Lys, have pK values of 12.5 and 10.8, respectively. Only the side-chain of His, with a pK value of 6.0, is ionized within the physiological pH range (6-8). It should be borne in mind that when the amino aicd are linked together in proteins, only the side-chain groups and the terminal α-amino and α-carboxyl gorups are free to ionize.
Pyridoxal 5’-Phosphate-Mediated Decarboxylation of an �-Amino Acid
Step 1: The amino acid reacts with enzyme-bound pyridoxal 5�-phosphate (PLP). An imine linkage (CoeN) between the amino acid and PLP forms, and the enzyme is displaced.
Step 2: When the pyridine ring is protonated on nitrogen, it becomes a stronger electron-withdrawing group, and decarboxylation is facilitated by charge neutralization.
Step 3: Proton transfer to the � carbon and abstraction of a proton from the pyridine nitrogen brings about rearomatization of the pyridine ring.
Step 4: Reaction of the PLP-bound imine with the enzyme liberates the amine and restores the enzyme-bound coenzyme.
The total chemical synthesis of a D-Enzyme experiment was conducted by R. C. deL. Milton, S.C. F. Milton, and S. B. H. Kent, which found enzyme enantiomers exhibiting reciprocal chiral specificity on peptide sequences. The concept of L-configuration of amino acids predominates in living organisms while the D-configuration remains biologically inactive; Milton et al. examined the ability of enzymes to distinguish and react with a specific enantiomer over the other.
The following properties of D-HIV PR and L-HIV PR were analyzed: covalent structure, physical properties, circular dichroism spectra, and enzymatic activity. After the total synthesis of D-HIV PR and L-HIV PR, the new synthesized L- and D- sequences of HIV PR were initially protected and then deprotected to allow the folding of their secondary and tertiary structures. The second method used reversed-phase high-performance liquid chromatography which resulted to identical retention rates of the two polypeptide sequences. It was further examined by ion-mass spectroscopy that both polypeptide sequences had the same molecular weight. This method found that both the D-HIV PR and L-HIV PR sequences had the same covalent structure.
Despite having the same covalent structure between D-HIV PR and L-HIV PR, differences arise within its chiral features; using a circular ion spectra proved the expected equal but opposite optical activity of the enantiomers. Within a fluorogenic assay containing a hexapeptide analog of a GAG cleavage site was used as a substrate to test the reactivity of the enantiomers. Both enzymes were equally active, yet exhibited reciprocal chiral specificity; reciprocal chiral specificity was apparent when L-enzyme degraded only the L-substrate and D-enzyme degraded only the D-substrate. In addition, reactivity of the D-HIV PR and L-HIV PR were further tested with enantiomers of an inhibitor called MVT101. As expected its corresponding enzyme determined the effectiveness of the inhibitor; L-MVT101 inhibited L-HIV PR but not D-HIV PR, and D-MVT101 inhibited D-HIV PR but not L-HIV PR.
The folding of the polypeptide chains into the three-dimensional structure holds importance to the specificity and catalytic activity of HIV-1 protease. D-HIV PR and L-HIV PR displayed mirror images of each other within the secondary, supersecondary, tertiary, and quaternary structure. In the primary structure, only one chiral amino acid was introduced in the synthesis of the polypeptide chain for D-HIV PR and L-HIV PR; the consequence of this one chiral amino acid in the polypeptide backbone resulted to mirror images of the secondary, supersecondary, tertiary, and quaternary structures.
The results of this experiment conclude that the two configurations of the enantiomer are reactive and should be reactive in vivo, yet due to evolution the L-proteins are prevalent in living organisms while D-proteins are biologically inactive.
del. Milton, R. C, S.C.F. Milton, and S.B.H Kent. "Total Chemical Synthesis of a D-Enzyme: The Enantiomers of HIV-1 Protease Show Demonstration of Reciprocal Chiral Substrate Specificity."Science. 256. (1992): 1445-1448. Print.
Nitrogen Fixation, or rather, the fixing of Nitrogen, is a process where N₂ is reduced into NH₃, either biologically or abiotically. The nitrogen in amino acids, pyrimidines, purines and other molecules all come from the N₂ in our atmosphere. The fixing of nitrogen can also be associated with the conversion of nitrogen into other forms, other than ammonia, such as nitrogen dioxide. The triple bond that is present in N₂ is very strong; it has a bond energy of 940 kJ/mol. Yet, it is thermodynamically favorable to form ammonia from hydrogen and nitrogen, yet the reaction is still very difficulty kinetically speaking since intermediates can prove to be unstable. It has been estimated that approximately 60 percent of the newly fixed nitrogen on Earth is produced by diazotrophic microorganisms, while lightning and ultraviolet radiation contribute another 15 percent and the rest 25 percent is done by industrial processes.
The main avenue for entry of nitrogen into the biosphere is nitrogen fixation. In the nitrogen fixation, we basically fix the dinitrogen, or nitrogen gas into ammonia. Also, fixation of nitrogen requires lots of energy because the triple bond of nitrogen gas is stable. However, breaking the triple bond to generate ammonia requires a series of reduction steps involving high input of energy.
Biologically speaking, the conversion of nitrogen into ammonia is usually done by bacteria and archaea. These organisms that are responsible for nitrogen fixation are called diazotrophic microorganisms. For example, the symbiotic Rhizobium bacteria, a diazotrophic microorganism, goes into the roots of leguminous plants to form root nodules where they fix nitrogen. Other examples include Cyanobacteria, Azotobacteraceae, and Frankia.
Industrial Processes of Nitrogen Fixation include Dinitrogen complexes, Ambient Nitrogen reduction, and the most common process is the Haber process, invented in 1910. The Haber process involves high pressure, high temperatures, possibly an iron or ruthenium catalyst to produce ammonia.
Nitrogen Fixation, in the biological sense, is run by an enzyme called nitrogenase. The reason why the nitrogenase complex is used is because it has multiple redox centers. In general though, nitrogenase complex contains two proteins. The first, a reductase, which provides electrons while the second part, nitrogenase, uses these electrons to turn nitrogen into ammonia. The transferring of electrons, from reductase to nitrogenase, in this process is coupled with the hydrolysis of ATP by the reductase.
The reaction for this process is N2 + 8 H+ → 2 NH3 + H2. The reason why this process is an 8 electron process and not simply a 6 electron process is due to the extra mole of Hydrogen that gets generated along with the generation of the ammonia. Often the microorganisms that carry out nitrogen fixation, contain the 8 electrons from the reduced form of Ferredoxin, which can be made from photosynthesis or oxidative processes. Also, this process is coupled by two ATP molecules for each mole, which in turn, equals 16 molecules. The reason for this is not that the ATP hydrolysis is making the reduction thermodynamically favorable since the process is already thermodynamically favorable, but rather allows the reaction to be kinetically possible.
Nitrogen fixing bacteria generally separate anaerobic nitrogen fixation from aerobic metabolism by one of several mechanisms. In the ocean and in the freshwater systems, cyanobacteria are the major nitrogen fixers. Within an ecosystem, nitrogen fixers ultimately make the reduced nitrogen available for assimilation by nonfixing microbes and plants. Besides, nitrogen fixation is extremely energy intensive; thus the rate of fixation usually fails to meet the potential demand of other members of the ecosystem.
When there are unneeded amino acids from either protein digestion or turnover, they are broken down into certain compounds. This process usually occurs in the liver.
In amino acid degradation the amino group is removed and turned into an α-ketoacids which is then modified so that the carbon chain could enter the metabolism and eventually become glucose or intermediates of the citric acid cycle.
The amino group is transferred to α-ketoglutarate which forms glutamate. Then the glutamate is oxidatively deanimated to form the ammonium ion NH4+
Aminotransferases catalyzes the reaction that turns the α-amino group from an α-amino acid to an α-ketoacid. These enzymes catalyze α-amino groups from a variety of amino acids to α-keto-glutarate for conversion to NH4+
Aspartate aminotransferase, catalyzes the transfer of the amino group of aspartate to α-ketoglutarate.
Alanine aminotransferase catalyzes the transfer of the amino group of alanine to α-ketoglutarate.
The nitrogen from the α-ketoglutarate in the transamination reaction is converted into an ammonium ion by oxidative deamination. This reaction is catalyzed by glutamate dehydrogenase.
This enzyme is special in that it is able to utilize either NAD+ or NADP+. The reaction dehydrogenates the C-N bond, and then hydrolyses of the Schiff base to make a ketoglutarate
The equilibrium for this reaction favors glutamate. But the reaction can be pushed forward by the consumption of ammonia. Glutamate dehydrogenase is found in the mitochondria. This compartmentalization prevents interaction with ammonia. In vertebrates, the activity of glutamate dehydrogenase is allosterically regulated.
NH4+ is converted into urea, which is then excreted as waste.
Overview
To synthesize amino acids, there must be a source of nitrogen that is in a form that can be easily used. Various microorganisms reduce inert nitrogen gas into two molecules of ammonia to provide for this source of nitrogen. On the other hand, the carbon backbone can be provided in three different ways--these include the citric acid cycle, the glycolytic pathway, and the pentose phosphate pathway.
Since amino acids are all chiral except for glyciene, biosynthesis of amino acides must generate the correct isomers efficiently. This is done by transamination reactions and high regulation of biosynthetic pathways, through feedback and other mechanisms.
Nitrogen Fixation
To reduce atmospheric nitrogen gas (N2) to ammonia (NH3, a process called nitrogen fixation, microorganisms require ATP. Nitrogen fixation is performed by nitrogenase complex, an enzyme that has many centers for redox. This enzyme is composed of a reductase and nitrogenase. The reductase provide electrons while the nitrogenase uses these electrons, reducing atmospheric nitrogen to ammonia in the following reaction:
N2 +8 e- + 8 H+ <--> 2 NH3 + H2
Most microorganisms that are capable of nitrogen fixation carry out this reaction by generating a reduced ferredoxin through photosynthesis, providng the electrons. Two molecules of ATP are then used to transfer each electron, meaning that 2x8=16 electrons are needed to generate the two molecules of ammonia. The total reaction for this can then be written as:
Then, through the amino acids glutamine and glutamate, ammonium ion (NH4+)is assimilated.
Chirality
Of the 20 amino acids, humans can synthesize 11 of them. These amino acids are referred to as nonessential amino acids. The remaining 9 amino acids are referred to as essential amino acids, and they must be provided for in the diet. Synthesizing the 11 nonessential amino acids require different intermediates, but one fact remains common among them--the gycolytic pathway, the citric acid cycle, and the pentose phosphate pathway provide intermediates that their carbon skeletons come from. Also, in all these amino acids, the same step ensures the correct chirality. This step is in a transamination reaction, and a quinonoid intermediate is protonated, forming an external aldimine. The direction the proton comes from dictates the amino acid's chirality.
Regulation by Feedback
The rate of amino acid biosynthesis depends on the amount of enzymes present and the activity of those enzymes. However, there are other ways of regulating the biosynthesis of amino acids.
Feedback Inhibition
The first reaction that is irreversible in the biosynthesis of amino acids is referred to as the committed step, and the feedback loop of amino acid synthesis is a negative one, with the product inhibiting the catalyst to the committed step. This indicates that the biosynthesis of amino acids is regulated by a negative feedback loop. There are variety of different feedbacks that regulate the synthetic pathway.
Branched Pathways
Branched pathways are more complex in that they involve more sophisticated regulation. They can involve both positive and negative feedback. In other words, reactions have both feedback inhibition and feedback activation. An example of this is the enzyme threonine deaminase. This enzyme converts threonine to alpha-ketobutyrate, and valine activates this process, while isoleucine inhibits it.
Branched pathways may also involve enzyme multiplicity, a phenomenon in which multiple enzymes regulate or catalyze one single reaction. These enzymes may all have different activities and different regulatory mechanisms. Lastly, in cumulative inhibition, multiple proteins are capable of inhibiting one enzyme's activity. Even if the inhibited enzyme is saturated with one protein, other inhibiting proteins can still continue to reduce its activity. An example of this is the cumulative feedback inhibition of glutamine synthetase in E. coli.
Enzymatic cascade is another form of regulation in branched pathways. An enzymatic cascade is a reaction that requires successive steps of enzymatic catalysis after initiation. The advantages of this process is that it can amplify signals and highly increase allosteric control. This is due to the fact that requiring different enzymes basically combines multiple regulations of the enzymes, so that the process, in entirety, will have all these regulations occurring. This extends the potential for more efficient accruing of nitrogen in the cell.
So What?
Why is the biosynthesis of amino acids important? Amino acids are not only the basic building blocks to all peptides and proteins. A wide variety of biomolecules are also derived from amino acids. Examples of these include the purine and pyrimidine bases in DNA and RNA, a vasodilating protein called histamine, the hormone thyroxine, and the hormone epinephrine, to name a few. Amino acids are also a part of other compounds in the body, such as buffers, antioxidants, and enzymes. Another molecule formed from amino acids is nitric oxide (NO). Nitric oxide is derived from arginine, and serves as a messenger in signal transudction.
As amino acids are involved in the synthesis of so many proteins and compounds within the body, lack of amino acids therefore has its consequences. Various inherited disorders may occur as a result of lack of a certain amino acid, or a certain compound derived from amino acids. An example is porphyrias. Thisdisorder may be inherited or acquired during one's lifetime, and it is due to a deficiency of heme pathway enzymes.
Source: Berg, Jeremy and Stryer, Lubert. Biochemistry: Fifth Edition. United States of America: W.H. Freeman and Company, 2002.
The formation of amyloid fibrils, protofibrils, and oligomers from β-amyloid peptides have been very crucial for the research of the disease, Alzheimers. However, determining the structures of these peptides has been a struggle. In the past five years, there has been new data obtained about these structures through electron cryo-microscopy and NMR which has enhanced scientists' understanding of a certain mechanism, Aβ aggregation and has paved new pathways of relevance of specific conformers in terms of neurodegenerative pathologies.
The β-amyloid (Aβ) peptide resides inside the human brain as a proteolytic fragment of the amyloid precursor protein, with an amphiphilic structure, possessing a hydrophilic N- and hydrophobic C terminus.
The two most studied Aβ alloforms are Aβ(1-40) and aβ(1-42), where they contain 40 and 42 residues, respectively. More than 10 single-site sequence variants have been connected to similar forms of Alzheimer's disease. These alloforms are important because since Aβ amyloid fibrils form the center of amyloid plaques inside the brain parenchyma, they are correlated to Alzheimer's disease.
Scientists have been trying to determine the structure of these alloforms, but they cannot be isolated or easily purified within the laboratories. Thus, there is no reliable structural information of Aβ amyloid fibrils. This provides a challenge for scientists who need this structural information to understand their biological properties.
Amyloid fibrils are fibrillar polypetide aggregates with a cross-β structure. In cross-β structures, the β-sheet plane ad the backbone hydrogen bonds connecting the β-strands are positioned parallel to the axis while the β-strands run perpendicular to the axis. Further study of these structures showed that these peptides hve things called steric zippers. Steric zipper are composed of a pair of two cross-β sheets with interlacing side chains. They're formed by many short peptide chins, like Aβ residues 37-42 or 35-40. Also, steric zipper's structure is similar to that of the spine of amyloid fibrils.
General topology and polymorphism of mature amyloid fibrils
TEM (transmission electron microscopy) and atomic force microscopy have observed that mature amyloid fibrils have a length greater than 1 um, whereas previously analyzed fibrils were thought to have a length of about 25 nm. Mature Aβ amyloid fibrils have one or more protogilaments. Amyloid protofilaments create the substructures of mature fibrils, found by TEM to show that these fibrils are twisted left-handed with polarity. Studying thes structures shows that there's a structural feature of structural polymorphism of amyloid fibrils. Structural polymorphism is the variability in peptide conformation of fibrils3D reconstructions of polymorphic amyloid fibrils have revealed that fibrils differ in:
(i) number of protofilaments
(ii) different internal protofilament substructures
(iii) relative protofilament orientation
In addition to structural polymorphism (or inter-sample polymorphism), study of Aβ fibril samples with single particle techniques has shown that there is a lot of intra-sample polymorphism. Such as, an analysis of Aβ(1-40) fibrils created in 50mM sodium borate with a pH of 9 has revealed variations in the fibril width (13 to 29 nm); however, most fibrils demonstrate crossover distances of 100 to 200 nm. Thus, there is a wide range of morphologies, especially when fibrils are grown under sodium or potassium chloride (buffer systems).
Structural deformations report on the nanoscale flexibility properties of amyloid fibrils
Structural deformation is another cause for heterogeneity of amyloid samples besides polymorphism. These deformations bend and twist themselves and although these can create more potential problems for structural analysis, they can be used to understand anoscale mechanical properties of amyloid fibrils.
Atomic structures of full-length Aβ fibrils have not been found because:
There has not been any fibril that creates a crystal suitable for X-ray crystallography
The fibrils are too large for NMR techniques.
However, solid-state NMR and cryo-EM have been found to possibly determine the structure of Aβ amyloid fibrils at atomic resolution.
Solid-state NMR can determine structural constraints like chemical shift values, bond angles, and/or specific interatomic distances, and thus, identify residues of Aβ amyloids interconnecting with the β-sheet structure of fibrils.
Cryo-EM can visualize the structure of the fibrils and can calculate their 3D density. Thus, the observation of individual fibrils can determine specific fibril morphologies.
The protofilament substructure of an Aβ fibril has been found by cryo-EM. The protofilaments have cross-sectional dimensions of 4 x 11 nm and a cross-sectional subdivision of quasi twofold symmetry (4 x 5 nm) with two peripheral regions. Aβ(1-40) fibril contains two protofilaments and Aβ(1-42) fibril contains only one protofilament. The single-protofilament in Aβ(1-42) fibril has two equally shaped peripheral regions, fully solvent-exposed and structurally disordered.
In contrast, the two-rotofilament Aβ(1040) fibril has an arch-shaped peripheral region at the protofilament-protofilament interface. The other peripheral region is the one that is solvent-exposed and structurally disordered.
The Aβ(1-42) peptide is more pathogenic than the Aβ(1-40) peptide. For example, when it is expressed in Drosophila melanogaster, the Aβ(1-40) peptide is very toxic and halves the life-span of the animal; however, Aβ(1-40) don't present a discernible phenotype. Although of this difference, their chemical properties are pretty similar (the first 40 residues are identical) which leads to similarities in their conformation proerpties. Some of the differences include the Aβ(1-42) peptide having additional two C-terminal residues and the higher aggregation propensity of Aβ(1-40). Also, Aβ(1-42) can affect aggregation mechanisms of Aβ(1-40) and thus prevents formation of matue Aβ(1-40) fibrils.
According to cryo-EM of these two peptides, it shows differences in their protofilament packing. Aβ(1-40) fibrils have either a single-protofilament arrangement or a two-protofilament arrangement with a hollow core. But, all in all, the protofilaments of these two fibrils are pretty similar. For example, they can both produce the same mPL values, cross-sectional areas and shapes, and the cross-sections of the protofilaments have a similar division at the one central and two peripheral regions. Thus, they have similar peptide folding.
Also, according to IR and NMR data, they both have concluded that both fibrils have a parallel β-sheet structure.
Proteins are important organic compounds that serve as structural elements, transportation channels, signal receptors and transmitters, and catalysts; they are the most versatile macromolecules found in living organisms. Protein compositions are made up of one or more polypeptides which are composed of combinations that are derived from the 20 different amino acid subunits. These polypeptides are linear polymer chains of amino acids that are bonded together by a peptide bond that is formed between carboxyl and amino groups of adjacent amino acid residues. Each amino acid has its own size, shape, and set of properties, and proteins have 50 to 2,000 amino acids connected end-to-end in many different combinations (Structures of Life 3). Proteins can have different functionalities and roles in the body due to many different possible structures and shapes. One specific characteristic of proteins is that only the L isomers of amino acids are found in nature and used in protein. There is no evidence that explains why this happens. Proteins have many different active functional groups attached to them to help define their properties and functions. Proteins perform a number of important functions, ranging from acting as very rigid structural elements to transmitting information between cells. In addition, complex assemblies are formed due to proteins reacting with each other and with other macromolecules. Proteins fold into secondary, tertiary, and quaternary structures based on the intramolecular bonding between functional groups and can take on a variety of three-dimensional shapes depending on the amino acid sequence.
One example of a protein is collagen, a fibrous structural protein that is the most abundant protein found in animals. The structure of collagen consists of a triple helix and consists of mainly three polypeptide chains held together by hydrogen bonds, similar to that of DNA's double helix. This structure of collagen was determined using the method of X-ray crystallography.
There are several important properties that enable proteins to perform a variety of crucial functions.
1. LINEAR POLYMERS: Proteins are built out of monomer units (amino acids): Based on the sequence of amino acids, proteins spontaneously fold up into three-dimensional structures.
2. CONTAIN A WIDE RANGE OF FUNCTIONAL GROUPS: Proteins contain functional groups such as alcohols, thiols, thioethers, carboxylic acids, etc. These functional groups are key to the variety of functions the protein can perform.
3. PROTEIN INTERACTION FOR COMPLEX ASSEMBLIES: Within complex assemblies, proteins act synergistically in order to achieve a specific function.
4. STRUCTURE: Proteins vary in flexibility. Rigid units of a protein can function as structural elements in the cytoskeleton of cells or in connective tissue. Protein structure Is divided into four categories and is a crucial element in the specificity of protein function.
Proteins are usually portrayed in 3D structures. They are usually categorized into 4 different characteristics and levels:
A picture of primary structure of protein.
Primary: The primary structure of a polypeptide is its amino acid sequence, from beginning to end. The primary structures of polypeptides are determined by encoding genes. Genes carry the information to make polypeptides with a defined amino acid sequence. An average polypeptide is about 300 amino acids in length, and some genes encode polypeptides that are a few thousand amino acids long.
Secondary:
The amino acid sequence of a polypeptide, together with the laws of chemistry and physics, cause a polypeptide to fold into a more compact structure. Amino acids can rotate around bonds within a protein. This is the reason proteins are flexible and can fold into a number of shapes. Folding can be irregular or certain regions can give a repeating folding pattern. Such repeating patterns are called secondary structures. The two types are the α-helix and β-sheet. In an α-helix, the polypeptide backbone forms a repeating helical structure that is stabilized by hydrogen bonds. These hydrogen bonds occur at regular intervals and cause the polypeptide backbone to form a helix. In a β-sheet, regions of the polypeptide backbone come to lie parallel to each other. When these regions form hydrogen bonds, the polypeptide backbone forms a repeating zigzag shape called a β-sheet.
One type of secondary sturcture, an alpha helix.Another type of secondary structure, a beta sheet.
Tertiary:
As the secondary structure becomes established due to the primary structure, a polypeptide folds and refolds upon itself to assume a complex three-dimensional shape called the protein tertiary structure. The tertiary structure is the three-dimensional shape of a single polypeptide. For some proteins, such as ribonuclease, the tertiary structure is the final structure of a functional protein. Other proteins are composed of two or more polypeptides and adopt a quaternary structure.
Quaternary:
Most functional proteins are composed of two or more polypeptides that each adopt a tertiary structure and then assemble with each other. The individual polypeptides are called protein subunits. Subunits can be identical polypeptides or can be different. When proteins consist of more than one polypeptide chain, they are said to have quaternary structure and are also known as multimeric proteins, meaning many parts.
These proteins bind in a specific shape through interactions such as hydrogen bonding, salt bridges, and disulfide bonds. The two major structure categories of proteins are fibrous and globular. An example of a fibrous protein is keratin, which is found in wool, hair, myosin and actin in muscles, fur, nails, and fibrinogen for blood clotting. Examples of a globular protein include insulin, hemoglobin, and most enzymes.
A picture of Hemoglobin, one of the most well-known quaternary structure of protein.
Several factors determine the way that polypeptides adopt their secondary, tertiary and quaternary structures. The amino acid sequences of polypeptides are the defining features that distinguish the structure of one protein from another. As polypeptides are synthesized in a cell, they fold into secondary and tertiary structures, which assemble into quaternary structures for most proteins. As mentioned, the laws of chemistry and physics, together with amino acid sequence, govern this process. Five factors are critical for protein folding and stability:
Protein functions such as molecular recognition and catalysis depend on their complementary binding sites. They also depend on specialized microenvironments that result from protein's tertiary structure. Such specialized microenvironments at binding sites eventually contribute to catalysis. Binding sites have a diverse distribution of charges which allow the substrates to bind.
As temperature is risen, a protein starts to denature.
Upon addition of heat, proteins begin to denature. Denaturation occurs in the tertiary and secondary structures. If denaturation occurs, this could lead to protein inactivity, or even cause the cell to die and no longer function.
The reason that heat is able to cause the protein to denature is because it disrupts the bonds due to the rapid vibrations that it causes in these molecules.
Heat effects the tertiary and secondary structures. The primary structure of a protein is just peptide bonds, and heat is not strong enough to break these peptide bonds, so heat doesn't have an effect on the primary structure.
Hyperphagia as well as elevated levels of insulin and leptin are found in obesity although leptin is supposed to be a feeding inhibitor while lowering insulin levels and suppressing insulin production. Leptin may not be functioning as predicted due to the correlation found that Hyperphagia may cause leptin resistance. This could be related to insulin resistance as well. Leptin is a strong modulator of biochemical pathways and metabolic fluxes which in turn causes a redistribution of glucose fluxes. Research suggests that if leptin secretion at an early time due to overeating may have a correlation with obesity and glucose intolerance. Over feeding decreases the rate of glucose infusion needed to maintain regular glucose levels. Due to this, the intake of carbohydrates was drastically altered because after 7 days of over eating the rate of glucose intake was decreased. Over feeding drastically decreased insulin’s inhibition of glucose production. Voluntary over feeding decreases the extent to which leptin affects food consumption. In an experiment with over fed rats and rat control group, this was proved by injecting leptin to both groups. The group of over fed rats had no response to the leptin therefore their food intake did not decrease but the control group was seen to have the expected outcome of leptin. In the control group, the leptin functioned as expected and inhibited food intake. The increase in body mass due to increase in food intake may be related to causing insulin resistance as well as early increase in glucose production during hyperphagia. Therefore, it is proved that the increase in food consumption plays a role in the paralysis/ decrease of the leptin system and a decreased action of insulin on carbohydrate metabolism.
It is a linear polymer: linking the alpha-carboxyl group of one amino acid to the alpha amino group of another amino acid => PEPTIDE BOND (covalent bond).
In some proteins, the linear polypeptide chain is cross-linked: Disulfide bonds.
The primary structure is a polypeptide, in which:
each amino acid in the peptide is a residue
there is a regularly repeating segment called the main chain or backbone, and a variable part, comprised of the side chain.
The primary structure of a protein is a linear polymer with a series of amino acids. These amino acids are connected by C-N bonds, also known as peptide bonds. The formation of peptide bonds produce water molecules as a by-product when an amino acid N-terminal loses hydrogen and another amino acid C terminal loses -hydroxyl group. Thus, polypeptide, or polypeptide chain, is a term that describes the multiple connected peptide bonds between numerous amino acids. Each amino acid in a polypeptide chain is a unit, commonly known as a residue. These chains have a planar backbone, as the peptide bonds have double bond characteristics due to the existence of resonance between the carbonyl carbon and the nitrogen where the peptide bonds form. The primary structure of each protein has been precisely determined by the specific genes. The C-N bond in an amino acid's chain has the character of a double bond. This bond has a short length and stable. It cannot be rotated. This double-bond character can be explained structurally, in that the R groups in amino acid chains avoid steric clash.
Amino acids are linked by peptide bonds to form polypeptide chain; each amino acid unit is known as a residue; a polypeptide chain constructed by the same unit is known as the main chain or backbone and a changing R group, side chains.
Protein structures are governed primarily by hydrophobic effects and by interactions between polar residues and other types of bonds. The hydrophobic effect is the major determination of original protein structure. The aggregation of nonpolar side chains in the interior of a protein is favored by the increase in Entropy of the water molecules that would otherwise form cages around the hydrophobic groups. Hydrophobic side chains give a good indication as to which portions of a polypeptide chain are inside, out of contact with the aqueous solvent.
Hydrogen bonding is a central feature in protein structure but only make minor contributions to protein stability. Hydrogen bonds fine tune the tertiary structure by selecting the unique structure of a protein from among a relatively small number of hydrophobically stabilized conformations.
Disulfide bonding can form within and between polypeptide chains as proteins fold to its native conformation. Metal ions may also function to internally cross link proteins.
Extreme temperatures will result in the unfolding of a polypeptide chain leading to a change in structure and often a loss of function. If the protein functioned as an enzyme denaturing will cause that protein to lose its enzymatic activity. As the temperature of a solution containing the protein is raised, the extra heat causes twisting and bending of bonds. As proteins begin to denature the secondary structure of the protein is lost and adopts a random coil configuration. Covalent interaction between amino acid side chains such as disulfide bonds are also lost.
At high or low pH levels the protein will denature due to the lose or gain of a proton and, therefore, will lose their charge or become charged, depending on which way the pH is changed and by how much. This will eliminate many of the ionic interactions that were necessary for maintenance of the folded shape of the protein. As a result the change in structure will cause a change or loss of function.
Determination of Primary Structure: Amino Acid Sequencing
After the polypeptide has been purified, the composition of the polypeptide should be established. To determine which amino acid and how much of each is present, the entire strand is degraded by amide hydrolysis (6N HCl, 1100C, 24hr) to produce a mixture of all free amino acid residues. The mixture is separated and its composition recorded by amino acid analyzer. The amino acid analyzer establishes the composition of a polypeptide by giving a chromatogram, which records the peaks of each amino acid presents in the sequence. However, the amino acid analyzer can only give the composition of a polypeptide, not the order in which the amino acids are bound to one another.
To determine the amino acid sequence, it usually starts from the determination of the amino terminal of the polypeptide. The procedure is known as Edman degradation, and the reagent employed is phenyl isothiocyanate.
Phenyl isothiocyanate
In Edman degradation, the terminal amino group adds to the isothiocyanate reagent to produce a thiourea derivative. Treating with mild acid, the tagged amino acid is turned into a phenylthiohydantoin, and the remainder of polypeptide is unchanged. Since the phenylthiohydantoins of all amino acid are known, the amino terminal of the original polypeptide can be identified easily. However, Edman degradation can only be used to identify the amino end of the polypeptides; therefore, for polypeptides that are made up by hundreds of amino acids, it is not a practical method in general. In addition, multiple degradation rounds will build up impurities which will seriously affect the yield of peptide. High yield means not completely quantitative, and with each step of degradation, incompletely reacted peptide will mix with the new peptide, resulting in a intractable mixture.
In other words, secondary structure refers to the spatial arrangement of amino acid residues that are nearby in the sequence. The alpha helix, and beta strands are elements of secondary structure.
Secondary structures of proteins are typically very regular in their conformation. They are the spatial arrangements of primary structures. Alpha Helices and Beta Pleated Sheets are two types of regular structures. An interesting bit of information is that certain amino acids making up the polypeptide will actually prefer certain folding structures. The Alpha Helix seems to be the default but due to interactions such as sterics, certain amino acids will prefer to fold into Beta pleated sheets and so on. For example, amino acids such as Valine, Isoleucine, and Threonine all have branching at the beta carbon, this will cause steric clashes in an alpha helix arrangement. Glycine is the smallest amino acid and can fit into all structures so it does not favor the helix formation in particular. Therefore, these amino acids are mostly found where their side chains can fit nicely into the beta configuration.
The structure of polypeptide main chains is mostly of hydrogen-bonding; each residue has a carbonyl group that is a good hydrogen- bond acceptor; nitrogen- hydrogen group, a good hydrogen- bond donor.
Alpha helix look like the outside of structure.
+ Right hand appeared in right bottom of Rachamanda plot often
+ Left hand (LOOP): rare on the left top of Ramachandran plot
An alpha coiled coil consists of two or more alpha helices intertwined, creating a stable structure. This structure provides support to tissues and cell, contributing to the cell cytoskeleton and muscle proteins such as myosin and tropomyosin. Alpha keratin consists of heptad repeats (imperfect repeats of 7 amino acid sequences). This facilitates bonding between the two or more helices.
II. Collagen
Collagen is another type of fibrous protein that consists of three helical polypeptide chains. It is the most abundant protein found in mammals, making up a large component of skin, bone, tendon, cartilage, and teeth. Wrinkles are also caused by the degradations of this protein. In the structure of collagen, every third residue in the polypeptide is glycine because it is the only residue that is small enough to fit in the interior position of the superhelical cable. Unlike normal alpha helices, each collagen helix is stabilized by steric repulsion of the pyrrolidine rings of the proline and hydroxyproline residues. However, the three strands intertwined are stabilized by hydrogen bonding.
Motifs are simple combinations of the secondary structure such as the helix-turn-helix, which consist of two helices separated by a turn. The helix-turn-helix motif are usually found in DNA-binding proteins.
II. Domains
Domains, or compact globulars, consist of multiple motifs.They are polypeptide chains folded into two or more compact regions connected by turns or loops. Their structure is spherical, which is beneficial for the protein because it conserves space. Generally, inside the globular protein consist of hydrophobic amino acids such as leucine, valine, methionine, and phenylalanine. The outside consists of amino acids with hydrophilic tendencies such as aspartate, glutamate, lysine, and arginine. An example of a globular protein is myoglobin, which is the oxygen carrier in muscle. It is an extremely compact molecule made of only alpha helices (70%) except for loops and turns (30%).
Transmembrane and Non-Transmembrane Hydrophobic Helix
Studying the topography of transmembrane and non-transmembrane helix have helped answer many questions about membrane protein insertion. Specifically, studying the sequence and lipid dependence of the topography provide insights into post-translational topography changes. Furthermore, studying topography has lead to the design of hydrophobic helices that have biomedical applications. For example, a tumor marker called pHLIP peptide has been designed.
Different tests have been used to show the various effects on the hydrophobic helices. For example, hydrophilic residues such as tryptophan and tyrosine destabilize the transmembrane state. The hydrophilic domains cannot cross the membrane so it blocks any transmembrane and non-transmembrane equilibration. Furthermore, charged ionized residues also destabilize the transmembrane state. Stabilization of the transmembrane is also achieved in helix-helix interaction. Moreover, anionic lipids promote membrane binding of hydrophobic peptides and proteins.
Alpha helices, beta strands, and turns are formed by a regular pattern of hydrogen bonds between the peptide N-H and C=O groups of amino acids that are near one another in the linear sequence. Such folded segments are called secondary structure.
The alpha-helix consists of a single polypeptide chain in which the amino group (N-H) hydrogen bonds to a carboxyl group (C=O) 4 residues away. The alpha - helix is a rod-like structure. The tightly coiled backbone of the chain forms the inner part of the rod and the side chains extend outward in a helical array. This results in a clockwise coiled structure, which is known as a "right handed" screw sense. This folding pattern, along with the beta-pleated sheets were actually proposed by Linus Pauling and Robert Corey half a decade before people could actually see it. Most of the alpha strands are located in the lower left corner or upper right corner of the Ramachandran diagram . Essentially, most of the alpha helices are found in the right-hand helices area. An alpha helix is especially suited for cross-membrane proteins because all of the amino hydrogen and carbonyl oxygen atoms of the peptide backbone can interact to form intrachain hydrogen bonds while its aliphatic side chains can stabilize in hydrophobic environment of cell membrane.
Alanine, leucine and glutamic acid (existed as glutamate at physiological pH) are the most common residues present in alpha-helices.
The alpha-helix content of protein ranges widely, from none to almost 100%.
In general, the alpha helix is the "normal" shape of a polypeptide chain; however, features of certain amino acids disrupt alpha helix formation and instead favor beta strand formation. Amino acids with branching at the beta carbon (i.e. valine, threonine, and isoleucine) are problematic because they crowd the peptide backbone. H-bond accepting/donating groups attached to the beta carbon (i.e. serine, asparagine, and aspartate) can bond with backbone amine and carboxyl groups, again interfering with alpha helix formation.
While individual amino acids may favor one form or another, predicting the 2° structure of even a short (<7 amino acid) peptide strand is only 60-70% accurate. Such variability suggests other factors, like tertiary interactions with amino acids further down the chain, influence the folding into its observed 3° structure.
Beta-strand is:
Around ʊ = 120° and ϕ = -120°
You have the angle, and you form the zigzag
The zigzag have the distance between amino acids is 3.5 Angstrom
In contrast to the alpha helical structure, Beta Sheets are multiple strands of polypeptides connected to each other through hydrogen bonding in a sheet-like array. Hydrogen bonding occurs between the NH and CO groups between two different strands and not within one strand, as is the case for an alpha helical structure. Due to its often rippled or pleated appearance, this secondary structure conformation has been characterized as the beta pleated sheet. The beta strands can be arranged in a parallel, anti-parallel, or mixed (parallel and anti-parallel) manner.
Anti-parallel Beta Strand
The anti-parallel configuration is the simplest. The N and C terminals of adjacent polypeptide strands are opposite to one another, meaning the N terminal of one peptide chain is aligned with the C terminal of an adjacent chain. In the anti-parallel configuration, each amino acid is bonded linearly to an amino acid in the adjacent chain.
Parallel Beta Strand
The parallel arrangement occurs when neighboring polypeptide chains run in the same direction, meaning the N and C terminals of the peptide chains align. As a result, an amino acid cannot bond directly to the complementary amino acid in an adjacent chain as in the anti-parallel configuration. Instead, the amino group from one chain is bonded to a carbonyl group on the adjacent chain. The carbonyl group from the initial chain then hydrogen bonds to an amino group two residues ahead on the adjacent chain. The distortion of the hydrogen bonds in the parallel configuration affects the strength of the hydrogen bond because hydrogen bonds are strongest when they are planar. Therefore, due to this distortion of hydrogen bonds, parallel beta sheets are not as stable as anti-parallel beta sheet (exp: formation of parallel beta sheet with less than 5 residues is very uncommon).
The side chains of beta strands are arranged alternately on opposite sides of the strand. The distance between amino acids in a beta strand is 3.5 Å which is longer in comparison to the 1.5 Å distance in alpha strands. Because of this, beta sheets are more flexible than alpha helices and can be flat and somewhat twisted. The average length of beta sheets in a protein is 6 amino acid residues. The actual length ranges from 2 to 22 residues.
Ramachandran Plot: Beta strands are found in the purple region
Beta sheets are graphically found in the upper left quadrant of a Ramachandran plot. This corresponds to ψ angles of 0° to 180° and Φ angles of -180° to 0°.
The schematic model of beta sheets
Visual representations in 3D models for beta sheets are traditionally denoted by a flat arrow pointing in the direction of the strand.
Loop is everything, but what is alpha helix and beta-strand does. It is related to secondary structure of protein.
Polypeptide chains can change direction by making reverse turns and loops. Alpha helices and beta strands are connected by these turns and loops. Most proteins have compact, globular shape owing to reversals in the direction of their polypeptide chains, which allows the polypeptide to create folds back onto itself. In many reverse turns, the CO group of residue i of a polypeptide is hydrogen bonded to the NH group of residue i+3. A turn helps to stabilize abrupt directional changes in the polypeptide chain. Loops are more elaborate chain reversal structures that are rigid and well defined. Loops and turns generally lie on the surfaces of proteins so they often participate in interactions between proteins and other molecules. In a loop, there are no regular structures as can be found in helices or beta strands.
Two hypotheses have been proposed for the role of turns in protein folding. In one view, turns play a critical role in folding by bringing together interactions between regular secondary structure elements. This view is supported by mutagenesis studies indicating a critical role for particular residues in the turns of some proteins. Also, nonnative isomers of X-Proline peptide bonds in turns can completely block the conformational folding of some proteins. In the opposing view, turns play a passive role in folding. This view is supported by the poor amino-acid conservation observed in most turns. Also, non-native isomers of many X-Pro peptide bonds in turns have little or no effect on folding.
A motif is when secondary structure elements combine in specific geometric arrangements. Beta hairpin turns are one type of arrangement; they are one of the simplest structures and then are found in globular proteins. Upon turning, the antiparallel strand can bind effectively through hydrogen bonding between the carbonyl carbon and the peptide backbone nitrogen. It has been shown that 70% of beta-hairpins are less than seven residues long; the majority being 2 residues long. There are two types of two-residue beta hairpin turns. The first, Type I, forms a left-handed alpha-helical conformation. This left-handed conformation has a positive phi angle due to the properties of the aforementioned amino acids. Glycine does not have a side chain to sterically interfere with the turned amino acid sequence. Asparagine and aspartate both readily form hydrogen bonds with the carbonyl oxygen as a hydrogen bond acceptor. The second amino acid in the Type I turn is usually glycine due to steric hindrance that would result using any amino acid with a side chain. In a Type II beta hairpin turn, the first residue can only be glycine due to steric hindrance. However, the second residue is usually polar, such as serine or threonine.
Fibrous protein such as alpha-keratin and collagen consist of two right handed alpha helix intertwined to form a type of left handed super-helix called an alpha coiled coil.
The two helices in this type of protein usually cross-linked by weak interaction such as Van der Waals forces force and ionic interaction. The side chain interaction can be repeat every seven residues, forming heptad repeats. Another form of fibrous protein, that of collagen, exists as three helical polypeptide chains. These chains are relatively long, ~1000 residues, and because of overcrowding, glycine appears once every three residues. While the helix is stabilized by the steric repulsions, the three strands are stabilized by hydrogen bonding.
These protein usually serve structural roles in organisms, alpha-keratin is commonly found in the cytoskeleton of a cell, as well as certain muscle proteins. Collagen is often found in teeth, skin, and tendons.
The science of predicting what polypeptide chain will conform to which secondary structure group (alpha-helix, beta-sheet/strand or turns/loops) is not particularly exact. However, various frequencies of secondary structure formation of certain amino acids have been recorded in actual scientific experimentation, and these values can allow scientists to predict the folding of a protein based on its amino acid composition with about 60-70% accuracy. Stretches of six or less residues can usually be predicted with this accuracy. Although, certain amino acids tend to fold in its preferred conformation, there are of course exceptions and so secondary structure prediction is not always accurate. Tertiary interactions, interactions with residues further apart from each other, can also determine the folding structures. Each amino acid has a preference for either secondary structure, but it normally is only a small preference towards one in comparison to another, therefore, this unfortunately does not mean much. Amino acids can appear in an alpha-helix in one protein and also in a beta-sheet in another. Due to the unpredictability of the secondary structure based on the sequence of amino acids, secondary structures are being analyzed and predicted in relations to a similar family of sequences.
Various techniques have risen throughout history in the study of secondary structural prediction. With the aid of computers, prediction has been a pursued research topic in bioinformatics and many approaches continue to be proposed. After Linus Pauling and Robert Corey discovered the periodic alpha helix and beta sheet structures within proteins in 1951, further elucidation of protein structure prediction began to grow. A major method in secondary structure prediction was the Chou-Fasman method; it yielded a 50-60% accuracy. This method based its predictions on assigning a set of prediction values to a certain amino acid residue and then applied an algorithm to that value. Shortly after, further improvements were made on this method, the GOR method, which was developed in the late 1970s and utilized information theory|entropy and information concepts for secondary structure prediction. When devised, the method was about 65% accurate, however, improvements have also been made to it. There are deductive techniques in which similar sequences are found in already identified proteins. This method is accomplished by having computer software search databases of identified proteins. Opposite of that would be the Ab initio method, which builds 3-dimensional models without looking at similar residue sequences. This method is based on hydrogen bonding principals and localization.
Other methods and factors of folding prediction include analyzing the basic chemical tendencies of the side chains of amino acids to determine its preference in secondary structure. The alpha-helix is taken as the default structure, thus amino acids that destabilize alpha-helices are often found in beta-pleated sheets or loops and turns. For instance, valine, threonine, and isoleucine will often destabilize the helix because of branching of the beta carbon. These three amino acid residues are more often found in beta-pleated sheets, where their side chains will lie in a separate plane than the main chain. There are also amino acid residues that prefer neither alpha-helices nor beta-pleated sheets, for example, Proline has a restricted phi angle of ~60° degrees and no NH group, all due to the fact that it is cyclic. This will disrupt both alpha-helices and beta-pleated sheets, thus is found mostly in loops and turns. A counter-intuitive example is glycine which, according to its small size, theoretically can fit in any structure easily, but in reality it tends to avoid alpha-helices and beta-sheets also. The folding definitely also relies on chemical interactions between the side chains so the surrounding amino group interactions also affect the tendency of folding. These tendencies are reflected in the frequencies of secondary structure for individual amino acids.
The relative tendencies of secondary structures for particular amino acids are listed below:
alpha-helix: Glu, Ala, Leu, Met, Lys, Arg, Gln, His
Torsion angles are also called dihedral angles. The torsion angle is the measure in degrees in bonds between atoms. Folding of proteins are influenced by the degree of rotation amino bonds can hold. There are two different types of torsion angles existing in polypeptide bonds. Phi, φ is the angle between the α-carbon and the nitrogen atom of a peptide bond. The other bond is called psi, ψ which is the angle between the α-carbon and the carbonyl group. To measure φ, one must look from the nitrogen atom towards the α-carbon to measure if the angle is negative or positive. The angle is negative if the α-carbon rotates counterclockwise and vice versa. Furthermore, to measure ψ, one must look from the nitrogen atom towards the carbonyl group. Likewise, the angle is negative if the carbonyl group rotates counterclockwise and vice versa.
The Ramachandran Diagram, created by Gopalasamudram Ramachandran, helps to determine if amino acids will form alpha helices, beta strands, loops or turns. The Ramachandran Diagram is separated into four quadrants, with angle ϕ as the x axis and angle ψ as the y-axis. The combinations of torsion angles will put the amino acids in specific quadrants, which determine whether it will form an alpha helix, beta strand, loop, or turn. Those that fall in quadrants 1 and 3 a few times in a row form alpha helices, and those that repeat in quadrant 2 form beta strands. Quadrant 4 is generally disfavored because of steric hindrance. Also, it is mostly impossible because the different torsion angles combinations in quadrant 4 can't exist because they cause collisions between the atoms of the amino acids. If the amino acids land in the different quadrants, with no repeats, then they become loops or turns. Furthermore, the principle of steric exclusion states that two atoms cannot occupy the same place simultaneously.
Myoglobin is one of example of tertiary structure. Myoglobin is an extremely compact molecule. It is oxygen carrier in muscle is a single polypeptide chain of 153 amino acids. The capacity of myoglobin to bind oxygen depends on the presence of HEME, a non polypeptide PROSTHETIC group consisting of protoporphyrin IX and a central iron atom.
The tertiary structure of a protein is the three-dimensional structure of the protein. This three-dimensional structure is mostly determined by the amino acid sequence, which is denoted by the primary structure of the protein, however the amino acid sequence cannot entirely predict on how the three-dimensional structure is formed. Another contributing factor to the final shape of the tertiary structure is based on the environment in which the protein is synthesized. The tertiary structure is stabilized by the sequence of hydrophobic amino acid residues in the backbone of the protein. The interior consists on hydrophobic side chains while the surface consists of hydrophilic amino acids that interact with the aqueous environment.
Tertiary structure is formed by interactions between side chains of various amino acids - in particular disulfide bonds formed between two cysteine groups. At this stage, some proteins are complete, while other proteins incorporate multiple polypeptides subunits which creates the quaternary structure.
Nucleation-condensation model. The tertiary folding process is very structured with key intermediates. When a protein starts to fold, localized areas of the protein first begin folding. Then, the individual localized folds come together to complete the tertiary structure. The key concept is that when a correct fold is achieved, that fold is retained until all other parts of the protein are also correctly folded. This folding process follows reason because a random trial and error folding process would not only take much more time to complete, but also would require much more input energy.
Tertiary structure refers to the spatial arrangement of amino acid residues that are far apart in the sequence and to the pattern of disulfide bonds. Tertiary structure is also the most important protein structure that is used in determining the enzymatic activity of proteins.
A lobster's exoskeleton is not an example of keratin (it is made of chitin, a polysaccharide).A dog's fur is an example of keratin.
Cysteine, an amino acid containing a thiol group, is responsible for the disulfide bonds that hold a tertiary structure together. In the tertiary structure, when two helices come together, they may be linked by these disulfide bonds. A tertiary structure with fewer disulfide bonds form less rigid structures that are flexible, but still strong and can resist breakage such as hair and wool. While tertiary structures that contain more crossed disulfide bonds, formed by cysteine residues, lead to stronger, stiffer and harder structures such has exoskeletons. Others examples of protein that contain more disulfide bonds include claws, nails, and horns.
A structure made of two a-helices such as keratin can be found in living organisms. Immunoglobulin, also known as antibodies, is an example of an all beta-sheet protein fold. It consists of approximately 7 anti-parallel beta-strands arranged in 2 beta-sheets. For instance, if a cysteine is mutated to another amino acid it can code to a different protein which would lead to incorrect folding.
Some polypeptide chains fold into several compact regions. These regions in a polypeptide chain are called domains and generally range from 30 to 400 amino acids. On average, domains contain roughly 100 amino acids. Each domain forms its own tertiary structure which contributes to the overall tertiary structure of the protein. These domains are independently stable. Stabilization is caused by metal ions or disulfide bridges that cause the folding of polypeptide chains. Different proteins may have the same domains even if the overall tertiary structure is different.
There are four types of domains:
All-α domains - Domains made purely from α-helices.
All-β domains - Domains made purely from β-sheets.
α+β domains - Domains made both of α-helices and β-sheets.
α/β domains - Domains made from both α-helices and β-sheets layered in a β,α,β fashion with a α-helix sandwiched in between 2 β-sheets.
In order for a protein to be functional (except in food), it must have an intact tertiary structure. If a tertiary structure of a protein is disrupted, it is said to be denatured. Once a protein is denatured, it will not be able to perform its intended or original function. A primary cause for an alteration of the tertiary structure is a mutation in the gene encoding a protein. The mutation in the gene can cause a domino effect that will lead to the degradation of the tertiary structure. Degradation can cause several diseases, one of which is called cystic fibrosis. Cystic fibrosis is brought about by a mutation of a genes called cystic fibrosis transmembrane conductance regulator (CFTR). This disease causes the exocrine glands to overproduce mucus. Most commonly, CF patients suffer from lung failure by the age of early 20-30. Diabetes insipidus, familial hypercholesterolemia, and Osteogenesis imperfecta are also diseases that originate from degraded proteins. A mutation in the tertiary structure itself, rather than from a mutation in the nucleotide sequence can also lead to diseases. Such mutated proteins can also aggregate and become insoluble deposits called amyloids, and therefore lose the ability to function. A common mutation is when a hydrophobic R group folds in, rather than out, in a hydrophobic environment. The inherited form of Alzheimer's disease is one disease that is caused by mutated tertiary structure. Another disease includes mad cow disease, which is caused due to a-helix (which are soluble) mutating into b-sheets (which are insoluble and cause amyloid deposits). [7]
The folding of a protein is dependent on the amino acid sequence laid out in the primary structure. It is also dependent on the environment in which the folding occurs. In a hydrophobic environment, the hydrophobic side chains of the amino acids of the protein fold out while the hydrophilic side chains fold in and vice versa for a hydrophilic environment. An example of a protein that is folded in a hydrophobic environment is Porin. Its hydrophilic side chains are folded in which creates a channel for water to pass through. Amino acids that have nonpolar/hydrophobic side chains such as leucine, valine, methionine, phenylalanine, and isoleucine would be folded out in the folding of the protein in a hydrophobic environment. Likewise, in a hydrophilic environment, amino acids with polar side chains such as glutamine and asparagine fold outwards and the hydrophobic side chains would fold inwards.
The tertiary structure of a protein is determined through X-Ray Crystallography and Nuclear Magnetic Resonance (NMR) Spectroscopy. X-ray Crystallography was the first method used to determine the structure of proteins. X-ray crystallography is one of the best methods because the wavelength of an x-ray is similar to that of covalent bonds found throughout proteins, creating a clearer visualization of a molecule's structure. The scattering of x-rays by electrons is analyzed to determine the structure of proteins. In order to use x-ray crystallography, the protein in question must be in crystal form. Some proteins crystallize readily, while others do not. For those proteins that do not crystallize readily, nuclear magnetic resonance (NMR) spectroscopy must be used to determine its structure. NMR spectroscopy uses the spin of nuclei with a magnetic dipole and chemical shifts to determine a molecule’s relative position.
Hemoglobin is one of example of quaternary structure. Hemoglobin, the oxygen-carrying protein in blood, consists of two subunits of one type (designated alpha) and two subunits of another (designated beta).
Atomic structure of the 50S Subunit from Haloarcula marismortui. Proteins are shown in blue and the two RNA strands in orange and yellow.[11] This is an example of the tertiary structure of the large unit of a ribosome
A quaternary structure refers to two or more polypeptide chains held together by intermolecular interactions to form a multi-subunit complex. The interactions that hold together these folded protein molecules include disulfide bridges, hydrogen bonding, hydrogen bonding interactions, hydrophobic interactions interactions and London forces. These forces are usually conveyed by the side chains of the peptides.
These polypeptide chains are the subunits of a protein, capable of taking part in a variety of functions such as serving as enzymatic catalysts, providing structural support in the cytoskeletons of cells, and even composing the hair on our heads.
The peptides of the protein can be identical or different. Insulin is a dimer consisting of two identical peptides, while Hemoglobin is a tetramer consisting of two identical alpha subunits and two identical beta subunits.
Computer-generated image of insulin hexamers highlighting the threefold symmetry, the zinc ions holding it together, and the histidine residues involved in zinc binding.
Structure of human hemoglobin. The protein's α and β subunits are in red and blue, and the iron-containing heme groups in green. From PDB1GZXProteopediaHemoglobin
Hemoglobin
Consists of 2 alpha and 2 beta groups
Has a globular shape
Has reverse turns that contribute to circular shape of the protein
Aquaporin
Made of 6 alpha helices
Form hydrophobic loops
Forms tetramers in the cell membrane with each monomer acting as water channels
The quaternary structure of a protein can be denatured by breaking the covalent and non-covalent forces that keep it together. Heat, urea or guanidinium chloride will denature a protein by disrupting the non-covalent forces, while beta-mercaptoethanol will break disulfide bridges by reducing the bridges.
A protein is never "half folded", at the point where the concentration of the denaturant is in between that of the folded and unfolded form of the protein, there are two structures that exist. Folded and Unfolded, at a ratio of 1:1
Proteins are either folded, or not. There does not exist a stage where a protein is "half-folded". This can be observed by slowly adding denaturant to a protein. This will result in a sharp transition, from the folded state to the unfolded state, suggesting there only exist these two forms. This is a result of cooperative transition.
For instance, if a protein is put in a denaturant where only one part of the protein is unstable, the entire protein will unfold. This is due to the domino effect where destabilizing one part of the protein will in turn destabilize the remainder of the structure. When a protein is in conditions which correspond to the middle of the transition between folded and unfolded, there is a 50/50 mixture of folded and unfolded protein, instead of 'half-folded' protein.
After all is said about being in one structure or the other, there must be something in between them on an atomic level. Unfortunately, this is an area that is still under development, and much research is still being done. Theories such as the condensation Nucleation Principle are concerned with this area of protein folding.
The properties of quaternary structure:
Polypeptide chains can assemble into multisubunit structure
Refers to the spatial arrangement of subunits and the nature of their interactions
If one takes each student in a class to be a different amino acid, each right hand to be an alpha-carboxyl group, each left hand to be an alpha-amino group, and the head to be the R group; then by joining right hands to left hands, the class will form a polypeptide. The "bonds" joining the hands will be peptide bonds.
This can be considered the primary structure of a protein.
If one then takes students and "attract" them to other students 4 "bonds" away, this structure will then fold into a secondary structure; namely the alpha-helix. If the students were put into lines and were attracted to respective students in another line, they would form a beta-pleated sheet.
Now imagine that the heads, or R groups, vary in areas such as personalities, or polarity, like will attract like. The people who are more compatible will then gather together, for instance, hydrophobic areas will usually gather together in the center while surrounded by hydrophilic areas. This makes up the tertiary structure.
Now add in a different class, the people from the new class would have their own tertiary structure, these new people will then come in and react with the original class to form quaternary structures.
Human attempt to manipulate protein assemblies (Quaternary Structures)
Controlling the quaternary structures is currently catching more and more interest in academics. There are many advantages in manipulating protein assemblies. Firstly, people are able to grow/synthesize enzymes that are beneficial to human. Yet, to get these enzymes to work is the hard part. For example, nitrogenase, the enzyme that can fix nitrogen gas to yield ammonia, can only work under aerobic environment and coupled with ATP as energy source. In addition, researchers have revealed that nitrogenase is compose of two proteins, one for ATP coupling electron source and the other is the reactive center for nitrogen fixation. The two protein assemble to work as a whole. Recently, scientists remove the ATP coupling protein and replace it with a Ruthenium complex. It turned out that Ruthenium complex can provide electrons with light exposure. Now scientists don't have to deal with the complicate chemistry of coupling ATP, but just shine lights on engineered nitrogenase to get it work! Secondly, protein assemblies can have a lot of clinical/material applications. Ferritin is a family of high-order protein assembly family, usually 12mers or 24mers. Previous researches showed it can absorb large amount of Fe ion. Many researchers are working to control the association and dissociation of Ferritins, seeking for solutions of drug delivery, gas storage, metal harvest and etc. Many approaches have been developed to control protein assembling. Some of them include the following:
1. Transition metal-directed. Metal centers in protein are important, not only because they are reactive centers, but also they help stabilize the shape of protein by coordination. Many amino acids are ligands by themselves. Cysteine, Histidine, lysine are the common ones. Plus, researchers can engineer inorganic ligands onto proteins by cysteine substitution. Thus, introducing inorganic ligands much broaden the horizon of protein assemblies.
the structure of Phenanthroline (inorganic ligand).the structure of Terpyridine (inorganic ligand).
Metal-ligand bonding has several properties. Most obviously, it is a strong interaction. It is stronger than hydrogen bond and weaker than covalent bond. Therefore metal-ligand bond is strong yet not so strong that it is still reversible. Spatially speaking, metals have its coordination orientation, mostly, octahedral and tetrahedral. This property provides human great convenience in arranging proteins spatially.
shown is the cartoon model of a dimer of two terpyridine-labeled proteins.shown is the cartoon model of a trimer of three phenanthroline-labeled proteins.
2. Hydrophobic interaction. In aqueous environment, amino acid with hydrophobic side chains tend to aggregate together to minimize the exposure to water. Researchers utilize this character and engineer certain matching pair of non-polar amino acids onto proteins to obtain protein oligomers in water solution.
3. Salt bridges. It is well known that amino acids have different pI's. So at certain pH, some amino acids are negatively charged, some are positively charged. If an area on a protein is occupied by mostly negatively charged amino acid and another area is occupied by positively charged amino acids, proteins can aggregate by electrostatic attraction. However, this technique is usually not so selective.
More technique to direct protein assemblies are being investigated, such as coiled-coil. Mankind's potential to control quaternary structures is promising.
In most archaebacteria, a protein coat is the primary structure that surrounds and shapes the cell. This coat of protein armor is composed of a paracrystalline array of “surface layer proteins.”
Half a million surface layer proteins line next to each other to form a shell that encloses the cell. Inside the shell, they bind to sugar chains on the cell surface, or in the case of archaebacteria, interact directly with the membrane. This protein coat provides protection, and it can also assist in the gathering of nutrients and attachment to targets in the environment.
Kern, J. et al. Structure of surface layer homology (SLH) domains from Bacillus anthracis surface array protein. J. Biol. Chem. 286, 26041-26049 (2011)
Protein folding is a process in which a polypeptide folds into a specific, stable, functional, three-dimensional structure. It is the process by which a protein structure assumes its functional shape or conformation
Proteins are formed from long chains of amino acids; they exist in an array of different structures which often dictate their functions. Proteins follow energetically favorable pathways to form stable, orderly, structures; this is known as the proteins’ native structure. Most proteins can only perform their various functions when they are folded. The proteins’ folding pathway, or mechanism, is the typical sequence of structural changes the protein undergoes in order to reach its native structure. Protein folding takes place in a highly crowded, complex, molecular environment within the cell, and often requires the assistance of molecular chaperones, in order to avoid aggregation or misfolding. Proteins are comprised of amino acids with various types of side chains, which may be hydrophobic, hydrophilic, or electrically charged. The characteristics of these side chains affect what shape the protein will form because they will interact differently intramolecularly and with the surrounding environment, favoring certain conformations and structures over others. Scientists believe that the instructions for folding a protein are encoded in the sequence. Researchers and scientists can easily determine the sequence of a protein, but have not cracked the code that governs folding (Structures of Life 8).
Early scientists who studied proteomics and its structure speculated that proteins had templates that resulted in their native conformations. This theory resulted in a search for how proteins fold to attain their complex structure. It is now well known that under physiological conditions, proteins normally spontaneously fold into their native conformations. As a result, a protein's primary structure is valuable since it determines the three-dimensional structure of a protein. Normally, most biological structures do not have the need for external templates to help with their formation and are thus called self-assembling.
Protein renaturation known since the 1930s. However, it was not until 1957 when Christian Anfinsen performed an experiment on bovine pancreatic RNase A that protein renaturation was quantified. RNase A is a single chain protein consisting of 124 residues. In 8M urea solution of 2-mercaptoethanol, the RNase A is completely unfolded and has its four disulfide bonds cleaved through reduction. Through dialysis of urea and introducing the solution to O2 at pH 8, the enzymatically active protein is physically incapable of being recognized from RNase A. As a result, this experiment demonstrated that the protein spontaneously renatured.
One criteria for the renaturation of RNase A is for its four disulfide bonds to reform. The likelihood of one of the eight Cys residues from RNase A reforming a disulfide bond with its native residue compared to the other seven Cys residues is 1/7. Furthermore, the next one of remaining six Cys residues randomly forming the next disulfide bond is 1/5 and etc. As a result, the probability of RNase A reforming four native disulfide links at random is (1/7 * 1/5 * 1/3 * 1/1 = 1/105). The result of this probability demonstrates that forming the disulfide bonds from RNase A is not a random activity.
When RNase A is reoxidized utilizing 8M urea, allowing the disulfide bonds to reform when the polypeptide chain is a random coil, then RNase A will only be around 1 percent enzymatically active after urea is removed. However, by using 2-mercaptoethanol, the protein can be made fully active once again when disulfide bond interchange reactions occur and the protein is back to its native state. The native state of the RNase A is thermodynamically stable under physiological conditions, especially since a more stable protein that is more stable than that of the native state requires a larger activation barrier, and is kinetically inaccessible.
By using the enzyme protein disulfide isomerase (PDI), the time it takes for randomized RNase A is minimized to about 2 minutes. This enzyme helps facilitate the disulfide interchange reactions. In order for PDI to be active, its two active site Cys residues needs to be in the -SH form. Furthermore, PDI helps with random cleavage and the reformation of the disulfide bonds of the protein as it attain thermodynamically favorable conformations.
Posttranslationally Modified Proteins Might Not Renature
Proteins in a "scrambled" state go through PDI to renature, and their native state does not utilize PDI because native proteins are in their stable conformations. However, proteins that are posttranslationally modified need the disulfide bonds to stabilize their rather unstable native form. One example of this is insulin, a polypeptide hormone. This 51 residue polypeptide has two disulfide bonds that is inactivated by PDI. The following link is an image showing insulin with its two disulfide bonds. Through observation of this phenomenon, scientists were able to find that insulin is made from proinsulin, an 84-residue single chain. This link provides more information on the structure of proinsulin and its progression on becoming insulin. The disulfide bonds of proinsulin need to be intact before conversion of becoming insulin through proteolytic excision of its C chain which is an internal 33-residue segment. However, according to two findings, the C chain is not what dictates the folding of the A and B chains, but instead holds them together to allow formation of the disulfide bonds. For one, with the right renaturing conditions in place, scrambled insulin can become its native form with a 30% yield. This yield can be increased if the A and B chains are cross-linked. Secondly, through analysis of sequences of proinsulin from many species, mutations are permitted at the C chain eight times more than if it were for A and B chains.
There are various interactions that help stabilize structures of native proteins. Specifically, it is important to examine how the interactions that form protein structures are organized. In addition, there are only a small amount of possible polypeptide sequences that allow for a stable conformation. Therefore, it is evident that specific sequences are used through evolution in biological systems.
Helices and Sheets Predominate in Proteins because They Efficiently Fill Space
On average, about sixty percent of proteins contain a high amount of alpha helices, and beta pleated sheets. Through hydrophobic interactions, the protein is able to achieve compact nonpolar cores, but they lack the ability to specify which polypeptides to restrict in particular conformations. As seen in polypeptide segments in the coil form, the amount of hydrogen boding is not lesser than that of alpha helices and beta pleated sheets. This observation demonstrates that the different kinds of conformations of polypeptides are not limited by hydrogen bonding requirements. Ken Dill has suggested that helices and sheets occur as a result of the steric hindrance in condensed polymers. Through experimentation and simulation of conformations with simple flexible chains, it can be determined that the proportion of beta pleated sheets and alpha helices increase as the level of complication of chains is increased. Therefore, it can be concluded that helices and sheets are important in the complex structure of a protein, as they are compact in protein folding. The coupling of different forces such as hydrogen bonding, ion pairing, and van der Waals interactions further aids in the formation of alpha helices and beta sheets.
By investigating protein modification, the role of different classes of amino acid residues in protein folding can be determined. For example, in a particular study the free primary amino groups of RNase A were derivatized with poly-DL-alanine which consist of 8 residue chains. The poly-Ala chains are large in size and are water-soluble, thus allowing the RNase's 11 free amino groups to be joined without interference of the native structure of the protein or its ability to refold. As a result, it can be concluded that the protein's internal residues facilitates its native conformation because the RNase A free amino groups are localized on the exterior. Furthermore, studies have shown that mutations that occur on the surface of residues are common, and less likely to change the protein conformation compared to changes of internal residues that occur. This finding suggests that protein folding is mainly due to the hydrophobic forces.
George Rose demonstrated that protein domains consisted of subdomains, and furthermore have sub-subdomains, and etc. As a result, it is evident that large proteins have domains that are continuous, compact, and physically separable. When a polypeptide segment within a native protein is visualized as a string with many tangles, a plane can be seen when the string is cut into two segments. This process can be repeated when n/2 residues of an n-residue domain is highlighted with a blue and red color. As this process is repeated it can be seen that at all stages, the red and blue areas of the protein do not interpenetrate with one another. The following link shows an X-ray structure of HiPIP (high potential iron protein) and its first n/2 residues on the n-residue protein colored red and blue. Furthermore, the subsequent structures shown in the second and third row show this process of n/2 residue splitting reiterated as shown where the left side of the protein has its first and last halves with red and blue while the rest of the chain colored in gray. Through this example, it is clearly seen that protein structures are organized in a hierarchical way, meaning that the polypeptide chains are seen as sub-domains that are themselves compact structures and interact with adjacent structures. These interactions forms a larger well organized structure largely due to hydrogen bonding interactions and has an important role in understanding how polypeptides fold to form their native structure.
Since the side chains inside globular proteins fit together with much complementary its packing density can be almost like that of organic crystals. As a result, in order to confirm whether or not this phenomenon of high packing density was an important factor in contributing to protein structure, Eaton Lattman along with George Rose attempted to verify if there was an interaction between side chains that was preferred in a globular protein. They analyzed a total of 67 well studied structures of globular proteins, and concluded that there were no preferred interactions. This experiment demonstrated that packing is not what directs the native fold, but instead the native fold is necessary for packing of a globular protein. This notion can be further supported as members of a protein family result in the same fold despite their lack of sequence similarity and distant relationships.
In addition, structural experimental data have shown that there are a variety of ways that a protein's internal residues can become compact together in an efficient manner. In an extensive study done by Brian Matthews based on T4 lysozyme, which is produced by bacteriophage T4, it was found that changes in the residues of the T4 lysozyme only affected local shifts and did not result in any global structure change. The following link gives an X-ray view of T4 lysozyme and a brief biochemical description of the structure. Matthews took over 300 different mutants of the 164 residue T4 lysozyme, and compared them with one another. Also, it was observed that the T4 lysozyme could withstand insertions of about 4 residues while still not having any major structural changes to the overall protein structure nor enzyme activity. Furthermore, by using assay techniques it was demonstrated that only 173 of the mutants in T4 of the 2015 single residue substitutions done had significant amounts of enzymatic activity diminished. Through these experiments, it is evident that protein structures are extremely withstanding.
Levinthal's paradox is a thought experiment, also constituting a self-reference in the theory of protein folding. In 1969, Cyrus Levinthal noted that, because of the very large number of degrees of freedom in an unfolded polypeptide chain, the molecule has an astronomical number of possible conformations. An estimate of 3300 or 10143 was made in one of his papers.
The Levinthal paradox observes that if a protein were folded by sequentially sampling of all possible conformations, it would take an large amount of time to do so, even if the conformations were sampled at a rapid rate . Based upon the observation that proteins fold much faster than this, Levinthal then proposed that a random conformational search does not occur, and the protein must, therefore, fold through a series of meta-stable intermediate states.
In 1969 Cyrus Levinthal calculated that if a protein were to randomly sample every possible conformation as it folded from the unfolded state to the native state it would take an astronomical amount of time, even if the protein reached 100 billion conformations in one second. Observing that proteins fold in a relatively short amount of time, Levinthal proposed that proteins fold in a fixed and directed process. We now know that while protein folding is not a random process there does not seem to be a single fixed protein folding pathway.This observation came to be known as the Levinthal paradox. This paradox clearly reveals that proteins do not fold by trying every possible conformation. Instead, they must follow at least a partly defined folding pathway made up of intermediates between the fully denatured proteins and its native structure.
The way out of the Levinthal Paradox is to recognize cumulative selection. According to Richard Dawkins, he asked how long it would take a monkey poking randomly at a typewriter to reproduce "Methinks it is like a weasel", Hamlet's remark to Polonius. A large number of keystrokes, of the order of 1040 would be required. Yet if we suppose that each correct character was preserved, allowing the monkey to retype only the wrong ones, only a few thousand keystrokes, on average, would be needed. The crucial difference between these scenarios is that the first utilizes a completely random search whereas in the second case, partly correct intermediates are retained. This also reveals that the essence of protein folding is the tendency to retain partly correct intermediates, although the protein-folding problem is much more difficult than the one presented to Shakespeare example above.
In order to correctly understand the protein-folding problem, we must consider certain characteristics of protein. Since proteins are only marginally stable, the free-energy difference between the folded and the unfolded states of a typical 1000-residue protein is 42 kJ mol−1 and thus each residue contributes on average only 0.42 kJ mol−1 of energy to maintain the folded state. This amount is less than the amount of thermal energy, which is 2.5 kJ mol−1 at room temperature. This meagre stabilization energy means that correct intermediates, especially those formed early in folding, can be lost. The interactions that lead to cooperative folding, nonetheless, can stabilize intermediates as structure builds up. Thus, local regions that have significant structural preference, though not necessarily stable on their own, will tend to adopt their favored structures and, as they form, can interact with one other, resulting in increased stabilization. Nucleation-condensation model refers to this conceptual framework in solving the protein-folding challenge.
Intramolecular Interactions Role in the Folding Mechanism
Proteins folding forms energetically favorable structures stabilized by hydrophobic interactions clumping, hydrogen bonding and Van der Waals forces between amino acids. Protein folding first forms secondary structures, such as alpha helices, beta sheets, and loops. Different amino acids have different tendencies for whether they are going to form Alpha Helices, Beta sheets, or Beta Turns based upon polarity of the amino acid and rotational barriers. For example, the amino acids, valine, threonine, isoleucine, tend to destabilize the alpha helices due to steric hindrance. Thus, they prefer conformational shifts towards Beta sheets rather than alpha helices. The relative frequencies of the amino acids in secondary structures are grouped according to their preferences for alpha helices, beta sheets or turns (Table 1).
Table 1: Relative frequencies of amino acid residues in secondary structures
These structures in turn, fold to form tertiary structures, stabilized by the formation of intramolecular hydrogen bonds. Covalent bonding may also occur during the folding to a tertiary structure, through the formation of disulfide bridges or metal clusters. According to Robert Pain’s “Mechanisms of Protein Folding”, molecules also often pass through an intermediate “molten globule” state formed from a hydrophobic collapse (in which all hydrophobic side-chains suddenly slide inside the protein or clump together) before reaching their native confirmation. However, this means all the main chain NH and CO groups are buried in a non-polar environment, but they prefer an aqueous one, so secondary structures must fit together very well, so that the stabilization through hydrogen bonding and Van der Waals forces interactions overrides their hydrophilic tendencies. The strengths of hydrogen bonds in a protein vary depending on their position in the structure; H-bonds formed in the hydrophobic core contribute more to the stability of the native state than H-bonds exposed to the aqueous environment.
Water-soluble proteins fold into compact structures with non-polar, hydrophobic cores. The inside of protein contains non-polar residues in center (i.e. - leucine, valine, methionine and phenylalanine), while the outside contains primarily polar, charged residues (i.e. - aspartate, glutamate, lysine and arginine). This way the polar, charged molecules can interact with the surrounding water molecules while the hydrophobic molecules are protected from the aqueous surroundings. Minimizing the number of hydrophobic side chains on the outer part of the structure makes the protein structure thermodynamically more favorable because the hydrophobic molecules prefer to be clumped together, when surrounded by an aqueous environment (i.e. – hydrophobic effect). Proteins that span biological membranes (i.e. - porin) have an inside out distribution, with respect to the water-soluble native structure, they have hydrophobic residue covered outer surfaces, with water filled centers lined with charged and polar amino acids.
In “Folding Scene Investigation: Membrane Proteins”, a paper written by Paula J Booth and Paul Curnow, the authors attempt to answer how the folding mechanisms of integral membrane proteins with α helical structures work.
Studying the folding of membrane proteins has always been difficult as these proteins are generally large and made of more than one subunit. The proteins posses a high degree of conformational flexibility—which is necessary for them to perform their function in the cell. Also, these proteins have both hydrophobic surfaces, facing the membrane, and hydrophilic surfaces, facing the aqueous regions on either side of the membrane. The proteins are move laterally and share the elastic properties of the lipid bilayer in which they are embedded. In order to study these proteins, Booth and Curnow believe that one must manipulate the lipid bilayer and combine kinetic and thermodynamic methods of investigation.
Reversible Folding and Linear Free Energy
The free energy of protein folding is measured by reversible chemical denaturation. The reversible folding of a protein depends on this free energy. For the α helix proteins that were being studied, it was proven that a reversible, two-state process is followed. bR (a α helical membrane protein called bacteriorhodopsin) reversibly unfolds if SDS (a denaturant which is an anionic detergent) is added to mixed lipid, detergent micells. The two-state reaction involves a partly unfolded SDS state and a folded bR state.
By comparing the logs of the unfolding and folding rate, and the SDS mole fraction, a linear plot was generated proving a linear relationship. This plot proved that bR had a very high stability outside of its membrane—proving that it was unexpectedly stable. Furthermore, bR was so stable outside of the membrane that it would not unfold during a reasonable period of time without addition of denaturant.
Comparison with Water-Soluble Proteins
Booth and Curnow studied the 3 membrane proteins about which the most information is held: bR, DGK (Escherichia coli diacylglycerol kinase) and KcsA (Sterptococcus lividans potassium channel). These three membrane proteins were compared to water-soluble proteins (which fold by 2 or 3 state kinetics).
The overall free energy change of unfolding in the absence of denaturant was the same for water-soluble proteins and membrane proteins of similar size. This proves that it is the balance of weak forces rather than the types of forces that stabilize the protein that determines its stability. It was proven that H-bonds in the membrane proteins were of similar strength to those of the water-soluble proteins, rather than being stronger in membrane proteins as was expected.
Mechanical Strength and Unfolding Under Applied Force
Dynamic force microscopy can be used to measure the mechanical response of a particular region of a protein under applied force. The unfolding force in this case depends on the activation barrier. This unfolding has nothing to do with the thermodynamic stability of a protein.
For unfolding under applied force, the membrane proteins (especially bR) seem to follow the rules of Hammond behavior. The energy difference between two consecutive states of this reaction is reduced and the states become similar in structure.
Influence of Surrounding Membrane
Membrane proteins are influenced greatly by the membranes they are surrounded by. If the lipids incorporate in detergent micells—-increasing the stability of the lipid structure—both the protein and its folding are stabilized. Different combinations of different lipids can result in different stabilities or folding of membrane proteins. The size of the membrane can also affect the membrane protein.
Different types of lipids cause different membrane properties. A type of lipids called PE lipids have higher spontaneous curvatures than a second type of lipid called a PC lipid. By adding PE lipids to PC lipids the monolayer curvature of the bilayer increases. Increasing the curvature of the lipid bilayer increases the stability of the protein folding.
In mitochondria, the proteins that are made from the ribosomes are directly take in from the cytosol. Mitochondrial proteins are first completely synthesized in the cytosol as mitochondrial precursor proteins, then taken up into the membrane. The Mitochondrial proteins contain specific signal sequence at their N terminus. These signal sequences are often removed after entering the membrane but proteins entering membranes that has outer, inner, inter membrane have internal sequences that play a major movement in the translocation within the inner membrane.
Protein translocation plays a major role in translocating proteins across the mitochondrial membranes. Four major multi-subunit protein complexes are found in the outer and the inner membrane. TOM complexes are found in the outer membrane, and two types of TIM complexes are found integrated within the inner membrane: TIM23 and TIM22. The complexes act as receptors for the mitochondrial precursor proteins.
TOM: imports all nucleus encoded proteins. It primarily starts the transport of the signal sequence into the inter membrane space and inserts the transmembrane proteins into outer membrane space. A Beta barrel complex called the SAM complex is then in charge of properly folding the protein in the outer membrane. TIM23 found in the inner membrane moderates the insertion of soluble proteins into the matrix, and facilitates the insertion of transmembrane proteins into the inner membrane. TIM23, another inner membrane complex facilitates the insertion inner membrane proteins comprised of transporters that move ADP, ATP, and phosphate across the mitochondrial membranes. OXA, yet another inner membrane complex, helps insert inner membrane proteins that were synthesized from the mitochondria itself and the insertion of inner membrane proteins that were first transported into the matrix space.
File:Translocation.jpg
The place where the protein chain begins to fold is a topic that is greatly studied. As the nascent chain goes through the “exit tunnel” of the ribosome and into the cellular environment, when does the chain begin to fold? The idea of cotranslational folding in the ribosomal tunnel will be discussed. The nascent chain of the protein is bound to the peptidyl transferase centre (PTC) at its C terminus and will emerge in a vectorial manner. The tunnel is very narrow and enforces a certain rigidity on the nascent chain, with the addition of each amino acid the conformational space of the protein increases. Co translational folding can be a big help in reducing the possible conformational space by helping the protein to acquire a significant level of native state while still in the ribosomal tunnel. The length of the protein can also give a good estimate of its three dimensional structure. Smaller chains tend to favor beta sheets while longer chains (like those reaching 119 out of 153 residues) tend to favor the alpha helix.
The ribosomal tunnel is more than 80 Å in length and its width is around 10-20 Å. Inside the tunnel are auxiliary molecules like the L23, L22, and L4 proteins that interact with the nascent chain help with the folding. The tunnel also has hydrophilic character and helps the nascent chain to travel through it without being hindered. Although rigid, the tunnel is not passive conduit but whether or not it has the ability to promote protein folding is unknown. A recent experiment involving cryoEM has shown that there are folding zones in the tunnel. At the exit port (some 80 Å from the PTC), the nascent chain has assumed a preferred low order conformation. This enforces the suggestion that the chain can have degrees of folding at certain regions. Although some low order folding can occur, the adoption of the native state occurs outside the tunnel, but not necessarily when the nascent chain has been released. The bound nascent chain (RNC) adopts partially folded structure and in a crowded cellular environment, this can cause the chain to self-associate. This self-association, however, is relieved with the staggered ribosomes lined along the exit tunnel that maximizes the distances between the RNC.
Generation of RNC for studies:
One technique of generating RNC and taking snapshots as it emerges from the tunnel is to arrest translation. A truncated DNA without a termination sequence is used. This allows for the nascent chain to remain bound until desired. To determining the residues of the chain, they can be labeled by carbon-13 or nitrogen-15 and later detected by NMR spectroscopy. Another technique is the PURE method and it contains the minimal components required for translation. This method has been used to study the interaction of the chains and auxiliary molecules like the TF chaperone. This method is coupled with quartz-crystal microbalance technique to analyze the synthesis by mass. An in vivo technique in generating RNC chain can be done by stimulating it in a high cell density. This is initially done in an unlabeled environment, the cells are then transferred to a labeled medium. The RNC is generated by SecM. The RNC is purified by affinity chromatography and detected by SDS-PAGE or immunoblotting.
By generating the RNCs, many experiments can be done to study more about the emerging nascent chain. As mentioned above, the chain emerges from the exit tunnel in a vectorial manner. This enables the chain to sample the native folding and increases the probability of folding to the native state. Along with this vectorial folding, chaperones also help in favorable folding rates and correct folding.
Protein Entering the Mammalian ER:
The endoplasmic reticulum (ER) is a main checkpoint for protein maturation to ensure that only correctly folded proteins are secreted and delivered to the site of action. The protein entrance to the ER begins with recognition of a N’ terminus signal sequence. Specially, this sequence is detected by a signal recognition protein (SRP) causing the ribosome/nascent chain/SRP complex bind to the ER membrane. Then, the complex travels through a proteinaceous pore called Sec61 translocon which allows the polypeptide chain enter the lumen portion of the ER.
Processes in Conflict During Protein Folding:
After the protein enters the ER, the proteins break up into an ensemble of folding intermediates. These intermediates take three different routes. They are either folded properly and sent to be exported out of the endoplasmic reticulum (ER) into the cytosol, aggregated or picked out for degradation. These three processes are in competition to properly secrete a protein. In order for a protein to be properly secreted, the competition between folding, aggregation and degradation must be in favor of folding, so that folding occurs faster than the other processes. This balance is termed proteostasis. The balance of proteostasis can be tipped in favor of folding by either using smaller molecules to stabilize the protein (called co-factors) or increasing the concentrations of folding factors. This ability to control proteostasis allows scientists the power to overcome some of the protein folding diseases such as cystic fibrosis.
The proteins that are folded properly are ready for anterograde transport, and secreted through the membrane of the ER into the cytosol by a cargo receptor that recognizes the properly folded protein. The proteins that are incorrectly folded are not secreted and are either targeted for degradation or aggregated. The aggregated proteins are able to re-enter the stage of protein ensembles ready to be folded so that they may try again at being folded properly.
Protein Folding in ER
Folding Factors in the Endoplasmic Reticulum:
Biochemical research on folding pathways has provided a comprehensive list of folding factors, or chaperones, involved with protein folding in the ER. Folding factors are categorized based on whether they catalyze certain steps or if they interact with intermediates in the folding pathway. General protein folding factors are typically separated into four different groups: heat shock proteins as chaperones or cochaperones, peptidyl prolyl cis/trans isomerases (PPIases), oxidoreductases, and glycan-binding proteins.
Many folding factors are great in that they are multi-functional. One folding factor can take care of different areas of the folding pathway. Unfortunately, this leads to redundancy due to different classes of proteins carrying out overlapping functions. This functional redundancy complicates the understanding of the specific roles of individual folding factors in aiding maturation of client proteins. Folding factors also prefer to act in concert during the maturation process, which further obscures the individual roles of each factor. Since these roles are not clear, it is difficult to confirm that even if one folding factor deals with a particular reaction in one protein, that same folding factor will carry out the same function in another.
In addition to aiding non-covalent folding and unfolding of proteins, folding factors in the ER sometimes delay interactions with the protein. This allows time for nascent proteins to fold properly and enables folded proteins to backtrack on its folding pathway, which prolongs equilibrium in a less folded state, preventing the protein from being held in a non-native state.
Folding after Endoplasmic Reticulum:
Although ER provides only correctly assembled proteins to be secreted, some examples exist in which proteins change conformation in the Golgi bodies and beyond. Typically, newly folded proteins are sensitive and prone to unfolding while in the ER but resistant to unfolding after exit. In an environment without chaperones and other folding enzymes, proteins are compact and relatively resistant to change after exiting the ER. However, this doesn’t necessarily mean that protein folding ends because some molecular chaperones like Hsp 70s and Hsp 90s continue to assist in protein conformation throughout the protein’s existence.
A strategy for studying the folding of proteins is to unfold the protein molecules in high concentrations of a chemical denaturant like guanidinium chloride. The solution is then diluted rapidly until the denaturant concentration is lowered to a level where the native state is thermodynamically stable again. Afterwards, the structural changes of the protein folds may be observed. In theory, this sounds simple. However, such experiments are complex, since unfolded proteins have random coil states in chemical denaturants. Moreover, analyzing the structural changes taking place in a sample may is difficult, since all of the molecules may have significantly different conformations until the final stages of a reaction. As such, the analysis would have to be performed in a matter of seconds rather than days or weeks that are normally allowed to deduce the structure of a single conformation of a native protein. To avoid this problem, the disulphide bonds can be reduced after the protein is unfolded and reformed under oxidative conditions. The protein can then be identified by standard techniques such as mass spectroscopy to draw conclusions about the structure present at stages of folding where disulfide bonds are formed.
Multiple techniques are used to monitor structural changes during the refolding. For instance, in circular dichorism, UV is used from far away to provide a measurement of the appearance of the secondary structure during folding. UV at a close distance monitors the formation of the close-packed environment for aromatic residues. NMR is also a useful technique for characterizing conformations at the level of individual amino-acid residues. It can also be used to monitor how the development of structures protect amide hydrogens from solvent exchanges.
Circular Dichroism: This type of spectroscopy measures the absorption of circularly polarized light since the structures of protein such as the alpha helix and beta sheets are chiral and can absorb this sort of light. The absorption of light indicates the degree of the protein’s foldedness. This technique also measures equilibrium unfolding of protein by measuring change of absorption against denaturant concentration or temperature. The denaturant melt measures the free energy of unfolding while the temperature melt measures the melting point of proteins. This technique is the most general and basic strategy for studying protein folding.
Dual Polarization Interferometry: This technique uses an evanescent wave of a laser beam confined to a waveguide to probe protein layers that have been absorbed to the surface of the waveguide. Laser light is focused on two waveguides, one that senses the beam and has an exposed surface, and one that is used to create a reference beam and to excite the polarization modes of the waveguides. The measurement of the interferogram can help calculate the protein density or fold, the size of the absorbed layer, and to infer structural information about molecular interactions at the subatomic resolution. A two-dimensional pattern is obtained in the far field when the light that has passed through the two waveguides is combined.
Mass Spectrometry: The advantages of using Mass Spectroscopy to study protein folding include the ability to detect molecules with different amounts of deuterium, which allows the heterogeneity of the protein folding reactions to be studied. It can also measure the conformation of folding intermediates bound to molecular chaperones without disrupting the complex. Mass spectrometry can also directly compare refolding properties, since mixtures of proteins can be studied without separation if the two proteins have sufficiently different molecular weights.
High Time Resolution: These are fast time-resolved techniques where a sample of unfolded protein is triggered to fold rapidly. The resulting dynamics are then studied. Ways to accomplish this include fast mixing of solutions, photochemical methods, and laser temperature jump spectroscopy.
Computational Prediction of Protein Tertiary Structure: This is a distinct form of protein structure analysis in that it involves protein folding. These programs can simulate the lengthy folding processes, provide information on statistical potential, and reproduce folding pathways.
Protein misfolding refers to the failure of a protein to achieve its tightly packed native conformation efficiently or the failure to maintain that conformation due to reduction in stability as a result of environmental change or mutation. It has been established that failure of protein folding is a general phenomenon at elevated temperatures and under other stressful circumstances. The two most common results of misfolded proteins are degradation and aggregation. When a polypeptide emerges from the cell, it may fold to the native state, degraded by proteolysis, or form aggregates with other molecules. Proteins are in constant dynamic equilibrium so even if the folding process is complete, unfolding in the cellular environment can occur. Unfolded proteins usually refold back into their native states but if control processes fail, misfolding leads to cellular malfunctioning and consequently diseases. Diseases associated with misfolding cover a wide array of pathological conditions such as cystic fibrosis where mutations in the gene encoding the results in a folding to a conformer whose secretion is prevented by quality-control mechanisms in the cell. About 50% of cancers are associated with mutations of the p53 protein that eventually lead to the loss of cell-cycle control and causing the growth of tumors. Failure of proteins to stay folded can result in aggregation, a common characteristic of a group of genetic, sporadic, and infectious conditions known as amyloidoses. Aggregation usually results in disordered species that can be degraded within the organism but it may also result in highly insoluble fibrils that accumulate in tissue. There are about twenty known diseases resulting from the formation of amyloid material including Alzheimer’s, Type II diabetes, and Parkinson’s disease. Amyloid fibrils are ordered protein aggregates that have an extensive beta sheet structure due to intermolecular hydrogen bonds and have an overall similar appearance to the proteins they are derived from. The formation of the amyloid fibrils are the result of prolonged exposure to at least partially denatured conditions.
Alzheimer's: This neurological degeneration is caused by the accumulation of Plaques and Tangles in the nerve cells of the brain.[2] Plaques, composed of almost entirely a single protein, are aggregation of the protein beta-amyloid between the spaces of the nerve cells and Tangles are aggregation of the protein tau inside the nerve cells. Tangles are common in extensive nerve cell diseases whereas neuritic plaque is more specific to Alzheimer's. Although scientists are unsure what role Plaques and Tangles play in the formation of Alzheimer's, one theory is that these accumulated proteins impede the nerve cell's ability to communicate with each other and makes it difficult for them to survive. Studies have shown that Plaques and Tangles naturally occur as people age, but more formation is observed in people with Alzheimer's. The reasons for this increase is still unknown.
Creutzfeldt-Jakob Disease (Mad Cow Disease): This disease is caused by abnormal proteins called prions which eat away and form hole-like lesions in the brain. Prions (proteinaceous infectious virion) were discovered to be proteins with an altered conformation. Scientists hypothesize that these infectious agents could bind to other similar proteins and induce a change in their conformation as well, propagating new, infectious proteins.[3] Prions are highly resistant to heat, ultraviolet light, and radiation which makes them difficult to be eliminated. In Creutzfeldt-Jakob Disease there is an incubation period for years which is then followed by rapid progression of depression, difficulty walking, dementia and death. Currently there is no effective treatment for prion diseases and all are fatal.[4]
Parkinson's disease:A mutation in the gene which codes for alpha-synuclein is the cause of some rare cases of familial forms of Parkinson's disease. Three point mutations have been identified thus far: A53T, A30P and E46K. Also, duplication and triplication of the gene may be the cause of other lineages of Parkinson's disease.Victims of Parkinson's disease have primary symptoms that result from decreased stimulation of the motor cortex by the basal ganglia, normally caused by the insufficient formation and action of dopamine. Dopamines are produced in the dopaminergic neurons of the brain. People who suffer from this disease have brain cell loss (death of dopaminergic neurons), which may be caused by abnormal accumulation of the protein alpha-synucleinbinding to ubiquitin in the damaged cells. This makes the alpha-synuclein-ubiquitin complex unable to be directed to the proteosome. New research shows that the mistransportation of proteins between endoplasmic reticulum and the Golgi apparatus might be the cause of losing dopaminergic neurons by alpha-synuclein.
Cystic Fibrosis: Francis Collins first identified the hereditary genetic mutation in 1989. The problem occurs in the regulator cystic fibrosis transmembrane conductance regulator (CFTR), which regulates salt levels and prevents bacterial growth, when the dissociation of CFTR is disturbed as a protein regulating the chloride ion transport across the cell membrane.[5] The deleted amino acid doesn't allow bacteria in the lungs to be killed thereby causing chronic lung infections eventually leading to an early death.[6] Scientists have used nuclear magnetic resonance spectroscopy (NMR) to study Cystic Fibrosis and its effects.
Normal and sickle-shaped red blood cells.
Sickle Cell Anemia: Sickle-shaped red blood cells cling to walls in narrow blood vessels obstructing the flow of blood define sickle cell anemia. The shortage of red blood cells in the blood stream in addition to the lack of oxygen-carrying blood causes serious medical problems. The defect in the Hemoglobin gene is detected with the presence of two defective inherited genes. The sickle cell shape is formed as hemoglobin give up their oxygen resulting in stiff red blood cells forming rod-like structures. Some symptoms include: fatigue, shortness of breath, pain to any joint or body organ lasting for varying amounts of time, eye problems potentially leading to blindness, and yellowing of the skin and eyes which is due to the rapid breakdown of red blood cells. Luckily, sickle cell anemia can be detected by a simple blood test via hemoglobin electrophoresis. Even though there is no cure, blood transfusions, oral antibiotics, and hydroxyurea are treatments that reduce pain caused.[7]
Huntington's Disease: Also known as the trinucleotide repeat disorder, Huntington's disease results from glutamine repeats in the Huntingtin protein. Roughly 40 or more copies of C-A-G (glutamine) will result in Huntington's disease as the normal amount is between 10 and 35 copies. During the post-translational modification of mutated Huntingtin protein(mHTT), small fractions of polyglutamine expansions misfold to form inclusion bodies. Inclusion bodies are toxic for brain cell. This alteration of the Huntingtin protein does not have a definite effect except that it affects nerve cell function.[8] This incurable disease affects muscle coordination and some cognitive functions.
Cataract in human eye
Cataracts: Eye lens are made up of proteins called crystallins. Crystallins have a jelly-like texture in a lens cytoplasm. The current leading cause of blindness in the world, cataracts occurs when crystallin molecules form aggregates scattering visible light causing the lens of the eye to become cloudy. UV light and oxidizing agents are thought to contribute to cataracts as they may chemically modify crystallins. In children, it has been observed that the deletion or mutation of αB-crystallin facilitates cataracts formation. The likelihood of developing cataracts exponentially increases with age. Pain, Roger H. (2000). Mechanisms of Protein Folding. Oxford University Press. pp. 420–421. ISBN019963788. Retrieved 2009-10-18. {{cite book}}: Check |isbn= value: length (help)
Protein misfolding caused by impairment in folding efficiency leads to a reduction in number of the proteins available to conduct its normal role and formation of amyloid fibrils, protein structures that aggregate, resulting in a cross-β structure that can generate numerous biological functions. Protein aggregation can come from different processes occurring after translation including the increase in likelihood of degradation through the quality control system of the endoplasmic reticulum (ER), improper protein trafficking, or conversion of specific peptides and proteins from its soluble functional states into their highly organized aggregate fibrils.
Structures
X-ray Crystallography
From X-ray crystallography, three-dimensional crystals of amyloid fibril structures were formed and the structure of the peptide formation and how the molecule is packed together were examined. In one particular fragment, the crystal was found to contain parts of parallel β-sheets where each peptide contributes one single β-strand. The β-strands are stacked and β-sheets formed are parallel and side chains Asn2, Gln4 and Asn6 interact with each other in a way that water is kept out of the area in between the two β-sheets with the rest of the side chains on the outside are hydrated and further away from the next β-sheet.
Solid State Nuclear Magnetic Resonance (SSNMR)
Through solid-state nuclear magnetic resonance (SSNMR) and the help of other methods such as computational energy minimization, electron paramagnetic resonance and site-directed fluorescence labeling and hydrogen-deuterium exchange, mass spectrometry, limited proteolysis and proline-scanning mutagenesis the structure of an amyloid fibril was suggested to be four β-sheets separated by approximately 10Å.
Through NMR with computational energy minimization, a 40-residue form of amyloid β peptide at pH 7.4 and 24˚Celius was determined to contribute one pair of β-strand to the core of the fibril which is connected by a protein loop. The amyloid β peptides are stacked on each other in a parallel fashion.
From experiments of site-directed spin labeling coupled to electron paramagnetic resonance (SDSL-EPR), the molecule was found to be very structured in the fibrils and in parallel arrangement. SDSL-EPR along with hydrogen-deuterium exchange, mass spectrometry, limited proteolysis and proline-scanning mutagenesis suggests that the structure has high flexibility and exposure to solvent of N-terminal side, but is rigid in the other parts of the structure.
Experiments through SSNMR with fluorescence labeling and hydrogen-deuterium exchange determined that the C-terminals are involved in the core of the fibril structure with each molecule contributing four β-strands with strands one and three forming one β-sheet and strands two and four forming another β-sheet about 10Å apart.
SmallAmyloidFibril
Further experimentation approaching the atomic level with SSNMR techniques resulted in very narrow resonance lines in the spectra, showing that the molecules within fibrils hold some uniformity with peptides that display extended β-strands with the fibrils.
Conclusion
The structures determined from X-ray crystallography or SSNMR were similar to previously proposed structures from cryo-electron microscopy (EM) formed from insulin. EM, which uses electron density maps, revealed untwisted β-sheets in the structure. The similarities of the structures found in these experiments suggest a lot of amyloid fibrils can have similar characteristics such as the side-chain packing, aligning of β-strands and separation of the β-sheets.
[9] Annu. Rev. Biochem. 2006.75:333-366. www.annualreviews.org. Retrieved 24 Oct 2011</ref>
Formation
The capability to form amyloidal protein structures that are considered to be genetic is from the findings that an increasing number of proteins show no signs of protein related diseases. It has been found that amyloidal proteins can be converted from its own protein that has a function rather than disease- related characteristics in living organisms.
In these protein mutations, different factors that affect the formation of amyloid fibril formation and different chains form amyloid fibrils at different speeds. In different polypeptide molecules, hydrophobicity, hydrophillicity, changes in charge, degree of exposure to solvent, the number of aromatic side chains, surface area, and dipole moment can affect the rate of aggregation of protein. It has been found that the concentration of protein, pH and ionic strength of the solution the protein is in as well as the amino acid sequence it is in determines the aggregation rate from the unstructured, non-homologous protein sequences.
As the hydrophobicity of the side chains increases or decreases can change the tendency for the protein to aggregate.
Charge in a protein can create aggregations through interaction of the polypeptide chain with other macromolecules around it.
Also, the low tendency for β-sheets to form along with the high tendency for α-helixes to form contributes in facilitating amyloid formation.
It was found that the degree in which the protein sequence are exposed to solvent tend to affect the formation of amyloids. Proteins that are exposed to solvent seem to promote aggregation. Even though some other parts of the protein that had a high tendency to aggregate were not involved in the aggregation, they seem to at least be partially unexposed to the solvent but other regions that were exposed to solvent that were not involved in the aggregation had a low tendency to form amyloid fibrils.
It has even been raised that protein sequences have evolved over time to avoid forming clusters of hydrophobic residues by alternating the patterns of hydrophobic and hydrophillic regions to lower the tendency for protein aggregation to occur.
[9]
The Affects of Sequence on the Formation of Amyloid Proteins
Amyloid formation arises mostly from the properties of the polypeptide chain that are similar in all peptides and proteins, but sometimes, the sequence affects the relative stabilities of the conformational states of the molecules. In that case, the polypeptide chains with different sequences form amyloid fibrils at various rates. Sequence difference affects the behavior of the protein aggression instead of affecting the stability of the protein fold. Various physicochemical factors affect the formation of amyloid structure by unfolded polypeptide chains.
Hydrophobicity of the side chains affects the aggregation of unfolded polypeptide chains. The amino acid in the regions of the aggregation site can change the ability of aggregation of a sequence when they increase or decrease the hydrophobicity at the site of the mutation or folding site. Over time, sequences have evolved to avoid creating clumps of hydrophobic residues by alternating hydrophobic areas of the protein.
Charge affects the aggregation of amyloid protein folding. A high net charge can have the possibility of impeding self association of the protein. Mutations in decreasing the positive net charge may result in the opposite effect of aggregate formation as increasing the positive net charge. It has been seen found that polypeptide chains can be run by interactions with highly charged macromolecules, displaying the importance of charge of a protein aggregation.
Secondary structures of proteins affect the amyloid aggregation as well. Studies show that a low probability to form α-helix structures and a high probability to form β-sheet structures are contributive factors to amyloid formation. However, it has been found that β-sheet formation is not particularly favored by nature since there are little alternation of hydrophilic and hydrophobic residue sequence patterns to be found.
The characteristics of the amino acid sequences affect the amyloid fibril structure and rate of aggregation. Different mutations, including changes in the number of aromatic side chains, the amount of exposed surface area and dipole moment, have been said to change the aggregation rates of lots of polypeptide chains.
Unfolded regions play vital roles in promoting the aggregation of partially folded proteins. Some regions that were found to be flexible or exposed to solvent were fond of aggregation. Other regions that are not involved in the aggregation were found to not be exposed, but rather half buried even though they have high possibility of aggregating while the exposed regions of the structure that are not involved in the aggregation have a low probability of aggregating amyloid fibrils. The fibrils tend to come together by association of unfolded polypeptide segments rather than by docking the structural elements.
Overall, it has been found that unfolded proteins have lower less hydrophobicity and higher net charge than that of a folded protein. Residues that tend not to form the secondary structure of β-sheet structured proteins seem to inhibit the occurrence of amyloid aggregation. Concentration of protein, pH and ionic strength were found to be associated with the amino acid sequence, which affects the rate of aggregation.
It is understood that the primary structure (the amino acid sequence) of a protein predisposes the protein for a specific three dimensional structure and how it will fold from the unfolded form to the native state. The concentration of salts, the temperature, the nature of the primary solvent, macromolecular crowding, and the presence of chaperones are all factors that affect the mechanism of folding and the ratio of unfolded proteins to those in the native state. More than anything, these environmental factors affect the likelihood of any single protein reaching the correct final structure.
Isolated proteins placed in proper environments (specific solvent, solute concentrations, pH, temperature, etc.) tend to “self-fold” into the correct native conformation. Altering any of these environmental characteristics can disrupt the structure and/or interfere with the folding mechanism. A pH outside the “normal” range of a given protein can ionize specific amino acids or interfere with both polar and dipole-dipole intramolecular forces that would otherwise stabilize the structure. Excess heat (cooking) proteins can break hydrogen bonds essential to the secondary structure of proteins.
Extreme environments or the presence of chemical denaturants (such as reducing agents that can break disulfide bonds) can cause proteins to denature and lose its secondary and tertiary structure, forming into a “random coil.” Under certain conditions fully denatured proteins can return to their native state. Intentional denaturing is used in various methods to analyze biomolecules.
The complex environments within cells often necessitate chaperones and other biomolecules for proteins to properly form the native state.
Protein is an essential part of living thing. The development of human body is needed to be parallel with the development of protein. But protein contains so many mysteries that we did not discovery yet. For example, that is protein folding. Folding is a necessary activity of proteins. They need to fold to continue their biological activity. Folding is also a process that very protein goes through to have a stable conformation. But sometimes this process is happened incorrectly, and the scientist call this problem is protein misfolding. The results of protein folding incorrectly are so many bad diseases happening for human, animals and living things such as Alzheimer’s disease and Mad Cow disease. Because of this reason, the researches about protein folding and misfolding become very important. During the process of discovering about protein, folding, misfolding and its affects, the scientists have been collecting many successes; the mystery about protein is unraveled gradually. As a scientist, W. A. (Bill) Thomasson records many importance things about protein in the article Unraveling the Mystery of Protein Folding; in this article, he make the points about Alzheimer’s disease and Mad Cow disease and some affects of protein misfolding beside the successes of science about them.
Dr Thomasson begins his article by introduce generally about protein folding and misfolding. First of all, proteins consists the sequences of amino acid. The scientists have discovered 20 amino acids appearing in proteins. The protein structure is known with 2 basic shapes which are α_helix and β_sheet. “Most of proteins probably go through several intermediate states on their way to a stable conformation” (Campbell and Reece, 79). Proteins need to fold to continue its activity. The scientists have listed 3 type of protein folding; the protein can be folded, partial folded or misfolded. In the process of folding, the “proteins called chaperones are associated with the target protein; however’ once folding is complete (or even before) the chaperone will leave its current protein molecule and go on to support the folding of another” (Thomasson). The author of the article records the very important conclusion of Anfinsen about protein misfolding. In his point of view, the misfolding is occurred in the process of folding when the folding goes wrong. The research of protein misfolding is focus on the temperature sensitive mutation; the scientists observe the bacteriophage P22 with the changing of temperature to cause the mutation. And they conclude that the mutant proteins are less stable than the normal. It means, they give a conclusion is that in the tailspike of bacteriophage the misfolded proteins is less stable than the correctly folded proteins and they are difficult to reach the properly folded state. When the protein misfolding occurs, it results many bad disease. The aggregation can appear along with the appearance of misfolding and it is at the brain to cause Alzheimer’s disease and Mad Cow disease as many scientists consider.
One affect of protein misfolding on human life that is Alzheimer’s disease. This is a disease of the elderly. According to the research of scientist, this disease is occurred when the amyloid precursor protein is misfolding. This protein is processed into a soluble peptide Aβ. The scientists have not known exactly the reason of this disease yet. But the main reason causing the misfolding is the protein apolipoprotein E (apoE) inside our blood stream. The protein apoE has three forms such as apoE2, apoE3 and apoE4. The affects of each form of apoE on the Aβ is not discovered yet but the scientists consider that the apoE can bind to the Aβ. In the process of misfolding, the β-amyloid is formed to make “neuritic plaque in the Alzheimer’s patient”. This disease is just happened with the older people because in the amyloid process, a nucleus is formed very slowly. The mutation of this protein is not stable and causes the disease. The studying about apoE is still a secret because some scientists show that one form of this protein is developing the disease but another form is decreasing the development of the disease. Finally, the research about Alzheimer’s disease is continued in order to affirm the results of protein apoE on Aβ and to find the treatment for this disease successfully.
Another affect from the protein misfolding is the Mad Cow disease. This is a very dangerous disease because it can be transmitted from animals to human. This disease causes by the misfolding of prions. The process of misfolding is the self-replicating of the prions. Prions are protein particles containing DNA and RNA. The mutation appear in the process of folding, the prions self-replicate and cause the misfolding of the proteins. They contain DNA and RNA. This is a special situation of the protein; it can be served as its chaperons. Because of the replicating, the prion was multiplied very quickly along with the increasing of normal proteins. This disease shows that the protein folding can be occurred without the genetics such as the experiment on the sheep.
Dr. Thomasson continues his article by some more information about the misfolding and the way of the scientist to prove the mystery. He gives the information about the protein p53 and its mutation. It can cause the cancer, it also one type of protein misfolding. The point Dr. Thomasson wants to make that is his idea about the drug that can make the protein misfolding becoming more stable and minimize the misfolding of protein. This idea seems very good but its results are like a mystery as the mystery of protein folding.
The research about the protein folding is very important to our lives. The misfolding is one of the main reasons causing so many dangerous disease but we did not have a successful treatment yet. The study of protein folding is more and more successful to help the human to be able to destroy the disease causing by misfolding. The disease caused by protein misfolding has become one problem of human that need to be solved.
Molecular Chaperones are known mainly for assisting the folding of proteins. Chaperones are not just involved in the initial stages of a protein’s life. Molecular Chaperones are involved in producing, maintaining, and recycling the structure and units of protein chaperones. Chaperones are present in the cytosol but are also present in cellular compartment such as the membrane bounded mitochondria and endoplasmic reticulum. The role or necessity of chaperones to the proper folding of proteins varies. Many prokaryotes have few chaperones and less redundancy in the types of chaperones and whereas eukaryotes have large families of chaperones containing some redundancy. It is hypothesized that some chaperones are essential to proper protein folding such as the example of the prokaryote which has less variations of a chaperone family available. Other chaperones play less of an essential role such as in eukaryotes where more variations within a family of chaperones exist and gradients of efficiency or affinity are produced. This redundancy or existence of less efficient chaperones may exist in one state but the effectiveness of chaperones is also a function of their environment. The pH, space, temperature, protein aggregation and other external factors may render a chaperone that was once ineffective into a more essential chaperone. These environmental factors show why it is important to simulate cellular in vivo conditions, or native states, in order to grasp the conditions that require use of chaperones. This briefly summarizes the difficulties in analyzing and comparing chaperone function in vivo vs. in vitro.
Simulating in vivo, or the environment within the cell, is important not just because of physical factors such as pH or temperature but also because the time in which the chaperone begins to conform the polypeptide. Some chaperones are nearby the ribosome and attach immediately to the polypeptide to prevent misconformation. Other chaperones allow the polypeptide to begin folding by itself and attach later on. Thus the role of each chaperone becomes specific to its vicinity to the polypeptide and time and place in which it assists folding. Recent research has implicated that chaperones within the nucleolus not only catalyze protein folding but also catalyze other functions important to maintain a healthy cell. These nucleolar chaperones are called Nucleolar Multitasking Proteins (NoMP's). Heat shock proteins, for example, not only help other proteins fold but also act during moments of stress to regulate protein homeostatis. Furthermore, there is evidence that chaperones work together in networks to oversee certain functions like dealing with toxins, starvation or infection.
The nucleolar chaperone network is divided into different branches that have specific functions. The network is dynamic and can vary in concentration or location of the network components depending on changes in the physiology and environment of the cell. Heat shock proteins (HSPs), which are classified based on their molecular weights, are integral components of the chaperone network. HSP 70s and 90s maintain proteostasis by ensuring that proteins are properly folded and preventing proteotoxicity, which is the damage of a cell function due to a misfolded protein. HSP70s help to fold recently synthesized proteins, while HSP90s help later in the folding process. The nucleolar network also contains chaperones that are part of ribosome biogenesis, or the synthesis of ribosomes in the cells. Proteins in the HSP70 and DNAJ families, which help to process pre-rRNA, are regularly found in protein complexes that process pre-rRNA in Saccharomyces cerevisiae (a species of yeast). Other HSPs are important in ribosome biogenesis as well, including HSP90 which works together with TAH1 and PIH1 to create small nucleolar ribonucleoproteins. The nucleolar chaperone network provide the organization and assistance needed to complete the biological taks necessary for cell survival, and if it does not function properly there can be many problems. For instance, when cancer cells have increased levels of rRNA synthesis, ribosome biogenesis is increased. Scientists are researching the compound CX-3543, which can stop nucleolin from binding with rDNA and impede RNA synthesis, leading to cell death. It is possible to potentially use drugs designed to target specific branches of the nucleolar chaperone network in malfunctioning cells. Other networks of chaperones include networks that specifically participate in de novo protein folding, meaning they help to fold newly made proteins, and the refolding of proteins that have been damaged. One chaperone network that exists in tumor cell mitochondria contains HSP90 and TRAP1, which protect the mitochondria and prevent cell death, allowing the cancer cells to continue to spread uncontrollably.[10]
HSP 70 is a protein in the Heat Shock Protein family along with HSP 90. It works together with HSP 90 to support protein homeostasis. It binds to newly synthesized proteins early in the folding process. It has three major domains, the N-terminal ATPase domain, the Substrate binding domain, and C-terminal domain. The N-terminal ATPase binds and hydrolyzes ATP, the substrate binding domain hold an affinity for neutral, hydrophobic amino acid residues up to seven residues in length while the c-terminal domain acts as a sort of lid for the substrate binding domain. This lid is open when HSP 70 is ATP bound and closes when hsp 70 is ADP bound. HSP70, or DnaK, are bacterial chaperones and can help in folding by clamping down on a peptide.[11]
GroEL and GroES, or 60kDa and 10kDa, are both bacterial chaperones. Both GroEL and GroES are structured so that they are a stacked ring with an empty center. The protein fits in this hollow center. Conformational changes within the chamber can then change the shape and folding of the protein.[11]
HSP 90 is a protein in the Heat Shock Protein family. This particular protein, however, is different from other chaperones in that HSP90 is limited in the folding aspect of molecular chaperones. Instead, Hsp 90 is vital to study and understand because many cancer cells have been able to take over and utilize the Hsp 90 in order to survive in many virulent surroundings. Therefore, if one were to structurally study and somehow target Hsp90 inhibitors, then there could be a way to stop cancer cells from spreading. Furthermore, many studies have been performed in order to test whether or not the Hsp 90 chaperone cycle is driven by ATP binding and hydrolysis or some other factor. But after much research by Southworth and Agard, there was enough evidence to state that HSP90 protein could conformationally change without nucleotide binding but rather the stabilization of an equilibrium is the factor that will change the Hsp90 to a closed or compact or open state. The three conformations of the Hsp90 were found through x-ray crystallography and also through single electron particle microscopy and by studying the three-state conformational changes in yeast Hsp90, human Hsp90 and bacteria Hsp 90 (HtpG) it was clear that there are distinct conformational changes for specific species. Overall, Hsp90 is a chaperone that is more involved with maintaining homeostasis within a cell rather than the involvement of protein folding. Hsp90 has rising potential in the area of drug development in the future since it plays such an essential role in aiding the survival for cancer cells.
This is the first chaperone to interact with the nascent chain as it exits the ribosome tunnel. Without the nascent chain, the TF cycles on and off but once the nascent chain is present, it binds onto the chain, forming a protecting cavity around. In order to do its function, TF scans for any exposed hydrophobic segment of the nascent chain and it can also re-associate with the chain. Folding is found to be more efficient in the presence of the TF, however, this is done at the expense of speed, it can stay with the chain for more than 30 seconds. The release of the chain is triggered when the hydrophobic portions is buried as the folding progresses toward the native state.
YidC, Alb3, and Oxa1 are proteins that facilitate the insertion of proteins in the plasma membrane. YidC is a protein that has only two polypeptide chains. The formation of its structure has been supported by particular phospholipids. YidC proteins can be found in Gram-negative and Gram-positive bacteria. Oxa1 can be found in the inner membrane of the mitochondria. Alb3 locates in the membrane of the thylakoid inside the chloroplast. Experiments showed that YidC protein actively contributes to the insertion of Pf3 coat protein. In addition, YidC also has direct contact with the hydrophobic segment of Pf3 coat protein. Although Oxa1 can only be found in the mitochondria it can also facilitate the insertion of membrane proteins in the nucleus. The role of YidC and Alb3 seems to be interchangeable because Alb3 can replace YidC in E. coli. Moreover, YidC, Oxa1, and Alb3 all support the insertion of Sec-independent proteins. Oxa1 only supports the insertion of Sec-independent proteins because the mitochondria in yeast cell do not have Sec proteins.
Nucleotide-binding domains that are leucine- rich (NLR) provide a pathogen-sensing mechanism that is present in both plants and animals. They could either be triggered directly or indirectly by a derivation of pathogen molecules via elusive mechanisms. Researches show that molecular chaperones like HSP90, SGT1, and RAR1 are main stabilizing components for NLR proteins. HSP90 can monitor the function of its corresponding clients that apply to NLR proteins in three practical ways: promotion of steady-state of functional threshold, activating stimulus-dependent activity, and raising the capacity to evolve.
Plants contain many NLR genes that considered being polymorphic in the LRR domain in order to be familiar with the highly diversified pathogen effectors. The NLR sensor stability will be the mechanism that will determine the pathogen recognition. The HSP90 system is advantageous for plants because it will couple metastable NLR proteins and stabilize them in a signaling competent condition. This will allow for the masking of mutations that would be detrimental.
Molecular Chaperone Mechanism for Substrate Binding in Protein Folding
It is known that chaperones work together to aid in the folding of protein in order to prevent misfolding. However, the mechanism of how chaperones help in protein folding was not fully understood. Recent studies on Hsp40 and Hsp70 have provided more insights into the mechanism of chaperones and their substrate. The Hsp40 family consists of many Hsp40 with different J-domain. Different J-domain will carry out different Hsp70 ATPase activities when Hsp40 binds to Hsp70. In protein folding, an unfolded polypeptide binds to a Hsp40 co-chaparone. From there, the J-domain of Hsp40 binds to the nucleotide-binding domain (NBD) of Hsp70. A conformation change in the Hsp70 substrate-binding domain occurs when the hydrolysis of ATP to ADP takes place on the HSP70 NBD. This causes Hsp70 to have a higher affinity for the polypeptide substrate and unbind the substrate from Hsp40. When ADP is exchange for ATP, the polypeptide substrate is released from Hsp40. Studies have shown that nucleotide exchange factors make changes to the lobe on the Hsp70 ATPASE domain in way that decreases Hsp70’s affinity for ADP. Once the polypeptide is released from Hsp70, it can fold to its native state or it can be refolded by the chaperones if there is a misfolding. If a polypeptide that is bounded to Hsp70 is recognized by E3 ubiquitin ligase CHIP, it will be degraded.[12]
Small Heat Shock Proteins & α-crystallins as Molecular Chaperones
It is known that small heat shock proteins (sHSPs) and the related α-crystallins (αCs) are virtually ubiquitous proteins that are strongly induced by a variety of stresses, but that also function constitutively in multiple cell types in many organisms. Extensive research has demonstrated that a majority of sHSPs and αCs can act as ATP-independent molecular chaperones by binding denaturing proteins and reversing denaturation. This approach thereby protects cells from damage due to irreversible protein aggregation. Many inherited diseases have been discovered to result from defects in sHSP/αCs, and these proteins accumulate in neurodegenerative disorders and other diseases linked to aberrant protein folding. sHSP/αC proteins range in size from ~12 to 42 kDa and is a C-terminally located domain of ~90 amino acids, known as the αC domain. sHSP-substrate complexes can be observed by size exclusion chromatography . They are large and heterogenous, and their size distribution depends on the ratio of sHSP/αC to substrate as well as the rate of substrate aggregation, which is affected by concentration and temperature. Substrate binding is generally facilitated by an increase in available hydrophobic surface on the sHSP/αC, which seem to occur without significant loss of defined sHSP/αC secondary and tertiary structure. There is no single, specific substrate binding surface on sHSP/αCs. It rather appears that many sites contribute to substrate interactions, and binding is probably different for different substrates dependent on the conformation of surfaces exposed when a substrate unfolds. However, some sHSP/αCs recognize almost any unfolding protein, which suggests that they act on any labile or damaged cellular component.
If proteins folded randomly and unpredictably, the amount of time taken to reach the native conformation would be much larger than the actual time it takes. The current theory on how protein folding occurs naturally and efficiently involves a "funnel" of sorts-the idea being that there exists not a step by step means of reaching the correct 3-D structure, but rather a number of paths that become progressively narrower from top to bottom. The funnel starts at the top and proceeds downward from energetically disfavorable folding at the top to energetically favoring proper folding at the bottom.
The experiment that sparked the idea of proteins relying on energetics and thermodynamics to reach their native folding was conducted by Christian Anfinsenf in 1961, when he discovered that ribonuclease could spontaneously refold into its proper structure after being denatured without the help of other molecules. Further theoretical proof that protein folding is not random is seen in Levinthal's Paradox, which states that it would take roughly 10^81 years for a protein 100 amino acids long to reach the proper conformation, when in reality, it takes anywhere from a millisecond to a day.
These funnel models (such as the Go-type model) show funnels with hills and bumps that represent the protein taking the path of least resistance when moving down the energy funnel. These bumps are termed "points of frustration". It is believed that funnels with the fewest frustration points or bumps fold into their native forms faster since fewer energy boundaries exist. Although these models are simplified attempts and do not account for misfoldings, they nonetheless prove accurate in the case of many proteins.
Another model that uses algorithms and computers is the empirical force field. This model uses hundreds of thousands of computers running idly to compute folding scenarios of proteins under 50 amino acids with surprising accuracy. However, these computer models will sometimes overestimate unlikely folding structures or produce folding patterns that are rarely or never seen. For example, some simulations/algorithms have a tendency of getting stuck in the local minima and are unable to reach the global minima, which is the correctly folded protein. Simple models such as Go-type models not only predict the folded protein, but also the transition states that determine the rate of the protein folding.
These models are just beginning to show the dynamics of the intermediate stages of protein folding. As such, this is an area under further investigation. The understanding of the kinetics of protein folding is less established, and the movement of proteins between initial amino acid strands and the final product is also an area under investigation. The energy landscape model also has trouble accounting for external factors like crowding and aggregates. One such example of external interaction, called "domino swapping", involves the swapping of monomers from one protein to another in order to activate the correct folding of both proteins.
Recent studies have combined human and computer power to correctly predict the protein conformation. Websites like fold.it, overseen by the University of Washington's Computer Science department, turn the folding problem into a video game, allowing people around the world to solve protein folding problems like puzzle games. Users are given partially folded proteins, usually those stuck in a locally favorable conformation that seems optimal to a computer, and asked to reconfigure the protein into a shape that looks more stable. Utilizing a computer's computing power and speed along with a human's ability to manipulate objects in space shows promise in helping to solve protein folding problems more efficiently.
The cooperative nature expressed in protein folding is one of the most remarkable aspects of protein folding. Contrary to the traditional viewpoint of complex and heterogeneous mechanisms involved in the folding of a protein, the cooperative two-state folding kinetics shown by many proteins is relatively simple. Due to its simplicity, efforts to understand what determine the co-operativity and the diversity of protein folding rates are made recently by means of applying the cooperative two-state folding kinetics.
The co-operativity of the protein is usually referred to the mechanism by which the presence of a structural region makes additional order more favorable in protein folding. As mentioned previously, the cooperative two-state folding kinetics of small globular proteins is relatively simple and become an interest of study of many scientists. The experiment that excites single molecule that is sensitive enough to allow estimation of transition time reveals two-state co-operativity.
The general trends revealed by two-state folding proteins may be summarized as the following two points. Firstly, more topologically complex proteins tend to fold more slowly than proteins with simpler, local topology; secondly, larger proteins tend to fold more slowly than smaller proteins. The largeness and smallness of a protein here are defined base on its chain length.
Protein folding kinetics is controlled by the free energy barrier determined by the gain of energy and the loss of entropy in the transition state. In describing the pattern, scientists introduce principle of minimum frustration of energy landscape theory. The theory refers to the concept that native-like structures have lower free energy than other random configurations during protein folding. Thus, native-like structures encourage fast folding of the protein and serve as a driving force toward native state, the functional form or the tertiary structure of the protein. This principle can be expressed by the funnel energy landscape.
Funnel Energy Landscape
Funnel energy landscape depicts the energy landscape of a folding protein as a rough funnel. The roughness comes from non-native contacts in protein folding process.The landscape is inherently many-dimensional, so funnel is a projection on the two-dimensional graph. The depth of the funnel represents the energy of a conformational state; the width of the funnel represents the measure of l entropy. The bottleneck of the funnel represents the transition state configuration of the folding protein, whereas the bottom of the funnel represents the native state of the protein. As the protein goes toward its native state, it experiences entropy loss and it achieves lower energy state. The funnel energy landscape serves as a convenient illustration for scientists to envision the thermodynamics and kinetics of the protein folding process.
φ (phi)value
Another concept that plays a role in the study of protein folding kinetics is the φ (phi) value. The value refers to the approximate measurement of native structure content in transition state configuration. The comparison with φ value serves as one of the ways to examine various models that studies protein folding kinetics.
General observations
The fist trend mentioned may be easily understood from an entropic point of view. More topologically complex proteins, or proteins that have long-range contacts, are expected to have higher entropic cost compared with proteins have short-range contacts in terms of folding. The second trend was recently confirmed by experiments focused on the influence of protein size on folding rates. It was found that simple model based only on chain length could roughly predict a protein’s folding rate and stability.
Go¯model
Coarse-grained topology models (Go¯model) are widely used to study the co-operativity and kinetics of protein folding, as it is noted that the topology of native protein determines the folding mechanism. Typical Go¯model simplifies the protein where there is only one interactions stabilizing the folding protein. Early models often examine the non-additive force acting in the protein folding, such as side-chain ordering and hydrophobic effects. Recently, more variety of Go¯models is used to study the protein folding kinetics.
Bulleted list item
The Go¯model (this refers to Eastwood and Wolynes’ model here) with nonpairwise-additive interactions between the native contacts of the protein demonstrates that short-ranged multi-body interaction can increase the free energy barrier and make the transition state configuration more localized.
The lattice Go¯model, on the other hand, demonstrates the coupling local and core burial interactions promoting co-operativity as well as increasing the correlation with contact order.
The Go¯model with pairwise-additive interactions, particularly the ones focusing on the effects of varying strength of three-body interactions and φ values, shows that three-body interactions increase energy barrier and increases the agreement with measured φ values.
In addition, solvent-mediated interactions are also introduced into Go¯model. Where the interactions between contacts are replaced by solvent separated minimum and desolvation barrier, it is observed that kinetics and co-operativity of protein function increase as a function of the height of desolvation barrier. The advantage of solvent-mediated Go¯model is that it is useful in distinguishing short-ranged contacts and long-ranged contacts and therefore differentiating proteins with simple topologies and the ones with more complex topologies. In study of solvent-mediated Go¯model the chevron plot is often used. The chevron plot is a way to represent protein folding kinetic datas in varying concentration of denaturation that disrupts the native structure of the protein.
Variational Go¯model improves co-operativity by excluding volume force between the residues that are in close contacts in native state. In this model it is achieved that a) the Co-operativity is stronger for long-ranged contacts; b) the range of calculated rate is broaden; c) the calculated φ values are improved. There is also Go¯models that entirely focus on the funnel aspect of the protein folding energy landscape and ignore the non-native contact effects.
Other model, such as capillarity model, assumes the volume of folding nuclei scales with number of monomers. In such model, it is shown that increased co-operativity tends to slow down kinetics and smooth the energy landscape.
Conclusion
The recent development of topological models with non-additive forces is becoming a more popular and reliable way to understand the co-operativity of protein folding rates. Refinement of this model has shown its promising future on a more explicit and through understanding of what determines protein folding rates and mechanism. Go¯models that enables long-ranged contacts become more cooperative, and φ values more accurate need further improvement and more attention in the study of protein folding kinetics and the folding mechanism.
Relationship between Protein Sequence, Structure, and Function
There have been several protein prediction methods developed in the past 20 years. A universal method has not been developed that applies to all proteins because each method has its advantages and disadvantages. The difficulty of developing such a method is due to our incomplete understanding of the highly intricate relationship between protein sequence, structure, and function.
The theory of correlating amino acid sequence to its structure was shown by Anfinsen. He demonstrated that a denatured (unfolded) protein could regain its native tertiary structure spontaneously. This method is also a useful contributor for assigning function to protein structure. A protein researcher could predict that hydrophobic substrates could potentially bind to hydrophobic regions of the protein and vice versa for charged regions. The problem with this method is that it doesn’t take into account certain factors such as atypical environmental conditions.
It was thought that similar sequencing implies related structures. This theory only holds true for a handful of proteins. Researchers saw that similarities in protein folds aren’t always related to its protein sequence. Due to these findings, the ‘Paracelsus Challenge’ was purposed in 1995. The theory behind the ‘Paracelsus Challenge’ was to develop two proteins that were more than 50% identical in sequence, but they both had completely different folds. The challenge was satisfied in 1997 by with two protein sequences that shared 88% sequence identity (GA88 and GB88). Recent studies show that as little as 3 mutations are enough to induce different folding patterns. Although the outcomes of the ‘Paracelsus’ challenge are very interesting, they rarely occur in nature.
Functional convergence causes problems in assigning a specific function to a structure. Various structures can adopt similar functions, but some can adopt very different functions as well. However, there is a significant correlation between certain folds and specific functions. There are two major variables in function prediction: (1) the locations of binding site, and (2) the range of functions at the site. Metal, ions, cofactors, and other proteins that contribute to functions must be taken into considerations as well. One problem that arises with these factors is when determining a structure via crystallography. The PROCOGNATE resource and PIDA database offers a solution to this problem.
A widely used method by which protein function is defined is derived from the Gene Ontology, which consists of three graph structures in which functional terms and relationships between them are defined. Limitations of gene ontology arise with proteins that are non-positional and when proteins have no defined relationship between ligand in its crystal structure. Other developments that attempt to bridge this gap includes The Protein Feature Ontology (PFO [29]) and The Distributed Annotation System (DAS[30]).
Two approaches are used to determine a functional site: (1) either with no knowledge of where the site is or what it binds, or (2) with prior knowledge of the interaction partner. The most highly used methods involve bioinformatics such as the SOIPPA method. A very important contributor to assigning function to protein is sequence conservation, but it is difficult to determine if residues are conserved for structural or functional reasons. Another method involves energy-based approach. A recent development is the ProFunc server, which combines methods such as InterProScan and BLAST search.
Predicting binding sites (which are immensely complex in its own nature) is only the first step of the puzzle. The next step is to determine the overall function in terms of biochemical function, and even more challenging is determining its biological role. The difficulties with analyzing protein function increased another magnitude of complexity when researchers came across the fact that protein function may not only depend on its final folded product. A protein could have functionalities in its partially denatured state and it fully denatured state. With all of this said, it is safe to say that there is still a lot to learn about the relationship between sequence, structure, and function of proteins.
The domain swapping that occurs in proteins may be important in the folding or misfolding process in proteins. Domain swapping occurs when two or more identical protein chains swap with each other. The domain swapping can be thought of as a mechanism for the interchanging of monomers and oligomers. What happens in oligomeric swapping is that one monomer from one protein will swap with another identical monomer from a different protein. This domain swapping mechanism has been observed in various proteins, more than 40 different proteins. The swapping mechanism is important for some protein functions. For a specific protein for example, p13suc1 it has been seen that the swapping and aggregation correlate meaning that they have a common mechanism. P13suc1 is required for cyclin-dependent kinase (Cdk) during the cell cycle progression. P13suc1 has two different states, one being a monomer and the other a swapped dimmer. The domain swapped part is a β strand is not an independently folded domain. While studying this, it was found that β4 has a critical role when in contact with β2 because they pair with each other early on in the folding process. Therefore, for p13suc1, it has been shown that the regions that have been interchanged are responsible for the folding and misfolding of the protein. There seems to be a competition between folding and misfolding in proteins because polypeptide chains can fold into structures or misfold into amyloid fibrils. What seems to be even more crucial in protein folding is the presence of a folding nucleus which forms part of the protein chain in the transition state. A correlation between residues involved in protein folding nuclei location and amyloidogenic regions have been found as well as important information that fibril formation and protein folding may contain key residues. By using the modeling of folding of proteins and looking at the exchangeable regions in the oligomeric form, the relationship can be seen as responsible for folding and misfolding. This may take researchers one step closer to solving the protein solving problem and understand how proteins get their folding instructions.
Reference: http://www.benthamscience.com/open/tobiocj/articles/V005/27TOBIOCJ.pdf
There are 4 subfamily structures in the death-fold superfamily. They consist of Death Domains (DDs), Death Effector Domains (DEDs), CAspase Recruitment Domains (CARDs) and PYrin Domains (PYDs). These subfamily structures are involved in the assembly of multimeric complexes which may be implicated in cell inflammation and death.
Structure and Function of a Death-Fold Domain
There are currently 102 known proteins that have death-fold superfamily domains. These domains contain homotypic interactions. These proteins consist of 39 DDs, 8 DEDs, 33 CARDs, and 22 PYDs. Although these domains have up to a 90% difference in sequence, they all have the characteristic death-fold. This fold consist of a "globular structure where 6 amphipathic alpha-helices are arranged in an anti-parallel alpha-helix bundle with Greek key topology" (Peter Vandenabeele et al., 2012). The difference between these death-domains which constitute either of the subfamilies is found in the alpha-helices length and orientation and the distribution of hydrophobic and charged residues along the surfaces of the complexes.
The believed function of the death-fold domains is to mediate the assembly of large oligomeric signaling complexes. At these complexes, caspases and kinases activity is increased. Before now, little was known about the structural conformation of protein assemblies with death-fold domains.
Three distinct Interaction Types
Type I Interaction:
Residues from helices 1 and 4 (Patch Ia) of one death-fold domain interact with residues from helices 2 and 3 (Patch Ib) of another death-fold domain.
Type II Interaction:
Residues from helix 4 and the loop between helices 4 and 5 (Patch IIa) of one death-fold domain interact with residues of the loop between helices 5 and 6 (Patch IIb) of another death-fold domain.
Type III Interaction:
Residues from helix 3 (Patch IIIa) of one death-fold domain interact with residues located on the loops between helices 1 and 2 and between helices 3 and 4 (Patch IIIb) of another death-fold domain.
Previous theory suggested that the three interaction types were conserved throughout the death-fold superfamily but it now seems that there are differences seen between interactions of the same type of death-fold domains.
Crystal Analysis of Death-Fold Domains
Only three DD complexes have had their crystal structure analyzed. They are PIDDosome, MyDDosome, and the Fas/FADD-DISC. The analyses of these structures have shown that DDs can engage in up to six interactions.
Death-Domains and Medicine
Death-domains have been shown to facilitate the assembly of multimeric complexes that lead to inflammation and cell death. Understanding of these structures can generate therapeutic benefit by preventing or triggering the formation of these oligomeric complexes. Diseases that may be affected by these interactions can include neurodegenerative and inflammatory disorders as well as many others that have characteristic of inflammation or excessive cell death.
While folding is typically a major contributor to protein function, some proteins do not fold into a specific structure, yet still possess a function. Instead of a specific structure, these proteins often shift between different forms and/or have disordered regions that do not hold to a particular shape.
Just as a protein's folding is determined by its amino acid sequence, non-folding proteins are non-folding because of their sequence. These proteins tend to have much less of certain amino acids than folding proteins, and much more of others. Specifically, non-folding have less of the amino acids that form the hydrophobic cores of folding proteins and more of the surface amino acids. The formation of a hydrophobic core is one of the first steps in most protein folds and, once formed, the core tends to provide the driving force for stable final structures. Without the amino acids to form a core, proteins are not driven towards a specific structure.
Several molecular chaperons that are fully folded and inactive under non-stress conditions have been known as conditionally disordered proteins. These chaperons have a partially disordered conformation when they exposed to distinct stress conditions. This disorder is very important because they are able to protect cells against stressors. The study of these disordered chaperons lead to more understanding of the functional role for protein disorder in molecular recognition.
X-ray crystallography is a useful technique that helps visualize the structures of the proteins. Based on this technique, over 95% of the entire molecule is represented by 25% of crystal structures and all others have missing electron density for more than 5% of their sequence due to the multiple conformations on these regions.
Proteins actually have some disordered conformation and these disordered proteins lie at one extreme part from very flexible to static structural states on a continuous spectrum. Either only a part of the protein or the whole complete polypeptide chain is found in this disorder. Therefore, investigating only some parts of the proteins would not help summarize the flexibility of the protein. The term “conditionally disordered” means the disorder of proteins may happen under some certain conditions and may not happen under other conditions.
It is very common to see the intrinsic disorder within proteins. For example, between 30% and 50% of eukaryotic proteins are estimated to have more than 30 amino acids that violate the defined secondary structure in vitro and many complete unstructured proteins have been predicted to exist too. It is still very challenging to verify the status of folding of proteins within the region of cells despite a lot of computational methods that have been used. There is a chance that many proteins which are seen either partially or fully folded happen to be unstructured in cells. The number of these chances is still uncertain.
It is however thought that the presence of the appropriate binding pairs would make the disordered proteins come into their folded state, which means that the percentage of intrinsically disordered proteins in vitro might be lower in the cell. The extent of the disorder might be decreased by the stabilizing interactions within the cells.
Through chemical shift, residual dipolar coupling, and paramagnetic resonance enhancement measurements, NMR serves as a good method to provide the detailed information on extent of disorder of the proteins.
There are two states of disordered proteins. One shows a high degree of flexibility and the other state is where the protein is found more ordered. Thus, in order to know the cause and effect relationships between disorder and function, it is essential to study both states. Many disordered proteins like DNA, proteins, and membranes refold once they find a partner to bind to. Also, order-to-disorder-to-order transitions can occur. Proteins that are involved into multiple binding are very good examples of conditional disorder. Binding surfaces that are disordered before binding are able to fold into distinct conformations with other partners better than the binding surfaces that are already well-organized. The ‘conformational selection hypothesis’ suggests that different members of conformational ensemble can be stabilized by the binding of different partners. On the other hand, the ‘folding upon binding’ model proposes that proteins may be able to fold into different conformations when they bind with different partners.
Predictions done on whole proteomes suggest that the frequency of disordered proteins in eukaryotes is much larger than in prokaryotes, with the frequencies in the two groups of prokaryotes, archaea and eubacteria, being similar. In mammals, about half of all proteins are predicted to have large unordered regions, with about a quarter being fully disordered.
Disordered proteins are prevalent in signaling and regulation, especially in interactions with biomolecules such as nucleic acids and other proteins. Molecular recognition and protein assembly and modification frequently involve proteins with disordered regions. The ability of these proteins to interact with multiple molecular partners means that they are also common in protein-protein networks, either as hub proteins or as proteins interacting with hub proteins.
Disordered proteins are implicated in a number of human diseases. In particular, the amyloid diseases, which involve the accumulation of misfolded proteins, seem to be associated with disordered proteins, probably because their variable regions make them more likely to have a structure that favors their accumulation. This category includes many neurodegenerative diseases, such as Alzheimer's and Parkinson's.
The Role of Computers in Determining Structure and Function of Proteins
The structure or folding of an amino acid and by extension its function can be analyzed and compared through its primary structure or amino acid sequence using computer algorithms. Comparisons of amino acid sequences of unknown folding patterns with similar amino acid sequences of known folding is enhanced using computers. A computer automated tool called Protein Basic Local Alignment Search Tool, or protein BLAST, is a free search tool open to the public that allows quick comparison of amino acid sequences in an online database. The output of this tool is the percent match of amino acids and the known properties of the sequence matches. Furthermore, because amino acid sequences are based on DNA sequences, three bases code for one amino acid, the protein under scrutiny can be analyzed on a DNA level using DNA BLAST. The integration of public databases of amino acid and DNA sequences along with computer algorithms has accelerated the genome and proteome field by allowing scientists around the world to share and analyze sequences.
In silico modeling studies have helped identify several characteristics of co-translational folding pathway. First, it was determined that in vivo protein folding is a vectorial process, which is a dispersion change. Second, co-translational vectorial folding of the developing polypeptide from its N-terminal end to its C-terminal end results in a sequential structuring of the distinct regions of the polypeptide emerging from the ribosomal tunnel. Third, attachment to the developing polypeptide chain to the ribosome during protein synthesis reduces the conformational space and the degrees of freedom of the growing chain. This limits the number of possible intermediates and reduces the number of possible folding pathways. Fourth, co-translational protein folding begins early during the process of polypeptide chain synthesis on the ribosome, with some elements forming inside the ribosomal tunnel. Fifth, folding catalysis and molecular chaperones interact with the growing developing chain as soon as it emerges from the tunnel. This accelerates the slow steps in protein folding and prevents misfolding of proteins.
↑11. Kersse K, Verspurten J, Vanden Berghe T, Vandenabeele P. The death-fold superfamily of homotypic interaction motifs. Trends in biochemical sciences. 2011;36(10):541–52. Available at: http://www.ncbi.nlm.nih.gov/pubmed/21798745. Accessed October 29, 2012.
12. Small heat shock proteins and α-crystallins: dynamic proteins with flexible functions.
Basha E, O'Neill H, Vierling E.
Trends Biochem Sci. 2012 Mar;37(3):106-17. Epub 2011 Dec 14
Conditional disorder in chaperone action. Bardwell JC, Jakob U.
Trends Biochem Sci. 2012 Sep 24. pii: S0968-0004(12)00127-2. doi: 10.1016/j.tibs.2012.08.006. [Epub PMID 23018052 [PubMed - as supplied by publisher]
"Molecular Biology of the cell." Fifth Ed-Alberts, Johnson, Lewis, Raff, Roberts, Walter. pg. 716-717
Cabrita LD, Dobson CM, Christodoulou J. Protein folding on the ribosome. Current Opinion in Structural Biology 2010, doi:10.1016/j.sbi.2010.01.005
A Keith Dunker, Israel Silman, Vladimir N Uversky, Joel L Sussman. "Function and structure of inherently disordered protein." Curr Opin Struct Biol. 2008 Dec;18(6):756-64
Booth Paula J, Curnow Paul. Folding Scene Investigation: Membrane Proteins. Current Opinion in Structural Biology 2009, doi:10.1016/j.sbi.2008.12.005
Kuhn, Andreas, Rosemary Stuart, Ralph Henry, and Ross E. Dalbey. "The Alb3/Oxa1/YidC protein family: membrane-localized chaperones facilitating membrane protein insertion?" TRENDS in Cell Biology 13 (2003): 510-16. 26 Oct. 2011 <http://www.cell.com/trends/cell-biology/abstract/S0962-8924(03)00196-X>
Table 1: Berg, Jeremy. Relative Frequencies of Amino Acid Residues in Secondary Structures. 2012. Biochemistry, New York . Print.
Voet, Donald, Judith G. Voet. Biochemistry 3rd ed. New Jersey: John Wiley & Sons, Inc, 2004. Print.
Original hard-sphere, reduced-radius, and relaxed-tau φ,ψ regions from Ramachandran, with -180 to +180 axesBackbone dihedral angles φ and ψ (and ω)
A Ramachandran plot, also known as a Ramachandran diagram or a [φ,ψ] plot, was originally developed by Gopalasamudram Ramachandran, an Indian physicist, in 1963. Ramachandran Plot is a way to visualize dihedral angles ψ against φ of amino acid residues in protein structure. Ramachandran recognized that many combinations of angles in a polypeptide chain are forbidden because of steric collisions between atoms. His two-dimensional plot shows the allowed and disfavored values of ψ and φ: three-quarters of the possible combinations are excluded simply by local steric clashes. Steric exclusion is the fact that two atoms cannot be in the same place at the same time is the powerful organizing principle that propels the use of the Ramachandran plot forward.
The two torsion angles of the polypeptide chain, also called Ramachandran angles, describe the rotations of the polypeptide backbone around the bonds between N-Cα (called Phi, φ) and Cα-C (called Psi, ψ). The Ramachandran plot provides an easy way to view the distribution of torsion angles of a protein structure. It also provides an overview of allowed and disallowed regions of torsion angle values, serving as an important factor in the assessment of the quality of protein three-dimensional structures.
Torsion angles are among the most important local structural parameters that control protein folding - essentially, if we would have a way to predict the Ramachandran angles for a particular protein, we would be able to predict its 3D structure. The reason is that these angles provide the flexibility required for folding of the polypeptide backbone, since the third possible torsion angle within the protein backbone (called omega, ω) is essentially flat and fixed to 180 degrees. This is due to the partial double-bond character of the peptide bond, which restricts rotation around the C-N bond, placing two successive alpha-carbons and C, O, N and H between them in one plane. Thus, rotation of the main chain (backbone) of a protein can be described as the rotation of the peptide bond planes relative to each other.
The Ramachandran Plot helps with determination of secondary structures of proteins.
Quadrant I shows a region where some conformations are allowed. This is where rare left-handed alpha helices lie.
Quadrant II shows the biggest region in the graph. This region has the most favorable conformations of atoms. It shows the sterically allowed conformations for beta strands.
Quadrant III shows the next biggest region in the graph. This is where right-handed alpha helices lie.
Quadrant IV has almost no outlined region. This conformation(ψ around -180 to 0 degrees, φ around 0-180 degrees) is disfavored due to steric clash.
Exception from the principle of clustering around the α-helix and β-strand regions is glycine. Glycine does not have a complex side chain, which allows high flexibility in the polypeptide chain as well as torsion angles, something normally not allowed for other amino acid residues. That is why glycine is often found in loop regions, where the polypeptide chain makes a sharp turn. This is also the reason for the high conservation of glycine residues in protein families, since the presence of turns at certain positions is a characteristic of a particular fold of a protein structure.
Another residue with special properties in terms of its torsion angles is proline. Proline, in contrast to glycine, fixes the torsion angles at values, which are very close to those of an extended conformation of the polypeptide (like in a beta-sheet). Proline is often found at the end of helices and functions as a helix disruptor.
Berg, Jeremy M., John L. Tymoczko, and Lubert Stryer. "Chapter 2: Protein Composition and Structure." Biochemistry. New York: W. H. Freeman, 2007. N. pag. Print.
The Protein Folding Problem is the obstacle that scientists confront when they try to predict 3D structure of proteins based on their amino acid sequence. Although it is known that a given sequence of amino acids almost always folds into a 3D structure with certain functions, it is impossible to predict, with high precision, the exact folding pattern. Understanding the speed of proteins folding, which occurs extremely quickly, has also become a challenge to scientists. To be able to understand any type of biochemical reaction requires isolation and structure determination of reactants, intermediates and products. In protein folding, the isolation of reactants, intermediates and products is complicated because most interactions in proteins are non-covalent and weak interactions which lead to rapid rates of interconversion between each reaction state. Therefore, the isolation of intermediates is not easily achieved and therefore inaccessible for X-ray crystallography. In addition, several advances in protein folding research have been made in characterizing reactants and intermediates. Based on the complexity of protein folding, there are 3 major problems of protein folding: The folding code, structure prediction and the folding speed and mechanism.
In the late 1980s, scientists discovered that there is a sequence of amino acid code that folds proteins in a particular way. The starting point of protein folding is indeed the primary structure (the sequence of amino acids), also known as denatured state of the protein. Even the smallest amount of the denatured state can activate nucleation and proliferation carried out through protein folding pathways. Characterization of these denatured states of proteins at physiological conditions is very difficult because it is necessary to unfold the proteins to their denatured states without the presence of denaturants [2, Travagilini-Allocatelli et al.].
Recent research has allowed the study of denatured states to reach new heights using the single-molecule approach. Researchers used single-molecule experiments to examine coil to globule transition of proteins and have demonstrated that the denatured state showed steady expansion as the concentration of denaturant was increased. Similarly, at low denaturant concentrations, the peptide chain of the protein collapsed in a sequence dependent manner [2, Travagilini-Allocatelli et al.].
Also there have been advancements to study intermediates in protein folding. For example, the denatured state of the engrailed homeodomian (En-HD) was engineered to be denatured in physiological conditions and Nuclear Magnetic Resonance (NMR) has shown that it resembles a folding intermediate. An additional study discovered that the specific section of the En-HD called the helix-turn-helix motif (HTH) behaves as an independent folding domain. When examining the full protein, the HTH motif represents a folding intermediate in the En-HD folding pathway [2, Travagilini-Allocatelli et al.].
Although the folding of protein is still an enigma, scientists have taken the advantage of these protein information to design new materials, such as medicine, reagents and inhibitors, to benefit the society.
Nowadays, researchers predict the structure of a protein by inputting the amino acid sequence into a computer. The advanced technology and modeling software allow scientists and researchers to form a predicted structure. However, the structure is not accurate, as there is always a small degree of errors present. Nevertheless, this can speed up discovery of new medications since the digital structure can be manipulated.
Secondary structure prediction
Secondary structure prediction is a set of techniques that aim to predict the secondary structures of proteins and RNA sequences based only on their primary structure which is amino acid or nucleotide sequence. For example, proteins, a prediction consists of assigning regions of the amino acid sequence as alpha helices, beta strands, or turns. The success of a prediction is determined by comparing it to the results of the DSSP (the DSSP algorithm is the standard method for assigning secondary structure to the amino acids of a protein, given the atomic-resolution coordinates of the protein) algorithm applied to the crystal structure of the protein; for nucleic acids, it may be determined from the hydrogen bonding pattern. Specialized algorithms have been developed for the detection of specific well defined patterns such as transmembrane helices and coiled coils in proteins, or microRNA structures in RNA.
Tertiary structure prediction
Experimental methods such as NMR spectroscopy or x-ray diffraction analysis are widely used in order to determine tertiary protein structures. But the rate at which protein structures can be determined by experimental techniques is much lower than the rate at which new genes are identified by the various genome projects.
Ab initio protein modelling methods have been used to build 3-D protein models. For example, based on physical principles rather than on previously solved structures. There are many possible procedures that either attempt to mimic protein folding or apply some stochastic method to search possible solutions (like, global optimization of a suitable energy function). These procedures require massive computational resources, and have thus only been carried out for tiny proteins. To predict protein structure for larger proteins will require better algorithms and larger computational resources like those afforded by either powerful supercomputers. Although these computational barriers are massive, the potential benefits of structural prediction make ab initio an active research topic.
Side-chain geometry prediction describes a computational approach that can make predictions for a series of coiled-coil dimers. This method comprises a dual strategy that augments extensive conformational sampling with molecular mechanics minimization.
Quaternary structure
In the case of complexes of two or more proteins, where the structures of the proteins are known or can be predicted with high accuracy, protein–protein docking methods can be used to predict the structure of the complex.
In 1968, Cyrus Levinthal pointed out that protein folding, with precision, happens in microseconds, which seems unrealistic and impossible. This is also known as the Levinthal's paradox. Nowadays, we have advanced methods such as mutational methods, which give us the value of phi and psi during folding, and hydrogen exchange methods, which allow us to see structural folding events. However, the dynamics and mechanism of protein folding still require additional research and understanding.
The dynamics and kinetics of unfolded polypeptide chain have been addressed by recent studies of loop formation by Keifhaber and coworkers. They used different model systems each representing different types of loops: end to end, end to interior, or interior to interior. Their experiments showed that end to interior and interior to interior loop formation formed slower than end to end loops. This discovery suggests that chain motion of one part of the unfolded polypeptide chain is coupled to other parts of the chain. These kinetics experiments also revealed that protein folding processes take place on different time scales and thus there is a hierarchy in loop formation[2, Travagilini-Allocatelli et al.].
Although additional research is necessary to understand mechanisms in protein folding, there are two different classical mechanisms that have been used to describe folding of single domain proteins. The first of the mechanisms is called the Diffusion-Collision Model. Proteins that follow this mechanism fold in a stepwise manner that involves growing secondary structure elements. These elements then collide, combine and strengthen. For example, there is evidence that the En-HD mentioned above follows the diffusion-collision model. The second mechanism is known as the Nucleation-Condensation Model. Proteins following this method have been seen to fold from an unstructured denatured state with simultaneous formation of secondary and tertiary structure. For example, a homologous protein of En-HD called hTRF1 has been shown to follow this model. However, there are many proteins that exhibit characteristic pathways of both diffusion-collision and nucleation-condensation models [2, Travagilini-Allocatelli et al.].
The starting point of protein folding: the denatured state
In the denatured state, the structure can trigger nucleation and propagation, which may carry through the folding pathway. Characterization of denatured states of proteins at physical conditions represents a hard task as needed to disfavor the population of native states without adding denaturants. Chemically denatured states may act like random-coil polymer at high denaturant concentrations.
Sherman and Haran used single-molecule experiments to analyze the coil to globule transition of protein L and showed that the denatured state of the protein increases as the denaturant concentration increases. Also Eaton and co-workers compared the size and dynamics of the denatured states of those two proteins, displaying a similar length of 64 and 66 amino acids.
Mechanisms of protein folding
There were two different mechanism used to describe the folding of single-domain proteins. Some proteins such as barnase, has been described to fold in a stepwise manner with rapid formation of distinct nuclei and also with their collision and consolidation. There are also other proteins, with chymotrypsin inhibitor 2 as an example of the nucleation-condensation model.
The folding pathway of the small alpha beta protein domain has been shown to be distinct from the pure nucleation-condensation and diffusion-collision, but still displaying the characteristics of both models.
Folding stability and function
The inherent stability of individual protein segment is a key factor in determining the folding mechanism of a given protein. Many times, cell’s life relies on the ability of its constituent proteins to fold into 3D structures that are crucial for their function. The amount of folded functional protein in a cell depends on several factors such as, rate of protein biosynthesis and degradation.
There was a question about whether the stability and folding of fully folded proteins can be related to their activity. Allostery can be the bridge where protein folding meets function. Allosteric effects involve communication between ligand binding sites which is critical to many physiological processes.
As allostery is a thermodynamic process, it should not only be considered by changes in conformation but also by changes in the dynamics of the mean conformation.
Therefore more research is necessary to fully comprehend the mechanism of protein folding and find a solution to the protein folding problem.
Ken A Dill, S Banu Ozkan, Thomas R Weikl, John D Chodera and Vincent A Voelz. The protein folding problem: when will it be solved?Current Opinion in Structural Biology 2007.
Carlo Travaglini-Allocatelli, Yiva Ivarsson, Per Jemth and Stefano Gianni. Folding and stability of globula proteins and implications for function Current Opinion in Structural Biology 2009, 19:3-7.
Mount DM (2004). Bioinformatics: Sequence and Genome Analysis. 2. Cold Spring Harbor Laboratory Press. ISBN 0879697121
Zhang Y (2008). "Progress and challenges in protein structure prediction". Curr Opin Struct Biol 18 (3): 342–8. doi:10.1016/j.sbi.2008.02.004. PMC 2680823. PMID 18436442
Although much work has been done on protein folding "in vitro", few research has significantly advanced the work contributing to "in vivo" protein folding. The importance of the latter comes as a consequence that protein folding is presumably guided by a molecular mechanism instead of a protein independently folding according to the lowest energy conformation. Although it has proven that proteins are highly successful at reaching their native state only by chaperone proteins, it seems that at the creation of a new protein, something must assist the development of the secondary and tertiary structure. The authors of a current opinion article in Structural Biology, Lisa D. Cabrita, Christopher M. Dobson, and John Christodoulou have published an update on the recent discoveries of how the nascent chains of a newly synthesized protein emerges in the article entitled, "Protein Folding on the Ribosome."
The place where the protein chain begins to fold is a topic that is greatly studied. As the nascent chain goes through the “exit tunnel” of the ribosome and into the cellular environment, when does the chain begin to fold? The idea of cotranslational folding in the ribosomal tunnel will be discussed. The nascent chain of the protein is bound to the peptidyl transferase centre (PTC) at its C terminus and will emerge in a vectorial manner. The tunnel is very narrow and enforces a certain rigidity on the nascent chain, with the addition of each amino acid the conformational space of the protein increases. Co translational folding can be a big help in reducing the possible conformational space by helping the protein to acquire a significant level of native state while still in the ribosomal tunnel. The length of the protein can also give a good estimate of its three dimensional structure. Smaller chains tend to favor beta sheets while longer chains (like those reaching 119 out of 153 residues) tend to favor the alpha helix.
The ribosomal tunnel is more than 80 ampere in length and its width is around 10-20 ampere. Inside the tunnel are auxiliary molecules like the L23, L22, and L4 proteins that interact with the nascent chain help with the folding. The tunnel also has hydrophilic character and helps the nascent chain to travel through it without being hindered. Although rigid, the tunnel is not passive conduit but whether or not it has the ability to promote protein folding is unknown. A recent experiment involving cryoEM has shown that there are folding zones in the tunnel. At the exit port (some 80 ampere from the PTC), the nascent chain has assumed a preferred low order conformation. This enforces the suggestion that the chain can have degrees of folding at certain regions. Although some low order folding can occur, the adoption of the native state occurs outside the tunnel, but not necessarily when the nascent chain has been released. The bound nascent chain (RNC) adopts partially folded structure and in a crowded cellular environment, this can cause the chain to self-associate. This self-association, however, is relieved with the staggered ribosomes lined along the exit tunnel that maximizes the distances between the RNC.
The current understanding of protein folding has come from in vitro studies of renaturation of proteins through a variety of different environments as well as in silico computer simulations. These studies can only help to extrapolate fractions of the in vivo process of protein formation. Protein folding is initiated following the synthesis of the nascent polypeptide chain as it is synthesized by the ribosome. The start of protein folding is therefore coupled with the continuing synthesis of the polypeptide chain.
Currently, protein folding is view as a process that takes place as a consequence of interactions been the amino acid of that protein which can take certain paths to achieve a lowest energy state, the native state. However, there are certain paths a protein may start to fold by and lead to a conformation that is of low energy but not the native state. The protein has not way of coming of this conformation without a significant amount of energy input. This non-native state is a way a protein can be misfolded and lead to aggregation. Another factor that can influence the likelihood of obtaining the native state is the fact that larger proteins have more possibilities of folding, this decreases the likelihood of forming the most energetically favorable state. Proteins us the "co-translational folding' to reduce the extent of conformational space available to the protein. Adding to this, molecular chaperones help to further assist proteins in achieving their native conformational state.
One technique of generating RNC and taking snapshots as it emerges from the tunnel is to arrest translation. A truncated DNA without a termination sequence is used. This allows for the nascent chain to remain bound until desired. To determining the residues of the chain, they can be labeled by carbon-13 or nitrogen-15 and later detected by NMR spectroscopy. Another technique is the PURE method and it contains the minimal components required for translation. This method has been used to study the interaction of the chains and auxiliary molecules like the TF chaperone. This method is coupled with quartz-crystal microbalance technique to analyze the synthesis by mass. An in vivo technique in generating RNC chain can be done by stimulating it in a high cell density. This is initially done in an unlabeled environment, the cells are then transferred to a labeled medium. The RNC is generated by SecM. The RNC is purified by affinity chromatography and detected by SDS-PAGE or immunoblotting.
By generating the RNCs, many experiments can be done to study more about the emerging nascent chain. As mentioned above, the chain emerges from the exit tunnel in a vectorial manner. This enables the chain to sample the native folding and increases the probability of folding to the native state. Along with this vectorial folding, chaperones also help in favorable folding rates and correct folding.
Ribosome Structure and Co-translational Protein Folding
In E. coli the 70S ribosomal particle is composed of 50 proteins and three RNA molecules. The most interesting structural feature in the 70S ribosomal particle in regards to protein folding is the ribosomal exit tunnel. This is a channel that links PTC(peptidyl transferase centre) with the cellular environment. The dimensions include a length of 80 angstroms, width between 10-20 angstroms. 70S is lined with a large RNA molecule and L4 and L22 ribosomal proteins. Also L23 serves as a docking point for other molecules to assist in the folding process. L4 and L22 proteins in the ribosomal exit tunnel have been shown by recent cryoEM studies that they can interfere with proteins synthesis along with other interactions with the nascent chain. In addition, arginine residues have been observes to stop the translation process by changing electrostatic potentials.
Although ribosomal exit tunnel is presumably to have a more or less rigid structure, it seems that it does partake to a degree support nascent chain folding. This is evidence by the fact that on average the tunnel is able to accommodate about 30-40 residues, which is considerably more than a polypeptide chain sequence that is fully extended. The degree to which a nascent chain folds seems to vary depending on the kind of protein being synthesized. Certain nascent chains transmembrane protein sequences appear to possibly already construct an alpha-helical structure inside the tunnel.
Studying nascent chains emerging from the ribosomal exit tunnel has proven to be a significant challenge for any of the current methods of structural and cellular biology. One idea presented in this paper is to take be able to have "snapshot" of the elongation process. In order to due this, translation must be arrested artificially which would involve engineering DNA strands that lacks a stop codon. Another issue is also in focusing on the particular residues of interest on the nascent chain within the sea of other residues form the ribosome.
Understanding Co-translational Folding by Biochemical and Biophysical Studies
Once examples illuminated in the article is using SDS-Page on the risbosomal bound nascent chains(RNCs) of influenza haemagglutinin which showed they can form disulfide bonds and undergo glycosylation. Also, using monoclonal antibodies, it has been discovered that there is variability in the emergence of the nascent chain from the tunnel. These examples among others demonstrate that not only can nascent chains acquire structure but also activity while still being attached to the ribosome. The speed of folding for nascent chains seems to be related to the number of stop and rare codons present. The reasoning is that a discontinuous translation rate will slow down the folding process. However, slower rates seem to produce more efficient folding since the nascent chain has more time to develop its native structure. Most of the biochemical and physical methods illuminating the understanding of co-translational folding has been eluded by x-ray crystallography because of the dynamic nature of the folding process which in crystallography is very difficult to obtain.
As the nascent chain starts emerging from the tunnel, it has to opportunity to interact with molecules that will assist in the folding process. These include molecular chaperones, peptide deformylase, and the signal recognition particle. The first molecule in assisting the nascent chain in folding is the 48kDa TF which docks on L23. This protein in the absence of a nascent chain will dock on and off however with the presence of the nascent chain its affinity to bind to L23 increases. TF undergoes a conformational change in a where a protective cavity is formed for the nascent chain. TF enables enough of the polypeptide chain to emerge such that a significant degree of folding can be achieved. It does this by binding to hydrophobic segments of the chain even after is has released from L23. Once hydrophobic regions of chain are no longer exposed, TF seem to unbind and allow further helper molecules to assist in protein folding. TF seems to increase folding efficiency but at the expense of being slower to fold. Protein translocation is then done by SRT which shuttles the TF to a heterotrimeric integral membrane protein. This then allow further processing and folding.
Ribosome subunit in prokaryote cells and eukaryote cells
The ribosomes catalyze peptide bond formation, in a process called peptidyl transfer catalysis, and synthesize polypeptides by reading the genetic code of the mRNA. The ribosome is composed of a large and a small subunit both in prokaryote and eukaryote cells. Prokaryotes have 70S ribosomes, each consisting of a small (30S) and a large (50S) subunit. Eukaryotes have 80S ribosomes, each consisting of a small (40S) and large (60S) subunit. Due to the differences in their structures, the bacterial 70S ribosomes are vulnerable to these antibiotics while the eukaryotic 80S ribosomes are not. Within the cellular structure, mitochondria have ribosomes similar to the bacterial ones; however, mitochondria within eukaryote cells are not affected by these antibiotics because they are surrounded by membrane around its organelle. The initiation of the translation process in bacteria was found to locate on 30s subunit. This process requires the increase of both the incubation temperature and ionic strength in order to assemble into the correct tertiary structure contained with its amino acid sequence. The research experiments done by Dr. Masayasu’s research on the synthesis of ribosomes and ribosomal components in E-coli, also found that the correct assembly of the ribosomal particles is locating in the structures of their own molecular component and not by other nonribosomal factors.
A ribosome is the essential contributing factor in protein synthesis where it is assembled on the translation initiation region (TIR) of the mRNA during the initiation phase of translation. The mRNA is decoded as it slides through the large ribosomal subunit and places the a polypeptide chain in the other subunit of the ribosome. Newly synthesized protein will then dissociate once the stop codon is reached in the ribosome. In the final ribosome recycling phase, the ribosomal subunits dissociate and the mRNA is released. The main events of the translation process are relatively similar in both prokaryotic and eukaryotic cells. Major differences in the detailed mechanism of each phase exist. Bacterial translation involves relatively few factors, in contrast to the more complex process in eukaryotes.
Many membrane proteins form multiple sub unit protein complexes. They possess integral and peripheral subunits. Enzymes known as Sec translocase and YidC insertase insert bacterial membrane proteins into the inner membrane. This process is assisted by YidC and the phospholipid phosphatidylethanolamine. Glycine zippers and other motifs also help transmembrane-transmembrane helix interactions that can form alpha helical bundles of membrane proteins. When membrane insertion occurs or when after membrane insertion occurs, the subunits of oligomeric membrane proteins have to be able to locate each other to construct the homo-oligiomeric and the hetero-oligomeric membrane complexes. Even though chaperones can serve as assembly factors to construct the oligomer, numerous protein oligomers seem to fold and oligomerize spontaneously. It has been shown by experiments that many of the subunits of hetero-oligomers are structured after a sequential and patterned pathway to create the membrane protein complex. If it so happens that the inserted protein folds improperly or the membrane protein is assembled incorrectly, quality control mechanisms can deactivate the proteins.
Membrane protein can do a large variety of functions inside the cell from metabolite exchange to cell signaling and nerve conduction. They can also function as ATPases, electron carriers, ion channels, and transporters, sheddases, and photosynthetic reaction centers. They are abundant in both the eukaryotic and prokaryotic cell and they comprise about 20 percent to 30 percent of the total amount of proteins.
Many of the integral inner membrane proteins are alpha helical bundles with alpha helical membrane spanning areas. Advanced research has shown that the structures of the membrane proteins possess not only membrane spanning helices that are straight, but also possess very curved helices that span the membrane partially through. Alpha helical membrane proteins can exist as monomer or as multimeric complexes.
In order to guarantee that membrane proteins behave and function properly, they must be instructed to their destined membrane in the cell and then inserted and folded to the appropriate structure. Membrane tageting in the eukaryotic cells is necessary and more complicated than in eubacteria. Eukaryotic cells must instruct at least 10 membranes while eubacteria must only instruct 1 or 2 membranes in the gram-positive and gram-negative bacteria, respectively. After targeting, membrane protein integration and topogensis are instructed by a coordinated process of topogenic sequences and translocases. While this process is occurring, the transmembrane segments and extramembranous loops are folded.
The process of bacterial inner membrane protein assembling into the membrane is very complex. In addition, the mechanisms that control the protein targeting and inserstion into the membrane, folding of the alpha helical bundles, and the assembly into oligomeric membrane protein complexes will be explored more in depth.
The targeting of nascent chains to the membrane happens initially during the protein synthesis. It happens very early, even before the appearance of the polypeptide from the ribosomes channel. These nascent chains can already send signals in the ribosomes, which is a requirement of the signal recognition particle. A signal recognition particle is made up of a protein component Fth and a 4.5S RNA. The SRP combines with a hydrophobic part of a membrane protein as comes out from the ribosome at the membrane surface. The SRP-interacting area is most commonly the first TM region, but it can also be further apart and distinct from the TM segments. By studying the structure, it has been shown that a groove in the SRP M domain binds to the apolar segment.
When the receptor FTsy of the SRP- ribosome nascent chain complex is targeted by this complex, a SRP/FTsy complex is formed. The deconstruction of the complex and the freeing of the targeted protein needs GTP hydrolysis. The SRP and the FSty start out GTP bound and afterwards they construct into a complex by the interaction of their NG domains. A common trait between Ffh and FTsy is that they both have two homologous domains and a distinct domain. By analyzing the structure if the Ffh and the FtsY NG doman complex, an interesting thing was found that there is a shared composite active-site area in the Ffh/FtsY hetereodimer, which is combined with two bound nucleotides. After the process of GTP hydrolysis, the membrane protein-nascent chain complex is sent to the SecYEG translocation channel, and the SRP and FtsY break apart from each other, which enables the SRP to recycl and interact in another round of SRP targeting. This sending of the nascent chain to the translocation channel is assisted by the interaction of the FtsY with SecY.
It is necessary for the enzymes Translocases and intertases to put the freshly synthesized proteins into membranes. In bacteria, the SecYEG translocase and the YidC insertase have been depicted and analyzed. It reveals that they both display their translocation and insertion function in reconstituted systems. In addition, they are necessary processes for the bacterial life.
The enzyme Sec translocase catalyzes the bacterial membrane protein insertion. The Sec translocase is made up of the membrane-embedded SeYEG and SecDFyajC complexes, in addition to the peripheral membrane component SecA. SecYEG supplies the protein-conducting channel. This is necessary for translocation and to make membrane protein insertion more efficient. Sec, which also known as the motor ATPase, is crucial for the translocation of preproteins through the membrane and for the translocation of particular hydrophilic areas of the membrane proteins. SecA utilizes ATP hydrolysis to propel the inserting polypeptide chain through the Sec channel 20 to 30 residues simultaneously.
A major important discovery in the protein export area of studies was that the structure of the SecY complex was determine from an enzyme called Methanoccoccus jannaschii. This enzyme is made up of SecYEBeta. SecBeta does not have sequence homology to the eubacterial SecG but it does have sequence homology to the eukaryotic Sec61Beta. The SecY channel contains an hourglass structure with hydrophoibic narrow parts that is about 3 to 5 A in size which is found in the center of the channel. The narrow constriction within the SecYEbeta splits the interior hydrophilic cavities on the periplasmic and cytoplasmic areas of the membrane. This narrow area is made up of a hydrophobic pore ring, which consists of 4 isoleucine residues, one valine, and one leucine residue. In addition, the aliphatic side chains of these amino acids are directed toward each other, which creates a hydrophobic collar through which the hydrophilic region of the polypeptide chain would be transport during translocation across the membrane.
Based on the crystal structure, the SecY channel is in sealed off state with the pore ring closed off by a helix on the luminal side. When the Sec channel opens up through signal peptide binding to the SecY TM2-TM7 region, the plug is relocated out of the channel site about 20 A away near the SecE helix.
Another important aspect of the SecY channel is the lateral gate. This is made to let the Tm regions of the inserting membrane proteins to be freed from the channel laterally and to split it into the lipid phase. The lateral gate is at the surface of SecY TM2 and TM7 of the Sec61alpha (SecY) which is found at the front side of the Sec channel. Before, TM2 and TM7 of the Sec61Alpha was thought to form the signal peptide-binding region because a signal peptide of preprotein can potentially be cross linked to these Tm parts during posttranslational translocation. When translocation of a polypeptide chain occurs, the lateral gate is opened up. The opening of this lateral gate is significant because locking the lateral gate by disulfide cross linking does not allow SecA-mediated preprotein translocation in Escherichia coli.
It is important to understand how the SecA operates with the SecY channel to translocate hydrophilic domains of membrane proteins across the membrane. The 4.5 A structure of the SecA/SecYEG from Thermotoga martima helps explain this process. First one copy of the SecA is attached to one copy of the SecY channel in the structure. The SecA is placed flat on the SecY channel about parallel to the membrane surface. It is important to note that the opening of the SecYEG channel has a two helix finger domain of SecA that can serve to transport substrates into the channel.
The YidC insertase is important because its job is to fit tiny proteins into the membrane. It was discovered that YidC influences membrane protein insertion. When the amounts of YidC is lessened in the cell, the insertion of Sec-independent proteins were slowed and discouraged. Before it was thought to be fit into the membrane spontaneously.
Through experiments it was thought that YidC affects the process of insertion of Sec-independent substrates. Photocross-linking studies that utilize a cell-free system displayed that membrane proteins that were stuck at different points of membrane protein insertion interact with YidC. Lipid vesicles that have YidC are enough to put the Sec-independent Pf3 coat protein and the ATP synthase subunit c. It was found that the Pf3 coat proteins sticks to the YiDC. This leads a significant conformational structure difference in the YidC protein.
Many of important membrane proteins span the lipid bilayer often. They span it in such a way that the sequential TM segments are in an alternating N to C and C to N orientation of the alpha helices. The TM segments are put together by cytoplasmic and periplasmic loops. These loops are primary hydrophobic and have differences in how big or small it as including the charge. Small loops put the tow helices together. On the other hand, the big and longer transform into different domains by folding. This plays a role in how the protein behaves and functions.
↑Ross E. Dalbey and Peng Wang and Andreas Kuhn(2011).[4]. "PubMed", p. 3-6.
Enzymes go through several mechanisms in order for it to survive and thrive in the biological world. The fact that proteins can fold amongst itself in their functional states after the process of synthesis is one of the most fascinating mechanisms ever studied by researchers.
In a living cell, protein folding occurs in a highly complex environment and uses different utility proteins for function. Some proteins' sole function is to protect the incomplete folding process from malfunctioning or the polypeptide chain from interactions other than folding. It is especially protective against factors that could lead to aggregation, folding catalysis or others that can slow down the process of protein folding in relation to isomerization or the forming of disulphide bonds. There are exceptions to the process of folding where auxiliary proteins are not needed to protect the sequence. Evidence shows that the code for protein folding is contained within the protein sequence. This is because studies have been shown where proteins undergo in vitro processes and can still function the same way as a protein supported by auxiliary proteins, as long as the in vitro occurs within conditional environments.
There have been a mass amount of studies performed on the mechanism of protein folding recently. Many researchers have also been receiving plenty of successful feedback on these conducted experiments. Many different types of applications, such as experimental and theoretical, have provided the basis for the main reason of studying protein folding in the first place.
One of the strongest cases of protein folding into new enzymes is known as the "stochastic process". The stochastic process is a random process that calculates different possibilities of pathways and conclusions to the final result of the experiment. The stochastic process is opposite to the deterministic process, which is having one initial possible result occur after an experiment is conducted. The stochastic process may initially start off with one possible result, but might end up with several different, plausible results, some more probable than others, after the experiment is completed.
Biased parties, nonetheless, believe that the original interactions between proteins are still more reliable and stable than newly-tested interactions and techniques. Studies have shown that the sequences of proteins can still be found in pristine condition even if the sequences live in very complex environments within a cell. However, when a protein folds on itself incorrectly or does not maintain to stay folded in the living cells, diseases of different types can occur.
An example of a possible group of diseases is called amyloidosis. Some common diseases that are derived from amyloidosis are Alzheimer's Disease and spongiform encephalophaties. These diseases occur when the protein is aggregated from failure of folding. An interesting fact about amyloidoses is that the formation of the aggregates show similarities to the property of polypeptides and not just a feature of proteins that suffer from poor or inadequate protein folding. It is not normal to find such amyloid aggregates in biological evolution, which begs the question if there are a variety of mechanisms that have been tampered with over time. In order to prevent such diseases from developing and to stop such mechanisms from mutating into insufficient mechanisms, the study of the folding of proteins is crucial to understanding the structure of a protein as well as the function to all living cells.
Issues and Possible Results of New Protein Folding Mechanisms
Although groundbreaking discoveries have been mass produced in the protein folding community, several issues arise. Tampering with the folding of a protein can alter the initial theory as to why humans should manipulate a natural occurring mechanism. Because of the high volume of magnitude and conformational changes done on a protein sequence, it is more likely that the experiment could lead to the stochastic process in producing several pathways and results. Also, due to a strong presence of heterogeneity at the end of the folding process, the changing of the protein folding sequence can alter desired results. According to Christopher Dobson, a researcher at Oxford Centre for Molecular Sciences in the University of Oxford, "there are two main approaches to try and overcome this issue".
The first approach lies with the use of biophysical techniques that can monitor the properties of the amino acid sequence as the folding takes place. Because the process of folding occurs in a rapid fashion, several outlets of methods are needed to map out the individual properties of the sequence. For example, an ultraviolet circular dichroism can be used to monitor the secondary structure of evolution and fluoresence microscopy can monitor the progress of the tertiary structure.
The second approach is to use protein engineering to study the mechanism of protein folding. Protein engineering is a particularly good method of studying the folding process because it can also map out the transition states of the protein sequence. Examination of the folding and unfolding parts of the mechanism takes place upon mutation of the individual amino acids in the sequence. By studying the intermediate steps of the folding process, the mechanism shows that there is a formation of native-like proteins surrounding a number of important amino acids. This provides evidence that for another mechanism called "nucleation-condensation", where the majority part of the protein sequence rapidly forms once the nucleus of the entire process has been found.
A Fibrous protein is a protein with an elongated shape. Fibrous proteins provide structural support for cells and tissues. There are special types of helices present in two fibrous proteins α-keratin and collagen. These proteins form long fibers that serve a structural role in the human body. Fibrous proteins are distinguished from globular proteins by their filamentous, elongated form. Also, fibrous proteins have low solubility in water compared with high solubility in water of globular proteins. Most of them play structural roles in animal cells and tissues, holding things together. Fibrous proteins have amino acid sequences that favour a particular kind of secondary structure which, in turn, confer particular mechanical properties on the proteins.
Collagen is a triple helix formed by three extended proteins that wrap around one another. Many rodlike collagen molecules are cross-linked together in the extracellular space to form collagen fibrils that have the tensile strength of steel. The striping on the collagen fibril is caused by regular repeating arrangement of the collagen molecules within the fibril.
Elastin polypeptide chains are cross-linked together to form rubberlike, elastic fibers. Each elastin molecule uncoils into a more extended conformation when the fiber is stretched and will recoil spontaneously as soon as the stretching force is relaxed.
alpha helix
beta pleated sheet
triple helix
Hydrogen bonding
Peptide -C=O----HN-, Intrachain between, and n+4 residues Parallel to helix axis
Peptide -C=O-----HN- , Interchain, Perpendicular to chain axis
Peptide, -C-----HN- and -C=O-----HO- (hydroxyl from side chain of Hyp), Interchain
Residues
Many types, Small or uncharged residues, such as Ala, Leu, and Phe, most common; Pro never found
Mostly Gly, Ala, and Ser
Many types, Gly every third residue; Pro and Hyp common
Covalent cross-linking
Interchain disulfide cross-link
None
Interchain lysine-derived cross-links
Chain direction and aggregation
Four parallel right-handed alpha helices form a left-handed supercoil.
Antiparallel chains
Three parallel left-handed helices form a right-handed supercoil.
Unfolded Protein Response (UPR) is a response to cellular stress that is related to the endoplasmic reticulum (ER) in mammalian species, but has also been found in yeast and worms.
When ER conditions are disrupted (such as alterations of redox state, calcium levels, failure to posttranslationally modify secretory proteins, etc.) or the chaperone proteins that assist protein folding is overcapacity (both are considered ER stress), the cell launches signals that try to deal with these changes and make a favorable folding environment. When the UPR is not sufficient to deal with this stress, apoptotic cell death happens.
The ER lumen's environment is made so that it favors the production of secretory and membrane proteins and a good amount of these proteins are rapidly degraded which is probably due to improper protein folding. This would pose a problem for the cell due to a possibility of misfolded protein buildup. This would be even more of a problem if the changes in this environment would occur. These changes will deter the overall ability to make properly folded proteins and more improper proteins will build.
UPR monitors and responds to changes in the ER protein folding environment. It monitors the protein-folding capacity of the ER and sends signals of cell responses to help maintain the folding capacity to prevent a buildup of unwanted protein products. For mammals, this response is the transient inhibition of protein synthesis to hinder the production of new proteins, followed by transcriptional induction of chaperone genes to initiate protein folding and induction of the activation of the ER-associated degradation system. If this process fails, then the UPR tells the cell to go to a destructive pathway. The UPR has three main signaling systems: (IRE1), PERK, and ATF6.
IRE1 is a type I transmembraned protein that contained serine/threonine kinase activity as a stress sensor. Once activated, the enodribonuclease activity in the carboxyl terminus of IRE1 catalyzes splicing of the HAC1 (which is responsible for inducing the expression of ER stress response genes) mRNA.
In yeast organisms, the IRE1 contains nuclear localization sequences in the carboxyl terminus, which can interact with components of nuclear pore complex and target IRE1 to the inner nuclear membrane. The result is that the COOH-terminal domain is now facing the inside of the nucleus and can now have access to nuclear mRNA. HAC1 then moves into the nucleus and binds to a promotor element to induce the expression of genes required for various reactions.
In mammals, the IRE1 pathway is like that of yeast, except that two IRE1 genes have been cloned. Alpha and Beta -IRE1. It does not contain nuclear localization sequences like in yeast IRE1. IRE1 has also shown to mediate cleavage of additional mRNAs targeted to the endoplasmic reticulum as well as cleavage of the 28S ribosomal subunit. This leads to the beliefe that IRE1 has a role in translation attenuation by degrading these mRNA transcripts and/or the ribosomal subunits.
When undergoing ER stress, the first response is transient global translation attenuation and this is mediated by PERK. PERK is a type I ER-resident transmembrane protein that detects stress though its lumenal domain. It also binds to chaperone protein Grp78, but when unfolded proteins start to build up during ER stress, this protein Grp78 starts to dissociate and PERK then autophosphorylates and dimerize. Once activated, PERK phosphorylates serine-51 of eukaryotic initiation factor 2α (eIF2α). eIF2α is unable start translation when phosphorylated, and this leads to inhibition of global protein synthesis. In reverse, phosphorylated eIF2α initiates translation of ATF4 mRNA. ATF4 upregulates ER stress genes. Translational recovery is mediated by the stress-induced phophatase growth arrest and DNA damage-inducible gene.
ATF6 exist in to isoforms (alpha and beta ATF6) . These have fairly balanced tissue distributions. ATF6 pathway activation involves a mechanism called regulated intramembrane proteolysis (RIP). In RIP, the protein translocates from the ER to the Golgi for proteolytic processing. The stress-sensing mechanism of ATF6 dissociates the Grp78 from its lumenal domain (This is similar to the processes of IRE1 and PERK pathways). Frp78 signals to two Golgi localization signals to allow ATF6 to enter the COPII vesicles to translocate the Golgi compartment. Disulfide bonds in ATF6 lumenal domain are also believed to keep ATF6 inactive. During ER stress disulfide bonds are reduced and an increase ability of ATF6 to exit arises.
The three UPR pathways do not only contribute to fixing of improperly folded proteins, it also as can contribute to a cell's apoptosis if the UPR fails to restore folding capacity.
Technology advances in sequencing and microarrays allow for us to better understand pre-mRNA splicing patterns in different cells. For example, cellular splicing changes when it is stimulated by factors such as DNA damage, neuron depolarization and or metabolic changes in cells. In the last few years, there have been more studies regarding patterns in mechanisms that relate cellular stimuli to downstream alternative splicing control. Some of these splicing events include degradation of splicing factors, altered nuclear translocation, and regulated synthesis of splicing factors.
What is alternative splicing and how does it work?
Alternative splicing is a process that occurs during gene expression and allows for the production of multiple proteins (protein isoforms) from a single gene coding. Alternative splicing can occur due to the different ways in which an exon can be excluded or included from the messenger RNA. It can also occur if portions on an exon are exclude/included or if there is an inclusion of introns. For example, if a pre-mRNA has four exons (A, B, C, and D) these fours exons can be spliced and translated in a number of different combinations. Exons A, B, and C can be translated together or Exons A, C, and D can be translated. This is what results in alternative splicing.
The pattern of splicing and production of alternatively spliced messenger RNA is controlled by the binding of regulatory proteins (trans-acting proteins that contain the genes) to cis-acting sites that are found on the pre RNA. Some of these regulatory proteins include splicing activators (proteins that promote certain splicing sites) and splicing repressors (proteins that reduce the use of certain sites). Some common splicing repressors include: heterogeneous nuclear ribonucleoprotein (hnRNP) and polypyrimidine tract binding protein (PTB). Proteins that are translated from alternatively spliced messenger RNAs differ in the sequence of their amino acids and this results in altered function of the protein. This is the reason why the human genome can encode a wide diversity of proteins. Alternative splicing is a common process that occurs in eukaryotes; most of the multi-exonic genes in humans are spliced alternatively. Unfortunately, abnormal variations in splicing are also the reason why there are many genetic diseases and disorders.
The splicing of messenger RNA is accomplished and catalyzed by a macro-molecule complex known as the spliceosome. The areas for ligation and cleavage are determined by the many sub-units of the spliceosome. These sub-units include the branch site (A) and the 5' and 3' splice sites. Interactions between these sub-units and the small nuclear ribonucleoproteins (snRNP) found in the spliceosome create a spliceosome A complex which helps determine which introns to leave out and which exons to keep and bind together. Once the introns are cleaved and removed, the exons are joined together by a phosphodiester bond.
As noted above, splicing is regulated by repressor proteins and activator proteins, which are are also known as trans-acting proteins. Equally as important are the silencers and enhancers that are found on the messngerRNAs, also known as cis-acting sites. These regulatory functions work together in order to create splicing code that determines alternative splicing. The cis-acting sites will be discussed here.
Splicing repression
Splicing silencers are regulatory sites that are found in pre-messengerRNA's and are where the splicing repressor proteins bind to. When the repressor binds to the silencer site, it reduces the chance that a site close-by will be chosen as a splicing junction. These silencer sites can be found on introns or on exons. When found on introns, these sites are known as intronic splicing silencers and on exons they are called exonic splicing silencers. The sequences found on these sites are numerous and that allows for different kinds of proteins to bind.
Splicing activation
On the other hand, splicing enhancers are regulatory sites where splicing activator proteins can bind to. When the activator protein binds to the enhancer site, it increases the chance that a site close-by will be chosen as a splicing junction. Just like the splicing silencers, these sites can also be found in introns and exons. In introns they are called intronic splicing enhancers and in exons they are called exonic splicing enhancers. However, unlike their silencer counterparts, enhancer sites usually allow the binding of activator proteins that belong to the family of SR proteins. These proteins are rich in arginine and serine.
How is alternative splicing regulated by some specific signals?
Alternative splicing has been recently revealed to occur in nearly all human genes. Most typically, a specific exon may be either included or excluded in different cell types or growth conditions when alternative splicing occurs. In each case, the pattern of splicing, the binding of regulatory proteins to cis-acting auxiliary sequences generally determines the pattern of splicing and these sequences in turn control where the binding occurs and/or how the enzymatic complex reacts at neighboring splice sites. (Combinatorial Regulation of Alternative Splicing)
Importantly, the open reading frame of the resultant mRNA or the presence of cis-regulatory elements that control mRNA stability or translation can be altered by any of these above differential patterns. Therefore, shaping the proteome of any given cell requires the precise control of alternative splicing , and how the cellular function responses to changing environmental conditions can also be significantly altered by changes in splicing patterns.
Representation of intron and exons within a simple gene containing a single intron.
Combinatorial Regulation of Alternative Splicing
The spliceosome is a macromolecular complex that catalyzes the removal of introns and the basic joining of extrons. The binding of various subunits of the spliceosome in order to sequence elements at the intron and extron boundaries in a pre-mRNA determines the precise sites of ligation and cleavage. Those subunits are the 5 splice site, the branch point sequence, a pyrimidine-rich track, and the 3 splice site. However, for mammals, the splice sites are poorly conserved; hence, they are typically not sufficient to bind the spliceosome with high affinity. The efficiency of spliceosomal binding via mechanisms can be impacted by proteins bound to non-splice site sequences within the exon or intron. Exonic or intronic splicing enhancers are the sequences that help promote spliceosomal recognition of an exon, while the splicing silencers are needed to inhibit recognition of the exon. Exon inclusion (green ovals) is promoted by the binding of the enhancers of members of the ubiquitously expressed SRSF protein family, while the exon usage is repressed by members of the hnRNP family of proteins via silencer elements (red ovals). FOX, CELF, neuro-oncological ventral antigen (NOVA) and muscleblind-like (MBNL) proteins are some other splicing regulators that are more tissue restricted and these regulators function equally as enhancers and repressors of splicing through mechanisms that are still largely undefined. Therefore, the ratio of mRNA isoform expression can frequently be altered by the binding of single regulatory proteins or the subtle changes in the balance of expression.
Neuro-oncological ventral antigen 1
Muscleblind-like splicing regulator 1
Post-Translational Modification of Splicing Proteins
Phosphorylation, acetylation, methylation, sumolylation and hydroxylation are involved in the modification of splicing regulatory proteins in many cases.
The phosphorylation of the extensive Arg-Ser dipeptides found within SR proteins is the best characterized modification.
The extensive post-translational modifications also includes the HnRNP proteins, along with other non-SR splicing factors.
An example of regulated degradation of a RNA-binding protein modulating alternative splicing.
Recently, technical tools such as deep sequencing and sensitive microarrays have opened up for more knowledge of alternative splicing events. Almost all human genes go through some sort of alternative splicing, which includes differential exclusion or inclusion of a specific exon, exclusion of a part of an exon, and inclusion of introns and exons. These differential trends can change the reading frame of the processed mRNA or alter any cis-regulatory factors that monitor mRNA translation or stability. For that reason, the regulation of alternative splicing is crucial in shaping the proteome of cells; alterations in splicing patterns can change functions in cells in response to environmental changes. Observations in heart tissue in its development stage, pre and post depolarization of neurons and cells before and after apoptosis have showed that alternative splicing events play a large role in the functional outcome of the signaling and developmental processes.
Since alternative splicing is generally determined by binding regulatory proteins to auxiliary sequences that control the location of binding and activity of the enzymatic complex at neighboring sites of splicing, it is used in response to DNA damage and T cell activation. One case for DNA damage includes the alternative splicing of the E3 ubiquitin ligase murine double minute-2 (MDM2). MDM2 specifically controls levels of p53, a tumor-suppressing gene, by targeting it for proteasomal degradation. Once DNA damage is perceived, Mdm2 exons are skipped to reduce the functioning of MDM2, thus allowing p53 to accumulate. This induced regulation of MDM2 provides an example of how splicing that is coupled with transcription as the exon skipping mimics the damaged DNA. In this case, cells show a "tight control of alternative splicing that helps regulate protein expression due to changing conditions in the cell."[3]
Altering the interactions of proteins is another method in which alternative splicing can be achieved. One demonstration of this is T cell activation. In T cell activation, alternative splicing is used similarly in DNA damage where the altered protein interaction with other proteins regulate the splicing of, specifically, the CD45 gene during T cell activation. In resting T cells, PSF, a RNA binding protein, is phosphorylated by the enzyme GSK3 and this causes the phosphorylated PSF to form a complex with TRAP150. As a result, the PSF cannot bind to the CD45 RNA. This prevents any possible exon exclusion and results in no participation in splicing. However, in an activated T cell, there is little to none GSK3 due to an inhibiting phosphorylation because an antigen binds to the T cell receptor and causes GSK3 activity to drop. Without the GSK3, PSF is not bound to the TRAP150 and is free to bind to the RNA. This is a major example of how splicing is controlled by signal-induced changes in transcription.
Altering the level of expression of a regulatory protein is the most simple way that can affect alternative splicing. A small change in the expression of one splicing factor can change the elements that determine exon exclusion or inclusion, due to the complex influences on a given transcript. The control of transcriptional activators such as nuclear factor-kappa B and nuclear factor of T-cells have been proven to be altered by signaling pathways. Therefore, signaling induce transcription of genes encoding SR proteins or other splicing regulators that can change the splicing of genes that respond to these factors. In one instance, it is proposed that stimulation of T cells trigger the splicing signal of the gene that encodes tyrosine phosphatase CD45. Furthermore, the proteins PTB-associated splicing factor and hnRNP L-like activate the elimination of CD45 exons 4 and 6. Interestingly enough, inducible changes in protein expression do not only result from transcription. As shown in the splicing regulatory protein CELF1, its increased protein levels is due to an increase in the stability and phosphorylation of CELF1, which then leads to the overall up-regulated steady-state levels. This increase in phosphorylation is also responsible for the protein kinase C activity in DM cells. Not surprisingly, the increase in protein stability also has other regulations; it is also controlled by miRNAs during heart development. The two coupled- mechanisms highlight the idea that regulating regulatory protein expression is important to keeping a proper splicing pattern required for functions in cells. [3]
In addition to the method of protein expression and stability mentioned above, alternative splicing can occur when signals are changed due to the localization of regulatory proteins. Many of the regulatory proteins, such as SR proteins and hnRNP mentioned above, have to travel to and from the nucleus and cytoplasm. As a result, the relative distribution of these regulatory proteins in the nucleus versus the cytoplasm can alter signaling pathways. These altered pathways will lead to splicing differences. Two regulatory proteins that have their distributions regulated include SRPK1 and hnRNP proteins (hnRNP A1 specifically). In the case of SRPK1, this regulatory protein is normally found in the cytoplasm due to interactions with heat shock proteins. However, when the cell undergoes osmotic shock the SRPK1 proteins move to the nucleus and cause phosphorylation of SR proteins. This phosphorylation results in different interactions between the proteins and their target genes and produces varying splicing patterns. In the case of hnRNP, osmotic shock actually has an opposite effect on the localization of this protein in relation to SRPK1. hnRNP is also normally found in the cytoplasm but as opposed to SRPK1, osmotic shock does not cause it to move to the nucleus. In fact, phosphorylation of hnRNP prevents it from entering the nucleus.
An example of a feedback loop in alternative splicing.
As all living things go through homeostasis, cells do the same. In order for cells to practice homeostasis, they must therefore turn off induced splicing signals once conditions are normal again. For example, these regulations can include getting rid of antigen, DNA repair and neurons repolarization. One way to reset gene expression is to deactivate signals by removing the initial receptors or signaling factors themselves. Of course, receptors such as phosphatases and kinases undergo autoinhibitory signal-induced alternative splicing. For instance, in response to T cell activation, alternative splicing of CD45 will reduce the sensitivity of the cell to receive antigen stimulation signals. In another example, molecules that encode kinases responsible for T-cell signaling activation such as the FYN proto-oncogene, signal-regulated kinase-1, and tyrosine kinase 2 beta protein, all go through alternative splicing due to T cell activation to lessen expression or change localization patterns.
Inducing expression of an opposing regulatory factor can help in resetting the induced splicing signals. Neuron chronic depolarization is an example of this, which results in increased skipping of exons controlled by CaRREs. Some of these CaRRE-reduced exons appear again in prolonged depolarization. This splicing pattern is related to CaMK-induced alternative splicing of FOX1 that encodes RNA-binding proteins. FOX1 regulates the splicing patterns of genes involved with synaptic activity. In addition, many genes controlled by CaRREs also have a FOX1 binding site which can have an antagonistic effect on exon inclusion like that of the CaRRE sequence. Since most studies only regulate a few genes, many further studies are needed to have a fuller grasp of alternative splicing that occurs in the downstream of a given pathway. [3]
Despite the stimulated factors mentioned above, the overall picture of how signaling pathways regulate alternative splicing is far from being complete. The study of these signaling pathways is still very much in progress. The methods introduced here usually correspond to the alternative splicing of only a few genes. As a result, more progress needs to be made in order to understand the alternative splicing of an entire pathway.
1. Black, Douglas L. (2003). "Mechanisms of alternative pre-messenger RNA splicing". Annual Reviews of Biochemistry 72 (1): 291–336.
2. Clark, David (2005). Molecular biology. Amsterdam: Elsevier Academic Press.
3. Heyd, Florian, and Kristen W. Lynch. DEGRADE, MOVE, REGROUP: signaling control of splicing proteins Philadelphia: Trends in Biochemical Sciences, 2011. Print.
4. Matlin, AJ; Clark F, Smith, CWJ (May 2005). "Understanding alternative splicing: towards a cellular code". Nature Reviews 6 (5): 386–398.
5. Nilsen, T.W. and Graveley, B.R. (2010) Expansion of the Eukaryotic Proteome by Alternative Splicing. Nature 463, 457-463.
6. Pan, Q; Shai O, Lee LJ, Frey BJ, Blencowe BJ (Dec 2008). "Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing". Nature Genetics 40 (12): 1413–1415.
To understand structure-function relationships, it is crucial to study the individual amino acid residues and each of their molecular interactions in protein structures. Experiments and work have been conducted, observing that residue networks created by a 3D protein structure provides more insight into structural and functional roles of interacting residues. There are software tools called the RINerator and RINalyzer to see the 2D visualization.
Protein structure visualization and residue networks
Viewing a 3D protein structure has been accessible by using X-ray crystallography and NMR spectroscopy. Although 2D visualization is very important in terms of observing structures of proteins, 2D representations of RINs have started to become popular.
RINs simplify the visual complexity of 3D protein structures and allows the scientist to focus on individual residues and their interactions within the molecular level. RIN is derived from 3D coordinates of a protein model. Each RIN is composed of nodes, representing amino acid residues. RINs can study residue interactions in many application scenarios, like, with regard to protein dynamics.
Recently, RINs have been applied to study protein-ligand interactions and to observe the structural and functional effects of residue changes under drug use or disease.
The RINalyzer (http://www.rinalyzer.de) is a software tool that provides versatile structural analysis tools for RINs and one can observe the structure in either 2D or 3D. Residue nodes of interest are automatically highlighted in the RINalyzer.
Cytoscape plugin structureViz (http://www.cgl.ucsf.edu/cytoscape/structureViz/) analyzes and supports the structural analysis of protein-to-protein interactions.
One software feature is the ability to perform analysis of residue interactions by comparing the residues with one another by loooking at the similarities and differences between two proteins. One can also observe the binding site similarities.
The RINerator module generates RINs from a 3D protein structure. This provides a more realistic visual by sampling contacts on the Van der Waals surface on each atom. By doing this, different residue interaction types can be observed and the strength of the interactions can be determined as well.
Protein binding sites are the region where proteins interact with each other. This region usually contains the specific part of the three-dimensional of the protein. If we can identify their biding sites, we can proceed to study their function and the protein-protein docking by docking algorithms.
Protein Data Bank (or PDB) functions as storage of protein complex structures. Biochemists always try to obtain the structure of specific proteins, but under experiment condition, protein structures are really hard to be obtained under the condition when it needs for crystallization. Because of the disadvantages of constructing the experiment, biochemist leads to the development of protein-protein docking.
Binding-site prediction and protein-protein docking
Protein-protein docking is a computational approaches to predict the three-dimensional structure of complex proteins. The success of this technique depends mostly on pre-knowledge of the protein-protein binding sites. In order to predict the structure, the computational approach must focus difference in binding sites between the interfaces of a set of proteins. Most of the time, there are some proteins interfacing at the same regions which then become a hotspots, whereas others might change.
With the requirement in the precision of the binding sites, biochemists developed the algorithm- which is used for predicting the protein binding sites by preserving the protein surface structure and the properties of the fundamental protein structures. We have to insert this algorithm to ProBiS which is a host to detect protein binding sites. The idea behind the algorithm is that most of the conversed parts of protein surface are somehow in accompanying with other proteins or ligands. In order to obtain the conserved part of protein surface, we have to find out the similar local surface between the concerned protein and other proteins.
To conduct the example of this method, we choose the two unbound interacting proteins: FKBP12 (immunophilin) and TBR-1 (a growth factor) with PDB codes of 1d6o and 1ias. Some of the proteins that seem to share the same similarities in structure with FKBP12 are: 1ix5, 1jvw, 1pbk, 1q6h, 1r9h, 1u79, 2awg, 2d9f, 2if4, 2ofn, 2pbc, 2uz5, and 3b7x; with TBR-1 are : 1ckj, 1kob, 1m17, 1o6k, 1o9u, 1u59, 1wak, 1yhv, 1yvj, 2b7a, 2bfy, 2csn, 2f4j, 2ivt, 2izs, 2j0l, 2jbo, 2pzy, 2qkw, 2qlu, 2qr7, 2v7o, 3bkb.
ProBis is now used to predict the binding sites. The fundamental protein has to interact with the polypeptide chain. Our goal is to find out the similar surfaces of these proteins, so we want to minimize the dissimilarities as much as we can. As you can see on the picture on the right, all the conversed regions are mapped over the other ones.
AutoDock 4.0 is then used for docking of protein FKBP12 to the protein TBR-1. This program requires computational interference since it workswith the whole protein structures, so it needs a precise image. The AutoDock uses a force field to give a stronger attraction between the atoms on predicted binding site. The success of docking depends on the comparison between the regions of predicted binding site residues with the corresponding ones. This force field affects the docking. As you can see in this chart, five time larger force field has the highest number of best docked structure.
This 5x force field has 9 different structures between the predicted and the actual binding site residues. The most preferable clustering also belongs to this one since it has the most best docked structure. This theory somehow states that the docking algorithm can be able to explain the structure of the complex protein.
Three scientist in the field of structural biochemistry from the University of California San Diego(Ruben E. Valas, Song Yang, Philip E. Bourne), have proposed a new method of protein classification. This idea comes as a consequence of the great breadth of macromolecular structures having been solved and the many, yet, to not have been illuminated. This poses a grave problem of assimilation of the large amounts of structural information available. Secondly, it seems that the present manner of classification seems insufficient to unveil the great network of structural lineages that evolution has paved and therefore, their strategy is to employ a reductionist approach to better interpret the evolutionary basis of protein structure and the lineage amongst the diverse populations of such structures.
Two methods of protein classification are readily used today:
The bottom-up approach uses algorithms to in an attempt to compare proteins based on geometry, the ability to superimpose using a root means-square deviation(RMSD), length of alignment, number of gaps, and a score of statistical significance. The end result is a proteins domain comparison which renders very little biological significance.
Because of the diversity of methods available, there is usually more than one result for each sequence of amino acids analyzed. One drawback to the bottom-up approach is that, since sequences of amino acids in their primary state do not reveal much about the biological function of the protein, it is impossible to decide which one of the results is the most biologically important one. The benefit to the bottom-up approach is that it is a useful bit of reductionism that does give a representative comparison of different protein domains, which can prove useful.
Top-down approaches are considered today's gold standards as exemplified by CATH and SCOP. These methods primarily utilize homogous sequence comparisons to reflect a relationship among different protein domains and as a result a biological context. The authors agree that this technique can be taken one step further based on the premise that structural classification is developed as a consequence the evolutionary links among species. Furthermore, the authors propose to incorporate issues of gene duplication, convergence versus divergence, and co-evolution in a functional context as ideas that should be used in the future for protein classification.
The protein domain: a good unit of structural classification?
Both the bottom-up and top-down approaches rely on protein domains as the units of comparison. Domains are complicated units. Some domains have similar sequences and are evolutionarily related, some domains are vaguely related, with similar structures but different sequences, and some domains are similar topologies, but not enough to establish an evolutionary connection. The basic problem is that a domain can be an evolutionary or non-evolutionary unit. Many proteins are multi-domain proteins, which further increases the complexity.
The presence of folds, which are considered discreet components in most top-down classifications, further complicate matters. Folds are not a direct result of evolution, but they do provide insight into evolutionary practices. Folds sometimes change during evolution; it is possible for an alpha fold to change into a beta fold through a secondary structural change. It is also possible to create two peptides with similar sequences but different folds, leading to completely different functions. There are also chameleon sequences that can take on multiple different folds. Because of the diversity of structural variation in regard to folds, folds are not suitable units of classification. In essence, whether or not two proteins are in the same fold is really semantics, whereas determining which one led to the other evolutionarily actually gives insight into the relationship between proteins. The reason it has not been widely used is simply that it is more difficult than clustering similar structures.
Valas et al. present the prevalence of evolutionary selection by give two examples that highlight this phenomenon. The first, Basu et al. found in the genomic analysis of 28 different eukaryotic cells, that there were 215 strongly promiscuous domains. Basu et al. define strongly promiscuous as those domains that occur in diverse domain architectures, where these architectures are represented as a linear combination of these domains. "Domain architectures arise through domain shuffling, domain duplication, and domain insertion and deletion leading to new functions." The degree of dmain promiscuity depends on the frequency of being with different domain partners. The second example is by Vogel et al. which found over-representation of 2-domain or 3-domain combinations which were coined, "supradomains" or macrodomains. These are structure that throughout proteins evolution have proven to have stable internal domains. Over 1400 of these macrodomains have been found which show a natural associativity which seems to be evolutionarily advantageous.
The protein domain has been the only manner of evaluating the of evolution protein structure. Although the evolutionary analysis of the protein domain alone has proven successful at evaluating protein structure, it seems that there needs to be other factors contributing the unknown pieces of the evolutionary network. Therefore, the authors propose using a pluralistic approach to protein structure classification which includes incorporating not just domains, but subdomains, macrodomains, and both convergent and divergent evolution. In regards to subdomains, the authors mention areas of subdomains that could be important components to connecting the evolutionary network of proteins.
There are many tools that can be used to compare proteins at the subdomain level. One database called Fragnostic facilitates analysis based on fragments from different proteins that share structural and/or sequence similarity. The edges of the fragments are ambiguous; that is, they are not defined as divergent or convergent evolution, but combined with other information the fragments can be tested for structural evolution.
Closed loops are another subdomain unit. Most protein structure consist of loops spanning 25-30 residues. Domain Hierarchy and closed Loops (DHcL) uses van der Waals energies to elucidate domains and closed loops from protein structures. Researches have discovered that fragments that correlate to closed loops were more likely to form large clusters, which have connections to one another. This description might represent a more detailed view of protein function. Similar closed loops in different structures can be evidence that those structures once shared a common ancestor.
Another subdomain unit is the functional site. Many different proteins can bind to the same ligand, which implies that perhaps they share a common ancestor that bound to the ligand in question. The proteins diverged in structure during evolution, but the functional site remained. SMAP can find such functional site that have both sequence and structural conservation, a perfect example of divergent evolution. On the other hand, different proteins can converge on the same ligand. The PROCOGNATE database uses information from the PDB to put together which proteins bind to which ligand. A combination of these methods could incorporate both divergent and convergent evolution.
Besides subdomains, macrodomains can also be used to aid in classification. Divergent evolution is evident in some protein–protein interaction sites (a macrodomain feature). In those cases, while the proteins differentiate over time, the domain interface stays the same. Many of the protein–protein interfaces in the PDB contain very similar interfaces in vastly different proteins.
In essence, a domain-based scheme would not be as efficient, as it would only be able to determine that the proteins evolved from a common ancestor, while an examination that includes analysis of both subdomains and macrodomains would provide an evolutionary hypothesis. One problem posing the pluralistic approach to protein classification would be convergent evolution. The fact that two proteins with completely different evolutionary lineages can come together to have very similar structures can pose a great problem for connecting the protein evolutionary network.
The authors argue that to obtain the last universal common ancestor(LUCA) of the protein, it is necessary to look at more than the amino acid sequence as has been done but incorporate other structural aspect to be able to mesh the evolutionary puzzle.
Protein studies involves various step of sample preparation. The main protocols of protein studies are as followed:
Protein Synthesis
Purification
Evaluation of purified protein
Determination of Amino Acid Sequence
Calculation of Protein's Mass
Determination of Protein's 3-D Structure
There are many methods used to study proteins, including its shape and structure. For instance, X-Ray crystallography is used to give scientists the structure of the protein. Such information is used extensively in determining the characteristics of the protein as well as how it functions and under which circumstances. Other methods used includes amino acid sequencing, fluorescence microscopy, mass spectrometry, NMR, etc.
Carbohydrate-Binding proteins (CBPs) are identified as important mediators for numerous different types of cellular events through interactions between carbohydrates and proteins. There are three main families of CBPs.
the C-type lectin family (including the Selectins)
Pure forms of lectins are used for blood typing. Specifically, lectins are used to identify some glycolipids and glycoproteins on an individual's red blood cells.
In the brain, PHA-L, a lectin from a kidney bean, helps to trace the path of efferent axons through the anterograde labeling method.
Galectins are also found in humans, mice, and rats as well. These CBPs are abundant in most organs such as muscles, hearts, lung, liver, lymph nodes, thymus, and colon, stomach epithelial cells, gastrointestimal, erythrocytes, skin, brain, Hodgkin's lymphoma, kidney, and lens. Roles include:
Two method of counting protein molecules have been used widely: stepwise photobleaching and ratio comparison to fluorescent standards.
Fluorescence takes place when light is given off from the fluorophore after light is absorbed, and GFP is able to fluorescence without enzymatic modification or a cofactor, which allows a single gene to be expressed in detectable emission in any organism. Counting the number of protein molecules in live cells allows researchers to determine the stoichiometry of functional protein complexes and to seek models of cellular structures. Since genome-wide studies may not recognize information about low-abundance proteins or local protein concentrations, single-molecule techniques, if successful, would be able to solve this problem.
Stepwise photobleaching is one of the fluorescence microscopy method for counting protein molecules, which “relies on the irreversible and stochastic loss of fluorescence from repeated exposure of fluorescent proteins (FPs) to a light source.” The sample would be continuously exposed to excitation light at low intensity to allow the sample to be “slowly bleached until its emission intensity reaches background level.” The number of florescent molecules present in the structure determines the suitable light intensity and exposure time. The missed bleaching events need to be minimized because it would show a step approximately twice the size of other steps. The bleaching method is only useful for low protein numbers as the probability of missed events increases exponentially with the number of molecules in a structure. “Das et al. estimated that a maximum of 15 bleaching steps can be directly detected without mathematical extrapolation, although they detected no more than seven steps in their experiments.” The maximum number of molecules that can be counted by photobleaching can be increased to approximately 30 molecules using mathematical aids. A background correction is needed to eliminate fluorescence from diffused proteins and calibrate the starting intensity. During photobleaching, regions of interest (ROIs) should be selected to avoid confusing multiple structures. It is also essential to filter the data to reveal the discrete drops as the raw data are noisy. For example, Chung-Kennedy filter is the most commonly used filter for quantification of the bacterial replisome. “It calculates the mean and standard deviation in two consecutive sets within the data from one photobleaching ROI, and reports the mean of the set with the lower standard deviation.” The number of averaged data points in the data set should be big enough to reduce the noise but small enough to make sure that few steps are missed.
Quantification by ratio comparison to fluorescent standards
This method involves the measurement of ratio of the fluorescence intensities of a protein sample to a standard. It uses a series of images of cells that express either the protein sample or the standard, with had obtained fluorescent properties by fusion with an FP. “If the standard can be distinguished from the protein of interest, it is desirable to include cells that express the standard and experimental fusion proteins on the same slide to ensure comparable illumination. If the standard is not distinguishable, images can be taken consecutively or another marker can be imaged separately to distinguish the control cells distant to eliminate Forster resonance energy transfer.” This method is advantageous in the way that a relatively larger number of protein molecules can be counted. Corrections need to be made to achieve more accurate measurements. For example, the uneven illumination in the microscope system needs to be corrected if the whole field is used. Also, if sample molecules are at different depths relative to the coverslip, calibrations on the effect of depth on intensity should be done using fluorescent beads. Different exposure times can be used to control the signal to noise ration and avoid saturation. Excitation intensity should be kept constant to avoid nonlinear changes to photon counts due to blinking molecules. “The background should be taken from a concentric area unles there are overlapping neighbouring signals or an inhomogenous cytoplasmic intensity.” It is important to use a trustworthy standard for this method. When proteins of different sizes or same structure proteins with very different intensities are compared, the sum of intensity of multiple z sections should be used. Additional verification using methods such as genomic DNA sequencing should be used to ensure accuracy of number measured. The number of molecules of each protein and their relative stoichiometries can be obtained using the ratio method at one or many time points.
Genetically encoded FPs should be used in order to generate a 1:1 stoichiometry with the protein sample, which may affect the maturation efficiency or proportion of unfolded FPs.
The best available FPs should be used by researchers to maximize the signal-to-noise ratio, especially for less abundant proteins. The folding and maturation efficiency, brightness, and photostability of the FPs that are going to be used in the fusions should be taken into consideration before constructing fusion proteins. Research conduced in the budding yeast Saccharomyces cerevisiae and the folded YFP in E. coli suggest that YFP maturation and folding efficiency are not major issues for counting proteins, in particular for proteins with low turnover rates.
It is advantages to use yeast, because fluorescent fusion protein can replace native protein using homologous recombination, which allows the functionality of the fusion protein to be determined. The functionalities for some proteins could be improved by using a flexible linker between the FP and protein sample. The fact that endogenous genes cannot be replaced with tagged version, alternative methods of protein counting need to be used. The local actin abundance in actin patches can be quantified by making corrections after immunoblotting. However, this method is only possible given the assumption that tagged and untagged actins are utilized with similar efficiency in actin patches. Engel et al uses stepwise photobleaching method in a mutant background to count exogenous tagged proteins in exogenous tagged proteins in green algae Chlamydomonas reinhardtii flagella. Since the endogenous genes do not localize, the ratio of tagged and untagged protein assumptions do not need to hold. The recent development of ‘genome editing’ techniques has allowed endogenous genes to be tagged in any model organism in which “the zinc finger nuclease or transcription activator-like effector nuclease genes can be introduced.”
The environment in which the number of proteins is measured is important. Early studies employed in vitro standards, where the effect of background on fluorescence intensity is unknown. This meant that immunoblotting or internal standards were needed to calibrate the fluorescence intensity inside the cell. Experiments were done recently to suggest that in vitro YFP/GFP is comparable to YFP/GFP in bacteria or yeast. Also, fluorescence quenching could take place if FPs were packed into very tight structures. The effects of quenching on counting proteins should be examined individually depending on the specific structures of interest. Fluorescence lifetime imaging with the aid of specialized equipment and analysis can be used to measure quenching due to environmental changes.
Validation of protein quantification by complementary approaches
Cellular concentrations should be authorized by a cell sorting device called the flow cytometry or fluorescence correlation spectroscopy fro a higher resolution. It is also important to ensure that protein concentrations from fluorescence microscopy are consistent with quantitative immunoblotting. In any protein counting experiment, suitable fluorescent protein genes, suitable standards and controls for environmental changes or the possibility of quenching will ensure appropriate interpretations of the data, which can then be confirmed with complementary experiments.
Future of counting proteins using fluorescence microscopy
Super-resolution microscopy techniques can produce high-resolution images of intracellular structures, which pinpoint exact locations of individual fluorescent molecules. For such techniques, it is most important to simplify the analysis of high-density images of FPs and minimize errors due to blinking or photobleach failure. Single-molecule techniques are now more commonly used due to the inability to observe stochastic events in average population behaviours. The advantage of using such techniques is that molecules can be counted directly without using collective images, or even determine different protein complexes that are within a diffraction limited area. Super-resolution imagining could lead to the quantification of higher numbers of proteins.
Counting proteins molecules in a cell is essential in determining structural models and protein function. In vitro, protein numbers help determine the reaction rate and also give more of an understanding to multiproteins. The two methods introduced are stepwise photobleaching and ratio comparison to a given standard. This maybe used in any laboratory with a fluorescence microscope to isolate a particular protein. Of course, there are many advantages and disadvantages in every method including this one. The properties of FPs is of high significance to both methods. There are other methods that will help validate the quantity of proteins such as electron microscopy. Fluorescence microscopy has help determine exact numbers of proteins and also their binding ranges.
Source: Coffman VC, Wu JQ. Trends Biochem Sci. 2012 Sep 1.
Is the term referring to "protein homeostasis" where a system of biological pathways leads to proper protein function. The system is called a proteostasis network, which will be responsible for successful protein transport, proper folding of proteins, and elimination of misfolded proteins. The factors responsible for in improper protein function are genetic diseases and environmental stress. More knowledge of the proteostasis network is still in need for development but researchers have studied some of the pathways to create pharmaceutical agents and provide therapy for such protein abnormalities. The pharmaceutical agents used to modify the network pathways are called protein regulators which affect a pathway in a specific manner. For example, the antibiotic geldanamycin is known to act as an inhibitor for the chaperone protein HSP90. The HSP90 chaperone is involved in network pathways for protein folding, the success of HSP90 in assisting protein folding results in cell proliferation. Cancer cells are more sensitive to HSP90 inhibitors, consequently, by using geldanamycin as a protein regulator to inhibit HSP90 function will lead to cancer cell death. More research on the effects of HSP90 inhibitors is still done to propose a therapeutic treatment for cancer. Although the number of pathways involved in protein regulation is great, detailed study of these pathways will result in a successful treatment to ensure proteostasis.
Some diseases that can be caused by protein homeostasis are Parkinson’s, Alzheimer’s and cystic fibrosis. These diseases can occur as the results of the proteostasis network’s decreased ability to cope with misfolding prone proteins, aging, or environmental stress.
The protein homeostasis network and its networks are also controlled by integrated signaling pathways. These signaling pathways have the ability to maximize the capacity of the network in order to ensure consistent and correct protein function. Some examples of signal pathways include those that regulate protein synthesis, aggregation, as well as the degradative pathways of proteostasis.
For the proteostasis network to function correctly and in a stable condition, there are many interactions that help monitor and facilitate the process of successful protein folding.
1. The proteostasis network is made up of ribosomes, chaperons, aggregases, and disaggregases that control protein folding. There are also special pathways like the ubiquitin-proteasome system, endoplasmic reticulum-associated degradation systems, proteases, autophagic pathways, etc. that deal with the degradation of proteins.
2. There are the signaling pathways like mitochondria, aging, heat shock response, and unfolding protein response that affect the process of protein folding within the proteostasis network. This is perhaps the most direct influence that can alter the folding and stability of the proteins.
3. Outside influences include metabolities, physiological stress, genetics, and epigenetics that affect the overall activity of the proteostasis network. These influences can also alter the process of protein folding but some, like metabolites and physiological stress, can be prevented by the use of pharmacological chaperones and proteostasis regulators.
Within the cell the surroundings are compacted with many compartments and the lack of space triggers aggregation. Aggregation is related to the levels of toxicity and has to be balanced most importantly when the cell deals with stresses that are chemical, physical and metabolically related.
The overall energy of a protein is impacted by the folding aspect of the proteostasis network. The energy level of a protein achieves a good distribution by utilizing folding enzymes and chaperones to decrease the aggregation and improve folding. Chaperones and enzymes that help fold attach to the intermediate molecules and transition state.
The state and functionality of the proteostasis network directly influences the protein’s functional performance and proteins usually acquire intracellular help for protein folding.
Pharmacologic Chaperones and Proteostasis Regulators
The proteostasis, as the “protein homeostasis”, must maintain a stable level of activity in order to function correctly within a cell. The proteostasis boundary refers to the folding energies that the protein must have in order to achieve some level of functionality in a given proteostasis network. This proteostasis boundary can be regulated by both pharmacologic chaperones and proteostasis regulators. By regulation, the proteostasis boundary can be expanded to envelop destabilized protein (known as the node) by proteostasis regulators or pharmacologic chaperones can move the node from outside of the proteostasis boundary to the inside in order to stabilize the node. If the proteostasis boundary is not regulated, there will be loss-of-function misfolding diseases, which could create potential life-threatening diseases.
The pharmacologic chaperones (otherwise known as the PCs) perform its regulation by binding to the outside destabilized node in order to stabilize it. After binding to the node, the PCs can move the now stabilized protein inside the proteostasis boundary, which then increases the function within the proteostasis, maintaining a stable level of activity. This stability then translates to less misfolding diseases. The PCs can correct a misfolding disease in three ways:
1. The destabilized node can be thermodynamically stabilized
2. The folding rate of the node can be increased in order to stabilize the transition state of folding
3. Decrease the misfolding rate by stabilizing the native state
On the other hand, the use of proteostasis regulators (known as PRs) allow for an expansion of the proteostasis boundary for a number of destabilized nodes (as long as the nodes all share the same proteostasis network). By expanding the proteostasis boundary, the PRs can favor folding of the proteins by adjusting composition, concentration, and capacity of the proteostasis network. Besides promoting a stable proteostasis for proteins to fold correctly, PRs can also prepare the proteostasis network to handle metabolic stress and aging. The expansion of the proteostasis boundary helps increase the protective capacity of the proteostasis, hence expanding helps prepare for future abuse.
The overall energy of a protein is impacted by the folding aspect of the proteostasis network. The energy level of a protein achieves a good distribution by utilizing folding enzymes and chaperones to decrease the aggregation and improve folding. Chaperones and enzymes that help fold attach to the intermediate molecules and transition state. Binding to the transition state helps stabilize the protein so that there is a decrease in wrong folding and aggregation.
Chaperones help encourage more folding and also plays a role of preservation in the cell due to increasing correct folding and decreasing aggregation and wrong folding. Chaperones are understood as a large molecule that attaches to exterior hydrophobic areas during aggregated mode. Chaperones are specific and different for different compartments.
In all, the use of pharmacologic chaperones and proteostasis regulators both aid the proteostasis network in preventing numerous loss-of-function misfolding diseases. However, the advantages of using either lies in whether it is to bring in one destabilized protein (via pharmacologic chaperones) or to bring in a collection of similar destabilized proteins by expanding the proteostasis boundary (via proteostasis regulators).
FoldEX and FoldFX are both models representing the proteostasis boundaries. FoldEX is a model that shows when a protein would get exported from the endoplastic reticulum, whereas the FoldFX model shows when proteins would have its function, hence where proteostasis working. FoldFX stands for Folding for the Function of Protein X. The models have three dimensions and they include the folding rate, the misfolding rate, and the stability of the protein.
The FoldEX model is important because it establishes a threshold for protein export. This boundary is characterized by the protein’s correct and wrong folding rate and its stability. Proteins will be exported if their energy level matches the energy level of the threshold.
In a healthy cell, all the proteins would be situated usually well within the boundaries of the FoldFX model and all the enzymes would be working. However, when there is a disease that affects protein folding or if proteostasis is not quite working well, there could be proteins represented that fall outside the boundaries, which would mean that the proteins are not functioning properly.
In conservative mutation the substitution that occurs does not have a heavy impact on the kinetics or thermodynamics of folding. It does not really affect the functional aspects that much because the replacement of a similar amino acid is not too different from the amino acid that was changed. In a slightly conservative missense mutation and elimination of an amino acid does affect the thermodynamics and kinetics of protein folding because the change of a base in the genetic sequence does not alter the functional aspect.
However, there are ways to correct this. One way is with the application of PC’s, or pharmacologic chaperones. Pharmacologic chaperones specifically target proteins that fall outside of the proteostasis boundary and push it within the boundaries giving it the ability to fold properly and function. It does so by either increasing the folding rate, decreasing the misfolding rate or stabilizing the structure of the protein. Another way to correct this is by way of PR’s, or proteostasis regulators. Proteostasis regulators can either expand or retract the proteostasis barrier allowing more or less proteins to be correctly folded.
Powers, T Evan. Morimoto, Richard. Dillin, Andrew. Kelly, W Jeffrey. Balch E William. Biological and Chemical Approaches to Diseases of Proteostasis Deficiency. 2009. Annual Review of Biochemistry
The most popular method to synthesize peptides of more than 50 amino acids in length is automated solid-phase peptide synthesis. R. Bruce Merrifield first developed this method, and it can be used for both DNA and RNA. To begin the process, the carboxyl-terminal amino acid of the desired sequence is anchored to polystyrene beads, and the peptide is synthesized backwards from the C-terminal end to the N-terminal end (contrary to the usual sequence from the N-terminal end to the C-terminal end). The t-Boc protected group of this amino acid is then removed by a wash with trifluoroacetic acid (CF3COOH) and methylene chloride (CH2Cl2), which does not break covalent bonds. The next amino acid with t-boc (di-tri-butyl dicarbonate), a protected N-terminal, and a DCC (dicyclohexylcarbodiimide)-activated C-terminal is added to the reaction column. After the formation of the peptide bond, the excess reagents and dicyclohexylurea are washed away with an appropriate solvent. For the elongation of the peptides, the next amino acids continue to be added in the same manner. At the end of the synthesis, the peptide is released from the polystyrene beads by adding hydrofluoric acid (HF), which cleaves the ester bond without destroying the peptide bonds. Protected groups on the reactive side chains, such as lysine or histamine, also are removed at this time. The huge advantage of this method, besides the fact it is automated, lies in the purification step. Because the impurities are not bound to the reaction column, they can be washed away without losing the synthesized product. In the laboratories, this technique is used to synthesize drugs, such as insulin.
It starts in the nucleus. It is very similar to the DNA replication process in which the DNA is "unzipped" by helicase, producing one nucleotide chain ready to be replicated.
Transcription 3 Steps summary –> Producing an RNA message from DNA
(A) Binding and Initiation
DNA transcription unit divided into TATA Box and Enhencer region. TBP is bind to TATA region, other transcription factors (a protein has bound to the region) such as TFIIA and TFIIB are bonded to TATA regions as well. The RNA polymerase cannot bind to the DNA directly unless a transcription factor is bind first. Transcription begins when RNA polymerases bind to the enhancer region( or called the initiation site), separate it into two strands by requiring ATP energy Initiation initiate the location of the DNA strand to begin transcription.
(B) Elongation
RNA polymerase moves along the DNA promoter region by performs two elongate steps:
1) it untwists (unwind) the double helix DNA about 10 bases at a time at 3.4 A.
2) adds nucleotides to the 3’ end of the growing RNA.
As the RNA polymerase moves along, the growing mRNA molecule was replicated base on base. Transcription goes about 60 nucleotides per second. DNA’s nucleotides Adenine will be complimentary to RNA’s Uracil base. DNA’s nucleotides Guanine will pair with Cytosine.
(C) Termination
Transcription proceeds until the RNA polymerase reaches a termination site. No more RNA nucleotides will be added and the mRNA is released. So, mRNA will move out of the nucleus into the cytoplasm for the further use in protein synthesis.
The mRNA codons translates to amino acid polypeptide chains in three steps.
3 steps general guidance of translation
Initiation
2. Small subunits ribosomal attaches to mRNA. Large Subunit of ribosome is bind to small subunit with A site (entry for tRNA.)and P site ( leaving door for tRNA.) first attach to a tRNA. anticodon( nucleotide triplet in tRNA) is attaching to A site (entry site) to paired with 3 nucleotide codons from mRNA. tRNA carries an amino acid. As shown by the graph below, tRNA. carry an amino acid on the top
Elongation
3. Initiator tRNA. then moved to P site and A site is opened for the second triplet coded tRNA. to enter along with another amino acid. After the second tRNA. is bind to A site. The amino acid is then bonded together by peptide bonds. Afterwards the third tRNA comes in right after the second tRNA. move to P site. (Moving along from 3’’ to 5’’)
4. ribosomal enzymes link the amino acid into a chain. The process will continue until the stop codon (UAA) is reached.
Termination
5. a stop codon is reached (UAA, UAG, or UGA). A protein called a release factor binds in the A-site to the termination codon. The ribosomes adds a water molecule to the end of the polypeptide chain.
6. ribosome dissociates into its component parts
Good yield and high purity. All reactions are carried out in the single vessel, eliminating losses caused by the repeated transfer of products. This method is good for synthesizing long chain of peptide (50 residues and above).
Peptides can be made synthetically by linking an amino group of one amino acid to the carboxyl group of another; this being an example of a condensation reaction. A condensation reaction is the reaction when two molecules come together, releasing water, to form one molecule.
Peptide synthesis can be specific; meaning specific/desired products can be formed. To make unique products and to prevent side reactions, protecting groups such as tert-butyloxycarbonyl (t-Boc) are used. T-Boc is used in the first step of the formation of simple peptides. This protecting group, in order to block the alpha-amino group, reacts with the alpha-amino group forming a complex [[Image:known as t-butyloxycarbonyl amino acid. The blocking of the amino group is followed by the activation of the carboxyl group of the same amino acid. The carboxyl group is activated by dicyclohexylcarbodiimide (DCC).
Now, with the alterations being done to the amino group and the carboxyl group of the first amino acid, a second amino acid can be linked to the first amino acid. The second amino acid has a free amino group, meaning not blocked, and it links to the activated carboxyl group of the first; forming a rigid peptide bond and releasing dicyclohexylurea. The carboxyl group of the newly formed dipeptide is activated with DCC and ready to react with a third amino acid which has a free amino group. Again, a new peptide bond is formed and dicyclohexylurea is released. This process can be performed continuously until the desired peptide is synthesized. To end the synthesis, dilute acid, which removes the t-Boc and leaves the peptide undisturbed, is added.
Dicyclohexylcarbodiimide (DCC)
Solid-phase method is used to form synthetic peptides that contain more than 50 amino acids. It involves binding the last amino acid's carboxyl group to polystyrene beads. The anchored amino acids t-Boc is removed, and the next amino acid with t-Boc protected amino group and DCC activated carboxyl group is added to the amino acid with polystyrene beads. The peptide bond forms, and the peptide with polystyrene beads is filtered and washed, so the peptide is pure before the synthesis is continued. The following amino acids are linked with the same process until the desired peptide is synthesized. Finally, the finished peptide is removed from the beads by using hydrofluoric acid(HF).
Peptide ligation is used to synthesize peptides with more than 100 amino acids. The long peptide is formed from two or more smaller sized peptides with no protecting groups on them. Native thiol ligation is the most powerful and widely used peptide ligation. The long peptide is formed from peptides with thioester on C-terminal carboxyl group and the other peptides with cysteine on N-terminal. The thioester on C-terminal carboxyl group of one peptide reacts with the cysteine on N-terminal of another peptide to form a thioester-linked intermediate. The intermediate is then rearranged(S->N acyl shift) to form a peptide bond. The small sized unprotected peptides are linked by this process to synthesize the long peptide.
Synthetic peptides are made for many purposes. These peptides can act as antigens, which will stimulate the immune system of the body to produce antibodies that target such peptide. These antibodies can then be used to isolate a protein. Peptides can also isolate receptors for hormones.
Synthetic peptides can also be used as drugs. Such example is the synthetic analog of Vasopressin, also known as 1-Desamino-8-D-arginine vassopressin. This synthetic peptide is used to treat patients with diabetes insipidus who lacks the peptide hormone vasopressin, which cause them to urinate excess liquid from their body. By using the analog of vasopressin to substitute for the natural vasopressin, such patients can be treated.
Lastly, synthetic peptides can be used to gain a greater understanding of the 3D structure of proteins. Using synthetic proteins to study the 3D structure of proteins is extremely helpful because such peptides can include many amino acids that are not found in normal proteins; meaning these peptides are not limited to just the 20 standard amino acids. This result in a much greater variety of structures.
Polypeptide synthesis can be automated, known as the Merrifield solid-phase peptide synthesis, which uses a solid support of polystyrene to support a peptide chain. Polystyrene is a polymer whose subunits are derived from ethenylbenzene.
Polystyrene
The beads of polystyrene are insoluble and rigid when they are dry; however, they swell in certain organic solvents, dichloromethane for example. Therefore, reagents are able to move in and out of the polymer matrix easily. The phenyl groups on polystyrene are functionalized by electrophilic aromatic substitution.
Dicyclohexylcarbodiimide (DCC) is used specifically in peptide synthesis in order to activate the electrophilicity of the carboxylate group. This allows the C-terminus to be more favorable as an attachment site for other amino acids. Then the negatively charged oxygen will act as a nucleophile which attacks the center carbon in DCC. This intermediate will eventually be converted into urea, a stable end product that is relatively unreactive throughout the remaining peptide synthesis process. In addition, DCC's activation ability may sometimes racemize peptide bonds if not monitored correctly, therefore sometimes triazoles may be used instead which do not racemize the stereochemistry of peptides.
The advantage of solid-phase synthesis is that the products can be isolated easily since all the intermediates are immobilized on polystyrene. Thus, the products can be purified by filtration and washing. Repetition of the deprotection-coupling process will be able to synthesize larger peptides. A machine, designed by Merrifield, is able to carry out the series of manipulations automatically.
Peptide bond can be formed from the carboxyl group and amino group on the main chains of amino acids. It also can be formed from the side chains to synthesize an undesired peptide. In order to synthesize a desired peptide, protecting groups are used to prevent the formation of undesired products. They also prevent the polymerization from the excess amino acids used in the reaction. Protecting groups also aid in ensuring that the stereochemistry of certain amino acids remain unchanged. Configurations of amino acids may have their stereoisomers changed or racemized if not properly protected as well.
It is used to protect the N-terminal amino groups as well as the side chains of lysine, arginine, asparagine, and glutamine. Di-t-butyldicarbonate reacts with the NH2 of amino acid to form a t-Boc-amino acid. t-Boc group can be removed under acidic condition. Typically, they are treated with strong acid or Trifluoroacetic acid(TFA), CF3COOH. In the lab, Boc-amino acids are also available to buy since it can be synthesized easily in large quantity. People who synthesize peptides do not have to make Boc-amino acid on their own. Solid phase synthesis is effective because it allows the protein to remain in a primary structured configuration rather than being complicated by secondary or tertiary intermolecular interactions.
Boc-group, synthesized and removedMechanism of how T-boc is added to the amino acidMechanism of how T-boc is removed from the amino acid using HClTrifluoroacetic acid used to remove t-Boc group
Solution-Phase Peptide Synthesis (Using Benzyloxycarbonyl(Z) as protecting group)
Benzyloxycarbonyl is used to protect the N-terminal amino groups as well as the side chains of lysine, arginine, asparagine, and glutamine. The synthesis starts at the N-terminus and ends at C-terminus. For example, here are steps to synthesize a simple peptide such as Ala-Val:
First Step: Benzyl chloroformate react with the N-terminus of alanine, forming benzyloxycarbonyl alanine (alanine with the N-terminus protected by Z-group). Typically, triethylamine is used as catalyst for this reaction.
Second Step: The protected alanine is treated with ethyl chloroformate. Carboxyl group of the alanine was activated by forming anhydride. It is sensitive to any nucleophilic attack from the N-terminus of Valine.
Third step: Valine is added to the protected, activated alanine. This forms peptide bond, connecting Valine and Alanine. We'll have the product of Z-Ala-Valine. Notice that the N-terminus is still being protected after this step.
Final Step: The Z-protected group was removed by hydrogenolysis under mild condition with metal such as Pd acting as catalyst. (check the image for detailed reactions in each step)
Synthesis of Ala-Valine, using solution-phase synthesis
In order to synthesize a larger protein, we have the repeat the second and third step. Activating the C-terminus and then, coupling the next amino acid. The advantages of this synthesis are it works very fast, and have a good percentage yield of the product. However, it can only be used for small protein chain. The yields become smaller with larger protein. Therefore, solid-phase is more preferred with large protein.
It is used to protect the N-terminal amino groups as well as the side chains of lysine, arginine, asparagine, and glutamine. Fmoc can be removed by piperidine/DMF.
Fmoc protecting groupPiperidine. Used to remove Fmoc group
They are used to protect the C-terminal carboxyl groups as well as the side chains of serine, threonine, tyrosine, glutamate, and aspartate. t-butanol or benzenol reacts with the hydroxyl groups or carboxyl groups of amino acids to form t-butyl or benzyl amino acid. t-butyl or benzyl can be removed by strong acid and catalytic hydrogenation.
Non ribosomal peptide synthesis is an alternative pathway that allows production of polypeptides other than through the traditional translation mechanism. The peptides are created here by enzymatic complexes called synthetases and the resulting peptides are generally short, 2-50 residues.[1] Non ribosomal peptide synthesis produces several pharmacologically important compounds including antibiotics and immunosuppressors. This biosynthesis pathway is found in many bacteria and fungi. Non Ribosomal Peptide Synthesis (NRPS) utilizes a large monomer pool including all the amino acids and several unnatural amino acids along with aryl acid substrates to produce small molecule metabolites by a series of loading and condensation of peptides. Peptides produced by NRPS show peculiar features compared to traditional proteins. First, they can contain standard as well as non-standard amino acids.[1] Secondly, amino acids are linked not only by an amino-peptide, but also by non-conventional links that form a non-linear peptide backbone.Non ribosomal peptide synthesis is a key mechanism responsible for the biosynthesis of bioactive metabolites in bacteria and fungi. Non ribosomal peptide synthetase genes, generally represent a part of multigene clusters, encode NRP synthetase which in turn, biosynthesize peptide products.[1] an NPR synthetase is generally composed of one or more modules and can terminate in a thioesterase domain that releases the newly synthesized peptide from the enzyme.[1] Unlike ribosomal peptide synthesis, they do not involve the translation of mRNA in order to begin the synthesis. Because of this there is a very large degree of diversity and gives rise to an extremely varied host of possible products. NRPS is especially relevant because many secondary metabolites produced by this process are of medical importance, creating numerous antibiotics, antibiotic precursors, and immunosuppressant drugs. NRPS is similar to polypeptide synthesis and fatty acid synthesis but NRPS multienzymes do not bind covalently to acyl carrier protein intermediates, instead utilizing only a peptidyl carrier protein (PCP). The PCP has a conserved serine group on an alpha helix replaced by a 4'-phosphopantetheine prosthetic group, which allows it to convert to the holo form, and consequently allows for the thiol group at the end of the prosthetic group to attach to other peptides. NRPS begins with the loading of an activated aminoacyl–adenylate onto the PCP, and then undergoes a process of adenylation and condensation until the thiostearase domain completes the polypeptide chain and the synthesis is completed.[2]
PCP: Thiolation and Peptide Carrier Protein with attached 4'-phospho-pantetheine (required in a module)
C: Condensation forming the amide bond (required in a module)
Cy: Cylization into thiazoline or oxazolines (optional)
Ox: Oxidation of thiazolines or oxazolines to thiazoles or oxazoles (optional)
Red: Reduction of thiazolines or oxazolines to thiazolidines or oxazolidines (optional)
E: Epimerization into D-amino acids (optional)
NMT: N-methylation (optional)
TE: Termination by a thio-esterase (only found once in a NRPS)
R: Reduction to terminal aldehyde or alcohol (optional)
After the peptide chain is synthesized, it can then be modified by halogenation, hydroxylation, acylation or glycosylation, which is typically carried out by an enzyme coded for in the same operon or gene cluster that was associated with the carrier protein. Since NRPS is similar to PKS and FAS, components of the other methods of metabolite synthesis are often cross-linked to each other and combine to form natural products.
--A08954805 (discuss • contribs) 22:32, 15 November 2011 (UTC)
The DNA has two strands, a sense strand and a template strand. The sense strand has the same sequence as the mRNA that will be transcribed, except the T on the DNA will be replaced with U’s on the mRNA. RNA Polymerase will make a complementary mRNA transcript from the template strand of DNA.
Initiation: RNA polymerase will move along the DNA, looking for the -35 region and -10 region of the sigma-70 promoter in E.Coli. Once it finds the promoter, RNA polymerase will bind to the promoter, loosely at first then more tightly once DNA starts to unwind. RNA polymerase will then add a ribonucleoside triphosphate (rNTP), usually a purine. This rNTP will be complementary to the nucleotide on the +1 position of the DNA template. [1]
Termination: The transcription termination site is located downstream from the translation stop codon. In bacteria, there are two types of terminations possible:
Rho dependent:
A Rho factor will bind to the RNA in a region, called the transcription terminator pause site-- this is rich in guanine and cytosine and is after the part of the gene that codes for protein. Rho will then wrap the downstream RNA (the RNA between where Rho binds and the RNA polymerase) around itself and slowly pull itself to the RNA polymerase, which is now paused. When Rho comes into contact with the RNA polymerase, termination occurs and the mRNA transcript and RNA polymerase are released from the DNA template. [1]
Rho independent-
A region of the mRNA transcript that is rich in guanine and cytosine forms a RNA stem loop that will hold onto the RNA polymerase and cause it to pause. During this pause, the poly-U and poly-A base pairs on the 3’ end of the mRNA is weak and therefore easy to melt. Transcription is stopped when the molecule is melted, and the mRNA transcript and RNA polymerase will be released. [1]
Initiation: For bacteria, initiation factors (IF) are involved in the initiation of translation. IF3 will bring mRNA and the 30S subunit of ribosome together. The ribosome binding site on the mRNA can then bind the complementary sequence on the 16S rRNA. IF1 will bind to the A site of the 30S ribosomal subunit and block that A site. IF2 that is attached to GTP can then bring the initiatior fMet-tRNA (N-formylmethionyl-tRNA) to the start codon on the P site of the 30S ribosomal subunit. With the attachment of the initiator tRNA, IF3 will be released and then the 50S subunit of the ribosome will be attached to the 30S. This leads to the hydrolysis of the GTP and therefore the release of the IF2 and IF1. The ribosome continue through translation. [1]
Termination: The ribosome will encounter a stop codon-- either UAA, UAG, or UGA, which appears in the A site of the ribosome. Instead of a tRNA binding, a protein release factor, either RF1 or RF2, will enter the A site of the ribosome. Peptidyltransferase will then cut the bond between the finished protein and the P site. Once the protein is released from the ribosome, RF3 will cause the protein release factor used to leave the ribosome. After, a ribosome recycling factor (RRF) and a bound EF-G will bind at the A site of the ribosome. GTP hydrolysis will take apart the 30S and 50S ribosomal subunit. IF3 will then bind to the 30S to remove any tRNA or mRNA left on the subunit. There is now a synthesized bacterial protein and ribosomal subunits that can help in further translations. [1]
Protein Purification is the process of separating proteins for individual analysis. Protein purification is the second step of studying proteins, the first being the process of an assay. An assay is a procedure to measure the activity enzyme activity thus confirming the presence of the protein or proteins in interest. Popular assays include Western Blotting and ELISA(Enzyme-linked immunosorbent assay). Before the purification process, Cell Disruption is utilized to homogenize the cell's content. After the cell has been opened up, the process of purifying proteins from one another and the other organelles can be approached in several different methods. Protein mixtures are normally separated multiple times, each based on a different property, such as:
Solubility
Size
Molecular Weight
Charge
Binding affinity
The intended reason for purifying a specific protein governs the level and degree of protein purification. At times, a sample of protein that is only moderately purified suffices for its intended application; however, other situations require a higher degree of purification, especially if the fundamental ambition is to study the characteristics and tendencies of the specific protein in interest. By considering solubility, size, molecular weight, charge, and binding affinity, the goal of the scientist that conducts protein purification is to find a level of purification necessary and create a protein yield that is ample for further research and application. This means using the fewest steps in order to keep the yield high, as each protein purification step incurs a degree of product loss. Therefore two factors serve as obstacles in protein purification: yield and purification level. The main goal of each protein purification project falls under two categories: analytical (for studying and research purposes) and preparative (for production and creation of commercial products).
Proteins are separated based on masses or densities by a centrifugal force. Centrifugation enables the separation of proteins in different cell compartments.
Different proteins precipitate at different salt concentration. When the concentration of salt increases, more proteins are able to separate
Large molecules flow more rapidly to the bottom of the column.
Proteins are separated according to its charge. Positively charged proteins bind to negatively charge bead, and negatively charge proteins are released. The negatively charged proteins flow through faster.
Many proteins have high affinity for specific chemical groups.
Proteins separate according to different levels of hydrophobicity.
Electrophoresis separate protein while the gel enhances the separation. Small proteins move more rapidly through the gel.
Different proteins have different pI (isoelectric point).
Proteins are separated horizontally based on pI and vertically based on mass.
Proteins are separated through a semi-permeable membrane. Since the dimensions of proteins are generally larger than the pores of the membrane, proteins do not pass through and separate.
Purpose:you have the protein in some cells. Then, you want to remove the other protein to get the one you one.
Differential centrifugation is a method used to separate the different components of a cell on the basis of mass. The cell membrane is first ruptured to release the cell’s components by using a homogenizer. The resulting mixture is referred to as the homogenate. The homogenate is centrifuged to obtain a pellet containing the most dense organelles. Compounds that are the most dense will form a pellet at lower centrifuge speeds while the less dense compounds will likely remain in the liquid supernatant above the pellet. Each time, the supernatant may be centrifuged at faster speeds to obtain the less dense organelles. Performing centrifugation in a stepwise fashion, in which the centrifugation speed is increased each time, allows the components to be separated by mass. The rather dense nucleus is most likely to be found after the first centrifugation step, followed by the mitochondria, then smaller organelles, and finally the cytoplasm, which may contain soluble proteins.[1]
The result of the centrifugation of blood- compounds are separated by their weight.
Equilibrium sedimentation uses a gradient of a solution to separate particles based on their individual densities (mass/volume). A pivotal aspect about this type of sedimentation is that it is completely independent of the shape of the molecule. It is used to purify the differential centrifugation. A solution is prepared with the densest portion of the gradient at the bottom. Particles to be separated are then added to the gradient and centrifuged. Each particle proceeds until it reaches an environment of comparable density. Such a density gradient may be continuous or prepared in an incremental fashion. For instance, when using sucrose to prepare density gradients, one can carefully float a solution of 40% sucrose onto a layer of 45% sucrose and add further less dense layers above. The homogenate, prepared in a dilute buffer and centrifuged briefly to remove tissue and unbroken cells, is then layered on top. After centrifugation typically for an hour at about 100,000 x g, disks of cellular components residing due to the change in density can be observed from one layer to the next. By carefully adjusting the layer densities to match the cell type, specific cellular components can be enriched.
Sedimentation equilibrium is quite useful because a pellet is not formed. The speed of rotation creates enough force to make the protein leave the rotor, but it doesn’t condense it into a pellet. This is because a gradient in the concentration of the protein is produced. Diffusion reacts to counter the creation of the gradient and after a certain amount of time, a perfect balance between sedimentation and diffusion is achieved.
Sedimentation equilibrium is also practical to study the interactions between proteins. In particular it is used to ascertain the native state or native conformation of the protein. The native state tells us the exact structure in three dimensions. This information includes if it is a monomer, dimer, trimer, tetramer, etc. A monomer is a protein made up of one subunit. A dimer is two protein subunits that are rotated 180 degrees. A trimer is three subunits etc. This type of experimentation also allows us to determine whether the proteins can form oligomers (identical polypeptide chains tha make up two or more units of a protein). Additionally, the use of sedimentation equilibrium is that it determines equilibrium constants for protein-protein and protein-ligand interactions. The value of this Kd is often between 1nM-1mM. This is calculated by measuring the equilibrium constant (Kd). A final use of this is to determine stoichiometric ratios between protein complexes. An example of this is a ligand and its receptor or an antigen-antibody pair
The process of "salting out" is a purification method that relies on the basis of protein solubility. It relies on the principle that most proteins are less soluble in solutions of high salt concentrations because the addition of salt ions shield proteins with multi-ion charges. Those charges help protein molecules interact, aggregate, and precipitate. The exact concentration resulting in precipitation varies from protein to protein, allowing for the separation of different proteins (as proteins will precipitate at different points with increases in salt concentration). Salting out can also concentrate dilute solutions of proteins; once the protein precipitates, the remaining liquid can be removed. However, the salt can pose a problem to the purity of protein.
"Salting in" refers to the observation that at solutions of low salt concentrations, the solubility of a protein increases with the addition of salt. As the solubility of the salt is higher than that of the protein, it is more likely dissolve and take up space in the solution; therefore, proteins aggregate and precipitate. By contrast, "salting out" requires high salt concentration for the precipitation of the protein. There are two ways of "salting out". In one method, proteins are exposed to high concentrations of salt solutions, and in the other, the proteins are exposed to a series of low concentrated solutions.
Proteins contain various sequences and compositions of amino acids. Therefore, their solubility to water differs depending on the level of hydrophobic or hydrophilic properties of the surface. Proteins with surfaces that have greater hydrophobic properties will readily precipitate. The addition of ions creates an electron shielding effect that nullifies some activity between water particles and the protein, reducing solubility as the proteins bind with each other and begin to aggregate. Generally, larger proteins require less ionic input than do smaller proteins with lesser weight.
In the process of using low concentrations of salt solutions, the proteins are precipitated early in the process. In order to extract the proteins from the solution, cold solutions of ammonium sulfate at a series of decreasing concentrations are used on the precipitate. In order to recover the extracted protein, it is then recrystallized by warming the cold solution to room temperature. This process has many advantages because depending on the extracted protein, the efficiency rate can run anywhere from 30-90%, and rarely fails.
Ammonium sulfate is common substance used to precipitate proteins selectively since it is very soluble in water, it allows high concentration about 4M. At this state, harmful effects of proteins like irreversible denaturation are absent and NH4+ and SO42- are both favourable, non-denaturing, end of the Hofmeister series. Ammonium sulfate provides quantative precipitation of one protein from the mixture. This method is very useful to purify soluble proteins from the cell extracts.4
While proving itself to be an efficient method of protein separation, salting out requires that the solubility of the protein to be calculated or known initially. Proteins have differing amino acid chains and solubility. In trying to change the salt concentration to the point where the protein becomes insoluble, different ions can either increase or decrease the solubility of the protein. Hence, one must be careful in selecting the correct ions to alter salt concentration. A protein is typically least soluble near its isoelectric point, pI, or where it contains minimal net charge. The precipitation by salting out results in fractionation. An amount of precipitated protein is collected at one salt concentration and another amount from a different concentration. This is because some parts of the protein may be more soluble than another region.
Proteins with different pI values can be separated with salting out techniques via dynamic pH values in varying salt concentration. Since proteins are least soluble near their isoelectric point (pI), it is possible to cause them to precipitate them out of solution by increasing the salt concentration. This is possible since the hydration shell surrounding the protein structure is displaced by the increasing ionic concentration in the solvent. Thus by replacing the hydration shell with other ions, the water networks that solubilize proteins become destabilized and allow for
aggregation at high salt concentration due to hydrophobic groups coming together. Ultimately proteins are precipitated with aggregation (or "crashed out"). This technique can be used to separate proteins that initially have similar precipitation points. By modifying the pH of the solution, one can increase or decrease the solubility of one protein without affecting the target protein. Furthermore, the solution can later be purified by using dialysis to remove the salt ions in solution.
The effectiveness of the different ions was established by Franz Hofmeister in 1888. The first ion in the anion and cation series is the most effective in precipitating a protein out (dubbed "kosmotropes": ions that interact well with water, forming H-bonds and dehydrating proteins), and the ions at the end are the least ("chaotropes": ions that free up water by breaking H-bonds between water molecules, increasing protein solubility). ^
The starting molecules strengthen hydrophobic interactions by decreasing solubility of the nonpolar molecules, thus salting out the system. However, the later molecules begin to denature the structure of the protein because of strong ionic interactions that disrupt hydrogen bonding. Although the later molecules can be salted out through solutions such as Ammonia Sulfate, certain molecules can also experience salting in, where the solubility of the protein increases through the later molecules of the list.
Dialysis is a protein purification process that separates proteins from other small molecules, such as salt, by using a semipermeable membrane. This membrane contain micro pores through which the small molecules will escape. Therefore, protein molecules having dimensions significantly greater than the pore diameter are retained inside the dialysis bag. The small molecules and salt will diffuse out through the membrane and into the dialysate outside of the bag. This technique is useful to remove salt ions and other small molecule but can not be used to distinguish proteins. To enhance the separation of the proteins in the bag from other impurities such as salt we can also take advantage of the equilibrium constants. In an aqueous environment the salt will flow through the plasma membrane until its concentration outside the dialysis bag is equal to the concentration inside the bag. At this point there is no net flow of salt through the membrane because equilibrium is reached. But if we add in a new solution of buffer, then the remaining amount of salt will then flow out of the dialysis bag until the concentration of salt in the new buffer equals the concentration in the dialysis bag. If we keep replacing the buffer solution this will enhance the purity of the proteins inside the dialysis bag because each time we replace the buffer the salt has to flow out inorder to attain its equilibrium constant. This principle can also be applied for other impurities that are able to escape through the membrane.
In kidney-compromised patients, dialysis is often used as a procedure for removing undesirable solutes in the blood. For example, the calcium, potassium, and urea concentration of the dialysate is kept at low concentrations, enabling the target solutes in the blood to diffuse across the semi-permeable membrane. However, this entails the dialysate to be constantly cleaned in order to prevent concentration equilibrium, which would ultimately lead to a rising concentration of unwanted solutes in the blood. In another case, solutes can also be introduced into the blood. For example, bicarbonate ions are in high concentration in the dialysate, which diffuse across the membrane. This is done to prevent metabolic acidosis.
Capillary Electrophoresis is a family of techniques that use narrow-bore capillaries to perform high efficiency separations of both large and small molecules. Using a high voltage power supply, the solution travels from the anode to the cathode through the capillary. By doing so, the solution passes through the detector and based on the flow of the molecules, the integrator computes the separation of the molecules from the original solution. There are five modes of capillary electrophoresis which include capillary zone electrophoresis, isoelectric focusing, capillary gel electrophoresis, isotachophoresis, and micellar electrokinetic capillary chromatography.
Capillary zone electrophoresis is a separation mechanisms that is based on the differences in the charge-to-mass ratio of the molecules. The homogeneity of the buffer solution as well as the constant filed strength are fundamental to the capillary zone electrophoresis process. It can be used to separate both large (DNA) and small (drugs) molecules. Capillary Zone Electrophoresis is the simplest form of capillary electrophoresis.
Isoelectric focusing is when the solution tested is run through a pH gradient where the pH is low at the anode and high at the cathode. Therefore, when a voltage is applied, the ampholyte mixture separates in the capillary.
Capillary gel electrophoresis is conducted in an anticonvective medium, oftentimes such as polyacrylamide or agarose gel. The composition of the media thus serves as a molecular sieve for size separations.
In isotachophoresis, there is zero electroosmotic flow with the heterogeneous buffer. In fact, the capillary is filled with a leading electrolyte with a higher mobility than any of the sample components as well as a terminating electrolyte where the ionic mobility of the electrolyte is lower than any of the sample components. As a result, the solution is separated based on the leading and terminating electrolytes.
In Micellar Electrokinetic Capillary Chromatography (MECC or MEKC), the use of micelle-forming surfactant solutions can give rise to separations that resemble reverse-phase liquid chromatography. Based on the hydrophobic and electrostatic interactions, the analytes are organized at the molecular level.
In comparison to HPLC which uses hydrodynamic flow, capillary electrophoresis is based on electroosmotic flow (EOF). The factors that influence the rate of electroosmotic flow are pH, voltage, temperature and the concentration of the buffer.
At neutral to alkaline pH, the electroosmotic flow is sufficiently stronger than the electrophoretic migration such that all species are swept towards the negative electrode. At high pH, the electroosmotic flow is large and the peptide is negatively charged; despite the peptide’s electrophoretic migration towards positive electrode (anode), the EOF is overwhelming and the peptide migrates towards negative electrode (cathode). At low pH, peptide is positively charged and EOF is very small, thus resulting in peptide electrophoretic migration and EOF towards the negative electrode. However, most solutes migrate towards negative electrode regardless of charge when buffer pH is above 7.0. Oftentimes, the pH selected is at least two units above or below pKa of the analyte in order to ensure complete ionization.
At high temperatures, the viscosity of the solution is lower and the electroosmotic flow increases as a result. However, some buffers are known to be pH-sensitive with temperature.
Wätzig, H., Degenhardt, M. and Kunkel, A. (1998), Strategies for capillary electrophoresis: Method development and validation for pharmaceutical and biological applications. ELECTROPHORESIS, 19: 2695–2752. doi: 10.1002/elps.1150191603
High Pressure Liquid Chromatography (also known as High Performance Liquid Chromatography, or simply HPLC) is an enhanced form of column chromatography that is commonly used in biochemistry to separate and purify compounded samples. Instead of the solvent dripping through the column as a result of gravity as is the case in other methods of chromatography, the solvent is pushed through with high pressures.
The column materials of HPLC are much more neatly and greatly divided, and so there are more interaction opportunities and greater resolving (separating) power. Since the columns are made of materials of better quality, constant pressure must be applied to the column to obtain acceptable flow rates. Therefore, the final result is high resolution and very fast separation.
HPLC was developed and improved with new column technologies in the mid-1970's, replacing the other primeval column chromatographic techniques which failed when it came to quantifying and purifying similar compounds. Pressure liquid chromatography proved to be much less time consuming than the old methods. Compared with classical column chromatography, in which the columns are powered by gravity and a separation can take hours or even up to days, HPLC was able to produce results as fast as five to thirty minutes.
HPLC was used frequently for the compound purification by the 1980's. Computers and other improved technology added to the convenience of HPLC. Improvements in the types of columns and consequently, reproducibility of HPLC, led to developments of micro-columns, affinity columns, and fast HPLC.
The past decade has seen a vast advancement in the development of micro-columns, now commonly used for HPLC, and other specialized columns. The diameter of the typical HPLC column is about 3-5 mm. But the usual diameter of micro-columns, or capillary columns, ranges from 3 µm to 200 µm, so it is considerably smaller. Fast HPLC utilizes a column that is shorter than the typical column, and so they are packed with smaller particles.
These days, one has the option of considering several types of columns for the Purification of mixtures, as well as a variety of detectors to work with the HPLC in order to get the best possible analysis of the compound.
A small volume of the sample is put into the High-Pressure Liquid Chromatography where a mobile phase will move it through the stationary phase. The mobile phase is usually a gas or a liquid and the stationary phase is immobile and immiscible. The stationary phase will slow down the flow of the sample because of it physical or chemical properties (size, net charge, or other differences depending on the type HPLC) where it will be filtered or purified. Because of the difference in how the stationary phase affects the impurities from the desired compound, the different components of the sample will come out at different times. The time that a component comes out of the column is called the retention time. The retention time should be unique to the component in the particular sample, so that no two components being analyzed elute at the same time and obscure each other. If solvent composition cannot be tweaked to effectively separate components in HPLC analysis, then a different type of chromatography might be better suited. HPLC, unlike other column chromatography techniques, uses pressure via pumps to push components through the more finely packed columns to speed up analysis and enable analysis of component and column combinations that take longer to elute on their own.
The mobile phase is a solvent or mixture of solvents that carries the sample through the stationary phase. As it moves through the stationary phase, molecular interactions between the sample's components and the column material determine the retention time of the different components. The components that have stronger interactions with the mobile phase than the column will "prefer" the mobile phase and elute quicker with a shorter retention time while components that have stronger interactions with the stationary phase than the solvent will "prefer" the column and elute slower with a longer retention time. This is how HPLC separates, filters, and aids in purification of the compound. There are different techniques in regards to mobile phases that are tweaked to optimize retention time, separation, and peak clarity. These are isocratic, gradient, and polytyptic.
Isocratic elution involves a constant mobile phase composition. For example, a mobile phase of 50% acetonitrile and 50% water for a reversed phase HPLC (RP-HPLC) run that remains unchanged through the entire analysis. A solvent system is chosen and it will be used for the entire duration of the HPLC run. The sample is injected as the mobile phase flows through, enters the HPLC at a constant flow rate, and passes through the chosen column. This method is generally used when the sample being analyzed is simple enough that all the components of the sample come out at different times with sufficient clarity, and do not have impractically long retention times.
Most samples are not so easy to work with. In these cases, a gradient elution method is set up. The mobile phase mixture will shift as the run proceeds, and the concentrations of the solvents are modified so that the run begins with the "weaker" solvent, and the "stronger" of the solvents will be the most concentrated at the end. One such example is a reversed phase HPLC run that begins with more mobile phase A, which is composed of a 95% water and 5% acetonitrile mixture, and will gradually increase mobile phase B, which is a 100% acetonitrile mixture, until at the end of the run the majority of mobile phase flowing through the column is mobile phase B. Usually for reversed phase HPLC, the mobile phase will begin with the more polar solvent combination and increase the concentration of the less polar solvent combination as the run proceeds. This is so that the less polar molecules (relative to the mobile phase and stationary phase being used) will eventually elute due to a higher concentration of a less polar solvent and the necessary run time for the analysis can be shortened. An isocratic mobile phase can have a polarity too close to the stationary phase, resulting in components eluting out together immediately and their peaks overlapping, or a polarity too different from the nonpolar stationary phase, resulting in nonpolar components taking too long to elute. This is why a gradient mobile phase is often used in analysis, where concentration of less-polar to more-polar solvents can be modified to obtain optimal peak separation.
The polytptic elution, also known as mixed-mode chromatography, involves the use of a special column that can switch modes of analysis depending on the solvent. The same column can perform size exclusion, ion exchange, or affinity chromatography depending on the type of solvent that flows through it.
Retention times depend on the interaction of the component of the sample, the mobile phase, and the stationary phase to each other. Therefore, a well-designed HPLC run relies on choosing the correct type of column for the analysis desired and the right combination of mobile phases for the analyte and the column.
Column efficiency describes how well the stationary phase filters or purifies, basically how packed it is and how well things move along it. There are a couple of ways to measure column efficiency but they all use the same formula:
N=atr2/W2
N=number of theoretical plates
a=constant that depends on the height of a graph
tr=retention time
W=width of a peak
Normal phase chromatography, or NP-HPLC is the first kind of HPLC developed. In this method a polar stationary phase and a non-polar mobile phase is used in order to separate analytes based on their polarity. Since the polar phase is stationary, polar analytes will bind to that phase. Their adsorption strength and elution time depend on the strength of the analyte polarity and the analyte’s steric factors. Since the elution time depends on steric clashes, it is then possible to differentiate and separate structural isomers since each isomer has a different steric clash. One can increase the elution time by adding a non-polar solvent to the non-polar mobile phase. One can also able to decrease the retention time of the analytes by adding polar substances to the non-polar mobile phase and even occupy the stationary phase surface preventing the polar analytes from binding to the polar surface.
In the past, this method is unfavorable due to the fact that water or protic organic solvents changed the hydration state of the media in the system. However, this problem was solved with another version of NP-HPLC called hydrophilic interaction liquid chromatography, which uses a variety of phases that had better retention times.
Reverse phase chromatography, as the name suggests, is the opposite of normal phase chromatography, where it now has a non-polar stationary phase and a polar mobile phase. Consequently, the non-polar analytes will bind to the non-polar phase, and its elution time will also depend on how non-polar it is. One can still also increase the elution time by adding a polar solvent to the mobile phase or decrease the elution time by adding a non-polar solvent to the same phase. However, unlike NP-HPLC, the method depends on hydrophobic interactions.
Some factors can influence hydrophobic interactions. One of those factors is surface area. An analyte with a larger hydrophobic surface area would consequently have a longer retention time since there would be more bonds interacting between the analyte and the non-polar surface. However, too large of an analyte surface won’t be able to enter the pores of the non-polar phase and have no interactions with the phase. This strengthening in bonds is also due to the force of water for “cavity-reduction” around the analyte, and the energy released in this process depends on the surface tension of the eluent, which in this case is water.
Another factor that can affect the hydrophobic interactions is the pH. An ideal environment is one that is uncharged. As a result, chemists use buffering agents, such as sodium phosphate, to regulate the pH and neutralize the charge on exposed media, which usually is composed of silica, on the stationary phase and the charge on the analyte.
Reverse phase columns are stronger than normal silica columns, but still have some weaknesses. Aqueous bases shouldn’t be used with columns consisting of alkyl derivatized silica particles since the base will destroy the underlying silica particle. Also, if an aqueous acid is used, it should be exposed too long to the column in order to prevent corrosion.
Gel-filtration chromatography separates proteins based on differing in size. The process involves a gel in a buffer solution that is packed into a column. This gel has many porous carbohydrate polymer bead-like particles. The size of the pores is selected so that it can only allow proteins with a certain size to diffuse through them. The movement of the molecules that are small enough to enter through the pores of the beads is then slowed down because it is forced to enter the stationary phase of the column. The larger molecules on the other hand, end up moving through the column faster because they cannot enter the internal volume of the beads.
The most important advantage of gel-filtration chromatography is its ability to separate the proteins in its original, non-denatured condition, giving you a sample that is in a suitable form for possible further analysis. Another advantage as well is the high resolution that is obtained by applying pressure into the column to get adequate flow. Improved resolution is achieved with slower flow rates. An optimum flow rate for protein fractionation of approximately 5mL/cm2/h is recommended for most gels.
Reference: Aguilar, Marie-Isabel. HPLC of Peptides and Proteins Methods and Protocols. volume 251. Humana Press.
Ion-exchange chromatography separates proteins based on their charge. It is efficient enough to be able to resolve proteins that differ only by one single charged group. It depends on the formation of ionic bonds between the charged groups on the proteins and an ion-exchange gel carrying the opposite charge in a column. Proteins that do not have an electrical charge and are neutral are removed by washing. Those proteins that can form ionic bonds, though, are recovered by elution with a buffer of either higher ionic strength or changing pH. An increase in oppositely charged ions (those of the protein being analyzed and those of the gel medium) increases the retention time, which is based on the attraction between the protein ions and charged ions of the gel medium.
There are two types of ion-exchangers. One is the anion exchanger, which has positively charged groups that are stationary in a gel-medium and will interact and bind to negatively charged ions in the protein. The other is the cation exchanger, which has negatively charged groups that are stationary in a gel-medium as well but interact and bind to positively charged ions in the protein.
The pH of the solution can also alter how the ionization process between the protein ions and the ions in the gel-medium. When the pH is equal to the isoelectric point of the protein (the point where the net charge is zero). However, when the pH is less than the isolectric point, the net electric charge on the protein will be positive and it will bind to the cation exchangers. Finally, if the pH is greater than the isoelectric point, the net charge on the protein will be negative and it will bind to the anion exchangers. Therefore, by controlling the pH of the solution we can control how the protein gets separated since it is these exchangers that separate the protein
Reference: Aguilar, Marie-Isabel. HPLC of Peptides and Proteins Methods and Protocols. volume 251. Humana Press.
Affinity chromatography is the method of the separation of biochemical mixtures, based on a highly specific biologic interaction. It is used to purify a molecule from a mixture and concentrate it into a buffering solution, and also to recognize what biological compounds bind to another molecule, like drugs. It was discovered in 1968 by Pedro Cuatrecasas and Meir Wilcheck.
The process involves the trapping of the target protein (or molecule) that one wants separated from the mixture onto a solid or a medium. A column is filled with beads that contain covalent glucose residues, which are chosen to correspond with the target protein. The proteins will travel down through the beads as they are poured into the column, and when the target protein is recognized, it will get trapped to the column by covalent bonds due to its affinity for glucose. The rest of proteins will run down the column and become successfully separated. The portion of buffer will be added to the column to wash out the unbounded protein. Lastly, a concentrated solution of glucose is added to separate the target protein from the column-attached glucose residues, resulting with the protein being completely purified out of the mixture.
Adsorption Chromatography
Adsorption, meaning the accumulation of solutes of the surface of a solid or liquid, chromatography is useful in separating a mixture of solutes based on their different polarities. It is based on the notion that polar solute will form a tighter bond with the polar stationary phase than a less polar solute will.
An insoluble, polar material like silica gel (a derivative of silica gel, Si(OH)¬4) is filled into a glass column, making it the stationary phase. The sample containing the mixture is the mobile phase, which can be a liquid or gas, is poured onto the glass column, where each solute with a different polarity will bind differently to the solute. The polar solutes will bind tightly to the stationary phase, the less polar ones will bind more loosely, and the neutral ones will pass right through the column. The solute can be eluted with solvents of progressively higher polarity, where the solutes will be eluted with increasing polarity. So, neutral solutes will pass right through the column, the less polar ones will be eluted first, and very polar solutes will be eluted last.
Reference: Principles of Biochemistry 4th Edition.Nelson, David L.; Cox,Michael M.W.H Freeman and Company. New York
Practical HPLC Method Development 2nd Edition. Snyder, Lloyd R.; Kirkland, Joseph Jack; Glajch, Joseph L. New York.
Handbook Of Pharmaceutical Analysis By HPLC. M. W. Dong. Elsevier.
A Gel Filtration column
Gel-filtration chromatography, also known as 'size exclusion chromatography', 'molecular exclusion chromatography' or 'molecular sieve chromatography' is the simplest and mildest technique that separates molecules based on their size difference (hydrodynamic volume). This approach allows each polypeptide to be purified from other different sized polypeptides by passing through a gel filtration medium packed into the column. Unlike ion-exchange or affinity chromatography, fractions passing through the column do not bind to the chromatography medium. The big advantage of Gel-filtration chromatography is that the medium can be varied to suit the properties of a sample for further purifications.
When an organic solvent is used as a mobile phase, chemists tend to call it Gel permeation chromatography. The buffer or organic solvents used as the mobile phase are chosen based on the chemical and physical properties of the specific protein sample. The stationary phase of the column is simply the carbohydrate polymeric beads and the mobile phase goes through the stationary phase at a different speed depending upon the size of the molecule. This technique is used to analyze the molar mass distribution of organic-soluble polymers. It was invented by Grant Henry Lathe and Colin Ruthren who were working at Queen Charlotte's Hospital in London, United Kingdom.
Gel-filtration chromatography can be applied in two different ways: for group separations and high resolution fractionation of biomolecules. The group separation technique separates compounds in a sample into groups based on the size range. This technique is used for purification of a sample from high or low weight contaminants. The high resolution fractionation of biomolecules is a more precise technique. It can be used for isolation of one or more components in a sample, separation of monomers from aggregates, to determine molecular weight, or to perform molecular weight distribution analysis. Gel-filtration chromatography is very suitable for biomolecules which are very sensitive to pH changes, concentration of metal ions, or co-factors.
Within the size range of molecules that are subjected to gel-filtration chromatography and are separated by a particular pore size of beads in the column, there is a linear relationship between the relative elution volume of a substance (i.e., the volume of the fractions in which the molecule is found)and the logarithm of its molecular mass (this is assuming that the molecules have similar shapes). If a given gel filtration column is calibrated with several proteins of known molecular mass, the mass of an unknown protein can be estimated by its elution position.
An analogy to understand (this is CONCEPTUAL, not even remotely a literal representation of what happens in ME chromatography) why gel filtration works is to picture several whiffle balls (or sponges or Swiss cheese-whatever cratered object works for you) suspended in a glass tank. Now imagine that you have a mixture of sand, small marbles, and golf balls in a bucket; you dump it in. As you watch, first the golf balls reach the floor of the tank, then the marbles, and finally a layer of sand settles. Why? Essentially all of the sand goes into the holes of the whiffle balls(or Swiss cheeses or sponges), and it tends to fall from the interior of one whiffle ball to the interior of another, significantly slowing passage of the sand to the bottom of the tank. The marbles are only slightly smaller than the holes in the whiffle ball, so they sometimes fall into the holes on the way down but also sometimes bounce off; again, the whiffle balls slow their progress, but to a lesser extent. The golf balls are way too big to fit the holes of a whiffle ball, and so they push straight through the whiffle balls—the fastest and most direct route.
Key: sand=small molecules; marbles=medium molecules; golf balls=large molecules; whiffle balls=porous beads; tank of water=column & aqueous solution
The gel medium packed into the column is a porous matrix that consists of spherical beads, which have stable physical and chemical properties such as non-reactivity and lack of adsorption. The small molecules can enter the beads but the larger one cannot. The small molecules are distributed in the aqueous solution both inside and between the beads where as the large molecules are located in the solution between the beads. These beads are not soluble and are normally made from highly hydrated polymers such as dextran, agarose, or polyacrylamide. For commercial purposes, Sephadex, Sepharose, and Biogel are used. These commercial beads are about 100 miciro-meters in diameter and are used to separate proteins based on sized. Also, silica or cross-linked polystyrene can also be used as material for the beads under higher pressures. The pores and space between the particles is filled with a liquid buffer, which fills the entire column. The liquid filling the pore space is called a stationary phase and the liquid in the space between particles is a called mobile phase. Once the sample has been applied to the top of the column, it passes through the column along with the mobile phase from the top of the column to the bottom. Smaller molecules are able to cross and go through these polymer beads but large ones are not able to. Therefore, small molecules in the column are both inside the polymer beads and between them, whereas large molecules can only travel between the polymer beads. Since less traveling space is allowed for the larger beads, they tend to move faster down the column and they emerge first at the end of the column. Think of it this way. The molecules traveling down the column represent a faucet. If the faucet has a smaller volume of space to allow the water to travel, the water will come out faster and with greater force. The same concept applies here as well. Since less volume is accessible to the bigger molecules, they move much faster through the column than smaller molecules do. So, since the small molecules are stuck inside the beads, they tend to move slower. Theoretically, molecules that have the same size should elute simultaneously. An elution diagram, or a chromatogram, can be constructed to verify complete separation. Before separation of unknown sample, solutions with known biomolecules can be run in order to make a calibration curve, which later can be used as a reference for identifying of unknown molecules.
Gel-Filtration Chromatography is commonly used for analysis of synthetic and biological polymers such as nucleic acid, proteins, and polysaccharides. A downfall to this technique is that the stationary phase may also interact in an undesirable way with a molecule and affect its retention time. A major drawback to this method is its difficulty in producing a high-resolution image. An alternative to this may be Discontinuous Electrophoresis. Disc electrophoresis uses gels with different pHs and the proteins produce sharp bands when they go from one gel to the other, which creates high-resolution images.[1] This technique requires three different gels: the sample gel, the stacking gel, and the running gel. The proteins moves through the stacking gel and between the sample and running gels before the proteins enter them. This compresses the proteins and increases the resolution.[2]
Gel-Filtration Chromatography should not be confused with gel electrophoresis, where electricity is applied to create an electric field to separate molecules through the gel towards the electrode (anode and cathode) depending on their electric charge. Besides, large molecules in Gel-filtration Chromatography migrate down the column first whereas small molecules in gel electrophoresis migrate down the gel first.
Viadiu, Hector. Biochemistry 114A Lecture. "Protein Techniques." 10/15/12
Purpose: To separate a specific protein from its mixture by using the property of ion-charges.
Ion Exchange Chromatography (IEC) is a purification method aimed at separating proteins based on charge, which is dependent on the composition of the mobile phase (a separation of mixtures that is dissolved). Adjusting the pH, or the ionic concentration, of the "mobile phase" allows for separation. For example, if a protein has a net positive charge of pH 7, it will bind to a column of negative charge beads. On the other hand a negatively charged protein would not.
For example, if a proton has a net positive charge at pH of 7 then it will bind to a column of beads that contain the carboxyl groups, where as a negatively charged proteins will not. Once bound, the protein is eluted by increasing the ion concentration. The movement of a protein depends on the density of the net charge; the proteins that have a low density of net positive charge will emerge first.
Proteins bind to ion exchangers due to the electrostatic forces between the surface of the protein charges and cluster of the charged group on the exchangers. A column is packed with a resin (usually cellulose or agarose) with a charged group bonded to it. This allows positively charged proteins, for example, to bind to the negatively charged beads on the column and the negatively charged proteins to flow through the column. Therefore ion exchange chromatography consists of cation exchange chromatography and anion exchange chromatography. In addition, a protein must displace the counterions and become attached; in other words, the net charge on the protein will be the same sign as that of the counterions displaced-therefore "ion exchange." The protein molecules in solution are neutralized by counterions also; the overall reaction must be electrically neutral. Whatever one wants to purify is known as the sample and the parts that are separated are known as the analytes. The sample is added to the top of the column and a buffered solution is used to elute it.
Anion-Exchange chromatography involves the use of positively charged beads. In the purification of acids, which often has the negative charge on its carboxyl group, anion-exchange chromatography is utilized. Anion-exchange chromatography mainly recollects biomolecules by the interaction of amine groups on the ion-exchange resin with aspartic or glutamic acid sidechains, which have pK of ~ 4.4. The mobile phase is buffered at pH > 4.4, below which acid sidechains start to protonate and retention declines.
Above pH 4.4, retention is fundamentally reliant on on the number of anionic sidechains existing in the protein. Proteins including the same number of anionic sidechains can often be separated by modification of the mobile phase pH between 7 and 10 where histidine is not protonated and lysine starts to deprotonate.
Delicate changes occur to proteins in this pH region which affect the interaction of the protein with the resin and which allow fine-tuning of the anion-exchange separation. A mobile phase, pH > 10, is not usually suggested because of possible protein deprivation, such as deamination, at higher pH's.
In cation exchange chromatography, a sample consisting of a certain protein that bears a net positive charge at a certain pH is a added to a column. In anion exchange chromatography, a sample with a protein that bears a net negative charge at a certain pH is added to a column. Recall that a net charge is the sum of partial charges for each amino acid's particular R group at a given pH. The columns have resin that consists of cellulose (or agarose) beads, which have a function group covalently bonded to it. For cation exchange a carboxylate group is used, and for anion exchange a diethylaminoethyl group is used. A buffer solution, also called a mobile phase, has its pH set between the pl or pKa of protein and the pKa of the beads on the columns. The buffer solution then runs the sample through the column. Molecules with no charge or the same charge as the beads will pass through, while molecules with the opposite charge will bind to the column of beads. Like a magnet, it'll stick and stay there. To elute the bound proteins, the column is flushed with a salt, usually excess NaCl. In cation exchange chromatography the Na+ ion will compete with the bound protein for the negative functional group, and in anion exchange chromatography, the Cl- ion will compete to bind the columns. Another way to flush the system would be with a low pH buffer. The more acidic conditions will lower the net charge (or make it more positive) of the protein. Since the protein now bears a positive net charge, it no longer feels compelled to be around the like-charged resin (since like charges repel), and thus will come out of the column pure. Knowing the isoelectric point (pI) of the protein sample can be helpful in ion-exchange chromatography. Recall that pI is the pH at which a compound's net charge is zero. So if we have a compound with a high pI, for example 10, then to get the pH gown to 7 would cause the compound to become positive. Conversely, if the pH of the solution is higher than the pI, the protein becomes negative overall, thus more anion formation. Thus, depending on the pI of the protein, different solvents at specific pH's can be targeted to purify protein. This also implies that proteins with two significantly different pI's are the most successful in ion-exchange.
If there are impurities in the sample that have a similar charge of the protein being isolated, a pH gradient buffer solution is needed. Unless the proteins have exactly the same amino acids, it is unlikely that they will have exactly the same charge at the same exact pH. Raising (or lowering) the pH, which is in effect causing more molecules to be deprotonated (or protonated), will cause the molecule to have a slight change in charge negatively (or positively). This will affect the ionic interaction between the molecule and the resin, causing some of the molecules to elute from the column. By changing the pH, different molecules will have different charge densities (or degree of negative charge; -2,-1,-3, etc.). So at a certain pH, a protein might have a higher or lower charge density and will thus bind to the resin differently, and those with a lower charge density will elute first.
For another example, say we are analyzing an air sample that has been collected onto an air filter and put through filter extraction (adding water to the filter, purifying by putting through another filter, and extracting the water to be the sample). The samples are then further prepared to put into the IC (ion chromatograph) by adding a given amount of the sample and a given amount of a water. A series of standard solutions and water are first put through the IC in order to calibrate the instrument. The standard solutions consist of certain cation or anion, depending on which ion chromatography is being performed, that are to be detected in the samples. Once all the samples have been put through the IC an ion chromatrogram (see image)is created for each standard and sample solution. In the ion chromatogram the analyte separation can been seen. Each analyte travels through the column at a different rate due to the positively or negatively charged resin. In the ion chromatogram the time at which it takes each analyte to pass through as well as the amount present can be seen. Each analyte will travel through the column at a consistent time in each sample thus each peak can be determined to be certain analytes.
Affinity chromatography is an applicable technique used to purify proteins. It is performed depending on the advantage of the high affinity of proteins for specific chemical groups. Affinity chromatography was discovered by Pedro Cuatrecasas and Meir Wilcheck in 1968.
This process is generally used to isolate interested protein from the pool of proteins. A column is filled with beads that contain covalently attached glucose residues. It is taken in consideration that these residues are chosen corresponding to the target protein. As the protein mixture is poured into the column, the proteins will travel down through the beads. The target protein will be recognized and get trapped to the column by covalent bond because of its affinity for glucose. The rest of proteins will run down to the column and be separated. The portion of buffer needed to be added to the column to wash out completely the unbounded protein. Lastly, a concentrated solution of glucose with be added to separate the target protein from the column-attached glucose residues.
The starting part included an undefined heterogeneous mixture of molecules in solution. The desired molecules will have defined property which can be exploited during the affinity purification process. The process is a setup in which the target molecule becoming trapped on stationary medium. The non-target heterogeneous mixture will not become trapped due to its unbounded ability. The solid medium can then be removed from the mixture, washed multiple times, and the target molecule released from the entrapment in a process known as elution with high concentration of specific chemicals or altering the conditions to decrease the binding ability. Also, it is important that the reaction is carried in an appropriate pH; otherwise, it may reduce the affinity and change the conformation of the proteins, preventing the target protein to bind to the residues as expected.
Affinity chromatography is a powerful means of isolating transcription factors, proteins that regulate gene expression by binding to specific DNA sequences. A protein mixture is percolated through a column containing specific DNA sequences attached to a matrix. Proteins with a high affinity for the sequence will bind and be retained. In this instance, the transcription factor is released by washing with a solution containing a high concentration of salt.
In general, affinity chromatography can be effectively used to isolate a protein that recognizes group X by:covalently attaching X or a derivative of it to a column, adding a mixture of proteins to this column, which is then washed with buffer to remove unbound proteins, eluting the desired protein by adding a high concentration of a soluble form of X or altering the conditions to decrease binding affinity. Affinity chromatography is most effective when the interaction of the protein and the molecule that is used as the bait is highly specific.
Affinity chromatography can also be used in combinatorial chemistry (in-vitro evolution), in which you can imitate the process of evolution by creating large sets of molecules and selecting for a specific function. In this case, you start from a diverse population of molecules, then select for particular proteins, and reproduce that molecule. For instance, starting with a randomized pool of RNA segments and an ATP affinity column, you would apply the RNA pool to the top of the column. Next, you would allow the selection of ATP-binding molecules to occur, eluting from the RNA pool all the segments that did not bind to the ATP. Then to elute the bound RNA molecules, you apply ATP to the top of the column. This isolates the selected RNA molecules that are bound to ATP. You can expand this selection by using different salt concentrations, with increased salt concentrations being more selective.
An example of immunoaffinity chromatography is by the use of blood antibodies. Blood antibodies can be purified by use of affinity purification form the blood plasma (serum). If there is antibodies in the blood plasma that are against some particular antigen we can use this for the antigen purification by using affinity.
A common example to see if an organism is immune against a GST-fusion protein by observing if it produces antibodies against GST tag and the fusion-protein. Foremost, the GST affinity matrix is allowed to bind to the blood plasma. Allowing the blood plasma to bind helps remove antibodies against the GST. Separation of the blood plasma form the solid helps it bind to the GST-fusion protein matrix which in turn traps the antigen that is recognized by the antibody in the solid support. Using low pH ( pH 3 ) buffers for elution helps obtain the desired antibodies. Collection of the eluate is mostly done in phosphate buffer to neutralized the low pH.
Immobilized metal ion affinity chromatography (IMAC)
IMAC is particular based on coordination with covalent bonds form amino acids to metals. The concept of this technique is to keep in the column proteins with affinity to the metal ions which get immobilized inside the column. Iron, gallium or zinc can be used to purify phosphorylated proteins or peptides. Common metals for binding histidine are copper, cobalt, and nickel. DNA recombinant technologies are use since many natural occurring proteins do not have affinity to metal ions.
Commonly, affinity chromatography will be done through column chromatography. First of all, the binding ability of protein must be studied. Then, the solid medium modified with the binding material is packed in a chromatography column. Then, the initial mixture that contained desired proteins was added through the column to allow binding to occur. A wash buffer was gradually added to the addition of mixture. The elution buffer subsequently removes unbounded protein from the column and collected.
There is no generally applicable elution methods for all affinity media. When substances are very tightly bounded to the affinity medium, it may be useful to stop the flow after applying eluent, usually 10 minutes to 2 hours is referred, before continuing the elution process. This extra time helps to improve recovery percentage of bounded protein.
Forces that maintain the complex of substrate and bound substances include electrostatic interactions, hydrophobic interactions, and hydrogen bonding. Agents that deteriorate these interactions may be expected to function as efficient eluting agents. The optimal flow rate to achieve efficiency may vary according to the specific interaction.
This is one of most common techniques that are used to remove bounded protein from the ligands. A change in pH alters the charged groups on the ligands and/or the bound protein. This change may directly affect the binding sites and reducing their affinity. On the other hand, a change in pH can cause indirect modification in affinity by altering in conformation of proteins. A sudden decrease in pH is one of the most common methods to elute bounded proteins. The chemical stability of the ligand and target proteins determines the limitation of pH change. The column should always return to neutral pH immediately after the elution to avoid irreversible denature of proteins.
Changing ionic strength of buffer solution will alter the specific interaction between the ligand and target protein. This method is a mild elution using a buffer with increased ionic strength usually sodium chloride, applied as a linear gradient or in steps.
Selective eluents are often utilized to separate substances on a specific medium or in the presence of high binding affinity of the ligand/target protein interaction. The eluting agent competes either for binding to the target protein or for binding to the ligand. This is an example of competitive inhibitors that occur in nature. Substances may be eluted either by a concentration gradient of a single eluent.
In this method, the concentration of competitive agents should be added equally to the concentration of the coupled ligand. However, if the free competing compound binds more weakly than the ligand to the target protein, use a higher concentration of competitive agent to achieve efficiency in elution.
For example of competitive affinity chromatography, There is R1a protein. The target R1a protein bind to cAMP resin. The interaction between R1a protein and cAMP would separate by using cGMP elution buffer. This cGMP compete with target protein, however the elution buffer which contains high concentration of cGMP would bind to resin more. The separated R1a protein will eluted out.
Conditions are used to lower the polarity of the eluent promote elution without inactivating the proteins. Dioxane or ethylene glycol are typical of this type of eluent.
In case of other elution methods fail, deforming buffer solution, which alters the structure of proteins, can be used to achieve separation of ligand and target proteins. Typical chaotropic agents are guanidine hydrochloride and urea. Although this method will yield the highest percentage of recovery, chaotropes method should be avoided whenever possible since they are to denature the eluted protein.
Affinity chromatography can be performed using a number of different protein tags. One of the common tag using in laboratory is poly-hisitidine. Its shortness in length prevents altering the conformation of the tagged protein. Histidine tagging is favorable because it is very specific, allowing for a high level of purification.
The gene which encodes for a specific protein is first modified to include the tag. A string of histidine residues may be added to the amino or carboxyl terminus of the expressed protein. The tagged proteins are then passed through a column of beads containing covalently attached, immobilized nickel(II) (Ni 2+.) This His-tag binds tightly to the immobilized metal ions because the side chain of Histidine, imidazole, has a specific binding affinity to metal ions (in this case, nickel II). As a result, the desired protein is binded tightly to the beads while other proteins flow through the column easily. Even other, non-desired proteins, that have Histidine side chains will flow through because they do not have as many as the desired, tagged protein, which would have about 6 adjacent Histidine residues. The protein can then be eluted from the column by addition of imidazole or some other chemicals that bind to the metal ions and displace the proteins. The presence of desired proteins can be verified through enzyme-linked immunosorbent assay (ELISA).
Histidine
Nickel resin regeneration
In recombinant DNA, histidine tag on the desired protein and Nickel resin are commonly used to purify desired protein via affinity chromatography. That is, histidine has strong affinity towards the nickel resin which does not flow through the column. Undesired proteins do not have the designed histidine sequences hence could not bind to Nickel resin; those protein flow though the column. During elution, we add a relatively high concentration of imidazole buffer. Imidazole compete with our desired protein to bind with the nickel resin.
In practice, Nickel resin is rather expensive. Regeneration of Nickel resin is essential. It involves several steps. First, there are possible left over protein remained on used Nickel resin; these left over protein are denatured and washed away using Guanidinium chloride and corresponding buffer. The Nickel resin is washed with Milli Q water and increasing concentration of Ethanol. One essential step in Nickel resin regeneration is recharging the Nickel. We first remove the Nickel with EDTA, which is a hexa-dendate compound that releases the Nickel ion. The Resin would then turn white without Nickel. Then the resin is recharged with high concentration of nickel salt to obtain our slightly green resin.
GST has an affinity for glutathione, which is available immobilized as glutathione agarose. An excess amount of gluthione is used to displace the tagged protein for elution. Together with the histidine tags, the purification of recombinant proteins like GST tags is the most common use of affinity chromatography.
Glutathione
GST are enzymes involving cellular defense against electrophillic compounds. It has hing affinity and specificity to bind with glutathione. The strength and selectivity of this interaction allow GST tagged proteins to be purified by the glutathione-based protein resins. The glutathione resins selectively bind to GST-tagged proteins effectively, allowing the specific protein of interest be separated from the mixture at high efficiency.
GST is a 35-KDa protein, it has small peptides. It is this characteristic which allows one to perform GST-protein purification quickly without degradation by proteases and minimize sample loss.GST will lose its ability to bind Glutathione resin when it is denatured, therefore, strong denaturant such as Guanidine-HCl and urea cannot be added in the buffers.
Lectin protein, for example concanavalin A which is originally extracted from the jack-bean Canavalia ensiformis, binds specifically to some certain structures in sugars. Lectin affinity chromatography is one kind of affinity chromatography in which the plant protein concanavalin A is purified by passing a crude extract through a column of beads containing covalently attached glucose residues. Since it has affinity to glucose, concanavalin A will bind to this type of column. A concentrated solution of glucose is then added to remove the bound concanavalin A from the column.
• Affinity chromatography is a fairly achievable technique because of the great selectivity of the glucose residues and the target protein, giving purified product with a high yield of recovery.
1• It can be a one step process in many cases.
2• The technique can be used for substances of low concentration.
3• Rapid separation is achieved while avoiding contamination.
4. Unlike Gel filtration chromatography and ion-exchange chromatography, affinity chromatography would be able to isolate one specific protein at a time, where other techniques will isolate proteins with similar characteristics.
• The interaction of proteins of interest and ligand has to be determined carefully. This process required expensive materials, time, and small amount of protein that can be processed at once.
Hydrophobic Interaction Chromatography (HIC) (or Hydrophobic Chromatography) is a method of separation by using salt gradients (i.e. ammonium sulfate) to generate hydrophobic interactionsbetweenprotein and the ligands on the solid phase support resin [1]. The purpose of this type of chromatography is to utilize the hydrophobic properties of specific proteins rather than their charges, which is used in ion-exchange chromatography. Therefore, the more hydrophobic a protein is, stronger it will cling to the column and elution proceeds with the least hydrophobic proteins emerging first from the column. The salt gradient is important because it increases hydrophobic interaction and stabilizes proteins [2]. During elution, other factors besides hydrophobicity still affect how proteins separate, such as ionic interactions, pH, temperature, salt concentration, solvent amount, buffer conditions, etc. These attributes also point to the similarities between HIC and reverse phase chromatography and affinity chromatography [3]. It is important to note that HIC is advantageous because it can be prepared for specific proteins and applied to different facets of protein purification. Conditions may be altered in minor ways to apply the test to many other situations for purification and study purposes, especially in cell membrane studies.
Column chromatography is another method used to separate proteins or molecules from each other. It is essentially an upside-down version of TLC (Thin Layer Chromatography) - relying on the same physical principles, except that while TLC is driven by capillary forces for moving the solvent, column chromatography allows gravity to drive down the eluent. In this method, a sample to be separated is applied to the top of a glass column. the glass column is then packed with a solid phase. The purpose of this solid phase is to separate the compounds in the sample into different zones. Silica gel (SiO2) and alumina (Al2O3) are common adsorbents. The one expected to percolate out of the column first is the component that has the least interactions with the silica gel, so therefore the one that is least polar. The eluent carries the soluble compounds with it. When the column is packed with Silica Gel, the band expected to percolate out of the column first is the component that has the least interactions with the silica gel, so therefore the one that is least polar. The eluent carries the soluble compounds with it. The polarity of the eluent can be progressively increased from a nonpolar solvent to a polar solvent because as the nonpolar component is collected first, the bands of components left in the column are more polar. A more polar solvent would be more efficient to carry the polar component left in the column. After column chromatography is used to separate the mixture according to their respective polarities, Thin Layer chromatography should be used to separate the mixtures to observe the fractions separation and combine any that have “climbed” the same distance. The compounds found to have the same polarities using TLC are combined and analyzed by taking their respective melting points and comparing them to literature values. The amount of sample is used to figure out the initial concentration of each in the original sample. If the compound is colored this is easy, however if the compound is a clear solution then the plates can be CAM stained (Iodine can also be used) or put under UV to track their location. After these bands are collected, the solution can be put under a rotovap to evaporate off the solvent and a clean compound can be obtained. If the solvent is volatile, it can be evaporated in the hood or over night. The samples can also be heated in a sand bath.
Once the column is packed with dry stationary adsorbent material (such as silica gel), there are generally two methods to load column chromatography: wet loading and dry loading. The ability of the mixture to dissolve in a polar or non polar solvent determines the method of the column chromatography.
dry Column Chromatography separation
In wet loading method, the adsorbent is suspended in solvent and the slurry is transferred into the column as the eluent. This method is most commonly used when the desired separating mixture is soluble in the least polar solvent or a non-polar solvent. If excessively polar solvent is used, then it will stay inside the column and increase local polarity, which can mix the separation on the column.
In dry loading method, mixture is first dissolved in a minimal amount of solvent and the adsorbent material. Once the solvent mixed with the mixture and adsorbent is evaporated, the dried compound can be added into the column. After the addition of the dried compound, the column is flushed with mobile phase (can be polar solvent with various polarity, but they should be added with increasing polarity), and the column is not allowed to run dry after the addition of mobile phase. This method is most commonly used when the mixture is only soluble in solvent that are more polar than the eluent of choice.
The chemical compounds are separated and collected within the column. The separated sample can then be tested for purity and other properties. As the sample is applied to the top of the column, it is also washed with a solvent. As the sample moves over the solid phase in the column, the different molecules or compounds in the sample will begin to separate from each other into zones. The compounds in the sample will bind to the solid phase, but then the sample will also release from the solid phase and then bind to the liquid solvent that passes over it. This is a continuous process. A compound will bind to the solid phase, then release and bind to the solvent. it will then rebind back to the solid phase, and again rebind to the liquid solvent. This process keeps occurring as the compound moves down the column. Different molecules in the sample will have a different binding affinity to the solid phase or the liquid phase, these differences in affinity is what allows the molecule in a mixture to travel at different speeds and separate from the other compounds.(NOTE :*The preceding section was a description of column chromatography for organic chemistry.) This method is sometimes called reverse phase chromatography in biochemistry.
The factors that determine the distance a compound travels are 1) the interaction between the solvent and adsorption layer, 2) the interaction between the solute and adsorption layer, 3) the polarity of the solute, solvent, and adsorption layer, and 4) the weight of the solution. To determine the distance a compound travels one may calculate the retardation factor (Rf). The Rf value can be found by looking at a TLC plate that is spotted with the fractions collected after running a column. The Rf value is the ratio of the distance traveled by the solute over the distance traveled by the solvent. The range of Rf is 0 to 1. If the calculated Rf value is higher than desired, then a less polar solvent should be used when running the column.
Organic Chemistry Laboratory third edition with Qualitative Analysis,By Bell Jr, Charles E; Taber, Douglas F.; Clark, Allen K. Harcourt College Publisher
Planar chromatography is one type of chromatography technique in which the stationary phase is on a flat plate and the mobile phase moves through stationary phase due to capillary action. This technique was used to separate the mixture. There are two types of planar chromatography:
A. Thin layer chromatography TLC.
B. Paper Chromatography
Though this is a different kind of chromatography, it still separates mixtures of substances into the individual components, molecules, even atoms. The size and concentration of the component is determinant of the component's rate. The stationary phase, which is either a solid or a liquid supported by a solid, is absorbed in a uniform manner in paper chromatography. On the contrary, the mobile phase, being gas or liquid, serves as the solvent. Compounds can travel as far as the solvent does when the paper is dipped in a container filled with solvent. These compounds travel at different rates and separated into distinctly colored dots on the paper. The solvent that is used can be either nonpolar or polar. These properties affect the solubility of the compounds and components in the particular mixture. Polar components will be attracted to the water molecules attached to the cellulose (paper) and not attracted to a nonpolar solvent. The chromatogram will not contain the polar components, given that it doesn't climb up the paper with the nonpolar solvent. These components spend more time in the stationary phase rather than the mobile phase therefore the rate of moving up the paper is slow. If it were the opposite and nonpolar components were in a polar solvent, then the same thing will occur. The mobile phase can be various organic solvents or mixture. The compound can be stained with iodine in order to visualize where they have traveled easily. [1]The stationary phase can be called a paper chromatogram. Usually, one will split the paper into individual lanes so that multiple trials can be done with one paper. Also it will allow the experimenter to compare the differences or similarities present in each lane depending on how far the compound has traveled. [1]
The paper is placed in a container with a shallow layer of a suitable solvent or mixture of solvents in it. Sometimes the paper is just coiled into a loose cylinder and fastened with paper clips top and bottom. Then the cylinder stands in the bottom of the container.
The container is covered to make sure that the atmosphere in the beaker is saturated with solvent vapor. Saturating the atmosphere in the beaker with vapor stops the solvent from evaporating as it rises up the paper.
As the solvent slowly travels up the paper, the different components of the ink mixtures travel at different rates and the mixtures are separated into different colored spots.
The distance travelled relative to the solvent is called the Rf value. Its formula is: Rf = distance traveled by compound (a.k.a. the solute) / distance traveled by solvent. Thus, the higher the Rf value, the further the compound has traveled up the paper. The main benefit of the Rf value is that we can now compare values similar values and conclude that they are indeed the same compound[1]
Thin layer chromatography (TLC) is an extremely valuable technique in the organic lab. It is used to separate mixtures, to check the purity of a mixture, or to monitor the progress of a reaction. The polarity of the solute, polarity of solvent, and polarity of adsorbent are crucial factors that determine the mobility rate of a compound along a TLC plate. This technique helps separate different mixtures of compounds based on their mobility differences. TLC can also be used to identify compounds by comparing it to a known compound
Thin layer chromatography (TLC): this technique was used to separate dried liquids with using liquid solvent (mobile phase) and a glass plate covered with silica gel (stationary phase). Basically, we can use any organic substance (cellulose polyamide, polyethylene, etc.) or inorganic substance (silica gel, aluminum oxide, etc.) in TLC. These substances must be able to divide and form uniform layers. On the surface of the plate, will be a very thin layer of silica which is considered the stationary phase. Then, add a small amount of solvent into a wide-mouth container (i.e. beaker or developing jar) just enough to cover the bottom of the container. Place the prepared TLC plate into the sealed container which has small amount of a solvent (moving phase). Due to capillary action, the solvent moves up to the plate and now we can remove the plate and analyze the Rf values.
Usually TLC is done on a glass, plastic, or aluminum plate coated with silica gel, aluminum oxide, or cellulose. This coating is called the stationary phase. The sample is then applied to the bottom of the plate and the plate placed in a solvent, or the mobile phase. Capillary action pushes the sample up the plate. The rate the samples move up the plate depends on how tightly the sample binds to the stationary phase. This is determined by polarity. The Rf values or the Retention Factors are then compared for analysis. The retardation factor of a solute is defined as the ratio between the distance traveled by a compound to that of the solvent in a given amount of time. For this reason, Rf values will vary from a minimum of 0.0 to a maximum of 1.0. However, this retardation factor for a given protein compound will vary widely with changes in the adsorbents and/or solvents utilized. In addition, the retardation factor can vary greatly with the content of moisture in the adsorbent. The Rf values or the Retention Factors are then compared for analysis. This Rf value can be quantified as such:
Rf = (Distance that compound has traveled)/ (distance that the solvent has traveled)
A light pencil line is drawn approximately 7 mm from the bottom of the plate and a small drop of a solution of the dye mixture is placed along the line. To show the original position of the drop, the line must be drawn in pencil. If it was drawn in ink, dyes from the ink would move up the TLC plate along with the dye mixture and the results would not be accurate. In order to get more accurate results, dot the TLC paper with the dye mixture a few times trying to build up material without widening the spots. A spot with a diameter of 1 mm will give good results. While dotting the TLC plate, be sure to not dot mixtures too close to one another because when the dye mixture rises up the TLC plate, it will clash with the other spots and the Rf values will be difficult to calculate.
When the spots are dry, the TLC plate is placed in a beaker, with the solvent level below the pencil line. Cover the beaker to ensure that the atmosphere in the beaker is saturated with solvent vapor. Line the beaker with some filter paper soaked in solvent because this will help in the process of separating the mixture. Saturating the atmosphere in the beaker with solvent vapor stops the solvent from evaporating as it rises up the plate.
As the solvent slowly travels up the plate, the different components of the dye mixture travel at different rates and the mixture is separated into different colored spots. The solvent is allowed to rise until it approximately 1-1.5 cm from the top of the plate. This gives the maximum separation of the dye components for this particular combination of solvent and stationary phase.
Once the maximum separation of the dye components for this particular solvent and stationary phase solvent is induced, the TLC plate is removed from the beaker and allowed to dry. Immediately after removing the TLC plate, use a pencil to mark the solvent front before the solvent begins to evaporate. The solvent front is the line where the solvent rose up to on the TLC plate. Then, let the solvent evaporate from the TLC plate. The separated compounds are circled/marked to indicate their position on the plate. In some cases, the compounds that have traveled up the TLC plate do not give off any noticeable appearance with the naked eye. In such cases, the TLC plate can be dipped briefly in a visualizing solution containing certain reagents that will react with the separated compounds to form a colored compound upon heating. Another way to visualize colorless organic compounds separated on a TLC plate is by placing them in iodide (I2) vapor to test their absorption of iodide vapor. These TLC plates with colorless marks are placed in a bath of iodine vapor prepared by placing a small amount of iodine crystals in a tightly capped jar. Colorless spots gradually gain a dark brown color after placing the TLC plates in the bath for approximately 10 minutes. For the reason that the colored spots usually disappear in a short period of time, they are outlined immediately with a pencil after the TLC plate is taken out of the iodine bath.
In addition to the visualization technique of an iodine bath, a fluorescent indicator can also aid in helping to determine the distance in which the separated compounds had traveled. A short- wave ultraviolet lamp is used to illuminate the adsorbent side of the plate in a darkened room/ area. Many compounds will decrease the intensity of the fluorescent. Using this UV light visualization technique, the separated compounds appear as dark spots on the fluorescent TLC plates. It is often easier to visualize the darkened spots with 365-nm light. These dark spots are outlined with a pencil while the plate is under the UV light source to give a permanent record of the location in which the analyzed compounds had traveled.
Some examples of interpretation of TLC plates under UV light:
1. TLC gives useful qualitative results and interpretations. For example, if an individual wants to compare the components in an unknown mixture to standard compound A and B, TLC can be ran and if the dark spots for unknown under UV light aligns with those of compound A and B, the unknown contains both A and B.
Example 1
2. If there is only one dark spot for the unknown and it is uncertain whether the spot for compound A is at the same level as the spot for the unknown, one can co-spot both compounds on the TLC plate for a quick check. Co-spot means to spot compound A on one area of the TLC plate and spot the unknown on the same area as the spot of compound A. If there is only one dark spot under the UV light for the co-spotting lane, the identity of unknown is A.
Example 2
The co-spot result for example 1 should contain only 2 spots where one spot represents compound A + one component in the unknown, and the other spot represents another component in the unknown mixture. Extra spots may indicate that one of the components in the unknown does not match with the standards.
3. How can someone tell the reaction between A+B actually occurs to give a new product C?
TLC can be used to check. Compound A and B are spotted on a TLC plate separately. The mixture of A+B (C ) is then spotted on the TLC plate and after each time period a new sample can be spotted (C2, C3, and so on).
Two spots on C1 align with A and B suggests just a mixture of A+B, not a new product. C2 and C3 still have one spot aligning with reactant A, but C4 has both spots that do not match with either reactant, where C5 only has one dark spot.
There are two possible interpretations:
1) C2 to C4 are intermediates to the new product in C5,
2) the desired product is actually C4 and it degrades to just having one component on the plate.
Example 3
Tips in a lab:
1) A capillary tube is used to transfer solution onto the TLC plate. Smaller origin spots will give smaller area and better separation of dark spots under UV light and this will make calculation of Rf easier and more accurate.
2) The container with TLC plate and solvent should always be on a flat surface in order to get a "straight lane" for the run.
The effect of increasing the polarity of a solvent, this leads to a greater separation
As you might already know, the TLC plates are made of silica gel, which is a polar compound, and is the reason why non-polar compounds tend to have a great separation on TLC plates.
As shown in the diagram, initially, the solvent used consisted of a 7:3 ratio of hexane to hexyl acetate. This means that a majority of the solvent reacting with the TLC plate will be nonpolar.
Due to the lack of polarity of the solvent, there is less competition between the spotted samples and the TLC plate, thus, the polar parts of the sample will readily react with the silica gel leading to less of a separation. Because there is nothing 'hindering' the sample from reacting with the silica gel, it reacts right away and its separation is 'bogged down.' Think of a dog walking down a pathway, if the dog stops to sniff at every tree on the way, its distance separated from the beginning is less than if it had just kept walking without being distracted by the surroundings. This is the same with these samples, if they are constantly reacting with the silica gel as they are moving they will not move as far.
Now when the ratios are switched, and there is more of the hexyl acetate(more polar), then all of a sudden there is competition for the reacting with the TLC plate. The sample wants to react with the TLC plate, but so does the solvent(since it is now more polar), thus there will be less reaction of the sample with the TLC plate. Obviously the solvent is trying to react with the TLC plate, leading to the sample not getting as much of a chance to "stop and sniff" so it is separated further. The sample reacts less with the TLC plate because now there is the solvent reacting with the same TLC plate, and this explains why there is a greater separation.
Now the 3d TLC plate in the diagram is a bit tricky. One might think that petroleum ether would be semi-polar due to the name(it has ether in it), but actually petroleum ether is a non polar compound which consists of many hydrocarbon molecules. This will not lead to any different separation.
Gas Chromatography Diagram.
Gas Chromatography is common type of chromatography which is used to analyze or separate volatile components of a mixture. This technique helps us to test the purity of a particular substance or separate different components of a structure. Basically, the mechanism of this technique is carried out by injecting syringe needle which contains a small amount of sample into the hot injector port of gas chromatography. The injector is set to the temp that is higher than the boiling points of the components so that the components will be evaporated into gas phase inside the injector. The carrier gas (normally is Helium) then pushes the gaseous components into gas chromatography column. The separation of components occurs here, form partition between mobile phase (carrier gas) and stationary phase (boiling liquid). More interestingly, gas chromatography column showed what’s inside, the maximum temperature along with the length and diameter due to the presence of metal identification tag on the column. Additionally, the column temperature is raised by the presence of heating element. The detector inside the gas chromatography will recognized the differences in partition between mobile and stationary phases. The molecules reach the detector, hopefully, at different intervals depending on their partition. The number of molecules that regenerate the signal is proportional to the area of the peaks.
Although gas chromatography has many uses, GC does have certain limitations. It is useful only for the analysis of small amounts of compounds that have vapor pressures high enough to allow them to pass through a GC column, and, like TLC, gas-liquid chromatography doesn't identify compounds unless known standards are available. Coupling GC with a mass spectrometer combines the superb separation capabilities of GC with the superior ID methods of mass spectrometry. GC can also be combined with IR spectroscopy. IR can help to identify that a reaction has gone to completion. If the functional groups of the product are depicted in the IR, then we can be sure that the reaction has gone to completion. This can also be depicted in the GC analysis. The presence of peaks that do not correlate with the standards may be due to an incomplete reaction or impurities in the sample.
The basic parts of a GC machine are as follows:
Source of high- pressure pure carrier gas
Flow controller
Heated injection port
Column and column oven
Detector
Recording device
A small hypodermic syringe is used to inject the sample through a sealed rubber septum or gasket into the stream of carrier gas in the heated injection port. the sample vaporizes immediately and the carrier gas sweeps it into the column. The column is enclosed in an oven whose temperature can be regulated. After the sample's components are separated by the column, they can pass into a detector, where they produce electronic signals that can be amplified and recorded.
The steps need to be followed to use Gas Chromatography:--Cherryblossom06 (discuss • contribs) 06:02, 22 November 2012 (UTC)
1. Wash syringe with acetone by filling it completely and pushing it out into a waste paper towel.
~Possible errors that can occur during Gas Chromatograpy can be due to the improper rinsing of the syringe. The syringe should be rinsed twice with acetone and once or twice with the sample. If improper rinsing ensues, unknown peaks can occur and
alter our analysis of the sample. This error can be easily avoided. About 1 micro liter of sample is needed.
2. Pull some sample into the syringe. Air bubbles should be removed by quickly moving the plunger up and down while in the sample.
3. Turn on chart recorder, adjust chart speed in cm/min, set baseline by using zero so that the baseline is 1 cm from bottom of chart paper ( set 0), turn on the chart.
4. Inject sample into either column A or column B and push the needle completely into the injector till we can’t see the needle, then we pull the syringe out of the port.
5. Mark the initial injecting time on the chart.
~The sample should be injected at exactly the same time as the 'start' button is pressed. Otherwise, take note of how long after injection recording started. If the sample is not injected at the exact time the button is pressed, retention times will be off in the calculations.
6. Clean the syringe immediately.The syringe should be rinsed with acetone before injecting a different sample. Rinse before any other sample is injected and after every sample.
7. Record current (in milliamperes), temperature (in Celsius).
Notes on Injection:
1. The injection site, the silver disk, is very hot.
2. The needle will pass a rubber septum so there will be some resistance. Some machines have a metal plate near the septum, so if there feels like metal resistance, the needle should be pulled out and tried again. The needle should be completely inserted into the injection point if done correctly.
3. Quick injection is needed for good results.
4. Take out the needle immediately after injection.
Liquid Chromatography is a separation technique in which the mobile phase is a liquid. This technique can be done on either a column or a plane. Nowadays liquid chromatography is done by high performance liquid chromatography.
In High Performance Liquid Chromatography, the sample is forced by the mobile phase, a liquid at high pressure, through a stationary phase column that is irregularly packed, has spherically shaped particles, or a porous monolithic layer.
Isoelectric point, also called the pI of the protein, is the pH at which the net charge of the protein is zero. Isoelectric focusing is a separation technique which separates peptides according to their isoelectric point, or how acidic and basic their residues are. A gel with a pH gradient is used as the medium. The pH gradient is made by adding polyampholytes, which are multi-charged polymers, with different pI into the gel. Then the sample is put onto the gel and a voltage is applied. The proteins will move along the gel until they reach their isoelectric points. In other words, each protein will move until it reaches a position in the gel at which the pH is equal to the pI of the protein. a protein band that forms at a given pH can then be removed and analyzed further. This process can successfully separate proteins that have a difference in net charge greater than or equal to 1.
Isoelectric point (pI): The pH at which the net charge on the protein is zero. For a protein with many basic amino acids, the pI will be high, while for an acidic protein the pI will be lower.
Isoelectric focusing is a type of zone electrophoresis, and it is usually performed in a gel, that takes advantage of the fact that a molecule's charge changes with the pH of its surroundings. A protein that is in a pH region below its isoelectric point (pI) will be positively charged and so will migrate towards the cathode. As it migrates, however, the charge will decrease until the protein reaches the pH region that corresponds to its pI. At this point it has no net charge and so migration ceases. As a result, the proteins become focused into the sharp stationary bands with each protein positioned at a point in the pH gradient corresponding to its pI. This technique is capable of extremely high resolution with proteins differing by a single charge being fractionated into separate bands.
Molecules to be focused are distributed over a medium that has a pH gradient (usually created by aliphatic ampholytes). An electric current is passed through the medium, creating a "positive" anode and "negative" cathode end. The negatively charged molecules migrate through the pH gradient in the medium toward the "positive" end while positively charged molecules move toward the "negative" end. As a particle moves towards the pole opposite of its charge it moves through the changing pH gradient until it reaches a point in which the pH of that molecules isoelectric point is reached. At this point the molecule no longer has a net electric charge (due to the protonation or deprotonation of the associated functional groups) and as such will not proceed any further within the gel. The gradient is initially established before adding the particles of interest by first subjecting a solution of small molecules such as polyampholytes with varying pI values to electrophoresis.
The method is applied in the study of proteins, which separate based on their relative content of acidic and basic residues, whose value is represented by the pI. Proteins are introduced into an immobilized pH gradient gel composed of polyacrylamide, starch, or agarose where a pH gradient has been established. Isoelectric focusing can resolve proteins that differ in pI value by as little as 0.01. Isoelectric focusing is the first step in two-dimensional gel electrophoresis, in which proteins are first separated by their pI and then further separated by molecular weight through SDS PAGE.
We can determine pI of each amino acid when we know its pKas by titration with NaOH. For example, glycine, the smallest amino acid, has two pKa values, which are 2.34 and 9.60, respectively.[14]
First, add strong acid and let glycine to become complete protonated form. Then gradually add NaOH until pH raises up to 2.34. At this point, we use 0.5 mol of NaOH equivalent to first protonated form of glycine. Also, There would be 0.5 mol of second protonated form generated in the solution. After using 1 mol NaOH equivalent to first pronated form, there would be solely second pronated form. We'll see that second protonated form of glycine is zwitterion, which is zero net charge molecule. Therefore pH at this point is called isoelectric point (pI) and equals 5.97. Continue adding NaOH once pH equals 9.60. At this point, 0.5 mol of third protonated form is present in the solution and total amount of NaOH is 1.5 mol. Back to pI, we see that
Then we can write in the general form:
Remark To determine pI of amino acid which has more than two pKa, we'll use two pKa values covering the range in which zwitterion would present in the solution.[15]
This technique is not usable because it takes time and not work well, not good for big molecules. Thus, it is not popular technique to use.
In dialysis a semipermeable membrane is used to separate small molecules and protein based upon their size. A dialysis bag made of a semipermeable membrane (cellulose) and has small pores. The bag is filled with a concentrated solution containing proteins. Molecules that are small enough to pass through the pores of the membrane diffuse out of the bag into the buffer solution, or dialysate. Dialysis is sometimes used to change buffers. The molecules go from an area of high concentration to low concentration. When the level of concentration is equal between the bag and the buffer, there is no more net movement of molecules. The bag is taken out and inserted into another buffer, causing the concentration to be higher in the bag relative to the buffer. This causes more diffusion of molecules. This process is repeated several times to ensure that all or most of the unwanted small molecules are removed (usually done overnight). In general, dialysis is not a means of separating proteins, but is a method used to remove small molecules such as salts. At equilibrium, larger molecules that are unable to pass through the membrane remain inside the dialysis bag while much of the small molecules have diffused out.
The technique of dialysis is used in everyday life for hospital usages. Dialysis mimics one of the functions of a bodily organ, the kidneys. It is used in procedures to filter out the blood's toxins and waste products during kidney failure. During kidney failure, there is a build up of nitrogen-containing waste products (such as urea or creatine) in the body called azotemia, which can be detected from the blood. Patients result to a dialysis when the waste product accumulates on the blood causes metabolic acidosis leading to illness. Two tests are executed through a blood sample and a full day's worth of urine sample. There are two chemicals in the blood that are measured, the urea nitrogen level and the creatinine level. If these two chemicals are found to be high in the blood, then it is an indication that the kidneys are not cleansing bodily waste products efficiently. Certain solutes such as potassium and calcium are carefully calibrated at a concentration similar to the concentration of healthy blood. Another solute is Sodium Bicarbonate which is used as a pH buffer introduced by elevating the solute concentration within the dialysis to neutralize some of the metabolic acidosis occurring within the blood.
Protein Purification is the process of separating proteins for individual analysis. Protein purification is the second step of studying proteins, the first being the process of an assay. An assay is a procedure to measure the activity enzyme activity thus confirming the presence of the protein or proteins in interest. Popular assays include Western Blotting and ELISA(Enzyme-linked immunosorbent assay). Before the purification process, Cell Disruption is utilized to homogenize the cell's content. After the cell has been opened up, the process of purifying proteins from one another and the other organelles can be approached in several different methods. Protein mixtures are normally separated multiple times, each based on a different property, such as:
Solubility
Size
Molecular Weight
Charge
Binding affinity
The intended reason for purifying a specific protein governs the level and degree of protein purification. At times, a sample of protein that is only moderately purified suffices for its intended application; however, other situations require a higher degree of purification, especially if the fundamental ambition is to study the characteristics and tendencies of the specific protein in interest. By considering solubility, size, molecular weight, charge, and binding affinity, the goal of the scientist that conducts protein purification is to find a level of purification necessary and create a protein yield that is ample for further research and application. This means using the fewest steps in order to keep the yield high, as each protein purification step incurs a degree of product loss. Therefore two factors serve as obstacles in protein purification: yield and purification level. The main goal of each protein purification project falls under two categories: analytical (for studying and research purposes) and preparative (for production and creation of commercial products).
Proteins are separated based on masses or densities by a centrifugal force. Centrifugation enables the separation of proteins in different cell compartments.
Different proteins precipitate at different salt concentration. When the concentration of salt increases, more proteins are able to separate
Large molecules flow more rapidly to the bottom of the column.
Proteins are separated according to its charge. Positively charged proteins bind to negatively charge bead, and negatively charge proteins are released. The negatively charged proteins flow through faster.
Many proteins have high affinity for specific chemical groups.
Proteins separate according to different levels of hydrophobicity.
Electrophoresis separate protein while the gel enhances the separation. Small proteins move more rapidly through the gel.
Different proteins have different pI (isoelectric point).
Proteins are separated horizontally based on pI and vertically based on mass.
Proteins are separated through a semi-permeable membrane. Since the dimensions of proteins are generally larger than the pores of the membrane, proteins do not pass through and separate.
After each purification steps, the types of protein that exist in the solution is expected to decrease while its specific activity is expected to increase. These two qualities are desirable because experiment done using a pure protein sample gives a more quantifiable result. One method used to check the purity of the sample is using a form of Gel Electrophoresis, such as SDS PAGE or native PAGE.
Purification can also be quantitatively evaluated by measuring total protein, total activity, specific activity, yield and purification level. Total protein is the quantity of protein present in a fraction and can be determined by measuring the protein concentration of a part of each fraction and multiplying by the fraction's total volume. Total activity is measured by the enzymatic activity in the volume of fraction used in the assay multiplied by the fraction's total volume. Specific activity is the total activity divided by total protein. The yield is the amount of activity retained after each purification step. The purification level is the increase of purity which can be measured after each purification step by dividing its specific activity by the specific activity of the initial extract.
a good purification takes into account both purification levels of yield. A high amount of purification and a poor yield give little protein to work with. on the other hands, a low purification and a high yield give contaminated protein in the experiment.
After purification is complete, how will you prove that you have successfully isolated the correct protein? Several techniques can be used to identify whether or not the isolated protein is the desired one, including immunological reactions.
Overview
Millions of antibodies are produced by the body, with each one tailored to recognize specific protein structures. The "Y" shaped antibody recognizes protein structures through its binding site, which is able to attach to antigens with the perfect fit by forming intermolecular bonds. After being exposed to a pathogen, organisms can churn out several different antibodies that will recognize this same pathogen for every subsequent exposure. These polyclonal antibodies attach to different areas on the same pathogen to counteract mutations that change a pathogen's surface proteins and render a specific antibody recognition site obsolete.
Monoclonal Antibodies
Though useful from an organism's standpoint, polyclonal antibodies prove to be messy and inefficient in the lab because the body does not produce them in exact ratios. Different antibody samples would consist of different relative amounts of several antibodies, each of which attach differently to the protein product. So how can a researcher force a model organism to create only one type of antibody for a particular protein? The solution was discovered by Cesar Milstein and Georges Köhler, who mixed anti-body producing cells with immortal cancer cells (Meyloma cells) capable of mass producing identical proteins over and over again. The hybrid cells capable of producing the desired antibody could then be selected and grown in mass culture or within the model organism itself as tumors.
Enzyme-linked Immunosorbent Assay (ELISA)
There are two types of ELISA, "Indirect" and "Sandwich." Both use a specific antibody to recognize the desired protein. This first antibody must be specially produced for each and every different protein. After unbound antibodies or proteins are washed away, a second antibody that contains an enzyme capable of producing a visual confirmation that the isolated protein is present is introduced to solution. This second antibody is a generic antibody that can be used regardless of the specific protein.
Indirect ELISA: 1) A container is coated with protein.2)The first antigen, specific to the protein, binds to the protein. 3)The container is washed. If the desired protein is not present, the antibodies will not bind and will be removed from solution. 4)The second antibody with an enzyme is added and binds to the first antibody. 5)Binding to the first antibody induces a chemical reaction that causes a visually identifiable change in solution (color change or fluorescence), indicating that the first antibody is present, which in turn indicates that the desired protein is also present. SEE FIGURE 1.
Figure 1. Basic Indirect ELISA steps.
Sandwich ELISA: 1) A container is coated with the monoclonal antibody. 2)The protein is added and will bind to the antibody only if it is the desired protein. 3) The container is washed. Only the desired protein and antigens will remain (if any). 4) A second antibody linked to an enzyme is added and will attach to the protein. 5) Attaching to the protein will induce a chemical change that allows for visual confirmation that the protein is present. Note that since the second enzyme is attaching directly to the protein, the rate of visual change can be used to determine the amount of protein present. SEE FIGURE 2
Figure 2. Basic Sandwich ELISA steps.
Western Blotting
1) After separating the desired protein from other proteins or molecular impurities via gel electrophoresis, the resulting protein bands are transferred from the gel to a thin polymer sheet. This makes the proteins more accessible to reactions. 2) The monoclonal antibody is added. Only the desired protein will react with the antibody, so only one band will have antibodies attached. 3) The polymer sheet is washed to remove unbound antibodies. 4)A second antibody linked to an enzyme attaches to the first. 5) A chemical reaction induces a visual change in the band containing the desired antibody. Or photographic film can overlay the sheet and record the protein band that contains the attached antibodies. SEE FIGURE 3.
A sample protein is dotted on the marked center of a cellulose acetate strip and the strip is placed in barbital buffer of a desired pH and voltage is applied across the strip. The proteins that migrate towards the anode have a pI greater than the pH of the buffer while proteins that migrate towards the cathode have a pI less than the pH of the buffer. Positively charged proteins migrate towards the cathode while negatively charged proteins migrate toward the anode.
Cellulose acetate electrophoresis can be useful in identifying multimeric proteins formed by different isoforms since each ratio of isoforms will have a different charge due to the different amino acid structure.
Knowing the quantity of a protein after each separation step is useful in checking the progress of purification and evaluating the technique's efficiency. Quantifying proteins also helps us understand how an organism functions as one. Severalchromatographytechniques rely on quantifying proteins by mass, with additional observables such as charge to provide further differentiation.
Because specific activity is a ratio of the enzymatic reactions of a particular protein to the total amount of proteins, quantifying a protein can be followed throughout a purification. The equation for specific activity can be modeled as: . Therefore, as the total amount of protein decreases per step, the specific activity should rise. Generally, an assay performed will give the rate of reaction, in units such as micromoles per second. Dividing this rate by the concentration of your enzyme preparation yields the specific activity of a protein.
Ideally, the end of purification should be consistent with a constant specific activity. The specific activity can be monitored and used to quantify a purification by analyzing several variables which are total protein, total activity, yield, and purification level.
The concentration of a protein can be measured by immunological techniques such as ELISA or Western Blotting (the former being able to measure the quantity of protein present because of the direct proportionalities of reagents to proteins).
In order to determine how much activity is retained after each successive purification step in the crude extract, the yield can be calculated as . In order to convert this to a percentage, multiply the yield by (100). Also, it is important to note that in most cases, the amount of initial activity is always 100 %.
By obtaining a value for the purification level, we are able to assess how much purity has increased. The purification level can be calculated by: (
Important note: a purification scheme turns is only successful when taking into account BOTH purification levels and percent yield. Experimentation can become fairly complex if there is a high yield with very little purification. This is because there is an indication that there are a vast number of contaminants/proteins that aren't of interest. On the other hand, a purification level is high while the percent yield is low, then it is fair to conclude that there isn't enough protein available to carry out the experiment.
In addition to electrophoresis and immunological assays, the use of ammonium sulfate, (NH4)2SO4, can also quantitatively evaluate a purification. Because ammonium sulfate is non-denaturing and very water soluble (it is high on the Hofmeister series), it is used to effectively precipitate proteins: at high concentration, the ammonium and sulfate ions absorb most of the water through hydroelectric attraction, leaving the proteins to aggregate and precipitate out. ^
The mass of a protein can be measured using the sedimentation-equilibrium technique. This method requires slow centrifugation of a sample in order to establish a balance between sedimentation and diffusion. Unlike SDS-Polyacrylamide Gel Electrophoresis, which gives merely an estimate of the mass of dissociated and denatured polypeptide chains, sedimentation-equilibrium provides accurate mass measurements without requiring denaturation, thereby allowing the native structure of multimeric proteins to be left intact. Furthermore, the number of copies of each polypeptide chain that are present in a multimeric protein can be determined based on the mass of the dissociated chains and the mass of the entire multimeric protein, as measured by SDS-polyacrylamide gel electrophoresis and sedimentation equilibrium, respectively.
Mass spectrometry is another accurate analytical technique for determining protein mass. In this technique, atoms are ionized through a machine and passed through a vacuum into the detector. In which then, the time of flight (TOF) in the electrical field is directly proportional to the mass of the protein (or the mass-to-charge ratio). Thus, the smallest protein in a protein mixture has the smallest TOF, whereas the largest protein has the largest TOF. This technique allows the identification and analyzation of molecules based on their size and mass. This approach however, does not entail too much information about the structure or conformation of a protein.
Gel electrophoresis is a technique used to display and assert that the purification scheme was effective by measuring the number of different proteins in a mixture. The basis of gel electrophoresis is the fact that molecule with specific net charge will move through an electric field. The speed of protein migration can be quantified as:
With E as magnitude of the electric field, z as net charge of a protein, and f as frictional kinetic coefficient.
Frictional coefficient, for spherical molecule, is determined as:
f = 6 π η r
with η as viscosity.
As its equation implies, the velocity of molecule traveling in the gel matrix depends on its size, shape, and the charge that it has. The smaller the molecule, the faster it will travel. Furthermore, Gels can be made in a variety of wt percents: 6%, 8%, 10%, 12% and 15%. Higher percentages are used primarily for smaller molecules and smaller percentages are used for larger sized samples. Theoretically, larger molecules can still be used with higher percents, but these gels may take a long time to develop. Charge can also be a factor in the speed and distance that a specific sample travels through the gel. Using a higher voltage will send the samples farther and faster. However, caution must be used with higher voltages as the heat it generates may melt the gels.
Gel Electrophoresis (SDS-PAGE; SDS-polyacrylamide Gel Electrophoresis) is a powerful tool to check the purity of the sample because because it can detect minuscule amount of protein. Different proteins appear as different bands on SDS-Polyacrylamide Gel after gel has been stained with Coomassie blue (visualize ~2pm of protein) or silver stain (visualize 0.02 µg of protein).
Native Gel Electrophoresis involves running gels with samples in its native state. In doing so, the charge of the molecule becomes a factor in addition to size. More specifically, more charged molecules will migrate faster and farther than less charged molecules of comparable mass. Likewise, larger molecules will migrate less and at slower speeds than another molecule of comparable charge. Native Gel Electrophoresis most often involves two types of gels - Agarose and Polyacrylamide. Agarose is a derivative of the cell membranes of red algae composed of polysaccharides agarose and agaropectin, and due to the larger size of the pores, agarose gels are better suited for protein samples larger than 200 kilodaltons. Polyacrylamide (poly 2-propenamide, is a readily-crosslinked polymer of the neurotoxin acrylamide. It's pores are more fine, and while agarose is most commonly used for most cases, polyacrylamide is the gel of choice for smaller sample masses.
SDS Page. The Molecular Marker is located in the left lane.
Electrophoresis involves the movement of particles, such as nucleic acids or peptides, through a medium due to forces experienced by charges in an electric field. Electrophoresis can exploit molecular size differences or charge differences to separate similar molecules, and the amount of separation may be refined by changes in applied voltage or the density of the stationary medium. SDS -PAGE is a technique used to separate proteins based on size, and size alone. Sodium dodecyl sulfate (SDS) is a detergent that binds to proteins at every 2 amino acids in its sequence, and as SDS is very negative on its own, it changes the overall charge of the molecule to a negative charge. This negative charge is proportional to the protein's mass on the basis that the amount of SDS bound to the molecule is based on how many doublets of amino acids are present. The negative charge put on the protein is much larger than the charge originally there, which allows for a similar charge-to-mass ratio between different proteins. When SDS binds to proteins, it also changes the conformation of the proteins into similar shapes by denaturing the proteins and changing its bonds. SDS allows gel electrophoresis to separate proteins based on their molecular weights since the mass-to-charge ratio is relatively uniform among the proteins. This is because the SDS gel has sieving properties (offers resistance to particles based on their size)and is a uniform environment. It increases the differential mobility. The mobility of these proteins are then linearly proportional to the logarithm of their mass. Using this information, we can conclude from their mobility the mass of the protein and can even distinguish proteins that have a 2% difference in mass. Thus, the largest molecules, the ones that have more SDS bound to them, will fall down the electric field slower than the ones that have a smaller mass, and less SDS bound to them. This principle is contrary to the one in size-exclusion (gel-filtration) chromatography, which causes heavier molecules to come down first while the lighter ones come out later.
Certain solvents, such as PEG, glycerol, ethanol, and isopropanol, have an effect of decreasing the hydrodynamic radius of the proteins by decreasing the amount of free water to provide hydration spheres for the proteins. The polar solvents will hydrogen bond with the water, decreasing the disorder around the proteins and as a result, reducing the size of the hydration sphere. In such case, proteins will be eluted at a later stage as if they were of smaller size.
After the process is complete, the proteins are stained with a dye, forming bands, which represent the layers of mobility of each protein. With each additional purification process, the electrophoresis yields less bands, but a single darker band, which consequently represents the increased presence of the protein being isolated.
The separation techniques of SDS-PAGE and isoelectric focusing can be utilized in conjunction to allow for 2DGE, which employs higher resolution and sensitivity in the separation of proteins. The first dimension of this powerful technique is isoelectric focusing (IEF) and the second dimension is polyacrylamide gel electrophoresis (PAGE). In the first dimension, proteins are separated according to their isoelectric point (pI). To do so, the gel is applied to the top of an SDS-polyacrylamide slab. Electrophoresis is then applied horizontally across the top of the gel and the proteins migrate into the second-dimension gel. Electrophoresis will then be applied again, this time vertically across the gel slab, and the proteins will migrate based on their molecular size. Heavier proteins will move shorter distances. Conversely, lighter proteins will move further.
While Two-Dimensional Gel Electrophoresis is a powerful technique that presents a higher resolution of separation, it does have its own limitations. 2DGE is a time-consuming and labor-intensive process, requiring manual gel polymerization, staining, and hours upon hours of separation. Furthermore, the technique is not without risk. Because heating of the gel may cause warping and diffusion of the molecules on the gel surface, 2DGE is difficult to reproduce.
DNA Fingerprint. Each sample has a different pattern of bands indicating these samples are from three different individuals.
DNA fingerprinting is a technique used to differentiate between different organisms based on the differences between each organism’s DNA configuration. DNA fingerprinting is often used by forensics labs to identify criminals by comparing a suspect’s DNA to the DNA found at a crime scene. DNA from the suspect is run through a gel electrophoresis and compared to a sample of DNA that was found at the scene. If the two samples produce identical band patterns in the gel, then confirmation that the suspect was at the scene of the crime can be made, since no two people possess identical patterns in their DNA.
In order to perform a fingerprint, a sample containing DNA must be obtained from each organism under evaluation. Examples of DNA samples include blood, urine, saliva, skin or hair. Before the samples can be analyzed, they must first be prepared. Preparation includes using restriction enzymes to separate the DNA into smaller pieces. Restriction enzymes are enzymes that cut DNA strands at specific nucleotides. These nucleotides are called restriction sites, and typically mark the end of a 4-8 unit sequence in nucleotides. The components and length of each restriction sequence vary from person to person, thus the use of restriction enzymes is an efficient way of separating an organism’s DNA into unique and specific sections. Additionally, certain amount of chemicals are also inserted as dye into the gel which will illuminate under UV light. This causes the bands to be much more visible when analyzing the sample protein.
Regions of DNA that contain many different short repeated sequences are called microsatellites. The lengths of these microsatellites vary greatly from person to person, which makes them prime locations for restriction enzymes to fragment the DNA. After treating the DNA samples with restriction enzymes, the DNA is now ready to be analyzed. The samples are loaded into the wells in a slab of gel, and an electric current is applied. Smaller fragments of DNA run through the gel faster, and will therefore be closer to the bottom, while larger fractions remain closer to the top. If two samples of DNA are run at the same time, the locations of the bands can be compared. If the patterns of bands between the two samples are identical, it means that the restriction enzymes partitioned each sample’s DNA at the same locations, indicating the two DNA samples had identical nucleotide sequencing. Identical nucleotide sequencing reveals the two samples are from the same organism.
DNA fingerprinting is also a useful technique to determine whether or not two people are related. Although no two people share the same DNA patterns, sections of microsatellites are passed down from parent to child. Not all of these sections are passed down, but offspring do not contain any pattern that their parents did not possess. A paternity or maternity test can be performed by comparing the DNA fingerprint of the individuals in question. If there are large groups of patterns that repeat in each sample’s fingerprint, it is likely that the individuals are related. The embedded image contains a three different DNA fingerprints, as indicated by the three different patterns of bands. Although these patterns represent fingerprints from different people, sample 2 shares similar patterns with both 1 and 3, which indicates that the person whose DNA is represented by sample 2 is likely to be the child of sample 1 and 3.
Maternal and paternal DNA fingerprinting tests are used to determine the probability of two people being related. These tests do not give definitive answers, and are not foolproof.
As most proteins are not directly visible on gels to the naked eye, a method has to be employed in order to visualize them following electrophoresis. The most commonly used protein stain is the dye Coomassie brilliant blue. After electrophoresis, the gel containing the separated proteins is immersed in an acidic alcoholic solution of the dye. This denatures the proteins, fixes them in the gel so that they do not wash out, and allows the dye to bind to them. After washing away excesse dye, the proteins are visible as discrete blue bands. As little as 0.1-1.0 µg of a protein in a gel can be visualized using Coomassie brilliant blue. A more sensitive general protein stain involves soaking the gel in a silver salt solution. However, this technique is rather more difficult to apply. If the protein sample is radioactive the proteins can be visualized indirectly by overlaying the gel with a sheet of X-ray film. With time (hours to weeks depending on the radioactivity of the sample proteins), the radiation emitted will cause a darkening of the film. Upon development of the film the resulting autoradiograph will have darkened areas corresponding to the positions of the radiolabeled proteins.
Another way of visualizing the protein of interest is to use an antibody against the protein in an immunoblot (Western blot). For this technique, the proteins have to be transferred out of the gel on to a sheet of nitrocellular or nylon membrane. This is accomplished by overlaying the gel with the nitrocellulose then has an exact image of the pattern that was in the gel. The excess binding sites on the nitrocellulose are then blocked with a nonspecific protein solution such as milk powder, before placing the nitrocellulose in a solution cantaining the antibody that recognizes the protein of interest (the primary antibody). After removing excess unbound antibody, the primary antibody that is now specifically bound to the protein of interest is detected with either a radiolabeled, fluorescent or enzyme-coupled secondary antibody. Finally, the secondary antibody is detected either by placing the nitrocellulose against a sheet of X-ray film (if a radiolabeled secondary antibody has been used), by using a fluorescence detector or by adding to the nitrocellulose a solution of a substrate that is converted into a colored insoluble product by the enzyme that is coupled to the secondary antibody.
An SDS gel being visualized under UVA gel apparatus
SDS-Polyacrylamide Gel Electrophoresis is a technique to separate proteins according to electrophoretic mobility - a function of polypeptide chain length or protein mass). SDS-Polyacrylamide Gel Electrophoresis can also be used to separate DNA and RNA molecules.
SDS stands for sodium dodecyl sulfate. "SDS is an anionic detergent that disrupts non-covalent interactions in native proteins." SDS is used to create denaturing conditions to separate proteins by molecular weight and also confers negative charge to the proteins in proportion to its mass. By denaturing the proteins with SDS, proteins can be separated by their mass alone; without SDS, other molecular properties, such as a charge and shape, would interfere with the separation process (proteins that are strongly negative, for example, would move faster down a gel, even if they were larger, without SDS). In addition, a loading dye is introduced that helps bind the protein to the gel and make it more recognizable when exposed by UV light.
SDS-PAGE gives an estimates of the mass of dissociated polypeptides by the anions of SDS binding to the main chains of the polypeptide at a ratio of one SDS anion for every two amino acid residues.
SDS-PAGE is unlike sedimentation-equilibrium technique because denaturing of the proteins is applied for SDS-PAGE for mass determination.
This technique is used to test the purity of interest proteins and the percentage of interested protein in the sample solution.
This technique is rapid, sensitive and capable of high resolution compared to Gel-Electrophoresis because it can give a distinct band with as little as 0.1 micrograms of the protein when stained with Coomassie Blue and proteins that differ by 2% can still be separated.
SDS-PAGE can also be combined with Isolectric Focusing to obtain very high resolution separations. Proteins are first isolated by their net charge accordingly, then simultaneously run a SDS-PAGE adjacent to the filtering compartment.
Detergents are widely used to interrupt the hydrophobic interactions which can then destroy the lipid bilayer. Detergents are the most common types of agents used to solubilize transmembrane proteins.
Detergents are small amphiphilic molecules that are more soluble in water than lipids. Sometimes their hydrophilic heads (polar side) can be charged as in SDS but can be nonionic like octylglucoside and Triton. Detergents are monomeric in low concentration but form micelles in high concentration, after overcoming the critical micelle concentration. In order to keep the detergent monomer concentration constant, individual detergents go in and out of micelles. Detergents are very condition specific because they depend on the pH, salt concentration, and the temperature. Therefore detergents are very complicated to study.
Detergents help break the lipid bilayer by acting as a substitute. When the detergents are mixed with the lipids, the hydrophobic part of the detergent attaches to the hydrophobic head of the lipid bilayer making them soluble. If the detergent concentration decreases, the protein would not remain soluble.If more phospholipid were to be introduced, membrane proteins would form liposomes. Since the opposite side of the detergent is polar, the binding brings the membrane proteins into the solution as detergent-protein complexes. In this sense, the detergents acts as a capsule/substitute for the lipid membrane.
SDS, a strong ionic detergent, can solubilize even the most hydrophobic membrane proteins by attacking the hydrophobic core itself, which ultimately denatures the protein and can be used in a procedure known as SDS polyacrylmide-gel electrophoresis. The study of the protein function seems almost frivolous with the protein denatured but studies have showed that the protein can be renatured once the detergents are removed. Detergents are used commercially today to remove stains or proteins that stained clothes. By making the protein soluble, it is able to remove direct and other proteins from the clothes.
Similar to SDS-polyacrylamide gel electrophoresis, blue native-polyacrylamide gel electrophoresis is another useful method of protein purification that has allowed scientists to analyze membrane protein complexes in mitochondria, chloroplasts, microsomes, and bacteria.[1]
An SDS gel being visualized under UVA gel apparatus
SDS-Polyacrylamide Gel Electrophoresis is a technique to separate proteins according to electrophoretic mobility - a function of polypeptide chain length or protein mass). SDS-Polyacrylamide Gel Electrophoresis can also be used to separate DNA and RNA molecules.
SDS stands for sodium dodecyl sulfate. "SDS is an anionic detergent that disrupts non-covalent interactions in native proteins." SDS is used to create denaturing conditions to separate proteins by molecular weight and also confers negative charge to the proteins in proportion to its mass. By denaturing the proteins with SDS, proteins can be separated by their mass alone; without SDS, other molecular properties, such as a charge and shape, would interfere with the separation process (proteins that are strongly negative, for example, would move faster down a gel, even if they were larger, without SDS). In addition, a loading dye is introduced that helps bind the protein to the gel and make it more recognizable when exposed by UV light.
SDS-PAGE gives an estimates of the mass of dissociated polypeptides by the anions of SDS binding to the main chains of the polypeptide at a ratio of one SDS anion for every two amino acid residues.
SDS-PAGE is unlike sedimentation-equilibrium technique because denaturing of the proteins is applied for SDS-PAGE for mass determination.
This technique is used to test the purity of interest proteins and the percentage of interested protein in the sample solution.
This technique is rapid, sensitive and capable of high resolution compared to Gel-Electrophoresis because it can give a distinct band with as little as 0.1 micrograms of the protein when stained with Coomassie Blue and proteins that differ by 2% can still be separated.
SDS-PAGE can also be combined with Isolectric Focusing to obtain very high resolution separations. Proteins are first isolated by their net charge accordingly, then simultaneously run a SDS-PAGE adjacent to the filtering compartment.
Detergents are widely used to interrupt the hydrophobic interactions which can then destroy the lipid bilayer. Detergents are the most common types of agents used to solubilize transmembrane proteins.
Detergents are small amphiphilic molecules that are more soluble in water than lipids. Sometimes their hydrophilic heads (polar side) can be charged as in SDS but can be nonionic like octylglucoside and Triton. Detergents are monomeric in low concentration but form micelles in high concentration, after overcoming the critical micelle concentration. In order to keep the detergent monomer concentration constant, individual detergents go in and out of micelles. Detergents are very condition specific because they depend on the pH, salt concentration, and the temperature. Therefore detergents are very complicated to study.
Detergents help break the lipid bilayer by acting as a substitute. When the detergents are mixed with the lipids, the hydrophobic part of the detergent attaches to the hydrophobic head of the lipid bilayer making them soluble. If the detergent concentration decreases, the protein would not remain soluble.If more phospholipid were to be introduced, membrane proteins would form liposomes. Since the opposite side of the detergent is polar, the binding brings the membrane proteins into the solution as detergent-protein complexes. In this sense, the detergents acts as a capsule/substitute for the lipid membrane.
SDS, a strong ionic detergent, can solubilize even the most hydrophobic membrane proteins by attacking the hydrophobic core itself, which ultimately denatures the protein and can be used in a procedure known as SDS polyacrylmide-gel electrophoresis. The study of the protein function seems almost frivolous with the protein denatured but studies have showed that the protein can be renatured once the detergents are removed. Detergents are used commercially today to remove stains or proteins that stained clothes. By making the protein soluble, it is able to remove direct and other proteins from the clothes.
Similar to SDS-polyacrylamide gel electrophoresis, blue native-polyacrylamide gel electrophoresis is another useful method of protein purification that has allowed scientists to analyze membrane protein complexes in mitochondria, chloroplasts, microsomes, and bacteria.[2]
Another method in determining protein size is zonal centrifugation. Also known as band or gradient centrifugation, this technique relies on the concept of the sedimentation coefficient. The sedimentation coefficient is an equation that quantifies the rate of movement through a liquid medium through the formula:
s = m (1-vp)/f
where s = sedimentation coefficient, m = mass, v = partial specific volume, p = density of the medium, and f = frictional ratio. The unit of this equation are Svedberg units (S), which is equal to 10-13 s. A smaller S value generally means that a molecule will move more slowly in a centrifugal field, as opposed to a higher S value.
Some important conclusions that can be drawn from this equation include:
Since the velocity of a particle depends on its mass, particles with higher mass will sediment faster than particles with less mass.
Shape also determines the rate of sedimentation since it affects viscous drag. Therefore a more compact particle will have a smaller frictional coefficient than that of an elongated particle with the same mass. This means that more compact particles will sediment faster than elongated particles (same mass).
The sedimentation velocity is dependent upon the density of the solution (p). Particles that have a vp value less than 1 will sink, while particles that have a vp value greater than 1 will float. Particles that have a vp value equal to 1 won't move.[3]
To use this technique, a density gradient is first created in a test tube (usually with sucrose) with the highest density at the bottom. The purpose of the density gradient is to prevent convective flow. A sample of proteins is then placed on top of the gradient and then centrifuged. The proteins separate accordingly to their sedimentation coefficient into bands which can then be collected by creating a hole at the bottom of the tube. [3]
Zonal Centrifugation
The diagram below illustrates a simplified version of this technique with DNA as a sample instead.
The amount of protein in a sample can be determined by constructing a standard curve with known masses of protein plotted against the absorbance value. The absorbance of the sample can then interpolated into the standard curve and the mass of protein in the sample can be determined.
The Bradford assay is advantageous because it offers high precision and fidelity. It also is compatible with most reagents although not with detergents or surfactants.
Disadvantages of the Bradford include that it is a slow assay to perform, it depends on a standard curve, and it destroys the sample of protein used.
Quantum dots are microscopic semiconductor crystals that are made of clusters of cadmium selenide, cadmium sulfide, indium arsenide, or indium phosphide and they radiate colors when are exposed to ultraviolet light. They are typically between two to ten nanometers long in diameter. Their small size allows for the visible emission of photons as they are excited, which produces wavelengths of color that people can see. They are used to visualize and track individual molecules and their movements inside cells. They are also known as “artificial atoms” because their behavior is analogous to that of single atoms. Quantum dots work based on the principle of quantum confinement, which states that when an object is confined to a small space, the object is only able to occupy certain discrete energy levels. This principle is equivalent to how electrons are only able to occupy discrete energy configuration known as orbital’s. In the case of Quantum Dots, electrons are forced to occupy discrete energy levels based on which wave functions "fit" inside the quantum dot. When electrons are excited from their lower energy levels, the transition from a high energy state to a low energy state emits a photon, just like when an electron makes an energy transition in an atomic transition.
Energy diagram
This property of quantum dots is useful for one especially important application, to tag molecules or proteins of interest as well as several other uses outside the field of biology. Some examples include applications in memory chips, quantum computation, quantum cryptography, in room-temperature quantum-dot lasers, just to name a few. The basic concepts underlie these artificial atoms include, but not limited to, the magic numbers in the ground state angular momentum, the spin singlet-triplet transition, the generalized Kohn theorem, and their implications, shell structure, single-electron charging, diamond diagram, etc. They are often used more than traditional organic compounds that are used to stain cells and make cells radiate because they are brighter and more versatile.
The problem of a single ideally two-dimensional electron in a circular dot with zero confinement potential in the presence of an external magnetic field was studied by Landau leading to the term Landau levels. Hybridization of Landau levels with the levels that arise from spatical confinement occurs at low values of the magnetic field (the magnetic length is larger than or comparable to the size of the confinement potential). As magnetic field increases (the magnetic length becomes much smaller than the radius of the confinement potential), free-electron behavior dominates that of spatial confinement. Therefore, a gradual transition from spatial to magnetic quantization that depends on the relative size of the quantum dots as compared to the magnetic length can be observed.
Using single-electron capacitance spectroscopy, gated resonant tunneling devices, conventional capacitance studies of dot arrays, transport spectroscopy, far-infrared (FIR) magneto-spectroscopy, and Raman spectroscopy the electronic properties of quantum dots are found. An oscillatory structure in the measured capacitance was attributed to the discrete energy levels of a quantum dot. In the presence of a perpendicular magnetic field, Zeeman bifurcation of the energy levels of a quantum dot was also observed. This splitting is believed to occur due to the interplay between competing spatial and magnetic quantization.
Capacitance spectroscopy has been widely used to study the density of states of low-dimensional electron systems. The measured capacitance (or the first derivative of the capacitance versus the gate voltage) reveals structures related to the zero-dimensional quantum levels. As a result, fractionally quantized states, similar to the fractional quantum Hall effect in a two-dimensional electron system, are observed.
The electronic ground state in a parabolic confinement potential has been observed in an experiment by Ashoori. The method involved in this experiment is known as single-electron capacitance spectroscopy, and allows direct measurement of the energy levels of a ne-electron dot as a function of the magnetic field. The capacitance was measured between an electrode on top of the QD (the gate) and a conducting layer under the dot that is separated from the dot by a thin tunnel barrier. When the dc gate voltage on the top electrode is varied the Fermi level in the bottom electrode can coincide with the Fermi energy of the dot. Electron tunneling through the thin barrier is observed. Charge modulation in the QD induces a capacitance signal on the gate because of its close proximity to the dot. The capacitance as a function of the gate voltage was found to exhibit a series of uniformly spaced peaks, with separation decreasing with increasing electron number. The peaks are results of the addition of single electrons to the QD. The remarkable aspect of the experiment is that they probed the addition spectrum starting with the very first electron in the dot.
The quantum dot structure was created either by etching techniques or field-effect confinement in this experiment. The samples were prepared from modulation-doped AsGaAs/GaAs heterostructures. For the quantum dots, an array of photoresist dots was created by a holographic double exposure. The rectangular 200nm deep grooves were then etched all the way into the active GaAs layer. Quantum dots can also be grown from seed crystals. Like how sugar crystals are grown to make rock candy, quantum dots can be grown layer by layer until the desired size is achieved in a process known as self-assembly. Field-effect confined quantum dots were prepared by starting from a modulation-doped GaAs-heterojunction. Electrons were laterally confined by a gate voltage applied to a NiCr-gate. A strong negative gate voltage depletes the carriers leaving isolated electron islands (quantum dots).
Previously, there had been functional problems with the ligands that were attached to the quantum dots. Scientists have instead utilized these ligands to their advantage; They are now used to cover up the spaces in between the quantum dots. This creates a structure in which there are spaces for the quantum dots to fit in. This allows for the use of a single-layered Quantum-dot Light-Emitting Device, enabling scientists to pass current directly through the quantum dots rather than in between them. Scientists are currently pushing for this new technology of Quantum-Dot LEDs to be used in computer and television displays.
Quantum dots is a technology which utilizes microscopic semiconductor crystals to label proteins and genes of interest. The crystals are less than a millionth of an inch in diameter and radiate bright colors when exposed to UV light. Different sized dots radiate with different fluorescent colors. Large dots emit a red color, while small dots emit a blue color. The size affects the color of the fluorescence due to the phenomenon of quantum confinement. As the size of the quantum dot decreases, the electron is forced into a tighter and tighter space. This means that the quantized energy levels of the electron get spaced further and further apart, increasing the energy difference between the excited and relaxed electron energy levels. This phenomenon is exemplified in the classical quantum mechanics problem of the infinite potential well. Choice of the quantum dot material also affects the characteristics of the emission spectra. Choosing a semiconductor with a high bandgap, the energy difference between the highest occupied energy level and lowest unoccupied energy level, results in higher energy photons being released (blue shifting). Also, quantum dots tend to be made from direct bandgap materials like GaAs, which results in more efficient energy transitions and less energy wasted as heat.
The dots are more useful than fluorescent markers because there are more variety in colors, and the light emitted from quantum dots are brighter and more versatile. Another advantage is that until flurophores and chromophores, they do not photobleach, meaning that repeated use does not diminish their capacity to function properly. Because quantum dots are made from inorganic materials, they can be functionalized easily with molecules and do not degrade easily, which maybe pose an environmental risk. They can visualize individual molecules or every molecule of a given type. Quantum dots show promise in allowing scientist to quickly analyze thousands of genes and proteins from patients with disease, such as cancer. They can then customize treatments to each patient’s own molecular profile. Quantum dots can also improve the speed, accuracy, and affordability of various diagnostic tests, whether it be HIV or common allergies. They can also give a specific dose of a drug to a certain type of cell. Compared to other fluorescent markers, they are smaller, more specific and allow further insight into the structure and inner working of a cell. Large scale use of quantum dots, however, may be limited due to the unknown hazards of using nanomaterials in living organisms.
In a cell there are two main groups of proteins, there are cytoplasmic proteins and membrane bound proteins. The cytoplasmic proteins are floating around in the cell and have a particular structure. This structure is the polar and hydrophilic amino acids all tend to be on the outside of the protein, and the non polar hydrophobic amino acids are buried inside the protein (Fig 1)Protein in membrane. The reason for this assembly is because the proteins have to be stable in the cytoplasm which is mostly water. Since the structure is not complex in terms of the positions of certain amino acids it is much easier to look at a crystal or an NMR spectroscopy of cytoplasmic proteins. The membrane bound proteins have a unique structure because of the position of the protein. The cell is bounded by a phospholipid bilayer, which is hydrophilic on the outsides and hydrophobic on the inside. This structure of the cell membrane has to be mimicked in the protein or it will not be able to stay stable in the membrane. Due to the more complex nature of the membrane bound proteins it is harder to purify and perform a NMR spectroscopy of these proteins.
In order to perform a NMR of a stable membrane protein, it would have to be place in an environment which mimics the phospholipid bilayer. Scientist then saw that detergents had a similar structure and that they formed micelles, with a hydrophobic inside and hydrophilic outside (Fig 2)Protein in detergent. The only problem with the detergents is that they move around and are hard to get an NMR because it created a lot of noise. Another problem is that the micelles don’t completely replicate the membrane in that they are not attached parallel to the proteins non polar region, instead they are perpendicular and so can cause distortions in the proteins functionally and shape. To solve these problems the scientists created a lipopeptide detergent of LPD for short. The LPD’s is a chain of 25 amino acids that for an alpha helix. On the second and 24th amino acids there is an attachment to two alkyl chains that are about eight to twelve carbons in length (Figure 3)Lipopeptide detergent.
The advantage of using a LPD is that unlike a micelle, the LPD’s are closer in function to a membrane since they attach parallel to the protein (Fig 4)LPD and protein. Another advantage is that they are rigid and don’t move around and so reducing the noise that is present during a NMR spectroscopy. Since these structures are rigid and span the entire hydrophobic region of the protein, there only has to be a few LPD’s in place to keep the membrane protein stable. The only problem with the use of an LPD is that it is financially expensive and so is used as a last resort when all detergents fail. When LPD’s are used in experiments detergents still have to be present to surround the protein at first. Then the LPD is inserted and since it is more structurally favored it replaces the detergent’s micelle and creates the “membrane” and then the detergent is centrifuged out of the solution. After a few rounds of this process one can assume that the proteins are purely surrounded by LPD’s
Mitochondria have been known to be the powerhouse of the eukaryotic cell, possessing the ability to produce ATP which is used as cellular energy for the cell. However, mitochondria also fulfill other roles within the cell such as in metabolic pathways, apoptosis, cellular differentiation, and control of the cell cycle. As a result, to these multiple functions, mitochondria have evolved to develop a double membrane that surrounds the mitochondria complex. This double membrane functions as a high-traffic zone for the cell, possessing the ability to control what molecules go into the mitochondrion and what have to go out. For example, low-energy metabolites such as ADP have to go inside while high-energy metabolites such as ATP have to go out. This function of funneling ADP into mitochondria and ATP out of mitochondria is controlled by an integral membrane protein known as the voltage-dependent ion channel (VDAC), or also referred to as the mitochondrial porin.
The structure of VDAC has been examined for quite some time after it was discovered in 1975. Many structures of VDAC were determined, but the spatial arrangement, the topology, of the structure for the beta-strand could not be determined. However, in 2008, three long-term efforts to determine the three-dimensional structure of VDAC-1 were determined at atomic resolution. Three structures of the isoform VDAC-1 were determined by different methods. One was determined by using NMR spectroscopy alone, another by X-ray crystallography alone, and the last one using a combination of both NMR spectroscopy and X-ray crystallography. The comparison of these three different structures of VDAC-1 is examined as well as the discussion of the importance of solution NMR to determine the structure of VDAC-1.
Structure of VDAC-1 and Comparison of Three Structures
The structure of VDAC-1 is very unique as it contains a very large beta-barrel. For all three structures, the number of strands in this beta-barrel and the spatial arrangements of molecules is the same. In studying the amino acid sequence of VDAC, it has been identified as being conserved from yeast to human. As a result, the overall folding pattern of the structure is known to be the same in all eukaryotes. In the three structures of VDAC-1, one of the structures is derived from a mouse while the other two structures are derived from humans. When comparing the mouse form of VDAC-1 to the human form of VDAC-1, the two forms are highly identical, differing in only four amino acids. Due to the very small changes in amino acid sequence between the mouse and human forms, the three-dimensional folded structures are very similar. To further confirm the beta-barrel structure of VDAC-1, denaturation of the VDAC-1 protein was performed to allow it to refold into the detergent LDAO. The refolded VDAC-1 structure was then placed into a different environment containing bicelles known as DMPC. By placing the refolded VDAC structure in a different solution environment, the same beta-barrel structure was observed again, and it was concluded that this beta-barrel structure of VDAC-1 is the same no matter what type of environment solution it is placed in.
The beta-barrel structure of VDAC-1 is fairly unique because it is the only structure that is observed in any eukaryotic membrane protein, and it is also the only known beta-barrel membrane protein that contains an odd number of strands. The rest of the beta-barrel proteins are observed to arrange into anti-parallel beta sheets, and because of this, an even amount of strands is needed to stabilize the entire beta-sheet structure through hydrogen bonding. It is unknown why a beta-barrel structure is stabilized with an odd number of strands as the folding mechanism of this protein is not fully understood. The structure of the beta-barrel is defined using two numbers that are the number of strands, n, and the shear number, S. The shear number in the beta-barrel can be identified as the pairs of alpha-carbon atoms in adjacent strands that lie on a helical trace across the surface of the beta-barrel. The side chains of the alpha-carbon atoms must be pointed to the same side of the sheet, and following the trace of the helix once around until it arrives back at the first strand a certain number of residues away from the starting point is known as the shear number of the beta-barrel. In beta-barrels, the shear number is always even in order to have the hydrophobic residues of the protein on the outside of the complex. Beta-barrel structures usually contain a shear number in the range of n and n+4.
Another comparison that is made to differentiate the three VDAC-1 structures is the residues that branch off the protein that is not part of the beta-barrel. The 1-23 residues are compared between each of the three structures, but through the use of NMR, only residues 6-10 have been identified to be an alpha-helical structure. Also, through the use of X-ray crystallography, the structure of mouse VDAC-1 was observed to contain three aliphatic residues: Leucine 10, Valine 143, and Leucine 150. From the crystallized structure, it was observed that Valine 143 and Leucine 150 are the only hydrophobic side chains that point to the barrel interior from the barrel wall. Residues 11-20 in the mouse structure and human structure appear to contain similar segments. However, the conformation of these segments differs between these two structures. Both structures were analyzed at cryogenic temperatures through the use of NMR. The conformational changes between these structures are exposed through the use of NMR because as the conformations change, residues of the proteins will end up interacting with other different neighboring residues. As a result, these conformational changes can lead to multiple resonance lines, reduced signal intensity, or line broadening on the NMR graphs for the structures.
Solution NMR in Determining Structure of VDAC-1 and Other Integral Membrane Proteins
In determining the beta-barrel structure of VDAC-1, researchers have stated that the combination of NMR and X-ray crystallography data were not enough in fully determining the structure. As a result, the use of solution NMR techniques was used instead to solve the beta-barrel structure of this membrane protein. In total, nine structures of integral membrane proteins have been solved using solution NMR. In using solution NMR, two important techniques are used in determining membrane protein structures such as VDAC-1: protein refolding and deuteration of the detergent micelle.
The use of protein refolding from a denatured state has a very low success rate for most membrane proteins, but if the refolding process is successful, there are many benefits that help to study the structure of membrane proteins with much greater ease. First, the process of protein refolding can lead to a very high yield of the newly folded structure of the protein. In the case of VDAC-1, an average of 40 mg of VDAC-1 was obtained in a 1 liter solution of an E. coli cell culture. Second, protein refolding helps to purify the membrane protein to an extremely high degree. This is extremely important in studying the structure of VDAC-1 as the data obtained from X-ray crystallography and NMR would be accurate in examining the true structure. Third, protein refolding has a high reproducibility, which goes together with the high purity. Fourth, because high yield and high reproducibility can be done from protein refolding, efficient perdeuteration and selective isotope labeling can be done. Finally, since the predeuterated protein goes through a denatured state in the in deuterated-water, all the amide compounds are readily protonated by the deuterated-water, and therefore, the structure of proteins such as VDAC-1 are much easier to identify due to the presence of D’s instead of H’s.
The use of deuterated detergent in solution NMR is the second technique that helps in identifying the structure of large membrane proteins such as beta-barrel of VDAC-1. From other NMR studies, compounds not placed in deuterated solutions produce very broad resonance lines due to the strong dipole-dipole interactions between different atoms, causing the spectral sensitivity to be reduced an extremely significant amount. By examining membrane proteins such as VDAC in deuterated solutions, a much more specific NMR graph is observed. For example, when using the Nitrogen-15-resolved-NOESY spectra, when a deuterated detergent was replaced with a protonated detergent, a decrease of 10-30% in sensitivity was observed. This decrease in sensitivity is clearly seen when analyzing the spectra of the methyl groups of the aliphatic residues of Isoleucine, Leucine, and Valine. The NOESY spectra of these groups did not produce a clear spectra identifying these compounds in a protonated detergent, but when a deuterated detergent was used, clear images of these groups were able to be identified, concluding that the use of solution NMR in a deuterated detergent proved to be a powerful method in determining the structures of integral membrane proteins such as VDAC-1.
Hiller, S. The role of solution NMR in the structure determinations of VDAC-1 and other membrane proteins. 2009, Current Opinion in Structural Biology. p. 396-401.
ANALYTICAL ULTRACENTRIFUGATION
In principle, analytical centrifugation is similar to differential centrifugation in that both techniques apply the principles of centrifugal acceleration to separate components of a sample based on shape and mass differences. They both require a rotor capable of spinning samples at speeds enough to generate forces up to tens of thousand times greater than the force of gravity. However, analytical centrifugation is able to perform analysis of the concentration of the sample during centrifugation through the incorporation of light detection devices into the system, and this is the key point that differentiates the two techniques.
Analytical centrifuges can perform at least two different types of hydrodynamic analysis: (1) sedimentation velocity; and (2) sedimentation equilibrium. These two techniques, along with some of their advantages and disadvantages, are discussed below.
An ultracentrifuge.
1) Sedimentation Velocity
This test is sensitive to both the mass and the shape of the molecules. To perform this test, a uniform sample is first loaded into the sample slots and subjected to high acceleration spinning. A typical velocity is anywhere between 40,000 and 60,000 rpm's. Due to the difference in force applied to the components caused largely by mass and shape differences, the components will separate out in layers, forming boundaries in solution. The boundary is basically a concentration gradient that forms as a result of the movement of the particles. Although the velocity of the individual particles resulting from the centrifugal force cannot be determined, a series of scans (such as absorbance or refractive index detection) is performed on the sample as it spins to record the movement of particle boundaries over time.
More specifically, the rate of movement of the boundary can be used to calculate the sedimentation coefficient (s). The sedimentation coefficient can be affected by at least the following factors:
• Molecular weight—heavier particles tend to sediment faster;
• Density—more dense particles tend to sediment faster;
• Molecular shape—unfolded proteins or a more highly elongated shape will experience more friction from solvent, so will tend to sediment slower;
• Solute concentration—higher solute concentration tends to lower the rate of sedimentation;
• Solvent concentration/viscosity—higher solvent concentration and viscosity will tend to increase friction and lead to a lower sedimentation coeffient; and
• Charge of the protein and how it interacts with polarity of the solvent—for example, a charged particle will travel more quickly through a polar solvent.
In addition to analyzing the rate at which the boundary moves (i.e., the sedimentation coefficient), the characteristics of the boundary itself can also provide information regarding the sample. The diffusion coefficient (D) can be determined by measurement of the spreading of a boundary. A homogeneous product will often produce a boundary that is sharper. In contrast, a heterogeneous sample can produce multiple boundaries or a very broad boundary. However, these are only general rules of thumb because characteristics of the sample can produce contradictory results. For example, a single boundary is not necessarily indicative of a homogeneous sample where it includes two molecules that have similar sedimentation coefficients that would result in a what appears to be a single boundary. Likewise, multiple boundaries do not necessarily result from a heterogeneous sample because a homogeneous sample can have several stable aggregation states that can produce multiple boundaries depending on how rapid the states introconvert.
An additional factor that can create complications in analyzing the characteristics of the boundary is a phenomenon known as self-sharpening. Self-sharpening occurs where the molecules at the "front" end of the boundary move in a higher concentration of solvent and are restricted, whereas molecules at the "back" end of the boundary are in a less concentrated portion of the solvent and move more quickly. This causes an artificial narrowing of the boundary.
Sedimentation velocity is a useful technique for a variety of analyses, including: (1) determining whether a sample is homogeneous; (2) determining whether a protein is a monomer, dimer or other multimer in its native state; (3) determining the overall shape of a protein (for example: is it spherical or more extended); and (4) quantifying the distribution of sizes of proteins in a sample that includes a range of sizes. A critical advantage of a sedimentation velocity procedure is that it can be performed in a relatively short amount of time (often as low as 3–5 hours), as opposed to sedimentation equilibrium (which can often take days). Another important advantage of sedimentation velocity is that it can be used to analyze samples over a broader range of pH, ionic strength, and temperature conditions. One disadvantage is that interacting systems (such as proteins that reversibly self-associate—see discussion above) can lead to data that is difficult to interpret if those systems change during the course of the testing.
2) Sedimentation Equilibrium
This type of analysis is sensitive only to the mass of a particle (not its shape), and is performed at slower velocities than those for sedimentation velocity. As the sample spins, the components separate out due to acceleration from the spinning while diffusion simultaneously provides an opposing force. Eventually, these forces balance each other out and the components in solution reach an equilibrium point. A series of scans (such as absorbance or refractive index detection) monitors the sample for this equilibrium point, which provides information on the molar weight of the component in sedimentation.
Sedimentation is still regarded by many as the best method to determine the molecular weights of macromolecules in a sample. Although sedimentation equilibrium is conducted at a lower velocity than sedimentation velocity, it must be conducted at higher velocities when analyzing lower molecular weight samples. Sedimentation can also be used to separate heterogeneous samples of different molecular weights. Higher molecular weight particles will move further toward the bottom of the cell, whereas lower molecular weight particles will collect near the top of the cell.
In combination, these tests are able to provide details on the purity of samples and information on molecular weights quite accurately. In particular, analytical ultracentrifugation becomes extremely useful for the analysis of molecular weights for large macromolecules which wouldn’t be able to undergo sequencing tests, such as polysaccharides. Additionally, sedimentation equilibrium is able to provide information on the attractive forces between components of a sample in solution without disturbing the solution, which makes this method very reliable and accurate. Although analytical ultracentrifugation techniques can be used in isolation, they are also used in combination with other analytical techniques to provide more clear and complete conclusions. For example, these techniques are often used in combination with cheaper techniques such as gel electrophoresis and other chromatographic techniques. In addition, they are often used in combination with other analytical techniques such as mass spectrometry, x-ray crystallography, and multidimensional nuclear magnetic resonance (NMR).
Sedimentation Velocity Patterns:
Ultracentrifugation studies of ATCase have shown two different graphs of Protein concentration versus migration distance. Native ATCase has one peak and the 6 catalytic and 6 regulatory subunits are gound together. When the enzyme is treadted with p-hydroxymercuribenzoate, the enzyme is dissociated into two subunits. A 2 regulatory subunit and a 3 catalytic subunit. These experiments have helped show that the interaction of the subunits in the native enzyme produce its regulatory and catalytic properties.
The origin of ultracentrifugation
Ultracentrigugation is one of the powerful techniques to determine structure proteins because this method can be used as preparative and analytical. Thus, it is common use in biology, biochemistry and polymer area. In 1923, the analytical ultracentrifuge was invented by Theodor Svedberg and three years later, he won a Nobel Prize in Chemistry for his research on using the ultracentrifuge on separating the collides and proteins. In 1946, Pickel designed the first model preparative ultracentrifuge that can reach the velocity of 40,000 rpm.
Analytical ultracentrifuge
Preparative ultracentrifuge
REFERENCES
[17][18][19][20]
Crosslinking is one method that is used to study the interactions in protein and is often called bioconjugation when referring to proteins. Crosslinking involves covalently attaching a protein to another macromolecule (often another protein) or a solid support via a small crosslinker. A crosslinker, or a crosslinking agent, is a molecule which has at least two reactive ends to connect the polymer chains. The crosslinkers are usually reactive toward functional groups common on proteins such as carboxyls, amines, and sulfhydryls.
Homobifunctional crosslinkers are molecules that have the same reactive groups on each end of the crosslinker. Homobifunctional crosslinkers can give a good idea of all the interactions between molecules present in a solution or cell, but it can also cause unwanted crosslinks. The reactive ends are impartial and may crosslink a protein to an identical protein when interactions between different proteins are desired. Homobifunctional crosslinkers often also create intramolecular crosslinks.[4]
Heterobifunctional crosslinkers are molecules that have different reactive groups on each end of the crosslinker. Heterobifunctional crosslinkers can be more selective in the crosslinks formed because the reactivity of each group can be chosen so that a specific protein will only bind to one end. A two-step process can also be set up to minimize undesired crosslinks. First a crosslinker is added to a solution with one particular protein and allowed to react. The protein with the crosslinker attached is then purified and added to a solution with a second protein that will form a crosslink with the other reactive group on the crosslinker. This new structure can then be analyzed using different techniques to see if the proteins connected, how many connected, or other desired information.[5]
There are a number of different reactive groups used in crosslinkers that are targeted towards different functional groups on proteins including carboxyls, amines, sulfhydryls, and hydroxyls. Crosslinkers are generally selected based on their reactivity, length, and solubility. Crosslinkers can also be spontaneously reactive upon addition to a sample or be activated at a specific time, generally through photo-reactive groups.
Although a crosslinker can be chosen to target only a certain type of functional group, most proteins contain several residues with each type of group. If multiple target sites are available for binding, the crosslinker will lose specificity and multiple crosslinked products will be formed. However, a crosslinker will only be able to bind if the target functional group is on the surface of the protein. Thus, protein folding will often block access to a number of possible reaction sites and allow for greater specificity in crosslinking.
NHS esters react with amines to give stable amide groups. As such, NHS esters are useful for linking to the N-terminus or lysine residues on a protein. The reaction is generally carried out in slightly alkaline conditions (pH 7.2-8.5). However, the desired reaction competes with hydrolysis of the NHS ester. The rate of hydrolysis increases with increasing pH, so the pH of the buffer solution must be closer controlled.
Imidoesters are reactive groups that form amidines with primary amines. Like NHS esters, imidoesters are useful for linking to the N-terminus of a protein or a lysine residue. The reactivity of imidoesters increases with pH and the reaction is generally carried out between pH 8 and 10. However, imidoesters become labile at higher pH, and are thus not as stable as NHS esters. Imidoesters are useful for linking membrane proteins and for probing lipid-protein interactions, as they are able to penetrate the cell membrane.
Carbodiimides are not traditional crosslinkers in that crosslinker itself does not become part of the protein-protein complex. Carbodiimides instead covalently link two proteins directly together by forming an amide bond between a carboxylic acid group of one protein and an amine group of another. Because of the mechanism of carbodiimide crosslinkers, they are by nature zero length (they do not become part of the molecule) and heterobifunctional crosslinkers. EDC (1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide) is the only well known carbodiimide crosslinker.[6]
Maleimides react with sulfhydryls at physiological pH to produce a stable thioether linkage, and are usually linked to a cysteine residue. Since there are often far fewer free sulfhydryl groups than amine groups on a protein, maleimides tend to be more specific than reactive groups that target amines. Also, since sulfhydryls are often involved in disulfide bonds, connection to a crosslinker often does not disturb the structure of the protein.
Like maleimides, haloacetyls react with sulfhydryl groups at physiological pH to give a thioether linkage. The most common haloacetyls are iodoacetyls and bromoacetyls, and they react via a nucleophilic substitution of the halide by the sulfur of the sulfhydryl.
Pyridyl disulfides react with sulfhydryls to form disulfide bonds. They are active over a wide pH range, but pH 4-5 is optimal. Because a disulfide bond is formed, the linkage can be cleaved with normal disulfide reducing agents.
Diazirines are an example of a photo-reactive group. While most other reactive groups are spontaneously reactive upon addition to a sample, a photo-reactive group will be inert until activated by exposure to ultraviolet light. Diazirine analogs of methionine and leucine are typically incorporated into a protein and then photo-activated in the sample solution or cell. When activated, the diazirine will react with any protein within a few angstroms of the photo-reactive analog. This allows for protein-protein interactions to be captured and studied in live cells.[7]
Despite the structural modifications, the proteins made from diazirine analogs of methionine and leucine are still viable, although their growth is slowed slightly. This allows for the creation of photo-reactive proteins that lack toxicity. The amino acid analogs and the proteins they are incorporated in can thus be used to study in vivo protein-protein interactions without greatly disturbing the cell.[8]
Crosslinking is a good technique to learn more about protein-protein interactions. Frequently the interactions between proteins are too weak to notice normally, but with crosslinking the interactions can be solidified and detected easily. This could be useful in determining if certain proteins in a cell interact with each other and could lead to more information on how a cell functions. Another use of crosslinking is to attach proteins to solid stations to immobilize them in order to analyze them more easily. Crosslinking can also be used to attach a tag to a protein to help detect its presence.[9][10]
Crosslinkers can be chosen with different lengths between the reactive ends which can help determine the distances between certain functional groups in a protein structure. For example: if several crosslinkers, with the same reactive ends but with different lengths, were added separately to a solution with the desired protein to be analyzed, then depending on which solution reacted one would be able to tell the distance between the functional groups of interest based on the crosslinker added.
Interactions between proteins can be determined using crosslinkers to essentially freeze two proteins together while they are interacting. This technique creates a stable protein pair that can be purified or studied via gel electrophoresis or Western blotting. It is most common to characterize protein-protein interactions in vivo, where crosslinking can be performed at different times after a desired interaction has been initiated. The resulting crosslinks can give an indication of the interactions taking place in a cell during a response to some stimuli.
Another way of using crosslinkers to study protein-protein interactions is to use cleavable, labeled, photo-reactive crosslinkers to label any protein that interacts with a "bait" protein. The label may be a radioactive isotope or mass variant. A heterobifunctional crosslinker is usually used, with one end being linked to the purified bait protein and the other end being a photo-reactive group. The photo-reactive end can be activated at different times by UV radiation, which will cause it to immediately react with the first group it encounters - hopefully a protein or co-factor that is interacting with the bait protein. The crosslinker can then be cleaved, transferring the label to the other protein or co-factor.
Crosslinkers can be used for the quantification of certain amino acids and for determining the number of and distances between subunits. Using multiple crosslinkers that vary only in length allows the distances between particular amino acid functional groups to be determined. This information can be used to determine the relative positions of amino acids in secondary, tertiary, and quaternary structures. Homobifunctional crosslinkers that bind sulfhydryl groups can also be used to replace the disulfide bonds in a protein with non-cleavable linkages.
Crosslinkers can be used to link a toxin molecule to an antibody that is targeted at tumor cells. The antibody will bind to antigens on the surface of the tumor cell and the crosslinked complex is taken up by the cell. Once inside, the toxin is released and activated, killing the cell. To be effective, the crosslinked antibody-toxin must be stable and able to locate and target the correct cells in vivo.
A cleavable crosslinker is needed for immunotoxins so that the toxin is released once inside a cell. SPDP is one of the most common crosslinkers used for immunotoxins and contains an NHS-ester and a pyridyl disulfide group. The NHS-ester is first linked to the antibody and then the pyridyl disulfide is linked to the toxin. Since many toxins do not have surface sulfhydryls, free sulfhydryls are created by reducing disulfide bonds. Some of the toxins used are ricin and abrin.
Protein-protein conjugates are used in a number of immunodetection methods such as ELISA and Western blotting. Usually, an enzyme is linked to an antibody specific for an antigen of interest. The antibody will bind to the antigen and the attached enzyme will catalyze a detectable reaction, indicating the presence of the antigen. Horseradish peroxidase and alkaline phosphatase are the most common enzymes used as they produce products that are easily detected by spectroscopy.
Small peptide antigens can also be conjugated to larger proteins for the production of immunogens. Immunogens are usually prepared by injecting animals, typically mice, with an antigen and collecting the antibody produced by the animal in response. Small peptides are often not large enough to produce an antigenic response in animals, so linkage to a larger protein can be necessary to create an effective antigen.
Crosslinkers can be used to attach proteins to solid supports by selecting a solid resin containing functional groups that one end of the crosslinker is specific for. Attaching a protein to a solid support allows for affinity purification and for protein analysis. Other biological molecules such as DNA can be attached to solid supports in a similar fashion. Although DNA crosslinking is hindered by the lack of functional groups usually targeted by crosslinkers, DNA can be modified by adding primary amines or thiols to specific bases to increase crosslinker activity.
The amino acid sequence of a protein is a valuable source of insight into its function, structure, and history.
1. The sequence of a protein can be compared with other known sequences to decide whether significant similarities exist. Using computers, a search for kinship between a new sequenced protein and millions of previous previously sequenced ones only takes seconds. If the newly isolated protein is a member of an established class of protein, we can infer information about the protein's structure and function.
2. Comparison of sequences of the same protein between different species teaches about evolutionary pathways. Genealogical relationships between species can be inferred based on differences in the sequences between their proteins. Assuming the mutation rate of proteins is constant, the analysis of sequences of different proteins from different species can provide information when these two evolutionary lines diverged. For example, comparison of serum albumins found in primates indicate that humans and African apes diverged 5 million years ago instead of 30 million years ago.
3. Amino acids sequences can be searched for the presence of internal repeats. Such repeats reveal the history of the protein, and many proteins have arisen from duplication of primordial genes followed by diversification.
4. Many proteins contain amino acid sequences that serve as signals for their destinations or controlling their processes
5. Sequences provide a basis for preparing antibodies specific for a protein of interest. Parts of an amino acid sequence will elicit an antibody when injected into a mouse or rabbit. These specific antibodies are useful for determining the amount of proteins in the blood.
6. Amino acid sequences are valuable for making DNA probes used for encoding its proteins. By knowing the primary structure, it permits the use of reverse genetics. DNA sequences that correspond to part of an amino acid sequence can be constructed on the basis of genetic code. These DNA sequences can be used as probes to isolate the gene encoding the protein so that the entire sequence can be determined. The gene in turn can provide information about the physiological regulation of the protein.
Determining the amino acid sequences
1. Hydrolysis:
The peptide is heated in 6M hydrochloric acid (HCl) at 110o C for 24 hours. This procedure is required in order to hydrolyze peptide chain into its amino acid.
2. Separation
Amino acids from the peptide is identified by eluting the mixture with buffers of increasing pH in an ion-exchange chromatography column on a sulfonated polystyrene. The volume of buffer used can be correlated to a specific type of amino acid. The most acidic side chain amino acid will emerge first, while the most basic side chain amino acid will emerge last. The amount of each amino acid (one, two, or three residues of a same type)can be determined based on the absorbances.
3. Quantitation
Amino acids from a peptide are quantified by reacting them with ninhydrin, which is used to detect a microgram of an amino acid. Most amino acids will give an intense blue color, except proline which gives a yellow color due to the secondary amino group in its structure. Furthermore, to detect a nanogram of amino acid, fluorescamine, which reacts with the alpha-amino group, can be used, yielding a highly fluorescent product. The concentration of amino acid is proportional to either the optical absorbance of the sample treated with ninhydrin or the fluorescence emitted by the sample treated with fluorescamine.
This method tells you only the composition of the proteins, not the sequence of the amino acids. Edman degradation is the one that provides the order of the sequence of amino acids in a protein.
Determination of composition of amino acid is then followed by determination of amino acid sequence. It can be done using 2 complimentary methods:
1.Edman Degradation
The reaction for Edman Degradation occurs through the use of phenylisothiocyanate and in acidic conditions
This method is done by cleaving the amino acid one by one from the amino terminal. The chemical used for this process is Phenyl isothiocyanate. Amino acids that react with this chemical will form phenylthiohydantoin (PTH)-amino acid (e.g. PTH-glycine). Under mildly acidic conditions,( PHT)-one termial residue is released. This compound is then identified using chromatographic procedures. Edman degradation is quite simple to perform (the sequencer is automated), but this method is not effective for long peptides (more than 50 residues) because it takes an hour to perform one cycle of degradation.
One example of chromatographic procedure is high-pressure liquid chromatography. In this procedure, the PTH-amino acid is separated into its components such that the amino acid's identity can be found by its absorbance and elution time.
Edman degradation is the most efficient technique used to sequence proteins without breaking the bonds between residues. Also, the development of automated sequencers has allowed for much quicker and efficient polypeptide sequencing.
Controlled cleavage using various chemical and enzyme
For listing of special enzyme and chemicals, please see [21]
Some chemicals and enzymes are known to cleave peptides at certain locations (e.g.: the amino or carboxyl end of certain amino acid). Using these chemicals and enzymes, peptides can be cut into fragments with sizes that can be analyzed using Edman Degradation. By combining 2 or more chemicals and enzymes that cleave peptides at different position, the resulting small fragments (whose amino acid sequence has been identified through Edman Degradation) can be put together in a manner similar to putting together a jigsaw puzzle.
Limitations of Edman Degradation
Even peptides less than 50 amino acids in length can become problematic when performing Edman degradation. One example of this is when the N-terminal of an amino acid is in an unfavorable position such as the inside of a protein, or when it is sequestered. In addition, Edman degradation may fail due to post-translational modifications of proteins such as glycosidation, acetylation, phosphorylation, and fatty acid addition. For example, the formylation of an amino acid will prevent reaction with phenyl isothiohydantoin. In particular, disulfide bridge between two cysteins (Cys) can complicate the sequencing by sequestering the N-terminus, or sterically hindering the cyclization of the phenyl thiourea intermediate. This could be modified by reducing the disulfides (with beta mercaptoethanol or DDT) and oxidizing the cysteine sidechains to their corresponding sulfonic acids with performic acid to prevent disulfide formation, and then performing the sequence as usual.
2. Mass Spectrometry
Mass Spectroscopy is another technique that can be used to determine protein sequence, but it can only be identified with the parent protein with the fragments cleaved by specific enzymes. The mass of ionized proteins can be obtained by measuring the time of flight of those ions as they are triggered by a laser beam and travel through the flight tube to the detector. The lighter massive ions will travel faster and arrive at the detector first due to Newton's second law (F = ma). The mass spectrum recorded is then analyzed and compared against a database of sequenced proteins. The sequences of protein fragments, therefore, can be determined in detail if the process is repeated with different enzymes; the fragments become smaller, and the overlapping fragments can thus be used to establish the order.
1-fluoro-2,4-dinitrobenzene is used in Sanger's reaction to determine amino acid sequenceNinhydrin is used to determine the presence of amino acid after hydrolysis
Limitations of Mass Spectrometry
One limitation of using Mass Spectrometry as a means to determine protein sequence is in situations in which more than one amino acid has a specific mass. For example, since Leucine and Isoleucine have the same molecular weight (131.17 g/mol), identical mass spectrometry data would be obtained for these two amino acids and in this case render the method ineffective.
Proteolysis is a process of breaking down proteins into simpler compounds under the aid of proteases. This process takes place throughout the body for a variety of purposes. For example, food is digested into compounds that the body can use for cellular processes. Proteolysis can also link to some disease processes such as the venom causes tissue death by breaking down proteins of the person whom is bitten by a snake.[1]
A protease is an enzymes that breaks downproteins through a process called proteolysis. Proteolysis breaks thepolypeptide bond that link amino acids together.
The selectivity in regulated proteolysis is controlled by generation and recognition of specific degrons on substrates.
Scientists have found that the N-end rule pathway is also a proteolytic system, in which N-terminal residues of short-lived proteins are recognized by recognition components (N-recognins) as components of degrons (called N-degrons). Substrates of N-recognins can be produced during the exposure of embedded destabilizing residues at the N terminus by protyolytic cleavage. In addition, N-degrons can also be produced through the modifications of posttranslationally exposed pro-N-degrons of stable proteins. The Modifications include oxidation, arginylation, leucylation, phenylalanylation, and acetylation. Proteolytic systems that base on generation and recognition of N-degrons have been seen in both prokaryots and eukaryotes. In general, the N-rule pathway regulates homeostasis of a number of physiological processes.[2]
The proteases are categorized by the pH the enzymes work best in. There are acid proteases, neutral proteases, and basic proteases.
A protease's function is to cleave peptide bonds. They are enzymes that work best under acidic
conditions. There actually many types of proteases; some examples include
glutamic acid proteases and threonine proteases. Although most proteases operate
under acidic conditions, there are also a few that operate under basic conditions, these
are called alkaline proteases.
Proteases occur in all organisms. The cleaving action
of a protease can either halt a protein's function or activate it. Proteases
catalyze this peptide cleaving reaction by hydrolysis in which a water group acts as a
nucleophile and attacks the peptide bond. Proteases can cleave other proteases. These enzymes
play a huge role in digestion in that they cut proteins into fragments so the body can
salvage and absorb the freed amino acids. The use of proteases in medicine is also popular as
the study of proteases have helped us better understand inflammatory conditions and immune regulation.
If a protein is part of a living cell and necessary for that cell to carry out interactions with other
cells then proteases will not cleave it because normal living cells contain an inhibitor mechanism that stops
the cleaving process. Protease deficiency can cause many health related problems. The acidity
created in the stomach through protein digestion; if there are not enough proteases then
this acidic equilibrium is disturbed causing an increase in alkaline character in the blood which could
lead to insomnia or anxiety.
The study of protease structure has also been useful in providing a means for a structure-based drug design strategy to develop new products. For instance, HIV protease is an enzyme that is crucial to the development of HIV. Inhibiting this enzyme would help prevent the HIV virus from spreading throughout the body. By studying the structure of this enzyme, researchers hoped to determine types of molecules with the capability of blocking HIV protease. Thus giving us more insight into the effect that this disease has on the immune system so we can strive to minimize or eliminate the fatal consequences of fully-blown HIV. This strategy would prove faster and more efficient than the typical trial-and-error process, which could be lengthy and unsuccessful. (The Structures of Life, U.S. Department of Health and Human Services, http://www.nigms.nih.gov)
Generation of N-degrons by conjugation of amino acids
In both eukaryotes and prokaryotes (bacteria), conjugation of destabilizing amino acids to pro-N-degrons is the main way of producing primary destabilizing residues in the N-end rule pathway. This process is interceded by evolutionary conserved aminoacyl tRNA transferases, which allows pro-N-degrons to be recognized by N-recognins under certain conditions.[3][4][5][6][7]
In eukaryotes, the N-terminal Arg is structurally favored degron for the UBR box of N-recognins. The degron Arg can be induced by ATE1-encoded arginyl R-transferases. The Arg from Arg-tRNA is transferred to the N-terminal ɑ-amino group of acceptor subatrates. In mammalian, the ATE1 gene expresses at least six isoforms through alternative splicing of pre-mRNA. The importance of protein arginylation has been confirmed by the discorvery in which mouse embryos die because of defects in cardiac and vascular development due to ATE1 deficient.[8][9][10]
Leucylation and phenyllanylation in prokaryotic N-end rule pathway
N-terminal Leu and Phe residues, primary destabilizing residues on bacterial proteins, can be induced by conjugation of destabilizing amino acids derived from aminoacyl tRNAs[11]. Two types of aminoacyl transferases are found to be intercede leucylation and phenylalanylation in the N-end rule pathway.
Experiments have shown that the aat encoded Escherichia coli L/f transferase transfers Leu or Phe to the acceptors Arg and Lys (type 1 primary residues in eukaryotes)[12].
↑Balzi E, Choder M, Chen WN, Varshavsky A, Goffeau A. 1990. Cloning and functional analysis of the arginyl-tRNA-protein transferase gene ATE1 of Saccharomyces cerevisiae. J. Biol. Chem. 265:7464–71
↑GracietE,HuRG,PiatkovK,RheeJH,SchwarzEM,VarshavskyA.2006.Aminoacyl-transferasesand the N-end rule pathway of prokaryotic/eukaryotic specificity in a human pathogen. Proc. Natl. Acad. Sci. USA 103:3078–83
↑Kaji H, Novelli GD, Kaji A. 1963. A soluble amino acid–incorporating system from rat liver. Biochim. Biophys. Acta 76:474–77
↑Kwon YT, Kashina AS, Varshavsky A. 1999. Alternative splicing results in differential expression, activity, and localization of the two forms of arginyl-tRNA-protein transferase, a component of the N-end rule pathway. Mol. Cell. Biol. 19:182–93
↑Shrader TE, Tobias JW, Varshavsky A. 1993. The N-end rule in Escherichia coli: cloning and analysis of the leucyl, phenylalanyl-tRNA-protein transferase gene aat. J. Bacteriol. 175:4364–74
↑Kwon YT, Kashina AS, Varshavsky A. 1999. Alternative splicing results in differential expression, activity, and localization of the two forms of arginyl-tRNA-protein transferase, a component of the N-end rule pathway. Mol. Cell. Biol. 19:182–93
↑Hu RG, Brower CS, Wang H, Davydov IV, Sheng J, et al. 2006. Arginyltransferase, its specificity, putative substrates, bidirectional promoter, and splicing-derived isoforms. J. Biol. Chem. 281:32559–73
↑Rai R, Kashina A. 2005. Identification of mammalian arginyltransferases that modify a specific subset of protein substrates. Proc. Natl. Acad. Sci. USA 102:10123–28
↑GracietE,HuRG,PiatkovK,RheeJH,SchwarzEM,VarshavskyA.2006.Aminoacyl-transferasesand the N-end rule pathway of prokaryotic/eukaryotic specificity in a human pathogen. Proc. Natl. Acad. Sci. USA 103:3078–83
↑Shrader TE, Tobias JW, Varshavsky A. 1993. The N-end rule in Escherichia coli: cloning and analysis of the leucyl, phenylalanyl-tRNA-protein transferase gene aat. J. Bacteriol. 175:4364–74
A mass spectrometer schematic
Mass spectrometry is an analytical technique used for identifying the mass of a compound based on the mass-to-charge ratio of charged particles. The ratio of charge to mass of the particles is determined by passing them through an applied electric field in a mass spectrometer, which has three main modules: an ion source, a mass analyzer, and a detector. In such procedure, a sample of protein for analysis is placed in the MS instrument. A laser beam is applied to allow the sample to become ionized at the ion source. Positively charged ions of different sizes result and move through the electric field through the analyzer. The lighter ions arrive at the detector first, which triggers a clock to record the time of flight (TOF). This is characteristic of MALDI-TOF mass spectrometry, which can determine the mass of individual components of large protein complexes.
Mass spectrometry is used to analyze the ionized forms of molecules in the gas phase. Mass data is obtained by measuring how fast the ion accelerates through an applied electric field and using Newton's third law, F = ma where F is force, m is mass, and a the acceleration, to calculate the mass since the applied force is known and the acceleration is the experimentally measured value.
Modern techniques for protein analysis include Matrix Assisted Laser Desorption-Ionization (MALDI) and Electrospray Ionization (ESI). Advances to this technique has now made it possible to determine protein masses with an accuracy of one mass unit or less in most cases.
The protein or peptide is co-precipitated with an organic compound that absorbs laser light in the matrix. The laser light causes the molecules to expel from the surface and capture electrons as it exits the matrix, leaving the molecules as negatively charged ions. This ionization is very necessary because only ions can be accurately measured. The ionizing laser pulse triggers a clock that measures the time of flight (TOF) for the ions. In time of flight (TOF) analysis, the ions are accelerated through the flight tube in an electric field toward the detector. The lighter ions will arrive first. One of the biggest benefits in using MALDI, as opposed to molecule ionization methods is that MALDI can give molecular fragments, which accurately represent the molecular mass of a protein or peptide. In general weights ranging from a few thousand to several hundred thousand Daltons can be measured.[1]
Electrospray Ionization is an ionization technique used for tiny amounts of large molecules such as polymers and proteins, and peptides. The apparatus usually utilizes a hollow metal tube with a sharply pointed end that faces in front of a plate. During the procedure, a sample solution is sprayed, as if from a syringe, from the metal tube, into a strong electric field with the assistance of warm nitrogen for dissolving. The solutions containing proteins or peptides flow through a fine metallic tip at a nonzero electrical potential which releases the solution as electrically charged droplets containing the protein and solvent that evaporates from the droplet, leaving the protein charged at the plate. A great feature of the ESI spectrum is the ability of the ions to carry multiple charges. This technique is often coupled with mass spectrometry for protein analysis.
The research on mass spectroscopy started long time ago, but it was not until the 20th century when the electospray ionization technique was developed. The ESI was developed by 2002 Chemistry Nobel Prize winner, Dr. John B Fenn. Together with two other scientists, Tanaka Koichi and Kurt Wuthrich, Dr. Fenn focused the research in the field of mass spectrometry. especially on the ESI technique. Dr. Fenn's research discovery was quickly put into practical use. The ESI brought many benefits during the different usages. For instance, it increased the speed with which complex new pharmaceutical compounds could be evaluated. With this usage, it led to the development of AIDS medications in the mid 1990s.
Properties that make ESI a method of choice for biological applications
1. The phage conversion process is "soft"--meaning that it can handle very fragile molecules to be ionized. Furthermore, in some cases, even noncovalent interactions can be put through Mass Spectroscopy.
2. Allows for the analysis of complex mixtures because the eluting fractions can be directly sprayed into the MS
3. Produces natural ions, allowing for the measurement of high-mass biopolymers
[2]
One of the three instruments used for electrospray ionization, Quattro II is the primary instrument used for LR ESI. This instrument is a quadrupole-hexapole-quadrupole mass spectrometer. It has a mass to change rate of 4,000 Da and equipped with an ESI source. During the procedure, samples are inserted into the Quattro II by loop injection or direct infusion through a syringe pump.
The LCQ instrument is also known as the LCQ Deca XP. This is also an electrospray ionization or ion trap mass spectrometer. It is equipped with the X calibur[check spelling] software that allows acquisition of photodiode array data and mass spectral data. This equipment has a great advantage compared to the other instruments, and it is the capability to perform multiple stages of mass spectrometry. The ability to do this allows an increase in the amount of structural information obtainable for a given molecule. The injection techniques of the LCQ is similar to that of the Quattro, but in slight variations. The LCQ can also be introduced by flow injection using an LC pump or injection valve. Another way is by the LC fitted with a colume[check spelling]. While the Quattro II is the primary instrument for LR ESI, LCQ is the primary instrument for LC/MS and LC/MSMS in ESI.
The Q-Tof is a hybrid quadrupole mass spectrometer with MS/MS capability. Compared to the other two instrument, Q-Tof has a very high resolution, sensitivity, and mass accuracy. With these properties, the Q-Tof is able to assist the mass measurement accuracy for peptides. At the mean time, it can also improved the charge state identification of multiply charged ions and greater differentiation of isobaric species. This instrument can also be equipped with different source. For example, when it is equipped with a nanospray source, it can help to analyze small samples and identify the proteins through semi or complete de novo sequencing. Just like Quattro II and LCQ, Q-Tof can be used by injecting samples through an infusion pump, loop injection, or even an HPLC column. Different from the other two instrument, the Q-Tof is the primary instrument for HR ESI.
Mass spectrometry is an analytical technique used to investigate protein complexes. Recent developments of electrospray ionization mass spectrometry (ESI-MS) have allows characterization of previously unavailable protein complexes, especially those in the gas phase. ESI-MS allows the study of the kinetics of protein complex assembly. Intermediates can be isolated as well as identified using this method. Furthermore, tandem mass spectrometry is used to determine the building blocks that form the global structure of protein complexes. Moreover, ESI-MS is used to identify equilibrium constants by comparing the relative intensity of the complex to the intensity of the subunits. Finally, ion mobility spectrometry mass spectrometry (IMS-MS) is used to analyze macromolecular assemblies. It gives information on the size, shape, mass-to-charge ratio, and number of subunits. This allows the identification of protein rings and protein complex dissociation.
Individual spectrometer elements or a single mass spectrometer are used in multiple stages of mass analysis separation corresponding to MS steps separated in space or time. In tandem mass spectrometry in space, individual spectrometer elements are separated physically, and these elements can be transmission quadrupole, sectors, or time-of-flight. While in tandem mass spectrometer in time, the separation step is done with multiple steps occurring over a range of time and with the ions trapped in the same place, or space. A quadruple ion trap or FTMS instrument can be applied to such analysis, as it performs analysis on a multiple scale. It is often known as MSn. n refers to the number of steps, hence MS3 refers to a separation composed of three steps.
There are two main ways for the mass spectroscopy analysis of proteins: the bottom-up approach and the top-down approach. In the bottom-up approach, an enzyme, such as trypsin, will be put together with proteins of interest for digestion. The "tryptic peptides" formed as a result of the digestion process are then analyzed by MS and tandem MS. On the other hand with the top-down approach, a protein molecule is analyzed by MS without prior digestion by enzymes. The bottom-up approach is more widely used for several advantages over the top-down approach:
1. Smaller protein ions are more uniform and easy to handle than a whole protein molecule;
2. Masses of smaller protein ions can be determined with higher accuracy;
3. During MS, smaller pieces of proteins are more readily to be reduced into fragments.
However, the one main disadvantage of this approach is the incomplete coverage of the protein molecule. The top-down approach appears to own this advantage while having a lot of other problems, including:
1. The difficulty in handling large protein molecules;
2. Issues about heterogeneity through a whole protein molecule;
3. The complex nature of MS.
Therefore, the top-down analyzes are limited to be applied to only low-throughput single-protein studies. To improve the results of such methods, scientists have come out with an intermediate "middle-down" approach for analyzing proteins that are larger than the smaller protein fragments, such as tryptic pedtides. Although this method is still in development, it is beginning to prove effective, as can be seen in the analysis of modifications on histone tails.[3]
Usually, the characterization and identification of protein is done by SDS-PAGE (sodium dodecyl sulfate-polyacrylamide
gel electrophoresis). However, SDS-PAGE cannot identify small amount of protein samples. MALDI-TOF MS has higher sensitivity
and resolution. MALDI-TOF MS can separate protein samples up to 100 kilo Daltons. Therefore, MALDI-TOF MS is used after
SDS-PAGE for more accuracy.
First, sample protein is separated by two-dimensional gel. Then, specific cleavage like trypsin cut the proteins. These
separated peptides are identified by MALDI-TOF MS. Heavy peptides will slowly move and lighter peptides will move fast due to
Newton's second law; F = ma. As peptides move along the tube, the peptides of different accelerations or velocities takes
different the amount of time take to reach end of tube. These separated peptides can be analyzed by matching with the computer
simulated database.
MALDI-TOF MS also can be used of DNA Hybridization, analyzing microorganism and sugars that bonds with proteins like
glycosaminoglycan.
MALDI has many advantageous properties:
1. Robustness
2. High Speed
3. Immunity to contaminants, biochemical buffers, and common additives[4]
Applications of mass spectrometry to lipids and membranes)
Lipidomics is an important part of metabalomics is concerned with he detailed depiction and analysis of the structure and function of lipids within a living system. Mass spectrometry has played an extremely crucial and significant role in the determination of the structure of lipids.
Introduction
The ever increasing number of completely sequenced genomes available have given biologists the challenge of connecting gene structure to gene function. This drive for understanding the function of genes has spurred scientists to analyze expression levels of the components the make up biological systems like mRNA and proteins. Metabolomics is the study of the endogenously synthesized intermediates called the metabolome and is thought to represent the final result of gene expression. Endogenously synthesized intermediates (metabolomes) are primarily composed of lipids and fats. Studying these lipids can provide information in determining the relationship between the role of lipids and various diseases like cancer, atherosclerosis, and chronic inflammation. In addition, many lipids have cellular membranes and studying their interactions with membrane associated proteins/enzymes can give information on the development of drugs to inhibit these interactions that diseases have.
The first mass spectrometer was constructed by Nobel Laureate Sir J.J Thomson and was used to analyze marsh gas. He observed that the mass to charge rations were 16 to 26, which was identified to be positive ions of methane and acetylene, respectively. Ever since mass spectrometry has been extremely important for the study of proteomics and metabolomics.
Lipid Definition and Classification
The categorization of lipids can be wide ranged.It can include organic compounds like fats, oils, waxes, sterol, and tryglycerides, and are insoluble in water but soluble in nonpolar organic compounds. They can also be defined as oily to touch. However, there are many lipids that are not always bound by these definitions. This has led to the official classification of 8 categories of lipids. They are fatty acids, glycerolipids, sphingolipids, sterollipids, prenol lipids, saccharolipids, and polyketides.
Analysis of membrane proteins is challenging because of their hydrophobicity, complex post-translational modifications (PTMs) and relatively low abundance, thus they are not accessible by traditional methods such as X-ray crystallography and NMR spectroscopy. However, with the recent advancements in technology and methodology (ie. better liquid chromatographic performance), mass spectrometry (MS) accelerates membrane protein analysis in a large degree; specifically towards the determination and understanding of the complete plasma membrane (PM) proteome, membrane protein topology, membrane protein-protein interactions and signaling networks originating at the membrane.
MS proteomics to determine the complete membrane proteome
The hydrophobicity and low abundance of membrane proteins, and the intricate post-translational modifications has made the analysis of complete membrane proteome very difficult, especially when using traditional approaches, because it is technically challenging to isolate hydrophobic and insoluble proteins. However, with mass spectrometry, coupling with compatible detergents, better instrumentation (such as Multidimensional protein identification technology (MudPIT), which helps to facilitate analysis by separating peptides based on their charge and hydrophobicity), and optimized liquid chromatographic performance, the process of identifying thousands of proteins in one single analysis and get a global overview of all the proteins in the membrane can be done easily and more accurately.
Membrane proteins are generally categorized into three different types:
1. Integral membrane proteins, which is membrane penetrating, is determined to have a few β-barrels (for example, maltoporin) and a majority in α-helical arrangement (for example, the insulin receptor), which can be further divided into four different types based on terminus of the protein and number of times the protein traverses the membrane.
2. Peripheral membrane protein is found to be attached to membrane as in-plane α-helix (such as microtubule-affinity-regulating kinase) or by electrostatic interactions (such as diphtheria toxin).
3. Lipid- anchored proteins is found to be attached to fatty acid, prenyl group or glycophophatidylinositol anchor by covalent bonding (for example the G proteins).
When dealing with detergent-resistant membrane domains and microdomains, mass spectrometry, coupling with multicomplexed fractionation and 1D and 2D gel-based approaches, can also effectively be used to analyze the protein components of the membrane. Studies have shown that these detergent-resistant membranes contain proteins that account for concentrated function.
Instead of using traditional methods such as X-ray crystallography and NMR spectroscopy, mass spectrometry can be used to determine the membrane protein structures, folding and topology at submolecular level, in conjunction with one of the following methods:
1. Hydrogen/deuterium (H/D-MS) exchange
This is based on the hydrogen atom exchange between proteins and the surrounding aqueous solution. And the rate of exchange is ruled by the solvent accessibility through the bilayer of the membrane. New findings using this method are that most of the hydrogen bonding interactions in the protein only moderately stabilized the folded state, and that there are several dynamic regions within the β2 adrenergic receptor (G-protein-coupled receptor).
2. Oxidative or hydroxyl radical probe mass spectrometry
This has the key advantage, over other approaches, in that hydroxyl radicals can be generated directly in solution and react permanently with minimal modification through the incorporation of a limited number of oxygen atoms at reactive residue side chains. See http://en.wikipedia.org/wiki/Protein_footprinting
3. Covalent tagging with regents such as carbodiimide diisopropylcarbodiimide (DiPC-MS)
This is also specific in the residues, Asp and Glu. From this method, it was shown that Glu269 serves an important role in substrate binding of the membrane protein lactose permease.
One main advantage of using MS-based topology approaches is that they are not limited by the type and the size of the membrane proteins.
Mass spectrometry is useful to discover exactly which proteins physically and functionally interact, thus further our understanding of the molecular function of the plasma membrane proteins.
Two approaches are generally used:
1. Isolate membrane protein complexes by antibody purification: Best for identifying multiprotein complexes
With this antibody purification approach, more receptors are identified, such as the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR), γ-aminobutyric acid and kainate receptors, in addition to the previously identified N-methyl-D-aspartate (NMDA) and 5-hydroxytryptamine (5-HT2C) receptors.
2. In vitro binding experiments followed by mass spectrometry: Best for finding direct high-affinity interactions such as ligand and receptor pairs
Plasma membrane is where all the signaling between cells takes place. Many surface receptors, for example, GPCPs, receptor tyrosine kinases, adhesion signaling molecules and channels, are embedded in the plasma membranes. To further understand membrane signaling, mass spectrometry is used to determine the changes in protein abundance or PTM such as phosphorylation, after treated the cell cultures with growth factor – ligand.
One example of membrane signaling would be the analysis of brassinosteroid (BR) membrane. Arabidopsis thaliana seedling were first treated with BRs. Then the membrane fractions were analyzed using 2D gel and mass spectrometer. The findings showed that membrane-associated kinases of BR transmembrane receptors are responsible for BR membrane signaling. Another example is that the epidermal growth factor signaling of phosphorylated proteins was investigated using MS with the use of stable isotope labeling and affinity capture. From these studies, thousands of proteins were successfully identified.
Quantitative proteomics is becoming a major part of biological research because it allows scientists to track the dynamic changes of proteins, including post-translational modifications and the formation of protein complexes. This technique often plays a large role in generating new insights and hypothesis for biological processes, which can then be validated by other research approaches. Understanding how proteins change and interact leads to greater understanding in cellular processes and disease progression. For example, it is known that histone regulates gene activation and DNA repair by undergoing methylation, acetylation, and other modifications that changes its interaction with DNA and nuclear proteins. Studying the structural changes of histone during transcription would thus be helpful in learning how to deactivate undesirable genes and amplify beneficial genes. Another heavily targeted area of study is the comparison of protein expression, modifications, and interactions between the diseased state proteins versus that of the normal condition. Understanding these differences can offer valuable insights into the mechanics of disease progression and provide guidance for structure-based drug design. Although tracking protein dynamics is difficult due to the complexity of the biochemical pathways and the drastic changes during different cellular states, the use of mass spectroscopy has greatly reduced these challenges.
Before peptide tagging was used to study protein dynamics, quantitative proteomics relied on two-dimensional polyacrylamide gel electrophoresis (2D-PAGE). After running 2D-PAGE, the spot intensities from two or more samples were compared and a mass spectroscopic analysis was used to identify and quantify the protein. Modern quantitative proteomics relies on isotope labelling and mass spectrometry. These methods include:
The protein of interest is purified from different cell states and immersed in labelling reagents that has a biotin tag and a linker region with either heavy or light isotopes. These reagents bind to cysteine-containing peptides, creating a labelled peptide. The proteins are then combined and digested, forming labeled and unlabelled peptide fragments. Combining the digestion process reduces variation in the procedure so that experimental error can be minimized. The labeled peptides are first selected through affinity chromatography based on the presence of the biotin marker, then analyzed with mass spectrometer. Proteins labeled with the light isotope show a lower mass to charge ratio. The relative abundance of the proteins can be determined by the ratio of the peak intensities.
Stable Isotope Labeling with Amino Acids in Cell Culture (SILAC)
Stable Isotope Labeling with Amino Acids in Cell Culture (SILAC)
Stable isotope labeling with amino acids in cell culture (SILAC) is a method of in vivo labeling of proteins for mass spectrometry analysis. The cell culture is grown in a “light” medium and a “heavy” medium. These mediums are given identical conditions except for one particular amino acid that contains a heavy isotope for the “heavy” medium and a light isotope for the “light” medium. Typical isotopes chosen to make the “heavy” medium include 2H, 13C, and 15N. The cells incorporate the heavy or light isotope amino acids naturally during metabolism. After a few generations, every instance of that amino acid will be replaced by the type of amino acid provided in the medium. The proteins grown in both the mediums are then extracted from the cells, purified, and combined together. The proteins are digested with a protease, separated by high performance liquid chromatography, and analyzed by mass spectrometry. As in ICAT, the relative abundance of the proteins can be determined by the ratio of the peak intensities.
Isobaric labeling is a technique that attaches same mass chemical groups to peptides and then using tandem mass spectrometry to determine the relative abundance of the isobaric tag, which represents the abundance of the respectively tagged peptide. Each isobaric tag is composed of a reporter group and a balance group. A peptide-reactive group is attached to the tag to increase peptide affinity to the tag. In each experiment, many isobaric tags can be used to label proteins. All the reporter groups are made to have different masses by varying the amount of 13C, 15N, 18O in the reporter molecule. The balance group is a carbonyl group that is also of varying masses. The overall mass of the reporter group and the balance group are constant for all of the labels. This is useful as an internal standard for mass spectrometry because all labeled proteins should appear in one peak since all the masses are the same. There are two types of isobaric labeling, both utilizing the method as described above:
iTRAQ (isobaric tag for relative and absolute quantification)
Hydrogen exchange mass spectrometry is a contemporary method to study protein dynamics, protein-solvent interactions, and protein complex interactions. This technique takes advantage of the exchanging behavior of amide hydrogen to label the protein with deuterium, which can be detected through mass spectrometry or NMR analysis. This method generates information for protein dynamics by describing the solvent accessibility and level of disorder for the various parts of the protein.
In solution, the hydrogen covalently bonded to the nitrogen of the peptide (called amide hydrogen) exchanges proton with the solvent. By replacing H2O solvent with D2O, the amide hydrogen can be incorporated into the peptide during the hydrogen exchange process. This is usually accomplished by diluting the protein in H2O buffer with the analog D2O buffer tenfold, thus surrounding the protein with mostly D2O and lessening its contact with H2O. Hydrogen exchange reaction is significantly affected by temperature and pH. Since the exchange process can be acid or base catalyzed, pH must be monitored carefully during the D2O incubation. To study proteins at the natural state, exchange condition is set at pH 7-8 to mirror the physiological environment of the proteins. At pH 2.6, the exchange reaction occurs the slowest. A standard exchange experiment involves immersing the protein for a set amount of time in the D2O buffer, then rapidly shifting the pH of the solution to 2.6, thus slowing the exchange reaction, a process called quenching. A set of exchanging experiments consist of similar exchanging conditions but each experiment is given varying reaction times, commonly ranging from 10 seconds to 3000 seconds or longer. Exchange reactions occur extremely fast at room temperature. To more accurately characterize the deuteration levels among the proteins samples at each of the time points, exchanges are performed at cold temperatures (around 4oC) to decrease the rate of exchange.
To analyze the deuterated proteins with mass spectrometry, the proteins are denatured and digested to form peptides. The peptides are separated by HPLC and analyzed by the mass spectrometer. Since the deuterium nucleus is heavier than a hydrogen nucleus because it contains a neutron as well as a proton, the deuterated peptide has a higher mass compared to the non-deuterated peptide. Based on this mass difference, mass spectrometer can accurately distinguish among the peptides with different deuteration levels. The rate of deuteration for each peptide can also be tracked by measuring the masses of identical peptides that are immersed in D2O for different time points.
Two factors govern exchange behavior for each peptide: solvent accessibility and hydrogen bonding. Peptides buried in the interior of the protein or surrounded by hydrophobic surfaces show less exchange behavior due to the lack of contact with the D2O solvent. Peptides that form the surface of the protein exchange rapidly and completely because they are in constant contact with D2O. Hydrogen bonding also determines exchange behavior because the hydrogen participating in bonding cannot spontaneously exchange without breaking the bond. Thus great rates of exchange can be attributed to lack of bonding or disorganization. Flexible parts of the protein that are not structured can be characterized by the incorporation of many deuterons. Hydrogen exchange mass spectrometry is also used to study the dynamics of protein complexes to determine which areas of the protein changes in flexibility or solvent accessibility upon binding. This can help identify the areas of the protein that are pertinent to protein-protein interaction and the areas that are dramatically affected by the interaction.
Fourier-transform mass spectrometry is a type mass spectrometry that takes advantage of ion-cyclotron resonance to select and detect ions. Fourier-transform MS was invented by Alan G. Marshall and Melvin B. Comisarow at the University of British Columbia in 1974. Fourier-transform MS, also known as Fourier transform ion cyclotron resonance MS has high resolution; therefore, it is used as determining the composition of molecules based on accurate mass. It is also capable of studying large macromolecules such as proteins with multiple charges due to its high resolution.
Ion-trap mass spectrometry exists in both linear and 3-D varieties. IT MS was invented by Wolfgang Paul. It uses constant DC and radio frequency oscillating AC electric fields to trap ions in a small volume. This 3-D trap consists of a ring electrode separating two hemispherical electrodes. A mass spectrum is obtained by changing the electrode voltages to eject the ions from the trap. Such technique gives its compact size and the ability to trap and accumulate ions to increase the signal-to-noise ratio of a measurement.
Magnetic-sector mass spectrometry uses a static electric or magnetic sector to affect the path and/or velocity or the charged particles. There are two main types of magnetic-sector MS, single focusing analyzers and double focusing analyzers.
Quantum Sniffer(the figure shown on the right) is an ion mobility mass spectrometry developed by Implant Science Corporation. It is a photonic, non radioactive ionization to detect nano particles in an aqueous solution. The molecular weight, shape, and size of the ionized particles would impact the ion mobility as it pass through drift region and reach the detecting point. The detection of the samples are collected by the vortex, ionized photonically, and analyzed via ion mobility spectrometery (IMS). The samples are collected through a vacuum space, then it is pump into the ion source with electrical current in the ion-molecule reaction region. Then, the ion molecule is then further analyzed based on its drift time, the time for it to go from one chamber to another based on its molecular weight. At the end of the result, computer was able to analyzed a resultant graph of detecting particles.
The emergence of advances in studying of structural genomics as well as proteomics can yield more information on soluble proteins and their complexes.
An example of a new type of mass spectrometry technique is called laser induced liquid bead ion desorption (LILBID). This technique allows can detect membrane proteins by generatingseries of microdroplets of proteins that then get radiated using a laser into an IR range. At lower intensities, complete complexes can be identified whereas at higher intensities, individual subunits can be detected. This new technique can also be thought of as a combination of another type of mass spectrometry often used, MALDI and electrospray. Despite the idea that LILBID could be a combination of these techniques, there are some key similarities and differences between LILBID itself and ES techniques. Both LILBID and Es are able to explain the stoichiometry of intact membrane protein complexes; however ES has the ability to determine small molecule binding that occurs directly within the complexes. In addition, mass spectrometry studies using this ES technique shows that micelles can indeed exist while in the gas phase. Since LILBID has a lower mass resolution compared to ES, it is unable to provide a more precise characterization of the mass differences between the intact complexes.
Although MS technologies have been greatly improved in the last two decades, further improvements are needed for the analysis of protein molecules:
1. Sensitivity of the instrument should be improved in order to analyze smaller number of cells or samples, which allows one to look at corresponding cellular components with their specialized functions;
2. Improved methods are needed to measure low abundance components within protein molecules despite the existence of higher abundance molecules;
3. Higher instrumental speed is needed to allow for deeper and more routine protein analyzes;
4. More robust instruments should be developed to enhance the usage of MS, since some biologists are not quite familiar with these instruments;
5. Improved techniques should be devised to prepare "frozen" samples that are easier to be analyzed.[7]
↑Chait, Brian T. "Mass Spectroscopy in the Postgenomic Era." Laboratory for Mass Spectroscopy and Gaseous Ion Chemistry, The Rockefeller University, New York, NY 10021; email chait@rockefeller.edu.
↑Annu. Rev. Biochem. 2011. 80:239-46 The Annual Review of Biochemistry is online at biochem.annualreviews.org
↑Chait, Brian T. "Mass Spectrometry in the Postgenomic Era." Laboratory for Mass Spectroscopy and Gaseous Ion Chemistry, The Rockefuller University, New York, NY 10021; email: chait@rockefeller.edu.
↑Richard Harkewicz and edward A.Dennis(5 April 2011). [6]. "AnnualReview", p. 2-6.
↑Savas, Jeffery, Bejamin Stein, Christine Wu, and John Yates. "Mass Spectrometry Accelerates Membrane Protein Analysis." Trends in Biochemical Sciences. 36.7 (2011): 388-396. <http://dx.doi.org/10.1016/j.tibs.2011.04.005>.
↑Annu. Rev. Biochem. 2011. 80:239-46 The Annual Review of Biochemistry is online at biochem.annualreviews.org
X-ray crystallography is an experimental technique that is used to determine the structure of a molecule. X-ray crystallography works because the x-ray radiation used to study the sample is of a wavelength that is short enough to be able to discern the features of the molecule. The sample of interest is isolated, purified, and crystallized, and then the crystal sample has a beam of x-ray radiation fired at it. The path of the x-ray photons is perturbed depending on the structure of the molecules making up the structure of the crystal, and the path of the x-rays is captured on a photosensitive paper behind the crystal sample. The patterns the x-rays make on the photosensitive paper are analyzed and the structure of the molecule can be deduced.
Long wave radiatonShort wave radiation
Recombinant Protein Refolding Methods used in conjunction with X-Ray Crystallography and NMR:[1]
Because x-ray crystallography and NMR require large amounts (on the order of milligrams) of a purified protein (often unattainable with complications in current purification techniques) to analyze the protein’s structure, recombinant techniques are usually employed whereby a host organism is manipulated to express the protein to be studied. Usually, protein refolding methods must then be used because the protein does not fold properly and abnormalities, known as inclusion bodies, in the protein’s structure develop. To attain the correctly folded protein, the abnormalities are removed; the protein is denatured and then refolded to its correct structure. As a result, the refolded protein can then be studied via x-ray crystallography or NMR. Structure and function analysis of known proteins confirms that this method is comparable to studying the protein directly from its native source.
A solution of proteins is placed in a magnetic field and the effects of different radio frequencies on the resonance of
different atoms in a protein are measured. Proteins must be small (~120 residues) and must be soluble for this method.
Ab initio methods, homology modeling, and fold recognition are also other popular methods used to determine the structure of tertiary proteins.
A visualization can be created by this method which takes place at or below liquid nitrogen temperatures. It is a fairly new technique that can create visualizations at an extremely high resolution. This method is a form of electron microscopy which utilizes the extremely low temperatures to reduce the occurrence of radiation damage to the specimen.
Ab initio methods is used to predict tertiary structure of protein from first principle. It bases on Thermodynamic hypothesis predicts that the native conformation of a protein corresponds to a global free energy minimum of the protein/solvent system.
Homology modeling is a class of methods for constructing an atomic-resolution model of a protein from its amino acid sequence. Its motivation is if sequence similarity is high, then structural similarity is probably high, too. Almost all homology modeling techniques rely on the identification of one or more known protein structures likely to resemble the structure of the query sequence, and on the production of an alignment that maps residues in the query sequence to residues in the template sequence. The sequence alignment and template structure are then used to produce a structural model of the target. Because protein structures are more conserved than DNA sequences, detectable levels of sequence similarity usually imply significant structural similarity.
Hard X-ray fluorescence (XRF) microscopy is a powerful method for structural visualization. It detects traces of metal distributions in biological systems like Cu and Zn. 10-15 elements are mapped at the same time, which leads to accurate elemental colocalisation maps. The trace elements are important for most life forms. Metals play an essential role in many proteins by catalyzing functions or for a structural role. Metals are also recognized to have an impact on human health and disease.
X-ray fluorescence is suited for quantifying trace elements. X-ray fluorescence can be excited through x-ray beams or through exposures to a particle like a proton or electron. This helps map the elemental content at high spatial resolution. X-ray microprobes have zone-plates, Kirkpatrick-Baez mirrors, compound refractive lenses, and tapered capillaries, with ranging probe sizes and photo intensities.
Tomography (known as slice imaging) is a two-dimensional technique that is performed on many neighboring slices together, resulting in a three dimensional reconstruction. XRF has also used full-field imaging and structure detector approaches. The technologies have mixed strengths and weaknesses with varied spatial resolutions, sensitivities, and different elemental contrast. Projection tomography uses projections of a specimen as input data to a tomographic reconstruction algorithm. However, x-ray fluorescence micrographs are not exactly equivalent to projection imaging because of self-absorption effects, which include the absorption of the incident beam and re-absorption of the fluorescence by the specimen. Heavier metals in thicker biological tissue have successfully used back projection without correction. Confocal tomography is a direct-space approach to scanning XRF tomography with axial resolution reaching below 5 micrometers. A collimator or lens confines the field of view of the energy-dispersive detector so that the signal derives from only a small portion of the illuminated column. The probe volume is then reduced to a spheroid and elemental distribution is mapped by scanning the specimen through the probe volume in three dimensions. The confocal geometry also allows direct access to a small region of specimen; however, it can be very difficult to target features of interest.
Self-absorption plays a significant role in XRF tomography, and so good correction algorithms are required to ensure image fidelity to expand the specimen size domain, to extend the elemental domain, and to maintain quantitative data accuracy.
In soft x-ray tomography, projection images of a specimen are collected at different angles around a rotation or “tilt” axis, which is mathematically capable of being computed to reconstruct the specimen. A problem that is encountered with three-dimensional tomography is that biological materials become damaged when exposed to intense light in an x-ray microscope from photons or from ultra violet illumination in a fluorescence microscope. However, with soft x-ray tomography, collections of images are acquired at 1-2 degree increments through 180-degree rotation. This causes the specimen to get structurally damaged by receiving a large dose of radiation. However, cryoimmobilization of the specimen avoids this problem. When cells are imaged at liquid nitrogen temperature, more than a thousand soft x-ray projection images can be collected without apparent signs of radiation damage.
The major advantage of water-window soft x-ray tomography is that it can be applied to any imaging investigation in cell biology such as imaging simple bacteria, to yeast and algae, to advanced eukaryotic cells and tissues. The images also keep eukaryotic cells in the native state without the use of stains or labels, which is something that can’t be done with light or electron microscopy.
Electron Tomography, also known as ET, is a novel visualization technique that does what other structure determination methods cannot do in that this method is able to bridge the gap between the atomic structural information of supramolecular structures and its cellular events. It allows visualization of not only the structure of a molecule but its association with other structures and organelles. For instance, electron tomography has opened doors to the observation of virus propagation and viral life cycle in the host cell. Tomography slices allow the examination of molecular architecture of virus as it attaches to cells, penetrate cells, move to replication sites, assemble progeny and transport them to the membranous regions, and exits the cell. Though this technique is still new, it has already been applied to human and simian immunodeficiency viruses (HIV and SIV) and other viruses that infect plant, animals, and humans.
The following are examples of cellular events that have been observed and elucidated through the use of Electron Tomography:
1) Virus Entry: ET shows the presence of "entry claws" which are characteristic architectures of virus coming into contact with the host cell. The entry claw is made up of five to seven rods and represents the interaction between the viral spikes and cell surface receptors. Furthermore, CT shows that some cells, such as vaccinia virus infected cells, shows clear change in shape of the vaccinia virus before and after intrusion of the cell.
2) Virus Factory: ET sheds more light on viroplasms, which are inclusion bodies found at the viral replication site responsible for virus assembly and replication. ET supported the discovery of core proteins P1, P3, P5, and P7 inside the viroplasm and outer capsid proteins P2, P8, and P9, and the formation of virus particles around the viroplasm. With this information, scientists were able to propose a three-step viral replication process: 1)formation of core particles inside the viroplasm, 2) core particle coating with outer capsids at the periphery of the viroplasms, and 3) transportation of mature virus to the membrane region by microtubules.
3) Transportation of Virions inside the Cell: The third step of the proposed viral replication process was observed and confirmed by ET. After the virus propagate at the initial site, they move to another site for secondary multiplication. This movement to another site occurs through virus utilization filamentous substances such as microfilaments and microtubules (MTs) composed of actin and beta tubulins. ET has allowed visualization of the mechanism of cytosolic transportation and strong suggestion that microtubules aid in the transportation of newly assembled virus to the membrane which leads to cell-to-cell spread of the virus. This suggests that microtubules assist in cell-to-cell spread rather than in the entry of viruses to the replication site. Finally, ET shows a gap between the virus and the microtubules indicating that the virus particles do not interact directly with microtubules. Instead, the gap is filled with a rod-like structure that might act as a plus end-directed motor.
In conclusion, ET has shown to have two advantages to the other structure determining methods. The first is that ET prevents misleading conclusions based on 2D structures. Second, observation of finer structures in cells and virus allow elucidation of the organization of detailed virus particles in association with microtubules. This has only been observed using ET.
In soft x-ray microscopes, third generation synchrotrons are used as the x-ray source. Soft x-ray projection images are a result of precision nanostructured x-ray lenses, high-efficiency direct detection CCD cameras, and well designed transmission X-ray microscopes. Images are generated using phase contrast techniques.
Soft x-ray microscopes are operated using photons with energies in the “water window,” which is the region of the spectrum that lies between the K shell absorption edges of carbon and oxygen. White cellular water remains transparent as the structures in the cell are visualized as a function of biochemical composition and density. Lipid droplets which are structures that absorb more than high water content organelles like vacuoles.
Electron Tomography of the supramolecular structure of virus-infected cells. Kenji Iwasaki and Toshihiro Omura.
NMM spectroscopy is unique in being able to reveal the atomic structure of macromolecules in solution, provided that highly concentrated solution can be obtained. This technique depends on the fact that certain atomic nuclei are intrinsically magnetic. The chemical shift of nuclei depends on their local environment. Furthermore, the spins of neighboring nuclei interact with each other in ways that provide definitive structural information. This information can be used to determine complete three-dimensional structures of proteins.
Nuclear Magnetic Resonance Spectroscopy (NMR) is an analytical technique which exploits the fact that certain nuclei possess a property called spin (I). NMR spectroscopy is possible due to the Zeeman Effect discovered first in the 1890s. The structures of proteins can be determined by NMR by first preparing the isotopically labeled samples. In the magnet, the "natural magnets" in the atoms of the sample line up with the magnetic field of the NMR magnet. Then, the sample is exposed to a series of radio wave pulses to disrupt the magnetic equilibrium of the atoms. The reaction of these nuclei can be observed to assess their chemical nature specifically, their chemical shift properties. Sets of experiments can be conducted to refine possible structures and present the average of the experiments as the final structure. It can have advantages over X-ray crystallography because the samples of proteins can be in solution rather than crystallized form and other properties of the sample protein, such as flexibility and interactions, can be determined while X-ray crystallography only reveals the structure of the protein.
It is the splitting of atomic energy levels when the atoms are placed in a magnetic field. This effect was later found to occur as a result of nuclear spin. In 1924, Wolfgang Pauli first suggested the theoretical basis of NMR when he concluded that some atomic nuclei should possess nuclear spin and these nuclei would split into degenerate energy levels when placed in a magnetic field. These nuclei have a magnetic moment, in and of themselves, which is called spin. Nuclei that possess spin are said to be NMR active. Some NMR active nuclei can be seen in Table 1, with italicized nuclei being those commonly studied in biological molecules.
Table 1 NMR active nuclei
Separately, Bolch at Stanford and Purcell at Harvard in 1946 each translated the Zeeman effect into the first NMR spectrum. In 1949, Proctor and Yu at Stanford measured the first chemical shift and spin couplings. Initially, commercial NMR spectrometers were continuous wave type and were only able to detect the most sensitive nuclei, 1H & 19F.
NMR ‘active’ nuclei are active if the nuclear spin, I, is non zero.
To determine spin of nuclei:
Mass # is even and # neutrons is even, I=0, therefore no spin
Mass # is even and # neutrons is odd, I= integer, therefore spin
Mass # is odd and # neutrons is even or odd, I=half integer, therefore spin
The cause of spin can be visualized with the following analogy. A rotating charged particle (e.g. hydrogen nucleus aka proton) will produce a magnetic field perpendicular to the direction of rotation which is analogous to the magnetic field of a bar magnet, as seen in Figure 1.
Figure 1
Absent an external magnetic field (B¬0) these active nuclei are oriented in random directions, but in the presence of an external magnetic field (B¬0) these nuclei orient themselves either with or against the field, as seen in Figure 2.
Figure 2
Nuclei are dispersed between states of different energies. The different energy levels are induced by the presence of the external magnetic field (B¬0). (For spin ½ nuclei two orientations are possible, with the higher energy orientation being ‘β spin state’ and the lower energy orientation being ‘α spin state’.) This is evident in the following equation where E=energy, h=Planck’s constant, ν=frequency, γ=gyromagnetic ratio (nucleus dependent), B0= external magnetic field:
ΔE= h ν = γ B0
To see this graphically see Figure 3.
Figure 3
These induced energy level differences are what allow the use of NMR. At equilibrium, nuclei will occupy both levels of energy, low and high. However, according to the Maxwell-Boltzmann Distribution, the lower energy level will be occupied by a greater number of nuclei. This difference in the number of nuclei, between low and high energy levels, is what NMR exploits. The energy required to cause a spin flip, transition from low to high energy state, corresponds to the radio frequency range of the electromagnetic spectrum.
ΔE= h ν
A nucleus will only flip spins, absorb energy, when the frequency of electromagnetic radiation just matches the change in energy, ΔE. This is called the point of resonance. Nuclei will have differing resonant frequencies based on their local electronic environment. This can be seen in “A handmade example of a proton (1H) NMR spectrum.” All nuclei under observation are irradiated with radio waves to the higher energy state and are then allowed to decay back to the lower energy state. This decay is what is observed. A free induction decay signal is collected as the nuclei fall back to the lower energy state, Figure 4 depicts a generic FID.
Figure 4
This FID is then subjected to a Fourier transform to convert from the time domain to the frequency domain, seen in Figure 5.
Figure 5
A change in the magnitude of the magnetic field will change the resonant frequency of the nucleus. Therefore, in order to compare spectra between instruments with varying size magnetic strength it is necessary to convert from frequency to parts per million, ppm. This can be done with the equation seen in Figure 6.
Figure 6
With the arrival of Fourier Transform instruments in the 1970s it became possible to acquire spectra of nuclei that are not naturally abundant, such as 13C which is only 1.1% naturally occurring. This requires the acquisition of multiple spectra and adding them together to get a greater sensitivity. The signal to noise ratio (S/N) will increase as acquisitions increase, because the signal magnitude increases linearly with acquisition number while the noise magnitude increases as the square of the number acquisitions. So, the S/N will increase as the square of the number of acquisitions as seen in Figure 7.
Figure 7
The Nobel Prize in Chemistry 2002 went partly to Kurt Wuthrich for “NMR Studies of Structure and Function of Biological Macromolecules,”[1] so NMR is important to structural biochemistry. Table 2 lists Nobel prizes awarded for discoveries related to NMR spectroscopy.
Table 2A handmade example of a proton (1H) NMR spectrum
NMR is a powerful technique that can be used to determine the structure of proteins in highly concentrated solution, by analyzing the relative positions of certain atoms (Shriner 136). Protein NMR exploits the absence of zero net nuclear spin of the most abundant isotopes of carbon and oxygen (12 and 16) as well as the abundant isotope of Nitrogen (14) with a net nuclear spin of 1. NMR cannot be performed on an atom with zero net nuclear spin. Since there are many distinct nuclei present in a single protein much overlap is expected in the chemical shifts produced through the NMR analysis therefore a multidimensional approach is applied. Magnetization is applied to the protein and nuclei through electromagnetic pulses that can magnetize nuclei in two ways. The first involving magnetization through nuclei bonding and the second through open space. Each approach offers a different way to analyze the protein such as the first approach allowing for distinct nuclei to be targeted whereas the second approach allowing for structure calculation distance restraints and targeting an unlabeled protein.
Intrinsically magnetic nuclei display spinning properties that produce a magnetic moment. When an external magnetic field is applied to the system the magnetic moment can adopt either an alpha or beta state. The strength of the magnetic field determines the proportionality of alpha to beta with a tendency to favor alpha as it is aligned with the field and has a lower energy. By supplying an electromagnetic pulse alpha spin nuclei will change to beta and produce resonance. By keeping a constant magnetic field on the system and adjusting the frequency of the electromagnetic pulses a spectrum of resonance can be obtained.
Currently, NMR spectrometers use the Fourier transform method of pulse radiation. To change the nuclei in alpha state to beta, a strong pulse of radiation is used. Then the pulse of radiation is removed and the nuclei go back to their original alpha state, giving a decay signal. This signal is converted by the computer to a frequency domain spectrum in a very fast way. By storing many signals a more intense spectrum is produced. (Shriner 136)
Electrons around a magnetic nucleus produces a local magnetic field opposite to the applied magnetic field. Magnetic nuclei can absorb the electromagnetic pulses at specific frequencies coined "chemical shifts" (signal positions) expressed in parts per million (ppm). These chemical shifts are designated relative to a standard compound such as a derivative of tetramethylsilane (TMS) (CH3)4Si (which has a chemical shift of 0.0) that is soluble in water. NMR spectra that are produced through this assay can be analyzed, and with the combination of a multidimensional analysis the structure of the protein can be determined.
When an atomic nuclei is influenced by a magnetic field, it creates a magnetic moment which will give an orientation of spinning to itself. Different orientation of spinning, spin states, exist in different states of energy - alpha-state and beta-state - where alpha-state is found to be more stable with a lower energy through experiments as it has the same direction with the magnetic field. In order to transits alpha-state to beta-state (excited state), pulse of electromagnetic radiation is needed to increase the frequency of the electromagnetic radiation (through irradiation) to have the spinning nucleus to overcome the energy barrier and change its orientation. (See Bohr Model for more information) Thus, resonance will be obtained. It was discovered by American physicist Edward Mills Purcell and physicist Felix Bloch from Switzerland in 1946.
The difference between energy levels split by a magnetic field corresponds to the energy of radio waves. So, the electronics required for the NMR spectrometer weren’t developed until WWII when the government invested heavily into radar technology. Radar is simply the use of radio waves to detect location and direction of moving objects. Better frequency synthesizers, amplifiers, receivers and signal processors all contributed to advancements in the field of NMR spectroscopy.
The components of a NMR Spectrometer can be seen in Figure 8.
Figure 8
Some common 2D experiments can be seen in Table 3:
Common 20 experiments (Table 3)
COSY
Correlated Spectroscopy Through bond H-H correlation
TOCSY
Total Correlation Spectroscopy Extended through bond H-H correlation
1D NOE
Nuclear Overhauser Effect Spectroscopy H-H through space correlation
NOESY
Nuclear Overhauser Effect Spectroscopy H-H through space correlation
ROESY
Rotating Frame Overhauser Effect Spectroscopy Similar to NOESY
The magnets used for NMR are very strong. Those used for high resolution protein structure determination range from 500 megahertz to 900 megahertz and generate magnetic fields thousands of times stronger than the Earth’s. Although the sample is exposed to a strong magnetic field, very little magnetic force gets out of the machine.
NMR magnets are superconductors, so they must be cooled with liquid helium, which is kept at 4 Kelvin. Liquid nitrogen, which is kept at 77 Kelvin, helps keep the liquid helium cold.
Only certain isotopes are capable for use in NMR. The most common isotope that is used in NMR is Hydrogen-1 (Proton NMR). Other commonly used isotopes are Carbon-13 and Nitrogen-15.
The Vector Model only holds for uncoupled spins. Nuclei that are active under NMR spectroscopy have associated with them a nuclear spin magnetic moment, which means that these nuclei produce a small magnetic field. When these nuclei are placed in a magnetic field the energy of the interaction depends on the angle between the field and the magnetic moment. The highest energy interaction is when the magnetic field and the magnetic moment point in opposite directions, and the lowest energy interaction is when the magnetic field and magnetic moment point in the same direction.
Before a magnetic field is applied to a population of nuclei the magnetic moments of these nuclei are oriented randomly due to random thermal motion. After a magnetic field is applied one has to wait for the spins to come to equilibrium, a process known as relaxation. Pointing in the direction of the magnetic field is slightly energetically favored, and hence only a very small number of spins will do so, forming the bulk magnetization vector. The sample is hence magnetized. At equilibrium the bulk magnetization vector will point in the direction of the magnetic field (we will assume this is the z direction) and will be stationery. If the magnetization vector was tilted away from the axis it would precess at the Larmor frequency (-gyromagnetic ratio * magnetic field).
The purpose of a pulse is to quickly change the direction of the magnetization vector. This can be done by creating a much weaker magnetic field in the direction one desires, but allowing it to oscillate at the aforementioned Larmor frequency. This will result in the magnetization vector pointing in the direction one desires. This occurs because when the B1 field (the weaker field) oscillates near the Larmor Frequency, the result is that the strength of the B0 field (the stronger field in the z direction) appears to be reduced. Hence the resulting magnetic field (the effective field) can point very close to the direction of the B1 field if the B1 field is much larger than the reduced field.
An on resonance pulse involves changing the direction of the magnetization vector by applying the B1 field in a certain direction for a specific time. The angle through which the magnetization vector is rotated is given as the product of the frequency of the effective field and the time the pulse is applied. By applying different times one can rotate the magnetization vector to different angles.
For a real NMR experiment there are going to be more than one resonance frequencies in the sample. If one makes the B1 field strong enough it is possible to bring the magnetization vector very close to the desired orientation, which means we can bring it close to the ideal case of an on resonance pulse.
The vector model can be used to understand simple NMR experiments.
Chemical shifts are the units used in Protein NMR analysis graphs. Chemical shift correlates to the different radiofrequency electromagnetic pulses sent through the protein sample. Each molecule that exhibits spin properties show different distinct chemical shifts. Chemical shifts can be observed by sending nuclear magnetic energy onto the protein sample. The electron clouds that surround each spin nuclei create a local magnetic field that opposes the applied magnetic field. When the electromagnetic pulse runs through the sample alpha spin nuclei that are hit with the correct amount of energy to switch to beta spin do so and subsequently cause resonance. This resonance can then be recorded and is reported for the different frequencies applied to sample as parts per million (ppm chemical shifts). For example, when the nuclear magnetic field strength sends out a frequency of 1 ppm (parts per million), CH3 reacts to it and exhibits beta spin behavior from which the spectroscopy catches this signal and records it.
Most hydrogen absorptions in 300-MHz 1H NMR fall within a range of 3000 Hz. Rather than record the exact frequency of each resonance, we measure it relative to an internal standard, the compound tetramethylsilane, (CH3)4Si. Its 12 equivalent hydrogens are shielded relative to those in most organic molecules, resulting in a resonance line conveniently removed from the usual spectral range.
A problem with these numbers, however, is that they vary with the strength of the applied magnetic field. Because field strength and resonance frequency are directly proportional, doubling or tripling the field strength will double or triple the distance (in hertz) of the observed peaks relative to (CH3)4Si. To make it easier to compare reported literature spectra, we standardize the measured frequency by dividing the distance to (CH3)4Si (in hertz) by the frequency of the spectrometer. This procedure yields a field-independent number, the chemical shift δ.
NMR spectra uses superconducting magnets to many metal nuclei. Carbon 13 NMR has become increasingly useful in facilitating due to its low natural abundance and low sensitivity for NMR experiment. The integration of NMR peaks of organometallic complexes can provide the ratio of atoms in different environments. Since the relaxation times of the carbon atoms vary in organometallics, a great deal of inaccuracy result from this. However it does have its advantages as well. For example, terminal carbonyl peaks are frequently in the range of 195 to 225ppm, which allows it to be easily distinguished from other ligands. The 13C chemical shift correspond with the strength of the CO bond. The stronger the bond, the smaller the chemical shift. 13C in cyclopentadienyl ligands have a wide range of chemical shifts in paramagnetic compounds whereas it's narrower for diamagnetic compounds.
In NMR, certain atomic nuclei must be magnetic by nature in order for this technique to be used. However, only a certain number of isotopes have this magnetic characteristic called spin. An example is hydrogen. When an external magnetic field is applied, hydrogen's spinning proton generates a magnetic moment which can take either the α or β spin state. The energy difference between the two states is directly proportional to the amount of external magnetic field applied. The α state is aligned with the applied field and has lower energy than the β state. When a pulse of electromagnetic radiation is applied to the α state, it can be excited to the β state, thus a resonance will be obtained.
The NMR is only able to detect certain atoms and only certain isotopes. For example, it can detect the Hydrogen molecules with a mass of 1 amu, but not the other isotopes. Most commonly the Hydrogen and Carbon NMR's are used. The information that can be determined by the NMR is immense. In the case of the Hydrogen NMR, one can use a certain amount of knowledge from a table to determine what each peak corresponds to which group within your sample. Interpreting NMR's can also be a little tricky in that one must understand splitting patterns. For example, the carbon atom you may be examining may have only one hydrogen attached, however if attached to a methyl group, it will appear as a quartet.
Protons in different chemical environments are shielded by different amounts. When a nucleus surrounded by electrons is exposed to a magnetic field of strength H0,these electrons move in such a way as to generate a small local magnetic field, hlocal, opposing H0. As a consequence, the total field strength near the hydrogen nucleus is reduced, and the nucleus is thus said to be shielded from the magnetic field strength by its electron cloud. The degree of shielding depends on the amount of electron density surrounding the nucleus. dding electrons increases shielding; their removal, consequently, would cause deshielding. Also, a proton is deshielded when the induced field reinforces the applied field. For example, the induced field can reinforce the applied field. As a result, these protons are deshielded and their chemical shifts are at a higher value of ppm.
J-coupling arises from the interaction of different spin states through the chemical bonds of a molecule and results in the splitting of NMR signals. This coupling provides detailed information about the connectivity of atoms and the structure of a molecule.
Spin-spin slitting is given by the N+1 Rule: 1) H that is neighbored by one H resonnate as a doublet.2) H that is neighbored by two set of equivalent Hs resonate as trplet. 3) H that is neighbored by three equivalents of hydrogens resonate as a quartet.
A multiplet can be shown when there is a mixture of couple patterns, some of which could be broken down to small parts (i.e. qd- quartet of doublet). But for the most part, if a coupling pattern shows complexity not easily countable by N+1 rule, such pattern is called a multiplet.
In addition, functional groups containing alcohol (-OH) such as alcohol, carboxylic acid have a broad band on NMR spectra. This is due to the fact that Hydrogens on alcohol can hydrogenbond easily, thus being able to couple in broad spectra than other H's. Thus just by looking at the type of the spectra (such as multiple peaks or broad peak), one can easily categorize the functional group to which the H is associated with.
The ratio of the splitting peaks is given by a mathematical mnemonic device called Pascal's triangle. Each number in this triangle is the sum of the two numbers closest to it in the line above. It is important to remember that nonequivalent nuclei mutually split one another. In other words, the observation of one split absorption necessitates the presence of another split signal in the spectrum. Moreover, the coupling constants for these pattern must be the same. Double and triple bond characteristic chemical environments show complex splitting peaks. An alkyne for example, can have hydrogen splitting patterns an extra adjacent carbon away. An alkene depending on where the hydrogen is located (cis or trans) to a relative chemical environment, can show a slightly distorted peak.
Integration is very useful in NMR spectrum in determining the structure of a molecule. The relative integrated intensity of a signal is proportional to the relative number of nuclei giving rise to that absorption. Normally, an NMR spectrum plot will tell you very complex integration numbers. However, integration doesn’t have to be exact. You can just divide each integration number by the smallest number in your NMR spectrum. The ratio will help you get a big picture of the relative number of H’s represented by a peak. Combining the chemical shifts and the peak integration, you might be able to determine the structure of a molecule by using the chemical shift table.
Researchers usually use NMR to check if the product they made from a reaction is what they really want. So they don’t have to figure out the structure of a molecule by looking at a complex spectrum. They know what the spectrum should look like if the reaction works. By comparing the actual spectrum to the ideal one, they are able to verify the molecule they want is in the product. Solvent used is usually CD2Cl2, since D would not show up in a H NMR spectrum. Combine with chemical shifts, they can be used to determine structure.
To interpret proton NMR, it is important to know where each type of proton lies. Below is a list of types of proton and their chemical shift on NMR.
It is important to know that proton NMR peaks only indicate the presence of protons (H). It does not show other atoms like Carbon, Oxygen...etc. As mentioned in the above section, proton peaks shows spliting because of coupling by the neighbor protons. The integration of each peak is the amount of proton relative to other proton on the NMR. When solving structure from the H NMR, it is important to write down the chemical shift, integration and the split of each peak. Then base on the chemical shift of the peak, write down the possible functional group and structure. Lastly, arrange and connect each structure so that it matches the split and the integration of proton NMR. Below are examples of hand drawn proton NMR and also the detail instruction to interprete proton NMR.
The interpretation of each proton NMR is given right below of each graph. Try not to look at the structure, and only look at the molecular formula and proton NMR to interpret the structure of the compound. Note that the examples given are the basic and simple compound. The NMR of protein structures is much more complex than these examples. The goal of these examples is to give you an idea how proton NMR are interpreted.
Base on the chemical shift of proton NMR, there should be a ketone functional group or a structure that contains a carbonyl group at peak around 2.0. The peak around 1.3 could be a primary or secondary alkyl group.
Now look at the splitting of the peaks at chemical shift 1.3 (C), the triplet (3 peaks) shows that the proton is probably split by 2 neighbor protons (base on the n+1 rule mention in integration section). If it is split only by 1 neighbor proton, then it would be a doublet (2 peaks). Next, look at the peak at chemical shift 2.0 (B). The singlet (single peak) means that this proton environment probably has no neighbor proton. The peak at chemical shift 4.2 (A) is a quadruplet (4 peaks). This means there are about 3 neighbor proton next to proton environment A. Since it is known that B has no proton right next to it, and A and C both has neighbor protons, it is safe to assume that proton environment A is right next to proton environment C, which could be a -CH2CH3.
The following possible structures deduced above are a ketone and an ethyl group.
Splitting analysis
Substructure
Splitting pattern
Singlet
Quartet and triplet
Analysis
C with no neighboring H
C with three neighboring H C with two neighboring H
Note that the there are two oxygen atoms from the molecular formula. One of the oxygen is included in ketone. The other oxygen must be binding onto one of these two structures. Take a look at peak A, the chemical shift is near 4.0, which means it might be connected to a heteroatom like oxygen. Place the oxygen next to the -CH2CH3 and connect the structure together, you will get ethyl acetate as indicated below the proton NMR. Notice that the proton B does not have any neighbor proton. Proton A is split into 4 peaks by proton C (n+1 rule, n = 3 neighbor protons). Proton C is split into 3 peaks by proton A (n = 2 neighbor protons). The CH2, secondary alkyl, base on the proton frequency table should be around the chemical shift ~1.2 ppm, but instead it is at ~4.2 ppm. The reason for this is that the CH2 is attached to oxygen a heteroatom, which is a electron withdrawing group. The result is that the chemical shift is pulled left field resulting in a chemical shift of 4.2 ppm. Another way to look at this is to assume there is an ester functional group at chemical shift of 4.2 ppm, and the primary alkyl group is attached to the ester.
The integration of the proton NMR also matches the structure well. The methyl CH3 next to the carbonyl group has 3 protons, and the primary alkyl next to the CH2 has 3 protons as well. This explains why peak B has the same height as peak C (same amount of protons). Peak A has the lowest height because it only has two protons.
This proton NMR example looks more complicated than the first one, but it is actually easier. One important information from this proton NMR is that there are peaks around 6.0~ 7.0 ppm (F,B,D,E). This always almost indicates the presence of aromatic ring. Noticed that there are about 4 group of peaks from 6.0 to 7.0 ppm? This means that there are four protons attach to 4 carbons on the aromatic ring. This leaves 2 carbons of the aromatic ring free to attach to two substituents. The position of these substituents is clearly an ortho and para position on the aromatic ring. The reason is that two different substituents on ortho and para position of the aromatic ring would give 4 proton environments. If there are only two proton environments around 6.0~ 8.0 ppm with the same integration (height), it means that the aromatic ring is probably symmetric on both sides with double para substituents. Now look at the peak (A) at 5.5 ppm. The peak is a broad peak instead of sharp peak like the rest. This means that the proton of this environment is attached to a heteroatom. The only heteroatom present in the sample base on the molecular formula given is an oxygen atom. This means that the proton is directly attached to oxygen, which would be a alcohol group -OH (Note that the proton on the alcohol group can have the chemical shift range from 0.5 to 5.0 ppm). It is important to remember that the proton that attached to a heteroatom is also known as exchangeable proton. Exchangeable proton sometimes do not show up on H NMR, so do not worry when an expected exchangeable peak doesn’t appear on HMNR.
The peak (C) at the chemical shift of ~3.9 ppm has the highest integration (height) and is a singlet. Usually when such a high singlet peak is observed around 3.9 ppm with oxygen in the molecular formula, there is a high chance that it is a ether group, -OCH3. Since there is no neighbor proton next to the CH3 of the ether, it is a singlet peak. In addition, the height of the peak is high compare to the other peak, which indicate a possibility of CH3 with at least 3 times more protons than other proton environment. Base on the interpretations above, these are the possible function groups:
Attach the alcohol group and the ether group in ortho- para position will result in the correct structure of the compound.
NOESY Spectrum of Codeine. Each peak correlates to a proton.
NOESY is a NMR technique that uses the Nuclear Overhauser effect to help understand the tertiary structure of proteins and other large molecules. The nuclear Overhauser effect is the interaction of nuclei at short distances from each other. The extent of the effect is approximately 5 Angstroms. This is a better analysis of tertiary structure for proteins that do not form crystalline structures and need to be analyzed in solution. Like in NMR, a magnetic field is produced around the molecule and RF waves at different frequencies are produced to produce resonance. The resonance is produced when the nucleus reverses spin. Unlike normal NMR, the aim of NOESY is to determine the effect of the resonance of one nucleus on the other nuclei in the area. The graph that is produced gives you an idea of where the nuclei are in relation to each other.
The NOESY experiment reveals the distance of protons relative to one another. Protons very close to each other, (about 3 Angstroms apart) will produce a very large signal. The limit of detection is about 5-6 Angstroms apart, and will produce only a very weak signal.
The graph has diagonal dots correspond to the placement of the nuclei on the one dimensional NMR. There are dots out side of this diagonal spectrum. These dots show what nuclei are in close proximity of each other. If a line was drawn horizontally and vertically from the outlaying dots, the dots, on the diagonal spectrum, that lay on the lines would be in close proximity to each other. Using the information from the NMR and the NOESY a tertiary structure of the macromolecules could be formed. However, the resulting tertiary structure would contain a family of structures. This happened for three reasons. The first is the approximation of the distance between the nuclei. Second, there may not have been enough constraints to produce a singular structure. Thirdly, the protein is in solution, and the solution contains many proteins.
This technique is mainly used for hydrogen atoms, because they are the most abundant atom in biological systems and are also the simplest.
An NMR machine is essentially a huge magnet. Many atoms are essentially little magnets. When placed inside an NMR machine, all the little magnets orient themselves to line up with the big magnet. By harnessing this law of physics, NMR spectroscopists are able to figure out physical, chemical, electronic, and structural information about molecules.
NMR relies on the interaction between an applied magnetic field and the natural “little magnets” in certain atomic nuclei. For protein structure determination, spectroscopists concentrate on the atoms that are most common in proteins: hydrogen, carbon, and nitrogen. What researchers seek to learn through NMR is how this chain of amino acids wraps and folds around itself to create the three-dimensional, active protein. Solving a protein structure using NMR is a series of experiments, each of which provides partial clues about the nature of the atoms in the sample molecule, such as how close two atoms are to each other, whether these atoms are physically bonded to each other, or where the atoms lie within the same amino acid. Other experiments show links between adjacent amino acids or reveal flexible regions in the protein. Each new set of experiments further refines possible structures until finally the scientists carefully select 10 to 20 solution that best represent their data and present the average as their final structure.
Sample Preparation for NMR
In order to study protein structure by NMR, highly purified samples of the protein of interest must be prepared. The protein can either be isolated from natural sources, or expressed in host organisms such as E. Coli through recombinant DNA techniques. The bacteria with the recombinant gene coding for a specific protein are grown in minimal media with 15NH4Cl being the only source of nitrogen for the bacteria, and 13C-glucose offering the only source of carbon for the bacteria to make proteins. This way, when the bacteria produce the protein of interest, these proteins are labeled with 15N and 13C, both of which are NMR active. The protein of interest is then purified through various protein purification techniques (See protein purification techniques). Finally, the protein, in an appropriate buffer, is loaded into an NMR tube and is ready to be placed in the NMR magnet.
Figure 1. General scheme for protein structure determination by NMR in solution phase.
NMR Experiments
2D ExperimentsThere are many different types of NMR experiments one can run to help to reveal information about the structure of the protein. The experiments usually differ in what correlations are detected. For example, a 2D [1H-1H]-NOESY experiment will reveal hydrogen atoms that are within 0.5 nm apart. Protons that exhibit this interaction will show up as a cross peak on the 2D spectrum (Figure 2).
Figure 2. Simplified example of 2D [1H-1H]-NOESY NMR spectrum. The dots on the diagonal represent the 1D spectrum of the molecule. There are two protons that are being examined, one at 9 ppm and the other at 8 ppm. The presence of the cross peak (circled) reveals that these two protons are within 0.5 nm apart.
Taking this information, theoretically, the protons at 8 ppm and 9 ppm on the 1D spectrum could be assigned to a certain residue in the amino acid sequence. For example, if Ala1 is the point at 8 ppm and Ala54 is at 9 ppm, and there is a crosspeak present, then the observation of this NOE reveals a circular structure for this polypeptide chain (Figure 3).
Figure 3. Circular structure of polypeptide chain revealed by presence of NOE on 2D NOESY spectrum.
Because proteins are much more complicated than this example above, the 2D NOESY spectrum is often cluttered and crosspeaks cannot be made out clearly because many of these points may stack on top of one another. Other experiments are required to reveal the necessary information to determine the 3D structure of the protein, such as 3D experiments.
3D Experiments
3D experiments use a third dimension to reveal peaks that may be stacked on top of one another in the 2D spectrum. An example is 3D 15N-correlated [1H-1H]-NOESY (Figure 4).
There are many other experiments to correlate different nuclei and their neighbors.
Figure 4. Example of 3D 15N [1H-1H] NOESY. The peaks that would be stacked on top of one another in the 2D [1H-1H] NOESY are now drawn out in the third dimension by relating its 15N chemical shifts. This cleans up the spectrum and helps in resonance assignments.
NMR is a good method in trying to solve the structure of a protein and the continued unanswered protein folding problem. However it is not the only method as crystallography is also a possible method for determining protein structure. However, NMR has its advantages over crystallography.
One instance is that it uses molecules in solution, so it is not limited to those that crystallize well since crystallization is a very uncertain and time-consuming step in X-ray crystallography. In addition, some proteins do not readily crystallize. Furthermore, although structures present in crystallized proteins very closely represent those of proteins free of the constraints imposed by the crystalline environment, structures in solution can be sources of additional insights. NMR also makes it fairly easy to study properties of a molecule besides its structure such as the flexibility of the molecule and how it interacts with other molecules. With crystallography, it is either impossible to study these aspects or it requires an entirely new crystal.
The 15 carbon compound viniferone found in grape seeds was isolated in 2004. Viniferone falls into a group of substances, grape seed, that are very active against radicals and oxidative stress. There was very little recovery of viniferone from an abundance of grape seeds (~40 mg of viniferone from 10.5 kg of grape seeds), so precautions had to be taken in the techniques used to obtain its structure. Most tests would destroy the minuscule amount proanthocyanidins t of viniferone obtained, so a combination of spectroscopic techniques were utilized (including NMR) to obtain viniferone's structure. Obtaining the 1H and 13C NMR data was critical in determining the structure of Viniferone. The variety of absorption signals at various chemical shifts helped determine the presence of alkenes and benzene rings. Correlation spectroscopy and eventually X-ray crystallography were used to verify the structural arrangement of Viniferone.[2]
↑Hiller, S., Abramson, J., Mannella, C., Wagner, G., and Zeth, K., "The 3D structures of VDAC represent a native conformation," Trends in Biochemical Sciences, 2010.
↑Schore, Neil E. (2011). Organic Chemistry Structure and Function 6th Edition. W. H. Freeman
If you would like to learn more about interpreting NMR peaks, I highly suggest this website. http://www.wfu.edu/~ylwong/chem/nmr/h1/ Berg, Jeremy M., Lubert Stryer, and John L. Tymoczko. Biochemistry. 6th ed. Boston: W. H. Freeman & Company, 2006. 98-101.
Shriner, Christine K. F. Hermann, Terence C. Morrill, David Y. Curtin, Reynold C. Fuson. The Systematic Identification of Organic Compounds. 8th ed. U.S.A. John Wiley & Sons, Inc. 2004. 136-142.
Vollhardt, K. Peter C., Schore, Neil E. Organic Chemistry - Structure and Function. 5th ed. New York: W. H. Freeman & Company, 2005. 398-432.
Keeler, James. Understanding NMR Spectroscopy. West Sussex, England. John Wiley & Sons. 51-77.
Clore, Marius G., Gronenborn, Angela M. “Chapter 34: Structures of Larger Proteins, Protein-Ligand, and Protein-DNA Complexes by Multidimensional Heteronuclear NMR.” Multidimensional NMR Methods for the Solution State. United Kingdom. John Wiley & Sons Ltd.
Wuthrich, Kurt. “Chapter 33: Biological Macromolecules: Structure Determination in Solution.” Multidimensional NMR Methods for the Solution State. United Kingdom. John Wiley & Sons Ltd.
National Institutes of Health, National Institute of General Medical Sciences, New York, 2007, 29-30, http://www.nigms.nih.gov.
[1]"Kurt Wüthrich - Nobel Lecture". Nobelprize.org. 25 Oct 2010 http://nobelprize.org/nobel_prizes/chemistry/laureates/2002/wuthrich-lecture.html
Deuterium Exchange Mass Spectrometry is a powerful tool with which protein/enzyme structure and interaction can be studied. This can also determine the location and orientation of protein and enzymes associated with phospholipids.
This occurs based on the principle of hydrogen exchange with solvent. The hydrogen atoms on a protein molecule can be divided into three groups (1):
1) Hardly ever exchange (H that is attached directly to C)
2) Exchange extremely quickly (H that is attached to the side chain atom)
3) Exchange rate depends on the local environment (Amide Hydrogen N-H) (1)
The hydrogen atoms on a protein molecule that undergo exchange reaction can be followed experimentally used deuterated water.
The third process listed above, the amide hydrogen exchange is described as follows:
1) Incubated proteins in deuterated water are reacted with probes/perturbations to shows how it can influence the accessibility to water and therefore affect amide hydrogen exchange rates. It will only proceed smoothly and efficiently if the amide hydrogen is reacted in solvent water. As the name implies, hydrogens exposed to hydrophobic regions will need more time to exchange, and have an increased chance of not exchanging at all since efficiency is due to the amide hydrogen being in the solvent water.
2) The deuterium atoms can be "locked in place" to prevent further exchange (1)
3) High powered liquid chromatography-mass spectrometry analysis then uses a protease, which is a catalyst, to digest/cleave the protein into its respective peptides, which are 5-15 amino acids in length.
4) Mass spectrometry is used to fragment those digested peptides into smaller pieces, which helps in identifying the peptide.
One limitation to this approach is that collision induced dissociation causes "scrambling," where the deuterium atom changes conformation within the peptide (1). A method that reduces the chances of scrambling is electron transfer dissociation.
DXMS studies with two potent, specific, and reversible lipid inhibitors. These inhibitors provide very specific methods in docking, and when docked, the precise binding conformations of the inhibitors became defined. One of the specific and reversible inhibitors is substrate, and when this inhibitor is docked, it can provide an image of the conformation of a phospholipid molecular species bound in the active site of an enzyme (1).
DXMS could also be used in conjunction with phospholipid bilayer nanosdiscs to analyze membrane protein conformation. There have been complications in the past with studying in vitro systems by way of detergent micelles or liposomes. Detergent micelles often denature the membrane proteins due to their detergent nature. Liposomes form a mixture of small, large, and multilamellar vesicles which lead to inconsistent results (2). Nanodiscs, when used with DXMS, offers a solution to studying membrane proteins.
Nanodiscs are made with membrane scaffold protein, lipids, and the membrane protein in question mixed in a phospholipid/detergent solution (2). Detergent is removed in order for the nanodiscs to form on their own. The nanodisc allows the protein to remain in its natural conformation. The nanodiscs are then purified with size exclusion chromatogoraphy and the purified nanodiscs are subjected to the deuterated buffer multiple times(2). The DXMS needs to be carried out quickly in order to minimize deuterium loss from quenching. Three methods helped to ensure the success of the nanodiscs in conjunction with DXMS (2):
1) nanodisc disassembly by adding cholate (cholate is effective because it increases membrane scaffold protein peptides and protein digestion)
2) zirconium oxide beads separates the phospholipids from the rest of the mixture
3) optimized chromatography used to separate the membrane scaffold protein peptides from the membrane proteins peptides
DXMS in conjunction with nanodiscs allows the analysis of a protein in its natural conformation and ultimately the protein’s natural conformation leads to the understanding of its function.
1. Annu Rev Biochem. 2011 Jun 7;80:301-25. Applications of mass spectrometry to lipids and membranes. Harkewicz R, Dennis EA. Source Department of Chemistry and Biochemistry and Department of Pharmacology, School of Medicine, University of California at San Diego, La Jolla, California 92093-0601, USA. rharkewicz@ucsd.edu
2. Anal Chem. 2010 July 1;82(13):5415-5419. Conformational analysis of membrane proteins in phospholipid bilayer nanodiscs by hydrogen exchange mass spectrometry. Hebling M, Christine.
X-ray crystallography
X-ray crystallography can reveal the detailed three-dimensional structures of thousands of proteins. The three components in an X-ray crystallographic analysis are a protein crystal, a source of x-rays, and a detector.
X-ray crystallography is used to investigate molecular structures through the growth of solid crystals of the molecules they study. Crystallographers aim high-powered X-rays at a tiny crystal containing trillions of identical molecules. The crystal scatters the X-rays onto an electronic detector. The electronic detector is the same type used to capture images in a digital camera. After each blast of X-rays, lasting from a few seconds to several hours, the researchers precisely rotate the crystal by entering its desired orientation into the computer that controls the X-ray apparatus. This enables the scientists to capture in three dimensions how the crystal scatters, or diffracts, X-rays. The intensity of each diffracted ray is fed into a computer, which uses a mathematical equation to calculate the position of every atom in the crystallized molecule. The result is a three-dimensional digital image of the molecule.
Crystallographers measure the distances between atoms in angstroms. The perfect “rulers” to measure angstrom distances are X-rays. The X-rays used by crystallographers are approximately 0.5 to 1.5 angstroms long, which are just the right size to measure the distance between atoms in a molecule. That is why X-rays are used.
Protein X-ray crystallography is a technique used to obtain the three-dimensional structure of a particular protein by x-ray diffraction of its crystallized form. This three dimensional structure is crucial to determining a protein's functionality. Making crystals creates a lattice in which this technique aligns millions of proteins molecules together to make the data collection more sensitive. It's like getting a stack of papers, measuring the width with a ruler, and dividing that length with the number of pages to determine the width of one piece of paper. By this averaging technique, the noise level gets reduced and the signal to noise ratio increases.[2] The specificity of the protein's active sites and binding sites is completely dependent on the protein's precise conformation. X-ray crystallography can reveal the precise three-dimensional positions of most atoms in a protein molecule because x-rays and covalent bonds have similar wavelength, and therefore currently provides the best visualization of protein structure. It was the X-ray crystallography by Rosalind E.Franklin, that made it possible for J.D. Watson and F.H.C. Crick to figure out the double-helix structure of DNA.
We use this procedure to grasp the cellular mechanism and the knowledge of the 3-D structure of enzymes and other macromolecules. It is critical that we can better understand how each chemical reaction that occurs in a cell needs a specific enzyme for it to happen. Two common techniques used for analysis of proteins structure are Nuclear Magnetic Resonance (NMR), and x-ray crystallography. X-ray crystallography can be used to analyze any different compounds up to a molecular weight of 106 (g/mol) for instance; where as NMR is restricted to biopolymers(polymers produced by a living organism such as starch, peptides, sugars) with a molecular weight no more than 30,000 (g/mol). It can also measure compounds that are very small because the appropriate size to measure the distance between atoms in a molecule is 0.5 to 1.5 angstroms. X-rays are used as the form of radiation because their wavelengths are on the same order of a covalent bond (~1 Å or 1 * 10−10m) and this is necessary to obtain a diffraction pattern that reveals information about the structure of the molecule. If the radiation had a wavelength much bigger or much smaller than the bond length of a covalent bond, the light would not diffract and no new knowledge of the structure would be obtained.
The process begins by crystallizing a protein of interest. Crystallization of protein causes all the protein atoms to be orientated in a fixed way with respect to one another while still maintaining their biologically active conformations - a requirement for X-ray diffraction. A protein must be precipitated out or extracted from a solution. The rule of thumb here is to get as pure a protein as possible to grow lots of crystals (this allows for the crystals to have charged properties, and surface charged distribution for better scattering results). 4 critical steps are taken to achieve protein crystallization, they are:
Purify the protein. Determine the purity of the protein and if not pure (usually >99%), then must undergo further purification.
Must precipitate protein. Usually done so by dissolving the protein in an appropriate solvent(water-buffer soln. w/ organic salt such as 2-methyl-2,4-pentanediol). If protein is insoluble in water-buffer or water-organic buffer then a detergent such as sodium lauryl sulfate must be added.
The solution has to be brought to supersaturation(condensing the protein from the rest of the solvent forming condensation nuclei). This is done by adding a salt to the concentrated solution of the protein, reducing its solubility and allowing the protein to form a highly organized crystal (this process is referred to as salting out). Other methods include batch crystallization, liquid-liquid crystallization, vapor diffusion, and dialysis.
Let the actual crystals grow. Since nuclei crystals are formed this will lead to obtaining actual crystal growth.
NOTES ON RECRYSTALLIZATION TECHNIQUE TO ACHIEVE A MORE PURE PROTEIN:
Recrystallization is an incredibly important technique used for the purification of substances. Understanding the solubility of the solid in a certain solvent is the key to recrystallization. One of the applications of this technique can be seen in pharmaceutics and in many other fields. For example, crystallographers use methods of nuclear magnetic resonance and x-ray diffraction to gain insight into different compounds. X-ray diffraction requires the formation of pure crystals in order to acquire accurate results. Crystallographers can gain insight into protein structure by using x-ray diffraction, but in order to be able to use x-rays to examine their crystals, they must first spend time forming pure protein crystals. It is very difficult to form protein crystals. It may even take years and incredibly specific conditions. Temperature, pH, and concentration have to be very specific to form larger crystals with a pure structure. Recrystallization in this process is vital to get rid of impurities in the crystal lattice. Scientists today use crystallography and recrystallization techniques to understand protein structure and help understand how a single abnormality in the protein's primary structure can cause diseases. All in all, purification techniques are vital in order to use x-ray diffraction to understand structure. In this experiment we explore the differences in micro and macro recrystallization.
The techniques employed in recrystallization include finding a good solvent to work in, gravity filtration, slow cooling, and vacuum filtration. The key to a successful recrystallization is a good solvent. We need a solvent that will not dissolve the sample at cool temperatures but will dissolve it at high temperatures. This allows the precipitation of the solute after the solution is dissolved in warm temperatures. Since the solute is only soluble in the warm solute, upon cooling, a precipitate forms. Gravity Filtration is used to remove insoluble impurities remaining in the solution before recrystallization and it is used to filter out the charcoal used to remove the color impurities. Gravity filtration is effective, but we must avoid crystallization during this process as to avoid losing pure crystals in the filter paper. Slow cooling is also essential to ensure the purity and size of the crystals. When the solution is allowed to cool slowly, the dissolved impurities have time to interact with the solvent instead of remaining trapped in the crystal lattice. During fast cooling, impurities may remain trapped in the crystal lattice because crystallization occurs to quickly and impurities do not have time to return to the solvent. After the crystals are put in an ice bath to ensure maximum recrystallization, the solution is filtered using a vacuum filtration to extract the pure crystals from the solution with the impurities. After it is vacuumed, the pure crystals are collected and weighed. Micro recrystallization differs from macro recrystallization in the instruments and techniques used for filtration of the pure crystals. Micro recrystallization involves using a Craig tube and centrifugation instead of vacuum filtration. It is used for a recrystallization of less than 300mg of solid.
For the next step, x-rays are generated and directed toward the crystallized protein. X-rays can be generated in four different ways,
by bombarding a metal source with a beam of high-energy electrons,
by exposing a substance to a primary beam of X-rays to create a secondary beam of X-ray fluorescence,
from a radioactive decay process that generates X-rays (Gamma rays are indistinguishable from X-rays), and
from a synchrotron (a cyclotron with an electric field at constant frequency) radiation source.
The first and last method utilize the phenomenon of bremsstrahlung, which states that an accelerating charge will give off radiation.
Then, the x-rays are shot at the protein crystal resulting in some of the x-rays going through the crystal and the rest being scattered in various directions. The scattering of x-rays is also known as "x-ray diffraction". Such scattering results from the interaction of electric and magnetic fields of the radiation with the electrons in the atoms of the crystal.
The patterns are a result of interference between the diffracted x-rays governed by Bragg's Law: , where is the distance between two regions of electron density, is the angle of diffraction, is the wavelength of the diffracted x-ray and is an integer. If the angle of reflection satisfies the following condition:
,
the diffracted x-rays will interfere constructively. Otherwise, destructive interference occurs.
Constructive interference indicates that the diffracted x-rays are in phase or lined up with each other, while destructive interference indicates that the x-rays are not exactly in phase with each other.
The result is that the measured intensity of the x-rays increases and decreases as a function of angle and distance between the detector and the crystal.
The x-rays that have been scattered in various directions are then caught on x-ray film, which show a blackening of the emulsion in proportion to the intensity of the scattered x-rays hitting the film, or by a solid-state detector, like those found in digital cameras. The crystal is rotated so that the x-rays are able to hit the protein from all sides and angles. The pattern on the emulsion reveals much information about the structure of the protein in question. The three basic physical principles underlying this technique are:
Atoms scatter x-rays. The amplitude of the diffracted x-ray is directly proportional to the number of electrons in the atom.
Scattered waves recombine. The beams reinforce one another at the film if they are in phase or cancel one another out if they are out of phase. Every atom contributes to a scattered beam.
Three-dimensional atomic arrangement determines how the beams recombine.
The intensities of the spots and their positions are thus the basic experimental data of the analysis.
The final step involves creating an electron density map based on the measured intensities of the diffraction pattern on the film. A Fourier Transform can be applied to the intensities on the film to reconstruct the electron density distribution of the crystal. In this case, the Fourier Transform takes the spatial arrangement of the electron density and gives out the spatial frequency (how closely spaced the atoms are) in the form of the diffraction pattern on the x-ray film. An everyday example of the Fourier Transform is the music equalizer on a music player. Instead of displaying the actual music waveform, which is difficult to visualize, the equalizer displays the intensity of various bands of frequencies. Through the Fourier Transform, the electron density distribution is illustrated as a series of parallel shapes and lines stacked on top of each other (contour lines), like a terrain map. The mapping gives a three-dimensional representation of the electron densities observed through the x-ray crystallography. When interpreting the electron density map, resolution needs to be taken into account. A resolution of 5Å - 10Å can reveal the structure of polypeptide chains, 3Å - 4Å of groups of atoms, and 1Å - 1.5Å of individual atoms. The resolution is limited by the structure of the crystal and for proteins is about 2Å.
Protein molecules are very large, thus their crystals diffract x-ray beams much less than crystals from smaller molecules. Because larger molecules have fewer crystals, diffraction scattering and hence intensity emitted is very weak. Proteins contain carbon, nitrogen, and oxygen, and so are lighter elements(that is they have fewer electrons/atom); this is important since electrons are responsible for the diffraction and intensity, and therefore they scatter x-rays weaker than heavy elements. Knowing this, protein crystallographers use high intensity x-ray sources such as a rotating anode tube or a strong synchrotron x-ray source for analyzing the protein crystals.
The number of electrons in an atom is proportional to the wave's amplitude. An example would be comparing a carbon atom and hydrogen atom, you would see that the carbon atom would scatter six times as strongly as the hydrogen atom.
If in phase the waves combine with one another at the film but if the waves are out of phase then they cancel out one another at the film.
The only thing that matters when looking at how scattered waves recombine is the atomic arrangement.
Energy of X-ray:
where f is frequency and λ is wavelength. The SI unit of energy is the joule (J).
X-rays have higher energy than visible light due to its small wavelength.
The interaction of X-rays with the electrons in a crystal gives rise to a diffraction pattern, which mathematically is the Fourier transform of the electron density distribution. The detectors used to measure the X-rays, however, can only measure the amplitude of the diffracted x-rays; the phase shifts, which are required to use the Fourier Transform and find the electron density distribution, are not measurable directly using this method. This is known in the physics community as the "Phase Problem". In simpler terms the phases cannot be found from the measured amplitudes of the X-rays. Other extrapolations must be made and additional experiments must be done in order to get an electron density map. Many times, the existing data on the compound's physical and chemical properties can help aid when there is a poor density map. Another method known as Patterson Synthesis is very useful to find out an initial estimate of phases and it is very useful for the initial stages to determine the structure of proteins when the phases are not known. The problem can be simplified by finding an atom, usually a heavy metal, using Patterson Synthesis and then using that atom's position to estimate the initial phases and calculate an initial electron density map that can further help in the modeling of the position of other atoms and improve the phase estimate even more. Another method is called Molecular Replacement; it locates the location of the protein structure in the cell. In addition to the molecular replacement method, the phase problem can also be solved by the isomorphous replacement method, the multiple wavelength anomalous diffraction method, the single-wavelength anomalous diffraction method, and direct methods.
Phase problem can be solved by having an atomic model that can compute phases. A model can be obtained if the related protein structure is known. However, in order to build this atomic model, the orientation and position of the model in the new unit cell needs to be determined. This is when the technique, molecular replacement (or MR) comes in.
Molecular Replacement, also known as MR, is a method to solve phase problems in x-ray crystallography. MR locates the orientation and position of a protein structure with its unit cell, whose protein structure is homologous to the unknown protein structure that needs to be determined. The obtained phases can help generate electron density maps and help produce calculated intensities of the position of the protein structure model to the observed structures from the x-ray crystallography experiment.
MR method is also effective for solving macromolecular crystal structures. This method requires less time and effort for structural determination, since heavy atom derivatives and collecting data do not need to be prepared. The method is straight forward and model building is simplified because it needs no chain tracing.
This method consists of two steps:
a rotational search to orient the homologous model in the unit cell or target
a translational target where the new oriented model is positioned in the unit cell
Patterson maps are interatomic vector maps that contain peaks for each related atom in the unit cell. If the Patterson maps were generated based on the data derived from the electron density maps, the two Patterson maps should be closely related to each other only if the model is correctly oriented and placed in the correct position. This will allow us to infer information about the location of the unknown protein structure with its cell. However, there is a problem with molecular replacement, it has six dimensions, three parameters to specify orientation and position. With the Patterson maps, it can be divided into subsets of the parameters to look at each part separately.
Rotation function has intramolecular vectors that only depend on the molecule’s orientation and not its position because even when the molecule is translated in the unit cell, all of the atoms are shifted by the same amount but the vectors between the atoms are the same. The Patterson map for the unknown protein structure is compared with the homologous known protein structure in different orientations
File:MR Rotation Function.gifThe figure shows the molecule in a random orientation (left) and together with the rest of the intramolecular vectors (right).This is a Patterson map of the above structure. The intramolecular vectors are shown in red.
To find the orientation, determine the rotation axis and rotation angle about that axis. Two parameters will be needed to define an axis (a vector from the center of the sphere to a point on the sphere surface). The rotation axis starts off parallel to the z-axis and is rotated around the y-axis with angle ᶱ, then the object rotates around the z-axis with angle ᶲ, and finally it rotates around the rotation axis with angle ᵠ. These specify a point on the surface of a unit sphere.
The ĸ/ᵠ/ɸ description is useful if looking for rotations with a particular rotation angle (ĸ). For instance, a 2-fold rotations will have ĸ=180°, while a 6-fold rotations will have ĸ=60°
The rotation function can be computed by comparing two Patterson maps or the peaks in those Pattersons. Rotation function can be computed much faster with Fourier transforms only if the Pattersons were expressed in terms of spherical harmonics.
In direct rotation function, the protein structure can be placed in the unit cell of the unknown structure and the Patterson for the oriented molecule is compared with the entire unknown structure Patterson.
Once the orientation of the known structure is known its model (electron density map) can be oriented to compute structure factors where a correlation function is used to determine the vector to translate the model on top of the homologous one within an asymmetric unit.
With the correct oriented and translated phasing models of the protein structure, it is accurate enough to derive the electron density maps from the derived phases. The electron density maps can be used to build and refine the model of the unknown structure.
X-Rays are generated in large machines called synchrotrons. Synchotrons accelerate electrons to nearly the speed of light and travel them through a large, hollow metal polygon-ring. At each corner, magnets bend the electron stream, causing the emission of energy in the form of electromagnetic radiation. Since the electrons are moving at the speed of light, they emit high energy X-rays.
The benefits of using synchrotrons is that researches do not have to grow multiple versions of every crystallized molecule, but instead only grow one type of crystal that contains selenium. They then have the ability to tune the wavelength to match the chemical properties of selenium. This technique is known as Multiwavelength Anomalous Diffraction. The crystals are then bombarded several times with wavelengths of different lengths, and eventually a diffraction pattern emerges which enables researchers to determine the location of the selenium atoms. This position can be used as a reference, or marker to determine the rest of the structure. The benefits of this allow researchers to collect their data much more quickly.
This method compares the x-ray diffraction patterns between the original protein crystal and the same type of crystal with an addition of at least one atom with high atomic number. The method was used to determinate the structure of small molecules and eventually that of hemoglobin by Max Ferdinand Perutz (1914–2002). A perfect isomorphism is when the original crystal and its derivative have exactly the same conformation of protein, the position and orientation or the molecules, and the unit cell parameters. The only difference that the crystal and its derivative have in a perfect isomorphism is the intensity differences due to the addition of heavy atoms on the derivative. These differences can be identified manually or by an automatic Patterson search procedure, such as SIR 2002, SHELXD, nB, and ACORN, and such information is important as to determine the protein phase angles. However, perfect isomorphism hardly occurs because of the change in cell dimensions. For the protein with heavy atom, its tolerable change in cell dimension is dmin/4, for dmin is the resolution limit. Other factors, such as rotation, also contribute to nonisomorphism.
Prepare a few derivatives of the protein in crystalline structure. Then, measure the cell dimension to check for isomorphism.
Collect x-ray intensity data of the original protein and its derivative.
Apply the Patterson function to determine the coordinates of the heavy atom.
Refine the heavy atom parameters and calculate the phase angle of the protein.
Calculate the electron density of the protein.
The derivatives are made through two different methods. The preferred method is to soak the protein crystal in a solution that is composed identically to the mother liquor, but with a slight increase of precipitant concentration. Another method is co-crystallization, but it is not commonly used because the crystal will not grow or grow nonisomorphously. The soaking procedure depends on how wide the crystal pores are. The pores should be wide enough for the reagent to diffuse into the crystal and to reach the reactive sites on the surface of all protein molecules in the crystal.
Multiple Wavelength Anomalous Diffraction (abbreviated MAD) is a method utilized in X-ray crystallography that allows us to determine the structures of biological macromolecules, such as proteins and DNA, in order to solve the phase problem. Requirements for the structure include atoms that cause significant scattering from X-rays; notably sulfur or metal ions from metalloproteins. Since selenium can replace natural sulfur, it is more commonly used. The use of this technique greatly facilitates the crystallographer from using the Multiple Isomorphous Replacement (MIR) method as preparation of heavy compounds is superfluous.
this method is used to solve phase problems, when there is no available data regarding scattered diffraction besides amplitudes. Moreover, it is used when a heavy metal atom is already bound inside the protein or when the protein crystals are not isomorphous which is unsuitable for MIR method. The method has been mostly used for heavy metallo solution, these metallo enzyme normally comes from the 1st transition series and their neighbors. it is important to have a source for a powerful magnetic field to carry out this experiment, environment such as underground should be considered. A particle accelerator called a synchrotron is also required for the method.
In comparison to multi-wavelength anomalous diffraction (MAD), single-wavelength anomalous diffraction (SAD) uses a single set of data from a single wavelength. The main beneficial difference between MAD and SAD is that the crystal spends less time in the x-ray beam with SAD, which reduces potential radiation damage to the molecule. Also, since SAD uses only one wavelength, it is more time-efficient than MAD.
The electron density maps derived from single-wavelength anomalous diffraction data do need to undergo modifications to resolve phase ambiguities. A common modification technique is solvent flattening, and when SAD is combined with solvent flattening, the electron density maps that result are of comparable quality to those that are derived from full MAD phasing. Solvent flattening involves adjusting the electron density of the interstitial regions between protein molecules occupied by the solvent. The solvent region is assumed to be relatively disordered and featureless compared to the protein. Smoothing the electron density in the solvent regions will enhance the electron density of the protein to an interpretable degree. This method is called ISAS, iterative single-wavelength anomalous scattering.
The direct method can help recover the phases using the data it obtains. Direct Method estimates the initial and expanding phases using a triple relation. Triple (trio) relation is the relation of the intensity and phase of one reflection with two other intensities and phases. When using this method, the size of the protein structure matters since the phase probability distribution is inversely proportionate to the square root of the number of atoms. Direct method is the most useful technique to solve phase problems.
Paul Peter Ewald and Max von Laue developed the idea to use crystals as a diffraction grating for X-rays in 1912. Von Laue proposed that compared to the larger wavelength of visible light, x-rays might have a wavelength close to the spacing of crystals' unit cells. He worked with Walter Friedrich and Paul Knipping to record the x-ray diffraction of a copper sulfate crystal onto a photographic plate. Von Laue developed a relation between the scattering angles and the size of the unit cell spacing and their orientation in the crystal, winning the Nobel Prize in Physics in 1914. As a result of von Laue's research, William Lawrence Bragg developed a law to connect a crystal's observed scattering and reflection from evenly-spaced planes in the crystal. This could be used to deduce atomic structure, and the significance of Bragg's Law to determining molecular structure was recognized immediately. In 1914, the first structure to be solved was that of table salt. Its electron distribution proved that not only covalent but also ionic compounds can form crystals.
In 1914, the structure of diamond was solved using x-rays, and it was shown that the length of the carbon-to-carbon single bond is 1.52 Angstroms.
X-ray crystallography was first used to determine protein structure in the late 1950s.
John Kendrew and Max Perutz while both at Cambridge used x-ray crystallography to discover the structure of hemoglobin & myoglobin(oxygen carrier in muscle) in 1945. They received the Nobel Prize in chemistry in 1962.
The first three-dimensional crystal structure of an enzyme determined via x-ray crystallography was a hen egg-white lysozyme. This was especially important as visual evidence of the transition-state theory because it was physical proof that the catalytic site was complementary to the transition-state geometry. Immediately following this first crystal structure, there was an upsurge in reports of the x-ray structures of many different enzymes.[3]
Many advances in drug discovery and medicine are due in large part by X-Ray Crystallography by identifying drug targets in many diseases that thrive today. In the late 80’s for example, scientists made a breakthrough in using X-Ray Crystallography to produce the structure of HIV Protease, an enzyme that was vital to the retrovirus’ life cycle. The enzyme cuts viral proteins strands that are main components of immature viral cells into separate, mature proteins that can continue on to form more mature and infectious viral particles. By looking closely at it structure, specifically its symmetry, researchers began making compounds that interacted with the active site of the enzyme, which is in the middle of its symmetric halves, to shut the enzyme down and prevent it from functioning properly. Amazingly, by the mid 90s, three HIV Protease inhibitor drugs were on the market, drastically reducing the death rate of the AIDS Virus.[4]
Not only is X-Ray Crystallography a useful tool for drug discovery, it is proven to be beneficial for making drugs better. For instance, current treatment for Parkinson’s disease involves inhibiting an enzyme called Monoamine Oxidase B [MAO B] that help recycle neurotransmitters by removing sometimes crucial molecular components that are left inactive. Although effective, such inhibitors cause undesirable side effects such as changes in heart rate, blood pressure, breathing, etc. However by determining the three dimensional structure of MAO B, along with seeing how some inhibitors attach to the enzyme, Dale Edmondson and his coworkers at Emory University have begun to contemplate methods of making new drugs that bind more specifically to the enzyme, in order to ultimately reduce the side effects.[5]
Additionally, X-ray crystallography has helped to explain how drugs work within the body, how they interact, what makes them work, and so on. A good example of such a case is the widely used drug aspirin. Aspirin has the ability to block the production of prostaglandins, messenger molecules that play various important roles in metabolism, by blocking the cyclooxygenase enzyme (COX) known to operate in the body's metabolic and immune systems. Scientists were able to study the COX enzyme and determine its structure via X-ray crystallography, and by doing so they got a clear picture of how the precise details of the enzyme's structure contribute to its overall molecular function. By determining the 3-dimensional structure of COX enzymes, we are able to understand how drugs like aspirin interact and block it.[6]
The MIR experiments, conducted by NASA, revealed that some proteins produce better quality crystals in a microgravity environment. DCAM, Ambient Diffusion-Controlled Protein Crystal Growth, was the procedure used in these experiments. DCAM uses a liquid to liquid diffusion method to grow protein crystals. These proteins were sent to the Mir Space Station by the Shuttle orbiter to crystallize. The protein crystals were later brought back to the ground for x-ray diffraction analysis. The results were promising. The largest crystals ever produced of certain proteins were produced by these experiments. These proteins include lysozyme, albumin and histone octamer. Bacteriorhodopsin, a membrane associated protein, produced crystals of improved size and quality. Furthermore, DCAM, performed in a microgravity environment, proved successful in providing unusually large protein crystals that could be analyzed for structure by neutron diffraction, a technology that cannot be utilized for smaller crystals grown on Earth. These promising results could produce numerous benefits for people on Earth. Protein crystals are needed to make therapeutic drugs. Scientists can develop new drugs to treat diseases by growing protein crystals in space that could not be grown in a large enough size on the Earth due to the limitations of gravitational forces.
Some of the advantages of X-ray crystallography are that the technique itself can obtain an atomic resolution structure even if the atomic structure is in solution. This is because the structure in crystal form is the same if it were in solution.[Citation needed] Another advantageous aspect is that atomic structure contains a huge amount of data pertaining to the crystallized pure protein. The information one could receive from the structure of the protein can provide more information then finding its niche in the cellular environment.
The utilization of x-ray crystallography to determine protein structure has led to huge and significant breakthroughs in the area of structural biochemistry.
Although researchers have not found a cure for AIDS, structural biology has greatly enhanced their understanding of HIV and has played a key role in the development of drugs to treat this deadly disease. HIV was quickly recognized as a retrovirus, a type of virus that carries its genetic material not as DNA but as RNA. Long before HIV, researchers in labs all over the world studied retroviruses, some of which cause cancers in animals. These scientists traced out the life cycle of retroviruses and identified the key proteins the viruses use to infect cells. When HIV was identified as a retrovirus, these studies gave AIDS researchers an immediate jump-start making the previously discovered viral proteins the initial drug targets.
Scientists also determined the X-ray crystallographic structure of HIV protease, a viral enzyme critical in HIV’s life cycle, in 1989. Pharmaceutical scientists hoped that by blocking this enzyme, they could prevent the virus from spreading in the body. Scientists could finally see their target enzyme. By feeding the structural information into a computer modeling program, they could spin a model of the enzyme around, zoom in on specific atoms, analyze its chemical properties, and even strip away or alter parts of it. Most importantly, they could use the model structure as a reference to determine the types of molecules that might block the enzyme. Such structure-based drug design strategies have the potential to shave off years and millions of dollars from the traditional trial and error drug development process. The structure of HIV protease revealed a crucial fact, the enzyme is made up of two equal halves. For most such symmetrical molecules, both halves have an active site, that carries out the enzyme’s job but HIV protease has only one active site in the center of the molecule where the two halves meet. Pharmaceutical scientists can take advantage of this feature by blocking the single active site with a small molecule, they could shut down the whole enzyme and theoretically stop the virus’ spread in the body by using the enzyme’s structural shape as a guide.
Celebrex was initially designed to treat osteoarthritis and adult rheumatoid arthritis and became the first drug approved to treat a rare condition called FAP or familial adenomatous polyposis that leads to colon cancer. A fortunate discovery enabled scientists to design drugs that retain the anti-inflammatory properties of NSAIDs without the ulcer-causing side effects. By studying the drugs at the molecular level, researchers learned that NSAIDs block the action of two closely related enzymes called cyclooxygenases: COX-1 and COX-2. COX-2 is produced in response to injury or infection and activates molecules that trigger inflammation and an immune response. By blocking COX-2, NSAIDs reduce inflammation and pain caused by arthritis, headaches, and sprains. In contrast, COX-1 produces molecules called prostaglandins that protect the lining of the stomach from digestive acids so when NSAIDs block this function, they foster ulcers.
To create an effective painkiller that doesn’t cause ulcers, scientists realized they needed to develop new medicines that shut down COX-2 but not COX-1. Through structural biology, they could see exactly why Celebrex plugs up COX-2 but not COX-1. The three-dimensional structures of COX-2 and COX-1 are almost identical except for one amino acid change in the active site of COX-2 that creates an extra binding pocket. It is this extra pocket into which Celebrex binds. In addition to showing researchers in atom by atom detail how the drug binds to its target, the structures of the COX enzymes will continue to provide basic researcher with insight into how these molecules work in the body.
Some of the drawbacks of X-ray crystallography are that the sample needs to be in a solid form, the sample must be present in a large enough quantity to be studied, and the sample is often destroyed by the x ray radiation used to study it. This means that nothing in the gas or liquid state can be analyzed via x ray crystallography. Also, rare or hard to synthesize samples may be difficult to study, because there may not be enough of the sample for the radiation to provide a clear image. Thirdly, studying biological samples can be problematic because the radiation used to study the samples is most likely going to harm or destroy the living tissues.
One must also consider that x-ray crystallography takes a huge amount of time to complete upon one protein structure. The rough time estimates for each step of the process goes as follows: Cloning and purification of a protein structure to take up to 3-6 months with perfect execution and 99% purification. Crystallization can take up to 1-12 months pertaining to the physical properties of the protein which is completely based on the favorable enthalpy in which crystal formation is induced and the given solvent that the crystal is induced in. In addition data collection on the protein crystallized structure can take up to a month. Lastly, phasing the structure in solution can take about 3 months to complete. With this said, the process is not quick and with that comes financial issues when taking up a large time span and utilization of various laboratory equipment.
To learn more about Protein X-ray Crystallography see Drenth, Jan: Principles of Protein X-Ray Crystallography 3rd ed.
↑U.S. Department of Health and Human Services. The Structures of Life. July 2007.<http://www.nigms.nih.gov>.
↑Viadiu, Hector. "Why do we need crystals?" UCSD Lecture. November 2011.
↑Kraut, Joseph. "How Do Enzymes Work?", Science, vol.242, 28 October 1988, Pg.534
↑National Institutes of Health, Structure Of Life, 2007, Pgs. 37-38, 40, 44.
↑National Institutes of Health, “Medicine By Design, 2006, Pg.31.
↑National Institutes of Health, “Medicine By Design, 2006, Pg.25-27.
↑U.S. Department of Health and Human Services. The Structures of Life. July 2007.<http://www.nigms.nih.gov>.
↑U.S. Department of Health and Human Services. The Structures of Life. July 2007.<http://www.nigms.nih.gov>.
Berg, Jeremy M., John L. Tymoczko, and Lubert Stryer. Biochemistry. 6th ed. New York: W. H. Freeman and, 2006. Print.
Cryo-Electron Microscopy specializes in interpreting and visualizing unstained biological complexes such as viruses, small organelle, and macromolecular biological complexes of 200 kDa or larger preserved in vitreous (i.e. glassy or non-crystalline) ice. The basic goal is to compare other electron microscopy techniques to use cryo-fixation to rapidly freeze the biological sample so as not to destroy its aqueous enviornment. This avoids ultrastructural changes, redistribution of elements, and washing away of substances. Specimens frozen in vitreous ice show a structure similar to the liquid state, or the native state. The near native imaging conditions allows three dimensional reconstruction of the cellular machinery. Using state of the art computer controlled, automated microscopes, image reconstruction software, and visualization tools, sub-nanometer resolution structures of large biological complexes can be achieved. In Cryo-Electron Microscopy, an electron beam, a stream of high energy particles bombards the sample. The image that is viewed is a result of the interaction of the sample with this beam. Most of the electrons that form the high resolution image appear due to elastic scattering, where only their trajectory has been changed, but their energy is unaffected. However, a small fraction of the electrons transfer some of their energy to the sample. This energy accumulates and can break apart molecular bonds, destroying the sample after some time. Therefore, for high-resolution imaging, low dose parameters require that the area to be imaged is not exposed until the picture is actually taken.
Cryo-electron microscopy can be performed by various methods of specimen preparation, two popular methods use thin film and vitreous sections of biological material. The thin film method requires biological material to be placed on an electron microscopy grid and is rapidly frozen close to liquid nitrogen temperatures. Larger samples (vitreous sections) can be vitrified by different methods including high pressure freezing. These samples can then be cut thinly and placed on the electron microscopy grid, similar to the thin film. These samples must remain at liquid nitrogen temperature to undergo the high vacuum and are exposed to the electrons.
One branch of Cryo-electron microscopy is Cyro-Electron tomography (CET). Cyro-electron tomography is performed at cryogenic temperatures as is cryo-electron microscopy; CET constructs a 3D sample from 2D images.
Cryo-electron microscopy is used in a variety of fields. Nanoparticle research relies heavily on electron microscopy for the visualization of small particles. Pharmaceutical companies doing drug research utilize electron microscopy to help predict the behavior of drugs and biological matter. In the case of pharmaceutical research a 3D visualization is extremely useful and at cyro-electron microscopy proposes the least damage to the sample to obtain a usable image.
Allows the examination of native and hydrated structural features of the biological sample. The sample is always in solution and never comes into contact with an adhering surface. Therefore, the shape that is observed is the true shape of the hydrated molecule in solution and has not been distorted by attaching itself and flattening against the supporting film.
Provides good preservation of biological structure in the microscope vacuum.
There are no stains or chemical fixatives to distort the sample. When stained, the sample can be damaged in many ways, such as flattening and twisting.
When the sample adheres to the carbon grid, it could stick in a preferential orientation. If this happens, then information will be missing from the final image set (a missing cone), and the resolution of the calculated model in that direction will be absent.
Provides a 2-5 fold reduction in radiation damage compared to similar sugar-embedded or freeze-dried samples at room temperature. The reason behind this is thought to be from decreasing the temperature-dependent rearrangement or diffusion of fragments resulting from bond-fracture. In the solid frozen state, rearrangement or diffusion is decreased and the protein conformation is more likely to be maintained up to higher levels of irradiation.
Can observe contrast between nucleic acids, proteins, and lipids to be distinguished.
Enables one to control the chemical environment so that examination of different functional states of molecules is possible.
Very low signal to noise ratio. Biological macromolecules are normally made up of carbon, hydrogen, oxygen, and nitrogen. The electron absorption of such molecules is very low. As a result, image contrast is also very low and it is hard to detect features when dealing with just a few images.
Difficult to obtain images from tilted specimen. The ice cross section of a tilted frozen sample is too thick to yield good images.
Charging is more widespread when imaging a tilted frozen sample.
More time consuming to generate samples. However, this is generally not a big problem, especially once a working protocol is designed and good samples are readily available.
If vitreous ice cannot be easily formed, the resulting cubic ice absorbs electrons very easily and the frozen sample is basically worthless.
Sample must be maintained at less than 135 degrees Celsius.
Preparation of Frozen-hydrated Biological Specimens
The following are some general procedures for preparing frozen-hydrated biological specimens:
1)Development of a thin layer of the biological specimen.
2)Rapid cooling of the specimen to the vitreous state.
3)Transfer the specimen to the electron microscope without rewarming above the devitrification temperature.
4)Observe the specimen below the devitrification temperature with an electron dose that is low enough to preserve the structure of the sample.
This is a basic depiction of Tomography. In Cryo-ET, the images are obtained by tilting the specimen along an axis; this picture does not display this.
Tomography uses the effects and differences that waves of energy have on a solid object to produce a three dimensional image of the internal structure. Cryo-Electron Tomography is a branch of Cryo-Electron Microscopy in which two dimensional projections of a frozen sample, at cryogenic temperatures, are recorded and used to reconstruct a three-dimensional structure by computed back projection. This is done using a transmission electron microscope to take successive images of a sample while tilting the sample around an axis. The “projection theorem” states that a 3D object can be retrieved from its projections along different directions. So to obtain a 3D description of an object, it must be projected along different directions; this is achieved by incrementally tilting the specimen. Due to the limitations with the transmission electron microscope (TEM), the specimen can only be tilted to +- 60-70 degrees, and not to 90 degrees which would be necessary to retrieve all the 3D information about the specimen.
Cryo-ET is a very accurate way to determine the three-dimensional structure of a specimen because the rapid freezing of the object and cryogenic temperatures gives a good preservation of the structure and good time resolution of certain processes. For example, the rapid freezing of cells and tissues at a certain point in cellular processes can give a good understanding of the structure and activity of those cells and tissues at a certain point in time during that particular cellular process. This type of tomography aids in the learning of cells and their organelles at a more dynamic level. Each organelle of a cell is produced in a different color, in order to facilitate the viewing process. The cells are frozen in order for the cell to retain its original structure. Freezing such specimens is done by placing them on a grid, blotting them in a thin layer of water and emerging them into ethane before storing them into liquid nitrogen. The use of cyro-electron tomography involves the study of almost all specimens, such as viruses. This tool can be helpful in understanding the replication states of viruses, as well as, the individual structures that viruses can become. A recent study has been done on the spikes of the viruses and the various structures the spikes affect the virus. Today, cyro-electron tomography is used to help find a cure for cancer by assessing the building blocks of the protein, cadherins, which aid in blocking cancerous tumors for spreading throughout the body. The information obtained through Cryo-ET can aid in comprehending and understanding the structural basis, and therefore, the function of many cellular processes.
There are limitations with Cryo-ET. The main limitation is the thickness of the specimen. The specimen must been thin enough for it to freeze well and so that it can be properly collected with the TEM. If the specimen is too thick, it must be cut into thinner slices while the temperature is still very low, so that re-crystallization does not occur. There are a couple ways to obtain the images, one of the is by fixed tilt increments, and the other is by graduated tilt increments. Graduated tilt increments are more favorable, this is when the tilt increment is proportional to the cosine of the tilt angle. Another issue with Cryo-ET is radiation damage. To prevent radiation damage, the specimen should be imaged under low electron dose conditions, leading to a more limited resolution in the 3D image obtained, and it also limits the specimen thickness needed for Cryo-ET.
Icosahedral Reconstruction refers to the application of cryo-electron microscopy in elucidating the structure of particles with appropriate (icosahedral) symmetry. The high internal symmetry of icosahedral specimens makes it easier to determine the positions of symmetry elements, thereby decreasing the amount of images required to determine the 3D structure of the specimen.[1] This may seem like an arbitrary and irrelevant solid to apply to microscopy, but in fact there is an enormous number of particles that contain such symmetry. Examples of icosahedral particles include the majority of human viruses, as well as some molecules such as dodecahydro-closo-dodecaborate ion (B12 H122-) and the buckminsterfullerene.
An icosahedron.
A number of virus structures have been predicted and subsequently experimentally determined through the use of icosahedral reconstruction. An icosahedron belongs to the high symmetry group Ih which contains 120 symmetry operations, possibly most unique being the six five-fold symmetry axes.[2] The properties of this symmetry group are essential for the application of cryo-electron microscopy.[3]
Helical Reconstruction is a method that takes advantage of Cryo-Electron Microscopy in order to develop a three-dimensional structure for certain "filamentous" biological structures. This method used the 2-D projection images from Cryo-Electron microscopy to produce these 3-d Images as long as there is helical symmetry. This method however cannot be applied to structures that contain "seams" or "pertubations". There is a new method known as asymmetrical helical reconstruction that can be applied to helical structures that contain "seams". Similar to conventional Helical reconstruction methods, Fourier transform images are used to produce the layer line data which are then used to produce the 3-d structures.
Helical reconstruction allows the formation of large groups by regular contacts of a single type of protein molecule. Helical symmetry can be found in filamentous viruses (e.g., Pf1), in the proteins of the actin, tubulin, or other cytoskeleton, or in the proteins that form 2-D crystals folded onto the surface of a cylinder, such as the acetyocholine receptor or CopA.
Electron crystallography is a form of microscopy that uses a beam of electrons to construct images of small solids such as proteins. This process is used to determine and predict the structure and arrangement of a protein from secondary structure crystals such as alpha helices or beta sheets based on electron scattering. It can be used to study both organic and inorganic matters, and also protein structures. Electron Crystallography complements X-ray crystallography in many ways but also succeeds where X-ray crystallography fails. For example, X-ray crystallography study requires the quaternary structure of proteins which is often hard to attain than secondary structures. Electron Crystallography presents a problem in that it can cause radiation damage to the proteins under analysis. This hinders the range and function of the microscopy process. In order to reduce radiation damage, cryofixation, in which the imaging takes place in very low temperatures such as that of liquid nitrogen, is implemented. This resource is especially valuable when a specific protein is easily denatured or damaged by the electrons from the microscope.
A crystal structure determination includes two steps: ‘solving’ which finds a model of the heaviest atoms within about 0.25 Å using EM-images; and ‘refine’, which finds all atoms within about 0.02 Å using Selected Area Electron Diffraction or Convergent Beam Electron Diffraction data.
The use of electron diffraction in order to study the structures of crystals began in Moscow in 1937-1938 among a group of scientist led by Pinsker and Vainshtein. Their study used their own electron diffraction cameras that had relatively low acceleration to record electron diffraction data of different materials. From this data, they were able to locate hydrogen atoms in crystal structures which can not be done using X-ray diffraction. In order to solve unknown structures, phase information is needed which was first introduced by Hauptmann and Karle in 1953 called the "direct methods". Combining the use of direct phasing methods with modern day computers, electron crystallography has made significance advances in structure determination of crystals and other molecules.
There are two different electron diffraction techniques: 1)Selected Area Electron Diffraction (SAED)which requires near kinematic condition and applies for unit cell dimension >10 Å and for thin specimens <200 Å; 2)Convergent Beam Electron Diffraction (CBED)which makes use of dynamical effects and applies for unit cell dimensions <10 Å and for thick specimens >200 Å
Why electrons? Electrons are used in favor of X-rays because it is 10^4-10^5 times stronger interaction with matter compared with X-ray; and their phases are present in high resolution electron microscopy images.
There are some key advantages of electron crystallography compared to X-ray crystallography. One of these advantages is that electron crystallography can analyze much smaller crystals. This is because electrons interact more dominantly with matter than X-rays do. Another advantage is that electron beams can be focused by magnetic lenses to create an image while X-rays cannot form an image. Because the mechanism by which electrons interact with matter is based on the electrons detecting potential distributions in crystals compared to the mechanism of X-rays which depends on the X-rays detecting electron density distribution, electron crystallography can be used in certain situations that X-ray crystallography cannot. For example, the oxidation states of atoms in a crystal.
Using electron crystallography to determine structure is important due to the ability for a protein to be observed in its natural form. By utilizing electron crystallography, one can observe a protein in a lipid-protein bilayer in the structure that it is found in, thus allowing for better determination of function.
The techniques used to reconstruct the 3 Dimensional images of the molecule from a collections of 2-D images is called electron microscopy. It presents to structural biochemists insight views in term of structural information of many biological molecules because of its easy-to-access features. In order to acquire 3D structure from this method, two requirement must met.
1. reasonable size of proteins to large macro-molecular assemblies without need to use crystals
2. the molecules must exist in many identical copies
The resolution produced by electron tomography have a low resolution and high noise. The main goal of the single particle electron microscopy is to determine the geometric relation between the collected projection images. In the year of 2008, scientist were able to made it possible to trace the backbone of the polypetide chains and build atomic models. Single Particles EM indeed have the capability to deliver structural information at near atomic resolution.
The 3D structure would be the result of these following steps:
1) Sample preparation. This is the step where sample is collected and place on (metal) plate to generate the best contrast. There are three technique used to prepare the samples for single particle EM.
a) Negative staining : molecules are adsorbed to a continuous carbon film in which molecules are put into a metal plate by drying
b) vitrification: sample is plunged into liquid ethane to preserve molecules in a native environment, it produced low contrast. (preserving them in a fully hydrated state)Vitrification is the best speciment preparation method, but not applicable to heterogeneous samples.
c) Cryo negative staining: high contrast image immersed in high ionic strength of saturated ammonium molybdate solution. It is good to study the small and heterogeneous samples.
2) Particle picking. This is one of the most tedious processes of all because electron microscopists have to classify and separate particles according to their similarities in orientations; all these works are done by hand in order to achieve maximum efficiency. Having said that automated picking programs have been developed, but they failed to perform the task thank to the low signal-to-noise ratio. Result of this step is collection of small individual images of particles.
3) Generate initial model. Individual images collected from previous step are used to build a preliminary model. RCT is the primary method that is being used to generate initial models.
Random Conial Tilt Reconstruction is being use throughout the process. Random Conical Tilt are the reconstruction of the 3 dimensional image one at high tilt angle and the other at untilt angle. The tilt angles allowd the testing samples to align in unique orientations. Please see the image at the right for the figure. Random Conical Tilt is common used in negatively or cryo-negatively stained speciments, in which works well with heterogenous particle solutions.
4) Refinement. The preliminary model is used to calculate for better alignment using Euler angles, in-plane rotated/unrotated shifts of particles. From refined data, a new 3D structure of a molecule is reconstructed. The risk of overrefinement happeneds when the testing negative temperature is applied.
Single-particle electron microscopy is having advantages ahead of electron crystallographic because its economic features. Unlike crystallographic, it does not require crystals – meaning samples don’t have to be pure - which take a lot more works to achieve. Another advantage is that single particle electron microscopy takes very little sample which always makes researchers happy.
One of the disadvantages of single particle electron microscopy, however, is that it is difficult to determine the resolution of density maps as well as their accuracy. Since there is no evidence of a method to check for accuracy, often the only thing that can be done is to repeat the process and compare results to previous results. Therefore, results can only be assessed on the basis of consistency rather than accuracy.
Electron crystallography has been used in the study of membrane proteins by analyzing arrays of samples called protein-lipid arrays. These arrays can be arranged in many different ways and offer many advantages and disadvantages. Two forms that have given the most useful density maps of membrane proteins are the two dimensional sheet like crystal arrays and the tubular like crystal arrays. These are so precise that they can reveal information about individual lipid molecules and the protein side chains due to averaging the many unit cells in the image of a sheet or tube like array enhancing the poor signal-to-noise ratio.
Over the last few decades there has been much advancement in the methods used to create these protein-lipid arrays both in tubes and sheets from detergent-solubilized purified proteins but no advancements in screens have been made like an easily manipulated robotic screen. Despite much research in the field they still lack the ability to quickly and reliably check the quality of the samples. Several laboratories have advanced the methods of analysis of these crystals making the process more efficient and more user friendly by enhancing the existing software.
The tubular crystals have not seen as extensive use as the sheet crystals even though their helical array symmetry allows for substantial advantages in determining structure. One image of a tube contains many different views of the same molecule which is enough to reconstruct it in three dimensions without the need for tilting. To correct for distortions tubes are processed in a similar way to sheets, where two repeat lengths are divided into shorter pieces and are then compared to a reference structure to determine the parameters needed to help identify the structure completely. This procedure traditionally uses Fourier-Bessel methods to assess the data, enabling them to analyze the extent of helical symmetry preservation and twofold symmetry perpendicular to the tube axis, which can correct for the focus changes at different levels of the structure. Another method has been developed that doesn’t employ the Fourier-Bessel methods and instead treats segments as strings of single particles. This alternative is becoming more popularly used for extracting structural information from poorly ordered helical polymers such as tubular protein-lipid crystals. This shows great potential for determining structures from tubes at the near atomic level of resolution.
Methods involved in electron crystallography include free-trapping to create different conformational states. To freeze-trap the specimen, the electron microscope grid is placed into liquid nitrogen-cooled ethane, which cools the specimen rapidly enough that thus allows for the trapping of a structure of a lipid-protein array which has a life-time of a millisecond or longer. The freeze-trapped protein can be activated through light or an appropriate ligand. Recent developments of helium-cooled top-entry freeze-trapping has resulted in a more clear image for data collection, and hopefully would allow for the gating mechanism of the protein-lipid bilayer to be described in more detail.
Additionally, molecular tomography is used to explore proteins in their functional context. A three-dimensional picture of an entire scene is possible to create though taking images from a series of tilt views, therefore creating a better three-dimensional image.
It is a method that observing dynamic behavior of single molecule to determine mechanism of action at level of an individual molecule, and to identify, sort and compare subpopulation and substructure within cell. In order to characterize the dynamics of molecular structures, scientists look to real-time trajectories of individual molecule; and by observing many of them, a histogram of the dynamical properties over the population could be figured.
X-ray crystallography or NMR, in comparison to single-molecule methods, provides detail structural view but limit by static molecular view and ensemble average.
1. Single-molecule manipulation: In this method, molecules are attached to an external probe which exerts defined forces or torques on molecule in order to characterize their mechanical properties. This method is also called atomic force microscopy (AMF). Because cell is seen as a factory in which many processes are carried out by specialized machinery which converts the chemical energy into force, torque and mechanical work - of which the attached probe will now come in to detect the dynamics and mechanism. This method is recently used to study the folding and unfolding of RNA molecules and the enzymes that catalyze these reactions, and to study RNA polymerase.
2. Single-molecule detection: The molecule is tagged with a fluorescent label in two locations in the form of a “donor” and “acceptor” that can undergo fluorescence resonance energy transfer (FRET). The trajectory of molecules then can be watched regarding to a change in the intensity of the florescence of the probe or regarding to the change in FRET. Another name of this method is fluorescence method. . This is a powerful method to study dynamic behavior of molecules, their stability and track particles’ movements in and outside cell. This method is used to study, for example, the multiple interactions during translation by the ribosome.
Between the two mentioned above, the fluorescence detection method is preferable to researchers because it requires less elaborate and complex instrumentations, but the down side is the photons collected by instrument is limited.
↑Jiang, Wen, Zongli Li, Zhixian Zhang, Christopher R. Booth, and Matthew L. Baker. "Semi-automated Icosahedral Particle Reconstruction at Sub-nanometer Resolution." Journal of Structural Biology . 136. (2001): 214-225. Print.
↑Miessler, Gary. Inorganic Chemistry. 3rd ed. Dehli: Pearson Education in South Asia, 2007. Print.
↑Fuller, S.D., and S.J. Butcher. "Three-Dimensional Reconstruction of Icosahedral Particles-The Uncommon Line." Journal of Structural Biology . 116. (1996): 48-55. Print.
Infrared Spectroscopy (IR Spectroscopy) is a common spectroscopic technique used to analyze the functional groups of a sample by measuring its absorbency through different IR frequencies. IR spectrometers can accept a wide range of samples, including solid, liquids and gases. The infrared region of the electromagnetic spectrum can be divided into three regions, the near-, mid, and far- infrared. Near IR has a range of about 14000 cm-1, mid-infrared ranges from approximately 4000-400 cm-1, and far-infrared ranges from 400-10 cm-1. [1]
Infrared Spectroscopy essentially is a method used to identify functional groups of an unknown structure by varying vibrational activity. Infrared ‘light’ or radiation hits the bonds in the molecule, absorb the energy of the infrared light and respond by vibrating. This vibrating action can be anything from simple bending to stretching, rocking and scissoring. According to the amount of vibrational activity that occurs in the varying functional groups, the functional group is localized at a certain wavenumber that is calculated as inverse centimeters. Each functional group has a unique peak at certain wavenumbers. Essentially literature values of these functional groups can be used to help identify an unknown structure once put through an IR spectroscoper. Given peaks can be matched with literature values and a structure can be solved.
↑Hsu, Sherman. "Infrared Spectroscopy." Handbook of Instrumental Techniques for Analytical Chemistry. Norwalk: Perkin-Elmer, n.d. 247-83. Www.prenhall.com. The Perkin-Elmer Corp. Web. <http://www.prenhall.com/settle/chapters/ch15.pdf>.
Protein crystal, (PCG) Protein Crystal Growth Porcine Elastase. This enzyme is associated with the degradation of lung tissue in people suffering from emphysema. It is useful in studying causes of this disease.Protein crystal, Malic Enzyme is a target protein for drug design because it is a key protein in the life cycle of intestinal parasites. After 2 years of effort on Earth, investigators were unable to produce any crystals that were of high enough quality and for this reason, the structure of this important protein could not be determined. Crystals obtained from one STS-50 were of superior quality allowing the structure to be determined. This is just one example why access to space is so vital for these studies.
Neutron Diffraction (also known as neutron scattering or neutron crystallography) is an experimental science that studies the spatial arrangement of atoms in proteins. Although neutron diffraction and X-ray scattering techniques use different radiation sources, the resulting diffraction pattern is analyzed using the same coherent imaging techniques. However, the use of neutron diffraction as an experimental technique is still a relatively new technique compared to X-ray and electron diffraction because the accumulation of free neutrons, the radiation source, can only be obtained from nuclear reactors.
Although neutron diffraction has been used as an experimental technique in physics since the early 1900s, its application in chemistry and biology did not start until the 1980s. In 1984, Wlodawer, Walter, Huber, and Sjolin collaborated to bring X-ray crystallography and neutron diffraction methods together to come up with novel methods of determining internal dynamics of protein molecules. Their experiment utilized the joint application of two methods to determine the structure of a new crystal form of bovine pancreatic trypsin inhibitor (BPTI). The project is the first to reveal the atomic position of proteins to a size within 0.1 nanometers (i.e. the diameter of a hydrogen atom), and it is the first detailed analysis of how protein structure is affected by molecular packing.
In 1994, Clifford G. Shull was awarded the Nobel Prize in Physics for developing
a new way to use neutron diffraction. From this point on,
neutron diffraction took new life with the new usage of scattering, a technique that
allowed scientists to observe that the dynamics of atom movement and excitation.
He found that when a material was shot with a beam of neutrons, the neutrons bounced
off of atoms and hit other atoms thus scattering light and making neutrons go in every which
way and direction. This created a general pattern that we can use to deduce the surrounding
substituents and atom placement.
By understanding these substituents and their placements we can develop more insight into the intrinsic nature
of molecules and atoms. According to Clifford, these new discoveries would pave the way for "better semiconductors, better microphones, better window glass, etc."
Shull worked as a professor at MIT for many years. When he won the Nobel prize award in 1994, he was already retired from
teaching but continued to conduct research. Ever since his innovative technique of
neutron scattering, he was hailed as the father of neutron scattering.
Fundamentally neutron diffraction relies upon the fact that free neutrons exhibit wave-like diffraction behaviors, and this happens when the radiation wave encounters obstacles (e.g. proteins) that is comparable with the wavelength. Generally, the effects of diffraction are more pronounced for waves where the wavelength is on the order of the size of the diffracting object. Neutron diffraction is a type of elastic scattering, meaning that the incoming energy of the neutron is equivalent to the outgoing energy after scattering occurs.
Additionally, the method is a unique tool for studying a wide variety of materials with magnetic properties because neutrons have magnetic moments that can interact with orbital and spin moments in magnetic atoms.
To study crystalline solids and molecules and their structure it is useful to free these neutrons and excite them the intensity pattern and formation of the excitation gives us information about the structure of a molecule. These neutrons are not found in nature at least
not for a very long period of time. Nuclear reactors can set these neutrons free
and we can study diffraction of these neutrons. By studying their wavelength and quantum properties we essentially create a sample, not unlike
x-ray diffraction. Neutron diffraction is similar and generates structural information much like electron diffraction but
neutron beams actually have a strong affinity to react with the internal nuclei of cells than do x-rays. Also when studying neutrons we have more insight into the positioning of protons. X-rays at most times can destroy
or denature a material under study due to their intensive X-rays in which case neutron diffraction can be very beneficial.
The scattering effect of the neutrons can be explained through two different phenomena. Firstly there is a
close proximity reaction between the neutron and the atomic nucleus because these neutrons have a natural affinity to do so.
This interaction is specific to each atomic number because the atomic nucleus is classified as a point scatterer, thus producing
isotropic scattering. The second interaction relates to the function of magnetism and spin. The magnetic moment of the neutron
is directly connected with the spin and orbital hybridization and arrangement of the molecule or atom. Detailed data can be obtained through this
magnetic approach study which is absent in other forms of crystallography.
With the knowledge that scattering patterns do not vary from atom to atom with the same atomic number, we can substitute different enriched isotopes to get a
more panoramic and holistic study of the molecule. Because neutrons are not charged, we do not have to worry
about the possibility of them interacting heavily with the electron cloud surrounding the atom. This solves the problem
of electron diffraction. Neutrons will only directly react with the nucleus.
How does neutron diffraction help in studying crystallized lattice structures? Inelastic neutron scattering studies
vibrational thermodynamics which elucidates the equilibrium structure. Lattice vibration can be induced with neutrons
that are low in energy, they can also be induced to release ponons[check spelling] or quanta. With this diffraction pattern, we can relate vibrational modes of each part of the crystal and calculate dispersion relations and then construct an idea
of its structure.
The essence of neutron diffraction, as noted by its "father," Clifford Shull, was to identify hydrogen atoms
and how they appeared to go about processes in biological materials or inorganic substances. Hydrogen containing
structures are very much present in our world and necessary for the development of drugs, resources, and other endeavors.
With the use of neutrons, we can exploit nucleus relations with protons and neutrons without being
disturbed by the electron cloud.
Neutron Diffraction can be used to determine the atomic structure of low atomic number molecules such as proteins because low atomic number materials have higher nucleus cross-sectional areas for neutrons to interact with. This method of crystallographic experiment is similar to X-ray diffraction; however, the fact that neutrons are scattered by the nucleus rather than electrons in an atom means that the effect of diffraction is independent of atomic number. Furthermore, this diffraction method works because neutrons have energies with equivalent wavelengths in the 0.1 nanometers and are therefore suitable for interatomic interference studies.
Using neutron diffraction to determine protein structures requires several steps. First, it requires careful preparation of the protein crystals, for without perfect crystals of protein it is impossible to carry out any crystallographic structural studies. The aim of protein crystallization is to produce well-ordered crystals that are large enough to diffract neutron beam. Therefore, the crystallization process is long and is often the rate-limiting step in the experiment. After the derivative protein crystals become available, the crystals are mounted and neutron diffraction images are taken.
Neutron diffraction in the industrial world has been used to probe structures and magnetism of condensed matter. Industrial interests such as mechanical behavior in materials.
Engineers must study strain on certain molecules to understand strain mapping. Of course, all
mechanical behavior starts with microscopic scales of structural features. Engineers must take into account
residual stress which can be studied through neutron diffraction. When applying neutron diffraction to engineering work,
it is referred to as engineering diffraction.
Neutron diffraction is very sensitive to small nuances in structure thus creating a multitude
of different peaks that depend on three factors. These three factors are peak position, peak width, and integrated intensity.
This allows engineers to access the texture, strain, and strain fluctuation of a sample. The principle of understanding
strain studies is based on Bragg's law.
So what kind of engineering situations would call for neutron diffraction? One practical application is that of welding. When welds have nonuniform expansion or shrinkage in heat directed zones during times of intense heat when the metal alloys as passed through a weld pass,
residual stress is observed. Residual stress can include cracking and change in shape. This could limit the quality of a product
and even corrode machinery. Neutron diffraction provides a 3-D spatial
distribution of the tensor of stress. This can be used in all kinds of weld
materials. Different materials must take into account different parameters and geometries. In short, engineers
depend on the uniformity, rigidity, and strength of their materials and when these things fail, scientific approaches
such as neutron diffraction can help identify the problem and innovate new ideas of the solution to create and facilitate
advances in engineering materials. Thus neutron diffraction is a precursor to all industrial and practice clauses.
Neutron Peaks and Diffraction, Applications in Industry and Engineering
Recently, the traditional view of the protein universe as a set of discrete secondary structures has been challenged by the proliferation of a complementary theory describing continuous protein structuring. This new theory does not replace or supersede the idea of a discrete protein universe, but is complementary to it, allowing for a wider range of observed phenomena to be explained. The relationship between a discreet and a continuous protein universe is similar in nature to the relationship between the particle theory and wave theory of electromagnetic radiation (light). Just as light is best described as both a particle and a wave, the protein universe can be best described as being discreet and continuous. The combination of these two views is known as the dual view. The following is a discussion of the two single views and their application, as well as the advantages of considering the dual view.
The discrete view of the protein universe was first established via x-ray crystallography. Upon the structural elucidation of myoglobin and hemoglobin, these two proteins were found to be very similar, despite their dissimilarity in primary structure. Secondary structures were the cause of their similarity, and thus the concept of ‘folding’ was introduced to describe discrete sections of the protein that exhibit a repeating structure. Examples of secondary structures include alpha helices, beta sheets, and turns. As more structures were solved, more types of secondary structural folds were established. This view was embraced due in part to perceived evolutionary relationships between discrete secondary structures. Homology is transitive with respect to discrete secondary structures.
The most compelling argument for the existence of a continuous protein universe is the growing evidence that almost any set of secondary structures is possible. The potential continuous structure is dictated by the rules of hydrogen bonding. Not all structures can be perfectly aligned with one another; with most structures, at least 40% alignment can be achieved. Unlike discrete structures, the continuous structure of a protein is not the result of evolutionary fine-tuning, but rather the result of simple H-bonds between corresponding structures that lie adjacent to one another in space. It then follows that continuous structure is also known as geometric structure. On a continuous scale, density is not constant. All proteins will have areas with high and low numbers of substructures. This concept is similar to that of a Ramachandran plot of peptide bond conformations. In both cases, many conformations are possible but only a select few will most often be observed.
A dual view of the protein universe allows biochemists to better categorize and ultimately understand protein structure. One such instance is centered on folding. Based on its definition, folding implies that attention should only be given to how a specific structure interacts with itself and keeps itself together. This means that potential functional connections with other folds will most likely be overlooked. For example, the study of most alpha helices is centered on the differences between angles that dictate helical packing. As a result, differences in helical surfaces would be deemed unimportant, even though they may likely elucidate protein function.
Another such instance is the tendency of over-classification of secondary structures under the discrete structural system. When only discrete structures are considered, any minute difference from one instance of a structure to another is enough to argue that the two structures are different. This leads to the “discovery” of too many new types of folds, which is ultimately inefficient when applied to better understanding the big picture. Alternately, the continuous view will help eliminate the mental inertia caused by this problem. Since almost any type of fold is possible under the parameters of the continuous structural view, “new” folds could simply be placed into existing categories based on the intrinsic properties of their secondary structures, and connections to adjacent structures could be categorized separately, and thus better studied.
Analyzing protein structure and function using ancestral gene reconstruction
Learning how protein sequence determines structure and function as well as learning the processes that generated the diverse structures and functions of extant proteins requires knowledge of the distribution of structures and functions through the multidimensional space of possible protein sequences. However, characterizing that distribution can be very difficult due to the vast number of possible sequences and the time required to experimentally generate and study them. One answer to this problem is to analyze the evolutionary record. Evolution is one enormous experiment involving the diversification and optimization of protein structure. The outcomes of that massive experiment are preserved in the sequences, structures, and functions of modern-day protein families. Evolutionary analysis of these families can provide key insights into the nature of protein sequence space and the determinants of protein structure and function.
One way to study protein families is to identify candidate amino acid differences between divergent family members using sequence-based or structural analysis. This can be followed by testing the functional role of these residues by exchanging them between family members using site-directed mutagenesis. This is known as the "horizontal" approach, which identifies residues that are important to one function because exchanging them will result in impaired or nonfunctional protein. However, this approach rarely identifies the set of residues sufficient to switch the function of one protein to that of another. Protein function evolves as mutations accumulated through time, or vertically, in ancestral protein lineages. On the other hand, horizontal comparisons of modern proteins involve only the tips of the evolutionary tree. The horizontal approach has two major flaws. First, it is inefficient in that many functionally irrelevant sequence differences may have accumulate during intervals in which the function of interest did not change. Second, lineage-specific sequence changes may lead to epistasis, or the interdependence between mutations that cause a single change to have different effects in different protein family members.
An explicitly phylogenetic approach to study functional diversity within the protein families can solve these issues. A vertical strategy would address mutations that occur along the branch in the family tree on which functional diversification occurred. This strategy is more efficient in that only mutations that occurred during limited period of evolutionary time need to be investigated. Furthermore, this vertical strategy can avoid the effect of epistatic interactions by using the protein background in which the sequence changes actually occurred. A vertical strategy will even identify restrictive and permissive epistatic mutations.
Studying evolution along a family branch can be difficult in that it requires access to the nodes on either end of the branch. However, ancestral sequence reconstruction (ASR), a new strategy for studying molecular evolution, can address this problem. ASR is a mature technique that has been used to study many protein families including GFP-like proteins, steroid receptors, opsins, etc. ASR first infers ancestral sequences from an alignment of extant protein sequences. The maximum likelihood sequence at any ancestral node on the phylogeny is the sequence with the highest probability of generating all of the sequence data in modern-day proteins. Once the ancestral protein sequence is uncovered, a DNA molecule coding for it can be synthesized. This allows the ancestral protein to be expressed and characterized experimentally. The following case studies demonstrate the effectiveness of ASR studies to quantitatively dissect the interactions that determine function, reveal multiple amino acids that underlie function, and determine the role of epistasis in shaping protein evolution.
Opsins: quantifying functional interactions
The study of the opsins, a family of G-protein coupled receptors that absorb light in the vertebrate visual system, demonstrates the benefits of using ASR to study the effects of function-switching mutations. All opsins use the same covalently attached chromophore. However, each opsin has a distinct wavelength of maximum absorption. Comparative studies in modern opsins are difficult to interpret due to the complexity of sequence determinants of wavelength of maximum absorption. However, researchers were able to use ASR to dissect these interactions and yielded results universally applicable to the family as a whole.
GFP-like proteins
The work by Mikhail Matz's laboratory on GFP-like proteins from scleratinian corals demonstrated effective use of ASR in identifying those residues. By using ASR to characterize ancient sequences throughout the family, Matz and colleagues found that GFP-like protein in the ancestral Faviina fluoresced in the green, followed by a variety of other colors. Matz and colleagues then determined to identify the mutations responsible for the evolution of red fluorescence from this green ancestor in the great star coral Montastrea cavernosa. By using ASR, they were able to identify mutations that would have been impossible to identify using a horizontal approach.
These ASR case studies demonstrate that ASR promises new insights into the physical-chemical determinants that have shaped protein evolution and historical determinants of protein architecture. Furthermore, ASR has build a bridge between mechanistic biochemistry and evolutionary biology, fields of study that have been largely separate.
Enzymes are powerful biological catalyst. Catalysts speed up a reaction but are not consumed in the reaction. Enzymes are thus essential for such bodily functions as digestion because otherwise, these reactions would occur at too high of temperatures for the body to handle. The catalysis process takes place at the active site. Enzymes are extremely selective with their reactants, or substrates, and the type of chemical reactions they are involved with. This is due to similar shape, charge, and characteristics between the enzymes and the substrates. Bringing together the enzymes and the substrates is called enzyme-substrate (ES) complexes.
It has been observed through experimental analysis that enzymatic transition states often use kinetic isotope effects to understand the bonding differences between reactants and the intermediates found in the transition states. Kinetic isotope effects refer to the ratio of the rates of reaction of two different isotopically labeled molecules in a given reaction. In addition to transition states, enzymes also play an important role as common pharmaceutical targets since many drugs act as enzyme inhibitors.
The most common feature of enzymes is that they are able to catalyze reactions and increase the reaction rates. They can also overcome larger single transition state energy barriers by breaking them down and creating multiple steps of smaller barriers. Reaction rates are also limited by conformational changes that occur in proteins as well as the rate at which reactant are released to yield product. In addition, the values of the kinetic isotope effects, often intrinsic, typically are the result of differences in the bond environments for atoms in the reactant state compared to the bond environments found in transition states.
The ability to determine enzymatic activity is extremely important to clinical chemistry because it allows for early diagnosis of various diseases and helps doctors determine the course of treatment for such diseases. The spectroscopic characteristics of enzymes and substrates change when they combine to form an ES complex. In order to measure this activity demonstrated by the enzyme, the following spectroscopic techniques are used: Fluorescence spectroscopy, UV/VIS Spectroscopy, Spectrophotometric Assays, and Infrared spectroscopy.
Fluorescence spectroscopy
Fluorescence spectroscopy reveals the existence of ES complexes and what they are made of. In Fluorescence spectroscopy, a compound is exposed to UV-light which excites certain molecules and causes them to emit light at a lower wavelength, which is typically in the visible light range. This phenomenon in which the molecule's absorption of a photon at one wavelength leads to the emission of another photon from the same molecule at a longer wavelength is known as fluorescence. In this spectroscopic technique, the fluorescence of the substrate is measured and compared to the fluorescence of the product, and it is in the difference of these two measurements that enzymatic activity is measured.
A typical procedure for light-extinction measurements is as follows. At predetermined time intervals, 5-7 measurement points are collected by measuring the light-extinction of the enzyme sample, where a light-absorbing substance is either consumed or produced during the reaction. This can be measured using a photometer. The mean value and standard deviation of these measurement points are then found and plotted versus time. Then a regression curve is drawn through these points. The enzyme activity can then be found from the slope of the regression curve at a particular time
Many impurities found in fluorescent compounds, when exposed to light, interfere with the spectroscopy, making this technique more sensitive than other assays.
Ultraviolet-visible Spectroscopy
A method that is complimentary to fluorescence spectroscopy is ultraviolet-visible spectroscopy in that fluorescence spectroscopy deals with transitions from the excited state to the ground state and ultraviolet-visible spectroscopy deals with transitions from the ground state to the excited state. UV spectroscopy uses light in the UV region where molecules are most likely to undergo electronic transitions. What is meant by electronic transitions is that when a molecule absorbs UV energy, this causes the electrons to become excited, meaning they quickly and unstably move into a higher energy orbital. The instrument used in UV spectroscopy is a UV/VIS spectrophotometer. This device measures the transmittance of light through a sample. The equation used to calculate the transmittance is A=-log(%T) where A is the absorbance and T is I/Io where I is the intensity of light passing through the sample and Io is the initial intensity of the light, before it is transmitted through the sample. Once the absorbance is calculated, it can be plotted versus the wavelength giving a UV/VIS spectrum.
Spectrophotometric Assay
Spectrophotometric assays can track the course of a reaction by measuring how much light the assay absorbs. When the light is absorbed in the visible light region (400-750nm), the assay will actually change colors. This is called a colorimetric assay. An example of a colorimetric assay is the MTT. In the MTT assay, Yellow MTT(3-(4.5-Dimethylthiazol-2-yl)-2.5-diphenyltetrazolium bromide) reduces to purple formazan in the mitochondria of cells, giving the reaction a purple color which allows for analysis of the enzyme. Some type of solution is used to dissolve the purple formazan product. A spectrophotometer is then used to measure the wavelength, which is usually between 500-600nm. The solvent that is used determines the maximum absorption wavelength.
Ultraviolet-visible Spectroscopy
Ultraviolet-visible spectroscopy is a commonly used spectrophotometric assay that examines photons in the UV-visible region. It is mainly used to determine the amount of a highly-conjugated organic compound or enzyme contained in a specific solution. The Beer-Lambert Law is used in Ultraviolet-visible spectroscopy to determine the concentration of the species that is absorbing the light. The Beer-Lambert Law states:
A=-log10(I/I0)
This equation shows the directly-proportional relationship between the solutions' concentration and the absorption of the solution, where A is the absorption of the solution and I refers to its concentration.
Infrared Spectroscopy
Another type of spectroscopy that can be used to obtain information about enzyme-substrate complexes is infrared spectroscopy. In enzyme-substrate complexes, there is well-organized binding modes, which is quantifiable using infrared methods. In analyzing infrared data, it is possible to identify binding modes and heterogeneity of ES complexes.
Detectors are needed to actually measure the enzymatic activity produced in the previously described processes. The following describes typical detectors used in industry.
UV/visible detectors
UV/visible light detectors are the most common used in industry because they are versatile, have a wide dynamic range, have a high sensitivity, and they are not affected by temperature and flow variations.
Fixed-wavelength detectors
Fixed-wavelengths detectors use lamps that emit light at certain wavelengths. To select a particular wavelength a cut-off filter can be used. Fixed-wavelength detectors are good because they don't produce a lot of noise, it doesn't cost very much to operate them, and operating them is relatively simple.
Variable Wavelength Detectors
Variable wavelength detectors are different from fixed-wavelength detectors in that they use a range of wavelengths, typically between 190-700 nm continuously instead of just one wavelength at a time. For wavelength selection in the variable wavelength detector a continuously adjustable monochromator is used. The lamp is typically deuterium or tungsten. Light from the lamp travels to mirrors that focus and steer the light into a diffraction grating, and the grating drive mechanisms are all part of the monochromator assembly. To monitor more than one wavelength at a time, the grating is rapidly adjusted between two different wavelengths.
One of the most recent developments for variable wavelength detectors is the Diode Array Detector (DAD). The DAD basically speeds up the entire process by setting multiple detectors next to each other on a silicon crystal and using a capacitor to convert light to electric charge. this makes it so that light from the grating can be detected much quicker than in a conventional spectrophotometer. The advantages to using a DAD over motor-driven monochromators are that they possess fewer moving parts and they are less likely to have irregular data at high flow rates.
Fluorescence detectors
Fluorescence detectors are used only when the compound can't be detected by the other methods and the compound must have fluorescence or can come to have fluorescence by reacting with a fluorescent compound. The intensity of light that is emitted is directly proportional to the power of exciting radiation, therefore fluorometric detection is much more sensitive than absorption. Usually the fluorescence detector is made up of a light source, a wavelength reflector and a single or dual-flow cell.
The light source is typically xenon, mercury arc, or quartz halogen. The wavelength reflector allows for the excitation spectra to be obtained for the compound. This information can be used for rapid method optimization and verification of separation quality. Lastly, the single and dual-flow cells take account the fluorescence of the mobile phase.
Refractive Index (RI) detectors
Refractive Index (RI) detectors measure the change in refractive indices in the reference and sample cells. The drawback to this method is that refracted indices between compounds can be very small, therefore the sensitivity of the RI detector is much smaller than for UV/VIS and fluorescence detectors. Another negative in using an RI detector is that the detector response is affected by the mobile phase composition, which means there is a lot of error in the final data collected. There are three types of RI detectors: deflection, fresnel, and interferometric, but the deflection type is the most commonly used.
In the deflection detector, a light beam passes between two parallel chambers in a glass prism, which acts as a reference. If the refractive index of the solution is equal to the refractive index of the reference, then the light beam is parallel to the incident beam. However, if the refractive indices are different, the beam is deflected and then measure by a differential photodiode.
Electrochemical detectors
Electrochemical detectors are most commonly used for biogenic amines because they are more sensitive and more selective than the previous methods. There are two types of electrochemical detectors: bulk property and solute property. The bulk property detectors are most commonly used and they measure the change in cell resistance. Solute property detectors monitor the change in potential or current as the solute passes through the cell. Solute is passed over an electrode which is held at a constant voltage. The current produced is proportional to solute concentration.
Kappe, Walter, Gotz-reinhard Lampe, and Harald Neuer. Method and apparatus for the determination of enzyme activity. Carl, Zeiss-stiftung, assignee. Patent 4055752. 1977.
After post-translation, proteins can be further modified by being attached to carbohydrate groups (sugars) by glycosidic bonds via the process called "glycosylation", and the newly formed molecule is called a "glycoprotein". There are two types of glycosidic bonds that can occur in this process, called N-linkage and O-linkage.
N-linkage: The nitrogen atom in the side chain of Asparagine is attached to the sugar. The sequence can be Asn-X-Ser or Asn-X-Thr, where X is any kind of amino acid except proline.
O-linkage: The oxygen atom in the side chain of serine or threonine amino acids is attached to the sugar.
Proteins are often glycosylated while secretion from a cell. The glycosylated proteins are mostly found in the blood serum. Being one of the components in the cell membrane, they are responsible for combining cells or even combining sperm and eggs.
Protein glycosylation is an enzyme-directed chemical reaction that takes place in the ER(Endoplasmic Reticulum) and in the Golgi Apparatus body of the cell.[22] General glycosylation within the ER helps with folding, and glycosylation in the Golgi body tells a protein where to go. The ribosomes sticking on the cytoplasmic surface of the ER membrane synthesize the protein. The peptide chain is then sent into the lumen of the ER. There are N-linked and O-linked glycosylation processes. This is determined by whether the sugars in glycoproteins are attached to the amide nitrogen on the amino acid asparagine, or to the oxygen on the side chains of either serine or threonine. The N-linkage glycosylations happens in both ER and in the Golgi complex while the O-linked glycosylation only occurs in the Golgi complex. In the Golgi complex, the glycosylated proteins derive the carbohydrates out, which change their shape to keep working in the Golgi complex. Proteins derived from the glycoproteins diffuse into vesicles and are transported into different places according to the signals instructed by the amino acid sequence and the three-dimensional structures.
N-linked glycosylation mostly takes place in eukaryotes and archaea, but rarely in bacteria. When a 14-sugar chain, including 2 N-acetylglucosamine molecules, 3 glucose, and 9 mannose, is attached to the asparagine amino acid in the target protein, dolichol molecle is carried by reaction and sent into the ER lumen. There are two kinds of N-linked oligosaccarides: High-mannose oligosaccharides, and complex oligosaccharides. High-mannose oligosaccharides is a combination of the 2 N-acetylglucosamine molecules and many numbers of mannose residues attached. This is the most common chain. The complex oligosaccharides is the combination of any number and of any kinds of saccharides attaching together. The modification of both two types depends on the accessibility of the modified proteins in the Golgi complex. If the oligosaccharides are not accessible, then the high-mannose will not be cleaved for further modification.
The cytoplasm is not a place for protein glycosylation, because sugars and complex enzymes are stored in the lumenal side of the ER, so the proteins are not glycosylated as they are above.
Glycosylation can avoid the incorrect folding of the original proteins. Many proteins do not fold correctly unless they undergo glycosylation. It also increases the stability of the protein structures in blood so that they will not degrade as quickly as those unglycosylated proteins. For example, glycoproteins linked at the amide nitrogen in asparagine in the protein have increased stability. N-linked glycosylation of this sort occurs when the protein sequence Asn-X-Thr or Asn-X-Ser is reached. X, in this case can be any amino acid except for proline. Glycosylation helps to adhere between cells. This mechanism of cell to cell adhesion is especially vital in cells of the immune system. [23]
Congenital disorders of Glycosylation is a type of disease caused by incorrect glycosylation. I-cell disease is an example of such congenital disorders. The lysosomes include undigested glycosaminoglycans and glycolipids because the responsible enzymes containing a mannose residue are missed to degrade them. In other words, the mannose residues are not being modified in the enzymes so that they cannot degrade the glycosaminoglycan and glycolipids. Urine and blood also contain high level of such enzymes. Because of this mistake, the carbohydrates and the glycosaminoglycans will accumulate more and more and finally cause patients in a pathological condition.
Glycosylation is a posttranslational modification to proteins which influences the tertiary structure based on the placement of glycols on the protein and the timing in the folding process when the glycols get introduced. "The magnitude of thermodynamic protein stabilization by glycosylation depends on the properties of both the carbohydrate and the protein moieties."[24] Glycosylation stabilization is dependent on the position of the glycol in the protein. Also, it is shown that the size of the oligosaccharide is not as important of a factor on the outcome in the protein structure as the properties of the oligosaccharide attached to the protein. In highly structured regions of proteins glycolysation destabilizes the section and in highly flexible regions the glycol stabilizes the region. Furthermore the glycols either keep areas expanded if they are added before protein folding commences or compact the size of the overall protein if added at a later time in the protein folding sequence. Glycolysation often results in reduced flexibility of the folded protein. The stabilization conferred by glycolysation is similar to that of molecular crowding and confinement yet has little influence on the folding transition temperature when compared to these other effects. However, it is hypothesized that the stabilization by glycolysation increases in crowded molecular environments, but this has yet to be tested. [25]
An example of a glycoprotein that has provided much effect in the medical field is Erythropoietin also known as EPO. This glycoprotein has improved the treatment for anemia particularly induced by cancer chemotherapy. It is secreted by the kidneys and stimulates the production of red blood cells. EPO is made of 165 amino acid. It is N-glycosylated at the asparagine residue and O-glycosylated on a serine residue. It is 40% carbohydrate by weight. The glycosylation enhances stability of the protein in the blood as compared to the unglycoslyated protein which only carries about 10% of the bioactivity. This is because protein is rapidly removed from the blood by the kidney. Although recombinant human EPO has aided the treatment of anemia it has also been misused by athletes to increase their red blood cell count and their oxygen carrying capacity. However modern drug testing can usually distinguish between this and natural EPO.
Comparison of Vertebrate Innate Immune Response to Vertebrate Coagulation:
Horseshoe Crab Coagulation vs. Vertebrate Coagulation
LPS
Horseshoe crabs produce a gel that traps small foreign antigens via a proteolytic cascade that occurs in which bacterial lipopolysachharide (LPS), a part of the outer membrane of a bacterial antigen, binds to a cell-surface protein Factor C on a granular hemocyte containing inactive hemolymph. LPS’s binding to Factor C triggers the exocytosis of defense proteins.[1]
The binding site of Factor C on hemocyte cell surface has a specific tripeptide motif of two basic residues interspersed by an aromatic amino acid. Analogously, mammalian innate immune response, mammalian coagulation factors are also contained in granules before exocytosis which activate platelets.[2]
Also, phagocytic cells in mammals are able to distinguish between host cells and foreign microbes via common microbial patterns that are recognized by host proteins, called Pattern Recognition Receptors (PRRs), which operate in a way analogous to the particular PRR Factor C on hemocytes of horseshoe crabs by recognizing certain functionality or motifs on invading microbes. For example, the Macrophage Mannose receptor in mammals binds to mannose residues prevalent on the surface of antigenic microorganisms.[3] From this comparison of the innate immune system of horseshoe crabs and vertebrate mammals, it is evident that the structural characteristics of LPS-recognizing proteins across different species are conserved.
Complement System
The complement system is a cascade involving proteases, proteins that cleave peptide bonds, that “complement” the ability of antibodies to clear pathogens from an organism. The complement system is a part of immune system, specifically the innate immune system.
Vertebrate complement system (complement cascade)
The Vertebrate complement system has several serine proteases, enzymes that cleave peptide bonds in proteins that have one amino acid at the active site as a serine, involved in parallel proteolytic cascades. Proteases in the system cleave specific proteins to release cytokines, which themselves in turn, cascade more cleavages. The proteases involved in vertebrate complement system can be classified into two families, the complement factor B (Bf) and the mannose-binding lectin-associated serine protease (MASP). The Bf family includes Bf and C3 proteases. The MASP family comprises the MASP-1, MASP-2, MASP-3, C1r, and C1s proteases. In addition, the vertebrate complement system includes the C3 family of proteins, which are not proteases. C3 proteins have an intramolecular thioester bond that is revealed after proteolytic cleavage and can react with the surface of invading microbes via a PRR mechanism. Altogether, the three families form an integral part of complement activation.
Invertebrate complement system
Proteins involved in invertebrate complement system vary depending on phyla and species. However, the vertebrate Bf, C3, and MASP complement families of proteins have been generalized to their vertebrate counterparts, in reference to their common ancestral origins. The invertebrate orthologs (genes in dissimilar species that have evolved from a common ancestral gene) can be classified according to phyla. A few examples are explored in the following.
Horseshoe crab complement system
Horseshoe Crab
Horseshoe crabs are used an example because its complement system has been characterized functionally and biochemically. They have CrC3 (C3 ortholog) and CrBf (Bf ortholog) proteins that are structurally and functionally like their vertebrate orthologs.
CrC3’s chemical structure indicates that it may be activated via a thioester bond in binding to pathogens for opsonization.
Protostomes
Protostomes are organisms having bilateral symmetry as well as three germ layers. Arthropods, nematodes, mollusks are examples. Protostomes have C3 and Bf proteases.
Ascidians
Ascidian complement system include the components Bf, MASP, glucose-binding lectin, GBL, ficolin, and integrin alpha and beta chains. These components are synthesized in its hepatopancreas. MASPs associated with GBL can proteolytically activate ascidian C3, however this activation is poor, indicating that ascidian MASP is not a major C3-activating enzyme, a distinct difference from the vertebrate complement system.
Adenylyl cyclases (ACs) are proteins that transduce a large variety of extracellular signals into intracellular responses, and therefore are important in signal transduction. ACs function to control the rate of conversion of ATP (substrate) into the second messenger cAMP (cyclic Adenine monophosphate), which in turn activates effector cells. In eukaryotes, activated effector cells proceed through a mechanism, depending on pathway, resulting in intracellular signal amplification.
There are six classes of ACs, one of which is the Class III AC. Class II AC possesses a characteristic a dimeric tertiary structure. The dimer has important functional significance, because it forms catalytic pockets at the dimer interface. Previous research suggests that regulation of Class III AC activity is achieved by shifts of the three-dimensional spatial orientation of the two monomers towards each other. Specifically, research has been done on the homologs, mammalian class III ACs and bacterial Class III ACs.
Mammalian class III ACs have two catalytic domains, called C1 and C2. The C1 and C2 domains are 25-30% identical, and they have low affinity for each other. C1 and C2 interact to form two distinct binding pockets at the interface. One of the pockets binds ATP and catalyzes the cyclization of ATP into cAMP. The cyclization is directed by four amino acid residues that stabilize the involved species. Two negatively charged aspartic acid residues on C1 bind to Mg2+ ion to stabilize the triphosphate present in the ATP substrate. The positively charged residues arginine and asparagine contributed by the C2 domain stabilize the transition state by neutralizing an excess negative charge at the alpha-phosphoryl. The other binding pocket is a docking site for the activator molecule, forskolin.
Forskolin
Evidence of Mammalian AC Regulation via Reorientation
Evidence for AC regulation by reorientation in mammalian ACs is seen in conformational changes induces by the binding of G proteins, which are transducers, to both C1 and C2 at non-catalytic sites. Regulation by reorientation occurs in mammalian ACs when the AC is activated by the G-protein, Gs-alpha. When the stimulatory Gs-alpha binds to C1 and C2, a conformational change of a 7° rotation leads to a closure of the catalytic site, which enhances the catalysis of ATP into cAMP occurs. When the inhibitory Gi-alpha binds opposite the G2alpha, it counteracts the rotation induced by Gs-alpha, thus effectively reducing the efficiency of ATP catalysis. Hence, G-proteins activate (Gs-alpha) and inhibit (Gi-alpha) the catalyzation by causing changes in the dimer interface conformation.
Bacterial class III ACs are homodimers. Note that this makes mammalian ACs and bacterial ACs orthologs. A few examples of bacterial AC isoforms and their reorientation mechanisms are presented below:
The mycobacterial AC Rv1264 is a homodimer whose conformation is regulated by pH. At pH = 6, it is in its activated state for the ATP substrate. At neutral pH, Rv1264 is in its inhibited state, in which alpha helix is extended disrupting the catalytic pocket. In the inhibited state of Rv1264, the monomers are pulled close to the regulatory platform such that massive reorientation involving the rotation of each catalytic domain by 55° and a translation by 6 angstroms. The lysine and aspartate residue on Rv1264 are essential for the substrate to bind.
Mycobacterial AC Rv1900c is regulated by the binding of the ATP substrate itself
In the absence of the substrate, the catalytic domain of Rv1900c is slightly asymmetric.
When the bacterial ATP-analog binds, the dimer closes and the asymmetry is heightened, rotating the monomers by 16.6° and a translation of 11.4 angstrom of one monomer. Owing to is dimeric tertiary structure, there are two binding pockets, but ATP-analog binds to only one, while the other is non-functional.
CyaC is activated when bicarbonate binds to the catalytic domain which causes a shift in a single alpha-helix while overall orientation of the monomers in the homodimer remains unchanged. This is an example of an activation mechanism in which a small regulator moiety (the bicarbonate) activates the cyclase by only minor structural rearrangements. As the CyaC is activated, the catalytic pocket folds.
A CyaC adenylyl cyclase. Note the identical subunits in the dimer.
Dimer interface of ACs regulate catalysis of ATP into cAMP, and therefore regulate signal transduction pathways that involve ATP. The dimer interface regulates cyclization of ATP into cAMP via conformational changes in the tertiary structure of the adenylyl cyclase arising from various biochemical mechanisms and factors. These conformational changes in structure of the dimer arise from the binding of G-proteins that stimulate or inhibit the catalyzation reaction in mammalian adenylyl cyclases. In bacterial adenylyl cyclases, biochemical conditions and factors such as pH, presence of a particular moiety (e.g. bicarbonate), and the binding of the substrate itself (ATP) may change the conformation of the tertiary structure, which in turn regulates the activity of the catalytic site by opening or closing it.
Transmissible spongiform encephalopathies (TSEs or prion diseases) are a rare group of deadly neurodegenerative disorders that affect humans and other mammals. TSEs are protein misfolding diseases that encompass the aggregation of abnormally accumulated form of the normal host prion protein. TSEs are unique in that they are transmissible. Characteristics of TSEs include that they replicate, are capable of selective evolution (can evolve drug resistance), have various strains of infectious agent that are related to unique phenotypes in vivo, and demonstrate strong species specificities; the same characteristic of many viral and bacterial pathogens.
Prion subdomain-Residues 125-228 are shown
There are 3 classes of TSE diseases in humans:
Sporadic – the most common form of TSE (e.g. Creutzfeldt Jakob disease)
Heritable – in which TSE is mutation within the prion protein
Acquired – in which TSE is a result of ingestion or inoculation of TSE contaminated materials.
Microscopic "holes" are characteristic in prion-affected tissue sections, causing the tissue to develop a "spongy" architecture.
Typical infectious agents use nucleic acids to spread and propagate, evident in bacteria and viruses. The prion hypothesis states simply that the specific diseases detailed above are solely caused by proteins, a sentiment that went against common knowledge at the time of its proposal. At first, it was thought that prions were a side-effect of some other type of infection. This theory does not hold up, however. Experimental evidence since the concept of prion-induced disease was proposed has fallen in favor of this theory, and the effects of prion replication have been witnessed in the lab. [1]
Prions and their infectious nature were first discovered in 1937 when scrapie was accidentally transferred to a sheep while attempting a viral inoculation. In later experiments, scrapie was purposefully transmitted to sheep and then mice to determine the nature of this molecule. Cannibalism in New Guinea was the source of infection by kuru in humans and in 1966 this disease was demonstrated to be transmissible to monkeys[1]. At this point, science was beginning to understand that the infectious nature of prions was different from viral or microbial infection. More recently, outbreaks of Bovine Spongiform Encephalopathy and the emerging variant Creutzfeldt-Jakob disease that was linked to consumption of infected meat were shown to be caused by prions.
Historically, studies of prions first determined that the method of infection was novel, and then determined that it was due to a misfolding of the protein chain. The first experiments destroyed nucleic acids with UV and ionizing radiation and found the infectious agent still present[1]. Then, the smallest molecular weight particles that were still infectious were determined to be on the order of protein weight.
Later on, protease-resistant prion protein (PrP) concentration was found to be proportional to the infectivity. Additionally, agents that destroyed protein structure were employed, reducing the infectivity of the PrP. PrP was found to be present in a normally functioning brain, demonstrating that the protein could exist both as a normal and as a malfunctioning protein[1]. Recently, studies have shown that infectious and non infectious proteins could be mixed and the infectious agent would propagate among non infectious molecules. This demonstrates that the protein folding error can propagate indefinitely.
TSE is a protein misfolding disease in that disease occurs due to conformational changes in host prion protein (PrP). PrP is a mammalian glycoprotein, 209 amino acids long. When the PrP becomes a TSE, in a process known as pathogenesis, a protease sensitive form of PrP (PrP-sen) refolds into PrP-res (a protease- resistant form of prion protein). PrP-res and PrP-sen have the same primary sequence but different secondary structures, with the PrP-sen featuring more alpha-helices. (In other words, PrP-res and PrP-sen are isoforms) PrP-res is the primary component of TSE. Hence it is imperative to research how the PrP-sen to PrP-res conversion occurs, in order to inhibit the formation of the PrP-res form.
Current knowledge of the PrPc to PrPSc conversion can be generalized into a two-step mechanism:
1) Ordered aggregates of pre-cursor “seed” PrPSc bind to PrPc.
2) PrPc experiences some conformational change that results in the propagation into more PrPSc. The mechanism of this step is largely a mystery. The converted PrPSc is added to the polymer, which eventually fragments and causes more PrPc to be converted. This fragmentation is thought to be the rate-limiting step of the reaction.[1]
In 2001, a process for laboratory replication of prions was developed. This process, known as Protein Misfolding Cyclic Amplication (PMCA), produces prions in-vitro which mimic prions replicated normally in-vivo. This process relies on the idea that prions are auto-catalytic, and can reproduce indefinitely given the correct surroundings. This technique has proven invaluable in the study of prions and the testing of the prion hypothesis. [1]
One area that is still unclear to scientists is the importance of co-factors in the replication process of prions. While it is now understood that there are outside factors that influence the success and rate of prion replication, these co-factors have not been fully discerned and represent an area for future research.
Evidence for the role of co-factors in prion replication has come from various studies performed on animal subjects. In a study done on transgenic mice, there was found to be some sort of factor affecting prion protein expression. This was termed "protein X", though the identity of the factor specifically as a protein was never discerned. [2]. In a separate study performed on hamster PrPc. When isolated and purified, hamster PrPc could not be converted when mixed with PrPSc. When brain homogenate was introduced to the sample, the prion conversion occurred, indicating that there was something in the homogenate that contributed to the conversion, whether as a catalyst or as an integral factor. Further testing showed that that RNA in hamsters acted as a catalyst for prion replication, but not in mice. Specifically how it does this is not clear, but there is a possibility that it helps to stabilize the comformation of PrPSc that is produced. The fact that it does not work on all mammals leads to the possibility of multiple co-factors, or species/organ specific ones.
Although the importance of co-factors is not fully understood, the ways in which co-factors might affect the conversion of prions fall into 5 categories.
Genetic Information
It is possible that there are certain co-factors that contribute to prion replication by helping to determine how these proteins fold. One study that supports this was performed on mice by injecting prions into different types of cells. In each cell, prion replication led to different prion strains, which could be a result of different co-factors in each cell type.
Certain co-factors might catalyze the PrPc to PrPSc conversion by attaching to PrPc and partially unfolding it. This would make it easier for PrPSc to induce a misfolding pattern on the protein, resulting in the conversion. This type of co-factor has been examined in the lab in vitro.
Conformational Stabilization
Some co-factors might aid in the stabilization of the new prion conformation. Examples of these include nucleic acids, proteins, metal ions, and other things. Many of these are charged species that can bind to the prion and help form a more compact structure.
Fragmentation
PrPSc polymers often fragment, which greatly speeds up the conversion of PrPc to PrPSc. Some co-factors may help in this fragmentation process, which creates new seeds and is essential to the replication process. An example of this is found in yeast, which rely on protein 104 (Hsp104). Removing this protein stops prion replication from occuring.
Biological Stabilization
For successful prion replication to occur, prions must first survive in the biological medium. Certain biological resistance, such as microglia cells which can perform phagocytosis, can inhibit their spread. A co-factor that reduces the ability of microglia to destroy these prions would allow them to replicate, and might prove to be essential. [1]
Primary and secondary structural components within at amino acids 108-189 of the PrP-sen proved to be important for conversion, as determined by NMR structure and in vivo studies. PrP-sen at the critical residues (108-189) include most of the folded domain including beta strands, alpha-helices, and most critical, the loops and turns. Slight variation with turns and loops are particularly noteworthy, as it has been determined that the loops are involved in the intermolecular interactions between PrP-sen and PrP-res.
While there is extensive knowledge of the PrP-sen tertiary structure, the same cannot be said for PrP-res. Part of the reason as to why there isn’t extensive knowledge regarding the mechanistic pathway of the conversion into PrP-res is that the PrP-res structure is unknown, as it has not been purified sufficiently for high-resolution structural studies. Because high-resolution techniques cannot be used, focus on ascertaining the structure of PrP-res has been confined to using lower resolution techniques such as electron microscopy to determine ultrastructure and the secondary structure of PrP-res.
One interesting case of prions can be found in yeast. While they are normally observed in mammals, it was found that a particular protein in yeast (Ure2) behaves and reproduces in the way a typical prion does. However, instead of killing its host cell it instead reproduces by inducing conformational change in other proteins and is inherited through cellular division. This special case has led to the discovery of other prions in fungus which also exhibit non-lethal behavior.[1]
File:MAVS Protein.jpgMAVS protein conformational change as a result of viral activity.
As found in the example of yeast prions, these misfolded proteins are not all inherently malicious and harmful to organic life. In one study conducted by scientists in UT Southwestern, it was found that certain prion-like proteins found within the body may help in the immune system. These mitochondrial antiviral signaling proteins (MAVS) were found to be in mitochondrial membranes, and actively defend them against infection. It was found that under threat of viral infection, MAVS proteins misfold and aggregate on the surface of the mitochondria, effectively shielding the organelle from attack. This deliberate misfolding and aggregation is interesting because it does not reflect the malicious and uncontrollable misfolding often caused by prions in cases such as Cretzfeldt-Jakob Disease and Bovine Spongify Esophagus. While the sequential activation of the system by the initial viral detection can be complex, the aggregation of MAVS proteins actually occurred quickly. [4]
The way PrP-res amplifies itself is highly similar to the way in which amyloid fibrils grow through seeded polymerization from a reservoir of precursor protein. Therefore amyloid fibril offers insight for which to hypothesize the PrP-res growth mechanism.
Amyloids are fibrous protein aggregates that harbor specific structural traits. The structure of amyloid fibrils features a characteristic cross-beta folding pattern in which beta-strands align to form sheets that are perpendicular to the fibril axis while hydrogen bonds between strands is parallel to the fibril axis. While this cross-beta quaternary structure is known in amyloids, a detailed structure of this scope has not been determined for PrP, which is by far, a much larger protein.
Like amyloid fibrils, PrP can be induced to form amyloid-like fibrils. A particularly significant model of this was that of the recombinant peptide, PrP23-144. (Recombinant peptide is the peptide encoded from its corresponding recombinant DNA). The PrP23-144 proved to have a high degree of conformational plasticity (ability to change conformation in various possible ways) and can be induced to form a wide range of fibril amino acids. That the PrP23-144 in this model could be differentiated into a wide range of conformational possibilities is an indication that the PrP isoform conversion is highly dependent on the structural compatibility between PrP-sen to PrP- res (for example, the aforementioned variations in turns and loops). Choice of solvent also plays a role in that certain structural elements are affected by electrostatic or hydrophilic effects.
As a result of the limited knowledge scientists have on prions, there is currently no full-proof method of treatment for those diagnosed with a prion related disease. One possibility of prion removal is through microglia and phagocytosis. It has also been found that lichens posses the ability to break down prions. [5] Though there are many avenues of research possible, there is no clinical solution for those suffering from a prion related disease.
There has been research in the past decade that has helped to identify PrPSc in the blood. Up until this point, there has been no reliable way to identify these misfolded proteins in brain tissue. Employing a new method known as SOFIA (surround optical fibre immunoassay), scientists have been able to identify the presence of prions in brain tissue at high precision (one part in one hundred thousand million). First, the sample with PrPSc is amplified and fluorescently labeled using an antibody. It is then put through a device that surrounds it with optical fibers that detect emitted light. A laser is used to excite the dye, and the emission from the sample is picked up by the detector.
This method was tested on sheep that were previously thought to be healthy, but eventually developed prion disease. Samples of their blood before they exhibited symptoms were analyzed, and it was found that the presence of PrPSc could be detected very early on. While this method does not cure or remove any of the problems, it can be used to quarantine and greatly reduce the risk of these prions spreading. [6]
↑Rubenstein, R., Chang, B., Gray, P., Piltch, M., Bulgin, M.S., Sorensen-Melson, S. & Miller, M.W "Detecting Prions in Blood", '[Microbiology Today]', 2010. Retrieved on 20 November 2012.
Immunology deals largely with the interactions between antibodies and antigens. However, immunological techniques can be used for protein research. Antibodies are highly specific to their target proteins, thus making it possible to tag a certain protein for further in depth studies.
Immunological techniques can be utilized to make the polyclonal and monoclonal antibodies used in protein identification.
An antibody (also known as immunoglobulin or Ig) is one of an animal's defense mechanisms. One type of antibody, Immunoglobulin G or IgG, has a Y-like structure consisting of two heavy chains and two light chains. The structures of antibodies are partially maintained by disulfide bonds, which link the heavy chains to heavy chains or heavy chains to light chains. The heavy chains always have the same sequence of amino acids while the light chains have varied sequences of amino acids. This variation is important to ensure that the antibody will be able to respond to various antigens, foreign substances that enter animal bodies (e.g.: foreign protein, polysaccharides, nucleic acids, etc.). Antibodies are produced by the B cell, which is a type of white blood cell. B cells will allow the immune system to remember and react faster for future exposures. There are various types of antibodies, which are categorized based on the isotopes that the chains possess. Approximately five different types are discovered in mammals which each carry a special function and response based on the different types of bacteria that they encounter throughout the body. Each of the classes has identical light chains which is matched with a different heavy chain. Each of these classes of immunoglobulin has a specific function. Immunoglobulin M (IgM) is the first class of antibody to appear after exposure to an antigen. IgM has ten combining sites which enables it to bind extra well with antigens containing multiple identical epitopes. An example of avidity, which is the strength of an interaction that contains multiple independent binding interactions between matching antibody-antigen complex. Immunoglobulin A (IgA) is the antibody that is involved in external secretions - tears and saliva - meaning it is the body's first line of defense against bacteria and viruses. The role of Immunoglobulin D (IgD) is still unknown. Immunoglobulin E (IgE) helps to protect against parasites. However, it also participates in allergic reactions. The release of granules containing pharmacologically active molecules is triggered when the IgE - antigen complex forms cross-links with receptors on the surface of mast cells. These antibodies are generated randomly using different gene segments which give a specific bind site for the antibody. They are also randomized by the mutations in the genes, making them even more complex and diverse. This relates to the humoral immune system (see below) that helps humans protect against the wide range of diversity in bacterias and viruses. There are two forms of antibodies: one that is soluble in the blood and fluids of the body and the other one that is attached to the B cell in general. The structure of the antibody includes a heavy chain and a light chain. The heavy chain has a constant region and a variable region and it is about 110 amino acids long. Antibodies first bind to the pathogens to prevent them from damaging cells. Second, they will try to remove pathogens using macrophages. Finally, they will stimulate other immune responses to further destroy the pathogens. The cells and proteins connected with this process are discussed below.
Immune Cell Response- Cells and Proteins Involved
The immune response system is composed of two corresponding systems, the humoral and cellular immune systems. The humoral immune system occurs primarily in body fluids, targeting bacterial infections and extracellular viruses. The humoral system can also react to individual foreign proteins. The cellular immune system attacks host cells infected by viruses as well as some parasite and foreign tissues. Both of these systems are prompted by a category of white blood cells called leukocytes, of which include macrophages (cells that ingest by phagocytosis) and lymphocytes (cells that release antibodies). In the cellular immune system, a class of T cells, called cytotoxic T cells, are the main cells involved with reception of foreign cells or parasites. These cells have T cell receptors that are on the surface of cells and extend through the plasma membrane. When an antigen is deteected, either a T cell receptor or antibody will bind to a specific molecular structure in the antigen. This structure is referred to as antigenic determinant or an epitope (see Epitopes under Protein Function). Helper T cells are also a part of the cellular immune system, generating cytokines, a type of soluble signaling proteins. In clonal selection, helper T cells are involved only indirectly, prompting the selective reproduction of the cytotoxic T cells and B cells that can bind to the certain antigen.
In performing this response, antibodies can either be:
Monoclonal
One type of antibody responding to one type of antigen by recognizing one antigenic determinant.
monoclonal antibody
Polyclonal
Several type of antibody responding to one type of antigen by cooperatively recognizing various antigenic determinants on the antigen.
polyclonal antibody
Epitopes
Epitopes are the sites on a antigen that the immune system recognizes. It is also called an antigenic determinant. Both the host and foreign protein can produce epitopes that can bind to the paratopes of the B and T lymphocytes. An epitope can be either linear or conformational epitope. Linear epitotes are recognized by long chain of amino acids, or their primary structure: the sequence of the animo acids. Conformational epitopes are recognized by the antibodies by the 3-D structures of the epitopes.
Antibody Folding
Recent studies have shown how antibodies fold. Foldings occur in the endoplasmic reticulum even before chains complete translation. Most of these studies are made by dissecting the antibody and letting it denaturalize and refold. IgGs are mostly used for these experiments. IgGs mostly form through adding a light chain to a heavy chain dimer between the CL and CH1 region. Dissection of IgGs light chain shows that there are three pathways of folding caused and limited to proline cis-trans isomerization. In general, most antibodies are formed through these three pathways after the C2-C3 sulfide bridge formed.
In equilibrium, most proteins have the accessible conformations, such as the native state, unfolded state, and non specific aggregates. However, At low pH (pH<3), Antibodies have tendency to adopt specific additional conformation, called “alternating folded state”( though most other proteins are unfolded in this environment). Through spectroscopic, the structure is different from the nature state. However, it exhibits characteristic of the folded state ( e.g. stability against unfolding). The concern about this process is significant in biotechnology because antibody manufacturing process often include low pH steps, which can easily perform the “alternatively folded state”.
For some antibodies, another accessible state is the fibrillar amyloid structure, that can lead to some protein-folding diseases. Fibrillar amyloid structure is a cross-beta structure in which fibrils are formed by beta-strand exchange of the individual subunits. Isolated LCs and truncated HCs form fibrils and are deposited in organs such as kidneys. The large quantity deposit can cause some fatal diseases when it interferes with physiological functions. E.g. A large deposit of monoclonal LCs prone to misfolding and the formation of amyloid deposit can cause a fatal disease [light chain amyloidosis (AL)].The fibrilization mechanism is still under study . However, the mechanism in production of variable domains might generate less stable domain, therefore, can pass ER quality control but have a propensity to misfold outside the cell.
B cell: a stage in which heavy and light chains are synthesized and expressed at cell surface via a transmembrance .
Heavy/light chain: constituent polypeptide chains of antibody molecules. Light chains are made up of two Ig domains. Heavy chains are made up of minimum of 3 Ig domains.
Ig domain: 100 amino acid-folding unit conserves twisted barrel-like-beta-sheet structure. It is stabilized by a buried intrachain disulfide bond.
Clonal Expansion
When the antibodies recognizes a specific antigen, the B cells clone itself and turn into 2 types of cells: the plasma cells and the memory cells.
The plasma cells are effector cells that produce a large amount of a specific soluble antibody that attacks the target antigen.
Memory Cells are long lived cells that can be quickly activated to produce antibodies when the specific antigen is observed.
During the primary Immune response, that is the first time antibody attacks an antigen, it takes a long time for the antibody to make a large amount of plasma cells and memory cells when compared to the secondary response. During the secondary immune response, there is only a short time lag and the rate of Antibody produced is a lot greater. This is due to the memory cells activating the production of antibodies.
Nelson, David L., and Michael M. Cox. Principles of Biochemistry. 5th ed. New York: W.H Freeman and Company, 2008.
Buchner, Johannes; Feige, Matthias J.; and Hendershot, Linda M. "How antibodies fold". Trends in Biochemical Sciences. Vol. 5, (4). doi:10.1016/j.tibs.2009.11.005.
Agglutination occurs when an antibody interacts with antigen, resulting in cross-linking of the antigen particles by the antibody. This eventually leads to clumping. "Agglutination" comes from the Latin root agglutinare. "Agglutinare" means, "to glue". Cross-linking, or cross-matching is done to determine matches. Agglutination may occur when an unideterminate, multivalent antigen interacts with a single antibody. It may also occur if a multi determinate, univalent antigen interacts with at least two distinct antibodies.
Cross Agglutination: when an antibody that is raised against a similar antigen agglutinates that antigen.
Group Agglutination: when a collection of similar organisms is agglutinated by an agglutinin that is specific for that collection.
Applications for the agglutination test
BLOOD TYPING
There are different blood groups like A,B, AB and O. The different blood types are differentiated according to the types of proteins on the surfaces of the red blood cells. Human red blood cells may either posses both of epitopes A and B on their surfaces. Individuals possessing only epitope A have anti-B antibody in their serum, while individuals possessing only epitope B on their red blood cells have circulating anti-A antibody. Some individuals have neither epitope and both antibodies present, while others have both epitopes and neither antibody.
Blood type determination can be performed by mixing a sample of the individual’s blood with w solutions: one containing anti-A antibody and the other containing anti-B antibody. Agglutination occurs if the antibodies match the epitopes of the blood cells. By observing the agglutination patterns, the blood type of the individual can be determined.
DETERMINING MATING TYPES IN ORGANISMS
Agglutination can be used to determine mating types in organisms such as Chlamydomonas. Extracts from cloned Chlamydomonas cells of either of the minus or plus type are added to cells of the opposite mating type as well as of the same mating type. What is observed is that, in the cells of similar type, no agglutination occurs. However, in the type with opposite mating type, agglutination does occur. Thus, this proves that the cells of different mating types are chemically different.
The first usage of antibodiesis identifying and locating intracellular and extracellular protein by flow cytometry technique.
In immnunoprecipitation, an antibody is used to separate its specific protein target and anything bound to them from other molecules in a cell lysate.
In a western blot, an antibody is used to identify proteins separated by electrophoresis. This technique is useful for mixtures of many proteins.
In immunohistochemistry or immunofluorescence, an antibody is used to examine protein expression in tissue sections or to locate proteins within cells with the assistance of a microscope.
Proteins can also be detected and quantified with antibodies, using ELISA and ELISPOT techniques.
Enzyme-linked Immunoabsorbent Assay (ELISA) is an analytical method utilizing various
antibodies to detect the presence of a compound in a wet or liquid sample. Enzyme linked to the antibodies react with substrates to produce a color change, signifying the presence of desired substance, usually antigen. The intensity of the color can be used to determine the concentration of substance of interest in a sample. Antibodies are assayed to form a pure line of monoclonal antibodies that only detect the desired antigen or protein.
If the antibody-labeled enzyme is specific to another antibody, the indirect ELISA method is used whereas if the antibody-labeled enzyme can directly react with the antigen, sandwich method is preferred.
Purpose: Detect the presence of a type of antibody.
Procedure
Coat sample well with antigen.
Add sample to coated well. If antibody of interest (Antibody A) is present in the sample, it will bind to the antigen. Wash out the remaining sample.
Add a second antibody (Antibody B) to the sample. Antibody B is an antibody that will specifically bind to antibody A and is linked to a special enzyme. Wash out the remaining antibody B.
Substrate is added to the mixture. This substrate will trigger a reaction with enzyme attached to antibody B to produce colored substance.
The rate at which the color changes is proportional to the amount of antibody in the solution.
In the Sandwich ElISA, or two site capture assay, two different antibodies are used. The wells are coated with an antibody specific for one region of the antigen, then the test solution containing antigen is added. Following washing, the second antibody, which recognizes a different epitope of the antigen, will be added. This antibody will have the enzyme attach.
Purpose: Detect the presence of a type of antigen.
Procedure
Coat sample well with monoclonal antibody (Antibody Z) that responds specifically to one type of antigen.
Add sample that might contain the antigen of interest (Antigen C) to the coated sample well. If sample contains Antigen C, it will bind to antibody Z. Wash out the remaining sample.
Add a second monoclonal antibody (Antibody Y), which also binds to Antigen C, to the well. This antibody is linked to a special enzyme.
Substrate, which reacts with enzymes on Antibody Y, is added. Reaction occurs and color change is observed.
Presence of colored product confirms the presence of Antigen C. The intensity of the colored product can be used to determine the concentration of Antigen C. Also the rate of color change of the solution is proportional to the amount of antigen present.
Application of sandwich ELISA: Pregnancy tests
In the pregnancy test, the reaction zone will contain the primary antibody (usually a monoclonal mouse antibody IgG) that recognizes a portion of a unique beta chain of the pregnancy hormone called Human Chorionic Gonadotropin (HCG). The secondary antibody conjugated with enzyme in the test zone recognizes the alpha chain. This chain can be found in luteinizing hormone (LH) which also present in non-pregnant women. It will display a color change in this test zone if the antigen was bound to both antibodies forming a sandwich. At last, a control zone contains antibody that binds to primary antibody (anti-IgG). A color change in this zone will show that our test was correctly preformed.
Reverse ELISA is a relatively new technique specifically created to investigate the West Nile virus envelope protein and how it is able to detect virus-specific antibodies. This newer technique uses an solid phase made up of an immunosorbent polystyrene rod with ogives. The entire device is immersed in a test tube containing the collected sample and the following usual steps such as washing, incubation in conjugate and incubation in chromogenous are performed through dipping the micro-wells with the prepared sample concentration.
The advantage of Reverse ELISA over other ELISA techniques is how sensitive the test can detect different reagents, which is beneficial for detecting different kinds of antibodies with their respective antigens with large target assays. The sample volume can be increased to improve the test sensitivity in clinical (saliva, urine), food (bulk milk, pooled eggs) and environmental (water) samples. One ogive is left unsensitized to measure the non-specific reactions of the sample. The use of laboratory supplies for dispensing sample aliquots, washing solution and reagents in microwells is not required, facilitating ready-to-use lab-kits and on-site kits. Based on the platform of ELISA, this method uses an antibody microarray to capture native antigens. Autoantibody reactivity is then evaluated by differentially labeling patient IgG and incubating these antibodies with the native antigens that are immobilized on the antibody microarray.
Procedure
Preparation: Prepare autoantibodies (IgG) separated and purified from serum. Label them either through radioactive tagging or a fluorescent dye.
Extraction: Label autoantibodies with native protein on array. Native antigens bound to antibodies on array are extracted from microarray with unique monoclonal antibodies. Array should be incubated with native protein extracts such as cells, tissue, or body fluid, etc.
Analysis: Analyze concentration based on intensity of dye appearing in the assay. When the process is complete, the assay is then evaluated by its intensity of color which is determined for each well. The amount of color produced correlates to the amount of primary antibody bound to the proteins on the bottom of the microwells
Western Blotting is another technique that utilizes antibodies to detect the presence of a specific protein in a sample. It is capable of detecting small amounts of protein within a cell or body fluid. Western blotting grants the ability to find proteins in a mixture.
Western blotting, which originated from the laboratory of George Stark at Stanford, is the technique for detecting a particular protein by staining with specific antibody. Its analysis can give us the information about the size of the protein or how much protein accumulated in cells. It does not mean that it can do for any protein since in the case the protein is degraded quickly; it is hard to detect it well.
The important thing in this technique is antibody since Western blotting is based on the use of a “quality” antibody as a probe to detect specific protein. In this technique, first we separate the protein by using SDS-poly-acryl-amide gel electrophoresis, which base on the protein's size. Later, those proteins on the gel are transferred to the surface of a polymer sheet by blotting. As soon as we add antibody that is specific for the desired protein to the sheet, it will bind to this protein since they have nothing else to bind to. Later, we wash unbound primary antibody, and this protein-primary antibody can be detected by secondary antibody. Those secondary antibody can only recognize the primary antibody and many of them bind to one primary antibody and give out the signal, which is a radioactive label on the secondary antibody produces a dark band on X-ray film.
Sample undergoes SDS PAGE (Sodium Dodecyl Sulfate - Polyacrylamide Gel Electrophoresis) in which SDS detergent is attached to proteins, giving them negative charge relative to size so that proteins migrate to positive charged cathode. Larger proteins move slower through the acrylamide gel matrix whereas smaller proteins can move faster resulting in a distinct gradient of bands separated based on speed of travel through the matrix and subsequently size.
The resulting bands of proteins are transferred onto a polymer sheet (nitrocellulose filter or polyvinylidene difluoride, PVDF) that binds proteins. This allows the proteins to bind to antibodies.
Since the polymer sheet binds all proteins, the remaining empty space on the sheet must be blocked so it does not react with the antibody. This is done by washing the polymer sheet in non-fat dry milk, which contains casein (a protein), which will cover the rest of the sheet.
The sheet is then incubated with the primary antibody specific for the target protein. After washed and diluted, the sheet is again incubated with the secondary antibody, which binds to the primary antibody. Usually, this secondary antibody is fuorescent or radioactive labeled. The sheet is again washed and diluted.
The sheet is incubated with the enzyme substrate that would make the secondary antibody to emit light. Photographic films will detect this light as dark bands. The radioactive labeled antibodies are detected with X-ray films.
Factors Influencing Transfer
The buffer
A buffer that is commonly used for transfer is Tris-glycine which has pH 8.3 with a low concentration of SDS and 20% methanol. Glycine is used instead of Cl because it has a lower charge density and less current means less heat production. At this pH, many proteins will have a slightly negative charge which will make proteins transfer our easier since they will tend to move towards the positive charged electrode. SDS favors elution from the gel because it covers protein with negative charge. But if the protein is too negatively charged the hydrophobic membrane will repel the protein hence the protein will be less likely to transfer onto the membrane. To prevent that, methanol will be use to remove SDS from proteins which covers hydrophobic sites on the protein which can then bind to the membrane. If there is no methanol, elution from the gel is favored but binding to the membrane is poor. If there is too much methanol then the SDS could all come off before proteins leave the gel and the exposed hydrophobic regions could aggregate forming large protein aggregates that would be trapped in gel hence cannot transfer out.
The Membrane
There are different kinds of membrane with different pore sizes. The most commonly used membrane has pore sizes around 0.45 micrometer but smaller pore sizes are necessary to efficiently trap proteins that has size less than 20kDa.
Voltage during transfer
The current is more critical than the voltage. One of the factors here is the heating. The second factor is that if the current is too high, proteins with low molecular weight will move through the membrane so fast that they do not have a chance to bind with the membrane.
Time to transfer
The migration of the proteins will depend on the sizes. The larger the size of protein the longer time it will take to migrate. Sometimes two layers of membrane are placed next to the gel to trap smaller proteins that might pass through the first membrane.
Protein charge
If a protein has positively charged after SDS removed then they will be difficult to transfer. Since their isoelectric points can be similar to the pH of the buffer system. Hence, the protein will not migrate toward the positive electrode and not transfer onto the membrane. We can solve this problem by raising the buffer system to higher pH.
•Western blotting is effective and useful method to detect and characterize proteins in small amounts, such as clock proteins. Moreover, clock proteins’ other properties like half-life, molar amounts can also be found using western blotting.
•Immunogenic responses from infectious agents (ex. viruses, bacteria) are easy to detect by this technique.
•Western Blotting utilizes not only antigens, but also antisera as a diagnostic tool. Antisera is widely used in the test for HIV presence.
•Compared to ELISA, Western blotting has higher specificity; the higher specificity, the more the method is independent of the specificity of antibodies.
•Polyvinylidene difluoride (PVDF), or Nylon, is often used as membrane in Western blotting, since it has a high protein-binding capacity and chemical stability. Even, some protein groups only bind to Nylon or favor strongly to it.
•Among three common enzyme substrates, Fluorescent and Chemiluminescent create light detectable through X-ray or scanners. This ability enables high levels of sensitivity and quicker processing time.
•A non-intended protein has a slight chance of reacting with the secondary anti-body, resulting in the labeling of an incorrect protein.
•Incidental phosphorylation or oxidation of proteins may result in multiple bands appearing after sample is processed.
•The appearance of bubbles may occur when transferring the sample from the gel/membrane sandwich and may also occur when incubating the sample with antibodies, resulting in a skewed band reading.
•If the transfer time is not sufficient when transferring proteins to the membrane, the larger proteins of higher molecular weight will not transfer properly, resulting in an incorrect or no band reading at all.
•Too much methanol in the transfer buffer decreases the transfer efficiency of proteins from the gel to the membrane; however methanol aids in protein binding to several different membranes, so a correct balance is required.
•Western Blotting is a very delicate process requiring the correct amounts of each component in order for successful identification of the presence of proteins. An imbalance in any step of the procedure may skew the entire process.
Far- western blotting is based on the Western blotting technique. The major difference is that far-western uses non-antibody proteins which can bind to the desired protein. Far-western is used to detect protein protein interactions.
Eastern blotting is an extension of western blotting and is used in the detection of protein post-translational modifications. Eastern blotting is used to detect lipoylated, glycosylated or phosphorylated proteins or proteins with other post translational modifications. Blotting is mostly done from a SDS-PAGE gel on to a PVDF or nitrocellulose membrane. Post translational protein modifications including phosphorylation, lipoylation, glycosylation or any other protein modifications are detected by specific probes.
Analyses lines separated by high-performance thin layer chromatography (HPTLC). Further analysis is prepared by being transferred form the HPTLC plate to a PVDF then by either performing enzymatic or ligand assays or mass spectrometry. A disadvantage to this procedure is the amount of downtime present in being able to prepare both a chromatography and a western blotting continuously since both require post experimental readings which have to be prepared separately.
Far-eastern blotting allows for the following techniques:
■ Purification of glycosphingolipids and phospholipids.
■ Structural analysis of lipids in conjunction with direct mass spectrometry.
■ Binding study using various ligands such as antibodies, lectins, bacterium, viruses, and toxins, and
■ Enzyme reaction on membranes.
• In addition, it is important to know that in most cases, the blot is a nitrocellulose or polyvinylidene membrane, which is mobile when submersed in gel and electrical current is applied. In order for the proteins to move along the gel, the proteins must be soluble. A lysis buffer is needed to accomplish this. (Reference: www.abscam.com/ps/pdf/protocols/WB-beginner.pdf).
• Western Blotting is used in the human immunodeficiency virus (HIV) test. A blot is used to detect anti-HIV antibody in a human blood sample. The cells that have been infected by HIV have their proteins separated and blotted on the membrane. The human blood sample is then applied in the antibody incubation step and the free antibody is washed away, followed by a secondary anti-human antibody is added. After x-ray analysis, the bands that are stained show which proteins the person's blood contains antibodies to.
http://shop.expedeon.com/products/75-InstantBlot-Western-Blot/345-InstantBlot-1-Kit/http://www.piercenet.com/Proteomics/browse.cfm?fldID=8259A7B6-7DA6-41CF-9D55-AA6C14F31193http://www.westernblotting.org/troubleshooting%20page.htmlhttp://www.virusmyth.com/aids/hiv/epwbtest.htm
Fluorescent markers, sometimes known as fluorophores, is any molecule with the ability to absorb light and emit it at some other well defined wavelength. Flurophores come in a variety of types ranging from simple organic compounds to large proteins like Green Fluorescent Protein. Fluorescent markers give the ability to investigate proteins in their biological environment. When light of a certain wavelength is directed at the molecule's chromophore, a photon is absorbed and excites an electron to a higher energy state. The electron then relaxes back to its ground state. The energy that is released is determined by the formula E = hν, where h is Planck's constant and ν is the frequency of the photon. This energy can then be related back to the wavelength of the emitted photon, which corresponds to a specific color on the visible spectrum. The difference between the absorption and emission wavelength is called the Stokes shift. Large Stokes shifts are generally desirable because the emitted light from the fluorescent tag can be filtered out more easily from the excitation light. Fluorescence educing molecules such as FITC (fluorescein isothiocyanate) are used to stain cells which give the ability to examine these cells under a fluorescence microscope. Multiple fluorescent markers can be used to stain different parts of cell. Fluorescence microscopy can aid in determining the location of specific proteins within a cell. One can even track the movement of these proteins and derive possible functions for these proteins of interest.
GFP attaching to target cell for fluorescence microscopy using the direct method
Green Fluorescent Protein (GFP) is a fluorescent protein that is found in the jellyfish, Aequoria victoria. In biochemistry, it is frequently used as a marker to monitor gene expression and cell division. The naturally-occurring GFP has a major absorbance peak at 395 nm and a minor absorbance peak at 475 nm. It has an emission peak of 508 nm. This means that when exposed to blue light, it emits photons that give off green light, hence giving it the name Green Fluorescent Protein.
The Green Fluorescent Protein is comprised of 238 amino acid. It has a barrel shape, called a β-can, with 11 β-strands forming the walls of the protein and a α-helix running through the center. The β-can has a diameter of 30Å and a length of 40Å. At the center of the molecule is the hydrophobic fluorophore, which is the part of the protein that is responsible for absorbing and emitting light. The fluorophore is comprised of three amino acids: serine, tyrosine, and glycine, at locations 65, 66, and 67 respectively.[1]
To observe a target protein using fluorescence microscopy, the gene for the production of GFP must be spliced into the RNA transcript that codes for the protein of interest. Wherever the gene is expressed to produce this protein, GFP will be produced along with it. These target proteins will now fluoresce when observed under a fluorescence microscope.
The fluorophore is generated by a sequential mechanism in the catalytic process. No co-factors is required for the activation for the catalysis reaction. The reaction is initiated by a rapid cyclization between Ser65 and Gly67 to form an imidazolin-5-one intermediate, which is followed by a much slower rate-limiting oxygenation of the Tyr66 side chain by O2 on a timescale of hours. Gly67 is required for formation of the fluorophore, no other amino acid can replace Gly in this role. The reaction here is thermosensitive. The yield of formation of the fluorophore decreases when the temperature increase higher than 30oC. Once GFP has produced, GFP turns into thermostable state.
When fluorescent molecules are introduced into a genome, they tend to be phototoxic which can cause death of cells when the fluorescent molecule is active. Majority of small fluorescent molecules tend to have some degree of phototoxicity such FITC (fluorescein isothiocyanate). Because GFP's tri-peptide fluorophore is activated by oxygen, the addition of an enzyme is unnecessary. This oxygen activated characteristic allows the target cell to be less disturbed which causes GFP to be less harmful when used in living cells. This allows GFP to be maintained in the genome.
Another method that can be used to locate a protein is to bind GFP to an antibody that will bind to the target protein. The direct method would bind the fluorescent antibody to the corresponding antigen on the target protein. The indirect method first binds a non-fluorescent antibody on the target protein. Then the fluorescent antibody is introduced which will bind to the non-fluorescent antibody. Although it is possible to stain specific parts of cell directly, indirect methods are preferred due to high affinity. Then using fluorescence microscopy, the target cell which is now fluorescent can be studied.
The use of GFP is not only limited to monitoring gene expression and cell regulation. Inorganic species such as zinc and nitric oxide are important agents that help drive physiological processes. Therefore, it is valuable to study and visualize how these inorganic agents interact and are processed within cells. For zinc, there are many turn-on sensors that offer good methods to visualize zinc ions inside cells. Most zinc indicators used are intensity-based sensors such that the response to zinc indicators establishes the intensity of fluorescence emission. Nitric oxide, on the other hand, are more difficult to monitor because of their gaseous nature, limited water solubility, and other limitations. One way to work around this challenge is to use genetic encoding of nitric oxide-sensitive proteins which have transition metal nitric-oxide reactive sites. For example, two mutant GFPs are fused together to be used as a nitric oxide indicator. In a sense, one can manipulate GFPs to use as indicators, thereby giving even more possibilities to understand organization and regulation of signaling networks.[2]
Green fluorescent proteins are also used in the in vivo technique of FRET (Fluorescent Resonance Energy Transfer), an imagining method used to understand interactions between proteins. In FRET, the energy transfer from a fluorescent protein (the "donor") to another fluorescent protein with a longer wavelength (the "acceptor") are measured. By measuring the inter-molecular and intra-molecular distances between the GFP bounded proteins, researchers are able to observe and take note of protein interactions. For example, FRET allows us to detect interactions between signaling molecules. By situating a protein that is sensitive to changes in conformations between the FRET donor and acceptor proteins, we can determine the activity of the pathway.[3]
Metal-based fluorescent probes have been developed as method for detecting the presence of NO in a system; NO is a prominent compound that is associated with signaling pathways in the living organisms. These metal-based fluorescent indicators are based on the metal Cu(II). When the Cu(II) ions react with NO, Cu(II) gets reduced into Cu(I). This process results in diminishing the quenching energy of the lone pair on the nitrogen atom of the nitric oxide; the reaction causes a decrease in the fluorescence intensity of the lone pair on the nitrogen atom. In addition, the react between Cu(II) and NO that takes place in the system also leads to a lowering of the PET quenching in the N-nitrosamine. Fluorescent probes based on Cu(II) are ideal for detecting NO that reside in environments that do not contain oxygen.
A specific fluorescein dye that utilizes Cu(II) ions for identifying NO is the CuFL1 platform. The fluorescein dye in this particular scaffold has an aminoquinoline,a derivative of quinoline, which binds to Cu(II) and forms a 16-fold emission turn on. This reaction converts the secondary amine of the aminoquinoline into an N-nitrosated product, which reinforces the fluorescence of the molecule; the increase in the intensity of fluorescence is based on the concentration of NO. The presence of NO in the Raw 264.7 macrophages and certain gram positive cells such as Bacillus subtilis and Bacillus anthracis has been identified with the CuFL probe.[4]
Fluorescent markers are often used in DNA studies to determine the sequences of bases in a strand. The upside to this method is that it does not utilize radioactive reagents as in the auto-radiography seen in Western blotting. As a result it has become a very popular method of DNA sequencing. In a method devised by Frederick Sanger and colleagues, DNA polymerase was used to create complementary strands to a single strand DNA molecule in four mixtures of radioactively labeled nucleotides. Incorporated into this synthesis was a mixture containing the 2',3'-dideoxy analog of a nucleotide, a different analog for each mixture. The 2',3'-dideoxy analog in the mixture causes the termination of DNA replication creating fragments. This is due to the analog having no 3' hydroxyl terminus to promote phosphodiester bonding. The mixtures then underwent electrophoresis allowing base sequences to be read through autoradiogram. With the use of fluorescent markers, a different colored tag is attached to the dideoxy analog of each base instead of having to be radioactively labeled. The fragments then undergo the same electrophoresis. The resulting bands are detected by their fluorescence and the sequence of colors subsequently translates into the sequence of the bases. With this method more than 1 million bases can be sequenced per day with modern sequencing tools.[5]
Strategies are being developed for fluorescent protein (FP) reporters that "speak" the language of the cell and allows scientists to understand complex networks of biochemical processes that they were unable to witness before. There are three main types of FP's that are made currently. The first are ones that are bound to a protein to report its location and turnover, the second are ones created to undergo changes in fluorescent signal to allow scientists to know when there are changes or intermolecular interactions with the protein they are studying. Lastly, there are FP's that contain a sensory element which detects the accumulation or degradation of small molecules. These FP reporters are then used to understand the behavior of tagged signaling molecules and their spatiotemporal organization.
In a normal signaling pathway receptors on the plasma membrane detect extracellular cues and mediate production of intracellular second messengers, these second messengers then regulate the activity of signaling enzymes and downstream transcription factors. These reporters allow scientists to see how a protein reacts in its natural environment, whether it diffuses across a membrane or needs a receptor on the membrane to send a message. These can be observed using a GFP whose fluorescence changes color with time or are photoactivatable, This method allows for direct showing of molecules within cells responsible for the action being observed. The best part of FP's is their ability to remain discrete and not disturb pathways of cell function.[6]
A modified version of the ELISA immunoassay, the ELISpot or enzyme-linked immunosorbent spot, allows for the detection of cells that secrete cytokines or antibodies. Unlike ELISA, ELISpot is able to detect a single cell that secretes a protein of interest making it one of the most sensitive cellular assays.
A quick and simplified procedure is listed below:
1) Cytokine-specific antibodies are placed onto an ELISpot plate
2) Cells of interest are then added
3) Cells become activated and begin to produce cytokine which binds to the antibodies
4) The cells are then removed whilst detection antibodies that can bind with the enzyme are added
5) Finally a substrate forms a colored spot the secreting cell
6) The cells are then counted through a microscope
"The ELISpot Method Explained." ELISpot Info. MabTech, n.d. Web. 6 Dec. 2012. <http://www.elispotinfo.com/?page=elispot-method-explained>.
Monoclonal antibodies are a homogeneous collection of antibodies that all bind to the same antigenic determinant. They were first prepared by Cesar Milstein and Georges Kohler.
Similar to working with impure proteins, working with impure antibodies(polyclonal antibodies) made it difficult to interpret data and to understand function. Because their specificity was not known, immunological methods (e.g. ELISA, Western Blotting) were not able to be applied.
The ideal method of obtaining monoclonal antibodies would be to simply isolate the desired antibody producing cell. However, the problem with that is that antibody producing cells isolated from the organism don't live very long. The solution to this problem would be to use a cancer cell (myeloma cell).
Multiple myeloma is a disorder of antibody producing cells characterized by the uncontrollable division of a mutated cell. Each time this cell divides, it produces an exact copy of itself. Because cell division is not being regulated, a large aggregation of cells are quickly formed. All of these cells are identical to one another, and are thus referred to as clones. Milstein and Kohler hypothesized that if a cancer cell of this type could be fused with an antibody producing cell, they could have a potentially infinite source of monoclonal antibodies.
Monoclonal antibodies are produced by the process outlined in the flowchart above. A mouse is first injected with an antigen that is known to stimulate the production of the desired antibody. After approximately three weeks, the spleen of the mouse is removed. Plasma cells from the spleen are fused in vitro with myeloma cells, giving rise to a colony of hybrid cells (hybridoma cells). These hybridoma cells are capable of producing an indefinite amount of the antibody obtained from the spleen.
How to obtain monoclonal antibodies
A mixture of hybridoma cells is often formed. This mixture must be separated in order to obtain a homogeneous collection of antibody producing cells. An assay that tests for specific antibody-antigen interactions can be used to achieve separation. The assays are repeated and the collection of cells is continually subdivided until a pure cell line is obtained. It is good to note that the spleen also contains numerous other cells that produce antibodies for different antigens. Purification is necessary is order to find the one type of hybridoma cell that produces the desired antibody. The isolated cells can either be grown in a culture or injected into a mouse in order to procure the desired antibody. Another option is to freeze the cells for later use.
The advantage of monoclonal antibodies is that they are identical antibodies with the same affinity for the same area of the same antigen, thus allowing better purification results. Polyclonal antibodies, on the other hand, are a mixture of different antibodies each recognizing a different site (epitope) of the same antigen.
Antibodies have the ability to bind to two antigens. This is possible since any antibody has the ability to bind to two antigens and can be recognized by many antibodies (monoclonal and polyclonal) as far as there is still exposed surface where the antibodies could bind.
Berg JM, Tymoczko JL, Stryer L. (2002). Biochemistry, 5th Edition (4.3.2)
Antibodies are made by injecting live animals with antigens. These animals are the sources of producing the antibodies. They can be rabbits, mice, goats, and other kinds of animals. Some antibodies that are produced from one animal can also react in antibodies that are found in another type of animal.
For example, goat anti-mouse antibodies means the antibodies are taken out from goat and injected into mice, the antibodies from goat can react in the mouse's antibodies.
Antibodies can also be multiplied in a cell culture dish.
Preparation of Monoclonal Antibodies:
An antigen is injected into a mouse, and after a few weeks its spleen is removed and plasma cells are extracted. The mouse's spleen cells are fused with myeloms cells to create hybrid cells called hybridoma cells. Each hybridoma cell indefinitely produces identical antibody, and the hybridoma cells are then screened using an antigen/antibody assay that will reveal which cells produce the desired antibody. The collection of selected hybridoma cells that produce the preferred antibody are re-screened multiple times until a pure line is isolated. These cells are grown in a culture and/or injected into mice to induce tumors. The cells can also be frozen and saved for later use.
The hybridoma method for producing monoclonal antibodies is useful because large amounts of specifically-tailored identical antibodies can be produced easily.
An antibody (which is a protein itself), is also called an immunoglobulin. It is synthesized by an animal’s body when there is a foreign substance (called an antigen). The synthesis of antibodies has high and specific affinities. An antibody can recognize a specific group or cluster of amino acids on a specific molecule (called an antigenic determinant or epitope).
To collect antibodies that recognize a specific protein, a protein is injected once into a rabbit, then a second time about three weeks later. The protein will then stimulate the reproduction of cells and produce antibodies that recognize it. When the blood is taken from the immunized rabbit several weeks later, it is centrifuged to separate the blood cells from the supernatant. Antibodies can be found in the supernatant (also called the serum) to all the antigens the rabbit was exposed to. Only some of them will actually be antibodies to the protein that was originally injected. The indication that cells produce many different antibodies that each recognizes a different surface feature of the same antigen means that the antibodies are heterogeneous, or polyclonal.
Dynamic light scattering (DLS) or Photon Correlation Spectroscopy is a well-known and well used technique for measuring particles in solution with sizes ranging from a few nanometers to a few microns. In this process, a coherent monochromatic light source is radiated upon a sample. The frequency spectrum of intensity of the resulting scatter is recorded and the sizes of the particles are determined. The shift in frequency is termed a Doppler shift or broadening, and it is related to the size of the particles causing the shift. As a result of their higher average velocity, smaller particles cause a bigger shift in the light frequency than larger particles. It is this difference in the frequency of the scattered light among particles of various sizes that is used to determine the sizes of the particles present in the fluid. Compared with other methods, DLS is fast and somewhat cheap process. It is mostly used to determine the characteristics of bacteria as well as proteins. Optometrists can use this method to detect development of cataract in the eyes. DLS is often used to analyze macromolecules like proteins. Protein Crystallography and nanotechnology application. The molecular mass and the concentration of the protein in the solvent is directly proportional to the light scattered by it.
The theory behind this technique is based on two conditions. The first condition is that the particles follow Brownian motion, a random motion in solution. This random motion follows a mathematical formula in which the probability function can be determined. The second condition is that the particles are relatively spherical and with a diameter of less than a half of a wave length of the incoming radiation.
There are different ways to determine the dynamics of a particle in Brownian motion. One such method is by using a laser as a light source. The laser passes through lens that would then hit the particles. Then the light is scattered and passes through another collimator lens. The resultant of this diffracted light is "collected" and read by the photomultiplier. The photomultiplier translates all the different intensity into the form of voltage readings. It is essential to note that two collimator lens are required; the first is to better focus the light to directly hit the cell, and to ensure that the area on the cell that the light hits is far enough away from the sides of the cell; and the second lens is to get just the right amount of scattered light to be collected by the photomultiplier. After the beam is measured by the photomultiplier, the signal gets amplified and all the information can be sent to and analyzed by a computer.
In order to ensure accurate measurements, it is essential to calibrate the instruments. It is important to make sure that the light beam is shining at a consistent linear path. In other words, it needs to be at the same height in its entire path. This is to ensure that the beam will pass right through the first lens and straight into the center of the cell. Another thing to note is that all other light sources should be blocked out, other than the scattered light from the laser source. This will also allow for more accurate measurements.
Structural Biochemistry/Site Prediction
Protein misfolding is a particularly insidious contributor to human disease. During the complex kinetic and thermodynamic choreography required for a protein to achieve its proper structure and function, missteps can occur
by a variety of mechanisms. Coupled with a breakdown in the protein quality control systems designed to correct or remove misfolded proteins (39), loss- or gain-of-function phenotypes can lead to a sobering number of diseases
(39, 40). Two aspects of protein misfolding are reviewed. The remarkable hair-trigger conformational reorganization essential to the mechanism of protease inactivation by the serpins can spontaneously misfire as graphically described by Gooptu & Lomas (41). Inappropriate autoreorganization enhanced by specific mutations can lead to inactive forms of the serpin, resulting in loss-of-function diseases, which in this system result in an increase in the activity of the corresponding protease. In certain cases, serpin polymers can form, resulting in toxic gain-of-function
diseases—serpinopathies. The structural clarity, and relative simplicity, of the serpin system lends itself as a model for understanding other diseases of protein aggregation. Approaches to design small-molecule inhibitors of serpin
polymerization are also emerging (41).
Proteins or peptides convert from their usual soluble form into fibrillar aggregates. This shift in conformation is detrimental to proteins as it gives rise to many pathological problems. Misfolding results when formation of thread-like aggregates called amyloid fibrils begin taking shape. Failure for polypeptides to remain in their functional conformational state results in misfolding. This misfolding impairment reduces the proteins’ ability to perform its task in a cell.
Reasons for Misfolding Impairment
Misfolding impairment usually arises from the degradation of the endoplasmic reticulum or the wrong transportation of that protein. But the most common reason for misfolding is due to the fact that proteins or peptide convert from their usual soluble form into fibrillar aggregates, or amyloid fibrils. Amyloid fibrils are formed in “cross-β” arrangements of their polypeptide chain.
Discovery of Components of the Amyloid Fibrils
Components of these amyloid fibrils have been extracted and purified in numerous experiments in order for scientists to understand the cause of the misfolding. These fibrils are imaged using electron microscopy and reveals that they usually consist of 2-6 protofilaments that twist together to from a rope-like conformation. Further x-ray diffraction data shows that these molecular are arranged in β-strands that run perpendicular to the long axis of the fibril. Researchers have found that metal ions, glycosaminoglycans, serum amyloid P component, apolipoprotein E and collagen make up the bulk of the protein component associated with amyloid diseases. In recent years, Solid-State NMR (SSNM) has been used to analyze the amyloid β structure. SSNMR has identified the region of the C-terminal of the protein that is involved heavily in the core of the fibril. Torsion angles and internuclear distances have also been able to be measured and it provided valuable insight on the degree of uniformity the fibrils possess, which has only been associated with crystalline materials.
High resolution X-ray Crystallography has also been used to determine the structure of these amyloid fibrils. Data collected from crystallography further supports the parallel β-strands alignment in the protein. The β-sheets in the protein suggests that initiate interaction could represent crystal formation rather than the protein in fribrillar state.
Amyloid Formation
Research heavily supports that amyloid fibril formation is due to nucleated growth mechanism. For example, addition of fibrillar species under aggregated conditions causes a lag phase to appear. It is clear that the lag phase is the stage in which β-rich species provide nuclei for formation of mature fibrils.
Diseases caused by Protein Misfolding
Some prevalent human diseases that arise from misfolding include Alzheimer's, Parkinson's, Huntington's, dementia and Type II diabetes. Conditions of these diseases are predominantly sporadic (85%), and hereditary (10%), although transmissible (5%) has been recorded as well.
With this in mind, scientists still have not developed a full understanding as to why protein misfolding occurs. Much more research is still needed in this field and solving this mystery could lead to rise in potential drug treatments for these disease.
Chit, Dobson. “Protein Misfolding, Functional Amyloid, and Human Disease. Annual Review of Biochemistry. Vol. 75: 333-366 (Volume publication date July 2006) DOI: 10.1146/annurev.biochem.75.101304.123901.
Translation is the process of conversion from genetic information (codons) to protein sequence(amino acids). Translation requires some components before synthesizing proteins. The components are the mRNA,tRNA,ribosomes,amino acids,and energy.
The mRNA (messenger RNA molecule) is prepared during the Transcription.
We are told that amino acid and codon work together by the genetic code. However, tRNA plays a big role between amino acid and codon. The tRNA carries the correct amino acid to its ribosome. To do this, tRNA will need to be able to recognize both the codon and the amino acid that it is carrying. The tRNA has a 3D structure. On one end, there are nucleotides that are complementary to the codon; also known as the anticodon. On the opposite, tRNA is bound to the amino acid that corresponds to the codon. Every tRNA has a nucleotide sequence that binds to the amino aicd. Also, each tRNA has a helper tRNA synthetate which is an enzyme that binds the amino acid to the tRNA using GTP (stands for guanosine 5'-triphosphate). This result in an aminocyl-tRNA complex.
Ribosomes are known as the factories of the cell which is the protein primarily responsible for transcription. Now, we are putting them into actions. Ribosomes are composed of two subunits, one large and one small, that they only bind together during protein synthesis. The purpose of the ribosome is to take the actual message and the charged aminoacyl-tRNA complex to generate the protein. To do so, they have three binding sites. One is for the mRNA; the other two are for the tRNA. The binding sites for tRNA are the A site, which holds the aminoacyl-tRNA complex, and the P site, which binds to the tRNA attached to the growing polypeptide chain.
Each amino acid in the protein is coded by a set of three DNA bases, called a codon. While each codon codes for only one amino acid, many amino acids are coded for by multiple codons due to the fact that there are 64 possible combinations of the DNA bases, but only 20 amino acids. Three of the amino acids are known as stop codons (UAA, UGA, UAG) and their function is to end transcription. There is only one start codon (AUG), which also codes for the amino acid methionine. The relationship between the codons and the amino acids is called the genetic code.
Synthesis begins with mRNA seeking out a small ribosome. They bind in the presence of initiation factors, and the small ribosome slides along the mRNA until it reaches a start codon (AUG).The initiation aminoacyl-tRNA complex, methionine tRNA (with the anticondon 5'-CAU-3'), base pairs with the start codon. At this point, the large ribosomal subunits joins the complex, completing the ribosome. The tRNA is in the P site at this point, because it is the only part of the growing polypeptide chain.
Bacterial IF1 functions mainly to prevent tRNA binding to the A site, stabilize the mRNA binding by the small subunit, and stabilize the binding of fMet-tRNA/IF2/IF3 to the small subunit of the ribosome. IF1 affects IF3 by increasing IF3’s antiassociation activity and coordinates with IF2 on the ribosome for their simultaneous release after initiation procedure. eIF1A is the archeal and eukaryotic equivalent of IF1 and is identical in structure with the exception of an extra tail at the N-terminal and helix at the C-terminal, the function of which is not yet fully understood.
IF2/eIF5B
Universally conserved IF2 in bacteria and eIF5B in archaea are primarily involved in joining of the small and large subunits of their respective ribosomes. After subunit joining, the GTPase associated center of the large subunit causes the IF2 or eIF5B hydrolysis reaction that releases IF2/GDP or eIF5B/GDP along with IF2 or eIF1A. IF2 has been proven to help locate the fMet-tRNA in bacteria while no such process has been proven of the eIF1A in archaea or eukaryotes.
The main role of IF3 is bacterial ribosomal association and dissociation as it works along with IF1 and IF2. IF3 is also involved in locating the start codon and helping to select fMet-tRNA. The carboxyl terminal section of IF3 at sufficient concentration can perform the function of the whole IF3 shown by cleaving the protein below the carboxyl domain. It acts as a competitor to subunit association by binding to the active site of the small subunit and inducing conformational changes.
eIF3
Although eIF3 is smaller and dissimilar in structure and sequence to IF3, it performs some similar functions. eIF3 is able to recognize AUG start codons and discriminate against non-AUG codons at its carboxyl end. It functions similarly to IF3 by binding to almost the site as the carboxyl domain of IF3 inducing dissimilar conformational changes. eIF3 differs from IF3 in that it does not help select Met-tRNA.
eIF2
eIF2 is a heterotrimer that is involved in the selection of initiator tRNA and binding of the Met-tRNA to the small subunit. An eIF2/GTP complex forms a ternary complex with Met-tRNA by direct binding whereas eIF2 with GDP or bound to a tRNA without the methionine residue is unlikely to form a strong ternary complex and dissociation is likely. The eIF2/GTP/Met-tRNA ternary compound binds to the 40S subunit of eukaryotic ribosomes and following recognition the eIF2 is detached and recycled by hydrolysis of GTP. This GTP hydrolysis in eukaryotes requires an additional eIF5.
eIF4G
eIF4G is a large protein that forms the scaffold for the construction of the cap binding complex. It is also responsible for the recruitment of the 43S IC in forming the 5-prime cap that is required for efficient translation. It has also been shown the eIF4G binds to eIF3.
Once the complex has been formed, the ribosome can slide along the mRNA, adding new amino acids as it goes. Hydrogen bonds form between the mRNA codon in the A site and the complementary tRNA anticodon. This fills the A site. We now have a charged aminoacyl-tRNA n both the A site and the P site. The enzyme, peptidyl transferase, uses the energy that was stored in the aminoacyl-tRNA complex when the amino acid was loaded (from GTP) to catalyze the formation of a peptide bond. The aminoacyl-tRNA used for this is the one in the P site. Now the tRNA in the P site is free, and there is still an aminoacyl-tRNA in the A site. This aminoacyl-tRNA has its own amino acid, which is now bound to a methionine. Translocation is necessary to add the next amino acid residue. The ribosomal assembly slides in a 5' to 3' direction along the mRNA. This moves the next codon into place in the A site. At the same time, the deacylated tRNA from the P site is moved to the E site displacing the previously deacylated tRNA and the aminoacyl-tRNA that is carrying out nascent chain moved from the A site to P site. The process is ready to begin again with an empty A site.
In bacteria, the aa-tRNA is bound to GTP and elongation factor EF1A as a ternary compound before reaching the ribosome. EF1A is nonspecific and will bind to most aa-tRNAs based on varying levels of affinity for the tRNA or amino acid. Notable exceptions that have weak binding to EF1A include the initiator tRNA fMet-tRNA and Asp-tRNA. The homolog elongation factor eEF1A in eukaryotes has similar characteristics to EF1A.
Non-complementary or non-cognate aa-tRNA/GTP/EF1A complexes have an equal chance of binding to the ribosome as the correct complementary
aa-tRNA ternary complex. There are two exclusion methods to ensure the correct matching for aa-tRNA complexes with the mRNA. First, conformational changes in the aa-tRNA complex and the ribosome allow for the codon and anticodon to make initial contact. Non-cognate ternary aa-tRNA complexes will dissociate quickly and GTP hydrolysis by EF1A is unlikely to occur. Base pairing is obeyed up until the third base pair and thus nearly cognate aa-tRNA complexes are excluded by the universally conserved nucleotides 530, 1492 and 1493.
After correct complementary matching of the aa-tRNA ternary complex and ribosome, the small subunit of the ribosome assumes a closed conformation that promotes GTP hydrolysis by EF1A. The second process of elimination of near cognate aa-tRNA occurs in the PTC (peptidyl transferase center). Near cognate aa-tRNA have a much lower rate of accommodation compared to rate of dissociation while cognate aa-tRNA have a very low dissociation rate compared to their association rate. These two methods of exclusion for near-cognate aa-tRNA combine to give very low percentages of mutation during elongation.
A second elongation factor, EF2/GTP attaches to the ribosome at the same site as the aa-tRNA/GTP/EF1A ternary complex and induces translocation of tRNA and mRNA one codon down. The acceptor site of the tRNA is thought to move first from the A to P site followed by the movement of the tRNA anticodon and mRNA codon with the small subunit of the ribosome rotating against the large subunit. EF1A/GDP is recycled by a guanine nucleotide exchange factor to reform EF1A/GTP while the dissociation rate of EF2/GDP is fast enough to allow EF2/GTP and EF2/GDP to exist in near equilibrium.
Translation has its own set of stop signs. If the codon in the A site is UGA, UAA, or UAG, it is known as a termination codon. Instead of a new aminoacyl-tRNA binding to the A site, a protein called released factor binds to the termination codon, causing a water molecule to be added to the polypeptide chain. The chain will then be released from the tRNA in the P site, and the two ribosomal subunits will dissociate and as well as increase the amount of protein that may be made from a single transcript, several ribosomes may translate a message at the same time. This is known as a polyribosome.
Due to prokaryotes' significantly smaller amount of DNA, translation happens only one protein at a time. However, because prokaryotes do not have a nucleus, translation occurs at the same time as transcription. In eukaryotes, one complete strand of mRNA can be translated by many ribosomes at once, thus drastically reducing the amount of time required to produce a feasible amount of proteins, but transcription and translation are separate events. Transcription occurs in the nucleus and the mRNA is exported to the cytoplasm before translation can occur.
Also, prokaryotic ribosomes are similar in structure to eukaryotic ribosomes, but not identical. Prokaryotic ribosomes are smaller (30S for the small subunit, 30S for the large, whereas for eukaryotes, it's 40S and 60S, respectively). Thus drugs that prevent bacterial infection by stopping translation can specifically target the bacteria and leave the host cells to function normally.
Translation in Eukaryotes is highly regulated and are regulated by universal and lineaged mechanisms. Recent discovery has yielded information that suggests that the mechanisms that regulate translation emerged at different times based on evolutionary need. The evolution of eukaryotes thus paralleled the evolution of translation. Some mechanisms evolved independently of translation but were later incorporated into it. The thinking now by scientists suggests that the mechanisms that regulate translation may have been involved in other cellular processes and were later incorporated into translation. This overall view has been dubbed by scientists as 'tinker' which involves co-opting and assembling molecules and regulatory mechanisms from other cellular processes.
Campbell and Reece. "Biology, 8th Edition"
Myristoylation is an irreversible, co-translational (during translation) protein modification found in animals, plants, fungi and viruses. In this protein modification a myristoyl group (derived from myristic acid) is covalently attached via an amide bond to the alpha-amino group of an N-terminal amino acid of a nascent polypeptide. It is more common on glycine residues but also occurs on other amino acids. The modification is catalyzed by the enzyme N-myristoyltransferase (NMT), and occurs most commonly on glycine residues exposed during co-translational N-terminal methionine removal. Myristoylation also occurs post-translationally, for example when previously internal glycine residues become exposed by caspase cleavage during apoptosis.
Myristoylation also plays a vital role in membrane targeting and signal transduction in plant responses to environmental stress.
Eukaryotic translation initiation occurs through a cap mediated mechanism where eIF4E recruits the 5’ capped mRNA to the small subunit. It was discovered in viruses that another mechanism exists through the use of internal ribosome entry sites (IRESs). The IRES is a segment of DNA that allows for translation initiation in the middle of a messenger RNA bypassing the 5’ cap dependent system.
They were first discovered by Nahum Sonenberg and Eckard Wimmer in 1988 in Poliovirus RNA and encephalomyocarditis virus RNA. They are located mostly in the 5’ UTR of RNA viruses and allow for cap independent RNA translation. This is useful for the virus because it ensures that viral mRNA is still translated even when host cell translation is shut off. Poliovirus secretes a viral protease that cleaves the eIF4E complex and inhibits cap dependent translation while translating its own mRNA through IRESs.
The mechanism of viral IRES function varies depending on the virus. Hepatitis C virus does not require scanning for the initiator codon because its IRES directly binds to the 40S subunit in a way that the initiator codon is already in the P site. They also do not require any eukaryotic initiation factors. Picornavirus IRESs do not bind directly to the small subunit but rather to the eIF4G and require additional proteins know as IRES trans acting factors (ITAF) to function.
Proteins are capable of executing numerous functions based on the versatility of their 20 different amino acids. Proteins are also covalently modified to change their functions via attachment of other groups besides amino acids. For example, to make proteins more resistant to degradation, acetyl groups can attach to the amino termini of proteins. Also, hydroxyl groups can attach to different proline residues which stabilizes fibers of newly synthesized collagen. Furthermore, many proteins, specifically those present on the surfaces of cells, acquire carbohydrate units from attachment of specific asparagine residues. Carbohydrates in the form of aspargine-linked (N-linked) or serine/threonine (O-linked) oligosaccharides are major structural components of many cell surface and secreted proteins. The process of attachment of sugar molecules (such as carbohydrate) to the protein is called glycosylation. Protein glycosylation is known as one of the major post-translational modifications with significant effects on protein folding, conformation distribution, stability and activity. This addition of sugars makes the protein more hydrophilic and therefore able to better participate in protein-protein interactions. Besides the addition of certain groups to amino acids, other special groups can also be generated by chemical rearrangements of side chains and the peptide backbone, for example by spontaneous formation and oxidation.
Reference: Berg, Jeremy M., John L. Tymoczko, and Lubert Stryer, eds. "Biochemistry." New York: W.H. Freeman and Co., 2007: 57-58.
http://www.piercenet.com/Proteomics/browse.cfm?fldID=7CE3FCF5-0DA0-4378-A513-2E35E5E3B49B
Palmitoylation is the covalent attachment of deprotonated fatty acid palmitic acid to a cysteine residue on a membrane protein. Palmitoylation has many different functions in proteins. For instance, it functions as membrane attachment regulation, intracellular trafficking, and membrane micro-localization.
Specifically, S-palmitoylation refers to the reversible thioester linkage, or bond between the sulfide on the cystine to the electron rich oxygen of the palmitate. N-palmitoylation is similar, however, when the cystine is located on the N-terminus of the protein, the palmitate temporarily bonds with the sulfur, like in S-palmitoylation, and then quickly binds to the amide of the cystine for stability. (4)
Palmitoylated cystines share a few common features that narrow which cystines will and will not be palmitoylated such as: they are located in the cytoplasmic region, exposed in a peptide sequence so that they can be easily palmitoylated; they are sequenced next to or closely by hydrophobic or basic amino acids; they are often, but not exclusively, adjacent to prenylation and myristoylation sites. (5)
Once a membrane protein contains a palmitate, the membrane protein can interact with other lipids and proteins in new ways, effectively altering some of the proteins functions such as intracellular sorting, membrane interactions, stability, and membrane micropatterning. (5) Palmitoylation enhances the hydrophobicity of proteins and contributes to their membrane association. Interestingly, S-palmitoylation is a reversible process, which allows for unique and complex modes of trafficking between membrane compartments. (4) This is in contrast to prenylation and myrisoylation, which are not irreversible. The specific function of palmitoylation depends on the particular protein being considered.
Palmitoylation also appears to play a significant role in subcellular trafficking of proteins between membrane compartments, as well as in modulating protein-protein interactions. The specific location of where palmitoylation occurs gives insight into what proteins are palmitoylated and for what reasons. A location with high amounts of palmitoylation occurring takes place near the golgi, where peripheral membrane proteins are palmitoylated. In post golgi compartments, specific substrates are palmitoylated for the purpose of regulation. (5)
An example of a protein that undergoes palmitoylation is hemagglutinin, a membrane glycoprotein used by influenza to attach to host cell receptors.
Since S-palmitoylation is a dynamic, post-translational process, it is believed to be employed by the cell to alter the subcellular localization, protein-protein interactions, or binding capacities of a protein.
The palmitoylation cycles of a wide array of enzymes have been characterized in the past few years, including H-Ras, Gsα, the β2-adrenergic receptor, and endothelial nitric oxide synthase (eNOS).
Another example of proteins that undergo palmitoylation is the cell's ion channels. Throughout an ion channels life cycle, palmitoylation plays a crucial role in controlling and regulating its signaling pathways. From assembly of the ion channel to the recycling and degradation of the it, palmitoylation is controlled by the family of acyl palmitoyltransferases and a particular number of thioesterases. The NMDA receptors are palmitoylated at the golgi to keep these proteins anchored to the goli whereas palmitoylation of calcium- and voltage-activated potassium channels contributes to their cell-surface delivery. This demonstrates how dynamic and unique palmitoylation is, occurring at the same location for different proteins but with considerably different results. (6)
Recently, scientists have appreciated the significance of attaching long hydrophobic chains to specific proteins in cell signaling pathways. A good example of its significance is in the clustering of proteins in the synapse. A major mediator of protein clustering in the synapse is the postsynaptic density (95kD) protein, PSD-95. When this protein is palmitoylated it is restricted to the membrane. This restriction to the membrane allows it to bind to and cluster ion channels in the postsynaptic membrane.
Also, in the presynaptic neuron, palmitoylation of SNAP-25 allows the SNARE complex to dissociate during vesicle fusion. This provides a role for palmitoylation in regulating neurotransmitter release.
Internal cellular palmitoylation reactions are monitored by a palmitoyl transferase known as aspartate-histidine-histidine-cysteine, or more commonly known as DHHC. Researches have indicated that there are approximately 20 DHHC proteins that are present in mammalian genomes and have proven that they are crucial in cell function and physiology and pathophysiology influence.
Studies regarding yeast have established interesting characteristics of the DHHC protein family. For one, some DHHC proteins require protein cofactors. Second, DHHC play a very important role in cellular activity. Moreover, DHHC palmitoyl transferases are mobile to specific intracellular membranes. More precisely, the DHHC-CR domain is a prominent feature of palmitoyl transferases, and genes that are associated with DHHC proteins are found in organisms from yeast to humans. Approximately seven DHHC proteins are relevant for yeast organism, whereas more than 20 DHHC genes are discovered for mammalians. Most of the mammalian DHHC proteins have the tendency to catalyze palmitoylation independently of other protein cofactors.
There is not sufficient amount of data that has proven that DHHC proteins have achieved their respective intracellular localizations. However, a general scope of analysis has shown that a removal of a phenylalanine residue in the C terminus of DHHC21 repositions the proteins from the Golgi to ER membranes. Furthermore, mutations that occur at the 16-amino acid motif in the C terminus of DHHC proteins of yeast organisms cause a mis-localization from the vacuole membrane to the lumen. Protein degradation is based on the yeast vacuole functionality in the sense that there is consistency in the decreasing expression levels of mutant protein.
The analysis of DHHC proteins and their presence in cellular palmitoylation can be seen in the systematic experiment of S.cerevisae where a proteomic investigation of substrate palmitoylation profiles in deficiency in DHHC yeast strains imply a substrate selectivity. For instance, depletion in Akr1 led to an enormous downfall in palmitoylation of Yck1 and Yck2. Furthermore, it caused a removal of palmitoylation of Meh11, Yp199c, and Yk1047w. Conclusively, this experimental study has underlie that individual yeast DHHC proteins express a preference towards specific substrates Akr1 target were soluble proteins that are exclusively palmitoylated; Erf2 substrates had a tendency to be modify by myristoyl or prenyl groups; and Swf1 contained a preference for cysteine residues.
Protein binding assays have determined locations of palmitoylated substrates that communicate with DHHC proteins. Studies have shown that short, minimal sequences from palmitoylated proteins process effective palmitoylation when expressed in cells. Even though yeast genetic studies have indicated characteristics that substrates are affiliated with the modifications of DHHC protein, this cannot be said about mammalian proteins. This is due to the fact that there is a lack of data that show the substrate interactions performed on mammalian DHHC substrates. Thus, DHHC protein-substrate specificity is a subject that is hard to generalize and contain many undeveloped areas of research.
1. Linder, M.E., "Reversible modification of proteins with thioester-linked fatty acids," Protein Lipidation, F. Tamanoi and D.S. Sigman, eds. (San Diego, CA: Academic Press, 2000).
2. Basu, J., "Protein palmitoylation and dynamic modulation of protein function," Current Science, Vol. 87, No. 2, (25 July 2004).
3. Smotrys J and Linder A. (2004) "Palmitoylation of Intracellular Signaling Proteins: Regulation and Function". Annu Rev Biochem 73.
4. Linder, M. and Deschenes. R., "Palmitoylation: policing protein stability and traffic," Nature Review (January 2007)
5. Salaun, C et al., "The inracellular dynamic of protein palmitoylation," J. Cell Biol. Vol. 191 No.7 (December 2010)
6. Shipston MJ, "Ion channel regulation by protein palmitoylation," J Biol Chem. 2011 March 18; 286(11): 8709–8716 (January 2010)
7. Greaves, Jennifer, Luke H. Chamberlain, DHHC palmitoyl transferases: substrate interactions and (patho)physiology, Trends in Biochemical Sciences, Volume 36, Issue 5, May 2011, Pages 245–253, ISSN 0968-0004, 10.1016/j.tibs.2011.01.003. (http://www.sciencedirect.com/science/article/pii/S0968000411000144)
Prenylation, isoprenylation, or lipidation is the addition of hydrophobic molecules to a protein. It is believed that prenyl groups (3-methyl-2-buten-1-yl) facilitate attachment to cell membranes membrane, similar to lipid anchor like the GPI anchor, though direct evidence is missing. Prenyl groups have been shown to be important for protein-protein binding through specialized prenyl-binding domains.
Protein prenylation involves the transfer of either a farnesyl or a geranyl-geranyl moiety to C-terminal cysteine(s) of the target protein. There are three enzymes that carry out prenylation in the cell.
Farnesyltransferase and geranylgeranyltransferase I
Farnesyltransferase and Geranylgeranyltransferase I are very similar proteins. They consist of two subunits, the α-subunit which is common to both enzymes, and the β-subunit whose sequence identity is just 25%. These enzymes recognise the CaaX box at the C-terminus of the target protein. C is the cysteine that is prenylated, a is any aliphatic amino acid, and the identity of X determines which enzyme acts on the protein. Work reported in the journal Genome Biology in 2005 reports refinement of computational detection methods for identification of protein prenylation motifs and establishment of an on-line analysis facility entitled "PrePS".
Proteins that undergo prenylation include Ras, which plays a central role in the development of cancer. This suggests that inhibitors of prenylation enzymes (e.g. farnesyltransferase) may influence tumor growth. Recent work has shown that farnesyltransferase inhibitors (FTIs) also inhibit Rab geranylgeranyltransferase and that the success of such inhibitors in clinical trials may be as much due to effects on Rab prenylation as on Ras prenylation.
Reference:
Sebastian Maurer-Stroh and Frank Eisenhaber (2005). "Refinement and prediction of protein prenylation motifs". Genome Biology.
Taylor J, Reid T, Terry K, Casey P, Beese L (2003). "Structure of mammalian protein geranylgeranyltransferase type-I".
Posttranslation modification is the process by which proteome complexity (the global collection of proteins) is built by diversification at both the mRNA level and after translation ofmRNAs into proteins by covalent modification of specific proteins. There are two broad categories of posttranslational modifications. The first is the covalent addition of one of more groups, such as phosphoryl, acetyl, or glycosyl, to one or more of the amino acid side chains in a particular protein. The second is the hydrolytic cleavage of one or more peptide bonds in a protein by protein called proteases (protein hydolases). There are more the 200 kinds of posttranslational modifications and almost all of them occur by covalent addition of groups to side chains in thousands of proteins carried out by enzymes. These enzymes are proteins with catalytic activity dedicated to effecting the posttranslational modifications. There are many types of catalytic posttranslations, with about 500 proteases encoded in the human genome and over 500 protein kinases for covalent phosphorylations of proteins. Also, there are nearly 150 protein phosphates opposing and balancing the action of protein kinases. Furthermore, there is a small subset of proteins which undergo automodifications- modifications without the help of ancillary catalysts to effect covalent change.
Scope of Posttranslational Modifications
The diversity of posttranslational modifications by the proteome can be plotted on multiple axis. One scope is the number of proteins modified and thus the number of modified proteins produced. These numbers can differ for a given protein. A second and third axis of scope of posttranslational modification is by the type of amino acid side chain modified and also the type of covalent chemical modification introduced by the posttranslational modification enzymes. In posttranslational modification, the chemical reaction is of enzyme catalyzed transfer of an electrophilic fragment of a co-substrate molecule onto a nucleophilic side chain of the protein undergoing modification. The chemical modification occurs when the attacking nucleophilic side chain of the protein transfers the electrophile. Therefore, common sites for posttranslational modifications are side chains of proteins that can potentially act as nucleophiles.
Reversible vs. Irreversible Posttranslational Modifications of Proteins
Some posttranslational modifications of proteins are irreversible, due to the nature of the biological function enabled by the modification. The most irreversible modifications are the proteolytic cleavages undergone by all proteins during their life cycles. The removal of N-terminal signal sequences of all proteins passing into the endoplasmic reticulum during the first stage of eukaryotic cell pathways is also irreversible. Also note that some posttranslational modifications are freely reversible in vivo but not in vitro.
Post-Translational Modifications in Circadian Rhythms
Circadian rhythms are the biological clocks that exist in living organisms. These clocks are produced by oscillations in gene expression. Transcriptions factors and negatively acting transcription factors control the expressions of certain genes in an organisms genome.
An example of a circadian rhythm in humans based on day and night.
These factors are proteins in themselves, and are thus subject to modifications after they have been folded (post-translational modifications). Modifications made to these transcription factors effect the rhythms of the genes on which they act. The effects of post-translation modifications can be studied on the positive transcription factors as well as on the negative factors. As reported by Dunlap et al., modification by phosphorylation, either by kinases or phosphatases, on the positively acting transcription factors result in its degradation.[1] The same can be said about the negatively acting transcription factors, except that there are possibly non-degradative effects as well. For example, the negatively acting transcription factors can be phosphorylated by kinases and transported by these negatively acting factors in order to phosphorylate the positively acting transcription factors. The positively acting transcription factors, in turn, become inactivated.
Non-transcriptional Oscillator (NTO)
Theoretical evidences and recent discoveries have proven that there is a possible connection between non-transcriptional oscillator (NTO) and transcriptional/translational feedback loops (TTFLs). TTFL regulates translational modifications and engages in many important cellular programming relative to the circadian rhythm. However, studies have shown that the TFFL is challenged by the emergence of NTO circadian rhythms in cyanobacteria. NTO in eukaryotes produce a a very influential rhythmic output that is present in all organisms. As a result, NTO and TTFL elements are paired cooperatively in circadian science.
NTO and TTFL oscillators have history in operating separately, but evidence suggest that the coupling of the oscillators to function as a whole will be an evolutionary advantage. Linking NTO and TTFL enhances overall circadian performance. Mathematical and experimental data proved that the oscillators feed back to each other under habitual physiological conditions.
Reference: Walsh, Christopher. "Posttranslational Modification of Proteins: Expanding Nature's Inventory." Roberts and Co. (2006): 7-24.
An intein is a segment of protein that is able of excising itself and rejoining the remaining portions that do code for information, exteins, through a peptide bond. Splicing of inteins occur after translation, so inteins are present in the DNA template and mRNA strand, but are cut out from the final protein product.
The precursor of the intein may from from one gene, or from two genes. In split inteins, the inteins come from two genes. An example of split inteins is in cyanobacteria where DNA E is coded by two genes, dnaE-n and dnaE-c. The dnaE-n has an N intein, while the dnaE-c has a C intein. The two inteins will both be spliced and the two extein regions, dnaE-n and dnaE-c, will be joined.
Slonczewski, Joan L. Foster, John W. Microbiology: An Evolving Science, Second Edition, W.W. Norton & Company. 2009.
↑Mehra A, Baker CL, Loros JJ, Dunlap JC. 2009. Post-translational modifications in circadian rhythms. Trends Biochem. Sci. 34(10):483–90
Adenylation, also known as adenylylation or AMPylation, is the process of attaching an AMP molecule to a protein side chain by covalent bonding. It has two main functions: 1) to regulate enzyme activity via post-translational modification and 2) to produce unstable intermediates of a protein, peptide or amino acids to allow reactions that are not thermodynamically favored to occur.
Adenylations has a particular application when it involves the adenylation of RNA. There is a known procedure that allows for RNA 5' adenylation using T4 DNA ligases. Basically this is an approach to adenylate RNA so that it becomes a 5',5'-adenyl pyrophosphoryl cap structure. This new adenylated RNA structure is desirable in the sense that it can investigate certain natural biochemical pathways that require such intermediates. The 5' adenylated RNA is also useful for a certain amount of in vitro selection procedures to identify nucleic acid enzymes that specificially use the 5' adenlyated RNA as a reactive RNA substrate. This is particularly favorable given that the adenylated RNA is considered a transition state molecule which has evolved to become the most favorable of states when it comes to enzyme substrate complexes. in other words, the adenylation of the 5' RNA allows for a better enzyme substrate complex due to its nature of being a transitional state substrate for biochemical pathways that wish to be observed. In addition to being a RNA substrate for selective catalysis reactions, there are also applications to RNA '5 capping and 5' labeling. It was shown in the "Efficient RNA5' adenylation by T4 DNa ligase to facilitate practical applications" paper that the protein enzyme T4 RNA ligase was used along with some DNA oligonucleotides to create a net transfer of the AMP group onto a monophosphorolyated terminus of the desired RNA substrate.
Carboxyl groups of small molecules or amino acids may be adenylated to form unstable intermediates as shown in the diagram to the left.
For many decades, the example of glutamine synthetase has been used to explain the principles of adenylation, but recent discoveries in adenylation of Rho and Rab GTPases have shown potential in revealing the other aspects of adenylation.
Itzen A, Blankenfeldt W, Goody RS. Adenylylation: renaissance of a forgotten post-translational modification. Trends Biochem Sci. 2011 Apr;36(4):221-8. Epub 2011 Jan 20.
Efficient RNA 5′-adenylation by T4 DNA ligase to facilitate practical applications, Yangming Wang, Scott K. Silverman
ADP-ribosylation is a posttranslational modification of proteins that involves the addition of one or more ADP and ribose moieties.
These reactions are involved in cell signaling and the control of many cell processes, including DNA repair and apoptosis.
This protein posttranslational modification is produced by ADP-ribosyltransferase enzymes, which transfer the ADP-ribose group from nicotinamide adenine dinucleotide (NAD+) onto acceptors such as arginine, glutamic acid or aspartic acid residues in their substrate protein. In humans, one type of ADP-ribosyltransferases are the NAD: arginine ADP-ribosyltransferases, which modify amino acid residues in proteins such as histones by adding a single ADP-ribose group.
These reactions are reversible; for example, when arginine is modified, the ADP-ribosylarginine produced can be removed by ADP-ribosylarginine hydrolases.
As well as the transfer of single ADP-ribose moieties, multiple groups can also be transferred to proteins to form long branched chains, in a reaction called poly(ADP-ribosyl)ation. This protein modification is carried out by the poly ADP-ribose polymerases (PARPs), which are found in most eukaryotes, but not prokaryotes or yeast. The poly(ADP-ribose) structure is involved in the regulation of several cellular events and is most important in the cell nucleus, in processes such as DNA repair and telomere maintenance.
ADP-ribosylation is also responsible for the actions of some bacterial toxins, such as cholera toxin, diphtheria toxin, and pertussis toxin. These toxin proteins are ADP-ribosyltransferases that modify target proteins in human cells. For example, cholera toxin ADP-ribosylates G proteins, which causes massive fluid secretion from the lining of the small intestine and results in life-threatening diarrhea.
Acetylation introduces an acetyl group to a molecule. More specifically, the reaction replaces a hydrogen from an alcohol group with an acetyl. An example is the synthesis of aspirin from salicylic acid:
Aspirin synthesis
Acetylation is an important post-translational protein modification and regulation. An example is the acetylation/deacetylation of histone which subsequently express/inhibit genes since histone binds to DNA itself. Histone Acetyltransferase catalyzes the acetylation of lysine from the histone tail with an acetyl group from Acetyl CoA.
The acetylation of lysine in histone removes the positive charged ammonium group and renders the side chain neutral, which decreases the histone tail affinity for DNA and loosens the histone complex.
The now-acetylated histone can interact with an acetyllysine-binding domain in many eukaryotic proteins called w:bromodomain, an 110 amino acids protein with a four-helix bundle and a peptide-binding site at one end.
Acetylation is the reaction in which an acetyl functional group is added to an organic molecule. In proteins, an acetyl group to either added to the N-terminus of proteins and at lysine residues as a post-translational protein modification.
N-alpha-Terminal Acetylation
The role of N-alpha-Terminal Acetylation is still relatively unknown and under research. However, it is known that this modification is actually widely prevalent in eukaryotes and yeast, though uncommon in prokaryotes, and is performed by a subgroup of acetlytransferases known as N-alpha-acetyltransferases (NATs). There are three major NATs (A,B,C) which perform the majority of the N-alpha-terminal acetylations of eukaryotes The reaction is begun with the cleaving of N-terminal methionine residues with small side chains containing, glycine, alanine, serine, cysteine, threonine, proline, and valine, by methionine aminopeptidases (MAP), Map1p and Map2p. Subsequently, the NATs recognize and acetylate specific sequences of the cleaved proteins.
Lysine Acetylation
One major area in which lysine acetylation is prevalent is in the acetylation of histones, which is performed by histone acetyltransferases (HATs) and deacetylases (HDACs). In both acetylation and deacetylation reactions that attach to the NH3+ tail from an acetyl group from Acetyl-Coenzyme A and remove the acetyl group from lysine onto Coenzyme A, respectively. The acetylation and deacetylation of histones partake in gene regulation. Histones are packages of strongly alkaline protein around which DNA is wound to allow DNA to be stored in an orderly fashion. Since the acetylation occurs at the NH3+ tail of lysine, the charge of the protein is affected. When acetylated, the positive charge of lysine is eliminated. This decreases the histones affinity to the negatively charged DNA strand, thereby loosening the strand, and the reverse results occur with deacetylation. However, Lysine acetylation isn’t limited to histone acetylation. Among other things, it is also involved in the modification and regulation of non-histones such as cytoplasmic enzymes and p35, a tumor suppressor and involved with signal transmittance and signaling.
Many of the drugs used today for common diseases require additional structuring in order for efficient metabolism in the body. Drugs that are significantly metabolized by acetylation include isoniazid, hydralazine, procainamide, phenelzine, and dapsone that are used to treat tuberculosis, cardiac failure/hypertension, ventricular arrhythmias, depression, and leprosy/skin infections respectively.
A Histone is an alkaline protein found in eukaryotic cells that orders the DNA stain and plays a role in gene regulation. In order for DNA to form into chromosomes the DNA coils around the histone protein to form chromatin, without this process life would not exist.
There are five major classes of Histones: H1/H5, H2A, H2B, H3, and H4. Histones H2A, H2B, H3, and H4 are known as the core histones, while histones H1/H5 are known as the linker histones.[1]
A nucleosome core particle is a combination of two of each of the core histones, and this core particle is responsible for winding up a section of the DNA, approximately 147 base pairs. The linker histone keeps the wound section of DNA in place by binding to the neucleosome and to the entry and exit sites of the DNA, locking it into place. The combination of DNA wound up in histones is called a chromatin.
Sometimes histone modifications affect the structure of chromatin, or they may act as a mark or a signal for protein effectors. [2]
Alterations of the Structure of Chromatin
High orders of chromitin structure can be directly affected by Histone
modifications. Removal of Histone tails by trypsin results in a decrease
of the ability of nucleosome arrays to comdense into 30 nanometer
chromatin fibers. Acetylation of H4K16 has also been experimentally shown
to inhibit the formation of 30-nm chromatin fibers. Many Histone
Acetyltransferase (HAT) complexes have the function of acetylating
multiple lysines of Histone H4, including H4K16. Histone acetylation occurs at the lysine residues and is recognized by the bromo domain proteins. It is important for chromatin decondensation and euchromatin formation. Euchromatin is considered to be open chromatin and is more accessible to proteins. Thus, acetylation leads to additional transcriptional factors binding and thus the DNA can be available for transcription. Acetylation of H4K16
serves an important part in DNA replication, gene splicing, and life-span
regulation of yeast.
Chromatin can also be altered by methylation at the lysines and the arginines by histone methyl transferases (HMT) which add on methyl groups. These are recognized by chromo domain proteins and is important for the condensation of chromatin and heterochromatin formation. Heterochromatin is considered to be tightly closed chromatin and is less accessible to proteins.
Histone modifications can also affect chromatin structures by acting as marks for the activity or the gathering of protein complexes that change and reconstruct the chromatin structure. These are ATP-dependent chromatin remodeling complexes which are a group of proteins that slide nucleosomes on DNA with the purpose of changing or getting rid of the histones.[3] The nucleosome remodeling complexes have one to twelve subunits that include multiple protein domains that interact with DNA. Often, these domains recognize histone modifications.
One of the first domains discovered to interact with an already modified histone and to recognize acetyl-lysine residues in the histones were bromodomains. These bromodomains found in subunits of the SWI/SNF family of chromatin-remodeling complexes play an important role in gene activation.
For example, acetylated histones can be evicted from the DNA complex and replaced with the SWI/SNF complex (Figure 1). This process occurs when sequence-specific DNA-binding transcription activators recruit the SAGA histone acetyltransferase to target the genes where SAGA acetylated a patch of nucleosomes on the promoter region. Then, the same activators can obtain the SWI/SNF nucleosome -remodeling complex to the same site and binds to the acetylated histones. Finally, the SWI/SNF complex uses the energy of ATP hydrolysis to displace the acetylated histones generating a neucleosome free region.[4]
Actively Transcribed Genes
Two of the histone modifications are involved in active transcription
-Trimethylation of H3 lysine 4 at the promoter of active genes, which is done by the COMPASS complex.[5] The role of this modification is not clear, but the level of this modification is correlated with the transcriptional activity of the gene.
-Trimethylation of H3 lysine 36 at the body of active genes. This modification is recognized by the Rpd3 histone, which deacetylates surrounding histones. This increases chromatin compaction, which prevents the likelihood of new transcription events from occurring while one is already in progress.[6] This helps to ensure that a transcription in progress is not interrupted.
Repressed Genes
There are three histone modifications that are associated with gene repression.
-Trimethylation of H3 lysine 27. This is done by the polycomb complex PRC2, which is usually bound with other proteins and it binds to the gene, causing chromatin compaction, which silences transcription activity.[7] PRC1 has also been known to aid PRC2 in the histone modification.
-Di and trimethylation of H3 lysine 9 is a well-known marker for heterochromatin, a tightly packed form of DNA. An RNA-induced transcriptional silencing complex (RITS) is responsible for this modification.[8]
-Trimethylation of H4 lysine 20 is a modification done by Suv4-20hmethyltransferase that is also associated with heterochromatin.[9]
↑Krogan NJ, Dover J, Wood A, Schneider J, Heidt J, Boateng MA et al. (2003). "The Paf1 complex is required for histone H3 methylation by COMPASS and Dot1p: linking transcriptional elongation to histone methylation.". Mol Cell 11 (3): 721–9. doi:10.1016/S1097-2765(03)00091-1. PMID 12667454.
↑Carrozza MJ, Li B, Florens L, Suganuma T, Swanson SK, Lee KK et al. (2005). "Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription.". Cell 123 (4): 581–92. doi:10.1016/j.cell.2005.10.023. PMID 16286007.
↑Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P, Reinberg D (2002). "Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein.". Genes Dev 16 (22): 2893–905. doi:10.1101/gad.1035902. PMC 187479. PMID 12435631
↑Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P, Reinberg D (2002). "Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein.". Genes Dev 16 (22): 2893–905. doi:10.1101/gad.1035902. PMC 187479. PMID 12435631
↑Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P, Reinberg D (2002). "Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein.". Genes Dev 16 (22): 2893–905. doi:10.1101/gad.1035902. PMC 187479. PMID 12435631
During cell differentiation, most multi-cellular organisms form their distinctive gene expression pattern. Over time, this pattern is maintained over the course of many cell divisions even though the initiating signal is gone. Actively transcribed regions are characterized by specific histone modifications. The role of histones is that it is the most important protein component of chromatin. These findings confirm that a histone code uses histone post-translational modifications to translate chromatin structures into the genome with a form of stability.
DNA (in eukaryotic cells) is wrapped around histone proteins, forming chromatin. Chromatin's basic unit is the nucleosome, which is formed by a histone octamer that consists of two molecules of each of the four core histones: H2A, H4, H3, H2B. The histone octamer also consists of 147 base pairs of DNA wrapped around it on the left-handed helix.
Individual nucleosomes pack against each other to form higher order chromatin structures to regulate DNA accessibility. The nucleosome core is created by the globular domains of histones. The nucleosome core doesn't need the N-terminal tails of histone molecules because the histone N-termini don't form crystals in crystallographic studies; but, they are extremely important to chromatin function. It's not completely proven that histone modifications are a cause or a consequence of the activity of a gene. To determine the role of histone modifications, there's results on their targeting to specific genomic loci, their stability and dependence towards one another. The histone code hypothesis was creating because histone-modifying enzymes were discovered as well as the genome-wide mapping of histone modifications.
Genome-wide mapping studies of histone modifications
The genome-wide map purpose is to detect similarities between histone modification patterns and also specific states of gene activity. For example, modifications including H3K4me2,3 and H3K36me2,3 are located in actively transcribed regions and overlap one another. With these overlapping areas with gene activity, this information was used to figure out regions of transcription for untranslated RNA molecules. In a different example, modifications like H3K27me3 and H4K20me3 are frequently mapped to regions where transcription is repressed. This shows that histone modifications can be landmarks for inactivity.
Transcription and DNA assembly are important in finding out when a modification pattern is created and copied. One of the first histone-modifying enzymes characterized at the molecualr level was HAT (acetyltransferase Gen5). HAT was characterized to be a transcriptional co-activator in yeast which linked histone acetylation to gene activation.
Genome-wide mapping studies show that acetylated histones can be found at most actively transcribed regions.
Histone modifications are deposited during chromatin assembly and it also observes modifications that occur during activation or represseion of a certain gene. This is extremely important because when newly synthesizes histones are formed and moved into the chromatin, the histone modifications need to maintain the chromatin structure.
Histones are synthesized in the cytoplasm and then it's moved to the nucleus. Most histone synthesis is coupled with S-phase progression to meet the needs of the increased histones. An exception if the histone variant H3.3, which is synthesized into a replication-independent manner. Right after synthesis, histones get a modification pattern and associate with histone chaperones. Histone chaperones help with the deposition of histones onto DNA and they are grouped in the H3/H4 binding factors like Asfl.
Like mentioned above, newly synthesized histones carry a specific histone modification pattern and lysine 5 and lysine 12 of H4 become acetylated and a fraction of H3 is acetylated at lysine 18 and lysine 1. During the transportation from the cytosol, then the nucleus, and then to incorporate new chromatin at the replication fork, histones receive more modifications like monomethylation of lysine 9.
An epigenetic code is beneficial to initiate the generate and inherit functional chromatin states. In order for this to happen, there are requirements that need to be met:
(i) the system needs to initiate the generation of the code
(ii)the system needs to translate the modifications into varied chromatin states
(iii) the system needs to allow copying of a particular modification pattern from old histones to the ones that are newly synthesized.
There needs to be a mechanism that copies histone modification patterns during cell division if slow turnover of histone methylations want to have the cell pass its epigenetic information to future generations. The turnover is slow because for lysine di- and trimethylations on histones, the reestablishment of methylations on newly sytnhesized histones is slow. This slow turnover allows the cell to have time (if it needs to) to change the modification pattern on a specific gene or copy the previously existing one.
Recent investigations has led many to believe that histone modifications can facilitate the stabilization of gene expression when there's no incoming signal(s). But, there are some modifications, like lysine acetylation, that integrates incoming signals.
Barth, Teresa K., and Axel Imhof. "Fast signals and slow marks: the dynamics of histone modifications." Trends in Biochemical Sciences 35.11 (2010) 618-626. Academic Search Complete. Web. 05 December. 2012.
Phosphorylation of protein side chains in posttranslational modification of proteins is incredibly evolved and is a method of creating diversity in the proteomes of eukaryotes. Protein phosphorylation also occurs in prokaryotes but is less pervasive in bacterial protein metabolism and regulation. Phosphorylation of proteins involves the addition of phosphate groups to a target protein via protein kinases to activate or inactivate a certain function in the body. Marking proteins by the addition of phosphate groups assigns the proteins a code, which can instruct the cell to perform a number of functions, such as to divide or grow.
Side chains of proteins that are phosphorylated (addition of a PO4 group) most commonly are serine, threonine, and tyrosine, which reflects the nucleophilic behavior of their -OH side chains. Also capable of phosphorylation is the imidazole ring nitrogen of histidine side chains in proteins. The process of phosphorylation turns protein enzymes on/ off, which can either cause or prevent certain diseases, such as cancer or diabetes.
Nearly one-third of all the potential 30,000 proteins in the human proteome are estimated to be substrates for phosphorylation at a particular stage in their life cycle of eukaryotic cells. Kinases are those ATP dependent phosphorylation enzyme catalysts, and protein kinases are the subset working on protein substrates. There are over 500 estimated kinases in the human proteome, which is termed the human kinome.
It is known that about 20-30% of the proteins in human body are phosphorylated. However, the problem is sorting out which of the 500 different kinases is responsible to the specific phosphorylation activity.
Protein kinase A is an enzyme that covalently attaches phosphate groups to proteins. It is also known as the cyclic AMP-dependent protein kinase. An extremely significant characteristic of protein kinase A is its ability to be regulated by the fluctuation of cyclic AMP levels within cells. Essentially, protein kinase A is responsible for all cellular responses due to the cyclic AMP second messenger. Cyclic AMP activates protein kinase A, which phosphorylates specific ion channel proteins in the postsynaptic membrane, causing them to open or close. Due to the amplifying effect of the signal transduction pathway, the binding of a neurotransmitter molecule to a metabotropic receptor can open or close many channels.
Protein kinase B regulates various biological responses to insulin and growth factors. Akt is another way to classify Protein Kinase B. Protein Kinase B is a serine-threonine-specific protein kinase that contributes to multiple cellular processes such as glucose metabolism, apoptosis, and cell migration. [2]
Protein kinase C catalyzes the process of signals mediated by phospholipid hydrolysis. It is activated by the lipid second messenger, diacylglycerol. This lipid second messenger serves as the key initiation for most protein kinase C's. Protein kinase C isozymes consist of a single polypeptide chain that possesses an amino-terminal regulatory region and a carboxy terminal kinase region. The isozymes are categorized into various groups: conventional protein kinase Cs which are regulated by diacylglycerol, phosphatidylserine, and Ca^2+ in addition to novel protein kinase Cs which are regulated by diacylglycerol and phosphatidylserine. Activation of GPCR's, TKR's, and non-receptor tyrosine kinases can lead to protein kinase activation by stimulation of either phospholipase Cs to yield diacylglycerol, or phospholipase D to yield phosphatidic acid and diacylglycerol. Additionally, conventional protein kinase Cs are regulated by Ca^2+.[3]
Phosphorylation is used in various enzymatic activities. For example, proteins p300 and CBP (CREB binding protein), when phosphorylated, increase the activity of acetyltransferase. Acetyltransferase then stimulates histone acetylation that promotes the transactivation of genes controlled by p300 and CBP activity. Conversely, phosphorylation can also be triggered by acetylation.
Protein Kinase Inhibitors in celling transduction and in clinical use
The mutation of protein tyrosine kinases (PTKs) can change the communication between cells to be more or less frequent and can cause the spread of diseases. These diseases include various forms of diabetes and cancers. These mutations either enhance or detriment the rate of phosphorylation within the different proteins.
The inhibition of PTKs will help prevent the spread of these diseases and is believed to not cause much harm to the normal cells. PTKs are inhibited through the usage of tyrphostins (TYRosine PHOSphorylation INhibitors) as they bind to the ATP or substrate of the PTKs. These inhibitors were not supported at first as they were believed to block functions that were needed for the cell. However, after extensive tests and trials, it was found that there were natural occurring inhibitors and were as selective as needed. Both ATP competitive and substrate competitive molecules are used to block the signals.
The first inhibitors used were natural occurring; these included quercetin, genistein, erbstatitin, and lavendustin. However, these natural PTK inhibitors were not very effective as they were not very selective and also inhibited Ser/Thr kinaeses. These served as the model for the design of more potent and selective PTKs. These were developed through the usage of ATP mimics and substrate mimics to test the competitiveness of the designed inhibitor. One PTK that was developed was STI-571, which is effective in treating certain tumors.
Gefitinib and Erlotinib are two PTKs that were developed to treat non-small cell lung cancel (NSCLC).
Death-associated protein kinases (DAPk) are kinases that regulate cell death and can also be used to induced cell death. DAPk has the ability to act as a tumor suppressor because it can sensitize cells to many signals that a cell encounters as it undergoes tumorigenesis. Its ability to suppress tumors shows that it plays a key role in tumor development. The study of how it’s expressed can function as a diagnostic tool to help scientists better evaluate disease in its severity, progression and other factors. However, excessive levels or increased activity of DAP kinases can be harmful and can contribute to diseases associated with the brain.
One of the structural components of DAPk is a death domain, located on the C terminus of the protein kinase, followed by a tail of 17 amino acids rich in serine residues. These serine residues are a key feature of death domain-containing proteins. Deletion of this tail was determined to produce a mutant that showed more killing potential than if the tail were not deleted however, the C terminus tail negatively regulates the cellular functions of DAPk. These functions demonstrate that the death domain competes with the full length kinase.
References: Walsh, Christopher. "Posttranslational Modification of Proteins: Expanding Nature's Inventory." Roberts and Co. (2006): 35-40.
Berg, Jeremy. Biochemistry . 6th. New York : W. H. Freeman and Company, 2006.
Burnett G, Kennedy EP. "The Enzymatic Phosphorylation of Proteins." J. Biol. Chem. (1954) 211 (2): 969–80.
Wen Wu, Cheng Lu, Siyu Chen, Niefang Yu "The signal transduction pathway of multi-target kinase inhibitors as anticancer agents in clinical use or in phase III"
Arvin C. Dar1 and Kevan M. Shokat, "The Evolution of Protein Kinase Inhibitors from Antagonists to Agonists of Cellular Signaling"
Levitzki, Alexander, and Mishani, Eyal. "Tyrophostins and Other Tyrosine Kinase Inhibitors." Annual Review of Biochemistry, 2006. 75:93-109.
Bialik, Shani and Kimchi, Adi. "The Death-Associated Protein Kinases: Structure, Function, and Beyond." http://www.ncbi.nlm.nih.gov/pubmed/16756490
A specific enzyme called Nucleoside Monophosphate kinase (NMP kinase) catalyzes the transfer of a terminal phosphate group (ATP in most cases) to the phosphate group on the Nucleoside Monophosphate (NMP). Oftentimes, the transfer of NTP to NMP competes with a hydrolysis reaction in which the phosphate group from NTP transfers to a molecule of water instead. However, the use of the induced fit model allows the enzyme to wrap around the substrate and change the overall conformation of the enzyme-substrate complex in order to solve this problem. The phosphorylation reaction takes on the general form:
ATP + NMP <=====> ADP + NDP
In this reaction, the enzyme NMP binds to the substrate ATP (by induced fit). NMP gains one phosphate group and becomes NDP, whereas the ATP substrate loses one phosphate group and thus becomes ADP.
Through X-ray crystallography, scientists and researchers discovered the structure of many different NMP kinases. Analyzing the three-dimensional structures revealed that these kinases are homologous proteins. Moreover, crystallographic data suggested that the NTP-binding domain was strictly conserved. This domain consists of two alpha helices wrapped around a beta sheet. One distinct feature of this domain is the formation of a loop between the beta strand and the alpha helix (P-Loop). These loops tend to wrap or "loop" around substrates, therefore enclosing them. The P-Loop is unique in the sense that it interacts with phosphate groups on bounded nucleotides.
Studies of NMP kinases and ATP substrates reveal that these kinases are only active in the presence of divalent metal ions such as magnesium or manganese. In this case, the ATP substrate binds to the divalent ion, forming a metal ion-nucleotide complex. This complex is ultimately the true substrate for enzymes like NMP kinase.
Binding of divalent ions like magnesium or manganese increases the enzyme specificity. These ions help stabilize negative charges on phosphate groups. The interactions between the divalent ion and the oxygen atoms in the phosphate group changes the conformation such that it can bind specifically to the enzyme. These divalent ions also produce an interaction between the true substrate complex and the enzyme, therefore increasing the binding energy.
By understanding the tertiary structure of adenylate kinase, scientists and researchers discovered that a large conformational change occurs when adenylate kinase binds to an ATP analog. The P-Loop wraps around the phosphate chain, reacting mostly with the beta phosphate group. This allows the domain of the enzyme to shift downward such that a lid forms over the bounded nucleotide. As a result, the gamma phosphate group is positioned directly next to the NMP binding site. This binding induces yet another conformational change.
[1]
The life cycle of protein kinase C is regulated by multiple phosphorylation as well as dephosphorylation. The maturation of protein kinase C involves three ordered phosphorylations. These phosphorylations are: 1) One at the activation loop, 2) COOH-terminal sites, but two of them. These processes lead to a signaling-competent enzyme. The process of dephosphorylation results in protein degradation.
Recent discoveries have illustrated a new family of protein phosphatases called PHLPP, which stands for “PH domain leucine-rich repeat protein phosphatase”. PHLPP terminates Protein Kinase B signaling through dephosphorylation of the hydrophobic motif on Protein Kinase B also known as Akt.
There are two isoforms of PHLPP called PHLPP1 and PHLPP2, which dephosphorylate the hydrophobic motif on PKC BII. This process brings Protein Kinase C to the detergent-insoluble fraction, which effectively terminates its life cycle.
Deletion mutagenesis elucidates the idea that PH domain is vital for the effective dephosphorylation of PKC BII by PHLPP in cells. However, for the PDZ-binding motif, which is necessary for Akt regulation, it is dispensable.
The depletion of PHLPP in colon cancer and normal breast epithelial cells results in an increase in conventional and novel PKC levels, revealing that PHLPP regulates the cellular levels of PKC by specifically dephosphorylating the hydrophobic motif. This also causes the destabilization of the enzyme, which promotes its degradation.
Protein phosphorylation acts as one of the most significant regulatory mechanisms in cell signaling. Vital cellular decisions such as death or survival and proliferation or differentiation are made depending on the phosphorylation state of the signaling molecules. Therefore, precise control of the balance between phosphorylation and dephosphorylation is crucial for living organisms to maintain normal physiological functions. The disturbance of regulation of the signaling pathways, which results in disturbing a sense of equilibrium leads to the development of diseases such as cancer and diabetes. This leads to a pathogenic state in which both kinases and phosphatase are identified as oncogenes or tumor suppressors.
References: The Phosphatase PHLPP Controls the Cellular Levels of Protein Kinase C; By: Tianyan Gao, John Brognard, and Alexandra Newton from the Department of Pharmacology and the Biomedical Sciences Graduate Program, University of California, San Diego
PKC is controlled by diacylglycerol and the amino phospholipid, phosphatidylserine. The molecular basis for the phosphatidylserine specificity was proposed to arise from the presence of a putative phosphatidylserine binding motif, localized in the C2 domain of PKC. In order to determine whether this motif mediates the interaction of PKC with phosphatidylserine, the carboxyl-terminal basic residues were mutated to Alanine in PKC BII. Additionally, the phosphatidylserine regulation of the mutant enzyme was observed.
Membrane binding and activity measurements revealed that the phosphatidylserine regulation for the mutant protein was indistinguishable from that of wild-type protein kinase C. Neither the apparent membrane affinity for phosphatidylserine-containing membranes in the presence or absence of diacylglycerol nor the phsphatidyl-serine-dependence for activation was affected by removal of the conserved basic residues at the carboxyl terminus of the consensus sequence.
The protein kinase C family of serine/threonine kinases transduces the multitude of extracellular signals that result in generation of the lipid second messenger, diacylglycerol. This lipid allows protein kinase C to translocate from the cytosol to the plasma membrane where it becomes activated by an additional interaction with phosphatidylserine. This is mediated by two membrane-targeting modules: the C1 and C2 domains. One of the methods to observe this process is through mutagenesis. With this, mutation of Lys 236 and Arg 238 to Alanine occurs in PKC BII. This is achieved utilizing PCR with wild-type PKC BII in pBlueScript as a template.
Another method used was Protein Expression and Cell Fractionation. In this process, recombinant baculovirus encoding wild-type or K236A/R238A PKC BII was incubated with Sf-21 cells for 4 hours and then diluted with media and incubated at 27 degrees Celsius.
Another method used was Western Blot Analysis of Expressed Protein Kinase C. In this method, the distribution of PKC in the detergent-soluble and detergent-insoluble fractions was analyzed by Western blot analysis. Samples of the extracts were separated on SDS-polyacrylamide gels and transferred to polyvinylidene difluoride membrane.
Another method used was through lipid observation in which sucrose-loaded large unilamellar vesicles were used and prepared by drying mixtures of lipids in chloroform under a stream of nitrogen, followed by evacuation under vacuum, suspension of lipids in 20 mM HEPES, pH 7.5, 170 mM sucrose, and then 5 freeze-thaw cycles followed by extrusion using a Liposofast microextruder.
Another method used was the Protein Kinase C Activity Assay and the Protein Kinase C Membrane-binding Assay in which its membrane affinity was determined by measuring the binding to sucrose-loaded vesicles. The fraction of sedimented was determined by assaying the activity of PKC in the supernatant and pellet under identical conditions using the cofactor-independent substrate, protamine sulfate. In this case, the membrane affinity was calculated as the ratio of free/bound protein kinase C divided by the total lipid concentration.
Lastly, Data Analysis was another technique used for observation. In this technique, free calcium was calculated using a program that takes into account concentrations of Mg2+, ATP, Na+, EGTA, EDTA, and pH.
Reference: Joanne E. Johnson, Amelia S. Edwards, and Alexandra Newton from Departments of Pharmacology and Chemistry and Biochemistry, University of California, San Diego
Cellular homeostasis requires balance between phosphorylation catalyzed by protein kinases and dephosphorylation catalyzed by protein phosphatases. Deregulation of this balance leads to pathphysiological states that drive diseases such as cancer, heart disease, and diabetes. The discovery of PHLPP, which stands for Pleckstrin Homology Domain Leucine-Rich Repeat Protein Phosphatase, contributes to the cast of phosphate-controlling enzymes in cell signaling. PHLPP isozymes catalyze the dephosphorylation of a conserved regulatory motif, the hydrophobic motif, on the AGC kinases Akt, PKC, and S6 kinase to inhibit cellular proliferation and induce apoptosis.
The frequent deletion of PHLPP in cancer, coupled with development of prostate tumors in mice lacking PHLPP1 identifies PHLPP as an important tumor suppressor. It dephosphorylates a key regulatory site on the C terminus of Akt, the hydrophobic motif, therefore inactivating the kinase.
PHLPP plays an important role in disease. It not only blocks tumorigenesis by inactivation of oncogenic pathways, but also sensitizes cancer cells to chemotherapy. With the loss of PHLPP1, this causes prostate tumors in mice and its genetic deletion or mRNA repression is prevalent in prostate cancer patients.
Reference: Noel A. Warfeland Alexandra Newton from Departments of Pharmacology and Chemistry and Biochemistry, University of California, San Diego
Peptide biosensors are used to determine phosphorylation activities in cells. A biosensor is a device that provides information about the composition, structure and function of biological analytes such as isolated enzymes, immonosystems, tissues, and organelles by converting a biological response into an electrical, thermal, or optical signals. The biosensors that are mainly used for the analysis of phosphorylation events incorporate the use of a synthetic fluorophore, the portion of a molecule that illuminates the color of the molecule. When the fluorophore interacts with a phosphorylated peptide or protein domain, the fluorescence properties of the complex changes. Fluorscence are dyes that emit a certain color when a molecule is exposed to light. In general, the emitted light is usually longer than that of the incident light. The complex causes an enhancement in fluorescence and this property is used to determine the phosphorylation event of an analyte.
[1]
An environmentally sensitive biosensor detects phosphorylation activities that occur between a solute and a solvent molecule that forms relatively weak covalent bonds. In general, a domain that has a high affinity for binding to a phosphospecific amino acid attaches to a phosphorylated peptide or protein. This complex changes the polarity of the solvent in the fluorophore which increases the fluorescence of the molecule and the complex illuminates a noticeable color.
A deep-quench biosensor utilizes a quencher that interacts with the fluorophore in the peptide. When this complex is phosphorylated, the biosensor employs a domain that has a high affinity for binding to a phosphospecific amino acid to separate the quencher from the fluorophore. This separation causes an increase in the fluorescence in the molecule.
A self-reporting sensors is a type of sensor that does not require the use of a domain that has a high affinity for binding to a phosphospecific amino acid in order to detect the presence of phosphorylated amino acid. This type of sensor is only applicable for amino acids that are aromatic, usually tyrosine. The pi bonds in the molecule enable the amino acid to quench the fluorophore in the peptide biosensor. When the molecular is phosphorylated, the aromatic amino acid cannot quench the flouorophore again.
Metal chelation-enhanced fluorescence detects the presence of a phosphorylation event via Sox, a gene that encodes for a transcription factor that binds to the DNA. In addition being a sequence known the HMG box, Sox is also a fluorophore which can be intensified by chelation. Due to this unique property, Sox is used to determined phosphorylation activities in a system. In addition, metal chelation-enhanced fluorescence do not require the use of an acid-binding domain that has a high affinity for a phosphoamino acid.
In the presence of a serine or threonine residue, Sox is capable of recruiting a phosphate group to gather, which elicits phosphorylation to occur. Once the serine or threonine residue has been phosphorylated via Sox, the peptide acquires a higher ability to bind to Mg2+ peptides. The binding of the Mg2+ to the phosphorylated serine or threonine residue generates a fluorescent signal. In addition, several probes such as PKC, Cdk2, and PKA have been created to increase the intensity of the fluorescent signal.
[5]
Signal transduction is the chain of events that occurs when a cell converts a message to ultimately a physiological response. The message comes in the form of a particular molecule in the cell’s environment. Some examples of signal transduction are as follows. Upon release of epinephrine in response to stress, the cells in the body receive the message, then responds by preparing to use stored energy and improving cardiac function. After a meal, insulin is released into the bloodstream, indicating to cells to take in the glucose. In a wound, epidermal growth factor is released and simulates certain cells to proliferate.
Signal transduction is an important process since it occurs through the communication between difference domains and coupling of information. It is also important to know that ligand induced conformational changes are important to many aspects of protein function. Its importance is demonstrated when an enzyme binds a substrate because the act of binding causes changes in the protein’s structure to enable catalysis.
Signal transduction is an important process since it occurs through the communication between difference domains and coupling of information. It is also important to know that ligand induced conformational changes are important to many aspects of protein function. The importance of these conformational changes is demonstrated when an enzyme binds a substrate; the process of binding causes changes in the protein’s structure to enable catalysis. In addition, the binding of allosteric ligands can lead to the occurrence of more conformational changes as well.
Ligand binding often leads to changes in both the structure and dynamics with changes in the dynamics often occurring at different locations on the same proteins. The binding of a ligand to one site can often influence the structure, dynamics, and binding affinity at another site on the same protein. This demonstrates that coupling can often be achieved through many different ways. Many times, the effects of ligand binding on protein activity can be explained in terms of more common thermodynamic concepts; the fact that the transition of an active versus resting state depends on several factors such as the free energy difference between the two states, the protein affinity of the ligand, and the ability of a ligand to induce a transition once fully bound to the protein. In the case when a ligand binds to both an activated and resting conformation, incomplete activation often results. A ligand is considered to trigger activation when it binds with higher affinity to an active versus an inactive state.
The free energy difference, ΔGgap, can also be modified by environmental factors such as membrane composition. An example of this is demonstrated by the fact that the voltage dependent potassium channels exist in equilbrium only in an “on” or “off” conformations. These differ not only in their conductance characteristics but also in the number of charged groups on the two sides of the electrically impermeable membrane.
The K+ channels and M2 proton channels both share some similarities in their thermodynamic and structural coupling properties. For example, they are similar in the sense that both channels have dual gates to regulate the ion diffusion into the channel versus through the channel. In addition, they are able to achieve a high selectivity by binding multiple copies of permeablet ions.
The entry of ions through channels is also very tightly regulated to prevent leakage; leakage can be harmful to the life of the organism.
Signal transduction relays a message to a certain physiological response by way of certain key steps. In the first step, an event or condition stimulates the release of the signal molecule, otherwise known as the primary messenger. Generally, primary messengers do not enter cells, and thus work by binding to the cell’s membrane protein on the extracellular side. In the second step, the primary messenger binds to a receptor protein that spans across the membrane, causing conformational change. In the third step, the signal is relayed into the cell by way of conformational change on the receptor protein. This initiates a change in concentration of certain small molecules inside the cell. The small molecules are called second messengers, and they relay the signal inside the cell by activating other receptor-ligand complexes within the cell. In the fourth step, the secondary messengers activate the effectors that directly produce a physiological response. This physiological response can be activation/inhibition of enzymes, membrane channels, or gene-transcription factors. In the final step, after the physiological response is completed, the signal is terminated
Second messengers provide certain advantages for the signal transduction. A signal can be amplified significantly by generating second messengers. Small amounts of membrane receptors can be activated, but large amounts of second messengers can be generated. Each activated receptor can produce many secondary messengers. Low concentrations of primary messengers in the extracellular environment can give rise to a large signal due to amplification by secondary messengers. Also, second messengers are able to influence other processes in the cell by diffusing to other compartments. In addition, a common secondary messenger can signal for multiple pathways. This is called cross talk.
Epinephrine produces signals by binding as a ligand to a membrane protein called β-adrenergic receptor, which from a class of receptors called the seven-transmembrane-helix receptors. These receptors contain seven helices that cross the membrane seven times, thus also referred to as the serpentine receptors. The binding of a ligand on the extracellular side induces a conformational change on the seven-transmembrane-helix receptor on the cytoplasmic side. The conformational change on the intracellular side of the receptor activates G protein. Activated G proteins then binds to and promotes the activity of adenylate cyclase, which is a membrane bound enzyme that converts ATP to cAMP. cAMP can then move across the cell as the secondary messenger to initiate physiological response. cAMP activates protein kinase A (PKA). Activated PKA activates other proteins that directly produce a physiological response.
Unactivated G protein is bound to GDP and exists as a heterotrimer protein, consisting of the α-, β-, and γ- subunit. The GDP is bound to the α-subunit. To activate the G protein, the GDP is released and GTP binds to the α-subunit. Once GTP binds, the α-subunit dissociates from the βγ dimer. The activated α-subunit then binds and activates adenylate cyclase. The α-subunit has an intrinsic GTPase that slowly hydrolyzes the bound GTP to GDP. Once hydrolyzed to GDP, the α-subunit is deactivated and reassociates with the βγ dimer. The deactivation of G protein is a time dependent process, based on the kinetics of the intrinsic GTPase.
Aside from activating the cAMP cascade, the seven-transmembrane-helix receptor can also activate the phosphoinositide cascade. There are different types of G proteins. The β-adrenergic receptor functions with the Gs protein. The angiotensin II receptor activates the Gq protein. The mechanism of activating the G protein is the same in both cases. However, with the Gq protein, the α-subunit activates the enzyme phospholipase C, which catalyzes the cleavage of phosphatidylinositol bisphosphate on the membrane. Inositol trisphosphate and diacylglycerol is formed. Inositol trisphosphate diffuses away from the cell membrane and bind to the endoplasmic reticulum membrane. The calcium ion channels are opened and calcium ions enter the cytoplasm. Calcium ions are signaling molecules and ultimately stimulate release of vesicles and contraction of smooth-muscles. Diacylglycerol remains in the cell membrane, where it helps activates protein kinase C. Calcium ions are also needed to activate the protein kinase. Once activated, protein kinase C activates certain proteins by phosphorylation to produce physiological responses.
Calcium ion is a secondary messenger in many signaling processes because of several properties. The changes in calcium ions in the cell are easily detected. The calcium ion concentration inside cells is kept at a low level to avoid precipitation. Once calcium ions are released from the endoplasmic reticulum, the concentration of calcium ion in the cell increases by several orders of magnitude. This increase is readily felt by the cell. Calcium ions also bind tightly to proteins and induce a conformational change. Calcium ion can coordinate with several negative charged amino acid residues and thereby inducing a conformational change to activate proteins.
PLC is an type of enzyme that binds to the inositol phospholipids in eukaryotes by hydrolyzing lipid phosphatidylinositol 4,5-bisphosphate and creating inositol 1,4,5-trisphosphate and diacylglycerol (DAG). The importance of PLC is its ability to stimulate hosphoinositide metabolism and calcium signaling.
PLC is complex and are capable of covering a wide domain of protein. There are three subtypes of PLC: β, γ, and δ. Studies have shown that the DNA structure of δ were first found in single-celled eurkaryotes which are now similar to yeast, fungi, and mold. On the other hand, the β and γ were found to be more similar between plants and animals.
These enzymes are used for catalysis and since PLC have modular domains, they form catalytic α/β barrels from the X and Y regions. At the end of the barrel, there is catalytic and hydrophobic residue that allow substrates to come in and out of the mouth of the barrel. PLC hydrolyzes oxygen and phosphate bonds that contribute to binding phosphoinositol to DAG. This is done by the substrate forming the cyclic 1,2-phosphodiester intermediate and from here, catalysis begins.
PLC regulates cellular activity such as the binding of G protein subunits, the Rho and Ras of the GTPases, lipids, and tyrosine kinases. These enzymes have certain properties that allow them to regulate protein. Its structure was made to target PLC isozymes which led to the ability to solely control what the PLC does during protein-protein or protein-lipid interaction. During this process,
Serine/Threonine Kinase Transmembrane Receptors TGF beta RI and II
Serine/Threonine kinase's are enzymes that catalyze the addition of a phoaphate group to a serine or threonine (which have similar side-chains). Many of these receptors are vital to signal pathways that result in the alteration of gene expression. These receptors have two parts that are separated when they are not in contact with their extracellular. Once the signaling molecule complexes to the correct part of the receptor, a conformational change in this part of the receptor enables it to complex to the second separated part of the receptor. This form of the receptor-signal complex also activates the enzymatic activity of the cytoplasmic part of the receptor which results in a signaling cascade. Figure 1 shows these steps for the particular example of Serine/Threonine Kinase Transmembrane Receptors TGF beta RI and II.
One particular set of Serine/Threonine Kinase Transmembrane Receptors are TGF beta receptors I and II (TGF beta RI and II). The signal for these transmembrane receptors is Transformation Growth Factor beta (TGF beta), a cytokine that controls many numerous cellular responses like proliferation, differentiation, apoptosis and migration. In this case, before the signaling molecule can bind its receptor it needs to be activated. Activation of TGF beta is necessary since it is usually secreted from the source cells as an inactive complex that is referred to as the large latency complex (LLC) composed of TGF beta, latency associated peptide (LAP), and latent TGF beta binding protein (LTBP). When TGF beta is within this complex it is not able to bind to its receptors, TGF beta RI and II.
One mechanism by which TGF beta is released from the LLC involves the aid of integrins and the extracellular membrane (ECM) component fibronectin (Figure 2). In this mechanism the LTBP anchors to fibronectin in the ECM. Then, the LAP portion of the LLC anchors to an integrin. After this, a pulling force generated by a part of the cytoskeleton that is associated to the integrin protein results in a conformational change of the part of the LLC that is bound to the integrin. This results in TGF beta to be released from the LLC and will eventually find its receptors TGF beta RI and II.
There is a class of signal-transduction cascade that uses a receptor that intrinsically contains a protein kinase. One example of this type of signal-transduction cascade is insulin. The insulin receptor is composed to two identical chains connected by disulfide bonds. The receptor has an α-subunit on the extracellular side of the plasma membrane. This receptor extends across the membrane, where the β-subunit lies in the intracellular side. Insulin binds to its receptor by interacting with the α-subunit. With the two ‘arms’ made from identical chains, the α-subunit essentially wraps around insulin. The β-subunit on the cytoplasmic side primarily consists of a tyrosine kinase, which transfers a phosphoryl group from ATP to tyrosine residues. The tyrosine kinase is intrinsic to the receptor, and thus the insulin receptor is often referred to as the receptor tyrosine kinase.
The insulin receptor is activated when the α-subunit wraps around insulin. When the α-subunit closes around insulin, it causes the β-subunits in the intracellular side to come together. When the two ‘arms’ of the β-subunits come to close proximity, the intrinsic tyrosine kinase becomes active. The tyrosine residues on the β-subunit become phosphorylated, causing a dramatic conformational change on the intracellular end of the receptor. Phosphorylating tyrosine on the receptor also serve to generate docking sites for other substrates, such as insulin-receptor substrates (IRS). Upon docking, the tyrosine residues on IRS are phosphorylated by the receptor. In this form, IRS works as an adapter protein, where IRS binds to lipid kinases and moves them to the membrane. The lipid kinase phosphorylates phosphoinositol bisphosphate to generate phosphatidylinositol trisphosphate. This phosphorylated lipid then activates a protein kinase PDK1, which then also activates another protein kinase: Akt. All of the kinases mentioned above are either anchored to the receptor or the membrane, except for Akt. Akt can move across the cell and cause the movement of glucose transporter GLUT4 to the cell membrane. Once at the membrane, GLUT4 can transport glucose from the extracellular environment into the cell.
To terminate the signal, the activated receptor is returned to its deactivated state. Specifically, the phosphorylated tyrosine residues on the receptors need to be have the phosphoryl group removed. However, the phosphorylated residues are stable and do not spontaneously hydrolyze back to their original form. Specific enzymes are used to hydrolyze phosphorylated proteins and convert the protein back to their inactive form.
Bacteria and archea utilize a two-component system for signal transduction. These systems are absent in animals, and serve as an interesting source for developing antibacterials. The two component system mainly contains a membrane-bound sensor histidine kinase in addition to a response regulator that targets which genes the bacteria expresses in response to certain stimuli. Furthermore, they are linear signal transducers, modifying and amplifying transductions by adding extra modules and using extra proteins called connector proteins. Signal transduction occurs with the phosphorylation of a histidine kinase residue.
The first step in phosphorylation is histidine's autophosphorylation. The γ-phosphate is attacked by the exposed histidine in the DHp domain, and forces the CA domain to undergo different positions with respect to the histidine residue. Also, histidine phosphorylation is prevented with the binding of the RR to the ATP lid. This fixes the lid's position in between the nucleotide and histidine, preventing phosphorylation. In the entire histidine kinase, there are 2 mobile phosphorylating domains (otherwise known as CA domains) and 2 phosphoacceptors histidine residues. Evidence for both cis-autophosphorylation and trans-autophosphorylation have been seen, showing that this reaction and undergo both types. Current assumptions on determining cis or trans is based on the length of the hinge between the DHp and CA domains and the connection between the helices in the DHp domain. This knowledge can be obtained by studying phosphotransfer systems lacking autokinase activity, also called histidine phosphotransferases (HPts). Observations studying this system can transfer over to histidine autophosphorylation because the active center for phosphoryl transfer is very similar in both systems. Therefore, the data obtained from this system can be applied to any two-component system.
The phosphatate reaction is when the histidine kinase component catalyze the dephosphorylation of the P~RR, essentially the reserve of RR-phosphorylation. Phosphotransfer and phosphotase reactions occur in different complexes due to their opposite nature. If they were to react in the same complex, they essentially cancel each other out and render the signal ineffective. Not all histidine kinases can catalyze this dephosphorylation, and those that cannot rely on a protein to assume this role.
Histidine kinases have antiparallel transmembranes helices that aids in signaling from the membrane to the cytoplasm. Signal receptors on the membrane can trigger the helices into a combination of movements that relays the signal further deeper into the cell. Many complexes also have additional domains such as PAS and HAMP modules in between. For example, the HAMP module is composed of 4 helices in a bundle that contributes to the movement of signals by altering its structure in helical rotation and twisting.
There remains much to be learned about two component systems, such as determining a fine definition of what causes cis and trans autophosphorylation. The roles of many different proteins in their structure also remains a mystery. In order to reach these discoveries, more detailed imaging techniques needs to be developed to view the structures on an atomic level.
Components of signal transduction pathways have become connected hubs, which bind to specific partners depending on factors such as affinity. One of the hubs is Ras and it is affected by change (no matter the pace and size of the change) which allows information to be carried out. Ras is very complex in recognizing its structure and it makes it difficult to create a model of Ras joining to its effector. By understanding its model, we are to understand the biological what Ras does such as how it specific about binding domains of its effectors.
A sirtuin is a very conserved group of proteins that can increase the life span of simple organisms, and control the metabolic and stress pathways. In mammals there are seven sirtuins of which 3 are located in the mitochondria.
Mitochondria are very versatile organelles that function as the primary site of oxidative phosphorylation and plays a role in apoptosis and intracellular signaling. Mitochondria can modify their functions, morphology and cellular proliferation depending on extracellular conditions. The mitochondria have their own set of DNA which is referred to as mtDNA and codes for proteins that are involved in electron transport and ATP synthesis.
Sir 2 or Silent information regulator and its orthologs SIRTS 1-7 are called sirtuins. The seven in mammals have been conserved in the sirtuin domain in DNA. The sirtuins are used as regulatory proteins in the mitochondria that bind to NAD+ which is a co-factor in the for the proteins the sirtuins are bound to. The activity level of sirtuins is directly related to the increase in levels of NAD+
“Apoptosis is the cellular process of programmed cell death. Mitochondria play an important role in apoptosis by the activation of mitochondrial outer membrane permeabilization, which represents the irrevocable point of no return in committing a cell to death (article).” There is no clear line on how sirtuins controls apoptosis but it can seen that when a cell does not have SIRT 3 then there is less likely that the cell will be stress induced apoptosis, showing that sirtuins are important to apoptosis.
Worthington JJ, Klementowicz JE, Travis MA (2010). TGF?: a sleeping giant awoken by integrins. Trends in Biochemical Sciences (in press, September 2010).
Integrin proteins are receptor molecules that are adhesive to cells that mediate attachment between a cell and the tissues surrounding it, which include other cells or the extracellular matrix. Integrin proteins get their name from their ability to integrate the extracellular and intracellular environments by binding to ligands outside the cell and signaling molecules inside the cell. They also play important roles in the immune system, cell signaling, phagocytosis, cell migration, extracellular matrix assembly, and regulating the cell cycle. Integrin proteins are known to transmit information about how the cell is doing internally to and from the extracellular matrix. This allows the rapid response to changes in the external environment, which allows the maintenance of homeostasis. A good example of this is blood coagulation by platelets. Usually, receptors inform the cell of molecules near its environments so that the cell can initiate a response, integrin proteins perform this outside-in signaling but they can also perform inside-out signaling. Inside-out signaling is when they transduce information from the extracellular matrix to the cell and also reveals the status of the cell to the outside allowing quick responses to changes in environment. Integrins are usually found in an inactive state where they are not currently binding to receptors until needed. There are many types of integrin proteins, and many cells have multiple types on their surface. Integrin proteins can be found in just about any animal cell, and they have been exhaustively studied in humans especially.
Integrins are heterodimeric molecules that are associated noncovalently. 18 α subunits and 8 β subunits form 24 known αβ pairs in vertebrates. This diversity accounts for the diversity in ligand recognition (Figure 1), binding to cytoskeletal components and coupling to downstream signaling pathways. The β2 and β7 integrins are only expressed on leukocytes, whereas the β1 integrins are expressed on a large variety of cells throughout the body.
Priming, also known as inside-out signaling, dynamically regulates the adhesiveness of integrins by receiving stimuli from cell surface receptors which detect chemokines, cytokines, and foreign antigens which results in intracellular signals to be sent which will alter adhesiveness for extracellular ligands. Also, ligand binding transduces signals form the extracellular domain to the cytoplasm in the outside-in direction. These properties are the key to their proper function in the immune system.
Half of integrin α subunits include a region of 200 amino acids known as inserted (I) domains, or a von Willebrand factor A domain. The α I domain is the major ligand binding site in the integrins which contain this region. This domain was the first domain to be crystallized. The α I domain favors the dinucleotide binding with α-helices surrounding a central β-sheet. Beta strands and alpha helices usually alternate in secondary structure resulting in alpha helices wrapping around the domain in a counter clockwise order when viewed from the top face. The top face of the domain is defined by a divalent cation-biding site which physiologically binds Mg 2+. The Mg 2+ becomes ligated by 5 side chains located in three different loops. The first loop contains three coordinating residues in a signature sequence of I domains: Asp, Ser, Ser. The second loop donates a coordinating Thr residue and the third loop donates an Asp. This site is called the MIDAS (metal ion-dependent adhesion site).
I domains have been crystallized in three conformations: Closed, Intermediate, Open. These demonstrate the coordination of the metal in MIDAS, the arrangement of the beta six and alpha seven loops, and the axial repositioning of the C-terminal alpha seven helix along the side of the I domain. Oxygen is donated to the primary and secondary coordination spheres surrounding the metal via the five residues and several water molecules. In the open conformation of MIDAS, two serines and one threonine occupy the primary coordination sphere and two aspartic residues occupy the secondary coordination sphere. The glutamic acid residue donates only negatively charged oxygen to the primary coordination sphere in the open conformation. A hypothesis exists which states that the strength of the metal-ligand bond is enhanced because of the lack of any charged group in the primary coordination sphere that is donated by the I domain. The closed conformation of the alpha I domain results in the threonine from the primary coordination sphere to switch with the aspartic acid residue from the secondary coordination sphere. The backbone and side chain rearrangements are followed by a 2.3 angstrom sideways movement of the metal ion away from the threonine and closer to the aspartic acid which is on the opposite side of the coordination cell. The closed and open structures follow the idea that an energetically favorable MIDAS requires at least one coordination to a negatively charged oxygen. When there is no ligand, a pseudoligand, and the remainder of the integrin ectodomain crystallize in the closed conformation. This closed conformation is the low energy conformation, which is verified computationally. However, through an engineered disulfide bond, the alpha L I domain is crystallized in the open confirmation and is stable in the absence of ligand-mimetic lattice contact. As a result, interactions with other integrin domains might be capable of stabilizing an unliganded I domain in the open conformation and priming it for ligand binding.
Small allosteric inhibitors provide further support for the role of the alpha seven helix in alpha I domain regulation. Alpha I allosteric antagonists are a class of small molecule inhibitors which binds underneath the C-terminal α-helix of the αL I domain. These antagonists stabilize the closed conformation of the I domain by stopping the downward axial movement of the alpha seven helix and preventing the MIDAS rearrangements necessary for efficient ligand binding. The action of these antagonists is confirmed by finding that a mutant alpha L I domain which stabilizes the open conformation of the C-terminal alpha seven-helix with an engineered disulfide bond is resistant to inhibition by alpha I allosteric antagonists.
Chemokine in leukocytes, thrombin in platelets, and T-cell receptor engagement in T-cells are certain examples that cause integrin activation. Once activated, there is an increase in calcium and diacyl glycerol (DAG) levels which causes activation of a guanine nucleotide exchange factor (GEF). Rap1 is then activated, causing GDP/GTP exchange. Once Rap1 has been activated, it works together with RIAM, an interacting adapter molecule which helps connect the membrane targeting sequences in Rap1 to talin, and talin is then bound to the plasma membrane where it causes the β-TM helix to tilt, leading to full integrin activation. A complex formed by the β-tail, talin and the cell membrane is an important part of inside-out integrin activation in the cell, and thus the formation of this complex is regulated to ensure that integrin activation is under control. Β-tail and talin affinity is also controlled by environmental conditions, as well as competition from other proteins that compete for interaction with the β-integrin tail.
A certain type of proteins called kindlins also aid in talin activation of integrins. Kindlins contain FERM domains that are similar to talins with their N-terminal Fo domain and a large flexible F1 loop. Unlike talin which binds to the first NpxY motif in β-integrin tails, Kindlins bind to the second motif. However, Kindlin has also been known to be able to also inhibit integrins as well.
Structural Basis of Integrin Regulation and Signaling
Bing-Hao Luo, Christopher V. Carman, and Timothy A. Springer
Annu Rev Immunol. Author manuscript; available in PMC 2007 August 27.
PMCID: PMC1952532
Published in final edited form as: Annu Rev Immunol. 2007; 25: 619–647. doi: 10.1146/annurev.immunol.25.022106.141618.
Anthis, Nicholas J. "The Tail of Integrin Activation." Trends Biochem Sci. n. page. Web. 19 Nov. 2012.
Signal transduction is the chain of events that occurs when a cell converts a message to ultimately a physiological response. The message comes in the form of a particular molecule in the cell’s environment. Some examples of signal transduction are as follows. Upon release of epinephrine in response to stress, the cells in the body receive the message, then responds by preparing to use stored energy and improving cardiac function. After a meal, insulin is released into the bloodstream, indicating to cells to take in the glucose. In a wound, epidermal growth factor is released and simulates certain cells to proliferate.
Signal transduction is an important process since it occurs through the communication between difference domains and coupling of information. It is also important to know that ligand induced conformational changes are important to many aspects of protein function. Its importance is demonstrated when an enzyme binds a substrate because the act of binding causes changes in the protein’s structure to enable catalysis.
Signal transduction is an important process since it occurs through the communication between difference domains and coupling of information. It is also important to know that ligand induced conformational changes are important to many aspects of protein function. The importance of these conformational changes is demonstrated when an enzyme binds a substrate; the process of binding causes changes in the protein’s structure to enable catalysis. In addition, the binding of allosteric ligands can lead to the occurrence of more conformational changes as well.
Ligand binding often leads to changes in both the structure and dynamics with changes in the dynamics often occurring at different locations on the same proteins. The binding of a ligand to one site can often influence the structure, dynamics, and binding affinity at another site on the same protein. This demonstrates that coupling can often be achieved through many different ways. Many times, the effects of ligand binding on protein activity can be explained in terms of more common thermodynamic concepts; the fact that the transition of an active versus resting state depends on several factors such as the free energy difference between the two states, the protein affinity of the ligand, and the ability of a ligand to induce a transition once fully bound to the protein. In the case when a ligand binds to both an activated and resting conformation, incomplete activation often results. A ligand is considered to trigger activation when it binds with higher affinity to an active versus an inactive state.
The free energy difference, ΔGgap, can also be modified by environmental factors such as membrane composition. An example of this is demonstrated by the fact that the voltage dependent potassium channels exist in equilbrium only in an “on” or “off” conformations. These differ not only in their conductance characteristics but also in the number of charged groups on the two sides of the electrically impermeable membrane.
The K+ channels and M2 proton channels both share some similarities in their thermodynamic and structural coupling properties. For example, they are similar in the sense that both channels have dual gates to regulate the ion diffusion into the channel versus through the channel. In addition, they are able to achieve a high selectivity by binding multiple copies of permeablet ions.
The entry of ions through channels is also very tightly regulated to prevent leakage; leakage can be harmful to the life of the organism.
Signal transduction relays a message to a certain physiological response by way of certain key steps. In the first step, an event or condition stimulates the release of the signal molecule, otherwise known as the primary messenger. Generally, primary messengers do not enter cells, and thus work by binding to the cell’s membrane protein on the extracellular side. In the second step, the primary messenger binds to a receptor protein that spans across the membrane, causing conformational change. In the third step, the signal is relayed into the cell by way of conformational change on the receptor protein. This initiates a change in concentration of certain small molecules inside the cell. The small molecules are called second messengers, and they relay the signal inside the cell by activating other receptor-ligand complexes within the cell. In the fourth step, the secondary messengers activate the effectors that directly produce a physiological response. This physiological response can be activation/inhibition of enzymes, membrane channels, or gene-transcription factors. In the final step, after the physiological response is completed, the signal is terminated
Second messengers provide certain advantages for the signal transduction. A signal can be amplified significantly by generating second messengers. Small amounts of membrane receptors can be activated, but large amounts of second messengers can be generated. Each activated receptor can produce many secondary messengers. Low concentrations of primary messengers in the extracellular environment can give rise to a large signal due to amplification by secondary messengers. Also, second messengers are able to influence other processes in the cell by diffusing to other compartments. In addition, a common secondary messenger can signal for multiple pathways. This is called cross talk.
Epinephrine produces signals by binding as a ligand to a membrane protein called β-adrenergic receptor, which from a class of receptors called the seven-transmembrane-helix receptors. These receptors contain seven helices that cross the membrane seven times, thus also referred to as the serpentine receptors. The binding of a ligand on the extracellular side induces a conformational change on the seven-transmembrane-helix receptor on the cytoplasmic side. The conformational change on the intracellular side of the receptor activates G protein. Activated G proteins then binds to and promotes the activity of adenylate cyclase, which is a membrane bound enzyme that converts ATP to cAMP. cAMP can then move across the cell as the secondary messenger to initiate physiological response. cAMP activates protein kinase A (PKA). Activated PKA activates other proteins that directly produce a physiological response.
Unactivated G protein is bound to GDP and exists as a heterotrimer protein, consisting of the α-, β-, and γ- subunit. The GDP is bound to the α-subunit. To activate the G protein, the GDP is released and GTP binds to the α-subunit. Once GTP binds, the α-subunit dissociates from the βγ dimer. The activated α-subunit then binds and activates adenylate cyclase. The α-subunit has an intrinsic GTPase that slowly hydrolyzes the bound GTP to GDP. Once hydrolyzed to GDP, the α-subunit is deactivated and reassociates with the βγ dimer. The deactivation of G protein is a time dependent process, based on the kinetics of the intrinsic GTPase.
Aside from activating the cAMP cascade, the seven-transmembrane-helix receptor can also activate the phosphoinositide cascade. There are different types of G proteins. The β-adrenergic receptor functions with the Gs protein. The angiotensin II receptor activates the Gq protein. The mechanism of activating the G protein is the same in both cases. However, with the Gq protein, the α-subunit activates the enzyme phospholipase C, which catalyzes the cleavage of phosphatidylinositol bisphosphate on the membrane. Inositol trisphosphate and diacylglycerol is formed. Inositol trisphosphate diffuses away from the cell membrane and bind to the endoplasmic reticulum membrane. The calcium ion channels are opened and calcium ions enter the cytoplasm. Calcium ions are signaling molecules and ultimately stimulate release of vesicles and contraction of smooth-muscles. Diacylglycerol remains in the cell membrane, where it helps activates protein kinase C. Calcium ions are also needed to activate the protein kinase. Once activated, protein kinase C activates certain proteins by phosphorylation to produce physiological responses.
Calcium ion is a secondary messenger in many signaling processes because of several properties. The changes in calcium ions in the cell are easily detected. The calcium ion concentration inside cells is kept at a low level to avoid precipitation. Once calcium ions are released from the endoplasmic reticulum, the concentration of calcium ion in the cell increases by several orders of magnitude. This increase is readily felt by the cell. Calcium ions also bind tightly to proteins and induce a conformational change. Calcium ion can coordinate with several negative charged amino acid residues and thereby inducing a conformational change to activate proteins.
PLC is an type of enzyme that binds to the inositol phospholipids in eukaryotes by hydrolyzing lipid phosphatidylinositol 4,5-bisphosphate and creating inositol 1,4,5-trisphosphate and diacylglycerol (DAG). The importance of PLC is its ability to stimulate hosphoinositide metabolism and calcium signaling.
PLC is complex and are capable of covering a wide domain of protein. There are three subtypes of PLC: β, γ, and δ. Studies have shown that the DNA structure of δ were first found in single-celled eurkaryotes which are now similar to yeast, fungi, and mold. On the other hand, the β and γ were found to be more similar between plants and animals.
These enzymes are used for catalysis and since PLC have modular domains, they form catalytic α/β barrels from the X and Y regions. At the end of the barrel, there is catalytic and hydrophobic residue that allow substrates to come in and out of the mouth of the barrel. PLC hydrolyzes oxygen and phosphate bonds that contribute to binding phosphoinositol to DAG. This is done by the substrate forming the cyclic 1,2-phosphodiester intermediate and from here, catalysis begins.
PLC regulates cellular activity such as the binding of G protein subunits, the Rho and Ras of the GTPases, lipids, and tyrosine kinases. These enzymes have certain properties that allow them to regulate protein. Its structure was made to target PLC isozymes which led to the ability to solely control what the PLC does during protein-protein or protein-lipid interaction. During this process,
Serine/Threonine Kinase Transmembrane Receptors TGF beta RI and II
Serine/Threonine kinase's are enzymes that catalyze the addition of a phoaphate group to a serine or threonine (which have similar side-chains). Many of these receptors are vital to signal pathways that result in the alteration of gene expression. These receptors have two parts that are separated when they are not in contact with their extracellular. Once the signaling molecule complexes to the correct part of the receptor, a conformational change in this part of the receptor enables it to complex to the second separated part of the receptor. This form of the receptor-signal complex also activates the enzymatic activity of the cytoplasmic part of the receptor which results in a signaling cascade. Figure 1 shows these steps for the particular example of Serine/Threonine Kinase Transmembrane Receptors TGF beta RI and II.
One particular set of Serine/Threonine Kinase Transmembrane Receptors are TGF beta receptors I and II (TGF beta RI and II). The signal for these transmembrane receptors is Transformation Growth Factor beta (TGF beta), a cytokine that controls many numerous cellular responses like proliferation, differentiation, apoptosis and migration. In this case, before the signaling molecule can bind its receptor it needs to be activated. Activation of TGF beta is necessary since it is usually secreted from the source cells as an inactive complex that is referred to as the large latency complex (LLC) composed of TGF beta, latency associated peptide (LAP), and latent TGF beta binding protein (LTBP). When TGF beta is within this complex it is not able to bind to its receptors, TGF beta RI and II.
One mechanism by which TGF beta is released from the LLC involves the aid of integrins and the extracellular membrane (ECM) component fibronectin (Figure 2). In this mechanism the LTBP anchors to fibronectin in the ECM. Then, the LAP portion of the LLC anchors to an integrin. After this, a pulling force generated by a part of the cytoskeleton that is associated to the integrin protein results in a conformational change of the part of the LLC that is bound to the integrin. This results in TGF beta to be released from the LLC and will eventually find its receptors TGF beta RI and II.
There is a class of signal-transduction cascade that uses a receptor that intrinsically contains a protein kinase. One example of this type of signal-transduction cascade is insulin. The insulin receptor is composed to two identical chains connected by disulfide bonds. The receptor has an α-subunit on the extracellular side of the plasma membrane. This receptor extends across the membrane, where the β-subunit lies in the intracellular side. Insulin binds to its receptor by interacting with the α-subunit. With the two ‘arms’ made from identical chains, the α-subunit essentially wraps around insulin. The β-subunit on the cytoplasmic side primarily consists of a tyrosine kinase, which transfers a phosphoryl group from ATP to tyrosine residues. The tyrosine kinase is intrinsic to the receptor, and thus the insulin receptor is often referred to as the receptor tyrosine kinase.
The insulin receptor is activated when the α-subunit wraps around insulin. When the α-subunit closes around insulin, it causes the β-subunits in the intracellular side to come together. When the two ‘arms’ of the β-subunits come to close proximity, the intrinsic tyrosine kinase becomes active. The tyrosine residues on the β-subunit become phosphorylated, causing a dramatic conformational change on the intracellular end of the receptor. Phosphorylating tyrosine on the receptor also serve to generate docking sites for other substrates, such as insulin-receptor substrates (IRS). Upon docking, the tyrosine residues on IRS are phosphorylated by the receptor. In this form, IRS works as an adapter protein, where IRS binds to lipid kinases and moves them to the membrane. The lipid kinase phosphorylates phosphoinositol bisphosphate to generate phosphatidylinositol trisphosphate. This phosphorylated lipid then activates a protein kinase PDK1, which then also activates another protein kinase: Akt. All of the kinases mentioned above are either anchored to the receptor or the membrane, except for Akt. Akt can move across the cell and cause the movement of glucose transporter GLUT4 to the cell membrane. Once at the membrane, GLUT4 can transport glucose from the extracellular environment into the cell.
To terminate the signal, the activated receptor is returned to its deactivated state. Specifically, the phosphorylated tyrosine residues on the receptors need to be have the phosphoryl group removed. However, the phosphorylated residues are stable and do not spontaneously hydrolyze back to their original form. Specific enzymes are used to hydrolyze phosphorylated proteins and convert the protein back to their inactive form.
Bacteria and archea utilize a two-component system for signal transduction. These systems are absent in animals, and serve as an interesting source for developing antibacterials. The two component system mainly contains a membrane-bound sensor histidine kinase in addition to a response regulator that targets which genes the bacteria expresses in response to certain stimuli. Furthermore, they are linear signal transducers, modifying and amplifying transductions by adding extra modules and using extra proteins called connector proteins. Signal transduction occurs with the phosphorylation of a histidine kinase residue.
The first step in phosphorylation is histidine's autophosphorylation. The γ-phosphate is attacked by the exposed histidine in the DHp domain, and forces the CA domain to undergo different positions with respect to the histidine residue. Also, histidine phosphorylation is prevented with the binding of the RR to the ATP lid. This fixes the lid's position in between the nucleotide and histidine, preventing phosphorylation. In the entire histidine kinase, there are 2 mobile phosphorylating domains (otherwise known as CA domains) and 2 phosphoacceptors histidine residues. Evidence for both cis-autophosphorylation and trans-autophosphorylation have been seen, showing that this reaction and undergo both types. Current assumptions on determining cis or trans is based on the length of the hinge between the DHp and CA domains and the connection between the helices in the DHp domain. This knowledge can be obtained by studying phosphotransfer systems lacking autokinase activity, also called histidine phosphotransferases (HPts). Observations studying this system can transfer over to histidine autophosphorylation because the active center for phosphoryl transfer is very similar in both systems. Therefore, the data obtained from this system can be applied to any two-component system.
The phosphatate reaction is when the histidine kinase component catalyze the dephosphorylation of the P~RR, essentially the reserve of RR-phosphorylation. Phosphotransfer and phosphotase reactions occur in different complexes due to their opposite nature. If they were to react in the same complex, they essentially cancel each other out and render the signal ineffective. Not all histidine kinases can catalyze this dephosphorylation, and those that cannot rely on a protein to assume this role.
Histidine kinases have antiparallel transmembranes helices that aids in signaling from the membrane to the cytoplasm. Signal receptors on the membrane can trigger the helices into a combination of movements that relays the signal further deeper into the cell. Many complexes also have additional domains such as PAS and HAMP modules in between. For example, the HAMP module is composed of 4 helices in a bundle that contributes to the movement of signals by altering its structure in helical rotation and twisting.
There remains much to be learned about two component systems, such as determining a fine definition of what causes cis and trans autophosphorylation. The roles of many different proteins in their structure also remains a mystery. In order to reach these discoveries, more detailed imaging techniques needs to be developed to view the structures on an atomic level.
Components of signal transduction pathways have become connected hubs, which bind to specific partners depending on factors such as affinity. One of the hubs is Ras and it is affected by change (no matter the pace and size of the change) which allows information to be carried out. Ras is very complex in recognizing its structure and it makes it difficult to create a model of Ras joining to its effector. By understanding its model, we are to understand the biological what Ras does such as how it specific about binding domains of its effectors.
A sirtuin is a very conserved group of proteins that can increase the life span of simple organisms, and control the metabolic and stress pathways. In mammals there are seven sirtuins of which 3 are located in the mitochondria.
Mitochondria are very versatile organelles that function as the primary site of oxidative phosphorylation and plays a role in apoptosis and intracellular signaling. Mitochondria can modify their functions, morphology and cellular proliferation depending on extracellular conditions. The mitochondria have their own set of DNA which is referred to as mtDNA and codes for proteins that are involved in electron transport and ATP synthesis.
Sir 2 or Silent information regulator and its orthologs SIRTS 1-7 are called sirtuins. The seven in mammals have been conserved in the sirtuin domain in DNA. The sirtuins are used as regulatory proteins in the mitochondria that bind to NAD+ which is a co-factor in the for the proteins the sirtuins are bound to. The activity level of sirtuins is directly related to the increase in levels of NAD+
“Apoptosis is the cellular process of programmed cell death. Mitochondria play an important role in apoptosis by the activation of mitochondrial outer membrane permeabilization, which represents the irrevocable point of no return in committing a cell to death (article).” There is no clear line on how sirtuins controls apoptosis but it can seen that when a cell does not have SIRT 3 then there is less likely that the cell will be stress induced apoptosis, showing that sirtuins are important to apoptosis.
Worthington JJ, Klementowicz JE, Travis MA (2010). TGF?: a sleeping giant awoken by integrins. Trends in Biochemical Sciences (in press, September 2010).
β-linked N-acetylglucosamine is also known as O-GlcNAc. It is an intercellular carbohydrate that dynamically modifies proteins in the nucleolus and cytoplasm on the Serine and Threonine residues (Hart). The regulation of O-GlcNAc is dependent on only two enzymes OGT and O-GlcNacase (Hart).
The differences between O-GlcNAc and many forms of protein glycosylation are:
a).O-GlcNAcylation occurs only in the cytoplasm and nucleus
b). It is not an elongated structure
c). It is attached and removed several times in the life of a (Hart).
It was discovered in 1983 (Hart). It is present in all multicellular organisms, but not in yeast, for example, Saccharomyces cerevisiae (Hart).
O-GlcNAcylation shares more similarities to phosphoylation than other forms of protein glycosylation. The interplay between O-GlcNAc and O-phosphate is fact, that protein phosphatase 1 catalytic subunit (PP1c). The enzyme that removes O-phosphate also regulates OGT. This suggests that the enzyme can remove the phosphate group and attach the O-GlcNAc. The monosaccharide B-N-Acetyl-glucosamine (GlcNAc) attaches to serine/threonine residues via an O-linked glycosidic bond.
O-GlcNAcylation regulation has been proven to been an important factor in Alzheimer Disease patients as shown in the article done by Department of Neurochemistry, New York State Institute for Basic Research in Developmental Disabilities. (F.Liu, K.Iqbal, I.Grundke-Iqbal, G.W. Hart, and Cheng-Xin Gong) A microtubule associated protein, Tau, is "hyperphosphorylated and aggregated into a neurofibillary tangle in Alzheimer diseased brain".[29]The protein tau is modified by O-GlcNAcylation through process described in previous paragraph. The phosphoryl site-specificity of tau is both regulated in vitro and in vivo. Experiments were ran to show that the inverse affect that occurs when levels of O-GlcNAcylation occurs.
This experiment tested the affects of decreased glucose leading to decrease in O-GlcNAcylation. The mice starved for 48h as a result decreasing brain supply of glucose. The environment of the mouse was in a cage with prevention of coprophagy. There is a reduction in cellular concentration of UDP-Glucose consequently the protein of O-GlcNAcylation. In the experiment a I(125) Western blot is ran with anti-GlcNAcyl antibodies.[30]The tau phosphorylation increases suggesting an overall inverse regulation of tau phosphorylation by O-GlcNAcylation. The mouse's brain neurofilaments had an increased level of tau phosphorylation due to the decrease of tau O-GlcNAcylation in the brain. This is the result of deficient glucose uptake and utilization.
The catalyst that attaches O-GlcNAc from the UDP-GlcNAc substrate to either a Serine or Threonine residue forming a β-glycosidic linkage is the O-GlcNAc transferase (OGT) (Hart).
Mammals seem to have only one gene that catalysis the OGT. There is a dependent relationship between OGT and viability of the embryonic stem cells. In the absence O-GlcNacylation in the mammalian cells is lethal (Hart).
The complex regularion of OGT has not been clearly defined. But, it has been discovered that OGT is O-GlcNAcylated and phosphorylated (Hart).
1-2.^ Fei Liu*, Khalid Iqbal*, Inge Grundke-Iqbal*, Gerald W. Hart†, and Cheng-Xin Gong*‡ "O-GlcNAcylation regulates phosphorylation of tau: A mechanism involved in Alzheimer’s disease", *Department of Neurochemistry, New York State Institute for Basic Research in Developmental Disabilities; and †Department of Biological Chemistry, The Johns Hopkins University School of Medicine, 2004.
3.Hart, Gerald and Akimoto, Yoshihiro. Essentials of Glycobiology. 2009.
Protein acetylation is the process where an acetyl functional group is incorporated a protein. Protein acetylation is crucial in the processes of chromatin structure regulation and transcriptional activity.
t-Butyl Alcohol, a tertiary alcohol
Proteins are modified by fatty acid on the cysteine residue which induces a hydrophobic interactions. This is a very important post translation modification of proteins that diversifies proteins by enabling protein protein interactions, signal transduction and many other protein functions. One of the widely recognized lipid linked protein is G-protein1.
The pathway that proteins follow is now being clearly understood. The discovery of multi-drug resistance (MDR) genes has changed the ways cells can target proteins. Another significant discovery is that of small lipopeptied that have been found on the surface of some proteins. Lipid modifications of proteins are important in eukaryotic cells, by increasing flexibility and increasing protein specificity.
There are three covalent lipid modification of eukaryotic cells: Myristoylation, Palmitoylation and Isoprenylation
Many cellular peptides are covalently linked to a rare 14-carbon chain (acylation) on a amid bond of glycine. Since this modification was mostly observed in the cytosol, the hydrophobic function was not the only function in this modification. The N-myristoyl transferase was purified from yeast and mammals. Due to the solubility and catalysis after the removal of the interior Met can be achieved using myristoyl-coenzyme A. The acyl chain plays a more important role in the utility by an enzyme compared to the hydrophobic region 2.
In vivo the substitution of the methylene group with S or O therefore making it more hydrophilic. Therefore this changes the distribution of proteins in the membrane. This can be used to target drug delivery 2.
This involves a thioester bond on the cysteine residue and primary palmitric acid. However, the mechanism that regulates this process is poorly understood due to the difficulty of purifying labile (membrane-bound palmitoyl transferases). Another challenge is that Palmitoyl-CoA is an excellent acyl donor, but no protein sequence specifications need to be fulfilied. Yet, it has been observed that sites of interest are located close to the transmembrane region on the cytoplasm side. Many of the proteins that are located near the cytoplasm region are palmitoylated 2.
This is a triptlet modifications at the C terminal, depending on the primary sequence of motif CAAX (C, cysteine; A, aliphatic aminoacid; X, any amino acid). The first step is to add to isoprenoid lipid farnesol to cysteine residue which will add a 15th cardon. The second step is to protolytycally remove the AAX and carboxyl-methylation of the a-carboxyl 2.
GPI-anchored proteins were discovered from Trypanosoma Variant Surface Glycoprotein (VSG) and mammalian Thy-1 antigen. It has now been seen on the surface of many eukaryotic cells including humans and yeast. A test for this bond was discovered by low using bacterial phosphatidylinositol-specific phospholipases C (PI-PLC), which releases the GPI-anchored. The amid c-terminal is linked to ethanolamine which is further linked by a phosphodiester bond to tri-mannosyl glucosaminyl core glycan. On the 6th position of the inositol of the tri-mannosyl glucosaminyl core glycan a phosphatidylinositol (PI) is linked on the outer layer of the bi-layer. There are many possible variations in the glycerol group: alkyl or acyl and may also differ in chain length. Another structural difference is that the glycan can have extra sugars some examples are aGal, /JGalNAc or aMan, and/or ethanolamine phosphate. Due to these many structural differences therefore we have many different functions that these proteins can fulfill 2.
Only four methods have been identified to modify eukaryotic proteins. However, this doesn't mean that there are no other lipid modifications of proteins. Using metabolic labeling and SDS-PAGE to identify proteins that are regulated by lipids it was discovered that about 10-50 proteins were observed. This may lead to a conclusion that about 10-50% are modified by lipids. One of the major functions of lipid modification of protein is localization. Analyzing lipid modification of proteins we may better understand cell function 2.










































































 HSP 70
HSP 70
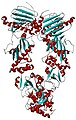 HSP 90
HSP 90

 Domain Swapping
Domain Swapping












 Neuro-oncological ventral antigen 1
Neuro-oncological ventral antigen 1 Muscleblind-like splicing regulator 1
Muscleblind-like splicing regulator 1






.jpg)
















































































 ||
||
.jpg)






















 This proton NMR example looks more complicated than the first one, but it is actually easier. One important information from this proton NMR is that there are peaks around 6.0~ 7.0 ppm (F,B,D,E). This always almost indicates the presence of aromatic ring. Noticed that there are about 4 group of peaks from 6.0 to 7.0 ppm? This means that there are four protons attach to 4 carbons on the aromatic ring. This leaves 2 carbons of the aromatic ring free to attach to two substituents. The position of these substituents is clearly an ortho and para position on the aromatic ring. The reason is that two different substituents on ortho and para position of the aromatic ring would give 4 proton environments. If there are only two proton environments around 6.0~ 8.0 ppm with the same integration (height), it means that the aromatic ring is probably symmetric on both sides with double para substituents. Now look at the peak (A) at 5.5 ppm. The peak is a broad peak instead of sharp peak like the rest. This means that the proton of this environment is attached to a heteroatom. The only heteroatom present in the sample base on the molecular formula given is an oxygen atom. This means that the proton is directly attached to oxygen, which would be a alcohol group -OH (Note that the proton on the alcohol group can have the chemical shift range from 0.5 to 5.0 ppm). It is important to remember that the proton that attached to a heteroatom is also known as exchangeable proton. Exchangeable proton sometimes do not show up on H NMR, so do not worry when an expected exchangeable peak doesn’t appear on HMNR.
The peak (C) at the chemical shift of ~3.9 ppm has the highest integration (height) and is a singlet. Usually when such a high singlet peak is observed around 3.9 ppm with oxygen in the molecular formula, there is a high chance that it is a ether group, -OCH3. Since there is no neighbor proton next to the CH3 of the ether, it is a singlet peak. In addition, the height of the peak is high compare to the other peak, which indicate a possibility of CH3 with at least 3 times more protons than other proton environment. Base on the interpretations above, these are the possible function groups:
This proton NMR example looks more complicated than the first one, but it is actually easier. One important information from this proton NMR is that there are peaks around 6.0~ 7.0 ppm (F,B,D,E). This always almost indicates the presence of aromatic ring. Noticed that there are about 4 group of peaks from 6.0 to 7.0 ppm? This means that there are four protons attach to 4 carbons on the aromatic ring. This leaves 2 carbons of the aromatic ring free to attach to two substituents. The position of these substituents is clearly an ortho and para position on the aromatic ring. The reason is that two different substituents on ortho and para position of the aromatic ring would give 4 proton environments. If there are only two proton environments around 6.0~ 8.0 ppm with the same integration (height), it means that the aromatic ring is probably symmetric on both sides with double para substituents. Now look at the peak (A) at 5.5 ppm. The peak is a broad peak instead of sharp peak like the rest. This means that the proton of this environment is attached to a heteroatom. The only heteroatom present in the sample base on the molecular formula given is an oxygen atom. This means that the proton is directly attached to oxygen, which would be a alcohol group -OH (Note that the proton on the alcohol group can have the chemical shift range from 0.5 to 5.0 ppm). It is important to remember that the proton that attached to a heteroatom is also known as exchangeable proton. Exchangeable proton sometimes do not show up on H NMR, so do not worry when an expected exchangeable peak doesn’t appear on HMNR.
The peak (C) at the chemical shift of ~3.9 ppm has the highest integration (height) and is a singlet. Usually when such a high singlet peak is observed around 3.9 ppm with oxygen in the molecular formula, there is a high chance that it is a ether group, -OCH3. Since there is no neighbor proton next to the CH3 of the ether, it is a singlet peak. In addition, the height of the peak is high compare to the other peak, which indicate a possibility of CH3 with at least 3 times more protons than other proton environment. Base on the interpretations above, these are the possible function groups:




























.jpg)




 Microscopic "holes" are characteristic in prion-affected tissue sections, causing the tissue to develop a "spongy" architecture.
Microscopic "holes" are characteristic in prion-affected tissue sections, causing the tissue to develop a "spongy" architecture.



















 Activation and inactivation mechanisms of PKA
Activation and inactivation mechanisms of PKA
 Crystal structure of Akt-1-inhibitor complexes
Crystal structure of Akt-1-inhibitor complexes
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
![[14]](http://philomath.exteen.com/images/11-20/18.jpg){kind=link}
![[15]](http://faculty.une.edu/com/courses/bionut/distbio/obj-512/Chap6-titration-lysine.gif){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}