Structural Biochemistry/Proteins/Protein Folding
Protein folding is a process in which a polypeptide folds into a specific, stable, functional, three-dimensional structure. It is the process by which a protein structure assumes its functional shape or conformation
Proteins are formed from long chains of amino acids; they exist in an array of different structures which often dictate their functions. Proteins follow energetically favorable pathways to form stable, orderly, structures; this is known as the proteins’ native structure. Most proteins can only perform their various functions when they are folded. The proteins’ folding pathway, or mechanism, is the typical sequence of structural changes the protein undergoes in order to reach its native structure. Protein folding takes place in a highly crowded, complex, molecular environment within the cell, and often requires the assistance of molecular chaperones, in order to avoid aggregation or misfolding. Proteins are comprised of amino acids with various types of side chains, which may be hydrophobic, hydrophilic, or electrically charged. The characteristics of these side chains affect what shape the protein will form because they will interact differently intramolecularly and with the surrounding environment, favoring certain conformations and structures over others. Scientists believe that the instructions for folding a protein are encoded in the sequence. Researchers and scientists can easily determine the sequence of a protein, but have not cracked the code that governs folding (Structures of Life 8).
Protein Folding Theory and Experiment
[edit | edit source]Early scientists who studied proteomics and its structure speculated that proteins had templates that resulted in their native conformations. This theory resulted in a search for how proteins fold to attain their complex structure. It is now well known that under physiological conditions, proteins normally spontaneously fold into their native conformations. As a result, a protein's primary structure is valuable since it determines the three-dimensional structure of a protein. Normally, most biological structures do not have the need for external templates to help with their formation and are thus called self-assembling.
Protein Renaturation
[edit | edit source]Protein renaturation known since the 1930s. However, it was not until 1957 when Christian Anfinsen performed an experiment on bovine pancreatic RNase A that protein renaturation was quantified. RNase A is a single chain protein consisting of 124 residues. In 8M urea solution of 2-mercaptoethanol, the RNase A is completely unfolded and has its four disulfide bonds cleaved through reduction. Through dialysis of urea and introducing the solution to O2 at pH 8, the enzymatically active protein is physically incapable of being recognized from RNase A. As a result, this experiment demonstrated that the protein spontaneously renatured.
One criteria for the renaturation of RNase A is for its four disulfide bonds to reform. The likelihood of one of the eight Cys residues from RNase A reforming a disulfide bond with its native residue compared to the other seven Cys residues is 1/7. Furthermore, the next one of remaining six Cys residues randomly forming the next disulfide bond is 1/5 and etc. As a result, the probability of RNase A reforming four native disulfide links at random is (1/7 * 1/5 * 1/3 * 1/1 = 1/105). The result of this probability demonstrates that forming the disulfide bonds from RNase A is not a random activity.
When RNase A is reoxidized utilizing 8M urea, allowing the disulfide bonds to reform when the polypeptide chain is a random coil, then RNase A will only be around 1 percent enzymatically active after urea is removed. However, by using 2-mercaptoethanol, the protein can be made fully active once again when disulfide bond interchange reactions occur and the protein is back to its native state. The native state of the RNase A is thermodynamically stable under physiological conditions, especially since a more stable protein that is more stable than that of the native state requires a larger activation barrier, and is kinetically inaccessible.
By using the enzyme protein disulfide isomerase (PDI), the time it takes for randomized RNase A is minimized to about 2 minutes. This enzyme helps facilitate the disulfide interchange reactions. In order for PDI to be active, its two active site Cys residues needs to be in the -SH form. Furthermore, PDI helps with random cleavage and the reformation of the disulfide bonds of the protein as it attain thermodynamically favorable conformations.
Posttranslationally Modified Proteins Might Not Renature
[edit | edit source]Proteins in a "scrambled" state go through PDI to renature, and their native state does not utilize PDI because native proteins are in their stable conformations. However, proteins that are posttranslationally modified need the disulfide bonds to stabilize their rather unstable native form. One example of this is insulin, a polypeptide hormone. This 51 residue polypeptide has two disulfide bonds that is inactivated by PDI. The following link is an image showing insulin with its two disulfide bonds. Through observation of this phenomenon, scientists were able to find that insulin is made from proinsulin, an 84-residue single chain. This link provides more information on the structure of proinsulin and its progression on becoming insulin. The disulfide bonds of proinsulin need to be intact before conversion of becoming insulin through proteolytic excision of its C chain which is an internal 33-residue segment. However, according to two findings, the C chain is not what dictates the folding of the A and B chains, but instead holds them together to allow formation of the disulfide bonds. For one, with the right renaturing conditions in place, scrambled insulin can become its native form with a 30% yield. This yield can be increased if the A and B chains are cross-linked. Secondly, through analysis of sequences of proinsulin from many species, mutations are permitted at the C chain eight times more than if it were for A and B chains.
Determinants of Protein Folding
[edit | edit source]There are various interactions that help stabilize structures of native proteins. Specifically, it is important to examine how the interactions that form protein structures are organized. In addition, there are only a small amount of possible polypeptide sequences that allow for a stable conformation. Therefore, it is evident that specific sequences are used through evolution in biological systems.
Helices and Sheets Predominate in Proteins because They Efficiently Fill Space
[edit | edit source]On average, about sixty percent of proteins contain a high amount of alpha helices, and beta pleated sheets. Through hydrophobic interactions, the protein is able to achieve compact nonpolar cores, but they lack the ability to specify which polypeptides to restrict in particular conformations. As seen in polypeptide segments in the coil form, the amount of hydrogen boding is not lesser than that of alpha helices and beta pleated sheets. This observation demonstrates that the different kinds of conformations of polypeptides are not limited by hydrogen bonding requirements. Ken Dill has suggested that helices and sheets occur as a result of the steric hindrance in condensed polymers. Through experimentation and simulation of conformations with simple flexible chains, it can be determined that the proportion of beta pleated sheets and alpha helices increase as the level of complication of chains is increased. Therefore, it can be concluded that helices and sheets are important in the complex structure of a protein, as they are compact in protein folding. The coupling of different forces such as hydrogen bonding, ion pairing, and van der Waals interactions further aids in the formation of alpha helices and beta sheets.
Protein Folding is Directed by Internal Residues
[edit | edit source]By investigating protein modification, the role of different classes of amino acid residues in protein folding can be determined. For example, in a particular study the free primary amino groups of RNase A were derivatized with poly-DL-alanine which consist of 8 residue chains. The poly-Ala chains are large in size and are water-soluble, thus allowing the RNase's 11 free amino groups to be joined without interference of the native structure of the protein or its ability to refold. As a result, it can be concluded that the protein's internal residues facilitates its native conformation because the RNase A free amino groups are localized on the exterior. Furthermore, studies have shown that mutations that occur on the surface of residues are common, and less likely to change the protein conformation compared to changes of internal residues that occur. This finding suggests that protein folding is mainly due to the hydrophobic forces.
Protein Structures Are Hierarchically Organized
[edit | edit source]George Rose demonstrated that protein domains consisted of subdomains, and furthermore have sub-subdomains, and etc. As a result, it is evident that large proteins have domains that are continuous, compact, and physically separable. When a polypeptide segment within a native protein is visualized as a string with many tangles, a plane can be seen when the string is cut into two segments. This process can be repeated when n/2 residues of an n-residue domain is highlighted with a blue and red color. As this process is repeated it can be seen that at all stages, the red and blue areas of the protein do not interpenetrate with one another. The following link shows an X-ray structure of HiPIP (high potential iron protein) and its first n/2 residues on the n-residue protein colored red and blue. Furthermore, the subsequent structures shown in the second and third row show this process of n/2 residue splitting reiterated as shown where the left side of the protein has its first and last halves with red and blue while the rest of the chain colored in gray. Through this example, it is clearly seen that protein structures are organized in a hierarchical way, meaning that the polypeptide chains are seen as sub-domains that are themselves compact structures and interact with adjacent structures. These interactions forms a larger well organized structure largely due to hydrogen bonding interactions and has an important role in understanding how polypeptides fold to form their native structure.
Protein Structures are Adaptable
[edit | edit source]Since the side chains inside globular proteins fit together with much complementary its packing density can be almost like that of organic crystals. As a result, in order to confirm whether or not this phenomenon of high packing density was an important factor in contributing to protein structure, Eaton Lattman along with George Rose attempted to verify if there was an interaction between side chains that was preferred in a globular protein. They analyzed a total of 67 well studied structures of globular proteins, and concluded that there were no preferred interactions. This experiment demonstrated that packing is not what directs the native fold, but instead the native fold is necessary for packing of a globular protein. This notion can be further supported as members of a protein family result in the same fold despite their lack of sequence similarity and distant relationships.
In addition, structural experimental data have shown that there are a variety of ways that a protein's internal residues can become compact together in an efficient manner. In an extensive study done by Brian Matthews based on T4 lysozyme, which is produced by bacteriophage T4, it was found that changes in the residues of the T4 lysozyme only affected local shifts and did not result in any global structure change. The following link gives an X-ray view of T4 lysozyme and a brief biochemical description of the structure. Matthews took over 300 different mutants of the 164 residue T4 lysozyme, and compared them with one another. Also, it was observed that the T4 lysozyme could withstand insertions of about 4 residues while still not having any major structural changes to the overall protein structure nor enzyme activity. Furthermore, by using assay techniques it was demonstrated that only 173 of the mutants in T4 of the 2015 single residue substitutions done had significant amounts of enzymatic activity diminished. Through these experiments, it is evident that protein structures are extremely withstanding.
The Levinthal Paradox
[edit | edit source]Levinthal's paradox is a thought experiment, also constituting a self-reference in the theory of protein folding. In 1969, Cyrus Levinthal noted that, because of the very large number of degrees of freedom in an unfolded polypeptide chain, the molecule has an astronomical number of possible conformations. An estimate of 3300 or 10143 was made in one of his papers.
The Levinthal paradox observes that if a protein were folded by sequentially sampling of all possible conformations, it would take an large amount of time to do so, even if the conformations were sampled at a rapid rate . Based upon the observation that proteins fold much faster than this, Levinthal then proposed that a random conformational search does not occur, and the protein must, therefore, fold through a series of meta-stable intermediate states.
In 1969 Cyrus Levinthal calculated that if a protein were to randomly sample every possible conformation as it folded from the unfolded state to the native state it would take an astronomical amount of time, even if the protein reached 100 billion conformations in one second. Observing that proteins fold in a relatively short amount of time, Levinthal proposed that proteins fold in a fixed and directed process. We now know that while protein folding is not a random process there does not seem to be a single fixed protein folding pathway.This observation came to be known as the Levinthal paradox. This paradox clearly reveals that proteins do not fold by trying every possible conformation. Instead, they must follow at least a partly defined folding pathway made up of intermediates between the fully denatured proteins and its native structure.
Cumulative Selection
[edit | edit source]The way out of the Levinthal Paradox is to recognize cumulative selection. According to Richard Dawkins, he asked how long it would take a monkey poking randomly at a typewriter to reproduce "Methinks it is like a weasel", Hamlet's remark to Polonius. A large number of keystrokes, of the order of 1040 would be required. Yet if we suppose that each correct character was preserved, allowing the monkey to retype only the wrong ones, only a few thousand keystrokes, on average, would be needed. The crucial difference between these scenarios is that the first utilizes a completely random search whereas in the second case, partly correct intermediates are retained. This also reveals that the essence of protein folding is the tendency to retain partly correct intermediates, although the protein-folding problem is much more difficult than the one presented to Shakespeare example above.
Nucleation-Condensation model
[edit | edit source]In order to correctly understand the protein-folding problem, we must consider certain characteristics of protein. Since proteins are only marginally stable, the free-energy difference between the folded and the unfolded states of a typical 1000-residue protein is 42 kJ mol−1 and thus each residue contributes on average only 0.42 kJ mol−1 of energy to maintain the folded state. This amount is less than the amount of thermal energy, which is 2.5 kJ mol−1 at room temperature. This meagre stabilization energy means that correct intermediates, especially those formed early in folding, can be lost. The interactions that lead to cooperative folding, nonetheless, can stabilize intermediates as structure builds up. Thus, local regions that have significant structural preference, though not necessarily stable on their own, will tend to adopt their favored structures and, as they form, can interact with one other, resulting in increased stabilization. Nucleation-condensation model refers to this conceptual framework in solving the protein-folding challenge.
Intramolecular Interactions Role in the Folding Mechanism
[edit | edit source]Proteins folding forms energetically favorable structures stabilized by hydrophobic interactions clumping, hydrogen bonding and Van der Waals forces between amino acids. Protein folding first forms secondary structures, such as alpha helices, beta sheets, and loops. Different amino acids have different tendencies for whether they are going to form Alpha Helices, Beta sheets, or Beta Turns based upon polarity of the amino acid and rotational barriers. For example, the amino acids, valine, threonine, isoleucine, tend to destabilize the alpha helices due to steric hindrance. Thus, they prefer conformational shifts towards Beta sheets rather than alpha helices. The relative frequencies of the amino acids in secondary structures are grouped according to their preferences for alpha helices, beta sheets or turns (Table 1). Table 1: Relative frequencies of amino acid residues in secondary structures These structures in turn, fold to form tertiary structures, stabilized by the formation of intramolecular hydrogen bonds. Covalent bonding may also occur during the folding to a tertiary structure, through the formation of disulfide bridges or metal clusters. According to Robert Pain’s “Mechanisms of Protein Folding”, molecules also often pass through an intermediate “molten globule” state formed from a hydrophobic collapse (in which all hydrophobic side-chains suddenly slide inside the protein or clump together) before reaching their native confirmation. However, this means all the main chain NH and CO groups are buried in a non-polar environment, but they prefer an aqueous one, so secondary structures must fit together very well, so that the stabilization through hydrogen bonding and Van der Waals forces interactions overrides their hydrophilic tendencies. The strengths of hydrogen bonds in a protein vary depending on their position in the structure; H-bonds formed in the hydrophobic core contribute more to the stability of the native state than H-bonds exposed to the aqueous environment.
Water-soluble proteins fold into compact structures with non-polar, hydrophobic cores. The inside of protein contains non-polar residues in center (i.e. - leucine, valine, methionine and phenylalanine), while the outside contains primarily polar, charged residues (i.e. - aspartate, glutamate, lysine and arginine). This way the polar, charged molecules can interact with the surrounding water molecules while the hydrophobic molecules are protected from the aqueous surroundings. Minimizing the number of hydrophobic side chains on the outer part of the structure makes the protein structure thermodynamically more favorable because the hydrophobic molecules prefer to be clumped together, when surrounded by an aqueous environment (i.e. – hydrophobic effect). Proteins that span biological membranes (i.e. - porin) have an inside out distribution, with respect to the water-soluble native structure, they have hydrophobic residue covered outer surfaces, with water filled centers lined with charged and polar amino acids.
Folding of Membrane Proteins
[edit | edit source]In “Folding Scene Investigation: Membrane Proteins”, a paper written by Paula J Booth and Paul Curnow, the authors attempt to answer how the folding mechanisms of integral membrane proteins with α helical structures work. Studying the folding of membrane proteins has always been difficult as these proteins are generally large and made of more than one subunit. The proteins posses a high degree of conformational flexibility—which is necessary for them to perform their function in the cell. Also, these proteins have both hydrophobic surfaces, facing the membrane, and hydrophilic surfaces, facing the aqueous regions on either side of the membrane. The proteins are move laterally and share the elastic properties of the lipid bilayer in which they are embedded. In order to study these proteins, Booth and Curnow believe that one must manipulate the lipid bilayer and combine kinetic and thermodynamic methods of investigation.
Reversible Folding and Linear Free Energy The free energy of protein folding is measured by reversible chemical denaturation. The reversible folding of a protein depends on this free energy. For the α helix proteins that were being studied, it was proven that a reversible, two-state process is followed. bR (a α helical membrane protein called bacteriorhodopsin) reversibly unfolds if SDS (a denaturant which is an anionic detergent) is added to mixed lipid, detergent micells. The two-state reaction involves a partly unfolded SDS state and a folded bR state. By comparing the logs of the unfolding and folding rate, and the SDS mole fraction, a linear plot was generated proving a linear relationship. This plot proved that bR had a very high stability outside of its membrane—proving that it was unexpectedly stable. Furthermore, bR was so stable outside of the membrane that it would not unfold during a reasonable period of time without addition of denaturant.
Comparison with Water-Soluble Proteins Booth and Curnow studied the 3 membrane proteins about which the most information is held: bR, DGK (Escherichia coli diacylglycerol kinase) and KcsA (Sterptococcus lividans potassium channel). These three membrane proteins were compared to water-soluble proteins (which fold by 2 or 3 state kinetics). The overall free energy change of unfolding in the absence of denaturant was the same for water-soluble proteins and membrane proteins of similar size. This proves that it is the balance of weak forces rather than the types of forces that stabilize the protein that determines its stability. It was proven that H-bonds in the membrane proteins were of similar strength to those of the water-soluble proteins, rather than being stronger in membrane proteins as was expected.
Mechanical Strength and Unfolding Under Applied Force Dynamic force microscopy can be used to measure the mechanical response of a particular region of a protein under applied force. The unfolding force in this case depends on the activation barrier. This unfolding has nothing to do with the thermodynamic stability of a protein. For unfolding under applied force, the membrane proteins (especially bR) seem to follow the rules of Hammond behavior. The energy difference between two consecutive states of this reaction is reduced and the states become similar in structure.
Influence of Surrounding Membrane Membrane proteins are influenced greatly by the membranes they are surrounded by. If the lipids incorporate in detergent micells—-increasing the stability of the lipid structure—both the protein and its folding are stabilized. Different combinations of different lipids can result in different stabilities or folding of membrane proteins. The size of the membrane can also affect the membrane protein. Different types of lipids cause different membrane properties. A type of lipids called PE lipids have higher spontaneous curvatures than a second type of lipid called a PC lipid. By adding PE lipids to PC lipids the monolayer curvature of the bilayer increases. Increasing the curvature of the lipid bilayer increases the stability of the protein folding.
Protein translocation in biological membranes
[edit | edit source]In mitochondria, the proteins that are made from the ribosomes are directly take in from the cytosol. Mitochondrial proteins are first completely synthesized in the cytosol as mitochondrial precursor proteins, then taken up into the membrane. The Mitochondrial proteins contain specific signal sequence at their N terminus. These signal sequences are often removed after entering the membrane but proteins entering membranes that has outer, inner, inter membrane have internal sequences that play a major movement in the translocation within the inner membrane.
Protein translocation plays a major role in translocating proteins across the mitochondrial membranes. Four major multi-subunit protein complexes are found in the outer and the inner membrane. TOM complexes are found in the outer membrane, and two types of TIM complexes are found integrated within the inner membrane: TIM23 and TIM22. The complexes act as receptors for the mitochondrial precursor proteins.
TOM: imports all nucleus encoded proteins. It primarily starts the transport of the signal sequence into the inter membrane space and inserts the transmembrane proteins into outer membrane space. A Beta barrel complex called the SAM complex is then in charge of properly folding the protein in the outer membrane. TIM23 found in the inner membrane moderates the insertion of soluble proteins into the matrix, and facilitates the insertion of transmembrane proteins into the inner membrane. TIM23, another inner membrane complex facilitates the insertion inner membrane proteins comprised of transporters that move ADP, ATP, and phosphate across the mitochondrial membranes. OXA, yet another inner membrane complex, helps insert inner membrane proteins that were synthesized from the mitochondria itself and the insertion of inner membrane proteins that were first transported into the matrix space. File:Translocation.jpg
Folding on Ribosome
[edit | edit source]The place where the protein chain begins to fold is a topic that is greatly studied. As the nascent chain goes through the “exit tunnel” of the ribosome and into the cellular environment, when does the chain begin to fold? The idea of cotranslational folding in the ribosomal tunnel will be discussed. The nascent chain of the protein is bound to the peptidyl transferase centre (PTC) at its C terminus and will emerge in a vectorial manner. The tunnel is very narrow and enforces a certain rigidity on the nascent chain, with the addition of each amino acid the conformational space of the protein increases. Co translational folding can be a big help in reducing the possible conformational space by helping the protein to acquire a significant level of native state while still in the ribosomal tunnel. The length of the protein can also give a good estimate of its three dimensional structure. Smaller chains tend to favor beta sheets while longer chains (like those reaching 119 out of 153 residues) tend to favor the alpha helix.
The ribosomal tunnel is more than 80 Å in length and its width is around 10-20 Å. Inside the tunnel are auxiliary molecules like the L23, L22, and L4 proteins that interact with the nascent chain help with the folding. The tunnel also has hydrophilic character and helps the nascent chain to travel through it without being hindered. Although rigid, the tunnel is not passive conduit but whether or not it has the ability to promote protein folding is unknown. A recent experiment involving cryoEM has shown that there are folding zones in the tunnel. At the exit port (some 80 Å from the PTC), the nascent chain has assumed a preferred low order conformation. This enforces the suggestion that the chain can have degrees of folding at certain regions. Although some low order folding can occur, the adoption of the native state occurs outside the tunnel, but not necessarily when the nascent chain has been released. The bound nascent chain (RNC) adopts partially folded structure and in a crowded cellular environment, this can cause the chain to self-associate. This self-association, however, is relieved with the staggered ribosomes lined along the exit tunnel that maximizes the distances between the RNC.
Generation of RNC for studies:
One technique of generating RNC and taking snapshots as it emerges from the tunnel is to arrest translation. A truncated DNA without a termination sequence is used. This allows for the nascent chain to remain bound until desired. To determining the residues of the chain, they can be labeled by carbon-13 or nitrogen-15 and later detected by NMR spectroscopy. Another technique is the PURE method and it contains the minimal components required for translation. This method has been used to study the interaction of the chains and auxiliary molecules like the TF chaperone. This method is coupled with quartz-crystal microbalance technique to analyze the synthesis by mass. An in vivo technique in generating RNC chain can be done by stimulating it in a high cell density. This is initially done in an unlabeled environment, the cells are then transferred to a labeled medium. The RNC is generated by SecM. The RNC is purified by affinity chromatography and detected by SDS-PAGE or immunoblotting.
By generating the RNCs, many experiments can be done to study more about the emerging nascent chain. As mentioned above, the chain emerges from the exit tunnel in a vectorial manner. This enables the chain to sample the native folding and increases the probability of folding to the native state. Along with this vectorial folding, chaperones also help in favorable folding rates and correct folding.
Protein Folding in the Endoplasmic Reticulum
[edit | edit source]Protein Entering the Mammalian ER: The endoplasmic reticulum (ER) is a main checkpoint for protein maturation to ensure that only correctly folded proteins are secreted and delivered to the site of action. The protein entrance to the ER begins with recognition of a N’ terminus signal sequence. Specially, this sequence is detected by a signal recognition protein (SRP) causing the ribosome/nascent chain/SRP complex bind to the ER membrane. Then, the complex travels through a proteinaceous pore called Sec61 translocon which allows the polypeptide chain enter the lumen portion of the ER.
Processes in Conflict During Protein Folding: After the protein enters the ER, the proteins break up into an ensemble of folding intermediates. These intermediates take three different routes. They are either folded properly and sent to be exported out of the endoplasmic reticulum (ER) into the cytosol, aggregated or picked out for degradation. These three processes are in competition to properly secrete a protein. In order for a protein to be properly secreted, the competition between folding, aggregation and degradation must be in favor of folding, so that folding occurs faster than the other processes. This balance is termed proteostasis. The balance of proteostasis can be tipped in favor of folding by either using smaller molecules to stabilize the protein (called co-factors) or increasing the concentrations of folding factors. This ability to control proteostasis allows scientists the power to overcome some of the protein folding diseases such as cystic fibrosis.
The proteins that are folded properly are ready for anterograde transport, and secreted through the membrane of the ER into the cytosol by a cargo receptor that recognizes the properly folded protein. The proteins that are incorrectly folded are not secreted and are either targeted for degradation or aggregated. The aggregated proteins are able to re-enter the stage of protein ensembles ready to be folded so that they may try again at being folded properly.

Folding Factors in the Endoplasmic Reticulum:
Biochemical research on folding pathways has provided a comprehensive list of folding factors, or chaperones, involved with protein folding in the ER. Folding factors are categorized based on whether they catalyze certain steps or if they interact with intermediates in the folding pathway. General protein folding factors are typically separated into four different groups: heat shock proteins as chaperones or cochaperones, peptidyl prolyl cis/trans isomerases (PPIases), oxidoreductases, and glycan-binding proteins.
Many folding factors are great in that they are multi-functional. One folding factor can take care of different areas of the folding pathway. Unfortunately, this leads to redundancy due to different classes of proteins carrying out overlapping functions. This functional redundancy complicates the understanding of the specific roles of individual folding factors in aiding maturation of client proteins. Folding factors also prefer to act in concert during the maturation process, which further obscures the individual roles of each factor. Since these roles are not clear, it is difficult to confirm that even if one folding factor deals with a particular reaction in one protein, that same folding factor will carry out the same function in another.
In addition to aiding non-covalent folding and unfolding of proteins, folding factors in the ER sometimes delay interactions with the protein. This allows time for nascent proteins to fold properly and enables folded proteins to backtrack on its folding pathway, which prolongs equilibrium in a less folded state, preventing the protein from being held in a non-native state.
Folding after Endoplasmic Reticulum: Although ER provides only correctly assembled proteins to be secreted, some examples exist in which proteins change conformation in the Golgi bodies and beyond. Typically, newly folded proteins are sensitive and prone to unfolding while in the ER but resistant to unfolding after exit. In an environment without chaperones and other folding enzymes, proteins are compact and relatively resistant to change after exiting the ER. However, this doesn’t necessarily mean that protein folding ends because some molecular chaperones like Hsp 70s and Hsp 90s continue to assist in protein conformation throughout the protein’s existence.

Techniques for Studying Protein Folding
[edit | edit source]A strategy for studying the folding of proteins is to unfold the protein molecules in high concentrations of a chemical denaturant like guanidinium chloride. The solution is then diluted rapidly until the denaturant concentration is lowered to a level where the native state is thermodynamically stable again. Afterwards, the structural changes of the protein folds may be observed. In theory, this sounds simple. However, such experiments are complex, since unfolded proteins have random coil states in chemical denaturants. Moreover, analyzing the structural changes taking place in a sample may is difficult, since all of the molecules may have significantly different conformations until the final stages of a reaction. As such, the analysis would have to be performed in a matter of seconds rather than days or weeks that are normally allowed to deduce the structure of a single conformation of a native protein. To avoid this problem, the disulphide bonds can be reduced after the protein is unfolded and reformed under oxidative conditions. The protein can then be identified by standard techniques such as mass spectroscopy to draw conclusions about the structure present at stages of folding where disulfide bonds are formed.
Multiple techniques are used to monitor structural changes during the refolding. For instance, in circular dichorism, UV is used from far away to provide a measurement of the appearance of the secondary structure during folding. UV at a close distance monitors the formation of the close-packed environment for aromatic residues. NMR is also a useful technique for characterizing conformations at the level of individual amino-acid residues. It can also be used to monitor how the development of structures protect amide hydrogens from solvent exchanges.
Circular Dichroism: This type of spectroscopy measures the absorption of circularly polarized light since the structures of protein such as the alpha helix and beta sheets are chiral and can absorb this sort of light. The absorption of light indicates the degree of the protein’s foldedness. This technique also measures equilibrium unfolding of protein by measuring change of absorption against denaturant concentration or temperature. The denaturant melt measures the free energy of unfolding while the temperature melt measures the melting point of proteins. This technique is the most general and basic strategy for studying protein folding.
Dual Polarization Interferometry: This technique uses an evanescent wave of a laser beam confined to a waveguide to probe protein layers that have been absorbed to the surface of the waveguide. Laser light is focused on two waveguides, one that senses the beam and has an exposed surface, and one that is used to create a reference beam and to excite the polarization modes of the waveguides. The measurement of the interferogram can help calculate the protein density or fold, the size of the absorbed layer, and to infer structural information about molecular interactions at the subatomic resolution. A two-dimensional pattern is obtained in the far field when the light that has passed through the two waveguides is combined.
Mass Spectrometry: The advantages of using Mass Spectroscopy to study protein folding include the ability to detect molecules with different amounts of deuterium, which allows the heterogeneity of the protein folding reactions to be studied. It can also measure the conformation of folding intermediates bound to molecular chaperones without disrupting the complex. Mass spectrometry can also directly compare refolding properties, since mixtures of proteins can be studied without separation if the two proteins have sufficiently different molecular weights.
High Time Resolution: These are fast time-resolved techniques where a sample of unfolded protein is triggered to fold rapidly. The resulting dynamics are then studied. Ways to accomplish this include fast mixing of solutions, photochemical methods, and laser temperature jump spectroscopy.
Computational Prediction of Protein Tertiary Structure: This is a distinct form of protein structure analysis in that it involves protein folding. These programs can simulate the lengthy folding processes, provide information on statistical potential, and reproduce folding pathways.
Protein Misfolding
[edit | edit source]Protein misfolding refers to the failure of a protein to achieve its tightly packed native conformation efficiently or the failure to maintain that conformation due to reduction in stability as a result of environmental change or mutation. It has been established that failure of protein folding is a general phenomenon at elevated temperatures and under other stressful circumstances. The two most common results of misfolded proteins are degradation and aggregation. When a polypeptide emerges from the cell, it may fold to the native state, degraded by proteolysis, or form aggregates with other molecules. Proteins are in constant dynamic equilibrium so even if the folding process is complete, unfolding in the cellular environment can occur. Unfolded proteins usually refold back into their native states but if control processes fail, misfolding leads to cellular malfunctioning and consequently diseases. Diseases associated with misfolding cover a wide array of pathological conditions such as cystic fibrosis where mutations in the gene encoding the results in a folding to a conformer whose secretion is prevented by quality-control mechanisms in the cell. About 50% of cancers are associated with mutations of the p53 protein that eventually lead to the loss of cell-cycle control and causing the growth of tumors. Failure of proteins to stay folded can result in aggregation, a common characteristic of a group of genetic, sporadic, and infectious conditions known as amyloidoses. Aggregation usually results in disordered species that can be degraded within the organism but it may also result in highly insoluble fibrils that accumulate in tissue. There are about twenty known diseases resulting from the formation of amyloid material including Alzheimer’s, Type II diabetes, and Parkinson’s disease. Amyloid fibrils are ordered protein aggregates that have an extensive beta sheet structure due to intermolecular hydrogen bonds and have an overall similar appearance to the proteins they are derived from. The formation of the amyloid fibrils are the result of prolonged exposure to at least partially denatured conditions.
Alzheimer's: This neurological degeneration is caused by the accumulation of Plaques and Tangles in the nerve cells of the brain.[1] Plaques, composed of almost entirely a single protein, are aggregation of the protein beta-amyloid between the spaces of the nerve cells and Tangles are aggregation of the protein tau inside the nerve cells. Tangles are common in extensive nerve cell diseases whereas neuritic plaque is more specific to Alzheimer's. Although scientists are unsure what role Plaques and Tangles play in the formation of Alzheimer's, one theory is that these accumulated proteins impede the nerve cell's ability to communicate with each other and makes it difficult for them to survive. Studies have shown that Plaques and Tangles naturally occur as people age, but more formation is observed in people with Alzheimer's. The reasons for this increase is still unknown.
Creutzfeldt-Jakob Disease (Mad Cow Disease): This disease is caused by abnormal proteins called prions which eat away and form hole-like lesions in the brain. Prions (proteinaceous infectious virion) were discovered to be proteins with an altered conformation. Scientists hypothesize that these infectious agents could bind to other similar proteins and induce a change in their conformation as well, propagating new, infectious proteins.[2] Prions are highly resistant to heat, ultraviolet light, and radiation which makes them difficult to be eliminated. In Creutzfeldt-Jakob Disease there is an incubation period for years which is then followed by rapid progression of depression, difficulty walking, dementia and death. Currently there is no effective treatment for prion diseases and all are fatal.[3]
Parkinson's disease:A mutation in the gene which codes for alpha-synuclein is the cause of some rare cases of familial forms of Parkinson's disease. Three point mutations have been identified thus far: A53T, A30P and E46K. Also, duplication and triplication of the gene may be the cause of other lineages of Parkinson's disease.Victims of Parkinson's disease have primary symptoms that result from decreased stimulation of the motor cortex by the basal ganglia, normally caused by the insufficient formation and action of dopamine. Dopamines are produced in the dopaminergic neurons of the brain. People who suffer from this disease have brain cell loss (death of dopaminergic neurons), which may be caused by abnormal accumulation of the protein alpha-synucleinbinding to ubiquitin in the damaged cells. This makes the alpha-synuclein-ubiquitin complex unable to be directed to the proteosome. New research shows that the mistransportation of proteins between endoplasmic reticulum and the Golgi apparatus might be the cause of losing dopaminergic neurons by alpha-synuclein.
Cystic Fibrosis: Francis Collins first identified the hereditary genetic mutation in 1989. The problem occurs in the regulator cystic fibrosis transmembrane conductance regulator (CFTR), which regulates salt levels and prevents bacterial growth, when the dissociation of CFTR is disturbed as a protein regulating the chloride ion transport across the cell membrane.[4] The deleted amino acid doesn't allow bacteria in the lungs to be killed thereby causing chronic lung infections eventually leading to an early death.[5] Scientists have used nuclear magnetic resonance spectroscopy (NMR) to study Cystic Fibrosis and its effects.
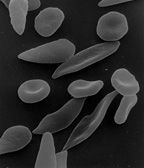
Sickle Cell Anemia: Sickle-shaped red blood cells cling to walls in narrow blood vessels obstructing the flow of blood define sickle cell anemia. The shortage of red blood cells in the blood stream in addition to the lack of oxygen-carrying blood causes serious medical problems. The defect in the Hemoglobin gene is detected with the presence of two defective inherited genes. The sickle cell shape is formed as hemoglobin give up their oxygen resulting in stiff red blood cells forming rod-like structures. Some symptoms include: fatigue, shortness of breath, pain to any joint or body organ lasting for varying amounts of time, eye problems potentially leading to blindness, and yellowing of the skin and eyes which is due to the rapid breakdown of red blood cells. Luckily, sickle cell anemia can be detected by a simple blood test via hemoglobin electrophoresis. Even though there is no cure, blood transfusions, oral antibiotics, and hydroxyurea are treatments that reduce pain caused.[6]
Huntington's Disease: Also known as the trinucleotide repeat disorder, Huntington's disease results from glutamine repeats in the Huntingtin protein. Roughly 40 or more copies of C-A-G (glutamine) will result in Huntington's disease as the normal amount is between 10 and 35 copies. During the post-translational modification of mutated Huntingtin protein(mHTT), small fractions of polyglutamine expansions misfold to form inclusion bodies. Inclusion bodies are toxic for brain cell. This alteration of the Huntingtin protein does not have a definite effect except that it affects nerve cell function.[7] This incurable disease affects muscle coordination and some cognitive functions.
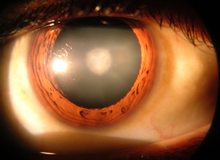
Cataracts: Eye lens are made up of proteins called crystallins. Crystallins have a jelly-like texture in a lens cytoplasm. The current leading cause of blindness in the world, cataracts occurs when crystallin molecules form aggregates scattering visible light causing the lens of the eye to become cloudy. UV light and oxidizing agents are thought to contribute to cataracts as they may chemically modify crystallins. In children, it has been observed that the deletion or mutation of αB-crystallin facilitates cataracts formation. The likelihood of developing cataracts exponentially increases with age. Pain, Roger H. (2000). Mechanisms of Protein Folding. Oxford University Press. pp. 420–421. ISBN 019963788. Retrieved 2009-10-18. {{cite book}}: Check |isbn= value: length (help)
Amyloid Fibrils
[edit | edit source]Protein misfolding caused by impairment in folding efficiency leads to a reduction in number of the proteins available to conduct its normal role and formation of amyloid fibrils, protein structures that aggregate, resulting in a cross-β structure that can generate numerous biological functions. Protein aggregation can come from different processes occurring after translation including the increase in likelihood of degradation through the quality control system of the endoplasmic reticulum (ER), improper protein trafficking, or conversion of specific peptides and proteins from its soluble functional states into their highly organized aggregate fibrils.
Structures
X-ray Crystallography
From X-ray crystallography, three-dimensional crystals of amyloid fibril structures were formed and the structure of the peptide formation and how the molecule is packed together were examined. In one particular fragment, the crystal was found to contain parts of parallel β-sheets where each peptide contributes one single β-strand. The β-strands are stacked and β-sheets formed are parallel and side chains Asn2, Gln4 and Asn6 interact with each other in a way that water is kept out of the area in between the two β-sheets with the rest of the side chains on the outside are hydrated and further away from the next β-sheet.
Solid State Nuclear Magnetic Resonance (SSNMR)
Through solid-state nuclear magnetic resonance (SSNMR) and the help of other methods such as computational energy minimization, electron paramagnetic resonance and site-directed fluorescence labeling and hydrogen-deuterium exchange, mass spectrometry, limited proteolysis and proline-scanning mutagenesis the structure of an amyloid fibril was suggested to be four β-sheets separated by approximately 10Å.
Through NMR with computational energy minimization, a 40-residue form of amyloid β peptide at pH 7.4 and 24˚Celius was determined to contribute one pair of β-strand to the core of the fibril which is connected by a protein loop. The amyloid β peptides are stacked on each other in a parallel fashion.
From experiments of site-directed spin labeling coupled to electron paramagnetic resonance (SDSL-EPR), the molecule was found to be very structured in the fibrils and in parallel arrangement. SDSL-EPR along with hydrogen-deuterium exchange, mass spectrometry, limited proteolysis and proline-scanning mutagenesis suggests that the structure has high flexibility and exposure to solvent of N-terminal side, but is rigid in the other parts of the structure.
Experiments through SSNMR with fluorescence labeling and hydrogen-deuterium exchange determined that the C-terminals are involved in the core of the fibril structure with each molecule contributing four β-strands with strands one and three forming one β-sheet and strands two and four forming another β-sheet about 10Å apart.

Further experimentation approaching the atomic level with SSNMR techniques resulted in very narrow resonance lines in the spectra, showing that the molecules within fibrils hold some uniformity with peptides that display extended β-strands with the fibrils.
Conclusion
The structures determined from X-ray crystallography or SSNMR were similar to previously proposed structures from cryo-electron microscopy (EM) formed from insulin. EM, which uses electron density maps, revealed untwisted β-sheets in the structure. The similarities of the structures found in these experiments suggest a lot of amyloid fibrils can have similar characteristics such as the side-chain packing, aligning of β-strands and separation of the β-sheets. [8] Annu. Rev. Biochem. 2006.75:333-366. www.annualreviews.org. Retrieved 24 Oct 2011</ref>
Formation
The capability to form amyloidal protein structures that are considered to be genetic is from the findings that an increasing number of proteins show no signs of protein related diseases. It has been found that amyloidal proteins can be converted from its own protein that has a function rather than disease- related characteristics in living organisms.
In these protein mutations, different factors that affect the formation of amyloid fibril formation and different chains form amyloid fibrils at different speeds. In different polypeptide molecules, hydrophobicity, hydrophillicity, changes in charge, degree of exposure to solvent, the number of aromatic side chains, surface area, and dipole moment can affect the rate of aggregation of protein. It has been found that the concentration of protein, pH and ionic strength of the solution the protein is in as well as the amino acid sequence it is in determines the aggregation rate from the unstructured, non-homologous protein sequences.
As the hydrophobicity of the side chains increases or decreases can change the tendency for the protein to aggregate.
Charge in a protein can create aggregations through interaction of the polypeptide chain with other macromolecules around it. Also, the low tendency for β-sheets to form along with the high tendency for α-helixes to form contributes in facilitating amyloid formation.
It was found that the degree in which the protein sequence are exposed to solvent tend to affect the formation of amyloids. Proteins that are exposed to solvent seem to promote aggregation. Even though some other parts of the protein that had a high tendency to aggregate were not involved in the aggregation, they seem to at least be partially unexposed to the solvent but other regions that were exposed to solvent that were not involved in the aggregation had a low tendency to form amyloid fibrils.
It has even been raised that protein sequences have evolved over time to avoid forming clusters of hydrophobic residues by alternating the patterns of hydrophobic and hydrophillic regions to lower the tendency for protein aggregation to occur. [8]
The Affects of Sequence on the Formation of Amyloid Proteins
Amyloid formation arises mostly from the properties of the polypeptide chain that are similar in all peptides and proteins, but sometimes, the sequence affects the relative stabilities of the conformational states of the molecules. In that case, the polypeptide chains with different sequences form amyloid fibrils at various rates. Sequence difference affects the behavior of the protein aggression instead of affecting the stability of the protein fold. Various physicochemical factors affect the formation of amyloid structure by unfolded polypeptide chains.
Hydrophobicity of the side chains affects the aggregation of unfolded polypeptide chains. The amino acid in the regions of the aggregation site can change the ability of aggregation of a sequence when they increase or decrease the hydrophobicity at the site of the mutation or folding site. Over time, sequences have evolved to avoid creating clumps of hydrophobic residues by alternating hydrophobic areas of the protein.
Charge affects the aggregation of amyloid protein folding. A high net charge can have the possibility of impeding self association of the protein. Mutations in decreasing the positive net charge may result in the opposite effect of aggregate formation as increasing the positive net charge. It has been seen found that polypeptide chains can be run by interactions with highly charged macromolecules, displaying the importance of charge of a protein aggregation.
Secondary structures of proteins affect the amyloid aggregation as well. Studies show that a low probability to form α-helix structures and a high probability to form β-sheet structures are contributive factors to amyloid formation. However, it has been found that β-sheet formation is not particularly favored by nature since there are little alternation of hydrophilic and hydrophobic residue sequence patterns to be found.
The characteristics of the amino acid sequences affect the amyloid fibril structure and rate of aggregation. Different mutations, including changes in the number of aromatic side chains, the amount of exposed surface area and dipole moment, have been said to change the aggregation rates of lots of polypeptide chains.
Unfolded regions play vital roles in promoting the aggregation of partially folded proteins. Some regions that were found to be flexible or exposed to solvent were fond of aggregation. Other regions that are not involved in the aggregation were found to not be exposed, but rather half buried even though they have high possibility of aggregating while the exposed regions of the structure that are not involved in the aggregation have a low probability of aggregating amyloid fibrils. The fibrils tend to come together by association of unfolded polypeptide segments rather than by docking the structural elements.
Overall, it has been found that unfolded proteins have lower less hydrophobicity and higher net charge than that of a folded protein. Residues that tend not to form the secondary structure of β-sheet structured proteins seem to inhibit the occurrence of amyloid aggregation. Concentration of protein, pH and ionic strength were found to be associated with the amino acid sequence, which affects the rate of aggregation.
Environmental Effects
[edit | edit source]It is understood that the primary structure (the amino acid sequence) of a protein predisposes the protein for a specific three dimensional structure and how it will fold from the unfolded form to the native state. The concentration of salts, the temperature, the nature of the primary solvent, macromolecular crowding, and the presence of chaperones are all factors that affect the mechanism of folding and the ratio of unfolded proteins to those in the native state. More than anything, these environmental factors affect the likelihood of any single protein reaching the correct final structure.
Isolated proteins placed in proper environments (specific solvent, solute concentrations, pH, temperature, etc.) tend to “self-fold” into the correct native conformation. Altering any of these environmental characteristics can disrupt the structure and/or interfere with the folding mechanism. A pH outside the “normal” range of a given protein can ionize specific amino acids or interfere with both polar and dipole-dipole intramolecular forces that would otherwise stabilize the structure. Excess heat (cooking) proteins can break hydrogen bonds essential to the secondary structure of proteins.
Extreme environments or the presence of chemical denaturants (such as reducing agents that can break disulfide bonds) can cause proteins to denature and lose its secondary and tertiary structure, forming into a “random coil.” Under certain conditions fully denatured proteins can return to their native state. Intentional denaturing is used in various methods to analyze biomolecules.
The complex environments within cells often necessitate chaperones and other biomolecules for proteins to properly form the native state.
Protein is an essential part of living thing. The development of human body is needed to be parallel with the development of protein. But protein contains so many mysteries that we did not discovery yet. For example, that is protein folding. Folding is a necessary activity of proteins. They need to fold to continue their biological activity. Folding is also a process that very protein goes through to have a stable conformation. But sometimes this process is happened incorrectly, and the scientist call this problem is protein misfolding. The results of protein folding incorrectly are so many bad diseases happening for human, animals and living things such as Alzheimer’s disease and Mad Cow disease. Because of this reason, the researches about protein folding and misfolding become very important. During the process of discovering about protein, folding, misfolding and its affects, the scientists have been collecting many successes; the mystery about protein is unraveled gradually. As a scientist, W. A. (Bill) Thomasson records many importance things about protein in the article Unraveling the Mystery of Protein Folding; in this article, he make the points about Alzheimer’s disease and Mad Cow disease and some affects of protein misfolding beside the successes of science about them. Dr Thomasson begins his article by introduce generally about protein folding and misfolding. First of all, proteins consists the sequences of amino acid. The scientists have discovered 20 amino acids appearing in proteins. The protein structure is known with 2 basic shapes which are α_helix and β_sheet. “Most of proteins probably go through several intermediate states on their way to a stable conformation” (Campbell and Reece, 79). Proteins need to fold to continue its activity. The scientists have listed 3 type of protein folding; the protein can be folded, partial folded or misfolded. In the process of folding, the “proteins called chaperones are associated with the target protein; however’ once folding is complete (or even before) the chaperone will leave its current protein molecule and go on to support the folding of another” (Thomasson). The author of the article records the very important conclusion of Anfinsen about protein misfolding. In his point of view, the misfolding is occurred in the process of folding when the folding goes wrong. The research of protein misfolding is focus on the temperature sensitive mutation; the scientists observe the bacteriophage P22 with the changing of temperature to cause the mutation. And they conclude that the mutant proteins are less stable than the normal. It means, they give a conclusion is that in the tailspike of bacteriophage the misfolded proteins is less stable than the correctly folded proteins and they are difficult to reach the properly folded state. When the protein misfolding occurs, it results many bad disease. The aggregation can appear along with the appearance of misfolding and it is at the brain to cause Alzheimer’s disease and Mad Cow disease as many scientists consider. One affect of protein misfolding on human life that is Alzheimer’s disease. This is a disease of the elderly. According to the research of scientist, this disease is occurred when the amyloid precursor protein is misfolding. This protein is processed into a soluble peptide Aβ. The scientists have not known exactly the reason of this disease yet. But the main reason causing the misfolding is the protein apolipoprotein E (apoE) inside our blood stream. The protein apoE has three forms such as apoE2, apoE3 and apoE4. The affects of each form of apoE on the Aβ is not discovered yet but the scientists consider that the apoE can bind to the Aβ. In the process of misfolding, the β-amyloid is formed to make “neuritic plaque in the Alzheimer’s patient”. This disease is just happened with the older people because in the amyloid process, a nucleus is formed very slowly. The mutation of this protein is not stable and causes the disease. The studying about apoE is still a secret because some scientists show that one form of this protein is developing the disease but another form is decreasing the development of the disease. Finally, the research about Alzheimer’s disease is continued in order to affirm the results of protein apoE on Aβ and to find the treatment for this disease successfully. Another affect from the protein misfolding is the Mad Cow disease. This is a very dangerous disease because it can be transmitted from animals to human. This disease causes by the misfolding of prions. The process of misfolding is the self-replicating of the prions. Prions are protein particles containing DNA and RNA. The mutation appear in the process of folding, the prions self-replicate and cause the misfolding of the proteins. They contain DNA and RNA. This is a special situation of the protein; it can be served as its chaperons. Because of the replicating, the prion was multiplied very quickly along with the increasing of normal proteins. This disease shows that the protein folding can be occurred without the genetics such as the experiment on the sheep. Dr. Thomasson continues his article by some more information about the misfolding and the way of the scientist to prove the mystery. He gives the information about the protein p53 and its mutation. It can cause the cancer, it also one type of protein misfolding. The point Dr. Thomasson wants to make that is his idea about the drug that can make the protein misfolding becoming more stable and minimize the misfolding of protein. This idea seems very good but its results are like a mystery as the mystery of protein folding. The research about the protein folding is very important to our lives. The misfolding is one of the main reasons causing so many dangerous disease but we did not have a successful treatment yet. The study of protein folding is more and more successful to help the human to be able to destroy the disease causing by misfolding. The disease caused by protein misfolding has become one problem of human that need to be solved.
Molecular Chaperones
[edit | edit source]Molecular Chaperones are known mainly for assisting the folding of proteins. Chaperones are not just involved in the initial stages of a protein’s life. Molecular Chaperones are involved in producing, maintaining, and recycling the structure and units of protein chaperones. Chaperones are present in the cytosol but are also present in cellular compartment such as the membrane bounded mitochondria and endoplasmic reticulum. The role or necessity of chaperones to the proper folding of proteins varies. Many prokaryotes have few chaperones and less redundancy in the types of chaperones and whereas eukaryotes have large families of chaperones containing some redundancy. It is hypothesized that some chaperones are essential to proper protein folding such as the example of the prokaryote which has less variations of a chaperone family available. Other chaperones play less of an essential role such as in eukaryotes where more variations within a family of chaperones exist and gradients of efficiency or affinity are produced. This redundancy or existence of less efficient chaperones may exist in one state but the effectiveness of chaperones is also a function of their environment. The pH, space, temperature, protein aggregation and other external factors may render a chaperone that was once ineffective into a more essential chaperone. These environmental factors show why it is important to simulate cellular in vivo conditions, or native states, in order to grasp the conditions that require use of chaperones. This briefly summarizes the difficulties in analyzing and comparing chaperone function in vivo vs. in vitro. Simulating in vivo, or the environment within the cell, is important not just because of physical factors such as pH or temperature but also because the time in which the chaperone begins to conform the polypeptide. Some chaperones are nearby the ribosome and attach immediately to the polypeptide to prevent misconformation. Other chaperones allow the polypeptide to begin folding by itself and attach later on. Thus the role of each chaperone becomes specific to its vicinity to the polypeptide and time and place in which it assists folding. Recent research has implicated that chaperones within the nucleolus not only catalyze protein folding but also catalyze other functions important to maintain a healthy cell. These nucleolar chaperones are called Nucleolar Multitasking Proteins (NoMP's). Heat shock proteins, for example, not only help other proteins fold but also act during moments of stress to regulate protein homeostatis. Furthermore, there is evidence that chaperones work together in networks to oversee certain functions like dealing with toxins, starvation or infection.
The nucleolar chaperone network is divided into different branches that have specific functions. The network is dynamic and can vary in concentration or location of the network components depending on changes in the physiology and environment of the cell. Heat shock proteins (HSPs), which are classified based on their molecular weights, are integral components of the chaperone network. HSP 70s and 90s maintain proteostasis by ensuring that proteins are properly folded and preventing proteotoxicity, which is the damage of a cell function due to a misfolded protein. HSP70s help to fold recently synthesized proteins, while HSP90s help later in the folding process. The nucleolar network also contains chaperones that are part of ribosome biogenesis, or the synthesis of ribosomes in the cells. Proteins in the HSP70 and DNAJ families, which help to process pre-rRNA, are regularly found in protein complexes that process pre-rRNA in Saccharomyces cerevisiae (a species of yeast). Other HSPs are important in ribosome biogenesis as well, including HSP90 which works together with TAH1 and PIH1 to create small nucleolar ribonucleoproteins. The nucleolar chaperone network provide the organization and assistance needed to complete the biological taks necessary for cell survival, and if it does not function properly there can be many problems. For instance, when cancer cells have increased levels of rRNA synthesis, ribosome biogenesis is increased. Scientists are researching the compound CX-3543, which can stop nucleolin from binding with rDNA and impede RNA synthesis, leading to cell death. It is possible to potentially use drugs designed to target specific branches of the nucleolar chaperone network in malfunctioning cells. Other networks of chaperones include networks that specifically participate in de novo protein folding, meaning they help to fold newly made proteins, and the refolding of proteins that have been damaged. One chaperone network that exists in tumor cell mitochondria contains HSP90 and TRAP1, which protect the mitochondria and prevent cell death, allowing the cancer cells to continue to spread uncontrollably.[9]
Example: Molecular Chaperone (HSP 70)
[edit | edit source]HSP 70 is a protein in the Heat Shock Protein family along with HSP 90. It works together with HSP 90 to support protein homeostasis. It binds to newly synthesized proteins early in the folding process. It has three major domains, the N-terminal ATPase domain, the Substrate binding domain, and C-terminal domain. The N-terminal ATPase binds and hydrolyzes ATP, the substrate binding domain hold an affinity for neutral, hydrophobic amino acid residues up to seven residues in length while the c-terminal domain acts as a sort of lid for the substrate binding domain. This lid is open when HSP 70 is ATP bound and closes when hsp 70 is ADP bound. HSP70, or DnaK, are bacterial chaperones and can help in folding by clamping down on a peptide.[10]
-
 HSP 70
HSP 70

Example: GroEL and GroES
[edit | edit source]GroEL and GroES, or 60kDa and 10kDa, are both bacterial chaperones. Both GroEL and GroES are structured so that they are a stacked ring with an empty center. The protein fits in this hollow center. Conformational changes within the chamber can then change the shape and folding of the protein.[10]
Example: Molecular Chaperone (HSP 90)
[edit | edit source]HSP 90 is a protein in the Heat Shock Protein family. This particular protein, however, is different from other chaperones in that HSP90 is limited in the folding aspect of molecular chaperones. Instead, Hsp 90 is vital to study and understand because many cancer cells have been able to take over and utilize the Hsp 90 in order to survive in many virulent surroundings. Therefore, if one were to structurally study and somehow target Hsp90 inhibitors, then there could be a way to stop cancer cells from spreading. Furthermore, many studies have been performed in order to test whether or not the Hsp 90 chaperone cycle is driven by ATP binding and hydrolysis or some other factor. But after much research by Southworth and Agard, there was enough evidence to state that HSP90 protein could conformationally change without nucleotide binding but rather the stabilization of an equilibrium is the factor that will change the Hsp90 to a closed or compact or open state. The three conformations of the Hsp90 were found through x-ray crystallography and also through single electron particle microscopy and by studying the three-state conformational changes in yeast Hsp90, human Hsp90 and bacteria Hsp 90 (HtpG) it was clear that there are distinct conformational changes for specific species. Overall, Hsp90 is a chaperone that is more involved with maintaining homeostasis within a cell rather than the involvement of protein folding. Hsp90 has rising potential in the area of drug development in the future since it plays such an essential role in aiding the survival for cancer cells.
-
 HSP 90
HSP 90

Example: Molecular Chaperone (TF)
[edit | edit source]This is the first chaperone to interact with the nascent chain as it exits the ribosome tunnel. Without the nascent chain, the TF cycles on and off but once the nascent chain is present, it binds onto the chain, forming a protecting cavity around. In order to do its function, TF scans for any exposed hydrophobic segment of the nascent chain and it can also re-associate with the chain. Folding is found to be more efficient in the presence of the TF, however, this is done at the expense of speed, it can stay with the chain for more than 30 seconds. The release of the chain is triggered when the hydrophobic portions is buried as the folding progresses toward the native state.
Example: Molecular Chaperone (YidC, Alb3, Oxa1)
[edit | edit source]YidC, Alb3, and Oxa1 are proteins that facilitate the insertion of proteins in the plasma membrane. YidC is a protein that has only two polypeptide chains. The formation of its structure has been supported by particular phospholipids. YidC proteins can be found in Gram-negative and Gram-positive bacteria. Oxa1 can be found in the inner membrane of the mitochondria. Alb3 locates in the membrane of the thylakoid inside the chloroplast. Experiments showed that YidC protein actively contributes to the insertion of Pf3 coat protein. In addition, YidC also has direct contact with the hydrophobic segment of Pf3 coat protein. Although Oxa1 can only be found in the mitochondria it can also facilitate the insertion of membrane proteins in the nucleus. The role of YidC and Alb3 seems to be interchangeable because Alb3 can replace YidC in E. coli. Moreover, YidC, Oxa1, and Alb3 all support the insertion of Sec-independent proteins. Oxa1 only supports the insertion of Sec-independent proteins because the mitochondria in yeast cell do not have Sec proteins.
NLR
[edit | edit source]Nucleotide-binding domains that are leucine- rich (NLR) provide a pathogen-sensing mechanism that is present in both plants and animals. They could either be triggered directly or indirectly by a derivation of pathogen molecules via elusive mechanisms. Researches show that molecular chaperones like HSP90, SGT1, and RAR1 are main stabilizing components for NLR proteins. HSP90 can monitor the function of its corresponding clients that apply to NLR proteins in three practical ways: promotion of steady-state of functional threshold, activating stimulus-dependent activity, and raising the capacity to evolve.
Plants contain many NLR genes that considered being polymorphic in the LRR domain in order to be familiar with the highly diversified pathogen effectors. The NLR sensor stability will be the mechanism that will determine the pathogen recognition. The HSP90 system is advantageous for plants because it will couple metastable NLR proteins and stabilize them in a signaling competent condition. This will allow for the masking of mutations that would be detrimental.
Molecular Chaperone Mechanism for Substrate Binding in Protein Folding
[edit | edit source]It is known that chaperones work together to aid in the folding of protein in order to prevent misfolding. However, the mechanism of how chaperones help in protein folding was not fully understood. Recent studies on Hsp40 and Hsp70 have provided more insights into the mechanism of chaperones and their substrate. The Hsp40 family consists of many Hsp40 with different J-domain. Different J-domain will carry out different Hsp70 ATPase activities when Hsp40 binds to Hsp70. In protein folding, an unfolded polypeptide binds to a Hsp40 co-chaparone. From there, the J-domain of Hsp40 binds to the nucleotide-binding domain (NBD) of Hsp70. A conformation change in the Hsp70 substrate-binding domain occurs when the hydrolysis of ATP to ADP takes place on the HSP70 NBD. This causes Hsp70 to have a higher affinity for the polypeptide substrate and unbind the substrate from Hsp40. When ADP is exchange for ATP, the polypeptide substrate is released from Hsp40. Studies have shown that nucleotide exchange factors make changes to the lobe on the Hsp70 ATPASE domain in way that decreases Hsp70’s affinity for ADP. Once the polypeptide is released from Hsp70, it can fold to its native state or it can be refolded by the chaperones if there is a misfolding. If a polypeptide that is bounded to Hsp70 is recognized by E3 ubiquitin ligase CHIP, it will be degraded.[11]
Small Heat Shock Proteins & α-crystallins as Molecular Chaperones
[edit | edit source]It is known that small heat shock proteins (sHSPs) and the related α-crystallins (αCs) are virtually ubiquitous proteins that are strongly induced by a variety of stresses, but that also function constitutively in multiple cell types in many organisms. Extensive research has demonstrated that a majority of sHSPs and αCs can act as ATP-independent molecular chaperones by binding denaturing proteins and reversing denaturation. This approach thereby protects cells from damage due to irreversible protein aggregation. Many inherited diseases have been discovered to result from defects in sHSP/αCs, and these proteins accumulate in neurodegenerative disorders and other diseases linked to aberrant protein folding. sHSP/αC proteins range in size from ~12 to 42 kDa and is a C-terminally located domain of ~90 amino acids, known as the αC domain. sHSP-substrate complexes can be observed by size exclusion chromatography . They are large and heterogenous, and their size distribution depends on the ratio of sHSP/αC to substrate as well as the rate of substrate aggregation, which is affected by concentration and temperature. Substrate binding is generally facilitated by an increase in available hydrophobic surface on the sHSP/αC, which seem to occur without significant loss of defined sHSP/αC secondary and tertiary structure. There is no single, specific substrate binding surface on sHSP/αCs. It rather appears that many sites contribute to substrate interactions, and binding is probably different for different substrates dependent on the conformation of surfaces exposed when a substrate unfolds. However, some sHSP/αCs recognize almost any unfolding protein, which suggests that they act on any labile or damaged cellular component.
The Energy Landscape for Protein Folding
[edit | edit source]If proteins folded randomly and unpredictably, the amount of time taken to reach the native conformation would be much larger than the actual time it takes. The current theory on how protein folding occurs naturally and efficiently involves a "funnel" of sorts-the idea being that there exists not a step by step means of reaching the correct 3-D structure, but rather a number of paths that become progressively narrower from top to bottom. The funnel starts at the top and proceeds downward from energetically disfavorable folding at the top to energetically favoring proper folding at the bottom.
The experiment that sparked the idea of proteins relying on energetics and thermodynamics to reach their native folding was conducted by Christian Anfinsenf in 1961, when he discovered that ribonuclease could spontaneously refold into its proper structure after being denatured without the help of other molecules. Further theoretical proof that protein folding is not random is seen in Levinthal's Paradox, which states that it would take roughly 10^81 years for a protein 100 amino acids long to reach the proper conformation, when in reality, it takes anywhere from a millisecond to a day.

These funnel models (such as the Go-type model) show funnels with hills and bumps that represent the protein taking the path of least resistance when moving down the energy funnel. These bumps are termed "points of frustration". It is believed that funnels with the fewest frustration points or bumps fold into their native forms faster since fewer energy boundaries exist. Although these models are simplified attempts and do not account for misfoldings, they nonetheless prove accurate in the case of many proteins.
Another model that uses algorithms and computers is the empirical force field. This model uses hundreds of thousands of computers running idly to compute folding scenarios of proteins under 50 amino acids with surprising accuracy. However, these computer models will sometimes overestimate unlikely folding structures or produce folding patterns that are rarely or never seen. For example, some simulations/algorithms have a tendency of getting stuck in the local minima and are unable to reach the global minima, which is the correctly folded protein. Simple models such as Go-type models not only predict the folded protein, but also the transition states that determine the rate of the protein folding.
These models are just beginning to show the dynamics of the intermediate stages of protein folding. As such, this is an area under further investigation. The understanding of the kinetics of protein folding is less established, and the movement of proteins between initial amino acid strands and the final product is also an area under investigation. The energy landscape model also has trouble accounting for external factors like crowding and aggregates. One such example of external interaction, called "domino swapping", involves the swapping of monomers from one protein to another in order to activate the correct folding of both proteins.
Recent studies have combined human and computer power to correctly predict the protein conformation. Websites like fold.it, overseen by the University of Washington's Computer Science department, turn the folding problem into a video game, allowing people around the world to solve protein folding problems like puzzle games. Users are given partially folded proteins, usually those stuck in a locally favorable conformation that seems optimal to a computer, and asked to reconfigure the protein into a shape that looks more stable. Utilizing a computer's computing power and speed along with a human's ability to manipulate objects in space shows promise in helping to solve protein folding problems more efficiently.
Co-operativity and Protein Folding Rates
[edit | edit source]The cooperative nature expressed in protein folding is one of the most remarkable aspects of protein folding. Contrary to the traditional viewpoint of complex and heterogeneous mechanisms involved in the folding of a protein, the cooperative two-state folding kinetics shown by many proteins is relatively simple. Due to its simplicity, efforts to understand what determine the co-operativity and the diversity of protein folding rates are made recently by means of applying the cooperative two-state folding kinetics.
The co-operativity of the protein is usually referred to the mechanism by which the presence of a structural region makes additional order more favorable in protein folding. As mentioned previously, the cooperative two-state folding kinetics of small globular proteins is relatively simple and become an interest of study of many scientists. The experiment that excites single molecule that is sensitive enough to allow estimation of transition time reveals two-state co-operativity.
The general trends revealed by two-state folding proteins may be summarized as the following two points. Firstly, more topologically complex proteins tend to fold more slowly than proteins with simpler, local topology; secondly, larger proteins tend to fold more slowly than smaller proteins. The largeness and smallness of a protein here are defined base on its chain length.
Protein folding kinetics is controlled by the free energy barrier determined by the gain of energy and the loss of entropy in the transition state. In describing the pattern, scientists introduce principle of minimum frustration of energy landscape theory. The theory refers to the concept that native-like structures have lower free energy than other random configurations during protein folding. Thus, native-like structures encourage fast folding of the protein and serve as a driving force toward native state, the functional form or the tertiary structure of the protein. This principle can be expressed by the funnel energy landscape.
Funnel Energy Landscape
Funnel energy landscape depicts the energy landscape of a folding protein as a rough funnel. The roughness comes from non-native contacts in protein folding process.The landscape is inherently many-dimensional, so funnel is a projection on the two-dimensional graph. The depth of the funnel represents the energy of a conformational state; the width of the funnel represents the measure of l entropy. The bottleneck of the funnel represents the transition state configuration of the folding protein, whereas the bottom of the funnel represents the native state of the protein. As the protein goes toward its native state, it experiences entropy loss and it achieves lower energy state. The funnel energy landscape serves as a convenient illustration for scientists to envision the thermodynamics and kinetics of the protein folding process.
φ (phi)value
Another concept that plays a role in the study of protein folding kinetics is the φ (phi) value. The value refers to the approximate measurement of native structure content in transition state configuration. The comparison with φ value serves as one of the ways to examine various models that studies protein folding kinetics.
General observations
The fist trend mentioned may be easily understood from an entropic point of view. More topologically complex proteins, or proteins that have long-range contacts, are expected to have higher entropic cost compared with proteins have short-range contacts in terms of folding. The second trend was recently confirmed by experiments focused on the influence of protein size on folding rates. It was found that simple model based only on chain length could roughly predict a protein’s folding rate and stability.
Go¯model
Coarse-grained topology models (Go¯model) are widely used to study the co-operativity and kinetics of protein folding, as it is noted that the topology of native protein determines the folding mechanism. Typical Go¯model simplifies the protein where there is only one interactions stabilizing the folding protein. Early models often examine the non-additive force acting in the protein folding, such as side-chain ordering and hydrophobic effects. Recently, more variety of Go¯models is used to study the protein folding kinetics.
- Bulleted list item
- The Go¯model (this refers to Eastwood and Wolynes’ model here) with nonpairwise-additive interactions between the native contacts of the protein demonstrates that short-ranged multi-body interaction can increase the free energy barrier and make the transition state configuration more localized.
- The lattice Go¯model, on the other hand, demonstrates the coupling local and core burial interactions promoting co-operativity as well as increasing the correlation with contact order.
- The Go¯model with pairwise-additive interactions, particularly the ones focusing on the effects of varying strength of three-body interactions and φ values, shows that three-body interactions increase energy barrier and increases the agreement with measured φ values.
- In addition, solvent-mediated interactions are also introduced into Go¯model. Where the interactions between contacts are replaced by solvent separated minimum and desolvation barrier, it is observed that kinetics and co-operativity of protein function increase as a function of the height of desolvation barrier. The advantage of solvent-mediated Go¯model is that it is useful in distinguishing short-ranged contacts and long-ranged contacts and therefore differentiating proteins with simple topologies and the ones with more complex topologies. In study of solvent-mediated Go¯model the chevron plot is often used. The chevron plot is a way to represent protein folding kinetic datas in varying concentration of denaturation that disrupts the native structure of the protein.
- Variational Go¯model improves co-operativity by excluding volume force between the residues that are in close contacts in native state. In this model it is achieved that a) the Co-operativity is stronger for long-ranged contacts; b) the range of calculated rate is broaden; c) the calculated φ values are improved. There is also Go¯models that entirely focus on the funnel aspect of the protein folding energy landscape and ignore the non-native contact effects.
Other model, such as capillarity model, assumes the volume of folding nuclei scales with number of monomers. In such model, it is shown that increased co-operativity tends to slow down kinetics and smooth the energy landscape.
Conclusion
The recent development of topological models with non-additive forces is becoming a more popular and reliable way to understand the co-operativity of protein folding rates. Refinement of this model has shown its promising future on a more explicit and through understanding of what determines protein folding rates and mechanism. Go¯models that enables long-ranged contacts become more cooperative, and φ values more accurate need further improvement and more attention in the study of protein folding kinetics and the folding mechanism.
Relationship between Protein Sequence, Structure, and Function
[edit | edit source]There have been several protein prediction methods developed in the past 20 years. A universal method has not been developed that applies to all proteins because each method has its advantages and disadvantages. The difficulty of developing such a method is due to our incomplete understanding of the highly intricate relationship between protein sequence, structure, and function.
The theory of correlating amino acid sequence to its structure was shown by Anfinsen. He demonstrated that a denatured (unfolded) protein could regain its native tertiary structure spontaneously. This method is also a useful contributor for assigning function to protein structure. A protein researcher could predict that hydrophobic substrates could potentially bind to hydrophobic regions of the protein and vice versa for charged regions. The problem with this method is that it doesn’t take into account certain factors such as atypical environmental conditions.
It was thought that similar sequencing implies related structures. This theory only holds true for a handful of proteins. Researchers saw that similarities in protein folds aren’t always related to its protein sequence. Due to these findings, the ‘Paracelsus Challenge’ was purposed in 1995. The theory behind the ‘Paracelsus Challenge’ was to develop two proteins that were more than 50% identical in sequence, but they both had completely different folds. The challenge was satisfied in 1997 by with two protein sequences that shared 88% sequence identity (GA88 and GB88). Recent studies show that as little as 3 mutations are enough to induce different folding patterns. Although the outcomes of the ‘Paracelsus’ challenge are very interesting, they rarely occur in nature.
Functional convergence causes problems in assigning a specific function to a structure. Various structures can adopt similar functions, but some can adopt very different functions as well. However, there is a significant correlation between certain folds and specific functions. There are two major variables in function prediction: (1) the locations of binding site, and (2) the range of functions at the site. Metal, ions, cofactors, and other proteins that contribute to functions must be taken into considerations as well. One problem that arises with these factors is when determining a structure via crystallography. The PROCOGNATE resource and PIDA database offers a solution to this problem.
A widely used method by which protein function is defined is derived from the Gene Ontology, which consists of three graph structures in which functional terms and relationships between them are defined. Limitations of gene ontology arise with proteins that are non-positional and when proteins have no defined relationship between ligand in its crystal structure. Other developments that attempt to bridge this gap includes The Protein Feature Ontology (PFO [29]) and The Distributed Annotation System (DAS[30]).
Two approaches are used to determine a functional site: (1) either with no knowledge of where the site is or what it binds, or (2) with prior knowledge of the interaction partner. The most highly used methods involve bioinformatics such as the SOIPPA method. A very important contributor to assigning function to protein is sequence conservation, but it is difficult to determine if residues are conserved for structural or functional reasons. Another method involves energy-based approach. A recent development is the ProFunc server, which combines methods such as InterProScan and BLAST search.
Predicting binding sites (which are immensely complex in its own nature) is only the first step of the puzzle. The next step is to determine the overall function in terms of biochemical function, and even more challenging is determining its biological role. The difficulties with analyzing protein function increased another magnitude of complexity when researchers came across the fact that protein function may not only depend on its final folded product. A protein could have functionalities in its partially denatured state and it fully denatured state. With all of this said, it is safe to say that there is still a lot to learn about the relationship between sequence, structure, and function of proteins.
Domain Swapping, Folding and Misfolding
[edit | edit source]The domain swapping that occurs in proteins may be important in the folding or misfolding process in proteins. Domain swapping occurs when two or more identical protein chains swap with each other. The domain swapping can be thought of as a mechanism for the interchanging of monomers and oligomers. What happens in oligomeric swapping is that one monomer from one protein will swap with another identical monomer from a different protein. This domain swapping mechanism has been observed in various proteins, more than 40 different proteins. The swapping mechanism is important for some protein functions. For a specific protein for example, p13suc1 it has been seen that the swapping and aggregation correlate meaning that they have a common mechanism. P13suc1 is required for cyclin-dependent kinase (Cdk) during the cell cycle progression. P13suc1 has two different states, one being a monomer and the other a swapped dimmer. The domain swapped part is a β strand is not an independently folded domain. While studying this, it was found that β4 has a critical role when in contact with β2 because they pair with each other early on in the folding process. Therefore, for p13suc1, it has been shown that the regions that have been interchanged are responsible for the folding and misfolding of the protein. There seems to be a competition between folding and misfolding in proteins because polypeptide chains can fold into structures or misfold into amyloid fibrils. What seems to be even more crucial in protein folding is the presence of a folding nucleus which forms part of the protein chain in the transition state. A correlation between residues involved in protein folding nuclei location and amyloidogenic regions have been found as well as important information that fibril formation and protein folding may contain key residues. By using the modeling of folding of proteins and looking at the exchangeable regions in the oligomeric form, the relationship can be seen as responsible for folding and misfolding. This may take researchers one step closer to solving the protein solving problem and understand how proteins get their folding instructions. Reference: http://www.benthamscience.com/open/tobiocj/articles/V005/27TOBIOCJ.pdf
-
 Domain Swapping
Domain Swapping

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Death-fold Superfamily[12]
[edit | edit source]There are 4 subfamily structures in the death-fold superfamily. They consist of Death Domains (DDs), Death Effector Domains (DEDs), CAspase Recruitment Domains (CARDs) and PYrin Domains (PYDs). These subfamily structures are involved in the assembly of multimeric complexes which may be implicated in cell inflammation and death.
Structure and Function of a Death-Fold Domain
There are currently 102 known proteins that have death-fold superfamily domains. These domains contain homotypic interactions. These proteins consist of 39 DDs, 8 DEDs, 33 CARDs, and 22 PYDs. Although these domains have up to a 90% difference in sequence, they all have the characteristic death-fold. This fold consist of a "globular structure where 6 amphipathic alpha-helices are arranged in an anti-parallel alpha-helix bundle with Greek key topology" (Peter Vandenabeele et al., 2012). The difference between these death-domains which constitute either of the subfamilies is found in the alpha-helices length and orientation and the distribution of hydrophobic and charged residues along the surfaces of the complexes.
The believed function of the death-fold domains is to mediate the assembly of large oligomeric signaling complexes. At these complexes, caspases and kinases activity is increased. Before now, little was known about the structural conformation of protein assemblies with death-fold domains.
Three distinct Interaction Types
Type I Interaction: Residues from helices 1 and 4 (Patch Ia) of one death-fold domain interact with residues from helices 2 and 3 (Patch Ib) of another death-fold domain. Type II Interaction: Residues from helix 4 and the loop between helices 4 and 5 (Patch IIa) of one death-fold domain interact with residues of the loop between helices 5 and 6 (Patch IIb) of another death-fold domain. Type III Interaction: Residues from helix 3 (Patch IIIa) of one death-fold domain interact with residues located on the loops between helices 1 and 2 and between helices 3 and 4 (Patch IIIb) of another death-fold domain.
Previous theory suggested that the three interaction types were conserved throughout the death-fold superfamily but it now seems that there are differences seen between interactions of the same type of death-fold domains.
Crystal Analysis of Death-Fold Domains
Only three DD complexes have had their crystal structure analyzed. They are PIDDosome, MyDDosome, and the Fas/FADD-DISC. The analyses of these structures have shown that DDs can engage in up to six interactions.
Death-Domains and Medicine
Death-domains have been shown to facilitate the assembly of multimeric complexes that lead to inflammation and cell death. Understanding of these structures can generate therapeutic benefit by preventing or triggering the formation of these oligomeric complexes. Diseases that may be affected by these interactions can include neurodegenerative and inflammatory disorders as well as many others that have characteristic of inflammation or excessive cell death.
Disordered Proteins
[edit | edit source]While folding is typically a major contributor to protein function, some proteins do not fold into a specific structure, yet still possess a function. Instead of a specific structure, these proteins often shift between different forms and/or have disordered regions that do not hold to a particular shape.
Just as a protein's folding is determined by its amino acid sequence, non-folding proteins are non-folding because of their sequence. These proteins tend to have much less of certain amino acids than folding proteins, and much more of others. Specifically, non-folding have less of the amino acids that form the hydrophobic cores of folding proteins and more of the surface amino acids. The formation of a hydrophobic core is one of the first steps in most protein folds and, once formed, the core tends to provide the driving force for stable final structures. Without the amino acids to form a core, proteins are not driven towards a specific structure.
= = CONCEPT = =
[edit | edit source]Several molecular chaperons that are fully folded and inactive under non-stress conditions have been known as conditionally disordered proteins. These chaperons have a partially disordered conformation when they exposed to distinct stress conditions. This disorder is very important because they are able to protect cells against stressors. The study of these disordered chaperons lead to more understanding of the functional role for protein disorder in molecular recognition. X-ray crystallography is a useful technique that helps visualize the structures of the proteins. Based on this technique, over 95% of the entire molecule is represented by 25% of crystal structures and all others have missing electron density for more than 5% of their sequence due to the multiple conformations on these regions. Proteins actually have some disordered conformation and these disordered proteins lie at one extreme part from very flexible to static structural states on a continuous spectrum. Either only a part of the protein or the whole complete polypeptide chain is found in this disorder. Therefore, investigating only some parts of the proteins would not help summarize the flexibility of the protein. The term “conditionally disordered” means the disorder of proteins may happen under some certain conditions and may not happen under other conditions. It is very common to see the intrinsic disorder within proteins. For example, between 30% and 50% of eukaryotic proteins are estimated to have more than 30 amino acids that violate the defined secondary structure in vitro and many complete unstructured proteins have been predicted to exist too. It is still very challenging to verify the status of folding of proteins within the region of cells despite a lot of computational methods that have been used. There is a chance that many proteins which are seen either partially or fully folded happen to be unstructured in cells. The number of these chances is still uncertain. It is however thought that the presence of the appropriate binding pairs would make the disordered proteins come into their folded state, which means that the percentage of intrinsically disordered proteins in vitro might be lower in the cell. The extent of the disorder might be decreased by the stabilizing interactions within the cells. Through chemical shift, residual dipolar coupling, and paramagnetic resonance enhancement measurements, NMR serves as a good method to provide the detailed information on extent of disorder of the proteins.
= = CONDITIONALLY DISORDERED PROTEINS = =
[edit | edit source]There are two states of disordered proteins. One shows a high degree of flexibility and the other state is where the protein is found more ordered. Thus, in order to know the cause and effect relationships between disorder and function, it is essential to study both states. Many disordered proteins like DNA, proteins, and membranes refold once they find a partner to bind to. Also, order-to-disorder-to-order transitions can occur. Proteins that are involved into multiple binding are very good examples of conditional disorder. Binding surfaces that are disordered before binding are able to fold into distinct conformations with other partners better than the binding surfaces that are already well-organized. The ‘conformational selection hypothesis’ suggests that different members of conformational ensemble can be stabilized by the binding of different partners. On the other hand, the ‘folding upon binding’ model proposes that proteins may be able to fold into different conformations when they bind with different partners.
Frequency
[edit | edit source]Predictions done on whole proteomes suggest that the frequency of disordered proteins in eukaryotes is much larger than in prokaryotes, with the frequencies in the two groups of prokaryotes, archaea and eubacteria, being similar. In mammals, about half of all proteins are predicted to have large unordered regions, with about a quarter being fully disordered.
Function
[edit | edit source]Disordered proteins are prevalent in signaling and regulation, especially in interactions with biomolecules such as nucleic acids and other proteins. Molecular recognition and protein assembly and modification frequently involve proteins with disordered regions. The ability of these proteins to interact with multiple molecular partners means that they are also common in protein-protein networks, either as hub proteins or as proteins interacting with hub proteins.
Diseases
[edit | edit source]Disordered proteins are implicated in a number of human diseases. In particular, the amyloid diseases, which involve the accumulation of misfolded proteins, seem to be associated with disordered proteins, probably because their variable regions make them more likely to have a structure that favors their accumulation. This category includes many neurodegenerative diseases, such as Alzheimer's and Parkinson's.
The Role of Computers in Determining Structure and Function of Proteins
[edit | edit source]The structure or folding of an amino acid and by extension its function can be analyzed and compared through its primary structure or amino acid sequence using computer algorithms. Comparisons of amino acid sequences of unknown folding patterns with similar amino acid sequences of known folding is enhanced using computers. A computer automated tool called Protein Basic Local Alignment Search Tool, or protein BLAST, is a free search tool open to the public that allows quick comparison of amino acid sequences in an online database. The output of this tool is the percent match of amino acids and the known properties of the sequence matches. Furthermore, because amino acid sequences are based on DNA sequences, three bases code for one amino acid, the protein under scrutiny can be analyzed on a DNA level using DNA BLAST. The integration of public databases of amino acid and DNA sequences along with computer algorithms has accelerated the genome and proteome field by allowing scientists around the world to share and analyze sequences.
Appendix
The Role of Computers
The scientists credited for creating the BLAST program are Webb Miller, David J. Lipman, Warren Gish, Eugene Myers, and Stephen Altschul from the NIH
Molecular Chaperones
Pain, Roger H. Mechanisms of Protein Folding. 2nd ed. 364-85
The Energy Landscape for Protein Folding
Cho, Samuel S. "Energy Landscapes for Protein Folding, Binding, and Aggregation: Simple Funnels and Beyond." UCSD Dissertation (2007).
Cheung, Margaret S. "Energy Landscape Aspects of Protein Folding Dynamics Relevant to Molecular Functions." UCSD Dissertation (2003).
Yang, Sichun. "Extending the Theoretical Framework of Protein Folding Dynamics." UCSD Dissertation (2006).
Intramolecular Interactions
Pain, Roger H. "Mechanisms of Protein Folding" 2nd ed.
http://www.nature.com/horizon/proteinfolding/background/importance.html
Berg "Biochemistry" 6 Edition
Co-translational protein folding
[edit | edit source]In silico modeling studies have helped identify several characteristics of co-translational folding pathway. First, it was determined that in vivo protein folding is a vectorial process, which is a dispersion change. Second, co-translational vectorial folding of the developing polypeptide from its N-terminal end to its C-terminal end results in a sequential structuring of the distinct regions of the polypeptide emerging from the ribosomal tunnel. Third, attachment to the developing polypeptide chain to the ribosome during protein synthesis reduces the conformational space and the degrees of freedom of the growing chain. This limits the number of possible intermediates and reduces the number of possible folding pathways. Fourth, co-translational protein folding begins early during the process of polypeptide chain synthesis on the ribosome, with some elements forming inside the ribosomal tunnel. Fifth, folding catalysis and molecular chaperones interact with the growing developing chain as soon as it emerges from the tunnel. This accelerates the slow steps in protein folding and prevents misfolding of proteins.
References
[edit | edit source]- ↑ "Alzheimer's Disease". Ohio State University Medical Center. 2009. Retrieved 2009-10-09.
{{cite web}}: External link in|publisher= - ↑ Lindquist, Susan (1999). "What is a Prion?". Retrieved 2009-10-09.
- ↑ "Mad Cow Disease and Variant Creutfeldt-Jakob Disease". eMedicine Health. Retrieved 2009-10-09.
{{cite web}}: External link in|publisher= - ↑ "Unraveling the Mystery of Protein Folding" (PDF). [Thomasson, W.A. "Unraveling the Mystery of Protein Folding]. Retrieved 2009-10-18.
- ↑ "Folding Away Cystic Fibrosis". [1]. Retrieved 2009-10-18.
{{cite web}}: External link in|publisher= - ↑ "Genetic Disease Profile: Sickle Cell Anemia". [2]. Retrieved 2009-10-18.
{{cite web}}: External link in|publisher= - ↑ "The Basics of Huntington's Disease". [3]. Retrieved 2009-10-18.
{{cite web}}: External link in|publisher= - ↑ a b c Invalid
<ref>tag; no text was provided for refs namedannu - ↑ Piotr Banski, Mohamed Kodiha and Ursula Stochaj (2010). "Chaperones and multitasking proteins in the nucleolus: networking together for survival?". Retrieved 2010-10-16.
- ↑ a b Joan L. Slonczewski, John W. Foster. "Microbiology: An Evolving Science."
- ↑ Summers, Daniel W., and Peter M. Dougla (2009). "Polypeptide Transfer from Hsp40 to Hsp70 Molecular Chaperones". Retrieved 2010-10-24.
{{cite web}}: CS1 maint: multiple names: authors list (link) - ↑ 11. Kersse K, Verspurten J, Vanden Berghe T, Vandenabeele P. The death-fold superfamily of homotypic interaction motifs. Trends in biochemical sciences. 2011;36(10):541–52. Available at: http://www.ncbi.nlm.nih.gov/pubmed/21798745. Accessed October 29, 2012.
12. Small heat shock proteins and α-crystallins: dynamic proteins with flexible functions. Basha E, O'Neill H, Vierling E. Trends Biochem Sci. 2012 Mar;37(3):106-17. Epub 2011 Dec 14
Conditional disorder in chaperone action. Bardwell JC, Jakob U. Trends Biochem Sci. 2012 Sep 24. pii: S0968-0004(12)00127-2. doi: 10.1016/j.tibs.2012.08.006. [Epub PMID 23018052 [PubMed - as supplied by publisher] "Molecular Biology of the cell." Fifth Ed-Alberts, Johnson, Lewis, Raff, Roberts, Walter. pg. 716-717
Braakman, Ineke, and Neil J. Bulleid. "Protein Folding and Modification in the Mammalian Endoplasmic Reticulum." Annual Review of Biochemistry. 80. (2011): 71-99. Web. 29 Oct. 2011. <http://www.annualreviews.org/doi/pdf/10.1146/annurev-biochem-062209-093836>.
Cabrita LD, Dobson CM, Christodoulou J. Protein folding on the ribosome. Current Opinion in Structural Biology 2010, doi:10.1016/j.sbi.2010.01.005
A Keith Dunker, Israel Silman, Vladimir N Uversky, Joel L Sussman. "Function and structure of inherently disordered protein." Curr Opin Struct Biol. 2008 Dec;18(6):756-64
Booth Paula J, Curnow Paul. Folding Scene Investigation: Membrane Proteins. Current Opinion in Structural Biology 2009, doi:10.1016/j.sbi.2008.12.005
Heijne, Gunnar Von. "Membrane Protein Folding and Insertion." Annual Review of Biochemistry 80 (2011): 157-60. 26 Oct. 2011 <http://www.annualreviews.org/doi/full/10.1146/annurev-biochem-111910-091345?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub%3dpubmed>
Kuhn, Andreas, Rosemary Stuart, Ralph Henry, and Ross E. Dalbey. "The Alb3/Oxa1/YidC protein family: membrane-localized chaperones facilitating membrane protein insertion?" TRENDS in Cell Biology 13 (2003): 510-16. 26 Oct. 2011 <http://www.cell.com/trends/cell-biology/abstract/S0962-8924(03)00196-X>
Table 1: Berg, Jeremy. Relative Frequencies of Amino Acid Residues in Secondary Structures. 2012. Biochemistry, New York . Print.
Voet, Donald, Judith G. Voet. Biochemistry 3rd ed. New Jersey: John Wiley & Sons, Inc, 2004. Print.