The cell is the most fundamental unit of living organisms, providing both structure and function. Different cells may take on different shapes, sizes, and functions, but all have the same fundamental properties. Within the cell are various organelles, which give the cell structure and function. The amounts and types of organelles found vary from cell to cell.[1]
There are two major types of cells: prokaryotes and eukaryotes. A prokaryotic cell, such as a bacteria cell, is one which lacks a "true" nucleus and membrane-bound organelles. The genetic information of a prokaryote is localized in the nucleoid region within the cytoplasm. On the other hand, eukaryotic cells store their genetic information in a membrane-enclosed nucleus. The larger of the two types, eukaryotes further differ from prokaryotes in that they contain membranous organelles, such as the mitochondria and the endoplasmic reticulum.
Two common types of eukaryotic cells are animal and plant cells. Plant cells have cell walls, which give them their rigid structure. Unique to plant cells, chloroplasts allows them to photosynthesize. These two structures are not found in animal cells. The absence of a cell wall enables animals cells to adopt a variety of shapes. A lysosome, which contains digestive enzymes, is found exclusively in animal cells. Another difference between animal and plant cells is the size of their vacuoles. Animal cells have several small vacuoles throughout the cytoplasm while plant cells contain just one large central vacuole.
The size of a cell is limited by diffusion. Though larger cells are able to contain more biochemicals, a smaller size that maximizes the surface area-to-volume ratio is most ideal. Simple geometry tells us that the area-to-volume ratio is given by 3/r, which is inversely proportional to the radius. Molecules, such as oxygen, that are required by the cell to function need to be able to reach all parts of the cell efficiently. Thus, a greater diffusion rate is promoted by a smaller cell size.[2]
One of the most traveled cells are blood cells. Red blood cells move passively as the bloodstream carries them. White blood cells protect people from viruses and bacteria so their movement needs to be quick to the site of infection. The feeling of pain is what happens when white blood cells move to the focused site. The white blood cells move like amoebae, where they move by stretching parts of themselves and the entire cell moves towards where said part was stretched. This process is repeated over and over again. [3]
The Cell Theory is that all living things are composed of cells and that all cells originate from pre-existing cells. Cells are relatively small because it reassures a big surface to volume ratio, which is need to facilitate a fast exchange of material with the extracellular environment.
Supramolecular complexes are composed of macromolecules, such as DNA, protein, and cellulose. Macromolecules are the rather large in size molecules in chemistry. The weak, non-covalent forces between macromolecules provide stability and structure to the supramolecular complex. The four major macromolecules are nucleic acids, proteins, sugars, and lipids. Macromolecules needs assistance, such as salts or ions, when dissolving. In the case of proteins, they will denature when the concentration is out of their range of concentration. Since they are so big, they will also effect the rate of equilibrium when there is a very high concentration of macromolecules.
Macromolecules can be further subdivided into their respective monomeric units. DNA is composed of nucleotide units, while protein is composed of amino acids. Carbohydrates are made of sugar subunits, and lipids are made of fatty acid subunits.
↑Nelson, David L. (2004). Principles of Biochemistry (4th Ed. ed.). W. H. Freeman. ISBN 0716743396. {{cite book}}: |edition= has extra text (help)
Pictorial representation of organelles in a typical animal cellCell
Structural biochemistry plays a vital role in the functions of an organism's cell through various means, one of them being the organelles in a cell. It is through the structure and functions of living molecules (and some non-living), such as nucleic acids, amino acids, purine, and lipids that life is even possible.
Some properties of living organisms include high degree of chemical complexity and microscopic organization, systems to extract, transform and use energy from the environment, self-replication and self-assembly, sensing and responding to changes in the environment, define functions for each component and regulation among them, and history of evolutionary change.
Organelles are the components of the cell that synthesize new materials, recycle old materials, transport molecules, and anything else that is essential to ensure the proper survival of the cell and its propagation. Organelles incorporate all broad ranges of organic molecules including nucleic acids, amino acids, carbohydrates, and lipids to produce a viable cell.
As discussed before, the "lipid bilayer" that forms the cell membrane contains membrane protein and cholesterol. The membrane protein plays a vital role in the membrane functions while the cholesterol performs the structural role within the membrane.
There are two types of protein membranes:
Integral membrane protein: lies within the membrane
Peripheral membrane protein: bound to membrane
The cell membrane is often referred to as a mosaic. Proteins in the membrane determine most of the membranes specific function. These proteins are categorized as integral and peripheral proteins. Integral proteins can perform a number of functions such as being transport proteins that provide a hydrophilic channel across the membrane. Integral proteins can also act a receptor sites for chemical messengers like hormones. Enzymes can also be found in the lipid bilayer with its active site exposed to substances in adjacent solutions. Elements of the cytoskeleton may also be bonded to the membrane proteins; a function that helps maintain cell structure.
The structure of these membrane protein can also be either alpha helices or beta sheets. Due to the hydrophobic interactions, the hydrophobic residues of the alpha helices will not exposed to the aqueous environment. The beta sheet forms a hollow cylindrical configuration where the inside is hydrophilic. The cylindrical structure is driven by the unsatisfied hydrogen bonding at the ends of the beta sheet. Wrapping around itself, the beta sheet is able to satisfy all hydrogen bonding.
The major functions performed by proteins of the plasma membrane are:
Transport: Some membrane proteins provide selective hydrophilic channels for exchange of substances.
Enzymatic activity: Membrane proteins may also be enzymes with their active sites exposed to the surrounding solutions.
Signal transduction: Membrane proteins may act as receptors with specific binding sites that allows perfect fit with chemical messengers, which can cause the protein to change shape and allow it to relay the message to the inside of the cell.
Cell-cell recognition: Some glycoproteins allow specific identification by membrane protein of other cells.
Intercellular joining: Membrane proteins of adjacent cells may join together in different junctions.
Attachment to the cytoskeleton and extracellular matrix (ECM): Membrane proteins can be non-covalently bound with elements of the cytoskeleton in order to maintain cell shape and stabilize the location of certain membrane proteins.
Cholesterol regulates the fluidity of the membrane in eukaryotic cells. The ability to incorporate cholesterol into the cell membrane with hydrophobic and hydrophilic interactions allows the cholesterol to disrupt the phospholipid interaction within the bi-layer. Since prokaryotic cells do not have cholesterol to regulate fluidity, these cells depend on the variation in the saturation level and length of the fatty acid chain. The shorter and more saturated the chain the more rigid the membrane will become (due to the fact that the longer and saturated chains can interact more closely with one another)
This model claims that the lipid layer has an important role in cell membrane. The cell membrane serves as the solvent for the integral membrane proteins, and it also serves as a barrier that separates the cellular activities within the cell from the extracellular space. The permeable barrier regulates what enters the cell. This Fluid Mosaic Model is regulated by the concentration of cholesterol and fatty acid chain mentioned above.
Ribosomes are the sites by which
nucleic acids are translated and proteins are synthesized. Ribosomes are about 20 nm in diameter and are composed of ribosomal RNA and proteins. They can be found freely floating in the cytosol and not attached to any organelles in prokaryote cells. In eukaryote, ribosomes may be found on the rough endoplasmic reticulum. The rough ER earns its name because of ribosomes on its surface, giving a studding appearance. The proteins produced by the ribosomes of the rough ER are sent through the lumen of the ER, where they are modified. The protein is then transported in a vesicle to the Golgi Apparatus, where the protein undergoes further modification.
First, the genetic code from DNA is transcribed into a complementary strand called messenger RNA (mRNA) (mRNA) by DNA polymerase. In prokaryotes, the mRNA moves away from the nucleoid and is bounded to free-floating ribosomes in the cytosol. However, in eukaryotes, mRNA is made in the nucleus and transported across the nuclear membrane and into the cytoplasm. This is called translocation. In the next step, known as translation, the mRNA is attached to the ribosome, and codons on the mRNA are matched with the complementary nucleotide bases (anticodons) located on a transfer RNA (tRNA) molecule. The enzyme aminoacyl tRNA synthetase matches the tRNA codons with the appropriate amino acids through a series of esterification reactions. Ribosomal RNA synthesizes the protein through use of RNA polymerase. This elongates the protein until a stop codon terminates the protein synthesis chain. The synthesis of proteins always moves in the direction of the N-terminus to the C-terminus. DNA replication also follows the 5' to 3' direction.
The cell membrane, as explained above, is a selectively permeable barrier of ions and molecules that move into and out of the cell. In other words, not all molecules are able to pass through the cell membrane. During the division of the cell, none of the membrane integrity is lost. As the cell grows, new lipid and protein molecules are placed into the cell’s plasma membrane.
Prokaryotes typically have no compartmentalized organelles. The cell's DNA and ribosomes are free-floating with the cytosol, which is surrounded by a cell membrane. A prokaryotic cell is generally one hundred times smaller than a eukaryotic cell.
In the nucleoid, the chromosomal DNA is wrapped around binding proteins. "Replication by DNA polymerase and transcription by RNA polymerase occur at the same time within the nucleoid." [1]
Pili (Fimbriae)is a thin structure that stick out from the surface. They are made out of a single protein called pilin. Pili's functions include DNA transfer, binding to surfaces, and motility. Pili has the ability to attach to a substrate. One type of pili called sex pili, it attaches a "male" donor cell to a "female" recipient cell for transfer of DNA. This process is called conjugation.
Eukaryotes include animals, plants, fungi, and protists. They are typically more complex than prokaryotic cells. There are many compartmentalized organelles enclosed in membranes within the cell which allow for various reactions to take place. The eukaryotic cell is typically much larger than the prokaryotic cell, usually by a factor of around 100. Each organelle in Eukaryotes has their own function in the cell.
The nucleus is one of the primary organelles that distinguish eukaryotic cells from prokaryotic cells. It is an organelle that enclosed compartment with a specific function. It contains chromatin. Nuclei contains nucleolus (where ribosome assembly is). Ribosomal RNA get together with ribosomal proteins to form the ribosomal subunits. The nucleus contains DNA that house the genes coding for the synthesis of proteins, antibodies and molecules that perform the basic functions of the cell. The nuclear membrane contains nuclear pore conplexes that allow for transport of material into and out of membrane. They also export mRNAs out of the nucleus. [2]
There are two types of endoplasmic reticulum: smooth ER and rough ER. The smooth ER is the site of lipid synthesis and some detoxification of noxious compounds. The rough ER is the site where transmembrane proteins or secreted proteins are translocated. Ribosomes are located in the rough ER instead of smooth ER because the protein has a hydrophobic signal sequence on its amino terminus. Endoplasmic Reticulum is where proteins can be modified too.
Mitochondria (Singular: Mitochondrion) involved in cellular energy production. It has a function in performing oxidative respiration and are found in nearly all eukaryotes. Mitochondria also produces ATP by oxidative respiration. It also has an outer and an inner membrane. Both DNA and ribosomes of mitochondria show similarities with DNA and ribosomes of bacteria.
Proteins that are not part of ER now move to the Golgi. Golgi complex has membrane stacks (cisternae) that each contain unique enzymes. Carbohydrates may be modified as proteins pass through the cisternae. Vesicles leaving the Golgi complex may fuse with the cell membrane.
Centrioles are involved in a process called nuclear division. They are small, self-replicating, and are located in the cytoplasm near the nucleus.
organelles present near nucleus of animal cells.
The cell wall, located outside the cell membrane, is a tough layer that provides the rest of the cell with structure support and protection. Cell walls are present in plants, fungi, algae, some archaea, and bacteria cells, but not in animal cells. The cell wall confers the shape and rigidity to bacterial cell and helps it withstand the intracellular turgor pressure that can build up as a result of osmotic pressure.
Chloroplasts are only found in photosynthetic eukaryotes. They convert Sun-derived light energy to ATP and reduced NADPH. Ancient cyanobacteria gave rise to chloroplasts. In other words, cyanobacteria are the ancestor of chloroplasts. Chloroplasts have an outer membrane, an inner membrane, and the thylakoid membrane. Some algal chloroplasts have more membranes outside these. Chloroplasts are typically found within the mesophyll cells, or the inner tissues of a leaf. Chloroplasts are discs that are typically somewhere between 2-7 micrometers in diameter and 1 micrometer thick. Chlorophylls a and b, which are the green pigments located within plant chloroplasts, give plants their typical green color. During photosynthesis, carbon dioxide enters the leaf through microscopic pores known as the stomata, and oxygen leaves as a byproduct through the stomata. The dense fluid found within the chloroplast is known as the stroma, and amongst this are several interconnected membranous sacs shaped in flat discs with their own compartments, known as the thylakoids. In plants, thylakoids are arranged in a stacked conformation known as grana. Most other photosynthetic organisms and some CTemplate:Sub plant chloroplasts have unstacked thylakoids. The space within these compartments are called the thylakoid space. The membranes of the thylakoids hold the cell's chlorophyll.
Vacuoles serve as the cell's storage centers of food and other necessary materials. The vacuole further functions in the removal of unwanted structural materials, the containing of several waste products and small molecules (which could involve the isolation of potentially harmful products in the cell), the exportation of unwanted materials from the cell, and the maintenance of an acidic internal pH and constant internal pressure. In plants, the central vacuole is typically the largest compartment of all the cell's organelles, occupying in itself the majority of the cell volume. This large compartment is enclosed by a membrane called the tonoplast, which is selectively permeable to certain solutes within the cytosol. The solution inside the vacuole is referred to as cell sap, which is of different composition from the cytosol. Some plant vacuoles contain pigments that color the cells, such as during pollination season by which the plant must attract different organisms to carry out its fertilization. Plants maintain its structure through the maintenance of internal pressure through the manipulation of water into and out of the vacuole. Through water osmosis, water diffuses into the vacuole, which places pressure onto the cell wall. If too much water were to be lost, this pressure against the cell wall would be lacking, and the cell would collapse onto itself. Thus, cells also serve to maintain the cell's size. Another important feature of the vacuole in plants is that an enlarged central vacuole may add a specific amount of pressure against the other compartments in the cell and push them towards the cell membrane, thereby giving a type of conformation that permits the absorbance of more solar energy.
Lysosomes are membrane enclosed organelles that help eukaryotic cells obtain nourishment from macromolecular nutrients. They contain hydrolytic enzymes. Lysosomal and phagocytosis digestion help the eukaryotic cell because they increase the membrane surface area. In eukaryotes, lysosomes allow intracellular digestion crosses the lysosomal membrane into cytoplasm. Lysosomes are the 'garbage bin' of the cell.
Peroxisomes are the centers by which the cell may be rid of potential toxins. The peroxisomes are in itself a receptacle of oxidative enzymes that remove hydrogen atoms from certain organic substances to produce peroxides, which in itself is harmful. This peroxide serves to oxidate the potentially harmful substances, such as alcohol.
During evolution, cells start to develop into two compartments: the outer and inner aqueous compartment. The advantage of having an inner aqueous compartment allows the better segregation of cellular organelles from the external environment. Thus, each organelle is able to develop and refine its structural and functional distinction inside this aqueous compartment throughout the course of evolution. The "lipid bilayer" (further discussed) existed as a protective wall that allows hydrophilic interaction in the external and internal compartment of the cells while maintain proficient rigidity with its hydrophobic interior structure. The inner aqueous compartment ultimately becomes the cytoplasm which serves the same purpose as it has been during evolution.
Liposomes are essentially lipid vesicles that are surrounded by a circular phospholipid bilayer. They form identical structure as other phospholipids vesicles: interior hydrophobic tails away from the aqueous solution, and external hydrophilic heads towards the aqueous solution. Liposomes are formed through the process called sonification that results in ions and solutes inside the enclosed compartments. They can be used to study the permeability of certain membranes and transportation of ions or solutes found in different cells.
Lipid bilayers form in a spontaneous and self-assembled manner in aqueous environment. Its unique properties allow the formation of enclosed compartments. The sheet-like bimolecular structure called lipid bilayers or energetically favored because of the hydrophobic interactions. As mentioned previously, phospholipids, an amphiphloic moiety as a major class of membrane lipids, exist in an aqueous solution. The hydrophobic tails from two phospholipid bilayers interact with each other to form a hydrophobic center. Meanwhile, the hydrophilic heads line up with each other, form a hydrophilic coating on each side of the bilayer, and isolate the inner compartment from the outer environment.
The lateral diffusion, or the motion of moving laterally, of the biological membranes illustrates the fact that the membranes are not rigid and static. In fact, the membrane is not stable. There is a technique known as Fluorescence Recovery After Photobleaching (FRAP)assists to visualize the lateral diffusion of membrane proteins. An example of the experiment is as follow:
1) Label a specific cell component with fluorescence.
2) Use a intense beam of laser light to bleach, or destroy, the small part of the florescence labeled cell surface.
3) The intensity of bleaching recovers as the lateral diffusion of unbleached membrane proteins move into the region that has been bleached.
Transverse Diffusion describes the movement of molecules from one side the membrane to the opposite side. In comparison to the rapid movement of lateral diffusion, the speed of transverse diffusion is rather slow. The reason for the preservation of membrane structural asymmetry is due to the greater energy barriers formed in order to travel across membrane from one side to the other.
Cell division is a process that a cell gets divided and then duplicated. In prokaryotes, cells are divided by binary fission. In eukaryotes, the process becomes more complicated that there are three steps or periods for division. The cell grows during the period of inter-phase, when it absorbs nutrients for mitosis and duplication of DNA. Then the cells comes to mitosis phase. During this mitosis, the cell splits into two different daughter cells. Moreover, the chromosomes in its nucleus will also be divided into two equivalent parts, each into separate nuclei. Finally, the cell finishes division during cytokinesis.
Aging is due to continuous damage to various molecules in our cells, including proteins, lipids and nucleic acids. There are internal and external reasons for the damage to happen. The example for external reasons is oxygen, which is regarded as the necessity for human beings. However, when the oxygen absorbs only one or two electrons, making itself reactive, this kind of oxygen molecules will damage lipids, mutate genes, and destroy proteins, all leading to cellular injury.
On the other hand, the internal reasons are related to cell retiring. According to the data, cells stop working when they divide about fifty times. This phenomenon might be traced to the period when a cell copies its chromosomes to its daughters. However, the very ends of the chromosomes will not be copied, so daughters’ chromosomes are shorter, compared to their maternal cell. Telomere, at the ends of cells’ chromosomes, supplies the genetic information, which is the same as that on the parts of maternal chromosomes left. Nonetheless, when a cell’s telomeres get shrunk to a minimum size, the cell will stop dividing and lose its function. The picture to the right shows the telomeres in 46 human chromosomes.
There are two kinds of cell death: apoptosis and necrosis.
Apoptosis is programmed cell death that cells that undergoes self-destruction by regulation. At first, a cell shrinks and escapes from its neighbors. Later on, the surface of the cell breaks into fragments and also the nucleus then collapses. In the end, the whole cell disassembles. There will be some organelles cleaning up the remains. During apoptosis, unneeded cells or retired cells are eliminated efficiently without pain. Apoptosis plays a significant role in our body, which is self-destruction. When some cells in our body become infected, apoptosis can help eliminate them to avoid the virus spreading the whole body. However, viruses have several solutions to prevent apoptosis, such as stop cells’ suicide by confusing them with a similar “off” signal as that sent by apoptosis. Therefore, further research in apoptosis can promote the development of clinical medicine.
On the other hand, Necrosis is unplanned cell death. It happens when the outer membrane of a cell is unable to control the liquid flowing across it. The cell is then shrink and burst. Finally, contents inside will flow out, blending into the tissues nearby. The reasons causing necrosis might be traumatic injury, infection, chemical poison, etc.
Now we understand that some cells are created and some are killed. The truth is that these processes are designed well to keep our body healthy. The unbalance between apoptosis and mitosis will cause cancer.
Endocytosis is the creation of internal membranes from the cell's plasma membrane lipid bilayer. It is a way for plasma membrane lipids and integral proteins to be brought inside the cell. It is thus the opposite of exocytosis. This allows cells to do things like regulate the sensitivity of cells to ligands since receptors can be removed from the cell surface by endocytosis.
Plasma membrane buds, called caveolae, are on little creices on the surface of many mammalian cells. These can make up almost a third of a cells surface area. Given their structure, they are often involved in endocytic events.
Another way of internalizing in cells is called phagocytosis. In this method, material can be take in when 'invaginations' are formed around particles to be engulfed while using or not using the growth of surrounding membrane extensions.
1. Berg, Jeremy M. Biochemistry. 6th ed. W.H. Freeman, 2007.
2. Campbell, Neil A. Biology. 7th ed. San Francisco, 2005.
3. "Inside the Cell" of U.S. DEPARTMENT OF HEALTH AND HUMAN SERVICES National Institutes of Health
National Institute of General Medical Sciences
4. "Mechanisms of Endocytosis". Doherty and McMahon, MRC Lab of Molecular Biology, Cambridge, UK.
5.Inside the Cell, U.S.DEPARTMENT OF HEALTH AND HUMAN SERVICES
National Institutes of Health
National Institute of General Medical Sciences
6. http://en.wikipedia.org/wiki/Cell_cycle
7. http://en.wikipedia.org/wiki/Mitosis
8. http://commons.wikimedia.org/wiki/File:Three_cell_growth_types.png
9. http://commons.wikimedia.org/wiki/File:Telomere.JPG
10. Slonczewski, Joan L. "Microbiology: An Evolving Science." 2009
Eukaryotes derive their name from the fact that they contain a nucleus. The nucleus is also often the most prominent feature of eukaryotic cells viewed under a microscope. The nucleus is an organelle, an intracellular membrane-enclosed compartment with a specific function. The nucleus also contains chromatin. Chomatin is a complex of DNA and proteins. The nuclear membrane consists two concentric phospholipid membranes. Nuclei contains a region called the nucleolus, where ribosome assembly begins. Besides, the nuclear membrane contains nuclear pore complexes that allow for transport of material into and out of the nucleus. The metabolites and small proteins can diffuse through the NCPs. Large proteins that need to enter the nucleus are actively transported in through the NPCs.
The nucleus is a membrane-enclosed organelle found in eukaryotic cells. It contains the genetic material of the cell, organized as multiple long linear DNA molecules that coil around to form chromosomes. The transcription of RNA also takes place in the nucleus.
The eukaryotic nucleus contains the genetic information of the cell, and insulates it from the cytosol. It is in the nucleus where the nucleolus is contained; the site of RNA synthesis and DNA replication. During the interphase stage of the cell cycle, the genetic material is found in the form of chromatin. It is during mitosis and meiosis that the chromatin coil together, using histone proteins, to form tightly packed sister chromatids which then replicate, forming daughter cells.
The nucleus is known as the information storage organelle. it is enclosed by a double membrane- "nuclear envelope". Inside of the nucleus there is DNA. The DNA is bound by proteins- "histones".There are 5 types of histone proteins out of which H2A,H2B,H3 and H4 ,each repeated twice form a core of 8 histone proteins around which wraps the DNA double helix linked by Histone H1 linker DNA proteins.this unit of 8 histones wrapped by DNA is called 'nucleosome'.A repitition of nucleosomes makes a nucleofilament that futher coils to form DNA coils and supercoils that form chromosomes. The dark spot of the nucleus is the nucleolus- site or rRNA synthesis. The nucleus has its own internal skeleton known as the nuclear lamina. It also has its own transport system called nuclear pores.
The Nuclear Envelope is a double layered membrane that surrounds the nucleus. It consist of nuclear pores that regulate the transportation of substances such as RNA into and out of the nucleus. The nuclear envelope consists of an outer nuclear membrane and an inner membrane. Both nuclear membranes are "lipid bilayers". The outer nuclear membrane is extensive and is connected to the rough endoplasmic reticulum or ER. The inner nuclear membrane is continuous with the nuclear lamina consisting of various lamins. The lamina is an attachment site for the chromosomes that also stabilize the structure of the nucleus. The inner nuclear membrane contains different kinds of membrane proteins. The inner and outer nuclear membranes are connected through nuclear pores.
There are some bound
ribosomes
attaching to the outside of the nuclear envelope. These bound ribosomes function on inserting protein into and secreting protein out of the membrane.
The cell nucleus is an important organelle because it is where genes and their controlling factors are formed. In order to do so, it needs the helps of the stored components:
Chromosomes: store and organize genes that allow cell division.
The nucleolus, or plural nucleoli, is normally a circular structure composed of proteins and nucleic acids. Nucleoli are not typical organelles for the reason that they have no lipid membrane, making it with of the few non-membrane bound organelles in the cell. The nucleolus is located within the nucleus of eukaryote cells and is in charge of producing ribosomal RNA and the arrangement of ribosomes. The structure of the nucleoli can be seen using electron microscopy and fluorescent protein tagging can be used to view the dynamics of the nucleoli.
The nucleolus is the nuclear subdomain that assembles ribosomal subunits in eukaryotic cells. The nucleolar organiser regions of chromosomes, which contain the genes for pre-ribosomal ribonucleic acid (rRNA), serve as the foundation for nucleolar structure. The nucleolus disassembles at the beginning of mitosis, its components disperse in various parts of the cell and reassembly occurs during telophase and early G1 phase. Ribosome assembly begins with transcription of pre-rRNA. During transcription ribosomal and nonribosomal proteins attach to the RNA. Subsequently, there is modification and cleavage of pre-rRNA and incorporation of more ribosomal proteins and 5S rRNA into maturing pre-ribosomal complexes. The nucleolus also contains proteins and RNAs that are not related to ribosome assembly and a number of new functions for the nucleolus have been identified. These include assembly of signal recognition particles, sensing cellular stress and transport of human immunodeficiency virus 1 (HIV-1) messenger RNA.
The purpose of the ribosome is to translate messenger RNA (mRNA) to proteins with the aid of tRNA. In eukaryotes, ribosomes can commonly be found in the cytosol of a cell, the endoplasmic reticulum or mRNA, as well as the matrix of the mitochondria. Proteins synthesized in each of these locations serve a different role in the cell. In prokaryotes ribosomes can be found in the cytosol as well. This protein-synthesizing organelle is the only organelle found in both prokaryotes and eukaryotes, asserting that the ribosome was a trait that evolved early on, most likely present in the common ancestor of eukaryotes and prokaryotes. Ribosomes are not membrane bound.
Ribosomes are also play a key role in the catalysis of two important and crucial biological processes. They are responsible for peptidyl transfer, in which they form peptide bonds during protein synthesis as well as peptidyl hydrolysis; in this process, the ribosome releases the completely formed protein from the peptidyl transfer RNA after completion of translation. Translation is the process of converting mRNA (from transcription) to functional proteins; The three steps involved are initiation, elongation, and termination. Proteins are translated one amino acid (three base pairs) at a time. During this process, tRNA assists the ribosome by bringing the complementary bases to the ribosome as translation proceeds.
Because ribosome synthesis is a major metabolic activity that involves hundreds of individual reactions in eukaryotes, errors will occur eventually during these processes. To deal with the error in ribosome synthesis if it occurs, eukaryotes will degrade the erroreous ribosomes. In addition, not only the erroneous ribosomes will be degraded but also the excess ribosomes. Recent researches show that eukaryote cells enhanced many strategies to recognize specific dysfunctional or functionally deficit ribosomes for degradation[1].
Along with having more complicated assembly regulations than bacterial ribosomes, eukaryotic ribosomes are triggered differently than bacterial ribosomes in translation initiation. Eukaryotic ribosomes use a scanning mechanism and require at least nine initiation factors.
[2]
In bacteria, ribosomes can identify the right reading frame and know where to attach to a mRNA stran, by finding the Shine Dalgarno sequence on the mRNA. This ribosome binding site is upstream from the AUG start codon on the mRNA. In the bacterium E. Coli, the Shine Dalgarno site is a purine-rich nucleotide sequence—5’ AGGAGGU 3’, on the mRNA that is located four to eight bases ahead of the start codon. This Shine-Dalgarno sequence is complementary to the sequence 5’ ACCUCCU 3’ on the 16S rRNA, which is found in the 30S subunit of the ribosome.
[3]
Ribosomes usually comprise 2 protein subunits and some component of rRNA. The protein componenet of the ribosomes is first synthesized in the cytosol much like any other protein. The newly synthesized protein subunits are then transported from the nucleus into the nucleolus, the center of ribosomal RNA transcription. RNA polymerase are then used to polymerize the rRNA needed for the synthesis of the ribosome. Cells that have a high rate of protein synthesis have many ribosomes.
In the past, scientists studied ribosome to get a better understanding about its components, which is consists of 30S, 50S, 70S, and 100S. All are made of by the same particles but different concentration of Mg ions. 30S and 50S are small and large subunits of 70S while 100S is a collection of 70S.
Ribosomal structure varies between eukaryotes and prokaryotes. Eukaryotes have 40S (small) and 60S (large) subunits, together the subunits form the 80S ribosome.[4] Prokaryotes have 30S (small) and 50S (large) subunits. These subunits work together during translation to synthesize proteins.
EColi 70S Ribosome. The red is the 50S large subunit and the blue is the 30S small subunit
In prokaryotes, the small subunit (30S) and the large subunit (50S) together make the 70S ribosome. The 30S subunit is composed of 21 ribosomal proteins and 16S rRNA. Its function is to decode and decipher mRNA to determine the corresponding amino acid to the three bases in codons. The small subunit consists of the body (5’), the platform region (central domain), and the head (3’). Each component can be formed separately of each other. The small subunit also contains a 3’-minor domain consisting of the last two helices (44 and 45) and the end of the 3’ end of the rRNA. The structural components of the small subunit have been shown to be structurally independent from each other suggesting that during protein synthesis they move and conform relative to one another. The long 44 helix stretches across the small subunit from the body to the head functioning as a potential relay for information across the entire subunit. Unlike the small subunit, the large subunit is mostly rigid with mobility restricted its peripheral regions. The 50S subunit consists of 33 proteins and the 23S and 5S rRNA.
The mRNA binding site is located along the neck of the small subunit while the large subunit contains the peptidyl transferase center (PTC) where aminoacyl and peptidyl-tRNA attach. The three binding sites are A, P, and E. Binding site A attracts aminoacyl-tRNA, binding site P attracts peptidyl-tRNA, and binding site E, the exit site, attracts deacylated tRNA. The anticodon stem loop is attached to its complementary codon on the mRNA binding site of the small subunit while the amino acid end of the tRNA attaches to A and P sites both located in the PTC. The GTPase-associated center and the sarcin/ricin loop are located on the large subunit and help to stimulate GTP hydrolysis needed for elongation of the protein.
Ribosome assembly consists of transcription, translation, the folding of rRNA and ribosomal proteins, the binding of ribosomal proteins, and the binding and release of the assembly components to make the ribosome. There are two intermediates in ribosome assembly, in vitro and in vivo. One method of in vitro assembly of the 30S is to only use free rRNA and ribosomal proteins. Another is to combine 16S rRNA with purified and recombinant proteins and precursor 16S rRNA. With the different ways to assemble proteins, the assembly of ribosomes also varies with different temperatures. At low temperature of 0°C -15°C, the reconstitution intermediate (RI) particle, composed of 16S rRNA and 15 ribosomal proteins, is formed and settled to 21S-22S. Then the RI particle is rearranged after getting heated to 40°C and settled to 25S-26S. After completing these two processes, the RI particle together with the remaining proteins can form the 30S subunit.
In contrast, the in vitro assembly of the 50S subunit requires more steps and harsher conditions compared to the assembly mechanism of the 30S. There are three reconstitution intermediates each dependent on different temperatures and ionic conditions. RI50 (1), the first intermediate, yields the 33S particle. As RI50 (1) is heated, RI50 (2) forms and sediments at the 41S-43S. The remaining proteins form the RI50 (3) which create the inactive 48S particle. In order for the 48S particle to become the active 50S subunit, the temperature and concentration of magnesium needs to be increased along with the involvement of the 5S rRNA.
In Vitro and In Vivo Ribosomal Assembly Mechanisms
Like the in vitro assembly mechanism, the in vivo assembly of the 30S subunit has two intermediates (p130S and p230S) and the 50S subunit has three intermediates (p150S, p250S, and p350S). However, the reconstitution intermediates are not the same as in vitro. The intermediates of the 30S subunit yield 21S and 30S particles while the intermediates of the 50S subunit yield 32S, 43S, and 50S particles. The intermediates in the in vivo assembly are precursor rRNA which is different from in vitro which uses matured rRNA. To complete the mechanism of ribosome assembly, these precursor rRNA gets transformed in the polysomes.
A eukaryotic ribosome is composed of a 40S and 60S subunits, they both have a solvent exposed side and a subunit interface. The solvent exposed side contains a higher concentration of protein and RNA elements that are only in eukaryotes compared to the subunit interface.
The eukaryotic specific proteins are responsible for 40S structure being stabilized by tertiary contacts. The majority of the eukaryotic specific proteins and extensions are to interconnect other proteins in the 40S and 60S subunit. The 40S subunit has 14 of these interconnected proteins located in the head. eukaryotic specific proteins and extensions play a bigger role in 60S subunits because their protein mediated connections are more extensive and reach across the subunit. The 60S specific extensions consist of beta sheets and long alpha helices which are crucial in long distance tertierary interaction, meaning they have the ability to interact across the subunit. This trait is unique for eukaryotes because protein-protein contact is very rare in the prokaryote ribosome.
The 40S and 60S subunits are joined by eukaryote specific inter-subunit bridges that connects them thus forming 80s. This is done by eukaryotic specific bridges forming at the 40S and the RPL19 interacts with ES6E and RPL24 interacts with rpS6. The other two eukaryotic specific bridges connect by 60S segment ES31 that connects to rpSA and another 60S segment, ES41 that connects with rp58 from the 40S subunit. [5]
Ribosomal cofactors help facilitate with improper folding during in vitro. The improper folding during in vitro is a cause which results in vitro being much slower than in vivo. Ribosomal factors can be used as a “check point” to make sure the assembly is correct. Examples of ribosomal cofactors are DEAD-box proteins, chaperones, and GTPases. DEAD-box proteins are factors which help with proper folding of the RNA. An over expression of DEAD-box proteins can suppress the deletion of another strain. DEAD-box proteins (DBPs) are characterized by a core of approximately 350 amino acids, each containing at least 9 conserved amino acid motifs. Studies have shown that these proteins use the energy from ATP hydrolysis to rearrange inter- or intra-molecular RNA structures or dissociate RNA-proteins interactions. In eukaryotes, DEAD-box proteins are fundamental in the life of a ribosome, but in E. coli, they are often unnecessary. To date, five different genes encoding DEAD-box proteins have been identified in E.coli – SrmB, CsdA, DbpA, RhlE, and RhIB – which are versatile cofactors that have helped further the understanding of cellular processes in many cells because of their roles in protein biosynthesis and ribosome assembly. For instance, CsdA and SrmB are essential for proper cellular growth; cells deficient in these proteins exhibit severe ribosomal effects.
E.Coli Cofactor interaction of various DEAD-box proteins
Ribosomes played a big role in our evolution. The origin of ribosomes are said to have roots in the RNA world, preceding the existence of proteins. Ribosomes evolved in structure and function, alongside the formation of complex and versatile peptides. A huge breakthrough in the scientific study of ribosomes began when Venkatraman Ramakrishnan and his group determined the crystal structure of the ribosome, and described it in atomic detail. Uncovering the structure of the ribosome allowed scientists a closer look at the mechanisms involved with ribosome function. More specifically for example, according to "The ribosome goes Nobel" article by Rodnina and Wintermeyer, in the "Trends in Biochemical Sciences" journal, knowledge of the structure of the ribosome revealed details of the translation process like "how the ribosome checks for proper codon–anticodon interaction by establishing specific contacts in the decoding site". More recently, ribosomes prove to have huge clinical potential as targets for antibiotic products. With the overuse of antibiotics and antibiotic resistance constantly on the rise, ribosomes could be the basis of new antimicrobials that would retaliate against the infamous antibiotic resistance.
Ribosomes found in the cytosol that are not bound to any other organelle are known as free ribosomes. These ribosomes are known to produce non-hydrophobic proteins that ultimately function in the cytosol. An example would be the catalysts for the first steps of sugar breakdown.
Ribosomes which are attached to the endoplasmic reticulum or ERare known as ER Ribosomes. These ribosomes are what give rough endoplasmic reticulum its "rough" appearance. Proteins that are produced from the ribosomes of the rough ER are sent through the lumen of the endoplasmic reticulum, modified within the ER, and then is exported in a vesicle to the cis face of the golgi apparatus, where the protein undergoes further modification.
The Peptidyl-Transfer Reaction Catalyzed by the Ribosome
Enzymes transform their substrates through the active site residues. This transformation can allow for the direct participation in chemical catalysis, such as facilitation of proton transfer reactions and covalent chemistry. These active-site residues can form the covalent bond acting as general acid-base or as nucleophiles. The ribosomes undergo general acid-base or nucleophilic catalysis. The residues can also be structural complement to the transition state (TS), dissolve or reorganize water molecules, and decrease the delta S (entropy) by using the binding energy.
In the elongation process, the ribosome PTC catalyzes the aminolysis of the ester bond: the alpha amino group of the A-site aminoacul tRNA acts as nucleophile and attacks the P-site peptidyl tRNA at the carbonyl carbon of the ester bond that connects the tRNA and the peptide. In order to efficiently form peptide bonds amines react with esters. This reaction is fast with the help of the ribosome because it makes the reaction rate increase by approximately 106 - 107 folds. Before the peptide bond is formed, the aminoacyl tRNA connects to the A site at a rate of 10 s-1 which is much slower than the rate of the tertiary complex forming a peptide bond. The slow rate of reaction allows for the minimal use of substrate analog which is short RNA fragments that is similar to the tRNA CCA 3’ end or the alpha amine substituted with a hydroxyl group. The minimal substrates consisted of puromycin (Pmn), C-puromycin (C-Pmn), and CC-puromycin (CC-Pmn). These substrates bind to the ribosome fast to react with the P-site substrate in order to allow for the direct analysis of their chemistry relationship.
Recently, methods of analysis using kinetic, biochemical, genetic, and computation allow for more progress in ribosome crystallography. However, there are still many questions regarding the proton transfer mechanism. Peptidyl transfer by the ribosome accompanies a decrease in the entropy activation, which provides more understanding of ribosome catalysis involves defining the molecular mechanism that make entropy of activation lower by taking into consideration the substrate, position of the active site, desolvation, and electrostatic shielding.
1)Peptidyl Transfer
2)Peptide Release
3)Structures of transition state analogs used to study the ribosome
Degradation of erroneous and excess ribosomes in eukaryotes
When cells are in stress, disease condition, and normal condition, RNA damage occurs. Exposing to ultraviolet light, oxidation, chlorination, nitration and alkylation result in chemical modifications to nucleobases, generating the potential of triggering ribosome degradation pathways.
There are several sources of errors in ribosome synthesis such as the alternation in rRNA sequences (occur in cis) and the failure to bind to or loss of an assembly factor or ribosomal protein (occur in trans). Beside these main sources, specific growth conditions can also cause certain effects in ribosome synthesis. For example, starvation requires excess ribosomes which are turned over efficiently[6].
Potential error-occurring process in Ribosome synthesis
mRNA decoding and peptidyl-transfer reaction are two subunites that have specialized function in ribosome translation[7]. Each eukaryote ribosome is composed of 4 rRNAs and ~80 ribosomal proteins. The processes of synthesizing, maturizing, transporting individual ribosomes and assembling into ribosomal subunits requires the contribution of ~200 proteins trans-acting cofactors and a large amount of small nucleolar RNAs (snoRNAs). These processes engage in hundreds of individual error-causing reactions[8][9].In addition, many ribosomal proteins also perform non-ribosomal functions, so the functions in these processes which are not directly connected to ribosome biogenesis are currently being assigned to ribosome synthesis factors (RSFs). RSFs include connections to cell cycle progression, pre-mRNA splicing, DNA damage response, nuclear organizing and telomere maintanance. Due to the large number of reactions that occur in ribosome assembling pathway, the possibility of error occurring is very high such as the potential harms for cell viability and human health. For example, inability to bind correctly to or loss of a synthesis factor can cause the production of erroneous ribosomes such as lacking part of ribosomal proteins or carrying misfolded rRNA which will affect on translation. As a result, cells develop control mechanism to avoid such problems. For example, instead of participating in later steps, synthetic factors that involved in late cytoplasmic ribosome assembly steps can first bind with pre-rRNAs at early stages, bringing pre-ribosomes to productive synthesis pathways[10].
Mutation in cis: non-funtional ribosomal RNA decay
There are many pathways that have been known to control the structural integrity of mature RNA molecules[11]. One of the pathways that control the decay of mRNAs is the "no-go" decay (NGD), which the mRNAs that cause the degradation of specific ribosomes[12].
LaRivie`re et al. introduced the way to test whether or not mutations in functional and non-functional ribosomal sites affect rRNA stability which is performing substitutions in the decoding site and peptidyl transferase centre at the position that are essential for ribosomal function in bacteria. This experiment gave result in the identification of non-functional rRNA decay, a pathway that detects and eliminates dysfunctional parts of mature ribosomes[13].
Mutations in trans: The TRAMP-exosome pathway and nucleolar surveillance
Aberrant nucleolar and nuclear pre-rRNAs are found to be degraded actively by a nuclear surveillance mechanism that involves the addition of unstructured oligoadenylate tails at the 3’-end of flawed pre-rRNAs. This is done with the TRAMP complexes’ polymerase activity, and after with their degradation by the RNA exosome. TRAMP conplexes consist of a poly(A) polymerase, a zonc-knuckle-containing and putative RNA-binding protein, and the DEVH helicase Mtr4. The pathway goes from having the addition of short poly(A) tails with the actual binding between TRAMP complex and RNA, and these two commit deviated molecules to degradation. This is done by separating them from the normal RNA and by stirring exosomal activity[14].
The decay of bulk ribosome and pre-ribosome by ribophagy and PMN
Growth-inhibiting conditions cause ribosome synthesis to shut down. The synthesis of complex molecules from simple molecules such as amino acids and sugars is obstructed and existing pre-ribosomes and mature ribosomes are directed towards bulk degradative pathways. To manage stress, and to adapt to a new environment, pre-ribosomes and mature ribosomes are passed on to be recycled for important cellular components. Ubiquitin, a small regulatory protein essential to 25S NRD (Nonfunctional RRNA Decay), is also responsible for bulk ribosome degradation.[15]
Autophagy is a catabolic mechanism that takes unnecessary or dysfunctional cellular components and performs cellular degradation through the lysosome, or the vacuole in yeast and plants.[16] Starvation conditions cause large portions of the cytosol, along with protein, to assemble and whole organelles, such as ribosomes and mitochondria, are recycled through two major types of autophagy: microautophagy and macroautophagy. Macroautophagy involves the development of a double membrane around the organelle, this together known as an autophagosome. [17] This isolates the organelle and delivers it to the lysosome, which engulfs the organelle through endocytosis for its breakdown and recycling. [18] In contrast, during microautophagy, the lysosome directly engulfs the cytoplasmic material, through the inward folding of the lysosomal membrane. [19] Because lysosomes and vacuoles contain non-specific enzymes for hydrolysis, autophagy can essentially degrade any biomolecule. Macroautophagy and microautophagy are both crucial to a cell's survival during starvation. Both mechanisms associate common trans-acting factors, and both are essentially bulk degradative processes. However, studies show that selective types of both channels exist. [20]
Ribophagy is the process in yeasts in which both small and large ribosomal subunits are are delivered to the vacuole through selective macroautophagy. This process is caused by prolonged nitrogen starvation and specific only to ribosomal subunits. [21] Small and large ribosomal subunit ribophagy depends on different and distinct pathways. The increased turnover kinetics of large subunits are dependent upon the Ubp3 ubiquitin protease and its activator Bre5. Increased turnover rates of small subunit are scantly affected when mutations arise in UBE3 or BRE5, indicating independent pathways of ribophagy. However, for small ribosomal subunit ribophagy, the effector remains unknown.[22] As Upb3 deubiquitylation necessary for 60S ribophagy, cells deficient of Upb3 have an increased ubiquitylation level of many ribosome-associated proteins which have yet to be identified. It is believed that the deubiquitylation of Upb3-Bre5 targets aids autophagosomes with the packing of ribosomes, or allows the maturation of autophagosomes and/or its merging with the vacuole. [23]
Alterations in RNA can cause ribosome degradation, some of which can lead to serious diseases. RNA damage are not only results of stress and disease situations, as they occur during normal cell growth as well. Exposure to ultraviolet light, chlorination, oxidation, alkylation and nitration can all result in the introduction of chemical modifications to nucleobases, as well as RNA-RNA and RNA-protein crosslinks, into RNA and RNPs. [24] Many believe that RNA oxidation could be associated with disease progression, and that many factors, including the extent of association with proteins or iron, may be causes to leaving RNA susceptible to oxidative damage. Some cases of ribosomal RNA damage inflicted by UV radiation and oxidation have lead to human neurodegenerative diseases, such as Alzheimer's, Parkinson's, and atherosclerotic plaques. [25] Reactive oxygen species (ROS) is developed by chronological aging, oxidative stress and other apoptotic stimuli. Yeast cells that are exposed to high levels of ROS fragment Mature 5.8S and 25S rRNA considerably. [26] Yeast cells that are given anti-metabolites and the chemotherapeutic agent 5-fluorouracil (5-FU) accrue polyadenylated pre-rRNAs. In exosome complexes that are hypersensitive to the drug, these accumulations are distressed, proposing that nucleolar surveillance is what is responsible for the degradation of 5-FU. Many nucleolar stresses, such as drug-based interference of pre-RNA processing and rRNA synthesis, have lead to nucleolar breakdown, p53 stabilization and cell cycle advancement defects and/or apoptosis. [27]
In the core of every ribosome, which is the protein factory, there are ribosomal proteins. Recent research indicates that these core proteins can differ between ribosomes under different growth conditions and that these differences confer the ability to more efficiently translate specific subpopulations of mRNA. Ribosomes exist in every living organism for the same purpose, to synthesize proteins from mRNA. Previously, it was thought that ribosomes were homogenous and lacked any structural diversity. Because function follows structure, these differences should be expected to alter a given ribosome’s affinity for more complementary mRNA strands. In addition to core variants, post-transcriptional modification of the ribosomal RNAs leads to additional structural differentiation, and therefore functional specialization. For ribosomes to be functionally specialized it requires that the producing cell be sensitive to changing environmental conditions such that the ribosomes being synthesized exhibit biochemical differences that affect translation.
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3056915/
Edward Ki Yun Leung, Nikolai Suslov, Nicole Tuttle, Raghuvir Sengupta, and Joseph Anthony Piccirilli. "The Mechanism of Peptidyl Transfer Catalysis by the Ribosome."
http://www.ncbi.nlm.nih.gov/pubmed/21548786
Sebastian Klinge, Felix Voigts-Hoffmann, Marc Leibundgut and Nenad Ban. "Atomic structures of the eukaryotic ribosome."
Zahra Shajani, Michael T. Sykes, and James R. Williamson. "Assembly of Bacterial Ribosomes."
http://www.ncbi.nlm.nih.gov/pubmed/21529161
Gilbert WV. "Functional Specialization of Ribosomes?"
Trends Biochem Sci. 2011 Mar;36(3):127-32. Epub 2011 Jan 16.
PMID: 21242088 [PubMed - indexed for MEDLINE]
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3056915/
↑ Steitz, T.A. (2008) A structural understanding of the dynamic ribosome machine. Nat. Rev. Mol. Cell Biol. 9, 242–253
↑Henras, A.K. et al. (2008) The post-transcriptional steps of eukaryotic ribosome biogenesis. Cell. Mol. Life Sci. 65, 2334–2359
↑Strunk, B.S. and Karbstein, K. (2009) Powering through ribosome assembly. RNA 15, 2083–2104
↑ Lafontaine, D. et al. (1995) The 18S rRNA dimethylase Dim1p is required for pre-ribosomal RNA processing in yeast. Genes Dev. 9, 2470–2481
↑Doma, M.K. and Parker, R. (2007) RNA quality control in eukaryotes. Cell 131, 660–668
↑Doma, M.K. and Parker, R. (2006) Endonucleolytic cleavage of eukaryotic mRNAs with stalls in translation elongation. Nature 440, 561–564
↑LaRiviere, F.J. et al. (2006) A late-acting quality control process for mature eukaryotic rRNAs. Mol. Cell 24, 619–626
↑Lafontaine, Denis. "Elsevier: Article Locator." ScienceDirect.com | Search through over 10 million science, health, medical journal full text articles and books.. N.p., n.d. Web. 20 Nov. 2012.
↑Lafontaine, Denis L.J. "A ‘garbage Can’ for Ribosomes: How Eukaryotes Degrade Their Ribosomes." Trends in Biochemical Sciences 35.5 (2010): 267-77.
↑Lin NY, Beyer C, Gießl A, et al. (September 2012). "Autophagy regulates TNFα-mediated joint destruction in experimental arthritis". Ann. Rheum. Dis.. doi:10.1136/annrheumdis-2012-201671. PMID 22975756
↑Česen MH, Pegan K, Spes A, Turk B (July 2012). "Lysosomal pathways to cell death and their therapeutic applications". Exp. Cell Res. 318 (11): 1245–51. doi:10.1016/j.yexcr.2012.03.005. PMID 22465226.
↑Mizushima N, Ohsumi Y, Yoshimori T (December 2002). "Autophagosome formation in mammalian cells". Cell Struct. Funct. 27 (6): 421–9. PMID 12576635.
↑Česen MH, Pegan K, Spes A, Turk B (July 2012). "Lysosomal pathways to cell death and their therapeutic applications". Exp. Cell Res. 318 (11): 1245–51. doi:10.1016/j.yexcr.2012.03.005. PMID 22465226.
↑Lafontaine, Denis L.J. "A ‘garbage Can’ for Ribosomes: How Eukaryotes Degrade Their Ribosomes." Trends in Biochemical Sciences 35.5 (2010): 267-77.
↑Kraft, Claudine, Anna Deplazes, Marc Sohrmann, and Matthias Peter. "Mature Ribosomes Are Selectively Degraded upon Starvation by an Autophagy Pathway Requiring the Ubp3p/Bre5p Ubiquitin Protease." Nature Cell Biology 10.5 (2008): 602-10.
↑Lafontaine, Denis L.J. "A ‘garbage Can’ for Ribosomes: How Eukaryotes Degrade Their Ribosomes." Trends in Biochemical Sciences 35.5 (2010): 267-77.
↑Kraft, Claudine, Anna Deplazes, Marc Sohrmann, and Matthias Peter. "Mature Ribosomes Are Selectively Degraded upon Starvation by an Autophagy Pathway Requiring the Ubp3p/Bre5p Ubiquitin Protease." Nature Cell Biology 10.5 (2008): 602-10.
↑Wurtmann, Elisabeth J., and Sandra L. Wolin. "RNA under Attack: Cellular Handling of RNA Damage." Critical Reviews in Biochemistry and Molecular Biology 44.1 (2009): 34-49.
↑Nunomura, Akihiko, Tim Hofer, Paula I. Moreira, Rudy J. Castellani, Mark A. Smith, and George Perry. "RNA Oxidation in Alzheimer Disease and Related Neurodegenerative Disorders." Acta Neuropathologica 118.1 (2009): 151-66.
↑Mroczek, S., and J. Kufel. "Apoptotic Signals Induce Specific Degradation of Ribosomal RNA in Yeast." Nucleic Acids Research 36.9 (2008): 2874-888.
↑Lafontaine, Denis L.J. "A ‘garbage Can’ for Ribosomes: How Eukaryotes Degrade Their Ribosomes." Trends in Biochemical Sciences 35.5 (2010): 267-77.
Ribosome synthesis is a multi-step, error-prone process. The ribosome is comprised of 2 subunits of unequal sizes. These subunits carry out specialized functions within translation such as mRNA decoding for the smaller unit and a peptidyl-transfer reaction for the larger subunit. In eukaryotes, the ribosomes consist of 4 rRNAs and about 80 ribosomal proteins. To synthesis, mature and transport the individual components of ribosomes and assemble them, requires the intervention of approximately 200 protein trans-acting factors as well as numerous amounts of small nucleolar RNAs (snoRNAs). These are involved in the hundreds of individual, error-prone, reactions. There are some functions in processes that don't connect directly to ribosome biogenesis and are then assigned to ribosome synthesis factors such as connections to cell cycle progression, or pre-mRNA splicing and DNA damage response. Because there are so many reactions within a ribosome assembly pathway, the possibility to introduce a mistake with potential deleterious consequences are immense. Such things such as failure to bind or loss of a synthesis factor could lead to a structurally defective ribosome that hold functional consequences in translation. In response to such problems, the cells evolved multiple quality control mechanisms. However, that is not to say that mutations only occur during synthesis as mutations can also occur as a consequence of exposure to genotoxic stress.
An example of creating a way to minimize defects would be in a late assembly step, the cell can bind pre-rRNAs at early nucleolar stages thus committing pre-ribosomes to productive syntheses pathways. Another case would be to bind pre-ribosomes to monitor and tether the structural integrity of ribosomal protein-binding sites. This case would be where there are trans-acting factors with partial homology to ribosomal proteins. This is because defects can delay the binding of trans-acting factors.
Eukaryotes have multiple methods in which they degrade their ribosomes. There are so many reactions in the ribosomal pathways that the possibility of mistakes and mutations are great. Many incidents can occur when mutations occur in cis such as the alteration of rRNA sequencing. Surveillance pathways monitor the structural and functional integrity of RNA. For mutations in the cis conformation, two possible pathways may occur depending on the size and type of the RNA.
There are multiple surveillance pathways that have been described that monitor the integrity of mature RNA molecules, one pathway that monitors mRNA is the "no-go' decay pathway or NGD. This is where mRNAs that induce stalled ribosomes are degraded. LaRivière et al. introduced substitutions in decoding sites and peptidyle transferase center at positions which are necessary in bacteria for ribosome functions. This is done to test if mutations in functionally relevant and conserved ribosomal sites affect the rRNA stability. As a result, the procedure led to the identification of non functional rRNA decay or NRD. In the mRNA NGD, stalled ribosomes triggers initiating endonucleolytic cleavage events on the defective mRNA at the pause site. Then followed by exoribonucleoytic digestion of the 5'- and the 3'- cleaved mRNA products. The RNA exosome digests the defective cleave at the pause site and the 5'->3' exoRNase Xrn1 at the 5'- and 3'- cleaved mRNA. RNA exosome is a conserved multiprotein 3'->5' exoRNase complex active in the synthesis, degradation and surveillance of most classes of cellular RNAs. For NGD, the key components include Dom34 and Hbs1. This however is not the case for 25S NRD, but is true for 18S NRD. As such it indicates two distinct pathways.
In 18S NRD, Dom34 acts together in the same pathway with Hbs1 and they interact in vitro and in vivo. The small molecule inhibitors of translation stabilize the 18S but not the 25S NRD substrates and as a result provides further evidence that 18S NRD activation requires elongating ribosomes and that 18S and 25S NRD are mechanically different. 18S NRD has been linked with cytoplasmic exoRNase Xrn1 and Ski7 as delting both Hbs1 and Ski7 enhances the stabilization of the 18S NRD substrates. Both 18S NRD and mRNA NGD accumulate in the P-bodies that are conserved RNA-protein cytoplasmic granules that contain untranslated mRNA's with a set of repressors. 25S NRD substrates however don't co-localize to P-bodies.
LaRivière's work allowed scientists to discover the "non functional rRNA decay" pathway or NRD. NRD emphasizes on detecting and removing developed ribosomes. NRD is similar to NGD however they both have different kinetics which can be attributed to the higher complexity and compaction of mature ribonucleoprotein particles. NRD has two pathways one in which focuses on the small ribosomal subunits and the other which modifies large ribosomal subunits. There are two main degradation systems for the larger ribosomal subunits: 1. ubiquitin-proteasome system (UPS) and 2. autophagy. UPS targets short lived proteins and involves ubiquitlation and autophagy that degrades long lived proteins.
When errors occur in the trans conformation it can signify a failure to bind to or loss of and assembly factor or ribosomal protein. The TRAMP surveillance revolves around the addition of unstructured oligoadenylate tails at the 3'-end of the destructive pre-rRNAs from a poly(A) polymerase activity. Afterwards this method of tagging leads to the degradation performed by the RNA exosome. The addition of these polyA tails allows one to distinguish between normally functioning RNA and then following with a stimulation by exosomal activity. It is suggested that defected RNAs will go through multiple rounds of TRAMP-mediated polyadenylation and then digested by the exosome to increase the degradation effect. There are certain cases that surveillance takes place in a specialized nucleolar domain called "No-body" that contains much TRAMP and exosomal components.
There are five pathways of rRNA decay described to date.(i)Nucleolar and nuclear pre-40S and pre-60S ribosomal units are monitored actively by the "TRAMP-exosome' pathway. If a misfolded pre-ribosomal unit is identified, TRAMP binds to the molecule followed by the polyadenylation of the 3' ends of defective rRNAs in a step that stimulates both the recruitment and decay of the exosome. It is still not understood how TRAMP detects defective ribosomes. Polyadenylation occurs both at normal and cryptic pre-RNA sites. Differences in RNA Polymerase I activity can activate cryptic cleavage sites. Cytoplasmic mature subunits carrying cis mutations are monitored by the NRD pathway.(ii) During 18S NRD, small subunits with errors along the mRNA are identified by Dom34 and Hbs1. These errors are cleaved endonucleolytically by an unknown activity (thought to be similar to mRNA NGD). The releases products are digested by Xrn1 and the RNA exosome assisted by its cofactor Ski7. (iii) During 25S NRD, defective 60S subunits are targeted for proteasomal degradation by Rtt101-Mms1-mediated ubiquitylation of unidentified associated ribosomal components. During conditions of starvation in the cell, excess ribosomes are turned over to ribophagy and PMN. (iv) Ribophagy is a type of macroautophagy that involves the engulfment of cytosolic fractions to the vacuole. Inside the vacuole, the components are recycled. (v) In PMN, a specific type of microautophagy, a portion of the nuclear envelope is pinched off by the vacuole. This creates a specialized organelle called the NVJ that matures into a vesicle. Finally, the mature vesicle is degraded by resident hydrolases.
RNA damage occurs under normal cell growth as well as during stress and in disease situations. Chemical modifications to nucleobases are introduced into RNA and RNPs from exposure to ultraviolet light, oxidation, chlorination, nitration, and alkylation. These alterations all constitute potential physiological triggers to ribosome degradation pathways. Neurodegenerative diseases such as Alzheimer's and Parkinson's have been correlated to damage in rRNA sequences because of exposure to UV light or oxidation. RNA oxidation has been speculated to be involved in disease progression and that RNA susceptibility to oxidative damage is influenced by various factors including the degree of association with protein protection. Mature 5.8S and 25S rRNA are fragmented heavily in yeast cells exposed to high levels of a reactive oxygen species generated by oxidative stress (including exposure to hydrogen peroxide and menadione), chronological aging, and other apoptotic cues. Yeast cells treated with the anti-metabolite and chemotherapeutic agent 5-fluorouracil (5-FU) accumulate polyadenylated pre-rRNAs. This accumulation is exacerbated in exosome mutants, that are hypersensitive to the drug. This suggests that the degradation of 5-FU containing RNAs happens through nucleolar surveillance. Nucleolar dysfunction in cells has been correlated with cancer. Various nucleolar stresses such as drug mediated interference of rRNA synthesis, pre-rRNA processing, or inhibition of ribosome synthesis factor function, lead to nucleolar disruption. Ultimately, the cell cycle becomes defective.
An interesting regarding this phenomena is the observation of an increased abundance of polyadenylated rRNA fragments in the gut of western honey bees infected with colony collapse disorder (CCD). The insect guts serves as a primary interference with the environment taking in all the pesticides from outside. CCD has been linked to picorna-like viral infections known to hijack cellular ribosomes. This in addition to the use of pesticides could trigger a ribosome degradation response.
Ribosome synthesis is a major cell activity that can enforce quick energy drain with little regulation. Control is exerted at the level of synthesis, assembly of the pieces, and surveillance of the final product. Ribosome synthesis has evolved to be fully integrated with complex nutrient sensing mechanisms. In the prescence of defective ribosomes, damaged ribosomes, or abundance of mature ribosomes, they are targeted for rapid breakdown and recycling. Many surveillance pathways exists that either select excess or defective ribosomes. The pathways are even intricate enough to survey large or small subunits of the ribosome. The most important concept is that we understand how all these pathways interconnect in the ribosome's synthesis.
Ribosomes are present in both eukaryotic and prokaryotic cells. However between the two exists differences in the functions and characteristics of the ribosomes. In eukaryotic cells ribosomes are assembled in the nucleus and transferred to the cytoplasm where they finish maturation. Maturation includes trans acting shutting factors, transport factors, incorporating the rest of the proteins in ribosomes, and the final step in rRNA step process. Recent research, for example on the large ribosomal subunit has confirmed that 60S subunit is transported from the nucleus using an inactive state. After the subunits reaches the cytoplasm it leads to events that cause it to be transitionally competent.
In cells the ribosome is responsible for the last step in decoding information from genes in proteins. Ribosomes are made of subunits, which are complex and made of RNA and proteins. The two subunits each have a distinct job they are responsible for. Small 40S subunit is used to decode and the large 60S is responsible for polypeptide syntheses. Using structural analyses of prokaryotic ribosomes provides detailed insights for the mechanisms of the ribosome functions, however what we know about the vivo assembly of ribosomes is still not as well known and is still rudimentary.
Biogenesis begins with the transcription of pre-RNA that undergoes co-transcriptional folding, modification and assembly with r-proteins forming the two subunits. In bacteria assembly of the subunits need a few trans-acting factors. However in eukaryotic cells it is a complicated process which requires all three RNA polymerase and over 200 trans acting factors. Thus helping the maturation and intercellular transport if the subunits.
Ribosomes in eukaryotic cells are assembled in the nucleolus at the rRNA transcription. The released pre-ribosomes although seeming preassembled required maturation steps in the nucleoplasm and the cytoplasm. In the 1970s Planta and Warner's work led to the identification of the first pre-ribosome, 90S particle. 90S is processed to give the smaller 66S and 43S particles. These being the precursors to the mature 60S and 40S subunits. The particles contain pre-rRNA, r-proteins and various amount of trans acting factors. In the early 1990s applying genetic approaches in buddying yeast allowed for the identification of various trans acting factors which led to a better and more clear understanding of the high ordered steps in the processes involved for rRNA. Even though these advances were accomplished, the composition and make up of pre-ribosomal particles still remains unknown and a mystery until the recent decade. With tandem affinity purification, TAP, in combination with mass spectrometry has allowed us to isolate and analyse the composition of maturing pre 60S and pre 40S in buddying yeast.The analyses helped in the ordering of the pre-ribosomal particles in the 60S and 40S pathways, allowing us to picture the highly complex assembly process.
In yeast, RNA polymerase I in the nucleolus helps transcribe 35S. The transcribed rRNA is methylated, pseudo-uridylated, and loaded with r-protein and trans acting factors which allows it to form 90S. 90S particle has r-proteins and trans acting factors which are important for the 40S biogenesis pathway. The cleavage of the rRNA at the A2 site thus releases the pre 40S particle which the maturation and biogenesis are independent of the 60S. Once pre 40S is released the remaining pre-rRNA assembles with large subunit r-proteins and the biogenesis factors form the pre 60S particles. Differently, pre 40S particles undergo few compositional changes as they move from the nucleoplasm. Compared to the pre 60S, pre 40S are exported to the cytoplasm. In contrary pre 60S particles are associated with about 100 trans acting factors along the biogenesis pathway and also change in composition as they move through the nucleoplasm to the nuclear pore complex.
In biogenesis there are different stages that take part in the nucleus and the cytoplasm. In the stages the trans acting factors are released from pre-ribosomal particles and are recycled for the new rounds of biogenesis. The events are caused by enzymes that consume energy that are associated with maturing pre-ribosomal particles. The site of these events of the enzymes and the precise function of them in ribosome maturation still remains unknown to us. Research in the field has shown that two large AAA-ATPases Rix7 and Rea1 implicates in maturation of the pre 60S subunit. Rix7 seems to strip Nsa1 fromt he subunit in the nucleolar transition, and Rea1 is believed to drive pre 60S particles to export competence by the removal of Rsa4. The AAA-ATPases is this believed to contribute directly to the sequential reduction of the complexity of the pre-ribosomal particles, before they are removed from the nucleus.
Once pre-ribosomal subunits are produced in the nucleus, they are transported into the cytoplasm through the NPC (Nucleaer Pore Complex). The NPC is the largest protein complex in the cell an dis responsible for the protected exchange of components between the nucleus and cytoplasm. It also serves to prevent the transport of material not destined to cross the nuclear envelope [1]. It is found that for the nuclear export of r-subunits, unique nucleoporins are needed to make the export of both subunits to happen. Nucleoporins are simply the proteins of the NPC. Also, researcher have discovered that Ran GTP-GDP cycle is required. Pre-40S and pre-60S particles are exported independently of each other, however, they both need to have the general nuclear export factor Xpo1 or Crm1 that directly recognizes nuclear export sequences.[2] Nuclear export sequences are amino acid sequenecs that label a protein for export into the cytoplasm from the nucleus. Nmd3 is the only known Crm1 adapter for the pre-60S particle. However, there are at least three NES-containing trans-acting factors that function as Crm1 adapters in pre-40S export. Those trans-acting factors include Ltv1 (in humans), DIM2 and RIO2.[2] An interesting fact is that there is an redundancy in 40S export adapters as the nature of Ltv1 is almost non-essential. In order to achieve efficient export of pre-ribosomal subunits requires multiple receptors. For example, in budding yeast, pre-60S particles use additional factors that assist the nuclear pore complex for its export business.
Maturation of pre-ribosomal subunits at the cytoplasmic level
During the early stage of biogenesis, many of the trans-acting factors that are related with pre-ribosomal particles are released from the nucleus and can be recycled back. This process happens before nuclear export. There is a few factors remain related to to the pre-ribosomal subunits as they go into the cytoplasm. We will look at the maturation of the pre-60S subunit and pre-50S subunit independently.
Pre-60S suniunits enter the cytoplams with a entourage of non-ribosomal factors that must be released by unique factors in the cytoplasm. It should be emphasized that several ribosomal proteins are needed to add functionality to the subunits. The steps in the maturation of the pre-60S subunit are shown as below.
Pre-60S particles exit the nucleus and enter the cytoplasm
In the cytoplasm, a third essential AAA-ATPase is introduced to pre-60S particles
The trans acting factors that are associated with pre-ribosomal particles in early biogenesis are released and recycled to the nuclease before nuclear exportation, but some factors are still associated with the particles as they enter the cytoplasm. By releasing and recycling the factors as well as the assembly of the remainder of the r-proteins and the finals processing of RNA constitute the "cytoplasmic maturation steps" in the ribosome biogenesis pathway. The steps needed are not only crucial for complete maturation of subunits but also because if it fails to recycle a factor the nucleus it leads to its depletion from the nucleolar, thus inducing lays in pre-rRNA processing, defects in assembly, and impaired nuclear export.
ATPases and GTPases do not dependent on each other and therefore the events driven by ATPases and GTPases can occur without a specific order. However, some evidence shows that coupling of these events could happen. Drg1 a type of AAA-ATPase, blocks Rlp24, Nog1, and Arx1 from recycling and also blocks Tif6 from recycling to an extent. With this information, it is possible to start ordering the events. First, Drg1 releases Rlp24, and the loading of Rlp24 into subunits brings Rei1. Arx1 is then released when Rei1 works with Jjj1 and Ssa1/Ssa2. Arx1 being in the subunit eventually hinders Tif6 from being released. Starting with Drg1 releasing Rlp24, a series of events takes place and eventually it gets to the release of Tif6.
Since it is extremely crucial that translation of the genetic code done correctly, it is reasonable to speculate that quality-control mechanisms are evolved by eukaryotic cells to monitor ribosome biogenesis. Few research proved that cytoplasmic maturation steps in 40S and 60S biogenesis pathways are done by activation of subunits. This is accomplished when inhibitory factors are removed and functionality is added. Transfer of only the functional ribosomal subunits is guaranteed with the control of these cytoplasmic maturation steps.
Ribosomally synthesized natural products (RNPs) are a class of peptides that are of interest to scientists because of their great diversity courtesy of post-translational modifications (PTMs) like hetero- or macrocy-clization, dehydration, acylation, glycosylation, halogenation, prenylation, and epimerization. These RNPs have great potential because PTMs with respect to structure and biological activity compared to the traditional 20 amino acids. They are also favorable because RNPs have relatively short biosynthetic pathways and like to interact with numerous other compounds.
A group among RNPs that is of particular interest is the lantipeptides. They are polycyclic peptides that contain the thioether cross-linked amino acids meso-lathionine (Lan) and (2S,3S,6R)-3-methyllanthionine (MeLan). Lantipeptides are all synthesized on the ribosome as a precursor LanA peptide, which contains both a leader peptide, the N-terminal portion of the precursor peptide important for production, but unmodified and cleaved from the final product and a core peptide, the C-terminal portion of the precursor peptide that becomes the final product after modification and leader peptide removal. There are four classes of lantipeptides: class I, class II, class III, and class IV. Class I lantipeptides perform serine/threonine dehydration and thioether cyclization by the dehydratase LanB and the cyclase LanC, respectively. Class II lantipeptides have LanM, a single bifunctional lantipeptide synthetase containing N-terminal dehydratase and C-terminal LanC-like cyclase domains. Class III lantipeptides are modified by a trifunctional synthetase with an N-terminal lyase domain, a central kinase domain, and a putative C-terminal cyclase domain. Finally, class IV lantipeptides have LanL, which contains N-terminal lyase and kinase domains as in class III, but its C-terminal cyclase domain is analogous to LanC.
Lantipeptides were originally thought to be only produced by a specific group of gram-positive Firmicutes, but genomic analysis has shown that other organisms also produce lantipeptides as well like actinomycetes, bacteroidetes, and chlamydiae. A recent study identified lantipeptide biosynthetic genes in 478 of 1,466 bacterial genomes. This shows how abundant lantipeptides are in biological organisms and can be found and harvested from a pool of organisms for researchers to use to do experiment involving these products.
Lantipeptides are interesting to scientists because they have antimicrobial properties, but researchers are finding numerous other uses for them as well. For example, there is interest in using lantipeptides as chemotherapeutics. This idea came from the use of the lantipeptide nisin as a food preservative for more than 50 years because of its use has not presented significant microbial resistance.
Lantipeptides also have a lot of potential in the pharmaceutical industry. They have been proven effective against gram-positive bacteria, including drug-resistant strains like Staphylococcus, Streptococcus, Enterococcus, and Clostridium, and certain gram-negative pathogens like Neisseria and Helicobacter. A few drugs made from lantipeptides are duramycin for the treatment of cystic fibrosis and a derivative of actagardine for the treatment of Clostridium difficle Mutacin 1140 is currently in the developmental stages for the treatment of gram-positive bacterial infections. Other than pharmaceuticals, lantipeptides have applications in agriculture, veterinary medicine, and molecular imaging, proving their diversity in biochemical activity.
Researchers have studied the mechanisms by which lantipeptides conduct biological activity and it has been observed that most antibacterial lantipeptides inhibit cell wall biosynthesis and/or disrupt membrane integrity through pore formation. Lantibiotics, lantipeptides with antimicrobial activity, target and bind to the essential cell wall precursor lipid II. This inhibits the reaction required to synthesize peptiodglycan, an essential part of the cell membrane.
The unique thioether cross-links of lantipeptides are added on post-translationally by biosynthetic enzymes via dehydration of serine or threonine to the α,β-unsaturated residues 2,3-didehydroalanine (Dha) or (Z)-2,3-didehydrobutyrine (Dhb), respectively, followed by Michael-type addition of a cysteinyl thiol to give a thioether bridge. The cross-links are essential for the activity of the compound and for maintaining stability against proteolysis and heat denaturation. This makes lantipeptides quite durable in biological conditions that would otherwise denature other peptides.
In order to synthesize class I lantipeptides, two separate enzymes carry out reactions forming thioether cross-links: dehydratase LanB and the cyclase LanC. LanB genes encode proteins of about 1,000 residues, while the lanC genes encode proteins of approximately 400 residues. It is believed that these reactions involve a zinc ion bound by a cysteine-cysteine-histidine triad at an active site.
Currently techniques are being developed to improve the pharmacological properties of lantipeptides, including potency, stability, and solubility. One approach is in vivo lantipeptide production in E. coli. This simplifies genetic manipulation and cuts costs because E. coli is relatively easy to grow and is a well-studied organism. A 2011 study demonstrated the versatility of E. coli production of modified LanAs. This study also represents the only report to date of class I lantipeptide production in E. coli. Two other E. coli expression systems reported in 2011 have utilized dedicated lantipeptide proteases to produce mature lantipeptides. One system produced a fourfold improvement in yield compared to the producing strain. Using E. coli appears to be a viable in vivo method for producing lantipeptides and further research will likely yield more effective methods.
In addition to using biological techniques to acquire lantipeptides, chemical methods have shown some promise as well. Before 2008 the only successful total chemical synthesis of a lantipeptide was the impressive solution-phase synthesis of nisin. However, recently the first first solid-supported total synthesis of a lantipeptide containing overlapping cross-links, the α-peptide of lacticin 3147, has been reported. This expands the possibilities for synthesizing lantipeptides and should be studied further in conjunction with bioengineering techniques.
Algal cells contain chloroplasts, membrane enclosed organelles of photosynthesis that evolved from a cyanobacterium. In Earth's biosphere, algae plus bacterial phototrophs feed all marine and aquatic ecosystems, producing the majority of oxygen and biomass available for Earth's consumers. Besides, the "green plants" include primary endosymbiont algae descended from a common ancestor containing a chloroplast. The chloroplasts of primary endosymbionts are enclosed by two membranes. They are the inner membrane and the outer membrane. The inner membrane is from the ancestral phototroph's cell membrane and the outer membrane is from the host cell membrane as it enclosed its prey. The chloroplast of the green algae and red algae have diverged from their common ancestor that use the pigments absorbing different ranges of the light spectrum.
Chloroplast
Chloroplasts are organelles responsible for photosynthesis or the conversion of CO2 to glucose in plant cells, some protists, and algae. There are three types of membranes in chloroplasts:
The smooth outer membrane that is permeable to molecules
The smooth inner membrane contains membrane proteins that selectively allow access to small molecules and proteins
The thylakoid membrane system
The fluid inside the chloroplast and surrounding the thylakoid system is known as the stroma.
Thylakoid membranes envelop a system of interconnected vesicles. Stacks of these thylakoid membranes are known as grana. The main function of the thylakoid membranes is to house the following protein assemblies:
Photosystem I
Photosystem II
Cytochrome b and f
ATP synthase
All of these carry out photosynthesis as a whole.
The thylakoid membranes also contain chlorophyll, which gives plants its green color. The function of the chlorophyll is to capture the light necessary for photosynthesis.
In the thylakoid membrane and compartment, the light reactions take place in the thylakoid membrane and the thylakoid compartment and are concerned with the initial conversion of light energy into chemical energy that stored in ATP and NADPH.
Besides, the ATP and NADPH feed into the Calvin cycle in the Chloroplast stroma where the ATP provides energy for the molecular rearrangments and also the electrons carried by the NADPH are transferred to the organic molecules involved in the Calvin cycle.
There are many herbicides on the market that advertise for their ability to kill weeds. In order to kill the weeds, a disruption of photosystem I or photosystem II is required. The basic idea is that the electrons are activated once they received sunlight. The activated electrons then moves from the chlorophyll into a series of cytochromes creating what is called an electron transport chain. These electrons that are transferred along the electron transport chain are later used for carbon fixing, hence resulting in photosynthesis. An interruption in photosystem II is a result of the inhibitors stopping the flow of electrons, whereas an interruption in photosystem I is initiated by an inhibitor that creates change in energy through electron diversion at the terminal photosystem. Both types of interruptions can stop the process of photosynthesis.
Some inhibitors of photosystem II are diuron and atrazine. Diuron blocks the active site of plastoquinone in photosystem II, and its blockage disrupts the steady flow of electrons to the plastoquinone. As a result, sunlight cannot be converted to energy, and ultimately leading to the death of weeds. Atrazine works in similar ways as diuron. An example of photosystem I inhibitor is paraquat. Paraquat converts the electrons that it receives from photosystem I to radicals. The radical then interacts with oxygen to form reactive oxygen substances. These substances, in turn, interact with the membrane lipids, which can cause damage to the membrane because the double bonds of the membrane lipids are altered.
The stroma is the fluid contents of the chloroplast, it contains the enzymes of the chloroplast such as RUBISCO, which is responsible for the dark reactions. The stroma is also home to the DNA of the chloroplasts that helps it to carry out its functions, some ribosomes of its own, and RNA. Due to the chloroplasts’ individual DNA, it has been hypothesized that the chloroplasts were once free living bacteria that became involved in symbiotic relationship with eukaryotes and eventually became permanently incorporated into the cell’s structure.
[12]
The Dark reaction, mainly the Calvin Cycle that take place in the substance surrounding the thylakoids. Carbon from the carbon dioxide is brought into the cycle as a source of carbon in the Calvin cycle.
Chloroplasts produces starches and sugars. In order to do so, the plants extract energy from the sun. The energy extraction from the sun is called photosynthesis. With the energy extracted, the chlorophyll in the plants can combine carbon dioxide and water together. Both the animals a plants uses chloroplast for energy and food. Animals also uses it for oxygen to breath.
Proteins have a lot of uses in the body. from cell structure to carrying out reactions. Proteins are in turn made of smaller building blocks called amino acids. There are a total of 20 amino acids.
The 20 Amino Acids
Proteins can be made from a lots of different combinations and number of amino acids. This creates an almost infinite amount of different proteins. Although the sequence of amino acids is a chain, the structure of proteins is far from similar. These amino acids interact with each other to form #D structures. Most common are the alpha helix and the beta strand.
A picture of an alpha helixAmino acids interacting to make a beta strand
Although it is possible to determine the 3D structure of a protein, it is not quite possible to predict protein structure from the amino acid chain. This is quite important because diseases such as Alzheimer's and Mad Cow Disease are thought to come from a misfolding of proteins. [1]
1. Nuclear semi permeable membrane 2. Nuclear pore 3. Rough endoplasmic reticulum (REM) 4. Smooth endoplasmic reticulum 5. Ribosome attached to REM 6. Macromolecules 7. Transport vesicles 8. Golgi apparatus 9. Cis face of Golgi apparatus 10. Trans face of Golgi apparatus 11. Cisternae of Golgi apparatus 12. Secretory vesicle 13. Cell membrane 14. Fused secretory vesicle releasing contents 15. Cell cytoplasm 16. Extracellular environment
Endoplasmic Reticulum is an extensive membranous network in the eukaryotic cells that is continuous with the outer nuclear membrane and composed of ribosome-studded (rough) and ribosome-free (smooth) regions.
The ER is responsible for the manufacture of the membranes and performs many other biosynethic functions.
The endoplasmic reticulum (also known as the ER) is made up of a wide system of membranes that make up over fifty percent of the total membrane in numerous eukaryotic cells, and consists of two sections that have different functions: the smooth endoplasmic reticulum and the rough endoplasmic reticulum. The endoplasmic reticulum consist of a system of membranous tubules and pouches called cisternae, which is held together by the cytoskeleton. The membrane segregates the inside section of the endoplasmic reticulum from the outside section (cytosol) . The internal section is called the lumen cavity, but is also known as the cisternal space. The name “endoplasmic” is defined as “within the cytoplasm,” and the Latin definition for the word “reticulum” is “little net.” The membrane of the endoplasmic reticulum is continuous throughout the nuclear envelope. Because of this, the area between the two membranes of the envelope is also continuous to the cisternal space.
The Endoplasmic Reticulum contains a quality control mechanism called chaperones. These special proteins attach themselves to newly synthesized proteins and assist them in folding into their native conformations. Charperones also keep mis-folded proteins in the ER to be broken down. These incorrectly folded proteins are degraded through the process of ER Associated Degradation (ERAD). These Chaperones can be affected by genetic diseases such as Cystic fibrosis. Cystic fibrosis is a genetic disease that causes the buildup of chaperones and their incorrectly folded proteins in the ER. This disease leads to mucus clogging the lungs and digestive track, which can lead to early death.
Under stress conditions, the normal function of the ER is impaired, causing the alteration of protein homeostasis and the corresponding accumulation of misfolded proteins in the ER lumen. This condition is known as 'ER stress'. The aglomeration of misfolded and/or unfolded proteins, detonates a dynamic response process called 'unfolded protein response' (UPR), which is the combination of several intracellular signaling pathways that have as a main purpose to reduce the load of misfolded proteins in order to re-establish protein homoestasis, adjusting to the current need in a dynamic fashion. In addition, the UPR carries out the transcriptional induction of genes expressing chaperones, ER-associated degradation (ERAD) components and autophagy regulators to the cytosol and the nucleus. If the upregulation of proteins involved in folding, quality control and trafficking is not enough to balance ER homoestasis, the UPR eliminates the damaged cell by apoptosis.
The UPR (mentioned above) is activated by three stress sensors, that detect the accumulation of misfolded or unfolded proteins in the ER. These sensors are:
Inositol-requiring enzyme 1α (IRE1α): It's a type I transmembrane protein with an N-terminal luminal region, a cytosolic kinase and RNase domains. Its activation has been explained by two different proposed models. The first one explains that immunoglobulin-binding protein (BiP), an ER-resident chaperone, inhibits oligomerization of IRE1α by binding to it. However, the aggregation of misfolded proteins causes BiP to dissociate from IRE1α, allowing it to interact with unfolded proteins and thus, to oligomerize. The second model affirms that unfolded proteins interact directly with IRE1α's luminal domain, inducing its oligomerization. These mechanisms are still under debate, although recent research data leads to the idea that both of them operate together in the regulation of IRE1α. The oligomerization process undergone by IRE1α leads to to trans-autophosphorylation of its kinase region which in turn engages its RNase activity. Activated IRE1α launches alternative splicing of the mRNA that encodes the transcription factor X-box binding protein 1 (XBP1), leading to the translation of a more stable spliced form of XBP1 (XBP1s), which in turn upregulates certain UPR-related genes associated with folding, ERAD and quality control.
Protein kinase RNA-activated (PKR)-like ER kinase (PERK): It's a type I transmembrane protein that contains both a cytosolic kinase domain and an N-terminal luminal stress sensing domain. Its activation is carried out by similar mechanisms to those of IRE1α. Activated PERK contributes to reducing overload synthesis of proteins in the ER.
Activating transcription factor 6 (ATF6): It's is a type II transmembrane protein, which under basal conditions associates with BiP. The accumulation of unfolded proteins initiates the dissociation of ATF6 from BiP, leading to the translocation of monomeric ATF6 to the Golgi and finally to the nucleus, where it upregulates the transcription of ER chaperones and ERAD-related genes.
The activation of the UPR Sensors is followed by a Four Step process in which the cell first attempts to repair the stressed ER. If this fails the cell will set itself on a path to apoptosis.
The First Step of the process attempts to decrease the amount of unfolded proteins on the ER. This is done by activating the previously introduced Protein kinase RNA-activated (PKR)-like ER kinase, or PERK. PERK directly decreases the amount of all protein synthesis in an effort to halt the production of proteins that are difficult to fold. Additionally Inositol-requiring enzyme 1α (IRE1α) plays a role in this stage by initiating macroautophagy, a process to destroy misfolded proteins to relieve stress on the ER.
The Second Step consists of a focus on the increase in quality of proteins being produced and restoring homeostasis to the ER. This is accomplished by two methods in no particular order. First, transcription factors XBP1s, ATF6f, ATF4, AXP1s, and ATF6f collectively work to folding and degradation capacity by upregulating chaperone activity,protein modification enzymes and the size of both the ER and Golgi. The second part of this step is ATF4 upregulating genes that change the redox state of the ER to provide a better environment for the folding of proteins. Both of these steps work to reduce the amount of misfolding proteins and increasing the precision with which new proteins are constructed to avoid producing more misfolded and unfolded proteins.
The Third Step begins to transition the cell to a state where apoptosis is more likely. This step has two contributing factors, both influenced by a protein called C/EBP homologous protein or CHOP. CHOP, acting downstream of AFT4 changes the redox state of the entire cell so that the cell is out of its natural state and therefore has trouble functioning. Additionally, again after attempts to slow protein synthesis, CHOP increases the protein synthesis of the cell further stressing it. Both of these factors will sensitize the cell to apoptosis.
The Fourth Step is the apoptosis of the cell. When ER stress results in apoptosis, the BCL2 protein family acts to bring the cell to apoptosis; all pro-apoptotic members of this family contain BH1, BH2, and BH3. Specifically, BH3 acts to trigger the activation of BAX and BAK. These then permeablilize the membrane of the cells mitrochonira and release cytochrome c and apoptosome. This moves the cell into a stage of apoptosis.
When discussing ER stress and human disease, it is important to recognize that nearly all data that suggests a linkage between ER stress is either correlative or based on in vitro evidence. Correlative evidence is flawed as it is unwise to assume that correlation always implies causation, and in vitro data is flawed as it is an isolated study rather than a comprehensive view of the organism in question.
Tumors often cause hypoxia in the tissues they affect. When this occurs, the ER begins a stress response to adapt to the new conditions. Under normal conditions the efforts of the ER stress response would be beneficial to the body, however, the result of the adaptation is that the tumorous cells are allowed to replicate, driving tumor growth rather than the death that would have occurred had ER stress not took place. Scientists believe that the IRE1α-XBP1 pathway involved in this process is a promising target for cancer treatment.
When glucose levels become excessive, an ER stress response is initiated. This response is thought to suppress insulin gene expression via IRE1α degradation of insulin mRNA. This downregulation of insulin is characteristic in type two diabetes.
The article "Endoplasmic Reticulum Stress and Type 2 Diabetes" by Sung Hoon Back and Randal J. Kaufman, they discuss how, due to certain lifestyles now, type 2 diabetes has vastly increased over the years. Type 2 diabetes is "characterized by increased levels of blood glucose owing to insulin resistance in adipose tissue, muscle, and liver and/or to impaired insulin secretion from pancreatic [beta]-cells." They discuss how beta-cells increase in response to obesity and insulin resistance, but it doesn't last forever and normally begins to fail. It is possible that beta-cell loss could be due to endoplasmic reticulum (ER) stress. Pancreatic beta-cells secrete insulin, which is a "blood glucose-lowering hormone." Proinsulin is synthesized in the ER and then stored in beta-cells until glucose levels increase and then insulin is released. The ER is in charge of many things and requires a certain environment, so when the environment changes, there are several stress responses that occur to try to keep the ER functioning properly. The main stress response is called the unfolded protein response (UPR). UPR includes "expansion of ER size, enhanced folding capacity, reduced protein synthesis through transcriptional and translational controls, and increased clearance of unfolded or misfolded proteins." IF this does not work and the ER does not return to homeostasis, then cell death is normally activated. It is believed that the ER sends signals for apoptosis is the stress is severe and unresolved by UPR. IRE1α can also mediate cell death pathways. ER stress can increase activation of IRE1α which leads to increased splicing of genes leading to cellular dysfunction and beta-cell death.
Correlative evidence shows that ER stress markers are present in deceased persons that suffered from Alzheimer's, Parkinson's, ALS, Huntington's, and Creutzfedt-Jabob disease. Scientists recognize however, that the role of ER response will be neurodegenerative diseases will be very difficult to determine.
The detailed study of persistent ER stress and the UPR is essential for the complete understanding of certain diseases, such as cancer, diabetes, neurodegenerative diseases (including AAlzheimer’s, Parkinson’s and amyotrophic lateral sclerosis) and inflammation-related diseases. What such diseases have in common is the accumulation of abnormal intracellular and/or extracellular protein aggregations and inclusions. Therefore, by the meticulous study of the enzymatic pathways followed by the UPR stress sensors, new pharmachological interventions could be developed for the mentioned illnesses.
The description of endoplasmic reticulum (ER) being "rough" comes from the organelle being studded with ribosomes. These ribosomes on the rough ER, are used to synthesize proteins which are destined to either be inserted into the membrane of the cell or transported out of the cell. Differences between ribosomes lying on the ER and those lying in the cytosol stem mainly from the destination the protein that is synthesized by the ribosome. With protein synthesis via ER ribosomes, proteins are synthesized as they normally would through translation of mRNA. After the protein is synthesized, called "posttranslation", it then enters the lumen of the endoplasmic reticulum, the inner compartment of the organelle. Although the secondary structure (i.e. beta sheets and alpha helixes) of the newly-synthesized peptide forms almost instantaneously as it is translated,its tertiary structure does not begin to form until it is in the lumen, where it will begin to fold into its specific 3-D conformation with the aid of chaperone proteins [1]. It is inside the lumen of the rough endoplasmic reticulum that N- and O- linked glycosidic bonds are formed between amino acids and sugars, thus this process is called "glycosylation" and the newly formed protein is called "glycoprotein".
The endoplasmic reticulum transports the proteins through the use of vesicles. Vesicles are membrane enclosed packages that contain protein. On the surface of the vesicles, there are coating proteins called COPI and COPII. The protein COPII allows the vesicles to be sent the Golgi and the COPII protein allow the vesicles to come back to the Rough ER. From inside the Golgi, proteins are again packaged into vesicles and transported to the cell membrane. Once at the membrane, the vesicle itself merges with and become part of the membrane as it is composed of the same lipid bi-layer. Its protein content either will remain on the membrane if it is to function as a membrane protein or be secreted outside of the cell [1].
The smooth endoplasmic reticulum, unlike its "rough" counterpart, does not package protein. Instead it carries out various metabolic reactions within the confines of its membrane. Functions range from carbohydrate metabolism, lipid synthesis (for example oils, new membrane phospholipids, and steroids), and even drug detoxification. Drug detoxification is usually accomplished by adding hydroxyl groups to drug molecules. This makes them more soluble in water and therefore easier to be flushed from the body. Another important function of the smooth endoplasmic reticulum is the storage of calcium ions. This is imperative for muscle contraction. When a muscle cell is stimulated by a nerve impulse, the calcium ions rush into the cytosol and trigger muscle contraction.
Structurally the smooth ER is not continuous with the cell's nuclear envelope. The smooth ER is involved in phospholipid synthesis.
1. Proteins are folded and modified in this organelle of the cell.
2. The internal volume of lumen in the endoplasmic reticulum (ER) is almost equal to the outside of the cell. This supports the idea that the ER evolved from the cellular membrane.
3. The ER contains an environment that is distinctly different from that of the cytosol in terms of solute and protein composition. This is because this environment is conducive to the ER’s role as a “staging post”; proteins stop at the this organelle to be packaged and modified before exiting the cell. The ER is also used as a checkpoint before the proteins are secreted, checking the protein quality and accuracy in terms of folding and modification before the protein is allowed to exit the cell.
4. How proteins are folded and modified within the ER:
When a protein first enters the ER, it is recognized by a signal sequence typically found at the N terminus of the protein. This sequence is recognized by a receptor located on the surface of the ER membrane called signal recognition particular (SPR). The molecule is then directed towards a pore, called the Sec61 translocon, in the membrane of the ER where it is allowed to pass through and into the ER. One interesting fact about protein folding is that when some proteins are passed through the ER, before entering the ER lumen, they are mere polypeptide chains. The folding of the protein actually sometimes happens entirely within the ER lumen! It is therefore important to note that when the polypeptide sequence enters the lumen of the ER and are subject to the modifications of the sidechains of its amino acids, this can alter the way the protein would fold. Thus, these modifications influence folding, and folding sometimes also influences modifications too. There are some exceptions to this, such as transmembrane proteins that have large cytosolic domains. These proteins fold within the lumen and within the cytosol of the cell. Once the protein is fully translocated into the ER lumen, it was finish the folding and modification that it needs. If the protein is part of a multisubunit complex, then the ER will assemble the protein into its native oligomeric structure. Modifications catalyzed by enzymes in the lumen of the ER greatly aid the maturation of the protein and bring it closer to its final desired outcome. Once this is complete, the finished protein is then put into a transport vesicle, which buds off from the ER and continues on its journey to either the golgi apparatus or for secretion. Some proteins, which do not end up being modified correctly can be consequently targeted for destruction and may leave the ER to be degraded by enzymes in the cytosol.
5. The UPR (mentioned above) is activated by three stress sensors, that detect the accumulation of misfolded or unfolded proteins in the ER. These sensors are:
Inositol-requiring enzyme 1α (IRE1α): It's a type I transmembrane protein with an N-terminal luminal region, a cytosolic kinase and RNase domains. Its activation has been explained by two different proposed models. The first one explains that immunoglobulin-binding protein (BiP), an ER-resident chaperone, inhibits oligomerization of IRE1α by binding to it. However, the aggregation of misfolded proteins causes BiP to dissociate from IRE1α, allowing it to interact with unfolded proteins and thus, to oligomerize. The second model affirms that unfolded proteins interact directly with IRE1α's luminal domain, inducing its oligomerization. These mechanisms are still under debate, although recent research data leads to the idea that both of them operate together in the regulation of IRE1α. The oligomerization process undergone by IRE1α leads to to trans-autophosphorylation of its kinase region which in turn engages its RNase activity. Activated IRE1α launches alternative splicing of the mRNA that encodes the transcription factor X-box binding protein 1 (XBP1), leading to the translation of a more stable spliced form of XBP1 (XBP1s), which in turn upregulates certain UPR-related genes associated with folding, ERAD and quality control.
Protein kinase RNA-activated (PKR)-like ER kinase (PERK): It's a type I transmembrane protein that contains both a cytosolic kinase domain and an N-terminal luminal stress sensing domain. Its activation is carried out by similar mechanisms to those of IRE1α. Activated PERK contributes to reducing overload synthesis of proteins in the ER.
Activating transcription factor 6 (ATF6): It's is a type II transmembrane protein, which under basal conditions associates with BiP. The accumulation of unfolded proteins initiates the dissociation of ATF6 from BiP, leading to the translocation of monomeric ATF6 to the Golgi and finally to the nucleus, where it upregulates the transcription of ER chaperones and ERAD-related genes.
Braakman, Ineke, and Neil J. Bulleid. "Protein Folding and Modification in the Mammalian
Endoplasmic Reticulum." Annual Review of Biochemistry 80.1 (2010): 110301095147058. Print.
Woehlbier, U., Hetz, C. Modulating stress responses by the UPRosome: A matter of life and death. Trends in Biochemical Sciences, June 2011, Vol. 36, No. 6.
Back, Sung H., Kaufman, Randal J. "Endoplasmic Reticulum Stress and Type 2 Diabetes." Annu. Rev. Biochem. 2012. 81:767–93
The Endoplasmic Reticulum (ER) is an important organelle in the cell that performs a wide variety of tasks including and not limited to protein folding, modification, and transfer of membrane proteins to and from Golgi apparatus. Also, the maintenance of ER homeostasis in insulin-secreting beta-cells is extremely important, because when the ER’s homeostasis is disrupted, the ER generates an unfolded protein response (UPR), to maintain homeostasis of this organelle. However, if homeostasis is not restored, the ER promotes death signal pathways. Observations have shown that type 2 diabetes is an important causal factor in the disruption of ER homeostasis and subsequent death of insulin secreting beta cells.
The frequency of Type 2 diabetes in society has dramatically increased due to unhealthy living and eating lifestyles with the consumption of foods rich in fat. Type 2 diabetes consists of a mixture of metabolic conditions characterized by increased levels of blood glucose. The disease develops in areas of the body that are resistant to insulin. Also it is important to note that the disease develops only when beta-cell dysfunction appears. Blood glucose and fatty acids that cannot be properly stored or excreted can overload the cell and disrupt ER and mitochondrial functions. High glucose and saturated fatty acids cause beta cell failure and subsequent oxidative stress through its metabolism.
Elevated levels of plasma free fatty acids (FFA) are associated with high fat diets and obesity in humans. Free fatty acids are considered to be vital mediators of apoptosis and dysfunction of pro insulin beta cells in type 2 diabetes. It is known that both unsaturated and saturated FFas inhibit proinsulin synthesis. The precise mechanism of Beta cell dysfunction and apoptosis is not yet fully understood. There is great evidence of long saturated fatty acid chains being responsible for the induction of ER stress and subsequent beta cell failure.
Insulin deficiency associated with insulin resistance is known to cause blood glucose levels to remain high (hyperglycemia). Over stimulated beta cells show a gradual decrease of glucose-induced insulin secretion and insulin gene expression and eventually impaired beta-cell function and survival. This phenomenon is known as gucotoxicity. As blood glucose levels continue to remain high, the glucose that originally served as fuel is now used to generate detrimental metabolites for beta cells.
1. Endoplasmic reticulum stress and type 2 diabetes.
Back SH, Kaufman RJ.
Golgi apparatus, also known as the Golgi complex, golgi body, or simply golgi, is an organelle found in most eukaryotic cells. The golgi can be thought of as the "packaging, modifying and shipping warehouse" of the cell.
The Golgi apparatus, also called the Golgi complex, is commonly found in eukaryotic cells. The Golgi complex can be identified by its unique structure which some say looks like a maze, but in fact the structure is made of stacks of flattened membranous sacs, or cisternae. Unlike the cisternae of the endoplasmic reticulum or ER, these membranes are not connected. The Golgi apparatus is responsible for the processing and packaging of protein and lipids, as well as processing proteins for secretion. The Golgi apparatus is often thought of as a post office; through the process of protein glycosylation, sugars are added to the protein which dictate to where the protein should travel. N-linked glycosylation is begun in the endoplasmic reticulum but is continued in the golgi complex. O-linked glycosylation is solely done in the golgi complex. After proteins have been modified in the golgi apparatus, they are usually sent to either the lysosomes, secretory granules, or plasma membrane depending on the signals encoded within the protein sequence and structure. For this reason, Golgi complex is recognized as "the major sorting center" of the cell.
Researchers in Yale show that the Golgi Apparatus is not just an outgrowth of another organelle, the endoplasmic reticulum, but an independent organelle on its own. The Golgi Apparatus is an organelle within a cell that has its own autonomy and so must grow and divide to keep pace with the growth and division that the cell inhibits. It has been found that even without the stacks of membrane compartments where secretory proteins pass, the Golgi Apparatus still functions.
Proteins referred to as matrix proteins that form a scaffold have been found to be responsible for its growth, division and partitioning.
The Golgi Apparatus has been observed to grow, divide and be inherited like other organelles as has been observed in the regrowth of the mother Golgi Apparatus in daughter cells with the use of matrix proteins.
The Golgi was discovered by Italian physician Camillo Golgi in 1898 during an investigation of the nervous system. [1] Although the discoverer denoted the structure as the "internal reticular apparatus," scientists changed it to the "golgi complex" in 1910.
Few doubted the discovery, claiming that the organelle was a mere illusion created by the optical instruments used that discovered them. Yet, soon after the advent of modern microscopes circa 1900's, scientists alike confirmed the presence of the Golgi.
The Golgi complex is made several flattened membranes sacs, but can be ultimately divided into two sections: the Cis Golgi and the Trans Golgi Network (TGN). The Cis Golgi functions as the receiving end for newly synthesized proteins from the lumen of the Endoplasmic Reticulum (ER). Vesicles containing proteins from the ER merge with the Cis golgi allowing the proteins to enter the Golgi complex. As the Cis golgi receives proteins from the ER, the proteins then begin their modification moving along membrane to membrane towards the TGN. At the other end of the golgi complex, the newly modified protein arrives at the TGN where it is then send off to different parts of the cell via a transport vesicle.
There are two prevailing theories as to the formation of the Golgi apparatus. The vesicular shuttle model postulates that Golgi cannot be made from scratch and that the vesicles of the endoplasmic reticulum are sent to the pre-existing Golgi. On the other hand, the cisternae maturation model suggests that vesicles from the ER fuse together to form the Golgi and as proteins are processed and mature they create the next Golgi compartment. New data suggest that perhaps neither model is completely correct. This will likely lead to yet another model. [2]
Cisternae Maturation Model shows that proteins from the ER join to make parts of the Golgi, so the Golgi can be made.
On the other side of the debate is Jennifer Lippincott-Schwartz of the National Institute of Child Health and Human Development (part of the National Institutes of Health) in Bethesda, Maryland. She says that the Golgi makes itself from scratch. According to her theory, packages of processing enzymes and newly made proteins that originate in the ER fuse together to form the Golgi. As the proteins are processed and mature, they create the next Golgi compartment. This is called the cisternae maturation model.
In this model, the cisternae of the Golgi apparatus move by being built at the cis face and destroyed at the trans face. The vesicles from the endoplasmic reticulum fuse together to form a cisterna at the cis face and this cisternae would appear to move through the Golgi stack when a new cisterna is formed at the cis face. This model is supported by the fact that structures larger than the transport vesicles were observed microscopically to progress through the Golgi apparatus. In summary, packages of processing enzymes and new proteins originating in the ER fuse together to form the Golgi and as the proteins are processed and mature, the next Golgi compartment is created. Vesicular transport is one of models of Golgi apparatus that says that Golgi cannot make from scratch and that vesicles in the ER are sent to pre-existing Golgi. [3]
On one side of the debate is Graham Warren of Yale University School of Medicine in New Haven, Connecticut, who argues that the Golgi is an architectural structure that cannot be made from scratch. He believes that newly made proteins are packaged in the rough ER and are sent for further processing to a pre-existing structure (the Golgi) that is made up of different compartments. This is called the vesicular shuttle model.
This model views the Golgi apparatus as a very stable organelle that is divided into compartments in the cis to trans direction. Membrane bound carriers transport material between the endoplasmic reticulum and the different compartments of the Golgi. Experimental evidence shows the abundance of small vesicles in close proximity to the Golgi apparatus. Actin filaments direct the vesicles by connecting packaging proteins to the membrane to ensure that they fuse with the correct compartment. To summarize, newly made proteins are packaged in the rough ER and are sent for processing to a pre-existing structure known as the Golgi which is made up of different compartments.[4]
As soon as modified proteins reach the Trans Golgi Network (TGN), the proteins are separated into several different transport vesicles for different areas of the cell.
Vesicle Type
Description
Exocytotic vesicles
These vesicles contain protein that are to be sent outside the cell membrane. These vesicles fuse with the plasma membrane, releasing their contents outside the cell. This process is called constitutive secretion.
Secretory vesicles
These vesicles are also to be sent outside the cell membrane. However what differs these from exocytotic vesicles is that secretory vesicles need a signal before they are released. When the signal is given, they will fuse with the plasma membrane to release the contents. This process is called regulated secretion.
Lysosomal vesicles
These vesicles contain protein that are sent to the lysosome to be digested.
The transport mechanism that proteins use to progress through the Golgi apparatus is still not clear yet, but there are a number of hypotheses that currently exist. Up until recently, the vesicular transport mechanism had been the favored mechanism for transport but there is now more evidence that supports the Cisternal maturation. The two models may work in conjunction with one another rather than being mutually exclusive. This is sometimes referred to as the combined model.
This is the process of adding sugar to a molecule. Glycosylation happens in the golgi apparatus and in the endoplasmic reticulum. Glycosylation can only happen when an amino acid group with a hydroxyl (-OH) attaches to a sugar molecule. Through a condensation reaction (water leaving) a sugar is added to the amino acid.
Inside the cell, U.S. Department of Health and Human Services
Viadiu, Hector
Reece, Jane (2011). Biology. Pearson. ISBN978-0-321-55823-7. {{cite book}}: Text "coauthors+ Lisa A. Urry, Michael L. Cain, Steven A. Wasserman, Peter V. Minorsky, Robert B. Jackson" ignored (help)
The purpose of the mitochondria in the eukaryote is to provide cellular respiration to the cell. The endosymbiotic theory asserts that the mitochondria came to be part of the eukaryote over time through a symbiotic relationship. The mitochondria consists of two membranes, the inner membrane and the outer membrane. It is speculated that the outer membrane came about when its ancestor was engulfed by the host celled via endocytosis, giving it a membrane in addition to the one the mitochondria ancestor already had. This endosymbiont theory would also explain why the mitochondria had its own DNA and why this DNA is circular. For some amino acids the genetic code of the mitochondria differ slightly from that of the nucleus (and the rest of the cell).
The mitochondria's energy from respiration is stored in an ion gradient across the organelle's double membrane, known as the "Mitchell Hypothesis".
Mitochondrial Biogenesis:
It’s a metabolic adaptation where mitochondrial mass increases which allow for conduction of glycolysis, oxidative phosphorylation, and ultimately result in higher mitochondrial metabolic capacity. With greater capacity to synthesize and transport fuels to mitochondria, the metabolic response would be faster, which can be beneficial for athletes during exercise. However, this improvement requires exercise and training.
Mitochondria produce ATP by oxidative respiration. A cell may contain tens or hundreds of mitochondria, depending on its energy needs. Mitochondria have two membranes: an outer membrane and an inner membrane. The inner membrane has many infoldings called cristae that increase its surface area. It is thought that as a result of the endocytosis of the bacterium, the inner mitochondrial membrane is derived from the larger eukaryote. Also, the inner membrane has structural characteristics of a prokaryotic cell membrane, while the outer membrane is similar to the host eukaryotic membrane. Mitochondria contains two distinct compartments: the matrix inside the inner membrane and the intermembrane space between the two membranes. The tricarboxylic acid cycle takes place inside the matrix, the metabolic equivalent of the prokaryotic cytoplasm. Furthermore, the fact that the mitochondria contain their own DNA and ribosomes lends further support to endosymbiosis, since these cell features would be a necessary part of any free-living organism. Both the DNA and ribosomes of mitochondria show similarities with the DNA and ribosomes of bacteria.
Inside the deepest compartments of the mitochondria is the mitochondrian matrix. It is in the matrix that cellular respiration occurs, where pyruvate (a product of glycolysis in the cytosol) is converted to Carbon Dioxide and water. The matrix is the site of the citric acid cycle, whereby the electron transport chain is used to setup a proton gradient between the inner and outer membrane of the mitochondria, known as the inter membrane space. The protons in the inter membrane space accumulate to a point that the concentration gradient causes the protons to flow back into the matrix.
It is the inner membrane that is studded with the proteins necessary for the electron transport chain, such as the cytochrome electron shuttles. Upon reentering the matrix, the H+ go through ATP synthase, which in turns powers the synthase to phosphorylate adenosine diphosphate (ADP) to adenosine triphosphate (ATP). The ATP can be used later on to be coupled with thermodynamically unfavorable reactions to allow those chemical reactions to proceed. The inner membrane is folded and convoluted which allows for a greater surface area to utulize for the electron transport chain. These convolutions are what make up the cristae.
Interestingly enough the matrix of the mitochondria are one of the few locations outside of the nucleus where genetic information can be found in the cell. Mitochondrial DNA is similar in appearance to that of bacterial DNA due to its circular shape. The matrix is also known to house tRNA and ribosomes, which further solidifies the theory that the mitochondria entered the ancestral eukaryotic cell as single celled organism.
The outer mitochondria membrane consist of a phospholipid bilayer], laced throughout with integral proteins. The lipid bilayer contains porins which allow the passage of molecules which are 10,000 Daltons or less. This permeability in the outer membrane allows for water, ions, and some proteins to flow freely into the inter membrane space.
It is well documented mitochondria produce the ATP necessary for life, and that a proton gradient and membrane potential are required in order for ATP synthesis to occur in the mitochondria. However, mitochondria is very dynamic in that it can reverse its process. Complex V is the enzyme responsible for final ATP synthesis at the end of oxidative phosphorylation. When the trans-membrane potential is insufficient for ATP production, complex V, or F1F0-ATP synthase, can reverse its process, instead hydrolyzing ATP in order to pump protons out of the mitochondrial matrix in order to restore the proper gradient.
In a normal mitochondria performing respiration, ATP is removed through the adenine nucleotide translocase, or ANT. This helps maintain the trans-membrane potential, favoring phosphorylation of ADP. However, ATP hydrolysis is favored when the ANT is reverse, shuttling in ATP from glycolysis.
The consumption of ATP by mitochondria can clearly be potentially lethal to cells in conditions of extreme proton gradient degradation. It can also serve as a potential mechanism used by pathogens in the case of diseases related to mitochondrial respiration inhibition, (such as lack of oxygen in stroke or heart attack, or in less extreme cases where mitochondria are affected such as Alzheimer’s or Parkinson’s).
This reversal process of complex V is directly affected by IF1, the inhibitory factor of F1F0-ATPase. In response to acidification of the matrix of the mitochondria, the IF1 protein inhibits the activity of complex V as ATPase, usually in conjunction with the halting of respiration in conditions such as hypoxia or ischaemia. There is still much to learn about the mechanisms of IF1, however it is the major factor in protecting cells from ATP depletion in hypoxic conditions.
The crystal structure of IF1 is known. The protein acts as a homodimer, inhibiting two F1-ATPase units synchronously. There are many residues in the protein complex that form many associations with subunits of the F1-ATPase. It is believed that full association of IF1 with the F1F0-ATPase occurs only during ATP hydrolysis. It is suggested that IF1 may loosely bind to the F1-ATPase even during normal respiration, and that it may also aid in the efficiency of oxidative phosphorylation by serving as a ‘coupling factor.’
The site of Reactive Oxygen Species production with in the cell is found in the mitochondria. Reactive oxygen species (ROS) are small, highly reactive molecules that are short-lived. An incomplete one-electron reduction of oxygen is how ROS is formed.ROS includes oxygen anions, free radicals, and peroxides. Autophagy is one of the signaling pathways of the redox regulation of proteins by levels of ROS. Stress conditions activate autophagy, but pathological conditions deregulate autophagy. A build up of ROS can lead to oxidative stress that causes cellular constituents to be oxidized and damaged. Non-enzymatic and enzymatic antioxidizing agents have been created by the cell to prevent oxidative stress. Antioxidants are natural downregulators of ROS inducing autophagy. Autophagy is inhibited by TIGAR.
Role in Signaling Pathway
In addition to being known for their toxic effects on the mitochondria, ROS are also found to help in cellular signaling pathways. For example, when the brain and body are low on oxygen (hypoxia), mitochondrial generation of ROS to turn on signaling pathways that regulate transcription, maintenance of calcium stores, and overall energy management. At the level of the organism, ROS helps to control and manage fluid absorption and exchange of oxygen in the pulmonary circuit. Studies have shown ROS to help with the level of cellular signaling but more research still needs to be done to determine if whether ROS processes are innate to the mitochondria or if other cellular factors are required.
Source:
Mitochondrial reactive oxygen species regulate cellular signaling
and dictate biological outcomes
Robert B. Hamanaka and Navdeep S. Chandel1,2,*
1 Department of Medicine, Division of Pulmonary and Critical Care Medicine, Northwestern
University Medical School, Chicago, IL 60611, USA
2 Department
Although reactive oxygen species (ROS) is commonly viewed as toxic byproducts of mitochondrial synthesis of ATP, ROS are crucial as intermediates of cellular signaling pathways; ROS has essential role in oxidative homeostasis and propagation of signaling pathway. There are certain pathways that affect or depend on the production of ROS. The level/accumulation of ROS also suggests biological outcomes.
How ROS are produced?
ROS are produced during electron transport chain (ETC) of mitochondrial synthesis of ATP
Through complex I, II, and III of ETC, molecular oxygen is reduced to superoxide anion, which is primary ROS produced by mitochondria.
ROS in the matric are produced through all complex I, II, and III
ROS in the intermembrane space are only produced through complex III
The rate of production of ROS depends on the concentration of electron carriers that are able to reduce molecular oxygen and type of electron carriers, which have different rate of electron release and supply among different electron carriers.
How level of ROS responses to hypoxia and mediates signaling pathways?
Under hypoxia, cells are exposed to low oxygen. Consequently, adaptive transcriptional program, reduction of cellular oxygen usage, and decrease in the consumption of ATP are promoted by the activation of signaling pathways due the increase in production of ROS.
In response to hypoxia, the production of ROS is increased through Q-cycle, which is a specific hypoxic generation of ROS through mitochondrial complex III. The increase in ROS then activates the signaling pathways.
Increase in production of ROS inhibits the activity of PHD. The inhibition of activity of PHD later enables the stabilization of HIF that eventually leads to the transcriptional regulation, such as erythropoiesis, glycolysis, angiogenesis, cell cycle, and survival
Increase in the production of ROS triggers the activity of AMPK that increase the production of ATP and minimize the cellular usage of ATP. The activation of AMPK also inhibits the activity of mTOR so that ATP is conserved by inhibiting protein translations that consumes lots of ATP. Eventually, the increase in the production of ROS also reduces the consumption of oxygen because the accumulation of ATP is maximized by activity of AMPK
How level of ROS responses to PI3-kinase pathway?
The activation of PI3 leads to the activation of Akt that would increase the accumulation of ROS by two ways.
Through metabolic pathway, mTOR is activated by Akt activity, and oxygen consumption and production ATP are increased. As a result, the production of ROS is increased
Akt activity inhibits ROS scavenging by inhibiting FOXOs, which are antioxidants that inhibit the activity of ROS
Important role of ROS
Stem cell population
Low level of ROS leads to quiescence, which is a state of inactivity, and maintenance of stem cell population. While the level of ROS increasing, population of stem cell is differentiating and proliferating
Oxidative homeostasis
ROS regulates activity of phosphatases that oppose the activity of kinases and possess a reactive cysteine, which enables oxidation of cysteine and the reduction of ROS
ROS maintains oxidative homoeostasis through regulating phosphatases
TNFα treatment
Outcomes of TNFα treatment depends on activity of two TNFR complexes
NF-ĸB → cell survival
JNK → cell death
The activity of TNFR complexes depend on cellular level of ROS
Increasing level of ROS → increasing activity of JNK and decreasing activity of NF-ĸB → cell death
Decreasing level of ROS → decreasing activity of JNK and increasing activity of NF-ĸB → cell survival
How ROS affects the cellular transformation?
Cellular transformation is caused by the activation of oncogenes and loss of tumor suppressor genes. As a result, there are signaling pathways controlling proliferation, survival and metabolism of cancer cells.
In normal cells, high level of ROS triggers the activation of tumor suppressor to enable the apoptosis and senescence
High level of ROS → high mutation rate and genomic instability → tumor growth
ROS as signaling intermediates: if Ras or Myc is expressed due to certain accumulation of ROS, the cellular transformation will occur.
Vicious cycle
lose of tumor suppressor
sustainability of higher level of ROS
proliferation, angiogenesis, and survival pathway of tumor
more accumulation of ROS
leads to #1
High level of ROS not only promote tumor growth but also increases possibility of metastasis, the spread of cancer cells from one part to another non-adjacent part
Misconception about ROS
Misconception #1: ROS are solely damaging agents
It is wrong because only when the accumulation of ROS is really high, the irreversible damages (cellular transformation, tumor growth, etc.) will occur. In fact, certain amount of ROS is required for the cell to proliferate, differentiate, and promote the fitness of the organisms.
Misconception #2: Because ROS contributes to human aging and pathologies, mortality can be increased through scavenging ROS by antioxidants.
It is not true because study has shown there is a correlation between lifespan extension and high oxidative stress. This correlation suggests the association between lifespan extension with increase in mitochondrial metabolism and production of ROS
Reactive oxygen species (ROS), the byproducts of synthesis of ATP in mitochondria, is harmful; ROS damage molecules in mitochondria; as a result, mitochondria are no longer functional. The accumulation of molecular damage would finally lead to cellular degeneration and death. The solution of the accumulation of ROS in mitochondria is the quality control (QC) mechanisms that keep mitochondria functional. Although different QC pathways minimize the harm of ROS in different ways, each QC pathway has its own capacity of regulating the ROS. Therefore, the mitochondrial QC work into a hierarchical surveillance network.
QC at molecular level
ROS scavenging
The first defense in a network of QC pathways
Counteract molecular damage when a critical threshold of molecular damage is reached
Key components: small molecules and enzymes
Decelerate the speed of molecular damaging
Although ROS scavenging pathways are effective in decelerating the space, they are unable to completely prevent molecular damage
Repair and Refolding
The second defense in a network of QC pathways
Repair specific modifications and restore the function of impaired function after damage has occurred
The homeostasis of mitochondria protein is controlled through protein degradation, de novo protein synthesis, and refolding of misfolded proteins back to their original 3-D structure
The repair of damaged mitochondrial DNA (mtDNA), which encodes essential subsets of proteins, the two RNA subunits of the mitochondrial ribosome and tRNAs needed for mitochondrial protein biosynthesis, is also important
Base excision repair
Direct reversal
Mismatch repair
Proofreading activity of polymerase during mtDNA replication
However, majority of proteins are not able to repair or refold efficiently.
Removal and replacement
The defense in a network of QC pathways after the repair and refolding of proteins
A molecular pathway of removing aberrant protein through protein degradation machinery when a critical threshold of unfolded or damaged protein is reached
Due to the limited capacity of repair and refolding of proteins, there is further decline of cellular function, removal and replacement of damaged proteins through proteolysis
Degradation of damaged proteins; cleavage of presequences and the maturation of proteins
replacement with newly synthesized functional proteins
Even though mitochondrial QC in molecular level can somewhat regulate the damage due to ROS, it is not sufficient enough to keep mitochondria functional and proliferating over time. Since several mitochondrial protein complexes are also encoded by nuclear genome, after the removal of damaged proteins, the coordinated expression of mitochondrial and nuclear genes and correct assembly of the proteins into macromolecular complexes is also essential for keeping mitochondrial functional. This last crucial step is dependent on the import of proteins from cytoplasm and a correct inner-mitochondrial sorting.
QC at the organellar level
Fission and fusion of mitochondria
The first QC pathway in organgllar level when the accumulation of damage molecules can not be solved by molecular QC pathways
Fission – content separation
Separation of larger organelle into multiple smaller organelles
Controlled by three proteins: Dnm1, Fis1 and Mdv1
Fusion – content mixing
Combination of multiple smaller organelles into larger organelle
Controlled by three proteins: Fzo1, Ugo1 and Mgm1
Through fission and fusion, mitochondria are able to maintain their functions due to degradation of damaged mitochondria; each mitochondria wouldn’t have too much damaged components since content are either mixing or separating from each other.
High stress → fission
Low stress → fusion
Mitophagy
The last step of mitochondrial QC pathway
The remaining damaged and dysfunctional mitochondria are eventually eliminated from the vital mitochondrial network
Mitophagy is a type of autophagy (“self-eating”) that the whole mitochondria are engulfed by autophagic membrane, leading the formation of autophagosomes
Works with fission and fusion
The mitochondria network is separated into individual mitochondria through fission. The damaged or dysfunctional individual mitochondria is then eliminated (“eaten”) through mitophagy. The remaining vital mitochondria are combined together into a new vital mitochondrial network through fusion.
It is highly selective process that is tightly regulated.
Mitophagy in mammalian cells is regulated via PINK 1 which is a serine/threonine kinase PTEN-induced putative kinase and the E3 ubiquitin ligase Parkin. In a healthy mitochondria PINK 1 is cleaved by PARL in the inner mitochondrial membrane. However, when there is sustained mitochondrial damage PINK 1 is able to accumulate in the outer mitochondrial membrane where it cannot be cleaved by PARL. This accumulation of PINK 1 attracts Parkin which in turn induces mitophagy.
In 1905, Mereschkowsky, a Russian biologist, published a paper on the theory that photosynthetic bacteria are the ancestors of modern day plant chloroplasts. Though this research was mostly ignored for several years, scientists came to see the similarities between isolated living bacteria and eukaryotic mitochondria.
It is now largely accepted that mitochondria are descendants of "free-living" bacteria that were engulfed and incorporated as organelles by eukaryotic cells. The endosymbiont theory was further confirmed when mitochondria were discovered to contain their own DNA. It was confirmed even more so with the discovery that the mtDNA made enzymes and proteins that were needed for its own functionalities. The fact that the mitochondria also contains a double membrane also depicts the notion that it was originally a free living organism that was later ingested into another host.[1]
Mitochondria are produced by other mitochondria that act as "structural templates". A cell's two DNA genomes are still not aware of how mitochondrial membranes are assembled, showing that a mitochondria's structure isn't dictated by our DNA but must be passed on to future generations.
Mitochondria are the energy processing organelle that is found in the cell. Alongside with chloroplast, mitochondria are part endosymbiont theory, as stated above. The endosymbiont theory clearly stated the following about mitochondria and chloroplasts: it is enclosed by a double-membrane, it is about the same size as bacteria, it has its own circular DNA, its ribosomes are bacteria-like, and it has prokaryotic activities such as respiration and photosynthesis (mitochondria and chloroplasts respectively). Focusing on the third point about having its own circular DNA, mitochondria DNA (mtDNA) was found to be able to self-translate and replicate itself.
Unlike nuclear DNA which is inherited from both mother and father, the mammalian mtDNA is only inherited from mother. The mitochondria in mammalian sperm are destroyed in the fertilized oocyte. However, the replication pathway of mtDNA is very similar to nuclear DNA. Before replication, mtDNA becomes unwind by TWINKLE, which is a protein used to undo the double-stranded DNA. After the double-stranded is undone, it is replicated at one end with the help of mtDNA polymerase. mtDNA polymerase starts to form another double-stranded starting at 5' end of the mtDNA. Another protein called mitochondrial single-stranded binding (mtSSB) helps stabilize the unwound conformation and stimulates DNA synthesis by the polymerase holoenzyme.
Unfortunately, mtDNA replication displays no strict phase specificity as in nuclear DNA synthesis. Therefore, segregation of heteroplasmic mtDNA mutation can occur as a cell divides.
The transcription mechanism in mitochondria is likely similar to transcription in nucleus. However, there are some differences between RNA synthesis in mitochondria and in nucleus.
The individual strands of the mtDNA molecules are denoted heavy strand (guanine rich) and light strand (guanine poor). This nucleotide bias explains why some codons are rare or absent in mitochondrial RNA.
The compact mammalian mtDNA genome lacks introns. The entire strand codes for either proteins, rRNA, or tRNA. Therefore, there is no need for slicing process in mitochondria
Each of the protein and rRNA genes is immediately flanked by at least one tRNA gene.
1. Albert Claude, who was a Belgian biochemist discovered in the first half of the last century discovered that Mitochondria catalyzed respiration. He did this by isolating them through centrifugation.
2. Scientists started from there and managed to map out the flow of electrons in cellular respiration in the past two decades.
3. Peter Mitchell then discovers that the key to the flow of free energy in respiration and photosynthesis is stored within the ion gradient across membranes. He receives a Nobel Prize for it in 1978.
4. Mitochondria is bordered by 2 membranes and, and holds one tenth of the cell’s proteins. Mitochondria also converts 10,000 to 50,000 times more energy per second than the sun does.
5. Mitochondria was also discovered to play a pivotal role in programmed cell death, or apoptosis. This shows mitochondria to thus also be part of the signal transduction network in the cell. For programmed cell death, Mitochondria first releases proteins called cytochromes into the cell’s cytoplasm. It is these signals that could potentially release proteases and nucleases onto the cell and trigger cellular suicide.
6. Isolated Mitochondria were discovered to produce their own proteins even though the identity of these proteins are yet to be determined.
7. It is a theory that Mitochondria evolved from bacteria, which explains why it is such an independent organelle and doesn’t seem to depend on other parts of the cell for survival. Another evidence for this rests in the fact that the mechanism for protein synthesis in mitochondria is similar to that in bacteria.
8. Mitochondria spread by growth and division of previously existing mitochondria. Mitochondria are thus able to tell building blocks for new mitochondria where to go and what to do.
9. Recent discoveries have revealed that mitochondria actually have a lot of extramitochondrial molecules that help regulate the expression of genes that turn into mitochondrial proteins. Peroxisomal-proliferator-actived receptor coactivator 1 (PGC1) plays a major role in this process.
10. The space in between the mitochondria’s membranes was recently discovered to be able to oxidize sulfhydryl groups to disulfide bridges even though that space is surrounded by highly reducing environments.
Scientists believe that there is a strong correlation between mitochondrial dysfunction. Mitochondrial dysfunction is one of mitochondrial diseases and is caused by reactive oxygen species (ROS). Reactive oxygen species cause oxidative damage that degrades the ability of mitochondria to make ATP. This means that mitochondria fail to carry out their metabolic functions, leading to cell death [2]. Since mitochondrial dysfunction is a factor of cell death, it is reasonable to believe that such a correlation between mitochondrial dysfunction and aging exists.
It should be noted before anything that regulation of complex protein-folding environment within the organelle is vital for keeping productive metabolic output. The reason for its necessity is that without efficient metabolic output, chemical wastes and heat produced in metabolic processes, which are potential harms to the cell, cannot be transported out of the cell. Despite the fact the cells do have such complex systems to maintain efficient metabolic output, several factors come into play to prevent this. We note 2 factors here;
1. It is inevitable that over a long period of time, dysregulation of protein homeostatis arises through stress caused by the accumulation of reactive oxygen species.
2. A failure at maintaining efficient metabolic output can also be induced by mutations in the mitochondrial genome introduced during replication.
These two reasons that deteriorate mitochondria's normal functions are dependent on time; the longer a mitochondrion lives, the magnitude of these time-related effects increases. Therefore, it is believed that damage incurred on mitochondria is deeply involved in aging.
Reactive Oxygen Species (ROS)
Having explained that reactive oxygen species are a crucial factor in aging, it is necessary to figure out what they really are. By definition, they are chemically reactive molecules containing oxygen. [3]
These are examples of some reactive oxygen species;
1. molecules like hydrogen peroxide
2. ions like hypochlorite ion
3. radials like hydroxyl radial (this is the most reactive of all kinds of reactive oxygen species)
4. ions like superoxide anion (this is both ion and radical)
[4]
These reactive oxygen species are generated by the electron transport chain.
Sources of ROS in living cells
Roles of the mitochondrial proteome in aging
The mitochondrial proteome sustains the cell's cellular metabolism.
Cellular metabolism inside mitochondria such as ATP production, apoptosis, and regulation of intracellular calcium. They are all essential elements to sustain life. However, the costs of maintaining such functions are the damaging effects of reactive oxygen species, as mentioned earlier. The mitochondrial proteome comprises mitochondrial and nuclear DNA-encoded proteins that needs folding and assembly within mitochondria. The two genomes that code for the structural requirements are damaged by accumulation of reactive oxygen species over time. The proteome of mammals is made up of between 1000 and 1500 proteins.
Here is a summary of protein production. The list shows how proteins made in the cell are transported into mitochondria in the cell.
1. proteins are encoded in the nucleus
2. proteins are translated in the cytoplasm
3. the unfolded state of the proteins is maintained and then is imported into mitochondria.
Unfolded proteins are needed to to construct ETC in mitochondria. To assist mitochondrial biogenesis and transferring of mtDNA and proteome, mitochondria must go through series of fission and fusion. Just like other organelles do, this organelle fission functions to multiply the number of mitochondria. It also serves to remove defective organelles for autophagic degradation [5]
Misfolded and misassembled mitochondrial proteins
Researchers have found that inhibition of mtDNA replication, accumulation of orphaned mitochondrial complex subunits or harmful protein aggregates and ROS all can create an excessive amount of misfolded mitochondrial proteins in yeast and Caenorhabditis elegans. It should be reasonable that accumulation of such misfolded and misassembled proteins generated by those factors lead to destruction of certain mitochondrial metabolic function and its dysfunction ultimately.
Here is a summary of how aging disease in mitochondria occurs
1. Reactive oxygen species accumulate inside the mitochondrion
2. This follows two possible consequences. One is that reactive oxygen species react with mtDNA and cause mtDNA mutations. It should be emphasized once again that reactive oxygen species are highly reactive agents. The other possible consequence is reactive oxygen species directly attack mitochondrial proteins. The proteins are distorted as a result.
3. mtDNA mutations caused by reactive oxygen species no longer encode for ordinary mitochondrial proteins. What encoded and translated from these mutated mtDNA are, in fact, misfolded proteins.
4. Keep in mind that proteins are used to build the complicated network of ETC. When misfold proteins are created, as long as they are present in the mitochondria, they will be used as building blocks of ETC. ETCs with misfolded proteins embedded in them no longer function properly. In other words, the mitochondria face ETC dysfunction.
5. ETCs with misfolded proteins cause to create more reactive oxygen species. As more reactive oxygen species are generated, this vicious cycle continues and misfolded proteins accumulate inside the mitochondria. Over time, the mitochondria die.
Non-native amino acids that damage the three dimensional structure of proteins are actively generated in the process of cytosolic translation of mitochondrial proteins [6].
Degrees of challenge of non-native amino acids derived from errors in cytosolic translation (under optimal conditions)
- nearly 10% of newly synthesized proteins are mistranslated
- 20-30 % of all nascent polypeptides are quickly denatured permanently due to folding errors
Organelle biogenesis and complex assembly
Complex I of ETC is named as the NADH-ubiquinone oxidoreductase. Complex I is known to possess about 45 subunits. And mutations or functional failures are known to be potential causes of neurodegenerative diseases. Such diseases include Parkinson's disease. Out of the 45 subunits of ETC, 7 of them are encoded by the genome in mitochondria. They need to be embedded into the mitochondrial inner membrane because that is where they build stoichiometric complexes with nuclear-encoded components. Suppose that one of the subunits was misexpressed due to mutation. Then the entire network of ETC is doomed to collapse. In other words, mutations or deletions of ETC's subunits (even just single mutation or deletion), have a tremendous effect on whole complex formation. This illustrates the significance of the coordination of genome of proper complex assembly and function.
Mitochondrial compartments
There are four compartments in mitochondria where protein folding and assembly happens. The four compartments are the outer membrane, intermembrane space, inner membrane and matrix [7]. It should be stressed that misfolded proteins can build up in those compartments. The interesting fact is that there exists a structure in the mitochondria that monitors these levels of accumulation. Compartment-specific QC machinery is the one which is responsible for monitoring the accumulating unfolded proteins. Normal fold proteins have hydrophobic amino acids buried inside them and their heads often stick out. Caperones and QC proteases then come into place to acknowledge these hydrophobic amino acid heads.
We should divide how the QC monitors the level of unfolded proteins into each compartment.
Outer Membrane
It seems from recent evidence that cytosolic chaperones and the ubiquitin-proteasome system are involved in the mechanisms that regulated quality control of mitochondrial protein import or outer membrane proteins. Nuclear-encoded proteins which are ordered to form mitochondria are translated in the cytoplasm. The chaperones are used to sustain precursor proteins that are unfolded or misfolded. They also serve to prevent protein accumulation during delivery to the translocase of the outer membrane channel
Damage done by different types of reactive oxygen species
-reactive oxygen species attack mitochondrial proteins and DNA during cell division
-superoxide anion is derived from dominantly from complexes I and III of the ETC during oxidative phosphorylaiton. Superoxide anion, having a strong negative charge upon it, can deteriorate all four mitochondrial compartments.
- storing up an excessive amount of reactive oxygen species could totally overwhelm mitochondrial ROS-detoxifying systems.
- reactive oxygen species may directly interfere with protein folding by changing its amino acid sequence, giving its secondary and tertiary structures
inevitable and irreversible changes
- reactive oxygen species may indirectly interfere with protein folding by introducing mutations in genes encoded by mitochondrial or nuclear DNA.
- note that mtDNA is very vulnerable to oxidative damage because it is located ner the site of reactive oxygen species production and does not have histone protection
Age-associated organelles damage
In the long run, the decrease in mitochondrial function caused by reactive oxygen species and mtDNA and others leads to the onset of progressive age-associated pathologies [8]
1. cancer
2. neurodegeneratin
3. hearing loss
It is should be aware that accumulation of destruction caused by free radical is one of the most commonly cited effects of age-related mitochondrial dysfunction.
Researchers have used knockin mouse strains that express error-prone mtDNA polymerases that ignore proof-reading activity. The mous matures normally, but showed properties that are determined to be accelerated aging. These symptoms included;
- apoptosis
- curvature of the spine
- reduced fertility
- weight loss
- early death
[9].
Recent research into aging has lead scientists to believe that damage done by free radical oxygen may be one reason why organisms die from old age. Reactive Oxygen Species, or ROS, are produced in greatest quantity at the mitochondria, so this organelle is the most likely to be damaged by the free radical oxygen. Increases in ROS damage the mitochondria in several ways. They can modify amino acids and mutate the genes in mitochondrial and nuclear DNA, all affecting proper protein folding. Furthermore, ROS causes oxidative damage to mitochondrial DNA (mtDNA), especially because mtDNA is near the ROS production area[10]. Mutations on the mtDNA could result in reduced ATP production, increased ROS production, and eventual apoptosis. The increased ROS production has the added effect of being potentially harmful to the cell hosting the mitochondria, which could cause mutation in the cell DNA.
Anti-aging research has shown in some model organisms that by genetically disrupting the function of mitochondria life span has been increased. This is because ROS production was decreased so that less damage was done to the mtDNA. Specifically, a reduction in the mitochondrial function of the electron transport chain (ETC) in c. elegans increased longevity of the organism.
Scientists hope to be able to apply this to extending human lifespan by somehow incorporating reduced mitochondrial function with a treatment of dietary restriction (DR). By reducing the amount of calories taken in, but not to the point of starvation, cellular processes would change such that emphasis is placed on maintaining existing cellular structures rather than generating new structures to replace old structures. This would cause cells to persist in the body longer and there would be fewer mutations due to gene replication during mitosis.
Recent studies have underscored the importance of mitochondrial quality control (QC) pathways such as chaperones and proteases in solving mitochondrial dysfunctions. The protease complex "Lon" contains a cofactor made of iron-sulfur clusters; it is therefore suspected that Lon plays a role in targeting proteins by making them susceptible to oxidative damage. Findings suggest that a lack of Lon in yeast and mammalian cells results in excess proteins and mtDNA deletions, as well as respiration loss[11]. These all suggest that Lon plays a role in quality control systems in the mitochondria. On the other hand, over-expression of Lon in the fungus Podospora anserina leads to greater cell life span[12]. This suggests that there can be ways to artificially enhance the activity of proteases in order to increase cell longevity. Although many studies have strongly implied the importance of chaperones and proteases in anti-aging properties, it is important to note that fewer studies have been done in mammals.
Although mitochondria have unique genetic and protein synthesis system, majority of mitochondrial proteins are synthesized as precursors in the cytosol and are imported to mitochondria from the cytosol. Now, five different protein import pathways have been distinguished. Since these pathways cooperate with each other and are connected to other systems that function in the respiratory chain, mitochondrial membrane organization, protein quality control and endoplasmic reticulum-mitochondria junctions, mitochondrial protein import is highly responsible for major mitochondrial functions.
Two classical import routes:
Classical import routes include presequence pathway and carrier pathway. Both pathways use the core of TOM (translocase of the outer mitochondrial membrane) complex, which is the main protein entry gate of mitochondria including Tom 40, to transport precursor from the cytosol to intermembrane space.
Presequence pathway targets proteins carry cleavable presequences, which are peptide extensions of about 10 to 60 amino acids residues located at the N-terminal end of the protein
Cleavable precursor proteins are recognized by the receptor Tom20 and Tom22 and are transported across the outer membrane from the cytosol to intermembrane space through Tom40 channel
Cleavable precursor proteins are then transferred to Tim23 complex (presequence translocase of the inner mitochondrial membrane) with the help of intermembrane space-exposed proteins, including Tim25 and Tim22. The membrane potential of the inner membrane enables Tim23 channel to translocate the preseqyebces across the inner membrane. The transport of protein into the matrix requires the ATP that powers the PAM (presequence translocase-associated motor).
Presequences-carrying precursors are inserted into the inner membrane by two different ways. A) Many presequence-containing inner membrane proteins are laterally released from the Tim23 complex as the inner membrane proteins. Some of the hydrophobic sorting signal of some presequences are removed by the inner membrane peptidase complex and released into the intermembrane space as the intermembrane space proteins. B)Other precursor proteins are first transported into the matrix and eventually integrated into the inner membrane through Oxa1 export complex.
Carrier pathway transports non-cleavable precursor proteins into inner membrane.
Non-cleavable precursors are recognized by the receptor Tom70 and are transported across the outer membrane from the cytosol to intermembrane space through Tom40 channel
The precursor proteins are then guided by small TIM chaperone, such as Tim9-Tim10 complex, to the Tim22 complex (the carrier translocase of the inner mitochondrial membrane). The membrane potential of the inner membrane enables Tim22 to insert precursor proteins into the inner membrane as the carrier proteins.
Two routes of inserting proteins into the outer membrane:
The outer membrane of mitochondria contains two classes protein: β-barrel proteins, which are probably derived from the bacterial ancestor of mitochondria, and proteins with α-helical transmembrane segments, which are probably derived from the eukaryotic cell. The two routes of inserting two different proteins are called β-barrel pathway (SAM) and α-helical insertion (Mim1) reprehensively. Although SAM and Mim1 complexes insert precursor proteins independently, they can cooperate with each other to assemble some outer membrane complexes, such as TOM complex, which consists of central beta-barrel protein Tom40 and some α-helical subunits.
β-barrel pathway inserts β-barrel protein via SAM complexes
β-barrel precursors are recognized by TOM receptors and are translocated to the intermembrane space by Tom40 channel.
β-barrel precursors are then guided by small TIM chaperone to the SAM complex, Sam50 and Sam35, and are converted to outer membrane β-barrel proteins, which is also linked to ER-mitochondria junctions due to the dual localization of Mdm10 in SAM and ERMES, ER-mitochondria encounter structure
α-helical insertion inserts most Tom proteins, which have a single α-helical transmembrane segment, and outer membrane proteins with multiple α-helical transmembrane segments via Mim1, which contains one α-helical transmembrane segment.
α-helical precursors are recognized by Tom and are transferred to Mim1.
α-helical precursors are then interacted with Mim1 and are inserted into the outer membrane. [The exact function of Mim1 and how Mim1 really inserts precursor proteins is still unknown]
Protein import into the intermembrane space:
Mitochondrial intermembrane space assembly pathway (MIA) targets the intermembrane space precursors, which are cysteine-rich.
The precursors are transferred from cytosol to intermembrane space though TOM complexes in a reduced state.
When the precursors are in the intermembrane space, the precursors are oxidized and are immediately bound with Mia40, forming a transient disulfide bond. Then, Mia40 forms the oxidative folding of proteins, Erv1, to function as disulfide relay. Erv1 catalyzed the formation of disulfide bonds in Mia40 that oxidizes cysteines in the precursor proteins. As a result, new disulfide bonds are formed. Electrons flow from Mia40 to Erv1 and eventually to respiratory chain. In other words, Mia40 acts as a receptors that recognizes the precursors and enables the translocation of precursors into intermembrane space.
↑“Mitochondrial protein quality control during biogenesis and aging” by Brooke M. Baker, Cole M. Haynes
↑“Mitochondrial protein quality control during biogenesis and aging” by Brooke M. Baker, Cole M. Haynes
↑“Mitochondrial protein quality control during biogenesis and aging” by Brooke M. Baker, Cole M. Haynes
↑“Mitochondrial protein quality control during biogenesis and aging” by Brooke M. Baker, Cole M. Haynes
↑“Mitochondrial protein quality control during biogenesis and aging” by Brooke M. Baker, Cole M. Haynes.
↑Brooke M. Baker, Cole M. Haynes, Mitochondrial protein quality control during biogenesis and aging, Trends in Biochemical Sciences, Volume 36, Issue 5, May 2011, Page 255, ISSN 0968-0004, 10.1016/j.tibs.2011.01.004. <http://www.sciencedirect.com/science/article/pii/S0968000411000156>
↑Brooke M. Baker, Cole M. Haynes, Mitochondrial protein quality control during biogenesis and aging, Trends in Biochemical Sciences, Volume 36, Issue 5, May 2011, Page 258, ISSN 0968-0004, 10.1016/j.tibs.21.01.00014. <http://www.sciencedirect.com/science/article/pii/S0968000411000156>
↑Brooke M. Baker, Cole M. Haynes, Mitochondrial protein quality control during biogenesis and aging, Trends in Biochemical Sciences, Volume 36, Issue 5, May 2011, Page 259, ISSN 0968-0004, 10.1016/j.tibs.2011.01.004. <http://www.sciencedirect.com/science/article/pii/S0968000411000156>
IF1: setting the pace of the F1Fo-ATP synthase. Campanella, M. Trends in Biochemical Sciences, Volume 34, Issue 7, 343-350, 25 June 2009.
Scherz-Shouval, Ruth, et al. Regulation of autophagy by ROS: physiology and pathology
Schatz, Gottfried. The Magic Garden, Annual Review of Biochemistry, 2007: 673-78.
Falkenberg, M., Larsson, N., & Gustafsson, C. M. (2007). DNA replication and transcription in mammalian mitochondria. Annual Review of Biochemistry, 679-699.
Mair, William, and Andrew Dillin. "Aging and Survival: The Genetics of Life Span Extension by Dietary Restriction." Annual Review of Biochemistry 77.1 (2008): 727-54. Web
Wager, Peter. Exercise Physiology. Aug 17,2011.
Becker, Thomas, Lena Böttinger, and Nikolaus Pfanner. "Mitochondrial Protein Import: From Transport Pathways to an Integrated Network." Trends in Biochemical Sciences 37.3 (2012): 85-91.
Hamanka, Robert B., and Navedeep S. Chandel. "MItochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes." Trends in Biochemical Sciences 35 (2010): 505-13.
Fischer, Fabian, Andrea Hamann, and Heinz D. Osiewacz. "Mitochondrial quality control: An integrated network of pathways." Trends in Biochemical Sciences 37 (2012): 284-92.
Slonczewski, Joan L. Microbiology "An Evolving Science." Second Edition
Cardiolipin is known as the signature phospholipid of mitochondria. It is responsible for a wide range of mitochondrial functions, including but not limited to ATP synthesis. Therefore, disorientation and disturbance in the metabolism of cardiolipin can cause pathological issues. Recently, there is a study on the enzymes that participate in cardiolipin biosynthesis and remodeling. After biosynthesis of cardiolipin, there are three remodeling enzymes that modify the acyl chain composition of cardiolipin, producing either a “dysfunctional” cardiolipin that is tied to michondrial dysfunction or a tissue-specific cardiolipin in its mature form. The following will discuss newly found molecules involved in cardiolipin metabolism as well as how human diseases are affected by the changes in cardiolipin metabolism.
Cardiolipin formation is key to the enzyme functionality of the mitochondria thereby with it malfunctioning mitochondria related diseases can occur. This compound is referred to as the “signature phospholipid” of the mitochondria because it is always present as well as is being formed there. It also appears to promote ATP formation which is the major role of the mitochondria.
Cardiolipin improves the efficiency of the OXPHOS machinery.
When cardiolipin is present in an organelle’s membrane, that organelle must be a mitochondrion. This is the reputation of cardiolipin, the signature phospholipid of mitochondria. There, there is no surprise that most of the cardiolipin in a cell is closely tied to the inner membrane of mitochondria. Furthermore, the cardiolipin synthesis actually takes place in the inner membrane of mitochondria. Unlike other phospholipids that are also synthesized in a special compartment of the cell, cardiolipin remains closely associated with the mitochondrial membranes and does not get distributed out to the other endomembrane system like other phospholipids do. Because of this close association between cardiolipin and mitochondria, it is hypothesized that cardiolipin plays a huge role in ATP production through oxidative phosphorylation. In addition to this, since cardiolipin is only found in membranes that has an electrochemical gradient, it therefore also supports that cardiolipin has something to do with ATP production in the powerhouse of the cell. In addition to oxidation phosphorylation, cardiolipin also play a role in many mitochondrial activities.
Cardiolipin contains two phosphatidyl groups connected by a glycerol. For this reason, cardiolipin is a lipid dimer that has four acyl chains instead of two as seen in other phospholipids. Out of all the biosynthetic enzymes except for cardiolipin synthase, they do not have any acyl chain specificity. Thus, the last acyl chain composition is not made during biosynthesis but rather in deacylation-reacylation/transacylation reactions. When added together, these reactions make the final form of cardiolipin cell/tissue-specific. In other words, different organisms can have different final molecular forms of cardiolipin and even in different tissues in the same organism. This proposes that the different forms of cardiolipin exist for the different tissues and cells function, mainly to accommodate the functions and energetic demands of the different tissues/cells. However, we are still unsure of whether having different acyl chain compositions will affect the function itself. We do know that the Barth syndrome is associated with the first inborn error of cardiolipin metabolism, since its cardiolipin is alternated in biosynthesis. So far, there are three known cardiolipin remodeling pathways as well as known proteins in the different stages of the cardiolipin biosynthetic pathways.
Cardiolipin biosynthesis takes place in the mitochondrion of eukaryotic cells. The enzymes of the dephosphorylation of phosphatidylglycerolphosphate and initiation of the cardiolipin remodeling cascade were recently found.
Phosphatidylglycerolphosphate phosphatase
Matrix IM protein import
The proteins Gep4 and PTPMT1 were identified in the phosphatidylglycerolphosphate phosphatases. Initially, Gep4 was detected in a screen in yeast as genetic interactors of prohibitions, which led to identifying Gep4 as a key player in the phosphatidylglycerolphosphate phosphatase of cardiolipin synthetic pathways. Indeed, a gep4 null has small amounts of cardiolipin and phosphatidylglycerol, which build up phosphatidylglycerolphosphate and destabilized respiratory supercomplexes. Furthermore, this is not able to grow on carbon sources that need a working OXPHOS system. In addition, recombinant Gep4 dephosphorylates phosphatidylglycerolphosphate to phosphatidylglycerol in experiments conducted in the laboratory.
Recently, the function of the substrates of PTPMT1 was discovered. PTPMT1 is part of the phosphatase and tensin homolog family that lives in the mitochondria. Knockout mices of PTPMT1 died in the uterus before embryonic day 8.5, showing that PTMPT1 is necessary for life. In observing the mouse embryonic fibroblasts of the PTPMT1 knockouts, slow growth, OXPHOS defects, reduced complex I levels, and altered inner membrane morphology were all seen. In addition, cardiolipin and phosphatidylglycerol levels decreased while phosphatidylglycerolphosphate accumulated. Like Gep4, recombinant PTPMT1 dephosphorylates phosphatidylglycerolphosphate to phosphatidylglycerol.
While Gep4 is only present in plants and fungi, PTPMT1 is conserved evolutionarily. Still, these two proteins play a major role in the inner membrane of mitochondria and catalyze the same reaction. Interestingly enough, PTPMT1 is able to rescue a Geo4 null. Even though cardiolipin is present on both sides of the inner membrane, the two proteins Gep4 and PTPMT1 reside in the matrix face of the inner membrane only.
Cardiolipin phospholipases
The removal of a single acyl chain leads to monolysocardiolipin, which initiates cardiolipin remodeling. In yeasts, tafazzin (Taz1) is the transacylase that participates in cardiolipin remodeling. It does so by taking an acyl chain from another phospholipid and connecting it to the monolysocardiolipin. In the upstream of Taz1, cardiolipin deacylase 1 (Cld1) resides there. Surprisingly, Δcld Δtaz1 yeasts does not have monolysocardiolipin and has normal level of cardiolipin. However, Cld1 is observed to be involved in more than one metabolic pathways. This is due to the growth phenotype of the Δcld1 Δtaz1 strain is more disastrous than just one mutant. Secondly, Cld1 can add water to phospholipids as well as cardiolipin.
This molecule proceeds through a maturation process before becoming functional following the steps:
Figure 2 Simplification of the Cardiolipin Maturation Process in the Mitochondria
The pathway for forming Cardiolipin requires many enzymes, 1 for each step. Problems with Cardiolipin formation may result in lowered functionality of the mitochondria implying the following roles of the Cardiolipin in the mitochondria:
Increasing efficiency of the OXPHOS- oxidative phosphorylation involved in ATP formation
Stabilized assemblies involved in this process
Traps protons resulting in changes in the gradient which is involved in the process that produces the most ATP in the cycle which results in an increasing gradient and a in turn a greater ATP production
Help in the process of cell death
Is key to other compounds being able to recognize and identify the mitochondria
An important aspect to proper activation of the Cardiolipin are the remodeling process of which there are three without which it does not function properly. There are three key enzymes that promote this remodeling process:
Tafazzin
Monolysocardiolipin acyltransferase 1
Acyl-CoA: lysocardiolipin acyltransferase 1
The major diseases involved in the malfunctioning of these include: Wilson’s disease and Barth syndrome.
While the studies in cardioipin metabolism and how it can lead to human diseases is still new, there is a consistent effort to conduct experiments. So far, we have the complete biosynthetic pathway and remodeling inventory. For future steps, the regulation of cardiolipin metabolism and how it relates to health can be further studied. With the increasing interest and models in search of answers, we are close to accumulating more knowledge for this signature phospholipid of mitochondria.
Claypool, Steven M., and Carla M. Koehler. "The Complexity of Cardiolipin in Health and Disease." Trends in Biochemical Sciences 37.1 (2012): 32-40. PubMed. Web. 3 Dec. 2012.
Zhang, J; Dixon JE (6-8-2011). "Mitochondrial phosphatase PTPMT1 is essential for cardiolipin biosynthesis". Cell Metab 13 (6): 690–700. doi:10.1016/j.cmet.2011.04.007. PMID 21641550.
Every animal cell has two small organelles called centrioles. Centrioles help the cell when it comes time to divide. Mitosis and meiosis both have centrioles involved in. A centriole is a small set of microtubules arranged in a specific way. Also, there are nine groups of microtubules. The centrioles are found in pairs and move towards the poles of the nucleus when it is time for cell division. During interphase, the cell is at rest. The centrioles move to opposite ends of the nucleus and a mitotic spindle of threads begins to appear during prophase.
Centrioles are cylindrical structures that composed of groupings of microtubules arranged in 9+3 pattern. They are usually exist in pairs that form centrosomes. Centrioles are found in animal cells and play a big role in cell division. While cell division, the centrosome divides and the centrioles replicate (make new copies). The result is two centrosomes, each with its own pair of centrioles. The two centrosomes move to opposite ends of the nucleus, and from each centrosome, microtubules grow into a "spindle" which is responsible for separating replicated chromosomes into the two daughter cells.
Centrioles contain delta-tubulin, a protein which is part of the structure of tubulin.
Centrioles are barrel-shaped organelles that are found in most eukaryotic cells, with the exception of vascular plants and fungi. Centrioles consist of nine triplets of microtubules and are involved in the mitotic spindling, as well as when the cytoplasm of a eukaryotic cell split to form two daughter cells.
The intracellular fluid inside a cell is called the cytosol. It is separate from certain cell organelles such as the nucleus and the mitochondria. In eukaryotes, the cytoplasm is the content within a cell membrane minus the content in the cell nucleus. It is where many metabolic reactions occur, there are still others within organelles. In prokaryotes, cytosol is where most metabolic chemical reactions occur. These reactions also occur in the membranes or in the periplasmic space but to a lesser extent.
A complex mixture of substances dissolved in water forms the cytosol even though water is the large majority of the mixture. Sodium and potassium concentrations are different in the cytosol compared with the concentrations in the extracellular fluid. The difference in ion levels are important for osmoregulation and cell signaling. Besides ions, the cytosol also has macromolecules.
There is often much confusion between the cytoplasm and the cytosol. The cytoplasm is the fluid contained within the cell that holds and surround the cell's organelles in a liquid environment which is necessary for many of the cell's vital functions to occur. Some of the organelles that are held within the cytoplasm include the mitochondria, the golgi apparatus, the endoplasmic reticulum, and other organelles. One thing to note is that the nucleus isn't considered to be part of the cytoplasm because it contains its own type of fluid-like material that is referred to as the nucleoplasm. The materials that are found within the cytoplasm are also typically found within the cell membrane. Typically, the cytoplasm contains materials that are known as cytoplasmic inclusions. These inclusions are typically starch granules, mineral crystals, or lipid droplets that are floating around within the cytoplasm.
When looking at the two, the cytosol is often confused as being the cytoplasm itself, and many individuals view the two as being synonymous. However, they cytosol is in fact just a part of the cytoplasm. The gel-like translucent fluid is what the cytosol really is. It is typically not enclosed within the organelles found within the cell. The cytosol is responsible for suspending the other elements contained within the cytoplasm, like the cytoplasmic inclusions and the cell organelles. Consisting of around 75% of the cell's total volume, the cytosol is composed of many different cellular components. Concentration gradients, cytoskeletal sieves, protein complexes, protein compartments, water, salts, organic compounds, and dissolved ions all consist of the contents of cytosol. Structurally, the cytosol consists mostly of water. However, it does house cytoskeleton filaments, which are necessary for maintaining the overall structure of the cell. In regards to function, the cytosol is also the location where many of the cell's chemical reactions occur. Transportation of metabolite and cell communication are among some of the important functions that occur within the cytosol.
Cytosol and Importing tRNA into Mitochondria
It has been observed that the mitochondria compensate for its lack of mitochondrial tRNA genes by taking in tRNA contained within the cytosol of the cell. The organelle does this by co-importing tRNA along with mitochondrial precursor proteins that intercepts the organelles pathway for traditional protein importing. This method also illustrates the complexity of mitochondrial protein importing, because specific modifications and mitochondrial precursor proteins are required to carry out imports for certain proteins; and with the variety of proteins that the organelle imports, this method possesses a unique set of problems. Another, more direct method, of obtaining more tRNA can be directly importing the tRNA without the help of mitochondrial precursors; a method that is seen in even earlier protozoa.
Macromolecules
Commonly seen, protein molecules that do not bind to cytoskeleton or cell membranes are simply because they dissolve in the cytosol. Since the amount of protein in cells approaches up to 200 mg/ml, it is known to be very high. Protein also occupies approximately 20-30% of the volume of cytosol. However, even with this many protein occupying cytosol, some proteins with the membranes or organelles in cells are known to be very weak and are eventually released into solution upon cell lysis.
If given ATP and amino acids, cells are able to synthesize proteins. This shows that many of the enzymes in cytosol are bound to the cytoskeleton in the cell. In cells such as prokaryotes cell, the cytosol within nucleoid contains the cell's genome. This is known to be an unusual mass of DNA and associated proteins that control the replication and transcription of the bacterial chromosomes and plasmids.
On the other hand, cells such as eukaryotes cell the cytosol containing the cell's genome is held within the cell nucleus, which then separates from the cytosol by nuclear pores that blocks the free diffusion of any kind of molecule that is larger than 10 nm in diameter.
The effect of macromolecular crowding is cause because of too high concentration of macromolecules in the cytosol. In other words, this happens when the effective concentration of other macromolecules is increased, since they have less volume to move in. This squishy effect can cause the position and rate of chemical equilibrium of the cytosol's reactions. Sometimes in the genome forming protein complexes or DNA binding proteins occurs to the favor the association of marcromolecules.
Schneider A., “Mitochondrial tRNA import and its consequences for mitochondrial translation.” Annu Rev Biochem. 2011 Jun 7;80:1033-53. Print
http://en.wikipedia.org/wiki/Cytosol
Lysosomes are spherical bodies, or vacuoles that are enclosed by a single membrane. The membrane serves as a protectorate to the cell, since lysosomes contain harsh digestive enzymes, which would cause significant damage if exposed to cell content. Lysosomes contain different hydrolytic enzymes, such as proteases, lipases, and nucleases that are capable of breaking down all types of biological polymers (e.g. proteins, nucleic acids, carbohydrates, and lipids) that enter the cell or are no longer useful to the cell. In all, lysosomes function as the digestive system of the cell. On another note, lysosomes avoid self-digestion by glycosylation of inner membrane proteins, which prevent their degradation.
Structure of Lysosome
Lysosomes are membrane-enclosed organelles that help eukaryotic cells obtain nourishment from macromolecular nutrients. The lysosomes contain many hydrolytic enzymes such as proteases, nucleases, and lipases). The lysosomes are formed vesicles containing hydrolytic enzymes and proton pumps bud off from the Golgi complex. Phagocytosis and lysosomal digestion help the eukaryotic cell because they effectively increase the membrane surface area over which nutrients can be absorbed. However, in eukaryotes, lysosomes allow for intracellular digestion, and digested material crosses the lysosomal membrane into the cytoplasm.
Lysosomes are manufactured and budded into the cytoplasm by the Golgi apparatus with enzymes inside. The enzymes that are within the lysosome are made in the rough endoplasmic reticulum, which are then delivered to the Golgi apparatus via transport vesicles. When lysosome reaches the cytoplasm, fusion forms a secondary lysosome.
Glycosidase: an enzyme that catalyzes the hydrolysis of glycosidic linkages in sugar molecules.
Protease: an enzyme that catalyzes the hydrolysis of peptide bonds.
Acid phosphatase: a phosphatase, an enzyme that catalyzes they hydrolysis of phosphate linkages in a variety of molecules.
Sulfatase: an enzyme that catalyzes the hydrolysis of sulfate ester linkages in molecules.
Lipase: an enzyme that catalyzes the hydrolysis of ester bonds in lipid molecules.
Amylase: an enzyme that catalyzes the breakdown of carbohydrates into sugars.
Nuclease: an enzyme that catalyzes the hydrolysis of phosphodiester bonds in nucleotide subunits of nucleic acids. Structure of Lysosome
Glucocerebrosidase: an enzyme which breaks down glucocerebroside
Phosphoric Acid monoesters
Lysosome enzymes are proteins that are synthesized in the endoplasmic reticulum and modified in the Golgi apparatus. Lysosomal enzymes are tagged for lysosomes by the addition of mannose-6-phopahte label. A shortage of any one of these enzymes will lead to lysosome diseases such as Tay-Sachs disease and Pompe’s disease.
In human cells, when lysosomes lack enzymes they can generate storage disease
Structure of Lysosome
. People with these defects are usually missing one or more of the Lysosomal hydrolysis enzymes.
Tay-Sachs Disease
Tay-Sachs disease is a disease that affects the nervous system. It occurs when the body lacks hexosaminidase A, a protein that breaks down ganglioside. Accumulation of ganglioside
Pompe’s disease
Pompe’s disease is a disease that affects the heart Structure of Lysosomeand skeletal muscles. It occurs when the body lacks alpha-glucosidase, an enzyme that breaks down glycogen.
Mucopolysaccharidosis I
MPS I is cause by the deficiency of the lysosomal enzyme alpha-L-iduronidase. Deficiency can effect both skeletal and mental development.
Gaucher’s disease
Gaucher’s disease is caused by the deficiency of the lysosomal enzyme glucocerebrosidase enzyme. A deficiency of this enzyme causes harmful substances to build up in liver, spleen, bones, and bone marrow.
Enzymes that are used by the lysosome are synthesized in the endoplasmic reticulum (ER) and are transported to the Golgi apparatus (GA). The ER formed transport vesicles that carry the enzymes and loads them by fusing them to the GA. Glycosylation may occur, which will determine the final destination of the proteins.
Lysosomes contain a variation of enzymes that hydrolyze proteins, nucleic acids, carbohydrates, and lipids. The enzymes present in the lysosomes work best at an acidic pH; thus they are referred to as acid hydrolases. Is it evident to note that the pH in a lysosome is about ~5 while the cytoplasm has a neutral pH of about ~7. In order for the lysosome to maintain an acidic pH, the lysosome must actively uptake concentrate H+ ions (protons). The concentrated H+ ions are actively equilibrated to reach a pH of about 5 by hydrogen ion pumps. In part with the pH level, enzymes within a lysosomes work best at an acidic pH.
Lysosomes digest unused and/or threatening content that is present in the cell. In retrospect, lysosomes recycle the cell’s organic material in a process known as autophagy. It also recycles waste products such as fats, carbohydrates, proteins, and other macromolecules into simple compounds, which are then utilized as new materials in the cytoplasm. The recycling of waste product is in large part due to about 40 different types of hydrolytic enzymes that are engineered by the endoplasmic reticulum and modified by the Golgi apparatus.
Autophagy (Autophagocytosis)
The term autophagy is derived from the Greek “auto” or oneself and “phagy” to eat. Autophagy is a process of self-eating and self-degradation. The degradation takes part when the cell content and organelles is consumed by lysosomes. The cell produces vesicles called autophagosomes that captures and deliver cytoplasmic material to lysosomes. Autophagosomes are formed when the ER wraps around damaged cell structures or cell content, of which the lysosome then joining the autophagosomes to destroy the content captured and delivers the broken down material to the cytoplasm. Overall, autophagocytosis preserves the health of cells and tissues by degrading unused and/or damaged cellular content to new ones.
Endocytosis
Endocytosis occurs when the cell membrane of a cell surrounds large nutritional molecules. This engulfing of a large nutritional molecule forms a endosome. Some of the things endosomes acquire are complex lipids, polysaccharides, nucleic acids, and proteins. Lysosomes then fuse with endosomes, whereby it transfers it enzymes to breakdown molecules. The broken down molecules are then delivered to the cytoplasm by proteins for later consumption.
Phagocytosis
Lysosome works in close relations with phagocytosis whereby it joins phagosomes to break down the object or bacteria with its enzymes. Phagosomes are produced when the cell membrane of a cell surrounds a disease-causing bacterium; toxic or unused content in/outside the cell.
Cell Structure. Letter i corresponds to the Peroxisome
They are theorized to have evolved from bacteria that formed a symbiotic relationship with their host cell. It was believed that the development of this relationship over generations led to bacteria evolving as an organelle inside the body. This view has recently been countered since it was found that cells without peroxisomes can restore these peroxisomes with a simple gene introduction. Therefore, the previous theory was challenged since the introduction of peroxisomes is not an evolutionary process and can easily be replicated.
Analyzing the structures of peroxisomes has helped figure their function and role in the biological world.
Peroxisomes are not derived from the endoplasmic reticulum and therefore are not a part of endomembrane system. They replicate by fission. This organelle is surrounded by a lipid bilayer membrane which encloses the crystalloid core. The bilayer is a plasma membrane which regulates what enters and exits the peroxisome. There are at least 32 known peroxisomal proteins, called peroxins, which carry out peroxisomal function inside the organelle.
The main function of peroxisomes is to break down long fatty acid chains through beta-oxidation and synthesize necessary phospholipids (such as plasmologen) that are critical for proper brain and lung function. Furthermore, they aid certain enzymes with energy metabolism in many eukaryotic cells as well with cholesterol synthesis in animals. Peroxisomes are also involved in germinating seeds in the glyoxylate cycle, photosynthesis in leaves, and oxidation of amines in various yeasts.
During catabolism of fatty acid chains in animal cells, peroxisomes break down long fatty acids into medium fatty acids which are then transported to mitochondria where the majority of catabolism happens. However, in yeast and plant cells, catabolism of fatty acid chains happens only in the peroxisome and the mitochondria is not involved. The catabolism of fats and fat-soluble vitamins, such as vitamin A and vitamin K, as well as the production of bile acids also takes place in peroxisomes. If this catabolism is not done properly, genetic disorders or skin disorders often result.
The synthesis of plasmalogens in animal cells also takes place in peroxisomes. These organelles are very important to the cell because production of plasmalogens is critical to proper functioning of the nervous system since a lack of plasmalogens causes abnormalities in the myelination of nerve cells.
Peroxisomal disorders may result due to abnormalities in single enzymes or groups of proteins that are necessary for normal peroxisome function.
These disorders can affect a range of organ systems, but problems with the nervous system are the most commonly observed. Along with brain damage, many of these disorders also lead to skeletal and craniofacial dysmorphism, liver dysfunction, progressive sensorineural hearing loss, and retinopathy.
Some examples of peroxisomal disorders:
1. Adrenoleukodystrophy: a fatal inherited X-linked disorder that leads to extensive brain damage and adrenal gland failure.
2. Zellweger syndrome spectrum (PBD-ZSD): a rare cerebrohepatorenal syndrome evolving due to a lack of functional peroxisomes.
3. Rhizomelic chondrodysplasia punctata type 1 (RCDP1): a rare brain disorder evolving due to shortening of the proximal bones.
Due to peroxisome's ability to "divide and import proteins post-translationally," it is suggested that it is similar to the mitochondria where an endosymbiotic relationship was formed in its origin.
Studies have shown that:
(39–58%) of peroxisome are of eukaryotic origin
(13–18%) are enzymes from the mitochondria
This lead to the conclusion that it did not have a endosymbiotic origin, but rather it used proteins from other eukaryotic cells.
The cytoskeleton provides support in a cell. It is a network of protein fibers supporting cell shape and anchoring organelles within the cell. The three main structural components of the cytoskeleton are microtubules (formed by tubulins) , microfilaments (formed by actins) and intermediate filaments. All three components interact with each other non-covalently.
Eukaryotic cells contain proteins called intermediate filaments, microfilaments, and microtubules that are collectively termed the cytoskeleton. Also, the cytoskeleton proteins are multifunctional and are also involved in whole-cell movements and movements of substances within the cell.
Actins and Tubulins are abundant cytoskeletal proteins which support diverse cellular processes due to their unique properties of filament-forming proteins. Recent evidences suggest that regulated degradation pathways exist for actin and tubulin collectively maintain the quality control of cytoskeletal proteins, ensuring the appropriate function of microfilaments and microtubules.[1]
Microtubules are polymers of tubulin. They help with cell transport. They also help with the cell shape because it resists compression. It also helps facilitate cell motility. There are two motor proteins that assist organelles to move along the microtubules:
Kinesin, which moves things away from the nucleus.
Dynein, which moves things towards the nucleus.
Microtubles have a larger diameter than microfilaments and intermediate filaments. The hollow microtubule structure consists of 13 tubulin dimers. They are one alpha-tubulin protein plus one beta-tubulin protein form one tubulin dimer. Microtubules also help movement of substances within the cell and are also involved in powering whole-cell movement by cilia and flagella. The microtubules provide tracks that can move vesicles from one organelle to the next in an efficient, directed fashion. Microtubules also segregate the duplicated chromosomes during mitosis.
Microfilaments are polymers of actin. They help with cell shape also because it bears tension in the cell. It is also involved in cell motility. There are three types of cell shape, which are microvilli, lamellipodia, and filopodia.
Microvilli are projections on surface that increase surface area.
Lamellipodia are membrane ruffles that help sense the environment and direct movement.
Filopodia are like microvilli but are less stable. They also sense the environment. They can turn into lamellipodia.
Microfilaments are formed when individual actin monomers polymerize, in a process fueled by ATP hydrolysis, to form chains of filamentous actin. Microfilaments are dynamic structures, growing and shrinking in a controlled manner. Some microfilaments play a structural role in the cell to maintain cell shape. These structural microfilaments have protein caps at both ends to prevent changes in microfilament length. Other microfilaments have functions that require dynamic changes in length. The microfilaments also mediate cytoplasmic streaming, a mixing of the cytoplasm that aids diffusion.
Intermediate filaments are polymers of keratin. They help with cell shape also because it bears tension in the cell. It is primarily involved in organelle anchorage.
The intermediate filaments also consist of various fibrous proteins that have a diameter of about 10 nm. Intermediate filaments often form a meshwork under the cell membrane and, in cells that lack a cell wall, help impart and maintain cell shape. Intermediate filaments are fairly stable and are not thought to undergo acute changes in length the way microfilaments.
Cytoskeletons are often synthesized based on the cell's needs; however, some protein fibers are permanent. The changeable nature of the cytoskeleton thus contributes to its five important functions.
The mechanical strength of the cell is due to protein scaffolding in the cytoskeleton. In some cells, protein scaffolding also determines the shape of the cell. Microvilli are supported by cytoskeletal fibers such as microfilaments. Microvilli also increase the surface area of the cell for the absorption of materials.
The cytoskeletal fibers of the cell help stabilize positions of organelles. However, the interior arrangement of the cell varies based on the cell's needs. The organelles are dynamic and change minute to minute.
The cytoskeleton has the ability to move materials not only in the cell but also within the cytoplasm, thus aiding in the movement of organelles as well. This function is important especially in the nervous system where materials are often transported over long intracellular distances.(3908283293)
Cells are connected to one another through the linking of the protein fibers of the cytoskeleton as well as the protein fibers in the extracellular space. In the process, materials outside the cell are stabilized as well. The assembly of cells not only contributes to the mechanical strength of the tissue but also allows information to transfer between cells from one to another.
The cytoskeleton of the cell allows the cell to move. For example, the cytoskeleton of white blood cells allows them to squeeze out of blood vessels. Growing nerve cells are also able to send out extensions that allow them to elongate. The microtubule cytoskeleton of cilia and flagella on the cell membrane allow them to move. In addition, motor proteins using energy from ATP can aid in the movement of cells and intracellular transport by sliding along cytoskeletal fibers. There are three types of motor proteins in the cytoskeleton that include myosins, kinesins and dyneins.Kinesins oftentimes brings vesicles from inside the cell out to the periphery. If the kinesins are mutated, then there is likely to be a neural disorder. KIF1β is one disorder resulting from a mutated kinesin. It causes lack of strengths in arms and legs. Dynein, on the opposite hand, brings the vesicles from the periphery back to the inside of cell. Motor proteins convert energy into movement and those found in the cytoskeleton use stored ATP. Myosins allow for muscle contractions. Kinesins allow for the movement along microtubules as dyneins help with the whiplike motion of the microtubule bundles of cilia and flagella. Many motor proteins consist of two heads (to bind to the cytoskeleton fiber), a neck and a tail region at the end to bind to organelles.
The evolution of cytoskeletal proteins required a novel biogenesis machinery. The cytoskeleton in eukaryotes enhances intracellular trafficking and cell division. These functions were once believed to be the distinguish elements of eukaryotes from prokaryotes because bacterial cytoskeleton also composed of proteins that similar to actin and tubulin. The actin-like and tubulin-like proteins in bacteria form filamentous structures which imply in the division of genetic material and maintenance of cell shape.[2] However, actins and tubulins in eukaryotes are distinct from similar prokaryotic proteins in the way that they maintain innovative properties which is critical for eukaryogenesis (the origin of the eukaryotic condition/the evolution of the eukaryotic condition).[3]
Both eukaryotes and bacteria have a cytoskeleton. The bacterial proteins homologous to actin are MreB and ParM, and the bacterial proteins homologous to tubulin are FtsZ. However, actin and tublin differ from MreB, ParM, and FtsZ in the sense that they have properties important for eukaryogenesis.
Actins and tubulins in eukaryotes formed microfilaments and microtubules which unite with their complementary molecular motors (myocin, kinesin, dynein) to be used for phagocytosis. Phagocytosis enables endosymbiosis and also cilia's development which supports mobility and sensory.[4] The inception of actins and tubulins in eukaryotes is an important factor in facilitating their efficient folding and assembly [5][6] which is called cytoskeletal protein biogenesis machinery (CPBM). The CPBM includes molecular chaperones which assist folding of chaperonin containing tailless complex polypeptide-1 (CCT), prefoldin (PFD) - phosducin-like proteins that regulate CCT functions, and five other cofactors. [7][8][9][10][11] In addition, post-translational modifications and proteasomal degradation are required in regulating the function of actins and tubulins which is also unique to eukaryotes.
On the other hand, in prokaryotes, the cytoskeletal protein biogenesis machinery is absent.
Actin and tubulin concentrations are strickly controlled due to their critical effects on cytoskeletal dynamics. In animal cells, tubulin synthesis is regulated by an autoregulatory feedback mechanism that can sense the concentration of tubulin heterodimer in order to regulate the stability of ɑ-tubulin, and β-tubulin mRNAs. [12][13] Similar to animals, the synthesis of tubulin in metazoans is also autoregulated due to its critical influences. Researches have shown that overexpression of β-tubulin in Saccharomyces cerevisiae leads to abnormal microtuble function and slow growth.[14]
Actin overexpression causes by an incompletely characterized feedback mechanism that is sensitive to the concentration of actin monomers is preventable by the presence of the 3' untranslated region of actin mRNA.[15]
The cytoskeleton protein biogenesis machinery is found in eukaryotes but not in prokaryotes. It includes chaperonin containing CCT (complex polypeptide-1) and PFD (prefoldin), phosductin-like proteins, and five cofactors. The chaperonin molecules help actin and tubulin proteins to fold. Challenges to the protein biogenesis machinery include: (1) the high concentrations of actin and tubulin in cells, (2) the tendency of actin and tubulin to self-associate, (3) the inability of actin and tubulin to fold without the help of other molecules, (4) the fact that actin and tubulin compete with one another for the same folding space.
Biogenesis of cytoskeletal proteins faces a lot of difficulty which mainly due to the abundant of actin and tubulin concentrations, self-associate tendency, and the inability in folding independently. In addition, the competing for access to limited folding space is also a challenge to biogenesis of cytoskeletal proteins.[16] Fortunately, the action of specific chaperonin cofactors can account for the regulation of chaperonin-mediated cytoskeletal protein folding.
Eukaryotic cytosolic chaperonin is a unique ability to assist the folding of actin and tubulin. Molecular chaperones can interact with the newly synthesized polypeptide chains to become stable during the folding process form a linear monomer amino acids chain into a more complex and functional protein [17]. There are many types of chaperones that direct the folding of new proteins, refolding of stress-denatured proteins, unfolding of proteins, and transporting proteins [18][19].
Chaperonin, an important family of molecular chaperones, has a barrel-like structure with two multimeric stacked rings of 60kDa (Dalton's atomic mass unit). Chaperonins undergo ATP-dependent conformational changes during its folding cycle: facilitate substrate binding, encapsulation and release[20]. In eukaryotes, the cytosolic chaperonin is the tailless complex polypeptide 1 ring complex (CCT) which is required for viability in yeast and worms.
CCT is crucial for the biogenesis of actin and tubulin. CCT is composed of eight related subunits - ɑ,β,ɣ,ẟ,ɛ,ʝ,ŋ,θ - which show up twice in each oligomer. CCT is closely related to archaeal chaperonin thermosome but quite different from bacterial chaperonin GroEL [21]. CCT is known to have a more specific binding profile than bacterial GroEL [22][23]. As a chaperonin, CCT undergoes ATP-dependent conformational changes during its folding cycle. Due on the copiousness of actin and tubulin, they occupy significant proportion of CCT complexes. Beside actin and tubulin, there are other CCT substrates also have key roles in progression of the cell cycle [24] Researches show that CCT function in vivo is regulated by several dedicated cofactors, including PFD and phosducin-like proteins.
PFD, a jellyfish-shaped molecular chaperone required for stabilization of new cytoskeletal proteins, is a CCT co-chaperone for the biogenesis of actin and tubulin [25][26].
Phosducin-like proteins, regulators of the folding of actin and tubulin in association with CCT, are thioredoxin domain-containing proteins with homology to phosducin, a regulator of retinal G-protein signaling [27].
Scientists believed that actin is released from CCT in a native, assembly-competent state, but cyclase associated protein might interact with and stabilize near-native or unstable forms of actin in close association with the chaperonin [28]. On the other hand, functional tubulin is an obligate ɑ-β heterodimer, and evolved in a folding pathway linked to dimer assembly[29].
Roles of each cofactor:
1. Tubulin cofactor A (TBCA): collects unassembled beta-tubulin
2. Tubulin cofactor D (TBCD): assists beta-tubulin down the assembly pathway
3. Tubulin cofactor B (TBCB): binds to alpha-tubulin after chaperonin release
4. Tubulin cofactor E (TBCE): receives alpha-tubulin from TBCB and processes it further
5. Tubulin cofactor C (TBCC): promotes GTP hydrolysis in beta-tubulin if in the presence of a stable supercomplex (formed by the joining of beta-tubulin-TBCD and alpha-tubulin-TBCE complexes)
a. also facilitates the release of native alpha-beta-tubulin heterodimer
Diseases linked to the cofactors:
It is believed that if TBCB is not degraded properly, a neurological disease called giant axonal neuropathy may result. This disease is related to decreased density of the microtubule of cells. Mutations of TBCE are associated with hypoparathyroidism (a developmental disorder), mental retardation, and facial dysmorphism (HRD).
Prefoldin (PFD) is a CCT co-chaperone required for stabilizing nascent cytoskeletal proteins. It consists of two alpha-type and four beta-type subunits which collectively make its structure resemble the shape of a jellyfish. The six subunits form a cavity shaped like a rectangle that attaches to newly formed actin and tubulin as the chains leave the ribosome. PFD then delivers the actin and tubulin to CCT, probably via a docking and substrate-release mechanism (supported by electron microscopy analysis of PFD-actin complexes). It is also possible that PFD improves the efficiency of actin and tubulin protein folding by navigating partially folded molecules back towards the CCT for more folding. This idea is supported by the fact that yeast cells without PFD were observed to fold actin and tubulin more slowly than wild-type cells. Furthermore, Pfd1 knockout mice showed signs of dysfunction of cytoskeletal proteins, neuronal loss, neuromuscular defects, and defective development of lymphocytes. These Pfd1 knockout mice were only viable for five weeks.
Phosducin-like proteins regulate the folding of actin and tubulin. Three such proteins have been termed PhLP1, PhLP2, and PhLP3 as they are CCT-binding proteins. PhLP1 assists in assembling heterotrimeric G-proteins by CCT; this process is regulated by PhLP1 phosphorylation. PhLP2 and PhLP3 are involved in the biogenesis of cytoskeletal proteins. It is believed that there is a specificity of PhLP2 for actin biogenesis and of PhLP3 for tubulin biogenesis. In other words, disruption of PhLP2 function caused severe actin cytoskeletal defects whereas disruption of PhLP3 function alters normal tubulin biogenesis.
When studied in vitro, it is observed that an excess of PhLP2 and PhLP3 prevents actin and tubulin from being folded via the CCT-mediated folding pathway. It is believed that this is due to the reduced activity of CCT ATPase. However, when the study in done on yeast cells, it appears that PhLP2 stimulates actin folding by purified yeast CCT. The researchers of this study concluded that amino acids of mammalian PhPL2 that were not present in yeast PhLP2 are responsible for preventing actin and tubulin from being folded. This also supports the idea that higher eukaryotes evolved with more regulation of cytoskeletal proteins.
For actin, post-translational modification is known to affect only folding. Tubulin, on the other hand, is affected by PTM in such a way that allows native proteins to turn on and off activity in a reversible and regulatory manner. Tubulin can be modified in a number of ways such as acetylation, detyrosination, and glutamylation. These tubulin modifications occur on microtubules, and it has been hypothesized (though not well-tested) that the free tubulin heterodimer is the substrate responsible for reversing the modifications. Much study, however, has been dedicated to Tubulin PTMs in general. Tubulin acetylation, for example, has been shown in recent studies to be linked to a human neurodegenerative disease called amyotrophic lateral sclerosis (ALS).
Part of the quality control process includes the degradation of proteins. Damaged proteins and misfolded proteins that cannot be refolded with the help of chaperones are removed in this process. Unfortunately, the pathway in which the degradation occurs is not as well studied as the biogenesis process.Invalid parameter in <ref> tag
Turn-over rates and steady-state concentrations of all cellular proteins is regulated in the following ways:
1. protein degradation through the ubiquitin-proteasome system (UPS)
2. lysosomal degradation
3. another proteolytic mechanism
“Proteostasis” is the regulatory process in which damaged or incorrectly folded proteins that cannot be repaired by charperones are removed by proteolysis. Not much work has been put into the study of actin and tubulin degradation, but tubulin has been known to rapidly degrade in the presence of microtubule-destabilizing drugs such as colcemid. Such drugs make tubulin more soluble.
Parkin is a ubiquitin-protein ligase important to tubulin degradation. Parkin is mutated in patients with autosomal recessive juvenile Parkinson disease (PD). Its normal function is to interact with HSP70-interacting protein (CHIP) to make stress-denatured proteins undergo ubiquitylation. Parkin is also believed to stimulate ubiquitylation and proteasomal degradation of alpha-tubulin and beta-tubulin. Cells that over-express mutant alpha-synuclein, a toxic inclusion-forming protein in Parkinson disease, reveal increased concentrations of alpha-tubulin and insoluble parkin. These two traits are also observed in patients affected by Lewy body disease.
E-like (COEL), a tubulin folding cofactor, is a protein that destabilizes tubulin. Cells without human COEL contain excess amount of stable microtubules while microtubule disassembly and the degradation of α-tubulin is observed in cells with excess COELs. The degradation of tubulin caused by the presence of COEL is countered by a negative regulator of microtubule called stathmin that isolates tubulins. Overall, COEL is important for three reasons: 1. it can remove misfolded tubulin 2. regulate the concentration of tubulin 3. control tubulin isotype.[30]
In metazoan cells, the concentration of tubulin is reduced when CCT or PFD does not function properly. The concentration of actin, however, is not affected significantly. This suggests that the quality control for actin differs from that of tubulin. Relative to the control tubulins, there seems to be less of a need to remove misfolded β-actins. Still, there are incidences when actin degradation is necessary. In the case of ischemic oxidative damage, α-actin specific to the heart is degraded by proteasome. It is also found that α-actins are degraded by lysosomes when muscle contraction in cardiomyocytes is inhibited by the use of drugs. It has been recently observed that TRIM32, an ubiquitin that ubiquitylates α-actin in vitro, when unusually expressed in the human embryonic kidney cells, reduces the concentration of cytoplasmic β-actins. TRIM32 mutations have been found in Bardet-Biedl syndrome and muscular dystrophy, although the actual role TRIM32 plays in these diseases remains unknown.[31]
Reece, Jane (2011). Biology. Pearson. ISBN978-0-321-55823-7. {{cite book}}: Text "coauthors+ Lisa A. Urry, Michael L. Cain, Steven A. Wasserman, Peter V. Minorsky, Robert B. Jackson" ignored (help)
Silverthorn, Dee Unglaub. "Compartmentation: Cells and Tissues." Human Physiology. Boston, MA: Pearson Custom Pub., 2007. Print.
↑Lundin , Victor , Michel Leroux, and Peter Stirling. "Elsevier: Article Locator." ScienceDirect.com | Search through over 10 million science, health, medical journal full text articles and books.. sciencedirect.com, n.d. Web. 17 Nov. 2012. <http://www.sciencedirect.com/science/article/pii/S0968000409002461#>.
↑J. Pogliano The bacterial cytoskeleton Curr. Opin. Cell Biol., 20 (2008), pp. 19–27
↑M.R. Leroux, F.U. Hartl Protein folding: versatility of the cytosolic chaperonin TRiC/CCT Curr. Biol., 10 (2000), pp. R260–264
↑T. Cavalier-Smith The phagotrophic origin of eukaryotes and phylogenetic classification of Protozoa Int. J. Syst. Evol. Microbiol., 52 (2002), pp. 297–354
↑S. Bertrand et al. Folding, stability and polymerization properties of FtsZ chimeras with inserted tubulin loops involved in the interaction with the cytosolic chaperonin CCT and in microtubule formation J. Mol. Biol., 346 (2005), pp. 319–330
↑S. Bertrand et al. Folding, stability and polymerization properties of FtsZ chimeras with inserted tubulin loops involved in the interaction with the cytosolic chaperonin CCT and in microtubule formation J. Mol. Biol., 346 (2005), pp. 319–330
↑S. Lacefield, F. Solomon A novel step in beta-tubulin folding is important for heterodimer formation in Saccharomyces cerevisiae
↑P.C. Stirling et al. PhLP3 modulates CCT-mediated actin and tubulin folding via ternary complexes with substrates
↑P.C. Stirling et al. Functional interaction between phosducin-like protein 2 and cytosolic chaperonin is essential for cytoskeletal protein function and cell cycle progression
↑E.A. Mccormack et al. Yeast phosducin-like protein 2 acts as a stimulatory co-factor for the folding of actin by the chaperonin CCT via a ternary complex
↑M. Lopez-Fanarraga et al. Review: postchaperonin tubulin folding cofactors and their role in microtubule dynamics J. Struct. Biol., 135 (2001), pp. 219–229
↑D.W. Cleveland et al. Unpolymerized tubulin modulates the level of tubulin mRNAs Cell, 25 (1981), pp. 537–546
↑M.E. Sellin et al. Global regulation of the interphase microtubule system by abundantly expressed Op18/stathmin Mol. Biol. Cell, 19 (2008), pp. 2897–2906
↑D. Burke et al. Dominant effects of tubulin overexpression in Saccharomyces cerevisiae Mol. Cell. Biol., 9 (1989), pp. 1049–1059
↑A. Lyubimova et al. Autoregulation of actin synthesis requires the 3′-UTR of actin mRNA and protects cells from actin overproduction J. Cell Biochem., 76 (1999), pp. 1–12
↑M.R. Leroux, F.U. Hartl Protein folding: versatility of the cytosolic chaperonin TRiC/CCT Curr. Biol., 10 (2000), pp. R260–264
↑F.U. Hartl, M. Hayer-Hartl Converging concepts of protein folding in vitro and in vivo Nat. Struct. Mol. Biol., 16 (2009), pp. 574–581
↑F.U. Hartl, M. Hayer-Hartl Converging concepts of protein folding in vitro and in vivo Nat. Struct. Mol. Biol., 16 (2009), pp. 574–581
↑E.T. Powers et al. Biological and chemical approaches to diseases of proteostasis deficiency Annu. Rev. Biochem., 78 (2009), pp. 959–991
↑C.R. Booth et al. Mechanism of lid closure in the eukaryotic chaperonin TRiC/CCT Nat. Struct. Mol. Biol., 15 (2008), pp. 746–753
↑C. Spiess et al. Mechanism of the eukaryotic chaperonin: protein folding in the chamber of secrets Trends Cell Biol., 14 (2004), pp. 598–604
↑C. Dekker et al. The interaction network of the chaperonin CCT EMBO J., 27 (2008), pp. 1827–1839
↑A.Y. Yam et al. Defining the TRiC/CCT interactome links chaperonin function to stabilization of newly made proteins with complex topologies Nat. Struct. Mol. Biol., 15 (2008), pp. 1255–1262
↑C. Spiess et al. Mechanism of the eukaryotic chaperonin: protein folding in the chamber of secrets Trends Cell Biol., 14 (2004), pp. 598–604
↑S. Geissler et al. A novel protein complex promoting formation of functional alpha- and gamma-tubulin EMBO J., 17 (1998), pp. 952–966
↑J. Martín-Benito et al. Structure of eukaryotic prefoldin and of its complexes with unfolded actin and the cytosolic chaperonin CCT EMBO J., 21 (2002), pp. 6377–6386
↑M. Blaauw et al. Phosducin-like proteins in Dictyostelium discoideum: implications for the phosducin family of proteins EMBO J., 22 (2003), pp. 5047–5057
↑E.A. McCormack et al. Mutational screen identifies critical amino acid residues of beta-actin mediating interaction between its folding intermediates and eukaryotic cytosolic chaperonin CCT J. Struct. Biol., 135 (2001), pp. 185–197
↑M. Lopez-Fanarraga et al. Review: postchaperonin tubulin folding cofactors and their role in microtubule dynamics J. Struct. Biol., 135 (2001), pp. 219–229
↑ Lundin, Victor, Loroux, Michel and Stirling, Peter. “Quality Control of Cytoskeletal Proteins and Human Disease” January 2010: 288-295.Retrieved on 20 November 2012.
↑ Lundin, Victor, Loroux, Michel and Stirling, Peter. “Quality Control of Cytoskeletal Proteins and Human Disease” January 2010: 288-295.Retrieved on 20 November 2012.
Berg, Jeremy "Biochemistry", Chapter 35 Molecular Motor. pp1018-1020. Seventh edition. Freeman and Company, 2010.
Slonczewski, Joan L. Microbiology "An Evolving Science." Second Edition.
All the different membranous organelles perform different tasks in the cell (animal cells and plant cells). However, they also communicate with each other though ions and lipids. In addition, some of the organelles have membrane that are connected. The others are related by transfer of membrane segments. As we know, structures determine functions in a lot of ways in nature. We can categorize eukaryotic organelles into four categories according to their general functions.
In this first category, nucleus, ribosomes, rough ER, smooth ER and golgi apparatus are included. They can perform this task because of they have metabolically active membranes. Some of them can also perform transportation of the products. Below is some examples of the manufacturing task these organelles do.
Nucleus--DNA and RNA synthesis
Ribosome--Polypeptide(protein) synthesis
Rough ER--Membrane protein synthesis, secretory proteins synthesis and hydrolytic enzymes synthesis; formation of transport vesicles
Smooth ER--Lipid synthesis; detoxification and carbohydrate metabolism in liver cells
Golgi apparatus--Modificaiton, temporary storage and transportation of macromolecules such as proteins; formation of lysosomes and transport vesicles
In this category, lysosomes, peroxisomes and vacuoles are included. They are all consist of single membrane sac that can be used to collect materials and/or break down unwanted/harmful materials.
Lysosomes--Digestion of bacteria, damaged organelle
Peroxisomes--breakdown of H2O2 by-product
Vacuoles--Digestion of waste products; collect excess water from the cell
In this category, chloroplasts and mitochondria are included. They both have metabolically active membranes and intermembrane compartments that can convert energy units to provide to the cells.
Chloroplast--Conversion of light energy to chemical energy in plant cells
Mitochondria--Conversion of chemical energy of food to chemical energy of ATP
In this last category, cytoskeleton(such as cilia, flagella and centrioles in animal cells), cell walls(in plants, fungi and some protists), extracellular matrix(in animal cells) and cell junctions are included. They are all responsible for the keeping cells in shape.
Cytoskeleton--Maintenance of cell shape; movement of organelles within cells; cell movement; mechanical transmission of signals from exterior of cell to interior
Cell walls--Maintenance of cell shape and skeletal support; surface protection; binding of cells in tissue
Extracellular matrix--Binding of cells in tissues; surface protection; regulation of cellular activities
Cell junctions--Communication between cells; binding of cells in tissues
Plants are eukaryotes, multicellular organisms that have membrane-bound organelles. Unlike prokaryotic cells, eukaryotic cells have a membrane-bound nucleus. A plant cell is different from other eukaryotic cells in that it has a rigid cell wall, a central vacuole, plasmodesmata, and plastids. Plant cells take part in photosynthesis to convert sunlight, water, and carbon dioxide into glucose, oxygen, and water. Plants are producers that provide food for themselves (making them autotrophs) and other organisms.
These are some of the parts common to plant cells:
•Cell Wall- smooth layer that provides DNA and protection from osmotic swelling.
•Cell (Plasma) Membrane- it is composed of a phospholipid lipid bilayer (including polar hydrophilic heads facing outside and hydrophobic tails facing each other inside) that makes it semipermeable and thus capable of selectively allowing certain ions and molecules in/out of the cell.
•Cytoplasm- it consists of the jelly-like fluid in and around the organelles.
•Cytoskeleton- is made up of microtubules, intermediate filaments, and microfilaments. It provides shape the shape of the cell and helps in transporting materials in and out of the cell.
•Golgi Apparatus (body/complex)- it is the site where membrane-bound vesicles are packed with proteins and carbohydrates. These vesicles will usually leave the cell through secretion.
•Vacuole- stores metabolites and degrades and recycles macromolecules.
•Mitochondria- is responsible for cellular respiration by converting the energy stored in glucose into ATP.
•Ribosome- contain RNA and proteins for protein synthesis. One type is embedded in Rough ER and another type puts proteins directly into the cytosol.
•Rough Endoplasmic Reticulum (roughER)- covered with ribosomes, it stores, separates, and transports materials through the cell. It also produces proteins in cisternae, which then go to the Golgi apparatus or insert into the cell membrane.
•Smooth Endoplasmic Reticulum (smooth ER)- it has no ribosomes embedded in its surface. Lipids and proteins are produced and digested here. Smooth ER buds off from rough ER to move newly-synthesized proteins and lipids. The proteins and lipids are transported to the Golgi apparatus (where they are made ready for export) and membranes.
•Peroxisome- is involved in metabolizing certain fatty acids and producing and degrading hydrogen peroxide.
•Nuclear Membrane (envelope)- the an extension of the endoplasmic reticulum that wraps around the nucleus. Its many gaps allow traffic in/out of the nucleus.
•Nucleus
- it contains DNA in the form of chromosomes or chromatin and controls protein synthesis.
•Nucleolus
- it is the site of ribosomal RNA synthesis.
•Centrosome- consisting of a dense center and radiating tubules, it organizes the microtubules into a mitotic spindle during cell division.
•Chloroplast- conducts photosynthesis and produces carbohydrates, oxygen, and internally ATP and NADPH from captured light energy.
•Starch Granule- temporarily stores produced carbohydrates from photosynthesis. Depending on the organism, it can be inside or outside of the chloroplast (if present).
The cell wall is a tough, usually flexible but fairly rigid layer that surrounds the plant cells. It is located just outside the cell membrane and it provides the cells with structural support and protection. A major function of the cell wall is to act as a pressure vessel, preventing over-expansion when water enters the plant cells. The strongest component of the cell wall is a carbohydrate called cellulose, a polymer of glucose.
The cell wall gives rigidity and strength to the plant cells which offers protection against mechanical stress. It also permits the plants to build and hold its shape. It limits the entry of large molecules that may be toxic to the cell. It also creates a stable osmotic environment by helping to retain water, which helps prevent osmotic lysis.
While the cell wall is rigid, it is still flexible and so it bends rather than holding a fixed shape due to its tensile strength. The rigidity of primary plant tissues is due to turgor pressure and not from rigid cell walls. This is evident in plants that wilt since the stems and leaves begin to droop and in seaweed that bends in water currents. This proves that the cell wall is indeed flexible. The rigidity of healthy plants is due to a combination of the cell wall construction and turgor pressure. The rigidity of the cell wall is also affected by the inflation of the cell contained. This inflation is a result of the passive uptake of water.
Cell rigidity can be increased by the present of a second cell wall, which is a thicker additional layer of cellulose. This additional layer can be formed containing lignin in xylem cell walls, or containing suberin in cork cell walls. These compounds are rigid and waterproof, making the secondary cell wall very stiff. Secondary cell walls are present in both wood and bark cells of trees.
The primary cell wall of most plant cells is semi-permeable so that small molecules and proteins are allowed passage into and out of the cell. Key nutrients, such as water and carbon dioxide, are distributed throughout the plant from cell wall to cell wall via apoplastic flow.
The major carbohydrates that make up the primary cell wall are cellulose, hemicellulose and pectin. The secondary cell wall contains a wide range of additional compounds that modify their mechanical properties and permeability. Plant cell walls also contain numerous enzymes, such as hydrolases, esterases, peroxidases, and transglycosylases, that cut, trim and cross-link wall polymers. The relative composition of carbohydrates, secondary compounds and protein varies between plants and between the cell type and age.
There are up to three strata, or layers, that can be found in plant cell walls:
The middle lamella, which is a layer rich in pectins. This is the outermost layer that forms the interface between adjacent plant cells and keeps them together.
The primary cell wall which is generally a thin, flexible layer that is formed when the cell is growing.
The secondary cell wall which is a thick layer that is formed inside the primary cell wall after the cell is fully grown. It is only found in some cell types.
The vacuole is essentially an enclosed compartment that is filled with water containing inorganic and organic molecules including various enzymes in solution. Vacuoles are formed by the fusion of multiple membrane vesicles and are effectively just larger forms of these vesicles. This organelle does not have a basic shape or size since its structure is determined by the needs of the cell. The functions of the vacuole in the plant cell include isolating materials that may be harmful to the cell, containing waste products, maintaining internal hydrostatic pressure within the cell, maintaining an acidic internal pH, containing small molecules, exporting unwanted substances from the cell, and allowing plants to support structures such as leaves and flowers. Vacuoles also play an important role in maintaining a balance between biogenesis and degradation of many substances and cell structures in the organism. Vacuoles aid in the destruction of invading bacteria or of misfolded proteins that are building up within the cell. They have the function of storing food and assist in the digestive and waste management process for the cell.
Most mature plant cells have a single large central vacuole that takes up approximately 30% of the cell's volume. It is surrounded by a membrane called the tonoplast, which is the cytoplasmic membrane separating the vacuolar contents from the cell's cytoplasm. It is involved in regulating the movements of ions around the cell, and isolating substances that may be harmful to the cell.
Other than storage, the main function of the central vacuole is to maintain turgor pressure against the cell wall. The proteins that are found in the tonoplast control the flow of water into and out of the vacuole through active transport, pumping potassium ions into and out of the vacuolar interior. Because of osmosis, water will flow into the vacuole placing pressure on the cell wall. If there is a significant amount of water loss, there is a decline in turgor pressure and the cell will plasmolyse. Turgor pressure exerted by the vacuole is required for cellular elongation as well as for supporting plants in the upright position. Another function of the vacuole is to push all contents of the cell's cytoplasm against the cellular membrane which helps keep the chloroplasts closer to light.
Plasmodesmata are microscopic channels that traverse the cell walls of plant cells enabling the transport and communication between the cells. Plasmodesmata enable direct, regulated intercellular transport of substances between the cells. There are two forms of plasmodesmata, primary ones that form during cell division and secondary ones that form between mature cells. They are formed when a portion of the endoplasmic reticulum is trapped across the middle lamella as a new cell wall is laid down between two newly divided plant cells and this eventually becomes the cytoplasmic connection between the two cells. It is here that the cell wall is thickened no further and depressions or thin areas known as pits are formed in the walls. Pits usually pair up between adjacent cells.
Plasmodesmata are constructed of three main layers, the plasma membrane, the cytoplasmic sleeve, and the desmotubule. The plasma membrane part of the plasmodesmata is an extension of the cell membrane and it is similar in structure to the cellular phospholipid bilayers. The cytoplasmic sleeve is a fluid-filled space that is enclosed by the plasma membrane and is an extension of the cytosol. The trafficking of molecules and ions through the plasmodesmata occurs through this passage. Smaller molecules, such as sugars and amino acids, and ions can pass through the plasmodesmata via diffusion without the need for additional chemical energy. Proteins can also pass through the cytoplasmic sleeve but it is not yet known just how they are able to pass through. Finally, the desmotubule is a tube of compressed endoplasmic reticulum that runs between adjacent cells. There are some molecules that are known to pass through this tube but it is not the main route for plasmodesmatal transport.
The plasmodesmata have been shown to transport proteins, short interfering RNA, messenger RNA, and viral genomes from cell to cell. The size of the molecules that can pass through the plasmodesmata is determined by the size exclusion limit. This limit is highly variable and is subject to active modification. There have been several models that have been proposed for the active transport through the plasmodesmata. One suggestion is that such transport is mediated by the interactions with proteins that are localized on the desmotubule, and/or by chaperones partially unfolding proteins which allows them to fit through the narrow passage.
Plastids are the site of manufacture and storage of important chemical compounds that are used by the cell. They often contain pigments used in photosynthesis and the types of pigments present can change or determine the color of the cell. Plastids are responsible for photosynthesis, storage of products like starch, and the ability to differentiate between these and other forms. All plastids can be traced back to proplastids, which happen to be present in the meristematic regions of the plant. In plants, plastids may differentiate into several forms depending on what function they need to play in the cell. Undifferentiated plastids, the proplastids, can develop into the following types of plastids:
•Chloroplasts: for photosynthesis
•Chromoplasts: for pigment synthesis and storage
•Leucoplasts: for monoterpene synthesis
The inside of a chloroplast.
Chloroplasts are the organelles that conduct photosynthesis. They capture light energy to conserve free energy in the form of ATP and reduce NADP to NADPH. They are observed as flat discs usually 2 to 10 micrometers in diameter and 1 micrometer thick. The chloroplast is contained by an envelope that consists of an inner and outer phospholipid membrane. Between these layers is the intermembrane space. The material within the chloroplast is called the stroma and it contains many molecules of small, circular DNA (though it is often found in branched linear form, such as in corn). Within the stroma are stacks of thylakoids, which are the site of photosynthesis. The thylakoids are arranged in stacks called grana. A thylakoid has a flattened disk shape and has an empty space called the thylakoid space or lumen. The process of photosynthesis takes place on the thylakoid membrane. Embedded in the thylakoid membrane are antenna complexes that consist of the light-absorbing pigments, such as chlorophyll and carotenoids, as well as the proteins that bind the pigments. These complexes increase the surface area for light capture and allows the capture of photons with a wider range of wavelengths. The energy of the incident photons is absorbed by the pigments and funneled to the reaction center of the complex through resonance energy transfer. From there, two chlorophyll molecules are ionized, which produces an excited electron which passes on to the photochemical reaction center.
Chromoplasts are responsible for pigment synthesis and storage. They are found in the colored organs of plants such as fruit and floral petals, to which they give their distinctive colors. This is always associated with a massive increase in the accumulation of carotenoid pigments. Chromoplasts synthesize and store pigments such as orange carotene, yellow xanthophylls and various other red pigments. The most probably main evolutionary role of chromoplasts is to act as an attractant for pollinating animals or for seed dispersal via the eating of colored fruits. They allow for the accumulation of large quantities of water-insoluble compounds in otherwise watery parts of plants. In chloroplasts, some carotenoids are used as accessory pigments in the process of photosynthesis where they act to increase the efficiency of chlorophyll in harvesting light energy. When leaves change color during autumn, it is because of the loss of green chlorophyll unmasking these carotenoids that are already present in the leaves. The term "chromoplast" is used to include any plastid that has pigment, mainly to emphasize the contrast with leucoplasts which are plastids that have no pigments.
Leucoplasts lack pigments and so they are not green. They are located in roots and non-photosynthetic tissues of plants. They can become specialized for bulk storage of starch, lipid or protein and are then known as amyloplasts, elaioplasts, or proteinoplasts, respectively. In many cell types, though, leucoplasts do not have a major storage function and are present to provide a wide range of essential biosynthetic functions, including the synthesis of fatty acids, many amino acids, and tetrapyrrole compounds such as haem. Extensive networkds of stromules interconnecting leucoplasts have been observed in epidermal cells of roots, hypocotyls and petals.
The heat stress response (HSR) in plants involves a complex network of different pathways which occurs in different cellular compartments. Scientists have been trying to figure out which is the main thermosensor that initiate the transcription of heat stress response gene in response to the heat stress.Up till now, scientists predicted that there are at least four major thermosensors in plants that activate a similar set of HSR gene which helps improving thermotolerance in plants, include a plasma membrane channel, an unfolded protein sensor in endoplasmatic recticulum, an unfolded protein sensor in cytosol, and a histone sensor in the nucleus. [1] However, the relationship between these four thermosensors remains unknown.
1. Heat plays tremendous roles, especially those of adverse, in plant development, growth, reproduction and fertilization. Plant tissues involved in reproduction are especially vulnerable to the exposure of heat.
2. Plants are sessile organisms that cannot escape heat. Their metabolism is unique as they adapt their metabolism to counterbalance this disadvantage.
3. Cells and organelles in plants are designed in a way to combat and prevent damages caused by heat.
4. Studies for heat stress in plants help agriculture. Necessary measures for heat damage may be prepared from these studies. Under heat stress, programmed cell death can be activated in plants and this can lead to the shredding of leaves, flowers not blooming, little to no production of fruits, or simply the death of plants. In addition, the reproductive tissues in plants are especially sensitive to heat. A difference of a few degrees in temperature can lead to no crops for the season. This can have an enormous and negative impact on the economy.[1] It is estimated that the 1980 and 1988 US heat waves resulted in overall damages of approximately 55 and 71 billion dollars[2]
Heat stress has certain effects on different components in plant cells such as the stability of membranes, proteins, RNA and skeleton structure in cells.It also can change the way chemical reactions are carried out such as enzymatic reactions in the metabolic system. Due to the changes in these components, metabolic process of plant cells becomes imbalance. The disrupted state of metabolites can result in unwanted by-products such as reactive oxygen species (ROS).[1]
In response to the changes of surrounding temperature, plants re-program their transcriptome, protome, metabolome and lipidome. This action basically induces changes in the composition of certain transcripts, metabolites and lipids which help the plants reset a new metabolic balance so that the cells can survive and function as normal even at very high temperature.[1] Moreover, when the temperature comes back to normal, plant cells can reverse the reprogram process and get back to the original metabolic balance that fit in with the current temperature. In addition, plants can also program cell death in response to the heat stress such as resulting in leaves shedding.
Several heat treatments have been applied on plants to study the heat response in plants. There are three major set of heat treatments as mentioned in the article "How do plants feel the heat" by Ron Mittler* : no priming, stepwise priming, and gradual priming.[1]
In the "no priming" treatment, the plants are introduced to a series of severe heat stress. For example, a plant that normally grows at 21oC is placed in an environment at 42-45oC for 0.5 to 1 hour. Plant survival is then measured 5-7 days after the treatment to determine the effect of heat stress.[1] Due to this series of severe heat applied, the plant dies very quickly (exponential decay). The plants ability to survive after such treatment is referred to as basal thermotolerance, the ability of plants to sense and adapt to extreme heat without any priming.
"Stepwise priming" subjects the plants to an environment of moderate heat stress. This is set up by first letting the plants grow at 21oC, and then subjecting them to a temperature of 36-38oC for a short period time of 1.5 hours. They are then recovered to the normal temperature of 21oC for 2 hours after to which they are subjected to a severe temperature of 45oC. Their survivability under such condition (after priming) is known as acquired thermotolerance.
"Gradual priming" is the process which the temperature was increased gradually and steadily until it reaches the temperature equal to extreme heat of 45oC. This is supposed to imitate the conditions that is seen in nature. Plants under these conditions also exhibit acquired thermotolorence. For Arabidopsis thaliana, gradual priming increase the survival rate by more than 10%. It is preferred over both no priming heat stress and stepwise priming temperature changes. [3]
The last type of heat treatment is "warming", which is not considered as one of the heat stress treatments. During warming treatment, plants are originally at a temperature lower than standard growing temperature of 12oC, instead of 21oC. They are then exposed to a temperature that is slightly higher than their ordinary growth temperature so instead of subjecting them to 21oC, they are grown at 27oC. This slight increase in temperature change from the norm is, however, not considered heat stress since plants actually do not express HSR markers under warming. In general, warming allows longer term adaptation and reprogramming of the development, which includes early flowering or shedding leaves.
The important message to take home from this experimental setup is that different heating treatments elicit different transcriptome responses. This suggests that there may be separate heat sensors and signaling pathways activating specific responses to rise in temperature.
When an A. thaliana experiences an increase in external temperature, its large surface-to-volume ratio makes sure that almost all macromolecules such as protein complexes, membranes and nucleic acid polymers in the plant cells to acknowledge the heat immediately. As it is true for every matter in the universe, the macromolecules' kinetic energy is raised by the introduction of heat into the system. This, consequently, results in reversible physical changes of the macromolecules. There are certain macromolecules that not only just "perceive" heat but also differentially trigger a unique signaling path that can specifically upregulate hundreds of HSR genes[4]
There are 2 basic routes plants use to mediate heat stress
1. Direct effect in a specific sensor: The specific macromolecule that behaves as a sensor molecule could be directly affected by heat. It is observed when temperature-induced changes in its quaternary and tertiary structures occur.
2. Indirect effect in a specific sensor: In this route, the specific macromolecule sensor is indirectly influenced by heat due to the effects of heat on other components of the cell. For example, temperature-induced changes in membrane fluidity could affect a membrane protein which is embedded in. It is indirect because the membrane protein is actually affected by heat but is only subject to change when the fluidity of plasma membrane varies.
The fluidity of the plasma membrane can increase when there is an increase in temperature. Scientists have found out that the increased fluidity of the plasma membrane will activate calcium channels, which will lead to an influx of Calcium ions into the cell. This is the primary heat sensing event in the moss Physcomitrella patens.[1] This is the mechanism of detecting heat. However, the actual heat stress sensor in plants cells in still unknown. The A. thalianagenome encodes over 40 calcium channels and many of them are known to be located in the plasma membrane. These presences of calcium channels strongly suggest that calcium ions are the ones, which are responsible for sensing heat.
Beside activating the Calcium channels, membrane fluidity change might also trigger lipid signaling due to the change of temperature.
How does a heat stress-induced inward calcium flux can control regulate multiple signaling pathways in plants?
In A. thaliana, the following 4 pathways are known to happen:
Mechanism of protein kinases
1. The calmodulin AtCaM3 signals heat stress and it itself is involved in the activation of different transcription factors such as WRKY39[5] and HSFs [6]
2. Chain reactions may be triggered by heat signaling. It is known that an inward flux of calcium activates unique calcium-dependent protein kinases or the ROS-producing enzyme NADPH oxidase. A protein kinases is a kinase enzyme that modifies other proteins by chemically adding phosphate groups to them.
3. AtCaM3 activated calcium/calmodulin-binding protein kinase. This protein kinase phosphorylates HSFA1a.
4. An HSP90/FKBP-dependent kinanse can also mediate HSF phosphorylation causued from calcium binding to calmodulin [7]
On top of ion channel activation in the plasma membrane, membrane fluidity changes caused by increase heat can also trigger lipid signaling. During heat stress, phospholipase D (PLD) and phosphatidylinositol-4-phosphate 5-kinase (PIPK) are activated. Lipid signaling molecules such as phosphatidic acid, D-myo-inositol-1,4,5-trisphosphate (IP3), and phosphatidylinositol-4-phosphate (PIP2) start to accumulate from heat stress and tigger the calcium channels open, resulting in calcium influx. On a side note, it should be noted that a reduction in phospholipase C9 activity is correlated with lower concentration of IP3, sHSP downregulation, and lower thermotolerance.
The mechanistic relationship between lipid signaling response to heat stress and plasma membrane channels (whether it is direct or indirect) is still unclear. The chronological order of events in heat stress sensing and signaling response also remains as a question to be answered. It is likely that the signaling pathways function downstream to what's responsible for sensing heat stress in plasma membrane.
Histones are proteins that help package DNA. As seen in wild-type plants, the increase in temperature causes a significant decrease in concentration of certain histones such as H2A.Z which contains the nucleosomes that might trigger the changes in transcriptome so that HSR genes are initiated. Scientists also found that the decrease in H2A.Z histone concentration might also cause the change in the expression of a specific transcription factor or other regulatory proteins which can initiate the response of transcriptome. After screening for mutants impaired in heat sensing, it was discovered that the ARP6 gene is possibly responsible for mediation of responses to temperature changes. ARP6 codes for a SWR1 subunit that is required in order for H2A.Z to be inserted into nucleosomes, instead of H2A. It was shown that warming in wild-type plants result in significant decrease in H2A.Z occupancy in nucleosomes where the transcription for warming-induced genes starts. Since less histones are in the way, this allows for more transcriptions of these warming-induced genes. Less H2A.Z occupancy in certain HSP promoters can also affect transcription factor and other regulatory proteins' expression and DNA binding ability, ultimately inducing transcriptome response. It is, however, unsure whether the occupancy of the histones is the cause for heat sensing related to acquired thermotolerance. [5].
Heat stress may activate the unfolded protein response (UPR) in both ER and Cytosol which can initiate heat stress response(HSR). The UPR is the weakening of protein stability. In the ER pathway, unfolded proteins can enter the nuclei and therefore affect the transcription of certain genes. This will lead to an accumulation of ER chaperone transcripts which will alter the metabolism of the cell in order to adapt to the heat.[1] Beside heat stress, UPR can also be activated by specific chemicals that cause UPR or changes in certain abiotic aspects.[8] Because few unfolded proteins and HSR chaperones are likely to exist under mild heat conditions, the UPR is considered to be not as sensitive as the PM heat response.[1]
Different metabolic pathways are likely to depend on enzymes with different responsiveness to unnecessary heat. From this reason. it has been proposed that heat stress might uncouple some metabolic pathways and cause the accumulation of unwanted by-products. An example of such by-product is reactive oxygen species (ROS). Studies have shown that the accumulation of ROS is mostly likely due to the change in fluidity of the plasma membrane.[9] This event is most likely a positive feedback loop since the accumulation of ROS will also open up more calcium channels in the plasma membrane and therefore lead to more calcium influx into the cell.[1] Therefore, it seems that the plasma membrane heat sensing is highly interlinked with ROS signaling. A large accumulation of ROS may lead to program cell death.[1]
Although scientists have proposed possible ways in which plants can sense heat, there is still much we don’t understand about the specific mechanisms and the order of events. How the signals from the speciated sensors integrate with each other within the network of signal transduction is also still of an important question that is yet to be answered. There could be additional pathways not yet discovered but crucial to heat stress response.
↑ abcdefghijkFinka, A., Goloubinoff, P. and Mittler, R.. “How Do Plants Feel the Heat?” Trends in Biochemical Sciences. March 2011: 118-125.
↑"Genetic engineering for modern agriculture: challenges and perspectives" by R. Mittler, E. Blumwald
↑Invalid <ref> tag; no text was provided for refs named Larkindale
↑"How do plants feel the heat?" by Ron Mittler, Andrija Finka, Pierre Goloubinoff
↑"Functional characterization of Arabidopsis thaliana WRKY39 in heat stress" from Mol. Cell, 29 (2010), pp 475-483) by S. Li
↑"The role of class A1 heat shock factors (HSFA1s) in response to heat and other stress in Arabidpsis" from Plant Cell Environment, 34 (2011), pp738-751 by H.C. Liu
↑"Arabidopsis ROF1(FKBP62) modulates thermotolerance by interacting with HSP90.1 and affecting the accumulation of HsfA2-regulated sHSPs" from Plant Journal 59 (2009), pp 387-399 by D. Meiri, A Breiman
↑ Moreno, A.A. and Orellana A. (2011) The Physiological role of the unfolded protein response in plants. Biol. Res. 44, 75-80.
↑ Königshofer, H. et al. (2008) Early events in signaling high-temperature stress in tobacco BY2 Cells involve alterations in membrane fluidity and enhanced hydrogen peroxide production. Plant Cell Environ. 31, 1771-1780.
↑Kumar, S.V. and Wigge, P.A. (2010) H2A.Z-containing nucleosomes mediate the thermosensory response in Arabidopsis. Cell 140, 136–147
Cellulosomes are a complex of multi-cellulolytic enzymes that break down cellulose and hemicelluloses found in the cell walls of plants. Cellulose and hemicelluloses are two organic polysaccharide compounds that are also two of the most abundant polymers on Earth. They are major sources of carbon and energy. Cellulases and hemicellulases participate in protein:carbohydrate [carbohydrate-binding modules (CBMs)] or protein:protein (dockerins) interactions that make it so hard to break down. Chemically, cellulose is relatively simple. However its crystalline structure makes it resistant to biological degradation. Deconstruction and digestion of cellulose and hemicelluloses will allow for carbon turnover and numerous biotechnological uses, including biofuels as an alternative fuel source. However, very few microorganisms have the capacity to digest cellulose and hemicelluloses, which are why cellulosomes are of great interest.
The first cellulosome was discovered in the early 1980s by Bayer and Lamed in their studies of the cellulolytic system of Closteridium thermocellum, an anaerobic bacterium. They described the cellulosome as a “discrete, cellulose-binding, multi-enzyme complex that mediates the degradation of cellulosic substrates.” They initially believed that the cellulosome only degraded cellulose, but it was later recognized that the cellulosome also degrade a large number of hemicellulases and even pectinases. Also following Bayer and Lamed’s studies were research in identifying the molecular mechanisms of how the enzyme complex assembles and how the cellulosome is present on the surface of the host bacterium. More recently, a range of anaerobic bacteria and fungi were shown to also produce cellulosome systems.
Scientists have argued that cellulosomes are more efficient in digesting cellulose and hemicellulases than corresponding enzyme systems produced by aerobic bacteria and fungi. It is possible that anaerobic environments imposed selective pressures that led to the evolution of cellulosomes. One hypothesis proposes that the integration of plant cell wall-degrading enzymes onto a macromolecular complex leads to a more efficient enzyme complex whose cellulosomal catalytic units interactions work together synergistically and further enhanced by enzyme substrate targeting through scaffoldin-borne CBM. Another hypothesis suggest that the function of the cellulosome on the surface of the bacterium, at least in C. thermocellum, enhances the capacity of the host bacterium to utilize mono- and oligosaccharide released from the cell wall.
The cellulosome was found to be composed cellulosomal catalytic components that contained noncatalytic modules called dockerins, which bind to cohesin modules. This receptor/adapter protein domain pair is responsible for cellulosome self-assembly. The recognition between cohesin and dockerin is type and/or species specific. Together cohesin and dockerin are located in a large noncatalytic protein that acts as a scaffoldin.
Dockerins consists of approximately 70 amino acids that contain two segments, each of about 22 residues, in the cellulosomal catalytic components. The first 12 residues resemble the calcium-binding loop of EF hand motifs in which aspartate or asparagines are highly conserved. The EF hand is a helix-loop-helix structural domain found in a large family of calcium-binding proteins. Calcium was shown to be necessary for dockerin stability and function. Without calcium, dockerins are unable to interact with cohesions. They are also present in a single copy at the C-terminus of cellulosomal enzymes.
Cohesins are 150 residue modules of tandem repeats in scaffoldins, which specifically bind to dockerin modules. The number of cohesin modules in scaffoldins varies between one and eleven, but is usually more than four. Scaffoldins can be defined as cohesin containing proteins, which play a role in cellulosome assembly. The scaffoldin subunits of the cellulosome function to organize and position other protein subunits into the complex and can also serve as an attachment device for harnessing the cellulosome onto the cell surface and/or for targeting its substrate because scaffoldins usually contain a noncatalytic CBM that anchors the entire complex onto cellulose.
More recently, researchers found that some cellulosome-producing microbes produce more than one type of scaffoldin. Scaffoldins that bind cellulases and hemicellulases are called primary scaffoldins. Proteins that bind primary scaffoldins are called anchoring scaffoldins.
Cell-surface attachment of cellulosomes is required for degradation of plant cell wall polysaccharides. CBMs are carbohydrate-binding modules that are separated into three types: Type A (interacts with crystalline polysaccharides), Type B (bind to internal regions of single glycan chains), and Type C (recognizes small saccharides). It was first thought that bacterial cellulosomes are bound to the cellulose by the “cellulose-binding factor,” but it was later realized that the attachment of the cellulosome to the cell wall is mediated by a family 3 CBM (CBM3) located in the scaffoldins. CBM3s are type A modules that bind tightly to the cellulose surface.
There are two major types of bacterial cellulosomes: those present in multiple types of scaffoldins and those that contain a single scaffoldin. Cellulosomes that assemble via a single primary scaffoldin are the simplest and contain six to nine catalytic components, depending on the number of cohesions in the primary scaffoldin. Bacterial cellulosomes are bound tightly to the cellulose and referred to as the “cellulose-binding factor.”
Bacteria expressing cell-surface cellulosomes contain a single primary scaffoldin and multiple anchoring scaffoldins. The majority of the anchoring scaffoldins contain SLH modules or SLH domains, which mediate the attachment of the structural proteins to the bacterial cell wall and may bind to the secondary cell wall polysaccharides.
Anaerobic fungi can hydrolyze many different polysaccharides, including cellulose and hemicelluloses. It is believed that anaerobic fungi are the initial invader of lignocelluloses and play a role in fiber digestion along with bacteria and other microorganisms. Anaerobic fungal plants have cellulosomes just like anaerobic bacteria. However, fungal cellulosomes are less characterized.
A number of dockerin sequences have been identified in different fungi strains. Fungal dockerins consist of a three-stranded beta-sheet and a short helix held together by two disulfides. Researchers also found that the amino acid sequences of fungal dockerins are unrelated to their bacterial counterparts. Genome sequencing of fungi will allow scientists to see which dockerin sequences present in proteins are associated with plant wall degradation. Another difference between fungal and bacterial cellulosomes is that fungal cellulosomes generally contain two copies of dockerin modules, each consisting of approximately 40 residues joined together by shorter linker sequences.
Researchers have also hypothesized that fungal cellulosome may not involve cohesin modules. However, more work and research is needed in characterizing fungal cellulosomes.
Studying cellulosomes and how they work would provide a large number of biotechnical benefits to society. There are many industrial applications, such as paper pulp production, that requires breakdown of plant cell walls in its process. Cellulosomes would be a great benefactor.
One of the biggest challenges facing the world today is the lack of alternative and renewable energy sources to the conventional fossil fuels. A potential, viable alternative can be found in the converting lignocellulosic biomass to fermentable sugars to produce renewable fuels, such as ethanol. Yet, the biggest rate limiting problems in converting lignocelluloses into biofuels, is the hydrolysis of structural polysaccharides, which require the development of a more efficient enzyme system. Extensive enzyme group is needed to overcome the recalcitrant and the plant cell wall. Cellulosomes could be the future solution.
↑
1. Fontes, Carlos and Harry J. Gilbert. “Cellulosomes: Highly Efficient Nanomachines Designed to Deconstruct Plant Cell Wall Complex Carbohydrates.” Annual Review of Biochemistry. 2011.
An animal cell is a type of cell that dominates most of the tissue cells in animals. Animal cells are different from plant cells because they don't have cell walls and chloroplasts, which are relevant to plant cells. Without the cell wall, animal cells can be in any sort of shape or size as they are instead surrounded by a plasma membrane.
One thing why animal cells are exclusive because they have centrioles and plant cells do not have centrioles. Centrioles are important for DNA segregation when the cell undergoes the process of mitosis, a process of dividing a cell. Centrioles are important in the structure of the spindles, which helps to pull the chromosomes apart.
Both animal and plant cells have vacuoles, however, in animal cells, vacuoles are very tiny or absent while the vacuole in plant cells are quite large. There are different kinds of animal cells such as- muscle cells, nerve cells, and many more.
Cell Membrane: The cell membrane is a fluid mosaic structure which is composed of a phospholipid bilayer and other important macromolecules such as proteins. The cell membrane separates the cell from the environment and allows the movement of materials in and out of the cell.
Animal Cell
Cytoplasm: the liquid within the cell where the different organelles are found. It is here where many functions occur. Including cell division and glycolysis.
Golgi Apparatus: The organelle in which proteins are modified, sorted, and sent to various parts of the cell. Modifications on the protein include but are not limited to, glycosylation.
Mitochondria: does the cellular respiration of the cell by converting glucose into ATP (cellular energy).
Ribosome: The mRNA from the nucleus are used by Ribosomes in a process called translation. Translation is when the Ribosome joins amino acids together according to the sequence of the mRNA. The more ribosomes in a cell, the proteins it synthesizes. They are located in two areas, on the ER or in the cytosol.
Rough Endoplasmic Reticulum:: is used to store and transport material through the cell. Proteins are produced here in the ribosomes bound to the rough ER.
Smooth Endoplasmic Reticulum: Functions in the synthesis of lipids, detoxification of drugs and poisons, storage of calcium ions, and metabolism of carbohydrates. In contrast to the Rough Endoplasmic Reticulum, the smooth ER is not studded with proteins.
Peroxisome: A specialized metabolic compartment bounded by a single membrane. Additionally, it possesses enzymes that transfer hydrogen atoms from substrates to oxygen, producing hydrogen peroxide as a by-product. Then, hydrogen peroxide is converted to water by another enzyme.
Nucleus:The nucleus is usually the largest organelle in a cell. It consists of different parts such as the nuclear envelope, chromosomes, and the nucleolus. The nuclear envelope surrounds the nucleus while segregating the chromatin from the cytoplasm and consists of two membranes each made of a lipid bilayer. The membranes have pores that regulate what goes in and out of the nucleus. Inside the nucleus is the nucleolus which holds the genetic material DNA. Using this DNA, transcription is carried out making mRNA.
Vacuole: the "storage space" that stores water, salt, and other important substances. There are also food vacuoles that are cellular organelles in which food is broken down by hydrolytic enzymes. These food vacuoles are the simplest digestive compartments. The process of intracellular digestion occurs inside vacuoles, which is the process of hydrolysis of food. This process begins after a cell engulfs food materials through phagocytosis (solid food) or pinocytosis (liquid food).
Lysosome: considered the "digestion compartment" of the cell. Lysosomes break down cellular wastes such as fats, proteins, or carbohydrates. The rid of the cellular materials that are no longer useful in the cell.
Cytoskeleton: is a structure made out of protein to give the cell its shape and structure. It also helps cellular motion with the use of flagella, cilia, or lamelllipodia.
Centrioles: are used through cell division. They organize the mitotic spindle during the end of cytokinesis. The centrioles are located within the centrosome and come in pairs. Each pair of centrioles are compiled of nine sets of triplet microtubules assembled into a ring. Prior to animal cell division, the centrioles replicate. Although centrosomes with centrioles may assist the organization of microtubule construction in animal cells, they are not crucial for this particular function in all eukaryotes; e.g. the fungi and the majority of plant cells lack centrosomes with centrioles, but still contain well-assembled microtubules. [1]
Schematic Diagram of the MSCs in a Eukaryotic Cell
Two organelles in a cell come in contact via membrane contact sites (MCS). Membrane contact sites govern the signaling and crossing of ions and lipids from one cellular organelle to the next. Even though research on MCS have been going on for over 50 years now, there is still so much more to uncover regarding their function, structure and regulation. To this date, there are multiple studies done on three main contact sites: the nucleus-vacuole junction, mitochondria-ER, and plasma membrane-ER contact sites. Having membrane- bound organelles is a special characteristic for eukaryotic cells, as it allows for specialized functions in different compartments of the cell. Two organelles exchange information through interorganellar or MCS when they are in close proximity. To visualize their communication, a technique such as electron microscopy was employed. This technique first arose in the 1950s when the close apposition of endoplasmic reticulum and mitochondria was first seen. Ever since then, similar electron microscopy techniques were used to visualize contact sites in cells between the endoplasmic reticulum and: mitochondria, vacuoles, Golgi, chloroplasts and plasma membrane. Since most studies were conducted without molecular markers, they usually refer to proximity of around 30 nanometers between two organelles’ membranes. Unfortunately, there is still no excellent 3D model visualization of MCS. However, in the recent years, there is an increase in the studies of the proteins that reside in the membrane sites and molecular determinants needed to create MCS.
The membrane contact site between the nucleus and the vacuole is known as the nucleus-vacuole junction (NVJ). The primary study is of the yeast cell Saccharomyces cerevisiae, in which the vacuolar membrane, inner nuclear membrane, and the ER membrane is bridged through NVJ. Nucleus-vacuole junctions are the best studied MCS to this date. NVJ is made of vacuolar protein Vac8 and the ER-localized protein Nvj1. If one of these two proteins is missing from deletion of genes, the MCS will not be made and there would not be contact between two organelles. : There have been two cellular functions suggested for the NVJ. The first one is piecemeal microautophagy of the nucleus. This is a process that marks a part of the yeast nucleus for degradation in the vacuole lumen. Piecemeal microautophagy of the nucleus is stimulated during nitrogen or carbon starvation and requires autophagic parts shared between the electrochemical gradient across the vacuolar membrane, the selective cytoplasm to vacuole, and unselective microautophagy. The second suggested function of NVJ is lipid biosynthesis. The two proteins that participate in lipid biosynthesis, Osh1 and Tsc13, reside in the NVJ. Osh1 comes from the yeast oxysterol-binding protein homology family whose role includes lipid sensors and lipid transfer. However, there is no evidence that shows NVJ regulates lipid biosynthesis because localization of osh1 in NVJ is exclusive to stationary phase. In addition, OSH1 is not needed for forming NVJs or for the piecemeal microautophagy of the nucleus in starved cells. The other protein resides in NVJ (Tsc13) is responsible for catalyzing the terminal step in biosynthesis of very-long-chain fatty acids(VLCFAs). These fatty acids can affect the structure and fluidity of membranes. Although we have pretty good understanding of these two enzymes, we haven't figured out why they reside in NVJ or if there are other proteins that are in the NVJ. : Similar contact sites between ER and the endosome/lysosome (for eukaryote cells) have been observed. Instead of osh1, late endosome-localized oxysterol-binding protein-related protein 1L(ORP1L) works as a cholesterol sensor and it sense level of cholesterol by changing its conformation. Through the change of conformation, ORP1L tells ER how much cholesterol there is in the cell. : Although NVJ is the most studied membrane contact site, we still don't know how much it can do for a cell.
The second most-studied MCS is the mitochondria-ER site. This site is primarily visualized via electron microscopy. The section of ER that co-purifies with the mitochondria, known as mitochondria-associated membrane fraction (MAM), is researched extensively on. The two main functions of this site is lipid biosynthesis and Ca2+ homeostasis. Early research showed that the MAM fraction encompasses enzymes that participate in phospholipid biosynthesis. In addition, the mitochondria and ER must work together to make certain lipids. For example, let’s take a closer look in the biosynthetic pathway of phosphatidylcholine. The substrate for this pathway is made in the ER, which then enters the intermembrane space of the mitochondria to produce phosphatidylethanolamine (PE), and then PE goes back to the ER to synthesize phosphatidylcholine. This process shows that the ER and mitochondria collaborate to produce certain lipids. On the other hand, ER-mitochondria MSCs is responsible for the transmission of Ca2+ . Homeostasis of mitochondrial Ca2+ is important because Ca2+ regulated the ATP production rate. Furthermore, an overload of Ca2+ can trigger cell death.
There are many contact sites of ER-PM in different cell types. The function of ER-plasma membrane contact sites is similar to that of the mitochondria-ER junctions, since Ca2+ homeostasis and lipid synthesis and trafficking take place there. As mentioned above, Ca2+ regulation is crucial as it is also responsible for protein synthesis, folding, and signaling. In excitable cells, the global calcium signal is generated by the coupling of PM depolarization and ER calcium release. In non-excitable cells, calcium influx is controlled by detecting luminal ER Ca2+ levels. Research has showed that the PM-ER contact site is involved in non-vesicular lipid trafficking. We know that lipids are insoluble in water. So to transfer lipid from its synthesis sites to its desired work place, one must shield the lipid. In fact, couple families of lipid transfer protein (LTPs) that can perform this task have been found on the contact sites. One of them is oxysterol-binding protein (OSBP) related proteins(ORPS). The oxysterol-binding protein related protein is of a big LTP family which is conserved from yeast to man. It binds to sterols and scientist believe it helps to transfer non-vesicular sterol between the ER and PM. Even though PM-ER contact sites is crucial for Ca2+ and lipid signaling, we still cannot identify the tether proteins that mediate such contact. One strong possible protein for PM-ER contact sites is a class of highly conserved protein called junctophilins. It can stabilize the junctions by anchoring ER/SR to the PM, giving a structual basis for physiological coupling between PM and ER/SR Ca2+ channels.
Further investigation is necessary to map the molecular machinery for the regulation and generation of PM-ER contact sites.
Functions of MCS was established first as a site of trafficking of lipids and ions. However, as more and more studies done on MCS, we know that MCSs are also responsible for signaling, effective transmission of metabolic cues within cell, organellar inheritance control mechanism. We are not clear on how MCS perform these tasks yet but better understanding of MCS would be achieved once we figure out its molecular components.
Much knowledge pertaining to cell architecture and its redevelopment during normal and disease filled processes has been derived from imaging. For centuries, the use of light microscopy techniques has reached its peak and has yielded a tremendous amount of information concerning cell and molecular dynamics. There exist two main types of microscopies, of which include fluorescence microscopy and transmission light microscopy. Though these two techniques have significantly advanced our understanding of cellular processes, they tend to provide limited details. Due to the low penetrating capabilities of electrons, it is not possible to examine the eukaryotic cell as a whole. Thus, most cells are sectioned into 60-500 nm slices. This tedious process requires initial fixation, dehydration, and plastic embedment as well as application of heavy metal stains to generate contrast. As can be seen, extensive work is required to obtain three-dimensional structural information of a whole cell, which is why most studies tend to be limited to small sections of cells.
All this leads to the emergence of X-ray imaging technologies, a crucial new tool for cellular imaging. X-rays are versatile, for they can penetrate thick cells and tissues, eliminating the need to section the specimen further. There are in fact three main types of X-ray imaging techniques, of which include soft X-ray microscopy, soft X-ray tomography, and coherent diffraction imaging. X-ray microscopy has the ability to penetrate thick cells deeply and initiate immediate structural development and outlook. Soft x-ray tomography can generate quantitative three-dimensional images of cells in the near-native state (at a better than 50 nm isotropic resolution). The most unique technique, which goes by the name of coherent diffraction imaging (the name speaks for itself), offers the possibility of high resolution x-ray imaging of cell using computation and high speed computers rather than an X-ray optic to phase the image. Let us take a closer look at the specifics of each X-ray imaging technique. Soft X-ray microscopy involves the use of particular zone plate lenses to allow the release of X-rays towards the cell of interest. These microscopes are operated using photons with energies in the ‘water window’, which is the region of the spectrum that lies between the K shell absorption edges of carbon and oxygen. However, lens-based imaging can also with energies outside the water window, where images can be generated using phase contrast techniques. To date, the most gripping images have been obtained using bright field, absorption contrast at a water window wavelength operation. To further specify the operations of each plate optic, it can be said that a condenser zone plate optic focuses X-rays on the cell itself and an objective zone late optic focuses the transmitted light onto the detector.
Soft x-ray tomography is in a sense similar to light and electron microscopes in that it can also generate two-dimensional representations of a three dimensional specimen. However, with the ability to collect projection images of the cell at angles around a rotation, it is possible to mathematically compute a three-dimensional reconstruction of the specimen of interest. It has been noted that all biological materials are damaged when exposed to intense light. This intensity can either come from ultra violet illumination in a fluorescence microscope or photons from an X-ray microscope. However, by cryosplicing, optical beams can be focused at a much lower temperature, thus avoiding the possibility of radiation cell damage altogether. As a matter of fact, when cells are imaged at liquid nitrogen temperatures, more than a thousand soft X-ray projection images can be retrieved without any sort of radiation damage. As stated before, it is crucial that specialized cryogenic tomography stages occur during this process of X-ray imaging. A key fact illustrating soft X-ray tomography’s versatility is that is can be applied to virtually any imaging investigation in cell biology, from imaging simple bacteria, to yeast and even algae. The first recorded evidence of X-ray tomographic reconstruction was algae. The distinctive ability to obtain high resolution images of intact eukaryotic cells is something that cannot be repeated by any sort of light or electron microscopy. In addition, x-ray tomography can be used to examine even the most complex cells, such as white blood cells and malaria-infested red blood cells.
The last, but certainly not least, technique of discussion is the method of coherent diffractive imaging. It is simply a method that offers the possibility of removing the need for a lens, thus avoiding the limitations imposed by the state-of-the-art in fabrication technology. As basic arrangement of a CDI experiment can be seen as a coherent beam which is used to illuminate the sample and its far-field diffraction pattern is measured. The beam will typically interact quite weakly with the sample, and the undiffracted component of the beam is held to be dominant and will damage the detector unless a beam-stop is initiated. The measurement of the beam at very low diffraction angles closely follows this previous process. Instead of a lens, a zone plate is used instead in coherent diffraction imaging, as it is used to create a focus and the sample is placed in the beam diverging from it. Sure enough, soft X-ray microscopy and soft X-ray tomography seem to be more in use at this time, but coherent diffraction imaging has great promise in the future. The procedural techniques must be sharpened further for it to play a bigger role in three-dimensional imaging. After all, CDI offers access to the quantitative amplitude and phase information that are not easily available through other forms of X-ray imaging. One thing is for sure: X-ray imaging techniques will seek development in the years to come.
Hard X-ray Fluorescence Tomography was a method originally created to detect the presence of very small amounts of transition metals in biological tissue, like copper, zinc, magnesium, etc. Metal ions like zinc and magnesium, for example, act as cofactors in many catalytic enzymatic reactions, and are hence very important for the cell. Using hard X-rays, a large number of transition metals can be detected and mapped all at once, since transition elements automatically fluoresce when hit by hard X-rays. Although this method is very powerful, there are and have been a number of technical problems with the usage of this method.
Hard X-ray tomography is a technique that uses a very large number of two-dimensional pictures of slices of a tissue in order to assemble a three-dimensional picture of the given sample of tissue. There are three main broad categories of tomography – full-field, projection tomography and confocal tomography. Full-field projection tomography is the process of utilizing the entire field of the specimen to carry out the tomography. However, this is still a young, developing technology with many problems. Projection tomography on the other hand, is moderately successful, and certain errors have to be accounted for in the final image produced. Confocal tomography is the most interesting of the three – it gives scientists the ability to analyze a very small portion of a specimen, although it is difficult to find the particular target using this method.
There are a few challenges being faced by scientists using this method to analyze biological tissues. The first is that the hard X-rays are very strong, and hence very harful to the tissue being studied. This results in only hard tissues like seeds being able to be studies, since other tissues die off so easily under exposure. Samples currently analyzable are freeze-dried or chemically fixed to endure the radiation. Scientists also face a number of time constraints due to speed limitations in dealing with the technical difficulties of using this technique.
Imaging cellular architecture with X-rays. Nugent and Carolyn A Larabell.
Hard X-ray fluorescence tomography —an emerging tool for structural visualization. Martin D de Jonge and Stefan Vogt.
Fluorescence is the emission of light by a substance that has absorbed light or other electromagnetic radiation. It is a form of luminescence. In most cases, the emitted light has a longer wavelength, and therefore lower energy, than the absorbed radiation. However, when the absorbed electromagnetic radiation is intense, it is possible for one electron to absorb two photons; this two-photon absorption can lead to emission of radiation having a shorter wavelength than the absorbed radiation. The emitted radiation may also be of the same wavelength as the absorbed radiation, termed "resonance fluorescence". The most striking examples of fluorescence occurs when the absorbed radiation is in the ultraviolet region of the spectrum, and thus invisible to the human eye, and the emitted light is in the visible region.
Fluorescent stains reveal structures within the cell dramatically and can allow us to see cellular structure better which can lead to better understanding.[1]
The decrease in the cell's ability to proliferate with the passing of time. Each cell is programmed for a certain number of cell divisions and at the end of that time proliferation halts. The cell enters a quiescent state after which it experiences cell death via the process of apoptosis.
Aging is inevitable as it is an accumulation of damage over time that eventually affects the function and survival of organisms. It is evident that this occurs because the accumulation of damages is contributed by the inability of the biological systems to maintain and protect the somatic tissues over a long duration of time. There are approaches that contribute to aging. One approach is outlined by the degradation of physiological systems output which directly leads to functional decline. Functional decline changes in cell number which affects metabolic record of the cell. This simple pathway serves as a framework for analyzing methods involved in aging.
Physiological and Metabolic Processes
All organisms’ functions are described as a set of physiological systems that interact with each other and the environment. Their system only communicates with other systems through “inputs” and “outputs”. The diagram on the left shows the network of the physiological system that gives a overview of organism’s function. System aging happens over time when output becomes inappropriate and impairs organisms’ function. The progressive dysfunction with aging of an organism is due to the decline influenced by some other outside forces.
Physiological Decline
From an evolutionary perspective, natural selection will try to maximize reproductive success in tissues in order to slow down mortality. Inversely, many physiological systems within an organism will wear out at similar rates. Similar rates of functional decline could occur because the output of the dysfunctioning system maybe directly be connected to other systems. Another possible explanation for analogous rates would be the existence of an aging process common to different physiological systems. For example, mutations in a single gene ameliorate many forms of aging-related damages. Furthermore, the evolutionary effects on aging raises the probably of similar aging process in different organisms.
Cell Number
Cell number change is directly correlated to aging. Change in the outputs of a system is influenced by alterations in their constituent cells. Even changes outside the extracellular matrix of exoskeleton can be ascribed to changes in cell function. Accumulation of extracellular debris such as fatty plaques can cause dysfunction of macrophages. Even age related modification such as glycation-mediated loss of elasticity of blood vessel wall or damage to lens protein may contribute to the effectiveness of cell function. Since modifications occur at same hierarchical level as cell changes, they inherently affect system function in much the same way as cell function. Therefore, the decline in physiological systems during aging is caused by changes in cell count and/or changes in noncellular component of the system.
Metabolic Control Analysis (MCA) and Aging
MCA has been used to describe the control and regulation of metabolic pathways and networks. Activity of each enzymatic step is varies very slightly and the effect of the overall flux is determined. Simple definition of the control is where the greater the change in flux, the greater the control of that enzyme over the flux. Thus, the control lover flux is a property of pathway that can share control and distribution of control as metabolic conditions are altered.
Further research is still required in this field of study but scientists have built a concrete foundation in which they can experimentally build upon.
There are three main forms of death that are widely recognized: Apoptotic death, Autophagic death, and Necrotic death. Apoptotic death is also known as the programmed cell death. It occurs when cells are no longer needed and they “commit suicide by activating a intracellular death program”. [4] Autophagic death is also another type of cell death, commonly known as autophagic Programmed Cell Death (PCD). [5] This type of death occurs by the delivery of autophagic vesicles into lysosomes [2]. Necrosis death is a more abrupt form of death that cells undergo. Necrosis is when the plasma membrane of a cell brakes. It is important to take into account that these types of deaths could not be exactly the cause of cell death but could happen before, or after.
Phagocytosis is important because it is a major homeostatic mechanism in multicellular organisms. For example, it defends against pathogens by removing defective cells. However, excessive or lack of cell death is harmful and can lead to pathology. Recent discoveries have shown that cells that are still considered viable can be marked for phagocytosis prematurely in a process known as phagoptosis. Prior to this discovery, phagocytosis was believed to only eat dead cell or cells that are close to death. However, recent studies show that healthy cells can be marked for phagocytosis if they possess a signal marking it for phagocytosis or lack a signal to prevent it from being eaten by phagocytosis. The phagoptosis of neurons is particularly concerning due to the limited capacity of the brain to replace these neurons. By the same logic, prevention of phagoptosis of cancer cells are also of concern.
Phagocytosis ( process of cell devouring) is started by the release of signals (‘ find me’ signals) which attracts macrophages that are close by to the cell. When the macrophage is close enough to the cell it recognizes signals of ‘eat me’ and ‘don’t eat me’ from its surface. The most common “eat-me” signal is generated when cell surface is exposed to phospholipid PS. PS exposure occurs when aminophospholipid translocase (enzyme that assists proteins to move across a membrane) is inhibited or phospholipid scramblase is stimulated or both. Healthy cells usually have aminophospholipid translocase that uses ATP to keep phospholipid PS in the inner plasma membrane. When this translocase is inhibited, PS flows out to the outer membrane and is exposed to cell surface, thereby creating “eat-me” signals. Moreover, any condition that activates phospholipid scramblase, which changes phospholipid distribution on the plasma membrane, can cause PS exposure. Conditions such calcium elevation, ATP depletion, oxidative stress can stimulate scramblase and inhibit the translocase. However, exposure of PS is not the sole indicator on whether or not a cell is marked by a phagocyte and additional environmental conditions must also be met. In addition of having an "eat-me" signal of exposed PS, the cell can also posses a "don't-eat me" signal which can be expressed by the protein CD47. Cells can expose this CD47 "do-not-eat" region causing the binding of certain residues to the protein, resulting in the phagocyte being unable to bind to the protein. Moreover, not all cells that are PS-exposed are sufficient to start phagocytosis. Some cells have strong “don’t eat me” signal or require other more potent signals to induce phagocytosis. Therefore, phagocytosis depends on the net exposure of both types of signals and the amount of PS or CD47 expressed determines if phagocytosis occurs. PS exposure can be reversible if and only if the induced eat me signal on the cell surface is quickly removed before macrophage finds the cell. [2]
The most applicable connection is to current cancer research. Unsurprisingly, cancer cells possess an overwhelming amount of "don't-eat me" or CD47 signals. It was recently shown that high levels of CD47 have a strong correlation to the capability of producing tumors in both mice and humans. Antibodies that identify CD47 have shown to induce phagocytosis of these cancer cells and show promise in future cancer treatment.[2]
LDS activates microglia to release MFG-E8 to bind onto exposed PS of the neuron cell marking it for phagoptosis.
Destruction of neurons can be a serious potential health risk if the natural process goes amiss. The cells responsible for the phagocytosis of living neurons are microglia, which are brain cell macrophages. Microglia cells become activated by lipopolysaccharide (LPS) to consume and destroy inflamed neurons via phagocytosis. This activation of the microglia cells, in essence, makes them hunt for inflamed neurons by discharging MFG-E8, a binding protein. When living neurons become inflamed, they bare the phospholipid phosphatidylserine (PS) on the exterior surface of the cell. Once the PS is out in the open, MFG-E8 binds to PS marking it for the microglia to come engulf and destroy the neuron.[3]
Scientists have been looking for a way to inhibit or block this targeting and destruction of healthy neurons. In one experiment they use mice that have been genetically altered to deactivate a specific gene that is used to make MFG-E8 binding proteins. Their results showed that the mice without the MFG-E8 binding protein did not have phagoptosis of regular inflamed neurons. However, by adding pure MFG-E8 binding proteins back into those mice, phagoptosis of the inflamed neurons resumed as usual. They also found that if LPS is inserted in close proximity to the neurons of a mouse that inflammation would occur and microglia targeted the neurons. Additionally to support their findings, when LPS was inserted into the MFG-E8 deficient mice neuron destruction was much less than it was in the normal mice. [3]
In conclusion, inflammation activates microglia cells to destroy healthy neurons but this can be averted by inhibiting the marking of neurons for destruction. This is not to say that microglia are troublesome, they actually help clean up dead and dying neurons to keep inflammation down. In turn, inflammation would make the microglia go on the hunt for living neurons as well as neurons that need to be disposed of. [3]
It has been found that cancers usually expose a great quantity of “don’t eat me” signals in order to not undergo cell death, this being the reason why cancer is so difficult to fight. The “don’t eat me” signal majorly exposed by cancers is the CD47 which has been found to have a correlation with tumourigenicity and its exposure amount. [2] The “don’t eat me” signal is usually neautralized by our bodies by the CRT(Cell Surface Calreticulin) “ eat me” signal, which is greatly exposed in the human body.
Necrotic cell death has been revealed to be more coordinated than random as was previously understood. Known as ‘programmed’ necrotic cell injury, this form of cell death is regulated by TNF receptors which are involved in apoptosis as well. Necrotic cell injury differs from apoptosis in that it is characterized by extensive swelling, plasma membrane rupture, and distinct biochemical components involved in the pathway-specifically RIP serine/threonine protein kinases. Apoptosis and programmed necrosis are therefore induced in response to similar cellular conditions with minor disparities in the degree of cell damage as cells communicate and coordinate the proper course of action through molecular cross talk involving structural modification at the cell membrane and signalling between cells. [6]
[1]Toward a Control Theory Analysis of AgingAnnual Review of BiochemistryVol. 77: 777-798 (Volume publication date July 2008) First published online as a Review in Advance on March 4, 2008DOI: 10.1146/annurev.biochem.77.070606.101605Murphy, Michael. Partridge, Linda.
[2]Eaten alive! Cell death by primary phagocytosis: 'phagoptosis'.
Brown GC, Neher JJ. Department of Biochemistry, University of Cambridge, Tennis Court Road, Cambridge CB2 1QW, UK.
[3]Brown, Guy C. and Neher, Jonas J..”Eaten alive! Cell death by primary phagocytosis: ‘phagoptosis’.” Trends in Biochemical Sciences 37.8 (2012): 325-332. Print.
[4]Alberts, Bruce. "Apoptosis Is Mediated by an Intracellular Proteolytic Cascade." Programmed Cell Death (Apoptosis). U.S. National Library of Medicine, 18 Feb. 0000. Web. 29 Oct. 2012. <http://www.ncbi.nlm.nih.gov/books/NBK26873/>.
Cell adhesion is fundamental in the formation of tissues due to the attraction between individual cells. These adhesions are due to the internal cytoskeleton, which determines the overall structure of the cell. The cell can be in two states - stable adhesion interactions and dynamic adhesive interactions. The stable adhesion interactions are typically made up of cell adhesion receptors (which usually are the glycoprotein that binds the ECM between cells), ECM (extracellular matrix, which are large proteins that interact with other cellular receptors on the site) proteins, and cytoplasmic plaque proteins.
One of the most significant cell adhesions that is required to form a stable tissue is called the Cadherins adhesion molecules, which are transmembrane receptors that depends on Ca2+ to recognize other cells during growth. One of the most studied cadherin molecules is called E-cadherin, which is known to fight epithelial cancer. Cadherin work by combining with catenins and the actin in the cytoskeleton. Catenins are cytoplasmic plaque proteins, which incorporates both a-catenins and b-catenins. a-catenins are responsible for the linkage between the cadherins to the actin in the cytoskeleton. b-catenins are responsible for the interaction between the a-catenin and the cadherin cytoplasmic domain. Based on the observations of b-catenin's interaction with growth factors and transformation, it can be concluded that b-catenin are simply acting as the regulatory component. Cadherins are also responsible for the regions of the cell known as the junctional localization, which are the stronger points of the cell that is capable for adhesion. The EC (extracellular matrix) domains are split into five sub-domains, EC1, EC2, EC3, EC4, and EC5. With the help of x-ray crystallography, the EC1 sub-domain can be used to form a dimer, more specifically called the strand dimer, which are monomers that are parallel to the adhesive binding surfaces and orient outwards.
Desmosomal junctions are primarily located in epithelia and cardiac muscles. They link with the intermediate filament cytoskeleton to form a network capable of withholding immense weight.
Gumbiner, Barry M (1996). "Cell Adhesion: The Molecular Basis of Tissue Architecture and Morphogenesis". Cell. 84 (3): 345–57. doi:10.1016/S0092-8674(00)81279-9.
Similar gene sequences between prokaryotes and eukaryotes suggest that they originated from a universal ancestor and evolved into separate domains billions of years ago. Prokaryote evolved to eukaryote through several stages. An ancestral anaerobic (without air) eukaryote cannot metabolize efficiently due to its inability to oxidized fuel completely. To improve its metabolism, the ancestral eukaryote ingests a bacterial genome that is aerobic (with air). An aerobic metabolism is more efficient because fuel is oxidized to carbon dioxide. Once the bacterium is engulfed by the eukaryote, it uses the cell for replication. This symbiotic system can now carry out aerobic catabolism; thus, transforming anaerobic eukaryote to aerobic eukaryote. There are three major changes that occurred as prokaryotes evolved to eukaryotes. First, the mechanism needed to fold DNA into compact structure containing specific proteins and the ability to divide equally between daughter cell during cell division became more elaborate. Since cell are now larger, system intracellular membranes developed to create a double membrane to surround the DNA. Lastly, early eukaryotes were incapable of carrying out photosynthesis or aerobic metabolism until an aerobic bacterium is introduced to form endosymbiotic and to eventually form plastids. They are similar in their metabolic reactions and in the way they produce energy, as well as in regards to what both prokaryotic and eukaryotic cells are composed of. To name some of their similarities, both have their cells surrounded by plasma membranes, both contain cytoplasm, and they both contain structures of RNA and protein called ribosomes. Though variations are present, their distinct differences result from DNA mutations that have occurred over time.
Eukaryotic Cells Evolved from Simple Precursors
Major changes of the simple cells lead to the development of Eukaryotic Cells
1. Cells were able to acquire more DNA, therefore mechanisms that required to fold DNA strands into complexes with more specific proteins and divide into daughter cells (cell division)
2. Growth of Cells- the cells grow larger in size allowed intramolecular membranes to develop, which led the development of double membrane surrounding DNA
-RNA synethsis on DNA template from cytoplasmic process of protein synthesis on ribosomes became possible
Cell membrane: phospholipid bilayer that encloses the cytoplasm, serves as attachment point for the intracellular cytoskeleton and cell wall.
Cell wall: rigid, outside of the plasma membrane. Its function is to determine the shape of the organism and to act as a vessel pressure, preventing over-expansion when water enters the cell.
Nucleoid: analog to nucleolus of eukaryotes, nucleoid contains DNA, genetic material of the cell, but it is not enclosed by any membrane.
Chromosomes: contains genetic information. Chromosomes make up nucleoid. Prokaryotic cells are haploid.
Flagella: tail-like organelles in charge of movements of cells.
Pili: shorter and thinner than flagella, used also for motility and adherence.
Morphology of prokaryotic cells
Prokaryotic cells have a variety of shapes. These shapes are to describe, classify and identify micro-organism. Some common shapes are:
Cocci: spherical shape
Bacilli: cylindrical or rod shape
Spirilla: a curves rod long enough to form spirals
Vibrio: a short curved rod (comma) shaped
Spirochete: long helical shape
Cell division
Prokaryotic cells reproduce through asexual reproduction. They usually are divided by binary fissions (breaking in half, forming two identical daughter cells) or budding (daughter cells grow out of the parent and gradually increase in size)
Prokaryotic cells have their genes passed out completely to their daughter cells through mitosis. Genome is stored in chromosome.
Energy intake
Bacteria and Archaea are the main branches of prokaryote evolution.
Generally, Bacteria and Archaea are quite similar in size and shape. They share the characteristics of prokatyotes but are different in many key structural, biochemical, and physiological characteristics.
Campbell NA, Reece JB. 2008. Biology. 8th ed. San Francisco (CA):
Pearson/Benjamin Cummings.
There are many difference between prokaryote and eukaryote cell.
In nuclear body of prokaryote, the nuclear body is not bounded by a nuclear membrane while eukaryotic cell have a nuclear that is bounded by a nuclear membrane having pores connecting it with the endoplasmic reticulum. In prokaryote, the cell is covered by cell envelope, a structure varies with type of bacteria, while in eukaryote cell, there is cell membrane to separate the cell from outside environment and regulates movements of materials in and out of the cells. Circular, unorganized DNA molecule is located in nucleoid of prokaryote; on the other hand, linear DNA that is organized by histones is located in nucleus of eukaryote which the protector of nuclear envelope. In prokaryotes, the nuclear body contains a circular chromosome with the lack of histones (unwounded DNA). There is no nucleolus eukaryotic chromosome but a nucleolus, which is present with one or more paired, linear chromosome containing histones. This says that the DNA of eukaryotic cells are organized in nucleus and the DNA of prokaryotes cells are unorganized and floats free in the nucleolus. Eukaryotes sex cell and prokaryotes cell both have flagella, organelle that helps the cell move. Prokaryote's flagella is consist of two protein building block while eukaryote's flagella is complex and consisting of multiple microtubules. Eukaryotes have many organelles in cells such as mitochondria, golgi, lysosomes.... besides ribosomes, there is no organelles in prokaryotes. Prokaryotic cell on average are usually ten times smaller than eukaryotic cell. Cell division in prokaryotic cell and eukaryotic cell is also different. In prokaryotic cell, the cell divided by binary diffusion and prokaryotic cell are haploid. In eukaryotic cell, cell division follows process of mitosis; haploid sex cells in diploid. Cell membrane in prokaryotic cell is a phospholipid bilayer which usually lacking sterols while eukaryotic cell membrane contains sterols. Eukaryotic cell membrane is capable of endocytosis and exocytosis while prokaryote cell is not. Cell wall is present in plant cell, algea, and fungi which belong to eukaryote. Cell wall of eukaryotic cell never composed of peptidoglycan. In prokaryotic cells, a few member of domain bacteria have cell walls which composed of peptydoglycan. Member of domain Archae have cell wall composed of protein or unique molecules resembling but not the same as peptidoglycan. In cytoplasmic structures of eukaryote, the ribosomes are composed of a 60S and a 40S subunit forming an 80S ribosome. Internal membrane-bound organelles such as mitochondria, endoplasmic reticulum, Golgi apparatus, vacuoles, and lysosomes are present. Chloroplasts serve as organelles for photosynthesis. A mitotic spindle involved in mitosis is present during cell division. A cytoskeleton is present. It contains microtubules, actin micofilaments, and intermediate filaments. These collectively play a role in giving shape to cells, allowing for cell movement, movement of organelles within the cell and endocytosis, and cell division. In prokaryote, the ribosomes are composed of a 50S and a 30S subunit forming an 70S ribosome. Internal membrane-bound organelles such as mitochondria, endoplasmic reticulum, Golgi apparatus, vacuoles, and lysosomes are absent. There are no chloroplasts. Photosynthesis usually takes place in infoldings or extensions derived from the cytoplasmic membrane. There is no mitosis and no mitotic spindle in prokaryote but fission and budding only. They may contain only actin-like proteins that, along with the cell wall, contribute to cell shape.
Plasma membrane: A lipid/protein/carbohydrate complex, providing a barrier and containing transport and signaling systems.
Mitochondrion: Surrounded by a double membrane with a series of folds called cristae. Functions in energy production through metabolism. Contains its own DNA, and is believed to have originated as a captured bacterium.
Cytoskeleton
Microfilaments
Intermediate filaments
Microtubules
Nucleus: double membrane surrounding the chromosomes and the nucleolus. Pores allow specific communication with the cytoplasm. The nucleolus is a site for synthesis of RNA making up the ribosome.
Nuclear envelope: doubled membrane, enclosing the nucleus.
Nucleolus
Chromatin: contains genetic information of cells (DNA)
Chromosomes: only visible during cell divisions.
Endoplasmic Reticulum (ER)
Rough ER: A network of interconnected membranes forming channels within the cell. Covered with ribosomes (causing the "rough" appearance) which are in the process of synthesizing proteins for secretion or localization in membranes.
Smooth ER: A network of interconnected membranes forming channels within the cell. A site for synthesis and metabolism of lipids. Also contains enzymes for detoxifying chemicals including drugs and pesticides.
Golgi apparatus: A series of stacked membranes.
Lysosome: A membrane bound organelle that is responsible for degrading proteins and membranes in the cell, and also helps degrade materials ingested by the cell.
Ribosome: Protein and RNA complex responsible for protein synthesis
Cell division
Energy intake
Eukaryotes that live in extreme environments tend to be microbial. Microbial life is present in all extreme environment with enough energy to support life. Eukaryote cells are very adaptable, but prokaryotes are a little bit more so. This is not conclusive due to many environments that have yet to be explored. Anaerobic environments lack oxygen, but eukaryote cells still manage to survive.
Thermophile eukaryote cells are resistant to high temperatures. These cells must adapt by modifying their lipid and cell walls. The heat is dangerous so modifications to resist heat are necessary.
Psychrophiles are cells that have adapted to extremely cold temperatures. An example would be Heteromita globosa, which
is a heterotrophic flagellate that lives in Antarctica. Psychrophiles face an extra challenge in that they must make modifications to the lipid and cell walls in order to maintain fluidity. The cold temperatures cause cell walls to become rigid.
Acidophiles are cells that can survive in extremely acidic conditions. Until recently, there were only four organisms, all eukaryotic that could survive in near 0 pH conditions. The way acidophiles can survive in such low pH levels is not known, but it is suggested that it could be due to a strong proton pump or low proton membrane permeability.
Alkalophiles are cells that can survive in high pH levels. Some of these Alkalophiles can also be stable at neutral pH also which leads us to believe that the internal chemistry is highly resistant to environmental influence. Possible compartmentalization of eukaryotic cells could help this.
Barophiles are cells that survive in high pressure conditions.
Xerophiles are cells that survive in dry conditions.
Halophiles are cells that live in high saline conditions. These cells must face the problem of drying out by osmotic pressure.
Eukaryotes such as flagellates thrive in such conditions.
The CRISPRs, or Clustered Regularly Interspaced Short Palindromic Repeats, functions as a prokaryotic immune system. It protects bacteria and archaea against mobile genetic elements. This defense system can continuously adjust its reach at the genomic level which implies that both gain and loss of information is inheritable. The CRISPR defense system consists of stretches of interspaced repetitive DNA and associated cas genes. The CRISPR system consists of three distinct stages:
Adaptation of the CRISPR via the integration of short sequences of the invaders as spacers;
Expression of CRISPRs and subsequent processing to small guide RNAs;
Exogenous DNA is processed into small elements by proteins encoded by CRISPR-Cas genes. These small elements are then inserted into the CRISPR locus near the leader sequence. RNAs from the CRISPR loci are constituitively expressed and processed by cas proteins to small RNAs composed of individual exogenously-derived sequence elements with flanking repeat sequence. The RNAs then guide other cas proteins to silence exogenous genetic elements at both the RNA and DNA level.
Stage 1 - CRISPR adaptation
Adaptation of the CRISPR is the recognition of alien DNA by dedicated cas proteins and/or host proteins, as well as the subsequent processing and integration into the chromosomal CRISPR locus. This stage has recently been demonstrated experimentally by Barrangou et al. by triggering virus resistance on the lactic acid bacterium Streptococcus thermophilus. The acquisition of new spacers occurred at the 5' end of the CRISPR as predicted. The fact that new spacers are integrated at the 5' side of the CRISPR suggests that they are a chronological record that reflects previous encounters with mobile genetic elements. Furthermore, spacers in both sense and anti-sense orientation turned out to be functional while only the leader strand is transcribed. This implies that the mechanism does not operate with a classical anti-sense mechanism at the level of mRNA, implying that DNA is the target.
Stage 2 - CRISPR expression
CRISPR expression is the transcription of the poly-spacer precursor crRNA which is followed by binding to a complex of Cas proteins and processing to mono-spacer crRNAs that serve as the guide sequences. It was discovered that pre-crRNA (longer transcripts) potentially covered the entire CRISPR and were step-wise processed to small crRNAs. Furthermore, only the leader strand of the CRISPR is transcribed and processed. Similar results has been obtained by expression studies of CRISPRS from both the bacterium Staphylococcus epidermidis and the archaea Archaeoglobus fulgidus and S. solfataricus.
Stage 3 - CRISPR interference
CRISPR interference involves the binding and/or degradation of the target nucleic acid (DNA). Studies of CRISPRs from S. thermophilus has provided experimental evidence that the CRISPR-Cas system has the ability to bring about specific viral resistance. Furthermore, experiments on the CRISPR-Cas system from S. epidermidis has showed that this bacterium lost its CRISPR-based immunity against a plasmid after a self-splicing intron was inserted into the proto-spacer of the plasmid. The intron is present at DNA level but spliced from mature RNA. This strongly suggests that DNA is the target of the CRISPR interference system.
Significance for evolution and possible applications
By implementing the CRISPR-Cas mechanism, bacteria are able to acquire immunity against certain phages and halt further transmission of targeted phages. Because of this, some researchers have suggested that the CRISPR-Cas system is a Lamarckian inheritance mechanism.
Several proposals to exploit CRISPRs-derived biotechnology include:
Artificial immunization against phages by adding matching spacers of industrially important bacteria, such as in the milk and wine industries.
Knockdown of endogenous genes by transformation with a plasmid which contains a CRISPR area with a spacer, which inhibits a target gene.
Distinguish different bacterial strains by comparison of the spacer regions.
The method prokaryotes use most often when responding to environmental changes is altering their gene expression. Expression is when a gene is transcribed into RNA and then translated into proteins. The two types of expression are constitutive—where genes are constantly being expressed—and regulated—where specific conditions need to be met inside the cell for a gene to be expressed. This sub-section focuses on how prokaryotes go about regulating the expression of their genes.
Transcription, when DNA is converted into RNA, is the first place for controlling gene activity. Proteins interact with DNA sequences to either promote or prevent the transcription of a gene.
Keep in mind that DNA sequences are not discernible from one another in terms of having features that a regulatory system would be able to register. Therefore, when regulating gene expression, prokaryotes rely on other sequences within their genome, called regulatory sites. These regulatory sites are most often also DNA-binding protein binding sites and are close to the DNA destined for transcription in prokaryotes.
An example of one of these regulatory sites is in E. coli: when sugar lactose is introduced into the environment of the bacterium a gene for encoding the production of an enzyme, β-galactosidase, begins to be expressed. This enzyme’s function is to process lactose so that the cell can extract energy and carbon from it.
lac regulatory site
The sequence of nucleotides of this regulatory site (pictured) displays an almost completely inverted repeat. This shows that the DNA has a nearly twofold axis of symmetry, which in most regulatory sites usually correlates to symmetry in the protein that binds to the site. When studying protein-DNA interactions, symmetry is generally present.
Furthering the investigation into the expression of the lac regulatory site and the protein-DNA interactions that take place there, scientists looked at the structure of the complex formed between the DNA-binding unit that recognizes the lac site and the site itself, which is part of a larger oligonucleotide. They found that the DNA-binding unit specific to the lac regulatory site comes from the protein lac repressor. The function of lac repressor, as the name suggests, is the repression of the lactose-processing gene’s expression. This DNA-binding unit’s twofold axis of symmetry matches the symmetry of the DNA, and the unit binds as a dimer. From each monomer of the protein an α helix is inserted into the DNA’s major groove. Here amino acid side chains interact (via very site-specific Hydrogen bonding) with the exposed base pairs in such a fashion that the lac repressor can only very tightly bind to this specific site in the genome of E. coli.
The helix-turn-helix motif is common to many prokaryotic DNA-binding proteins
After discerning the structures of many prokaryotic DNA-binding proteins, a structural pattern that was observed in many proteins was a pair of α helices separated by a tight turn. These are called helix-turn-helix motifs and are made of two distinct helices: the second α helix (called the recognition helix) lies in the major groove and interacts with base pairs while the first α helix is primarily in contact with the DNA backbone.
In Prokaryotes, DNA-Binding Proteins Bind Explicitly to Regulatory Sites in Operons
Looking back at the previous example with E. coli and β-galactosidase, we can garner the common principles of how DNA-binding proteins carry out regulation. When E. coli’s environment lacks glucose—their primary source of carbon and energy—the bacteria can switch to lactose as a carbon source via the enzyme β-galactosidase. β-galactosidase hydrolyzes lactose into glucose and galactose which are then metabolized by the cell. The permease facilitates the transport of lactose across the cell membrane of the bacterium and is essential. The transacetylase is on the other hand not required for lactose metabolism but plays a role in detoxifying compounds that the permease may also be transporting. Here we can say that the expression levels of a group of enzymes that together contribute to adapting to a change in a cell’s environment change together.
An E.coli bacterium growing in an environment with a carbon source such as glucose or glycerol will have around 10 or fewer molecules of the enzyme β-galactosidase in it. This number shoots up to the thousands, however, when the bacterium is grown on lactose. The presence of lactose alone will increase the amount of β-galactosidase by a large amount by promoting the synthesis of new enzymes rather than activating a precursor.
When figuring out the mechanism of gene regulation in this particular instance, it was observed that the two other proteins galactoside permease and thiogalactoside transacetylase were synthesized alongside β-galactosidase.
An operon is made up of regulatory components and genes that encode proteins
The fact that β-galactosidase, the transacetylase, and the permease were regulated in concert indicated that a common mechanism controlled the expression of the genes encoding all three. A model called the operon model was proposed by Francois Jacob and Jacques Monod to explain this parallel regulation and other observations. The three genetic parts of the operon model are (1) a set of structural genes, (2) an operator site (a regulatory DNA sequence), and (3) a regulator gene that encodes the regulator protein.
In order to inhibit the transcription of structural genes, the regulator gene encodes a repressor protein meant for binding to the operator site. In the case of the lac operon, the repressor protein is encoded by the i gene, which binds to the o operator site in order to prevent the transcription of the z, y, and a genes (the structural genes for β-galactosidase). There is also a promoter site, p, on the operon whose function is to direct the RNA polymerase to the proper transcription initiation site. All three structural genes, when transcribed, give a single mRNA that encodes β-galactosidase, the permease, and the transacetylase. Because this mRNA encodes more than one protein, it is called a polycistronic (or polygenic) transcript.
The lactose operon
The lac repressor protein in the absence of lactose binds to the operator and blocks transcription
The lac repressor (pictured bound to DNA) is a tetramer with amino- and carboxyl-terminal domains. The amino-terminal domain is the one that binds to the DNA while the carboxyl-terminal forms a separate structure. The two sub-units pictured consolidate to form the DNA-binding unit. When lactose is absent from the environment of the bacterium, the lac repressor binds to the operator DNA snugly and swiftly (4x10^6 times as powerfully to the operator as opposed to random sites on the genome). The binding of the repressor precludes RNA polymerase from transcribing the z, y, and a genes which are downstream from the promoter site and code for the three enzymes. The dissociation constant for the complex formed by the lac repressor and the operator is around 0.1 pM and the association rate constant is a whopping 10^10 M-1s-1. This suggests that the repressor diffuses along a DNA molecule to find the operator rather than via an encounter from an aqueous medium.
When it comes to the DNA-binding preference of the lac repressor, the level of specificity is so high that it can be called a nearly unique site within the genome of E. coli. When the dimers of the amino-terminal domain bind to the operator site, the dimers of the carboxyl-terminal site are able to attach to one of two sites within 500 bp of the primary operator site that approximate the operator’s sequence. Each monomer interacts with the bound DNA’s major groove via a helix-turn-helix unit.
Ligand binding can induce structural changes in regulatory proteins
Now let’s look at how the presence of lactose changes the behavior of the repressor as well as the expression of the operon. All operons have inducers—triggers that facilitate the expression of the genes within the operon—and the inducer of the lac operon is allolactose, a molecule of galactose and glucose with an α-1,6 linkage.
In the β-galactosidase reaction, allolactose is a side product and is produced at low levels when the levels of β-galactosidase are low in the bacterium. Additionally, though not a substrate of the enzyme, isopropylthiogalactoside (IPTG) is a powerful inducer of β-galactosidase expression.
In the lac operator, the way the inducer prompts gene expression is by inhibiting the lac repressor from binding to the operator. Its method of inhibition is by binding to the lac repressor itself thus immensely reducing the affinity of the repressor to bind to the operator DNA. The inducer binds to each monomer at the center of the large domain, causing conformational changes in the DNA-binding domain of the repressor. These changes drastically reduce the DNA-binding affinity of the repressor.
The operon is a common regulatory unit in prokaryotes
Numerous other gene regulation complexes within prokaryotes function analogously to the lac operon. An example of another network like this one is that which takes part in the synthesis of purine (and pyrimidine to a certain extent). These genes are repressed by the pur repressor, which is 31% identical to the lac repressor in sequence with a similar 3D structure. In this case, however, the pur repressor behaves opposite from the lac repressor: it blocks transcription by binding to a specific DNA site only when it is also bound to a small molecule called a corepressor (either guanine or hypoxanthine).
Transcription can be stimulated by proteins that contact RNA polymerase
While the previous examples of DNA-binding proteins all function by preventing the transcription of a DNA sequence until some condition in the environment is met, there are also examples of DNA-binding proteins that actually encourage transcription.
A good instance of this is the catabolite activator protein in E. coli. When the bacterium is grown in glucose it has very low amounts of catabolic enzymes whose function it is to metabolize other sugars. The genes that encode these enzymes are in fact inhibited by glucose, an effect known as catabolite repression. Glucose lowers the concentration of cAMP (cyclic AMP). When the concentration of cAMP is high, it stimulates the transcription of these catabolic enzymes made for breaking down other sugars. This is where the catabolite activator protein (CAP or CRP, cAMP receptor protein) comes into play. CAP, when bound to cAMP, will stimulate the transcription of arabinose and lactose-catabolizing genes. CAP, which binds only to a specific sequence of DNA, binds as a dimer to an inverted repeat at the position -61 relative to the start site for transcription, adjacent to where RNA polymerase binds (pictured).
This CAP-cAMP complex enhances transcription by about a factor of 50 by making the contact between RNA polymerase and CAP energetically favorable. There are multiple CAP binding sites within the E. coli genome, therefore increasing the concentration of cAMP in the bacterium’s environment will result in the formation of these CAP-cAMP complexes, thus resulting in the transcription of many genes coding for various catabolic enzymes.
Regulatory Circuits Can Result in Switching Between Patterns of Gene Expression
In investigating gene-regulatory networks and how they function, studies of bacterial viruses—especially bacteriophage λ—have been invaluvable. Bacteriophage λ is able to develop via either a lytic or lysogenic pathway. In the lytic pathway, transcription takes place for most of the genes in the viral genome which leads to the production of numerous virus particles (~100) and the eventual lysis of the bacterium. In the lysogenic pathway, the bacterial DNA incorporates the viral genome where most of the viral genes stay unexpressed; this allows for the viral genetic material to be carried in the replicate of the bacteria. There are two essential proteins plus a set of regulatory sequences within the viral genome that are the cause for the switch between the choice of pathways.
λ repressor is one of these key regulatory proteins which promotes the transcription of the gene that encodes the repressor when levels of the repressor are low. When levels of the repressor are high, it blocks transcription of the gene. It is also a self-regulating protein. While the λ repressor binds to many sites in the λ phage genome, the one relevant here is the right operator, which includes 3 binding sites for the dimer of the λ repressor in addition to 2 promoters within an approximately 80 base pair region. The role the first promoter plays is driving the expression of the λ repressor gene, while the other promoter is responsible for driving the expression of a variety of other viral genes. The λ repressor binds to the first operating site with the most affinity; and when it is bound to this first operating site, the chances of a protein binding to the adjacent operating site increase 25 times. When the first and second operating sites have these complexes bound to them, the dimer of the λ repressor inhibits the transcription of the adjacent gene whose purpose is to encode the protein Cro (controller of repressor and others). The repressor dimer at the second operating site can interact with RNA polymerase so as to stimulate the transcription of the promoter which controls the transcription of the gene encoding the λ repressor. This is how the λ repressor facilitates its own production.
λ repressor fusions can be used to study protein-protein interactions in E. coli. There are two different domains in λ repressor: the N-terminal (DNA binding activity) and the C-terminal domain (dimerization). In order to have an active repressor fusion, the C-terminal domain should be replaced with a Heterodimers domain and form a dimer or higher order oligomer. However, inactive repressor fusions cannot attach to the DNA sequences and affect the expression of phage or reporter.[1]
A circuit based on lambda repressor and Cro form a genetic switch
We can see in the above picture how the λ repressor blocks production of Cro by binding to the first operating site with the most affinity. Cro meanwhile blocks the production of the λ repressor by binding to the third operating site with the most affinity. This entire circuit is the deciding factor as to whether the lytic or lysogenic pathway will be followed: if λ repressor is high and Cro is low, the lysogenic path will be chosen; if Cro is high and the λ repressor is low, the lytic path will be chosen.
Many prokaryotic cells release chemical signals that regulate gene expression in other cells
Some prokaryotes are also known to undergo a process where they release chemicals called autoinducers into their medium (quorum sensing). These autoinducers, which are most of the time acyl homoserine lactones, are taken up by the surrounding cells. When the levels of these autoinducers reach a certain point, receptor proteins bind to them and activate the expression of several genes, including those that promote the synthesis of more autoinducers. This is a way for prokaryotes to interact with one another chemically to change their gene-expression patterns depending on how many other surrounding cells there are in their medium. Communities of prokaryotes that carry these mechanisms of quorum sensing out are collectively called a biofilm.
Gene Expression Can Be Controlled at Posttranscriptional Levels
Though most of gene expression regulation happens at the initiation of transcription, other steps of transcription are also possible targets for regulation.
Exploring the genes of the tryptophan operon (abbreviated as trp operon) in order to study the regulation of tryptophan synthesis shows two types of mutants. One type of mutant involves structural gene mutations and the other a regulatory mutant. The mutants that involve structural gene mutations are auxotrophic for tryptophan and need tryptophan to growth. To convert the precursor molecule chorismate to tryptophan, the trpE, trpD, trpC, trpB, and trpA genes codes for a polycistronic message and the mRNA will be translated to the enzyme that carries out the conversion.
Tryptophan
The second type of mutants is able to constitutively synthesize the enzymes necessary for the synthesis of tryptophan. The trpR gene codes for the tryptophan repressor. The gene mapped in another quadrant of the E. coll chromosome compared to the trp operon. The trpR gene cannot regulate the synthesis of tryptophan efficiently.
Studies on the dimeric trp repressor protein show that it does not function alone. The repressor must bind the last product of that metabolic pathway in order to regulate the synthesis of tryptophan. Thus, tryptophan is a corepressor for its own biosynthesis. This process is called feedback repression at the transcriptional level.
When the concentration of tryptophan is high enough, then the repressor binds to tryptophan to make a repressor-tryptophan complex. This complex will attach to the operator region of the trp operon and prevents RNA polymerase to bind and initiate transcription of the structural genes. Also, when the concentration of tryptophan is low in the cell, due to lack of tryptophan-complex RNA polymerase is able to bind to the gene and transcribe the structural genes. Therefore, tryptophan will be biosynthesized.[2]
Attenuation is a prokaryotic mechanism for regulating transcription through the modulation of nascent RNA secondary structure
While studying the tryptophan operon, Charles Yanofsky discovered another means of transcription regulation. The trp operon encodes 5 enzymes that convert chorismate into tryptophan, and upon examining the 5’ end of the trp mRNA he found there was a leader sequence consisting of 162 nucleotides that came before the initiation codon of the first enzyme. His next observation was that only the first 130 nucleotides were produced as a transcript when the levels of tryptophan were high, but when levels were low a 7000-nucleotide trp mRNA which included the entire leader sequence was produced. This mode of regulation is called attenuation, where transcription is cut off before any mRNA coding for the enzymes is produced.
Attenuation depends on the mRNA’s 5’ end features. The first part of the sequence codes for a leader peptide of 14 amino acids. The attenuator comes after the open reading frame for this peptide—it is an RNA region capable of forming a few alternate structures. Because transcription and translation are very closely coupled in bacteria, the translation of say the trp mRNA begins very soon after the synthesizing of the ribosome-binding site.
The structure of mRNA is altered by a ribosome, which is stalled by the absence of an animoacyl-tRNA necessary for the translation of the leader mRNA. This allows RNA polymerase to transcribe the operon past the attenuator site
↑Leonardo Mariño-Ramírez, Lisa Campbell, and James C. Hu. Screening Peptide/Protein Libraries Fused to the λ Repressor DNA-Binding Domain in E. coli Cells. 2003; 205: 235–250.
↑Arkady B. Khodursky, Brian J. Peter, Nicholas R. Cozzarelli, David Botstein. DNA microarray analysis of gene expression in response to physiological and genetic changes that affect tryptophan metabolism in Escherichia coli. 2000 October 24; 97(22): 12170–12175. Published online 2000 October 10.
The Control of Gene Expression in Eukaryotes
Chromatin are a combination of eukaryotic DNA and histones. The eukaryotic DNA binds tightly to the histones, which are basic proteins. Changes in the structure of chromatin are largely responsible for the regulation of gene expression. Other regulators of gene expression include interactions between proteins and translation.
In Eukaryotes, as compared to prokaryotes, gene regulation is a lot more complex. This is because the Eukaryotic genome is a lot larger and therefore encodes for a lot more proteins. There are also many different types of cells in eukaryotes, such as liver cells, pancreatic cells, and etc. The genes that are in these highly specialized cells have a huge difference in expression. Another reason for the complexity is that eukaryotic genes that encode proteisn are usually spread throughout the enormously large genome. The final reason is that eukaryotic transcription and translation are not coupled, and this negates some of the gene regulation mechanisms that prokarotes utilize.
Chromatin Structure
Chromatin is composed of units that repeat. Each of these units are composed of 200 base pairs of DNA, and two copies each of the four histones H2A, H2B, H3, and H4. This unit is called the histone octomer, and the repeating units are referred to as nucleosomes. The five main histones present in chromatin are H2A, H2B, H3, H4, and H1, but H1 is not part of the histone octomer. Each histone also has a flexible amino tail that have various lysine and arginine residues and extends beyond the core. These tails are very important because covalent modifications of them alter the DNA affinity of the histones. When chromatin is digested, it yields only 145 base pairs of DNA binding to the histone octomer, and this smaller unit is called the nucleosome core particle. The DNA that connects these nucleosome core particles in undigested chromatin is referred to as linker DNA. This is where H1 binds.
The three dimensional structure of the nucleosome is composed of eight histones arranged into a tetramer, and a pair of dimers. When the tetramer comes together with the dimers, they form a superhelical ramp that is left-handed, and DNA wraps around it forming a left-handed superhelix. The contact between the superhelical ramp and the DNA superhelix occurs primarily along the phosophodiester backbone and minor groove of the DNA. The winding of this DNA reduces its length by packing it together very tightly.
Chromatin Remodeling
The DNA adjacent to actively transcribed genes in chromatin are more sensitive to being cleaved, indicating that those sites contain less compact DNA. Additionally, some other sites, usually within proximity to the start of an actively transcribed gene, are also more sensitive to cleavage by nucleases. These sites are referred to as hypersensitive sites, and they either have fewer nucleosomes, or nucleosomes that are in an altered conformation. These hypersensitive sites specific to different cell types and are developmentally regulated. This indicates that a prerequisite for gene expression lies within chromatin.
Chromatin structure differs between active and inactive genes, and this indicates that some form of modification must be done to modify chromatin structure. Enhancers are DNA sequences that increase the activity of many promoters, even when they are thousands of base pairs away. Enhancers work by binding certain regulatory proteins, and they are only effective in the unspecific type of cell that express those regulatory proteins. These proteins may disrupt chromatin structure, exposing a gene and/or regulatory sites and thus influence transcription. This accounts for its ability to function at a distance from the gene being expressed.
DNA Modification
Gene expression can be inhibited by modifying DNA. This conclusion was drawn by studying sequences in the mammalian genomes. Lots of sequences in mammalian genomes have cytosines that are methylated at the C5 carbon. These cytosines have differing distribution throughout the genome depending on cell type.
Transcriptional Activation and Repression
Interactions between proteins mediate much transcriptional activation and repression. Eukaryotic transcription factors recruit proteins, which build large complexes that interact with and thus activate or repress transcription. This type of regulation is extremely advantageous because depending on the different proteins present in the cell, the regulation can have different effects. This is referred to as combinatorial control, and it is responsible for generating different types of cells.
Hormone receptors on the nucleus recruit various proteins to the transcription complex. These proteins are generally coactivators and corepressors. Coactivators are proteins that contain three repeated sequences within a central region of 200 amino acids. The repeated sequence is Leu-X-X-Leu-Leu, where X can be any amino acid, and they form short alpha helices that bind to the hormone receptors on the nucleus, inducing a change in conformation that enables the receptors to recruit the entire coactivator. Corepressors repress transcription. Sometimes repression can be done without binding a ligand, such as the receptors for retinoic acid and thyroid hormone. When unbound, the receptors bind to corepressors. The site in which the corepressor binds overlaps with the binding site for the coactivator and thus serves to repress transcription.
So What?
Tamoxifen and raloxifene are drugs used in the treatment of breast cancer. Tamoxifen inhibits the activation of gene expression by blocking coactivators from binding. This is important because cancer is such a prevalent disease in today's society, and by studying and understanding the mechanisms of gene activation and repression, new drugs can be produced to combat the disease by altering gene expression.
Source: Berg, Jeremy and Stryer, Lubert. Biochemistry: Fifth Edition. United States of America: W.H. Freeman and Company, 2002.
Gene expression is the process that transfers genetic information from a gene made of DNA to a functional gene product made of RNA or protein. Genetic Information flows from DNA to RNA by the process of transcription, and then from RNA to protein by the process of translation. In order to ensure that the proper products are produced, gene expression is regulated at many different stages during and in between transcription and translation. In eukaryotes, the gene contains extra sequences that do not code for protein. In these organisms, transcription of DNA produces pre-mRNA, which must be spliced into mRNA that lacks these intervening sequences or introns, before translation begins. Splicing can be regulated so that different mRNAs can contain or lack regions called exons, in a process called alternative splicing. Alternative splicing allows more than one protein to be produced from a gene [1], and is an important regulatory step in determining which functional proteins are produced from gene expression. Splicing of pre-mRNA has been proven to be an important mechanism to controlling human development, and misregulation of splicing can lead to disease [2].
Alternative splicing is a 2 step process carried out by the splicesome that ligates the 5’ splice site of an upstream exon to the 3’ splice site of a downstream exon. The splicesome is primarily composed of 5 small nuclear ribonuceloproteins (snRNP’s). snRNP’s carry out splicing through the recognition of 5’, 3’, and a branch point sequences in an intron.
Steps of Alternative Splicing:
1.) U1 snRNP recognizes GU of 5’ splice site, while U2 snRNP recognizes branch point bulged Adenosine.
2.) The A-complex is formed when U1 and U2 position the branch point near the 5’ splice site.
3.) The B-complex is formed when the tri-snRNP U4-U5-U6 bind to the A complex.
4.) The activated B-complex is formed when U1 and U4 leave, forming the catalytic splicesome.
5.) The C-complex is formed after the 1st step of splicing when the branch site adenosine attacks the 5’ splice site, creating an intron lariat.
6.) The 2nd step of splicing is carried out by the C-complex when the 5’ splice site attacks the 3’ splice site, resulting in the ligation of both exons. [3]
Regulation of Alternative Splicing by the rate of RNAPII Transcription
Mechanisms that influence splice site selection are important in regulating alternative splicing, and ensure production of the proper proteins at the right time. Many genes have been proven to be spliced co-transcriptionally [4], and the rate of RNA Polymerase II transcription has been suggested to be a regulatory mechanism of co-transcriptional Alternative splicing [5][6]. Aebi and Weissman’s “first come, first served” model [7] shows how rate of transcription can influence the inclusion of internal exons. Slow recognition of the first intron or rapid recognition of the second intron creates a transcription dependent competition for 3' splice site selection. Slow transcription through the second intron could allow recognition of the first intron before the second is even synthesized, while fast transcription through the first two introns could increase the ability of the second intron to compete with the first intron for recognition. Formation of the “A Complex” (figure 2) is a step in alternative splicing which creates a commitment to splice site selection [8], and is completed by pairing of the U2 snRNP with the branch point [9]. This data shows control over the rate of transcription could be a very powerful tool in regulation of 3’ splice site selection.
One method eukaryotes use to regulate the rate of transcription is sequence based arrest sites located in DNA [10]. Using the thermodynamic theory of DNA-dependent transcriptional arrest, Artificial Arrest Sites (ARTAR sites) have been created and shown to affect rates of RNA Polymerase II transcription [11]. When placed downstream of both 3’ splice sites, the ARTAR site will have no effect on alternative splicing. If placed in a location in between the first and second branch points, the ARTAR site can increase inclusion by decreasing 3’ splice site competition.
↑Douglas L. Black (2003) Mechanisms of Alternative Pre-Messeneger RNA Splicing. Annual Review of Biochemistry, 72: 291-336
↑Mo Chen, James L. Manely (2009) Mechanisms of alternative splicing regulation: insights from molecular and genomic approaches. Nature, 10: 741-754
↑Douglas L. Black (2003) Mechanisms of Alternative Pre-Messeneger RNA Splicing. Annual Review of Biochemistry, 72: 291-336
↑Pandya-Jones A., Black DL (2009) Co-Transcriptional Splicing of constitutive and alternative exons. RNA, 10: 1896-908
↑Kenneth James Howe, Caroline M. Kane, Manuel Ares Jr. (2003) Perturbation of transcription elongation influences the fidelity of internal exon inclusion in Saccharomyces Cerevisiae. RNA, 8: 993-1006
↑Cramer P., Caceres J.F., Cazalla D., Kadener S., Muro A.F., Baralle F.E., Kornblihtt A.R. Coupling of transcription with alternative splicing: RNA pol II promoters modulate SF2/ASF and 9G8 effects on an exonic splicing enhancer (1999) Molecular Cell, 4 (2), pp. 251-258.
↑Aebi, M. and Weissman, S.M. 1987. Precision and orderliness in splicing. Trends Genet. 3: 102-107
↑Lim SR, Hertel KJ (2004) Commitment to splice site pairing coincides with A complex formation. Mol. Cell 15: 477–483.
↑Qin Li, Ji-Ann Lee, Douglas L. Black (2007) Neuronal regulation of alternative pre-mRNA splicing. Nature Reviews Neuroscience 8: 819-831
↑Spencer CA, Groudine M. Transcription elongation and eukaryotic gene regulation. Oncogene. 1990; 5:777–785.
↑Dmitry Kulish, Kevin Struhl (2001) TFIIS Enhances Transcriptional Elongation through an Artificial Arrest Site In Vivo. Mol Cell Biol. 13: 4162-4168
Transition metals like zinc, iron, and copper are common components in a many different types of proteins. These metals are crucial for living; however, surplus amounts of these metals are detrimental to cell growth and viability. Luckily there are many mechanisms that help regulate this excess of metals when required. In response to these changes of metal levels, certain genes encodes transporting of metals and storage proteins that help maintain ideal levels of each metal. Deficiencies of these metals within cells can also cause health problems. Through many different types of complementary mechanisms, cellular metal homeostasis is achieved. Although not all transcriptional factors have been understood completely, they give researchers a sense of idea that could potentially explain the linkage between metal levels, health, and diseases.[1]
Metal levels can also be altered by different types of cancers, and even by neurodegenerative disorders such as Alzheimer’s and Parkinson’s diseases. Thus imbalanced metal levels can further cause severe implications when diagnosed with these illnesses. [1]
Zinc
Zinc one of the vital transition metals in organisms, is important for the accurate folding of certain protein structures. It is the cofactor for hundreds of enzymes; however, in excess, zinc is very toxic to growth, and can wrongfully bind to unfitting locations in proteins and damage their function.[1]
Iron
Iron is another type of transition metal that is vital to life, acting as an electron acceptor or donor in physiological reactions. Like zinc, iron is also used as cofactors in enzymes required for oxygen transportation, DNA synthesis, ribosome biosynthesis, photosynthesis and many other functions. Like other transition metals, in excess, iron is very hazardous to growth and viability. Thus many organisms have evolved specialized mechanisms that regulate cellular iron levels. [1]
Copper
Found in a variety of eukaryotes, copper is a fundamental metal that is necessary for life in many organisms. Being a redox active metal, copper is a cofactor in many different proteins, including Cu/Zn superoxide dismutase 1 and cytochrome c oxidase. Like zinc and iron, when copper is in excess, it is highly toxic to cells and viability. Thus in order to maintain ideal level of copper, many eukaryotes developed transcription factors that regulate copper levels by controlling the genes that encodes for copper uptake and elimination.[1]
Changes in gene expression do not only occur in excess of zinc, but in deficiencies as well. In a variety of eukaryotic species, different sequence-specific DNA-binding factors have been found that are necessary for the variations in gene expression. Studying these zinc-regulated factors can provide a means to obtain certain types of proteins and different mechanisms that can be used to detect cellular zinc limitations in other organisms not known.[1]
Found in budding yeast Saccharomyces cerevisiae, the transcription factor Zap1, a zinc-responsive protein, can detect zinc levels. When zinc deficiency occurs, Zap1 can initiate the expression of about 80 genes, targeting genes that are require for zinc uptake and genes that can help survival during extreme zinc deficiencies.
Zap1 can detect cellular zinc levels by a variety of domains. Zinc finger pair is a regulatory domain that overlaps with AD2, or activation domain 2. During zinc deficiencies, zinc binds to the zinc fingers within the AD2, which allows AD2 to undergo a conformational change causing exposures of residue that initiates the domain function. The zinc can rapidly switch from one of the zinc finger pair to the other, which allows the possibility of Zap1 to detect cellular zinc level.
AD1, another activation domain within Zap1, is independently controlled by cellular zinc levels-activating gene expression in response to deficiencies. When AD1 is combined with AD2, it allows Zap1 to recruit more coactivators under precise stress situations. [1]
In another type of organism, more than one transcription factor are required for regulating genes during zinc deficiencies. Found in Arabidopsis thaliana, bZip19 and bZip23 activates the expression of genes required for zinc uptake.[1]
Changes in gene expression do not only occur in excess of iron, they also occur in response to iron limitations as well and have been observed in many different eukaryotes such as green algae, fungi and plants. Most of the information that is known about gene transcription that has been affected by iron limitation is by the studying of yeasts. [1]
Found in Saccharomyces cerevisiae, Aft1 and Aft2, are transcription factors that responds to iron deficiencies by increasing the expression of about 40 genes. These genes can encode for certain proteins that require iron uptake, iron searching, or intracellular iron transport. Aft1 and Aft2 also regulate the expression of CTH1 and CTH2, genes which are iron-dependent, which help cells to conserve iron. Aft1 and Aft2 senses cellular iron deficiency by using a signal produced by the mitochondrial Fe-S cluster machinery. This signal however is still presently unknown, but studies revealed that Aft1 has the ability to sense these iron signals due to certain cytoplasmic proteins. [1]
Found in fission yeast Schizosaccharomyces pombe, Php4, is another iron-responsive transcription factor that helps cells fight against iron deficiency. Similar to CTH1 and CTH2, Php4 controls the flux of iron through iron-depending pathways when iron is present. This is due to Php4 regulating genes that encodes for proteins that bind to iron or those found in metabolic pathways that needs iron. Transcription factor Grx4 is necessary to deactivate Php4 in high levels of iron concentration.[1]
In many different eukaryotes such as yeast, plants, green algae, and fruit flies, cells can activate certain genes that encodes copper uptake when deficiency of copper occurs. Many of the transcription factors required for gene activation during copper limitation have been identified; however, how they function to detect deficiency have not been identified yet. Although only a few is shown here, there are often homologs of these transcription factors that provides the same role in a variety of other eukaryotes. Thus, there are many different proteins that have evolved using the equivalent copper responsive domain to detect multiple levels of coppers needed for homeostasis. [1]
Mac1 is a transcription factor found in the yeast Saccharomyces cerevisiae, becoming active when copper deficiency occurs and regulating the expression of genes required for copper uptake. In order for Mac1 to become activated, it requires both the Mac1 DNA-binding and transactivation domains. The transactivation domain comprise of cysteine-rich domains that are able to bind four copper ions. In copper excess, over binding of copper to this domain disables the Mac1 transcription factor. Mac1 DNA binding activity can become maximized with addition of Sod1, a rich copper-binding protein, during copper cellular deficiency. Thus Sod1 might have some unknown ability in copper sensing.[1]
In another species, Chlamydomonas reinhardtii, the green algae uses a different transcription factor that regulates copper limitation called Crr1. When copper levels are low, Crr1 can activate the expression of over 60 genes to achieve needed copper levels. Like Mac1, Crr1 also contains copper regulated domains, one is a cysteine-rich metal-responsive domain, and the other is SBP DNA binding domain. [1]
In another species, A. thaliana, copper levels are balanced by transcription factor SPL7, which is a homolog or descendant of the same ancestor, of Crr1. In order for copper homeostasis to become obtained, SPL7 expresses genes that are required for copper uptake and intracellular copper organization during copper deficiency. Different miRNAs can become activated by SPL7 that allows targeting of mRNAs that encodes for copper-binding proteins.[1]
In many multicellular organisms, there are distinct systems that detect zinc excess and deficiencies. MTF1 is a transcription factor that provides protection against zinc excess, which is very toxic to cells.[1]
MTF1 is a transcription factor found in mammals and fishes that helps protects cells from excess of zinc, activated when high concentration is present. It binds to metal-response elements which activates the target gene expression. Similar to Zap1, MTF1 senses cellular zinc excess by using regulatory zinc-finger domains. In contrast, the zinc fingers controls the binding of MTF1- only when zinc are in excess.[1]
Studies obtained in a range of eukaryotes such as yeast, nematodes, flies and mammals, have noticed a variety of changes in gene expression due to excess of iron. Only in fungi however, have researchers identified transcription factors that are able to detect high levels of iron. Iron homeostasis is usually controlled at a post-transcription level within multicellular organisms.[1]
Found in Saccharomyces cerevisiae, transcription factor Yap5 protects cells from toxicity by expressing CCC1 activation when iron excess is present. Once Yap5 binds to the CCC1 prometer, CCC1 provides protection by transporting iron into an iron storage vacuole. CCC1 isn’t active unless high iron levels have been reached. Yap5 also comprises of cysteine-rich domains that are required for regulation of iron.[1]
This transcription factor is expressed when excess levels of iron have been reached. Found in S. pombe, Fep1 protects cells by stopping the expression of genes that acquires for iron when iron are at high concentration. Fep1 also regulates the expression of transcription factor Php4, controlling the flow of iron through metabolic pathways. Transcription factor Grx4 is required to deactivate Fep1 when iron levels are low, which is also required to deactivate Php4 when iron levels are high, which is likely the same signal that determine the switch of cells between both iron levels.[1]
In order to protect eukaryotes species such as fungi and flies from copper toxicity, certain transcriptional factors are able to detect this high level of copper, and express regulatory mechanisms that can help protect cells. These factors can directly bind to copper and gene expressions are regulated. [1]
Found in Saccharomyces cerevisiae, Ace1 is a transcription factor that can bind directly to copper ions when copper is in excess. The binding to copper allows the activation of genes that help defend cells from toxicity that arises due to surplus of copper. A variety of other fungi have homologs of Ace1 that can also provide the same function by protecting against excess of copper. [1]
dMTF-1, a homolog of MTF1, regulates the expression of genes that are required for copper levels to become balanced when copper levels are in excess or in deficiency. dMTF-1 also have a cysteine-rich domain that functions in sensing copper toxicity by binding to four Cu+ ions.[1]
In cell biology, pluripotency is defined as "the potential of a cell to develop into more than one type of mature cell, depending on environment"[1]. So if a cell is pluripotent, it has the potential to transform itself into a lung cell, heart cell, etc. Pluripotency describes a cellular state of specific cells within the early embryo [2].
Stem cells represent a category of unspecialized cells that possess the unique capability of developing and maturing into a variety of specific types of cells with various functions.[3] Through the maturation and cell division processes, stem cells can specialize, grow, and replace unhealthy cells within living organisms. After cell replication, these cells have the ability to either mature and specialize in function, or remain an unspecialized stem cell, which preserves the cell’s potential to specialize as it is needed in the body. Not only are these cells able to specialize in function, but they are also capable of joining together to replace damaged tissues. The two types of stem cells scientists are currently studying are: embryonic stem cells and adult stem cells. In 1981, scientists first discovered methods to derive embryonic stem cells from mouse embryos, and in 1998, scientists made further developments and developed methods to derive and harvest stem cells from human embryos. Since stem cells are capable of self- replication, they offer great potential for treatment of diseases such as diabetes and heart disease. The use of these cells for treatment of certain diseases is called reparative medicine, and is currently a topic of much debate in present society and culture. Stem cells also offer the potential for use in screening new drugs as well as the study of how and why certain birth defects develop. Stem cell therapy offers a great deal of insight into medicine as well as human growth and development, and research is currently underway in order to find methods to fully implement all the advancements these cells could provide to modern medicine.
Stem cells are characterized by their ability to differentiate into a diverse range of specialized cells and to grow indefinitely. They have two properties:
Self-renewal: The ability to undergo cell division indefinitely while maintaining their undifferentiated state. Stem cells are capable of doing so due to having active telomerase.
Potency: The ability to differentiate into specialized cell types in response to a signal. Skin cells, heart cells, and neurons all have the same genome, but are all different cells.[4] This is due to differentiation and different RNA/protein combinations.
Stem cells are often classified into one of two categories: embryonic stem cells, or adult stem cells.
Totipotency is defined as a cell's ability to transform into all cell types of the organism[5]. Both oocyte (the female germ cell involved in reproduction. This is an immature ovum) and zygote (this is the initial cell created after tow gamete cells are merged by means of sexual reproduction) do not possess the potential to be totipotent. However, the fertilized egg becomes equipotent blastomeres from its specialized cell type with a limited fate. Blastomeres are a type of cell created by cleavage of the zygote after fertilization [6].
What researchers have found through a series of transplantation experiments
1. Maternal proteins and RNAs are taken into the oocyte and deposited there
2. Those maternal proteins and RNAs have the capability to reset the epigentic state of somatic chromatin
3. They also serve to turn the oocyte into totipotent blastomeres after fertilization
4. So far, it has not been achieved to make totipotent cell lines in vitro.
5. This may be impossible due to the fact that in vivo, that is within living body, the transient nature of the totipotent is extremely different. The pre-embryonic stage of embryonic development offers blastomeres that are not actually self-renewin but they are generated by means of cleavage
Slowly, a restriction in development potency become apparent as cleavage of the early embryo approaches to the 16-cell stage. Then the cells undergo two different lineages, the trophoblast lineage and the inner cell mass. The question is how the cell differentiation begins. Differentiation begins as blastomeres flatten and as they strengthen cell-to-cell contact. This process is called compaction.
Embryonic stem cells are cells that are derived from embryos that have typically been harvested from eggs through in-vitro fertilization.[7] Embryonic stem cells are known as pluripotent, or having the ability to differentiate into every cell line within the organism. The embryos are 3–5 days old and consist of collections of hollow balls of cells that are called blastocysts.
In order to produce these blastocytsts, the embryonic stem cells must be harvested. The process of growing these stem cells is called cell culture, and is performed in the laboratory. The inner cell mass of human embryonic stem cells are placed onto a culture dish that has been coated with a medium that provides an adhesive surface to which the inner cell masses can stick. This medium is called a feeder layer, and is oftentimes composed of mouse skin cells. The human embryonic stem cells are then allowed to divide, spread, and attach to the medium of the culture dish in order to create a colony of embryonic stem cells. If the stem cell colony grows, divides and survives, the cells are plated onto new, clean culture dishes many times for the duration of many months in order to yield millions of embryonic stem cells, which is called a stem cell line. A sign that a stem cell line has developed successfully is if the cells have remained unspecialized for the duration of at least six months. If this is the case, the stem cell line can then be used for further testing and experimentation.
Throughout the harvesting process the embryonic stem cell colony must undergo a process called characterization, which involves a series of tests performed by scientists to see whether or not the cells have in fact developed correctly. One such test involves harvesting and examining the stem cell colony for many months in order to verify that the cells are healthy and capable of successful renewal. These cells are also analyzed to make sure they remain unspecialized. A way to test whether or not the cells have specialized is to test for the presence of transcription factors that are produced by unspecialized cells. Two examples of these transcription factors are Nanog and Oct4. The presence of these transcription factors indicate the cells have remained unspecialized and that they are capable of self replication and renewal.
Another test performed on embryonic stem cell cultures involves freezing, thawing and re-plating the cells to see whether or not they are capable of self renewal and re-growth.
An important test to examine whether or not the cells are functioning properly involves injecting a collection of embryonic stem cells into a mouse with a suppressed immune system followed by tests that indicate the formation of teratomas, which are benign tumors. If the injected stem cells successfully form a teratoma, scientists can be confident that the cells are capable of growth and differentiation, since teratomas contain various specialized cells.
Embryonic stem cells (ESC) are harvested from a blastocyst, which is an early stage in embryonic development(approximately four to five days in humans) that consists of 100-150 cells. ESC give rise to all three primary germ layers during development: ectoderm, endoderm, and mesoderm; which means they can develop into more than 200 specialized cell types. They can grow indefinitely under the right conditions due having active telomerase. Telomerase is inactive in adult cells, and as a result, cannot grow indefinitely.
Embryonic stem cells have proven to be usable in different situations:
Embryonic development: Understanding ESC aids scientists in further understanding embryonic development.
Drug testing: Certain cells cannot be cultured because they will die if removed from an organ or tissue. For example, in designing drugs for the heart, scientists cannot culture heart cells because once the heart cells are removed, they die. However, ESC can be cultured, and the ones that are differentiated into heart cells can be sustained in a culture to be used to test the drug.
Regenerative Medicine: This is not something ESC are currently being used for, but something that scientists are striving towards. The theory, or goal, is to be able to remove the nuclei in an embryonic stem cell, insert a patient's DNA into the cell, and have it differentiate according to the patient's needs. For example, Parkinson's disease is a degenerative disease of the central nervous system, and in theory, by adding back new neurons, it should be able to cure Parkinson's.
Paralysis: In addition, those who suffer from paralysis and unmovable body parts could regenerate body motor control through the study of stem cell research.
Leukemia: Patients suffering from Leukemia who may be in need of bone marrow transplant or cord blood transfusion can benefit from these stem cell researches. These products could be reproduced without having the need to constantly find a matchable donor. Although this may not treat the disease, it can diminish the symptoms greatly.
The purpose of harvesting embryonic stem cells is to create a colony of self replicating cells that have the potential to specialize into specific cells of interest that can be used to treat diseases.While they are being grown, the cells remain unspecialized but once they are allowed to clump together under certain specific conditions, they begin to form various specialized cells and tissues. The challenge scientists are currently facing is figuring out how to control the specialization to create specific cells of interest as opposed to spontaneous specialization that can result in the formation of various cells that carry unwanted specific functions. One such way to control specialization is by subjecting the colonies of embryonic stem cells to changes in chemical composition within the culture medium, changing the surface of the culture dish, or inserting specific genes into the culture to change the composition of the harvested cells. By transplanting the successfully specialized embryonic stem cells into humans, scientists can formulate treatments for certain diseases such as Parkinson’s Disease, diabetes, spinal cord injury, heart disease as well as vision and hearing loss.
Adipocytes are cells specialized for energy storage and so when the balance of energy intake versus expenditure is shifted so that more energy is taken in than is burned, the process of adipogenesis begins. This process involves adipocyte hyperplasia, the excessive recruitment of stem cells to become adipocytes, thereby increasing the number of adipocytes. In order to accomplish this, pluripotent mesenchymal stem cells (MSCs) become preadipocytes then are further developed into mature adipocytes.
The pluripotent MSCs located in the vascular stroma of adipose tissue and bone marrow first become preadipocytes, a process initiated by bone morphogenic proteins BMP4 or BMP2, then after several rounds of mitotic clonal expansion, they become adipocytes. While this process is activated by the BMP factors, other factors such as Hh provide inhibitory signals if the process becomes unnecessary[8].
For stem cells, transcription factors are important to their functioning and can be categorized into three central groups; 1. ones affecting development and its hierarchy (Developmental Core Module), 2. ones controlling cell growth and homeostasis (Cell Growth Module), and 3. ones that control signaling pathways of the other two groups (Signaling Module). These three groups mostly function independently, though there is some interaction present.
The best understood of transcription factors controlling cell pluripotency is Oct4 which is an octamer class protein that specifically recognizes the sequence ATGCAAAT.[9] Oct4 is unique apart from the other transcription factors in that it is crucial for epigenetic reprogramming (differentiation of cells during development). If it is inhibited or silenced in any way, ESCs will instantly stop self-renewing and differentiation into trophoblast-like cells. Oct4 will interact with many cofactors in the form of transcription factors. These cofactors tend to interact with DNA and include Sall4, Sp1, Hdac2, Nanog, Dax1, Nac1, Tcfp2l1, and Essrb. Their interactions do not form one protein nexus but multiple complexes all with different composition of proteins. This differing of composition and complexes may be the reason for different states of pluripotency.
Sox2 is another transcription factor involved in development and cell pluripotency. It has an expression pattern that is, to some extent, similar to Oct4. However, when inhibited or silenced, this will mainly affect the later stages of embryogenesis. Sox2 silencing will only affect a small portion its target genes and will lead to trophoblast-like cells. This is mainly because Sox2 loss can affect the amount of Oct4 levels in the cells which is the main reason for the creation of the trophoblast-like cells.
Nanog is also a member in the core developmental module and is a homeodomain-containing DNA-binding factor that is not associated with the normal ones. Nanog expression alone is able to help keep cell pluripotency in extreme conditions where factors are missing. It has been shown in studies to have a role of starting cell pluripotency rather than maintaining it. Nanog has cofactor proteins it interacts with which includes Smad1, Small3, Nr0b1, Nac1, Essrb, Zfp281, Hdac2, and Sp1.
This is the gene network that controls and maintains the growth and proliferation of pluripotent stem cells.
The proto-oncogene c-Myc is one member of this group is associated with activating transcription and opening chromatin. Its expression allows ESCs to self-renew even in extreme conditions. If it is inhibited ESCs will shut off their self-renewing mechanism. c-Myc also controls transcription of genes from multiple unconnecting functions such as cellular metabolism, control of cell cycle, and manufacture of protein. It has an inhibition factor also and will prevent the expression of Gata6 which is a prodifferentiation factor.[9] It also has been found to overlap with functions from genes of the developmental module such as Tip60 and p400. It has also seen to function in some cases of cancer growth.
c-Myc expression is regulated by the transcription factor Ronin (Thap11) which is part of a family of proteins containing an N-terminal THAP domain. It is well conserved between mice and humans which means its sequence and target sequence is very similar for both mammals. Ronin inhibition will led to cell apoptosis while overexpression will lead to an abundance of mESCs.
There are several signaling pathways that will directly promote self-renewal and/or preserve pluripotency by blocking or signaling normal differentiation prompts.
Lif (Leukemia inhibitory factor) stimulates the self-renewal of mESCs. It does not directly increase the growth rate of mESCs but rather increases the probability that the cells with self-renew rather than differentiate. The Lif-signaling effect is controlled by a heterodimeric complex that is made of a Lif receptor (LifR) chain and the protein gp130. It has been shown to signal three pathways which are the Jak/Stat3, the PI3K-Akt, and Mapk pathways.[9] Of the three, only the Jak/Stat3 pathway completely depends on Lif for its signal. The other two pathways require multiple signals from multiple sources in other to start the process. Signaling of the Jak/Stat3 pathway and PI3K pathways will promote expression of Klf4 and Tcf3 which lead to the creation of greater transcription of Sox2 and Nanog which as stated earlier are involved in differentiation of stem cells and development of the body. For the Mapk pathway, its signaling will lead to less amounts of Tbx3 which will cause a decrease of transcriptional activity.
FgfR1 (Fibroblast growth factor signaling) is the most ample receptor of ESCs in the body. It is involved in signaling the Jak/Stat, phophoinositide phospholipase C-y, PI3K, and Erk pathways. A similar protein, Fgf2 is important for the maintenance of hESC. For example, in mouse feeder cell cultures a hot amount of Fgf2 is required to maintain its self-renewal and prevent differentiation.
Wnt signaling is very important during embryonic development and controls differentiation of stem cells. Its can induce expression of mesodermal and endodermal markers that, for mESCs, includes Barchyury, Flk1, Foxa2, Lxh1, and Afp. Wnt signaling also maintains cell stem pluripotency by controlling the expression of Oct4, Sox2, and Nanog which are all transcription complexes involved in maintaining pluripotency and development.[9]
It should be noted that every cell has a unique chromatin structure. Chromatin is the combination of DNA and proteins that make up the contents of the nucleus of a cell[10]. In pluripotent cells, heterochromatic-associated foci seems to be more spaciously distributed. The associated histones are usually hyperacetylated. There exists chromatin-modifying enzymes that play a role with the transcriptional modules. They do so by controlling genes that encode key enzymes capable of covalently modifying proteins[11]. For instance, the transcription of the H3K9 methyltransferase SetDB1 is controlled by Oct4. It is known that histone labels characteristic of this enzyme. When this happens, the following things occur;
1. Polycomb group proteins are introduced
2. Polycomb group proteins trigger subsequent events which involve methylation of H3K27 residues
3. Then further additional chromatin-associated modifications are enabled.
The largest potential source of blastocysts for stem cell research is from in vitro fertilization (IVF) clinics. The process of IVF requires the retrieval of a woman's eggs via a surgical procedure after undergoing an intensive regimen of "fertility drugs," which stimulate her ovaries to produce multiple mature eggs.
When IVF is used for reproductive purposes, doctors typically fertilize all of the donated eggs in order to maximize their chance of producing a viable blastocyst that can be implanted in the womb. Because not all the fertilized eggs are implanted, this has resulted in a large bank of "excess" blastocysts that are currently stored in freezers around the country. The blastocysts stored in IVF clinics could prove to be a major source of embryonic stem cells for use in medical research. However, because most of these blastocysts were created before the advent of stem cell research, most donors were not asked for their permission to use these left-over blastocysts for research.
The IVF technique could potentially also be used to produce blastocysts specifically for research purposes. This would facilitate the isolation of stem cells with specific genetic traits necessary for the study of particular diseases. For example, it may be possible to study the origins of an inherited disease like cystic fibrosis using stem cells made from egg and sperm donors who have this disease. The creation of stem cells specifically for research using IVF is, however, ethically problematic for some people because it involves intentionally creating a blastocyst that will never develop into a human being.[12]
The process called nuclear transfer offers another potential way to produce embryonic stem cells. In animals, nuclear transfer has been accomplished by inserting the nucleus of an already differentiated adult cell-for example, a skin cell-into a donated egg that has had its nucleus removed. This egg, which now contains the genetic material of the skin cell, is then stimulated to form a blastocyst from which embryonic stem cells can be derived. The stem cells that are created in this way are therefore copies or "clones" of the original adult cell because their nuclear DNA matches that of the adult cell.As of the summer of 2006, nuclear transfer has not been successful in the production of human embryonic stem cells, but progress in animal research suggests that scientists may be able to use this technique to develop human stem cells in the future.
Scientists believe that if they are able to use nuclear transfer to derive human stem cells, it could allow them to study the development and progression of specific diseases by creating stem cells containing the genes responsible for certain disorders. In the future, scientists may also be able to create "personalized" stem cells that contain only the DNA of a specific patient. The embryonic stem cells created by nuclear transfer would be genetically matched to a person needing a transplant, making it far less likely that the patient's body would reject the new cells than it would be with traditional tissue transplant procedures.
Although using nuclear transfer to produce stem cells is not the same as reproductive cloning, some are concerned about the potential misapplication of the technique for reproductive cloning purposes. Other ethical considerations include egg donation, which requires informed consent, and the possible destruction of blastocysts.[12]
Adult stem cells are known as mesenchymal stem cells. Just like embryonic stem cells, adult stem cells are unspecialized cells capable of renewal and specialization, but unlike embryonic stem cells, adult stem cells are typically found in tissues surrounded by other specialized cells. This makes the stem cell only multipotent; it can only differentiate into certain cell lines as opposed to pluripotent embryonic stem cells which can differentiate into almost any cell line. The role of adult stem cells is to replenish damaged cells within tissues and organs when needed. Adult blood-forming stem cells from bone marrow have been used for transplants, and current research is underway to determine whether or not differentiation of stem cells found in the brain and heart can be controlled and used for various other forms of transplantation.
Adult stem cells are found in areas called “stem cell niches” in various organs and tissues. These cells remain dormant until there is a need for them, which could include a requirement for new cells to replace damaged tissue due to injury or disease. Like embryonic stem cells, scientists are developing methods to extract these adult stem cells, harvest them in colonies, and manipulate them to create colonies of specialized cells that can be used for treatments of certain diseases. The challenge arises because once extracted from the human body, these cells have limited capacities to grow and reproduce.
A way scientists identify adult stem cells is first by extracting cells from a living organism. Next, the cells are labeled in a cell culture and transplanted back into a different organism. If the cells reproduce in the organism, scientists can verify the presence of properly functioning stem cells. Scientists have been conducting research to identify various differentiation pathways, which are the processes by which adult stem cells differentiate and convert into highly specialized cells with specific functions.
Different types of adult stem cells include the following:
Hematopoietic stem cells: develop into bone cells, cartilage cells, fat cells
Neural stem cells: develop into neurons, astrocytes and oligodendrocytes
Skin stem cells: develop into epidermal stem cells and hair follicles
Adult stem cells are found in developed organisms and have the ability to divide and create another cell like itself, or create a cell more differentiated than itself.
They have a few differences from embryonic stem cells, which include:
They cannot grow indefinitely due to having inactive telomerase. Some can actually only double once in their cellular life span.
Adult stem cells have a limited ability do differentiate, unlike ESC.
They exist in much smaller quantities than ESC do. There exists about 1 adult stem cell for every 10,000-15,000 cells in an adult human.
There also exists less ethical issues regarding adult stem cells because they are extracted from adults who are able to make their own decisions and give consent.
Potential exists for adult stem cells and regenerative properties, such as:
Healing organs: In the event of a heart attack, heart cells die and are replaced by scar tissue. Adult stem cells have been used to partially heal the scar tissue in animals by removing the damaged heart tissue, culturing adult stem cells in a petri dish, and putting them back in the damaged heart.
Organ regeneration: In 1999, Dr. Anthony Atala, Urologist and Director of the Wake Forest Institute for Regenerative Medicine, was studying patients with a bladder defect. He took a small piece of bladder, removed the stem cells, and actually grew it into a new bladder. He discovered that he could stick them back in the original patient, and seven years later, the patients were either better, or like normal. The bladder he was able to create was functional enough to keep the patients healthy and alive.
CD34+ cells contain the protein CD34. CD34 protein is a glycoprotein found on human cell surfaces and functions as an adhesive for cells to interact. It could also have a function in attaching stem cells to bone marrow or connective tissue cells. The protein is expressed early in hematopoietic cells. Another function is that it is needed for T cells to enter lymph nodes. CD34+ cells are commonly found in as hematopoietic stem cells in umbilical cords and bone marrow.
To isolate CD34+ cells from blood samples, immunomagnetic or immunofluorescent techniques are used. These hematopoietic stem cells can be purified for research or clinical use in bone marrow transplant, by using antibodies. These cells can differentiate into different blood cells. CD34+ cells can be injected into patients to treat diseases such as Liver Cirrhosis, Peripheral Vascular disease, and spinal cord injury.
Induced pluripotent stem cells or iPSCs are adult somatic cells that have been reprogrammed to a state similar to embryonic stem cells. Although iPSCs are theoretically pluripotent cells, it remains unknown if they differ at all from embryonic stem cells. Induced pluripotent stem cells were first produced in 2006 from mouse cells and human cells followed in 2007.
iPSCs are produced through a transfection which is process of introducing nucleic acids into a cell.[13] In the case of iPSCs, this done through retroviruses. Other methods do exist in the production of iPSCs such as the use of adenoviruses and plasmids.
Adenoviruses differ from retroviruses because it does not incorporate its own genetic information into the adult cell. Adenoviruses are used to transport the necessary transcription factors into the DNA of the adult cells. Adenoviruses present some benefits such as the fact that they avoid any potential for mutagenesis that may stem from the insertion as well as the fact that adenoviruses only need to be present for a small period of time to reprogram the adult cell.
Plasmids can also be used to reprogram the adult cell and in this method, two plasmids are required to carry the necessary transcription factors. The main benefit of using plasmids is the simple fact that viruses do not have to used. Two main problems exist for the use of plasmids though and they are the fact that plasmids demonstrate low efficiency as well as the fact that plasmids have a continued risk of insertional mutagenesis and use cancer-promoting genes during reprogramming.
IPSCs have been created successfully by fusing EGCs (embryotic germ cells) or ESCs (embryotic stem cells) with somatic cells.[9] Somatic cell nuclei can be reprogrammed by EGCs and mESCs after they are fused with thymic lymphocytes. After reprogramming, the procedure leads to tetraploid cells that resemble EGCs where the female cells have their inactive X chromosomes became active again. These tetraploids can then be injected into blastocyst to form chimeric embryos. Specifically for humans, this can, in the future, be used for both bone marrow cells and brains cells in the central nervous system that have been isolated. These cells can then be fused with mESCs in culture to create artificial cells that are similar to real ones in morphology and characteristics.
iPSCs can be also be obtained through reprogramming somatic cells (body cells of an organism) into pluripotent stem cells.[9] This procedure is performed by the use of transcription factors which are proteins that bind to specific DNA sequences. The four transcription factors that are used in mouse fibroblasts via retroviral infection are Oct4, Sox2, Klf4, and c-Myc. For humans, different combinations of transcription factors have been used which includes Nanog, Lin28, or Nr5a2. The exogenous expression of the transcription factors is followed by selection in a mESC (mouse embryotic stem cell) agent to finally lead to the iPSCs. The resulting iPSCs have characteristics of normal ESCs (embryotic stem cells) such as reactivation of both X chromosomes in female cells, expression of endogenous pluripotency indicators, and the ability to create chimeric animals. The molecular events are still poorly understood, but how reprogramming occurs can be investigated. During reprogramming, epigenetic marks are expansively altered. Studies have shown that iPSCs clones to be impossible to tell apart from ESCs though there are still small differences that need to be tested before they can be used in regenerative medicine.[9]
Induced pluripotent stem cells promise the almost infinite possibilities of embryonic stem cells without the nagging moral implications of the destruction of human embryos, and for this reason offers a promising place for research for regenerative medicine. iPSCs also hold the promise of other benefits over other types of stem cells. The main one being the fact that iPSCs can be developed from a patients own cells which mean that any treatments derived from the iPSCs should avoid any immunogenic responses. Even with the enormous possibilities of iPSCs in a clinical setting, a major concern hovers over the future which is the fact that iPSCs have an increased tendency to form tumors. From experiments, it was discovered that iPSCs injected into mice formed teratomas which are tumors with tissue or organ components inside. Moving toward the future, research is focused on not only coming up with potential treatments using iPSCs but on producing methods with higher efficiency as well as decreased occurrences of tumors using recombinant proteins.
Both embryonic and adult stem cells have the potential to provide both treatments for various diseases as well as insight into human growth and development. One specific area of stem cell therapy is the use of these cells to treat people with type I diabetes. Type I diabetics are unable to produce insulin in the pancreas. Current research is underway to determine whether or not embryonic stem cells can be selectively specialized to give rise to insulin producing colonies that can be transplanted into diabetic patients. In order for such a procedure to be successful, stem cells must be capable of reproducing and generating enough of the tissue/cells of interest. They must also be able to survive in the recipient host, function properly, and maintain the capability to reproduce, while leaving the recipient unharmed. All of these requirements must be met without the recipient rejecting the cells, which is a challenge researchers are presently trying to overcome.
Human stem cells could also be used to test new drugs. New medications could be tested for safety on differentiated cells generated from human pluripotent cell lines. Cancer cell lines for example are used to screen potential anti-tumor drugs. Using stem cells scientists are also developing new ways to grow brain cells in the laboratory that could be used to treat patients with Parkinsons disease. Among the various approaches one is to grow stem cells into brain cells and transplant them into patients. If stem cells can be cultivated to become dopamine-producing nerve cells, researchers believe that they could replace the lost cells.
By using stem cells one could observe the specailization of specific cells such as nerve and muscle cells. By observing this process some of the most serious medical conditions, such as cancer and birth defects, are due to problems that occur somewhere in this process of specialization. Another potential application of stem cells is making cells and tissues for medical therapies.
Cancer cells have the ability to divide indefinitely and spread to different parts of the body during metastasis. Embryonic stem cells can self-renew and, through differentiation to somatic cells, provide the building blocks of the human body. Embryonic stem cells offer tremendous opportunities for regenerative medicine and serve as an excellent model system to study early human development.
There is much ethical controversy regarding embryonic stem cell research. The number one being how ESC are harvested because once ESC are retrieved, the blastocyst dies. There is no way to harvest them without destroying the embryo. This brings to the table the question of when does life begin? There are many different beliefs as to when life begins, whether it be scientific, religious, etc., and so it is difficult to set a definition that everyone can agree upon.
Most of the blastocysts that are used for stem cell research come over from excess invitro fertilization. People who have fertility issues and decide to have invitro fertilization. This is where sperm and an egg are combined in a test tube, which results with a number of embryos, some of which are implanted in the mother. The left over embryos are then frozen for a variety of reasons; whether the first implantation doesn't take, or the couple decides to have a second child later on. Some decide to discard the embryos, but a vast majority are left frozen in labs. Another controversial question that arises is that whether it is alright to use the frozen embryos for ESC.
Political controversy in research arises as well:
late '90s: The first embryonic stem cell line was created.
2001: Bush outlaws the creation and studying of more embryonic stem cell lines with the use of federal money. There were 21 distinct ESC lines at the time.
2009: Obama creates the National Institute of Health (NIH) panel that will approve whether or not federal funding can be used to study certain stem cell lines. Established guidelines state that the stem cells must come from embryos that were created through invitro fertilization but were frozen and not going to be used anymore. Parents must provide consent for their frozen embryos to be used for stem cell research, and once approved by the NIH federal money can be used to study them. However, federal money cannot be used to create new stem cell lines. They must be created through private funding, and once they are created, federal funding can be used to study it.
An issue of the overall safety of the use of stem cells exists as well. Researchers do not necessarily know what will happen when stem cells are inserted into a person, they know what they WANT to happen, but cannot guarantee what will. Scientists have yet to be able to precisely control what cells an embryonic stem cell will differentiate into.
↑Takahasi, Kazutoshi, and Shinya Yamanaka. "Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors." Cell 126 (2006): 663-76. Invalid <ref> tag; name "tak" defined multiple times with different content
↑“Pluripotency and Nuclear Reprogramming” From Annual Review of Biochemistry Vol. 81 737-765, by Marion Dejosez and Thomas P. Zwaka
↑^ Blastomere Encyclopædia Britannica. Encyclopædia Britannica Online. Encyclopædia Britannica Inc., 2012. Web. 06 Feb. 2012.
↑Oliver Dreesen and Ali Brivanlou.Stem Cell Reviews and Reports,Humana Press Inc.
↑ abcdefgDejosez, Marion, and Thomas P. Zwaka. "Pluripotency and Nuclear Reprogramming." Annual Review of Biochemistry 81 (2012): 737-65. Annual Reviews. Web. 6 Dec. 2012.
↑Bellin, Milena, Maria Marchetto, Fred Gage, and Christine Mummery. "Induced Pluripotent Stem Cells: The New Patient?" Nature Reviews Molecular Cell Biology 13 (2012): 713-26.
There is only one type of cell that is completely generic—its gene expression is tuned so broadly that it has unlimited career potential to become any kind of cell in the body. These undifferentiated cells exist a few days after conception and form the blastula, consisting of three layers, the ectoderm, endoderm, and mesoderm and about 100 cells. Embyonic stem cells have an almost unlimited capacity to divide due to high levels of telomerase that prevents telomare degradation in aging cells. Shortly after, a wave of growth hormones such as Testosterone and Sonic Hedgehog, induce the blastocyst to change from a spherical structure and begin to take on the rough morphology that the animal has.
Due to their almost unlimited capacity to divide and their potential to divide into any kind of cell in the body, embryonic stem cells are being looked at as therapies for many different types of neurodegenerative therapies such Parkinson's, Alzheimer's, and Huntingtons in which the neurons in an aging brain or body die and are not replaced. by attempting to induce stem cells to differentiate into these types of neurons and repopulate the affected regions; the progress of the disease can be halted or even reversed.
Embronic stem cells are also being considered for organ transplantations. If Embryonic stem cells are saved at birth from the placenta and umbilical cord. These cells can theoretically then be used to grow organs with the same blood type and identifying proteins in them, eliminating the need to wait for an organ donor which reduced the organ shortfall already present, and ensuring that the organ is a perfect match with the recipient's body, eliminating the chance of rejection and the need for anti rejection drugs.
Because embryonic stem cells are so few, numbering only a hundred or so per embryo, they are very expensive and difficult to harvest and maintain. This process inevitably destroys the developing embryo leading to charged political debate versus the cost of sacrificing a potential life versus the enormous benefits these types of cells could bring.[1]
Although researchers have been studying stem cells from mouse embryos for more than 20 years, only recently have they been able to isolate stem cells from human embryos and grow them in a laboratory. In 1998, James A. Thomson of the University of Wisconsin, Madison, became the first scientist to do this. He is now at the forefront of stem cell research, searching for answers to the most basic questions about what makes these remarkable cells so versatile. Although scientists envision many possible future uses of stem cells for treating Parkinson’s disease, heart disease, and many other disorders affected by damaged or dying cells, Thomson predicts that the earliest fruits of stem cell research will be the development of powerful model systems for finding and testing new medicines, as well as for unlocking the deepest secrets of what keeps us healthy and makes us sick.[1]
Long after our embryonic stem cells have differentiated, we all still harbor other types of multitalented cells, called adult stem cells. These cells are found throughout the body, including in bone marrow, brain, muscle, skin, and liver. They are a source of new cells that replace tissue damaged by disease, injury, or age.
Researchers believe that adult stem cells lie dormant and largely undifferentiated until the body sends signals that they are needed. Then selected cells morph into just the type of cells required. Like embryonic stem cells, adult stem cells have the capacity to make identical copies of themselves, a property known as self-renewal. But they differ from embryonic stem cells in a few important ways. For one, adult stem cells are quite rare. In addition, adult stem cells appear to be slightly more “educated” than their embryonic predecessors, and as such, they do not appear to be quite as flexible in their fate.
However, adult stem cells already play a key role in therapies for certain cancers of the blood, such as lymphoma and leukemia. Doctors can isolate from a patient’s blood the stem cells that will mature into immune cells and can grow these to maturity in a laboratory. After the patient undergoes high-dose chemotherapy, doctors can transplant the new infection-fighting white blood cells back into the patient, helping to replace those wiped out by the treatment. Although researchers have been studying stem cells from mouse embryos for more than 20 years, only recently have they been able to isolate stem cells from human embryos and grow them in a laboratory. In 1998, James A. Thomson of the University of Wisconsin, Madison, became the first scientist to do this. He is now at the forefront of stem cell research, searching for answers to the most basic questions about what makes these remarkable cells so versatile. Although scientists envision many possible future uses of stem cells for treating Parkinson’s disease, heart disease, and many other disorders affected by damaged or dying cells, Thomson predicts that the earliest fruits of stem cell research will be the development of powerful model systems for finding and testing new medicines, as well as for unlocking the deepest secrets of what keeps us healthy and makes us sick.
Cells manage a wide range of functions in their tiny package — growing, moving, housekeeping, and so on — and most of those functions require energy. Cellular nutrients come in many forms, including sugars and fats. In order to provide a cell with energy, these molecules have to pass across the cell membrane, which functions as a barrier — but not an impassable one. Like the exterior walls of a house, the plasma membrane is semi-permeable. In much the same way that doors and windows allow necessities to enter the house, various proteins that span the cell membrane permit specific molecules into the cell, although they may require some energy input to accomplish this task.
Complex organic food molecules such as sugars, fats, and proteins are rich sources of energy for cells because much of the energy used to form these molecules is literally stored within the chemical bonds that hold them together. Scientists can measure the amount of energy stored in foods using a device called a bomb calorimeter. With this technique, food is placed inside the calorimeter and heated until it burns. The excess heat released by the reaction is directly proportional to the amount of energy contained in the food.
In reality, of course, cells don't work quite like calorimeters. Rather than burning all their energy in one large reaction, cells release the energy stored in their food molecules through a series of oxidation reactions. Oxidation describes a type of chemical reaction in which electrons are transferred from one molecule to another, changing the composition and energy content of both the donor and acceptor molecules. Food molecules act as electron donors. During each oxidation reaction involved in food breakdown, the product of the reaction has a lower energy content than the donor molecule that preceded it in the pathway. At the same time, electron acceptor molecules capture some of the energy lost from the food molecule during each oxidation reaction and store it for later use. Eventually, when the carbon atoms from a complex organic food molecule are fully oxidized at the end of the reaction chain, they are released as waste in the form of carbon dioxide.
Cells do not use the energy from oxidation reactions as soon as it is released. Instead, they convert it into small, energy-rich molecules such as ATP and nicotinamide adenine dinucleotide (NADH), which can be used throughout the cell to power metabolism and construct new cellular components. In addition, workhorse proteins called enzymes use this chemical energy to catalyze, or accelerate, chemical reactions within the cell that would otherwise proceed very slowly. Enzymes do not force a reaction to proceed if it wouldn't do so without the catalyst; rather, they simply lower the energy barrier required for the reaction to begin.
The living systems are highly ordered and utilize energy. This energy is not created by the living system. It is instead, obtain from the environment, and then processed into usable forms.
Metabolism is a series of chemical reactions beginning with a particular molecule and converting it into another molecule or molecules. It has many defined pathways in the cells which are interdependent and their activity is coordinated very sensitively by means of communication in which allosteric enzymes are predominant.[1]
The overview of the process is that through photosynthesis, carbon dioxide and water, with the help of light, is converted into organic molecules, or food in our sense. Through cellular respiration, the organic molecules are converted back into carbon dioxide and water. Metabolism is the total chemical reaction occuring in the body, and it involves molecular interaction. It is highly regulated and yields a change in energy content of the reactants. The metabolic pathways is a series of reaction controlled by multiple enzymes.
Organisms transform energy, and energy is the capacity to do work. There are kinetic energies and potential energy. Kinetic energy is the energy of motion, it can be used to perform work. Examples of kinetic energy are light, heat, and electricity. Potential energy is stored energy. An example is electrochemical gradients. Light can be transformed into chemical bonds. Chemical bonds can be transformed to be used for mechanical work. Energy transformation must follow the two thermodynamic laws. The first law -conservation of energy - is that energy can neither be created or destroyed; it can only change forms. The Universe has a constant form of energy. The second law of that energy transformation yields an increase in the entropy of the universe. Entropy is a measure of disorder. But this is referring to the a closed system, one in which matter is isolated from the surrounding. So as long as the entropy of the system and surrounding increases, entropy of the system itself may decrease.
ATP is an activated carrier of phosphoryl groups because phosphoryl transfer from ATP is an exergonic process. The use of activated carriers is a motif in biochemistry and most function as coenzymes:
1. Activated Carriers of Electrons for Fuel Oxidation. In aerobic organisms electrons are not transferred directly to O2 despite being the ultimate electron acceptor. Rather, fuel molecules transfer electrons to special carriers which are either pyridine nucleotides or flavens. The reduced form of these carriers then transfer their high-potential electrons to O2.
2. Activated Carrier of Electrons for Reductive Biosynthesis. High potential electrons are required in most biosyntheses because the precursors are more oxidized then the products. Hence, reducing power is needed in addition to ATP.
3. An Activated Carrier of Two-Carbon Fragments. Coenzyme A, another central molecule in metabolism is a carrier of acyl groups. Acyl groups are important constituents in both catabolism, and anabolism. The terminal sulfhydryl group of CoA is the reactive sites. Acyl groups are linked to CoA by thioester bonds resulting derivative called acyl CoA. The hydrolysis of thioester is thermodynamically favorable that that of an oxygen ester because the electrons of C=O cannot form resonance structures with C - bonds, making acetyl CoA have a high acetyl-group transfer potential because transfer of acyl group is exergonic. Acetyl CoA carries an activated acetyl group, just as ATP carries an activated phosphoryl group.
Use of activated carriers illustrates that Kinetic stability of these molecules in the absence of specific catalysts is essential for their biological function because it enables enzymes to control the flow of free energy and reducing powers. Secondly most intercharges of activated groups in metabolisms are accomplished by rather small set of carriers such as ATP, NADH and NADPH.
[2]
Almost all activated carriers that act as coenzymes are derived from vitamins. Vitamins are organic molecules that are needed in small amounts in the diets of some animals. They play the same roles in nearly all forms of life, but higher animals have lost the capacity to synthesize the in the course of evolutions.[3]
[14] Vitamin B6,coenzyme pyridoxal phosphate. Typical reaction type is group transfer to or from amino acids. Deficiency can lead to depression, confusion and convulsion.
Metabolic reactions must be to rigorously related but at the same time must be flexible to adjust metabolic activity to constantly changing external environment cells. Metabolism are regulated through:
1. Controlling the Amounts of Enzyme. Amount of particular enzyme depends on both its rate of synthesis and its rate of degradation.
2. Controlling Catalytic Activity. Catalytic activity of enzymes are controlled in several ways:
(a.) reversible allosteric control
(b.) feedback inhbition
(c.) reversible covalent modification
(d.) hormones coordinate metabolic relations between different tissues
(e.) energy status of cell --> energy charge
3. Controlling the Accessibility of Substrate. Compartmentalization segregates opposed reactions and Controlling the flux of substrates.[5]
The current thinking is that RNA was an early biomolecule that in an early RNA world would have served as a catalyst and information-storage. Activated carries such as ATP, NADH, FADH2 and coenzyme A contain adenosine diphosphate units evolved from early RNA catalyst. Non-RNA units such as isoalloxazine ring may have been recruited to serve as efficient carriers of activated electrons and chemical units, which were functions not performed by RNA itself. Then when a more versatile proteins replaced RNA as the major catalysts, the ribonucleotide coenzymes stayed essentially unchanged because they were already well suited to their metabolic roles. With the advent of protein enzymes, these important cofactors evolved as free molecules without losing adenosine diphosphate vestiage of their RNA-world ancestry explaining why molecules and motifs of metabolism are common to all forms of life.[6]
Thermodynamically unfavorable reactions can be driven by a thermodynamically unfavorable reaction when it is coupled. This is because a pathway must satisfy two criteria: (1.) the individual reactions must be specific and (2.) the entire set of reactions that constitute the pathway must be thermodynamically favored. A reaction that is specific will yield only one particular product or set of products from its reactants due to enzyme specificity.
Thermodynamics of metabolism is most readily approached in relation to free energy, which states that a reaction can occur spontaneously only if is negative. This is due to the formation of products C and D from subtrates A and B given by the formula
Thus depends on the nature of the reactants and their concentrations, which leads to thermodynamically fact that overall free-energy change for a chemically coupled series of reactions is equal to the free-energy changes of the individual steps.[7]
↑Berg, Jeremy (2007). Biochemistry, 6th Edition. New York, New York: Sara Tenney. pp. 410–411. ISBN978-0-7167-8724-2. {{cite book}}: Unknown parameter |coauthors= ignored (|author= suggested) (help)
↑Berg, Jeremy (2007). Biochemistry, 6th Edition. New York, New York: Sara Tenney. pp. 420–422. ISBN978-0-7167-8724-2. {{cite book}}: Unknown parameter |coauthors= ignored (|author= suggested) (help)
↑Berg, Jeremy (2007). Biochemistry, 6th Edition. New York, New York: Sara Tenney. p. 423. ISBN978-0-7167-8724-2. {{cite book}}: Unknown parameter |coauthors= ignored (|author= suggested) (help)
↑Berg, Jeremy (2007). Biochemistry, 6th Edition. New York, New York: Sara Tenney. pp. 425–427. ISBN978-0-7167-8724-2. {{cite book}}: Unknown parameter |coauthors= ignored (|author= suggested) (help)
↑Berg, Jeremy (2007). Biochemistry, 6th Edition. New York, New York: Sara Tenney. pp. 428–429. ISBN978-0-7167-8724-2. {{cite book}}: Unknown parameter |coauthors= ignored (|author= suggested) (help)
↑Berg, Jeremy (2007). Biochemistry, 6th Edition. New York, New York: Sara Tenney. p. 429. ISBN978-0-7167-8724-2. {{cite book}}: Unknown parameter |coauthors= ignored (|author= suggested) (help)
↑Berg, Jeremy (2007). Biochemistry, 6th Edition. New York, New York: Sara Tenney. pp. 410–411. ISBN978-0-7167-8724-2. {{cite book}}: Unknown parameter |coauthors= ignored (|author= suggested) (help)
Adenosine triphosphate (ATP) is a nucleotide that consists of an adenine and a ribose linked to three sequential phosphoryl (PO32-) groups via a phosphoester bond and two phosphoanhydride bonds. ATP is the most abundant nucleotide in the cell and the primary cellular energy currency in all life forms. The primary biological importance of ATP rests in the large amount of free energy released during its hydrolysis. This provides energy for other cellular work, such as biosynthetic reactions, active transport, and cell movement. ATP is used in cellular metabolism in plants. It involved with light to create energy for plant. Besides, ATP is also one of components of DNA.
ATP also known as adenosine 5'-triphosphate. It is formed from adenosine diphosphate (ADP) and orthosphosphate (Pi). When fuel molecules are oxidized in chemotrophs or when light is trapped by phototrophs. This nucleotide is tremendously important since it is the most commonly used energy currency. The energy is released from the cleave of the triphosphate group is used to power many cellular processes.[1]
ATP is composed of an adenine ring, ribose sugar, and three phosphate groups (triphosphate). The groups of the phosphate group are usually called the alpha (α), beta (β), and gamma (γ) phosphates. It is typically related to a monomer of RNA called adenosine nucleotide. Gamma phosphate group is the primary phosphate group on the ATP molecules that is hydrolyzed when the energy is needed to drive anabolic reactions. Basically gamma phosphate is typically located the farthest from the ribose sugar and has a higher energy of hydrolysis than either that of the alpha and beta phosphate. The bonds that are formed after hydrolysis or the phosphorylation of a residue by ATP are lower in energy than that of the phosphoanhydride bonds of ATP.
ATP is very soluble in water and is a quite stable solution that has a pH of 6.8-7.4, but is rapidly hydrolysed at extreme pH. Thus, ATP is best stored as an anhydrous salt.
Although, ATP is quite stable in solution, it is an unstable molecule in unbuffered water. This is because, once ATP gets in contact with unbuffered water, it hydrolyses to ADP and phosphate due to the strength of the bonds between the phosphate groups in ATP is commonly seen to be less than the strength of the hydrogen bonds (hydration bonds) between its products (ADP + phosphate) and water. Therefore, if ATP and ADP are in chemical equilibrium in water, almost all the ATP will form into ADP because of the reaction that will occur. Gibbs free energy is when a system is far from equilibrium and it is able to do some kind of work. It is seen that typical living cells maintain the ratio of ATP and ADP at a point ten orders of magnitude from equilibrium. However, this may only occur if ADP is thousand fold lower in concentration than that of ATP. This shows that hydrolysis of ATP in cells usually release a large amount of free energy in reaction.
However, even with releasing a large amount of free energy during reaction, any unstable system of potentially reactive molecules could potentially serve as a way of storing free energy. This is only if the cells maintain their concentration far from the equilibrium point of the reaction. However, the idea of both energy-release and entropy-increase always occur during the breakdown of RNA, DNA, and ATP into simpler monomers.
In an ATP molecule, two high-energy phosphate bonds called phsophoanhydride bonds are responsible for high energy content of this molecule. Based on biochemical reaction, these anhydride bonds are often referred to as high-energy bonds. Also the released of hydrolysis of the anhydride bonds can happen in the energy stored ATP.
Rossmann fold is a type of protein fold that some proteins and ATP bind together as. This characteristic protein fold is a general nucleotide-binding structural domain that can also bind the coenzyme NAD. Kinase is the most common ATP-binding protein. They share a small number of common folds and there biggest kinase superfamily all share common structural features specialized for ATP binding and phosphate transfer.
ATP also requires the presence of a divalent cation that is almost as magnesium as a metal used. This metal binds to the ATP phosphate groups. This metal ion can also serve as a mechanism for kinase regulation. The presence of magnesium greatly decreases the dissociation constant of ATP from its protein binding partner without even affecting the ability of the enzyme to catalyze its reaction once the ATP has bound.
Intracellular ATP hydrolysis is catalyzed by intracellular ATPases. For example, the (Na+-K+)-ATPase located in the plasma membranes of higher eukaryotes drives active transport of Na+ and K+ coupled to ATP hydrolysis, and generates electrochemical gradients across the cell membrane. Another important intracellular ATPase is myosin. The myosin heads form the cross-bridges to thin filaments in intact myofibrils and its ATP-powered movement is responsible for muscle contraction.
ATP is also present in extracellular spaces in nanomolar to micromolar concentrations, which are 3-6 orders of magnitude lower than intracellular ATP concentration (1, 2). ATP is released from cells to extracellular spaces by regulated exocytosis or plasma membrane channels (Figure 1). Regulated exocytosis is an important process used to release substances such as hormones or neurotransmitters from the cell and is triggered by an increase in cytoplasmic Ca2+ concentration (2, 3). ATP efflux also occurs through plasma membrane conductance channels, transporters, or constitutive secretory pathways as residual cargo products (2). Extracellular ATP acts as a neurotransmitter and an autocrine/paracrine chemical messenger in non-neural tissues. Its effects are mediated by the P2 purinergic receptors and elicit a variety of physiological responses, such as neurotransmission, regulation of secretion, modulation of immune functions, pain transmission, apoptosis etc.
P2 receptors consist of two major subfamilies, P2X and P2Y. P2X receptors are ligand-gated ion channels and P2Y receptors are G protein-coupled receptors. The concentration of extracellular ATP is regulated by its hydrolysis that is catalyzed by extracellular ATPases. Thus the physiological responses mediated by the purinergic receptors are modulated by extracellular ATPases (Figure 2). For example, Sesti et al. reported that ATP modulates norepinephrine release from cardiac sympathetic nerve endings and this action of ATP is controlled by purinergic receptors in cardiac synaptosomes and modulated by extracellular ATPases (4). Di Virgilio et al. reported that a potent platelet aggregating factor is ADP and its amount is regulated by the activity of extracellular ATPases on endothelial cells (5). However, the precise relationship of multiple P2X and P2Y receptor subtypes to extracellular ATPases remains to be determined.
The role of ATP is an energy-rich molecule because its triphosphate unit contains two phosphoanhydride bonds. Large amounts of free energy is liberated when ATP is hydrolyzed to adenosine diphosphate (ADP) and orthosphosphate (Pi) or when ATP is hydrolyzed to adenosine monophosphate (AMP) and pyrophosphate (PPi). The precise for these reactions depend on ionic strength of the metal such as Mg 2+. The free energy is liberated in hydrolysis of ATP is harnessed to drive reactions that require an input of energy for muscle contraction. The formation of ATP from ADP and Pi is known as the ATP-ADP cycle is the fundamental mode of energy exchange in biological systems. It is intriguing to note that although, all nucleotide triphosphates are energetically equivalent, ATP is the primary cellular energy carrier. Under cellular conditions, the hydrolysis of ATP shifts the equilibrium of a coupled reaction by a factor of 108[2]
ATP has a particularly efficient phosphoryl-group donor that can best be explained by features of the ATP structure:
Resonance Structures. ADP and Pi have greater resonance stabilization than does ATP. Orthophosphate has multiple resonance forms of similar energy whereas the phosphoryl group of ATP has a smaller number due to its unfavorability of the positively charged oxygen atom that is adjacent to a positively charged phosphorus atom.
Electrostatic Repulsion. At pH 7, triphosphate unit of ATP carries four negative charges which repel one another due to their close proximity. The repulsion between them is reduced when ATP is hydrolyzed.
Stabilization Due to Hydration. More water can bind effectively to ADP and Pi than can bind to the phosphoanhydride part of ATP, stabilizing the ADP and Pi by hydration.
[3]
The large amounts of energy provided by the hydrolysis of ATP are necessary to overcome the large free energy changes necessary to create the large macromolecular proteins. The cleavagle of the phosphoanhydride bonds in ATP provides the source for free energy to make biological reactions spontaneous (negative free energy). Because the amount of entropy of the universe is continually increasing it is unfavorable for large macromolecules to form without the use of ATP. Because of this, the free energy generated by the ATP is always immediately consumed by nearby endergonic (energy-reguiring) biological reactions. The exergonic reaction of the ATP is only able to proceed if it is coupled to an endergonic reaction, otherwise thermodynamic equilibrium would not be obtained. The consumption of ATP proceeds with the first step of having an enzyme attache an amino acid to the a-phosphate of ATP. This results in the release of a pyrophosphate. This release is called an aminoacyl-adenylate intermediate. The reaction then proceeds to the enzyme catalyzing transfer of an amino acid to one of two -OH locations on the ribose portion of the adenosine residue. ATP is able to release energy into cells because cells maintain a concentration of ATP that is far higher above the equilibrium concentrations. The high concentration of ATP allows it to be the main provider of driving endergonic reactions in cells. This coupling of energy releasing and consuming systems through a common intermediate is vital to energy exchange in living systems.
[4]
= Lehninger | firs = Albert | authorlink = |Nelson, David L. and Michael M. Cox | title = Lehninger principles of biochemistry, 4th ed | publisher = W.H. Freeman & Co | date = 2007 | location = New York, New York | pages 22–25 | isbn = 0-7167-4339-6 }}</ref>
ATP is a principal immediate donor of free energy in biological systems meaning that it is consumed within a minute of it formation. The carbon in fuel molecules such as glucose and fats are oxidized to CO2 and the energy released is used to regenerate ATP from ADP and Pi. Oxidation in fuel takes place one carbon at a time and the carbon-oxidation energy is used in some cases to create compounds with high phosphoryl-transfer potential and other cases to create ion gradient as well with the end formation of ATP.[5]
ATP is coupled with oxidation of carbon fuels directly and through the formation of ion gradients. Energy of oxidation is initially trapped as high-phosphoryl-transfer potential compound and then used to form ATP. In ion gradients the electrochemical potential, produced by oxidation of fuel molecules or by photosynthesis, which ultimately powers the synthesis of most ATP in cells. ATP hydrolysis can be used to form ion gradients of different types and functions.
Described by Hans Krebs the three stages in generation of energy from oxidation of foodstuffs:
1. Large molecules in foods are broken down into smaller units in a process known as digestion. Proteins are hydrolyzed to their 20 different amino acids, polysaccharides are hydrolyzed into simple sugars and lastly fats are hydrolyzed to glycerol and fatty acids.
2. Numerous small molecules are degraded to a few simple units that play a central role in metabolism. Sugars, fatty acids, glycerol and several amino acids are converted into the acetyl unit of acetyl CoA. Some ATP is generated but not a substantial amount.
3. ATP is produced from the complete oxidation of acetyl unit of acetyl CoA. Final stage consist of citric acid cycle and oxidative phosphorylation which are the final pathways in oxidation of fuel molecules. Acetyl CoA brings acetyl units into the citric acid, where they are completely oxidized to CO2. Four pairs of electrons are transferred for each acetyl group that is oxidized. Then a proton gradient is generated as electron flows from the reduced forms of these carriers to O2 and the gradient is used to synthesize ATP.[6]
↑Berg, Jeremy (2007). Biochemistry, 6th Edition. New York, New York: Sara Tenney. pp. 110–111. ISBN978-0-7167-8724-2. {{cite book}}: Unknown parameter |coauthors= ignored (|author= suggested) (help)
↑Berg, Jeremy (2007). Biochemistry, 6th Edition. New York, New York: Sara Tenney. pp. 413–415. ISBN978-0-7167-8724-2. {{cite book}}: Unknown parameter |coauthors= ignored (|author= suggested) (help)
↑Berg, Jeremy (2007). Biochemistry, 6th Edition. New York, New York: Sara Tenney. p. 415. ISBN978-0-7167-8724-2. {{cite book}}: Unknown parameter |coauthors= ignored (|author= suggested) (help)
↑Biochemistry, 6th Edition. New York, New York: Sara Tenney. 2007. p. 110. ISBN978-0-7167-8724-2. {{cite book}}: |first= missing |last= (help); Unknown parameter |= ignored (help); Unknown parameter |coauthors= ignored (|author= suggested) (help)
↑Berg, Jeremy (2007). Biochemistry, 6th Edition. New York, New York: Sara Tenney. p. 417. ISBN978-0-7167-8724-2. {{cite book}}: Unknown parameter |coauthors= ignored (|author= suggested) (help)
↑Berg, Jeremy (2007). Biochemistry, 6th Edition. New York, New York: Sara Tenney. pp. 419–420. ISBN978-0-7167-8724-2. {{cite book}}: Unknown parameter |coauthors= ignored (|author= suggested) (help)
1. Schwiebert, E. M. ABC transporter-facilitated ATP conductive transport. Am. J. Physiolo., 1999, 276, C1-C8.
2. Lazarowski, E. R., Boucher, R. C., and Harden, K. T. Mechanisms of release of nucleotides and integration of their action as P2X- and P2Y-receptor activating molecules. Mol. Pharmacol., 2003, 64, 785-795.
3. Theander, S., Lew, D. P., and Nüße, O. Granule-specific ATP requirements for Ca2+-induced exocytosis in human neutrophils. Evidence for substantial ATP-independent release. J. Cell Sci., 2002, 115, 2975-2983.
4. Sesti, C., Broekman, M. J., Drosopoulos, J. H., Islam, N., Marcus, A. J., and Levi, R. Ectonucleotidase in cardiac sympathetic nerve endings modulates ATP-mediated feedback of norepinephrine release. J. Pharmacol. and Exp. Ther., 2002, 300, 605-611.
6. Di Virgilio, F., Chiozzi, P., Ferrari, D., Falzoni, S., Sanz, J. M., Morelli, A., Torboli, M., Bolognesi, G., and Baricordi, O. R. Nucleotide receptors: an emerging family of regulatory molecules in blood cells. Blood, 2001, 97, 587-600.
Acetyl coenzyme A, or better known as acetyl-CoA, is an important molecule used in metabolic processes. It is primarily used by the body for energy production through the citric acid cycle, or Krebs cycle.
Acetyl-CoA is a product of the oxidation of several amino acids, pyruvate and fatty acids.
It is formed after pyruvate enters the mitochondria via active transport. The oxidation transformation that converts pyruvate into acetyl-CoA is known as a pyruvate dehydrogenase reaction. In this process, each pyruvic acid molecule loses one carbon atom, and combines with oxygen to produce carbon dioxide. This carbon dioxide is then exhaled out of the cell. This process is catalyzed by a complex of three enzymes through a process known as pyruvate dehydrogenase complex (PDC). [1]
The main function of Acetyl-CoA is to carry acyl groups or thioesters. It is the precursor to HMG CoA, an important part of cholesterol and ketone synthesis. It can also be found as a vital reagent in the synthesis of fatty acids and sterols, as well as the oxidation of fatty acids as well as the breaking down of many amino acids. [2]
Acetyl-CoA is well known as the junction between Glycolysis and the Citric Acid Cycle as well as an essential component in balancing between carbohydrate and fat metabolism. Acetyl-CoA has also been a central metabolite that is involved in many metabolic transformations within the cell. The acetyl group of the acetyl-CoA is used to oxidize via the TCA cycle to reduce NAD+ and FAD to NADH and FADH2, respectively. These products are then used to fuel ATP production through the electron transport train.
In May 2011, Ling Cai et al. found that Acetyl-Coa functioned as a carbon-source rheostat that signals the initiation of the cellular growth program by promoting the acetylation of histones specifically at growth genes. By intimately coordinating with nutrient availability, the acetyl-CoA was concluded to play as a critical signal for metabolic growth and proliferation.
Activation of acetyl-CoA: Some acetyl groups within the cell are tagged with coenzyme A (CoA). Acetyl-CoA molecules are tagged at the back end by a process called condensation with carbon dioxide, catalyzed by acetyl-CoA carboxylase in order to redirect acetyl groups into fatty acid biosynthesis.
Glycolysis/Gluconeogenesis is the set of reactions that converts glucose into pyruvate. Glycolysis is a compilation of ten reactions (with 1 being an isomerization of 2 products into one another) of glycolysis take place in the cytoplasm.
One glucose molecule will go under group-transfer reaction with 1ATP (Adenosine Triphosphate) - which is catalyzed by hexokinase - to become Glucose 6-phosphate (Glu 6-P) and produce 1ADP (adenosine diphosphate) molecule.
Glu 6-P, under the activity of the enzyme phosphoglucose isomerase, becomes another isomer, Fructose 6-Phosphate (Fru 6-P).
Then, the group-transfer reaction between Fru 6-P and ATP, catalyzed by the enzyme Phosphofructokinase-1 (known as PFK-1), produces 1 fructose 1,6-biphosphate (Fru 1,6-P) molecule and another ADP molecule.
Fruc 1,6-P (after ring opening), after binding to the enzyme aldolase, produces two new molecules known as:
Glyceraldehyde 3-phosphate (GAP)
Dihydroxyacetone phosphate (DHAP)
Note that GAP can isomerize to DHAP and vice versa in the reaction catalyzed by the enzyme Triose Phosphate Isomerase.
GAP, which is constantly supplemented by the isomerization of DHAP, continues the process of glycolysis by reacting with 2 inorganic phosphorus molecules and 2 nicotinamide adenine dinucleotide (NAD+, oxidized form) molecules, under oxidation-reduction reactions catalyzed by the enzyme Glyceraldehyde Phosphate Dehydrogenase, to produce 2 1,3-bisphosphate glycerate molecules, 2 protons, and 2 NADH molecules (reduced form of NAD+). Note that from this stage on, reactions are carried out by 2 molecules of the previous products to produce 2 new molecules. Therefore, it is assumed in the next stages, "1,3-biphosphate glycerate reacts with some compound A" means 2 1,3-bisphosphate glycerate molecules react with 2 molecules of compound A to produce 2 new molecules.
Next, 1,3-bisphosphate glycerate undergoes group-transfer reaction, catalyzed by phosphoglycerate kinase, with ADP to produce 3-phosphoglycerate and ATP.
Then, 3-phosphoglycerate, catalyzed by phosphoglycerate mutase, becomes 2-phosphoglycerate.
Catalyzed by the enzyme enolase, 2-phosphoglycerate produces phosphoenolpyruvate (PEP) and water.
In the last step, PEP reacts with ADP in a group-transfer reaction catalyzed by phosphopyruvate kinase, to produce pyruvate and ATP.
In total, glycolysis consumes 1 glucose molecule, 2 ATP molecules, and 2 NAD+ to produce 2 pyruvate molecules, 4 ATP molecules, and 2 NADH molecules.
Therefore, in overall, 1 Glu + 2 NAD+ → 2 Pyruvate + 2 NADH + 2 ATP
After glycolysis, the products, depending on the appearance of O2, will undergo either aerobic reaction (with O2) to continue the metabolic pathway into the citric acid cycle (also known as Kreb's cycle), or anaerobic reaction (without O2) to start a new process known as fermentation to produce lactic acid (mostly in human's muscular cells) or ethanol and carbon dioxide (CO2) in microbes such as yeasts.
Lactose, sucrose, and maltose are disaccharides that can be hydrolyzed into monosaccharaides and then enter the glycolysis. Latcase, sse, and maltase are enzymes(in epithelial cells) that can hydrolyze the disaccharides respectively.
Maltose is hydrolyzed into two β-D-glucopyranose molecules. These monosaccharaides can readily enter glycolytic pathway.
Add caption here
Hydrolyzing sucrose produces α-D-glucopyranose and β-D-fructofuranose. However, β-D-fructofuranose must be converted to a form in order to enter glycolytic. This conversion is facilitated by the enzyme hexokinase to form β-D-fructofuranose-6-phosphate or by the fructokinase to form β-D-fructofuranose-1-phosphate. Both of these phosphorylation reactions consume ATP.
Add caption here
Constituent monosaccharaides residues of lactose are β-D-glucopyranose and β-D-galactopyranose. β-D-glucopyranose is ready to enter into glycolytic process but β-D-galactopyranose needs to convert to a form to enter glycolytic pathway. Since galactose is a epimer of glucose at the C-4 position, it can form glucose-6-phosphate (an intermediate reactant) during glycolysis. [1]
The brain uses a very large amount of energy, and neurons and astrocytes are mainly responsible for the consumption of oxygen and glucose. The brain is only ~2% of the human body weight, though it consumes over 20% of the body’s oxygen and glucose.
When nitric oxide inhibits respiration in neurons of rats, ATP concentration quickly decreases along with a sharp decrease in mitochondrial transmembrane potential, followed by spontaneous apoptosis or cell death. Astrocytes respond to respiration inhibition by instead increasing metabolism through glycolysis and using the generated ATP to maintain the mitochondrial transmembrane potential. This is done through an increase in the enzyme activity of PFK1 or 6-phosphofructo-1-kinase, independently of cGMP, at a level twice that of normal neuron conditions. Levels of fructose-2,6-bisphosphate rise in astrocytes as well, which also activates PFK1 allosterically. However, levels of this enzyme are NOT raised in the neuron cells. The maintenance of the potential prevents cell apoptosis.
The mechanism that increases the levels of these enzymes in astrocrytes during conditions of respiration inhibition is quick phosphorylation of the enzyme AMPK, or 5’-AMP-activated protein kinase and the gathering of fructose-2,6-bisphosphate. This shows that when respiration is inhibited a ‘crisis’ has occurred causing a cascade of events: 5’-AMP concentration increase, causing phophorylation of AMPK, which activates fructose-2,6-bisphosphatase.
Neurons however are unable to block cell death through an increase in glycolytic activity due to this lack of 6-phosphofructo-2-kinase and fructose-2,6-bisphotase. Instead the metabolism of glucose is achieved via the pentose phosphate pathway, which regenerates reduced glutathione. [2]
Anaerobic respiration is the formation of ATP without the presence of oxygen. This method uses the electron transport chain without the presence of oxygen as the electron acceptor. Although oxygen is highly oxidizing, it is only used during aerobic processes. In anaerobic repiration, less oxidizing molecules such as sulfate (SO42-), nitrate (NO3-), or sulfur (S) are used as electron acceptors. Thus, less energy is formed per molecule of glucose during anaerobic respiration. Most prokaryotes that live under environmental conditions that lack oxygen uses aerobic respiration, although humans too, use it sometimes as well (Lactic Acid Fermentation).
In this process, specifically with the absence of Oxygen during respiration process, organisms have evolved with mechanisms to recycle Nicotinamide Adenine Dinucleotide (NAD+) for glycolysis to continue in order to synthesize Adenosine Triphosphate (ATP) molecules, known as "energy currency" of cells.
This process evolves into two different mechanisms, although both share the name of "fermentation":
Ethanol Fermentation occurs in bacteria, yeast,...
Lactic Acid (or Lactate) Fermentation occurs in animals (humans,...)
After glycolysis, without Oxygen, pyruvates are carried into the ethanol fermentation cycle:
With cofactors such as pyruvate decarboxylase enzyme, TPP (thiamine pyrophosphate), pyruvate is converted into acetaldehyde and carbon dioxide.
Acetaldehyde, under oxidation-reduction reaction catalyzed by the enzyme ethanol dehydrogenase, is reduced to ethanol, with NADH being oxidized to NAD+.
NAD+ is then used for glycolysis to synthesize ATP, and if Oxygen is still absent, fermentation continues. Therefore, ethanol and CO2 are waste products of this fermentation process.
Note that because of this reaction, when using yeast or some bacteria in food processing, there are a slight smell of alcohol (from ethanol) and gas coming off the mixture (from CO2)
Similarly, in animals (humans,..), pyruvates must go into fermentation cycle if no oxygen is carried into the cells. However, there is one difference:
Under reduction-oxidation reaction catalyzed by the enzyme lactate dehydrogenase, pyruvate is reduced into lactate, with NADH being oxidized to NAD+.
Note that in animals, overworked muscles, whose cells cannot receive sufficient amount of oxygen to compensate for the loss of ATP, produce a lot of lactic acid as waste product of this fermentation process.
Fermentation is diverse. There are two types of fermentation: homofermentative and heterofermentative. Homofermentative include lactic acid, ethanol, and carbon dioxide. However, heterofermentative include propionic acid, acetic acid, carbon dioxide, hydrogen gas, butyric acid, butanol, acetone, isopropyl alcohol, and succinic acid.
Furthermore, in lactic fermentation there are homolactic versus heterolactic. The heterofermentative lactic acid bacteria lack aldolase and that the first steps of heterofermentative pathway are from the Pentose-Phosphate Pathway. An example of lactic fermentation is an organism called Lactobacillus acidophilus. This organism is a gram positive, it is homofermentative, it is belong to Firmicutes group, and it can be found in the normal human flora.
Sometimes, glucose may not be available for cellular respiration. Without this essential fuel, cellular respiration would stop completely and death would occur. So there must be backup storage that keep fuel just in case glucose levels are insufficient. When glucose is low, the body uses carbohydrates, such as glycogen stored in the liver and muscles. When carbohydrates deplete, fat is used next and, as last resort, protein is used. When fats and proteins are used, they must first be converted to glucose or some derivative. Fats are broken down to fatty acids and glycerol. Fatty acids can turn into acetyl-CoA by beta oxidation. Glycerol can turn into an intermediate of glycolysis. Proteins are broken down into amino acids. They are modified into -keto acids then converted into the various intermediates of Kreb's cycle.
In the aerobic respiration is a release of energy from glucose or another organic substate in the presence of Oxygen. Also, aerobic respiration is in the absent of air. Aerobic respiration is a process of cellular respiration that uses oxygen in order to break down molecules, which then release the electrons and also create the energy. It creates substances known as ATP. ATP stands for adenosine triphosphate. ATP's role is to store and carry most of the energy to other body cells. Besides, there are two main byproducts of aerobic respiration. They are water and carbon dioxide. Aerobic respiration is also contains three stages: glycolysis, Kreb's cycle, and the third stage is the electron transport phosphorylation.
The pyruvate produced in glycolysis undergoes further breakdown through a process called aerobic respiration in most organisms. This process need oxygen and yields much more energy than glycolysis. Aerobic respiration is separated into two processes: the Krebs cycle, and the Electron Transport Chain, which produces ATP through chemiosmotic phosphorylation. The energy conversion is as follows:
The Krebs cycle occurs in the mitochondria of a cell. Before entering the Krebs cycle, the pyruvic acid molecules are altered. During the process, the pyruvic acid molecule is broken down by an enzyme, one carbon atom is released in the form of carbon dioxide, and the remaining two carbon atoms are combined with a coenzyme called the coenzymes.
After the glycolysis takes place in the cell's cytoplasm, the pyruvic acid molecules travel into the interior of the mitochondrion. Once the pyruvic acid is inside, carbon dioxide is enzymatically detached from each three-carbon pyruvic acid molecule to form acetic acid. The enzyme then syndicates the acetic acid with coenzyme A to produce acetyl CoA. Once acetyl CoA is formed, the Krebs cycle begins. The cycle is split into eight steps, picture of this cycle is included in this link: http://notesforpakistan.blogspot.com/2011/02/krebs-cycle-or-citric-acid-cycle-or-tri.html
Prior to entering the Krebs Cycle, pyruvate must be converted into acetyl CoA (pronounced: acetyl coenzyme A). This is achieved by removing a CO2 molecule from pyruvate and then removing an electron to reduce an NAD+ into NADH. An enzyme called coenzyme A is combined with the remaining acetyl to make acetyl CoA which is then fed into the Krebs Cycle.
1. Citrate is created when the acetyl group from acetyl CoA combines with oxaloacetate from the previous Krebs cycle.
2. The citric acid molecule experience an isomerization. A hydroxyl group and a hydrogen molecule are detached from the citrate structure in the form of water. The two carbons form a double bond until the water molecule is added again. Only now, the hydroxyl group and hydrogen molecule are reversed with respect to the original structure of the citrate molecule. Thus, isocitrate is formed.
3. Isocitrate is oxidized to form the 5-carbon α-ketoglutarate. This step releases one molecule of CO2 and reduces NAD+ to NADH2+.
4. The α-ketoglutarate is oxidized to succinyl CoA, yielding CO2 and NADH2+.
5. Succinyl CoA releases coenzyme A and phosphorylates ADP into ATP.
6. Succinate is oxidized to fumarate, converting FAD to FADH2.
7. An enzyme adds water to the fumarate molecule to form malate. The malate is formed by adding one hydrogen atom to a carbon atom and then adding a hydroxyl group to a carbon next to a terminal carbonyl group.
8. Malate is oxidized to oxaloacetate, reducing NAD+ to NADH2+.
Unlike anaerobic respiration, in this process, where oxygen appears in sufficient amount, pyruvates are transported into mitochondria, where the largest ATP-producing facilities will provide energy for the cell.
Aerobic Respiration consists of three main steps:
Pyruvate dehydrogenase complex
Krebs Cycle (Nitric Acid Cycle)
Oxidative Phosphorylation Cycle (Electron Transport Chain cycle)
This process involves with the conversion of pyruvate molecule into compound called acetyl coenzyme A, or acetyle CoA. This step is the junction between glycolysis and the Krebs Cycle (Citric Acid cycle) and is accomplished by a multi-enzyme complex that catalyzes three reactions:
Pyruvate's carboxyl group (COO–), which is fully oxidized and is removed to release CO2
The remainning two-carbon is oxidized and form a compound named acetate. An enzyme transfers the extracted electrons to NAD+, storing energy in the form of NADH
Finally, coenzyme A(CoA), a sulfur-containing compound derived from a B vitamin, is attached to the acetate by an unstable bond and this makes the acetyl group become very reactive. acetyl CoA has a high potential energy will undergoes the Citric Acid cycle to release energy to make ATP.
Other name for citric acid cycle is tricarboxylic acid (TCA) cycle or the Krebs cycle. The citric acid cycle is the central metabolic core of the cell. It is the final common pathway for oxidation — in other words harvesting high energy electrons--fuel molecules such as carbohydrate fatty acids, and amino acids by entering the cycle as Acetyl Coenzyme A (CoA). This reaction takes place inside of mitochondria. It is very efficient because it can generate large amounts of NADH and FADH. The citric acid cycle provides the majority, 90 percent, of energy used by aerobic human cells. By acting as the first stage of cellular respiration, the generation of high energy electrons from the citric acid cycle, in turn, are used in oxidative phosphorylation to reduce O2, generate proton gradient, and, later, the synthesis of ATP.
Citric Acid Cycle Links to Glycolysis by Pyruvate Dehydrogenase
Carbohydrates are mostly processed by glycolysis into pyruvate. Depending on the organism, the pyruvate is converted into either lactate or ethanol under anaerobic conditions. Through a specific carrier protein embedded in the mitochondrial membrane, pyruvate is transported into the mitochondria under aerobic conditions. Then, the pyruvate is oxidatively decarboxylated by the pyruvate dehydrogenase complex in order to form the acetyl CoA in the mitochondrial matrix. The reaction is shown in the following:
Pyruvate + CoA + NAD+ → acetyl CoA + CO2 + NADH + H+
This is an irreversible reaction which links glycolysis and the citric acid cycle together. As shown, CO2 is produced by pyruvate dehydrogenase complex and it captures high-transfer-potential electrons in the form of NADH. Therefore, the pyruvate dehydrogenase relates to the reactions of the citric acid cycle itself. The pyruvate dehydrogenase complex is composed of three different enzymes. Its complex is composed of members of a family of homologous complexes which include citric acid cycle enzyme a-ketoglutarate dehydrogenase complex. These complexes are very large, even bigger than ribosomes, with its molecular mass to be in between 4 million to 10 million daltons.
Acetyl-CoA, main product of the Pyruvate Dehydrogenase Complex in aerobic respiration, starts the Krebs cycle. (An irreversible reaction that is link between glycolysis and the citric acid cycle.) The mechanism of the synthesis of acetyl coenzyme A from pyruvate requires five coenzymes and three enzymes. It is a very complex mechanism which many enzymes and coenzymes. Cofactors, which function as substrates, are divided into two different cofactors which are catalytic cofactor and stoichiometric cofactor. The catalytic cofactor includes coenzymes such as thiamine pyrophosphate (TPP), lipoic acid, and FAD. The stoichiometric cofactor includes coenzymes such as CoA and NAD+. Pyruvate converts into acetyl CoA in three distinct steps which include: decarboxylation, oxidation, and transfer of the resultant acetyl group to CoA.
Pyruvate results in formation of acetyl-CoA by a three step reaction:
1. Decarboxylation: TPP is combined with the pyruvate and decarboxylated in order to yield hydroxyethyl-TPP. Of the pyruvate dehydrogenase component, TPP is known as the prosthetic group which the carbon atom between the nitrogen and sulfur atoms in the thizaole ring is more acidic than most double bonded carbon groups with pKa values near 10. This reaction is catalyzed by the (E1) pyruvate dehydrogenase component of the multienzyme complex. The carbon center located in the TPP is ionized to form a carbanion which is added to the carbonyl group of pyruvate. As part of decarboxylation, a positive charged ring of TPP stabilizes the negative charge which was transferred to the ring. Finally, the protonation yields hydroxyethyl-TPP.
2. Oxidation: In order to form an acetyl group, the hydroxyethyl group which is attached to TTP is oxidized. Simultaneously, the hydroxyethyl group is transferred to lipoamide which is lipoic acid derived that links to the side chain of a lysine residue by an amide linkage. This creates the formation of an energy-rich thioester bond. In this reaction, the disulfide group of lipoamide acts as an oxidant and is reduced to the disulfhydryl form. This reaction is catalyzed by the pyruvate dehydrogenase component (E1) as well and yields the acetyllipoamide.
3. Formation of Acetyl CoA: Formation of Acetyl CoA: In this step, acetyl CoA is formed when acetyl group is transferred from acetyllipoamide. This reaction is catalyzed by dihydrolipoyl transacetylase (E2). As the acetyl group is transferred to the CoA, the energy-rich thioester bond is preserved. Thus, the fuel for the citric acid cycle, acetyl CoA has been generated from pyruvate for use. Until the dihydrolipoamide is oxidized to lipoamide, the pyruvate dehydrogenase complex cannot complete another catalytic cycle.
CoA + Acetyllipoamide --> Acetyl CoA + Dihydrolipoamide
4. Formation of NADH: The final step in this reaction occurs when the oxidized form of lipoamide is regenerated by dihydrolipoyl dehydrogenase (E3).Two electrons are transferred to first an FAD prosthetic group of the enzyme and then to NAD+. This process of transferring electron is very unusual because FAD are known to receive electrons from NADH, not transfer them. Within the enzyme, the electron-transfer potential of FAD is increased by its chemical environment which enables it to transfer electrons to NAD+. Flavoproteins are proteins which are tightly associated with FAD or FMN also known as flavin mononucleotide.
1. The first reaction of the cycle is condensation of acetyl-CoA with oxaloacetate to form citrate. In this reaction, acetyl group is joined to the carbonyl group of oxaloacetate. The reaction between acetyl-CoA with oxaloacetate is necessary to form an active site closed citryl CoA complex for hydrolysis because the active site of acetyl-CoA with hydrolysis is a wasteful process.
Oxaloacetate + Acteyl-CoA --> Citryl-CoA + H2O -->Citrate + CoA
2. Formation of Isocitrate via cis-Aconitate: Enzyme, aconitase catalyzes the reversible transformation of citrate to isocitrate. This is actually a 2 steps mechanism, which interchange an hydrogen with an hydroxyl group. First citrate is dehydrated to form the intermediary formation of the tricarboxylic acid cis-aconitate. Then by using the same enzyme, aconitase, isocitrate is formed. Aconitase is an iron-sulfur protein that participate in the dehydration and rehydration of the substrate.
3. Oxidation of ioscitrate with NAD+ to α-Ketoglutarate, CO2 and NADH. Isocitrate dehydrogenase catalyzed oxidative decarboxylation of isocitrate to form α-ketoglutarate. Mg2+ is used to interact with the carbonyl group of oxalosuccinate. The rate of formation of α-ketoglutarate determines of overall reaction in the citric cycle. The intermediate, oxalosuccinate, is a very unstable β-ketoacid.
Reaction with Intermediate:
4. Oxidation of α-ketoglutarate to succinyl-CoA and CO2. This is another oxidative decarboxylation. Alpha-ketoglutarate is converted to succinyl-CoA and CO2 by the action of the α-ketoglutarate dehydrogenase complex. NAD+ is used as electron acceptor and CoA as the carrier of the succinyl group. This reaction is virtually identical to the pyruvate dehydrogenase reaction. It has three enzymes participating in this step. This is similar to the Pyruvate Dehdrogenase Complex reaction.
5. Conversion of Succinyl-CoA to Succinate: Succinyl-CoA has a thioester bond. Thioester bond retains a lot of energy (ΔG° = -33.5 kJ mol -1. Energy is released by breaking the thioester bond. The enzyme that is used in this step is called succinyl-Co synthetase or succinic thiokinase. The cleavage of the bond is coupled with GDP getting phosphorylated.
Succinyl CoA + Pi + GDP --> succinate + CoA + GTP
6. Oxidation of succinate to fumarate: the succinate formed from succinyl-CoA is oxidized to fumarate by the enzyme called succinate dehydrogenase. FAD, which is attached to histidine side chain, acts as the electron acceptor by removing two hydrogen from succinate. FADH2 will pass its electrons to coenzyme Q, which will be use in the electron transport chain.
His-FAD + succinate <--> His- FADH2 + fumarate
7. Hydration of fumarate to malate: fumarate is then converted to malate by using fumarase. This enzyme is highly stereospecific; it catalyzes hydration of the trans double bond of fumarate. The reaction with add H+ and OH- to make L-malate.
Fumarate + H2O --> L-Malate
8. In the final step, Malate (C4), in a oxidation-reducation reaction catalyzed by malate dehydrogenase, is oxidized to oxaloacetate (C4) with NAD+ being reduced to NADH. The reaction is very positive with ΔG° = +29.7 kJ mol -1.
Malate + NAD+ <--> oxaloacetate + NADH + H+
9. Then, the Krebs cycle restarts again as long as Oxygen is transported into the cell.
Note that the number of Carbon is going from the sum of 4 and 2 from the beginning to 6, 6, 5, 4, 4, 4, and back to oxaloacetate (C4) again.
Overall, there are 6 NADH, 2 FADH2, and 4 CO2 being produced by two acetyl-CoA molecules from the Pyruvate Dehydrogenase Complex.
Since all the complex pyruvate dehydrogenase structure is known, the atomic model was able to be formed so that its activity could be understood from it. The center of the complex is formed by the transacetylase component E2. Transacetylase contains eight catalytic trimers which are gathered to form a hollow cube. There are three major domains for each three subunits that form a trimer. The amino terminus has a small domain that contains a bound flexible lipoamide cofactor which is attached to a lysine residue. This domain is homologous to biotin-binding domains like pyruvate carboxylase. The lipoamide domain is followed by a small domain that interacts with E3 within the complex and the larger transacetylase domain completes an E2 subunit. E1 is considered to be α2β2 tetramer, and E3 is considered to be an αβ dimer. E2 is surrounded by multiple copies of E1 and E3. These three distinct active sites work together through the long, flexible lipoamide arm of the E2 subunit that carries substrates from one active site to the other. The process of moving lipoamide between different active sites are:
1) In the active site of E1, pyruvate is decarboxylated and forms a hydroxyethyl-TPP intermediate while the CO2 leaves as the first product. The active site is connected to the enzyme’s surface through a long hydrophobic channel within the E1 complex.
2) The lipoamide arm of the lipoamide is inserted by E2 into the deep channel in E1 which leads to the active site.
3) The transfer of the acetyl group is catalyzed by E1 to the lipoamide. The acetylated arm leaves E1 and enters the E2 cube in order to visit the active site of E2. This is located deep in the cube at the subunit interface.
4) The acetyl moiety is then transferred to CoA. The second product which is the acetyl CoA, leaves the cube. The reduced lipoamide arm then swings to the active site of the E3 flavoprotein.
5) The lipoamide is oxidized by the coenzyme FAD in the E3 active site. The reactivated lipoamide is ready to start a new reaction cycle.
6) NADH, the final product, is produced through the reoxidation of FADH2 to FAD.
This coordinated catalysis of a complex reaction is possible because of the structural integration of three different kinds of enzymes and the long, flexible lipoamide arm. The overall reaction rate is increased and the side reaction is minimized due to the proximity of one enzyme to another. Throughout the reaction sequence, all the intermediates in the oxidative decarboxylation of pyruvate remain bound to the complex. This is readily transferred to the flexible arm of E2 calls on each active site in turn.
Although glucose can be formed by pyruvate, the irreversible step of the formation of acetyl CoA from pyruvate causes the acetyl CoA to be unable to convert back into glucose. The oxidative decarboxylation of pyruvate to acetyl CoA commits the carbon atoms of glucose to one of two principal fates: oxidation to CO2 by the citric acid cycle, with the concomitant generation of energy, or incorporation into lipid. The activity of the pyruvate dehydrogenase complex is stringently controlled. The reaction can be inhibited by the high concentration of reactions: by binding directly, the acetyl CoA inhibits the transacetylase component E2 while the NADH inhibits the dihydrolipoyl dehydrogenase E3. High concentrations of NADH and acetyl CoA inform the enzyme that the energy needs of the cell have been met or in order to produce the acetyl CoA and NADH, the fatty acids are being degraded to produce because most pyruvate is derived from glucose by glycolysis. Covalent modification is very important in regulating the complex in eukaryotes. The phosphorylation of the of the pyruvate dehydrogenase component (E1) by pyruvate dehydrogenase kinase I (PDK) switches of the activity of the complex. The deactivation is reversed by the pyruvate dehydrogenase phosphate.
In animal cells, the rate of citric acid cycle is regulated to fitted the required needs for ATP. The two enzymes that allosteric control the cycle are isocitrate dehydrogenase and α-ketoglutarate dehydrogenase, which both generate high-energy electrons in cycle.
Isocitrate dehydrogenase is stimulated by ADP that increase the affinity of enzyme for substrates. Since isocitrate, NAD+, Mg+, and ADP are cooperative, the binding of these substrates are regulated. If energy is unnecessary, there will be more NADH and ATP that will compete for the binding of enzyme with NAD+ and ADP respectively. Because the reaction requires positive energy or electron acceptors, the reactions is slowed until high energy like ATP and NADH are needed.
α-ketoglutarate dehydrogenase is another allosteric enzyme that regulated the rate of citric acid cycle for ATP. This enzyme is inhibited by succinyl CoA and NADH, which are the product that also compete for binding with reactants. α-ketoglutarate is also inhibited by high energy electron.
Between mitochondrial matrix and intermembrane space, within the innermembrane, there are 5 complexes involved in the oxidative phosphorylation cycle, also known as electron transport chain cycle
At complex I - NADH dehydrogenase: NADH is oxidized to NAD+: a process that releases electrons, which is transported by FMN (flavin mononucleotide to the Fe-S center (similar to heme group of hemoglobin) and reduce Q (ubiquinone) to QH2 (ubiquinol). Besides, 4 protons are pumped from matrix to intermembrane space.
At complex II - Succinate dehydrogenase: succinate is oxidized to fumarate: a process that releases electrons to reduce FAD to FADH2, which carries the original electrons to reduce Q (ubiquinone) to QH2 (ubiquinol). Again protons are pumped into the intermembrane space
At complex III - Ubiquinone Cytochrome c oxidoreductase: Q-cycle transfers electron from QH2 to cytochrome c (cyt c).
At complex IV - cytochrome oxidase: cyt c transfer electrons to oxygen O2 (by the support of Cu-S center and hemes). This is where water is released and proves the role of oxygen in aerobic respiration. Again protons are pumped into the intermembrane space
At complex V - ATP Synthase: with protons pumped into intermembrane space now return to the matrix, a rotary complex carries out the job of combining ADP and inorganic phosphate in mitochondria into ATP for cellular energy.
While the electron transport chain transports electrons and pumps H+ ions into the intermembrane space, the process of ATP synthesis does not occur until the ATP Synthase. The other part of the oxidative phosphorylation, after the electron transport chain, is the ATP synthase. ATP synthase is a transport protein that is consists of four parts, the Stator, the Rotor, the internal rod, and the Catalytic knobs.
The H+ ions in the intermembrane space pumped by the electron transport chain will flow down their gradient first through the stator. The stator is anchored in the membrane.
The H+ ion then binds onto the rotor, which is shaped somewhat like a waterwheel. This binding causes the rotor to change its shape and thus, makes it spin within the membrane.
The spinning of the rotor causes the internal rod to spin, which leads the catalytic knob to spin. The spinning of the catalytic rod causes the catalytic sites in the rod to transform ADP and inorganic phosphate group into ATP in the mitochondrial matrix.
Overall, the ATP synthase functions like a waterwheel. When the concentration of H+ ions in the intermembrane space becomes higher than that of the matrix, the H+ ions will go down the ATP synthase and facilitate the rotor of the ATP synthase. This causes the other components of the ATP synthase to spin. As a result, ATP can be generated from the catalytic sites in the catalytic rod.
Chemiosmosis is the movement of ions across a membrane down its gradient. It couples the electron transport chain and chemiosmosis in order to create ATP. In electron transport chain, the multiprotein structure pumps out H+ ions into the intermembrane space. As the H+ ions get pumped out, the concentration of H+ in the intermembrane space gets higher. As a result, H+ ions will start flowing down back to the chromosomes matrix through the ATP molecule. Through this movement, the cardiac rotor of the ATP synthase transforms the ADP into ATP.
Gluconeogenesis pathway with key molecules and enzymes. Many steps are the opposite of those found in the glycolysis.
Gluconeogenesis is a process, in which Pyruvate (a product of Glycolysis) is backward-converted into sugar, glucose in particular. Which can then be stored in the form of glycogen in animals' cells or starch and cellulose in plants' cells.
There are three basis steps involved in Gluconeogenesis:
Pyruvate to Phosphoenolpyruvate (PEP):
The enzyme pyruvate carboxylase converts pyruvate into oxaloacetate by adding the CO2 from the bicarbonate ions with the support of ATP and the coenzyme biotin.
Then oxaloacetate is converted into phosphoenolpyruvate (PEP)by the enzyme PEP carboxykinase in a process that uses GTP (guanosine triphosphate) as energy and releases CO2 as a waste product. Note that the released CO2 is actually the same CO2 molecule from the bicarbonate ion at the previous step.
The next steps are just the reversed processes of glycolysis going from PEP back to Fructose 1,6-bisphosphate, with released products in glycolysis being reactants/cofactors in gluconeogenesis.
Fructose 1,6-bisphosphate (Fru 1,6-P) to Fructose 6-Phosphate (Fru 6-P): This is an irreversible hydrolysis reaction catalyzed by the enzyme Fructose 1,6-bisphosphatase (FBPase-1). This reaction is heavily regulated by Fructose 2,6-bisphosphate.
Glucose 6-Phosphate to Glucose: Fru 6-P in the previous step is converted into Glucose 6-Phosphate by reversed step of glycolysis. Glucose 6-phosphate is converted into glucose by a hydrolysis reaction.
The main regulatory factor is the concentration of Fructose 2,6-bisphosphate (Fru 2,6-P), which controls glycolysis (catabolic pathway) and gluconeogenesis (anabolic pathway).
First, Fru 2,6-P is the conversion of Fructose 6-Phosphate (Fru 6-P) by the enzyme Phosphofructokinase-2 (PFK-2), which uses ATP as an energy source. This is a reversible reaction, thus the reversed reaction gives back Fruc 6-P. The reversed reaction is catalyzed by the enzyme Fructose 2,6-bisphosphatase (FBPase-2) in a process releasing an inorganic phosphate.
Second, how can Fru 2,6-P regulates glycolysis and gluconeogenesis?
In a breakdown pathway, because Fru 6-P is readily phosphorylated into Fru 1,6-P by the enzyme Phosphofructokinase-1 (PFK-1), a high concentration of Fru 2,6-P will accumulate and promotes the activity of PFK-1 and inhibits FBPase-1. Note that PFK-2 is active and FBPase-2 remains inactive to stimulate the production of Fru 2,6-P
In the synthesis pathway, Fru 6-P concentration rises, inhibits the activity of PFK-2, and stimulates FBPase-2 to reduce the concentration of Fru 2,6-P. As the concentration of Fru 2,6-P decreases over time, the activity of PFK-1 also decreases, thus stimulating FBPase-1 to convert Fru 1,6-P into Fru 6-P,which is later ultimately converted into glucose.
Note that in the case of low glucose level in the blood: the enzyme glucagon from liver can stimulate the production of an enzyme called cAMP-dependent protein kinase, which stimulates the conversion of the PFK-2 (active)/FBPase-2 (inactive) into PFK-2 (inactive)/FBPase-2 (active). This process will inhibit glycolysis and promote gluconeogenesis to pump more glucose from liver into the blood.
Phosphofructokinase-1 (PFK-1) is a major factor in process known as glycolysis. PFK-1 catalyzes the committed step, or begins an irreversible enzymatic reaction, which is the point of no return. PFK-1 also regulates triacylglycerol synthesis, which helps decrease the amount of fat storage. By preventing PFK-1 from doing its job, other glycolytic intermediaries decrease such as 2,3-bisphosphoglycerate.
Faulty PFK-1 can cause a build up of glucose, glucose-6-phosphate, and other glycolytic intermediaries. The end result of this is glycogen synthesis, which can lead to increased solute concentration in red blood cells, enlarged spleen, a swelled heart, and ultimately death.
Phosphofructokinase-1 (PFK-1) is a putative controlling enzyme in glycolysis. Cancer cells have defective mitochondria, which forces them to depend on glycolysis as their main source of energy. Ascorbate (commonly known as Vitamin C) has been shown to inhibit PFK-1 and Lactate Dehydrogenase (LDH). In the muscle cells, when the muscle is active, PFK-1 and LDH form a complex with contractile proteins in the muscle, protecting them from inhibition allowing for glycolysis (glucose is degraded and ATP is made. When the muscle is at rest, it is hypothesized that the complex is not formed, and PFK-1 and LDH are inhibited by ascorbate, glycolysis does not occur and glucose is stored as glycogen. Glycogen storage mainly occurs in the liver and muscle tissue. It is also hypothesized that because cancer cells depend mainly on glycolysis for energy, that PFK-1 in cancer cells will not be inhibited by ascorbate. In order to test this hypothesis it is important to be able to purify PFK-1. Because aldolase (Ald) protects from inhibition, the purified PFK-1 must not have aldolase in it in order to properly test the hypothesis.
Rabbit muscle was used as a source of PFK-1. Preparation of three assays (PFK-1 assay, LDH assay, and aldolase assay) were necessary to test the enzyme activity after each step using a spectrophotometer. This is done to keep track of what is removed and what stays after each step.
Preparation
Rabbit muscle was blended in a solution of ethylenediaminetetraacetic acid (EDTA), dithiothreitol (DTT, and sodium fluoride (NaF), then centrifuged and washed in order to remove soluble proteins. This was done multiple times to try and remove some LDH and aldolase.
Heat Step
The pellet containing the PFK-1 was then resuspended in a solution of Tris, MgSO4, EDTA, ATP, and DTT and then submerged in a heat bath and then placed in an ice bath. This was done to denature the other proteins while solubilizing and keeping safe the PFK-1 by using ATP and magnesium. The suspension was centrifuged and the supernatant saved.
Ion-exchange and Dye Columns
The supernatant was then placed on four different columns, all standardized with 40TP8 and DTT. The four columns were: blue agarose, brown agarose, blue dextran, and DEAE-Sephacel. The supernatant was allowed to drip through the columns by gravity and then collected to be tested. Theoretically, the PFK-1 should be stuck to the columns at that point. Solutions of 40TP8/ DTT were then added to the columns and collected in fractions. And then solutions containing an increased concentration of TP8 were added and then collected in fractions. The purpose of adding increasing concentrations of TP8 was to wash off any other proteins with a weaker affinity for the beads in the columns first. An additional method using a vacuum filtration system was tried, but would pull the PFK-1 and aldolase through the column too quickly, not giving PFK-1 enough time to stick to the column, making this method counterproductive.
Gel Electrophoresis
Samples taken from the column were then tested for purity by polyacrylamide gel electrophoresis (PAGE)
The DEAE Sephacel column seemed to have the highest yield of PFK-1 activity out of the four columns, but when tested using PAGE there was a lot of contamination. As mentioned before, there shouldn’t be aldolase because it inhibits PFK-1, which would make testing the affects of ascorbate on PFK-1 very hard to do. The blue agarose, while it didn’t have as high of a yield as DEAE Sephacel, showed to be very pure when tested by PAGE. The brown agarose showed potential at producing high yields of PFK-1, but after continuous testing, while it was good at removing aldolase, the percent yield of PFK-1 was too low.
Blue agarose so far has shown to be the best choice out of the four, but continuous testing/adjusting must be done before using this process on cancerous tissue. This is because while blue agarose removes aldolase well, the yield is still not high enough for it to be efficient to use with cancer tissue.
1. Queja, A., Marshall, A., Willaims, A., & Russell P.J. (2012, June). Micro-purification of Rabbit Phosphofructokinase-1. Poster presented at the biennial symposium of the Intercultural Cancer Council (ICC), Houston, TX.
2. Russell P, Williams A, Marquez K, Tahir Z, Hossein B, Lam K. 2008. Some characteristics of rabbit muscle phosphofructokinase-1 inhibition by ascorbate. J Enzyme Inhib & Med Chem; 23: 411-417
3. Vassault, A. 1983. Methods of Enzymatic Analysis, Enzymes I: Oxidoreductases, Transferases Vol. III, Verlag Chemie, Basel, pp. 118–126.
Light Reaction converts light to ATP and NADPH, an electron carrier. This reaction occurs in the thylakoid, and it requires visible light for the chlorophyll (a type of molecule known as pigment that absorbs light) to absorb all visible light except for green. Chlorophyll are organized in the thylakoid membrane (in the photosystems).
Then the electron gains energy from light and jumps up to a higher electron shell; this excited state is unstable and temporary, and so it jumps back to the original position. As it returns to its ground state, it gives off energy in the form of heat, and this heat is then absorbed by a neighboring chlorophyll, which passes on the heat to another neighboring chlorophyll and so on until the heat reaches the reaction center chlorophyll. At this point, the reaction center chlorophyll's electron gets excited and passed on to the primary electron acceptor, and the reaction center chlorophyll results with a missing electron that is regained from H2O (electron donor). Before this stage, H2O is converted to 2e- + 2H + 1/2 O2 and this is how it is able to donate an electron. The primary electron acceptor now has an extra electron, which is donated to the electron transport chain that has H+ pumping. The ATP synthase pumps the H+ back in to the stroma and converts ADP + P → ATP. All of this occurs in Photosystem II. Then, the final electron acceptor is the reaction center chlorophyll of PhotoSystem I. PS I receives an electron from PS II, and the primary electron acceptor of PS I donates the electron to another electron transport chain. This electron transport chain has no H+ pumping and no ATP; instead, it generates NADPH from NADP+. Thus, the purpose of the light reaction is to convert light to chemical energy in the form of ATP and NADPH [3]
Photosystem II and I
There are three steps in converting Light energy to ATP and NADPH
To capture the light energy from the sun
Undergo Photosystem II and I to convert ATP and NADPH
The converted ATP and NADPH is used to reduce CO2 to sugar
The photosystems undergo in the internal membrane called Thylakoid membrane. Thylakoid membrane is membrane full of collections of pigments and proteins. There are four types of pigments they are: chlorophylls, carotenes, xanthophylls and phycobilins. The light reaction uses the light power to create ATP and NADPH2 to provide reducing energy to undergo the chemical reaction to reduce the CO2 to sugar.
[4]
Calvin Cycle is also known as the dark reaction part of the photosynthesis in which reduction of carbon atoms from carbon dioxide to a reduced state of hexose occurs by utilizing ATP and NADPH produced by the light reactions. Another reason why Calvin Cycle is known to be the dark reaction is because unlike light reactions, this reaction is independent of the presence of light. This cycle was first formed by Melvin Calvin. The Calvin Cycle uses sunlight as an energy source to synthesize glucose from carbon dioxide gas and water for photosynthetic organisms. This introduces all the carbon atoms used as a fuel source and as backbones of biomolecules in life. There are a lot of similarities between the Calvin Cycle and the Pentose Phosphate Pathway. Like mirror images of each other, the pentose phosphate pathway generates NADPH by breaking down the glucose into carbon dioxide. Similarly, the Calvin Cycle reduces the carbon dioxide to generate hexoses using NADPH.
Calvin Cycle Intermediate
Biochemists tried to figure out the mechanism of carbon dioxide fixation, believing that agricultural photosynthesis could be made more efficient. In each "turn" of the cycle, one molecule of carbon dioxide is condensed with the five-carbon sugar. The resulting six-carbon intermediate splits into two molecules of 3-phosphoglycerate. Besides, the water and the phosphate group are recycled during biosynthetic assimilation of G3P [An Evolving Science].
The stages of Calvin Cycle occurs in the stroma of chloroplasts, the photosynthetic organelles.
Three stages include:
1) Two molecules of 3-phosphoglycerate formed by fixation of carbon dioxide by ribulose 1,5-bisphosphate
In the beginning of this process, the ribulose 1,5-bisphosphate is converted into a highly reactive enediol intermediate. With the enediol intermediate, the carbon dioxide molecule is condensed into an unstable six-carbon compound. Rapidly, this unstable compound is hydrolyzed to two molecules of 3-phosphoglycerate. This reaction is highly exergonic with the Gibbs free energy equalling to -51.9 kJ/mol. This is catalyzed by rubisco which is also known as ribulose 1,5-bisphosphate carboxylase / oxygenase, an enzyme found in the stromal surface of the thylakoid membranes of chloroplasts. This reaction is very important because it is the rate-limiting step of the hexose synthesis. The structure of rubisco in chloroplasts contains eight large subunits (L, 55-kd) and eight small subunits (S, 13-kd). Each of the L subunits have a regulatory site and a catalytic site. Each of the S chains enhance L chains’ catalytic activities. Rubisco is known to be one of the most abundant enzymes and even the most abundant protein in the biosphere. Due to its slowness, rubisco must have large amounts present for the catalysis to work.
- Rubisco: For activity, it requires a bound divalent metal ion, commonly magnesium ion. By stabilizing a negative charge, the magnesium ion serves to activate a bound substrate molecule. It requires a carbon dioxide molecule other than the substrate to conclude the assembly of the magnesium ion binding site in rubisco. This carbon dioxide molecule is added to the uncharged ε-amino group of lysine 201 which forms a carbamate. Then, the negatively charged adduct binds to the magnesium ion. Although the formation of the carbamate will form spontaneously at a lower rate, it is enabled by the enzyme rubisco activase. Magnesium ion plays an important role in binding ribulose 1,5-bisphosphate and activating it to react with carbon dioxide. Magnesium ion and ribulose 1,5-bisphosphate bind together through its keto and adjacent hydroxyl group. The complex forms an enediolate intermediate through deprotonation. This reactive species couples with carbon dioxide and forms a new carbon-carbon bond. Including the newly formed carboxylate, the product is coordinated to the magnesium ion through three groups. An intermediate is formed when H2O is added to β-ketoacid which cleaves to form two molecules of 3-phosphoglycerate.
- Rubisco also causes catalytic imperfection by catalyzing a wasteful oxygenase reaction. Instead of reacting with carbon dioxide, the magnesium ion sometimes reacts with O2 which catalyzes a deleterious oxygenase reaction. The resulting products of this reaction are 3-phosphoglycerate and phosphoglycolate. Just like the carboxylase reaction, this oxygenase reaction requires the lysine 201 to be in the carbamate form. However, rubisco is prohibited from catalyzing the oxygenase reaction when carbon dioxide is not present because the carbamate only forms when carbon dioxide is present.
2) Hexose sugars formed by the reduction of 3-phosphoglycerate
The resulting product of rubisco, 3-phosphoglycerate, is converted into fructose 6-phosphate which isomerizes to glucose 1-phosphate and glucose 6-phosphate. Mixture of three phosphorylated hexoses is known as hexose monophosphate pool. The reaction of this conversion is very similar to the gluconeogenic pathway, except that glyceraldehyde 3-phosphate dehydrogenase is specific for NADPH rather than NADH which generates glyceraldehyde 3-phosphate (GAP). Carbon dioxide is brought up to the level of a hexose by the product catalyzed by rubisco and these reactions. Then, carbon dioxide is converted into a chemical fuel at the expense of NADPH and ATP which are generated from the light reactions.
3) Fixation of more carbon dioxide through the regeneration of ribulose 1,5-bisphosphate
The last phase of the Calvin Cycle is the regeneration of ribulose 1,5-bisphosphate, which is the acceptor of carbon dioxide in the first phase. From six-carbon and three-carbon sugars, a five-carbon sugar must be constructed. In the process of rearranging the carbon atoms, transketolase and aldolase play a major role. The transketolase transfesr a two-carbon unit from a ketose to an aldose by utilizing the coenzyme thiamine pyrophosphate (TPP). On the other hand, aldolase catalyzes an aldol condensation between an aldehyde and dihydroxyacetone phosphate (DHAP). Although this enzyme agrees with wide variety of aldehydes, it is very specific for dihydroxyacetone phosphates. In sum, when forming the five-carbon sugars, transketolase converts the three carbon and the six carbon sugars into a five carbon sugar and a four carbon sugar. The next process is when aldolase combines the four carbon sugar and a three carbon sugar to form a seven carbon sugar. The final step is that the seven carbon sugar reacts with another three-carbon sugar in order to form two more five carbon sugars. When the process for forming five carbon sugars are complete, ribose 5-phosphate is converted into ribulose 5-phosphate by the phosphopentose isomerase. Meanwhile, xylulose 5-phosphate is converted into ribulose 5-phosphate by phosphopentose epimerase and ribulose 5-phosphate is converted into ribulose 1,5-bisphosphate by phosphoribulose kinase. The following reaction shows the overall sum:
Fructose 6-phosphate + 2 glyceraldehyde 3-phosphate + dihydroxyacetone phosphate + 3 ATP → 3 ribulose 1,5-bisphosphate + 3 ADP
Calvin cycle requires six rounds to be completed since in each round, one carbon atom is reduced. In order to phosphorylate 12 molecules of 3-phosphoglycerate to 1,3-bisphosphoglycerate, 12 molecules of ATP are expended. In order to reduce 12 molecules of 1,3-bisphosphoglycerate to glyceraldehyde 3-phosphate, 12 molecules are NADPH are consumed. This is the net reaction of the Calvin cycle:
6 CO2 + 18 ATP + 12 NADPH + 12 H2O → C6H12O6 + 18 ADP + 18 Pi + 12NADP+ + 6H+
Below shows a diagram of the net reaction of the Calvin cycle:
Roles of Hexose
In plants, there are two major storage forms of sugar which include starch and sucrose. Starch is very similar to its animal counterpart glycogen but has less branches since it has a smaller proportion of α-1,6-glycosidic linkages. Also, the activated precursor is ADP-glucose, not UDP-glucose. Starch is commonly known to be a polymer of glucose residues which is synthesized and stored in chloroplasts. Distinctly, sucrose, a disaccharide, is synthesized and stored in the cytoplasm. Plants are able to transport triose phosphates from the chloroplasts to the cytoplasm, but they lack the potential to transport hexose phosphates across the chloroplast membrane. In exchange for a phosphate through the phosphate translocator, the triose phosphate intermediates cross into the cytoplasm. From the triose phosphates, fructose 6-phosphate is formed which joins the glucose unit of UDP-glucose. This forms the sucrose 6-phosphate. The phosphate hydrolyzes and yields sucrose which is stored in many plant cells.
Regulation occurs when the stromal environment alters by the light reactions. pH increases in the light reactions and concentrations of magnesium ion, NADPH, and reduced ferredoxin. These changes help couple the Calvin cycle to the light reactions. Specifically, rubisco gets activated when the concentration of these molecules increases and the pH increases. Activity of rubisco increases because light creates the carbamate formation which is a necessity in enzyme activities. In the stroma, when the concentration of magnesium ion increases, the pH also increases from 7 to 8. From the thylakoid space, the magnesium ions are released in order to create the influx of protons into the stroma. Carbon dioxide is added to the rubisco’s deprotonated form of lysine 201 while magnesium ion is bound to the carbamate in order to generate enzyme’s active form. Therefore, the light generates the regulatory signals, ATP, and NADPH.
Thioredoxin
One of the important molecule in regulating the Calvin cycle is known as thioredoxin. When thioredoxin is oxidized, it contains a disulfide bond. This disulfide bond is converted into two free sulfhydryl groups when the thioredoxin is reduced with the reduced ferredoxin. Reduced form of thioredoxin can cleave disulfide bonds in enzymes which activates some of the Calvin cycle enzymes and inactivates some of the degradative enzymes. Examples of enzymes that are regulated by thioredoxin include: rubisco, fructose 1,6-bisphosphatase, glyceraldehyde 3-phosphate dehydrogenase, sedoheptulose 1,7-bisphosphatase, glucose 6-phosphate dehydrogenase, phenylalanine ammonia lyase, phosphoribulose kinase, and NADP+-malate dehydrogenase.
C4 Pathway
By having a high concentration of carbon dioxide at the site of the Calvin cycle, plants are able to prevent very high rates of wasteful photorespiration when growing in hot climates. The process behind this is that C4 (four carbons) compounds carry carbon dioxide from mesophyll cells. Carbon dioxide is concentrated by the ATP in mesophyll cells in the bundle-sheath cells. This decarboxylation of C4 compounds in the bundle-sheath cells have the ability to maintain high concentrations of carbon dioxide in the Calvin cycle. The remaining three carbons are returned to the mesophyll cell to proceed another round of carboxylation. The transportation of the carbon dioxide in the C4 pathway begins inside the mesophyll cell when the carbon dioxide and phosphoenolpyruvate is condensed to form oxaloacetate. This reaction is catalyzed by the phosphoenolpyruvate carboxylase. At times, by an NADP+ linked malate dehydrogenase, oxaloacetate may be converted into a malate. This malate enters the bundle-sheath cell and is decarboxylated inside the chloroplasts. By condensing the ribulose 1,5-bisphosphate, the released carbon dioxide enters the Calvin cycle. In the last process, pyruvate by pyruvate-Pi dikinase forms the phosphoenolpyruvate. This is the C4 pathway net reaction:
CO2 (mesophyll cell) + ATP + 2H2O -> CO2 (bundle-sheath cell) + AMP + 2 Pi + 2 H+
References
Berg, Jeremy M., John L. Tymoczko, and Lubert Stryer. Biochemistry. 7th ed. New York: W.H. Freeman, 2012. Print.
As we all have known that cellular energy is inevitably important for metabolism of the body in all organisms, one important factor controlling all those metabolic pathways is enzymes. Most enzymes are proteins, which are synthesized by ribosomes in the cytoplasm. The precursor for protein synthesis are amino acids, which are obtains from the translation of RNA's, which come from the transcription of DNA. Therefore, DNA and RNA play major roles in metabolism. In order to synthesize DNA and RNA, there is one important precursor, which is Pentose Phosphate. There is a pathway which produces Pentose Phosphate and NADPH (which has a phosphate group at 2'-C of NADH instead of the hydroxyl group)
It starts with Glucose 6-Phosphate, Glu 6-P, which comes from other pathways, glycolysis for example.
Instead of using the enzyme glucose 6-phosphate isomerase to isomerize the original reactant into fructose 6-phosphate for glycolysis, cells use another enzyme, glucose 6-phosphate dehydrogenase, and one important cofactor, NADP+, to oxidize the Glu 6-P into 6-phosphoglucono-δ-lactone with NADP+ being reduced to NADPH.
Next, the enzyme lactonase hydrolyzes 6-phosphoglucono-δ-lactone into 6-phosphogluconate.
In this step, cofactor NADP+ is used again as an oxidizing agent to oxidize 6-phosphogluconate to ribulose 5-phosphate in a reaction catalyzed by the enzyme 6-phosphogluconate dehydrogenase with the reduced NADPH as another product and the release of carbon dioxide.
Note that in every step of the pathway, an addition of Magnesium cation helps stabilizing the reactions, which involves releases of electrons and protons.
In a ketose-aldose reaction catalyzed by the enzyme phosphopentose isomerase, ribulose 5-phosphate is isomerized into ribose 5-phosphate, a precursor for later important reactions, such as DNA synthesis.
The Pentose Phosphate pathway produces to important precursors for basic building blocks of life:
The production of ribose for the synthesis of some cofactors, DNA, RNA.
The production of NADPH for the synthesis of fatty acids, steroids, and other oxidation-reduction reactions (some occur in photosynthesis, especially in the Calvin cycle)
The human body is approximately divided into different categories of systems:
Nervous System
Circulatory System
Respiratory System
Muscular System
Endocrine System
Digestive System
Integumentary System
Other systems can include, lymphatic, immune, excretory, skeletal, or reproductive systems. The lymphatic system is a system that is designed to move fluids and nutrients through the body. Also, it is used to generate disease fighting antibodies. The immune system is the system that protects our bodies from foreign invaders. Helps prevent infection and diseases by employing white blood cells and antibodies. The excretory system is for excretion of urine and waste, while the skeletal describes how bones, tendons and ligaments all interact. Finally, the reproductive system is allow for the reproduction of offspring through gametes like eggs and sperm. [1]
The human nervous system.A nerve cell.The propagation of an action potential.
The nervous system is a network of specialized cells that coordinate the actions of an animal and send signals from one part of its body to another. These cells send signals either as electrochemical waves traveling along thin fibers called axons, or as chemicals released onto other cells. The nervous system is composed of neurons and other specialized cells called glial cells (plural form glia).
In most animals the nervous system consists of two parts, central and peripheral. The central nervous system contains the brain and spinal cord. The neurons of the central nervous system are interconnected in complex arrangements and transmit electrochemical signals from one to another. The peripheral nervous system consists of sensory neurons, clusters of neurons called ganglia, and nerves connecting them to each other and to the central nervous system. Sensory neurons are activated by inputs impinging on them from outside or inside the body, and send signals that inform the central nervous system of ongoing events. Motor neurons, situated either in the central nervous system or in peripheral ganglia, connect neurons to muscles or other effector organs. The interaction of the different neurons form neural circuits that regulate an organism's perception of the world and its body and behavior.
Nervous systems are found in most multicellular animals, but vary greatly in complexity. Sponges have no nervous system, although they have homologs of many genes that play crucial roles in nervous system function, and are capable of several whole-body responses, including a primitive form of locomotion. Radiata, including jellyfish, have a nervous system consisting of a simple nerve net. Bilaterian animals, which include the great majority of vertebrates and invertebrates, all have a nervous system containing a brain, spinal cord, and peripheral nerves.
A human nerve cell is composed of various components: the soma, or cell body (which has a nucleus), the axon (by which nerve signals travel), the myelin sheath, which provides conductivity and allows electrical signals to travel through nerve cells, dendrites, which receive signals from other nerve cells, and axon terminals, which nerve cells use to communicate with each other via the release and binding of neurotransmitters.
Neurons communicate with each other using neurotransmitters, which travel across synapses (the space between axon terminals of one nerve cell and the dendrites of another nerve cell) and bind to their appropriate receptors. However, inter cellular communication between nerve cells depends on action potentials, which are voltage differences across membranes. Action potentials are initiated by the movement of charged ions, such as potassium and sodium, across the cell membrane through voltage dependent ion gates. These gates are opened by binding of neurotransmitters to post-synaptic cells. Thus, when a neurotransmitter binds and causes the voltage dependent ion gates to open, ions flow across the membrane, causing a voltage difference which results in an action potential.
These action potentials travel along the axon, and axon terminals and dendrites allow these potentials to move through various nerve cells. Action potentials function on the all or nothing principle. In other words, if a particular stimuli or neurotransmitter concentration does not reach required levels, no action potential will occur. Thus, if a mosquito lands on your hand, you may not feel it because the pressure changed caused by the mosquito landing on you is not significant enough to generate an action potential. However, the pressure of, for example, a handshake, does, and therefore generates an action potential, causing you to feel the other hand. Under the all or nothing principle, action potentials either occur or they do not- an amplitude difference is irrelevant so long as the threshold for an action potential is reached. Instead, the varying feeling you get depends on the rate, or frequency, of action potentials. In other words, if someone threw a pencil at you, it would hurt less than if someone hit you with a car not because the amplitude of the action potential is higher when you are hit by a car, but because nerves are transmitting action potentials much faster.
The myelin sheath surrounding axons is critical to the propagation of action potentials. It essentially serves to maintain conductivity; without it, action potentials would travel much more slowly (so, for example, you would not be able to feel something hit you until after several seconds). This also improves efficiency and decreases the amount of energy required for nerve signaling. Multiple sclerosis is an example of disease caused by the degradation of the myelin sheath in nerve cells. The degradation of this sheath prevents nerve cells from communicating with each other by reducing the effect and velocity of action potentials. Because many important functions depend on a healthy nervous system, such as speech, movement, coordination, sensation, and vision, the degradation of the myelin sheaths can have a debilitating effect.
All neurons exhibit a resting membrane potential which is the membrane potential of a resting neuron. Recall the definition of electricity, there is a voltage difference between the inside of the neuron and the extracellular space. The difference, resting potential, is usually about -70 mV between inside and outside of the neuron. However, the system then would want to equilibrate to 0 mV. Neurons use selective permeability to ions and the Na+/K+ ATPase to maintain a negative internal environment.
The neuron also has a plasma membrane that is fairly impermeable to charged species. Ions are unlikely to cross the non-polar barrier, because it is energetically unfavorable. Inside the neuron, the concentration of potassium ion is high and concentration of sodium ion is low. Outside of the neuron has the opposite condition. The negative resting potential is generated by both permeability of the membrane to potassium ion compared with sodium ion. If potassium ion is more permeable and its concentration is higher inside, it will diffuse down its gradient out of the cell. In terms of charge movement, potassium ion is positively charged, so its movement out of the cell results in a cell interior that is negative. If we assume that the membrane starts at zero, and we take away a positive one, we end up with a negative one on the inside of the cell. Sodium ion cannot readily enter at rest, so the negative potential is maintained.
The Na+/K+ ATPase is important for restoring the gradient after action potentials have been fired. They transport three Na+ out of the cell for every two K+ into the cell at the expense of one ATP with a Na+/K+ pump. ATP is qualified as active transport. Each time the pump works, it results in the inside of the cell becoming relatively more negative, as two positive charges are moved in for every three that moved out.
Are neurons the only cells with the resting membrane potential?
No. All cell have the resting membrane potential. Neurons and muscle tissues are unique in using the resting membrane potential to generate action potentials.
Modeling of the Resting Potential
• Resting potential can be modeled by an artificial membrane that separates two chambers: --1. The concentration of KCl is higher in the inner chamber and lower in the outer chamber. --2. K+ diffuses down its gradient to the outer chamber. --3. Negative charge builds up in the inner chamber.
• At equilibrium, both the electrical and chemical gradients are balanced.
• The equilibrium potential (Eion) is the membrane voltage for a particular ion at equilibrium and can be calculated using the Nernst equation:
Eion = 62 mV (log[ion]outside/[ion]inside)
• The equilibrium potential of K+ (EK) is negative, while the equilibrium potential of Na+ (ENa) is positive.
• In a resting neuron, the currents of K+ and Na+ are equal and opposite, and the resting potential across the membrane remains steady.
Neurons are the cell of the nervous systems. Neurons carry electrical signals and communicate with each other via junction called synapse. Neurotransmitters, mainly hormones (epinephrine), are chemical that is released at synapse. Neurons uses membrane portention. The resting neuron membrane potential is -70mV. A neuron is composed of a soma - the cell body, and lots of dendrites. There are three types of neurons: Sensory neurons, interneurons, and motor neurons. Sensory Neurons are found in the Peripheral Nervous System. They communicate the signals from the external and internal environment to the Central Nervous System. Interneurons can only communicate between neurons. These are found in the Central Nervous System. Interneurons integrate signals and synapse with other neurons. Motor neurons carry out the signals from the Central Nervous System out to the effectors.
Glial cells are supporting cells. Types of glial cells include astrocytes, oligodendrocytes, schwann cell, and myelination. Astrocytes are associated with capillary beds. It prevents capillary beds from leaking. It forms the barriers between the blood and the brain and there is great selectivity. Oligodendrocytes form insulating myelin sheath in the Central Nervous System. Schwann cells insulate myelin sheath associated with axon, it is found in the Peripheral Nervous System. Myelination wrap around axons and serves as electrical insulation. Multiple Sclerosis is a disorder caused by the deterioration of the myelin sheath.
Action potentials are a means of communicating between neurons. An action potential is started when the membrane is depolarized to a certain threshold. Action potentials are all or none, so depolarizations that do not meet the threshold do not do anything for the neuron. The threshold is a point when the charge is -55 mV. The resting potential of a neuron, which is the charge at equilibrium, is around -70 mV. The threshold is 15 mV above the resting potential, and only when the cell depolarizes to this point will the action potential initiate.
The action potential starts when voltage gated sodium channels are activated. These channels allow an influx of sodium. Potassium channels also open up, which causes an efflux of potassium ions. The efflux of potassium ions causes the membrane to hyperpolarize
(makes more negative) the cell. If the current of potassium exceeds the current of sodium, then the voltage of the cell returns to -70 mV, which is the resting potential. If the voltage increases past the threshold level, then the sodium current is larger than the potassium current. This induces a positive feedback, where more sodium channels are opened (slowly) from this effect, and even more sodium ions enter the cell. This sharp increase in the flow of sodium ions causes the cell to depolarize rapidly, which results in the cell “firing”, which produces an action potential.
The rapid depolarization is ended when the sodium channels open all the way. This causes the membrane voltage to reach a maximum. The voltage shuts the sodium channels off, and the channels are inactivated. At the same time, the voltage opens voltage gated potassium channels. The result of these two actions is the repolarization of the membrane. As potassium ions leak out and sodium channels can no longer diffuse across the membrane, the cell is brought to its equilibrium potential.
An action potential consists of various phases.
(1) At the resting potential, voltage-gated sodium channels are closed. Some potassium channels are open, but most voltage-gated potassium channels are closed.
(2) When the membrane depolarizes, some voltage-gated sodium channels open, allowing the influx of sodium ions. The sodium ion influx causes further depolarization, which more voltage-gated sodium channels will open, causing more sodium ions to flow in.
(3) Once threshold is reached, an action potential will occur. The positive-feedback cycles of opening of sodium channels depolarize the membrane rapidly
(4) Voltage-gated sodium channels will inactive after opening, stopping the influx of sodium ions. Voltage-gated potassium channels will open and potassium ions will flow out.
(5) The gated potassium channels eventually close, and the membrane potential returns to the resting potential.
(6) The refractory period is a result of the closing of the sodium channels, which cannot be opened again until the refractory period is over.
Information transmitting occurs at the synapses. There are two types of synapses - Electrical synapses and Chemical synapses. At the electrical synapses, the electric current flows from one neuron to another neuron through gap junction. At the chemical synapses, chemical neurotransmitter carries the information through the synaptic cleft.
The central nervous system consists of the brain and the spinal cord. The spinal cord is a long nervous tissue that extends along the vertebral column from the head to the lower back. It is composed of many distinct structures working together to coordinate the body. The most important is the brain, which has several components:
The cerebrum is the largest portion of the brain and it controls consciousness. It is in control of voluntary movement, sensory perception, speech, memory, and creative thought.
The cerebellum helps to fine-tune voluntary movement, but is not directly involved in it. It makes sure that movements are coordinated and balanced.
The brainstem is a part of the medulla oblongata and is responsible for the control and regulation of involuntary functions. These functions include breathing, cardiovascular regulation, and swallowing. The medulla oblongata is needed to sustain life and processes a great deal of information.
The hypothalamus is the source of posterior pituitary hormones and releasing hormones acting on the anterior pituitary. It is also responsible for homeostasis maintenance, which includes regulation of temperature, hunger, thirst, water balance, generation of emotion, as well as roles in sexual and mating behaviors.
cerebrum lobes
Brain
It is responsible for integration of sensory information, coordination of motor movement, and cognition. The myelination that we saw around axon is also present in the brain. Its presence allows us to distinguish between gray matter, which is unmyelinated, and white matter. The brain of all vertebrates develops from dividing it into the forebrain, the mid-brain, and the hindbrain.
-Forebrain is the most recently acquired part of the CNS in terms of evolutionary development. It is further broken down into the telencephalon and diencephalon. Telencephalon consists of a pair of large left and right hemispheres that can be further sectioned into the frontal, parietal, occipital and temporal lobes. A group of structures located deep with the cerebrum that makes up the diencephalon. A large portion of the diencephalon is the cerebral cortex, a region of highly convoluted gray matter that can be seen on the surface of the brain. The cortex is responsible for the highest-level functioning in the nervous system, including creative thought and future planning. It also integrates sensory information and controls movement. Each hemisphere is independent, however, they do communicate through a large connection called the corpus collosum.
-Midbrain serves as a relay point between more peripheral structures and the forebrain. It passes sensory and visual information to the forebrain, while receiving motor instructions from the forebrain and passing them to the hindbrain.
-The hindbrain contains three “main structures” that are medulla oblongata, pons, and cerebellum. Together, they make the brainstem. Medulla oblongata is the most highly conserved part of the brain that is responsible for modulating ventilation rate, heart rate, and gastrointestinal rate. “The pons seems to serve as a relay station carrying signals from various parts of the cerebral cortex and cerebellum. The pons also participates in the reflexes that regulate breathing.” The cerebellum is a quality control agent that checks the motor signal sent from the cortex is in agreement with the sensory information coming from the body. It is what prevents us from falling over when we trip. It rapidly realizes that the motor signal to take a step was not successfully carried out, as we tripped. Instead of letting us fall on our faces, the cerebellum helps the cortex to adjust to the new situation so that we catch ourselves.
The Spinal Cord
The spinal cord is divided into 4 distinct regions. These distinct regions in the spinal cord organize neurons segmentally. Within a single segment, neurons are grouped and located according to their function throughout the rest of the body. These four segments are the cervical region (8 segments), thoracic region (12 segments), lumbar region (5 segments), and the sacral region (5 segments). Each region is responsible for different parts of the human body. For example, the cervical region is responsible for controlling the arms and the lumbar region is responsible for controlling the legs. Axons usually enter the nerves near each segment in this innervated structure (e.g. fibers that innervate the arm run in the cervical spinal nerves while fibers that innervate the leg run in the lumbosacral spinal nerves). Sensory and motor neurons lie in separate portions of the spinal cord. In general, cells in the dorsal spinal cord and axons in the dorsal spinal nerves serve an afferent function (sensory fibers), while the cells in the ventral spinal cord and axons in the ventral spinal nerves serve as motor in function (efferent fibers). In specific, however, position of ascending pathways carrying fine touch, pressure, and information about the position of muscles and joints are found in the dorsal and lateral columns of each spinal segment. The dorsal column pathway runs on the same side of the spinal cord and crosses at the brain stem, goes through the medial lemniscus, and travels to the cerebral cortex. The position of fibers carrying pain information and pressure information are found in the anterolateral pathway. Information is carried in contralateral anterolateral tracts: the spinothalamic and spinoreticular tracts. Spinoreticular tract ends in the brain stem while spinothalamic tract ends in the thalamus. The anterolateral pathway crosses at the spinal cord and travels to the cerebral cortex. For motor information (efferent information), positions of descending fibers are found in the dorsolateral and ventromedial columns.
The Peripheral Nervous System
This system consists of a sensory system that carries information from the senses to the central nervous and then back to the body. Vertebrate PNS structurally consist of left and right pairs of cranial and spinal nerves and associated ganglia. The cranial nerves start from the brain and end mostly in organs of the head and upper body. The spinal nerves start in the spinal cord and spreads into parts of the body below the head. It also consists of a motor system that branches out from the central nervous system, so it targets certain muscles or organs. The motor system can be divided into the somatic system and the autonomic system.
The Somatic Nervous System
The somatic nervous system is responsible for voluntary movement. We described the interface between the neuron and muscle as the neuromuscular junction. Release of acetylcholine fom the nerve terminal onto the muscle leads to contraction. The acetylcholine binding to its receptor on the muscle ultimately leads to muscle depolarization. The somatic nervous system is also responsible for providing us with reflexes, which are automatic. They do not require input or integration from the brain o function. There are two types of reflex arcs: monosynaptic and polysynaptic. Reflexes usually serve a protective purpose. For example, we’d pull our hand away from a hot stove before our brain processes that it is hot.
-Monosynaptic
In a monosynaptic reflex arc, there is a single synapse between the sensory neuron that received the information and the motor neuron that responds. An example will be the knee jerk reflex. When the patellar tendon is stretched, information travels up the sensory neuron to the spinal cord, where it interfaces with the motor neuron to contract the quadriceps muscle. The net result is a straightening of the leg, which lessens the tension to the patellar tendon. However, this reflex is responding to a potentially dangerous situation. If the patellar tendon is stretched too far, it may tear, damaging the knee joint. This reflex helps to protect us.
-Polysynaptic
In a polysynaptic reflex arc, there is at least one interneuron between the sensory and motor neuron. An example will be your reaction to stepping on a tack, which involves the withdrawal reflex. The foot that steps on the tack will be simulated to jerk up (monosynaptic reflex). However, if we are to maintain our balance, we need out other foot to go down and plant itself on the ground. For this to occur, the motor neuron that controls the opposite leg must to be stimulated. Interneuron in the spinal cord provide the connection from the incoming sensory information on the leg being jerked up to the motor neuron for the supporting leg.
On the other hand, the autonomic system controls tissues other than skeletal muscles, such as cardiac muscle, glands, and organs. This system controls processes that are involuntary, such as heartbeat, movements in the digestive tract, and contraction of the bladder. Autonomic neurons are able to either excite or inhibit target muscles or organs. This nervous system is subdivided into sympathetic division and parasympathetic division. These 2 systems work antagonistically and usually have opposite effects.
The Autonomic Nervous System
The autonomic nervous system is a part of the peripheral nervous system that controls visceral functions. The autonomic nervous system affects heart rate, digestion, respiration rate, salivation, perspiration, diameter of the pupils, urination, and sexual arousal. Although most of this system's actions are voluntary, some actions, such as breathing, are involuntary. The autonomic nervous system is divided into the sympathetic nervous system and the parasympathetic nervous system. These two subsystems work together to produce homeostasis. Both subsystems are made up of a two-neuron chain between the central nervous system and the peripheral nervous system.
In the sympathetic and parasympathetic nervous systems, pre-ganglionic neurons are in the central nervous system, but they synapse to post-ganglionic neurons in the peripheral nervous system. These post-ganglionic neurons will then synapse to the target cells.
In the sympathetic nervous system, the preganglionic transmitter is acetylcholine, and they bind to nicotinic cholinergic receptors on post-ganglionic cells. Post-ganglionic cells transmit nor-epinephrine, which bind to adrenergic receptors on target cells. The parasympathetic nervous system releases acetylcholine as well as the pre-ganglionic transmitter, and bind to nicotinic cholinergic receptors on post-ganglionic cells. The transmitter from these cells is acetylcholine, and they bind to muscarinic cholinergic receptors on target cells.
Increased activity in the sympathetic nervous system includes increase of heart rate, increase in blood pressure of smooth muscles, and decrease of gut motility. Parasympathetic activity decrease heart rate and increases gut motility.
Reflex Arcs
Occurs when a signal from the peripheral nervous system travels straight to another site in the body, triggering an involuntary reaction with the signal entering the central nervous system and traveling to the brain for processing before a reply signal is sent back. This results in a much faster reaction time and is typically used in situations where bodily harm is imminent.
For example, when you pull your hand back after touching a hot surface, you don't have to think about it. In this case a reflex arc was activated allowing you to remove your hand from a damaging heat source faster, resulting in less damage to the cellular structure of your skin.
Sympathetic Nervous System
The sympathetic nervous system controls the body's resources under stress, otherwise known as the fight-or-flight response. However, the sympathetic nervous system is constantly active in order to maintain homeostasis. An example would be that when heart beats faster, the liver would convert glycogen to glucose, bronchi of the lungs would expand to support increased gas exchange.
Examples of sympathetic system action.
Organ
Effect
Eye
Dilates pupil
Heart
Increases rate and force of contraction
Lungs
Dilates bronchioles
Digestive tract
Inhibits peristalsis
Kidney
Increases renin production
Penis
Promotes ejaculation
Parasympathetic Nervous System
The parasympathetic nervous system controls activities that occur when the body is at rest such as salivation, lacrimation, urination, digestion, and defecation. It's actions are often described as "rest and digest." It works in conjunction with the sympathetic nervous system to maintain homeostasis in the body. An example would be the increase activity in parasympathetic division decreases heart rate would increase glycogen production and enhance digestion.
Examples of parasympathetic system action.
Organ
Effect
Eye
Constricts pupil
Heart
Decreases rate and force of contraction
Liver
Glycogen synthesis
Digestive system
Increases activity
Kidney
Increases urine production
Tear Glands
Secretion
What is Mental Inertia and what causes this symptom. It is the involuntary or the unwillingness to perform something. In the other hands, we can say it is slacking in people’s mind to think of something or come up with a plan. People usually call that in a normal way is laziness that is hidden somewhere inside each of us. And based on each person’s function, it will display different level of the laziness. Therefore, the immunity is also different. So when we can break this slacking and laziness, we can create the impulse. There are many types that that cause by mental inertia:
- By incorrectly established result
- By adherence to a faulty technique
- By the incorrect understanding of mechanism of action
- By improper controls
Now we know what causes this symptom so we can find a way to overcome it. Here is just an example of how to overcome it. There are several ways to handle it. By mentally, try to see the result of our action and capture it. Then we will start moving to physically and let our brains follow the suit. We just try to start very slow to see the actual result from the small step. The most important thing is just believe in ourselves that we can do everything. It is very easy to break and control once we know how to overcome it.
Depression is a disorder characterized by depression mood, for example, appetite, sleep and energy level. There are two forms of depressive illness: major depressive disorder and bipolar disorder. Major depressive disorder maybe last many months with no pleasure and no interest. Bipolar disorder involves swings of mood form high to low and affects very rarely to people.
Schizophrenia is a very rare yet severe mental disturbance characterized by psychotic episodes in which patients have a distorted perception of reality. People with this illness usually suffer from hallucination and delusions.
Alzheimer’s disease is a mental deterioration, or dementia characterized by confusion and many other symptoms. This disease is progressive, with patients gradually becoming less able to function. The disease is mainly associated with the development of senility as a result of a buildup in β-amyloid plaques. However, the causes for the disease are not only genetically related, but they can stem from any damage to the axonal transport system which itself might arise from structural changes or even traumatic brain injury. The resulting damage incurred into the axonal transport system has the potential for vesicles containing chemical precursors to build up and create damaging plaques, as is the case with Alzheimer’s disease. Various other defects in axonal transport systems or mutations in gene coding can cause this neurodegenerative disorder. Which could lead to the death of neurons in many areas of the brain.
Parkinson’s disease is characterized by difficulty in initiating movement and slowness of movement. Its symptoms result from the death of neurons in the midbrain. At present, there is no cure for Parkinson’s disease.
Gorazd B. Stokin and Lawrence S.B. Goldstein, "Axonal Transport and Alzheimer's Disease". Annual Review of Biochemistry Vol. 75: 607-627 (Volume publication date July 2006) Print
Parkinson's disease is a progressive disorder of the nervous system which affects one's movement. Parkinson's disease is a motor system disorder in which is is the result of the loss of dopamine-producing brain cells.
Parkinson's disease is typically known for a common symptom of a tremor. However, patients can show signs of stiffness or slowing of movement.
Parkinson's disease is caused by slow progression of deterioration of neurons in the substantia nigra. The substantia nigra is known for producing dopamine. Dopamine is known to be a messenger in which is allows the substantia nigra and the corpus striatum to communicate with one another to initiate smooth and balanced muscle movement.
Although experts know what causes Parkinson's disease, there is no known reason for the impairment of the neurons. However, experts have an idea of may play a role in causing the impairment. These factors are either genetics or environmental factors.
Parkinson's signs and symptoms can vary from person to person. A younger patient can show particular signs and symptoms of the disease at an earlier age, while a much older patient can show those same exact symptoms later on.
It is common for symptoms to begin on one side of the body and slowly start effecting the other side of the body.
Common symptoms include:
Tremors in a hand, arm, or leg (typically the first symptom that most people notice)
Currently, there are no precise tests or exams to diagnose Parkinson's disease. However, a doctor may diagnose a patient based on their medical history, reviewing of signs and symptoms, and a neurological and physical examination. The physician may order certain tests to be done in order to rule out other possible diseases with similar signs and symptoms.
Currently, there are no cures for Parkinson's disease. However, medications can be prescribed to manage with certain symptoms. By providing certain medication, it can increase the brain's supply of dopamine.
Generally, a doctor may prescribe these medications:
The Circulatory System is an organ system that transfers the body's essentials such as blood cells, nutrients, gases, and etc. to and from the cells in order to maintain homeostasis. Under the circulatory system, there are two systems: cardiovascular system and the lymphatic system. The cardiovascular system consists of the heart, blood, and blood vessels, while the lymphatic system is composed of the lymph, lymph nodes, and lymph vessels.
Not all organisms require a circulatory system. Its primary purpose is to distribute nutrients and essential elements like oxygen throughout the cell. Smaller organisms, or those with a high surface area relative to their volume, do not need a circulatory system because transfer can take place directly across their cellular membranes. Flatworms are an example of this; due to their size and shape, cells can obtain nutrients and remove waste without the need for an extensive circulatory system because diffusion is sufficient. Human beings, however, require a circulatory system due to their size; for example, oxygen would not be able to effectively diffuse into organ cells without a circulatory system because oxygen would have to go through our skin and into organ cells for them to receive oxygen. In such a case, an internal circulatory system, a network of blood vessels, veins, and arteries, is beneficial because nutrients and oxygen can be gathered at a central location and distributed throughout the body.
Open/Closed Circulatory system
In open circulatory system is the circulatory fluid bathes the organs directly. In these animals, the circulatory fluid, called hemolymph is the same as interstitial fluid. The heart pumps hemolymph through vessels into sinuses, fluid-filled spaces where materials are exchanged between the hemolymph and cells. Arthropods and most mollusks have an open circulatory system.
A closed circulatory system circulates blood entirely within vessels, so the blood is distinct from the interstitial fluid. One or more hearts pump blood into large vessels that branch into smaller ones and the interstitial fluid bathing the cells. For example, annelids, such as earthworms have closed circulatory system. This is often called cardiovascular system.
Arteries, veins, and capillaries are the three main types of blood vessels. Within each type, blood flows in only one direction.
(1) Arteries carry blood away from the heart to organs throughout the body.
(2) Veins carry blood back to the heart.
(3) Capillaries are microscopic vessels with very thin, porous walls. Capillaries also touch every organ in the body.
The hearts of all vertebrates contain two or more muscular chambers. The chambers that receive blood entering the heart are called atria. The chambers that pump blood out of the heart are called ventricles.
The blood passes through the heart once in each complete circuit. In single circulation, blood that leaves the heart passes through two capillary beds before returning to the heart. When blood flows through a capillary bed, blood pressure drops substantially. Fish undergo single circulation, they have a two-chambered heart and single circuit. The blood must pass through two capillary beds. The capillary bed is where exchange takes place while oxygen is loaded and carbon dioxide is being unloaded. The blood collects in the first capillary bed and continues onto the second capillary bed. Two capillary beds are problematic because blood pressure decreases as it branches off into smaller branches and this occurs twice in single circulation causing blood pressure to decrease dramatically. The contraction of muscles and movement helps the system get blood back to the heart. This is why single circulation works for fish because they are constantly moving.
Pulmonary circulation is the portion of the circulatory system which carries oxygen-depleted blood away from the heart, to the lungs, and then returns oxygenated blood back to the heart. The pathway that blood takes in the pulmonary circuit starts at the right section of the heart. Oxygen-depleted blood from the body enters the heart through the right atrium where it pumped through the right atrioventricular valve and into the right ventricle. The blood is then pumped into the two pulmonary arteries, one for each lung, and travels into the lungs. The pulmonary arteries carry the deoxygenated blood to the lungs while the pulmonary veins carry oxygenated blood to the red blood cells. There they release carbon dioxide and then pick up oxygen during respiration. Now that the blood has been oxygenated, it leaves the lungs via the pulmonary veins and it returns to the left heart, completing the pulmonary cycle. The blood enters the left atrium and is pumped through the left atrioventricular valve and into the left ventricle. From here, the blood is distributed to the body via the systemic circulation before once again returning to the pulmonary circulation to pick up more oxygen.
Systemic circulation, as indicated with the word "system" in the title itself, is all throughout the body. This circulation's key role is to provide nourishment to all of the tissues located throughout your body. However, it does not benefit the heart and lungs because they have their own systems. Nonetheless, systemic circulation is a vital supporting piece of the circulatory system as a whole. The blood vessels, which consist of the arteries, veins, and capillaries, are responsible for the delivery of oxygen and nutrients to the tissue. Oxygen rich blood enters the blood vessels through the heart's major artery called the aorta. There exists the application of forceful contraction of the heart's left ventricle which compels the blood to flow through the aorta and into smaller arteries to distribute throughout the body. As a matter of fact, the inside layer of an artery is very smooth, thus allowing blood to flow rather quickly. As the atrium contracts in the heart, more blood is filling the ventricles. Eventually, the atrioventricular valve closes. The ventricles undergo isovolumetric contraction, which is when the ventricles contract but the volume does not change. Once the pressure in the ventricles exceeds the pressure in the aorta will that aortic valve open. In comparison to the inside layer of the artery, the outside of the artery is relatively strong, allowing blood to flow forcefully. As the blood flows through the arteries, the velocity of flow is fast. This is because the cross-sectional area of the arteries is small, and linear velocity is inversely proportional to the cross-sectional area. Once blood reaches the capillaries, the velocity decreases because capillaries have a large cross-sectional area. The second to last step of this systemic process involves oxygen-rich blood entering the capillaries where the oxygen and nutrients are then released. The process concludes with the collection of waste products and waste-rich blood that flow into veins to be taken back to the heart. The rest of the work is left to pulmonary circulation, which is mentioned in the section above. Other key aspects of systemic circulation include the fact that blood passes through the kidneys, which is known as renal circulation. During this phase, the kidneys filter as much of the waste from blood as possible. Blood also makes its way through the small intestine, which is known as portal circulation. In this phase, the blood from the small intestine collects in the portal vein which then passes through the liver. To explain with more specificity, the liver filters sugars from blood, storing them for later.
While pulmonary and systemic circulation provide oxygen and nutrients for the rest of the body, it is crucial not to forget about the heart's everlasting desire for nutrients as well. This is pursued through coronary circulation which refers to the movement of blood through the tissues of the heart. Despite the fact that blood fills the chambers of the heart, the muscle tissue of heart which is referred to as myocardium, is so thick to the point that it requires blood vessels to deliver blood deep into it. These blood vessels, responsible for delivering oxygen-rich blood to the myocardium, are known as coronary arteries. The vessels responsible for the removal of deoxygenated blood from the heart muscle are known as cardiac veins. The coronary arteries, when healthy, are capable of autoregulation to maintain coronary blood flow at levels appropriate to the needs of the heart muscle. The critical nature of coronary arteries are illustrated by means of classification as the "end circulation" because they represent the only source of blood supply to the myocardium.
The coronary circulation of the heart consists of blood (a fluid that carries materials between the outside world and the cells of the body), a lot of blood vessels (carrying blood throughout the body to deliver those nutrients to the various cells), and the main component of the coronary circulation system, the heart (which forces blood into the blood vessels. The pressure that the heart produces to push blood through the blood vessels is called the blood pressure. If the blood pressure becomes inadequate to produce a large enough force for the blood to move into the vessels, certain cells become starved for Oxygen, a condition referred to as ischemia.
In coronary circulation, in order to understand how the heart and the blood vessels can respond to autonomic and endocrine input and how they change their behavior, it is essential to look carefully at each of the components in the cardiovascular system. The heart consists of two separate pumps, joined together. These two halves of the heart are connected by parallel sets of blood vessels in series with one another. The right heart receives blood from most of the body and pumps the blood into the vessels of the lungs (the pulmonary circulation); the left heart receives blood from the lungs and pumps the blood into vessels into the rest of the body (the systemic circulation). Both the right and left heart are separated in themselves into two subsections, the atria and ventricles. Veins leading to the right heart are connected to the right atrium at first. The bloods that enters the right atrium is then pumped into the right ventricle, which is separated from the right atrium by the bicuspid valve. Once in the right ventricle, the blood circulates through arteries and veins in the pulmonary circulation into the left atrium. Once in the left atrium, the blood is allowed into the left ventricle as the mitral (tricuspid) valve opens and closes due to blood pressure differences in the two compartments.
After release into the left ventricle, blood is propelled past the aortic valve to flow to the rest of the body. For the reason that the left ventricle must produce enough force to send the blood through the rest of the body, the muscles of the left ventricle are much stronger than the muscles of the right ventricle.
The output of blood in the two halves of the heart must be exactly equal to one another on a minute-to-minute basis or else blood will be backed up in either the systemic or pulmonary circulation, which leads to major problems for the entire circulatory system. Damage to the heart results in a heart pump with less efficiency. In congestive heart failure, the left heart forces more blood than the right heart, a condition that is referred to as systemic edema. This condition can be treated with drugs that increase fluid excretion and that increase the strength of the heart as a whole. In left heart failure, the right heart pumps more blood than its counterpart, a condition referred to as pulmonary edema. This condition interferes with gas exchange in the lungs and can produce suffocation. If the heart becomes extremely weak overall, a condition referred to as congestive heart failure occurs. In congestive heart failure, residual blood pressure in the heart causes the heart to become abnormally full, greatly stretching the heart muscles. Under this condition, the heart becomes unable to even contract strongly and is unable to force any blood into the great arteries. This situation must be remedied immediately or death follows.
Body fluids are primarily distributed among three major compartments in the circulatory system. These three compartments are the volume inside of the cells (the intracellular compartment), the volume inside the circulatory system itself (the plasma compartment), and the volume that lies between the circulatory system and the intracellular compartment (the interstitial compartment). Under normal conditions, the three different compartments are in osmotic equilibrium with one another, but they do contain different distributions of solutes. There is a lot of organic anion (mostly proteins) inside cells, essentially none in the interstitial fluid, and a little in the plasma. Sodium and potassium ions are distributed with the inverse concentration profiles across cell membranes. The total number of millimoles of solute is equal in each of the three compartments. Cell membranes separate the intracellular and interstitial compartments. Capillary walls separate the interstitial and plasma compartments. Materials that must be exchanged between the different compartments must cross these barriers in order to reach the other side.
In all cases, the basic techniques of measuring body fluids consists of diluting an appropriate marker molecule (Evan's blue, inulin, H2O) into the volume that you would like to measure, allowing it to mix thoroughly in the compartment, and then measuring the concentration of the marker. The basic calculation is based on the equation volume = (quantity/concentration). There are, however, several possible sources of error tin this method.These errors include an inadequate mixing of the marker within the compartment, loss of the molecule marker by metabolism or excretion, and leakage into other compartments. an ideal marker substance for these measurements has the characteristics of going only into the compartment that is measured, is broken down or excreted very slowly compared to mixing time, and is lost as a simple exponential function of time (therefore allowing the extrapolation back to its initial concentration from the data. the plasma volume is measured using Evan's blue, which binds to blood cells and plasma proteins. The interstitial volume is measured using molecules that equilibrate between the plasma and the interstitial fluid, but that won't enter cells (inulin). Total body water is measured by deuterated or tritiated H2O, and with subtraction an intracellular volume is measured.
Body fluid compartments are separated by layers composed of cells that are arranged side-by-side with one another. The layer of cells that solutes and water traverse as they move between compartments are epithelia cells or endothelia cells. Chemical species move across there cell walls driven by concentration gradients or pressure gradients. These chemical species may traverse the capillary walls by either a transcellular pathway of through a paracellular pathway, or even both. Most cells of the epithelial layer are polarized, meaning that the properties of the cell membrane facing the interstitial fluid are different from the properties of the cell membrane of the very same cells facing the lumen of the tube. Chemical species can cross the paracellular pathway if there is a driving force on them and if they aren't too big. Passage through the transcellular pathway is more complex in that a transporter molecule may be needed to facilitate diffusion across the cell membrane.
The composition of mammalian blood is 55% plasma and 45% cellular elements.
Plasma
Plasma is about 90% water, the dissolved salts are an essential component of the blood. Some of these ions buffer the blood, which in humans normally has a pH of 7.4.
Cellular elements
Two classes of cells: red blood cells, which transports oxygen, and white blood cells, which function in defense. Blood also contains platelets, fragments of cells that are involved in the clotting processes.
One complete sequence of pumping and filling is a cardiac cycle. The contraction phase of the cycle is called systole while the relaxation phase is called diastole.
For an adult human at rest with a heart rate of about 72 beats per minute, one complete cardiac cycle takes about 0.8 second.
(1) During when atria and ventricle are in diastole, blood returning from the large veins flow into the atria and ventricle through the AV valves.
(2) A brief period of atrial systole then forces all blood remaining in the atria into the ventricles.
(3) During the remainder of the cycle, ventricular systole pumps blood into the large arteries through the semi-lunar valves.
Depolarization is passed down the bundle of Hiss to the apex of the heart.
Depolarization spreads upward from the apex, through the bundle branches. The bundle branches divide into left and right bundle branches.
The message is passed through the Purkinje fibers, causing the ventricles to contract.
The SA node is called the pacemaker of the heart because it has the fastest rhythm, about 80beats per minute. Every time the SA produces an action potential, the message is relayed the heart as described above. If the SA node becomes damaged and the message is not passed down, the AV node will take over as the heart's pace maker. The AV node beats a lot slower, only about 40beats per minute. Although this will be enough to keep a person alive, contraction will be too slow for strenuous activity. Doctors will recommend putting in an artificial pace maker in such a case.
The electrical activity of the heart, as described above, is measured with an electrocardiogram, or an ECG. This measurement is taken using electrodes on the skin, which are capable of picking up electrical fields from signals conducted within the heart. Because body fluids are saline, they can conduct signals well. ECG's are recorded from limb or chest leads. A typical ECG consists of a P wave, QRS wave (complex), and a T wave. ECG's are small potentials and are only a few millivolts in amplitude. The P wave correlates to atrial depolarization, which occurs just before atrial contraction. The QRS complex correlates to ventricular depolarization, which occurs just before ventricular contraction. The final T wave is due to ventricular repolarization which occurs during ventricular relaxation. There is a temporal relationship between the cardiac action potentials and the ECG record.
ECG's are valuable because they are non-invasive methods to monitor the condition of the heart in a relatively simple way. Vector analysis is used to analyze the heart as well and can indicate the position of damage to the heart.
There are several issues that can occur in the conducting paths of the heart, which overall cause an ECG to look much different:
In a first-degree block, there is a delay in the depolarization of the ventricles, therefore making the P-R interval prolonged.
In a second-degree block, the P waves are sometimes not followed by the QRS-complex or could be completely missing.
In a third-degree block, the P waves and QRS complex occur completely independent of each other.
In atrial fibrillation, the waves can all be out of order, but this is not as big of an issue because the ventricles are still functioning normally and are simply having the wrong atrial impulses. This would make an irregular heartbeat occur. The
In ventricular fibrillation, the waves are scattered all around and this is a fatal heart problem if not taken care of immediately.
Pacemaker cells help initiate the rhythmic depolarization of the heart. It is what leads the heart to beat in a rhythmic pattern. The heart does not receive transmitters from the nervous system to function it to contract. Instead, the heart has a combination of pacemaker cells and conducting fibers to maintain autonomic depolarization. The two pacemaker cells are sino-atrial node and atrio-ventricular node. Conducting fibers such as the Bundle of His and Purkinje Fibers help take the electrical current to all the ventricular muscle system. This process is important because the heart contracts synchronously and without the transmission of electrical current through the conducting fibers, the heart would not function properly. Pacemaking cells also contribute to the pacemaking potential, in which the depolarization occur differently than normal cardiac action potential.
The SA node in the heart is made up of a group of cells which is also called a pacemaker because it sets the timer at which all the cardiac muscles contract. The pacemaker generates a wave of signals that conduct through the atria and causes both atria to contract simultaneously. Signals travel from the SA node to the AV node, another group of cells that receives the current from the first node. There is a delay that takes place at the AV node because the atria must empty completely and all the blood must flow in before the ventricles are able to contract. After the delay, the AV node is now able to send the current through the bundle branches into the apex of the heart where it spreads throughout the ventricles of the heart through Purkinje fibers triggering a contraction.
There are three properties including cross sectional area, blood flow velocity and blood pressure that that explain the pattern of blood flow from arterioles into the capillaries. Blood flow from arteries to arterioles to capillaries slows due to the increase in total cross sectional area. The number of capillaries is extremely high making the cross sectional area much greater in the capillary beds than in arteries or any other part of the circulatory system. Blood flow here is also slow because the exchange of material is taking place. The increase of cross sectional area correlates to the decrease in the speed of blood flow because arteries must transfer blood to many capillaries. However, it is noted that there is a slight increase when the blood enters the vein again. This is because the veins and venules have a slightly larger cross sectional area than capillaries. As a result, the change in diameter aids in speeding up the blood flow into the veins. As blood enters the capillary bed, the capillaries have narrow diameter which produces resistance to blood flow. This resistance causes much of the pressure that is generated by the heart to disperse. Arteries have the ability to keep blood pressurized at all times. The pressure in the arteries during ventricular systole is called systolic pressure and is the highest pressure in the arteries. Pressure in the arteries during diastole is called diastole pressure, which in comparison to systolic pressure is much lower.
In the capillary bed, there are two forces that are in charge of driving the diffusion of water outward or inward. These two forces are known as blood pressure and osmotic pressure, opposing forces in the capillaries. Osmotic pressure is established by the protein albumin which is always present in the blood. Albumin is too large of a protein to be transferred out of the blood, as a result the osmotic pressure is always constant because ultimately, the proteins cannot leave the capillary beds and they are the ones that maintain this pressure. The blood pressure is established by systolic and diastolic pressure in the arteries. When the blood pressure is higher than the osmotic pressure, there is an outward flow of fluid. On the other hand, when the osmotic pressure is higher than the blood pressure, there is an inward flow of fluid. There is usually more exchange leaving the capillary then coming back. Much fluid is lost that does not return to the circulatory system.
Disorders of the heart and blood vessels. Cardiovascular diseases range from a minor disturbance of vein or heart valve function to a life-threatening disruption of blood flow to the heart of brain.
One side of the heart is weaker than the other, causing unequal pumping of blood from the two halves of the heart. People can be born with the disease, but usually occurs when one side of the heart is damaged.
Right heart failure occurs when the left ventricle pumps more blood than the right ventricle. Blood becomes backed up and collects in the body, leading to a condition called systemic edema. The accumulation of excess fluid causes the arms to be swollen constantly. Fortunately, right heart failure is not very serious or life threatening. They can easily be treated with drugs that increase cardiac contraction.
Left heart failure occurs when the right ventricle pumps more blood than the left ventricle. This causes blood to collect in the lungs, leading to pulmonary edema. The collection of blood can lead to suffocation, and possible death. Unlike right heart failure, left heart failure is very serious and must be treated immediately.
Both the left and right ventricles must be contracting the same amount of blood at all times. Even though left ventricle contraction is stronger than the right, the same amount of blood is pumped.
The leading causes of heart failure are diseases that damage the heart such as coronary heart disease (CHD), high blood pressure, and diabetes. Many of these risk factors cannot necessarily be changed or reversed. Leading risk factors that are capable of being changed in a lifetime include, tobacco use, unhealthy blood-cholesterol levels, physical inactivity, and being overweight/obesity. Leading risk factors that cannot be changed include heredity, aging, being male and ethnicity.
Tobacco use harms the cardiovascular system in many different ways, such as by damaging the lining of the arteries and reducing the "good" cholesterol in the body. Nicotine in specific raises the heart rate and the blood pressure of the body. Depletion of the oxygen supply in the blood is caused by the inhalation of carbon monoxide. Those who smoke and those who are exposed to smoke are capable of experiencing heart-related problems.
In the United States, heart failure is acknowledged as a common condition and affects about 5.8 million Americans. Heart failure has no cure however many treatments such as pharmaceutical medications and lifestyle modifications can help people relieve some symptoms and help them live longer. Current treatment options for advanced heart failure include the implantation of left ventricular assist devices (LVAD) to help the heart mechanically continue pumping blood. Currently, LVAD implants are being used as destination therapy or bridge to transplantation of artificial hearts.
The hardening of the arteries by accumulation of fatty deposits. Healthy arteries have a smooth inner lining that reduces resistance to blood flow. Damage or infection can roughen the lining and lead to an inflammation. Leukocytes are attracted to the damaged lining and begin to take up lipids, including cholesterol.
The hardening process often accompanies the natural progression of aging. High blood cholesterol due to unhealthy lifestyle as caused by a diet high in fat content, alcohol use, and limited exercise can accelerate the hardening of arteries. Other risk factors for atherosclerosis include diabetes, high blood pressure, smoking, and family history of atherosclerosis.
Diagnosis include a series of medical tests such as magnetic resonance arteriography (MRA), angiography (the use of x-rays to view the inside of arteries), and ultrasound doppler tests.
Also called a myocardial infarction, is the damage or death of cardiac muscle tissue resulting from blockage of one or more coronary arteries. Since the coronary arteries are small in diameter, they are especially vulnerable to obstruction. Such blockage can destroy cardiac muscle quickly because the constantly beating heart muscle cannot survive long with oxygen.
A heart attack may be caused by blood platelets sticking to tears in the plaque and from a blood clot that blocks blood flow to the heart. Heart attack symptoms include chest pain, anxiety, cough, fainting, light-headedness, shortness of breath, and sweating.
A stroke is the death of nervous tissue in the brain due to a lack of oxygen. Strokes usually result from rupture of blockage of arteries in the head. The effects of a stroke and the individual's chance of survival depend on the extent and location of the damaged brain tissue.
Strokes are also medically known as cerebrovascular disease, cerebral infarction, or cerebral hemorrhage. There are two main types of stroke called ischemic stroke and hemorrhagic stroke.
ischemic stroke: This type of stroke occurs when a blood vessel that supplies blood to the brain is blocked by a blood clot. The clot maybe in an already narrow artery and is called a thrombotic stroke. Another pathway by which a clot can form is through a dislodged clot from another blood vessel or another part of the body and travels up to the brain. This type of stroke is known as embolic stroke. These clots consist of fat, cholesterol that create plaque.
In ischemic stroke, there are two types of stroke that can occur; embolic and thrombotic. In embolic stroke, blood clots travel from somewhere in the body to the brain. In thrombotic stroke, caused by thrombi, blood clots form where an artery has been narrowed by atherosclerosis.
hemorrhagic stoke: This type of stroke occurs when a blood vessel in the brain is weakened and as a result, burst open. Blood starts to leak into the brain. People who already have defects in blood vessels of the brain are more likely to experience hemorrhagic stroke.
In hemorrhagic stroke, there are two different types of stroke that can occur; subarachnoid hemorrhage stroke and intra-cerebral hemorrhage stroke. In subarachnoid, there is a bleed in between the brain and the skull which usually develops from an aneurysm. In intra-cerebral hemorrhage, there is a bleed in the brain from a blood vessel which is caused from high blood pressure and its damaging effects on arteries.
The risk factors for stroke include family history, high cholesterol, age, race, unhealthy lifestyle and diet, and diabetes. Women over 35, smoke, and take birth control pills are at a very high risk of stroke.
Overtime, arteries that are narrowed by disease can still open enough to deliver blood to the heart. However, during exertion, the heart requires more oxygen to function properly, than the narrowed arteries can process successfully. The result is called angina pectoris, which is severe chest pain. The pain can occur in the chest, shoulder, neck, arm, hand or back and usually is expressed as a tightness in one of those areas or heavy pressure. Angina can be controlled via drugs or surgical treatment but overtime can become a blocked artery or even lead to cardiac failure.
When an artery becomes blocked, it may become non-functional or could affect the overall blood supply to the heart. In this case, a vein would be grafted into an artery at the point above and the point below the obstructed segment.
The excretory system in organisms is a system that helps with the homeostasis of water and fluid regulation within each organism by absorbing useful fluids such as water and excreting out wastes and excess fluids. Although various animals have distinct and unique excretory systems, most systems carry out the same processes of filtration, reabsorption, and finally secretion.
Most vertebrate organisms contain an organ called the kidney that helps with both osmoregulation as well as the excretion process. Fluids enter the kidney where fluids and nutrients are reabsorbed, and the waste product is stored in the kidney until the organism is ready for excretion. Each kidney then pushes out the stored waste product (urine in vertebrates) into ducts known as ureters. These ureters then drain the waste product into urinary bladder. The urine stored in the bladder is then finally excreted through the urethra out of the organism’s body system.
The kidneys are the major organ that helps aid in osmoregulation/excretion. The kidneys themselves are separated into various segments. The renal cortex and renal medulla are constantly provided with blood via the renal artery, in which the blood is drained out of the kidney via the renal vein. Upon filtering and reabsorbing the essential nutrients, the kidneys produce the urine waste, which is transferred to the renal pelvis via excretory tubules, until the urine is finally released into the ureters. Within each kidney are many functioning units of the kidney known as nephrons, which weave back and forth through the renal cortex and renal medulla.
Major Kidney Functions
Regulate volume of fluid in the body
Regulate content of body fluid through osmotic concentration, ionic content, acid/base balance
Excrete wastes (especially nitrogenous, but also water-soluble waste)
A single kidney is made up of several layers including the renal cortex (the outer layer), the renal medulla (the inner layer) and the renal papillae (strands that connect to the renal medulla. In order to carry out the specific functions named above, the kidneys work to filter out a large amount of blood and process the filtrate. The kidneys make up a profound part of the body, especially in function.
In vertebrates, nephrons are the functioning unit of the kidney that performs the various tasks associated with the excretory system. They are responsible for the filtration, reabsorption, and secretion steps associated with excretion. The nephrons function starts with the glomerulus, which is a capillary cluster that delivers blood/fluid to the nephrons. The glomerulus is encompassed by Bowman’s capsule; and blood pressure squeezes fluids out from the blood in the glomerulus out to Bowman’s capsule, and the filtrate then travels to the proximal tubule.
The proximal tubule marks the beginning point of reabsorption. The proximal tubule then utilizes methods of active and passive transport to reabsorb nutrients from the filtrate. Active transport is utilized to reabsorb salts and other nutrients, while passive transport allows for the reabsorption of potassium ions, water, and bicarbonate. After the filtrate passes through the proximal tubule, it is then transported into the loop of Henle, which is divided into two distinct portions. The filtrate first passes through the descending loop of Henle, in which water is passively transported out of the nephron back into the body (in the outer medulla). Upon looping back up, the ascending loop of Henle passively transports salts in the inner medulla, and actively transports it in the outer medulla. Upon shooting up on the ascending loop of Henle, the filtrate enters the distal tubule, in which salts and carbonate are actively transported out, and water is passively diffused outwards. In the final part of the nephron, remaining salts are finally transported outward to the body actively, as well as traces of urea and water.
Arterial pressure drives fluid and small solutes across the walls of the glomerular capillaries. From this, filtration occurs. The most proximal part of the nephron is where filtration occurs. All of the filtered material enters the Bowman's capsule through special filtration slits between structures called pedicels. Reabsorption and secretion occur in the proximal tubule.
Capillary exchange is where an exchange between blood and extracellular fluid occurs. It contains small precapillary sphincters by the arteriole end:
- When sphincters are relaxed, there is more blood flowing through the capillary bed.
- When sphincters are contracted, there is less blood flowing through the capillary bed.
- Constriction and dilation also impact capillary blood flow.
Hydro-static pressure tends to be higher inside the capillary at the arteriole end and lower at the venous end. This causes fluid to move out of the capillaries at the arteriole end. On the other hand, osmotic pressure tends to be lower outside of the capillary at the arteriole end and higher at the venous end. This causes the fluid to enter the capillaries again at the venous end. The plasma proteins creates osmotic pressure and are unable to pass through the capillary wall, making the blood more concentrated.
Erythrocytes (red blood cells)
- Erythrocytes do not have any type or organelles. They transport oxygen and carbon dioxide and use glycolosis for metabolism. They look like a flat biconcave disk, which maximizes the surface area to increase diffusion of gases. Their life span is 3-4 months and a spherical shape is observed when they are ready to die.
Leukocytes (white blood cells)
There are different kinds of white blood cells:
Lymphocytes: play a role in defense and immunity, responsible for the production of antibodies;
Monocytes: also participate in defense and immunity;
Eosinophils: respond to allergic reactions;
Neutrophils: fight against infections and are usually inert;
Basophils: participate in the secretion and storage of histamine;
Platelets (fragments which trigger blood clotting)
Blood clotting is an example of a positive feedback loop. It is a mechanism that converts prothrombin to thrombin which in turn converts fibrinogen to fibrin. The main requirement for this mechanism is the presence of vitamin K and calcium.
Plasma (The plasma is made up of ions, plasma proteins, nutrients, water, hormones, and respiratory gases. The proteins found in the plasma include albumin, fibrinogen, and immunoglobulins).
The respiratory system's function is to allow gas exchange to all parts of the body. The space between the alveoli and the capillaries, the anatomy or structure of the exchange system, and the precise physiological uses of the exchanged gases vary depending on the organism. In humans and other mammals, for example, the anatomical features of the respiratory system include airways, lungs, and the respiratory muscles. Molecules of oxygen and carbon dioxide are passively exchanged by diffusion between the gaseous external environment and the blood. This exchange process occurs in the alveolar region of the lungs.
Muscle action changes the volume of the rib cage and the chest cavity, and the lungs match these volume changes. The inner layer of the lung's double-walled sac adheres to the outside of the lungs, and the outer layer adheres to the wall of the chest cavity. A thin space filled with fluid separates the two layers. Because of surface tension, the two layers are like two plates of glass stuck together by a film of water: The layers can slide smoothly past each other, but they cannot be pulled apart easily. surface tension couples movement of the lungs to movement of the rib cage.
Lung Volume increases as a result of contraction of the rib muscles and the diaphragm, a sheet of skeletal muscle that forms the bottom wall of the chest cavity. contraction of the rib muscles expands the rib cage by pulling the ribs upward and the breastbone outward. At the same time, the chest cavity expands as the diaphragm contracts and descends like a piston.All these changes increase the lung volume, and as a result, air pressure within the alveoli becomes lower than atmospheric pressure. Because gas flows from a region of higher pressure to a region of lower pressure, air rushes through the nostrils and mouth and down the breathing tubes to the alveoli. During exhalation, the rib muscles and diaphragm relax, lung volume is reduced, and the increased air pressure within the alveoli forces air up the breathing tubes and out of the body.
A More in Depth look at the Anatomy of the Respiratory System
The main purpose of breathing is in fact to simply supply the body with sufficient oxygen and remove excess amounts of Carbon Dioxide. The process of respiration can be observed at two levels, one large and one small. At the lungs (large level), gas exchange occurs through the process of breathing. At a more cellular level (small level), oxygen performs its role of transforming the food we eat into must needed energy, producing carbon dioxide as a certain by-product.
The picture above gives a detailed look at the nose. It is important to note that the nose is not simply a passageway to our throat, but that the nose is a critical aspect of the skull that provides and extensive passage array with inner walls covered by mucus. These walls are supplied with heat-radiating blood vessels. As a matter of fact, the nose can be looked at as an air-conditioner that prepares prepares the air before being taken into the lungs. There are three processes that occur when air is brought in through the nose, which include warming, filtering, and humidifying.
Warming: The air is warmed by blood vessels, so that it is always at the right temperature before entering the lungs. As labeled on the figure above, the three turbinates (superior, middle, and inferior), serve a critical function in the nose: They are shelves at the side of the nose that warm inhaled air before it enters the lungs. They are also covered by millions of cilia that defend the body against irritants in the inhaled air. They seem to enlarge when someone catches a cold, but no matter how trouble these turbinates may cause, it is crucial not to just get rid of them.
Filtering: The dust particles taken in are trapped by tiny hairs in the nose in addition to mucus, so that the air is rather clean when taken into the lungs. The nasal septum, as shown above, actually separates the left and right airways of the nose, dividing the two nostrils that are involved throughout nose filtering.
humidifying: The air is 100% humidified by the mucus in the nose. After these three processes occur, the "treated" air is passed through the lungs and into the trachea.
The inner lining of the bronchial tubes is a membrane called the bronchial mucosa. In terms of function, these tubes carry air into the tiny branches and smaller cells of the lungs after the air has passed through the mouth, nasal passages, and windpipe (trachea). There exists a layer of smooth muscle which is capable of contracting, thus producing airway narrowing. Mucus glands inside the layers of the bronchial tubes can produce a large amount of thick mucus when stimulated.
At the end of the small bronchioles exist millions of air sacs or alveoli. They have large gas space in between with thin blood space provide surface area and is also necessary for air to diffuse. This is explained by the ratio between surface area to volume ratio where when dividing large area into small volumes, the surface area increases. In addition, the thin membrane allow for better diffusion. In a way that is easier to understand, these air sacs can compared to grapes hanging off a stem. The one cell thick walls of the alveoli allow oxygen and carbon dioxide to pass through. Outside the lungs exist a network of air sacs called capillaries which also have walls one cell thick. The alveoli contain oxygen rich air, but carbon dioxide depleted air. The blood in the capillaries, however, contain more carbon dioxide than oxygen. Gas tries to equalize pressure levels, as oxygen passes into the carbon dioxide rich capillaries. At the same time, carbon dioxide passes from the blood and into the alveoli. This allows for gas equilibrium. The next step involves the exit of carbon dioxide through exhalation. This particular gas exchange occurs at the large (lung) level. Oxygen is then bound to hemoglobin in the bloodstream, as it travels from the lungs to the heart. Hemoglobin then performs its role of being a very efficient oxygen carrier, as it is pumped throughout the body supplying oxygen to oxygen-depleted tissues and muscles.
In contrast to the large (lung) level, tissue cells have high levels of carbon dioxide and low levels of oxygen. Though the process of equilibrium is the same, the tissue cells use oxygen to make energy and as a result, carbon dioxide and water are produced. They pass from the cell to the bloodstream and oxygen enters to produce energy. This particular gas exchange cycle repeats more than 60 times a minute at both large and small levels.
Asthma is a lung disease that inflames and narrows the airways or bronchial tubes.
Asthma causes periods of wheezing, chest tightness, shortness of breath, and coughing. In an asthmatic person, the muscles of the bronchial tubes tighten and thicken, and the air passages become inflamed and mucus filled, making it difficult to breathe. Factors that trigger asthma are allergens found in dust, animal fur, mold, dustmites, pollen from, grass, and flowers. Other factors are irritants such as cigarette smoke, air pollution, chemicals or dust in the workplace, and sprays such as hairspray also trigger asthma symptoms.All these factors contribute to constriction of our airways.
The airways of the lungs called bronchi (or bronchial tubes), are tubes with muscular walls that contract when irritated. Along the lining of the Bronchi there are cells with microscopic structures called G protein coupled receptors. These G protein coupled receptors are called beta-2 adrenergic receptors and cholinergic receptors. The beta-2 adrenergic receptors respond to chemicals such as epinephrine to make the muscles relax and thus opening the airways and increasing airflow. Cholinergic receptors on the other hand respond to a chemical called acetylcholine making the muscles contract, thereby decreasing airflow.
During an Asthma attack constriction of the Bronchi are caused by abnormal sensitivity of the cholinergic receptors, which cause the muscles of the airways to contract when they should not. There are cells in the Bronchi called mast cells which are held responsible for this cellular response. These mast cells release substances such as histamine and leukotrienes, which activate the cholinergic receptors cellular response for smooth muscle contraction, mucous buildup, and white blood cell migration to certain areas. Eosinophils, a type of white blood cell is found in the airways of asthmatic people, also release substances that contribute to airway constriction.
There’s no cure for asthma, however there are Albuterol inhalers for keeping asthma symptoms in a controlled state. There are several inhalers such as Albuterol, Ventolin and Proventil. Albuterol inhalers also known as a bronchodilator is a quick-relief or rescue medication used to decrease asthma symptoms.
Albuterol is a beta2-adrenergic receptor agonist How albuterol works is that when Albuterol is inhaled to the lungs, it acts on the Beta2-adrenergic receptors which activates the Gs protein. The Gs protein then activates adenylyl cyclase. Adenylyl cyclase then converts ATP to cyclic AMP. Cyclic AMP then activates a protein kinase A which continues the rest of the cyclic AMP pathway.
The pathway is continued until the cell responds by relaxing the smooth muscles in the Bronchi, thus reducing inflammation and allowing oxygen to be inhaled
The digestive system deals primarily with the breakdown of food polymers into smaller molecules to provide energy for the body.
1.) The entire digestive process can be simplified through the individual processes of ingestion, digestion, absorption, and elimination. Through analysis of each of these processes, an effective comprehension of the organs that make up the digestive system can be observed. During the process of ingestion, an organism utilizes mechanical digestion by chewing the particular food of interest. In this situation, the mouth, also known as the oral cavity, acts as the source of increasing the food’s surface area by creating it into a bolus, a smaller and more circular version of the food that will subsequently be swallowed down the esophagus. Besides these organs, there are also accessory glands that contribute to ingestion as well such as the tongue and the teeth. Through observing the digestion portion of the digestive system, enzymatic hydrolysis can be witnessed in which the esophagus transfers food down from the oral cavity and towards the stomach in which hydrochloric acid and pepsin allow the chemical breakdown of the food. Next, in the absorption portion of the digestive system, nutrient molecules enter body cells within the small intestine. Lastly, the elimination process of the digestive system is closely connected with the excretory system in which the undigested material of an organism becomes eliminated from the body.
2.) With the general idea and characteristics about the digestive system recently observed, the structure and function of the digestive system of three different types of organisms can be examined: a human, an invertebrate, and a non-mammal vertebrate.
Firstly, in the human digestive system, there is an alimentary canal, also known as a complete digestive tract, which represents a digestive tube extending between two openings, a mouth and an anus. The mouth acts as the initiating factor of the digestive tract and digestion initiates with the very first bite of any particular food type in which subsequent chewing breaks the food into pieces that are more easily digested. Simultaneously, saliva mixes with food to begin the process of breakdown into a form that the body can more readily absorb. Generally, humans are considered to be omnivorous, consuming both meat and plant-like objects.
Secondly, the digestive system of the invertebrate octopus will be observed. An octopus also possesses a two way digestive system consisting of a mouth and an anus. In contrast the typically omnivorous human diet, the octopus is a carnivorous creature with a diet generally consisting of lobster, crab, and shrimp. Utilizing their incredibly strong beaks to kill and consume their prey, the food will be digested in the stomach and digestive sac and then released from the anus.
Thirdly, as for a non-mammal vertebrate, a turtle’s digestive system can be observed. The digestive system of turtles is similar to that of other vertebrates. Unlike amphibians, they do not have mucous glands, but do have salivary glands. Turtles swallow their food whole or in large chunks and as this occurs, the salivary glands empty into the mouth and moisten the food to aid in swallowing. Although omnivorous in general, most turtles are at least partly carnivorous which cause them to possess strong digestive enzymes. The turtle's tongue is broad and flat while being firmly attached to the bottom of the mouth, preventing the tongue from moving. Alligator snapping turtles have small worm-like appendages on their tongues. These appendages can be filled with blood and are allowed to wiggle. With the turtle's mouth open and appendage wiggling, the alligator snapper can attract small fish to catch more effectively. The walls of the turtle's digestive tract are made of smooth muscle tissue and the muscles contract, pushing food down the esophagus and intestines towards the stomach.[1]
Ingestion
The act of ingestion begins the process of the whole digestive system. Ingestion begins when an organism puts a consumable in their mouth, and mechanical digestion begins in the form of chewing and breaking down the food. Performing mechanical digestion allows for the consumed food to increase in surface area, which allows for easier digestion and absorption of nutrients. During this time, the food turns into a bolus to allow for easier swallowing. Also during mechanical digestion, saliva is secreted in order to protect the lining of the mouth from the mechanical digestion. Saliva contains several enzymes, including amylase which protects the lining of the mouth, as well as mucin, which aids in swallowing. After mechanical digestion by chewing, and chemical digestion with saliva, the bolus of food is pushed into the esophagus, and moves down the throat by peristalsis.
Digestion
There are two important forms of digestion where molecules are broken into smaller pieces for absorption, known as mechanical and chemical digestion. Mechanical digestion occurs first in the oral cavity and begins with the chewing of food. Polysaccharides and sugars are only broken down in the oral cavity. Chemical digestion then follows as the salivary glands produce saliva and brings it through ducts to the oral cavity where it helps the breakdown of food. There is an enzyme in saliva called amylase that hydrolyzes polysaccharides and disaccharides into simple sugars in a process known as enzymatic hydrolysis. The tongue helps shape the food into a bolus and aid with swallowing. Once swallowing has occurred, the bolus of food reaches the pharynx, a region in that throat that opens to up to either the trachea (which leads to the lungs) or the esophagus (which leads to the stomach). During swallowing, there is a cartilage flap called the epiglottis that covers the glottis, an entryway that leads to the trachea so that food will not go down the wrong way and end up blocking the windpipe. Because this entryway is blocked, the larynx is able to guide the bolus of food to the esophagus entryway. There is an esophageal sphincter that regulates the movement of material in and out of the esophagus. This sphincter contracts when person is not swallowing and contrastingly, it relaxes when swallowing occurs so that the bolus of food can travel down the esophagus to the stomach. Chemical digestion is also prevalent in the stomach. The bolus of food is further broken down by the stomach’s gastric juices, which is composed of HCl and pepsin. The strongly acidic HCl helps to break down the extracellular matrix in the bolus of food, while the pepsin, being a protease, aids in breaking down peptide bonds and digesting proteins in the food. As the bolus is mixed with the gastric juices, the stomach constantly churns until the bolus turns into chime. Various other organs assist in the digestion of food, with the liver aiding in lipid breakdown, and the pancreas secreting bicarbonate to neutralize the HCl in gastric juice.
Chemical Digestion in the Stomach The stomach utilizes strong chemicals and enzymes that would normally destroy organic matter easily. The stomach is able to protect itself from its own gastric juices by carefully synthesizing the chemicals together to efficiently digest the bolus of food and turn it into chyme. The entire inside of the stomach is lined with a layer of mucus, as well as other layers of epithelial cells that are replaced every few days. The stomach utilizes hydrochloric acid as a means to destroy harmful bacteria as well as to denature the proteins in the bolus. Because hydrochloric acid can cause high damage to the stomach, the H+ ions and Cl- ions are kept separate until it is needed for digestion. Separate parietal cells in the stomach keep the ions separate, until they use ATP to drive the H+ and Cl- ions out, where they react in the stomach lumen to form the HCl. HCl is also able to activate pepsinogen into pepsin, a useful enzyme that specializes in protein cleaving and digestion. Stomachs that do not have the proper defensive ways to utilize the HCl and pepsin within the gastric juice often get damaged stomachs, leading to gastric ulcers.
Absorption
Absorption of nutrients begins in the small intestine. The small intestine has massive surface area lined with villi and microvilli, which help aid in absorbing the nutrients from the chyme. Afterwards, the chyme goes through the large intestine, which is divided into three parts: the colon, cecum, and appendix. The cecum aids in fermenting the ingested food, and the colon reabsorbs water and other nutrients that the body might’ve used during the digestive process. As the ingested material passes through the intestines by peristalsis, usable nutrients are absorbed, while waste is left behind in the form of feces.
Excretion
The final stage in the digestive system is excretion, in which the ingested food is now formed as a (usually) solid waste known as feces. The feces is excreted out of the body via the anus, ending the digestive process for the particular ingested food.
There are gastric glands located on the interior surface of the stomach. The gastric glands have three different types of cells that are responsible for secreting the parts that make up the gastric juice such as mucus, pepsin and hydrochloric acid. The parietal cells in the gastric gland secrete hydrochloric acid (HCl). The chief cells secrete an inactivate form of pepsinogen that is not activated to pepsin until it comes into contact with hydrochloric acid in the lumen of the stomach. Lastly are the mucus cells that secrete mucus, a substance that protects the lining of the stomach. Gastric juice does not destroy stomach cells that make it because all these ingredients remain inactive until they are released into the lumen. Pepsinogen and HCl are first secreted into the lumen of the stomach. Hydrochloric acid converts the pepsinogen (inactive form) into pepsin. And pepsin, the activated form begins a chain reaction, activating more pepsinogen. Gastric juice begins the process of chemical digestion of proteins in the stomach.
The small intestine is the longest compartment of the alimentary canal and is the major organ of digestion and absorption. This is the site where the rest of the digestion and enzymatic hydrolysis processes takes place, followed by absorption. First portion of the small intestine is the duodenum, where acid chyme combines with the gastric juices of the pancreas, liver, gall bladder.
When food arrives at the stomach, it causes the walls to stretch and this stimulates the release of the gastrin hormone from the stomach. Gastrin will travel through the bloodstream and come back to target the stomach, causing the stomach to release gastric juices.
Once chyme passes from stomach to duodenum, it triggers the release of hormones, secretin and cystokinin (CKK) because of the fatty acids and amino acids present in the chyme. The secretin works positively on the pancreas to stimulate the release of bicarbonate, which will help break down fats into fatty acids and neutralize acid chyme. Meanwhile CCK stimulates the gallbladder to release bile, which is a detergent that breaks down fats into smaller fats. CCK also stimulates the pancreas to release more digestive enzymes that can hydrolyze fats.
The secretin hormone and CKK both have inhibitory responses and act negatively on the stomach to slow down digestion and the production of chyme.
The jejunum and ileum are parts of the small intestine that are strictly responsible for absorption. The large surface area of the small intestine is due to the villi and microvilli that are exposed to the intestinal lumen. This large microvillar surface increases the rate at which nutrients are absorbed. The villi and small epithelial cells make up and line the border of the small intestine. Nutrients can be transported across the epithelial cell through passive or active transport. Sugar moves through passive diffusion down the concentration gradient into the epithelial cell, it then gets collected in small blood capillaries and travels through the circulatory system. Meanwhile amino acids, peptides, vitamins and glucose are pumped actively against the concentration gradient by the epithelial cells and then circulated throughout the bloodstream by capillaries. Fats, on the other hand, need to be reassembled in the lymphatic system and transported differently.
Absorption of Fats
In the lumen, the fat molecules, called triglycerides undergo enzymatic hydrolysis during which the enzyme, lipase breaks down the fat molecules into fatty acids and monoglycerides. The smaller molecules made up of fatty acids and monoglycerides diffuse into the epithelial cell. In the epithelial cell, the small molecules revert back to being triglycerides which are mixed with cholesterol and coated with protein to form chylomicrons. These molecules leave the epithelial cells and are transported into the lacteals.
While the primary function of the small intestine is to absorb nutrients and water, the large intestine functions to absorb mainly water. The colon is a part of the large intestine that connects to the small intestine and leads to the rectum and anus. The colon functions to recover the water that enters the alimentary canal. The cecum connects where the small and large intestine meet and helps in the fermentation of plant material. In humans, the cecum also has an extension known as the appendix. The appendix does not play a large part in immunity. Feces are any wastes from the digestive tract. As they move through the colon, they become more solid and ultimately pass through the rectum and leave out through the anus.
There are hormones secreted by tissues and organs in the body that then are transported through the bloodstream to the satiety center, a region in the brain that triggers impulses that give us feelings of hunger or aid in suppressing our appetite. Ghrelin is a hormone that is released by the stomach and targets the pituitary, signaling to the body that it needs to eat. PYY is a hormone that is released by the small intestine and counters ghrelin. It is released by the hypothalamus and signals that you have just eaten and helps to suppress our appetite. The pancreas releases the hormone insulin, which targets the hypothalamus and also aids in suppressing our appetite after we have just eaten and there is a rise in blood glucose levels. The last hormone is leptin which also helps to suppress appetite. Leptin is produced by adipose fat tissue and targets the hypothalamus.
The intestinal barrier is composed of a mucin (glycoproteins) layer and tight junctions between epithelial cells as well as by regular epithelial cell replacement which protects the intestine from mechanical stress produced by the passage of digested food through the gastro intestinal (GI) tract, molecular breakdown from digestive enzymes, and foreign invaders such as bacteria and viruses. While providing protection, the mucin layer must allow the passage of digested molecules through to be absorbed by epithelial cells. Unraveling the depth of protection by the mucin layer to all damaging vectors can develop a comprehensive understanding of mucin’s potential role in many intestinal diseases[1].
Mucin (glycoproteins) In GI Tract
Up to 20 mucin genes have been identified and are broadly classified as either secretory or membrane-associated large glycoproteins[1]. Recent studies have shown that the mucin layers in the GI tract vary in mucin protein composition, mucin layers, and mucin thickness[2]. The mucin layer has been shown to prevent digestive enzymes from entering the wall of the intestines[3]. The mucin layer can be compromised if there is a structural abnormality or production of the glycoprotein is altered. This results in poor protection of the intestinal wall and can lead to systemic damage to the body.
For example, shock is a physiological process that still kills many individuals through multiple organ failure. Recent studies have shed light on the role the intestine plays in this process. The intestinal mucosal barrier becomes compromised during shock and no longer performs as a barrier[4]. When this occurs, the luminal contents of the intestine, including cytotoxic free fatty acids (FFAs), may enter the wall of the intestine causing damage both to it and other organs[3][1].
A complete comprehension of mucin’s role as a protective agent can lead to a greater understanding of many GI diseases and complications and the role mucin has in each. Diseases such as necrotizing enterocolitis, Crohn’s Disease, and Irritable Bowel Syndrome could be treated more efficiently since mucin plays a role in each. Shock victims would also benefit since many of the complications arise from the breakdown of the intestinal barrier[5].
Reece, Jane B., and Neil A. Campbell. Biology. Boston, MA: Cummings, 2011. Print.
↑ abcMalin E. V. Johansson, Daniel Ambort, Thaher Pelaseyed, André Schütte, Jenny K. Gustafsson, Anna Ermund, Durai B. Subramani, Jessica M. Holmén-Larsson, Kristina A. Thomsson and Joakim H. Bergström, et al. Composition and functional role
of the mucus layers in the intestine. Cellular and Molecular Life Sciences, 2011.
↑Atuma, C., V. Strugala, A. Allen, and L. Holm: The adherent gastrointestinal mucus gel layer: thickness and physical state in vivo. Am J Physiol Gastrointest Liver Physiol 280: G922–G929, 2001.
↑ abMarisol Chang, Tom Alsaigh, Erik B. Kistler, and Geert W. Schmid-Schönbein: Breakdown of Mucin as Barrier to Digestive Enzymes in the Ischemic Rat Small Intestine. PLoS ONE 7(6): e40087. doi:10.1371/journal.pone.0040087.
↑Chang M, Kistler EB, Schmid-Schönbein GW. Disruption of the mucosal barrier during gut ischemia allows entry of digestive enzymes into the intestinal wall. Shock. 37(3):297-305, March 2012.
↑Alexander H. Penn and Geert W. Schmid-Schönbein: The intestine as source of cytotoxic mediators in shock: free fatty acids and degradation of lipid-binding proteins. Am J Physiol Heart Circ Physiol 294: H1779–H1792, 2008.
Lactose Intolerance, commonly known as milk intolerance or dairy product intolerance, is an inability to digest lactose in small intestines. Lactose, a type of disaccharides sugar consisting of galactose bound to glucose, is commonly found in milk and other dairy products. The absorption necessitates the hydrolysis of lactose to it's components by the enzyme lactase. This is the enzyme not present or the absorption does not occur correctly in those with lactose intolerance. [1]
Lactose structure
Cause
Lactose intolerance is very common among people. The cause is the deficiency of lactase and it is because of both genetic and environmental induced factors
Dairy products
If one has diagnose with lactose intolerance, it is better to not eat dairy food. Here is a list of common dairy food that should be avoided:
Milk
Butter
Cheese
Casein
Yogurt
Gelato
Ice Cream
Management
The main strategy employed by those with lactose intolerance is to restrict their diet by not consuming products containing dairy. This can be harmful for it can lead to a diet without the recommended daily dose of calcium, though this can be easily supplemented. Symptoms can be slightly reduced when dairy is taken in with other nutrients. [1]
Reference
↑ abTimothy J. Wilt, et al. "Systematic Review: Effective Management Strategies For Lactose Intolerance." Annals Of Internal Medicine 152.12 (2010): 797-803.
Another disease that involves the digestive system, Crohn’s disease, causes inflammation within the digestive tract. The end part of the small intestine called the ileum is the most commonly affected towards the Crohn’s disease. Normally, the movement of muscles in the GI tract and the release of hormones and enzymes allows for the digestion of food. However, in Crohn’s disease, inflammation occurs in the inner lining of the affected part of the GI tract and the subsequent effect of inflammation and swelling can cause pain and can cause emptiness within the intestines, resulting in diarrhea. A long lasting type of inflammation may produce scar tissue that builds up inside the intestine to create a stricture, a narrowed passageway that can decrease the movement of food through the intestine, causing pain or cramps. Crohn’s disease is considered an inflammatory bowel disease, the general name for diseases that cause inflammation and irritation in the intestines. Crohn’s disease can be difficult to diagnose because its symptoms are similar to other intestinal disorders, such as ulcerative colitis. For example, ulcerative colitis and Crohn’s disease both share the characteristics of abdominal pain and diarrhea.[1]
Crohn's disease is a chronic inflammatory process primarily involving the intestinal tract. Although it may involve any part of the digestive tract from the mouth to the anus, it most commonly affects the last part of the small intestine (ileum) and/or the large intestine (colon and rectum). Crohn's disease is a chronic condition and may recur at various times over a lifetime. Some people have long periods of remission, sometimes for years, when they are free of symptoms. There is no way to predict when a remission may occur or when symptoms will return.
Because Crohn's disease can affect any part of the intestine, symptoms may vary greatly from patient to patient. Common symptoms include cramping, abdominal pain, diarrhea, fever, weight loss, and bloating. Not all patients experience all of these symptoms, and some may experience none of them. Other symptoms may include anal pain or drainage, skin lesions, rectal abscess, fissure, and joint pain (arthritis).
Common Crohn's symptoms:
•Cramping - abdominal pain
•Diarrhea
•Fever
•Weight loss
•Bloating
•Anal pain or drainage
•Skin lesions
•Rectal abscess
•Fissure
•Joint pain
Who does Crohn's disease affect?
Any age group may be affected, but the majority of patients are young adults between 16 and 40 years old. Crohn's disease occurs most commonly in people living in northern climates. It affects men and women equally and appears to be common in some families. About 20 percent of people with Crohn's disease have a relative, most often a brother or sister, and sometimes a parent or child, with some form of inflammatory bowel disease. Crohn's disease and a similar condition called ulcerative colitis are often grouped together as inflammatory bowel disease. The two diseases afflict an estimated two million individuals in the U.S.
The exact cause is not known. However, current theories center on an immunologic (the body's defense system) and/or bacterial cause. Crohn's disease is not contagious, but it does have a slight genetic (inherited) tendency. An x-ray study of the small intestine may be used to diagnose Crohn's disease.
Initial treatment is almost always with medication. There is no "cure" for Crohn's disease, but medical therapy with one or more drugs provides a means to treat early Crohn's disease and relieve its symptoms. The most common drugs prescribed are corticosteroids, such as prednisone and methylprednisolone, and various anti-inflammatory agents.
Other drugs occasionally used include 6-mercaptopurine and azathioprine, which are immunosuppressive. Metronidazole, an antibiotic with immune system effects, is frequently helpful in patients with anal disease.
In more advanced or complicated cases of Crohn's disease, surgery may be recommended. Emergency surgery is sometimes necessary when complications, such as a perforation of the intestine, obstruction (blockage) of the bowel, or significant bleeding occur with Crohn's disease. Other less urgent indications for surgery may include abscess formation, fistulas (abnormal communications from the intestine), severe anal disease or persistence of the disease despite appropriate drug treatment.
While it is true that medical treatment is preferred as the initial form of therapy, it is important to realize that surgery is eventually required in up to three-fourths of all patients with Crohn's. Many patients have suffered unnecessarily due to a mistaken belief that surgery for Crohn's disease is dangerous or that it inevitably leads to complications.
Surgery is not "curative," although many patients never require additional operations. A conservative approach is frequently taken, with a limited resection of intestine (removal of the diseased portion of the bowel) being the most common procedure.
Surgery often provides effective long-term relief of symptoms and frequently limits or eliminates the need for ongoing use of prescribed medications. Surgical therapy is best conducted by a physician skilled and experienced in the management of Crohn's disease.
What is a colon and rectal surgeon?
Colon and rectal surgeons are experts in the surgical and non-surgical treatment of diseases of the colon, rectum and anus. They have completed advanced surgical training in the treatment of these diseases as well as full general surgical training. Board-certified colon and rectal surgeons complete residencies in general surgery and colon and rectal surgery, and pass intensive examinations conducted by the American Board of Surgery and the American Board of Colon and Rectal Surgery. They are well-versed in the treatment of both benign and malignant diseases of the colon, rectum and anus and are able to perform routine screening examinations and surgically treat conditions if indicated to do so.
Scientists have recently conducted new research into the genetics of inflammaroy bowel disease such as Crohn's disease and ulcerative colitis. These research studies have recently reveals new insights into the origin of this set of illnesses.
Researchers have presented information of linked genetic variations in 163 regions of the human genome that are at heightened risk of developing inflammatory bowel disease (IBD). Of those discovered regions, 71 are newly discovered. IBD includes various automimmune digestive disorders that affect nearly 2.5 million people worldwide. Symptoms include abdominal pain and diarrhea. Patients with IBD typically have to undergo lifelong treatment with medications and other forms of drug therapy.
In recent studies, researchers analyzed data from about 34,000 people who took part in 15 previous studies of either Crohn's disease or ulcerative colitis. They also examined more than 41,000 DNA samples data from genome-wide scans of patients of Crohn's disease and ulcerative colitis. These samples were collected around the world from 11 different centers.
Since 163 regions were found to develop IBD, 71 were newly found and the other 92 regions were confirmed. Both groups were confirmed to be prone to inflammatory bowel disease. The way these regions were analyzed was done by overlapping those linked with other autoimmune diseases, suggesting IBD results from overactive immune defense systems that can evolve into serious bacterial infections.
Sceintists have stated, "Until this point we have been studying the two main forms of IBD -- Crohn's disease and ulcerative colitis -- separately," co-lead author Judy Cho, professor of gastroenterology and genetics at Yale School of Medicine, said in a Yale news release. "We created this study based on what seems to be a vast amount of genetic overlap between the two disorders." The new studies reveal "a genetic balancing act between [the immune system] defending against bacterial infection and attacking the body's own cells," co-lead author Jeffrey Barrett of the Wellcome Trust Sanger Institute in Cambridge, England, said in the news release. "Many of the regions we found are involved in sending out signals and responses to defend against bad bacteria. If these responses are over-activated, we found it can contribute to the inflammation that leads to IBD."
Cirrhosis is the final stage of chronic liver disease in which the liver begins to slowly deteriorate and malfunction. With cirrhosis, blood flow through the liver is blocked from scar tissue replacing healthier liver tissue. The scar tissue hinders the liver's everyday functions to control infections, remove bacteria and toxins from the blood, process nutrients, hormones and drugs, make proteins that regulate blood clotting, and produce bile to help absorb fats and fat-soluble vitamins.
Common causes of cirrhosis, especially in the United States, are long-term Hepatitis C infection and long-term alcohol abuse.
Other potential causes are: autoimmune inflammation of the liver, disorders of the drainage system of the liver, long-term Hepatitis B infection, medications, metabolic disorders of iron and copper, nonalocoholic fatty liver disease (NAFLD), and nonalcoholic steatohepatitis (NASH).
Typically in the earlier stages of this liver disease, people will not show symptoms. But gradually, as the disease progresses, some may show symptoms of:
During a doctor's visit, if the physician feels that a risk factor for cirrhosis, such as alcohol or obesity use, is present, the physician will conduct a physical examination in which they will ask the patient about their medical history and if they have displayed any symptoms of this disease.
The physician may request for a computerized tomography (CT) scan or magnetic resonance imaging (MRI) of the liver to find signs of enlargement or reduced blood flow.
A liver biopsy can confirm the diagnosis of cirrhosis, but it is not necessary to determine if a patient is diagnosed with cirrhosis.
Cirrhosis patients should make changes to their lifestyles to cope with the disease by:
Ceasing intake of alcohol
Limiting salt and eating nutritious diet
Getting vaccinated for influenza, hepatitis A & B
Some patients may be eligible for liver transplants if they have reached the end-stage of cirrhosis. Typically, patients at this stage are still facing complications regardless of treatments.
Muscle cells consist of myofibrils which are in turn made up of repeating subunits called sarcomeres. Sarcomeres each contain two Z-bands which make up the walls of the sarcomere to which the actin molecules are connected. In between each pair of actin, bands is a myosin molecule. When the sarcomere contracts, myosin, and actin pull the two z bands closer together shortening the overall length of the sarcomere. The distance that the sarcomere is capable of contracting is called the I band and is the space in between the z-band walls of the sarcomere, and the start of the myosin molecules.
When muscles are stressed i.e. exercise, the number of sarcomeres and myofibrils in each myocyte are increased, instead of the myocyte numbers themselves. This leads to a stronger contractile strength for each individual myocyte without increasing the overall cell population.
There are three main muscle types: cardiac, smooth, and skeletal.
Cardiac: Only found in the heart and works involuntarily throughout the human body. These muscles are controlled by the medulla oblongata, which is located at the lower section of the human brain. The heart cells come in long strips with a single nucleus in each cell. They are located at the walls of the heart. Their main function is to propel blood into circulation. Contraction of the cardiac tissue is caused by an impulse sent from the medulla oblongata to the SA nerve located at the right atrium of the heart.
Smooth: Work involuntarily. Our internal organ muscles are mostly made up of smooth muscles, such as the stomach-hyper link, throat-hyper link, and small intestine-hyper link. Maintains homeostasis in our body. only the heart is not a smooth muscle. Smooth muscles are spherical in shape and contain one nucleus.
Skeletal: Also known as striated muscle tissue. The structure involves a parallel network of fibers of actin and myosin and the formation of actin myosin crossbridges. The movement of skeletal muscle is described by sliding filament theory in which actin filament slide against myosin heads during the process of contraction. Energy is induced from the ATP released by myosin heads when they slide against the actin filaments, changing its conformation from "cocked" to its "resting" state. This process is also known as powerstroke. When calcium level is high in the sarcoplasmic reticulum and ATP is available, contraction of muscles continues. Work voluntarily for our body. They are the muscles that move the bones and show external movement. Skeletal muscles contain multiple nuclei, which account for their large size. Skeletal muscle cells also contain multiple nuclei in order to synthesize actin and myosin efficiently. They measure up to several feet in length.
Skeletal muscles do not work alone. When muscle is attached to the skeleton, the connection will determine the force, speed, and range of movement, which is produced by contraction of a muscle and modified by attaching the muscle to a lever. A lever is a rigid bar, such as the bone, that moves on a fixed point called fulcrum. Each bone is a lever, and each joint is a fulcrum. The fulcrum helps to support the lever. Levers can change the direction of applied force when body in exercise, the distance and speed of movement affected by the force, and the effective strength of the applied force. Therefore, the movement of the muscle is based upon the type of joint. Joint helps skeleton muscles to expand or contract the muscles.
(1) The myosin head is bound to ATP and is in its lower-energy configuration
(2) The Myosin head hydrolyzes ATP to ADP and inorganic phosphate and is in its high-energy configuration
(3) The myosin head binds to actin, forming a cross-bridge
(4) Releasing ADP and an inorganic phosphate, myosin returns to its lower-energy configuration, sliding the thin filament
(5) Binding of a new molecule of ATP release the myosin head from actin, and a new cycle begins
Important structural domains of myosin include the motor region, the lever, and the tail. Heavy chain subunits form the major structural units of the motor region, lever and tail, with light subunits working to stabilize the major components of the myosin functional unit. Each of the over 35 myosin subclasses has minor structural variations on this theme, but in most cases, the light chains simply serve to stabilize the structure.
As its name suggests, the motor function confers the mobility to the myosin motor domain and is involved in forcibly moving the lever domain. Regions that bind actin are divided by a cleft, that when closed, initiates a strong bond with actin elements. During an active powerstroke, the myosin changes from an up conformation to a down conformation. A central region dubbed the transducer region strained under certain conditions and a relaxation of this strain contributes to the generation of the powerstroke.
The sequence of the powerstroke involves several steps. First, the myosin head binds to the actin filaments, then the conformational shift from up to down is accomplished, then a phosphate group is released, triggering a return to the up conformation. ATP can be hydrolyzed only in the up conformation, a step that leads to lever priming and firing. The conformational shift of the lever that forms the basis of the powerstroke is thought to be the rate-determining step, a process that does not occur when there has been no bind to actin.
(1) Aceylcholine are released at synaptic terminal diffuses across synaptic cleft and binds to receptor proteins
(2) Action potential is propagated along plasma membrane and down T tubules
(3) Action potential trigger calcium ions to be released from the sarcoplasmic reticulum
(4) Calcium ions bind to troponin complex in thin filament
(5) Proteins bound along the actin strands shift position
(6) Myosin binding site exposed
(7) Myosin cross-bridges alternately attach to actin and detach, pulling thin filament toward center of sarcomere; ATP powers sliding of filaments
(8) Cytoslic calcium ion is removed by active transport into sarcoplasmic reticulum after action potential ends
(9) tropomyosin blockage of myosin-binding sites is restored
(10) contraction ends, and muscle fiber relaxes
Bones interact with muscles through tendons. Movement happens when muscles contract and pull the attached bones to bend joints. Vertebrates have three kinds of muscles:
Skeletal muscles are also called striated muscles. They are associated with the skeletal system and are primarily involved in voluntary movement. A vertebrate has conscious control over these muscles. Each skeletal muscle cell contains many nuclei. They make up the bulk of muscle in the body and constitute about 40% of total body weight. They are responsible for positioning and moving the skeleton. Skeletal muscles are usually attached to bones by tendons made of collagen. The origin of a muscle is the end of the muscle that is attached closest to the trunk or to the more stationary bone. The insertion of the muscle is the more distal or more mobile attachment.
When the bones attached to a muscle are connected by a flexible joint, contraction of the muscle moves the skeleton. If the centers of the connected bones are brought closer together when the muscle contracts, the muscle is called a flexor, and the movement is called flexion. If the bones move away from each other when the muscle contracts, the muscle is called an extensor, and the movement is called extension. Most joints in the body have both flexor and extensor muscles, because a contracting muscle can pull a bone in one direction but cannot push it back. Flexor-extensor pairs are called antagonistic muscle groups because they exert opposite effects.
Muscles function together as a unit. A skeletal muscle is a collection of muscle cells, or muscle fibers, just as a nerve is a collection of neurons. Each skeletal muscle fiber is a long, cylindrical cell with up to several hundred nuclei on the surface of the fiber. Skeletal muscle fibers are the largest cels in the body, created by the fusion of many individual embryonic muscle cells.
The fibers in a given muscle are arranged with their long axes in parallel, and each skeletal muscle fiber is sheeted in connective tissue. Groups of adjacent fibers are bundled together into units called fascicles. Collagen, elastic fibers, nerves, and blood vessels are found between the fascicles. The entire muscle is enclosed in a connective tissue sheath that is continuous with the connective tissue around the muscle fibers and fascicles and with the tendon holding the muscle to underlying bones.
Smooth muscle is found in the walls of the internal organs. These organs include the stomach, intestines, and urinary bladder. It is an involuntary muscle. Although skeletal muscle has the most muscle mass in the body, cardiac and smooth muscle are more important in the maintenance of homeostasis. Smooth muscle is found predominantly in the walls of hollow organs and tubes, where its constriction changes the shape of the organ. Often smooth muscle generates force to move material through the lumen of the organ. For example, sequential waves of smooth muscle contraction in the intestinal tract move ingested material from the esophagus to the colon.
Smooth muscle is noticeably different from striated muscle in the way it develops tension. In a smooth muscle twitch, contraction and relaxation occur much more slowly than in either skeletal or cardiac muscle. At the same time, smooth muscle uses less energy to generate a given amount of force, and it can maintain its force for long periods. By one estimate, for example, a smooth muscle cell can generate maximum tension with only 25~30 % of its crossbridges active.
In addition, smooth muscle has low oxygen consumption rates yet can sustain contractions for extended periods without fatiguing. This property allows organs such as the bladder to maintain tension despite a continued load. It also allows some smooth muscles to be tonically contracted and maintain tension most of the time. The esophageal and urinary bladder sphincters are examples of tonically contracted muscles whose function is to close off the opening to a hollow organ. These sphincters relax when it is necessary to allow material to enter or leave the organ.
Until recently, smooth muscle had not been studied as extensively as skeletal muscle for many reasons:
1. Smooth muscle has more variety.
2. Smooth muscle anatomy makes functional studies difficult.
3. Smooth muscle contraction is controlled by hormones and paracrines in addition to neurotransmitters.
4. Smooth muscle has variable electrical properties.
5. Multiple pathways influence contraction and relaxation of smooth muscle.
Smooth Muscle vs. Skeletal Muscle
Smooth muscles are small, spindle-shaped cells with a single nucleus, in contrast to the large multinucleated fibers of skeletal muscles. In neurally controlled smooth muscle, neurotransmitter is released from autonomic neuron varicosities close to the surface of the muscle fibers. Smooth muscle lacks specialized receptor regions such as the motor end plates found in skeletal muscle synapses. Instead, the neurotransmitter simply diffuses across the cell surface until it finds a receptor.
Most smooth muscle is single-unit smooth muscle (unitary smooth muscle), so called because the individual muscle cells contract as a single unit. Single-unit smooth muscle is also called visceral smooth muscle because it forms the walls of internal organs (viscera), such as blood vessels and the intestinal tract. All the fibers of single-unit smooth muscle are electrically connected to one another, so an action potential in one cell will spread rapidly through gap junctions to make the entire sheet of tissue contract. Because all fibers contract every time, no reserve units are left to be recruited to increase contraction force. Instead, the amount of calcium that enters the cell determines the force of contraction.
Multi-unit smooth muscle consists of cells that are not linked electrically. Consequently, each individual muscle cell must be closely associated with an axon terminal or varicosity and stimulated independently. This arrangement allows fine control of contractions in these muscles through selective activation of individual muscle cells. As in skeletal muscle, increasing the force of contraction requires recruitment of additional fibers.
Multi-unit smooth muscle is found in the iris and ciliary body of the eye, in part of the male reproductive tract, and in the uterus except just prior to labor and delivery. Interestingly, the multi-unit smooth muscle of the uterus changes and becomes single-unit during the final stages of pregnancy. Genes for synthesis of gap junction connexin proteins turn on, apparently under the influence of pregnancy hormones. The addition of gap junctions to the uterine muscle cells synchronizes electrical signals, allowing the uterine muscle to contract more effectively while expelling the baby.
Cardiac muscle makes up the heart. These muscles are also involuntary and can contract without stimulation from the nervous system. Cardiac muscle shares features with both smooth and skeletal muscle. Like skeletal muscle fibers, cardiac muscle fibers are striated and have a sarcomere structure. However, cardiac muscle fibers are shorter than skeletal muscle fibers, may be branched, and have a single nucleus (unlike multinucleate skeletal muscle fibers). As in single-unit smooth muscle, cardiac muscle fibers are electrically linked to one another. The gap junctions are contained in specialized cell junctions known as intercalated disks. Some cardiac muscle, like some smooth muscle, exhibits pacemaker potentials. In addition, cardiac muscle is under sympathetic and parasympathetic control as well as hormonal control.
Muscles can be thought of as participators in the nervous system. Nerves will send messages to muscles through voluntary impulse or involuntary instinct. Muscles take these messages and convert them into movement by either contracting or relaxing.
Skeletal muscles have neuromuscular junctions, which is a synapse of an axon terminal of a motorneuron. The NMJ is responsible for the movement of action potentials across the neurons to initiate a physical reaction from the muscles.
The process begins with the release of acetylcholine from synaptic vesicles of an alpha motor neuron into the synaptic cleft. Vesicles containing acetylcholine fuse to the cell membrane, and by exocytosis, releases the chemical. Acetylcholine then diffuses throughout the synaptic cleft, and binds to acetylcholine receptors that are located on the motor end plate. These are nicotinic receptors, and are ligand-gated ion channels which means they respond when a ligand binds to it. The binding of the acetylcholine signals the opening of the channels, and sodium ions flow in while potassium ions flow out. This results in a neuron depolarization, which spreads across the muscle fiber's t-tubules. The depolarization causes activates voltage gated calcium channels (dihydropyridine receptors) in the T tubule membrane. Dihydropyridine receptors interact with calcium-release channels (ryanodine receptors) in the sarcoplasmic reticulum. This causes a release of calcium ions from the sarcoplasmic reticulum, which is the holding cell for calcium. The release of calcium results in a muscle contraction when calcium binds to troponin of skeletal muscle.
Enzymes called acetylcholinesterase degrade acetylcholine by hydrolysis and the neurotransmitter is diffused away. This causes the ligand gated receptors to close.
When signaled to, a synaptic terminal nearby the muscle fiber releases Acetylcholine (ACh) into the synaptic cleft. This neurotransmitter binds to receptor proteins on the muscle fiber’s plasma membrane, creating an action potential. This action potential is sent along the membrane and down through T tubule found on and within the membrane. The action potential triggers the sarcoplasmic reticulum (SR) to release Ca2+ into the cytosol of the muscle. This occurs when the plasma membrane depolarizes and an action potential sweeps along the membrane, the depolarization moves into the T-tubules and activates integral membrane proteins that are confined to T-tubule membranes in skeletal muscle fibers. The activated proteins are called dihydropyridine receptors (DHP receptors), which mechanically interact with particular proteins in the membranes of the sarcoplasmic reticulum. These proteins in the membranes of the ER are called ryanodine receptors.
These ryanodine receptors are Ca2+ channels that when activated by DHP receptors, open and allow Ca2+ to diffuse out of the SR and into the cytoplasm, where it can bind to troponin. Within the myofibril, the calcium ions bind to the troponin complex, causing the tropomyosin, which is covering the myosin binding sites, to shift, and thus exposing the myosin binding sites of the thin filament. With the aid of ATP, the myosin is able to form cross-bridges by binding to the actin. The attachment and detachment from the actin cause the sliding of the filaments, and thus the contraction of the muscle. When the motor input stops, and it is time for the muscle to relax, the filaments slide back to their original positions, with the tropomyosin blocking the myosin binding sites. The calcium ions are pumped back into the SR by transport proteins. The calcium ions accumulate in the SR until it is needed to respond to the next action potential.
Once the Ca2+ is able to bind to the troponin on the muscle fibers, the "sliding filament" theory takes place. This theory describes the mechanism of muscle contraction. In this theory, the myofilaments slide past one another when myosins bind to actins and "row" along the thin filaments (actin). When a myosin is bound to actin, the hinge between the head and the straight rod of the myosin molecule bends, which in turn releases ADP and Pi. As a result of this bending, the myosin molecule pulls itself along the actin, but actins are bound to Z-lines, so when an actin moves along myosins, they pull the Z-lines toward one another, shortening the sacromere, leading to contraction of the entire muscle as a whole. When the muscle is to relax, ATP attaches to the myosin head again, which breaks the thin filament to thick filament bonds. ATP binding is therefore the basis for relaxation of muscles.
The bound ATP is hydrolyzed to ADP and Pi, returning the hinge between the head and the rod of the myosin back to its original conformation, continuing the cycle of muscle contraction and relaxation. At any one time, many myosin heads are attached to each actin filament. The amount of tension that is exerted during a contraction is proportional to the number of connections between actin and myosin heads.
The unitary response of a muscle fiber to stimulation is called a twitch (a brief contraction). A twitch occurs in response to a depolarization of the muscle membrane, either in response to electrical stimulation or as the result of and excitatory post-synaptic potential it the neuromuscular junction. These twitches sum to produce long, strong contractions at their tetanus points. As the frequency of a stimulation increases, the frequency of twitches increases up to a maximum. Each summed twitch increases the magnitude of contraction because the elastic properties of the muscle are not sufficient to allow the muscle to return to resting length in the time between twitches. The maximum contraction in which there is no time for relaxation between stimuli is called tetanus. Therefore, the strength of a whole muscle contraction depends on the number of fibers in a muscle, because more fibers will bring a stronger contraction, the number of neurons that are active, and the frequency of contractions.
Summary of Muscle Contraction
Mechanism
When an action potential reaches the neurotransmitter junction, Acetylcholine gets released into the synaptic cleft. This results in the opening of nicotinic acetylcholine receptors and entrance of sodium ions. An action potential is generated in the T-tubule, then calcium is released from sarcoplasmic reticulum. This marks the step where contraction occurs.
Highlights
Calcium is removed by calcium pumps that pump the ion back into the sarcoplasmic reticulum. This is when relaxation occurs.
T-tubule contains voltage gated protein channels that open in response to depolarization.
Sarcoplasmic reticulum has several calcium channels which open when there is stimulation by T-tubules. In addition, it contains many calcium pumps that maintain a high concentration of calcium inside the cell so that ions are able to flow out during the process of contraction. The flow of calcium activates troponin, which stimulates a power stroke and results in sarcomere contraction.
There are two types of contractions:
A- Isotonic contraction which occurs when the muscle contraction is equal to the load on muscle. For example, when you lift an object and hold it in place.
B- Isometric contraction which occurs when the load on muscle exceeds the force of contraction. For instance, when you try to lift a very heavy object (the muscle contracts at a maximum rate)
Oxidative fibers rely mostly on aerobic respiration. It is made this way to make sure a steady energy supply. It has many mitochondria, a rich blood supply, and a large amount of hemoglobin.
Glycolytic fibers uses glycolysis as their primary source of ATP. It has a larger diameter and less myoglobin than oxidative fibers and it fatigue much more readily.
Fast-twitch fibers are used for brief, rapid, powerful contractions. Fast-twitch fibers are made up of white muscles, which depend on anaerobic glycolysis for energy. Although glycolysis is very quick, it is also inefficient at producing ATP. Glycolysis produces lactic acid as a byproduct, which leads to fatigue. The use of glycogen cycle is the reason why fast-twitch muscles tire out quickly.
Slow-twitch fibers are used to maintain posture. A slow fiber has less sarcoplasmic reticulum and pumps calcium more slowly than a fast fiber. Since calcium remains in the cytosol longer, a muscle twitch in a slow fiber lasts about five times as long as one in a fast fiber. Slow-twitch fibers are usually found in the red muscles. The red muscles use oxidative phosphorylation to obtain ATP. Oxidative phosphorylation occurs in the red muscles because the process requires a lot of oxygen, and the red muscles contain high amounts of myoglobin. The process is slower than glycolysis, but much more efficient, which is why slow-twitch muscles do not tire easily.
Although it is still being debated, scientists believe that individuals are born with a set amount of fast-twitch fibers and slow-twitch fibers. Sprinting everyday will not convert some of the slow-twitch muscle into fast-twitch muscle, and vice versa. Marathon runners become marathon runners because they naturally have more slow-twitch fibers, allowing them to be effective in the sport. Likewise, sprinters are born with more fast-twitch muscles than others. Although this may be true, exercise can make both fast and slow-twitch muscles bigger, leading to better fitness results.
Human Physiology: An integrated approach (Fourth edition) by Dee Unglaub Silverthorn
Endocrine System
The endocrine system consists of ductless glands that work together and secrete different hormones to regulate the human body. As an information signal system, hormones regulate many functions of an organism.
There are many types of signaling involved in the endocrine system including: autocrine, paracrine, and juxtacrine. Autocrine hormones act on the secreting cell itself, paracrine hormones act only on neighboring cells, and juxtacrine hormones act either on the emitting cell or adjacent cells.
The number of glands that signal one another in a sequence is called an axis. The typical endocrine glands are the pituitary, thyroid, adrenal glands, pancreas glands, ovaries, and testes.
Also known as the hypophysis, the pituitary gland is about the size of a pea weighing 0.5g and is located at the bottom of the hypothalamus at the base of the brain and rests in a small, bony cavity (sella turcica) covered by a dural fold (diraphragma sellae). It is considered the master gland as it secretes hormones regulating homeostasis. It is functionally connected to the hypothalamus by the median eminence.
Anterior pituitary (adenohypophysis) synthesizes and secretes important hormones such as: ACTH, TSH, PRL, GH, endorphins, FSH, and LH and are released under the influence of the hypothalamus through the hypothalamic-hypophyseal portal system. Hypothalamic hormones travel along axons until they’re released from terminal arborizations into the blood vessels of the hypothalamic-hypophyseal portal system. They are then carried into the adenohypophysis where cells are stimulated with hormone-specific receptors which trigger a release of a certain hormone. The anterior pituitary gland is divided into regions known as the pars tuberalis, pars intermedia, and pars distalis.
Posterior pituitary (Neurohypophysis) stores and releases oxytocin and antidiuretic hormone (ADH and AVP). Oxytocin has a positive feedback loop and is released from the paraventricular nucleus in the hypothalamus. ADH (vasopressin) and AVP (arginine vasopressin) is released from the supraoptic nucleus in the hypothalamus. ADH is controlled by the blood osmolarity. As blood osmolarity increases, the signal leads to increased ADH release from supraoptic nucleus. If a concentrated solution is perfused near the supraoptic and paraventricular nuclei, that can also signal an increase in ADH release.
The process by which hormones are released in the neurohypophysis and the andenohypophysis are quite similar. In the neurohypophysis, the hormones are synthesized in the supraoptic and paraventricular nuclei of the hypothalamus (in the brain- most of it is synthesized in the supraoptic nucleus). These hormones are then transported to the posterior pituitary where it is stored in and released from the terminals of these neurons whose cell bodies are in the hypothalamic nuclei. These terminals are like the synaptic terminals between neurons, except for the fact that they release their "transmitter" into the bloodstream rather than onto other neurons. Finally, release of these hormones occur when electrical activity increases in the hypothalamic cells that project into the neurohypophysis. In the adenohypophysis, the release of hormones synthesized and stored by the cells is controlled by local hormones produced by cells in the hypothalamus. These hypothalamic hormones (called release hormones or release-inhibit hormones) are all peptides. These releasing hormones are transported along axons of the hypothalamic neurons, where they are released from terminal arborizations that resemble synapses. The synapses end on the blood vessels of a portal system in the median eminence of the hypophyseal stalk (a portal system being a collection of veins that connect capillary beds in two organs rather than connecting the capillary bed in an organ with the rest of the circulation). The releasing hormones are then carried by the blood to the adenohypophysis where they stimulate cells with hormone-specific receptors to release their stored hormones.
Intermediate lobe produces melanocyte-stimulating hormone (MSH).
Functions:
The pituitary hormones help control: growth, blood pressure, breast milk production, sex organ functions in both sexes, thyroid gland function, metabolism, water regulation, secretion of ADH to control water absorption in the kidneys and temperature regulation.
Located in the neck below the thyroid cartilage, the thyroid controls the rate at which the body burns energy, makes proteins, and controls sensitivity to hormones. The thyroid produces its own hormones including thyroxine (T4) and triiodothyronine (T3) which regulate the rate of metabolism and affect the growth and functional rate of other systems in the body.
Controlled by the hypothalamus and pituitary gland, the thyroid gland gets its name from the Greek "shield" after the shape of the thyroid cartilage.
The most common problems involving the thyroid gland include: hyperthyroidism and hypothyroidism. Hyperthyroidism is an overactive thyroid while hypothyroidism is an underactive thyroid.
The important hormone is parathyroid hormone (PTH) and is regulated by calcium in blood. Its main function is to raise blood calcium level. It is secreted by parathyroid glands, causes bone to release Ca2+ in the kidneys. PTH also stimulates the kidneys to activate vitamin D, promoting intestinal uptake of Ca2+ from food.
Also known as the suprarenal gland, the star-shaped adrenal gland sits on top of the kidneys and is responsible for releasing hormones in conjunction with stress through the synthesis of corticosteroids and catecholamines using cortisol and adrenaline, respectively.
The adrenal cortex is vital to the synthesis of corticosteroid hormones from cholesterol. The source of cortisol and corticosterone synthesis is the hypothalamic-pituitary-adrenal axis. Under normal unstressed conditions, the human adrenal glands produce the equivalent of 35–40 mg of cortisone acetate each day. They also have other functions which include producing androgens (like testosterone)and regulating water and electrolyte concentrations via secretion of aldosterone. The adrenal cortex is regulated by neuroendocrine hormones secreted by the pituitary gland renin-angiotensin system, and hypothalamus.
The three layers of the adrenal cortex are: zona glomerulosa, zona fasciculata, and zona reticularis. Zona glomerulosa is the site for production of mineralocorticoids which affect the body's sodium homeostasis. Zona fasciculata produces glucocorticoids in humans. Cortisol secretion is simulated by adrenocorticotropic hormone (ACTH) from the anterior pituitary. In the absence of ACTH, zona fasciculata secretes a basal level of cortisol. Zona reticularis produces mainly dehydroepiandrosterone (DHEA) and DHEA sulfate.
The adrenal medulla is the core of the adrenal gland and is surrounded by the adrenal cortex as it releases secretions directly into the blood stream. The chromaffin cells of the medula are the main source of circulating water-soluble catecholamines derived from the amino acid tyrosine: adrenaline (epinephrine) and noradrenaline(norepinephrine). This is party of the fight-or-flight response initiated by the nervous system.
Arteries and veins supply blood to the adrenal gland. The superior suprarenal artery is provided by the inferior phrenic. The middle suprarenal artery is provided by the abdominal aorta and the inferior suprarenal artery is provided by the renal artery. The right suprarenal vein drains the inferior vena cava and the left suprarenal vein drains the left renal vein also known as the left inferior phrenic vein. The suprarenal veins may form anastomoses ( the branching out and reconnection of blood vessels) with the inferior phrenic veins.
Situated deep within the abdomen between the stomach and the spine, it is known as both the double gland and the mixed gland because it has both endocrine and exocrine functions. As an endocrine gland, it secretes hormones into the bloodstream. When it comes to its function as an exocrine gland, it plays a pivotal role in the secretion of enzymes to help break down carbohydrates, fats, proteins, and acids in the duodenum. More importantly, the pancreas gland secretes the insulin hormone, which regulates sugar metabolism. Too little insulin would result in high blood sugar levels in addition to muscle and body weakness (a condition known as diabetes).
Observing the functionality of the pancreas gland reveals its involvement in the production of chemicals that are critical to
proper digestion and blood sugar regulation. Besides its role in the secretion of the body's important enzymes, the pancreas gland
also creates digestive juices as a member of the exocrine system. These fluids play a key role in breaking down the nutrients that
the stomach acids were incapable of metabolizing. The juices initially are alkaline in the pancreas but as they make their way
through the stomach where they meet other substances, they become acidic.
The ovaries, situated in the pelvis with each ovary on opposite sides of the uterus, are a pair of female reproductive organs. To give you a sense of the shape and size of an ovary, we can consider it quite similar to the size and shape of an almond. Ovaries specifically have two functions: they produce eggs and just like the other glands of the endocrine system, they produce female hormones.
The ovaries are in fact the main source of female hormones, which include estrogen and progesterone. These hormones play a pivotal role in controlling the development of female body characteristics, such as the breasts, body shape, and body hair. More importantly, the ovaries also regulate the menstrual cycle and pregnancy.
The testes is the male reproductive organ held responsible for the production and storage of millions of tiny sperm cells. The testicles are oval-shaped and can grow to about five centimeters in length and 3 centimeters in diameter. The testes do indeed play a pivotal role in the endocrine system due its production of hormones, namely testosterone. As a young man reaches puberty, his testes actually produce more and more of this hormone. More importantly, this hormone causes boys to have deeper voices, bigger muscles, grow facial and body hair, and stimulate the production of sperm.
It is a portion of the brain containing some small nuclei with a variety of functions. It plays a central role in integrating the endocrine and nervous systems. The hypothalamus is located below thalamus, just above the brain stem. The hypothalamus receives information from nerves throughout the body and from other parts of the brain. Signals from the hypothalamus travel to the pituitary gland, which is located at the base. Some produce directacting hormones that are stored and released from the posterior pituitary. Other hypothalamic cells produce hormones that are transported by portal blood vessels to the anterior pituitary. These hormones either promote or inhibit the release of hormones from the anterior pituitary.
The way in which the hypothalamus promotes or inhibits the release of hormones from the anterior pituitary is through the use of a portal vein system and hormone-releasing hormones. Hormone-releasing hormones include, Growth hormones, Prolactin, Folicle-stimulating hormone, Luteinizing hormone, Thyroid-Stimulating Hormone, and ACTH. These hormones are released by the anterior pituitary upon promotion of their release via hormone-releasing hormones such as antidieuretic hormone, ADH. Once these hormones reach the anterior pituitary gland via the portal vein connected the hypothalamus and the gland, the hormones are secreted into the bloodstream and taken up by their specific receptors throughout the body.
The effect of any hormone on a cell depends on the cell's intracellular machinery. Hormones only affect the specific receptors to their appropriate receptor molecules in their membranes. Their is little effect of these hormones on other types of cells due to receptor specificity.
An example of this is that in the smooth muscle fibers of the blood vessel walls, catecholamines produce contraction but in the liver, catecholamines make it possible for the breakdown of glycogen and the release of glucose into the bloodstream.
Tropic hormones
It is a hormone that regulates the function of endocrine cells or glands. It is also produced and secreted by anterior pituitary that target endocrine glands.
Tropic hormones include TSH, follicle-stimulating hormone (FSH), luterinizing hormone (LH), and adrenocorticotropic hormone (ACTH). The main target ofFSH and LH are testes or ovaries.
Nontropic hormones include prolactin (PRL) and melanocyte-stimulating hormone (MSH).
Growth hormone is secreted by the anterior pituitary and stimulates growth through tropic and nontropic effects. The major targets are liver, bones, and other tissues.
Some hormones have very specific targets while others affect essentially all cells of the body. Most hormones belong to one of a few basic families: the peptides, the amino acid derivatives, and the proteins. Many endocrine glands secrete more than one hormone each. When this is the case, the hormones secreted by a gland belong to the same family. In general, each family operates by a characteristic mechanism to regulate the function of target cells. All of them must first bind to receptor molecules that specifically recognize hormones. Protein and peptide hormones bind to membrane surface receptors and through a series of reactions involving small cytoplasm molecules called "second messengers" and enzymes called protein kinases. These hormones modify the activity levels of proteins within the cytoplasm by causing them to be phosphorylated on particular amino acid residues. Also, these peptides and protein hormones are charged at normal pH and cannot cross cell membranes. These hormones therefore must bind to the extracellular domain of g-protein coupled receptors. On the other hand, steroid hormones and thyroid hormones are hydrophobic. Due to their physical properties, steroid hormones and thyroid hormones must bind to intracellular receptors, which allow them to move into the nucleus bound to their receptors, directly affecting the transcription of genes.
When level of hormone is too high, the normal effects of hormones are exaggerated. This effect is known as hypersecretion. The causes of hypersecretion include benign and malignant tumors of the endocrine glands. Imbalance of hormones in the body can also come from outside of the body, known as exogenous hormones or agonists. The introduction of exogenous hormones in the body is known as iatrogenic. For example, when certain hormones such as cortisol in the body is introduced, it causes the shutdown of the secretion of hypothalamus and anterior pituitary glands. This causes the loss of cell mass, called atrophy. In contrast, hormone deficiency in the body is called hyposecretion. The main cause of hyposecretion is from atrophy of the gland due to a certain disease process.
The specificity of the response to any hormone lies in the properties of the receiving cells and not in the delivery of those hormones to the cells. Only the cells with the appropriate receptor molecules respond, even to the steroid hormones that will dissolve through the membranes of all cells. The effect that any hormone has on any particular cell depends upon the cell's own intracellular machinery. When a hormone binds to its specific receptors on different kinds of cells, the effect depends upon the nature of the cell.
Hormonal control involves a negative feedback loop and thus homeostasis within the body. Many types of hormones exist in the body and thus many different feedback loops exist for the systems. Some of them are very simple, others are very complex.
A simple example of hormonal control is the feedback loop for Insulin. Insulin is released into the bloodstream via blood cells of the pancreas, and it controls the rate at which most cells remove glucose from the bloodstream. Insulin is synthesized and stored in pancreatic beta cells. The synthesis and the release of insulin are triggered by the increase of glucose in the bloodstream. Other signals such as an increase in amino acid concentration in the blood, the presence of several other hormones, and neural control, have an effect on the increase of insulin into the bloodstream. Hormone signals are always terminated by either the binding of the hormone to its specific receptor and the breakdown of the hormone via bloodstream enzymes.
Ovarian cancer is a type of cancer which abnormal cells begin to grow in one or both of a woman's ovaries. An ovary is a small gland, located on both sides of the uterus; it is known for producing and storing eggs.
Unfortunately, ovarian cancer is often not detected in its earlier stages. However, some tests are done if a patient is showing signs of ovarian cancer.
Some exams that are used to diagnose ovarian cancer include:
Biopsy
Physical exams, including a pelvic exam and a Pap test
Cancer antigen 125 (CA-125) test to measure amount of protein found on cancer cell surfaces
Graves Disease is an autoimmune disorder that leads to over activity of the thyroid gland (hyperthyroidism).
What is the thyroid gland?
The thyroid gland is an important organ of the endocrine system. It is located in the neck, below the voice box. This gland releases the hormones thyroxine (T4) and triiodothyronine (T3), which control body metabolism. Controlling metabolism is critical for regulating mood, weight, and mental and physical energy levels.
Graves disease is caused by an abnormal immune system response that causes the thyroid gland to produce too much thyroid hormone. Graves disease is most common in women over age 20. However, the disorder may occur at any age and may affect men as well.
One normal immune system response is the production of antibodies designed to target a specific virus, bacterium or other foreign substance. In Graves' disease — for reasons that aren't well understood — the body produces an antibody to a particular protein on the surface of cells in the thyroid, a hormone-producing gland in the neck.
Normally, thyroid function is regulated by a hormone released by a tiny gland at the base of the brain (pituitary gland). The antibody associated with Graves' disease — thyrotropin receptor antibody (TRAb) — can essentially mimic the action of the regulatory pituitary hormone. Therefore, TRAb overrides normal regulation of the thyroid and results in overproduction of thyroid hormones (hyperthyroidism).
Rapid or irregular heartbeat (palpitations or arrhythmia)
Restlessness and difficulty sleeping
Shortness of breath with activity
Tremor
Weight loss (rarely, weight gain)
Thick, red skin usually on the shins or tops of the feet (Graves' dermopathy)
Graves' ophthalmopathy
About half the people with Graves' disease show some signs and symptoms of a condition known as Graves' ophthalmopathy.
In Graves' ophthalmopathy, inflammation and other immune system events affect muscles and other tissues around your eyes.
The resulting signs and symptoms may include:
Bulging eyes
Excess tearing
Dry, irritated eyes
Gritty sensation in the eyes
Pressure or pain in the eyes
Puffy eyelids
Reddened or inflamed eyes
Light sensitivity
Double vision
Limited eye movements, resulting in a fixed stare
Blurred or reduced vision (rare)
Ulcers on the cornea (rare)
Graves' dermopathy
An uncommon manifestation of Graves' disease, called Graves' dermopathy, is the reddening and thickening of the skin, most often on your shins or the top of your feet.
Family history. Because a family history of Graves' disease is a known risk factor, there is likely a gene or genes that can make a person more susceptible to the disorder.
Gender. Women are much more likely to develop Graves' disease than are men.
Age. Graves' disease usually develops in people younger than 40.
Other autoimmune disorders. People with other disorders of the immune system, such as type 1 diabetes or rheumatoid arthritis, have an increased risk.
Emotional or physical stress. Stressful life events or illness may act as a trigger for the onset of Graves' disease among people who are genetically susceptible.
Pregnancy. Pregnancy or recent childbirth may increase the risk of the disorder, particularly among women who are genetically susceptible.
Smoking. Cigarette smoking, which can affect the immune system, increases the risk of Graves' disease. The degree of risk is linked to the number of cigarettes smoked daily — the larger the number, the greater the risk. Smokers who have Graves' disease are also at increased risk of developing Graves' ophthalmopathy.
The purpose of treatment is to control the overactive thyroid gland. Beta-blockers such as propranolol are often used to treat symptoms of rapid heart rate, sweating, and anxiety until the hyperthyroidism is controlled.
Hyperthyroidism is treated with one or more of the following:
Antithyroid medications
Radioactive iodine
Surgery
If you have radiation or surgery, you will need to take replacement thyroid hormones for the rest of your life, because these treatments destroy or remove the gland.
Some of the eye problems related to Graves disease usually improve when hyperthyroidism is treated with medications, radiation, or surgery. Radioactive iodine can sometimes make eye problems worse. Eye problems are worse in people who smoke, even after the hyperthyroidism is cured.
Sometimes prednisone (a steroid medication that suppresses the immune system) is needed to reduce eye irritation and swelling.
You may need to tape your eyes closed at night to prevent drying. Sunglasses and eyedrops may reduce eye irritation. Rarely, surgery or radiation therapy (different from radioactive iodine) may be needed to return the eyes to their normal position.
The primary treatment goals are to inhibit production of thyroid hormones and lessen the severity of symptoms.
Hoarseness from damage to the nerve leading to the voice box
Low calcium levels from damage to the parathyroid glands (located near the thyroid gland)
Scarring of the neck
Eye problems (called Graves ophthalmopathy or exophthalmos)
Heart-related complications, including:
Rapid heart rate
Congestive heart failure (especially in the elderly)
Atrial fibrillation
Thyroid crisis (thyrotoxic storm), a severe worsening of overactive thyroid gland symptoms
Increased risk for osteoporosis, if hyperthyroidism is present for a long time
Complications related to thyroid hormone replacement
If too little hormone is given, fatigue, weight gain, high cholesterol, depression, physical sluggishness, and other symptoms of hypothyroidism can occur
If too much hormone is given, symptoms of hyperthyroidism will return
A diagnosis of Graves' disease is based primarily on your answers to the doctor's questions and findings from a physical exam.
He or she may also order laboratory tests to confirm a diagnosis or gather more evidence if a diagnosis isn't clear.
Diagnostic procedures may include:
Physical exam. Your doctor examines your eyes to see if they're irritated or protruding and looks to see if your thyroid gland is enlarged. Because Graves' disease increases your metabolism, your doctor will check your pulse and blood pressure and look for signs of tremor.
Blood sample. Your doctor may order blood tests to determine your levels of thyroid-stimulating hormone (TSH), the pituitary hormone that normally stimulates the thyroid gland, as well as levels of thyroid hormones. People with Graves' disease usually have lower than normal levels of TSH and higher levels of thyroid hormones. Another laboratory test measures the levels of the antibody known to cause Graves' disease. This test usually isn't necessary to make a diagnosis, but a negative result might indicate another cause for hyperthyroidism.
Radioactive iodine uptake. Your body needs iodine to make thyroid hormones. By giving you a small amount of radioactive iodine and later measuring the amount of it in your thyroid gland with a specialized scanning camera, your doctor can determine the rate at which your thyroid gland takes up iodine. A high uptake of radioactive iodine indicates your thyroid gland is overproducing hormones.
Imaging tests. If the diagnosis of Graves' ophthalmopathy isn't clear from a clinical assessment, your doctor may order an imaging test, such as computerized tomography (CT), a specialized X-ray technology that produces thin cross-sectional images. Magnetic resonance imaging (MRI), which uses magnetic fields and radio waves to create either cross-sectional or 3-D images, may also be used.
Addison's disease is a disorder that occurs when the adrenal glands produces insufficient amounts of particular hormones. Typically, with Addison's disease, the adrenal glads to not produce enough cortisol and aldosterone.
Addison's disease is also known as adrenal insufficiency. It can affect people of all ages and both genders. It can also be found life-threatening.
The three hormones produced by the cortex are: the glucocorticoid hormones, the mineralocorticoid hormones and sex hormones. Glucocorticoid hormones such as cortisol, maintain glucose levels and decrease immune response, while helping the body to respond to stress. Mineralocorticoid hormones such as aldosterone help regulate and balance potassium and sodium levels. Sex hormones such as androgens in males and estrogens in females, affect sex drive and work with the body during sexual development.
Addison's disease is caused by damage to the adrenal cortex, glands located just above the kidneys. The damaged adrenal cortex produces less hormones.
Addison's disease occurs when there is damage to the adrenal cortex, affecting the adrenal glands. The damage to the adrenal cortex causes the cortex to produce less of its hormones. This damage could be due to a number of incidences. For example, the immune system may mistakenly attack the adrenal glands, developing an autoimmune disease. Infections such as tuberculosis, HIV, or fungal infections may also occur. Tumors and the use of blood-thinning drugs (anticoagulants) may also cause damage to the cortex.
Typically, a physician will go over with their patient about their medical history and signs and symptoms. If the doctor suspects that a patient may have Addison's disease, the patient may have to undergo some tests, such as:
A patient with Addison's disease will usually be treated with oral corticosteroids to control the symptoms of the disease. Unfortunately, the patient must take these drugs for the rest of their life. A physician may change the dosage due to infection, injuries, stress, or surgery.
If one has an extreme form of adrenal insufficiency, hydrocortisone should be injected into a patient.
There are five major sensory systems that all humans exhibit those being: olfaction, taste, vision, hearing, and touch. Our senses provide us with the ability to understand and detect the environment around us. Each of these primary sensory systems contains specialized sensory neurons that transmit information through the form of nerve impulses or action potentials to the central nervous system. The action potentials that reach the brain from sensory neurons are called sensations. As the brain receives the information of sensations, the brain interprets them into colors, smells, sound, and tastes, which are also called perceptions, and gives stimuli. Sensations and perceptions are detected through sensory receptors. In human body, there are different specialized sensory receptors for different stimuli coming from inside and outside of the body. The sensory receptors that detect stimuli coming from outside of the body are called exteroreceptors, where as those that detect stimuli coming from inside of the body are called interoreceptors.
Sensory Transduction is the conversion of stimulus energy into receptor potential, which is the change in membrane potential of a sensory receptor. As the ion channels in the sensory receptor's plasma membrane opens and closes, the ionic permeability of the membrane is changed, and results in receptor potentials. These sensory receptors can detect the smallest stimulus possible, which makes them really sensitive.
As the information is received in the CNS, through summation the receptor potentials are then being delivered to sensory receptors at different parts of the body are integrated. One type of integration is called adaptation, where it decreases the responsiveness of stimuli so we don't feel every stimuli that is there.
Sensory Transduction is the conversion of stimulus energy into receptor potential, which is the change in membrane potential of a sensory receptor. As the ion channels in the sensory receptor's plasma membrane opens and closes, the ionic permeability of the membrane is changed, and results in receptor potentials. These sensory receptors can detect the smallest stimulus possible, which makes them really sensitive.
As the information is received in the CNS, through summation the receptor potentials are then being delivered to sensory receptors at different parts of the body are integrated. One type of integration is called adaptation, where it decreases the responsiveness of stimuli so we don't feel every stimuli that is there.
Olfaction is very closely related to the sense of taste, since it has the ability to detect odors. A lot odorants are detected when they are carried as vapors into the nose. Many of these odorants are small organic molecules, which account for the smell of many familiar smells.
Odorants are detected in a specific region of the nose, called the main olfactory epithelium, which lies at the top of the nasal cavity. Approximately 1 million sensory neurons line the surface of this region. The property that is responsible for the smell of these molecules is the shape of the molecule and not necessarily the physical properties. The shape is the factor that matters the most because it determines how well it can bind to a specific surface, usually a protein receptor. Take for instance the molecule Carvone ((5R)-2-methyl-5-(prop-1-en-2-yl)cyclohex-2-en-1-one) which is chiral. Each enantiomer produces a different smell yet the only difference in the molecule is the way it is oriented, with no other physical differences. The (R) enantiomer produces the spearmint smell while the (S) enantiomer produces the smell for the plant caraway.
Biochemical research in olfactants suggested that G proteins played a role in detecting smells, and therefore it was determined that 7TM receptors were the cause. The seven-transmembrane-helix (7TM) receptors are responsible for transmitting information initiated by signals as diverse as photons, odorants, tastants, hormones, and neurotransmitters. Several thousand such receptors are known, and the list continues to grow. As the name indicates, these receptors contain seven helices that span the membrane bilayer, they are the largest class of cell receptors. The receptors are sometimes referred to as serpentine receptors because the single polypeptide chain “snakes” through the membrane seven times.
There are neurons that line the upper part of the nasal cavity called olfaction receptor cells. When odorant molecules bind to these receptors, it triggers a signal transduction pathway that sends action potentials straight to the olfactory bulb of the brain. The odorant molecules only bind to specific receptor proteins. The different odors are determined by the binding of distinct odorants to selective receptors.
The sense of vision is based on the absorption of light by photoreceptor cells in the eye. Photoreceptor cells are sensitive to light in the region between 300-850nm in the electromagnetic spectrum. There are two types of photoreceptors, they are called rods and cones. A person’s retina contains around 3 million cones and 100 million rods cone receptors are responsible for colors and only function in bright light. Rods function in dim light and do not perceive color. The human retina contains two types of photoreceptors called rods and cones. Rods are light sensitive but cannot distinguish color. Cones, on the other hand, are able to distinguish color but are not as light sensitive as rods. Rods and cones both have visual pigments that contain the retinal, a light-absorbing molecule. Rods specifically contain rhodopsin, a pigment that is made up of the retinal attached to a particular opsin. When rods are exposed to light, the retinal molecule absorbs the light and it changes shape. In the retinal inactive state, the rod stays in the cis isomer. After the retinal of the rod absorbs light, it changes from cis isomer to the trans isomer. In the trans isomer, it also becomes unbound from the opsin. The outer segment of the rod is made up of disks that are stacked together. The rhodopsin is located within these disks. Once retinal absorbs light, this triggers a signal transduction pathway. First the rhodopsin gets activates by the absorption of light. The active rhodopsin activates transducing, a G protein. This G protein then activates the enzyme, phosphodiesterase. The activated phosphodiesterase hydrolyzes cGMP to GMP, which allows the cGMP to become removed from the sodium channel. Once the cGMP is removed, the sodium channels close and the rod hyperpolarizes as the membrane’s permeability to sodium decreases.
There are three types of neurons that aid in information processing in the retina. Ganglion cells are neurons that send signals from bipolar cells to the brain and contribute in long range signaling. Horizontal cells and amacrine cells help integrate information before it gets transmitted to the brain. Horizontal cells and amacrine cells participate in local signaling. The horizontal cells receive information from rods or cones and will reduce the effect of surrounding rods and cones, which helps to sharpen our image by increasing the contrast. Amacrine cells are activated by the bipolar cells and reduce the effect of surrounding bipolar and ganglia cells. Lateral inhibition is a specific type of integration that aids in creating a greater contrast in image. The optic chiasm is located near the cerebral cortex and is the area where the optic nerves meet and axons cross. The axons from the left visual field, in both the left and right eye, join and travel to the right side of the brain. Vice versa, the axons from the right visual field, join and travel to the left side of the brian.
Usually when one has nasal congestion one can have difficulty tasting food. This is due to the fact that taste is a combination of senses that function by different mechanisms. The sense of taste can therefore be called the sister sense to olfaction. They are however very different in many ways. Tastants fall into five groups: sweet, salty, umami, bitter, and sour.
The simplest of the tastants, the sodium ion, is perceived as salty while the hydrogen ion is considered sour. The salty taste is not caused by 7TM receptors, it is actually detected by the passage of sodium ion through channels expressed on the surface of cells in the tongue. Like salty tastes, sour tastes arise from the effect of hydrogen ions on channels. The taste called umami is caused by the amino acids glutamate and aspartate. Usually one encounters glutamate as a salt called monosodium glutamate (MSG), which is used as a flavor enhancer. Glutamate and aspartate are the only amino acids that cause taste. The umami taste they emit is mediated by a heterodimeric receptor related to the sweet receptor. On the other hand, bitter and sweet tastants are caused by a wide range of different molecules. Many bitter tasting compounds are associated with being toxic. Most sweet compounds are carbohydrates, which are rich in energy and easily digestible. Some non-carbohydrate compounds such as saccharin and aspartame also taste sweet. Just as in olfaction, research pointed to the involvement of G proteins which, also meant that 7TM receptors detect the bitter and sweet taste.
Hearing is based on the detection of mechanical stimuli. People can detect frequencies ranging from 200 to 20,000Hz, corresponding to times of 5 to 0.05ms. Our ability to hear noise is enhanced by the ability to detect where the noise is coming from. It the ability to detect time delay between the arrival of sound in one ear to the time it takes to reach the other. Sound waves are detected inside the cochlea, which is a fluid-filled membranous sac that is coiled like a snail. It is located in the inner part of the ear. Each cochlea has approximately 16,000 hair cells, which are responsible for sound detection. Within each hair cell there is approximately 20 to 300 hair-like projections called stereocilia.
Not only do human ears can detect sound, the inner ear of humans can also detect body position and balance. hair cells in the utricle in inner ear respond when there is changes in head position due to gravity or movement. Different body positions can cause different hair cells and sensory neurons to be stimulated and send different message to the body. This is why some people get motion seasickness with the sudden change in movement or gravity.
The sense of touch like other senses is a combination of many factors. Touch, detected by skin, senses pressure, temperature, and pain. Although currently biochemist do not necessarily know what exactly causes the sense of touch, it has been determined that specialized neurons, termed nociceptors transmit signals from skin to pain processing centers in the spinal cord and brain. It was determined that nociceptors play a role when studying spicy food. The molecule capsaicin activates nociceptors and is the molecule responsible for the “hot” taste. The capsaicin receptor, also called VR1, functions as a cation channel that initiates a nerve impulse.
↑Von Lintig, Johannes, Philip D. Kiser, Marcin Golczak, and Krzysztof Palczewski. "The Biochemical and Structural Basis for Trans-to-cis Isomerization of Retinoids in the Chemistry of Vision." Trends in Biochemical Sciences 35.7 (2010): 400-10. Print.
The Immune System
The world is teeming with such a wide variety of parasites, bacteria, and viruses, and many of these can be potentially very harmful or devastating to the human body. However, humans have a remarkable ability to defend against unfamiliar organisms, and they owe their immune systems to this defense. The immune system is the body's way of identifying between itself and what is foreign. The human body can make more than 108 different antibodies and 1012 T-cell receptors. These all represent a different foreign invader the body can bind to and begin to destroy.
The Immune System Adapts
The human immune system can essentially take on a limitless number of different pathogens. It does this by transforming its immune cells. This huge degree of variability and flexibility is rooted in the principles of evolution. Generations of reproduction and selection has resulted in this powerful system, which is composed of two interrelated systems.
The humoral immune response utilizes antibodies, which are secreted by plasma cells in B lymphocytes. Antibodies, also known as immunoglobulins, are soluble proteins that recognize and bind to foreign invaders, marking them for destruction. The site on the foreign invader to which the antibody binds is referred to as the epitope or antigenic determinant. If the body gives an immune response when the antibodies bind the epitopes of the invaders, then the invader molecule is an immunogen.
The cellular immune response utilizes cytotoxic T lymphocytes, which are also referred to as killer T cells. These cells destroy cells that have foreign markers on their surfaces. Cytotoxic T lymphocytes have special receptors on their surfaces that mediate this immune response.
Helper T lymphocytes are a specific kind of T cell that are involved in both the humoral and cellular immune response. They stimulate the specialization and propagation of B lymphocytes and cytotoxic T lymphocytes.
The Humoral Immune Response
Immunoglobulin G is the most common antibody in serum. Because it has two binding sites, it is able to cross-link multiple antigens. It also has segmental flexibility, or flexibility in regions of polypeptide that allow angle variation. This allows the antibody to bind an antigen in multiple binding sites.
Immunoglobulin M is the first kind of antibody to enter serum after the body is exposed to an antigen. It is able to bind many multivalent antigens that immunglobulin G is unable to. Immunoglobulin A is the major antibody in saliva, tears, and other external bodily secretions. It is first in the body's defense when it comes to bacteria and viruses. Immunoglobulin E protects against parasites but is also responsible for allergic reactions.
Antibodies have domains, or common sequences that adopt the immunoglobulin fold structure. Immunoglobuiln G has 12 immunoglobulin domains that consist of a pair of antiparallel beta sheets held together by disulfide bonds and hydrophobicity. There are two particularly important structures in the structure. Firstly, at one end there are three hypervariable loops that provide the mechanism for generating antibodies and T-cell receptors. The loops allow antibodies to bind to specific molecules. Secondly, the amino and carboxyl groups at the ends of the structure allow domains to form chains together. The immunoglobulin domains together result in the immunoglobulin fold structure, a structure so common that more than 700 genes encode for proteins that have at least one.
The humoral immune system is also referred to as the antibody-mediated system. Firstly, macrophages, a special blood cell, ingest the foreign invader through phagocytosis. They digest the invader and then display bits of it on their cell membranes. Helper-T cells, upon recognizing this, multiply quickly. This is referred to as the activation phase. The helper-T cells then contact B-cells through chemical signals, and the B-cells also begin to multiply quickly. This is called the effector phase. The B-cells divide and their daughter cells may be plasma cells, which produce great amounts of antibodies to signal the destruction of antigens, or B memory cells, which last in the body, keeping it immune to that same antigen for years to come.
The Cellular Immune Response
Many viruses and mycobacteria are not easy to detect. Appropriate T-cell receptors can recognize harmful foreign invaders, and recognize when cells should undergo apoptosis, or cell death. This prevents damage to surrounding cells. The cellular immune response is also referred to as the cell-mediated system. It involves cytotoxic T cells that recognize when other cells have been infected by a foreign invader. As a result, they ingest and destroy the infected cell. Cytotoxic T cells are also vital to the immune system in that they can recognize and destroy larger invaders such as parasites.
Like B cells, T cells too produce memory cells. The body's combination of T and B memory cells enables it to more quickly respond should the same foreign invader enter the body. This is referred to as a secondary response.
So What?
The immune system is obviously vital in maintaining the body's health in a world full of foreign invaders, bacteria, and viruses. However, the body's ability to create T and B memory cells provide uses in the field of health. Today, more and more shots have been created to make the body immune to the different diseases throughout the world. This holds great potential for the diseases that are still rampant in society today. Perhaps one day there will be an immunization for cancer.
Source: Berg, Jeremy and Stryer, Lubert. Biochemistry: Fifth Edition. United States of America: W.H. Freeman and Company, 2002.
Out of all of the organs in the human body, the largest one is the integumentary system.
The integumentary system consists of the skin and the skin’s derivatives, such as hair, nails, glands, and receptors.
The overall purpose and functions of the integumentary system is to protect the body’s internal living tissues from foreign particles, damage, and water dehydration.
The integumentary system acts as a receptor for touch, pain, pressure, heat and cold.
It also protects the underlying tissues and organs, the body from water loss and against abrupt changes in temperature, protects the body against the invasion by infectious organisms; helps dispose waste materials and store water, fat, and vitamin D.
The structure and functions of the integumentary system varies depending on certain organisms.
For humans, the skin has two main layers: the epidermis and dermis.
The outer layer of the skin is the epidermis, which is made up of epithelial cells.
The epidermis contains many types of cells, such as squamous cells, which are the flat cells that lie on the surface of the skin, and melanocytes which gives the skin it’s color.
The second layer that lies beneath the epidermis is the dermis, which includes the secretion glands, blood vessels, hair follicles, and most of the receptors.
The dermis has two layers, the upper papillary, which has receptors that communicate with the central nervous system, and the lower reticular layers which houses hair follicles, nerves, and certain glands.
The dermis contain glands such as the sebaceous glands, which secretes natural oils in order to keep the skin and hair moist, the sudorferous glands, which secrete sweat in order to regulate the temperature in the body, and the ceruminous glands, which secrete wax in order to prevent dust from entering the ear.
On every part of the body, except for the palms and soles, there is hair, which helps maintain body temperature.
Underneath the dermis, is the subcutaneous tissue, which is the “fatty cushion below the skin that separates the dermal layers from the underlying tissues”.
In invertebrates, such as the lobster, its body is covered by a strong and impermeable exoskeleton. Its exoskeleton is composed of chitin and layers of protein, and lobsters shed their exoskeletons and secrete a larger external skeleton in a molting process.
As for a non-mammal vertebrate, such as the bird, its integumentary system includes the skin, feathers, and appendages (beak and claws). Birds have three tissues, the epidermis, dermis, and hypodermis. The epidermis consists of three separate layers: the outermost layer being the horny cell layer, the transitional layer joins both the outer layer to the inner layer, and the innermost layer is the columnar layer. The main component of the dermis is collagen and it has a thin and uniformed structure. The hypodermis is more loosely arranged compared to the dermis and also contains fewer cells. Feathers are the epidermal derivatives, and each feather follicle undergoes telogen, which is the mechanical process where the molting of the feather generates the growth of a new feather.
Similarities and differences between humans, invertebrates, and vertebrates
The human, invertebrate, and non-mammalian vertebrate share both differences and similarities in their integumentary systems.
The similarities include the integumentary system of a human and bird, where the functions of their epidermis and dermis provide a thermal insulation in order to regulate body temperature, their nerve endings are sensed through the central nervous system, and their skin produces vitamin D in the presence of sunlight. Similarities that the invertebrate and non-mammalian vertebrate shares are the molting process of their exoskeleton and feathers.
Differences are that the human skin is made up of glands, hair, and nails, whereas a vertebrates integumentary system is comprised of skin, scales, feather, hair, and glands, and a strong and permeable external skeleton protects the internal organs of an invertebrate.
Interesting facts about the integumentary system in humans
Asides from being the largest organ system of the human body, the integumentary system has many fascinating aspects to it. Some interesting aspects are that a whole new layer of skin is produced every month, forty pounds of skin is shed in an average lifetime, and an adult has approximately twenty square feet of skin. Also, melanin absorbs and reflects ultraviolet radiation from the sun, and the skin has the ability to produce vitamin D just in the presence of sunlight.
Out of the many diseases that are associated with the integumentary system, such as acne, herpes, blisters, and melanoma, two diseases that are typically known are alopecia areata and athletes foot.
Alopecia areata is a non-contagious autoimmune disorder where the immune system attacks the hair follicles on the human body. Hair loss may be in a few or all regions of the body, but is most commonly lost on the scalp, which results in baldness. “In males and females, the spot baldness condition affects 0.1% - 0.2% in early childhood to young adulthood, however, the disease affects people of all ages”.
As for the more commonly known disease known as athlete’s foot, the disease is a fungal infection that causes scaling, flaking, and itching in various areas of the skin. Although this condition typically affects the feet, it can spread to other areas such as the groin. This condition is transmitted in moist areas where people typically walk barefoot such as in the shower, and prevention of athlete’s foot is to maintain good hygiene.
The reproductive system or genital system is a system of organs within an organism which work together for the purpose of reproduction. Many non-living substances such as fluids, hormones, and pheromones are also important accessories to the reproductive system. Unlike most organ systems, the sexes of differentiated species often have significant differences. These differences allow for a combination of genetic material between two individuals, which allows for the possibility of greater genetic fitness of the offspring.
The major organs of the reproductive system includes, the external genitalia (penis and vulva) as well as a number of internal organs including the gamete producing gonads (testicles and ovaries). Diseases of the human reproductive system are very common and widespread, particularly communicable sexually transmitted diseases.
Most other vertebrate animals have generally similar reproductive systems consisting of gonads, ducts, and openings. However, there is a great diversity of physical adaptations as well as reproductive strategies in every group of vertebrates.
In the animal kingdom, there are two type of reproduction. Sexual reproduction occurs when haploid gametes form a zygote, a diploid cell. Both the male and females have gametes. The gamete in males is called the sperm which is motile. Female gamete is the egg which remains stationary, it is not mobile like the sperm. The other type is asexual reproduction which does not involve the sperm and egg. Asexual reproduction occurs on mitotic cell division. Majority of animals reproduce sexually however there are invertebrates that undergo asexual reproduction.
Sexual reproduction is advantageous because it allows for the production of varied genotypes that result from recombination during meiosis. The varied genotypes increases reproductive success because in the case of pathogens or other environmental factors, this will not wipe out the entire population. Asexual reproduction makes genetically identical copies that live better in stable, unchanging environments.
Fission and budding are two forms of asexual reproduction. Fission occurs when a parent organism divides into two daughter organisms. Sea anemones undergo fission as the parent organism divides equally into two smaller organisms. These two daughter organisms are genetic copies of the parent. Budding happens when an organism develops from the outgrowth of an existing one. Budding is observed in corals where buds are formed. There's another form of asexual reproduction that requires two steps, fragmentation and regeneration. Fragmentation is when the body splits into several parts and then regeneration takes place as the body parts that got released become restored and grow back. Reproduction occurs when the parts that spilt off grow and are capable of evolving into a complete animal. Many animals including sponges, bristle worms, sea squirts and cnidarians undergo asexual reproduction in two steps. One example are n annelid worms, they are able to break their body into several parts and each part is able to undergo regeneration and become a complete worm in a very short amount of time. Parthenogenesis is another form of asexual reproduction where eggs develop without the need for fertilization. The offspring for parthenogenesis may be either haploid or diploid.
Hormones that control the reproductive cycle are regulated by environmental cues such as seasonal temperature, lunar cycles or the length of days. Ovulation is a part of the reproduction cycle that takes place midway during which mature eggs are discharged. These cycles occur in both sexual and asexual reproduction. The Daphnia water flea produces two kinds of eggs. One egg undergoes fertilization while the other uses the process of parthenogenesis where no fertilization is necessary for the development. However, asexual reproduction can only happen if environmental settings are agreeable.
The two sets of labia make up the external part of the female reproductive system. The labia covers the opening of the vagina and clitoris. The gonads, where reproductive hormones and eggs are produced, and system of ducts make up the internal part. These ducts store the embryo and fetus as well as transporting gametes. Gonads in the female are a set of ovaries that are made up of follicles. The ovaries are also where the oocytes, an egg cell that has not been fully developed yet. The oocyte are protected by supporting cells when eggs are being developed. During the menstruation cycle, ovulation occurs where a follicle reaches maturation and releases an egg. After this has occurred, a follicular tissue develops and forms the corpus luteum in the ovary. The corpus luteum releases the hormones, estradiol and progesterone. The corpus luteum will deteriorate if the egg is not fertilized. The vagina is a tubular chamber through which the penis enters during sexual intercourse. It also serves as the birth canal through which babies are delivered. The opening to the vagina is the vulva which is protected by the labia majora.
The scrotum and penis make up the external part of the reproductive system. The gonads, where reproductive hormones and sperm are produced, accessory glands and ducts make up the interna; part. Gonads in the male are the testes where sperm is produced. Sperm can only be produced in mammals when the testes are cooler than body temperature. When ejaculation occurs, the sperm travels from the epididymis duct through the vas deferens. Semen is the result of ejaculation and this is made up of secretions from the accessory glands that mix with sperm to ultimately make this fluid.
The mammary glands are not part of the reproductive system but they play an important role in reproduction. Mammary glands in female produce milk. There are epithelial tissues inside the glands that secrete milk. This milk goes through several ducts and leaves through the nipple. Female breasts are made up of fatty tissues that are connected to one another. Male breasts usually are small because they have the hormone, estradiol which reduces the development of connective adipose tissues.
In most animals, individuals are either definite males or definite females. However, in some species, individual organisms are both male and female. Hermaphroditism is when one organism has both sexes. Earthworms and garden snails always have both male and female organs, and when, for example, two earthworms mate, they fertilize each other. A special variation on the theme is sequential hermaphroditism, in which an organism changes sex during its life. If an organism is female first and later changes to male, that organism is protogynous, and if the organism is male first and changes to female, it is said to be protandrous. In different species, sequential hermaphroditism can be influenced by the organism’s age or size or by various environmental/climatic factors.
Cell Signaling is an important facet of biological life. It allows cells to perceive and respond to the extracellular environment allowing development, growth, immunity, etc. Additionally, errors in cell signaling may result in cancer growth, diabetes. By understanding the processes that govern these pathways, scientists may understand the flow of information and transmission thereby allowing humans to treat diseases and grow tissues.
There are many different ways for cells to communicate with each other and the outside environment. They may communicate directly through juxtacrine signaling, over short distances through paracrine signaling and over large distances through endocrine signaling. Additionally, some cells require cell-to-cell contact in order for communication to occur. For this there are gap junctions which connect the cytoplasms of two cells together. In most cases, a molecule carries the signal from one cell and receptors on the other cell bind to the signal molecule thereby allowing communication. Afterwards, many pathways occur which ultimately trigger a cellular response.
Juxtacrine signaling are reactions when proteins from the inducing cell interact with receptor proteins of adjacent responding cells. The inducer does not diffuse from the cell producing it. There are three types of juxtacrine interactions. In the first type, a protein on one cell binds to its receptor on the adjacent cell. In the second type, a receptor on one cell binds to its ligand on the extracellular matrix secreted by another cell. In the third type, the signal is transmitted directly from the cytoplasm of one cell through small conduits into the cytoplasm of an adjacent cell.
Paracrine signaling is a form of cell signaling in which the target cell is near the signal-releasing cell. Some signaling molecules degrade very quickly, limiting the scope of their effectiveness to the immediate surroundings. Others affect only nearby cells because they are taken up quickly, leaving few to travel further, or because their movement is hindered by the extracellular matrix. Growth factors and clotting factors are paracrine signaling agents. The local action of growth factor signaling plays an especially important role in the development of tissues.
Endocrine signaling can be contrasted with two other modes of signaling: neural signaling and paracrine signaling. The different modes of signaling are schematized in the figure.
A key difference is the distance that the regulatory molecule travels to reach its target. Neurons are connected to their target cells via synapses. A neurotransmitter crossing a synaptic cleft will travel between 10 and 20 nanometers. A paracrine will travel only a few millimeters before it is broken down, so it can only act on nearby cells. By contrast, hormones travel via the circulation to reach their targets, which may be multiple tissues that are far apart, and distant from the endocrine cells. Thus, hormones could be said to have systemic effects.
Note that the timing involved in endocrine signaling also differs markedly from neural signaling. Neural signaling is brief and discrete, generally beginning and ending in less than a second. The timing of endocrine signaling is longer: the hormone takes more time to reach its target, the response of target cells takes longer, and hormones are more stable and capable of signaling over longer times.
Protein acetylation is just one of the many examples of a cell signaling pathway. Protein acetylation plays a crucial role in the regulation of chromatin structures and transcriptional activity. Many of the transcriptional coactivators that are found in the body possess intrinsic acetylase activity while transcriptional corepressors posses deacetylase activity. Deacetylation or acetylation complexes both are associated with DNA-bound transcription factors that are in direct response to many signaling pathways. For example, histone hyperacetylation conducted by acetyltransferases are associated with transcriptional activity whereas Histone deacetylation is not. Histone acetylation stimulates transcription by remodeling of the higher order chromatin structures, that then weaken histone-DNA interactions and allow for binding sites for transcriptional activation complexes containing proteins that contain bromodomians which can then bind to acetylated lysines. Histone deacetylation on the other hand represses transcription through an inverse mechanism involving the assembly of higher order chromatin and exclusion of bromodomain-containing transcription activation complexes. At the organism level, acetylation plays many important roles in immunity, circadian rhythmicity and memory formation rather than just the Histone DNA interaction mentioned earlier. Because of these important roles protein acetylation has, it is now a favorable target in the drug design for numerous disease conditions.
In juxtacrine interactions, proteins from the inducing cell interact with receptor proteins of adjacent responding cells. The inducer does not diffuse from the cell producing it. There are three types of juxtacrine interactions:
1) A protein on one cell binds to the corresponding receptor on the cell right next to it.
2) A receptor on one cell binds to its ligand on the extracellular matrix given off by another cell.
3) The signal is transmitted from the cytoplasm of a cell through the cytoplasm to an adjacent cell.
Juxtocrine signaling is a type of intercellular communication that is transmitted by oligosaccharide, lipid or protein components of a cell membrane. Many juxtocrine signals affect the emitting cell or the adjacent cells nearby. A juxtocrine signal occurs between neighboring cells that have extensive patches of closely opposed plasma membranes linked by transmembrane channels known as connexons. Unlike other types of cell signaling, like paracrine and endocrine, juxtacrine signaling requires physical contact between the two cells involved.
There are three types of signaling modes of juxtacrine interactions:
The Notch Pathway
The Extracellular matrix
Gap Junctions
The Notch Pathway
Notch proteins are activated by cells that express the Delta, Jagged or Serrate proteins in their cell membranes and is present in most multicellular organisms. A Notch protein extends through the cell membrane and has an external compartment exposed to the outsides, which is where it contacts Delta, Jagged or Serrate proteins that are protruding out from an adjacent cell. When attached to one of these ligands, Notch proteins undergo a conformational change that enables it to be cut by a protease. The cleaved portion enters the nucleus and binds to an inactive transcription factor of the CSL family. When bound to the Notch protein, the CSL transcription factors activate their target genes.
There exists four different notch receptors in mammals: NOTCH1, NOTCH2, NOTCH3, and NOTCH4. The notch receptor is a single-pass transmembrane receptor protein.
Discovered in 1917 by Thomas Hunt Morgan, the Notch gene was noticed in the wingblades of a strain of the fruit fly Drosophila melanogaster. Further analysis was conducted as the molecular analysis and sequencing took place in the 1980s.
The signaling pathway of a Notch protein is important for cell-cell communication which takes place during embryonic life and in adults. It plays a role in:
1.) Neural function and development
2.) Cardiac valve homeostasis along with other repercussions in disorders involving the cardiovascular system
3.) Cell lineage specification of the endocrine and exocrine pancreas
4.) Regulation of cell-fate in mammary glands at several development stages
5.) stabilization of arterial endothelial fate and angiogenesis (the growth of new blood vessels from pre-existing vessels).
6.) regulation of crucial cell communication ev ents between endocardium and myocardium during the formation of the primordial valve and the ventricular development and differentiation.
7.) influencing of binary fate decisions of cells- between secretory and absorptive lineages in the stomach
8.)expansion of the hematopoietic stem cell compartment during bone development and participation in the osteoblastic lineage inferring potential therapeutic role for Notch in bone regeneration and osteoporosis
Disease involving Notch signalling include: T-ALL (T-cell acute lymphoblastic leukemia), CADASIL (Cerebral Autosomal Dominant Arteriopathy with Sub-cortical Infarcts and Leukoencephalophy), Multiple Sclerosis (MS), Tetralogy of Fallot, Alagile syndrome as well as other disease.
The Extracellular Matrix as a Source of Critical Developmental Signals
The extracellular matrix consists of macromolecules secreted by cells into their immediate environment. Macromolecules form a region of noncellular material in the regions between cells. The extracellular matrix is made up of collagen, proteoglycans and a variety of specialized glycoprotein molecules such as fibronectin and laminin. These two glycoprotein molecules are responsible for organizing the matrix and cells into an ordered structure.
Fibronectin is a large glycoprotein dimer synthesized by numerous cell types. It’s function is to serve as a general adhesive molecule linking cells to one another and to other substrates such as collagen and proteoglycans. It has several distinct binding sites and their interaction with appropriate molecules result in proper alignment of cells with extracellular matrix.
File:PBB Protein FN1 image.jpg
Laminin along with Type IV Collagen is a major component of a type of extracellular matrix called basal lamina. Laminin plays a role in assembling the extracellular matrix, promoting cell adhesion and growth, changing cell shape and permitting cell migration. The ability of a cell to bind to Laminin and Fibronectin depends on its expression of a cell membrane receptor for the cell-binding site of these large molecules. Fibronectin receptor complexes bind fibronectin on the outside of the cell and bind the cytoskeleton proteins on the inside of the cell. Fibronectin receptor complexes span the cell membrane and unite two types of matrices. On the outside, it binds to fibronectin of the extracellular matrix, while on the inside it serves as an anchorage site for actin microfilaments that move the cell. These receptor proteins are known as integrins because they integrate extracellular and intracellular scaffolds, allowing them to work together. On the extracellular side, integrins bind to an arginine-lysine-aspartate (RGD) sequence, while on the cytoplasmic side, integrins bind to talin and alpha actin, two proteins that connect to actin filaments. The dual binding enables cells to move by contracting actin microfilaments against a fixed extracellular matrix. The binding of integrins to the extracellular matrix can stimulate RTK-Ras pathway. When an integrin on a cell membrane of one cell binds to the fibronection or collagen secreted by a neighboring cell, integrins can activate tyrosine kinase cascades through an adaptor protein-like complex that connects the integrins to a Ras G protein.
Direct Transmission of Signals through Gap Junctions
Gap junctions, also called nexus, are made up of connexin proteins and serve as communication channels between adjacent cells. Six identical connexins in the membrane group make up one connexon (hemichannel) and two connexons makes up one gap junction. The channel complex of one cell connects to the channel complex of another cell, enabling cytoplasm of both cells to be joined. When two identical connexons come together to form a gap junction, it is called a homotypic gap junction. When there is one homomeric connexon and one heteromeric connexon that come together or two heteromeric connexons join it is called a heterotypic gap junction.
Properties of gap junctions include:
1.) They allow for direct electrical communication between cells
2.) They allow for chemical communication between cells through transmission of small second messengers
3.) They allow molecules smaller than 1,000 Daltons to pass through
4.) Ensure that molecules and currents passing through gap junction do not leak into the intracellular space.
Below is an example of the autocrine versus juxtacrine signaling modes. In step 1 of the autocrine signaling, the signaling is regulated by the removal of the prepro-extension from the membrane-anchored ligand, following by its controlled release from the membrane in step 2. Orientation restrictions are responsible for the release requirement. On the other hand, in step 1 of the juxtacrine signaling, the prepro-extension release is required, following by the binding to the auxiliary molecule on a neighboring cell in step 2. Furthermore, autocrin ligands bind to the cell that produced them, while, juxtacrine ligands bind to a neighboring cell.
Like "para" which means near, paracrine signaling is a form of cell signaling in which the target cell is near or adjacent to the signal-releasing cell while autocrine signaling acts on surface receptors on the same cell that produced the signal.
Paracrine is the secretion of a hormone by an organ other than an endocrine gland.
Below is a figure of the Oncogenic Ras of the paracrine signaling that promotes the growth of tumor.
File:Paracrine signaling.gif
Oncogenic Ras promotes the transcription of cytokine genes (the double helix), leading to elevated levels of secreted cytokines (the blue spheres) that act to modulate the immune system (yellow cells), promoting angiogenesis (purple capillaries), activating tumor stroma, the brown cells.
Paracrine signaling is used in the signaling through the epithelial estrogen receptor which allows for proliferation and morphogenesis in the mammary gland. Estradiol is a major regulator of postnatal mammary gland development. It uses paracrine signaling to exert its effects through the estrogen receptor and is expressed in the mammary gland stroma and epithelium. Estradiol stimulates the secretion of prolactin by the pituitary gland and suppresses the secretion of other hormones called gonadotropins. These facts help understand that impairment of mammary gland development may be due to the mammary glands inability to react to the estradiol itself.. The signaling failure is usually a result of inactivation of the ER alpha (estrogen receptor alpha).
More conclusive tests will be needed in order to more clearly understand the mechanism behind this process, but it is clear that the ER alpha expression in the epithelium is a heterogenous process that involves using ductal cells which are triggered by estradiol processes. The ER alpha cells proliferate and act on a subtype of cells which releases paracrine signals that permit other epithelial cells to participate in growth.
Two paracrine signaling agents are growth factor and clotting factors. Growth factor signalling affects the development of tissues. An example is in insects where Allotostatin controls growth through paracrine action on the corpora allata.
In mature organisms, paracrine signaling is involved with allergen responses, tissue repair, formation of scar tissue, and blood clotting.
Paracrine Signaling with the ER Alpha-Sonia Mallepell, Sndree Krust, Pierre Chambon, Cathrin Brisken
Estrogen Target and SourceEndocr. Rev. 2006 27:677-706-U. Ohnemus, M. Uenalan, J. Inzunza, JA Gustafsson, R. Paus
Biology online (2009), viewed 15th of November 2014, http://www.biology-online.org/dictionary/Autocrine
The word endocrine is actually from the Greeks and the endo definition means "within" and krine definition means "to separate or to secrete." The term endocrine means “secreting internally,” and specifically refers to secretions that are distributed in the body by way of the bloodstream. Endocrine cells are made up of ductless glands that produce chemical messages called hormones, which are released into the internal environment of the body. These Endocrine secretions are distinguished from exocrine secretions, which are released to the external environment. Thus, endocrine signaling occurs when endocrine cells release hormones that act on distant target cells in the body.
Endocrine signaling can be distinguished from two other types of signaling: neural signaling and paracrine signaling. The key difference between these types of signaling is the distance that the regulatory molecule travels to reach its target. Neurons are connected to their target cells via synapses. A neurotransmitter crossing a synaptic cleft will travel between 10 and 20 nanometers. However, paracrine will travel only a few millimeters before it is broken down, so it can only act on nearby cells. On the other hand, hormones travel via circulation to reach their targets, which are distant from the endocrine cells. Thus, hormones could be said to have systemic effects. The following figure depicts the differences between endocrine, neural and paracrine signaling.
In addition, the timing involved in endocrine signaling differs significantly from neural signaling. Neural signaling is brief and discrete, and usually starts and finishes in less than a second. Endocrine signaling takes longer because the hormone takes more time to reach its target, the response of target cells takes longer. To account for this significant timing difference, hormones are more stable and capable of signaling over longer times.
There are two main categories of hormones: either proteins or steroids. Only sex hormones and those derived from the adrenal cortex are to be considered steroids, everything else in the rest of the body are made from proteins or derivatives or proteins.
Hormone Mechanism
Hormones that are transported via the blood throughout the whole body but only affect particular cells. The ones that to give a response to particular hormones have receptors that are very specific for that hormone. This is analogous to the lock-and-key mechanism. Essentially if the hormone, which is the key, fits into the lock or the specific receptor then the door will be unlocked. When hormones bind to the receptors there will essentially be an effect or action that occurs. When the hormones cannot bind to the receptor sites for various reasons, no reaction will occur. Essentially all the cells that have these receptor sites for the hormone compromise of the target area for the hormone. Sometimes the target tissues will be an centralized area such as in a gland or organ, but sometimes the target tissues disperses all over the body and this is how multiple areas may become affected at the same time. Hormones influence cells by altering the activity of cells. Steroid-type hormones will generally target the receptors which are located inside a cell. Protein-type hormones typically react with receptors that are located on the surface of cells, since this is so easily accessible the events that lead to completion occurs relatively accelerated rate. On the contrary, steroid hormones takes a bit longer because proteins have to be synthesized.
Hormone Control
Hormones are extremely powerful in that minute concentrations will have very intense effects on processes of metabolism. With this great power, hormones must have strict parameters to follow so that equilibrium in the body can be maintained. A lot of hormones would be regulated by negative feedback. Glands initiating the hormone release from another gland will gradually be turned off- this is to avoid hormone imbalance and from production of way too much hormone. An example of this regulation occurs when hypothalamus, which secretes TRh and this prompts the pituitary to release TSH, which then prompts the thyroid gland to secrete T4 or also known as the thyroid hormone. After the body as sufficient amounts of thyroid hormone in the bloodstream, it is the T4 that goes back to the hypothalamus and pituitary to initiate the decrease of TRH and TSH secretion. Similar mechanisms of negative feedback also occurs in adrenal gland and the ovaries and testes.
Chemical Classes of Hormone
1. Polypeptides(proteins and peptides)
2. Amines
3. Steroids
Pituitary gland is a minuscule gland approximately 1 centimeter in its diameter. It is essentially encompassed by bone since it sits right in the sella turcica. This gland is actually connected right to the hypothalamus of the brain with what is known as the infundibulum. This gland is split up into two regions: one is the anterior lobe or also known as the adenohypophysis and the posterior lobe which is known as the neurohypophysis. The hormones that are released by the hypothalamus is what controls the adenohypophysis activity and posterior lobe is controlled by stimulation of nerves.
Hormones of Adenohypophysis
Growth hormone, which is a protein, promotes the growth of muscles, bones, and even other organs and is done by creating new proteins. This hormone is what controls the height of an individual whether that person is tall or short. Having not know growth hormone as a infant, that person will be small and a dwarf. Too much hormone on the other hand will cause the person to be abnormally gigantic.
Thyrotropin which is also known as thyroid-stimulating hormone effects the glandular cells of the thyroid to give off the thyroid hormone. When too much of thyroid-stimulating hormone is secreted then the thyroid gland will become bigger and follow suite by secreting an ample amount of thyroid hormone as well.
Gonadotropic hormones will bind to the receptor sites which are located in the gonads, ovaries, or testes. Their role is to manage growth, development, and function of those three organs.
Prolactin hormone is what initiate the glandular tissue development in female breast at pregnancy and this also promotes the production of milk after the infant is already born.
Adrenocorticotropic hormone binds to the receptors in the cortex of the adrenal gland and this promotes the release of cortical hormones, but mostly cortisol.
Hormones of Neurohypophysis
The hormone that is localized here is the antidiuretic hormone(ADH) and this prompts water reabsorption in the kidney tubules and so not as much water is loss in urine. The overall goal is to keep the water within the body. if one does not have enough of ADH, there will be a lot of water loss through urine.
Pineal gland is a small structure that looks like a cone and extends toward the back of the brain. This gland encompasses neurons, neuroglial cells, and pinealocytes. It is the pinealocytes that give off the hormone melatonin and inputs it right into the cerebrospinal fluid and then directly into the bloodstream. This hormone alters physiologic cycles and also reproductive development.
Thyroid Gland
The thyroid gland is an organ centralized in the neck. It is made up of two lobes, where each is on one side of the trachea or right below the voice box. Isthmus is what connects these two lobes. Inside of the gland there are follicles, which is what produces the thyroxine and triiodothyronine hormones. Iodine is incorporated into these hormones.
Roughly 95% of the thyroid hormone that is active is thyroxine and the last percentages will be triiodothyronine. To be created, both of them require iodine. The secretion of thyroid hormone is controlled by negative feedback and this includes the hormone circulating around, hypothalamus, and adenohypophysis. Not having enough iodine, thyroid will not make enough hormone. When this occurs, the anterior pituitary is stimulated to release thyroid-stimulating hormone which affects the thyroid gland by increasing its size to attempt to produce more hormones and will obviously be ineffective. The hormones cannot be produced any more since one of the essential elements iodine is not present. This thyroid enlargement is what's called an iodine deficiency goiter.
Calcitonin is given off by the parafollicular cells in the thyroid gland. This has an opposite effect to the parathyroid gland by decreasing the calcium levels in the bloodstream. If the calcium in the blood is too high, calcitonin is given off all the way until the calcium ion levels decreases back to normal.
Parathyroid Glands
On the back surface of the thyroid glands and in the connective tissues are four tiny pieces of epithelial tissue, which is known as the parathyroid gland. From this gland what is secreted is the parathyroid hormone or parathormone. Parathyroid hormone regulates how much calcium is in the blood. This hormone will be give off when there is low calcium levels in the blood and the hormone's role is to increase these levels.
Not having enough parathyroid hormone that is being secrete causes nerves to be more excitable. The effects of low calcium in the blood is that this causes random and unceasing nerve impulses and this promotes the contraction of muscles.
The adrenal gland is what sits on top of both of the kidneys. Each glad is divided into two parts: outer cortex and then the inner medulla. Both of these parts of the adrenal gland are synonymous to there being two parts of the pituitary, since it is produced from dissimilar embryonic tissues and will secrete different hormones. Actually only the adrenal cortex is necessary to the body but if body can do without the medulla and have no serious adverse effects.
Both parts of the adrenal gland are controlled by the hypothalamus. Negative feedback is what regulates the adrenal cortex and includes the hypothalamus and adrenocorticotropic hormone. The hypothalamus will give nerve impulses to help control the medulla portion of the adrenal gland.
Hormones of Adrenal Cortex
Even within the adrenal cortex, it is separated into three distinctive regions as well, and each region produces its own characteristic type of hormone. In chemical form, the cortical hormones made up of steroids. Mineralcorticoids is what's secreted by far outside of the adrenal cortex. A primary mineralocorticoid would be aldosterone, which influences or increases the conservation of sodium and water that is in the body. The middle region secretes glucocorticoids. Its primary type is cortisol which acts to increase the glucose concentration in the blood. The third or last group, which is in the far inside region is called the gonadocorticoids or also known as sex hormones. Androgens, which is male hormones and estrogens, which is female hormones, are actually secreted by both females and males in minute amounts. The reason why it seems as if they do not have an effect is that hormones derived from the testes and ovaries will cover up their effects.
Hormones of Adrenal Medulla
Neural tissues create the adrenal medulla and it is here that two hormones are secreted- epinephrine and norepinephrine. During a sympathetic response, usually during times that are stressful both of these hormones will be secreted. Secreting too much of this hormone may cause extremely long or perennial sympathetic responses. Having too little of this hormone does not have any substantial effects.
The pancreases is a long organ that lies crosswise the back of the abdominal wall, to the back of the stomach, and spans from the duodenum to the spleen. The pancreas has two portions an exocrine portion that gives off digestive enzymes for food through to the duodenum and an endocrine portion, which is made up of pancreatic islets and secretes both insulin and glucagon.
There are two types of cells in the islets and they are alpha cells and beta cells. Alpha cells will secrete the glucagon hormone when there is low concentration of glucose in the bloodstream. Beta cells will react in the reverse, when there is high glucose concentration in the bloodstream the insulin hormone will be secreted.
Gonads are the principle reproductive organs, where the testes are for men and ovaries in the female. Not only do they secrete sperm and eggs, but since they are both viewed as endocrine glands they secrete hormones as well.
Testes
Androgens when are collectively as sex hormones for male are known as androgens. The primary androgen is the testosterone, which is released from the testes. The adrenal cortex also produces a very minute amount of this hormone. Testosterone creation starts during fetal development and stops shortly after birth. Production is nearly none during fetal development and commences after puberty. This hormone has a number of responsibilities including development of male reproductive structures, increases growth of muscles and skeletons, widening of the larynx and changing of voices, distribution of hair on the body, and lastly an sexual drive for males that is increased. This testosterone production is managed by negative feedback which includes releasing hormones from both the hypothalamus and gonadotropins, which is from the anterior pituitary.
Ovaries
There are two types of groups of hormones made in the ovaries- both estrogen and progesterone. These hormones are of the steroid type and aid in the development of the female reproductive organs. When puberty commences, estrogens initiate breast development, allocation of fat to the hips, legs, and the breasts, and maturing of the reproductive organs such as the vagina and the uterus. The hormone progesterone brings about the thickening of the uterine and this is done to prepare for pregnancy. Both the progesterone and estrogen induce the changes in the uterus during the menstrual cycle.
Endocrine in Organs
Aside from the big endocrine glands, there are other organs that utilize hormones as part of their way to get things done. Organs that use hormones include thymus, stomach, small intestines, and heart.
Development of the body's immune system is greatly helped by thymosin from the thymus gland. In the stomach's lining known as the gastric mucosa creates a hormone known as gastrin, which is activated by food in the stomach. Gastrin promotes the production of hydrochloric acid and along with the pepsin enzyme- all of which aids in digestion. WIthin the mucosa of the small intestine has two hormones secretin and cholecystokinin. Secretin prompts the pancreas to produce bicarbonate so that the stomach acid can be neutralized. Cholecystokinin prompts the gallbladder to compress it and bile is released. The heart is called an endocrine organ. Certain cells in the atria produces atrial natriuretic hormone.
Diseases and disorders of the endocrine system can be grouped in various ways. Typically endocrinologists will take a target one or two endocrine diseases to focus on.
Diabetes
People that have abnormally high levels of sugar in their bloodstream. There have been research done that shows that manager the amount of sugar in the blood aid in preventing adverse effects stemmed from diabetes. Some of the problems include problems with the nerves, kidneys, and eyes, and gradually may lead to blindness or even amputation. Diabetes patients are treated with a new diet and drugs including insulin. Patients need to help keep their blood sugar low and be monitored so that other health problems may be avoided.
Type 1 diabetes - insulin-dependent diabetes, is an autoimmune disorder in which the immune system destroys the beta cells of the pancreas. Therefore, a person cannot produce insulin.
Type 2 diabetes - non-sinulin-dependent diabetes, is caused by a failure of target cells to respond normally to insulin.
Thyroid
People with adverse thyroid circumstances a lot of the time have lack of energy. Not only this but also conflicts in strength of muscles, controlling weight, very emotional, and also withstanding the hot or cold weather. Typically their problem is that the amount of thyroid hormone is either over-abundant or immensely lacking, which is resultant from either too active or not to active thyroid. The solution for this would be to reach back to a hormone equilibrium by either restoring or obstructing the thyroid hormone. Other potential problems with thyroid include thyroid cancer and also to have an overtly big thyroid gland.
Bone
Problems with bones include rickets- the softening of bones or osteoporosis- making fragile bones, can both be treated with the endocrine system. There are particular hormones that are designed to guard bone tissue. Having anomalies in hormone levels, calcium may be depleted from bones and then become less strong. Losing function of the testicle in men or menopause in women puts the population at higher risk for bones to break. Other problems that affect bones include an over-abundant parathyroid hormone and using steroids such as prednisone for a while.
Reproduction or Infertility
There is a staggering statistic that approximately 10% of couples in America are infertile. Endocrine hormone imbalances may lead in infertility and knowing the source the reproductive conflicts may be addressed. Reproductive endocrinology may be used to work with patients who have irregular periods, premenstrual syndrome, and impotence.
Obesity and Overweight
People that are overtly obese have their source of problems as metabolic or hormonal. Problem in pathways of thyroid, adrenal, ovarian, or pituitary can lead to obesity. More specific reasons associated with obesity include resistance to insulin and also problems in genes.
Pituitary Gland
Pituitary gland manages all of the other glands and so it is sometimes called the master gland. The pituitary do make a lot of important hormones. Too much or too little production of these hormones can lead to imbalance and then cause infertility, disorders in growing, and possibly too much cortisol. These conditions are helped through drugs and sometimes might require surgery.
Growth
Not having enough growth hormone result in children that have disorders in growing and remain short. Growth hormone problems that occurs in adults, make them often feel tired and have emotional distress. Growth hormone replacement therapy can help to manage this imbalance.
Hypertension
High blood pressure could cause eventual problems with the heart. About 1/10 of people will have hypertension as a result of an over-abundance of aldosterone- which is produced from adrenal glands. About 1/2 of the cases the initiate these types of growth can be taken out through surgery. Other smaller factors that can lead to hypertension include a metabolic syndrome or pheochromocytoma and this causes hormonal imbalance which then induces hypertension.
Lipid Disorders
Lipid disorders cause problems with up-keeping the right levels of body fats. Hyperlipidemia, an example, is having super saturate levels of overall cholesterol, low-density lipoprotein, triglycerides in the bloodstream. Having copious amounts of these fats are very associated with heart disease, strokes, and troubles with circulation in legs. Potential causes to disorders caused by lipids could be to less thyroid hormone, steroid drug use, or problems in metabolic or gene regulation. Solutions to help fix this problem may include certain diets, exercise, and drugs.
Endocrine Disruptors are various types of chemicals that can be both natural or made by humans that disrupts the endocrine system and has detrimental effect on humans, fish, and other organisms. They interfere with the creation or hormone activity of the endocrine system and is damaging to health.
How Endocrine Disruptors work
Endocrine disruptors have three main ways in which they work. The first one is to simulate the hormones, which occur naturally in the body such as androgens or the male sex hormone and has the ability to overstimulate. Another way is to bind to receptors in that cell and then block the real hormone from binding. Without the proper signals, the body cannot respond in a proper manner. Examples could include anti-androgens that block the real hormones from binding. Lastly, disruptors affects the way hormones are produced or receptors and their control mechanisms. An example is blocking the metabolism of hormones in the liver.
Endocrine Disruptors examples
There are numerous substances that are probable of causing endocrine disruption. Chemicals that are for certain endocrine disruptors include DES, PCBs, DDT, and other types of pesticides. Examples of probable ones are pesticides and plasticizers like Bisphenol A, which is from animal studies.
Exposure to Endocrine Disruptors
Exposure is very widespread. It can be anything from food and beverages to cosmetics and drugs. Exposure usually happens through either air, diet, or skin.
G-Protein coupled receptors (GPCRs) are a group of seven transmembrane proteins which bind signal molecules outside the cell, transduct the signal into the cell and finally cause a cellular response. The GPCRs work with the help of a G-Protein which binds to the energy rich GTP.
Also known as heptahelical receptors, serpentine receptors, and G protein-linked receptors. These proteins make up transmembrane receptors whose purpose is to find molecules on the outside of the cell and initiate the signal transduction pathways. The signal transduction pathways are the processes by which a cell changes the form of one signal into the stimulus or a signal of another. These processes are carried out by enzymes. As the number of proteins and molecules increases, the size of the signal cascade increases rapidly, allowing for a large response, to a relatively small initiation factor.
G protein linked receptors are activated by ligands in the form of hormones, proteins, or other signaling molecule. This in turn leads to the activation of an intracellular G-protein by way of a certain interaction with the receptor. The G proteins act like relay batons to pass messages from circulating hormones into cells and transmit the signal throughout the cell with the ultimate goal of amplifying the signal in order to produce a cell response. Firstly, a hormone such as an epinephrine encounters a receptor in the membrane of a cell then a G protein is activated as it makes contact with the receptor to which the hormone is attached. Lastly, the G protein passes the message of a hormone to the cell by switching on a cell enzyme that triggers a response (Medicines by Design 46).
In addition to signaling, they have other physiological roles:
-Sense of smell-the olfactory epithelium receptors bind odorants and pheromones
-Mood regulation-receptors in the brain bind neurotransmitters (dopamine)
-Immune system regulation- deals with inflammation and response to foreign bodies
-Nervous system transmission-proteins control blood pressure, heart rate, and digestive processes
-Cell density sensing
Another example of G-protein-coupled receptors (GPCRs) in the body has to do with the sense of taste. Five different tastes have been recognized by science: sweet, salty, sour, bitter and umami. Umami, the flavor of meaty foods, was the taste most recently added to the list, as it was identified by a Japanese scientist in 1908. GPCR taste receptors are found on our taste buds, and there are several different types.
Close up view of taste buds on the tongue
The receptors associated with the umami taste are given the titles T1R1 and T1R3. These are class C GPCRs, which means they have an N-terminus outside of the cell. In this case, these receptors’ extracellular domain is quite large and the shape of the active site contains two halves; such domains have been dubbed Venus flytrap (VFT) domains due to this characteristic shape. The active site of the VFT of the T1R1 and T1R3 receptors binds amino acids, especially aspartate and glutamate (two amino acids with one and two CH2 groups in its R chain, respectively).
Sweet tastes are recognized by receptors in the same family as the umami tastes: T1R2 and T1R3. The VFT domains on these GPCRs have many active sites that can bond many ligands. This is why these particular receptors can not only recognize carbohydrates, like sugars, but also some types of amino acids, peptides, proteins, and the ligands of artificial sweeteners.
While the sweet and umami tastes are recognized by class C receptors, the unpalatable bitter sensation is acknowledged by class A GPCRs, which do not have the large N-terminus of the class C. There are more than 30 different types of GPCRs for the bitter taste, dubbed the T2R receptors. Like the T1R3 sweet/umami receptor, these T2Rs can each detect many chemicals with the bitter taste. Compared to the two or three types of GPCRs for the sweet and umami tastes, thirty different kinds seems like an unnecessary amount. The explanation, however, seems to be one of evolution. Bitter tastes are associated with toxins or poisons produced by plants and insects, so those mammals that recognized the vile taste and no longer consumed the toxin had the opportunity to procreate.
G-protein coupled receptors are only found in eukaryotic cells and are found in a variety of sizes.
There are two main pathways that the G-proteins follow. The first is the cAMP pathway, and the second is the Phosphatidylinositol signal pathway.
The cAMP pathway has five players that aid in the signal transduction: a hormone that stimulates the receptors, a regulative g protein, Adenylyl cyclase, protein kinase A, and cAMP. The stimulative hormone is a receptor that binds with the stimulative signal molecules. The g-proteins is linked to the simulative hormone receptor and its alpha subunit can stimulate activity. Adenylyl cyclase is a transmembrane glycoprotein that catalyzes ATP to form cAMP with the help of a cofactor, usually Magnesium or Manganese ions. Protein kinase A is an enzyme used for cell metabolism.
The cAMP transfers the effects of hormones which are unable to pass directly through the cellular membrane. It also regulates the effects of adrenaline and glucagon in addition to regulating the passage of Calcium ions through the ion channels. cAMP activates PKA (protein kinase A) which is usually an inactive tetramer. cAMP binds to the regulatory subunits of the kinase and causes the dissociation of the regulatory and catalytic subunits. This dissociation activates the catalytic units and allows them to phosphorylate the substrate proteins.
The phosphatidylinositol signal pathway has the signal molecule bind with the G-protein receptor. This activates the phospholipase C which is located in the membrane. Lipase hydrolyzes phosphatidylinositol in to two messengers with bind with the receptors in the membrane. This opens up a Calcium ion channel and activate protein kinase C which causes the cascade of signals.
Studies have shown that GPCR consists of transcriptional, post-transcriptional and post-translational mechanisms and recently, it has been observed that by splicing the pre-mRNA regulates GPCR activity by targeting GPCR Secretin of the exon on a 14 amino acid sequence.
Inside the cell, on the plasma membrane, G Protein binds GDP when inactive and GTP when active. When the GPCRs binds to a signal molecule, the receptor is activated and changes shape, thereby allowing it to bind to an inactive G Protein. When this occurs, GTP displaces GDP which activates the G Protein. The newly activated G Protein then migrates along the cell membrane until it binds to an enzyme and changes the enzyme's shape and conformation. This change in the enzyme structure leads to the next step in the pathway and generates a cellular response. After transduction, G Protein functions as a GTPase and hydrolyzes the bound GTP which causes a phosphate group to fall off. This regenerates GDP and inactivates the G Protein and the cycle repeats.
Membrane-bound receptors are peptide hormone receptor. The membrane-bound receptors are proteins located on the plasma membrane. The receptors can be single polypeptide chains or have up to four subunits. Some may have up to seven transmembrane domains. Some hormones that these receptors bind to are prostaglandins, ACTH, glucogon, catecholamines, parathyroid hormone, etc. The receptor initiates signal transductions when a hormone is bind to it. Many of the signal transductions involve second messengers. Second messengers are compounds that are activated or produced when the receptors bind to the ligands. The second messengers are normally cyclic adenosine monophosphate (cAMP) molecules or cyclic guanine monophosphate (cGMP) molecules, Ca2+, or protein kinase C (PKC). During the signal transduction, many compounds form complexes and either activate or inactivate other molecules inside the cell. These reactions are coupled with G-proteins. Other receptors used are tyrosine kinase, serine kinase, and guanyl cyclase domains known as Enzyme-Linked receptors.
Seven-transmembrane domain receptor
Seven-transmembrane domain receptor has three cytoplasmic loops: loops I, II, and III. Loop III and the carboxyl tail end have a kinase activity, which enables them to autophosphorylate. This receptor is coupled with Guanine Nucleotide Binding Protein (G-Protein). G-protein is a heterotrimeric, meaning it has three non-identical subunits (alpha, beta, and gamma subunits). The alpha subunit has a GDP bound to it when inactive and has a GTP bound when active.
The structure of most G-Protein Coupled Receptors are not very well known. The typical method of determining protein structure is by x-ray crystallography of the protein once it has been crystallized. However, due to the membrane environment, flexibility, and dynamic shifting of GPCRs, it is difficult to form crystals of them. Some have been crystallized by mutating certain amino acids to stabilize the structure, but there is no universal way to study them all.
Although it is tough to find exact information about the structure there are a few known traits of the proteins. G-proteins are integral membrane proteins that make up transmembrane helices. The parts of the receptor can be glycosylated. Of the seven transmembrane proteins, each one may or may not contain an ion channel. These glycosylated loops are made of two cysteine residues that form disulfide bonds which help stabilize the receptor structure.
Conformational change
The receptor molecule exists in equilibrium between the active and inactive states. The ligand binding pushes the equilibrium towards the active sites. There are three types of ligands that bind to the g-proteins. The first are agonists, ligands that shift the equilibrium towards the active states. Inverse agonist shift the equilibrium towards the inactive states, and neutral antagonists are ligands that do not change the equilibrium.
G-Protein Coupled Receptors are widespread in their use. In the eyes, Opsins, a GPCRs, translate electromagnetic radiation into cellular signals thus allowing visual perception. In the nose, olfactory epithelium binds odorants and pheromones which allow for the sense of smell. However, there are problems associated with GPCRs. Many human diseases, including bacterial infections, involve the GPCRs where bacteria produce toxins which interfere with the function of the G-Protein. Examples of such diseases include cholera, whooping cough, botulism, etc.
Here is a closer view of how altered G-protein affects cholera and whooping cough. When there is a β subunit binding to Gαs gangliosides and a catalytic subunit entering the cell, then choleragen, a toxin resulted from cholera, forms. The catalytic subunit alters the Gαs ganglioside in which α part of the protein is adjusted via attaching ADP-ribose to arginine. As a result of this alteration, Gαs ganglioside become stable, meaning that now Gαs ganglioside is in its active form. The active Gαs ganglioside then does its job to activate protein kinase A, denoted PKA. The chloride channel is by PKA, PKA enters and thus there is no more absorption of Na+. This is saying that there is a huge loss of NaCl and water in the body as seen in the symptoms of cholera. In treating cholera, the most effective way is to rehydrate the body using glucose-electrolyte solution.
Whooping cough, on the other hand, is different from cholera in which the ADP ribose moiety is added by the toxin. In this case, Ca 2+ chanel is closed whereas the K + channel is opened by Gαs ganglioside. The result is that the Gαs ganglioside is in its inactive form and ultimately ending up with uncontrollable coughs.
In addition, Pharmacologists estimate that 60% of all medicines achieve their effect by acting on G Protein pathways. Since G protein is a switch molecule which passes the message inward (like relay baton), it can be turned on only when needed, then shut off. Some illnesses like cholera, occur when a G protein is errantly left on. Discovery about Gprotein switches and its structure which is made into 3 subunits (alpha, beta, gamma) will help us understand how we can inhibit the transmission or increase with some other ligand. G-protein coupled receptors are trans-membrane receptor proteins that when activated by ligands (hormones, proteins or other signaling molecules), they lead to the activation of an intracellular G-protein through a specific interaction with the receptor. The G-protein in turn transmits the signal to other proteins within the cell to ultimately amplify the signal and produce a cellular response. Understanding the structure and dynamics of the receptor could clarify the specific interactions the receptor makes with the ligand on the outside and the Gprotein on the intracellular side, thereby leading to the understanding of how the receptor works. Consequently, drugs (agonists or antagonists) can be designed to bind the receptor and control its response, whether it's to transmit the signal to the G-protein or inhibit the transduction. Moreover, monitoring the effects of such signals
can help in understanding the type of induced cellular responses and potentially uncover diseases that are proliferated in this manner.
Cyclic adenine monophosphate (cAMP) is a secondary messenger used in intercellular signal transduction. The synthesis of cAMP from ATP is done by adenylyl cyclases located in the plasma membrane. The understanding of this signaling mechanism is attributed to Earl W. Sutherland whose work won him the Nobel Prize in 1971.
cAMP is used for intracellular signal transduction and affects a number of cellular processes. For example, cAMP stimulates the production of ATP for muscle contraction. In other cells, cAMP enhances the degradation of storage fuels, increases the secretion of acid by the gastric mucosa, leads to the dispersion of melanin pigment gradules, and diminishes the aggregation of blood platelets. It is also involved in the activation of protein kinases and regulates the effects of adrenaline and glucagon. In addition, cAMP regulates the passage of metal ions, like Ca2+, through ion channels.
cAMP works by activating protein kinase A (PKA). PKA is a normally inactive tetrameric holoenzyme, consisting of two catalytic and two regulatory units (C2R2), with the regulatory units blocking the catalytic centers of the catalytic units. cAMP binds to specific locations on the regulatory units of the protein kinase, and causes dissociation between the regulatory and catalytic subunits, thus activating the catalytic units and enabling them to phosphorylate substrate proteins. Binding of four cAMP molecules causes the release of free and active catalytic subunits, which may phosphorylate serine and threonine residues on target proteins. The figure below shows a cAMP/PKA signaling pathway:
The active subunits catalyze the transfer of phosphate from ATP to specific serine or threonine residues of protein substrates. The phosphorylated proteins may act directly on the cell's ion channels, or may become activated or inhibited enzymes. PKA can also phosphorylate specific proteins that bind to promoter regions of DNA, causing increased expression of specific genes.
When a signal is first transducted inside the cell, an enzyme called adenylyl cyclase is activated which converts ATP to cAMP. Then the cAMP activates protein kinase A by binding to the regulatory units in protein kinase A, thereby allowing the disassociation between the catalytic and regulatory subunits in protein kinase A. The newly activated protein kinase A allows for the phosphorylation of serine and threonine from ATP which generates cellular response. This secondary messenger amplifies the signal many times over through the synthesis of many molecules of cAMP by adenylyl cyclase. However, the cAMP molecules do not last long as another enzyme, phosphodiesterase, converts cAMP to AMP which means another signal is necessary in order for another response to occur.
Inositol
Inositol Phosphates are a type of mono-to-polyphosphated inisitols[check spelling] which act as secondary messengers in signal transduction. They play roles in cell growth, apoptosis, endocytosis, etc. There are several types of Phosphoinositol with the most prominent being inositol triphosphate (IP3).
This molecule is used in conjunction with diacylglycerol (DAG) in cell signaling as a secondary messenger. However, whereas DAG stays on the lipid bilayer, inositol triphosphate is soluble in the cytoplasm with the cleaving of phosphatidylinositol 4,5-bisphosphate (PIP2) which attaches it to the lipid bilayer. Once free, inositol triphosphate activates the IP3 receptor on the endoplasmic reticulum triggering the release of calcium ions. The increase in calcium ions raises the level of calcium in the cytoplasm, activating the next protein in the signaling chain until a cellular response is achieved.
Calcium is a common signaling mechanism, because once it enters the cytoplasm it exerts allosteric regulatory affects on many enzymes and proteins. Calcium can also be a second messenger caused by indirect signal transduction pathways such as g-protein coupled receptors.
There are two major properties that allow Calcium (Ca2+) Ion to work effectively as a signaling mechanism
Ca2+ levels inside the cell are readily detectable. This is because the levels of Ca2+ are highly regulated by transport systems that expel Ca2+ from the cell. The level of Ca2+ in the cytoplasm is approximately 100nM, which is several orders of magnitude lower than outside the cell. This must be regulated in order to keep salts from forming between Ca2+ and carboxylated and phosphorylated compounds, as the salts formed are mostly insoluble. Because of all these factors, any small increases of the Ca2+ inside the cell as a signal is readily and quickly detectable, making it a useful signaling mechanism.[1]
Ca2+ can readily bind to proteins and cause conformational changes. Ca2+ is attracted to the negatively charged oxygen atoms in the side chains of glutamate and asparagine, and the uncharged oxygens in both the side chains and main chains of glutamine and asparagine. Ca2+ is readily able to cause large conformational changes due to the fact that it can form ligands with up to eight oxygen atoms. This can lead to cross linking of amino acids in a protein that did not exist before Ca2+ was introduced.[2]
Calcium influx within a cell is a fundamental process within many types of cells, namely, nerves and muscle cells. In nerves, calcium entry drives the release of neurotransmitters, while in muscle cells, calcium is directly involved in actin-myosin sliding filament theory[1]. However, merely stating that calcium influx causes these responses would be undermining the process of calcium signal integration cells undergo.
There are three modes upon which a cell can integrate a calcium oscillation signal: frequency, amplitude, and spatial. Realizing that Calcium oscillations follow sinusoidal waveforms makes it easier to see what these characteristics refer to. The frequency of oscillation is integrated by Protein Kinase C, and Ca/calmodulin-dependent protein kinase II. Here, frequency refers to how many cycles per second the oscillation occures, namely Hertz. Additionally, the amplitude of the signal is a main attribute of the oscillation that is integrated by the cell. To accomplish this, calcium receptors of varying affinities are located within the cell. Since amplitude refers to the magnitude of calcium oscillation, different magnitudes of calcium will activate specific receptors. This allows essentially allows specific cellular functions to taker place based upon the magnitude of calcium spark [2].
A more recently discovered signal integration method involves spatial layout of the calcium flux. Since calcium enters the cell via membrane channels, it is inevitable that on a small timescale, calcium concentration will vary spatially throughout the cell. The amplitude of calcium spark will decay radially from the channel's location. The versatile calmodulin is responsible for integrating these Calcium micro-domains and global-domains signals. Here micro-domains refer to calcium sparks in a small volume relative to the cytoplasm, where global-domain refers to complete cytoplasmic calcium concentration changes. Calmodulin's amino and carboxyl terminals collaborate to carry out spatial integration [2].
[1] Silverthorn, U. D. Human Physiology. Pearson, 2010. pg 400-444.
[2] Parekh, A. B. Decoding Cytosolic Calcium Oscillations. Cell Press. TIBS-804. pg 1-10.
Picture By: Jjbeard
The /calcium signaling/Calcium Signaling mechanism begins when an agonist molecule binds to its receptor, which is located on a G protein. This docking dissociates GDP from the G protein complex, leading to the dissociation of G-alpha subunit and G-beta/G-gamma complex. The G-alpha is bound to GTP to form the G-alpha-GTP complex, which then moves to PLC (phospholipase C).
The docking of G-alpha-GTP on PLC causes conformation change of PLC, activating an enzyme that is involved in hydrolyzing PIP2 into DAG and IP3. The IP3 moves to the ER and docks on the receptor of IP3, opening the calcium channel and allowing calcium ions exit the ER. Calcium ions then bind to buffer, or enter Mitochondria through calcium uniporter. They could also simply become suspended in the cytoplasm.
IP3 can be phosphorylated or hydrolyzed, increasing the amount of IP3. When Calcium ions in ER are nearly depleted, a signal is sent out of the ER and to activate the SOC channel, which then opens and allows calcium ions from extracellular fluid to enter the cytoplasm. These Calcium ions can enter either ER through the SERCA pump or exit the cell through the PMCA pump.
When the agonist leaves the receptor on the G protein, the recovery process begins. GTP on G-alpha-GTP complex is hydrolyzed to GDP and reassociates with G-alpha, G-beta/G-gamma attaching on G-protein. Calcium ions enter the mitochondria via ion exchanger with sodium ions. SERCA and PMCA pumps slowly allow Calcium ions to move in and out. Consequently, the amount of calcium ions in the ER increases.
Here is a link to an animation of calcium signaling. Open animation link with Internet Explorer. Calcium Signaling Animation
↑Berg, Jeremy (2007). Biochemistry, 6th Edition. New York, New York: Sara Tenney. pp. 389–390. ISBN978-0-7167-8724-2. {{cite book}}: Unknown parameter |coauthors= ignored (|author= suggested) (help)
↑Berg, Jeremy (2007). Biochemistry, 6th Edition. New York, New York: Sara Tenney. pp. 389–390. ISBN978-0-7167-8724-2. {{cite book}}: Unknown parameter |coauthors= ignored (|author= suggested) (help)
Protein kinases are not enzymes that can not function by phosphorylating other proteins to assign the proteins a code. The codes instruct the cell to do work, such as divide or grow. Phosphorylation is a common way of activating or inactivating enzymes, and protein kinases are thus often found in signaling pathways. As many as 30% of all human proteins are modified by phosphorylation, making kinases extremely important in cellular regulation. Since one kinase molecule can act on many proteins, the effect of activating a kinase is amplified in the number of target proteins activating. If a kinase acts on another kinase as part of a signaling pathway, the effect of activating the kinase is amplified several times, resulting in a signaling cascade. A cascade can be stopped by removal of phosphate groups by protein phosphatases[1].
An example of a signaling cascade can be seen in life which remodels the extracellular matrix that causes tumor cells to intervene. Heparanase adds onto to the signaling cascade which results in more phosphorylation of specific protein kinases and producing a tumor due to more gene transcription.
Both kinases and phosphatases are regulated by complex signaling pathways. The activation of a kinase at the start of signaling cascade can come from several sources, including cyclic adenosine monophosphate (cAMP) and calmodulin. cAMP release is activated by hormones such as epinephrine.
Cyclic adenosine monophosphate
Cyclic adenosine monophosphate
Calmodulin 3D structure
Example: the binding of cAMP to the regulatory subunit of PKA (protein kinase A) relieves its inhibition of the catalytic subunit. In short, cAMP activates the kinase.
Kinases generally function by bringing an ATP molecule together with the protein to be modified in a hydrophobic pocket to exclude water. A phosphate group is then transferred from ATP to an amino acid on the protein being activated. Phosphorylation usually takes place on the hydroxyl group of serine, threonine, or tyrosine, but there are also kinases that act on other amino acids such as histidine, aspartate, and glutamate. In a signaling cascade, the newly activated protein might also be a kinase that can then phosphorylate other proteins.
The binding of a phosphate group can affect the activity of a protein in two ways. Phosphorylation can directly affect activity by causing a conformational change in the protein. Alternately, the phosphorylated amino acid can be recognized by and bind to specific proteins or other substrates[2].
The carboxyl-terminal includes a phosphotyrosine residue, which can contact with SH2 domain. also, SH2 domain and the Kinase domain is bound by the SH3 domain. in this case the kinase become in its inactive conformation. the protein can be acteivated by three basic steps 1. the phosphotyrosine residue bound in the SH2 pocket, which can be replaced by another phosphotyrosine that containing higher affinity polypeptide for that SH2 domain. 2. in Tyrosine the phosphoryl gp can be removed by a phosphatase and finally 3. the liker can be replaced from SH3 domain by a high affinity polypeptide.
Proteins that have been phosphorylated by a protein kinase can be dephosphorylated by protein phosphatases. Phosphatases bring in water to hydrolyze the phosphodiester bond to release the dephosphorylated protein and orthophosphate. The signaling cascade as a whole is stopped when the initial trigger signaling molecules disappear, such as when a hormone is broken down. Some cascades are self terminating, as the activated kinase phosphorylates enzymes that catalyze the breakdown of the trigger molecule, as in the case of cAMP.
Since kinases can quickly cause very large changes in a cell because of the amplification of a signal, their activity is tightly regulated. Many kinases are themselves controlled by phosphorylation and some kinases phosphorylate themselves, in a process called cis-phosphorylation or autophosporylation. Other means of kinase regulation include the binding of inhibitor or activator proteins or small molecules and controlling their location within the cell.
Mitogen-Activated Protein Kinase (or MAP kinase) is well known to play an important role in keeping oocytes in an arrested state until fertilization. Scientists believe that MAP kinase signaling is one of very important cellular communication streams. In mammalian eggs, which arrest in metaphase II of meiosis, active MAP kinase keeps the eggs from completing meiosis until after fertilization. Like other protein kinases, it transfers phosphate group from ATP to serine, threonine or tyrosine amino acids on recipient proteins. This phosphorylation will change the activity and function of their target proteins. MAP kinase phosphorylates a variety of cytoplasmic proteins and transcription factors that carry out the downstream effects of the enzyme. MAP kinase is regulated by another protein kinase called MEK which specifically phosphorylates Thr 202 and Tyr 204 in the MAP kinase amino acid chain and turns on the protein kinase activity of MAP kinase. Other functions of MAP kinase include instructing immature cells how to grow up to be specialized cell types or how to die. Since they are keys to many important cell processes, many researchers want to use them as good targets for drugs.
Evidently, MAP kinases activate in response to almost any change in the equilibrium of the environment, and its regulation is very crucial in maintaining the health of the organism. Incorrect MAP kinase signaling can lead to complications such as metabolic syndrome and type 2 diabetes. Metabolic syndrome is the term used to describe various side effects of disruptions in metabolic processes, such as insulin resistance and obesity.
Stress-activated protein kinase (SAPK) signaling is responsible for regulating cellular responses to environmental changes such as development, transformation, inflammation, and physical stress. SAPK pathways are similar to MAPK pathways due to common characteristics such as substrate specificity and dual phosphorylation. Improper regulation of SAPK signaling may lead to complications such as cardiovascular disease.
As we know, kinases is the enzyme that phosphorylate proteins. Serine, tyrosine and threonine kinases are the three most common. Especially, the receptor tyrosine kinases (RTK) play an important role in the cell cycle, cell migration, cell metabolism and many other substantial cell functions. In animal, receptor tyrosine kinases is the membrane receptors that recognize hydrophilic ligands. In plant, instead of tyrosine kinases, serine-threonine kinases, called plant receptor, were used.
Receptor tyrosine kinases is a transmembrane structure that anchors into the membrane. The structure consists of ligand-binding domain in the outside and the kinase domain in the inside of the cell. When received the signal, the inside domain adds phosphate group to tyrosine. The ligand-binding domain is made from two receptor complexes that phosphorylate each other in a process called autophosphorylation. RTKs are activated by autophosphorylation. The propagation of the signal in the cytoplasm is triggered. Tyrosine kinase started to phosphorylate intracellular targets. Depending on what response proteins the cell has inside its cytoplasm, the cell would have very different response to the signal.
The signal was transferred from the receptor into the cytoplasm via proteins that bind to phosphorylated tyrosines. Then, those binding-proteins will convert the signal into response inside the cell. Below is the two common classes of proteins that can bind to phosphotyrosines:
1. The Insulin Receptor: Insulin is the hormone that control constant level of blood glucose. It works by binding to RTK and lowering blood glucose. When the blood glucose level is high, the insulin response protein is phosphorylated when it binds to the receptor. Then, this protein binds to additional protein, activating the enzyme glycogen synthase. Glycogen synthase can catalyze the reaction converting glucose to glycogen, making the reaction goes much faster. Therefore, the blood glucose level was maintained.
2. Adapter Proteins: Those proteins function as connection between the receptor and proteins that respond to the signal. A good example of adapter proteins’ functions is the activation of Ras protein. Adapter protein binds to the receptor. Then, it is able to activate Ras protein, which involve in cellular signal transduction.
Mitogen-activated Protein (MAP)- One Important Class of Cytoplasmic Kinases
Mitogen stimulates cell divition by triggering mitosis. It functions by initiating the normal division-control pathways. Kinase cascade (sometimes called phosphorylation cascade) can activate MAP kinase. Kinase cascade is a signaling module that phosphorylates each other. At the end of the process, it initiates MAP kinase (see the mapping below)
In each step, one enzyme can act with many substrates, resulting in the formation of many final products. Consequently, the original signal was amplified through each step. This is also the main function of kinase cascade. How the cell response depends on the MAP kinase.
In order to maximize the function of kinase cascades, a type of proteins, called scafford proteins, rearrange the kinase cascade into a protein complex. The function of each cascade is optimized because of their spatial organization. Even thougha the arrangement helps increase the effect of the cascade significantly and allows segregation of signal modules in different locations, this arrangement also has disadvantage. The amplification of the original signal was decreased since the kinase cascades are not allowed to move freely to bind new substrate.
Groups of MAP kinase
A total of six different forms of MAP kinase have been derived from mammals.
1. Extracelluar-signal regulated kinases (ERK1, ERK2)-ERK1/2 produce long lasting changes in cell signaling. These signals are usually activated in response to proteins or steroids that are capable of stimulating cellular growth (growth factors) and the organic compound phorbol ester also known as tumor promoter. This is important for the human cell because it regulates both cell proliferation and also cell differentiation.
2. c-Jun N terminal kinases (JNKs) also known as Stress Activated Protein Kinase (SAPK). JNK are activated by cytokines, certain ligands for GPCR, agents that interfere with DNA and Protein synthesis, etc. Three of the JNK genes (1,2,3) have been derived from humans. Studies performing knockout of these genes have revealed that these kinase are associated with apoptosis (the cellular signaling leading to the deal of potentially cancerous cells) and immune response.
3. p38 isoforms- similar to JNKs,function as signaling pathways like the other kinase. They are involved in apoptosis like (JNKs) and cell differentiation. Some of the things these isoforms are responsive to are cytokines, stress stimuli, ultraviolet irradiation, heat shock, and osmotic shock.
4. ERK5 (MAPK7)- Further performs in cell proliferation and is activated by both growth factors and stressful stimuli. It has been found that this kinase has a key role in cardiovascular development and neural differentiation. Also, it has unique properties compared to ERK1/2 including its carboxyl-terminal half which has a unique function that is different from ERK1/2.
5. ERK3/4 which also is MAPK6 (ERK3) and MAPK4 (ERK4)- these are considered cytoplasmic proteins that bind, translocate, and finally activate MK5. Furthermore, the stability of ERK3 and 4 are different in that ERK4 is more stable than ERK3.
6. ERK7/8 (MAPK15)- it performs similar to the other pathways in that it is activated by various conditions and molecules leading to signaling. A similarity between ERK3/4 is that it contains a long C terminus as well.
Every receptor must be regulated to make the cell function properly. Too many activations of one receptor may result in the hinder of response to other signal. Therefore, RTKs must be inactivated at some point. The cell uses two mechanisms to deactivate the receptor: dephosphorylation and internalization by endocytosis. Dephosphorylation is the reverse reaction of phosphorylation, making the signal stop being transmitted. Internalization by endocytosis is when the receptor was transferred into the cytoplasm to be degraded.
The Activation of Tyrosine Kinase Receptor, Leading to Cellular Response
1.) Before the binding of the signaling molecule, the receptors act as individual units called monomers. Each of these individual monomer units have an extracellular ligand-binding site, an alpha-helix in the membrane, a ligand binding site, and an intracellular tail with many tyrosine amino acids. The monomers are inactive until reaction with a signaling molecule that binds to its ligand-binding site.
2.) The ligand binding site is susceptible to the binding of various signaling molecules which causes two receptor monomers to work hand-in-hand with each other, forming a complex called a dimer. This process is called dimerization.
3.) The process of dimerization activates the tyrosine kinase region of each individual monomer. Both of these tyrosine kinases adds a phosphate from an ATP molecule to a tyrosine on the tail of the other monomer.
4.) After this process, the receptor is completely activated and it is then recognized by specific relay proteins located within the cell. Each protein binds to a specific phosphorylated tyrosine, leading to a structural alteration which activates the bound protein. Each of these activated proteins trigger a transduction pathway, causing the desired cellular response. [1]
Signal transduction in cells occupies an essential position during the process of cellular metabolism, segmentation, differentiation, biological behaviors and cell death. Kinase inhibitors are used to restrain specific receptors, blocking the signal transductions and causing the disease cells to die.
Most protein kinase inhibitors were discovered in the past 10 years, in order to treat cancer and inflammatory disease, and most of them are multi- target kinase inhibitors, which can restrain multiple positions of kinase at same time, blocking the signal transduction more efficient.
There are three type of Protein Kinase Inhibitors:
Type I- a small molecule that binds to the active conformation of a kinase ATP pocket.
Type II- a small molecule that binds to the inactive conformation of a kinase ATP pocket.
Cancer chemotherapy has been one of the major medical advances in the last few decades. However, the drugs used for this therapy have a narrow therapeutic index, and often the results produced are only just palliative as well as unpredictable. In contrast, targeted therapy that has been introduced in recent years is directed against cancer-specific molecules and signaling pathways and thus has more limited nonspecific toxicities. Tyrosine kinases are an especially important target because they play an important role in the modulation of growth factor signaling.
Cell growth and cell cycle pathways are constitutively activated in cancer cells. The standard controls exerted by the kinase/phosphatase enzymes don’t function anymore. The main characteristic of cancer cells is their capability to replicate in the nonexistence of external signals such as growth factors.
Growth factors are involved in the initialization and regulation of cell cycles. The type of growth factor determines its effects on the cell. There are 3 primary growth factors that narrate to tyrosine kinase. The receptors of these growth factors are members of the Receptor Tyrosine Kinase family. Epidermal growth factors (EGF) help regulate cell growth and differentiation. Platelet-derived growth factor (PDGF) regulates cell growth and development. Vascular endothelial growth factors (VEGFR) are involved in the creation of blood vessels.
Tyrosine kinase inhibitors (TKIs) are a class of chemotherapy medications that inhibit, or block, the enzyme tyrosine kinase. TKIs were created out of modern genetics- the understanding of DNA, the cell cycle, and molecular signaling pathways- and thus represent a change from general to molecular methods of cancer treatment. This allows for targeted treatment of specific cancers, which reduces the risk of damage to healthy cells and increases treatment success.
Scientific research is being focused on Tyrosine kinase inhibitors because of their unique traits compared to previous methods. All medicines for chemotherapy seek to discontinue cells division and growth. They also attempt to kill cancerous cells without destroying the healthy cells. An inherent weakness in cancerous cells is that a failure of cell repair mechanisms is what turned the cells cancerous. The cell is therefore unable to repair damaged or changed DNA effectively.
Multifaceted kinases that are vital to mitotic cell cycle include BUB1 and BUBR1. They primarily are known for their ability to construct the spindle assembly checkpoint. By doing so, the presence of protein kinases BUB1 and BUBR1 assist in allows for the high fidelity of chromosome separation by slowing down the onset of anaphase until the chromosomes are bi-oriented on the mitotic spindle. Even though both protein kinases are essential in the mitotic cycle, they serve different functions in the spindle assembly checkpoint (SAC).
BUB1 is important for chromosome congression and for maintaining the stability of bipolar attachment to spindle microtubules. If this protein kinase was removed, the rate of chromosome missegregation and possibly a slow growth of chromosome, whereas extreme cases include elevated chromosome loss entirely.
BUBR1 is highly affiliated with the misattached kinetochores and provides stability to kinetochore-microtubule attachment in chromosome alignment. Moreover, BUBR1 monitors the Prophase 1 arrest, which is critical in the meiosis I in the production of fertilizable eggs. In doing so, it accumulates to chromatids that will generate unrepaired DNA double-strand breaks.
Many cancer-affiliated mutations are present throughout the human BUBR1 and BUB1 sequence. BUB1 and BUBR1 also have a very functional role in mutating cancer cells. Chromosome destabilization and cancer occurs with the existence of the BUB1 and BUBR1 sequences. BUB1 plays a significant role in oncogenesis; variations in mutations of the BUB1 gene and protein sequence can encourage the growth of cancer tissues and cells. Mutations in the BUBR1 sequence have identified to be related to a family of mosaic-variegated aneuploidy, in other words, a syndrome that is presented with microcephaly and mental retardation. Additionally, gastric cancer progressions have shown great relevance to excess BUBR1 expression. Such mutations in the sequences of the BUB1 and BUBR1 have led to the development of cancer treatments. In fact, recent research has discovered that BUBR1 could be used as an expression marker of inadequate survival of various types of human cancer. The protein kinases result in the impairment of mitotic checkpoint function. Studies have also shown that weakening the SAC gives leverage for the survival of cells that are associated with anticancer therapy.
Since last century 80s, key point of research of anticancer transfers from restraining syntheses of DNA to restraining catalysis activity of kinase. The following anticancer drugs are already used in clinical.
Imatinib, used to treat chronic myelogenous leukemia Gastrointestinal stromal tumors.
Imatinib, discovered by Novartis Company in 1990s, was approved by FDA in May 10, 2001. By using multiple hydrogen bond, Imatinib occupies ATP pockets of ABL protein kinase, preventing the combination of ATP and ABL kinase, which restrained the activity of protein kinase and stopped the lower signal transduction. Imatinib is used to treat chronic myelogenous leukemia Gastrointestinal stromal tumors. Imatinib also can restrain PDGFR kinase on membrane and c-Kit kinase. Imatinib is no harm to normal human cells.
Gefitinib is used to help speed organ growth.
Gefitinib, discovered by AstraZeneca Company, was approved by FDA in May 5, 2003. Gegitinib mainly focuses on EGFR which belongs to the HER receptor family. In normal cells, EGFR helps organ growing by adjusting the speed of forming and differentiation. However, in cancer cells, EGFR is overexpression. Compared to Imatinib, Gefitinib has a relatively narrow spectrum, which means that Gefitinib is more selective than Imatinib. Recent research proves that Gefitinib has really high combination ability of EGFR in nonsmall cell lung cancer cells.
Sorafenib, used to treat certain forms of carcinoma.
Sorafenib, discovered by Bayer Company, was approved by FDA in December 20, 2005. Sorafenib uses to treat Renal cell carcinoma and Hepatocellular carcinoma. It uses both Hydrogen bonding and Van der Waal forces to restrain RAF kinase, occupying both ATP pockets and hydrophobic pockets of RAF. B-RAF kinase fails to phosphorylate under this condition, which loses its activity and restrains the signal transduction.
Erlotinib, used to treat pancreatic cancer.
Erlotinib, discovered by OSI Company, was approved by FDA in November 18, 2004. Erlotinib is the kinase inhibitor for EGFR-TK, belonging to small molecule compound. Erlotinib occupies the ATP pockets of ErbB, stopping the phosphorylation process of ErbB Tyrosine. Erlotinib also has strong effects on LOK, ABL, FLT, AND SLK. Erlotinib uses to treat Pancreatic cancer.
Sunitinib is used to treat Imatinib-resistant gastrointestinal stromal tumors.
Sunitinib, discovered by Pfizer Company, was approved by FDA in January 26, 2006. The process of inventing sunitinib is actually the process of transferring sample target to multi-target. Compare to other compound in the early stage of invention, the long chain of sunitinib increases the solubility. This could be the second reason why the effect of sunitinib is better than earlier drugs. In the experiment of measuring kinase spectrum, sunitinib shows broad spectrum level and strong restrainment of kinase activity. Sunitinib mainly restrain VEGFR2、PDGFRs、FLT3 and c-Kit protein kinases, controlling 3 lower signal transductions: PI-3K/AKT、Ras/Raf/MEK and PKCs. Sunitinib uses to treat patients who have Renal cell carcinoma or Imatinib-resistant gastrointestinal stromal tumor.
Nilotinib, used to treat Imatinib-resistant chronic myelogenous leukemia.
Nilotinib, discovered by Novartis Company, was approved by FDA in October 29, 2007. Nilotinib is the improvement of imatinib. Similar as imatinib, Nilotinib using multiple hydrogen bonds to occupied ATP pockets of ABL kinase, stopping the combination of ATP and ABL, restraining the activity of kinase, controlling the signal transductions. The different part of Nilotinib is that Nilotinib also reacts with hydrophobic pockets, which could be the reason why Nilotinib can restrain more abnormal ABL kinase than imatinib. Nilotinib uses to treat Imatinib-resistant chronic myelogenous leukemia.
Lapatinib, used to treat breast cancer.
Lapatinib, discovered by Glaxosmithkine Company, was approved by FDA in March 13, 2007. There are four different RTKs in human EGFR family: EGFR, HER2, HER3 and HER4. Lapatinib restrains both EGFR and HER2 Tyrosine kinase. EGFR is overexpression in HNSCC, nonsmall lung cancer, colon cancer, and breast cancer. At same time, it may help secret TGF-α, keeping activating signal transduction. HER2 also cause increasing signal transduction and its overexpression is related to women breast cancer. Lapatinib has a narrow spectrum level, but the activity of restrainment of kinase is stronger than other kinase inhibitors. Lapatinib controls 2 signal transductions: Ras/Raf/MEK and PI-3K/Akt. Lapatinib uses to treat HER2+ breast cancer.
Pazopanib, used to inhibit tumor growth.
Pazopanib was approved by FDA in 2009. Pazopanib is a multi-target kinase inhibitor, which retrains VEGFR-1, VEGFR-2,VEGFR-3,VEGFR-α/β and C-KIT kinase, stopping the signal transductions and slowing down the growth of tumor. Pazopanib uses to treat Advanced renal cell carcinoma.
Dasatinib, used to treat certain types of leukemia.
Dasatinib, discovered by Bristol-Myers Squibb Company, was approved by FDA in June 8, 2006. By using hydrogen bond to combine with receptor kinase, Dasatinib can restrain multi-target at same time, which include ABL. In the experiment of measuring kinase spectrum, sunitinib shows very broad spectrum level, mainly restraining DDR、EPHA、EPHB kinase receptor families. Dasatinib has highest percentage of restraining kinases in whole drug market. During the process of signal transduction in the cell, Dasatinib restrains TCR receptor on the membrane, Src kinase in the cell and the activity of BCR-ABL Tyrosine to stop 3 signal transductions: Ras/Raf/ MEK、JAK/STAT and PI-3K/Akt, controlling formation of cancer. Dasatinib uses to treat Chronic myelogenous leukemia and Philadelphia chromosome positive acute lymphoblastic leukemia.
The three-tiered Raf-MEK-ERK is a common cytosolic kinase cascade triggered by the downstream of the small GTPase Ras and plays a critical role in cellular generation. The analysis of protein-protein interactions in the pathway has given sufficient information on temporal and spatial pathway regulation. This three-tiered array allows for a large increase in cumulative signal strength, and diversifies the signal needed for temporal modulation as it makes its way down the pathway.
Amongst other MAPK cascades that function in vertebrates, the Raf-MERK-ERK pathway was the first to be introduced and remains the one of most interest. Generally, the pathway is inflicted downstream with growth factor receptors through the trade of GTP for GDP occurring on the membrane-associated small G protein Ras. Ras that are GTP-bound drafts at the entrance of kinase Raf to the membrane, where it is then initiated by the complex. As a result, Raf phophorylates MEK, a dual specificity kinases which targets only the extracellular signal-regulated kinase, ERK. In contrast, ERK monitors targets that are distributed in different subcellular locations –such as metabolic enzymes, transcription factors, and structural proteins. Simply, regulation can be accomplished through direct binding of the components with other components within the pathway. This will cause the scaffolding of proteins and accommodation of localization signals.
The partnerships of dimers with the Raf-MEK-ERK have shown advantages and disadvantages. Dimerization is a regulatory mechanism in signal transduction; in this particular case, it can activate kinases (like in Raf), regulate negative feedback control (like in MEK), and permit concurrent binding to substrates (like in ERK). Dimerization in the Raf pathway promotes the heterodimerization of B-Raf with the protein scaffold KSR1, C- Raf, or A- Raf. The heterodimers contain high levels of MEK kinase activity and signify MEK activating unit. Phosphorylation of B-Raf that is ERK regulated cause the dissociation of Raf heterodimers and prevents the re-alliance of stable MEK1-MEK2 heterodimers.
Wen Wu, Cheng Lu, Siyu Chen, Niefang Yu "The signal transduction pathway of multi-target kinase inhibitors as anticancer agents in clinical use or in phase III"
Arvin C. Dar1 and Kevan M. Shokat, "The Evolution of Protein Kinase Inhibitors from Antagonists to Agonists of Cellular Signaling"
Vascular endothelial growth factor stimulates protein kinase C{beta}II expression in chronic lymphocytic leukemia cells
Blood June 3, 2010 115:4447-4454
The MPS1 gene was found in a budding yeast, called the Saccharomyces cerevisiase. The gene (MPS1) after being cloned was at first named RPK1, but due to the MPS1 being the name that was used in the publication first, it stayed that way [1]. In the early years it was shown that MPS1 was a protein kinase by the use of glutathione S-transferase tagged MPS1, which resulted in strong autophsphorylation and substrate phosphorylation of other common kinase substrates. A phenotype analysis was done on the original yeast MPS1 mutants and as a result many critical functions were identified of the kinase.
MPS1 protein kinase function & importance in cancer
MPS1 protein kinases are involved in different steps in mitosis such as chromosome attachment and functions at the centrosomes. In addition, they participate in development and in multiple signaling pathways after mitosis. This large family of MPS1 protein kinases can be identified by a conserved C-terminal domain that appears in all the MPS1 protein kinases even if the N-terminal domain varies. The over-expression of MPS1 protein kinases in mitosis plays a role in cancer growth and has lead to the discovery and development of MPS1 protein kinase inhibitors.
This protein has a bilobe architecture, where the N-terminal is made up of a five-stranded β-sheet and an α-helix. In MPS1 specifically, there is an additional β-strand present. The two lobes of the protein are joined by Glu603-Gly605 (a hinge loop). The C-terminal is made up of a two-stranded β-sheet and next to this lobe are seven α-helixes, the catalytic loop and the activation loop. Polyethylene glycol (used in protein crystallization) was found to be present by the catalytic portion of the kinase and created a second pocket that isn't present in other kinases. This unique feature can be potentially used for the creation of inhibitors specific for this kinase.
Insulin is a hormone released by pancreatic beta cells in response to elevated levels of nutrients in the blood. Insulin causes cells in the liver, muscle, and fat tissue to take glucose from the blood, promoting the storage of these nutrients as glycogen in the liver and muscle and stop using fat as an energy source. When fails to control the insulin levels, diabetes will result. Patients with Type 1 diabetes is characterized by the inability to produce the hormone internally, whereas in Type 2 diabetes, the body becomes resistant to the effects of insulin presumably due to the failure of controlling glucose levels. As a peptide hormone, insulin consists of 51 amino acids and has a molecular weight of 5808Da. Produced in the islets of Langerhans in the pancreas, insulin's name stems from Latin insula for "island."
The receptor of insulin is a dimer of two identical subunits that spans the cell membrane. Each of the two subunits is made of one α-chain and one β-chain, connected together by a single disulfide bond. The α-chain lies on the exterior of the cell membrane, while the β-chain spans the cell membrane in a single segment, and with the exception of this segment lies on the inside of the cell membrane.[1]
The two α-chains on the exterior of a cell move together when insulin is detected and fold around the insulin. This action moves the β-chains together, thus making the β-chains an active tyrosine kinase. The tyrosine kinase catalyzes the transfer of phosphoryl groups from ATP to tyrosine in the activation loops of the β-chains. The phosphorylated activation loop then drastically changes conformation, causing the kinase to become fully active.[2]
Insulin-Receptor Substrates are a special group of proteins that are attracted to the phosphorylated sites on the activated Insulin Receptor. These sites act as a docking point for IRS proteins. Each IRS molecule has four sequences that are approximately Tyr-X-X-Met. This reoccurring sequence is responsible for the IRS molecules' affinity of the receptor tyrosine kinase. The tyrosine kinase then phosphorylates these Tyrosines, causing the IRS molecules to activate. Activated IRS proteins act as adaptor proteins. Adaptor proteins bring a kinase and its substrate together rather than activating a kinase. An example would be an activated IRS protein binding to a lipid kinase, thus attracting the lipid kinase to lipid membrane, its substrate.[3]
SH2 is a domain present in many signal-transduction proteins. SH2 domains are host to specific phosphotyrosine sequences, such as those in activated IRS proteins. This is a specific process, so each SH2 domain has a binding preference to an approximate sequence including phosphotyrosine. A large group of lipid kinases have SH2 domains that are attracted to IRS proteins. These lipid kinases react at the 3-position of inositol in phosphatidylinositol 4,5-bisphosphate (PIP2) and add a phosphoryl group. By attracting the lipid kinases to IRS proteins, the reaction moves the kinases to PIP2 located on the membrane where it can phosphorylate them to phosphatidyl-inositol 3,4,5-trisphosphate (PIP3) This causes a cascade which activates the protein kinase PDK1, which phosphorylates the protein kinase Akt, thus activating it as well. Akt is a free moving kinase inside the to cytoplasm, and moves around phosphorylated targets within the cell, such as proteins that control the movement GLUT4, a glucose transporter, bringing it to the cell surface. This is only one of many pathways that begin with the activation of IRS by an insulin receptor.[4]
The phosphorylation of the insulin receptor starts up serine-threonine phosphorylation of a series of proteins. They are coupled to 3 additional protein kinase signal systems. The protein kinase signal systems include: 1) The pathways signaling through PI3-kinase and phosphatidylinositol (3,4,5)P3 (PI-3 kinase and protein kinase B/Akt), 2) Mitogen-activated protein kinases (MAPKinases), 3) Possible interaction via kinases not coupled to IRS proteins.
Insulin binds to a-subunit and changes the conformation causing autophosphorylation of tyrosine residues. Those residues are then picked up by phosphotyrosine-binding domains known as PTB, such as insulin receptor substrate, SHC and Cbl. Activated receptors phosphorylate tyrosine residues on the receptors. These receptors interact by signaling molecules through their SH2 domains and activates many other pathways such as PI 3-kinase signaling, MAPK activation and Cbl/CAP complex activation. Regulation of glucose, lipids and protein metabolism are the results of these pathways.
•Protein synthesis
Insulin stimulates the uptake of amino acids into the cell, stops protein degradation and promotes protein synthesis.
•Regulation of lipid synthesis
Insulin stimulate the uptake of fatty acids and lipid synthesis, as well as inhibiting lipolysis. Lipid synthesis requires a lot of transcription factor steroid regulatory element-binding proteins (SERBP)-1c. Insulin decrease the concentration of cAMP through the activation a cAMP specific phosphodiesterase in adipocytes to inhibit lipolysis, lipid metabolism.
•Mitogenic responses
Another substrate for the insulin receptor is SHC. The phosphorylation of SHC is related to GRB2 which can activate the MAPK pathway independently of an insulin recptor substrate. Other signal transduction proteins interact with GRB2, an insulin receptor substrate that contain the SH3 domains which is related to the guanine nucleotide exchange factor son-of sevenless and promotes the activation of the MAPK pathway, then reaches mitogenic responses.
↑Berg, Jeremy (2007). Biochemistry, 6th Edition. New York, New York: Sara Tenney. pp. 392–393. ISBN978-0-7167-8724-2. {{cite book}}: Unknown parameter |coauthors= ignored (|author= suggested) (help)
↑Berg, Jeremy (2007). Biochemistry, 6th Edition. New York, New York: Sara Tenney. pp. 392–393. ISBN978-0-7167-8724-2. {{cite book}}: Unknown parameter |coauthors= ignored (|author= suggested) (help)
↑Berg, Jeremy (2007). Biochemistry, 6th Edition. New York, New York: Sara Tenney. pp. 393–394. ISBN978-0-7167-8724-2. {{cite book}}: Unknown parameter |coauthors= ignored (|author= suggested) (help)
↑Berg, Jeremy (2007). Biochemistry, 6th Edition. New York, New York: Sara Tenney. pp. 394–395. ISBN978-0-7167-8724-2. {{cite book}}: Unknown parameter |coauthors= ignored (|author= suggested) (help)
An epidermal growth factor or EGF is a type of growth factor that controls the proliferation, differentiation and survival of a cell. It is found in all living organisms.
The purpose of EGFs is to protect all living tissue by sending out signals for other cells to multiply or release certain chemicals. EGFs are secreted by cells and released into the external environment where they may bind to epidermal growth factor receptors or EGFRs. Upon binding, the EGFs can dimerize with themselves (homodimer) or dimerize with other growth factors (heterodimer). Once dimerized, the inner side of the EGFR that contains a protein kinase adds a phosphoryl group (donated from ATP) to tyrosines on neighboring chains. In doing so, proteins attach to the newly phosphorylated tyrosines thus activating a signaling cascade that ends with DNA synthesis and cell growth.
The modular structure of the EGF receptor: the amino acid position of the EGF receptor proves that he EGF-binding site is outside the cell, one transmembrane helix domain, one intracellular tyrosine kinase domain, and the one tyrosine-rich site at the last carboxyl group.
The EGF signaling is stopped by protein phosphatases and the actions of Ras (GTPase activity). Important phosphatases kick off the phosphyl groups from the tyrosine residues on the EGF receptor and from the serine, threonine, and tyrosine component on the protein kinases that are involved in the signaling mechanism. The activation of the phosphatases is the result of the whole signaling process. Therefore, the signal activation must start the signal termination as well.
Examples of Ras Superfamily of GTPases
Subfamily
Function
Ras
Regulates cell grown via the serine-threonine kinases
Rho
Reorganizes the cytoskeleton via the serine-threonine kinases
Arf
Activates the ADP-ribosyltransferase of cholera toxin A subunit, regulates vesicular pathway, activates phospolipase D
Rab
Helps the secretory and endocytotic pathways
Ran
Operates in the transport of RNA and protein into and out of the nucleus
Epidermal Growth Factors and Receptors were found by Nobel Prize winner Stanley Cohen. While at Washington University in the 1950s, Cohen had been researching nerve growth factors in mice when he discovered EGFs. Research since then has shown promise for anti-cancer drugs and with cosmetics.
EGF structure is critical to cell response. EGFs cause the replication of targeted cells and thus need a specific structure to ensure that non-targeted cells are targeted. Note that cancer cells are capable of self-replication without the use of EGFs.
The most important structure in EGFs are disulfide bonds that result from long chains of cysteine residues. These disulfide bonds make it possible for the differentiation between the many types of EGFs.
Amphiregulin (AR-promotes growth of the normal epithelial cells and it inhibits growth of aggressive carcinoma cell lines.
Epiregulin (EPR)
Epigen
Betacellulin (BTC)
Neuregulin-1 (NRG1)-development of the nervous system and the heart.
Neuregulin-2 (NRG2-induces growth and differentiation of epithelial, neuronal, glial, and other cell types.
Neuregulin-3 (NRG3)-stimulates tyrosine phosphorylation on the receptor tyrosine kinase ErbB4.
Neuregulin-4 (NRG4).
General form:
CX7CX4-5CX10-13CXCX8GXRC
X represents an amino acid
Ras proteins are a family of genes involving cellular signal transduction as one of its many functions. They are binary molecular switches that control intracellular signaling networks. However, they can also exert control over processes such as actin cytoskeletal integrity, proliferation, differentiation, cell adhesion, apoptosis, and cell migration. These signals also affect cell growth, differentiation, and cell survival.
The signals are sent from outside the cell to the nucleus. Mutations in RAS genes can cause inappropriate transmission inside the cell in the absence of extracellular signals.
Ras is a guanosine-nucleotide-binding protein with 6 beta sheets and 5 alpha helices. Ras binds to a magnesium ion to help coordinate nucleotide binding. In other words, it is a G-protein, where the G comes from the guanosine in the protein. G proteins function have “on” and “off” states, and as might be expected they have different functions and properties in these two different states.
Off state: In the "off" state Ras is bound to the nucleotide guanosine diphosphate (GDP).
On state: In the "on" state, Ras is bound to guanosine triphosphate, (GTP). There is the obvious difference that GTP contains one more phosphoryl group than GDP, which can be ascertained from the name itself.
The extra phosphate in the “on” state is very important because it holds the two switch regions in a specific configuration. When released from this configuration, the switch regions relax – this causes a change in the conformation which leads to the activate state. It becomes clear that the activation and the deactivation of the Ras is controlled by the properties of GTP and DTP.
However, Ras can be separated from GDP but only with the use of GEFs. The balance between GEF and GAP activity determines the guanine nucleotide status of Ras, thereby regulating Ras activity. In the GTP-bound conformation, Ras has high affinity for numerous effectors that allow it to carry out its functions. Ras can attach to the membranes of the cells in our body, and this is very important because this binding is responsible for many pathways which couple growth factor receptors to downstream mitogenic effectors involved in cell proliferation or differentiation.
H-ras structure with alpha helices and beta strands shown.
First identified by Edward M. Scolnick and his team at the National Institutes of Health (NIH) as transforming ocogenes responsible for the cancer-causing activities of the Harvey (HRAS oncogene) and Kirsten (KRAS) sarcoma viruses, Ras genes were originally discovered in rats during the 1960s. Therefore the name Rat sarcoma was formed. In 1982 Geoffry M. Cooper at Harvard, Mariano Barbacid and Stuart A. Aaronson at the NIH and Robert Weinberg of MIT discovered transforming and activated human RAS genes in human cancer cells.
A third human RAS gene known as NRAS was discovered later on. It is named NRAS for its initial identification in human neuroblastoma cells.
All three human RAS genes have 188-189 amino acid proteins designated as H-Ras, N-Ras, and K-Ras4a and K-Ras4B. K-Ras4a and K-Ras4B arise from alternative gene splicing.
RAS affects cell growth and division. Therefore, cancer and oncogenesis can arise. Activating mutations are found in 20-25% of all human tumors and up to 90% in specific tumor types.
Cancer causing genes are called oncogenes.And that of the normal cellular genes are called proto-oncogenes.A proto=oncogene becomes an oncogene when a genetic change that leads to an increase either in the amount of the proto-oncogene's protein product or in the intrinsic activity of each protein molecule. The genetic changes that convert proto-oncogenes to oncogenes fall into three main categories: movement of DNA within the genome, amplification of a proto-oncogene, and point mutations in a control element or in the prot-oncogene itself. In most cases, cancer cells are frequently found to contain chromosomes that have broken and rejoined incorrectly, translocating fragments from one chromosome to another.If a translocated proto-oncogene ends up near an especially active promoter (or a control element), its transcription may increase, making it an oncogene.The proteins encoded by many proto-oncogenes and tumore-suppressore genes are components of cell-signaling pathways.One of the genes involving in such activities is the ras gene.Ras gene and the ras protein are important for a lot of things, but more particularly for regulating the growth of cells. Normal cells need to have a good ras gene in order to grow, in order to make new DNA, to time it all right so they don't grow out of control. Moreover, the ras gene occurs in virtually all living things.Mutations in the Ras are being attributed as a main reason for cancer. As of now, Ras mutations have been found in 20% to 30% of all human cancer outbreaks which is a significant amount. In the future, it might be possible that monitoring Ras activity could prevent certain cancer types from occurring. Ras inhibitor trans-farnesylthiosalicylic acid (FTS, salirasib) exhibits profound antioncogenic effects in many cancer cell lines. It is believed that inappropriate activation of the Ras gene is responsible for cancer as well as other misshapes and diseases. Inappropriate activation of the gene has been shown to play a key role in signal transduction, proliferation and malignant transformation.
[1][2]
ROS, which stands for reactive oxygen species, oxidizes DNA, lipids, and proteins. They are molecules that are highly reactive, but small. They modulate activities of oxidized targets when they are controlled tightly. Reactive oxygen species is one of many cell-signaling processes. At first it was once thought to be toxic byproducts of aerobic metabolism, but now it is known that ROS plays an important role in the complicated signaling network of cells in different kinds of organisms. ROS is generated from different kinds of sources like chloroplasts, peroxisomes, cell membrane, germin-like oxalate oxidases, and amine oxidases, etc. ROS can activate autophagy, which is one of their essential roles. They can cause cells to either survive or die. As of the moment, ROS and its involvement with autophagy are not completely understood. Autophagy's redox regulation has a focus on mitochondria's role as one of the sources of ROS as well as clearance of ROS by mitophagy.
1. Singlet oxygen (O2)
As the first excited electronic state of O2, it is formed during photosynthesis. However, singlet oxygen can cause violent damage on PSI and PSII in the photosynthetic mechanism. Singlet oxygen is more acting as a signal to activate stress-response pathways instead of acting as a toxin like other ROS.
2. Superoxide radicals (O2•-)
Superoxide radicals is moderately reactive ROS, which can protonate or donate electron to iron(3+) result with a iron(2+) product, the product will then lead to reduce hydrogen peroxide(H2O2). Photosynthetically active chloroplasts generates superoxide radicals to active genes in signaling pathways.
3. Hydrogen peroxide (H2O2)
Hydrogen peroxide is also moderately reactive with longer half-time life(1ms) than other ROS (2-4μs). Hydrogen peroxide can oxidize enzyme’s thiol groups and lead to cell death. In low concentration, hydrogen peroxide is a signal molecule that triggers tolerance to biotic/abiotic stress. In high concentration, hydrogen peroxide will lead to programmed cell death.
4. Hydroxyl radical (OH•)
Hydroxyl radical is the most reactive ROS so far. As a product from superoxide radicals and hydrogen peroxide, hydroxyl radicals can be formed at neutral pH with catalyst. Hydroxyl radical can react with protein, nucleic acid, and lipids. Just like hydrogen peroxide, high concentration of hydroxyl radical will lead to programmed cell death.
Autophagy, also known as autophagocytosis, is involved in degradation of cells when they are dysfunctional or unnecessary by lysosomes. Autophagy is a catabolic basic mechanism.
Incompleteness in oxygen one electron reduction forms ROS molecules that are small, highly reactive, and short-lived. Oxygen anions and free radicals are included as a ROS.
[1]Regulation of autophagy by ROS: physiology and pathology.
Scherz-Shouval R, Elazar Z.
Trends Biochem Sci. 2011 Jan;36(1):30-8. Epub 2010 Aug 20. Review.
[2]L.-J. Quan, B. Zhang, W.-W. Shi, H.-Y. Li, Hydrogen peroxide in plants: a versatile molecule of the reactive oxygen species network, J. Integrat. Plant Biol. 50 (2008) 2e18.
[3]Gill SS, Tuteja N,. 2010. Reactive oxygen species and antioxidant machinery in abiotic stress tolerance in crop plants. Plant Physiology and Biochemistry. 48. 909–930.
[4]S. Bhattachrjee, Reactive oxygen species and oxidative burst: roles in stress, senescence and signal transduction in plant, Curr. Sci. 89 (2005) 1113e1121.
Cancer can occur in many different ways. The overall idea of cancer is that a cell grows and divides erratically on its own, even without a cell signal, resulting in uncontrolled cell growth. The cell cannot die either, causing a build up of excess cells in a certain area c. Such an excess cells is known as a tumor. Cancer is due to multiple cell signaling breakdown. Uncontrolled cell communication also leads to the incorporation of the blood vessels to grow into the tumor, taking up a majority of the nutrients and causing the tumor to grow even larger. More cell signaling allows the cancerous cells to move to other parts of the body.
Many mechanisms maintain healthy cell growth. External signals direct cell division, cell-to-cell signaling occurs, and enzymes can repair damaged DNA. Apoptosis is programmed cell death, which is needed to maintain the shape of the organism. When a cell is not repairable, it signals its own death.
When cell communication breaks down, uncontrolled cell growth can occur and often lead to cancer. Cancer often begins when a cell gains the ability to grow and divide even in the absence of a signal from the environment. Ordinarily, the unregulated growth triggers a signal for self destruction, otherwise known as apoptosis. However, when a cell also loses the ability to respond to self-destruct signals, the cell divides uncontrollably and, consequently, forms a tumor.
The signal transduction pathway is a chain of events that converts the message of a molecule present in the extracellular environment to a physiological response.
The signal transduction pathway is illustrative of a molecular circuit.
Primary messenger release is triggered by a stimulus such as a wound or food.
The primary messenger, also referred to as a ligand, is received by a receptor, forming a receptor-ligand complex. Proteins that span the cell membrane act as receptors that bind the ligands, transmitting extracellular information from the environment to the cell's interior.
Smaller molecules inside the cell known as secondary messengers relay the information from the protein receptor. Examples of secondary messengers are GMP and calcium ion. Some undesired effects of secondary molecules are the freedom of secondary messengers to diffuse within the cell, which then influences other process within the cell. "Cross talk" alters the concentration of common secondary messengers when multiple receptor signals are processed, and a single activated receptor can yield a large intracellular response through the activation of a generation of secondary messengers.
The effect of the signaling pathway is to activate or inhibit pumps, enzymes and gene transcription factors, all of which produce physiological changes.
Termination of the signal is crucial after the response is completed. The signaling process must be terminated or the cell loses responsiveness to new and important signals. One such signal is the signal to self-destruct, which prevents undesirable effects such as uncontrollable cell division and growth.
An example of a virus that causes a type of cancer is Rous Sarcoma Virus. The Rous Sarcoma Virus is a retrovirus that causes sarcoma, a cancer of tissues of mesodermal origin such as muscle or connective tissue.
The sarcoma virus carries a gene referred to as v-Src and is an oncogene which means that the gene causes a generation of cancer-like characteristics in cell types.
The v-Src gene is similar to a proto-oncogene referred to as the c-Src gene that encodes for a signal transduction that regulates cell growth. The difference between the v-Src gene and the c-Src gene that causes cancer is the substitution of a completely different set of 11 amino acids that lack the key tyrosine residue that is phosphorylated to inactivate the c-Src cell growth signal. Therefore, v-Src is always active. Small differences in the amino acid sequences between the proteins encoded are responsible for the product "oncogene" being swetched into the "on" position. The encoding proteins that are responsible are called protooncogene and oncogene.
Tumors are usually caused by commonly mutated genes. An example is the gene encoding for Ras protein which is mutated so that it loses the ability to hydrolyze GTP, keeping the Ras protein to stimulate continuous cell growth.
Overexpressed receptors also lead to tumors. For example EGFR, epidermal growth factor receptor, in human epithelial cancers such as breast and ovarian cancer. The overexpression of the receptor increases the likelihood that a grow and divide signal is sent to the cell. In breast cancer, Her2 may be overexpressed and is breast cancer patients are now being offered testing for this EFGR family member that may stimulate cell proliferation during overexpression.
Apoptosis occurs when a cell is damaged beyond repair, infected with a virus, or undergoing stressful conditions such as starvation. Damage to DNA from ionizing radiation or toxic chemicals can also induce apoptosis via the actions of the tumor-suppressing gene p53. The "decision" for apoptosis can come from the cell itself, from the surrounding tissue, or from a cell that is part of the immune system. In these cases apoptosis functions to remove the damaged cell, preventing it from sapping further nutrients from the organism, or halting further spread of viral infection. In addition to its importance as a biological phenomenon, defective apoptotic processes have been implicated in an extensive variety of diseases. Excessive apoptosis causes hypotrophy, such as in ischemic damage, whereas an insufficient amount results in uncontrolled cell proliferation, such as cancer.Scientists believe that too much apoptosis is at least partly to blame for some neurodegenerative diseases, such as Alzheimer’s, Parkinson’s, and Lou Gehrig’s. On the other hand, unchecked mitosis can lead to cancer. In treating some cancers radiation and chemicals are used to induce apoptosis to stop the continual growth of cancerous cells. Apoptosis is nevertheless necessary for proper healthy development and needed to destroy cells that threaten the organism. Apoptosis works as a balance to mitosis to keep us healthy for example,our skin and hair cells are renewed via a continuous
cycle of apoptosis and mitosis.
In apoptosis the cell shrinks and pulls away from its neighbor. Apoptosis causes the cells develop bubble like beads on their surface. The DNA in the nucleus condenses and breaks into regular sized fragments, and soon the nucleus itself, followed by the entire cell, disintegrates. The chromatin in the cell begins to degrade and mitochondria break down with the release of cytochrome c.
Some viruses associated with cancers use tricks to prevent apoptosis of the that they have transformed. Several human papilloma viruses(HPV) have been implicated in causing cervical cancer. One of them produces a E6 protein that binds and inactivates the apoptosis promoter p53. Mutations in the p53 gene are often found in cancer cells where apoptosis is not promoted to begin. Some viruses such as a type causing common colds make proteins mimic the off switch of cellular apoptosis fooling cells. Other viruses such as HIV have an enzyme that can disable a key component of the pathway, bringing the death march to a screeching halt.
A. normal cell division B. cancerous cell division 1. damaged cell 2. apoptosis
Apoptosis Inducing Factor:
Apoptosis Inducing Factor (AIF) is a protein that triggers chromatin condensation and DNA degradation in a cell in order to induce programmed cell death. The mitochondrial AIF protein was found to be a capasase-independent death effector that can allow independent nuclei to undergo apoptotic changes. The process triggering apoptosis starts when the mitochondria releases AIF, which exits through the mitochondrial membrane, enters the cytosol, and finally ends up in the cell nucleus where it signals the cell to condense its chromosomes and fragment its DNA molecules in order to prepare for cell death. Recently, researchers have discovered that AIF is in fact dependent upon the type of cell, the apoptotic insult, and its DNA binding ability. AIF also plays a significant role in the mitochondrial respiratory chain as well as in metabolic redox reactions.
Synthesis of AIF:
The AIF protein is located across 16 exons on the X chromosome in humans. AIF1 (the most abundant type of AIF) is translated in the cytosol and is sent in the direction of the mitochondrial membrane and the intermembrane space by the C-terminus of an MLS protein. AIF is transported with the assistance of its bipartite N-terminal MLS protein into the inner and outer mitochondrial membrane enzymes to allow it to enter the organelle. Inside the mitochondria, AIF folds into its functional configuration by the help of the co-factor, flavin adenine dinucleotide (FAD). A protein called Scythe, which is used to regulate organogenesis, can increase the AIF lifetime in the cell. As a result, decreased amounts of Scythe lead to a quicker fragmentation of AIF. The x-linked inhibitor of apoptosis (XIAP) has the power to influence the half-life of AIF along with Scythe. Together, the two do not affect the AIF attached to the inner mitochondrial membrane, however they influence the stability of AIF once it exits the mitochondria.
Role of AIF in Mitochondria:
Researchers believed that if a recombinant version of AIF lacks the first 120 amino acids of the AIF protein, then AIF would function as an NADH and NADPH oxidase. They discovered however, that recombinant AIF that do not have the last 100 N-terminal amino acids have limited NADP and NADPH oxidase activity. Therefore, researchers concluded that the AIF N-terminus may function in interactions with other proteins or control AIF redox reactions and substrate specificity.
AIF Respiratory Chain Complex I:
Mutations of AIF due to deletions have stimulated the creation of the mouse model of complex I deficiency. Complex I deficiency is the reason behind over thirty percent of human mitochondrial diseases. For example, complex I mitochondriopathies mostly affect infants by causing symptoms such as seizures, blindness, deafness, etc. These AIF-deficient mouse models are important for fixing complex I deficiencies. The identification of AIF-interacting proteins in the inner mitochondrial membrane and intermembrane space will help researchers identify the mechanism of the signaling pathway that monitors the function of AIF in the mitochondria.
Unlike apoptosis necrosis is the premature death of cells and living tissue. Necrosis is often caused by external factors, such as infection, toxins or trauma. During necrosis, the
cell’s outer membrane loses its ability to control the flow of liquid into and out of the cell. Necrosis is a form of traumatic cell death that results from acute cellular injury. Apoptosis in contrast to necrosis, confers advantages during an organism's life cycle. For instance during the development of the fetus in the mother, the differentiation of fingers and toes occurs because cells between the fingers apoptose with the end result that the digits are separate. Approximately between 50 billion and 70 billion cells die each day due to apoptosis in the average human adult. In a year, this amounts to the proliferation and subsequent destruction of a mass of cells equal to an individual's body weight. Apoptosis differentiates from necrosis as the processes associated with apoptosis in disposal of cellular debris do not damage the organism in apoptosis.
Transforming growth factor beta (TGFβ) is a small cell-signaling protein molecule (cytokine), secreted by the glial cells of the nervous system as well as some cells of the immune system. They are responsible for cell proliferation (cell growth), cell differentiation, apoptosis (cell death), and cell migration.
There exists three different isoforms of TGFβ, TGFβ-1, TGFβ-2 and TGFβ-3. The three forms of TGFβ are expressed by many different cells of the body and likewise most cells in the body also possess the TGFβ receptor sites for binding.
TGFβ is always produced in the form of an inactivated complex. In order for TGFβ to bind to its receptor and act on its function, it must be activated. Integrins which acts like a mediator between cells and tissues surrounding it are actually responsible for the activation of the inactive TGFβ complex to an active complex. The reason TGFβ is produced in its inactive form is so that its effects only occur at the right time and place. The function of active TGFβ is to bind to its receptor, which leads to a signaling cascade, and results in the activation or repression of gene transcription [1]. These pathways and the biochemical mechanisms that regulate them are of interest because of their importance to human health and role in diseases. Further understanding of the integrins that control TGFβ activation could lead to new therapeutics. Examples of such beneficial therapeutics include treating cancer because TGFβ has abnormal function during tumor formation [1].
Each of the three TGFβ isoforms contains a 25kDa N-terminal propeptide, which is called latency-associated peptide (LAP) as well as a 12.5 kDA C-terminal active TGFβ. The N-terminal LAP and C-terminal TGFβ are encoded by separate genes and together, form a homodimeric LAP- TGFβ propeptide complex. A propeptide is a short sequence that dictates how its associated protein folds [3]. In order for TGFβ to function, the LAP and TGFβ must be cleaved in the Golgi. However, the TGFβ receptors are blocked by cleaved LAP- TGFβ, leaving TGFβ in its inactive form [1]. The LAP- TGFβ complex is called the small latent complex (SLC). The covalent association of latent TGFβ and SLC forms the large latent complex (LLC). This has important effects when TGFβ localizes in the extracellular matrix and is activated by cells [1]. Studies have shown that some hydrophobic regions in LAP play an important role in the formation of latent TGFβ [4]. Because of its control over TGFβ, it is crucial that LAP does not contain mutations, because these can lead to disease when TGFβ function is not properly controlled [1].
Many different processes have been advanced to account for the activation of TGFβ, these include heat, acidic pH, reactive oxygen species, various proteases, the membrane glycoprotein thrombospondin-1 and shear stress, but however strong compelling arguments have come to show that integrins are responsible for the activation of TGFβ. Integrins are part of a cell adhesion and signaling receptor family. They are composed of an α and a β subunit that together create a heterodimeric transmembrane receptor I. There are a total of 24 integrin receptors in mammals formed by 18α and 8 β subunits. [5].
Once activated TGFβ binds to its transmembrane receptors, TGFβRI and TGFβRII. These two receptors form complexes with TGFβ to enable its signaling. A simple pathway of TGFβ signaling can be shown on the figure below. Figure 1 shows that when TGFβ binds to TGFβRII, which subsequently binds with TGFβRI, intracellular signals are triggered and lead to activation or repression of transcription. TGFβ must be in its active form to bind with TGFβ RII. When TGFβ binds to TGFβ RII, TGFβRII phosphorylates TGFβRI. Then, Smad2 or Smad3 is phosphorylated by TGFβRI. Phosphorylated Smad2 or Smad3 can associate with Smad4. The resulting complex can translocate to the nucleus to activate transcription or induce apoptosis [1].
Activation of TGFβ by different intergrins can change the function of TGFβ. In mammals, six out of the 24 integrin can bind to latent TGFβ through a tripeptide RGD integrin-binding motif in the Latency-associated peptide (LAP) region in the latent complex. Integrins αvβ3 and αvβ5, when expressed in certain fibroblastic cells, activate TGFβ to cause lung fibroblast cells to differentiate into myofibroblast cells. αvβ6-mediated TGFβ activation helps to maintain the immune homeostasis in epithelial cells. Activation of TGFβ by αvβ6 has been proposed to play a role in cancer progression through tumor invasion via up-regulation of MMP9, but further studies are required to understand the process. TGFβ activated by αvβ8 regulates the brain vascular developement and, and when αvβ8 activates the isoform TGFβ1 and TGFβ3, the cytokin helps to control neurovascular development. Integrin αvβ8 activated TGFβ in dendrite cells also play a role in controlling self-harful T-cell responses. The function of TGFβ activated by integrins αvβ1 and α8β1 have yet to be determined, and requires further study.[1]
Integrins can bind to TGFβ in two different ways. Integrins (like αvβ5 and αvβ6) bind to the LAP region outside the cell, and from the generation of a pulling force by the actin cytoskeleton connected to cytoplasmic domain of the integrin, there is a conformational change. This conformational change of the TGFβ complex also requires the binding of the LTBP1 to ECM, which creates a holding force for the conformational change. The mechanism of the conformational change of the latent complex can be improved by pathways that enhance cell contraction, such as thrombin. This will lead to the activation of TGFβ. There is little knowledge past this extent of the mechanism of the conformational change since there is no structural information currently available for an integrin-latent TGFβ complex.[2]
The second way an integrin can bind to the TGFβ is through cell-specific mechanisms. For example, in lung and airway cells, αvβ8 binds to TGFβ when the cell surface metallprotease MT1-MMP proteolytically cleaves the LAP region. This protease-dependent mechanism has not been tested in other αvβ8-expressing cell types that are known to activate TGFβ, so it is unsure if this is the mechanism αvβ8 induced TGFβ activation.[3]
↑Worthington, John J., Joanna E. Klementowicz, and Mark A. Travis. Cell Press, Jan. 2011..
↑Worthington, John J., Joanna E. Klementowicz, and Mark A. Travis. Cell Press, Jan. 2011..
↑Worthington, John J., Joanna E. Klementowicz, and Mark A. Travis. Trends in Biochemical Sciences. Vol. 36. Iss.1. ScienceDirect.com. Cell Press, Jan. 2011. Web.
3. Wang J, Wang D, Mei ZH, Liu , Yu HW. Applied Microbiology and Biotechnology. Volume 96. Number 2. 2012. Pubmed.gov. Web.
4. Walton, K.L. et al. (2010) Two distinct regions of Latency-associated Peptide coordinate stability of the Latent Transforming GrowthFactor-b1 complex. J. Biol. Chem. 285, 17029–17037
Hormone is an important signal molecule that is used by both]]]]
plants and animals. “Hundreds of hormones can regulate a wide variety of physiological functions, including growth and development, rates of body processes, concentrations of substances, and responses to stress and injury” (Becker, et al. 416). Distance traveled by hormones can vary depending on hormones’ life span. Some hormones can last only seconds in blood stream, indicating short distance regulation. Hormones that are long distance traveler can have life spans ranging from minutes to even hours. Hormones also have different chemical properties. Generally, “hormones can fall into four different categories: amino acid derivative, peptides, proteins, and lipid-like hormones such as steroids” (416). Difference in their chemical properties allows hormones to bind to different receptors thereby regulating different pathways. For example, adrenergic hormones bind to a family of G proteins to increase the concentration of glucose in muscle cells (416-417).
Hormonal signaling follows the following steps:
Biosynthesis of a particular hormone in a particular tissue
Storage and secretion of the hormone
Transport of the hormone to the target cell(s)
Recognition of the hormone by an associated cell membrane or intracellular receptor protein.
Relay and amplification of the received hormonal signal via a signal transduction process: This then leads to a cellular response. The reaction of the target cells may then be recognized by the original hormone-producing cells, leading to a down-regulation in hormone production. This is an example of a homeostatic negative feedback loop.
Apoptosis or programmed cell death is an important biological process. It helps body to get rid of cells that are superfluous or potentially harmful. In addition, it helps to shape our feature by removing cells that are no longer needed. When a cell undergoes apoptosis, its DNA is cleaved into fragment by an “apoptosis-specific DNA endonuclease” known as DNase. “Eventually, cell is dismantled into small pieces called apoptotic bodies” which will be engulfed by the specialized cells nearby (Becker et al. 419-420). The process is carried out by chromatin condensation and fragmentation; it is executed by the formation of multi-protein complexes that involve the binding of extracellular death ligands to death receptors. The complexes generate and activate initiator caspases, which then trigger and cleave effector caspases that then target and focus on specific cellular substrates for the process of proteolysis. Caspases is the key enzyme involved in apoptosis and it is catalytically active only during this time.They are directly inhibited by members of the IAP family. The inactivated form of this enzyme is called procaspases which is activated by a different caspases through proteolytic cleavage. Once it is turned on, caspases will cleave the inhibitory protein on DNase to induce cell death (420).
There are two major types of signals that can trigger apoptosis in human, death signals and withdrawal of survival signals (420). In the death signal pathway, a cell that is infected with virus or bacterial will accumulate CD9 or similar proteins on its surface. These proteins are called death signals that will attract procaspases to cell’s surface. Once procaspases is turned on, it will activate more procaspases to initiate apoptosis. In the survival factor pathway, mitochondria play an important role in programming cell death. Proteins called anti-apoptotic proteins reside on the outer membrane of mitochondria. The function of those proteins is to prevent apoptosis as long as the cells are exposed to survival factors (421). Another type of proteins called pro-apoptotic proteins which can promote apoptosis exist in balance with anti-apoptotic proteins. Once the survival factor is withdrawn from cell, this balance will be broken, causing equilibrium to shift more toward pro-apoptotic proteins. As a result, there is an increasing in chance for cell to undergo apoptosis (421).
All neurons have suicide genes, and these genes are influenced by brain chemistry and activation patterns.
Fetal development is a very competitive process because at this stage, the nervous system massively overproduces cells (produces more than 50% the amount that survive). As this huge amount of cells compete for connections, those who are not able to make a connection die off because their suicide genes are activated. This competition for connections consists of axons fighting for space on the Post-Synaptic cell: axons begin by branching widely and connecting to many sites, and the few that are strengthened through positive feedback (in this case, recurring signals between the axon and the post-synaptic cell) are the ones that are maintained. As a result, each cell makes fewer and more selective connections as prenatal development progresses. On the other hand, the others who lose the competition and fail to receive positive feedback go through apoptosis (suicide genes are activated)
Apoptosis and necrosis differ in the sense that necrosis results in messy byproducts from injury or toxins, which can cause contamination or inflammation. This sort of cell death is injurious and deleterious, whereas apoptosis is an essential and neat process where cell death does not leave byproducts or unnecessary material behind.
Caenorhabditis elegans, or C. elegans, are a type of nematode where research is undergone to better understand apoptosis in multicellular organisms. Research can be done with the C. elegans by the fact that throughout the C. elegans' development, the cell numbers are regulated and controlled in about all adult worms. Therefore, all the adult worms contain about the same number of cells in each mature organ. The executioner caspase in C. elegans is CED-3, which in turn triggers the apoptosis.
The apoptosis in C. elegans rank similar to apoptosis in higher organisms because the CED-3 caspase in C. elegans contain homologs to the caspases in mammals, e.g. caspase -3 and caspase-8. CED-3 is synthesized as an inactive zymogen; dimerization and autoproteolysis generate the active components (the large and small subunits) from the N-terminal prodomain. The oligomerized CED-4 bring together and active the before inactive CED-3 monomers, a parallel process of mammalian caspase-9 activation.
A way for C. elegans regulates the activation of CED-3 and apoptosis is recently found to be CSP-3; a cytoplasmic protein that mimics the CED-3 small subunit, binding to and sequestering the CED-3 zymogen. Before, CSP-3 was found to be a negative regulator of apoptosis in cells that should survive, but it did not block the appropriate induction of apoptosis in cells that should die. Therefore, CSP-3 was considered a caspase-like gene and scientists were still clueless on the true origin of regulation. Recent research shows on the other hand that the CSP-3 is able to bind the CED-3 zymogen in vitro and in vivo, and that CSP-3 is a modulator of CED-3 activation instead of a true caspase inhibitor. This demonstrates that CSP-3 does prevent inappropriate CED-3 dimerization and autoactivation. On a side note, CED-4 is a gene that can override the CSP-3 and its effects on CED-3 and apoptosis.
ATP production is not the only invaluable skill mitochondria possess. They are a vital component of the cell’s cellular transduction network that is capable of triggering apoptosis, or programmed cell death. The primary proteinic signal that initiates this process is cytochrome-c, an electron transporting membrane protein, which releases chemical messengers that activate caspases and lead the cell to following an apoptotic program that shuts it down. So, even though mitochondria are constantly providing the cell with energy through maintaining an ionic gradient that converts ADP into ATP by way of ATP Synthase, the organelle is also capable of inflicting cellular suicide by over-expressing the same membrane proteins that perform electron transport to maintain that ionic gradient. To prevent unintentional initiation of apoptosis, the organelle relies on the expression of bcl-2 to counteract the effects of cytochrome-c by preventing its translocation from the mitochondria.
Autophagy is a process described as a pro-survival pathway that is considered essential for cellular homeostasis and stress responses. It accompanies cell death and is capable of leading to cell death under certain circumstances. This process is carried out by the Atg genes; during this period, intracellular contents are consumed by either double or multi-membrane vesicles called autophagosomes. The suppression of the mammalian target of rapamycin kinase is necessary for the progression of autophagy because the rapamycin kinases release its inhibitory effects on the ULK1 kinase complex. Autophagy is in charge of the turnover of long-lived proteins, the elimination of misfolded proteins and some damaged organelles, and also the recycling of cellular building blocks after nutrient depravation. Therefore autophagy is an essential pro-survival pathway in cellular homeostasis and also stress because it can actually carry out cell death.
Necrosis was originally categorized as accidental cell death, but due to recent research, it is now recognized as a genetically controlled event. This process involves various characteristics such as: cell swelling, organelle dysfunction, and cell lysis. Necrosis does not have precise mechanisms identified, but a few regulators such as c-Jun N-terminal kinase, apoptosis inducing factor, death-associated protein kinase, and reactive oxygen species have been identified and associated with necrosis. A sub-category of necrosis is known as necroptosis, which is triggered by death receptors in cells that are apoptosis incompetent.
Picture of Apoptosis/Autophagy/Necrosis
Click Link Below
Although AIF is one of many regulators for programmed cell death (PCD), it has other unique functions that allows itself to be differentiated from other apoptosis factors shown in the picture. It was at first discovered as a caspase-independent death effector but later studies have shown that it is directly related to programmed cell death pathways that particular types of cells take. AIF performs its deadly function when it is signaled to be released from mitochondria and is translocated initially to the cytosol then to the nucleus of the targeted cell. At the nucleus, it can induce apoptosis related features such as DNA fragmentation and chromatin condensation. AIF’s effect is entirely dependent on its intrinsic DNA binding abilities as well as the cell type of the target cell because AIF has later been discovered to have crucial mitochondrial roles in healthy cells that are not tagged to have programmed cell death. AIF has a role in DNA binding as well. These studies lead to many questions as to AIF’s essential role in the body and if proper functions can be carried out in its absence.
Mitochondrial AIF proteins, as mentioned above, have a lethal purpose to facilitate programmed cell death. It does so upon mitochondrial outer membrane permeabilization (MOMP), after which it will be released into the cytosol and eventually to the nucleus where it condenses chromatin and degrades DNA fragments to carry out PCD. Primary AIF transcripts go through very tissue-specific splicing and create two exons: exon 2a and 2b. The two exons allow for production of two splice variants and create two isoforms that show a small and limited difference found in the inner membrane-sorting signal (IMSS). The N-terminal mitochondrial localization signal (MLS) is absent in these variants, but the C-terminal domain remains unaffected. The C-terminal domain is where the pro-apoptotic segment is located, and upon transfection (or more commonly termed transformation) the variant genes (AIFsh) are expressed in the nucleus and activate apoptosis. There are variations of the AIFsh that include isoforms that do not have the C-terminal domain with the apoptotic features or another form that lacks the mitochondrial localization signal and essentially is rendered to have similar functions as the AIFsh lacking the C-terminal domain.
X-ray crystallography has provided pictures of the AIF structure that has led to another way that AIF carries out its function. The structure shows that AIF possesses positively charged amino acids that are distributed throughout its surface; this distribution of positive charge is very similar to that of DNA-binding histones. This discovery explains and is consistent with the observation of AIF’s intrinsic DNA-binding capabilities and how effective it is as an apoptosis regulator. A few of the positive residues are required for AIF-nucleic acid binding and for inducing nuclear apoptosis due to the overexpression of AIF. The recombinant AIF genes induces DNA condensation through direct, sequence-independent interactions with the DNA, whether it be single- or double-stranded. The DNA condensation can be amplified by adding reduced pyridine nucleotides, NADPH, or NADH.
Other regulators of apoptosis come from the same larger mitochondrial flavoprotein family that AIF belongs too. They have common structural and functional features in eukaryotic versions of the protein. But AIF has a slightly different biosynthesis and finalizing processing than the other members of the family. The AIF gene consists of over 16 exons and is located on the human chromosome X. AIF1, variant AIF gene with the C-terminal domain, is the most common and abundant transcript. It is translated in the cytoplasm and sent to the mitochondria of healthy cells. Its N-terminal part is required for the import of the AIF protein through the membrane (outer and inner) of the mitochondria. This N-terminal part is encoded by the first exon of 35 amino acids that make up the gene. As the fully processed AIF protein is inserted in the inner membrane, it matures and folds into its quaternary structure using its co-factor flavin adenine dinucleotide (FAD). Because of AIF’s dependence on its cofactor, it has similar folds as the bacterial NAD and contains two FAD-binding segments. These features were conserved through the evolution of these different proteins.
AIF can be both triggered and inhibited (or regulated) by other metabolic processes or proteins. AIF expression has been correlated to the sensitivity of anti-cancer drugs. Its reduced expression has also been seen when hepatocyte growth factors have been added to the cell, which increases cisplatin resistance. Increased cisplatin sensitivity is often seen with increased expression of AIF. In mouse embryonic fibroblasts that lack FAK (focal adhesion kinase), it is observed that there’s a smaller amount of HGF which exhibit the aforementioned correlations. Most of these studies are only clinical and have been performed on only mice.
The amount of AIF proteins are also regulated after translation. Its concentration is positively regulated by scythe, also referred to as BAT3. BAT3 is a protein used in the regulation of cell death and cell proliferation. Scythe has been known to prolong the half-life of AIF, and when its levels are low it promotes AIF breakdown. Other inhibitors of apoptosis have been known to trigger the degradation of AIF. However, these occurrences have little to no effect on the AIF proteins found in the inner mitochondrial membrane which have yet to be modified to be released into the cytosol.
Lack of AIF expression leads to a defect in the mitochondrial respiratory chain, especially in complex I. This observation again was only experimented on mice. A significant detection of malfunction in the respiratory chain was located in various tissues such as the cerebellum, cortex, retina, and skeletal muscles. Malfunction, however, could not be detected on tissues of the heart, liver, and testis of male mice. Researchers are still perplexed at how tissue-specific the defect is. Analyses do show that the deficiency in complex I activity in mouse and human cells that lack AIF is due to a decreased expression of various other nuclear-encoded protein subunits found in the respiratory chain. There are 46 total protein subunits that make up complex I, and 39 of which are nuclear-encoded to be translated int the cytosol and taken into the mitochondria. It has been shown that overexpression of a mitochondrion-localized AIF fixes the malfunctions in complex I subunits that are caused by endogenous AIF proteins. Although this evidence shows valuable insight, mechanisms that lead to inefficiencies of complex I are still up to greater debate and research. Results of various recent experiments have shown inconsistent observations that may or may not link AIF directly to the complex I.
Many hypotheses have been made about AIF’s relation to complex I:
AIF levels affect the high level of complex I proteins through post-transcriptional mechanisms
AIF is an integral part of the mitochondrial respiratory chain complexes, not just related to complex I
Physical interactions of the AIF proteins with the respiratory chain subunits could affect their assembly or stability
AIF could be a part of the maintenance of the active complex I due to its redox-regulated activity which is inherent because of AIF’s outer and reactive positive charge distribution
Although AIF has been known for years of its function to induce apoptosis, it also has essential functions in cell survival because of its role in the metabolism of cells and in the mitochondria. As of recently, studies of AIF’s role in survival have been exclusively done on mice. Having low continuous amounts of AIF throughout an aging adult’s lifetime can have dire health consequences. An approximate 80% decrease in AIF gene expression in mice has shown continual neurodegeneration that affects the mice’s various brain regions. When a complete absence of AIF proteins in the mice was observed, apoptosis still occurred in its usual fashion. This leads to the conclusion that AIF in terms of apoptotic function is dispensable, especially during embryonic neural tube closure.
Rather than using AIF, the triggering of programmed cell death was done by defective activity of the mitochondria’s respiratory chain complex I. Lack of AIF expression in the brain is directly linked to the inefficiencies of the mitochondrial respiratory chain. Because of its malfunction, the midbrain and cerebellum of the mice at early stages of brain development were observed to have defects such a cerebellar hypoplasia. The Purkinje precursor cells of these embryos undergo apoptosis even in the absence of AIF as well. Granule cells also show defective cell cycle transitions (particularly from G1 to S steps). AIF in skeletal muscle and the myocardium causes organ-specific complex I malfunction. Although these mice look normal for the first 2 months since birth, they develop muscle atrophy, dilated cardiomyopathy, and have a dramatic reduced body weight. Decreased expression of AIF leads to oxidative phosphorylation complications that affect the body’s entire metabolism. Mice with absence of AIF in either their muscles or life (called AIF knockout) display higher glucose tolerance, increased sensitivity to insulin, and overall reduced fat mass. These observations imply that no AIF expression in one organ leads to it being fully resistant to obesity and diabetes even when put on a diet high of lipids.
Becker, Wayne M, et al. The World of the Cell. 7th ed. New York: Pearson/Benjamin Cummings, 2009. Print
Klein, Stephen B. "Biological Psychology". New York: Worth Publishers, 2006. Print
Brady, Graham F, Brady G.F., Duckett C.S. "A caspase homolog keeps CED-3 in check". (2009) Trends in Biochemical Sciences, 34 (3), pp. 104-107.
Schatz, Gottfried. "The Magic Garden." Annual Review of Biochemistry 76.1 (2007): 673-78. Print.
Jie Yang, Xuesong Liu*, et al. "Prevention of Apoptosis by Bcl-2: Release of Cytochrome c from Mitochondria Blocked" Science 21 February 1997: Vol. 275 Print
"Developmental Apoptosis in : C. Elegans: : a Complex CEDnario : Abstract : Nature Reviews Molecular Cell Biology." Nature Publishing Group : Science Journals, Jobs, and Information. Web. 30 Nov. 2011. <http://www.nature.com/nrm/journal/v7/n2/abs/nrm1836.html>.
RAGE is a central signaling molecule found in the immune system and is involved in enduring and complicating responses toward inflammation. RAGE is also a receptor for products of glycation and acts as a pattern recognition receptor that recognizes common characteristics instead of specific ligands. New information about RAGE’s extracellular structure led to this discovery. Experimental procedures of x-ray crystallography and NMR show that ligand binding is driven by electrostatic interactions between ectodomains and ligands, which are positively charged and negatively charged, respectively.
RAGE is part of the immunoglobulin superfamily of cell surface receptors.
This receptor, RAGE, is part of a superfamily of cell surface receptors known as immunoglobulin (Ig). It is a major molecule in the development of severe chronic diseases including diabetes, inflammation, atherosclerosis, neurodegeneration and even cancer. Animals that are healthy have low expressions of the RAGE molecule but expression increases as the state of health or a disease worsens. Increased expression in the RAGE molecule also increases the expression of ligands attached to the RAGE molecule. Examples of RAGE ligands include AGEs, which are advanced glycation end products and members of the S100 protein family. AGEs are also part of many other groups including the high mobility group protein box-1 (HMGB1), β amyloids, and fibrous protein aggregates. Attachment of ligands to RAGE receptors activates various signaling pathways that depend either on the ligand, environment or cell type. These pathways can be:
p38 mitogen-activated protein (MAP) kinase pathways
cAMP response to element-binding (CREB) protein
activation of transcription family (STAT3)
When an inflammation response is in process, the RAGE-ligand attachment leads to an increase in the expression of the RAGE molecule. This is a type of positive feedback loop that results in prolonged activation of NF-κB, which is a nuclear transcription factor allowing for the conversion of a positive inflammatory response into a chronic physiological state. Researchers of this phenomenon realize that in order to prevent the RAGE molecule from inhibiting inflammatory responses, the positive feedback loop created by the RAGE-ligand and RAGE molecule, should be disrupted by blocking the receptor-ligand interaction site. However, this would require extensive knowledge of the mechanism of RAGE-ligand interaction.
X-Ray crystallography and NMR spectroscopy have allowed for studying the structure of RAGE as well as the mechanism of RAGE-ligand recognition. RAGE has a large positive surface charge allowing it to create electrostatic traps for ligands that have negative charges. The molecular organization of RAGE plays an important role initiating signals for ligand interaction. By studying fluorescence-labeled receptors, it was evident that RAGE did not exist as a single molecule in the plasma membrane of cells but collects in assemblies of receptors.
RAGE has a single transmembrane helix that connects the ectodomain with the short cytoplasmic domain and an extracellular component that is required for the ligand to recognize and bind. This extracullar component has three immunoglobulin-like (Ig) domains. The N-terminal Ig domain is assigned to the V-set of Ig-like molecules and is known as the V domain of RAGE. The other two Ig domains are part of the C1 and C2 set. The N-terminal V domain is located far away from the plasma membrane but the C2 domain lies close to the membrane. V and C1 domains can be joined together to form an elongated structure. V and C1 domains can be fixed and become the VC1 domain, which can connect to the C2 domain by several amino acids that have no secondary structure, which allows the VC1 and C2 domains to link. NMR spectroscopy studies have shown that VC1 moves as a single unit and can combine with the C2 domain.
The V domain consists a large amount of arginine and lysine, which carry positive charges at neutral pH. RAGE V domain has more arginine and lysine than the V-set of Ig domains. Arginine and lysine residues form large positively charged patches on the surface of V and C1 domains. Meanwhile, C2 domain has mainly acidic residues on its surface and is negatively charged. Because the two domains are oppositely charged, the extracellular component of RAGE is subdivided. This subdivision is reflected in the ligand binding properties of the different domains since ligands do not bind to C2 due to charge repulsion. Most ligands tend to bind to the V domain or the VC1 domain since ligands are negatively charged. There is only one case where a ligand has bound to the C2 domain. Nonetheless, charge-charge interactions are important for the formation of the receptor-ligand complex and suggested that the positively charged ligand-binding domain of the RAGE molecule can recognize certain arrangements of negative charges of ligands and can recognize these as common features for the ligands.
Surface Plasmon Resonance allowed for the revelation that ligands with multiple subunits have long periods of ligand-receptor interaction.
Positron emission topography (in vivo) and in vitro have allowed for the study of binding of ligands. In vitro studies are conducted by monitoring the cellular response of RAGE ligands as well as protein-protein interactions. It was concluded that RAGE-ligand binding causes cells to respond immediately. Signaling of the ligand and its RAGE molecule increase in strength when the bonding affinity between the RAGE and ligand is high. The signal strength also increases when the signaling complex is activated for longer periods of time. There are two phases of the process of ligand binding. First, the dissociation phase reveals the lifetime of the signaling-competent complex. Ligands that dissociate slowly in vitro can induce longer and more intense receptor activations. Surface plasmon resonance (SPR) is a method for observing these activations since it examines the real-time interaction of the receptor, which can become immobilized and its ligand, which can bind. The thermodynamics of binding including the affinity of the RAGE-ligand attraction as well as the kinetics of the dissociations can be observed. The SPR technique has also revealed that multimeric, or consisting of multiple subunits, ligands have prolonged periods of binding to their receptors.
Rage ligands are very diverse and have common features that allow for the activation of the RAGE molecule. The following are examples of RAGE ligands:
AGEs (Advanced glycation endproducts)
These molecules are heterogeneous and are derived from the condensation and oxidation of proteins, peptides and sugars. The first step in forming AGE is the reaction of the aldehyde group of sugars with amine groups from lysine or arginine amino acids. This is a nonspecific and non-enzymatic process and it is called glycation. When glycation is high and frequent in individuals, it leads to diabetes and other complications. Oxidation of AGEs allows for the increase in overall negative charge on the ligand by carboxymethylation of the amino group. AGEs and RAGE have high affinities for one another and initiate a high level of endurance for pro-inflammatory responses. AGE and RAGE interaction has been observed as the main cause of diabetes complications and cardiovascular disorders.
S100 Proteins
This family of proteins includes more than 25 members and each displays different patterns and functions. These proteins are small and acidic and have two different calcium bindings that are connected by a flexible loop. S100 proteins form homodimers and sometimes heterodimers and oligomers such as tetramers and octamers. S100 proteins can be found in vertebrates mainly and are localized in the cytoplasm, acting as calcium sensors. These proteins can display intracellular calcium signaling and binding that causes the S100 proteins to change in conformation and shape. In the extracellular space, S100 proteins are loaded with calcium since the calcium concentration is high, so they can bind to RAGE easily.
Amyloid β and amyloid fibrils
Beta Amyloid structure
Alzheimer disease is caused by the extracellular deposition of amyloid β peptide, originating from proteoloytic cleavage of amyloid precursor protein. Amyloid β peptides contains between 40 and 42 amino acids. These peptides have acidic and hydrophobic regions that are prone to accumulation of amyloid fibrils that build up amyloid plaques in the brain, causing Alzheimer’s disease. Mouse models of Alzheimer’s disease have shown that RAGE binds amyloid β and moves it around the blood stream to the blood-brain barrier in the central nervous system (CNS). Analyzing this using in vitro revealed that RAGE and amyloid β have high affinity for binding the soluble amyloid β peptide. RAGE can also bind amyloid fibrils.
High mobility group box-1 protein (HMGB1)
Structure of high mobility group box-1 protein (HMGB1)
This protein has an extracellular regulatory function and acts as a proinflammatory activator. HMGB1 has three domains: two N-terminus domains that are involved in DNA binding and one C-terminus domain that consists of various acidic amino acids as well as directs the binding to the RAGE molecule. HMBG1 binds to RAGE and Toll-like receptor 9 (TLR9), which forms the HMGB1-RAGE-TLR9 complex that activates B cells. DNA can bind with RAGE with high affinity and form a stable complex without the HMGB1 protein, but DNA-RAGE interaction has not produced noticeable results yet.
RAGE can recognize common characteristics in different ligands. RAGE ligands have a net negative charge at their surfaces. They also usually have neutral pH, but S100 proteins have acidic pH. Also, AGEs do not usually have their surfaces negatively charged until they transform during glycation and oxidation. Most ligands also oligomerize. AGE modifies proteins, which leads to covalent bonds to higher molar mass molecules. S100 proteins also form large assemblies of tetramers or octamers.
Fritz, Gunter. RAGE: a single receptor fits multiple ligands. Trends Biochem Sci. 2011 Dec(12): 625-32. Epub2011 Oct19.
Mobile zinc and the gaseous nitric oxide radical are two prominent examples of inorganic compounds that are found in many signaling pathways in living systems.[1] To detect the presence of mobile zinc and nitric oxide, fluorescent probes is used. Fluorescent probes which are dyes that give off light when exposed to light, are excellent tools for detecting zinc and nitric oxide with high spatial and temporal resolution. To monitor biological processes in real time with high spatial resolution, using fluorescent imaging offers an excellent platform. It is known that fluorescent probes maintain good water solubility and membrane permeability.
1- Fluorescent Sensors For Mobile Zinc
The divalent zinc cation, Zn2+, influences a widely metabolic spectrum. Mobile zinc plays many physiological roles in zinc-enriched tissues, such as hippocampus, pancreas and prostate.[2] Mobile zinc bioimaging is currently dominated by fluorescent probes. That is due to the technical advances in microscopy instruments and fluorescent sensors, which have been proved to be sensitive and versatile tools for zinc detection in living cells and tissues. In fluorescence microscopy experiments, zinc indicators depend on an increase or rarely a decrease in fluorescence emission intensity.
2- Fluorescent Sensors For Nitric Oxide
Nitric oxide is a free radical gas produced endogenously by a variety of mammalian cells.[3] Nitric oxide plays various roles as the signal transmitter and it is involved in many physiological and pathological processes. Nitric oxide is synthesized from arginine by a complex reaction, while catalyzed by nitric oxide synthase. Nitric oxide synthase is an enzyme that catalyzes the conversion of l-arginine, NADPH, and oxugen to citrulline, nitric oxide and NADP+.[4] There are two strategies in developing the small-moleculed fluorescent probes for nitric oxide. This strtegies are conducted by Pluth M.D et al. in their paper entitled, "Biochemistry of Mobile Zinc and Nitric Oxide Revealed by Fuorescent Sensors". The first is based on organic probes and they often result in bright emission enhancement in the presence of nitric oxide under aerobic conditions. this strategy is widely used. the second strategy is using transition metals to mediate reactivity of an emission dye with nitric oxide. it was found that these probes can be less bright than purely organic probes.[5]
3- Interactions between Zinc and Nitirc Oxide
In “central nervous system”, Zinc and NO as well as their derivatives are essential inorganic species to make the system function well. Otherwise, “neurotoxic disease” may occur, such as “Alzheimer’s”, “Parkinson’s”, and “Huntington’s” diseases. When the “hippocampal neurons” are exposed under “NO donors” in an extent that beyond the nonneurotoxic level, Zn(Ⅱ)in brain tissue becomes more active and collective. As a result, more and more Zn(Ⅱ) accumulate in brain, leading to the increase of O2−, which finally causes the dysfunction of mitochondrial, the rise in ONOO−, as well as the change of the signaling pathway. Moreover, this process may also bring cells to death. By using the techniques of probes for NO and Zn, scientists might be able to understand the link between nitric oxides and zinc better and their quantitative properties on cell death.
Besides for their important roles in nervous system, zinc and nitric oxide also cooperate to defend diseases in the immune system. Less amount of zinc can weaken the immune system, while higher amount of NO can be caused by inflammation. By experiments, scientists have found out that adding 10 μM of Zn(Ⅱ) lowers the production of NO, which agrees with the anti-inflammatory property of Zn(Ⅱ).Therefore, the development in various fluorescent probes for zinc and nitric oxide will continuously help scientists explore how zinc and nitric oxides work in nervous, cardiovascular and immune system and also be applied into pathology field.
Nitric oxide is a unique gas that serves as a chemical messenger in the human body. In contrast to proteins and neurotransmitters, nitric oxide is rather difficult to analyze due to its volatile nature (Proteins and neurotransmitters are readily extracted and remain intact for hours; whereas, nitric oxide vanishes in seconds).
Nitric oxide is very important in the physiological aspects of the body, including cell to cell communication in the nervous system, as well as functioning in the immune system. It is particularly important in the physiology of the brain. [1]
The role of Nitric Oxide in cells is as a chemical messenger. NO, however, vanishes in seconds, such volatility renders NO a molecule with extraordinary diversity. Because of its nature, nitric oxide can perform specific functions such as open blood vessels, help pass electrical signals between nerves, and most importantly, fight off infections. For example, nitric oxide is the active substance in INOmax, a drug that is delivered by inhalation. The drug is a pulmonary vasodilator, meaning it will increase the flow of blood in the lungs by relaxing the muscles and opening the blood vessels wider.
The production of this gas is primarily controlled by an enzyme called nitric oxide synthase (NOS). Tiny and hard to study in the laboratory, NO eluded scientists for many other years because other molecular messenger such as neurotransmitters and larger proteins can be relatively easily extracted from body fluids and studied where they can remain intact for minutes or even hours. Scientists and researchers used X-ray crystallography to understand the shape and structure of NOS, and how it binds to specific substrates.
From this, they were able to determine how to boost or inhibit the activity of nitric oxide. However, the production of nitric oxide must be controlled in order for it to be most effective. For instance, opening the blood vessels too widely can result in a deadly shock in which there is not enough blood flowing through to fill the organs. At high levels it can cause neurological stress as well as cell death. This shows that NO can also be a foe, too much or too little of this gas can be harmful. An overactive immune response, fired up by NO, can produce a painful syndrome called inflammatory bowel disease. With all this at stake, the body works hard to stringently control production of this powerful gas. [1]. Fatal symptoms can also include hypertension, irregular heart beatings, and drop in blood pressure. [2].
Nitric oxide has become an extremely popular drug in the bodybuilding world. Taking nitric oxide supplements leads to increased blood flow to the muscles, which will let a greater amount of nutrients be delivered to the muscles while working out. Taking nitric oxide also affects the endocrine system's gonadotroptin releasing hormone, as well as the release of adrenaline.
Nitric oxide supplements usually contain the amino acid Arginine. Taking too much of arginine can lead to weakness, diarrhea, nausea, and feelings of tiredness. It is important to monitor the amount of nitric oxide supplements being taken in order not to overdose. [4]
↑ abJelena Bašić, et al. "Nitric Oxide - Mediated Signalization And Nitrosative Stress In Neuropathology." Journal Of Medical Biochemistry 31.4 (2012): 295-300.
↑ Berg, Jeremy M., ed. (2002), Biochemistry (6th ed.) New York City, NY: W.H. Freeman and Company,
↑U.S. Department of Health and Human Services. Chemistry of Health. October 2006.<http://www.nigms.nih.gov>.
Heparan Sulfate molecules are found on the surface of the extracellular matrix of most animal tissues. It occurs as a proteoglycan (HSPG) in which two or three HS chains are attached in close proximity to cell surface or extracellular matrix proteins. A proteoglyan is a protein that is heavily glycosylated Heparan Sulfate Proteoglycans achieve are involved in development, homeostasis, and pathological processes. For example the mediation of cytokine-induced cell signaling. Their effect is achieved by interaction with different types of ligands.
The synthesis begins with the formation of a polysaccharide-protein linkage region. A four-unit sugar is attached to a serine residue at the core of the protein. The sugar is then extended by the addition of either d-acetylglucosamine or d-acetylglactosamine and d-glucuronic acid residues. This forms Heparan Sulfate structures.
One of the most important roles of Hepran Sulfate is the meditation of growth factors and their cognate receptors. It is proposed that heparan sulfate participates in the signaling process by acting as a fine-tuner for the process, this has been prototipically demonstrated in the fibroblast GF, or FGF, family. In this family heparan sulfate acts as a necessary coreceptor.
Somatic overgrowth can occur in patients with mutations in the HS polymerase genes. It is thought that because of the interactions with HS and growth controls, HS might act as tumor suppressor if it is functioning properly. Mutations affecting the biosynthesis of heparan sulphate proteoglycans (HSPGs) are the cause of several human
hereditary diseases.
It has been shown that a member of the glypican family of membrane proteoglycans, GPC3, is mutated in human patients that suffer from Simpson–Golabi–Behmel syndrome (SGBS), an X-linked disorder that is associated with developmental tissue overgrowth and with a high incidence of neuroblastomas and wilm's tumors. On the basis of an analysis of GPC3 mutations, it has been proposed that SGBS is probably caused by a lack of functional GPC3 protein, which is supported by the defects shown in GPC3-deficient mice. Although the mechanism by which changes in glypican function lead to cancer is unknown, tumour progression has been found to be associated with changes in the expression of GPC3, and increases in the expression of another member of the glypican family, GPC1, have been observed in cervical and pancreatic cancers.
In this disease, benign tumors derive from the growth plates of endochondral bones. These tumors later become malignant. Interestingly, the cause is the mutations of EXT1 or EXT2 genes that encode the HS polymerase genes.
Major growth factors involved in skin wound repair are HB-EGF and FGF-2. This is achieved by the binding of HS to the FGF-2. When tissue is injured, neurotil heparanase is excreted the wound area to degrade the inhibition of the heparin sulfate chains. These HS chain inhibition is eliminated, HSD is liberated and stimulates FGF-2 to promote repair.
1) http://www.ncbi.nlm.nih.gov/pubmed/10872465
Functions of cell surface heparan sulfate proteoglycans.
Bernfield M, Götte M, Park PW, Reizes O, Fitzgerald ML, Lincecum J, Zako M.
Division of Developmental and Newborn Biology, Children's Hospital, Harvard
Medical School, Boston, Massachusetts 02115, USA. bernfield@a1.tch.harvard.edu
2)
Cell signaling pathways are very essential for skeletal muscle remodeling. Skeletal muscles are composed of heterogeneous muscle fibers (myofibers), which are different in their metabolic functions. There are four main types of myofibers: type I, type IIa, type IId/x, and type IIb. Type I and type IIa are specialized in oxidative metabolism, and type IId/x and type IIb are specialized in glycolysis. These skeletal muscles can be remodeled depending on the signaling due mainly to calcium concentrations in the human body and also due to various transcription factors. Muscles can change by transforming the myofibers to consist of more type I or more type IIb depending on how often exercise is done. If a person continually exercises, his/her skeletal muscles will contain more type I myofibers, which increases the overall oxidative metabolism in the body. This in turn keeps the person healthier in multiple ways, mostly in preventing bodily disorders, such as diabetes, muscular dystrophy, etc. These myofibers can be transformed when certain signaling pathways are activated or inhibited.
One of the main component responsible for the transformation of myofibers is the signaling of calcineurin. Calcineurin is a protein phosphatase, which promotes the expression of muscle-specific genes. Calcineurin is activated by myocyte enhancer factor 2 (MEF2), which is a type of transcription factor that will recognize DNA sequences that are rich in adenine-thymine nucleotides. There is also a class II HDAC (histone deacetylase) that controls the activity of MEF2. When the class II HDAC binds to MEF2, MEF2 is inhibited so then it cannot activate the muscle specific genes. To prevent inhibition of MEF2 by HDAC, a HDAC kinase is activated and it phosphorylates the class II HDAC, which gives it a signal/message to leave the nucleus and leave the MEF2 uninhibited.
Phagoptosis is a term proposed by Guy. C. Brown and Jonas J. Neher to describe death of working cells by phagocytosis (the engulfing of a cell by a macrophage). The word is derived from two words in ancient Greek: 'phagein' which means to devour and 'ptosis' which means to fall or die.[1] Thus the term, phagoptosis means death by being swallowed. Phagoptosis is also known as "primary phagocytosis".
Simply stated, phagoptosis is caused by the exposure of “eat me” signals or the loss of “don’t eat me” signals on the cell surface. The exposure and loss of these signals is reversible, thus cell death can be prevented by preventing phagocytosis. [1].
Phagoptosis is a nascent term, combining the ideas of phagocytosis and cellular apoptosis (autonomous cell death). While not yet a confirmed phenomenon in macroorganisms, phagoptosis has been proposed as an explanation for various homeostatic and pathological processes in the body that induce the engulfing of healthy or unhealthy cells by a macrophage. Phagoptosis has been proposed, therefore, to be both a contributor to cellular degenerative diseases such as Parkinson’s or Alzheimer’s, whereby healthy neurons are destroyed, and a natural aging tool used by the body to rid itself of unhealthy cells, such as the consumption of old erythrocytes.
Since the 1880s, the work of Russian biologist Ilya Ilyich Mechnikov has led us to believe that phagocytosis, a form of cell destruction, was beneficial because only dead or dying cells were targeted. However, modern-day scientists have found that sometimes healthy, viable host cells can also be marked for cell death; this process is called phagoptosis (or primary phagocytosis).
[1].
1. Autophage: mediated by autophagic in which the cell is eating itself to cause its own death.[1].
2. Apoptosis: mediated by caspases and it is a program of cell death (PCD).[1].
3. Necrosis: cell injuries causes cell death within high stress level of cell structures. [1].
4. Cornification: cell death due to conversion of proteins
5. Shedding:cell death caused by replacement in cell structures.
6. Phagoptosis: cell death caused by phagocytosis with macrophages.
Apoptosis
Apoptosis is sometime referred to as “programmed cell death.” It is when the cell relatively speaking commits suicide. It generally occurs in response to certain signals in the body It is better than necrosis because it follows a structured routine and is not random like necrosis. When a cell undergoes apoptosis, the protein caspases will be calld upon to break down the any cellular components necessary for survival. The enzyme DNases is also released to destroy any DNA in the nucleus of the cell, thus eliminating any existence of that cell in that area. Aside from the production of caspases and DNases, cells that goes through apoptosis will release a signal to macrophages telling them to clean up any remaining debris. The macrophages clean everything up giving no opportunity to the dead cell to damage nearby cells.
Apoptosis is actually important to human development as it gives human the fingers and toes feature that human possesses. Originally the 10 fingers are all connected to a web-like feature, but apoptosis is what causes that web to be broken down and destroyed, leaving human with 10 fingers and toes instead of four web like hands and feet. [1].
Necrosis
Necrosis is when cells and tissues die randomly, meaning it is not programmed. It is caused by inflammation, injury, cancer, toxins, etc that may harm the body. The problem with necrosis is that when cells die they do not send signals to nearby phagocytes that order them to clean up the dead cell. Therefore it is harder for the body’s immune system to react and clean up the dead cell if it goes through necrosis, causing build up of dead tissue or cell debris a that location. Cells that goes through necrosis may also release harmful chemicals that may hurt or kill nearby cells. [1].
Phagoptosis
Phagoptosis is a form of cell death when phagocytes is the primary cause of cell death, which is provoked by cells displaying “eat-me-signals” instead of “don’t eat me signals.” Initially the cell produces “find me signals,” that triggers chemotaxis of phagocytes. Upon arrival the cell will either produce “eat me signals” or “don’t eat me signals” and the will ultimately determine if the cell will live or die. The most abundant form of eat me signal is the phospholipid PS found on the inner leaflet of the plasma membrane of the cell. The eat me signal comes out of the cell as a result of calcium-activated phospholipid scramblase that causes the phospoholipid to shuffle the inner leaflet from the outer leaflet, exposing the eat-me-signal. There are different ways PS can be exposed onto the cell such as, calcium elevation, ATP depletion, oxidative stress, fusion of intracellular vesicles with plasma membrae, necrosis, and apoptosis. All of these can cause a cell to release PS signals and hence result in their removal by phagocytes.
A way a cell can protect itself from phagocytes is producing “don’t-eat-me-signals”. A type of “don’t-eat-me-signals” that a cell displays is CD47, CD200, etc. CD47 is the most abundant “don’t-eat-me-signal” expressed by a cell. Though if the “don’t-eat-me-signal” is disrupted then phagoptosis may occur. [1].
Even though Phagoptosis support cell to shape development in cell structures, removes excess and defective cells, and protect cells from pathogens and other cancerous cells, over-excess process of Phagoptosis can cause diseases that might harm the cell replication, production and its natural cycle. Due to the malfunction and irregularity of cell death can lead to lack of important cells in our bodies. For example, excess PS exposure in brain cell structure can terminate not only old and phagocytic neurons but also important neurons that supports our brain cells. Neurons cells are phagocytised by Microglia (Microphage in brain cells) which is caused due to Inflammation Activation. Microglia can eat apoptic neurons to reduce debris or inflammation to help cell circulation and stay in good shape for brain cells[1].; However, it also can destroy viable neurons and neuronal processes which disintegrate the healthy process of cell, neurons in brain; thus, affecting other healthy brain cells and causing cerebral diseases or disorders like Frontotemporal Degeneration (FTD). Frontotemporal Degeneration (FTD) is heterozygous, inactivating mutation in the progranulin gene due to lack of neurons in brain cells. [1]. It is caused when there is lack of neurons in brain cell due to excess activities of Phagocytosis by Microglia. Many neurons were phagocytised, so it caused inactivating mutation that malfunction the role of progranulin genes in DNA. This FTA disorder is also contributed to causes of Alzheimer's disease and Parkinson's disease. [1].
Cell Stress causes several difference responses including phagoptosis, but the response depends on the amount of cell stress present. The different responses include: adaptation, phagoptosis, apoptosis, and necrosis. These lowest stress level would cause adaptation and as the stress level rises the other responses follow accordingly.
File:Cell StressThe cell has several different responses to cell stress. Different levels of stress cause different responses.
Numerous conditions may cause cell stress. Temperature shifts such as heat shock could cause proteins in the cell to denature. Heavy metals can change the conformation of the protein and therefore the function. Free radicals can cause proteins to be fragmented. Fragmentation of proteins render them ineffective. Solvents such as ethanol can cause translation errors. Cell stress response can be the upregulation of some genes and the downregulation of others. In essence, response to cell stress can either cause some proteins to be produced more and others to be produced less depending on the situation. Regulation of cell stress is different in prokaryotes and eukaryotes mainly due to codon differences. [2]
Cell-In-Cell phenomena is most likely seen in tumor cells, especially those are very strong and metastatic. Tumor cells can engulf immune cells which eventually kills tumor cells; On the other hand, tumor cells can also engulf other tumor cells for survival or protection from other harms. Some tumor cell can go through cannibalism process (Cell-In-Cell Phenomena) to protect themselves from immune cells; and Entosis process can happen to both tumor and normal cells.
Entosis is caused when tumor cell invade each other; it is caused when detached from Extracelluar Matrix. This Entosis process can cause either survival of the cells or death of invading cells by Lysosome Digestion. Entosis mostly happen in animal cells because animal cells are mostly attached to extracellular matrix for their survival. When some cells are detached from ECM (Extracellular Matrix), they lose the adhesion and cells start to push into their neighboring cells; thus, sometimes, Entosis is considered as interaction between two neighboring cells. Interestingly, cells which are locked due to the result of Entosis are alive and they can divide themselves inside another cells. Entosis is also referred to "cell-in-cell structures" which indicates the loss of attachment from extracellular matrix. In other word, It is process by which tumor cells invade each other when detached from matrix and it is relatively common in malignant cancers. [1].
Cannibalism is caused when cells eating up each other for survival and protection due to damages or infection. For example, Wang et al found out some tumor cell can go proceed apoptic cell death. From the bodies of patients who have Huntington's disease, immortalised lymphoblasts phagocyotes are eating up each other to survive in tough condition or to replenish and protect themselves. However, whether restriction of phagocytosis will be able to avoid cell death in cell-in-cell phenomena is still questioned. [1].
What can cause a macrophage to consume a cell? Many intercellular interactions occur via chemical signaling; the cell displays certain compounds outside of the plasma membrane, to which other cells respond thanks to receptors on the exteriors of their plasma membranes. While some of these compounds invoke phagoptosis, others repulse it and it is the net amount of these "eat me" versus "don't eat me" signals that determine the final fate of the cell. Three main compounds have been proposed to signal macrophages to engulf a cell, each with its own signaling pathway that causes the compound to be expressed or suppressed on the plasma membrane.
1. PS (phosphidatylserine)—normally suppressed by an ATP-operated inhibitor called aminophospholipid translocase, which actively pumps PS to the inner membrane of the cell, PS has been shown to, under certain circumstances, lead to phagocytosis of so-called “activated” T cells. PS binds to the receptor Tim-4 on phagocytes, which signals the phagocyte to engulf the cell. If there is no ATP to run the translocase or if there is an overabundance of free calcium in the cytoplasm, the translocase stops running, and another membrane protein called scramblase randomizes the exposure of PS in the cell membrane, potentially exposing it to free phagocytes for endocytosis. This process can happen even in healthy cells.PS exposure can also occur due to oxidative stress which activates the scramblase and prevents translocase. Another result of PS exposure is when the intracellular vesicles fuse with the plasma membrane.
In summary, there are 5 ways that the PS “eat me” signal can be exposed: elevation in calcium levels, lack of ATP, oxidative stress (which activates scramblase and stops translocase), fusion of intracellular vesicles with plasma membrane, necrosis, or apoptosis. Another requirement for phagocytosis to be carried out is the presence of macrophages that are capable of attaching to PS.
In order for macrophages to recognize PS, different receptors must be activated. Resting macrophages express the following PS receptors: Tim4, stabilin-1, stabilin-2, and BAI1. On the other hand, macrophages that are activated express the protein MFG-E8 and its receptor, vitronectin, in addition to the MerTK receptor.
Furthermore, it is important to note that PS exposure does not always equate to cell death; the cell type and surrounding conditions also plays a role. For example, when purified PS is added to 3 different cell lines, the viable cells show an elevation of PS on their surfaces. This stimulated phagocytosis as expected, but as soon as the PS was “internalised”, phagocytosis was inhibited. In other cases such as that of lymphoma cells, PS exposure is not enough to even initiate phagocytosis because either the cells also expressed “don’t eat me” signals or they required another signal to co-stimulate phagocytosis.
[1] However, PS exposure on viable cells can be reversed. When neutrophils are activated they have galectins that induce PS on the surface of the cell. If the galectin is removed before the macrophages detect it, then phagoptosis will not occur.
2. CRT (cell-surface calreticulin)—CRT is abundant in cells, particularly in the endoplasmic reticulum. Its transport to the exterior of the cell (exocytosis) can cause it to behave as an attractive signal to phagocytes, which have the CRT receptor LRP (lipoprotein receptor-related protein). Rather than actively displaying CRT as a self-destructive signal, a stressed cell will decrease the repulsive chemical signal CD47 (discussed in the subsequent section), after which a phagocyte will become attracted to excess CRT on the external surface of the target cell. This is an especially prevalent mechanism in cancer cells, which actively attempt to display the CD47 signal. Cancer cells require strong exposure of "don't-eat-me" signals in order to prevent phagoptosis. While CRT is tied to the surface of a cell, it can also send signals to bind proteins, PS, or C1q on target cells. This will stimulate phagocytosis through the LRP, which is on the phagocyte.
An “eat me” signal similar to CRT is thrombospondin 1 (TSP). Like CRT, TSP1 activates phagocytosis with the use of lipoprotein receptor-related protein (LRP) on the phagocyte.
[1]
3. MFG-E8 (milk fat globule EFG-like factor-8)—This chemical signal binds to the phagocytotic initiator MerTK (Mer tyrosine kinase) on the phagocyte with the help of several connecting or “bridging” compounds, including Gas-6, protein S, galectin-3, tubby and Tulp 1. These can interact with PS and other receptor proteins in complicated pathways to induce phagocytosis on white blood cells called neutrophils.[1]
In addition to these attractive signals, there are repulsive signals that cells can display to actively prevent phagocytosis. While the attractive signals may be displayed as a result of cellular stress, repulsive signals are altogether more specific. Some repulsive signals include:
1. CD47 (Cluster of Differentiation 47: CD47 is the inhibitory membrane protein expressed on the surface of the majority of cells. It binds to signal-regulatory proteinα (SIRP-α) on the phagocyte. Examples of cells that use this protein as a 'don't eat me' signal include erythrocytes (red blood cells), cancer cells, and platelets, T-cells. In clinical studies performed on mice with cancerous growths, it has been found that blocking of this protein results both reduction of tumor size and spread throughout the body as a result of phagocytosis. [1][2]
2. Sialic-Acid Derivatives: These react with a variety of receptors, such as cofactors to prevent phagocytosis. Modifications of sialic-acid on cell surfaces can stop C3b and C1q from binding and signaling nearby macrophages. Sialic acid can be removed from the cell surface by implementing neuraminidase and doing so can induce phagocytosis. Siglec-11, a receptor on brain microphages (microglia), can prevent inflammation and phagocytosis of neurons by binding to their surface. However, Siglec-11 requires polysialylated proteins to be present on the cell surface. [1]
3. PAI-1: PAI-1 (or plasminogen activator inhibitor-1) is another major repulsive signal in cells. Neutrophil cells are an example of a cell type that mainly uses PAI-1 as its repulsive signal.[1]
4. CD200 (Cluster of Differentiation 200): This is a protein expressed on the membrane of certain cells. In the case of myeloid cells, it can prevent phagocytosis by releasing an inhibitory signal.[3]
How Attractive and Repulsive signals Work Together
1. However, it is important to note that the display of an “eat me” signal alone may not be sufficient to induce phagocytosis. Despite having “eat me” signals on its surface, a cell may not be attacked by phagocytes because it also displays “don’t eat me” signals. The proportion of “eat me” signals such as PS to “don’t eat me” signals such as CD47 plays a major role in determining how the cell will be recognized by macrophages. Furthermore, the type of cell as well as its environment also influences the degree of influence that signals monitoring cell death will have. Multiple cell types require a cooperative protein or PS oxidation to undergo phagocytosis. [1]
Much of the information about cell death has been obtained from model organisms, such as Caenorhabditis elegans (a species of transparent nematode) and Drosophila melanogaster (a species of fly commonly used in laboratories).[1]
Caenorhabditis elegans, a nematode around 1mm in length.
In C. elegans it has been found that a combination of loss-of-function mutants in ced-1 as well as in ced-3 (two transmembrace receptors in C.elegans)[4] led to reduced apoptosis as well as reduced phagocytosis, indicating that both are important factors in programmed cell death during development of the species. This also prevented some death from mutation and toxins. From this it was concluded that light stress on the cell (such as weak activation of caspase, which is involved in apoptosis) is not enough to cause a cell to die, but in conjunction with PS exposure could cause phagoptosis. By removing the srgp-1 gene, which prevents phagocytosis of PS-exposed cells, it was found that rates of phagocytosis in normally apoptotic cells and other stressed cells increased. [1]
Drosophila melanogaster, a species of fly often used for genetic testing.
In D. melanogaster, developmental cell death is caused by 3 proteins: Hid (head involution defective), Rpr (Reaper), and Grim. All of these induce apoptosis of the cell by binding to the caspase inhibitor DIAP1. This leaves developmental phagocytosis intact, which seemed to remove most of the cells normally lost in development. This indicates that while apoptosis might not be essential for development of D. melanogaster, phagocytosis might be. One of the mechanisms through which phagoptosis occurs in D. melanogaster is the ER protein pretaporter, which is expressed externally on the surface of cells. There is also competition between cells of different genotypes within D. melanogaster through induced phagoptosis of surrounding cells. [1]
Erythrocytes are red blood cells. The highest rate of cell death in the body is caused erythrophagocytosis, the cell destruction of red blood cells. Two million red blood cells, or erythrocytes, are produced every second in the human body. They live for about 120 days and then are destroyed by macrophages in the spleen, liver, and bone marrow in such a way that equals its rate of production. Instead of undergoing apoptosis, red blood cells display “eat me” signals such as PS. A loss of CD47 “don’t eat me” signals in old red blood cells is enough to cause rapid phagocytosis.
Older erythrocytes tend to get phagocytized more because they contain more "eat-me" signals such as PS, phosphidatylserine. When PS is exposed on the cell’s surface, a macrophage senses this and phagocytizes the red blood cells. This can be reversed by other signals such as CD47 on the cell’s surface, binding to the macrophage’s SIRPα receptors. As erythrocytes become older, they lose CD47 signal, causing phagocytosis of erythrocytes. It was found that older erythrocytes live longer when there were depletion of macrophages, which concludes that phagoptosis was the reason for red blood cell's turnover. [1]
The phagocytosis of red blood cells is mediated by “eat me” signals such as PS as well as “don’t eat me” signals such as CD47. Erythrocytes are particularly excellent for studying the regulation of phagocytosis because they are destroyed at the same rate they are produced, giving them the highest rate of cell death in the human body. Every second, about 2 million red blood cells are formed in the bone marrow. Each cell has a life span of approximately 120 days. As erythrocytes get older, they begin to display more “eat me” signals and fewer “don’t eat me” signals on their surface. Signals that prevent phagocytosis such as CD47 act on the SIRPa receptors of macrophages, preventing them from attacking viable erythrocytes. Having more “don’t eat me” signals or having fewer macrophages will prolong the survival of older erythrocytes. [1]
Neutrophils, which are the most plentiful type of white blood cell, are quite different in comparison to erythrocytes. Like red blood cells, neutrophils are also produced in bone marrow and destroyed in the spleen, liver, and bone marrow. They are created at a rate of 0.5-1 million per second in humans, with a lifespan of about 5 days. What’s peculiar about these white blood cells is that the younger cells are just as susceptible to phagocytosis as the older cells. When isolated, neutrophils undergo spontaneous apoptotic cell death. For neutrophils, the protein PAI-1 is a “don’t eat me” signal. When PAI-1 is blocked, CRT becomes the main “eat me” signal. Neutrophils are the only leukocytes to expose CRT without the need of a stimulus, which would explain their fast renewal rate in the body. They also have the ability to eat activated platelets and small cells such as bacteria.
Also, phagocytosis is not the only cause of turnovers in neutrophils like it is for erythrocytes; instead spontaneous apoptosis occurs when neutrophils are isolated. [1]
When CRT is expressed on neutrophil’s surface, it signals the drive of phagocytosis by macrophages. In contrast, PAI-1 expresses the signal that prohibits phagocytosis, but depletion of this signal causes increasing chance of it being phagocytized. This can be reversed by adding PAI-1 proteins that binds to the white blood cell’s surface, barring phagoptosis.[1]
Neutrophils can also act as phagocytes, but only devouring smaller bacterias.[1]
Like erythrocytes, neutrophils are also made in the bone marrow. However, neutrophils only have a lifespan of 5 days before they are phagocytised. Another noteworthy difference between neutrophils and erythrocytes is that unlike for an erythrocyte, the phagocytosis of a neutrophil is not concurrent with its age. “Younger neutrophils are just as likely to be phagocytised as older neutrophils. (p. 328)” The protein PAI-1 acts as the primary signal preventing phagocytosis on neutrophils. When this “don’t eat me” signal is removed, viable neutrophils are attacked by macrophages. Like PS, the PAI-1 protein also exhibits reversible binding. Adding the protein back to the surface of neutrophils will decrease their rate of phagocytosis. The phagocytosis of neutrophils is also influenced by antibodies that block PAI-1. When PAI-1 is blocked, the “eat me” signal CRT can induce phagocytosis of neutrophils that are still viable leading to a decreased immune response. Although CRT acts as the primary eat me signal on neutrophils, neutrophils can also be stimulated to bring PS to their surface, encouraging attack by surrounding macrophages. The phagocytosis of red blood cells is mediated by “eat me” signals such as PS as well as “don’t eat me” signals such as CD47. Erythrocytes are particularly excellent for studying the regulation of phagocytosis because they are destroyed at the same rate they are produced, giving them the highest rate of cell death in the human body. Every second, about 2 million red blood cells are formed in the bone marrow. Each cell has a life span of approximately 120 days. As erythrocytes get older, they begin to display more “eat me” signals and fewer “don’t eat me” signals on their surface. Signals that prevent phagocytosis such as CD47 act on the SIRPa receptors of macrophages, preventing them from attacking viable erythrocytes. Having more “don’t eat me” signals or having fewer macrophages will prolong the survival of older erythrocytes. [1]
T-Cells play an active role in adaptive immunity. These cells activate themselves by attaching to part of antigens and proliferate to leave memory T-cells for secondary immune responses. The activation of T-cells leads to PS exposure and recognition by the Tim-4 receptor, which leads to phagocytosis; thus, carrying out its role to destroy the body's foreign invaders. The Tim-4 receptors on phagocytes are blocked when antigens are introduced during immunization or during infection with influenza virus. This decreases the production of antigen-specific T cells which in return will increase immune responses to those antigens. Similar to erythrocytes, neutrophils, platelets, the turn over of T-cells is regulated by the CD47 signal. Once the T-cell loses that signal, it will be devoured by macrophages. [1]
Phagoptosis has been linked to multiple inflammatory and immune disorders, where viable blood cells are attacked by macrophages. A reduction in red or white blood cell count is known as cytopenia. Individuals with cytopenia often have compromised immune systems and are vulnerable to infection. Inflammation can induce the increased phagocytosis of red blood cells by causing these cells to display “eat me” markers, primarily PS, on their surface. Inflammation also increases the capability of macrophages to target red blood cells. [1] Hemophagocytosis is difficult to treat because inflammation plays a vital role in the immune response by isolating foreign substances which may potentially be harmful so they can be destroyed by phagocytes. [5]
There is still much research to be done with phagoptosis, but early experiments indicate that certain instances of cell death such as erythrocytosis, neutrophil “cannibalism,” and unnatural neurodegeneration, which were once attributed to apoptosis, may be the result of this more complicated process. Further research may be able to illicit specific solutions to problems that apoptosis models could not adequately describe, which in turn, could provide an opportunity to develop new treatments for degenerative diseases.
While there are many different health problems that arise with unnecessary phagoptosis, one of the most serious of these comes from loss of neurons in the brain. This is particularly harmful because neurons are not able to be regenerated. Therefore, permanent damage may result from microglia, the macrophage in the brain responsible for consumption of neurons among other tasks, engulfing any neuron that exposes inflammation, whether or not that neuron is viable. It has been recently discovered that a major cause of frontotemporal degeneration (FTD) is linked to inactivation of the progranulin gene which inhibits phagocytosis. This suggests that Parkinson's disease, Alzheimers, and amyotrophic lateral sclerosis may be controlled by phagocytosis control in the brain. Controlling phagocytosis in the brain can be done, for example via PS blocking, which should stop all loss of viable neurons without having to inhibit inflammation.[1]
Although there are problems with unnecessary phagoptosis in the brain, phagoptosis can also be potentially beneficial in the brain as well. Microglia, or brain macrophages, can also devour apoptotic neurons (neurons which have been programmed to die), reducing the debris and inflammation. There are some cases where the microglia's ability have been impaired due to inflammation, which impairs its ability to classify which neuron to devour, mistaking viable neurons for apoptotic neurons during phagocytosis.[1]
Phagoptosis performs many beneficial functions in the body including defense against harmful pathogens and regulation of the inflammatory response. However, recent studies indicate that it may be a primary culprit of diseases associated with frontotemporal degeneration. FTD is caused by phagoptosis of viable neurons, which is normally prevented by a protein known as progranulin which regulates phagoptosis in the brain. Mutations in the gene that codes for progranulin are associated with Alzheimer’s, Parkinson’s, and other neurodegenerative diseases. [1]
It has been discovered that cancer cells contain high quantities of exposed CD47 protein which contributes to their low rate of consumption by macrophages. Potential research may find inhibitors or antibodies against CD47 that may help induce natural death of cancer cells. For example, in leukaemic cells, it has been discovered that the addition of CD47 antibodies is enough to eradicate several types of leukemia from model organisms such as mice.[1]
Several applications are examined to illustrate the idea of phagoptosis:
Example
Eat me signal
Don't eat me signal
C elegan
"PS exposure: reversed by ATP-dependent translocase tat-1, disruption of tat-1 exposes PS
on surface of viale cell induce phagocytosis. PS exposure Downregulated by srgp-1"
Drosophila
Death induced by Hid, Rpr and Grim protein, bind to DIAP1, adtivate caspases to induce apoptosis. Knockout of IAP of DIAP1 prevent apoptosis, but doesn't affect phagocytosis--apoptosis not required for Drosophila,while phagocytosis required. Pretaproter act as eat me signal
"
Erythrocytes red blood cells"
no apoptosis, old erythrocytes phagocytosed by "eat me signal" PS and altered Band 3 transporter.
Phagocytosis inhibited by CD 47 on erthroyte surface, acts on macrophage SIRP receptor. Expression of CD47 reduce in old eryghrocytes--promote phagocytosis. Knockdown of PS-receotors stabilin-1 and stabilin-2 block phagocytosis
Neutrophils (white blood cell)
"phagocytes by spleen, liver and bone marrow. Young neutropils are as likely to be phagocytised neutrophils undergeo spontaneous apoptosis when isolated (contrast to Erythrocytes)neutrophil turnover is independent of apoptosis(similar to erythrocytes)""eat me"" signal: CRT (via LRP) when PAI1/CD47 blocked Uniqueness: exposeCRT constitutively--turnover depleted by CD47-blocking antibodies""eat me"" signal: oxidised PS and lyso-PS"
"don't eat me" signal: PAI-1 on neutrophils, PAI-1 can inhibit interaction between CRT and LRP"
Platelets
platetes are phagocytosed by neutrophils cayse downregulating blood clotting via phago
Don't eat me signal: CD47.CD47 expression on platelets, such that platets lacking CD47 are rapidly cleared
T cells
Antigen recognition causes PS exposure on surface of activated T cells. T cell surface PS is recognized by PS receptor Tim-4, mediating phagoptosis. During immunisation with antigen blocking Tim-4 on phagocytes, increase subsequent immune responses to those antigen. expressing Tim-4 in phagocytes decrease # of T cells
"don't eat me" signal: CD47 regulate T cell turnover. Bax/Bak double knockout mice, can't perform apoptosis, have large expansion of T and B cell, so apoptosis contributes to turnover.
Hemophagocytosis entosis and cell cannibalism
found in infectious and inflammatory disorder activatedmacrophage engulf viable blood cells inflammation increase both phagocytic capacity of macrophage and PS exposure of leukocytes, so inflammation induced hemophagocytosis and cytopenia.
Neurons and inflammatory neurodegeneration
viable neurons are phagocytised by microglia in brain. Inflammatory activation caused the microglia to become highly phagocytic and release phagocytic adaptorprotein MFG-E8, which induce reversible PS exposure on neurons and lead to neuronal phagocytosis.blocking exposed PS, MFG-E8 prevent neuronal loss without inhibiting inflammation. Inflammation in brain causemicroglia to eat viable neurons, can be prevented by blocking phagocytic signaling. microglia can spontaneously phagocytise viableneurons, which increase by removal of surface sialic acid residue
Progranulin inhibit phagocytosis of apoptotic and PS-exposed cells in vivo, so neuronal loss in FTD is caused by phagoptosis that progranulin normally suppresses
Cancer
Cancer can be limited by antibody and complement-dependent phagocytosis of cancer cells, or by induction of "eat me" signals by macrophage and T cells. "eat me" signal is CRT: drive phagoptosis one CD47 is neutralised.
cancer cell overexpress "don't eat me" signal CD47 antibodies against CD47 induce phagocytosis of leukaemic cells by macrophage--completely clear leukaemia.
Necrosis is a type of cell death, distinct for apoptosis and autophagy, which has commonly thought to be an uncontrolled form of death. However, recent research has shown that the process and initial start may actually be regulated. Necrosis is typified by distinguishable signs such as generation of reactive oxygen species and ATP depletion. Necrosis is considered a more inflammatory form of cell death that might contribute to antiviral immunity. Additionally, it has been found that inhibition of proteins that are involved in apoptosis or autophagy lead to necrosis.
There are three types of mammalian cell death that can be distinguished by criteria of the morphological kind. While apoptosis is characterized by plasma membrane blebbing, among other criterion, and autophagy by accumulation of autophagic vacuoles, necrosis has been commonly defined negatively in that it is death lacking the aforementioned characteristics. Necrosis is associated with the loss of cells in many pathologies and is linked to local inflammation. This is through immune system alerting factors that are released. Furthermore, necrotic cells are cleared through a macropinocytotic mechanism whereby only parts of the cell are phagocytosed.
Necrosis is the combine of cell changes after localized cellular death through a process called autolysis. Necrosis consists of five distinctive morphological patterns. The five are listed below:
Coagulative necrosis- characterized by the formation of a gelatinous (gel-like) substance in dead tissues.
Liquefactive necrosis- also known as colliquative necrosis and is characterized by the digestion of dead cells to form a viscous liquid mass.
Caseous necrosis- combination of coagulative and liquefavtive necroses that is usually caused by mycobacteria.
Fat Necrosis- specialized necrosis of fat tissue.
Fibrinoid necrosis- special form of necrosis that caused by immune-mediated vascular damage containing antigen and antibodies.
Besides these five distinctive morphological patterns, there are also other clinical classifications of necrosis such as gangrene, gummatous, and haemorrhagic. Often time, spider bites may also lead to necrosis.
Experimental evidence in different species with similar necrosis with early plasma membrane rupture with little signs of apoptosis or autophagy has shown several traits leading to a sequence of events that are specific to necrotic cell death. These include mitochondrial dysfunction through production of reactive oxygen species (ROS) and swelling, depletion of ATP, lack of Ca2+ homeostasis, perinuclear organelle clustering, protease activation such as calpains and cathepsins, lysosomal rupture, and plasma membrane rupture. This is further confirmed by the existence of a similar pathway in plant cells. Although research remains to be done to completely elucidate the molecular and chronological components of this pathway, it is possible that necrotic cell death is part of an organized programmed cascade to self-destruction.
While there are necrotic processes that are poorly defined and cannot be regulated, such as that from harsh external conditions as freeze-thawing and detergents, there are programmed necrotic happenstances in its occurrence. Evidence for this includes developmental necrosis such as that limiting the growth of bones and that of intestinal epithelial cells. Additionally, certain plasma membrane receptors, if triggered by attachment of their physiological ligands, can initiate necrosis, suggesting that there are signal transduction pathways connected to the induction of necrosis specifically. In addition, certain types of genetic and epigenetic factors can increase susceptibility to necrotic death, as seen in mouse brain ischemia. Furthermore, the inhibition of specific enzymes can prevent necrosis, indicating that certain enzymes play a vital role in the necrotic pathway. Also telling is that inhibition of caspases can change the type of cell death from that of apoptosis or autophagy to necrotic cell death.
The idea that necrotic cell death is the default pathway for cell death is supported by observations that inhibition of apoptosis as well as autophagy leads to necrosis in a variety of cell models. In a model using baby mouse kidney epithelial cells, suppression of both mitochondrial membrane permeabilization and transfection with Akt protein kinase changes the type of cell death from apoptosis to autophagy to necrosis. Simultaneous inhibition is required.
It has also been seen that inhibition of caspases induces necrosis, as shown in mice interdigital cells. By adding Z-VAD-fmk, a caspase inhibitor, the cells from the limb anlage undergo a non-apoptotic, non-autophagic cell death. What is notable is that this necrosis undergoes that same spatial and temporal pattern as what apoptosis what produce, and leads to the same formation of normal digits. Here, necrosis is a substitute for a lack of apoptosis. Furthermore, caspase inhibition can lead to sensitization of the cells for necrosis, leading to a reduced dosage of tumor-necrosis factor-α needed for necrosis. This indicates that elements of the apoptotic signaling cascase might inhibit necrosis.
Other signs include the depletion of ATP favoring necrotic cell death (due to reducing optimal activation of caspases), presence of nitric oxide (inhibiting caspases), and inactivation of atg1 gene for autophagy. As such, it would appear that necrosis can occur as a part of signal transduction cascades or upon inhibition of other cell death pathways.
The occurrence of necrosis upon the inhibition of other pathways indicates its potential emergence early on in evolution as primordial. Apoptosis and autophagy may have been added later on. One hypothesis is that a primordial necrotic pathway existed. Addition of apoptosis or autophagy came later and was recruited for optimal dismantling of cells. However, this pathway would appear after the point of no return of cell death has already been reached. Autonomization of that pathway would arrive later, where two mechanisms are available for cell death, either apoptosis or necrosis. Both happen independently and the point of no return is not shared. Exclusivity of one mechanism, such as apoptosis, would come later in certain species, where necrosis is no longer a viable cell death pathway and cell death relies exclusively on apoptosis, such as in C. elegans.
Studies have shown that there may be certain molecules that are specifically required for necrosis. Some potential ones are described below. However, unlike apoptosis where genes necessary for its course have been identified, in necrosis, genes identified are not as specific.
In the L929 mouse fibrosarcoma cell line with TNFα, necrosis is induced through a signal transduction pathway involving the TNF receptor 1 and the recruitment of Fas-associated death domain to that receptor. This is accompanied by a rapid burst in ROS production by the mitochondria. Here, necrosis is blocked by rotenone, an inhibitor of the respiratory chain complex I, and lipophilic antioxidants. Additionally, knocking down expression of RIP1, a kinase, prevents these mitochondrial manifestations. RIP1 knockout cells are refractory to the propagation of necrosis, indicating the role of RIP1 in necrotic signaling.
In certain types of cell death signaling involving activation of caspase-8, RIP1 is cleaved by caspase-8, inactivating any pro-necrotic activity. By inhibiting caspase-8, cell death follows a necrotic-like pattern which is dependent on RIP1 expression. RIP1 has been implicated in other necrotic signaling such as that induced by DNA alkylation. The mechanism for this, however, is not completely elucidated, and more research is needed. However, it appears that RIP1 is a necrosis contributor as an upstream signal.
Since RIP1 activates signaling pathways other than necrosis, it suggests that there are additional mechanisms existing to regulate its pro-necrotic function. RNA interference screens have identified a kinase known as RIP3, which showed that RIP3 over-expression prompts apoptotic and necrotic cell death. This kinase serves as an inducer of programmed necrosis. The assembly of the pro-necrotic RIP1-RIP3 complex is during the TNF-induced programmed necrosis. The complex is mediated through the RHIM (RIP homotypic interaction motif), which is a representation of an emerging protein-protein interaction motif. Evidence favor RIP1 over RIP3 as the upstream kinase activator in necrotic signaling cascade. RIP1/RIP3-dependent programmed necrosis may be important in other inflammatory diseases as well as cancer.
Cyclophilin D (CypD) is a mitochondrial matrix protein which interacts with certain proteins that result in the removal of the transmembrane potential of the inner mitochondria. The strongest stimulus is Ca2+, but ROS condtions or ATP depletion can also favor this permeability. However, knockout of the CypD gene gives resistance to necrotic cell death in many cell types. Furthermore, cyclosporin A, which targets CypD, has been shown to reduce the loss of cells induced by necrotic stimuli such as TNFα.
However, CypD is not the cause of permeabilization mediated by the Bax/Bak genes, suggesting two different mechanisms of MMP, one necrotic and the other apoptotic. Cyclosporin A, strangely though, has been shown to inhibit apoptotic death in some situations, indicating some cross over between the two pathways. As involved in necrosis as CypD may be, it is not involved in all necrosis cases and is not specific to necrosis either.
Calpains, cysteine proteases that are Ca2+-dependent, and lysosomal cathepsins have been shown to be the likely executors of cell death through cleavage of the plasma membrane sodium/calcium exchanger, resulting in the inactivation of homeostasis. In C. elegans cases where there is a deg-3(d) allele, lysosomal proteases appear to be important as lysosomes fuse together and eventually result in necrosis, possible due to calpain activation. It would appear that altered ion homeostasis results in calpain activation and eventually cell death by necrosis.
To therapeutically inhibit necrosis may be desirable. One way to do this would be to block surface receptors that are linked to necrosis. Studies have shown that inhibition of PARP, RIP1, CypD, calpains, and cathepsins lead to necrosis inhibition in in vivo settings. On the other side, we may want to initiate necrosis in cells such as tumors that are resistant to cell death. Switching from necrotic cell death to the non-inflammatory apoptosis may also be a desirable deviation. By activating caspases, necrosis may be attenuated and inflammation decreased.
The most likely desirable outcome is the prevention of cell death completely. Knowing that several cell death pathways exist and are inter-related, inhibition of all of the pathways would prevent cell death. Combined inhibition of caspases, apoptotic proteins and necrotic factors would truly prevent cell death.
Golstein, P., & Kroemer, G. (2007). Cell death by necrosis: towards a molecular definition. Trends in biochemical sciences, 32(1), 37–43. doi:10.1016/j.tibs.2006.11.001
Golstein, P. and Kroemer, G. (2005) Redundant cell death mechanisms as relics and backups. Cell Death Differ. 12 (Suppl. 2), 1490–1496
Xu, K. et al. (2001) Necrotic cell death in C. elegans requires the function of calreticulin and regulators of Ca2+ release from the endoplasmic reticulum. Neuron 31, 957–971
Moquin, David, and Francis Ka-Ming Chan. The Molecular Regulation of Programmed Necrotic Cell Injury. Worcestor: Cell Press, n.d. PDF.
Noise can be defined as random fluctuations of a signal. Noise caused by stochastic fluctuations in genetic circuits (transcription and translation) is now appreciated as a central aspect of cell function and phenotypic behavior. Noise has also been detected in signaling networks, but the origin of this noise and how it shapes cellular outcomes remain poorly understood. The noise in signaling networks results from the intrinsic promiscuity of protein-protein interactions (PPIs), and that this noise has shaped cellular signal transduction. Features promoted by the presence of this molecular signaling noise include multimerization and clustering of signaling components, pleiotropic effects of gross changes in protein concentration, and a probabilistic rather than a linear view of signal propagation.
Genetic noise is caused by the random fluctuations of signals during the processes of transcription and translation in DNA replication. The noise can cause genes to express differently. For example, two genetically identical cells can behave differently due to the influence of noise. This random gene expression may allow the cell to suddenly be able to resist a substance that would originally have killed it if the noise did not cause the cell to change.
Noise in signaling is caused by numerous protein-protein interactions(PPI) owing to its promiscuous nature. Since proteins interact with many other types of proteins in a short period of time, many signals are being produced. The promiscuity of PPIs that create signal noise may help the recovery of cell functions through homologous pathways. This signaling noise may also be important for increasing robustness of signaling between cells by dampening incorrect events. Increasing robustness of signals can be due to multimerization, functional selectivity, and also pleiotropic effects. There has been research that shows that noise plays a role in epigenetic memory by affecting the central switches of cell functions. There is also speculation that changes in levels of signaling noise may be related to human diseases. By understanding how signaling noise affects cell function, researchers and scientists can finally get a better idea on its affect on diseases or also on propagation of epigenetic memory.
Noise creates a threshold for signals to cross before a signal can create an effect. This threshold prevents many irrelevant signals due to the promiscuous nature of PPI to cause an effect. A single signal from a receptor cannot overcome the noise threshold by itself. To overcome this threshold, multiple receptors must come together to create a signal large enough to overcome the signal. The process of which multiple receptors come together is called multimerization.[1]
Multimerization is the clustering of multiple receptors which together create a noise which surpasses the background noise in the cells. This is the first method of overcoming the noise threshold so that signals can be sent appropriately and accordingly. When the receptors sending the same signal join together, the signal is then amplified which allows it to be sent. In contrast, when there is just one or a few receptors, the combined noise of their signals will less likely be greater than the threshold, resulting in no signal being sent.
Functional selectivity is another way to allow signals to be sent through a "proof-reading" method. If the components of the signal are all present and in the exact order in which the signal consists of, then the signal will be sent. If there are some but not all of the components of the specific signal, then that signal will not be sent. This method can be referred to as "proof-reading" because the body makes sure that the signal components are all correct in order for the process to proceed. The body will "proof-read" for any mistakes and correct them to allow signals to proceed.
Pleiotropic effect is when the body produces more of one specific molecule that will lead to the surpassing of the noise threshold which results in a signal being sent. This effect does not necessarily mean that the number of receptors will change (like in multimerization). Instead, the concentration of a molecule is what changes in order to increase the chances of sending a signal. The body can also reduce the chance of a sending a harmful signal by reducing the concentration of a molecule related to the harmful signal. This way the noise level of the harmful signal will not surpass the noise threshold and will not be sent.
The human interactome is a large intricate network of thousands of protein-protein interactions (PPIs). It contrasts the classical idea that protein interactions follow a linear signaling pathway. Within the human interactome, proteins not only interact with one other protein, but they interact with many different proteins. This could also be seen as proteins having multiple protein partners to communicate with. The interaction of several proteins is what makes up the promiscuity of these PPI networks. The human interactome is a huge discovery because it allows researchers to search deeper into its relationship with diseases and genetics.
There are three different ways to see whether interactome networks have been established.
The first is through a compilation of interactions from published work. There are usually some established literature-based work on the physical or biochemical interactions done by other researchers which can reassure that the interaction network does exist. The second way is through computational predictions which are based on the structural information of a protein, sequence and gene-order, and the existence of genes in genomes that transfer interactions between organisms (via orthology mapping). Though this way might be quick, the lack of experimental proof due to indirect 'orthogonal' information makes this a not so strong approach. The last approach is through systematic mapping strategies applied towards large groups of genomes and proteomes. Because of the advance in technology, researchers and scientists are able to put together interactomes in shorter periods of time (than before).
Many pathways have been evolved from metazoans, or animals, that regulate organ as well as organism size. The main pathway that is described here is the Salvador-Warts-Hippo pathway, known more commonly as Hippo. Scientists have done research on the Hippo pathway for the past 8 years and have noticed a complex network of signals and levels of modularity. There is a large amount of the WW module in the core kinase of Hippo, the components of upstream regulation as well as the nuclear proteins involved in the downstream movement. This WW domain can help predict new pathways and their components.
Drosophila melanogaster, a species of fly, was used in the lab to examine their tissues and observe the Hpo pathway.
The Hippo pathway, abbreviated as Hpo pathway, was discovered about a decade ago. From observing Drosophila tissues that had mutations, scientists discovered that there was a set of genes that could be isolated where the mutated tissues could grow to larger sizes. The proteins that encode these genes were included in two kinases, the Hpo and Warts (Wts), as well as a protein involved in scaffolding known as Salvador (Sav). These three proteins interacted with one another and made up the core composition of the Hippo pathway that will be later referred to as Hpo kinase cassette. The pathway also included a nuclear effector called Yorkie, a downstream transcription factor called Scalloped, and upstream regulators such as receptors of membrane spans. The Hpo pathway is mostly found in flies and mammals. Hpo pathways have also been researched in the fields of homeostasis, stem cells and cancer.
A real microscopic view of apoptosis in cells.
Even when it was first discovered up until today, the Hpo pathway has been quite the topic of research. The Hpo pathway has been determined as a tumor suppressor pathway that can restrict the growth of cancer cells and promote apoptosis or cell death of these unwanted cells. Hpo kinase cassette can be regulated by many upstream regulators or proteins. An example of an upstream regulator protein is the complex of membrane span receptors, Ft and Ds. The external areas of these proteins can be exerted onto cells that are next to the proteins and the cell and protein can interact. Once a downstream signal is activated, the protein is stabilized and the cell becomes localized. The Ft protein can be cleaved into two fragments with one fragment homodimerizing and the other fragment heterodimerizing. Ft, however, opposes a myosin protein that activates Yorkie, the nuclear effector and destabilizes the Wts (Warts protein) which is one of the major components of the Hippo pathway. There are other proteins that can act similarly to Ft and Ds. These are known as Kibra, Ex and Merlin (Mer) which make up the KEM complex. Kibra is involved mainly in the WW domain, which is a domain that consists of two tryptophans that can bind peptides that are abundant in proline. On the other hand, Ex and Mer are involved in the FERM domain, which is involved in localizing proteins to the plasma membrane. These proteins, Kibra, Ex and Mer can be localized in the apical junction of flies (Drosophila) and their epithelial cells. The proteins can also interact with one another, as suspected. The KEM complex, however, can use part of the Hpo pathway to activate its proteins towards the apical membrane of the fly. KEM complex proteins can also interact or signal proteins that are involved in the upstream pathway of Hpo such as Ft and Ds.
This is a phosphorylated molecule containing a serine end. This molecule is important in protein phosphorylation.
Ultimately, the Hpo pathway has four core proteins: Hpo, Wts, Sav and Mats and are considered the conserved Hpo kinase cassette. Hpo and Wts are kinases that are attracted to serine and threonine. Their enzymes can be activated by phosphorylation. Also, autophosphorylation can activate Hpo which then phosphorylates Wts, Sav and Mats in their respective order. Sav is a protein that can scaffold or assemble Hpo and Wts together in order to facilitate the process of phosphorylation of Wts by Hpo. This Hpo core kinase cassette is conserved in eukaryotes ranging from yeast to humans.
Hpo pathways depend on their nuclear effectors which are different for flies and humans. For flies, the effector is Yorkie, but for humans the effectors are YAP (Yes kinase-associated protein) and TAZ (transcriptional coactivator with PDZ-binding, also known as WW domain containing transcription regulator (WWTR1)). Yorkie can be phosphorylated by activating Wts in flies; meanwhile YAP can be phosphorylated by LATS 1/2 which are large tumor suppressor kinases. Because Yorkie and YAP are transcriptional co-activators, they can be inhibited by mediated cytoplasmic retention when they are phosphorylated. LATS 1/2 phosphorylation of YAP begins at the serine end of the molecule and recruits a ligase that triggers the degradation of YAP. When Yorkie is not in the cytoplasm, it can activate transcription factors such as Sd, Homothorasx (Hth), and Teashirt (Tsh). In humans, YAP can activate Sd orthologues in the TEA domain and the internal region of a mammalian homolog of avian erythroblastosis virus oncogene receptor.
The structure of Merlin, one of the proteins of the KEM complex.
The Merlin (Mer) protein is one regulator that can shuttle from the cytoplasm to the nucleus and Mer can then exert signals that suppress proliferation of cells. This can have a function in the KEM complex. The Hpo pathway in humans was suspected to possibly be regulated by Merlin just as expected in flies. However, there was no regulation by Merlin in humans and the MST2 signal was never present in the extra cellular signal regulated kinase pathway. MST2, derived from Merlin, might not act as a tumor suppressor but just as a positive regulator of proliferation unlike the Hpo proteins.
Modularity means that homologous structures are being reused by individuals as well as species. A module is not only a structure but can also be a process or pathway that is characterized by internal integration. The WW domain mediates protein interactions and can be used by components of the Hpo pathway.
This domain is quite small, and probably one of the smallest modular protein domains existing. The domain has a pattern of imperfect repitition of 38 amino acids in a spliced isomeric form of YAP called YAP2. These 38 amino acids were added by the process of splicing to YAP, which already had a copy of the semiconserved sequence. This sequence showed that there were two conserved tryptophans that were about 22 amino acids apart. There have been approximately one hundred WW domains identified in humans. This domain can fold as a compact beta sheet and is stable when there are no ligands or cofactors. Some ligands can be present causing instability. These ligands were proline rich. Furthermore, there is an upstream complex that is made of kibra and merlin in humans but known as KEM in flies that can integrate signals that have not been categorized into the Hpo kinase cassette. There are many modular protein domains that can determine the Hpo pathway and its components.
The cJun N-Terminal Kinase (JNKs) is kinases that are associated with Mitogen-Activated protein kinase and responsible for activation of insulin resistance and responses to stress like ultraviolet irradiation, heat irritation, shocks in osmosis process. The cJun NH2 terminal kinase isoforms JNK1 is associated with cause of activating insulin resistance, which eventually results in obesity in physical body. According to the scientists who have analyzed the physical effect of JNK in genes, JNK on insulin can be differed from it on obesity. JNK1 can affect regulation of insulin resistance such as gene expression, cytokine production, and lipid metabolism. [1]
JNK1 is responsible for insulin resistance that can cause lacking insulin-obesity, which results in diabetes. Risk factors of JNK1 signaling activation in body can cause serious obesity, such as sedentary lifestyle and diet, are seen as major threats to human health since early 2000s. [2]
Due to the activation of JNK1, obesity can be occurred in physical body that can grow into diabetes, a chronic disease in which blood contains excess amount of sugar, causing health-threatening symptoms, such as blindness, damage in nerves, pains in skins and organs, and weight loss. The JNK signaling protein groups can be encoded by JNK1, JNK2 ubiquitously and JNK3 limitedly. Out of all three JNKs, chronic JNK1 has been responsible for activating its own signaling that can be a direct cause of insulin resistance. Such somatic relation between JNK1 Signaling and Obesity-Insulin Resistance can contributed to development and improvement of drug industries, giving an insight of JNK1's mechanism in human bodies.
The regulation of JNK1 on insulin resistance can be taken at the level of resistance to HFD-induced obesity. It is concluded that JNK1 can contribute to HDF-induced insulin resistance regardless of obesity effects on human bodies. Therefore, excess of amount of JNK1 activation can cause obesity and diabetes; yet, in some case, JNK1 can be independent of obesity in body. For example, scientists have concluded that feeding mice with HFD (high fat diet) that have deficient amount of JNK1 can still cause obesity with its normal level consumption on cells and can shows increasing reactivity on insulin compared to controlled mice in the experiment.[3]
Adipose tissue, JNK1 controls insulin resistance in cell, especially adipocytes. Adipose tissue takes an essential role in storing fuel of molecules in cell. JNK1 in adipocytes cause knockdown of JNK expression in adipocytes in vitro. [4] IRS phosphorylation can contributed to increasing reactivity of insulin resistance within JNK1 signals. JNK1-deficiency in adipocytes implicated to failure of witnessing increasing expression of Adipokin Interleukin 6 (IL6) in blood when human are fed with HFD. IL6 is responsible for hepatic insulin resistance by increasing adapter protein, SOCS3, which can contributed to degradation in the adipocytes. IL6 can be an important role in meditating insulin signaling in the brain. Increasing the concentration of IL6 in the blood can increase diabetic tissues and causes sensitivity of hepatic insulin in the system. [5]
JNK1 in adipocytes stimulates the IL6 that affects the SOCS3 to activate insulin resistance. Therefore, JNK1 can be independently affect the insulin resistance directly or can affect it dependently via IL6 and SOCS3. The following picture in the left clearly shows that JNK in adipocytes can act either way to cause insulin resistance in human bodies.
Adipose tissue is not the only site for storage of energy; skeletal muscle tissue also participates in the regulation of glucose levels and insulin sensitivity. Muscle can thus also be where JNK1 resides. An experiment where mice with JNK1-deficient muscle and control mice were fed a HFD was conducted to test this hypothesis. Results showed that the mice suffered from similar forms of obesity; although the muscle tissue of variable mice were protected against insulin resistance, adipose tissue was not. The reasoning behind this protection against HFD-induced insulin resistance is undetermined, but may be attributed to a decrease in IRS1 phosphorylation, also seen in the experiment on mice with JNK1-deficient adipocytes.
Mice with JNK1-deficient muscle tissue experience an increase in the concentration of triglycerides in the bloodstream. A decreased concentration of muscle lipoprotein lipase (LPL) also contributes to increased amounts of triglyceride in the blood while sending some triglyceride to non-muscle tissue. Redistribution of triglycerides to other tissues results in disorders such as hepatic steatosis (liver disease) and adipose tissue inflammation, as well as an increase in insulin sensitivity in muscle tissue and a decrease in insulin sensitivity in tissue receiving the triglycerides. This leads to the conclusion that LPL-deficiency may be a contributing factor to the protection against insulin resistance in muscle JNK1-deficient mice. [6]
The liver plays a key role in regulating a wide range of homeostatic processes, including glucose homeostasis. It is therefore a candidate for regulation by JNK1. However, studies show that JNK1 in the liver does not contribute to insulin resistance; in fact, the opposite occurs. Mice with JNK1-deficient liver tissue experience insulin resistance when fed a HFD. [7]
Since the hypothalamus is responsible for regulation of food intake and physical activity, the central nervous system is a strong candidate for regulation by JNK1. An experiment was conducted where mice with JNK1-deficiency in the nervous system were fed a HFD. Results showed that removal of JNK1 from the nervous system helped prevent obesity and increased insulin sensitivity. The hypothalamus reacted to the HFD by decreasing food intake and increasing physical activity. However, JNK1-deficiency in the nervous system led to a change in hormone levels regulated by the hypothalamic-pituitary-thyroid axis. Such changes may be a contributing factor to resistance against HFD-induced obesity. In conclusion, mice with JNK1-deficient nervous systems are protected against obesity and insulin resistance due to responses regulated by the hypothalamus. [8]
To activate JNK1 in Insulin tissues, such as muscle, liver and fat, addition of HFD (High Fat Diet) should be applied in human.[9] Mitogen-activated protein kinase kinases (MMK4 and MKK7) activates JNK1. Four main mechanisms for JNK1 activation to be processed: 1) exposure of cell to fatty acids (lipids); thus results in stress in ER (Endoplasmic Reticulum) of protein response pathway to JNK1 activation, 2) saturated fatty acids acts as ligands in cell structures that activates JNK pathway, 3) Fatty acids also activates the JNK pathway by protein kinase C-mediated activation that is medicated by proteins, and 4) High Fat Diet-induced insulin resistance is implicated with low expression of inflammatory cytokines---causing activation of JNK, including necrosis of tumor.
During the development of insulin resistance on human body, JNK1 in macrophages can influences on the absorption of adipose tissue by macrophages in cells and also can alter the inflammatory cytokines such as tumor necrosis factors. Two main roles of JNK1 in macrophages are following:
1) Transplantation of bone marrows in skeletal tissues can be contributed by JNK deficient hematopoietic system; surprisingly, in this case, the system is independent from HFD-induced insulin resistance. [10] 2) Insulin resistance is independent of containment of JNK1 in cell. For example, both controlled mice with JNK1-deficiency can have insulin resistance in their bodies.[11]
In immune system, the signal for cells leads to activation of different cell specific immune activities. Ligands that binds with receptors on the cell-membrane of immune systems to trigger reactions, signal transductions. Cytokines are secreted by immune cells in response to cellular signaling, and bind to specific membrane receptors, which then signal the cell via second messengers, often tyrosine kinases, to alter cellular activity (gene expression). Interleukins comprise the largest class of cytokines, and are manufactured by one leukocyte to act on other leukocytes as signaling ligands. Cytokines are often produced in cascades.[1] Immunes signaling serves a variety of functions. There are two types of immune systems, including innate immunes system and adaptive immune system.
The innate immune system, known as non-specific immune system and first line of defense, defends the host from infection by other antigen in a non-specific manner. It means that the cells of the innate system recognize and respond to pathogens in a generic way. Innate immune systems provide immediate defense against infection.
Pattern recognition receptors (PRR) are a class of innate immune response-expressed proteins that respond to pathogen-associated molecular patterns (PAMP) and endogenous stress signals termed danger-associated molecular patterns (DAMP). The evolutionarily more recent adaptive immune response employs diverse surface receptors that display decremental binding affinities for epitope stimuli.
An antibody is made up of two heavy chains and two light chains. The unique variable region allows an antibody to recognize its matching antigen.
Unlike the innate immune system, it confers long-lasting or protective immunity to the host. Antigens act as ligands for BCR, while epitope peptide•MHC complexes act as ligands for TCR. Hematopoietic growth factors stimulate cell division in immune and blood cell lines.[3]
The RIG-I-like receptor family (RLRs) is comprised of RIG-I (retinoic acid inducible gene 1), MDA5 (melanoma differentiation associated gene 1), and LGP2 (laboratory of genetics and physiology 2) which functions as an intracellular PRR (pattern recognition receptor) sensor that act as the first line of defense against RNA viruses through the detection of viral replication by direct interaction with dsRNA. These RLRs are in most tissues that initiate immune activation in cell types such as myeloid cells, epitheal cells, and cells in the central nervous system. They are able to control infections by detecting RNA located in the cytoplasm through immunity and inflammation. RLRs usually exist in low levels, but increase when exposed to IFN and viral infections. The RLR family or SF2 subfamily is also related to mammalian Dicer and motor proteins involved in gene silencing.
RIG-I is a 925 residue, 106kDa protein composed of two N-terminal tandem caspase activation and recruitment domains (CARD), a Zn2+-containing regulatory C-terminal domain (CTD), and a central DECH-box RNA helicase. N- and C-terminal RecA-like domains (Hel1 and Hel2 respectively) contain conserved sequence motifs implicated in ATP/nucleic acid binding and ATP hydrolysis, indicating that RIG-I functions as a dsRNA-dependent ATPase. RIG-I and MDA-5 share a conserved helicase core as well as similar signaling pathways and adaptor molecules whereas LGP2 lacks N-terminal CARDs.
RIG-I surrounds viral RNA and encloses it within the central cavity of the protein. Hel1 domain contains seven α helices and seven β strands that face the minor groove of the RNA and binds the RNA backbone of the dsRNA. The insert domain of Hel2 (Hel2i) interacts with the minor groove in the RNA backbone and also plays an important role in RNA recognition through Q511 and 2’OH of G5 residue on RNA bottom strand.
The CTD is most notable for its 5’triphosphate (5’-ppp) electrostatic binding via RNA loop-binding groove. The helicase interacts with the 3’ strand while CTD interacts mainly with the 5’ extremity, which includes 5’ppp. CTD is flexibly linked to the helicase and without strong interactions with the remainder of the protein, it acts as a sensor for 5’ppp dsRNA due to its higher affinity and longer off-rate for 5’ppp dsRNA compared to 5’OH ds-RNA.
Structural Domains of RLR Family and IPS-1 Adaptor Protein
Since the RNA binding occurs via 3 separate protein domains, large conformational changes are induced upon binding to dsRNA. Free RIG-I has an extended, multipart shape that collapses into a compact variant upon binding. Oligomerization of RIG-I is thought to be essential for activation and is triggered by RNA binding and dependent on dsRNA length.
Autoinhibited RIG-I is held in closed configuration by casein kinase II phosphorylation and via interactions with repressor domain in the C-terminus, prevents CARD binding. Engagement of 5’ppp RNA and K63-linked polyubiquitination through TRIM25 and RING finger protein allows for RIG-I activation through helicase/CTD enclosure of RNA and the outward exposure of CARD.
RIG-I is activated by both positive-strand and negative-strand RNA viruses such as Rabdoviridae, Orthomysoviridae, Paramyxoviridae, and Hepacivirus genera. In vitro activation of RIG-I requires either blunt-ended, base-paired region of 18-20 nucleotides with a 5’ppp end or dsRNA of longer length (>200bp). RNA binding to RIG is thought to be mediated by both the CTD and helicase domain. The presence of 5’PPP end on RNA substrates acts as a non-self marker or pathogen-associated molecular patterns (PAMPs) that can be differentiated from autoantigens. The CTD binds blunt-end 5’ppp-dsRNA and induces conformational changes that exposes CARDs and allows polyubiquitination of Lys 172 via E3 ligase, which recruits IPS-1 adaptor and allows for induction of type I IFN production.
Sequence composition of RNA ligands may also contribute to activation of RIG-I-dependent signaling as well. Preferential IFN signaling has been noted in response to polyuridine motifs containing interspersed C nucleotides (poly-U/UC) such as that in hepatitis C viral genome. Also, RNA cleavage products generated by 2’, 5’-linked oligoadenylate-activated RNase L, which produce 3’ monophosphates instead of 5’ppp, can trigger RIG-dependent IFN production. It is thought that PAMP RNA sequence composition in tandem with 5’ppp are vital determinants for RIG-I binding.
RLR signaling program relies on its recruitment of IPS-1 adaptor protein and assembly of IPS-1 signalosome that drives downstream activation of IFN transcriptional responses. IPS-1 associates with TNR-receptor-associated factor 3 (TRAF3) through its C-terminal TRAF domain. TRAF3 recruits and activates two IKK-related kinases, TANK-binding kinase 1 (TBK1) and inducible IκB kinase (IKKε), which further phosphorylate IRF-3 and IRF-7. Phosphorylation of IRF-3 and IRF-7 induces formation of IRF homodimers/heterodimers which translocate to the nucleus and bind IFN-stimulated response elements (ISRE), resulting in expression of type I IFN genes and IFN-inducible genes. Also, FAS-associated death domain-containing protein (FADD) interacts with caspase-8, caspase-10, and IPS-1 which further activates NF-κB downstream and induces proinflammatory cytokines.
Regulation of the inflammatory response is vital to prevent uncontrolled interferon production that may lead to autoimmunity or immune toxicity. RIG-I, an essential antiviral PRR, is regulated through various mechanism such as its C-terminal portion which contains a repressor domain and inhibits RIG-I signaling in steady state. LGP2, which may function as both a positive and negative regulator, encodes a functional repressor domain that can suppress RIG-I signaling through interactions with RIG-I and MDA5.
NLRX1, a member of the Nod-like receptor (NLR) family, acts as a negative regulator of RLR-induced antiviral response by disruption of IPS-1 interaction with RLR signaling. Furthermore, NLRC5, another member of the NLR family, interacts with RIG-I and MDA5 to inhibit IFN production by direct blockage of IKKα and IKKβ phosphorylation and its subsequent NF-κB transcriptional activation.
Posttranslational modifications such as ubiquitination and deubiquitination also control both positive and negative regulation events. TRIM25 mediates K63-linked polyubiquitination at Lys 172 and stabilizes interactions between RIG-I and IPS-1. K63-linked polyubiquitination binding is thought to act as a second ligand for RIG-signaling activation. However, phosphorylation at Ser 8 or Thr 170 residues inhibit the TRIM25-RIG-I interface. RNF125 is another ubquitin ligase that acts as a negative feedback loop in concert with E2 ligase HbcH5c to conjugate K48-linked ubiquitin to RIG-I for proteasomal destruction.
Bowzard, J. B.; Davis, W.; Jeisy, V.; Ranjan, P.; Gangappa, S.; Fujita, T.; Sambhara, S. PAMPer and tRIGer: Ligand-Induced Activation of RIG-1. Trends in Biochem. Sci. 2011, 36, 314-319.
Kowalinksi, E.; Lunardi, T.; McCarthy, A. A.; Louber, J.; Brunel, J.; Grigorov, B.; Gerlier, D.; Cusack, S. Structural Basis for the Activation of Innate Immune Pattern-Recognition Receptor RIG-I by Viral RNA. Cell. 2011, 147, 423-435.
Loo, Y. M.; Gale, M. Immune Signaling by RIG-I-like Receptors. Immunity. 2011, 34, 680-692.
Luo, D.; Ding, S.; Vela, A.; Kohlway, A.; Lindenbach, B.; Pyle, M. A. Structural Insights into RNA Recognition by RIG-I. Cell. 2011, 147, 409-422.
Takeuchi, O.; Akira, S. MDA5/RIG-I and Virus Recognition. Curr. Opin. in Immun. 2008, 20, 17-22.
The nucleotide binding domain and leucine-rich-repeat-containing-proteins or NLR family are a type of cytoplasmic pattern recognition receptors (PRR) responsible for initial proinflammatory and antiviral responses. 22 members of the human NLR protein family have been reported and 2 main subfamilies within the NLR family can be distinguished, NLRC and NLRP. Other members of the NLR family include NLRA or CIITA (class II major histocompatibility complex transactivator), NLRB or NAIP (NLR family apoptosis inhibitory protein), and NLRX.
All NLR proteins contain a conserved NOD motif and up to 2 other characteristic domains. An N-terminal effector binding domain is responsible for signal transduction and activation of inflammatory response. Examples of amino-terminal domains in NLRs are the acidic transactivation domain, caspase activation and recruitment domain (CARD), pyrin domain (PYD), TOLL/interleukin-1 receptor (TIR) or baculoviral inhibitor of apoptosis repeat (BIR) domain. The central NOD or NACHT domain is a conserved, intermediary NTPase which shares similarities with NB-ARC motif of the apoptotic mediator APAF1. A C-terminal leucine-rich repeat (LRR) domain is responsible for ligand sensing and modulation of NLR activity.
PAMPs (pathogen-associated molecular pattern) or DAMPs (danger-associated molecular pattern) detected by the C-terminal LRR motif causes conformational rearrangement of the NLR, which triggers oligomerization. The newly exposed N-terminal effector domain induces the recruitment and activation of CARD and/or PYD-containing effector molecules which enhances spatial proximity and further oligomerization. NLRC proteins such as NOD1 and NOD2 are distinguished by NACHT-LRR-CARD domains whereas NACHT-LRR-PYD domains are characteristic of the NLRP family. PYD domains in particular drive caspase activation and pro-inflammatory cytokine processing in NLRP proteins. Oligomerization of NACHT domains upon ligand sensing is thought to be critical for activation and formation of high molecular weight inflammasome complexes.
Structure of Apaf-1, an apoptosis-related protein with domains similar to NLR family proteins
In the NLRC family, NOD1 and NOD2 act as intracellular microbial sensors that identify and bind peptidoglycan fragments released from bacterial cell walls. NOD1 recognizes N-acetyl glucosamine-N-acetyl muramic acid disaccharide linked to an tripeptide with an N-terminal meso-diaminopimelate (mDAP). This component is characteristic of most Gram-negative bacteria. For NOD2, broad range recognition of muramyl dipeptide (MDP), which is found in both Gram-negative and Gram-positive bacterial peptidoglycan, allows for detection during cell wall synthesis or degradation of bacterial components after lysozymal activity.
In the NLRP family, a wider repertoire of PAMPs and DAMPs such as microbial toxins, cytosolic dsDNA, and uric acid may activate inflammatory signaling. PYD domains in NLRP proteins are shown to associate with apoptosis associated speck-like protein (ASC), which is an adaptor molecule that interacts with CARD of pro-caspase-1 and leads to the formation of the inflammasome. The presence of N-terminal PYRINN-PAAD-DAPIN domain (PYD) and C-terminal CARD on ASC adaptor molecule facilitate signaling with NLR protein members such as homotypic PYD-PYD or CARD-CARD interactions in order to stimulate and/or regulate caspase-1, NF-κB activation, and secretion of IL-1β and IL-18.
NOD1 activation may result in apoptosis through a number of protein-protein interactions. Notably, NOD1 has been shown to bind to pro-caspase-9, leading to caspase-mediated cell death. NOD1, via CARD may interact with the CSN6 component of the COP9 signalosome, which may synergize in the apoptotic pathway. However, NOD1 has also been shown to interact with both receptor-interacting serine-threonine protein kinase 2 (RIP2) and pro-caspase-1 as well for the enhancement of pro-IL-1β processing. NOD2 has been shown to be associated with mitochondrial antiviral signaling protein (MAVS) for the induction of type I interferons. Both NLRC proteins NOD1 and NOD2 interact with RIP2 through CARD-CARD interactions to induce nuclear factor-κB (NF-κB) and mitogen-activated protein kinase (MAPK) signaling. RIP2 interacts with regulatory NF-κB subunit NEMO/IKKγ, triggering IκB phosphorylation and NF-κB activation. NOD2, on the other hand, also interacts with TGF-β-activated kinase 1 and GRIM-19 which allows for the activation of IFN-β and NF-κB, subsequently upregulating production of chemokines and antimicrobial peptides.
The inflammasome is a signal platform that is significant in the NLR family and its main purpose is for the processing and maturation of proinflammatory cytokines, IL-1β and IL-18. It is a multiprotein oligomer comprised of NLRP proteins, ASC adaptor molecule, caspase-1 and in some cases, caspase-5. Recruitment of the inflammasome complex permits binding from ASC molecule to caspase-1 p45 precursor, pro-caspase-1, which is autocatalytically cleaved into p10 and p20 subunits. Caspase-1 is then assembled into an active form consisting of p10/p20 heterodimers. Ultimately, caspase-1 is involved in inflammatory signaling through proteolytic cleavage of pro-IL-1B and pro-IL-18 into biologically active IL-1βp17 and IL-18p18, respectively. Several known inflammasomes exist composed of varying NLR and PYHIN proteins such as NLRP1, NLRP3, NLRC4 and AIM2.
NLRP1 inflammasome consists of ASC adaptor protein, caspase-1 and caspase-5. NLRP2/3 inflammasome is composed of ASC adaptor protein, NLRP2, NLRP3, CARDINAL and caspase-1. NLRP3 inflammasome is the most characterized and studied inflammasome model to date. In particular, 3 models for NLRP3 activation are currently debated. One model suggests that extracellular ATP acts as an agonist for P2X7 receptor which triggers K+ efflux and pannexin-1 mediated membrane pore formation. This is thought to allow the entry of extracellular factors for direct NLRP3 activation as well as NLRP3 detection in K+ efflux and loss of membrane integrity. The second model implies that lysosomal destabilization initiated by the presence of DAMPs may cause rupture and release of lysosome content into the cytosol. Detection of lysosome components such as lysosomal protease cathepsin B may prompt activation as a direct NLRP3 ligand. The third model for NLRP3 activation suggests production of reactive oxygen species or ROS may be caused by NLRP3 agonists and detection of ROS via thioredoxin-interacting protein (TXNIP), a ROS-sensitive NLRP3 ligand may induce activation.
The effects of inflammation and apoptosis must be regulated in the event of NLR stimulation in order to be prevent adverse effects in local tissue and systemic applications. Certain NLRs are complexed with ubiquitin ligase-associated protein SGT1 (suppressor of G2 allele of SKP1) and HSP90 (heat shock protein 90 kDa) which keep the receptors in an inactive but signal receptive state. Also, NLRP12 acts to silence NF-κB and MAPK activation by inhibiting the phosphorylation of interleukin-1 receptor-associated kinase 1 (IRAK-1). Anti-apoptotic proteins Bcl-2 and Bcl-XL have been shown to bind and inhibit NLRP1 through the prevention of ATP binding to NLRP and inhibition of oligomerization via Bcl-XL.
Kanneganti, T. Central Roles of NLRs and Inflammasomes in Viral Infection. Immunology. 2010, 10, 688-698.
Kaparakis, Maria; Philpott, Dana J.; Ferrero, Richard L. Mammalian NLR proteins; discriminating foe from friend. Australasian Society for Immunology Inc. 2007, 85, 495-502.
Proell, M.; Riedl, S. J.; Fritz, J.H.; Rojas, A. M.; Schwarzenbacher, R. The NOD-Like Receptor (NLR) Family: A Tale of Sim. and Diff. PLoS ONE. 2008, 3, 1-11.
Schroder, K. ; Tschopp, J. The Inflammasomes. Cell. 2010, 140, 821-832.
IL23R gene (interleukin 23 receptor) is one of the members of IL gene family. The genes in the IL family give instruction for making interleukins and interleukin receptors. Interleukins are the subset of cytokines, small proteins which participate in the communication between cells. They are involved in immunity, inflammation, and hematopoiesis.
IL23R provides instructions for the production of the protein “interleukin 23 receptor” (IL-23 receptor). This protein is composed of interleukin 12 receptor-beta-1 chain (IL-12RB1) and interleukin 23 alpha subunit p19 ( IL23A ). It anchors in the cell membrane of T cells, natural killer cells, monocytes, and dendritic cells.
Interleukin 23 receptor interacts with a cytokine, interleukin 23, which produces a complex somewhat like a lock-key model. When there is an interaction between IL-23 and its receptor, chemical signals are triggered which regulate inflammation and the immune systems response to extracellular bacteria.
When IL-23 receptor is stimulated, it would lead to gene transcription of genes which encodes the anti-apoptotic proteins and pro-inflammatory cytokines through the activation of STAT nuclear kappa factors.[1][2]
Below is the pathway of the IL23 signaling, which is important in maintaining a population of Th17 cells, a recently discovered T-cell subset. Th17 cells are mainly involved in antimicrobial immunity and are create inflammation and tissue damage in many autoimmune diseases.
1) When interleukin 23 binds to its receptor, there will be conformation change in the cytoplasmic tails of the receptor.
2) The component of IL-23 receptor, interleukin 23 alpha subunit p19, associates with Janus kinase (Jak2) which is protein implicated in signaling by type II cytokine receptor family, a signal transducer and the activator of transcription STAT3. Another component of IL-23 receptor, interleukin 12 receptor-beta-1 chain (IL-12RB1), interacts with tyrosine kinase 2 (Tyk2). The activation of STAT3 induced by IL-23 makes Interleukin-17 (IL-17) and Interleukin 17F promoters to bind to phosphorylated STAT3.
3) STAT3 regulates the expression of a Th17 transcriptional regulator, Retinoic Acid Receptor-Related Orphan Receptor Gamma-T (ROR-gamma). It is important as it is involved the expression of IL-17 and IL-17F.
4)JAK2 activation induced by IL-23 triggers different pathways, including phosphoinositide-3-kinase (PI3K)/RAC-alpha serine/threonine kinase (AKT) and Nuclear factor kappa B (NGkB). These pathways are necessary for the production of IL-17 production.
5) The IL17 promoter binds to V-rel reticuloendotheliosis viral oncogene homolog A, nuclear factor of kappa light polypeptide gene enhancer in B- cells 1 (p105) (NF-kB1 (p50) and in B-cells 3 (p65) (RelA (p65 NF-kB subunit) . This binding regulates the IL17 expression
6) Suppressor of cytokine signaling 3 (SOCS3) inhibits the activity of JAK2. It thus decreases the IL17 and IL17F expression induced by IL23.[3][4]
The pathway above is important in regulating Th17 population. Th17 contributes much to inflammatory disease and they are very in mucosal defense. They differenciate in the presence of nuclear hormone receptor RORγt and other transcription factors. They produce cytokines which stimulate the production of antimicrobial proteins at epithelial cells and hence defending against microbe.[5]
It can be caused by several variations in or near IL23R gene. It is a chronic disorder which affects the digestive system. Symptoms include diarrhea, rectal bleeding, abdominal cramps, and the need of mobbing bowels. It can be caused by a combination of certain genetic variations, including ATG16L1, IL23R, IRGM, and NOD2 in chromosome 5 and chromosome 10 . As these genes give instructions for making proteins involved in immune system, variations can make the intestine not being able to response to bacteria appeared in the intestinal walls. This can lead to Crohn disease.[6][7]
2. Ankylosing spondylitis
It is a joint inflammation which affects the spine the most. Symptoms include fatigue, low back pain and stiffness, arthritis in joints. Eventually the bones and spine will fuse together which is called ankylosis. Similar to Crohn disease, ankylosing spondylitis can be caused by combination of genetic variation. The genes include HLA-B, ERAP1, IL1A, and IL23R. However, the variation of genes which cause ankylosing spondylitis still requires further researches and studies. [8][9]
8. http://ghr.nlm.nih.gov/condition/ankylosing-spondylitis
Apoptosis inducing factor (AIF) is a protein that plays a part in starting a caspase-independent pathway to apoptosis. Apoptosis inducing factor carries this out by causing chromatin condensation and DNA to fragment. Apoptosis inducing factor can also act as a NADH oxidase. Another function of apoptosis inducing factor is to regulate how permeable the mitochondrial membrane is upon apoptosis. Normally apoptosis inducing factor is found on the outer membrane of the mitochondria and is separated from the nucleus. However, in the case when the mitochondrion is damaged, the apoptosis inducing factor moves into the cytosol and to the nucleus.
Apoptosis Inducing Factor is a protein that triggers DNA degradation and chromatin condensation in a cell which induces programmed cell death. Mitochondrial apoptosis inducing factor protein is a caspase-independent death effector that cause nuclei to undergo apoptotic changes. The apoptosis process begins when the mitochondria releases apoptosis inducing factor. The apoptosis inducing factor then leaves through the mitochondrial membrane and enters the cytosol. Eventually it ends up in the cell nucleus where it signals to the cell causing chromosome condensation and DNA fragmentation. This will eventually lead to cell death or apoptosis. The apoptosis inducing factor is different depending on the type of cell, the apoptotic insult, and its ability to bind to DNA. Apoptosis inducing factor also plays a large and important role in the mitochondrial respiratory chain and metabolic reduction-oxidation reactions.[10]
In humans the apoptosis inducing factor is located across 16 exons on the X chromosome. A apoptosis inducing factor called AIF1 is translated in the cytosol and sent to the mitochondrial membrane and to the intermembrane space by the C-terminus of an MLS protein. Apoptosis inducing factor is transported with help from the N-terminal MLS protein to the inner and outer mitochondrial membrane enzymes which allows it to enter into the organelle. Once inside the mitochondria, apoptosis inducing factor folds into the functional configuration with assistance of the co-factor, flavin adenine dinucleotide. Then a protein called Scythe increases the apoptosis inducing factor's lifetime in the cell. So a decrease in amount of Scythe would lead to a quicker fragmentation and degradation of apoptosis inducing factors. The x-linked inhibitor of apoptosis can affect the half-life of apoptosis inducing factor similar to Scythe. The two don't affect the apoptosis inducing factor attached to the inner mitochondrial membrane but they influence the stability of the apoptosis inducing factor once it leaves the mitochondria.[10]
Recombinant apoptosis inducing factors that do not have the last 100 N-terminal amino acids have limited NADP and NADPH oxidase activity. It is determined that the apoptosis inducing factor N-terminus can function in many other interactions with other proteins or control apoptosis inducing factor reduction-oxidation reactions and substrate specificity.
Mutations of apoptosis inducing factor can occur due to deletions. This has caused the creation of the mouse model of complex I deficiency. Complex I deficiency is the cause of over 30% of human mitochondrial related diseases. These apoptosis inducing factor-deficient mouse models are useful in the study for finding cures for complex I deficiencies. The identification of apoptosis inducing factor-interacting proteins in the inner mitochondrial membrane and intermembrane will allow researchers to better identify and understand the mechanisms in the signaling pathway that monitors the function of apoptosis inducing factor in the mitochondria.[10]
Adiposity and adipocyte hyperplasia are promoted by an excessive intake of calories without allowing energy expenditure to rise. This rise in the number of adipocyte is caused by signaling factors that are induced by mesenchymal stem cells conversion, also known as MSC. Mesenchymal stem cells are recruited from adipose tissues of vascular stroma. This provides adipocyte precursors an unlimited supply. Producing preadipocytes are committed by stem cell mediators from the members of Wnt and BMP families. Following this commitment, growth-arrested preadipocytes are exposed to inducers of differentiation. These inducers are cAMP, which are cyclic AMP, glucocorticoid, and IGF1, which is insulin-like growth factor. They can trigger replication of DNA and reenter into the cell cycle of mitotic clonal expansion. This expansion needs a cascade of transcription factor, followed by adipocyte gene expression. Phosphorylation of transcription factor gives rise to the activity of DNA-binding.
Origin: Adipocytes are developed pluripotent stem cells called MSCs. MSCs originated in adipose tissues as well as bone marrow, where development initially causes these cells to develop into preadipocytes. Upon evolving into preadiposites, the pre-specialized cells then go through mitosis several times via mitotic clonal expansion. With this expansion, there is a generation of many preadipocytes which eventually differentiate into fully functional adipocytes. One of the most studied preadipocytes is 3T3-L1, which is reliable in showing the historical transformation of the preadipocyte from a stem cell.
Adipocyte function: The adipocyte has endocrine-like functions such as the secretion of hormones and cytokines that directly influence bodily metabolism functions. These hormones include leptin, which helps the body limit energy storage once adipose tissues are at their maximum capacity, as well as adiponectin, which influences the hypothalamus in terms of digestive and food intake. Because adipocytes directly influence adipose tissue, adipocyte hormones also have an indirect influence over sympathetic nervous system hormones in the central nervous system, controlling hormones that regulate fat production (a cascading pathway).
The stroma of adipose tissues contains a vast culture of MSC stem cells that have the ability to differentiate into functional adipocytes. Among the MSCs that can differentiate into adipocytes are C2H101/2, which also has the capability to differentiate into myocytes and osteocytes.
Inhibitors and Promoters of Differentiation: BMP4 and BMP2 are bone morphogenetic proteins that have been known to help progress the MSC stem cells from developing into adipocytes (or at least preadipocytes). BMP4 allows for stem cells to turn into adipocytes by interfering with the DNA methylation process of stem cells, allowing for commitment of differentiation into adipocytes. On the other hand, Wnt10b inhibits the MSC stem cells from becoming an adipocyte due to the fact that Wnt10b differentiates the stem cell into osteogenesis/myogenesis (although some Wnt proteins have been known to help the stem cell commit into forming into an adipocyte). Another adipocyte formation inhibitor has been known to be Hh ligands, which performs Hedgehog signaling. Hedgehog signaling inhibits the formation of adipocytes by decreasing the amount of fat that can be stored in the cytoplasm, resulting in reduced fat mass and less of a need for new adipocytes.
Induction: Preadipocytes are ready to fully differentiate into adipocytes when they reach their “growth arrest” state, also known as the G1 phase in the normal cell cycle. Inducers and inducing conditions, such as high concentrations of insulin, dexamethasone, and low IGF1 levels, then further differentiate the cell by activating a series of pathways, such as cAMP-signaling pathways, as well as glucocorticoids. With inducers, the “growth arrested” preadipocyte is forced back into the normal cell cycle, and continue to differentiate. By going through the cell cycle, the preadipocyte perofmes mitotic clonal expansion, further differentiating the cell. Upon completing the cell cycle once more, the preadipocyte loses their fibroblasts, and obtain cytoplasmic triglyceride as well as common traits as well as structures for a fully fledged adipocyte.
Adipogenesis: from stem cell to adipocyte. Tang QQ, Lane MD. Annu Rev Biochem. 2012;81:715-36. Epub 2012 Mar 29. Review.
Nitric oxide (NO) plays an important role in the biological system as a vital signaling molecule. In mammalian physiology, this gaseous molecule functions as the primary activator of soluble guanylate cyclase (sGC) in the cyclic guanosine monophosphate (cGMP) pathway. When coupled with enzyme nitric oxide synthase (NOS), synthesis of NO is derived from L-arginina and oxygen (O2). NO binds to the heme cofactor of SGc after diffusing across the cell membrane. From this, sGC can only form a stable structure with NO and carbon monoxide (CO), but not with O2. The binding of sGC with NO results in a substantial increase in cGMP levels in the system. The second messenger then modifies phosphodiesterases (PDEs), gated-ion channels, or cGMP-dependent protein kinases to maintain physiological tasks , such as platelet aggregation, vasodilation, and neurotransmission. In pursuit of therapeutic intervention in diseases concerning the NO/cGMP-sginaling pathway, many studies have been centered on the explication of sGC activation/deactivation. This article condenses the contemporary knowledge of sGC form and function as well as recent works in NO signaling.
Following the discovery of the nitric oxide/cyclic guanosine monophos-phate (NO/cGMP) pathway in the 1980s, cGMP production has stimulated with the clinical administration of organic nitrites, specifically glycerol trinitrate (GTN). These compounds relieve the pain following angina by soothing vascular smooth muscle, leading to vasodilation. During the past years, studies concerned the mechanism of smooth muscle relation by these compounds, which gave rise to the discovery of NO as a signaling molecule. In addition, this also led to the discovery of the enzymes that synthesize NO and cGMP.
It has been shown that both cytosolic and particulate fractions of mammalian tissue exhibit guanylate cyclase activity. Within these particulate fractions are membrane-bound particulate guanylate cyclases that are activated by natriuretic peptides (reviewed in References 3 and 4). In contrast, cytosolic fractions contain soluble guanylate cyclases (sGCs) that are activated by NO. NO-responsive guanylate cyclase activity is also exhibited within the cell membranes of certain tissues, such as skeletal muscle and brain, as well as in platelets (5-7). Most tissues contain Guanylate cyclases, while the protein distribution in these tissues are isoform specific. Because localized groups of the signaling compound can be synthesized within specific types of tissues and in closeness to either soluble or membrane bound cGMP receptors, this provides another reason to regulate cGMP-dependent responses. Therefore, specific tissues can control cGMP levels by expressing unique GC isoforms, which have distinct peptide receptors/ligand activators. In addition, during human and mouse vascular homeostasis (8), a reciprocal communication between particulate guanylate cyclase and sGC has been. It is likely that communication between these two pathways is performed through several processes involving cGMP. In eukaroytic individuals, NO signaling is marked by the initial release of calcium, then the binding of a calcium/calmodulin complex to nitric oxide synthase (NOS), which causes the enzyme to be activated. Then, following NO synthesis, it diffuses into target cells and binds to the heme in sGC. sGC is a histidine-ligated hemoprotein that binds NO and carbon monoxide (CO), but not oxygen (O2). As a result, cGMP synthesis increases several hundredfold.
Over time, development of a cost-effective technique for cGS purification has progressed little, but several techniques have been developed to yield about a microgram of the homogeneous product. Primarily, cGS extraction derived from rat and bovine tissues. In the 1980s, purifies sGC for studies was obtained from rat lung, liver as well as bovine lung. Regardless, it was observed that sGC could be purified without the heme cofactor, depending on the purification method. The use of ammonium sulfate precipitation and solubilizing agents can lead to misfolding protein synthesis. Heme reconstruction of this misfiled protein produces a unique sGC that is biochemically different from the native protein. Currently, the bovine lung sGC method is the most effective and efficient method of isolating heme-bound protein from source tissue. For one kilogram of lung, these is a 1 mg protein yield. In further advancement, sGC production advanced from the progress of heterologous expression systems for recombinant sGC expression. COS-7 cells was used for the first successful heterologous expression system. The establishment of sGC as an obligate heterodimer containing both alpha 1 and beta 1 subunits was achieved despite the low sGC from COS-7 cells. In addition, COS-7 cells were used to examine truncations and mutants of sGC through lysate activity assays. The first procedure to separate pure recombinant protein was the overexpression of ratsGC inside insect cells with the Sf9/baculovirus expression system. sGC expression in insect cells became successful without an affinity tag, but recent techniques require a His tag to accomplish the purification process. Most of the protein is insoluble. From this method, there is a 0.2-0.4 mg yield of pure soluble protein per liter of culture. Up to date, this is now commonly used to obtain purified rat and human of pure protein per liter of culture. Another method using an E. coli expression system was used for heterodimer, Manduca sexta. It produces (0.5-1.0 mg/liter) of only partially pure protein of full length. Higher yield were obtained using the truncation of the C terminus of the alpha one and beta one subunits. Although the resulting heterodimeric proteins cannot cyclize GTP, they can still be purified to homogenitiy.
The structure of heterodimeric sGC contains two homologous subunits, alpha and beta. Most studies focus on the isoform, alpha-one beta-one protein although alpha-two and beta-two subunits have also been discovered. Initially, these proteins existed only in mammals, but also in insects like Drosophila melanogaster and M. sexta, and in fish. In mammals, humans, rats and cows, the localization of the alpha and beta subunit has been studied. By techniques of Western blotting and quantitative polymerase chain reaction analysis, the alpha-two subunit is less available than the alpha-one and beta-one counterparts. It is highly present in the brain, colon, heart, spleen lung, placenta and unteres. Studies have shown that with purified protein, the alpha-two beta-one heterodimer has ligand-binding character that is completely similar the alpha-one beta-one heterodimer, but a spliced variant of the alpha-two subunit combines to form a dimer with the beta-one subunit to make an chemically inactive complex.
Currently, several studies in mice show the importance of various sGC isoforms for physiological function. It seems that the mice lacking the sGC beta-one subunit displayed high blood-pressure, low heart rate and gastrointestinal contractility disorder. In addition, removal of the beta-one subunit within smooth muscle cells causes loss of the protein in these cells like the hypertension in the knockout mice. In general, deletion of the beta-one subunit tends to be viewed as a global sGC knockout because the alpha-one and beta-one are not compatible heterodimers with beta-two.sGC alpha-one and alpha-two knockout mice were also made. Here, both proteins were discovered to be vital for long-term potentiation, and vasodilation, the contraction of blood vessels in smooth muscle tissue. Studies have shown that the alpha-one subunit-deficient mice have had both alpha-subunits to be participation of colon tissue.
Structure and regulation of soluble guanylate cyclase. Derbyshire ER, Marletta MA. Annu Rev Biochem. 2012;81:533-59. Epub 2012 Feb 9. Review.
There are many hormones within the human body which regulate and control bodily functions. Among them, one key member is Secretin. Secretins are hormones which regulate normal secretion into the duodenum and manipulate homeostasis throughout the human body. Duodenums are found in the intestinal gland found contained within the epithelial lining of the small intestine in the human body. Here, S cells, which are responsible for the creation of secretin, can be found. Secretins are found to participate in vital features of osmoregulation in several of the body's functional organs such as the kidney.
A more specific feature for the functionality of secretins is their ability to regulate pH of the contents within the duodenum. This functionality can be carried out because the secretin hormones are able to manipulate specific secretion of gastric acid and control it's pH properties by using various chemical buffers. Typically, the chemical buffers used must be something the human body can have readily and not require additional intake of nutrients. The compound used by this the specific secretin to buffer pH then is bicarbonate. Bicarbonate can be found from the spindle-shaped cells called centroacinars within the pancreas. In addition to being discovered in the pancreas, bicarbonate can also be found in the intercalated ducts.
An interesting fact about secretins are that they were the first hormone to be discovered and identified correctly.
Secretins were first discovered in the early 1900s by two english physiologists, Ernest Starling and William Bayliss. The experimental design which for their project was to target how a nervous system responded and regulate digestion processes. Because of earlier discoveries, they knew that the pancreas was the main component in secreting compounds which aid in digestion. This process can occur as food enters the duodenum through the pyloric sphincter. The two scientists were able to contradict this earlier discovery during this experiment when they found that the nervous system, in fact, did not control digestive functions. Rather, they discovered substances now known as secretin produced in the intestines is the actual factor which controls the digestive system.
The chemical composition of Secretin involves several key components. The structure is first created initially with a precursor which contains the a spacer, the secretin hormone itself, an N-terminal signal peptide, and a C-terminal peptide which contains 72 amino acids. The entire precursor to the hormone is 120 amino acids long in total; however, the secretin portion is actually only between the residues of 28 and 54.
In addition to the precursor peptide, there also contains a mature secretin peptide. This peptide is composed of 27 Amino Acid residues and is arranged in an linear sequence. It is characteristic in having a molecular mass of 3055. The 3-D structure of this mature linear secretin peptide shares its structure to some other common compounds. Similar to that of glucagon, glucose-dependent insulinotropic peptides, and vasoactive intestinal polypeptide, the secretin takes on the shape of a helix. This is most prominent in positions of amino acids five to thirteen. The amino acid breakdown of this helical chemical structure is as follows. Seven of the amino acid residues in mature secretin can also be found in the vasoactive intestinal polypeptides, fourteen can be found in similar placement as those found in glucagon, and ten amino acids residues in secretin can be found in corresponding positions in the glucose-dependent insulinotropic peptides.
In addition to the precursor and mature secretin. Another special property of the secretin hormone is that it has an amidated carboxyl-terminal amino acid, valine.
Therefore, the corresponding sequence for the secretin hormone is:
H2N–His-Ser-Asp-Gly-Thr-Phe-Thr-Ser-Glu-Leu-Ser-Arg-Leu-Arg-Asp-Ser-Ala-Arg-Leu-Gln-Arg-Leu-Leu-Gln-Gly-Leu-Val–CONH2.
T2SS: The Type II Secretion System helps regulate the amount of hydrolytic enzymes as well as expelling toxins from the periplasm outwards of the cell. Bacteria that have been known to carry T2SS was Escherichia coli and Vibrio cholera, bacteria which are known for cholera and diarrheal diseases. The toxins that are associated with those diseases have been correlated to the T2SS protein.
T4PS: The Type IV Pili System is a secretin that helps with the production and dismantling of extracellular fibers found on pathogenic/environmental bacteria. This secretin is made of many types of type IV pilin; T4aP and T4bP. These type IV pili are mainly responsible for motility, protein secretion, and attaching the secretin to cells. Some bacteria that have T4PS are Neisseria gonorrhoeae and pseudomonas aeruginosa.
T3SS: The Type III Secretion System is a pathway that transfers viral proteins to the cytoplasm. Also known as injectisomes, T3SS allows for pathogens to transfer effectors into the host and directly altering normal cell behavior. Pathogenic responses from T3SS could lead to physiological cell responses such as inflammation, phagocytosis, and even cell apoptosis. Bacteria that utilize T3SS is Salmonella, enteropathogenic Escherichia coli, and Shigella flexneri.
Secretin hormones are first created in the S-cells. Specifically, in the cytoplasmic secretory granules portion of this cell. Furthermore, S cells can also be found in the mucous membranes which are linings covered in epithelium, a product which plays a key component in absorption and secretion. In addition, these linings can also be found in the duodenum as well as the jejunum portion of the smaller intestines. However, the S-cells and the secretin count in the small intestines are in smaller portions here.
Depending on differing species, secretin usually produce different responses based on the pH range of the environment of the duodenum. Generally, when the duodenal pH reaches an acidic function of around 2 to 5, the secretin hormone will be activated and released into circulation. Another case of secretin release could also occur within the intestinal lumen. However, both cases are in response to the pH change into the lower spectrum. In addition to pH control, the secretin secretion is can also be maximized when amino acid residue decomposition produces products which are contained in the mucosa of the smaller intestine, usually in the upper region.
Duodenum and Pancreas
Duodenums usually create their acidic environment based on the amount of hydrochloric acid(HCl) contained in their chambers. Masses of semi-digested food, or chyme, is jettisoned by the stomach and into the duodenum. As a result of this, hydrochloric acid is carried through the region of the stomach which connects the stomach and duodenum, called the pylorus sphincter. Once the HCl has reached the duodenum, it will acidify the environment and cause a pH drop, and in turn activate the release of secretin. Once secretin is finally released, it is used to target the glandular organ in the digestive and endocrine system, the pancreas. Once the pancreas receives a certain amount of insulin, the organ can then produce and expel a bodily fluid which is rich in bicarbonate. This solution then flows through the corresponding intestines. The pancreas needs to produce bicarbonate because the individual ions produced by this polyatomic ion can be used to neutralize the acid which is already present. With the bicarbonate present, the body can now utilize this to create a pH favorable environment so that it can carry out actions of the digestive enzymes in the intestinal tracts. A key feature of having bicarbonate is to neutralize the acid and prevent acid burns. The pancreas can also introduce bicarbonate as a response to the secretin by produce fatty acids and various bile salts. Both of these compounds can in turn produce bicarbonates as a product and be put in combination with the initial set of bicarbonate and placed in the small intestines.
A method in which the secretion of secretin can be inhibited is by the addition of a class of drugs used to block the action of histamine in the cells on the stomach region. These are called the H2 receptor antagonists. These compounds function by reducing the gastric acid secretions. Due to the lowered amount of gastric acid in the duodenum, the pH cannot be maintained at a lowered level. Thus, once the pH of the duodenum has increased above 4.5-5, the secretin can no longer be produced and released. And the functions of the corresponding organs can be successfully shut down.
The main function of the secretin hormone is to increase the bicarbonate solution from the bile as well as the pancreatic duct epithelium. The cells which are stationed in the pancreas have receptors which recognize secretin hormones once the secretin reaches its plasma membrane. Once the hormone has attached itself to the receptor, it can actively energize and produce adenylate cyclase activity. The adenylate cyclase activity is used to convert adenosine triphosphate, ATP, to Cyclic adenosine monophosphate or cAMP. In other words, an energy storing molecule to a messenger important in biological processes. It is worth noting that Cyclic AMP is used as a secondary messager to transmit signals throughout the cell and promote the increase in releasing more water carbonate ions. In addition, the production of more Cyclic AMP can be used to promote the growth and structurally stabilize the pancreas.
Picture of Cyclic AMP
Further functions of secretin include counteracting the effects of an increase in blood glucose concentration spikes that is produced by the pancreas. When the pancreas decreases its release of insulin, the blood glucose concentration within the human body begins to spike haphazardly. By releasing secretin into the body, it can postpone and eliminate the effects of such blood concentration spikes by triggering the insulin release.
Another function which secretin takes place in the human body is that it can actively delay and stop the gastrin release from the stomach. Secretin can preform this function because it can reduce the acid secreted from the stomach and inbits the gastrin release from G cells.
This functionality aids in neutralizing the pH of the products of digestion entering the duodenum. It has been shown that enzymes produce digestive functions at an optimal state that is a pH with slight basicity.
In addition, a function that secretin serves in the human body is that it can actively increase the pepsin secretion from cells in the stomach which releases pepsinogen, chymosin, and gastric lipase, or chief cells. This active stimulation of chief cells can help decompose proteins during the process of food digestion. Secretin can achieve this functionality because it stimulates the release of somatostatin release-inhibiting factor and glucagon.
Picture of somatostatin
Finally, the last function of secretin in addition to maintaining the additional acidic chyme passed through the duodenal organs, secretin also has other functions as well. One of these is that secretin ameliorates the effect the peptide hormone cholecystokinin and induce extra secretion of certain enzymes needed in digestion from the pancreas. Another effect produced by secretin in this function is that it can also increase bile produced within the gallbladder, a small organ which aids in fat digestion.
There are a variety of sectors in industry which secretin can be used. One unique function that secretin serves is that it can increase the pancreatic secretion of a human. This is extremely important in the medical field as scientists whom monitor pancreatic functions and testing must require the amplified effect produced the secretin hormone. Several methods can be used to introduce secretin into the body. Two main pathway in which secretin can be introduced to the human body is either through a direct injection into the duodenum or through a normal injection
The result of certain secretin tests have been used to prove and provide insight into abnormalities in the pancreas.
Finally, it has been shown that secretin can provide potential treatment to autism and other brain-based diseases.
Secretins assist in secreting proteins into the extracellular environment of gram-negative bacteria (which are bacteria that have the ability to gain resistance to antibiotics [9]). In bacteria, they make up three different, outer membrane channels. [10]
The T2SS consists of three subassemblies: the outer membrane complex, a filamentous pseudopilus, and the inner membrane platform.These are responsible for secreting toxins from the periplasm (the area between plasma membrane and membrane bordering the cytoplasm [11]).
Secretion occurs in two steps. The proteins are produced with N-terminal signal peptides. This is followed by removal of the signal peptide along with the folding and release of the mature proteins into the periplasm. They may undergo further modifications before they are secreted across the membrane through the T2SS.[13]
The T4PS help move the bacteria across surfaces without the using a flagella by assembling and disassembling the fibers on the surfaces of bacteria. These system consists of two subclasses of pilus: T4aP and T4bP. [10]
The fiber is a three-layered helical structure of alpha-helices surrounded by a beta-sheets. These two inner layers are covered with the C-terminal regions of the surface. The N-terminal amino acid sequence forms the innermost coil of alpha-helices. This hydrophobic packing and the flexibility of α-helices allow pili to bend and to adopt twisted or bundled conformations. The middle layer of β-sheet is continuous from one monomer to the next, and the β-sheet hydrogen bonding which provide much of the stability. It is generally believed that the pilus is assembled from its base, as a pool of pilin is found in the cell membrane. As there is no channel in the center of the fiber, assembly from the tip is excluded.[14]
These are also known as injectisomes. They are used to transport bacterial effectors.[10]
It has a basal body, a needle structure, and a tip. [12]
The tip is secreted into the surrounding environment by the bacteria and comes in contact with host cells. The needle structure forms a pore in the host cell membrane. The effector protein can then pass into the host cell via needle.[12] This would eventual lead to the spread of the bacteria within the host.
Secretins are generally produced in the outer membrane by a lipoprotein called pilotins. One known example is MxiM, a pilotin commonly found in T3SS. MxiM is composed mainly of B-sheets with one alpha-helix, combining together to form a cone-like structure. The hydrophobic portion of MxiM’s B-sheets act as the binding portion for lipids or itself (the N-terminus can bind with the C-terminus, making a circular-linked structure). After formation, the pilotin binds with the outer membrane and secretin is assembled. The way that the pilotin forms with each other (or with other lipids or pilotin) help dictate its structure, which thus resembles the function of the pilotin. Different structured pilotin thus lead to the formation of different secretin specific for different functions.
Another notable example would be the pilotin PilF, which is essential for the production of PilQ, a T4PS secretin commonly found in the bacterium P. aeruginosa. PilF often binds with another pilotin from a similar bacterium N. meningitides; PilW, allowing for the creation of a multimer secretin. With regards to function following form, both pilotins have a concave structure on the surface, which many researchers have argued could be a potential binding site for the production of T4aP necessary to create T4PS secretins.
Upon production of the different secretin systems by the pilotins, the secretion systems bind to the inner membrane of the cell and forms a secretin multimer. These secretin multimers are composed of many secretins that come together in order to form a specific secretin system. Examples of such secretin include GspCEpsC and GspMEspM, which are two fluorescent green proteins that are commonly found in the T2SS system. These two proteins have been known to help with localization of the secretin system inside the membrane as well as the actual production of the entire system as a whole.
Microscopy is a technique that is oftentimes used to study the cell (inside and outside). The two most common types of microscopy are light microscopy and electron microscopy.
The three types of light microscopy are fluorescence, phase-contrast, and confocal.
1) Fluorescence shows the location of molecules in the cell by tagging them with fluorescent dyes or antibodies.
2) Phase-contrast enhances contrast in cells by amplifying variations in density.
3) Confocal uses lasers and special optics to optically section fluorescently-stained cells.
The two types of electron microscopy are scanning and transmission.
1) Scanning shows a 3D image of the surface of a cell or specimen.
2) Transmission is used to section through a cell or specimen.
The difference between light microscopy vs. electron microscopy is that election microscopy uses a beam of electrons instead of light.
The key to excellent microscopy is magnification and resolution (clarity) to be able to see what you're trying to find in a cell.
Scanning Electron Microscopy works by having an electron beam scan over the entire object. This process can be related to a 3D scanner that takes the positioning data of each atom on the object to create a computer generated model that reflects the data. This positioning data is retrieved by the detecting the collision behavior of the particles when an incoming electron causes the ejection, scattering, or excitation of other electrons. Other data such as the velocity and amount of x-ray packets that are produced by electron scattering can also be used to provide more details for the computer model.
In order to provide maximum magnifying capabilities for the microscope, an electromagnetic field is maintained by fluctuating voltage contained in copper coils surrounding critical portions of the beam. This magnetic is used to maintain a very precise and linear beam of electrons to scan the object with. These “scanning” coils are also what allows the beam to direct and adjust itself accordingly to properly scan the specimen.
Electron microscopy is particularly popular amongst scientists visualizing minute specimens, some of which are smaller than the smallest frequency of light physically smaller. It is this inherent problem of light microscopy that prevents normal microscopes from viewing nanoscopic structures, since the “light” used to too big to view anything that small. Given that the de Broglie wavelengths of electrons are capable of being created thousands of times shorter than normal light, electron beams are the preferred wave particles for observing molecular structures.
Atomic force microscopy (AFM) is a type of high-resolution imaging with resolution on the nanometer scale. Interestingly, this is a resolution that is 1000 times more than the expected optical diffraction limit. The original AFM is a derivative of the scanning tunneling microscope that was engineered by Gerd Binnig and Heinrich Rohrer in Zurich, Sweden in the 1980s. Today, the AFM is widely used to investigate on the subcellular, nanometer scale.
The AFM has a cantilever with a sharp tip at the end. This tip is used to scan the surface of interest, which is mounted on a glass surface. The cantilever is brought close to the surface of the sample. Tiny forces act on the principle of springs (Hooke's law) caused by interaction between the tip and the sample through chemical interactions such as van der waals, capillary forces, electrostatic forces, and magnetic forces to name a few. These various forces causes a measurable deflection of the cantilever. As the cantilever tip taps along the surface of the sample, the components of the cantilever is continuously measured in the x, y and z direction to create an image that is computationally constructed based on the amplitude, phase, and other features of the cantilever tip movement. From the tracking of the cantilever movement, the surface of the sample can be analyzed and 3D images can be produced such as amplitude and phase traces.
Atomic Force Microscopy is widely used in the academic lab setting. A research group at the University of California, Davis headed by Dr. Tina Jeoh focuses on using atomic force microscopy to explore the surface chemistry and structure of cellulose, a polysaccharide component of the cell wall in plants. With the investigations of the cellulose structure, the aim is to access the mechanisms by which enzymes such as cellulases act on the cell wall to release soluble, fermentable sugars in order to optimize efficiency of converting the sugars into second generation cellulose-based biofuels. The group uses chemical vapor deposition with methyltrimethoxysilane (MTMS) and centrifugation to adhere purified macro-algae based cellulose to silanized glass. The wet cellulose is imaged under AFM along with cellulase. The time-lapse AFM gives an insight to the mechanism by which cellulase breaks down cellulose into beta glucan rings through imaging.
Transmission Electron Microscopy operates similarly to scanning electron microscopy, but rather than letting an electron beam individually scan the surface of a specimen, the microscope takes electrons and transmits them through an ultra-thin specimen. The resulting electron collisions are then detected by electron detectors to create a model of the imaged specimen. Fundamentally, a Transmission Electron Microscope is similar to the Scanning variant, they only differ in how the image is created. A disadvantage for this system is that it requires the creation of very thin specimens for the electron beam to pass through, which is a very meticulous and laborious process. There is also the risk of damaging thin biological specimens with the beam.
The electron source used to image the specimen with. They come in two types.
Thermoionic:
This type of electron source utilizes thermal energy in a filament to eject electrons from.
Field emission:
Uses an electrical field to eject electrons
The lenses of a scanning electron microscope are the components that concentrate the electrons coming from the gun into a fine linear beam. These lenses use magnetism rather than optical prisms to fine tune the electron beam.
The case for practically the entire electron microscope. A vacuum is required to operate the electron microscope because anything less would cause unintended electron collisions and interference for the electron beam.
Reece, Jane (2011). Biology. Pearson. ISBN978-0-321-55823-7. {{cite book}}: Text "coauthors+ Lisa A. Urry, Michael L. Cain, Steven A. Wasserman, Peter V. Minorsky, Robert B. Jackson" ignored (help)
Endocytosis is a process in which substances enter the cell without passing through the cell membrane, but by forming an intracellular vesicle. This process allows the entry of large polar molecules that are not able to get into the hydrophobic plasma membrane. Also, it plays a important role in regulation of intracellular signaling. The intracellular vesicle is formed by the plasma membrane surrounding the potential food molecule. In essence, the cell engulfs the whole molecule in order to ingest it. A part of the cell is "invaginated" and can forms a vesicle or endosome that contains the molecule ingested. The different type of molecule that is associated with endocytosis is given specific names such as phagocytosis. The opposite of endocytosis is exocytosis which is the expulsion of such molecules.[1]
Endocytosis in animal cells:
Phagocytosis: a cell engulfs a particle by surrounding it with pseudopodia and infusing it into the food vacuole, which contains hydrolytic enzymes. Only occurs in specialized cells such as the amoeba. Known as cell eating and can be used as an immune system defense! The endosomes attributed to phagocytosis is so large that they are usually referred to as a vacuole or phagosome.
Pinocytosis: the cell produces droplets of extracellular fluid into tiny vesicles that also fuse with lysosome containing enzymes, which breaks down the particles. Known as cell drinking. The amount of liquids entering are usually very small. Almost all cells undergo pinocytosis and they do so continuously.
Receptor-mediated endocytosis: membrane embedded proteins with specific receptor site are exposed to the extracellular fluid in order to bind with specific ligands. Then, vesicles are formed containing the ligand molecules. The material is then ingested and liberated from the vesicle.
In the mechanism of clathrin-mediated endocytosis, triskelion structure clathrin perform self-assembly to form a regular lattice. At the mean time, adaptor proteins such as epsin, SNX9 then bind to the membrane receptor protein to form CCV (clathrin-coated vesicle). Disassembly immediately begins after the formation of the vesicle neck, and it is carried out by Hsc70 and its cofactor auxilin. The uncoated CCV can undergo further reaction in the cell.
Since many cells don't have the required cytoplasm sequences to turn into clarthin-coated vesicles, there is a significant clathrin-independent endocytosis. There are several different CIE mechanisms.
1) Caveolar Endocytosis: This is used for the uptake of glycophingolipids and some viruses. It involves the caveolin coat and dynamin.
2) This is used for uptake of toxins from bacteria,GPI anchored proteins, and fluid-phase markers. This mode depends on actin, CDC42, ADP-ribosylation factor but is independent of dynamin.
3) This is used for uptake of integral membrane proteins that do not have the adaptor protein recognition sequences. It is independent of dynamin but is dependent on ARE6 GTPase.
All these forms of Clathrin-independent endocytosis require free cholesterol. Clathrin-independent endocytosis is also used prominently by proteins and lipids that are in sphingolipid-rich lipid raft membranes.
After the uptake of molecules, they are snet to distinct vesicles and are transferred to the early endosome. In the early endosome, there is a mixing of clathrin-independent and clathrin-dependent endocytosis cargo and then finally, recycling happens.
Endocytic membranes require high membrane curvature, but membranes were mostly found to be sheet-like structure. However, in recent study some small G proteins were observed to be capable of membrane curvature generation. In the process that CCP(clathrin-coated pit) is developed to form CCV (clathrin-coated vesicle), an amphipathic helix is introduced into lipid monolayer by epsin. By doing so, the helix remains at the glycerol backbone of the lipid, and thus make the phospholipid moieties bent. Membrane curvature generation can be induced by the splay of the phospholipid. Also, cytoskeleton is important to membrane curvature regulation by promoting dynamin's fission ability.
r== Endocytic Recycling ==
Recycling of old endocytosis material is necessary when endocytic uptake occurs in order to maintain the shape and size of the cell and the plasma membrane. Recycling contributes to many processes such as nutrient intake, movement of cells, cytokinesis, and intracellular signaling. Recycling of materials depends on whether endocytosis was Clathrin dependent or Clathrin independent.
Early Endosome
The early endosome receives and sorts material that comes from CIE and CDE. Since the lumen of the early endosome is acidic, confirmational changes in proteins for ligand release from receptor occur. To enter the fast recycling pathways the following must happen: 1) membrane proteins and lipids must be separated from luminal content 2) generation of membrane tubules. Otherwise, the material can enter endocytic recycling compartment where recycling endosomes emerge.
Rapid Recycling Route
The fast recycling route is used for the transport of TFR and glycosphingolipids. A few studies have shown, RAB4 is important for recycling these materials. However, RAB4 inhibits rapid recycling and increases slow recycling. It seems that small interfering RNA might knock down RAB4 and increase rapid recycling. Recent evidence suggests that RAB5 might be the regulator of rapid recycling by localizing to the plasma membrane and to early endosomes.
Slow Recycling Route
This route is used for transporting cargo proteins from early endosome to ERC and then to the plasma membrane. In many cells, ERC is localized and is near the Golgi complex and the microtubule organizing center. However, in polar cells, the early endosome extends tubules that become the ERC. This model of transformation has been supported by live imaging studies.
ERC traffic
One of the reasons that endocytosis material is moved from early endosome to ERC might be to make sure that the material does not enter degradative compartments. In mammalian cells, sorting nexin 4 connects the early endosome and ERC. If nexin 4 is not present, TFR would be sorted to late endosome where it would be degraded.
ERC to the Plasma Membrane
Early explanations of recycling included ARF6 associated tubular endosomes that extend from ERC and carry material to the plasma membrane. The tubular endosomes would align with the microtubules and recycling would depend on both microtubules and actin. ARF6 would activate Phospholipase D2 (PLD2) which is present on tubular recycling endosome. PLD2 products (phosphatidic acid and diacylglycerol) take part in the recycling. Phosphatidic acid promotes membrane fission and might also cause the release of recycling carriers. Meanwhile, diacylglycerol promotes membrane fission and fusion. Thus, it might promote the fusion of the carriers with the plasma membrane again.
ARF6 also activates PtdIns (4)P5K enzyme which generates PtdIns(4,5)-bisphosphate. PtdIns(4,5)-bisphosphate is present on cell surface and on tubular endosome. PtdIns(4,5)-bisphosphate is also responsible for recruiting proteins to the plasma membrane which result in cell spreading, cell migrating, and wound healing.
Evidence of ARF6 ivolvement in CIE sorting and recycling:
1) Recycling of syndecan 1 and FGFR require PtIns(4,5)P2 (which is produced by ARF6 activation of PtdIns(4)P5K) and synthenin. When mutant synthenin is introduced which cannot bind to PtIns(4,5)P2, recycling of synedcan 1 and FGFR does not occur. This shows that without ARF6, synthenin cannot alone do the job of recycling and impairs cell spreading.
2) A cytoplasmic acidic cluster that is present on an inwardly rectifying potassium channel, Kir 3.4 binds to ARF6 GEF. This leads to activation of ARF6. Furthermore, there is an increase of Kir 3.4 on the plasma membrane suggesting that recycling has taken place and the material has been moved to the plasma membrane again.
ARF6 Inhibition
Extracellular signal regulated kinase (ERK) inhibits AFR6 activation. This inhibition causes a buildup of CIE tubular recycling endosomes which stops recycling. Other signaling molecules such as Ras, Rac, and Src proteins on these tubules might also change AFR6 activity and stop recycling.
Regulators of Recycling
1) ERC can be detected by the presence of RAB11 and other proteins. Since slow recycling occurs at the ERC, manipulating RAB11 could stop recycling and change the place of ERC in the cell.
2) RAB8 seems to be important in the early endosome to ERC transport and may also interact with ARF6. Thus manipulation of this protein could lead to inhibition of ARF6 and thus recycling.
3) ALIX was found to be a RME-1-binding protein that is needed for recylling TFR. Thus, destabilizing of ALIX could regulate recycling.
Grant, Barth D., and Julie G. Donaldson. "Pathways and Mechanisms of Endocytic Recycling." Nature Reviews Molecular Cell Biology. N.p., Sept. 2009. Web. 28 Oct. 2012.
Actin is a protein complex that forms the majority of a cell’s cytoskeleton. The globular portions of action (G-actin) are bound together in the long, thin microfilaments of F-actin. The F-actin microfilaments are double-stranded in a double helix formation. It is the actin-related protein (Arp2/3) complex that facilitates a process called nucleation, which is the formation of a new actin microfilament branch. The daughter filament stems from the parent filament in a y-shape at an expected angle of about 70 degrees. Actin has been observed to have many different functions like muscle contraction, maintaining cell shape, mobility of the cell, division of the cell and also in cell signaling. The function of interest here is the role of actin in the many steps of endocytosis.
Endocytosis is the method by which cells uptake materials by engulfing them with a vesicle made from the cell membrane itself. There are different kinds of endocytosis: clatharin-mediated, alveolae, macropinocytosis, and phagocytosis. The best understood method in mammals is clatharin-mediated endocytosis (CME). Clatharin is a protein component in the cytosol, which helps to form a coated pit on the inner side of the cell membrane. This pit buds into a Clatharin-Coated Vesicle (CCV) that is characteristic of this form of endocytosis. In mammals, it is understood that actin plays a role in this process by helping to pinch off the neck of the vesicle during CME.
Actin Assembly Model for Endocytosis in Mammalian Cells
The figure shows a basic actin assembly model that is particular to helping the endocytosis of mammalian cells, although the model for yeast actin assembly is quite similar. In yeast it has been proven that actin is always required for endocytosis, but this mammalian endocytosis only needs actin under certain conditions: for instance when the thing being ingested by the cell is very large or when the location of endocytosis is already rich with actin microfilaments.
This specific model is for actin assembly during the process of Clathrin-Mediated Endocytosis. The clathrin and other specific adaptors (FCHo1/2 and FBP17/TOCA-1) are the first to start the curvature of the cell membrane in this process. Then the compound Arp2/3 is introduced, whose purpose is the regulation of actin filament nucleation. The actin filaments continue to grow longer through polymerization and this process at the barbed ends of actin helps to force the neck of the vesicle to elongate. The entire infolding of the vesicle gets longer and eventually membrane fission occurs with the help of additional amphipysins and dynamin. Lastly, the proteins cofflin and coronin are employed to dissemble the actin filaments once the job of endocytosis is completed and the vesicle is well within the cell. The entire process of the cell membrane invagination, elongation and fission in the process of endocytosis is mediated by the formation and subsequent pushing actions of these actin filaments.
Advancing our understanding of the Endocytic Process
Improvements in technology and methodology using the light and electron microscope have been significant in advancing the understanding of molecular mechanisms. Fluorescent proteins especially green fluorescent proteinss (GFPs) have allowed the individual components to be studied and follow the movement of endocytic structures. The development of pHluorin as a GFP derivative is sensitive in luminescence with pH levels. It is a useful tool to mark the time of scission of the endocytic vesicle from the plasma membrane.
Great improvements in signal-to-noise rations have been provided by the development of confocal microscopy, both laser scanning and spinning disk, and by implementation of total internal reflection fluorescence microscopy. This has improved the visualization of faint fluorescent signals.
There have been advances in electron microscopy especially in the ability to visualize clathrin assemblies and actin filaments. This has benefited from the use of freeze-etch, platinum replica, and tomographic approaches.
Quantitative measurements of GFP fluorescence can be converted to absolute numbers of molecules by comparison with standards in cells. Computers track these individual entities such as endocytic vesicles and clathrin-rich sites providing data on their fluorescence intensity. Computers have given the ability to analyze hundreds to thousands of these entities.
Most of our knowledge of the role of actin in the process of endocytosis comes from the exploration of this process in yeast and not in mammals. Thusly the process in mammals and the true nature of the actin microfilaments are largely unknown. Some important further issues arise, such as determining exactly where and how the actin creates the force to cause vesicle fission at the neck of the invagination. Another potential field of interest is discovering why actin is not always necessary for the endocytic process in mammals. It would be useful to know how the cell signals its need for actin and the exact signaling process that leads to this actin assembly. Overall, the study of this process must continue to take place more in mammalian cells in order to gain more knowledge of actin’s functioning in the cell.
Akin , O. and Mullins , R. D. Capping Protein Increases the Rate of Actin-Based Motility by Promoting Filament Nucleation by the Arp2/3 Complex. Cell 133, 841–851 (2008).
doi: 10.1016/j.cell.2008.04.011
Even though viruses are not complex and quite simple in terms of their structure and their components, the way they interact with host cells are quite complex. In order to enter the cell, animal viruses utilize a wide variety of cellular processes that deal with numerous cellular proteins. Even though some viruses are able to go into the cytosol through the plasma membrane, the majority of the viruses rely on endocytic uptake, vesicular transport via the cytoplasm, and the transportation to the endosomes and the other intracellular organelles. This process of taking in the viruses are associated with clathrin-mediated endocytosis, macropinocytosis, caveolar/lipid raft-mediated endocytosis, or other mechanisms. There are many ways and endocytic mechanisms that animal viruses utilize to allow a virus to enter a cell.
Initially viruses attach to the cell surface of proteins, carbohydrates, and lipids. The interactions virus receptors usually are specific and have at least 3 valences. In addition, these interactions result in the activation of cellular signaling pathways. Cells react by incorporating the viruses utilizing a few endocytic mechanisms. After the viruses go in the lumen of endosomes or the endoplasmic reticulum, they obtain signals which are in the form of being exposed to low pH, proteolytic cleavage, and the initiation of viral proteins. These cause modifications in the viral proteins, and then they are able to penetrate the vacuolar membrane. After they penetrate the vacuolar membrane, they pass the viral genome, the caspid, or the viral particle that is kept together into the cytosol. Afterwards, the majority of RNA viruses replicate at a variety of positions within the cytosol. In contrast, most DNA viruses continue through their passage towards to the nucleus. A process that consists of steps that dissemble and uncoat operate parallel with the movement of the virus and viral capsids deeper into the cell. This whole process results in a regulated dispersion of the genome and accessory proteins in a replication-competent form.
It is important to study and understand the virus-host cell interactions for a variety of reasons. Number one, the threat and potential damage that pathogens and viruses can cause are growing exponentially due to many factors like the world population growing larger, the immense increase in international trade and travel, and global warming. Therefore, it is extremely important and helpful to obtain as much information that can assist in the fight against viruses that are currently present or viruses that can potentially come up in the future. The study of the interactions between viruses and the cell is a growing field with many undiscovered details that can assist in developing strategies to deal with viruses.
Another important reason to study the interaction between viruses and cells is that viruses can be utilized as instruments in molecular medicine. Viruses have developed to the point where they can enter cells and bring in foreign genes and macromolecules. This is why they are very useful in gene therapy and transporting macromolecules and drugs into cells. In addition, they viruses of have the capacity to perhaps be able to pinpoint cancer cells and exterminate them. Viruses are an ever increasing important instrument that scientists can utilize and manipulate to develop new strategies and concepts of in the field of molecular, structural and cell biology.
Morphological, genetic, and biochemical studies of bacteriophages have shown that the mechanisms of infection are extremely complicated. They also showed that the formation of coliphages T4 and T2 are made like hypodermic syringes with a tail that is capable of contracting. This creates a machinery that has the ability to strike through the two membranes of a gram-negative bacteria and penetrate the cytosol to send in the DNA. It was discovered that attachment to host cell receptors have the function of being a signal that activates the process of injection. Because the host cells of animal cells do not have the outer membrane and cell wall, they do not require complex and elaborate tools and mechanism to enter the cell. In contrast, bacteria has these cell wall and membranes that inhibit the pathway to the plasma membrane. In addition, animal cells give endocytic mechanisms that send viruses that come in advantages that bacteriophages do not possess. Endocytic vesicles carry viruses that come in from the outer edges to the perninuclear area of the host cell, where the conditions for infection are encouraged and the distance is minimized towards the nucleus. In effect, this enables viruses to cross through obstacles freely that deal with cytoplasmic bunching up and the complex construction of microfilaments in the cortex. The ability to travel through the endocytic vesicles is especially crucial for viruses that infect neurons, where the separate axons from the cell body have very long distances. Also the maturation of endosomes that have slowly changing conditions, like lowering of pH or the switching of a redox environment, enables viruses to detect their position within a cell and the passage. This will also allow the endosomes to utilize this information to put a time of penetration and uncoating. When certain proteases like furin and cathepsins are present they give the required proteolytic activation of speciic viruses. In addition, when animals viruses are endocytosed, they are able to avoid leaving evidence of them being exposed on the plasma membrane.It is important to avoid leaving evidence of them being exposed on the plasma membrane because then it would result in a delay in being detected by immunosurveillance. Combined as one, endocytosis contains numerous advantages that cause viruses, like herpes simplex virus 1 and human immunodefiency virus 1 to utilize endocytic pathways for productive entrance rather than entering directly.
The study of viral endocytosis has been combined with reviews on endocytoic pathways. It is important understand both of these aspects in these topics of wide range.
Animal viruses have sizes that range from approximately 30 nm for parvoviruses to 40 nm for poxviruses. The majority of viruses lie in the 60 to 150 nm range. Even though viruses are typically shaped like a sphere, there are other viruses like Filovirus, Paramyxovirus, and influenza virus that can be fibrous and very stretched apart. When they attach to cells, viruses do not become disfigured. On the other hand, the plasma membrane changes shape usually to adjust to the shape of the virus. There are a few instances were invagination is necessary for endocytosis. The outer layer of viruses is usually covered proteins that attach to receptors in the form of capsid proteins formed in an icosahedral grid or as spike glycoprotein that span the entire viral envelope. The separate interactions with the receptors are usually weak, but interaction with many different receptor cause the activity to increase really high which causes the binding to cells almost impossible to reverse. Multivalent binding causes receptors to cluster, which may cause an association with lipid domains and activation of signaling pathways.
At the time when virus particles arrive at the endosomes, the virus particles are usually similar in size to the intralumenal vesicles. When they are too large to fit into the narrow tubular extensions, they are usually localized to the bulbous, vacuolar areas of the endosomes and are organized to the degradative pathway.
Viruses usually in the past have been utilized as model cargo in endocytosis and membrane trafficking studies. By using an electron microscopy one can easily recognize these viruses and they can be identified with fluorescent groups or proteins, which enable single-particle location and monitoring in live cells. By giving a focused source of light, the center of mass of fluorescent viruses can be accurately outlined using a point-spread function. Because infection causes amplification, when viruses have successfully entered the cell, it can be easily measured, even with very small amounts of virus. Also the many studies of virology has created numerous tools, like virus mutants, fluorescent viruses, antibodies, expression systems, and modified host cells.
Lipid raft function to control the signaling, fluidity and receptor functions on the membrane. These are usually rich in cholesterol and sphingolipids. Viruses that use the Caveolar/Raft-dependent pathways form primary endocytic vesicles that are dependent on cholesterol, lipid rafts, and complex signaling pathways. These ways use tyrosine kinases, phosphatases, and glycophingolipids. The process is started by the ligands and the site of penetration for these viruses is the ER. One of the most studied virus that uses this pathway is the polyomavirus. These viruses make use of the multiple receptors available in order to bind better.
Even though some viruses utilize receptors that have defined endocytic receptor functions, like transferrin and low-density lipoprotein receptors, the majority of molecules that viruses attach to deals with different functions like cell to cell recognition, ion transpotation, and attachment to the extracellular matrix. Most of the time the carbohydrate moieties serve as a important function in the binding of viruses.
It is important and helpful to distinguish the difference between attachment factors that simply attach viruses and therefore assist them to focus the viruses on the top area of the cell, and virus receptors that function as a trigger to modify the viruses, promote cellular signaling, or activate penetration. One objective that a lot of receptors have in common is that they encourage endocytosis and they assist the virus into the cell. A lot of the time, when the viruses enter, they begin with binding to attachment factors. Afterwards they interact with other receptors. In reality, it is hard to distinguish the difference between attachment factors and receptors because both of them play a role in the effectiveness of the infection.
The attachment factor that is most seen is called glycosaminoglycan chains in proteogylcans. Attaching to these negatively charged polysaccharides is typically electrostatic and somewhat not specific. There has been identification that in some instances viruses evolve to utilise GAGs when they adjust to the development in tissue culture. Another common category of carbohydrates consist of sialic acids, to which many viruses attach to. Similar to the situation with influenza and polyomaviruses, the majority of the time, this attachment is very specific and deals with lectin domains or lectin sites.
Because binding is very specific, this is a significant cause in determining tropism and species specificity and therefore one can tell the nature of viral diseases. One other dimension of receptor specificity is that it can identify the choice of endocytic pathway and intracellular routing that the viruses there are coming in will travel through. In this specific instance, parvoviruses that bind to the transferrin receptor utilize clathrin-mediated uptake passage. They are also capable to recycle to cell surface with their receptor. In contrast, not as significantly present groups like rhinoviruses that bind to the LDL receptor break apart from the receptor in early endosomes and taken to the late endosomes.
There are a few receptors that have the job of promoting modifications the virus that allows attachment of the virus to a coreceptor, promotion of endocytic uptake, or transformation to configuration that is membrane fusion-active. The most suitable defined case is HIV-1. This is because in this case two receptors are necessary to promote conformational modifications to promote the fusion. Adenoviruses 2 and 5 contain two receptors that promote conformational modifications and encourage endocytosis. For the avian leukosis virus, the signals necessary to entrance are receptor binding in conjunction with low levels of pH.
How viruses hijack cell regulation
SLiMs
Virus' life cycle is shaped by the interactions they have with cellular proteins. They can take over and use proteins for their own interest for their mechanisms. However, viruses are compact spatial constrained with tiny genomes, yet they can command so many pathways and processes. One of the reason they are able to do so is by extensive mimicry of host protein short linear motifs (SliMs).
The Human papilloma virus E7 oncoprotein mimic of the LxCxE motif (red) bound to the host Retinoblastoma protein (dark grey)(PDB 1gux.
When viruses enter the cell, they have their own DNA and RNA polymerases, helicases and proteases, which enable replication, expression, and maturation. But, the virus still needs the cell to help with viral replication. Compact non globular protein interaction interfaces known as SliMs are present in eukaryotic proteomes. They have a wide variety of specific functions, such as the ability to encode a functional interaction interface in a short sequence, participation in a region of proteins, and capacity to function independently, and convergently evolve. Their functions include targeting proteins to specific destinations, acting as recognition sites for proteolytic cleavage, and provide specific modifications., signaling, recognition, rewiring pathways, molecular switching.
There are a diverse set of host pathways targeted and their functionality. Some of them include the host target 14-3-3 by viral protein Rep68 with Virus AAV with the Motif RsxSxP. Another example is host target ALIX by viral protein Gag for HIV with motif LYPxxxL. Thus, there are many different mechanisms by how viruses use the motifs.
One of the mechanism is cellular transport. Directing proteins to destinations require large complexes or cytoskeletal structure of micro tubules. Consequently, viral proteins use the vast cellular infrastructure by copying the motifs. For example, signal mimicry is the driving force of the essential regulatory protein Rev of HIV.
Another mechanism is signal transduction. SliMs have low binding affinities and fast off-rates, which promotes fast reaction in response to variation stimuli. Thus, viruses copies the signaling motifs and deregulate their pathways.
A 3rd mechanism is controlling cellular protein levels. Viral proteins play roles as scaffolds, by redirecting substrate specificity to unconventional host protein targets. For example, in ubiquity ligage recruitment, the virus hijacks the cell resulting in polyubiquitylation of the host, followed by proteasonal degradation. By doing so, the cellular protein is altered, which enables the viral proteins to survive and replicate their genome. Destruction motifs can also be used to inhibit the destruction of the proteins as well, which can prolong the lives of pro viral proteins.
A 4th mechanism is transcriptional regulation, which is a very tightly controlled cellular process. Even so, even its regulation can be degraded by motifs mimicry.
A 5th mechanism is to modify the host proteome by deregulation of endogenous modification processes. Viral proteins can act as scaffolds between host enzymes and viral targets. A single modification can ultimately result in modifying an entire genome.
A 6th mechanism is their own modification. Viral proteins can also direct their own modification by mimicking docking and modification sites of host enzymes.
Viruses must use motifs because the regular function of the cell is largely controlled by SliMs. They are evolution's results to rewire cellular regulatory pathways, which makes them equally vulnerable to the faster evolving viral proteins, allowing the viral proteins to deregulate the pathways.
SliMs length, which are 3-10 residues long, allows it to convergently evolve. Usually, only one or two mutations are necessary for the creation of a new motif. RNA viruses usually go through 10^-5 – 10^-3 mutations in each position in a generation, while DNA viruses usually go through 10^-8 – 10^-5. Convergent evolution of these viral motifs can be examined through viral strains and isolations. Phenotype variations of the viral proteins are due to the adaption of motif specificity.
The size of viral genomes are largely restricted, yet they still interact extensively with the host cell. Once the viral protein acquires the interaction with the protein interface, it alters a pre-folded region, but the host cannot operate under multiple binding sites that are close to each other, so they do not have the ability to interact with a number of these distinct partners because they are spatially restricted. Viral proteomes usually have folded structures. Generally, a globular interface is not needed for the viral to take over a process, but nevertheless, multiple contrasting mechanisms are available to manipulate a specific pathway or protein.
Eukaryote regulatory systems are robust, due to the cellular networks cooperation. Viruses can also use the mechanisms to also be robust. The small sizes of motifs also enable viral proteins to be robust against the mutations and adaptions. Mimicry of SliMs are also robust.
Mitotic spindles are filaments of microtubule that move the chromosomes towards opposite poles during metaphase of mitosis. Mitotic spindles compose of actin and myosin filaments. Cables of actin stretch from pole to pole of the mitotic spindle and myosin control spindle length and shape. Mitotic spindles are attached to a centrosome, the site of microtubule organization and anchor of the spindles. Metaphase plate forms when the chromosomes are aligned in the center of the cell with the mitotic spindles attached to the kinetochores, site of microtubule-chromosome attachment, of chromosomes. Mitotic spindles play a key role in acting as the "fishing line" to move the chromosomes to opposite poles during cell division. In addition, centrosomes are the "reel" that draws the spindle fibers closer to the poles. Disruption of the mitotic spindle formation or arrangement can have huge consequences such as leaving one cell with too many or too little chromosomes.
Mitotic spindles in the process of controlling the chromosomes during mitosis
The mitotic spindle is known as a director that moves chromosomes during mitosis. It is currently unknown how the microtubule fibers move the chromosomes, although it is believed that motor proteins act like carriers moving the chromosomes in different directions. Others tend to believe that the fibers grow and shrink and in doing so move the chromosomes back and forth. Issues arise when the spindle makes mistakes. Sometimes the meiotic spindle doesn't separate correctly and therefore an abnormal amount of chromosomes are present which could cause Down syndrome.[1]
Machalek M Alisa. http://publications.nigms.nih.gov/insidethecell/insidethecell.pdf "Inside the Cell" The National Institute of General Medical Sciences (2010): 36–37.
Pili--plural for pilus--are hairlike appendages found on the surface of many bacteria. Pili are fragile and need to be constantly replaced.
The pili (plural) or pilus (singular)is a biological molecule that is on the surface of many bacteria. It is sometimes described as a hair structure. The term pilus comes from the latin word thread or fiber. All of the pili are made up of oligomeric proteins. The pili is used for different purposes in the cell. It is attached on the surface of cells in order to transfer DNA from cell to cell, attach to surfaces and for general cell-cell adhesion. There are two different kinds of Pili. Short and long. The short is very plentiful around the cell and help the cells colonize environmental surfaces and stick to other cells. This also helps the cells resist potential flushing! The long pili have to do with sex or transfering DNA. This is called the conjugation pili because it helps with conjugation or sex. Conjugation is how a cell transfers DNA to another by contact with the cell. Pili also have implications with bacterial infection. The pili help the bacteria not get flushed by adhesion, and therefore stick to cells and infect. This causes illness due to pili helping bacteria bind to cells. [1]
During the process of bacterial conjugation, conjugative pili allow the transfer of DNA between bacteria. They are sometimes called "sex pili", in reference to sexual reproduction, because they allow for the exchange of genes via the formation of "mating pairs".
Type IV pili, generate motile forces. The external ends of the pili adhere to a solid substrate--either the surface to which the bacteria are attached or to other bacteria--and when the pilus contracts, it pulls the bacteria forward, like a grappling hook. Movement produced by type IV pili is typically jerky, but can be a gliding motion as well.
Attachment of bacteria to host surfaces is required for colonization during infection or to initiate formation of a biofilm. A fimbria is a short pilus that is used to attach the bacterium to a surface. Fimbriae are either located at the poles of a cell, or are evenly spread over its entire surface. Mutant bacteria that lack fimbriae cannot adhere to their usual target surfaces and, thus, cannot cause diseases.
Type IV pili is a type of pili that uses twitching motility as a means of bacterial movement rather than the more common swimming motility. These pili are generally located at the poles of a bacterial cell. The usage of type IV pili as a means of transportation is predominantly seen in bacterial colonies in which twitching motility is present. Isolated cells that are not located to any inert surfaces have not been observed to use type IV pili. Movement reversals often times are related to alternating usage of type IV pili at opposite poles of the bacteria cell. This is better known as twitching motility.
Movement resulting from type IV pili is often observed to be sporadic and erratic rather than the smoother swimming motility. The principle force behind twitching motility results from pili retraction. Pili retraction on a cell are independent from each other. Movement via type IV pili is described as being similar to a “grappling hook” because type IV pili are too flexible to be used to push and propel a cell forward. Twitching motility is predominantly observed in a colonial setting. Colonies of cells use type IV pili and the proximity of neighboring cells as a means to move as a whole.
Type IV pili bind to inert surfaces through ambiguous and unclear adhesion at the tip of the structure. The adhesion resulting from the contact of type IV pili and a surface are not significant, weak, and only occur at the tips of the pili. On the other hand, type IV pili bind to mammalian cells and other cell types through specific receptors. These receptors bind with the tip of the pili. However, there are many structural variations of type IV pili, which results in different binding specificities. Essentially, binding varies based on the structural variation of the pili itself as well as the receptors of that which it is attempting to bind to. Pili retraction can be attributed to a protein that is specific to type IV pili known as PilT. It is believed that when pili retract, the pili are partially disassembled into smaller subunits. This action, the disassembly of the pili, is controlled by PilT. Likewise, PilT is responsible for pilin degredation. Type IV pili can bind to either surfaces or other cells. Specifically, type IV pili form a sort of “grappling hook” for the bacteria cell, in which the cell can pull itself around using the curved pili. As such, a proximity to other objects is an important aspect of using type IV pili. Studies of P. aeruginosa have indicated that the retraction of type IV pili seems to be partially reflexive. Specifically, exposure to an attachable surface has resulted in the retraction of the type IV pili, even if the pili was not directly attached to the cell.
Type IV pili are 5-7 nanometers in width and multiple micrometers in length. Type IV pili are predominantly composed of a small subunit known as pilin. Pilin is a protein that is 145-160 amino acids in length, depending on the species. The majority of the pilin is hydrophilic and experience structural variation. Structural variation of type IV pili is thought to occur as a result of changes in the environment, changes in the behavior and the needs of the host cell, changes in selection pressure from bacteriophages, and evolutionary drift. Through analysis of three-dimensional models of the crystal structure of different bacteria, pilin is shown to be extremely assymetrical. Pilin contain an alpha helical spine, which contain a sugar portion, an anti-parallel beta sheet, and a c-terminal beta sheet. This structure is stabilized through the existence of disulfide bridges and the attractive close-range forces of nearby residues. Another key characteristic of type IV pili is the fact that the pilin of one bacteria is often times interchangeable with the pilin of another bacteria. Though variation occurs in type IV pili, all type IV pili have extremely similar quarternary structures and fairly similar tertiary structures.
Flagella (singular=flagellum), are extensions of the cell commonly found in many Bacteria and Archaea, and even in some Eukaryotic cells. Cells that have flagella often use their flagella as their main method of motility. Most bacterial cells use their flagella to travel randomly, a process called random walk, unless there is a chemical signal present, in which the cell would travel to the attractant (or away from a repellant) through chemotaxis.
There are three major types of flagella found among cells
Peritrichous flagella are a series of flagella that appear around the cell in all directions. Some examples of cells that have peritrichous cells are E.coli cells and salmonella cells.
Lophotrichious flagella are a group of flagella that are bundled together on one side of the cell.
Monotrichous flagella have only one flagellum attached to one side of the cell
Bacteria/Archaea cells: The primary function of flagella is to allow for cell motility. The flagella are comprised of protein monomers called flagellin. The flagella are able to perform this by spinning in an either clockwise or counterclockwise direction. When the flagella (or flagellum, for monotrichous cells) are spinning in the counterclockwise direction, the flagella is driving the cell forward towards the attractant or away from the deterring signal. When the flagella is rotating clockwise, the entire cell stops the forward movement in order to change direction. The flagella is able to rotate in either direction by generating torque in the basal body. Within the basal body is a specialized protein called the mot protein, which generates torque to propel the circular flagellar motion. The basal body also acts as an anchoring system for the flagella, consisting of a set of rings known as the L, P, MS, and C rings. The flagella is able to switch between the clockwise and counterclockwise directions through Fli proteins, which senses where the attractant/deterrent is, and switches the direction of rotation for the flagella in response to the chemical signal (therefore performing chemotaxis).
Eukaryotic cells: Eukaryotic cell flagella differ from bacterial cells in terms of how the flagella move in order to propel the cell forward. Because eukaryotic cells are much bigger than bacterial cells, instead of a clockwise/counterclockwise direction, most eukaryotic cell flagella move in a “whip-like” motion which is fueled by ATP hydrolysis in the cell. Eukaryotic cells with flagella also have many more microtubules in the membrane to help aid in support during cell chemotaxis.
Aging is when an organism, such as a cell, has accumulated an excess amount of damage over its lifetime. The resulting damage ends up having an effect on the overall survival and status of the organism. As an organism ages, “degradation of their outputs leads to functional decline and death as a result of aging” (1). This degradation of outputs is related to the metabolic history of the cell which affects the cell’s function.
The concept of aging has led to the accepted idea that aging is due to the accumulation of damage an organism as acquired over its lifetime resulting in the inability to protect, maintain, and repair itself. Experiments are being conducted that are trying to determine what kind of damage, related to aging, contributes to the loss of function for an organism. This is difficult as there are many factors dealing with damage that can vary across models and individuals such as amount of damage, type of tissue, age, and simply the kind of organism in question.
To begin, exactly what does an organism have to do in order to survive? First, the organism must be able to find food, shelter, and to fight off infections or predators. The organism must be able to avoid death. Even if these factors are eliminated, death and loss of function still occur with age. Nonetheless, no genes have evolved to cause death. An aging organism is said to reduce the genetic contribution of an individual for the next generation. In other words, it is disadvantageous.
In Murphy’s paper, “Control Theory of Aging,” he states that “the genome and how it is expressed constrain mortality and life span.” However, there is still a problem with this idea for life span varies across all organisms, even if they are genetically similar.
It is concluded that it is indeed a combination of genetic determination, variation in the environment, and other events that occur in the organism’s life that contribute to death at the age to which the aforementioned factors can affect. As a result of a lifetime of low-dose exposure to external factors such as ultraviolet radiation and gamma-ray irradiation, the body's ability to carry out homeostatic mechanisms begins to fail and aging becomes apparent. It also has been proposed that aging is a reflection of cellular senescence, an irreversible halt in the cell's ability to self-replicate and grow.
An obstacle involves mutations and environmental interventions that hinder a number of functions which proves difficult to see what the cause for aging is. As mentioned before, there can be a variety of reasons why an organism may have died. It is important to see that one must look across all the possible biochemical and physiological entities and to observe each one independently. This will help narrow down the cause responsible.
Hierarchical Framework for Considering Organismal Aging
The top level of hierarchy is the organism’s functions and a set of physiological systems that are involved in the interaction with one another and the surrounding environment. Each system interacts with each other and also the environment in some manner by inputs and outputs.
Hierarchy of physiological systems.
In the figure above, for simplicity, interacting physiological systems are shown as two separate systems. They are affected by inputs from the environment and from other systems. For example, the mortality of an organism increases as the various outputs of its systems diminish over time due to the aging of the organism. Damage to the organism can also cause dysfunction which can result in inappropriate system outputs.
In general, systems within an organism usually decline with aging. However, it is clear that mortality in different species is not the same.
One theory involves natural selection that may influence an organism’s process of aging. The surviving generations of organisms can display characteristics that can be conserved by evolution. This hints at the possibility of a similar aging process in different organisms from flies, mice, yeast, and worms. Nonetheless, the biological age is not reliable as this also varies across species.
Other aspects of aging can be organ specific and manifest in the appearance of skin aging. For an independent system, it is difficult to know for certain whether it is the effect of defective inputs or intrinsic damage to that system that result in the cell’s dysfunction.
All the aforementioned provides reasons why it is difficult for scientists to pinpoint whether if it is the decline of all physiological systems or just one that results in aging and eventual death of the organism.
2. Changes in Cell Number, Function, And Phenotype
Cells can undergo many changes but the changes that affect the functional outputs of physiological systems will influence aging. Dysfunction of a system is due to the decrease in number of cells and their outputs. According to Murphy, “the changes to cells are caused by their metabolic history and are due to nonspecific damage and to changes in signaling pathways and gene expression. These in turn lead to effects on cell function and on cell number.”
Tissues undergo many changes as the organism ages. The aging causes a change in the cell number. The decline in cell number results in a disruption of mechanisms that maintain the cell.
In some mitotic tissues can replenish lost cells from other differentiated cells. But as the organism ages, mammalian stem cells are less effective at replenishing lost cells.
In order to determine whether a physiological system is impaired by the decline in cell number, it is crucial to record the cell loss or gain that occurs before and after the impairing is seen in the system.
An example of a decline in cell function includes the synaptic transmission in neurons and contractions of musculoskeletal motor units. In mammals, studies have shown that the increase in age results in a decline in motor and neurological function due to decreased numbers of synaptic connections and conductions.
Longevity linked to animals under dietary restrictions becomes the organism's response to food availability and their reaction to nutritional changes in the environment. These changes are measured with its energy at its cellular level when it's activated. Changes in feeding patterns and foraging behavior are also indicators of its adaptive phases. These nutrient-sensing pathways also gives the organism time to detect and respond to those changes in its resource availability. However, because this kind of longevity depends on daf-16, which means the nutrient sensing of these neurons aren't necessary.
We now look at the individual cell where cell dysfunction and death are attributes to cell’s metabolic history. These attributes will affect system function and thus morality of the organism.
The initial state of a cell is due to its genome which results in developmental history, physical niche occupied within the organism, and epigenetic factors affecting genomic expression.
A cell’s metabolic history can result in changes in DNA sequence which will in turn affect gene expression. This also influences the cell’s proliferation, dysfunction, and death over its lifetime.
All these topics are related to each other and to the process of aging.
Dysfunction of a system occurs due to the changes in cell number and their function. What causes the changes in cell number and function is caused by the cell’s metabolic history that involves mainly nonspecific damage and changes in gene expression. The result from these changes in the metabolic history results in changes in cell number and function.
The important and contributing factors of a cell’s metabolic history which influences aging are nonspecific damage and changes in gene expression and signaling pathways.
Different types of nonspecific damage done to the organism contribute to the loss of cell function and dysfunction. Some damages include oxidative damage, radiation, or chemical reactions. The nonspecific damage end up damaging and hindering molecules as time passes on.
The cell tries its best to survive and to protect itself against the damage. The cell initiates damage repair in cases where it can fix a misfolded protein but damages such as a fixation of a DNA mutation cannot be resolved. Such nonspecific damages that are also irreversible will greatly impair the cell and its function. The question arises whether the damage done to the cell affects outputs of the system enough for aging.
In regards to nonspecific damage, it can affect gene expression and cell signaling of the cell. What the gene expresses that was affected by nonspecific damage will also affect its outputs of the system that affects mortality during normal aging.
There is evidence of there being a great amount of changes in gene expression that may occur during normal aging but their contribution in the aging process will vary across systems and overall the organism.
Metabolic Control Analysis (MCA) – an experimental approach developed to understand how control is distributed within metabolic pathways and networks
In order to determine what is involved with the process of aging, there must be a set limit on systems being analyzed and the measurable variables being observed. Once these factors are defined and limited, the physiological system in question can be tested and manipulated so a relationship between said variable and the step it is controlling can be seen. The extent of the variable’s control will also be determined during this experiment of MCA.
With the use of MCA, there has been powerful mathematical formalism developed in order to see the entire system and the following: extent of control to be determined quantitatively, uncovering of all controlling sits, and influence of controlling steps. Though it has been shown that extreme and large changes in biochemical pathways and mechanisms can increase or decrease life span, there is not enough evidence that shows they influence normal aging.
Comparing alongside metabolic pathways and aging, MCA can tell one the important information of steps in controlling of a particular process in a system.
The process of MCA and it being applied on a system needs to be conducted a certain way in order to obtain results. Since the topic is the aging of organisms, this requires mortality readings that relate to organismal aging. As mentioned in the previous section, small changes to factors of a system can be made and by utilizing mathematics, quantifiable results can be obtained.
The read out of the mortality needs to be measured in response to the small changes made in the system. Recall that those changes in a factor are due to the belief that it may control aging. In order for a factor to affect aging, it must alter the corresponding morality rate and contribution to aging.
The MCA experiments are conducted with populations of a diverse group of animals ranging from flies to mice. The mortality readout can then be measured and compared to see if there is a similarity across the species.
In the graph shown above, the morality readout is shown by plotting it against the variable in question for aging that has been both increased and decreased. The shaded dark blue area indicates how the variable alters in normal aging. The graph also displays different situations for normal aging. The effect of the variable can be decreased or increased which affects mortality as well. This is an example of a graph that can be plotted after an MCA experiment to showcase one’s findings and results. The main idea is that if the right variable is chosen that does affect normal gaining, the curve of mortality will have a slope that is measurable.
Chromatin is first formed in an organism during embryogenesis. This genetic material is susceptible to modification via DNA methylation and different types of histone modification; these changes do not affect the nucleotide sequence of DNA. The accumulation of changes over an organism's lifetime results in stochastic, random, structure. In an experiment with Danish twins, it was observed that variations in relatives is a result of genetic shift from chromatin changes. This supports the idea that One theory cites an experiment with Danish twins and proposes that aging occurs as a result of the accumulation of changes in the chromatin structure(2). There are several proteins associated with chromatin modification. These proteins directly affect transcription regulation, but also affect the structure and the expression of the genes in chromatin.
CDKN2a regulates the retinoblastoma protein, which suppresses tumors, by preventing CDK4/CDK6 from phosphorylating retinoblastoma protein. Increased expression of CDKN2a is associated with cellular senescence and aging, which is linked to increased levels of stress on cells. Because of these associations, it has been suggested to use this protein as a biomarker for aging. In an experiment with murine BubR1 haploinsufficiency, it was found that subjects that eliminated CDKN2a-positive cells had an extended lifespan and less age related degeneration. The experiments also indicate that by removing CDKN2a-positive cells in old mice improved deterioration due to aging and prolonged these effects. CDKN2a expression is controlled by trithorax (TrxG) and polycomb (PcG), which turn the gene on and off respectively.
SIRT1 and SIRT6 , both NAD+-dependent lysine deactylases, play roles in DNA repair and aging. A homologous protein to SIRT1 and SIRT6, Sir2 has been found and experimented on in yeast. This protein silences rRNA and telomeres. Without the expression of this gene, then rDNA arrays are removed from the genome to form circular DNA and the lifespan of the yeast declines. Similarly, SIRT1 and SIRT6 affect the cell's response to DNA repair and stress levels. These two proteins influence the aging process through DNA repair processes and transcription regulation.
When the DNA undergoes oxidative damage, SIRT1 is dispersed to all repair sites where H1K26 is deacetylated to increase DNA repair. However, when SIRT1 is recruited to damaged areas of the genome, ataxia telangiectasia-mutated pathway activation is required, which deacetylates Nijmegan breakage syndrome protein (NBS1), which controls the pathway to DNA repair, but also damage-dependent transcriptional deregulation. Therefore, while SIRT1 maintains DNA, it also plays a critical role aging. Increased SIRT1 expression does not increase an organism's lifespan, but it does increase its tumor resistance, improve its metabolism and prevents diabetes. SIRT1 also plays a role in stimulating non-histone proteins such as p53 (tumor suppressor gene), FOXO (transcription box), and Ku70 (DNA repair factor).
An overexpression of SIRT6, however, does increase the lifespan of the mouse (especially in males). SIRT6 is a regulatory protein that affects the nuclear factor kappa-light-chain-enhancer of activated B-cells-dependent inflammatory response (NF-kB) by attaching to its active site and inhibiting its transcription. Therefore, decreased SIRT6 expression results in increased NF-kB response. Humans lacking SIRT6 are more susceptible to attack by DNA-attacking agents. SIRT6 is also involved in homologous recombination, regulating response to DNA damage, and promote aging by increasing telomere malfunction.
Overall, the development of MCA methods has helped immensely the degree of understanding what influences and affects the normal aging process. MCA methods should be applied alongside with experimental models testing for aging because it will allow us to address the possible factors that affect aging.
O'Sullivan, Roderick and Jan Karlseder. "The Great Unravelling: Chromatin as a Modulator of the Aging Process". ("http://www.sciencedirect.com/science/article/pii/S0968000412001132"), 'Trends in Biochemical Sciences', 1 November 2012 (Vol. 37, Issue 11, pp. 466-476)
Viruses are known to be able to infect their host cells and even “hijack” them, taking control of normal cell metabolism and reproduction/replication. Viruses are able to change the host cell’s functionality by introducing their own DNA/RNA; as well as accompanying replication/transcription enzymes; such as DNA polymerase, and helicases. One way that the virus is able to take massive control of its host cell’s processes is through taking over the cell’s SLiM (short linear motifs). SLiMs are minimotifs that are existent in all eukaryotic cells/proteomes. SLiMs help regulate many of the cell’s processes, such as proteolytic cleavage, post-translational adjustments, and also aids in substituting as binding sites for cell signaling. They are also very unique in that they allow for encoding for very short translations for smaller proteins. With such diversity in functions of the cell, it has been argued that SLiMs are perhaps the most crucial element in the evolution of cells in the past and for the future. With such high evolutionary influence, SLiMs are also a very vulnerable as well as preferable portion of the cell for an invading virus to victimize in order to hijack the entire cell and its processes.
Due to the fact that SLiMs are very short in terms of length (only around 3-10 residues), as well as the variability in them, there is much room for evolutionary change, plasticity, and mutation. Upon mapping out an evolutionary history of known SLiMs, the only actual portion that remained conserved was the physicochemical properties. Most viruses are also known to have high mutation rates, which easily leads to convergent evolution; making viral motif mimicry relatively easy. Due to the high plasticity of most cellular motifs, the SLiMs are thought to be the weakest link for the cell in terms of vulnerability towards viruses.
Virus’ Influence on SLiM Transcriptional Regulation
A virus take-over of the SLiM can lead to severe effects on normal regulation of transcription. One example of a SLiM that controls transcriptional regulation would be the LxCxE motif, which binds to RB and controls the E2F transcription factor. Upon introducing a virus that would interfere with normal function of the LxCxE motif by forcing the cell to go (or stay) in S-phase. This stasis in the S-phase nullifies immune system response, and allows for more efficient virus proliferation. Other viruses such as HPV E7, a viral protein, invades the LxCxE motif by binding to RB pocket-binding domains, which overrides the cell’s usual regulatory checking system.
Carcinogenesis has been known form as a secondary effect of viruses taking over the cell and SLiMs’ mechanics. Carcinogenesis is induced due to the deregulating effect that viruses have on cell’s cellular pathways; resulting in often irregular protein productions, in which the proteins/RNA responsible is ingrained into the replication process for the infected cell. The most notable viruses that cause carcinogenesis in cells would be human papillomavirus (HPV), hepatitis B/C, herpes (HHV), and HIV co-infections). These sexually transmitted viruses make up for about 12% of the developed cancer cases in humans. One example that correlates viral invasions to the creation of cancerous traits would be EBC LMP1, which forces proliferation and blocks the signals for natural cell apoptosis; two traits that are highly prominent in cancerous cells.
Although taking direct control of a cell’s motif system is a common and easy method for viruses to be able to thrive inside it’s host cell, it can take control by other means as well. One method is by indirectly influencing the motif control utilizing globular proteins and domains. Some examples of domains that do this are EBV BHRF-1, which binds specifically to BH3 motifs. Another example of this particular method would be STD viruses such as HIV or the bovine papillomavirus’ E6 oncoprotein. These particular viral proteins are able to bind onto paxillin, a cellular cytoskeletal protein.
Another mechanism of indirect motif control would be a viral infection leading to loss of the motif upon viral transferring into the host cell. The virus is able to do this by continually replicating itself and changing its genetic code via mutations until the sequence of the clone viral protein is virtually useless for the host cell. By becoming useless, the viral proteins are thus able to decouple from normal host cell regulation, allowing the virus to freely interact within the host cell. With massive mutations in the virus, the viral sequence cannot be identified by normal host cell enzymes such as FBW7 ubiquitin ligase adaptors.
Phagocytosis refers to the cellular ingestion and digestion of bacteria and other foreign substances. This word refers to "cellular eating" whereas pinocytosis refers to "cellular drinking". Phagocytosis enables macrophages and dendritic cells to present antigens to and stimulate helper T cells, which in turn stimulate the B-cells whose antibodies contribute to phagocytosis. Phagocytosis can also be witnessed through amoebas and other protists engulfing smaller organisms or food particles. Additionally in phagocytosis, a cell engulfs a particle by wrapping pseudopodia around it and packaging it within a food vacuole. The particle will be digested after the food vacuole fuses with a lysosome containing hydrolytic enzymes.
Phagocytosis is an effective nonspecific immune response. Phagocytosis recognizes alien cells and particles. For the phagocytosis to proceed, macrophages and neutrophils must first recognize the surface of a particle as foreign. When a phagocyte surface interacts with the surface of another body cell, the phagocyte becomes temporarily paralyzed. So, paralysis allows the phagocyte to evaluate whether the other cell is friend or foe, self or nonself. During phagocytosis, the cytoplasmic membrane of the phagocyte flows around, and then engulfs, the bacterium, producing an intracellular phagosome. Overall, phagocytosis is selective for particles recognized as foreign to the body. Oxygen-independent and oxygen-dependent mechanisms of killing are initiated by fusion between a lysosome and a bacteria-containing phagosome.[1]
Primary phagocytosis (or known as phagoptosis) has recently recognized as a form of cell death caused by phagocytosis of viable cells, which causes cell destruction. Viable cells either loss their 'don't eat me' signals or exposing the 'eat me' signals, which causes their phagocytosis by phagocytes.[2]
Beside destroying microorganism, phagocytes also release regulatory molecules that diffuse to other cells to help coordinating the overall response to infections.[3]
What Happens
A pathogen is engulfed into a macrophage or neutrophil to become a phagosome. This is basically a container for the pathogen. Then lysosome is incorporated into the vessicle due to the destructive contents of lysosome. The outcome is a phagolysosome. The microbes inside the phagolysosome are destroyed by many factors. Some relevant factors are oxygen radicals, nitric oxide, anti-microbial peptides/proteins, binding proteins and hydrogen ion transport. These all contribute to the destruction of the microorganism.
The phagocytosis process can be sped up through the use of opsonins. These are molecules that bind to the surface of microorganisms to mark them for faster and more effective attacks. Some special bacteria present problems to the phagocytosis process. For example, the tuberculosis bacteria prevents the lysosome from binding to the phagosome, thus preventing destruction by phagocytosis. Other bacteria have a capsule around them which protects against phagocytosis.[4]
Cytokines, regulatory molecules released from phagocytes, regulate an immune response. The majority of cytokines are small proteins and mainly released from white blood cells and their relatives such as macrophages. Cytokines act similar to pancrines, regulatory molecules that released by one cell and diffuse locally to neighbor cells. Cytokines (TNFɑ and IL1) released from macrophages are important because they help coordinate an immune response.[5]
↑Brown, Guy C.; Neher, Jonas J. (2012). "Eaten alive! Cell death by primary phagocytosis: 'phagoptosis'". Trends in Biochemical Sciences. 37 (8): 325–32. doi:10.1016/j.tibs.2012.05.002. PMID22682109.
Slonczewski, Joan L. Microbiology: An Evolving Science. Second Edition.
In pinocytosis, the cell “gulps” droplets of extracellular fluid into tiny vesicles. It is not the fluid itself that is needed by the cell, but the molecules dissolved in the droplets. Because any and all included solutes are taken into the cell, pinocytosis is nonspecific in the substances it transports. In contrast to phagocytosis referring to "cell-eating", pinocytosis refers to "cell drinking". The hydrolysis of food inside vacuoles, called intracellular digestion, begins after a cell engulfs solid food by phagocytosis or liquid food by pinocytosis. In the process of pinocytosis, the plasma membrane forms an invagination and any substance found within the area of invagination is then brought into the cell. Generally, this material will be dissolved in water and thus this process is also referred to as "cell drinking" to indicate that liquids and material dissolved in liquids are ingested by the cell. This is opposed to the ingestion of large particulate material like bacteria or other cells or cell debris.
In essence, pinocytosis adsorbs extracellular fluids for the nutrients and material broken down in the fluid. This process is takes energy in the form of adenosine triphosphate. It is also a method of ingestion that is less selective than phagocytosis. It is less selective in that it basically just adsorbs all fluid in its immediate environment. Basically, pinocytosis is unspecific to what it transports, while phagocytosis is careful in choosing food particles to engulf. [1]
Cytokines are important in growth and differentiation of cells. They are signaling molecules that lead to long-term genetic effects by activation of transcription factors. Cytosine receptors bind tightly to tyrosine kinases, the JAK kinases, which are members of a family of cytosolic protein. JAK kinases directly phosphorylate and activate transcription factors members of STAT (Signal Transduction and Activation of Transcription) family. Activation of cytokine receptors initiates the JAK/STAT pathway.
Erythropoietin and formation of red blood cells
Cytokines Influence the Development of Different Cell Types
Cytokines form a small family of secreting signaling molecules that contain about 160 amino acids that control different parts of growth and differentiation of specific types of cells. Interleukins are cytokines that are essential for proliferation and functioning of T cells and antibody-producing B cells of the immune system. Interferons are another family of cytokines that are produced and secreted by cells after virus infections and act in nearby cells to induce enzymes that give these cells more resistance to virus infection. Many cytokines induce formation of important types of blood cells. Erythropoietin (Epo) for example, triggers the production of red blood cells by inducing the production and differentiation of erythroid progenitor cells in the bone marrow. Erythropoietin is synthesized by kidney cells and monitors the concentration of oxygen in the blood. A major function for erythropoietin is to transport oxygen to hemoglobin. Kidney cells respond to low amounts of oxygen by synthesizing large amounts of erythropoietin and delivering it to the blood through HIF-1α, which is an oxygen-sensitive transcription factor. When erythropoietin levels increase the level of erythroid progenitors increase and producing more red blood cells.
All cytokines have a similar tertiary structure that consists of four long α helices that are folded together in a specific orientation. Cytokine receptors have the similar structures as well. They have an extracellular domain that is made of two subdomains. Each domain contains seven β strands folded together. An example of a cytokine binding to its receptor is the interaction of erythropoietin molecule with two identical erythropoietin receptor (EpoR) proteins.
Cell response to a certain cytokine depends on the cell expressing the correct receptor. Even though cytokine receptors activate similar intercellular signal transduction pathways, cell response to a cytokine signal depends on the cell’s transcription factors, chromatin structures and other proteins responsible for the cell’s development. Eventually all activated pathways will lead to activation of transcription factors, which cause an increase or decrease in expression of some target genes.
JAK2 kinase is tightly bounded to the cytosolic domain of all cytokine receptors. It contains an N-terminal receptor-binding domain, a C-terminal kinase domain, and a middle domain, which regulates kinase activity by an unknown mechanism. A mouse study showed that JAK2, erythropoietin, and the EpoR are all required for formation of adult-type erythrocytes. Embryonic mice lacking functional genes encoding the EpoR don’t form adult-type erythrocytes and die due to the inability to transport oxygen to the fetal organs. Similar results were seen in mice lacking functional genes encoding either Epo or JAK2.
Since erythropoietin binds to simultaneously to extracellular domains of two EpoR monomers in the cell surface, JAKs are brought close together in order for one to be phosphorylating the other on a tyrosine located in a region of the protein known as the activation lip. Phosphorylation of the activation lip leads to a conformational change that reduces the Km for ATP or leads to the substrate to be phosphorylated and increases the kinase activity.
When the JAK kinases are activated they phosphorylate many tyrosine residues on the cytosolic domain of the receptor. The phosphotyrosine residues act as binding sites for SH2 domains (Src homology 2 domain), which are part of signal-transduction protein, including the STAT group of transcription factors. SH2 domains have three-dimensional structures, which bind to different sequences of amino acids that surround a phosphotyrosine residue. Differences in the hydrophobic socket in the SH2 domains of different STATs allow them to bind to phosphotyrosines adjacent to different sequences.
All STAT proteins contain an N-terminal DNA binding domain, an SH2 domain that binds to a specific phospotyrosine in a cytokine receptor’s cytosolic domain, and a C-terminal tyrosine that is phosphorylated by an associated JAK kinase. This ensures that in a specific cell only STAT proteins with an SH2 domain that can bind to a particular receptor protein will be activated. When a phosphorylated STAT dissociates spontaneously from the receptor, and two phosphorylated STAT proteins form a dimer, the SH2 domain on each binds to the phosphotyrosine in the other. Since dimerization also exposes the nuclear-localization signal (NLS), STAT dimers travel into the nucleus. It is here where they bind to specific enhancers controlling target genes.
Different STATS activate different genes in different cells. For example, stimulation of erythroid progenitor cells by erythropoietin (Epo) leads to activation of STAT5. Bcl-xL is a major protein induced by active STAT5, which prevents apoptosis of progenitors. They then proliferate and differentiate into erythroid cells. In a normal state, when Epo levels are low, bone marrow stem cells continuously create progenitor erythroid cells that are quickly destroyed. This process allows the body to respond quickly to the need for more erythrocyted in response to a rise in Epo levels.
3. Pellegrini S, Dusanter-Fourt I., The structure, regulation and function of the Janus kinases (JAKs) and the signal transducers and activators of transcription (STATs). PMID: 9342212 [PubMed - indexed for MEDLINE]
Ras-ERK and PI3K-mTOR pathways
Ras-extracellular signal-regulated kinase (Ras-ERK) is and phosphatidylinositol 3-kinase-mammalian target of rapamycin (PI3K-mTOR) signaling pathways are important for many major mechanisms in the cell.
Ras-ERK Pathway
ERK is a type of mitogen-activated protein kinase that plays a role as an effector for the Ras oncoprotein. The ERK-MAPK consists of Ras-GTPase, Raf kinase, MEK, and ERK. This pathway is activated by growth factors, polypeptide hormones, neurotransmitters, chemokines, and phorbol esters. When ERK is activated, it phosphorylates cytoplasmic signaling proteins and end-point effectors. Then, one of the cytoplasmic signaling proteins, p90 ribosomal S6 kinase, proceed to go phosphorylates other cytoplasmic targets and transcriptional regulators. Another pathway of ERK is nuclear targets, which are ternary complex factor transcription factors that functions to start the expression of immediate early genes. The end results of the pathways are to promote cell survival, cell division, and cell motility. Other Ras-ERK signaling pathways lead to the inhibition of cyclin-dependent kinase and trigger cell arrest.
PI3K-mTOR pathway
The PI3K-mTOR pathway is activated by growth factors, energy status, amino acid levels, and cellular stress. Growth factors turn on lipid kinase P13K by either recruiting the kinase protein directly or docking proteins insulin receptor substrate or GRB2-associated binder. Then, P13K phosphorylates photidylinositol 3,4,5 triphosphate (PIP3), which recruits protein kinase AKT to the plasma membrane. (AKT also phosphorylates many other factors involved survival, proliferation, and motility). Next, 3-phosphoinositide-dependent kinase 1 and mTOR complex are activated. The next step phosphorylates eukaryotic initiation factor 4E binding protein (4E-BP) and p70 ribosomal S6 kinase that start ribosome biogenesis and the translation of cell growth and proteins division. 4E-BP inhibition allows the assembly of cap-binding complex and translation initiation. Amino acids activate the pathway result in the activation of Rag GRPases. On the other hand, glucose deprivation and hypoxia causes AMP concentration to be increased that leads to AMP kinase to be turned on. This leads to the suppression of the pathway itself.
Other Information
The strength of the stimulus and the feedback loops affect the intensity and the duration of the pathway activation. In addition, some of the pathways initiation factors are the same. For example, insulin and insulin growth factors can activate the pathways, but it is a stronger P13K-mTORC1 pathway activator. Growth factors’ degree of strength depends on its quantity, the expression and cell surface localization of RTKs, and the co-expression of receptor family members and docking proteins. Positive and negative feedback loops also influence on the signal pathway dynamic. A positive loop is the GAB docking proteins that binds to RasGAP, Src homology 2-domain-containing protein tyrosine phosphatase, P13K, and PIP3. Consequently, GAB1 intensify P13K pathway. On the other hand, negative feedback loops suppress Ras-ERK and P13K-mTORC1 signaling pathways. An example is S6K phosphorylation of IRS and RICTOR that reduce AKT and mTORC1 activity.
Some research found clues that the two pathways can be integrated. One of the integrated pathways is cross-inhibition, where the two pathways regulate negatively regulate each other’s activity. An example is MEK inhibitors increase epidermal growth factor that starts AKT activation. However, this caused phosphorylation of GAB1, which suppresses the Ras-ERK pathways. Another is the pathway cross-inactivation. This integrated pathway is where Ras-ERK pathway causes the activation of P13K-mTORC1 pathway.
References: Mendoza, Michelle C., E. Emrah Er, and John Blenis. "The Ras-ERK and PI3K-mTOR Pathways: Cross-talk and Compensation." Trends in Biochemical Sciences (2011): 1-9. Print.
Bacteria are very adaptable to their environments. They divide by binary fusion, preceded by replication of bacterial chromosome. Bacteria proliferate rapidly and cannot increase genetic diversity by meiosis and fertilization. They have low mutation rate, but can have a significant effect on genetic diversity because of rapid rate of proliferation. Also, genetic recombination also adds genetic diversity to a population.
Genetic recombination can produce new bacterial strains. Genetic recombination for bacteria is combining DNA from two individuals into a single individual. For example, arg +trp- and arg-trp+ strain. Genetic recombination can occur by transformation, transduction, and conjugation.
Transformation is the alternation of a bacteria’s genotype by the uptake of a naked, foreign piece of DNA. Foreign DNA can be incorporated into bacteria’s chromosome by crossing over at homologous (similar) sequences. If foreign DNA is plasmid DNA, it will remain independent. Example: Calcium chloride treatment stimulate E. coli to take up small pieces of foreign DNA.
Transduction is when phages carry DNA from one bacterium to another.
Generalized transduction – DNA from infected cell is accidentally packaged into a phage, and transferred to another bacterium by infection. This occurs in lytic cycle.
Specialized transduction – when prophage (lysogenic cycle) is excised from bacterial genome. It takes a small amount of bacterial DNA with it. This DNA is then package into a phage and also transferred to another bacterium by infection.
Conjugation is the direct transfer of DNA between bacterial cells that are temporarily joined.
F factor is a piece of DNA conferred as “maleness”. F factor can exist in bacterial chromosome or as a plasmid. It encodes genes required for formation of sex pili. More specifically bacteria with F factor are F+. The “female” cells are F-.
“Male” bacteria form sex pili which attaches to “female” bacteria. If plasmid is just F factor, plasmid is transferred. If F factor is integrated into bacterial chromosome, bacterial DNA is also transferred. If F factor integrates into bacterial chromosome, bacteria is a Hfr (High frequency of recombination) cell.
Transposon is a piece of DNA that can move from one point to another in a bacterial cell’s genome. It doesn’t exist independently. It can move DNA within bacterial chromosome or from one plasmid to another (i.e. to give multiple druge\ resistance).
Cut and paste transposition – some transposons jump from one location to another.
Replicative transposition – transposon is copied, and the copy inserts in the new location
Insertion sequences are the simplest transposons. They contain one gene, transposase, which catalyzes transposition.
Composite transposons contain addition genes. These additional genes are stuck between two insertion sequences that travel together.
Mechanism: The DNA is cut in staggered fashion by transposase. Insertion sequence is inserted (also by transposase). The DNA polymerase and ligase fill in DNA and ligate ends. The DNA next to the insertion sequence thus contains direct repeats.
There are two levels of metabolic control. Cells can vary the numbers of specific enzyme molecules made and regulate gene expression. Cells can adjust the activity of the enzyme already present. This occurs quickly, since it does not require transcription.
Tryptophan Example: If the cell is growing in presence of tryptophan, it does not need to synthesize it. When tryptophan is present, it inhibits the first enzyme involved in synthesizing tryptophan. The presence of tryptophan also causes cell to stop making the enzymes needed for tryptophan synthesis (occurs at the level of transcription).
In bacteria, the genes for a particular pathway are clustered together on the chromosome. A single promoter serves all the genes of the operon. The clustered genes constitute a transcription unit (one long mRNA is made). The long mRNA is translated into separate polypeptides because the mRNA contains separate start and stop codons for each polypeptide. An operon has a single on-off switch that controls the expression of all the genes. This switch is termed an operator. The operator is located within the promoter or between the promoter and the genes. It controls access of RNA polymerase to the genes. The cluster of genes, promoter and operator, is termed an operon. The operon can be switched off by a protein call a repressor. The repressor is the product of a regulatory gene, which is not part of the operon and has its own promoter. The regulatory genes are transcribed continuously at a low rate. Many repressors are allosteric molecules, with two shapes: active and inactive. Corepressor is a small molecule that interacts with a repressor to switch an operon off.
Trp operon is an example of a repressible operon. It is normally transcribed. When tryptophan is present, it binds with the trp repressor, triggering an allosteric change. The trp repressor with bound tryptophan binds to the operator, shutting off transcription of the trp operon. The tryptophan is a corepressor.
Trp operon is repressible because transcription is inhibited by a specific small molecule (i.e. tryptophan) interacts with a regulatory gene. Inducible operons are stimulated when a specific small molecule interacts with a regulatory protein.
Lac operon is an example of an inducible operon. Lac operon encodes enzymes needed for metabolism of lactose (milk sugar). lacI is the regulatory gene. It encodes an allosteric repressor that binds to the operator in the absence of lactose. When lactose is present, an isomer, allolactose, binds to the repressor and causes a conformational change so that it can no longer bind to the operator. Lac operon is then transcribed. Allolactose is an inducer, as it induces transcription of operon. This is also an example of negative control, because the operon is turned off by the repressor.
Transcription of lac operon requires that lactose be present and that glucose be in short supply. If glucose levels are high, cell does not need to synthesize enzymes to catabolize glucose. Glucose levels are detected by interaction of an allosteric protein with a small organic molecule. Cyclic AMP accumulates when glucose is absent. cAMP receptor protein (CRP) binds cAMP, and is an activator of transcription. CRP binding site is next to the promoter of lac operon. CRP plus cAMP bind this site, and makes it easier for RNA polymerase to bind to the promoter and start transcription.
If the DNA mutation changes the amino acid sequence of the protein, then it can have one of these functional consequences (morph = form).
Gain-of-function are typically a missense or translocation that changes the promoter.
Hypermorph – increased expression
Ex. A mutated protein that signals continuously and can’t be shut off.
Neomorph – the protein is expressed somewhere ‘new’
Ex. In a new location on the organism - perhaps the neomorph is now expressed in the eyes, where it was normally expressed in the feet. At a different time during development – perhaps the neomorph is now expressed during adulthood, when normally it is expressed only during childhood, such as lactose tolerance.
Loss-of-function
Hypomorph – reduced, or “leaky” expression; usually the result of a missense mutation, since that is a less drastic change
Amorph – no expression, or null; usually the result of a more drastic mutation (deletion, nonsense, frameshift)
Antimorph – against the form/protein, dominant negative; a poisonous protein, the antimorph will actually harm the normal protein when both are present in heterozygotes; usually this is a situation where the protein forms a multi-subunit complex.
Gene product activity: different, or same but in different location
Recessive/Semi-dominant/Dominant: semi-dominant or dominant
History of Genetics
Gregor Johann Mendel is a German-Czech Augustinian monk is referred to as the father of genetics. Mendelian genetics was founded based on his work with pea plants. His research with the nature of inheritance in plants was first published in 1865 in his paper "Experiments on Plant Hybridization" in the Society of Plant Hybridization journal.
The main idea of Mendelian and classical genetics is that the pattern of inheritance can be described through simple rules and ratios. These rules were derived from Mendel's analysis of seven characteristics of pea plants that include the following traits:
1. Color and smoothness of the seeds
2. Color of the cotyledons
3. Color of the flowers
4. Shape of the pods
5. Color of unripe pods
6. Position of flowers and pods on the stems
7. Height of the plants
Mendelian genetics is founded on two laws. The first law is the law of segregation. The law of segregation states that each individual that is a diploid has a pair of allele (copy) for a particular trait. Each parent passes an allele at random to their offspring resulting in a diploid organism. The allele that contains the dominant trait determines the phenotype of the offspring. In essence, the law states that copies of genes separate or segregate so that each gamete receives only one allele.This occurs during the natural process of meiosis that occurs during sexual reproduction in eukaryotes. The second law states is known as the Law of Independent Assortment. It states that the genes that are for separate traits are passed to the offspring independently of other genes. In other words, the inheritance pattern of another gene has no influence on the inheritance pattern of the gene of interest. Alleles of different genes assort independently during gamete formation. In Mendelian experiments, dihybrid crosses showed that the 9:3:3:1 ratio table is just two genes is independently inherited with a 3:1 phenotypic ratio.
During Mendel's time, it was believed that genetic information was passed down through the concept of blending inheritance. This is the idea that the offspring is the result of the combination or blend of genetic traits inherited from the parents. For example, the flower color of an offspring pink when a white flower and red flower are crossed. This theory was disproved by Mendel who showed that traits are combinations of distinct genes rather than a gradient or continuous blend of traits from the parents.
Significance of Mendel's Work
Gregor Mendel performed a series of breeding experiments on pea plants. Mendel studied how pea plants inherited two observable traits: flower color (white or purple) and the texture of the peas (smooth or wrinkled). Mendel bred many generations and learned that these characteristics were passed on to the next generation in orderly and predictable ratios. He cross bred purple flowered pea plants with white flowered ones and found that the next generation had only purple flowers but directions for making white flowers were hidden somewhere in the peas because when the second generation of only purple flowered peas were bred with each other, some of their offspring had white flowers. The second generation plants displayed the colors in a predictable pattern: 75% purple, 25% white. The same ratio persisted even when the experiment was repeated over and over. Mendel reasoned that “factors,” each of which determined a specific trait, must exist in order to reproduce physical material because they passed from parent to offspring.
Mendel’s experiment was groundbreaking because later on these “factors” were found to be genes and his mathematical rules for inheritance were applied not just to peas but to all plants, animals, and people. It revealed that a common, general principle governed the growth and development of all life on earth.
Once Mendel's work was recognized the next goal was to determine the molecules in the cell that was responsible for the control of genetic information. In 1910, Thomas Hunt Morgan discovered that genes are present on chromosomes through his experiments with fruit flies and monitoring sex-linked white eye mutations in Drosophila.
Once genes were known to exist on chromosomes which are made up of proteins and DNA, the question scientists were inclined to answer is whether protein or DNA is responsible for the inheritance of genetic material. In 1928, scientist Frederick Griffith showed that dead bacteria could transfer genetic material to "transform" other living bacteria. This interesting theory was reassured through the experiments of Oswald Theodore Avery, Colin McLeod and Maclyn McCarty. These three scientists discovered the molecule that was responsible for the bacterial transformation is DNA. In 1952, the Hershey and Chase experiment showed that DNA is the genetic material of bacteriophages, or viruses that infect bacteria.
Now that DNA had been identified as the genetic material, scientist sought to solve the structure of DNA and to identify mechanisms by which genetic material is inherited. James D. Watson and Francis Crick, back in 1953, determined the structure of DNA using the x-ray crystallography image of a DNA helix imaged by Rosalind Franklin and Maurice Wilkins. They discovered that the structure of DNA is highly adapted to allow for its function especially for DNA replication and for following a set genetic code. After the years of these scientists, modern genetics started to evolve.
Now it is known the genetic information can be passed down through multiple patterns of inheritance including single gene inheritance such as recessive or dominant autosomal inheritance, and recessive or dominant x-linked inheritance. Other patterns of inheritance include multi-factorial inheritance in which genetic factors and environmental factors contribute to the passing of genetic information. A third pattern of inheritance is mitochondrial inheritance. Mitochondria have circular chromosomes that are inherited from the mother. Diseases acquired though mitochondrial inheritance are often concerned with the functionality of heart, skeletal muscle, kidneys, and liver as these organs use large amounts of energy processed by the mitochondria.
6. http://www.uvm.edu/~cgep/Education/Inheritance2.htmlLaw of segregation- every individual possess a pair of alleles for any particular trait and that each parent passes a randomly selected copy (allele) of only one of these to its offspring.
Law of independent assortment- separate genes for separate traits are passed independently of one another form parents to offspring.
Chromosomal theory of inheritance- inheritance patterns may be generally explained by assuming that genes are located in specific sites on chromosomes.
Genome- 1 set of all chromosomes
Gene- all the DNA needed to make one protein or RNA
Dominant- allele that is fully expressed in phenotype when heterozygous
Recessive- hidden allele when heterozygous
Co-dominant- both alleles affect the phenotype in separate and distinguishable ways
Incomplete Dominance- phenotype in between two parents
Loci- position of a gene on a chromosome (locus=singular)
Allele- variations of a gene at a locus
Genotype- list of alleles (genes)
Phenotype- expression of alleles "traits"- what you observe
Heterozygous- alleles that are different
Homozygous- alleles that are the same
Telomere- ends of DNA
Epistasis- gene at one locus alters the phenotype of the gene at another locus; method to determine which gene comes first in a pathway.
3’ to 5’ phosphodiester bond- chemical linkage between adjacent nucleotides
Solenoid- circular ring of nucleosomes
Central Dogma- DNA to RNA to Proteins
Mutagens- physical or chemical agents that cause mutations
Types of mutations in substitutions- silent mutations, missense mutations, nonsense mutations
Insertions/Deletions- frameshift
More accurate replication- Nucleotide selection, DNA proofreading, Mismatch repair
Robertsonian Translocation- Short arm of an acrocentric chromosome is exchanged with the long arm of another creating a large metacentric chromosome + a fragment that fails to segregate and is lost.
Character or Characteristic- An attribute or feature
Syndromes- Down (age of mother) chromosome 21, Patau chromosome 13, Edward chromosome 18, Turner XO, Triplo-X XXX or (XXXX: extra X chromosome can be inactivated, not necessarily active), Klinefelter XXY (or XXXY), Jacob XYY (gigantism)
Nondisjunction- failure of the chromosome pairs to separate properly usually during anaphase stage.
Crossing Over- exchange of genetic material between chromosomes
Synapsis- pairing of two homologous chromosome during meiosis
Parts of chromosome- telomere, centromere, kinetochore, spindle microtubules, tetrad, centrioles
Vector- DNA that self replicates and inserts/transfers DNA to host cells.
Parthenogenesis- The development of a person from an egg that was never fertilized.
Oncogenes- Genes that are associated with cell cycle and cell reproduction. A mutation in these can cause a cancerous tumor.
Multifactorial- The idea that multiple factors contribute to the end product such as genetic factors and environmental factors contributing to make one person.
Ligase- An enzymatic protein that participates in cell repair or gluing.
Probe- A single strand of DNA that is labelled radioactively or in other methods to be identified later.
Pedigree- An organized diagram of the hereditary passing of one gene. Follows the gene through generations to see how the gene is passed on.
Nonsense Mutation- A stop codon is coded for early in the sequence resulting in an unfinished protein.
[1]
1.Reece, Jane (2011). Biology. Pearson. ISBN978-0-321-55823-7. {{cite book}}: Text "coauthors+ Lisa A. Urry, Michael L. Cain, Steven A. Wasserman, Peter V. Minorsky, Robert B. Jackson" ignored (help)
Example of Structural Biochemistry in Genes.
DNA is far from perfect. It generates mutations more frequently than we think of, but there are proteins that help correct these mutations so the cell isn't as affected by these mutations. one big complex of molecules that work together incorporate several different proteins, each with their own "job". The first protein is called BRCA1. When there is a mutation in a DNA strand, the strand breaks. When this happens, one of two things follow, a protein called 53BP1 comes in and chops the pieces of DNA in order to avoid making a bad copy. this unfortunately means that that chromosome is not working anymore and depending on how important that was for the life of the cell it could kill the cell. This can hopefully be avoided by having BRCA1 bind to that site making impossible for 53BP1 to come in and chop the piece of DNA. Once BRCA1 is in place a series of other small proteins attach to help correct the mutation. one of the protein is PalB2. The major job of this protein is to provide a binding site for two proteins that can interact together. one protein is RAD51. Every chromatid has a sister chromatid that is identical. RAD51 is essentially the scanner of the protein complex, looking for the sister chromatid and scanning it so the complex knows how to fix the mutation. finally BRCA2 is what actually fixes the mutation taking what RAD51 "scanned" and staying where the mutation with the help of the binding proteins PalB2 and BRCA1. This is how a lot of mutations are corrected to avoid problems that could pose very negative results to the cell and the organism as a whole.<ref name="Links between genome integrity and BRCA1 tumor suppression">[18],
Mendelian Genetics/
Mendelian genetics hereditary patterns include the idea of dominant and recessive alleles that were determined using his monohybrid and dihybrid crosses. The basics of Mendelian inheritance is that the dominant genotype is expressed in individuals that are homozygous or heterozygous for the dominant allele. The recessive phenotype is only expressed in individuals homozygous for that allele.
Geneticist Gregor Mendel studied how pea plants inherited two different traits that had two different possibilities each: flower color (white or purple) and pea shape (smooth or wrinkled). When he crossbred a purple-flowered pea plant with a white-flowered pea plant—known as the P generation—he obtained an F1 generation of all purple-flowered pea plants. After crossbreeding two F1 purple-flowered pea plants, he obtained an F2 generation with a ratio of 3 purple-flowered pea plants to 1 white-flowered pea plant.
Non-Mendelian Genetics/
Non-Mendelian Genetics is the branch of inheritance patterns that do not follow the basic recessive and dominance laws that is characteristic of Mendelian genetics. One example is incomplete dominance. In this type of inheritance, both types of alleles contribute to the phenotype, resulting in an appearance that is in between the phenotypes of the two parents.
In co-dominance, both alleles are expressed fully. For example, human blood groups ABO are from the three alleles, IA, IB, and IC. If an individual has the alleles IA and IB, they will have AB type blood.
Sometimes, one gene is responsible for more than one phenotype and this is referred to as pleiotropy. Pleitropy is observed in PKU (phenylketonuria)disease in which one gene causes mental retardation as well as reduced pigmentation in skin, among other complications.
Another type of inheritance is sex-linked inheritance. In human females, two X chromosomes are present, and in human males, one X and one Y chromosomes are present. Because genes on the X chromosome is not present in the Y chromosome, the inheritance pattern is significantly different from any other patterns of heredity. For recessive traits, males only need one copy of the allele on the X chromosome to display the recessive phenotype, whereas females still need two recessive alleles on both their X chromosomes to have the recessive phenotype. A standard example of such recessive sex-linked inheritance is hemophilia.
Due to inheritance of alleles on mitochondria genes
Trait is passed exclusively from mother to all progeny (eggs provide the bulk of cytoplasm for the zygote, and mitochondria are located in the cytoplasm)
Trait is not passed from father to progeny (sperm provide essentially no cytoplasm to the fertilized zygote)
During mitosis and meiosis chromatin fibers coil and condense into chromosome structures. Each chromosome is made up of chromatin, a complex of proteins and DNA. The chromosomes are long pieces of DNA that are found in the center of the nucleus inside the cells.
Centromeres are a constricted region on the chromosome, which determines its appearance. Centromeres can be placed along different points along the length of the chromosome classifying it as metacentric, submetacentric, acrocentric or telocentric depending on centromere location. Chromosomes are cellular structure carrying genetic material located in the nucleus of eukaryotic cells and contain DNA molecules and proteins. Somatic cells from the same species contain identical number of chromosomes, which represents the diploid number (2n). Chromosomes exist in pairs; referred to as homologous chromosomes, one homologous chromosome is inherited from the organism’s mother and one from the father. Homologous chromosomes share genetic similarities such as a locus, an identical gene site along their length. On the other hand, bacteria and viruses contain only one chromosome. A normal human cell contains 46 chromosomes in its nucleus. Sex cells contain 23 chromosomes in humans. Haploid number (n) of a chromosome is one half the diploid number. During meiosis, diploid number of chromosomes are converted to haploid number as gametes are formed. Sex determining chromosomes X and Y are not often homologous. Females carry two homologous X chromosomes while males carry one Y and one X chromosome, which are not homologous but contain homologous regions and behave homologous in meiosis.
However, "the remaining chromosomes are refered to the autosomal chromosomes and they are known as chromosome pairs 1 through 22" (University of Maryland).
Chromosome mutations, occur when change in the total number of chromosomes are effected by deletion or duplication of genes or chromosome segments and rearrangement of genetic material within or among chromosomes. Aneuploidy is a condition in which one or more chromosomes are present in extra copies or are deficient in number but not a complete set. To be more specific, the loss of a single chromosome from a diploid genome (diploid cell-cell containing two set of chromosomes (2n), one set inherited from each parent) is called monosomy (2n-1). The gain of one chromosome is called trisomy (2n+1). A condition with complete set of haploid chromosome is called euploidy. There are events in which organisms have more than two sets of chromosomes; triploids for example are organisms with three sets of chromosomes. Tetraploids have four sets of chromosomes.
Chromosome mutations result in serious problems such as Turner syndrome, a monosomy in which females may contain all or part of an X chromosome. Monosomy for autosomes is usually lethal in humans and other animals. Klinefelter syndrome, a trisomy genetic disorder in males caused by the presence of one or more X chromosome. The effects of trisomy are similar to those of monosomy. Down syndrome is the only autosomal trisomy in humans that has a substantial number of survivors one year after birth. Trisomy in chromosome 21 is the cause of Down syndrome and it affects 1 infant in every 800 live births.
Polyploidy, is an occurrence in which more than two sets of haploid chromosomes are present. This condition is rare in animal species but common in lizards, amphibians, fish, and very common in various plants. Polyploidy originates in two ways: (1) autopolyploidy the addition of one or more extra sets of chromosomes of the same species; (2) allopolyploidy is the combination of chromosome sets from different species. Autopolyploidy have sets of chromosomes identical to parent species. Autotriploids for example can be represented as AAA and can arise from the failure of all chromosomes to separate during meiotic division, which result in a diploid gamete. The fertilization of the gamete by a haploid gamete results in the production of a zygote with three sets of chromosomes. Autotriploids often produce genetically unbalanced gametes with odd numbers of chromosomes. Autotetraploids can be represented by AAAA and are most likely to produce balanced gametes in sexual reproduction.
National Human Genome Research (USA)National Human Genome Research (USA)
Deletion in chromosomes occurs when a chromosome breaks in one or more places and a portion is lost or missing. Deletions can occur either near one end or within the chromosome, which are known as intercalary deletions. Deletion loops are unpaired regions of the normal homolog which occurs when synapsis between a chromosome with intercalary deletion and a normal homolog. The size of the chromosome deletion can determine the survival of the organism and unlike large deletions, small deletions can result in survival. Duplications in chromosomes occur when a large piece of chromosome is present more than once in the genome. They can arise from unequal crossing over between synapsed chromosomes during meiosis.
In the 1902, Walter Sutton and Theordor Boveri proposed that how the chromosomes are transmitted is the same as Mendel's Theory of how genes are transmitted. Genes are located on chromosomes. Chromosomes segregate and assort independently during meiosis. The evidence comes from Morgan's experiment with fruit flies. In 1910, Thomas Hunt Morgan started his work with Drosophila melanogaster, a fruit fly. He chose fruit fly because they can be cultured easily, are very prolific, have short generation time, and have only four pair of chromosomes that can be easily identified under the microscope. They have three pair of autosomes and a pair of sex chromosomes. At that time, he already know that X and Y have to do with gender. He used normal flies with red eyes, and mutated flies with white eyes and cross breed them. He was able to conclude that gene for eye color was on the X chromosomes. Males have only 1 X, Y does not carry gene for eye color.
Cambel, Neil,Jane B Reece, et all. Biology Eigth Edition., Klug, William, Michel Cummings, et all. Esssential of Genetics Custom Edition for UCSD
http://www.umm.edu/ency/article/002327.htm
Image source: wikimedia-commons
Derived from the Greek, epigenome means "above" the genome. The epigenome consists of chemical compounds that modify, or mark, the genome in a way that tells it what to do, where to do it and when to do it. The marks, which are not part of the DNA itself, can be passed on from cell to cell as cells divide, and from one generation to the next.
The protein-coding parts of your genome, called genes, do not make proteins all of the time in all of your cells. Instead, different sets of genes are turned on or off in various kinds of cells at different points in time. Differences in the types and amounts of proteins produced determine how cells look, grow and act. The epigenome influences which genes are active — and which proteins are produced — in a particular cell. So, the epigenome is what tells your skin cells to behave like skin cells, heart cells like heart cells and so on.
Epigenetics involves heritable gene functions that are not attributed to changes in the cell’s DNA sequence. Some known examples of epigenetic modifications include DNA methylation and histone modification.
DNA methylation of a DNA strand and its complimentary strand
DNA methylation is the process of adding a methyl group to a DNA sequence. This reaction is catalyzed by an enzyme known as DNA methyltransferase (DNMTs). There are two types of DNMTs: De novo DNMTs and Maintenance DNMTs. De novo DNA methyltransferase enzymes initially attaches the methyl group to a DNA strand. Maintenance DNMTs copy the methylation from an existing DNA strand onto the complimentary strand after replication. DNMTs recognize regions where a cytosine nucleotide is placed next to a guanine nucleotide along the base sequence. This region is known as CpG, where a phosphate group binds a cytosine to an adjacent guanine.
DNA methylation processing is used to repress gene expression. The methylation blocks the promoter site at which transcription factors bind to, thus stopping the gene expression. Though DNA methylation is very stable, the reaction can be reversed by a different group of enzymes.
Alterations to DNA methylation patterns play a major role in the onset of cancer. In fact, all tumors that have been studied exhibit changes in DNA methylation. Early studies seem to suggest that widespread demethylation is characteristic of cancer. One type of methylation pattern is called de novo methylation. In this process, the polycomb complex (PRC2) targets genes that, under normal conditions, have unmethylated CpG island promoters. These genes are repressed due to the methylation of H3K27. Methylation of H3K27 is mediated by the histone methylase EZH2. The methylated H3K27 then binds to the chromodomain-containing complex (PRC1), thereby giving rise to a type of heterochromatin. During normal development, targeted de novo methylation can take place at certain sites on DNA. Similarly, it can occur abnormally in cancer. The explanation offered for this phenomenon suggests that the histone methylase EZH2 recruits Dnmt3a and Dnmt3b. Some studies show that the polycomb complex PRC2 may be released from the methylated islands. This event results in a more stabilized form of methylation-mediated repression, which occurs instead of the flexible polycomb repression mechanism.
Many believe that de novo methylation is a completely random process. This theory helps to explain the presence of methylated tumor suppressor genes in some cancer types. However, more recent research reveal DNA methylation in cancer to involve hundreds of CpG islands that are methylated in a more pre-determined way. Under the older theory, it is believed that DNA methylation causes genes to become inactivated. In contrast, the newer theory revealed that the genes targeted for methylation have already been inactive. Many of these targeted genes have a polycomb complex nearby, which may explain how the methylases are recruited to carry out the gene modification. Thus, the newer theory based on programmed methylation suggests that de novo methylation in cancer occurs on gene sites that were targeted regardless of whether the genes are active or inactive. The target sites are also independent of whether the genes affect tumorigenesis. Nonetheless, studies on human colon cancer reveal the possibility that DNA methylation can cause a more permanent repression mechanism than that of the normally flexible polycomb repression mechanism; this observation links DNA methylation to tumorigenesis.
Fragile X Syndrome is a developmental disease that eventually causes mental retardation. This disease occurs when abnormal de novo methylation causes the repression of the FMR1 gene early in development. Abnormal de novo methylation is believed to be the cause of Fragile X syndrome due to the fact that inactivation of the FMR1 gene is observed to occur alongside a process known as H3K9me3 heterochromatinization. From these studies, scientists believe that the programming of all abnormal modifications may proceed through a single mechanism in which histone methylases mediate random DNA methylation and repression.
Active demethylation occurs in a step-wise manner that results in a modification of 5-methylcytidine (5mC). This modification allows the molecule to be recognized for removal and replacement by repair; the modified 5-methylcytidine is replaced with unmethylated cytosine. The following steps depict the pathway suggested for active demethylation:
Hydroxylation: An enzyme known as ten-eleven-translocation (Tet) catalyzes the hydroxylation of the methyl group on 5-methylcytidine, thereby forming 5-hydroxymethylcytidine (5hmC).
Deamination: Activation-induced deaminase (Aid) or Apobec family proteins replaces the amine group of 5-hydroxymethylcytidine with a doubly bonded oxygen atom. This new molecule is called 5-hydroxymethyluridine (5hmU).
Glycosylation: A glycosylase (Mbd4 or Tdg) essentially removes the 5-hydroxymethyluridine structure from the sugar-phosphate backbone. What is left is an apyrimidinic acid residue.
Repair: The apyrimidinic acid residue is replaced with cytosine (C) through a repair process such as nucleotide excision repair (BER or NER).
The hypothesis of the histone code, a recent postulation by Strahl and Allis,[1] suggests that it is a number of histone modifications at a particular locus that is involved in determining what genes are expressed or suppressed. This expression is a heritable epigenetic factor that allows regulatory proteins to have an effect on gene expression. By changing the charge of histones or the addition of structural proteins, genes can be essentially turned on or off. But the question of whether it is the combination of histone modifications working in concert or a cumulative effect of numerous modifications is a topic of current debate.[2] Histone modifiers consist of residue specific proteins and reader proteins allow for these epigenetic changes to be transmitted post-translationally.
Acetylation of histones has been associated with increased transcription of specific genes.[2] Several acetyltranserfase compounds have been documented that allow acetylation of histone complexes and increase genetic transcription. Using genome mapping techniques, it has been shown that the most acetylated regions are responsible for the highest rates of transcription. However, the acetylation is relatively dynamic, often only lasting for several minutes.[2] This suggests that it is the frequent acetylation that has a role in the promotion of a genetic region rather than simply the presence of acetylated histones.
Lysine methylation in histones that leads to decreased transcriptional activity is much longer lived in relation to acetylation. Mono, di, and tri-methylation is part of an indexing system that is involved in epigenetic control and each manner of methylation has a different longevity.[2] Additionally, methylation has a longer turnover rate than phosphorylation.
Serine, Threonine, and Tyrosine can be phosphorylated as another impermanent epigenetic control factor that occurs on histone tails. During mitosis, core histone tails are occasionally phosphorylated in a process that is initiated then spreads throughout the genome.[2] Phosphorylation has been linked to acetylation and other post transcriptional modifications.
As chromatin is assembled, histones are constructed in the cytoplasm and transported through the nuclear membrane to be deposited on DNA. Generally, histone assembly and placement is concurrent with S-phase progression. Histones are initially modified on their journey into the nucleus and placed at the replication fork of the chromatin then further modifications are made within the first few minutes of placement. However, further modifications like the methylation of lysine occur after longer periods of time, indicating the importance of epigenetic changes.[2]
For histone modifications and the epigenetic code to be heritable, the cell must be able to initiate modification, alter modifications, and transmit modifications to future copies of the genetic code. As mentioned previously, acetylation of histones has a rapid turnover, which does not strongly suggest a lasting or heritable epigenetic control. Even in situations where acetylation is observed for longer durations, it is usually seen to attract proteins responsible for increasing levels of acetylation, and is not likely responsible for the heritability of epigenetic codes.[2] Methylation, however has been observed to have a prolonged binding and increased stability of reaction and could be responsible for a transmittable epigenetic code. Although it is still to be determined if there is a “histone code” that carries epigenetic changes as DNA is copied, a code based on methylation of histones is the most likely type of histone modification to be involved.
Cancers are caused by a combination of changes to the genome and the epigenome.
Adding or removing methyl groups can switch genes involved in cell growth off or on. If such changes occur at the wrong time or in the wrong cell, they can wreak havoc, converting normal cells into cancer cells that grow wildly out of control.
For example, in a type of brain tumor called glioblastoma, doctors have had some success in treating patients with a drug, called temozolomide, that kills cancer cells by adding methyl groups to DNA. But that's only part of a very complex picture. Cells also contain a gene, called MGMT, that produces a protein that subtracts methyl groups — an action that counteracts the effects of temozolomide. In some glioblastomas, however, the switch for the MGMT gene has itself been turned off by methylation, which blocks production of the protein that counteracts temozolomide. Consequently, glioblastoma patients whose tumors have methylated MGMT genes are far more likely to respond to temozolomide than those with unmethylated MGMT genes.
Changes in the epigenome also activate growth-promoting genes in stomach cancer, colon cancer and the most common type of kidney cancer. In other cancers, changes in the epigenome silence genes that normally serve to keep cell growth in check.
To come up with a complete list of all the possible changes that can lead to cancer, the National Institutes of Health (NIH) has started a project called The Cancer Genome Atlas. Beginning with glioblastoma, these researchers are comparing the genomes and epigenomes of normal cells to those of cancer cells. They are looking for any changes in the DNA sequence, called mutations; changes in the number and structure of chromosomes; changes in the amounts of proteins produced by genes; and changes in the number of methyl groups on the DNA.
As mentioned in the main page, the epigenome is made up of chemicals that tell your genome what to do. Your genome is basically the same in all of your cells, so what makes your cells different in the way they behave is epigenome which controls them.[1] An example of epigenomics would be to look at a set of monozygotic twins. Their genome are identical, yet due to exposure to different environmental factors as they grow up, they will experience different epigenetic changes, which may make one of the twins more susceptible to developing certain diseases that the other would be less likely to get.[2] This is why many find it important to study the epigenome and there are many programs funded to catalogue the human epigenome, in hopes that we can understand and learn what genes are being turned on and off and how that affects us.[3] Right now there are multiple ways that are being used to read the epigenome. The PHD finger is one of the ways.
In the article “The PHD finger: a versatile epigenome reader” written by Roberto Sanchez and Ming-Ming Zhou, they discuss how the plant homeodomain (PHD) zinc fingers that they studied were found to able to read complex histone sequences. Activation and silencing of gene transcription is controlled by modifications of histones, H2a, H2B, H3, and H4. Histones are proteins that pack and organize DNA into nucleosomes. Histones can be modified by adding a methyl group to the amino acid lysine, or adding an acetyl group to the amino acids lysine or arginine. PHD fingers have evolved to recognize when, in the histone, those amino acids have been methylated or acetylated. Main cites that are methylated on the histone H3 tail are K4 and R2.[4]
Out of the four modifications to histones mentioned before, PHD fingers (a small protein domain whose structure is stabilized by zinc atoms) can read histone H3. PHD does this by binding to the first six N terminal residues of the histone. Ligands are molecules that bind to a central metal. This is important because bromodomain PHD finger transcription factor (BPTF) is a PHD finger bound to its ligand, and it has the ability to bind to H3 when K4 is methylated and R2 is not. Most of these PHD fingers have an aromatic cage that helps with binding. Those that don’t however, have the ability to bind to K4 when it is not methylated. There are many different types of PHD fingers that will bind depending on what amino acids are methylated and/or acetylated. When they bind, the conformation changes slightly, allowing one to distinguish between them.[4]
Further study is still required to completely understand how PHD fingers work.[4]
Mitosis is the process by which two identical daughter cells are produced after nuclear divison and cytokinesis. It is the process by which cells replicate and occurs during the normal cell cycle, and should not be confused with meiosis. The following outlines the stages of mitosis.
Interphase
Interphase is technically not part of mitosis, but it is important because it is discussed as part of the cell cycle and precedes the process of mitosis. In interphase, the cell is in preparation for nuclear division. It is also the beginning stage, where DNA is synthised and proteins are made. C The cell undergoes the G1, S, and G2 phases of the cell cycle during interphase. G1 is recognized as the stage before synthesis of DNA. S phase is when DNA is synthesized, and the G2 phase is the stage immediately preceding the first stage of mitosis (prophase) during which the cell synthesizes proteins and grows.
Prophase
Prophase is the next step in the cell cycle and during prophase, the chromatin in the nucleus becomes compact and the nucleolus breaks down. The centrioles, or microtubule organizing centers for plant cells, move to opposite ends of the cell. Fibers extend from the centrioles to create the mitotic spindle.
Prometaphase
During this stage, the nuclear membrane degrades and proteins attach to the centromeres creating a new complex called kinetochores. The microtubules attach at this complex and the chromosome start moving toward the middle of the cell.
Metaphase
During metaphase, the chromosomes are aligned at the middle of the cell with the help of spindle fibers. This line known as the metaphase plate is a method of organization to ensure proper division of the cell and that each new daughter cell receives exactly one copy of each chromosome.
Anaphase
At this stage, the paired chromosomes are split by separation at the kinetochores to opposite sides of the cell. The chromosomes are now called sister chromatids.
Telophase
The chromatids are fully separated as they are at opposite ends of the cell. New membranes form around the perimeter of the daughter nuclei and the chromosomes are dispersed. The spindle fibers start to degrade and prepare for cytokinesis.
Cytokinesis
Cytokinesis is the process by which the cell partitions to make two complete, and identical daughter cells. The process is slightly different for animal and plant cells. In animal cells, cytokinesis is completed through the help of actin that pinches the cell into two equivalent cells. However, in plant cells a cell plate is formed in the middle of the cell to result in two equivalent daughter cells. This extra step is due to the rigid cell wall that is a characteristic of plant cells.
Although components that actually partake in cell division is very important, there are other components involved in regulating and signaling mitosis that are just as crucially important and are often overlooked.
G1 Checkpoint
Before a cell enters the S phase, it must pass the G1 checkpoint. This checkpoint decides whether the cell should undergo cell division, also known as mitosis, or enter into a rest phase and delay division. Whether the cell goes into the resting stage or cell division depends on its type. For example, liver cells only pass the G1 checkpoint to undergo cell division twice a year. If the cell stops at the G1 checkpoint, it will go into the G0 stage which is known as the resting stage. The G1 checkpoint in eukaryotes is controlled by the CDK inhibitor p16, or CKI p16. The purpose of this protein is to inhibit CDK4/6 which then prevents its interactions with cyclin D1. Inhibited interactions between CDK4/6 and cyclin D1 will prevent a cell from entering the cell cycle. When growth is induced, the cell proceeds from G0 to G1 to S phase due to the increased expression of cyclin D1 which then competitively binds to CDK4/6. When CDK 4/6-Cyclin D forms, this complex phosphorylates retinoblastoma, denoted Rb. Rb allows transcription factor E2F to express cyclin E, and the cyclin E interacts with CDK2 to allows cells to transition from the G1 phase to S phase. 6
G2 Checkpoint
Once the cell has passed the first checkpoint, G1, it must pass the G2 checkpoint before actually going into mitosis. G2 checkpoint occurs at the end of the G2 phase, prior to the M phase or the mitotic phase. This checkpoint is primarily to make sure that the cell is ready for mitosis. At this checkpoint, the cell is checked to see if it has a number of factors that determines if it is ready to undergo cell division. The CDKs present at the G2 checkpoint are initiated by the phosphorylation of the CDK via MPF, which is called the “Mitosis Promoting Factor”. At the G2 checkpoint, an activating phosphatase called CDC25 removes phosphates from MPF so that the MPF may promote mitosis. However, DNA tends to be damaged prior to mitosis. Therefore, the cell cycle is put in arrest by inactivating CDC25. It is important to inactivate the CDC25 so as to prevent the transmission of damaged DNA to daughter cells. 5
Mitotic Checkpoint of Spindle Assembly (SAC)
An important process that regulates the cell cycle is called the spindle assembly checkpoint, also known as SAC. SAC is a cell cycle checkpoint that cells encounter before going into anaphase. Checkpoints are important to the cell cycle because they control the rate and extent to which a certain phase occurs. SAC specifically helps chromosomal stability and prevents cases of aneuploidy. 4
Protein Kinase involved in SAC: BUB1 and BUBR1
BUB1 (budding uninhibited by benzimidazole-1) and BUBR1 (budding uninhibited by benzimidazole-related 1) are proteins that play a key role in the establishment of central mitotic checkpoint. BUB1 and BUBR1 consist of three main regions. One of these regions is the C-terminus. This region is a catalytic serine/threonine protein domain. The other region is the N-terminal region which is conserved in BUBR1 and BUB1, as well as their homologs. The N-terminal region contains the kinetochore localization domain. The last region is a non-conserved region where BUB3 is known to bind.3 Both BUB1 and BUBR1 are directed to the kinetochores via blinkin. Blinkin is a multiprotein macromolecular complex that works as a connecting dock between the kinetochores and BUB1/BUBR1. 7
BUB1 Functions
Although similar in structure, BUB1 and BUBR1 are paralogs because they have different functions and roles in the mitotic checkpoint. The role of BUB1 is primarily: 1) to establish and/or maintain the efficient bipolar attachment of spindle microtubules to the kinetochore of chromosomes and 2) for chromosome congression.
The primary role of the SAC is to ensure that chromosomes are being passed on to the next generation in a reliable manner by serving as the central surveillance mechanism. The SAC halts metaphase to anaphase transition as long as the kinetochore lacks bipolar attachment to the microtubule. Because of this, a highly sensitive signaling pathway is extremely crucial. BUB1 comes into play by acting as the master regulator of forming and signaling SAC. There are several other proteins (such as MAD1, MAD2, MAD3/BUBR1, BUB3, MPsp1) that are also a part of the checkpoint, but many of these proteins are known to interact with BUB1.
Because BUB1 is a multi-domain protein kinases, it has several domains that may function independently of one another. Aside from all the other functions, one of BUB1’s functions is to transport phosphate from ATP to different molecules. Thus, once SAC is activated, BUB1 phosphorylates APC/C’s coactivator called Cdc20. This causes a decrease in activity of APC/C, which is responsible for the metaphase to anaphase transition. APC/C acts in turn by priming BUB1 for degradation so that it can exit mitosis. The N-terminus is extremely vital for an efficient SAC because studies involving a mutation or a deletion of an exon that code for the N-terminus have led to chromosome segregation errors as well as chromosome instability and meager spindle checkpoint responses. The deletion of BUB1 in yeast cells increased the rate of chromosome missegregation, which verifies the role of BUB1 in SAC.3
BUBR1 Function
BUBR1 has a vastly different role. Its roles are associated with fixing incorrectly attached or unattached kinetochores. BUBR1 also comes into play for chromosome alignment. BUBR1 helps stabilize the attachment of microtubules to the kinetochore on a chromosome so that segregation may occur efficiently.3
Connections to Cancer
BUB1 and BUBR1 are important components in helping regulate cell division and the overall cell cycle. Any damages to BUB1 or BUBR1 would lead to a disturbed mitotic checkpoint. A disturbed mitotic checkpoint has been linked to many forms of cancer. The reason for this is because a mutation in the spindle checkpoint leads to cases of aneuploidy and chromosomal instability. Specifically, a reduced gene expression of the BUB1 gene or a mutation of the gene has been proven to correlate with the formation of tumors in the colon, breast, gastric, esophageal, and melanoma. Animal experiments have also pointed to the possibility of BUB1’s involvement in tumor formation. For example, mice with low BUB1 expression have shown an increase for tumor susceptibility. 3
Meiosis is the process in which a cell reduces its cell from being a diploid, having two sets of chromosomes, to being a haploid, having one set of the twenty-three chromosomes, when creating eggs and sperm. A haploid cell will combine with another haploid cell (one from an egg and one from a sperm) to form a diploid cell. This will result in the right number of chromosomes, 46. During this process, abnormalities of the number of chromosomes usually occur in humans.
This process happens in the cell nucleus, where there is a pair of chromosomes from each parent. Chromosomes replicates when the chromosomes from both parents are copied and paired to exchange portions of their own DNA. Chromosomes, that are similar, will pair up. The paired chromosomes will swap portions of their DNA, creating a mix of new genetic material in the offspring's cell. When the nucleus divides up into daughter cells, the chromosome pairs are divided. The daughter nuclei will divide again, resulting in further division of chromosomes. The daughter nuclei will end up having single chromosomes and a new mix of genetic material.
Unlike mitosis, meiosis undergoes two replications rather than one. It is because of these two divisions that allow for the resulting daughter cells to end up with only half of the number of chromosomes, or haploid, from the total amount of chromosomes, or diploid. The steps goes as follows:
Meiosis I
Prophase I: just like in mitosis, during the prophase step, DNA is condensed into very thick like rodes. In additon the nuclear membrane or envelope along with the nucleoli dissappear which allows for free DNA roaming. The spindle apparatuses begin to form ready for the metaphase step. One difference between mitosis prophase and meiosis prophase I is that as the DNA condenses, the chromosomes are visible as tetrads, in other words, homologous DNA pairs, which is not seen in mitosis.
Metaphase I: The tetrads of chromosomes are lined up on the equator of the cell with the spindle apparatus having already been completely formed. It is during prophase I and metaphase I where genetic recombination occurs. When the homologous pairs of DNA, or tetrads, are first lined up against each other, a event called crossing over occurs at the chaismata. Crossing over events happen between neighboring tetrads which swap genetic information with each other. More genetic variation comes from metaphase I specifically in the sense that tetrads can line up completely randomly on the equator of the cell, also known as independent assortment. It is these two events that derive most of genetic variation.
Anaphase I: The tetrads of chromosomes that were previously lined up on the equator are pulled apart in this phase. They are pulled apart in such a fashion that the spindles that are attached to the centromere of all the tetrads lined up on the equator and are pulled apart to each pole on either side of the cell, towards the centrioles. The resulting chromosomes are now two chromatids.
Telophase I: In this phase the chromosomes with two chromatids that have moved to eeither side of the cell decondense and a nuclear envelope begins to form around the genomic material.
Meiosis II
Prophase II: The chromosomes that have two chromatids from the previous meiosis I cycle recondense, with their nuclear envelope and nucleoli disappear similar to prophase I. In addition the spindle forms in this phase.
Metaphase II: The chromosomes with the two chromatids line up at the equator similar to how they lined up in metaphase I. Although genetic variation is seen in this phase as well as it was seen in metaphase I, the degree is very much diminished. In prophase I, crossing over occurred, however in prophase II, since homologous chromosomes are non existent in this phase, the chromatids cannot react with any neighboring pieces of DNA. The only source of variation in this phase is independent assortment of genes on the equator of the cell. The possibility of chromatids either going left or right on the cell and being chosen for fertilization is the source of variation here.
Anaphase II: The chromosomes split by the same mechanics explained in Anaphase I except only one chromatid head moves towards each pole of the cell.
Telophase II: This phase has the same exact mechanics explained in Telophase I. The only difference is that instead chromatids being de-condensed with a nuclear envelop developing around it, it is solely a chromatid head. The cell is now ready to develop into sperm or eggs at this stage.
According to Angelika Amon, a molecular biologist from the Massachusetts Institute of Technology in Cambridge, the leading cause of human birth defects and miscarriages are the mistakes in dividing DNA between daughter cells during meiosis. Miscarriages occur when embryos have an incorrect number of chromosomes and do not go to full term.
The likelihood that chromosomes will not be apportioned properly increases with age in women. Studies have shown that one out of every eighteen babies born to women who are over the age of forty-five has three copies of chromosomes 13, 18, or 21 instead of the normal amount of two. This can lead to birth defects or mutations. An example is that Down Syndrome is caused by three copies of chromosome 21.
Amon studies yeast cells, which separate their chromosomes almost exactly the same way cells of humans do. The exception is that yeast cells' chromosomes separate much faster. A yeast cell copies its DNA and produces daughter cells in about half an hour. Humans cells, on the other hand, takes about a whole day.
Homeobox genes are a large family of similar genes that direct the formation of many body structures during early embryonic development. A homeobox is a DNA sequence found within genes that are involved in the regulation of patterns of anatomical development morphogenesis in animals, fungi, and plants. In humans, the homeobox gene family contains an estimated 235 functional genes and 65 pseudogenes, which are structurally similar genes that do not provide instructions for making proteins. Homeobox genes are present on every human chromosome, and they often appear in clusters. Many classes and subfamilies of homeobox genes have been described, although these groupings are used inconsistently.
They were discovered independently in 1983 by Ernst Hafen, Michael Levine, and William McGinnis working in the lab of Walter Jakob Gehring at the University of Basel, Switzerland; and by Matthew P. Scott and Amy Weiner, who were then working with Thomas Kaufman at Indiana University in Bloomington, Indiana.
Homeobox genes contain a particular DNA sequence that provides instructions for making a string of 60 protein building blocks (amino acids) known as the homeodomain. Most homeodomain-containing proteins act as transcription factors, which means they bind to and control the activity of other genes. The homeodomain is the part of the protein that attaches (binds) to specific regulatory regions of the target genes.
A homeobox is about 180 base pairs long. It encodes a protein domain (the homeodomain) which when expressed (i.e. as protein) can bind DNA. The following shows the consensus 60-residue chain corresponding to homeobox domain, with typical intron positions noted with dashes:
Homeobox genes encode transcription factors that typically switch on cascades of other genes. The homeodomain binds DNA in a sequence-specific manner. However, the specificity of a single homeodomain protein is usually not enough to recognize only its desired target genes. Most of the time, homeodomain proteins act in promoter region of their target genes as complexes with other transcription factors. Such complexes have a much higher target specificity than a single homeodomain protein. Homeodomains are encoded both by genes of the Hox gene cluster and by other genes throughout the genome.
The homeobox domain was first identified in a number of drosophila homeotic and segmentation proteins, but is now known to be well-conserved in many other animals, including vertebrates.
Genes in the homeobox family are involved in a wide range of critical activities during development. These activities include directing the formation of limbs and organs along the anterior-posterior axis (the imaginary line that runs from head to tail in animals) and regulating the process by which cells mature to carry out specific functions (differentiation). Some homeobox genes act as tumor suppressors, which means they help prevent cells from growing and dividing too rapidly or in an uncontrolled way.
Scientists found this short sequence of DNA called the homeobox present in several genes. When geneticists find very similar DNA sequences in the genes of different organisms, it pretty much means that these genes must do something important and useful that evolution keeps using the same sequence over and over and permit very few changes in structure as new species evolve. Hundreds of homeobox genes have been identified in many organisms and the proteins they make turn out to be involved in the early stages of development of many species. Researchers have found that abnormalities in the homeobox genes can lead to extra fingers or toes in humans.
Because homeobox genes have so many important functions, mutations in these genes are responsible for a variety of developmental disorders. For example, mutations in the HOX group of homeobox genes typically cause limb malformations. Changes in PAX homeobox genes often result in eye disorders, and changes in MSX homeobox genes cause abnormal head, face, and tooth development. Additionally, increased or decreased activity of certain homeobox genes has been associated with several forms of cancer later in life.
"What are the homeobox genes?." Genetics Home Reference: Your Guide to Understanding Genetic References. N.p., Oct. 2012. Web. 20 Nov. 2012. <http://ghr.nlm.nih.gov/geneFamily/homeobox>.
Genes can be deemed “on” or “off” depending on whether they are expressed or not. In some cases expressed genes exhibit a specific phenotype or function thus if the phenotype or the function is not observed then the gene is probably not expressed and is therefore “off.”
Chromatin control access to genes within cells. Histones on chromosomes contain chemical tags that act as switches to control access to DNA and turn genes on and off. A direct measure of gene activity is to monitor the production of mRNA by that gene as with microarray chips.
Microarrays are also used to deem whether a potential gene is on or off. A microarray is a collection of DNA spots that measure expression levels of genes. Due to the nature of microarrays, large numbers of genes may be tested at once. The DNA spots are microscopic and contain a specific gene code that is hybridized with cDNA or cRNA or mRNA. The more matches, the more tightly the DNA becomes hybridized. When the non relevant interactions are washed away we are left with hybdridized DNA that can suggest what genes are on or off. Gene expression is shown through hybridization of mRNA with DNA. [11]
Some people that are exposed to HIV, never end up developing AIDS, this leads to the question of why that happens? Researchers discovered a rare genetic variant that protects people from getting AIDS. Scientists think that the rare variant of a gene called CCR5 originally may have been selected during evolution because it made people resistant to an organism unrelated to HIV. However, CCR5 gene variant’s ability to protect against AIDS may contribute to keeping it in the human gene pool in terms of evolution.
The CCR5-delta32 gene codes for protection against the HIV virus in the human blood. The HIV virus binds to blood cells that have normal receptors on the surface. The CCR5-delta32 gene protects by changing the receptors into a smaller version that is in the inside. This makes the HIV virus unable to bind to the blood cell and is therefore rendered useless. The fact that almost 20% of Caucasians have one copy of the CCR5-delta32 gene shows that it is selected for evolutionarily. The gene came out of mutation, but became fixed in the gene pool due to a benefit. Resistance to HIV is not logical because HIV is a relatively new phenomenon, and therefore can not be the cause of this evolutionary selection. The current thought is that the CCR5-delta32 gene provided protection from previous epidemics. That would explain why it has such a high prevalence in northern Europe. Also, there is almost no CCR5 gene in the Asian or African population. This suggests that the CCR5-delta32 gene was not for the prevention of HIV due to high prevalence populations lacking the gene. [4]
↑U.S. Department of Health and Human Services. The New Genetics. October 2006.<http://www.nigms.nih.gov>.
A structural variation (SV) is a alteration of the genome in an organism's chromosome. These variations may be deletions, duplications, copy-number variants, inserts, inversions or translocation. Regions of alteration can range from as small as 1Kb to 3Mb in size.
In chromosomal duplication, extra copies of a region in the chromosome are formed. If these duplications are side by side, this is known as tandem duplication. If they are separated, however, they are said to be displaced.
If an organism has several copies of the same gene, its phenotype may be altered. As the gene segments are multiplied, so are the chances of that gene being transcripted, translated and expressed. As a result, people may produce more protein than others. This may play either a detrimental or beneficial role in the organism's physiology.
Organisms may also benefit from this anomaly in that it gives them extra genes that may mutate into other vital genes necessary for evolutionary fitness.
Deletion is a mutation in the chromosome that results in a loss of DNA sequences. Depending on the size of the deletion, the organism may experience various phenotypes. Similar to duplication, deletion may alter gene dosage.
Deletion can be extremely harmful to an organism, depending on the size and location. Some phenotypes require two genes to be present for normal expression. Haploinsufficiency is the term used to describe a mutant phenotype that only has one of the two genes.
Amos-Landgraf, J. M., et al. Chromosome breakage in the Prader-Willi and Angelman syndromes involves recombination between large, transcribed repeats at proximal and distal breakpoints. American Journal of Human Genetics 65, 370–386 (1999)
Christian, S. L., et al. Large genomic duplicons map to sites of instability in Prader-Willi/Angelman syndrome chromosome region (15q11-q13). Human Molecular Genetics 8, 1025–1037 (1999)
Cancer is a set of diseases in which cells escape from the control mechanisms normally limiting their growth. Cancer cells ignore the normal signals that operate the cell cycle and affect the body by dividing uncontrollably and invading surrounding tissues. The gene regulation systems that go wrong during cancer turn out to be the very same systems that play important roles in embryonic development, the immune response, and many other biological processes.The genes that normally regulate cell growth and division during the cell cycle include genes for growth factors, their receptors, and the intracellular molecules of signaling pathways. mutations that alter any of these genes in somatic cells can lead to cancer.
Nearly all cancers are caused by abnormalities in the genetic material of the transformed cells. These abnormalities may be due to the effects of carcinogens, such as tobacco smoke, radiation, chemicals, or infectious agents. Other cancer-promoting genetic abnormalities may be randomly acquired through errors in DNA replication, or are inherited, and thus present in all cells from birth. The heritability of cancers is usually affected by complex interactions between carcinogens and the host's genome. New aspects of the genetics of cancer pathogenesis, such as DNA methylation, and microRNAs are increasingly recognized as important.
Cancer is simply the gain of oncogenes and the loss of tumor suppressor genes. Genetic abnormalities found in cancer typically affect two general classes of genes. Cancer-promoting oncogenes are typically activated in cancer cells, giving those cells new properties, such as hyperactive growth and division, protection against programmed cell death, loss of respect for normal tissue boundaries, and the ability to become established in diverse tissue environments. Activated oncogenes result form different types of mutations including point mutations, chromosomal translocations, promoter translocations, and amplifications. Point mutations increase proteins' activity and prevent it from being turned off. Chromosomal translocations encode fusion proteins that are hyperactivated or inappropriately regulated or localized. Promoter translocations drive abnormally high levels of expression. Amplifications increase the copy number and expression of a gene. Tumor suppressor genes (TSG) are then inactivated in cancer cells, resulting in the loss of normal functions in those cells, such as accurate DNA replication, control over the cell cycle, orientation and adhesion within tissues, and interaction with protective cells of the immune system. Tumor suppressor genes can be inactivated by missense mutations that decrease the protein's activity, deletions, frameshifts, and promoter methylation.
Diagnosis usually requires the histologic examination of a tissue biopsy specimen by a pathologist, although the initial indication of malignancy can be symptoms or radiographic imaging abnormalities. Most cancers can be treated and some cured, depending on the specific type, location, and stage. Once diagnosed, cancer is usually treated with a combination of surgery, chemotherapy and radiotherapy. As research develops, treatments are becoming more specific for different varieties of cancer. There has been significant progress in the development of targeted therapy drugs that act specifically on detectable molecular abnormalities in certain tumors, and which minimize damage to normal cells. The prognosis of cancer patients is most influenced by the type of cancer, as well as the stage, or extent of the disease. In addition, histologic grading and the presence of specific molecular markers can also be useful in establishing prognosis, as well as in determining individual treatments.
The word cancer comes from the Greek physician Hippocrates. He used the Greek words "carcinos" and "carcinoma", meaning crab in Greek, to describe cancer because he believed that tumors resembled crabs. The two words were combined into one: "karkinos".
The oldest record of cancer is from the Egyptians around 1500 B.C. The Edwin Smith Papyrus documents 8 cases of breast tumors treated with cauterization, a method of destroying tissue with a hot instrument called a "fire drill". Records indicate that there were no treatments for the disease. In addition, archaeological finding has discovered mummies with fossilized bone tumors suggestive of bone cancer. Also in the Papyrus, it is suggested that Egyptian physicians could differentiate malignant tumors from benign tumors.
Later, the city of Constantinople became the Medical Center of the World. During this time period, the cause of cancer was attributed to an excess of black bile. This was based on Hippocrates's idea that the human body was composed of four different types of bodily fluids: blood, phlegm, yellow bile and black bile. Any excess or deficiencies in either fluid would cause disease, and in this case, excess of black bile was believed to cause cancer. This was the knowledge for the next 1400 years, through the Middle Ages, and went unchallenged since religious superstition at the time prohibited doctors from performing autopsies.
In the 15th Century Renaissance, physicians began to acquire more knowledge on human physiology. Giovanni Morgangni, in the 16th century, began to do autopsies on the bodies of the deceased in order to discover a pathological relationship between disease and death. Also, during this time, a Scottish physician named John Hunter suggested that cancerous tumors may be removed by surgical means. In the 17th Century, the Black Bile theory was replaced with another theory called the Lymph theory. The idea was that cancer was caused by degenerating and fermating lymph.
In the 19th Century, Rudolf Virchow, who is considered the father of cellular pathology, linked the clinical course of illnesses with pathological findings. This discovery allowed physicians to not only assess the damage of a particular cancer on the body, but also laid the foundations of cancer surgery. Tissues removed from the infected area could rapidly and quickly assessed to determine the type of cancer and also to elucidate whether or not the tumor was completed excised.
The 20th Century saw a rise in cancer knowledge. Research on carcinogens, radiation therapy, and better means of identification were discovered. There was tremendous scientific improvement on the understanding of cell growth and division mechanisms.
There are many different kind of cancer diverse by the type of cell that made up the tumor and therefore the tumor is known as the origin of cancer. A benign tumor is a lump of abnormal cells that do not spread to other cells or other parts of the body, but rather remains at the original location that it was formed. However, a malignant tumor is a more dangerous form because it is able to invade and damage the functions of other cells and organs.
When cancers are derived from the epithelial cells whose genome has altered or damaged, we called it Carcinoma. It is the most common type of cancer occurring in humans and it begins in a tissue that lines the inner or outer surfaces of the body. The cells will begin to exhibit abnormal malignant properties. The most common example is the breast, lung, prostate cancers.
The second type is called Sarcoma which the cancers are arising from the connective tissue. It develops develop from cells originating in mesenchymal cells outside the bone marrow. Therefore Sarcoma includes tumors of bone (osteosacrcoma), fat (liposacrcoma), muscle (leiomyosarcoma) and vascular. There are two classes of cancer arise from the blood forming cells.
Leukemia is a type of cancer of the blood or bone marrow characterized by an abnormal increase of white blood cell. The cause of such cancer is also by mutation in the DNA that can trigger leukemia by activating oncogenes or deactivating tumor suppressor genes.
Lymphoma is a cancer that develops in the immune system. It usually presents to be a solid tumor in the lymphoid cells. The malignant cells often originate in the lymph node and presents as an enlargement of a tumor. It will also affect other organs such as skin, brain, bowels and bone.
Cancer also appears in the pluripotent cell in the testicle or the ovary. This is called the germ cell tumor. There are two types of germ cell tumor either cancerous or non-cancerous. It may cause by error in development of the embryo.
The last type is the Blastoma which is developed from immature embryonic tissue. This is most common in children.
Uncontrolled Cell Division Cancer cells ignore signals that would regulate cell growth and division in the body. Since the cells grow and divide uncontrollably, it leads to the production of more and more cancer cells within the body.
Evading Apoptosis Cancer cells are able to avoid the process of apoptosis for normal cells. Apoptosis is the process of programmed death carried out in normal cells, but cancer cells are able to evade the process and can therefore continue to progress.
Independent from Growth Regulation Cancer cells are self-sufficient and do not require external signals to regulate its growth and division. Cancer cells are also capable of ignoring negative signals from its neighbors and can therefore continue to grow and divide on its own.
Angiogenic The cancer cells that make up the tumor need a system to provide nutrients and dispose of waste. Therefore cancer cells have become angiogenic, in which the tumor attracts blood vessels to grow into the tumor mass and nourish the cancer cells.
Immortality Cancer cells have developed the ability to proliferate indefinitely. By doing so, cancer cells have become immortalized and are capable of indefinite growth and cell division.
Invasion and Metastasis Cancer cells are capable of entering the stage of metastasis and invade surrounding cells and tissues. By developing the ability invade and metastasize, cancer cells are able to spread and establish the disease in other areas of the body and not just the original location.
In 1910, an American researcher, Peyton Rous, discovered that the Rous sarcoma virus (Rous virus)could cause cancer and he studied how this cancer could spread in chickens. He performed three experiment to test how this cancer spread.
He took out cancerous cells from one chicken and injected into a healthy chicken. He observed that the healthy chicken became afflicted with cancer.
He purified the cell to create cell-free extracts. He then injected the extract into a healthy chicken. Again, the chicken contracted cancer.
He used a special filter, that had holes too small for viruses to pass through, to filter the cells. He inject this extract into healthy chickens and discovered that they did not contract cancer, and remained healthy.
From his experiments, he concluded that viruses can cause cancer.
Later, scientists discovered that the Rous virus is a retro-virus. A retro-virus can insert its gene into a host DNA and uses host protein mechanisms to reproduce. The viral DNA inside of the host cell causes uncontrollable cell division, cell growth, etc.
Oncogenes are genes that have the potential to cause tumors. [1]
Classification
Although there are several different systems for classifying the oncogenes, these classifications are yet to be accepted standard. Some are grouped spatially or chronologically. The first category, growth factors (mitogen), include cancer genes such as fibrosarcomas, osteosarcomas, breast carcinomas, and melanomas. These genes induce cell proliferation. The growth factors from specific cells trigger cell proliferation not only in themselves but also nearby and distant cells. The Receptor tyrosine kinases cause breast cancer, non-small-cell lung cancer and pancreatic cancer. These receptor tyrosine kinases genes transduce signals for cell growth. The receptor tyrosine kinases add phosphate groups to other proteins. This can either turn the proteins permanently on or off. The cytoplasmic tyrosine kinases category include breast cancers, melanomas, ovarian cancers, head and neck cancers, blood cancers and brain cancers. These genes are in charge of the responses back and forth from active receptors of the cells that mediate proliferation, migration, differentiation and survival. The cytoplasmic serine/threonine kinases are responsible for malignant melanoma, colorectal cancer, and ovarian cancer. These genes are involves in cell cycle regulation, cell proliferation, cell survival, and apoptosis. More than 125 of the human protein kinases are serine/threonine kinases. The regulatory GTPases genes are involved in leading pathways to cell proliferation. An example of the regulatory GTPases include the Ras protein. The Ras hydrolyses GTP into GDP and phosphate and is responsible for adenocarcinomas of the pancreas and colon, thyroid tumors, and myeloid leukemia. Lastly, the transcription factors regulate the transcription of genes that trigger cell proliferation. For example, the myc gene can cause cancers such as small cell lung cancer, breast cancer, acute myeloid leukemia and malignant T-cell lymphomas.
Cancer cannot be diagnosed accurately by one single test. A complete and thorough history and physical examination along with several diagnostic tests must be performed in order to evaluate whether the patient has cancer or other conditions are being misinterpreted as symptoms of cancer.
An effective procedure of testing can be used to confirm or exclude the presence of cancer, determine the disease process and preliminary plan for treatments. Tests needed to be repeated if the patient’s symptoms have changed or the testing sample is not qualified or the test results show abnormality.
Diagnostic procedures for cancer may include imaging, laboratory tests, tumor biopsy, and endoscopic examination.
Diagnostic imaging is the process of obtaining pictures of body structures and organs. It is used to detect tumors and other abnormalities and their extent. It is also the most applicable way to determine the effectiveness of the treatments.
Transmission imaging In transmission imaging, a beam of photons with high energy is generated and allowed to pass through the body structure being examined.
X-ray X-rays use electromagnetic energy beams to produce images of internal tissues, bones, and organs on film. Based on the images, tumor or cancer cells can be located.
Computed Tomography scan (CT scan or CAT scan) A CT scan is more detailed than general X-ray scan. By combining x-rays and computer technology, images of bones, muscles, fat, and organs in the body are showed with more details.
Bone scan Bone scans are pictures of X-ray or CT scan taken of the bone after a dye has been injected to bone tissue. These scans are used to detect tumors and abnormalities in the bone structure.
Lymphangiogram(LAG) Lymphangiogram is an image that can detect cancer cells or abnormalities in the lymphatic system and structures after a dye is injected into the lymph system.
Mammogram A mammogram is an x-ray image of the breast. It is used to detect and diagnose breast disease in women by locating abnormal area. A biopsy is required for further diagnosis.
Reflection Imaging In Reflection Imaging, images are produced by high-frequency sounds bouncing off of the surface of body tissues and structures at varying speeds, depending on the density of the tissues present. The bounced sound waves are then analyzed by a computer and a visual image is produced.
Emission Imaging MRI combine a large magnet and a computer to produce detailed images of the heart, brain, liver, pancreas, male and female reproductive organs, and other soft tissues.
Blood Tests Blood tests are used to check the levels of substances that are indicative of how healthy the body is and whether infection is present. Other tests check for the presence of electrolytes such as sodium and potassium that are critical to the body's healthy functioning.
Urinalysis Urinalysis breaks down the components of urine to check for the presence of drugs, blood, protein, and other substances.
Tumor markers Tumor markers are substances released by cancer cells or substances created by the body in response to cancer cells.
Prostate-specific Antigen (PSA) An elevated PSA level in the blood may indicate prostate cancer, but other conditions such as benign prostatic hyperplasia (BPH) and prostatitis can also raise PSA levels.
CA 125 Ovarian cancer is the most common cause of elevated CA 125, but cancers of the uterus, cervix, pancreas, liver, colon, breast, lung, and digestive tract can also raise CA 125 levels.
Prostatic acid phosphatase (PAP) In addition to prostate cancer, elevated levels of PAP may indicate testicular cancer, leukemia, and non-Hodgkin’s lymphoma, as well as some noncancerous conditions.
Human chorionic gonadotropin (HCG) If pregnancy is ruled out, HCG may indicate cancer in the testis, ovary, liver, stomach, pancreas, and lung.
Carcinoembryonic Antigen (CEA) Colorectal cancer is the most common cancer that raises this tumor marker. Several other cancers can also raise levels of carcinoembryonic antigen.
Alpha-fetoprotein (AFP) In men, and in women who are not pregnant, an elevated level of AFP may indicate liver cancer or cancer of the ovary or testicle. Noncancerous conditions may also cause elevated AFP levels.
CA 19-9 Elevated levels of CA 19-9 may indicate advanced cancer in the pancreas, but it is also associated with noncancerous conditions, including gallstones, pancreatitis, cirrhosis of the liver.
CA 27-29 Cancers of the colon, stomach, kidney, lung, ovary, pancreas, uterus, and liver may also raise CA 27-29 levels. Noncancerous conditions associated with this substance are first trimester pregnancy, endometriosis, ovarian cysts, benign breast disease, kidney disease, and liver disease.
CA 15-3 Elevated levels of CA 15-3 are also associated with cancers of the ovary, lung, and prostate, as well as noncancerous conditions such as benign breast or ovarian disease, endometriosis, pelvic inflammatory disease, and hepatitis. Pregnancy and lactation also can raise CA 15-3 levels.
Neuron-specific enolase (NSE) NSE is associated with several cancers, but it is used most often to monitor treatment in patients with neuroblastoma or small cell lung cancer.
A biopsy is the removal of tissues or cells from the patient’s body for examination under a microscope. Biopsies are usually performed to determine whether a tumor is cancerous or just an infection or inflammation.
Endoscopic biopsy This type of biopsy is performed through a fiberoptic endoscope (a long, thin tube that has a close-focusing telescope at the end for closeup observation) through a natural body orifice or a small incision. The endoscope is used to view the organ in question for abnormal or suspicious areas, in order to obtain a small amount of tissue for study.
Bone marrow biopsy This type of biopsy is performed either from the sternum or the iliac crest hipbone. A needle is inserted into the marrow, and cells are taken for study.
Excisional biopsy This type of biopsy is often used when a wider or deeper portion of the skin is needed. Using a scalpel, a full thickness of skin is removed for further examination.
Fine needle aspiration (FNA) biopsy This type of biopsy involves using a thin needle to remove very small pieces from a tumor. FNA is not used for diagnosis of a suspicious mole, but may be used to biopsy large lymph nodes near a melanoma to see if the melanoma has metastasized (spread).
Punch biopsy Punch biopsies involve taking a deeper sample of skin with a biopsy instrument that removes a short cylinder of tissue.
Skin biopsy Skin biopsies involve removing a sample of skin for examination under the microscope to determine if melanoma is present.
An endoscope is a small, flexible tube with a light and a lens on the end used to look into the esophagus, stomach, duodenum, colon, or rectum. It can also be used to take tissue from the body for testing or to take color photographs of the inside of the body.
Colonoscopy
Colonscopy involves inserting a colonoscope, which is a long, flexible, lighted tube, in through the rectum up into the colon. It allows the physician to view the entire length of the large intestine, and can often help identify abnormal growths as well as inflamed tissue, ulcers, and bleeding. The physician can also remove tissue for further examination.
ERCP is a procedure that allows the physician to diagnose and treat problems in the liver, gallbladder, bile ducts, and pancreas. The procedure combines x-ray and the use of an endoscope. The scope is inserted through the patient's mouth and throat, then through the esophagus, stomach, and duodenum.
Esophagogastroduodenoscopy (EGD)
An EGD is a procedure that allows the physician to examine the inside of the esophagus, stomach, and duodenum. An endoscope is guided into the mouth and throat, then into the esophagus, stomach, and duodenum. The endoscope allows the physician to view the inside of this area of the body, as well as to insert instruments through a scope for the removal of a sample of tissue for biopsy.
Sigmoidoscopy
A sigmoidoscopy is a diagnostic procedure that allows the physician to examine the inside of a portion of the large intestine, and is helpful in identifying the causes of diarrhea, abdominal pain, constipation, abnormal growths, and bleeding. A short, flexible, lighted tube, called a sigmoidoscope, is inserted into the intestine through the rectum. The scope blows air into the intestine to inflate it to have a better view of the inside.
Cystoscopy
An examination in which a scope is inserted through the urethra to examine the bladder and urinary tract for structural abnormalities or obstructions, such as tumors or stones.
Surgery is one of the initial treatments used to operate on cancer cells. Cancer surgery attempts to prevent the spread of cancer cells by locating the tumor and removing it along with any possible lymph nodes that are near the source location. This method is commonly used for cases with benign tumors because the mass of cancer cells are typically located in one area. Surgery may be the only treatment needed for some cancer cases, but if a few cancer cells have broken off or get left behind, then it would only be a matter of time before the disease returns and more treatments are needed.
Chemotherapy is another treatment used in addition to surgery if cancer cells still remain in the body. Chemotherapy is used to treat cancer cells that have entered the stage of metastasis, in which the cells have spread from their original location. The treatment utilizes drugs that are toxic to interfere and kill cells that divide. The goal is to kill the cancer cells faster than the normal dividing cells in the body because cancer cells divide more rapidly. Even though chemotherapy has been proven to be effective, the side effects include hair loss and nausea because the treatment is blasting every cell being divided including the normal cells.
Radiation therapy for cancer utilizes a beam of high-energy particles to kill off the cancer cells located in the body. This treatment targets the cancer areas by marking the skin, thus enabling the beam of high-energy particles to directly hit and destroy the cancer cells. The radiation from this therapy can shrink the tumors and also relieve the symptoms caused by the cancer. Although this therapy benefits cancer patients, it still has side effects and risks of causing new problems for these patients. The side effects caused by the radiation depends on the area of the body that undergoes the therapy; and this area is also the main location of the side effects.The radiation focuses on the tumor, but since the ray of high-energy particles targets the skin as well, a common side effect caused from radiation therapy is that the skin that was marked turns red and gradually may look more dark or tan over the years. The skin may also dry and flake, like a burn to the skin, during the period of recovery after the radiation therapy.
Monoclonal antibodies serve as a method in cancer therapy to enforce the immune system and aid in diagnosis. Monoclonal antibodies are created by injecting human cancer cells into mice so that they are able to produce antibodies against the foreign antigens invading their immune system. From there, the murine cells producing the antibodies are then removed and combined with laboratory-grown cells. This combination creates hybrid cells called hybridomas, which in turn is capable of producing large quantities of these pure antibodies so that the human body is able to process and use these antibodies to fight off the cancer cells.
In a prokaryote that divides by the use of a Z-Ring, such as E. coli, the Z-Ring would form at the interface between the two dividing daughter cells. The dotted lines above would be the site of the Z-Ring in E. coli.
What are Minicells
Minicells are small non-chromosomal "cells" that result from abnormalities during cell division in prokaryotes. They do not contain any of the original DNA present in its larger sister, but may contain proteins and copies of transformed plasmids that were present in the are in the original cell when the minicell formed. First discovered over 70 years ago, minicells are becoming of interest to researchers in their potential as anti-tumor agents1. In order to understand how minicells form, it is important to understand how most prokaryotes mediate cell division. During the cytokinesis stage of cell division, a framework of proteins form at the site of division. The main protein involved in this process, FtsZ, forms what is called a Z-Ring around the septum of division. FtsZ is "tethered" to the membrane by a series of other proteins involved in the process of cytokinesis. The Z-Ring usually aligns itself at the middle of the enlarged cell as a systems of proteins inhibit the formation of the Z-Ring at the poles of the cell. Abnormalities in this regulatory system for cytokinesis results in the formation of the Z-Ring near the poles of the cell. This results in two dimorphic daughter cells: one larger with all of the bacterial DNA, and another smaller one with no chromosomal DNA. It is important to note that not all prokaryotes divide in cytokinesis by the use of a Z-Ring, and the mechanism by which they divide is still unknown. Minicells can form under certain growth conditions or from mutations. E. coli is usually used to make minicells.
Using Minicells to Treat Tumors
Brahmbhatt et al. reported that minicells can be used in treating drug resistant tumors by using siRNA and cytotoxic drugs2, what follows is a summary of their findings. The goal of the research was to demonstrate that minicells could be targeted to tumor cells, and release cytotoxic drug and siRNA specific to drug resistant genes. It is known the RNA interference caould be a powerful tool in fighting cancer, however there is a need to find an efficient method to transfer siRNA or microRNA to cancerous cell in vivo without them being degraded or causing adverse effects. It was observed that minicells had the ability to uptake cytotoxic drugs and siRNA. The minicells could also contain plasmids encoding siRNA by harvesting them from bacteria (E. coli) transformed with that specific plasmid. Minicells have the ability to uptake siRNA and to contain it for long periods of time. Specific antibodies (BsAB) could also be incorporated into the minicell to help target tumor cells. They had showed previously that minicells could bind to the tumor cells via specific antibodies, and initiate endocytosis via receptors on the cell surface. Once inside the cell, the drug contained in the minicells could be released. The same idea was again applied to siRNA with the ability to interfere with drug resistance genes in the tumor cells. The minicells would contain siRNA, or siRNA encoding plasmids, and would use the same antibody to bind to the receptors of tumor cells. It was shown that in vitro, the siRNA delivery via minicells could diminish drug resistance in cells containing a drug resistance gene.
siRNA is capable of inhibiting the expression of a certain gene by splicing the transcribed mRNA that originates from that gene. The siRNA discussed here interferes with the mRNA of MDR1 (multi drug resistance gene.)
This was done by delivering the minicells full of siRNA to a type of colon cancer cell overexpressing MDR1 (multi drug resistance gene), and then treating the cells with drugs in order to measure toxicity of the drug to the cell. This resulted in a highly significant increase of toxicity. In vivo experiments were done on human cancer xenografts in mice and showed that the tumor cells treated with a wave of siRNA containing minicells, experienced a knockdown of the drug resistance gene which was targeted. The results were confirmed by performing a Western Blot on protein in the cells.
The next step was to show the efficacy of first treating mice with a wave of siRNA and the cytotoxic drug using minicells. Mice with the Caco-2/MDR1 (colon cancer cells with multi drug resistance gene) were treated siRNA containing mincells to reverse drug resistance by knocking out the MDR1 and thereafter treatment with drug containing minicells. Their analysis showed that all the mice which had under gone this dual treatment exhibited a significant inhibition of cancerous growth and complete survival. This result also showed that after the initial minicell treatment with siRNA, the tumor cells were still able to process the next wave of minicells (no adverse effects on the cellular machinery). However, the mechanism by which siRNA is loaded with minicells or possible effects on the immune system are not yet known, especially since they originated from bacteria. No adverse effects were reported in the in vivo studies, however. It was also concluded that overall, using this sequential method of treating the tumors with minicells, a smaller amount of drug is needed than for the case in which the drug is administered without the siRNA containing minicells.
PKM2 is an enigmatic enzyme that catalyzes the last step of glycolysis, converting phosphoenolpyruvate to pyruvate and phosphorylating ADP to Atp. There are four types of PK in mammals, all encoded by these two genes: PKLR and PKM2.
PK has not much evolved during evolution, as the four isoforms in mammals are very similar in sequence. In cancer cells, PKM2 takesover the PK isoform until it becomes the main isoform, which supports the idea of tumorigenesis. A study was then in which PKM2 was replaced with PKM1, and tumor growth was delayed, supporting the hypothesis. However, tumor cells can also switch the expression of PKL/R to PKM isoforms, but this hypothesis has not been scientifically validated. So the specific PKM isoform in primary tumors still need to be identified. PKM2 can jump between two states: a tetrameric form and a dimer. The dimer is present in cancer cells. The tetramer may in fact be a dimer of the dimer. Though PKM2 is highly present in cancer cells, it may not be the only PK isoform in cancer, and it may not be cancer specific.
Other isoforms are allosterically regulated. In PKM2, it is the main PK isoform, and the ratio between the active and inactive forms mold the usage of glucose for energy production or anabolic precursors. PKM2 is complex regulator. It has been shown to also be phosphorylated on tyrosine residues in the low activity state. In the high activity state, PKM2 also forms complexes with other glycolytic enzymes such as GADPDH, PGAM, and LDH. It can also control pyruvate and induce lactate production. Tyrosine phophorylated polypeptides can also inhibit PKM2 binding, releasing activator FBP. These events are seen often in cancer and can control glucose metabolism. There are many mechanisms to inhibit PKM2, which is useful for survival under cell stress and can explain why PKM2 is the main enzyme in cancer cells. PKM2 also is regulated by nutrients and other growth factors, which allows it to meet the demands metabolcity of cancer cells.
PKM2 is also present in the nucleus, due to a nuclear localization signal in the C0terminal domain. The functions of nuclear PKM2 are not clear. It is required for cell survival after interleukin stimulation, but is also necessary for agoptosis. Nuclear PKM2 also can interact with transcription factors and epidermal growth factors. PKM2 binds to phophorylated beta catenin and promotes transcription in the nucleus. It is activated by HIF-1. However, in cancer cells, a regulatory feedback mechanism seems to occur where instead, PKM2 activates HIF-1 instead, leading to activation of genes for glucose transporters and glycolytic enzymes.
In cancer treatment, PKM2 is a target, so an understanding of its regulation can improve pharmaceutical drugs aimed to counteract or mimic its effects. PKM2 is highly expressed in cancer cells, but many post-translational modifications also suggest that inhibiting PKM2 may promote cell proliferation. Therefore, both PKM2 inhibiting and activating drugs have been synthesized to target tumor cells. A study suggested that replacing PKM2 isoform with PKM1 may also result in a significant reduction of cancerous cell growth, but no evidence of PKM2 activators providing a efficient treatment have yet been performed. Thus, many challenges remain in this field of research today.
↑Wilbur, Beth, editor. The World of the Cell, Becker, W.M., et al., 7th ed. San Francisco, CA; 2009
2. "Sequential treatment of drug-resistant tumors with targeted minicells containing siRNA or a cytotoxic drug", 2009, Nature, MacDiarmid, J.A., Amaro-Mugridge, N.B., Madrid-Weiss, J., Sedliarou, I., Wetzel, S., Kochar, K., Brahmbhatt, V.N., (...), Brahmbhatt, H.
3."Assembly Dynamics of the Bacterial MinCDE System and Spatial Regulation of the Z Ring", Joe Lutkenhaus
4. Campbell NA, Reece JB. 2008. Biology. 8th ed. San Francisco (CA): Pearson/Benjamin Cummings.
8. "Caring with the Patient with Cancer at Home". <http://www.cancer.org/treatment/treatmentsandsideeffects/physicalsideeffects/dealingwithsymptomsathome/caring-for-the-patient-with-cancer-at-home-radiation-therapy>. Web 28 Oct 2012.
Many cancers are caused by the defective repair of DNA and by mutations in genes associated with growth control. Defects in DNA-repair systems increase the overall frequency of mutations and, hence, the likelihood of cancer-causing mutations. Indeed, the synergy between studies of mutations that predispose people to cancer and studies of DNA repair in model organisms has been tremendous in revealing the biochemistry of DNA-repair pathways.
A DNA repair mechanism.
Genes for DNA-repair proteins are often tumor-suppressor genes; that is, they suppress tumor development when at least one copy of the gene is free of a deleterious mutation. When both copies of a gene are mutated, however, tumors develop at rates greater than those for the population at large. People who inherit defects in a single tumor-suppressor allele do not necessarily develop cancer but are susceptible to developing the disease because only the one remaining normal copy of the gene must develop a new defect to further the development of cancer.
Cancer cells often have two characteristics that make them especially vulnerable to agents that damage DNA molecules. First, they divide frequently, and so their DNA replication pathways are more active than they are in most cells. Second, cancer cells often have defects in DNA-repair pathways. Several agents widely used in cancer chemotherapy, including cyclophosphamide and cisplatin, act by damaging DNA. However, because chemotherapy drugs are largely non-specific, this results in damage to healthy cells and other side effects.
There are a few human disorders characterized by defects in DNA repair.
1. Xeroderma pigmentosm – the patients with XP have clinical sun sensitivity, a lot of freckle-like lesions due to sun explosion and about 1000-fold increase of developing skin cancer. Several of XP patients have progressive neurologic degeneration. XP cells are very sensitive to UV and have defected DNA repair. The defective genes in XP are involved in nucleotide exclusion repair.
Nucleotide exclusion repair (NER) is a process that is carried out by proteins; these proteins' codes are in the genes, XPA-G, in which are the genes that are affected by mutations that are linked with XP cases. This process (NER) is a method by the cells to get rid of dangerous injuries created in the genetic components by UV rays from the sun. So, for patients with XP, this process does not work correctly. [1]
2. Cockayne syndrome – the patients with this syndrome have sun sensitivity, short stature, and progressive neurologic degeneration. Unlike XP, Cockayne syndrome patients are very sensitive to killing by UV and have defective DNA repair of actively transcribing genes. It also contains two complementation groups. The defective genes also are involved in both nucleotide exclusion and transcription like XP, yet the precise function is unknown.
Roughly half of CS patients will display a changed cellular response to UV rays as a result of mutations in the genes, CSA and CSB. The genes CSA and CSB both code proteins that are related in repairing a particular portion of the DNA that is in current use by the cell.[1]
3. Trichothiodystrophy – the patients with TTD have photosensitivity, short stature, mental and growth retardation and sulphur deficient brittle hair. It does not associate with cancer. TTD patients’ cells are also very sensitive to killing by UV and have defective DNA exclusion repair. The defective genes in TTD are found in one of the XP complementation group.
About half of TTD patients, who display sun sensitivity, have a defect in their nucleotide excision repair. This is a result from mistakes in one of three genes, which are the following: XPB, XPD, and TTDA. With recent findings, there are patients with non-photo-sensitivity, due to a defect in the gene C7orf11(TTDN1). [1]
BRCA1 is a gene that is involved in DNA repair via homologous recombination. When one allele of this gene is mutated, there is a 85% risk for breast cancer and 30-50% risk for ovarian cancer. BRCA1 produces the BRCA1 tumor suppressor protein, which is made up of 1863 amino acids. This protein can be divided into several domains: RING, exon 11, coiled-coil doman, and BRCT.
RING - This domain consists of the first 150 amino acids of BRCA1. BRCA1 heterodimerizes with BARD1, BRCA1-associated RING domain protein.
Exon 11 - This domain is the middle 60% of the protein.
Coiled-coil domain - This domain is associated with PalB2 and its interactions with BRCA2, another tumor suppressor protein. PalB2 connects BRCA1 and BRCA2 together. It is also active in homologous repair of BRCA1 and BRCA2.
BRCT - This domain interacts with phosphoserine residues on binding partners and facilitates in DNA double-strand breaks localization.
BRCA1 and BRCA 2 are proteins that are closely associated with breast and ovarian cancer. The gene’s main job is to repair any double stranded breaks (DSB) and fight against cancer caused by mutations on the DSBs. BRCA1 and BRCA2 do not work independently to repair the breaks, but have specific jobs and work through a protein network.
BRCA1 acts as a coordinator of all the proteins necessary to repair the break because of its and damage sensing abilities. It first binds to the DSB using RING (Really interesting new gene), which is encoded and forms the BARD1 complex. If the RING part of the BRCA1 protein is mutated or disabled, BRCA1 becomes unable to bind to a DSB and the likelihood of cancer increases.
BRCA1, the coordinator, then recruits PalB2 that in turn attaches BRCA2 to the complex. BRCA2, unlike BRCA1, has a large role in repairing DSBs and is known as an effector that uses Rad51 to help use homologous recombination to repair the DSBs. When the protein complex fails to use homologous recombination, Non-homologous end joining (NHEJ) is used- often leading to further complications.
Ultimately, to best repair DSBs, the BRCA1-PalB2-BRCA2 complex needs to be fully functioning.[2]
Cancers are classified in two ways: by the type of tissue in which the cancer originates (histological type) and by primary site, or the location in the body where the cancer first developed. This section introduces you to the first method: cancer classification based on histological type. The international standard for the classification and nomenclature of histologies is the International Classification of Diseases for Oncology, Third Edition (ICD-O-3).
By primary site of origin, cancers may be of specific types like breast cancer, lung cancer, prostate cancer, liver cancer renal cell carcinoma (kidney cancer), oral cancer, brain cancer etc.
Cancers can also be classified according to grade. The abnormality of the cells with respect to surrounding normal tissues determines the grade of the cancer. Increasing abnormality increases the grade, from 1–4.
Cells that are well differentiated closely resemble normal specialized cells and belong to low grade tumors. Cells that are undifferentiated are highly abnormal with respect to surrounding tissues. These are high grade tumors.
Grade 1 – well differentiated cells with slight abnormality
Grade 2 – cells are moderately differentiated and slightly more abnormal
Grade 3 – cells are poorly differentiated and very abnormal
Grade 4 – cells are immature and primitive and undifferentiated
Cancers are also classified individually according to their stage. There are several types of staging methods. The most commonly used method uses classification in terms of tumor size (T), the degree of regional spread or node involvement (N), and distant metastasis (M). This is called the TNM staging.
For example, T0 signifies no evidence of tumor, T 1 to 4 signifies increasing tumor size and involvement and Tis signifies carcinoma in situ or limited to surface cells. Similarly N0 signifies no nodal involvement and N 1 to 4 signifies increasing degrees of lymph node involvement. Nx signifies that node involvement cannot be assessed. Metastasis is further classified into two – M0 signifies no evidence of distant spread while M1 signifies evidence of distant spread.
Stages may be divided according to the TNM staging classification. Stage 0 indicates cancer being in situ or limited to surface cells while stage I indicates cancer being limited to the tissue of origin. Stage II indicates limited local spread, Stage II indicates extensive local and regional spread while stage IV is advanced cancer with distant spread and metastasis.
Carcinoma, the most common type of cancer that occurs in humans, is the classification of cancer that is derived from epithelial cells. It refers to the tumor tissue that is composed of epithelial cells that have altered or damaged genomes that lead the cell to exhibit detrimental and malignant properties.
Adrenocortical carcinoma, which arises from the adrenal cortex, the outside layer of the adrenal gland. The cells in the adrenal cortex make hormones that help the body work properly, such as steroids and hormones important for sexual development and maturation. When cells in the adrenal cortex become cancerous, they may make too much of one or more hormones, which can cause symptoms such as high blood pressure, weakening of the bones, or diabetes. If male or female hormones are affected, the body may go through changes such as a deepening of the voice, growing hair on the face, swelling of the sex organs, or swelling of the breasts.
Thyroid carcinoma: arises from the thyroid, a gland located in the neck that manufactures hormones that affect heart rate, body temperature, energy level, and also control the level of calcium in the blood.
Nasopharyngeal carcinoma: affects the nose and pharynx.
Malignant melanoma: a cancer of the skin.
Incidence:
Contrary to what occurs in adults, where carcinomas are the most common cancers, among children, particularly before the adolescent years, carcinomas are extremely rare. In the United States, about 1,050 children and adolescents are diagnosed with carcinomas each year. Of these, about 350 (35.5 percent) are thyroid carcinomas and 300-350 (30.9 percent) are melanomas. Adrenocortical carcinomas (1.3 percent), nasopharyngeal carcinomas (4.5 percent), other skin carcinomas (0.5 percent), along with other and unspecified (27.3 percent) make up the rest. All of the carcinomas represent only 9 percent of all cancers in children.
Nearly 75 percent (2,047) of the childhood carcinomas occur in adolescents (15-19 years old), including 75 percent of the thyroid carcinomas, 80 percent of the melanomas, 63 percent of the nasopharyngeal carcinomas, and 74 percent of the other and unspecified carcinomas.
Influencing Factors:
The most well established risk factor for thyroid carcinoma is exposure to ionizing radiation, from both environmental and therapeutic sources. The primary risk factor for melanoma is sun exposure.
Nasopharyngeal carcinoma appears to be associated with infection by the Epstein-Barr virus.
Survival Rates:
Outcome for patients with adrenocortical carcinoma that can be completely resected is usually very good. However, when the tumor cannot be completely removed, or when it comes back after surgery, the prognosis is much worse. For patients with nasopharyngeal carcinoma, treatment with chemotherapy and radiation therapy usually provides survival rates of 70 to 75 percent. The outcome for patients with thyroid cancer is also excellent with appropriate therapy.
Treatment Strategies:
For adrenocortical carcinoma, surgery is the primary treatment. When tumors cannot be removed or have already spread to other sites, chemotherapy is recommended. However, adrenocortical carcinoma is not very sensitive to chemotherapy, therefore the prognosis for these patients is poor. For patients with nasopharyngeal carcinoma, surgery is not possible due to the location of the tumor and the fact that it infiltrates the tissues and lymph nodes of the neck. These patients are treated with chemotherapy and radiation therapy to the nasal and pharyngeal cavities as well as the entire neck. Thyroid carcinoma can usually be treated with surgery alone. When thyroid cancer has spread to the lymph nodes of the neck or to the lungs, treatment can be given with iodine molecules that have a radioactive component.
Current Research:
For adrenocortical carcinoma, St. Jude is currently leading a study in collaboration with American and Brazilian institutions, in which a comprehensive approach to the treatment of this cancer is proposed, including new surgery techniques and uniform chemotherapy regimens.
Researchers at St. Jude are growing tumor cells in cultures to study new drugs for treatment.
St. Jude participated in the discovery of a new gene mutation in some patients. This unique mutation provides new insights into the mechanism for cancer development. An epidemiological genetic study is being held to understand more about this gene mutation and its role to the incidence, early diagnosis and treatment of disease. Epidemiology is a branch of medical science that deals with the incidence, distribution, and control of disease in a population.
Carcinoma can be defined as a cancer that expresses characteristics that include malignant tumors, but it is still classified as a "cancer of unknown primary origin," also known as CUP. Despite the fact that the origin and developmental lineage is still unknown, the cells affected by carcinoma still exhibit specific cellular, histological, and molecular characteristics that are deemed as normal within epithelial cells. These characteristics include keratin pearls, intermediate filaments, tissue architectural motifs, and intercellular bridge structures.
Typically, carcinomas can be diagnosed through the process of biopsy, which may include subtotal removal of a single node, core biopsy, and fine-needle aspiration. After the biopsy, a pathologist usually plays the role in identifying the cellular, molecular, and tissue properties of the epithelial cells that are to be examined.
Carcinoma in situ (CIS) is defined by a regional, miniature carcinoma that has not gone through the stages of invading the membrane of an epithelial cell. It is usually described as a pre-invasive cancer and not necessarily detrimental to the whole system of cells. If left untreated, the CIS will almost always continue to go through the regular process of penetration into the membrane and other structures, thus making it a truly invasive carcinoma. The lesion of the carcinoma can typically be removed through surgery, before the invasion process occurs. On some occasions, the CIS lesion may exhibit less aggressiveness and return to the more normal molecular characteristics.
Sarcoma refers to the cancer that is derived from connective tissue that develop from cells that come from mesenchymal cells. Examples of tissue that this type of cancer arises include cartilage, bone, nerves, muscle, and fat tissues. Unlike carcinomas, sarcomas are rare.
Tissue:
Sarcomas are given a number of different names based on the type of tissue from which they arise. For example, osteosarcoma arises from bone, chondrosarcoma arises from cartilage, liposarcoma arises from fat, and leiomyosarcoma arises from smooth muscle.
Grade:
In addition to being named based on the tissue of origin, sarcomas are also assigned a grade (low, intermediate, or high) based on the presence and frequency of certain cellular and subcellular characteristics associated with malignant biological behavior. Low grade sarcomas are usually treated surgically, although sometimes radiation therapy or chemotherapy are used. Intermediate and high grade sarcomas are more frequently treated with a combination of surgery, chemotherapy and/or radiation therapy.[1] Since higher grade tumors are more likely to undergo metastasis (invasion and spread to locoregional and distant sites), they are treated more aggressively. The recognition that many sarcomas are sensitive to chemotherapy has dramatically improved the survival of patients. For example, in the era before chemotherapy, long term survival for patients with localized osteosarcoma was only approximately 20%, but now has risen to 60-70%.
Typically sarcomas are diagnosed through the process of biopsy, in which the suspected tissues are extracted and examined by the pathologist. Fine needle aspiration, core needle, and surgical biopsy are all methods used to diagnose sarcoma. Imaging tests are also utilized before the biopsy in some scenarios, but is usually done once the diagnosis of the sarcoma is made. Methods of imaging tests include chest x-rays, computed tomography scans, magnetic resonance imaging scans, ultrasound, and positron emission tomography scans. Sarcomas typically originate within the arms or legs, and the physical characteristic shown is usually a lump that has appeared over a period of time.
Surgery is the most significant treatment that is used for most sarcomas. Limb sparing surgery is used instead of amputation. Other treatments include chemotherapy and radiation therapy.
Only 15,00 new cases of sarcomas occur per year within the United States, thus making sarcomas take part as around one percent of the 1.5 million new cancer diagnoses. The most common form of sarcoma is the gastrointestinal stromal tumor, which occurs at around 3000 cases per year in the U.S. Sarcomas also tend to affect people of all ages.
Borden EC, Baker LH, Bell RS, et al. (Jun 2003). "Soft tissue sarcomas of adults: state of the translational science". Clin Cancer Res. 9 (6): 1941–56. PMID 12796356
Lymphoma refers to the cancer that is derived from lymphocytes, which is the type of cell that makes up part of the immune system. Lymphoma is usually shows presence within solid tumors of lymphoid cells. The malignant cells formed typically originate within lymph nodes and shows presence as an enlarged node (usually a tumor). Common sites of lymphoma include the skin, brain, bone, and bowels.
Usually, an excisional biopsy is utilized to test for lymphoma. The biopsy sample is then observed by a pathologist to determine if there are any signs of lymphoma. The biopsy is usually obtained from the lymph node, but there may be cases in which the patients experience lymphoma effects in areas such as the skin, brain or stomach. Needle aspiration tests are also performed for diagnosis. When the lymphoma is diagnosed, further scans and image tests are utilized to observe how far the cancer has spread.
These warnings can be subtle and may take some time before serious affects occur.
Painless lumps in your neck, armpits or groin
This is the most common noticeable symptom and often the only one. The painless lumps are actually enlarged lymph nodes. A doctor should observe these lumps as these enlarged nodes may not always be a sign of lymphoma.
Weight loss
This can occur rapidly for no apparent reason. An individual can lose up to ten to 15 pounds over a period of a couple months.
Fever
Continuous fever or a reoccurring fever over a period of time may be a good sign of lymphoma symptoms, as long as they don't seem related to a urinary or chest infection. Fevers related to node swelling occur often with infections, which individuals may mistake as lymphoma at an early stage. Individuals with Hodgkin lymphoma undergo a characteristic fever called a Pel-Ebstein fever.
Excessive sweating at night
Waking up in the middle of the night drenched in sweat without any apparent reason can be a symptom of lymphoma. Excessive itchiness all over ones body can also occur. These symptoms are due to the secretion of some special chemicals from the lymphoma cells.
Loss of appetite
A loss of appetite can occur as the lymphoma cancer cells spread within an individuals body, often leading to weight loss.
A feeling of weakness
A feeling of weakness occurs due to the growing of cancer cells. These cells use up the body's nutrients, leaving the body with little energy to work with.
Lymphoma can occur in any organ, causing pain in the affected area. For example if the lymphoma was found in the stomach, pain may occur in the abdomen. If a lymphoma was found in the brain, serious headaches or leg weakness could occur as well.
Treatment of lymphoma include chemotherapy, radiotherapy, and/or bone marrow transplantation. These processes do not cure lymphoma, but have the tendency to alleviate the symptoms of it. Lymphoma can be curable depending on the histology, type, and stage of the disease in the individual.
Leukemia is defined as the classification of cancer in which the bone marrow or blood contains an irregular increase of mature white blood cells, which are also known as “blasts.” It serves as part of the broader group of diseases that affect the lymphoid system, blood, and bone marrow. Approximately 30% of children with cancer are diagnosed with leukemia, making it the most common type of cancer seen in children. [1] 90% of the cases of leukemia can be witnessed in adults. [2]
The first major classification of leukemia is the fact that it appears in both acute and chronic forms:
Acute leukemia refers to the characteristic in which there is an abnormally fast increase of malignant blood cells that are immature, which inevitably creates crowding in areas like the bone marrow. The crowding that occurs prevents healthy blood cells from being formed, hence making it detrimental to the body system. Acute leukemia requires immediate treatment due to the fast paced accumulation and progression of these immature blood cells, which eventually leaks into the blood stream and affects several organs of the body. This is the most common form of leukemia seen in children.
Chronic leukemia is leukemia in which there is a massive build up of white blood cells, and these cells are still deemed malignant. Even though the process of the excessive build up can take approximately months or years, these cells are still produced at an abnormally high rate, which results in an irregularly high number of blood cells as well. Unlike the acute form of leukemia, the chronic form is usually monitored for a period of time before an actual treatment in order to achieve maximum effectiveness. This form of leukemia can be seen mainly in older people.
In addition, these forms of leukemia can be subdivided into lymphocytic and myelogenous forms.
Lymphocytic leukemia, also known as lymphoblastic leukemia, is marked by a cancerous change that occurs within the type of marrow cell that usually proceeds into the form of lymphocytes. Lymphocytes are defined as immune system cells that fight infections. Lymphocytic leukemia tends to also involve B cells, which is a common subtype of the lymphocyte.
Myelogenous leukemia, also known as myeloid leukemia, is marked by the cancerous change that occurs within the type of marrow cell that proceeds to form red blood cells, platelets, and other forms of white blood cells.
The two classifications listed above enable four central categories of leukemia to be made. In addition to these four main categories, there are also some other rare types of leukemia that is exhibited.
Acute lymphoblastic (ALL) leukemia serves as the most frequently appearing type of leukemia in children. The survival rates for children tend to be approximately 85% while the survival rate for adults is approximately 50%. Other examples and subtypes are not limited to but include precursor B acute lymphoblastic leukemia, Burkitt’s leukemia, and acute biphenotypic leukemia.
Chronic lymphocytic leukemia (CLL) typically affects adults over the age of 55, and almost never affects children. Men appear to be more prone to this type of leukemia and the five-year survival rate is approximately 75%. As of now, this type is incurable but there are several treatments that are effective. A subtype of this is B-cell prolymphocytic leukemia.
Acute myelogenous leukemia (AML) typically occurs more frequently in adult men. It is typically treated with chemotherapy and the five-year survival rate s approximately 40%. Subtypes of this leukemia include acute myeoblastic leukemia, acute promyolocytic leukemia, and cute megakaryoblastic leukemia.
Chronic myelogenous leukemia (CML) usually occurs in adults. The treatment to CML is typically imatinib, and the five-year survival rate is 90%. A subtype of CML would be chornic monocytic leukemia.
Hairy cell leukemia (HCL) is often considered a subtype of chronic lymphocytic leukemia, but this kind of classification is very rough. 80% of the affected population are adult men. This type of leukemia is incurable but easily treatable. The survival rate is between 96% and 100% at 10 years.
T-cell prolymphocytic leukemia (T-PLL) is a rare and aggressive leukemia that typically affects adults. It appears more in men than women. Even though it is rare, it is the most common type of T cell leukemia that has matured. It is not easily treated and the survival rate usually lasts a few months.
Large granular lymphocytic leukemia involves T-cells and/or NK cells. It is a rare leukemia that is not aggressive.
Adult T-cell leukemia occurs due to the human T-lymphotropic virus (HTLV), which is a virus that can be relatable to HIV. HTLV infects the CD4+ T-cells and replicates within them.
Mutations in the DNA typically act as one of the causes of leukemia and other cancers. Specific mutations can then result in leukemia by deactivating tumor suppressor genes or activating oncogenes. These series of events then lead to the disruption of differentiation, cell death, or division.
Viruses may also have an effect on certain forms of leukemia. Retroviruses have been identified as relevant to some cases of leukemia, and the first human retrovirus that goes under this classification is the human T-lymphotropic virus or HTLV-1.
There are cases in which chromosomal irregularities correlate with the greater risk of leukemia. Down syndrome serves as an example that correlates with an increased risk of the development of acute myeloid leukemia, and Fanconi anemia creates an increased risk of acute myeloid leukemia as well.
Due to the high number of immature and malignant white blood cells, a high number of normal bone marrow cells are consequentially displaced, which can cause significant damage to the bone marrow. This damage ultimately creates a lack of blood platelets, which creates difficulty for the usual blood clotting process. This signifies the fact that people with leukemia tend to be easily bruised, develop pinprick bleeds, and can have excessive bleeding.
Individuals with leukemia may also have a hard time fighting off simple infections throughout the body, due to the fact that their white blood cells are either suppressed of dysfunctional. Those affected with leukemia frequently experience infection, which can affects body systems that result in sores within the mouth, infection in the tonsils, and diarrhea.
In some cases, the malignant cells of leukemia affect the central nervous system, which results in headaches and other neurological symptoms.
The diagnosis of leukemia is processed by bone marrow examinations and repeated blood counts after acknowledging the common symptoms of leukemia. There are however, some cases in which blood tests do not show indicate the existence of leukemia due to the fact that it is in early stages. Lymph node biopsies are also performed to diagnose specific forms of leukemia.
After a diagnosis is completed, blood tests are utilized to observe the degree of damage that has occurred onto the liver and kidney. X-rays, MRI, Ultrasound, and CT scans are also used to witness visible damage that is caused by leukemia.
A germ cell tumor is characterized by the abnormal increase of germ cells, also known as a neoplasm of germ cells. Typically, germ cells are found within the gonads. When germ cell tumors originate outside of the gonads, it is usually due to birth defects, which results from errors from embryo development.
The germ cell tumor is usually classified by its histology, and is split into two classes:
Germinomatous germ cell tumors (GGCT), also known as seminomatous germ cell tumors (SGCT) include mainly germinoma, dysgerminoma, and seminoma. The survival rate of the GGCT tend to be higher due to the fact that they are sensitive to radiation and chemotherapy.
Nongerminomatous germ cell tumors (NGGCT), also known as nonseminomatous germ cell tumors (NSGCT) include the other germ cell tumors that can either be pure or mixed. Nongerminomatous tumors develop faster than germinomatous tumors and have an earlier mean age at the time of diagnosis. Individuals affected by NGGCTs have an average of 5 year survival rate.
Ovarian: Ovarian germ cell tumors are hard to detect and typically grows to a relatively large size before symptoms can be seen. Swelling in the abdomen is a possible sign in the later stages of this cancer.
Testicular: Testicular tumors can be detected during the early stages due to the fact that they are noticeable in the scrotum and can cause pain. [1]
Mediastinum Tumors: Mediastinum tumors are located within the cavity that contains the heart, trachea, connective tissues, thymus, and large blood vessels. These tumors usually cause chest pain, coughing, fever, and breathing problems.
Presacral Tumors: Presacral tumors are located in areas above or on the dorsal side of the sacral bone of the hip. This type of germ cell tumor usually appears as a mass in the lower abdomen of a child. It can cause difficulty in passing urine or enabling bowel movement. In some cases, it can create difficulty in walking.
Pineal Gland Tumors: The pineal gland is a small-sized gland located in the middle of the brain, usually causing symptoms by pressing directly on parts of the brain or interfering with the normal flow of the fluids within it. Symptoms of this germ cell tumor include headache, nausea, vomiting, memory loss, lethargy, difficulty walking, an inability to look upward, double vision, and uncontrolled eye movements. [1]
Sacrococcygeal Tumors: Sacrococcygeal tumors are located near the tailbone or the distal end of the spinal column. It is the most common tumor affecting newborns, and can sometimes be visible from the outside of the body.
A majority of patients diagnosed with a germ cell tumor is treated with combination chemotherapy for 3 cycles. The most frequently used chemotherapy regimen for germ cell tumors is known as PEB, and contains antineoplastic, etoposide, and bleomycin.
Women with germ cell tumors are usually cured through the process of oophorectomy or ovarian cystectomy. Staging surgery is usually done for those with epithelial ovarian cancer.
Blastoma is a type of tumor that is caused by precursor cells or blasts. Blasts are defined as immature or embryonic tissue that usually contain differentiated or precursor cells. Blastoma serves as the type of cancer that usually occurs in children. Some common forms of blastomas include hepatoblastoma, medulloblastoma, nephroblastoma, neuroblastoma, pancreatoblastoma, pleuropulmonary blastoma, reteinoblastoma, and glioblastoma multiforme.
The symptoms of blastomas typically vary depending on the area of the body that is affected. It can affect areas such as the brain, kidneys, liver, nervous system, retina, and bones. A list of signs and systems include pain, weight loss, lethargy, impaired vision, impaired hearing, neurological symptoms, enlarged liver, enlarged spleen, and asymptomatic in early stages.
Like many other cancers, blastoma has been linked with the mutation in tumor suppressor genes and oncogenes. These mutations enable proliferation of incompletely differentiated cells, and hence create variations between patient to patient.
Oligonucleotide mutagenesis is a process in which a specific DNA sequence is incorporated into a vector (such as a plasmid or lambda phage) to introduce a new function into that molecule. Mutagenesis allows the insight into how proteins fold, catalyze reactions, act as substrates and process information. Cloned gene proteins can be used medically in a number of ways. For instance, insulin and interferon can be obtained from production of bacteria. DNA probes may be manufactured for discovery of genetic diseases, cancer, and bacterial infections. Two main forms are used to produce these modified proteins, Site directed mutagenesis and oligonucleotide-directed mutagenesis. The later is used if restriction sites are absent.
Mutagenesis is the process of changing or creating genetic information. This process can occur naturally or made by using different methods. There are more approaches involving mutagenesis methods. One method is a method of site-directed mutagenesis using mismatched oligonucleotides. Second method is cassette mutagenesis by annealing complementary oligonucleotides. Third method is PCR by generating a mutant fragment starting from a double-stranded DNA template using mismatched oligonucleotides. Mutagenesis is also used by experimenters to analyze DNA. Site mediated mutagenesis mutates one sequence of amino acids to determine that specific sequences overall purpose once it stops working. This is an example of experimenters using Mutagenesis.
In site-directed mutagenesis, individual amino acids in the primary structure of a protein are replaced by changing the DNA sequence of a cloned gene. The process involves removal of a DNA segment and replacement of that segment with a chemically synthesized segment identical to the original but with the desired change. The steps are as follows: A recombinant plasmid containing the gene of interest is treated with a restriction endonuclease to cleave the sequence of interest. The synthetic DNA fragment with the specific base pair change is inserted into the plasmid. This is done using a DNA ligase. The plasmid then contains the gene with the desired nucleotide base pair change.
Site directed mutagenesis is also useful for testing the validity of an enzymatic mechanism.
This form of mutagenesis creates a specific DNA sequence change. This method makes point mutations, differing in a single nucleotide. In this process, a DNA strand is synthesized with the specific nucleotide base pair change and annealed to the copy of the gene in a plasmid. Increasing the temperature allows for the mismatched base pair to be substituted in order for this reaction to occur. This annealed strand acts as a primer for the complementary strand to be synthesized in the plasmid. Oligonucleotide-directed mutagenesis uses DNA polymerase, dNTPs and DNA ligase. The result are two types of progeny: one with the original sequence and another with the new sequence.
Cassette Mutagenesis is the replacement a region of DNA with new sequences. Any length and sequence can be replaced by this method. Therefore, more variations of proteins can be made. By taking advantage of this, new mutant sequences can be made.
The Cassette Mutagenesis uses the original gene containing restriction sites. This site allows cleavage of the gene and produce the region where the new DNA is inserted. After appropriate restriction sites are identified in a vector, the vector is cleaved at the two sites by using a endonucleotide. The new sequence is inserted and ligated in the region produced. The new sequence allows a variety of studies on protein structures or nucleic acid sequences that have not been explored before.
Polymerase chain reaction is used for recombination of sequences and mutagenesis. This method is very useful because it is a fast reaction and it is not limited by any restriction sites. In the PCR, oligonucleotide primers are incorporated into the ends of the product DNA. The 5' ends of these primers can contain any desired sequence. two PCR products are added by PCR. They are mixed, denatured, and reannealed. There are two different sequences produced. One is 5' overlapping strands which is not productive. 3' overllapping strand is produced and reactive. The polymerase extends the 3'strand. In this process, the 3' strand acts as primers. The whole process are repeated by producing synthetic sequences. This overlapping by joining two DNA fragments together can produce mutatation in PCR fragment.
M.J. McPHERSON, Directed Mutagenesis
The Ames test was named after its developer, Bruce Ames, and is a method for determining if a chemical is a mutagen, an agent that causes mutations. This test is used by many cosmetics companies, pharmaceutical companies and other industries to prove that their products will not cause cancer or harmful in that way in humans. The use of this test is based on the assumption that any substance that is mutagenic may also turn out to be a carcinogen, to cause cancer. A positive test shows that the chemical can act as a carcinogen. If the substance is a carcinogen, it will cause mutation in the bacterium as the cell divide. The mutant cells will have the enzyme to form colonies.
In the Ames test, a thin layer of agar containing about 109 bacterial of a specially constructed tester strain of Salmonella is placed on a petri plate. T hese bacteria are unable to grow in the absence of histidine, because a mutation is present in one of the genes for the biosynthesis of this amino acid. The addition of a chemical mutagen to the center of the plate results in many new mutations.
The bacterium used in the test is a strain of Salmonella typhimurium, which carries a defective gene making it unable to synthesize the amino acid histidine from the ingredients in its culture medium. Yet, some of mutations can be reversed with the gene regaining its function. These are able to grow on a medium lacking histine. Since cancer is often linked to DNA damage, the test can also quickly estimate the carcinogenic potentical of a compound as it is hard to determine whether standard carcinogen examination on animals were successful.
A small proportion of them reverse the original mutation, and histidine can be synthesized. These revertants multiply in the absence of an external source of histidine and appear as discrete colonies after the plate has been incubated at 37 Celsius degrees for 2 days. A series of concentrations of a chemical can be readily tested to generate a dose-response curve. These curves are usually linear, which suggests that there is no threshold concentration for mutagenesis.
Some of the tester strains are responsive to base-pair substitutions, whereas others detect deletions or additions of base pairs. The sensitivity of these specially designed strains has been enhanced by the genetic deletion of their excision-repair systems. Potential mutagens enter the tester strains easily because the lipopolysaccaride barrier that normally coats the surface of Salmonella is incomplete in these strains.
A key feature of this detection system is the inclusion of a mammalian liver homogenate. Since some potential carcinogens such as aflatoxin are converted into their active forms by enzyme systems in the liver or other mammalian tissues, bacteria lack these enzymes. So the test plate requires a few milligrams of a liver homogenate to activate this group of mutagens.
This test is extensively used to hep evaluate the mutagenic and carcinogenic risks of a large number of chemicals. This rapid and inexpensive bacterial assay for mutagenicity complements epidemiological surveys and animal tests that are necessarily slower, more laborious, and far more expensive. The Salmonella atest for mutagenicity is an outgrowth of studies of gene-protein relations in bacteria.
The picture shows the Ames test. A suspension of a his- strain of Salmonella typhimurium has been plated with a mixture of rat liver enzymes on agar lacking histidine. The disk of filter paper has impregnated with a carcinogen. The mutagenic effect of the chemical has caused many bacteria to regain to grow without histidine to form the colonies around the disk. The scattered colonies on the disk represents spontaneous revertants.
http://upload.wikimedia.org/wikibooks/en/5/5f/AmesTest.gif
Tumors, which are neoplasms, are solid or liquid filled lesions that may or may not be formed and grown in size due to neoplastic cells. The tumor cells are part of a tissue that continually grows abnormally. It can be malignant or benign, but these cells may look like "swellings" and grow to bigger proportions. Although tumor cells that are malignant maybe confused with cancer cells, they do not invade neighboring tissues. These cells can grow and become pestering problems that affect mobility, breathing, and circulatory of bodily functions. They cells are triggered in growing by the mutations in DNA, and may have a higher chance of growing with age. (ISCID 2011)
Tumor suppressor proteins protect cells from being cancerous. Tumor suppressors play a crucial rule in not allowing the cell to divide and multiply if there is damage to the cell’s DNA; this will occur until the DNA of the cell is repaired. In the event DNA cannot be repaired, tumor suppressors play a role in the process called “apoptosis”, which is the process of programmed cell death. If tumor suppressors do not function properly in a cell with damaged DNA, the cell continue dividing and replicating damaged DNA, which eventually leads to cancer .[1]
p53 is a protein that is responsible for preventing cell division of cells that have damaged DNA
p63 is part of the p53 family and has a similar structure to bother p53 and p73. However, the function of p63 seems to be more similar to p73 than p53.
p73 is a protein that is part of the p53 family and is structurally and functionally similar to p53
PTEN stands for phosphatase and tensin homologprotein and is involved in the pathway that signals for a cell to stop dividing and triggers apoptosis
Retinoblastoma is a protein that is responsible for repressing the transcription of genes regulated by E2F, which include cyclins that regulate the cell cycle.
APC Adenomatous polyposis coli (APC) or also known as deleted in polyposis 2.3 (DP2.5) is a protein found in humans that are coded by the APC gene.
Joerger & Fersht, Adreas C & Alan R. (2008). "Structural Biology of the Tumor Suppressor p53". The Annual Review of Biochemistry. Retrieved 2011-11-30.
Phosphatase and tensin homolog (PTEN) is a tumor suppressor gene located on chromosome 10 in region 10q23 that is part of the protein tyrosine phosphotase (PTP) gene family. It provides the genetic instructions for producing the tumor suppressor protein of the same name (PTEN) [1]
The PTEN protein is involved in the pathway that signals for a cell to stop dividing and triggers apoptosis.[1] It has been found that PTEN is often deleted in multiple types of advanced cancers. Cells without PTEN have higher levels of phosphatidylinositol 3,4,5-trisphosphate and protein kinase B, which is a signal that prevents apoptosis. This signal may also cause the cell cycle to continue.[2] Both of these two actions are characteristic of tumor cells. PTEN also plays a role in preventing cell spreading, thus preventing metastasis, by inhibiting focal adhesion. If the growth cells are unable to attach to something while they growth, they will not be able to survive and continue to spread.
Crystallographic structure of human PTEN. The N-terminal phosphatase domain is colored blue while the C-terminal C2 domain is colored red.[3]
The PTEN protein is made up of 403 amino acids. It contains the HCXXGXXR motif that is characteristic of the active sites of the PTP family. At its 190-amino acid N-terminal, the protein is similar to two other proteins, tensin and auxilin. At the C-terminus, there is a 220-amino acid sequence whose function has not yet been figured out, but it is known that both domains are necessary for proper activity and function of the protein.[3] Recent studies of the C-terminus have scientists believe that the region is necessary for PTEN stability and enzymatic activity.[4] In an experiment, the 50-amino acid tail was deleted and was confirmed to be necessary for stability. However, the activity of the PTEN proteins, with the deleted tail, increased. Phosphorylation sites were found to be located on the tail and the results of the experiment suggest that it is possible that the phosphorylation status of PTEN determines the levels of activity.
Using X-ray crystallography, the crystal structure of the PTEN protein has been determined. It has been revealed that in the structure, the phosphatase domain is similar to protein phosphatases but it has an enlarged active site that is also able to interact with a phosphoinositide substrate.[3] This property of the phosphatase domain is the reason that PTEN is able to dephosphorylate lipids which is essential for it to function as a tumor suppressor.[4]
PTEN also has a C2 domain that allows the protein to bind to membrane phospholipids and prevent the growth of tumor cells.[3]In virto studies have shown that the C2 domain has an affinity for phospholipid membranes which suggests that the C2 domain may be responsible for positioning the catalytic domain of PTEN to interact with cell membrane.[4]
Since the 1980s, there had been evidence of a tumor suppressor located on chromosome 10.[4] Cytogenetic and molecular analysis showed that in brain, bladder and prostate cancer, partial of all of chromosome 10 had been lost.[4] Then, LOH analysis was used to identify the region 10q23 as the most common region that was lost in prostate cancer. As a ressulf of this finding, many experiments were performed to test the role of chromosome 10 and the region 10q23. The wide-type chromosome was reintroduced into tumor cells and had the effect of reducing the ability of these cells to cause tumors in mice[4]. Then in 1997, Li et al used representational difference analysis (RDA) on breast tumors to generate a probe that would match to chromosome 10q23.[4] Yeast artificial chromosomes that contained the probe were present on the sequence-tagged site were isolated and helped identify deletions in breast xenografts. Then exon-trapping analysis was use to identify two exons that created an open reading frame of 403 amino acids that generated a protein with a region that was homologous in both chicken and cows.[4] Another group was able to identify the same candidate tumor-supporessor gene on 10q23 by performing high-density scans on glioma cells lines. From these scans, it was found that the same deletions occurred across the cells lines and exon trapping was used to identify the exons of the gene.[4] A separate study searching for protein tyrosine phosphatases used PCR to identify this gene. Primers that bind to the phosphatase catalysic domain were used to screen human cDNA libraries and found PTEN. The scientists were able to confirm the role of the gene as a phosphatase.[4] It is recorded that PTEN was officially discovered in 1997[5]
Integrins are receptor proteins that are involved in signaling for cell migration and invasion. PTEN shares structure characteristics with tensin, a cytoskeletal protein that is very involved in integrin-signaling complexes. Integrins are involved in tumor growth because malignant tumor cells are invasive and metastatic and carry out this behavior by interacting with the integrin family of cell surface receptors [6] It has been shown that Focal Adhesion Kinase (FAK) co-localizes with integrins at focal adhesions and FAK may play a central role in the signal transduction pathway that integrins trigger.[6] FAK has increased expression in invasive and metastatic tumors and is active when tyrosine phosphorylated upon integrin activation.[6]
It has been shown that PTEN interacts to dephosphorylate FAK and thus prevent it from causing integrin-mediated cell spreading, migration, invasion and cytoskeleton organization.[6] PTEN functions to inhibit FAK, and thus prevent the progression of tumor growth and spread.
PTEN may also have a role apoptosis, but the role is not universal in all cell types.[7]Apoptosis is a process where a cell genetically determines whether it goes through cell self-destruction. It is activated by either a presence of a stimulus or by a removal of a stimulus or a suppressing agent[8] It is a normal process that eliminates damaged DNA or unwanted cells. When this process is stopped, it may result in uncontrolled cell growth and tumor formation. In vivo studies has shows that PIP-3 is the main substrate of PTEN. PIP-3 is stimulated by growth factors and if there is accumulation of PIP-3 at the membrane, then it starts to recruit proteins to bind to it.[9] Akt is one of the targets of this recruitment and is a well-known survival factor that is responsible for preventing apoptosis by preventing to release of genes necessary for apoptosis to occur. Therefore, the role of PTEN is to regulate the levels of PIP-3 and keep the level low. In mouse models, it has been observed that loss of PTEN causes PIP-3 and Akt to be hyperactivated which has a dramatic effect on cell survival and proliferation.[9]
It has been observed that when PTEN was deleted in mouse embryonic firbroblasts, it causes spontaneous DNA double-strand breaks that are unable to be repaired by the cell.[10] PTEN is believed to act on chromatin and regulate Rad51, which is the gene responsible for preventing spontaneous double-strand breaks.[10] Others have argued that regardless of the presences of PTEN, the sensing and repair of DNA double-strand breaks is all the same. However, these experiments used different cell lines, so it is possible that the role of PTEN in controlling DNA double-strand breaks depends on cell line and assays used.[10] More study needs to be done in order to determine the exact role of PTEN.
Another possibility of PTEN being involved in DNA damage repair is through the nucleotide excision repair (NER) pathway which is a highly conserved pathway in eukaryotes that is responsible for repairing DNA lesions caused by UV. It has been observed that mice with downregulated PTEN that are exposed to low suberythemal UV radiation are more predisposed to skin tumorigenesis and in humans, PTEN is is downregulated in premalignant and malignant skin lesions.[10] It is believed that PTEN is responsible for promoting XPC transcription in keratinocytes. XPC is a critical protein for the NER pathway and without it, repair of the damaged DNA cannot take place.
Another interaction PTEN has been found to be involved in is with Chk1, an important signal transducer in the cell cycle checkpoint pathway that is necessary for the cell cycle to continue after stalled DNA replication.[10] Even thought Chk1 regulates PTEN, PTEN levels can also have an effect on CHk1. Loss of PTEN has been observed to cause impaired Chk1-mediated checkpoint activation.
PTEN may also play a cooperative role with another tumor suppressor, p53. PTEN located in the nucleus of the call may interact with p53 to arrest cells upon oxidative damage.[10] PTEN has been found to play a regulating role by controlling the DNA binding activity of p53. Up-regulation of PTEN increases the level of p53 and causes G2/M arrest and apoptosis.[10]
As PTEN mainly functions to inhibit cell growth and signals for apoptosis to occur.[6] It mainly works through dephosphorylation of phospholipids that are signals for cell growth and survival. by inhibiting these phospholipids (such as PIP3), PTEN functions to prevent the cells from any further activity.[6] By keeping PIP-3 levels lows, PTEN is able to regulate the growth and proliferation of cells and prevent them from becoming oncogenic.
the structural component of PTEN that is responsible for for its tumor supressive properties is its activity as a lipid phosphatase. In an experiment, the lipid phosphatase activity of PTEN was ablated while the protein phosphatase activity was kept intact. When wild-type PTEN and the mutant PTEN were introduced into a PTEN-null glioblastoma cell line, the cells with wild-type PTEN showed induced growth suppression, while cells with the mutant PTEN did not show such results.[4]
↑ ab"PTEN." Genetics Home Reference. National Institutes of Health, National Library of Medicine, n.d. Web. 11 Nov. 2012. <http://ghr.nlm.nih.gov/gene/PTEN>
↑Lodish, Harvey. "Oncogenic Mutations Affecting Cell ProliferationFactor Genes Can Autostimulate Cell Proliferation." National Center for Biotechnology Information. U.S. National Library of Medicine, 18 Dec. -0001. Web. 11 Nov. 2012. <http://www.ncbi.nlm.nih.gov/books/NBK21513/>.
↑ abcdLee, Jie-Oh; Yang, Haijuan; Georgescu, Maria-Magdalena; Di Cristofano, Antonio; Maehama, Tomohiko; Shi, Yigong; Dixon, Jack E; Pandolfi, Pier; Pavletich, Nikola P (1999). "Crystal Structure of the PTEN Tumor Suppressor". Cell. 99 (3): 323–34. doi:10.1016/S0092-8674(00)81663-3. PMID10555148.
↑Uzoh, Christopher C.; Perks, Claire M.; Bahl, Amit; Holly, Jeff M.P.; Sugiono, Marto; Persad, Raj A. (2009). "PTEN-mediated pathways and their association with treatment-resistant prostate cancer". BJU International. 104 (4): 556–61. doi:10.1111/j.1464-410X.2009.08411.x. PMID19220271.
↑ abcdefTamura, M.; Gu, J.; Tran, H.; Yamada, K. M. (1999). "PTEN Gene and Integrin Signaling in Cancer". Journal of the National Cancer Institute. 91 (21): 1820–8. doi:10.1093/jnci/91.21.1820. PMID10547389.
↑Allocati, N.; Di Ilio, C.; De Laurenzi, V. (2012). "P63/p73 in the control of cell cycle and cell death". Experimental Cell Research. 318 (11): 1285–90. doi:10.1016/j.yexcr.2012.01.023. PMID22326462.
p53 is one of the most extensively studied tumor suppressors in multicellular organisms. It was discovered in 1979 and over 60,000 papers have been published about it.[1] It is nicknamed "Guardian of the Genome" and in 1993 it was named the "Molecule of the Year" by Science Magazine.[1] It is also known as “protein 53” or “tumor protein 53” and is part of the small p53 family which also includes p63 and p73[2]. This tumor suppressor is regulated by a gene called “TP53 gene”. p53 works by sensing oncogenic cytotoxic and genotoxic stress signals and responds by signaling for cell-cycle arrest as well as apoptosis in order to prevent the cells from further growing. Several experiments have shown that loss of p53, due to inactivation, rearrangement or some other change that causes the protein to lose it's function, promotes the growth and survival of cancer. [3]. It is estimated that 50% of the people who have cancer have a mutation in the p53 protein [1]
Cartoon representation of a complex between DNA and the protein p53 (described in Cho et al. Science 265 pp. 346, 1994 [11])
The tumor suppressor p53 in humans is a polypeptide of 393 amino acids that take up five domains. p53 is active as a tetramer that has four identical chains of 393 residues. The N-terminal region consists of disordered transcription domains and a proline rich region. At the C terminus, p53 contains the regulatory domain of unfolded basic amino acids that binds DNA nonspecifically as a transcription factor. [4]
The protein is normally found in the cytoplasm of the cell. If the cell is properly functioning and the p53 protein is not needed, then it is degraded by the MDM2 protein.[1] If the cell is stressed or there is something wrong, then p53 is passed to the nucleus where is dimerizes and eventually tetramerizes to act as a transcription factor.[1].
p53 is a protein which regulates the cell cycle. There are three domains in the p53 protein that are responsible for directly DNA repair or cell death.[1] The three domains are responsible for binding to DNA, recruiting proteins (other transcription factors) to the DNA, and the third domain is a regulatory domain in the C-terminus that regulates the binding of p53 to DNA. [1] The wild-type p53 is a labile protein that is comprised of folded and unstructured regions which function in a synergistic manner.[1]
The N terminus of p53 has unfolded regions, but has secondary residual structures that contain hydrophobic resides. The acidic TAD, the main body, which has 2 ill-defined subdomains (TAD1 & TAD2), has binding sites for multiple interactions with proteins that deal with transcription machinery and transcriptionary coactivators. Thanks to the intrinsic disorder of TAD it can facilitate the binding of diverse proteins with high specificity. The n terminus of p53 deals with post translation modification where multiple phosphorylation of serine and threonine residues, by several protein kinases, happen which shift the affinity for different proteins that compete to bind with p53. The proline rich region that is in the n terminus links the TAD to the DNA binding domain of the humans. The PXXP motifs, which are in p53 help modify and mediate the protein to protein interactions. [4]
The DNA binding site for p53 is 20 nucleotides long. As the protein is a tetramer, this means that each dimer binds to 10-basepair sequences that have the general pattern Pu Pu Pu C A/T A/T G Py Py Py from the 5' to 3'direction.[1] Thanks to the crystal and solution structure of p53C, the structures have been found in which there are complexes within the p53C that bound to domains of signaling protein. This consists of immunoglobulin beta sandwich that is able to provide a scaffold for DNA-binding surfaces. There is a loopsheet helix motif which dock to the DNA major grooves including loop L1, and beta strands of S2 & S2'. Although these loops are large, they are stabilized by a zinc ion. If this zinc is lost, there will a be a decrease in the thermodynamic stability and a increase in the aggregation tendencies. [4]
The C terminus consists of regions in the protein that help tetramerization. With the help of 3 residues, it forms the central hydrophobic core of the dimer. Formation of stable dimers are through high protein concentrations in the solution. Some extreme C terminus may be intrinsically disordered through local disorders to order transitions from binding to nonspecific DNA or proteins. Some motifs from residues may affect the confirmation. The C terminus is required for efficient binding and transactivation of target genes in large molecules of DNA. [4]
Mechanism
p53 plays an important role in cell cycle control and apoptosis. The defective p53 could also allow abnormal cells to proliferate, which then results in cancer. When the DNA is damaged, it will trigger the increase of p53 proteins, which have three major functions. They are DNA repair, growth arrest, and apoptosis (cell death). The cellular concentration of p53 must be tightly regulated. However, the high level of p53 may accelerlate the aging process by excessive apoptosis.
Role of p53 in Disease
The tumore supression is reduced when the p53 is reduced. p53 also damaged in cells by mutagens (viruses, chemicals, or rediation). Also, p53 in itself can inhibit normal p53 (Blagosklonny, 2002).
p53 regulates the gene transcription binding to double-stranded DNA sequences of its choices. In other words, the p53 protein acts as a transcription factor. Each half-site that the p53 can be described as an inverted pentameric quarter site. The human genome is found to have a high probability binding loci that contains half sites without insertion. Within p53C domains, there are 4 spots that bind to response elements which increase upon binding to full length proteins. The teramers also demonstrate different areas of binding which is formed through a symmetrical dimer. The interface is stabilized within the loops that bind by hydrophobic and water mediated polar contacts. Weak protein-protein interactions are observed within the dimers that can reflect to the response elements within the different spacers. The key residues in the DNA interface make direct contact with DNA half sites inside the DNA minor groove. When making contact, it makes salt bridges that help bind. Although the specific contacts with DNA is important, the residue in the DNA domain are disordered sometimes. [4]
Although the p53 may help in repressing tumor, some mutations of the TP53 gene is found in some human cancers. These cancers may have over 17,000 cases of somatic p53 missense mutations. Some causes may be the common ancestor found between the p53 genes. There are different mutant classes amongst the mutants. Contact mutations remove DNA-contact residues, while structural mutations affect residues that are essential in the big picture of the DNA binding surface. The equilibrium unfolding and denaturation allow response elements to find even more mutant classes. B sandwich mutations are found in 1/3 of cancer cases in which the B sandwich cannot collapse with the surrounding structure. The mutation destabilizes the protein losing the hydrophobic interactions. [4]
Most of the mutations in p53 are missense mutations that affect the DNA binding domain region of the protein. When this happens, the molecule is not longer able to bind to DNA which results in a loss of function.[5] In other cases it has been found that the molecule still tetramerizes and is still capable of binding to DNA, but it no longer can function as a transcription factor. [1]
In the case of mutations that result in the protein losing its ability to bind to DNA, there is a subgroup of mutations that actually result in a "gain of function" and p53 now has an oncogenic role[5]. It this case, it is believed that p53 binds to the DNA binding domain of p73, thus inactivating p73 by preventing it from binding to its targets. As a result, this inhibits that pathways p73 is responsible for, transactivation and apoptosis, thus further promoting cancer[5] This inactivation of p73 by an alread inactive p53 further results in a decreased response to chemotherapy[5].
P53 inhibits tumor formation through the use of Cell Cycle Arrest, Apoptosis and Senecence. This gene has the ability to induce cell apoptosis as part of its role as a tumor suppressor. P53 becomes activated by damage in the cell and is suppressed by MDM2 and when P53 becomes too activated to be suppressed it is possible for it to activate the apoptosis pathway in the cell, among other pathways. Some of the proteins that activate p53 are the MAPK proteins, ATR, TP53RK, and p14ARF. The acetylation sites on p53 play a key role in controlling the specific p53 activation sites and if both are mutated the mutation causes the loss of ability for p53 to induce cell cycle arrest and apoptosis.
MDM-2 (murine double minute 2) is the negative regulator of p53 and acts by binding to the first 42 N-terminal residues that contain a transactivation domain.[6] When MDM-2 binds to p53, is causes a α-helix conformation to form which is successful in blocking the transcription and ubiquitinating lysine resides in the C-terminal domain region. When stress signals or abnormal cell division is sensed, the ARF gene is transcribed and its purpose is to bind to MDM-2 and inhibit its activity and allowing p53 levels to rise.[6]
Determining the 3D structure of p53 has been very difficult due to the very complicated folding, especially in the C- and N- termini. The best way to study the three dimensional structure of this interesting protein is to look at the structure of each of its domains separately. The domains structure that have been solved so far are the tetramerization domain; DNA binding domain in complex with DNA, a complex with a transactivation domain; and some parts of the regulatory domain in complex with proteins.[4]
↑ abcdefghijViadiu, Hector. "Gene Recognition by the P53 Protein Family." CHEM 114A Lecture. University of California, San Diego, La Jolla. 14 Nov. 2012. Lecture.
↑Andrea Bisso1, Licio Collavin1, and Giannino Del "p73 as a Pharmaceutical Target for Cancer Therapy" Laboratorio Nazionale CIB, Area Science Park, Padriciano 99, 34149 Trieste, Italy. 2Dipartimento di Scienze della Vita, Universitàdi Trieste, Via L. Giorgieri 1, 34100, Trieste, Italy/>
↑ abBelyi, Vladimir A., Prashanth Ak, Haijian Wang, Wenwei Hu, Anna Puzio-Kuter, and Arnold J. Levine. "The Originals and Evolution of the P53 Family of Genes." Cold Spring Harbor Perspectives in Biology, n.d. Web.
p63, also known as TP63 (tumor protein p63), is encoded by the TP63 gene. It is also part of the p53 family along with p53 and p73. It was discovered in 1998 as a p53-related ortholog in mouse and human cells.[1] Similarly to p53 and p73, p63 also has domains that are highly conserved across the family. Because of this homology, it is believed that the three genes in the p53 family have the same functions. However, after studying the genes more specifically and carefully, it was been shown that even though the genes may performs some of the same functions, they each have unique physiological roles.[2] p63 has been associated more with have a developmental role rather than a tumor suppressing role.[3] All three genes can cause cell cycle arrest and apoptosis, but like p73, there is alternative splicing for p63 that produces different isoforms of p63.[2] α forms contain the sterile alpha motif (SAM) that is responsible for mediating protein-protein interactions.[1]
C-terminal domain of the p63 protein PDB rendering based on 1rg6.
The structure of p63 seems to be more similar to the structure of p73 than to p53.[1] p63 has a long transactivation domain (TA), a DNA binding domain (DBD) and an oligomerization domain (OD) that are all highly conserved with the corresponding domains in p53 and p73.[2] The protein is composed of 15 exons and various isoforms of protein exist. Isoforms containing the long transactivation domain as known as TA forms and are the full-length protein TAp63, while isoforms that do have only the short (~15-18 amino acid residues) transactivation domain are called ΔN forms, ΔNp63. Each form can undergo alternative splicing that produces three different C-termini forms, α, β, or γ [1] Additionally, both TAp63α and ΔNp63α also have a sterile alpha motif (SAM) interacting domain at its C-terminus that is responsible for mediating protein-protein interactions.[4]
The ΔNp63 isoform of p63 (like the ΔNp73 isoform of p73) does have only the short N-terminal TA domain and this p63 isoform seems to be the main negative inhibitors of TAp63 isoforms as well as other family members of the p53 family, similar to role of the ΔNp73 isoform.
↑ abcdLittle, Natalie A; Jochemsen, Aart G (2002). "p63". The International Journal of Biochemistry & Cell Biology. 34 (1): 6–9. doi:10.1016/S1357-2725(01)00086-3.
↑ abcGraziano, Vincenzo; De Laurenzi, Vincenzo (2011). "Role of p63 in cancer development". Biochimica et Biophysica Acta. 1816 (1): 57–66. doi:10.1016/j.bbcan.2011.04.002. PMID21515338.
↑Nekulova, Marta; Holcakova, Jitka; Coates, Philip; Vojtesek, Borivoj (2011). "The role of P63 in cancer, stem cells and cancer stem cells". Cellular and Molecular Biology Letters. 16 (2): 296–327. doi:10.2478/s11658-011-0009-9. PMID21442444.
p73 is part of the p53 family of transcription factors that also includes p63. The family is well known for their ability to regulate cell cycle arrest and apoptosis. p73 and p63 are both structurally and functionally similar to p53, and it is believed that they are also tumor suppressor proteins[1]. The monomer of p73 has a 50% homology with p53 causing the folding in both proteins to be very similar.[2] Even thought both proteins have a very similar structure and function, while 50% of cancers have a mutated p53 protein, but only 0.5% of cancers have a mutated p73 protein which is of great interest to scientists.[2]
The p73 protein has a DNA domain structure very similar to p53. The p73 protein is larger than the p53 protein as it has a 636 amino acid sequence.[2] Like p53, there are three structural domains that include a transactivation domain (TA) that is a proline-rich sequence following the N terminus of p73, a central DNA-binding domain (DBD) and a C terminus with an oligomerization domain (OD).[3] Because there are from the same protein family, p73 shares a 50% degree of sequence homology with p53, especially in the DBD.[2] The similarity in sequence leads to a similar folding structure which allows p73 to function in a very similar way to p53, such as recognizing and regulating targets of the p53 protein.[2] However, the oligomerization domains are less conserved between the two proteins. In this area, p73 is more similar to p63 as it can hetero-oligomerize with p63, but not with p53. [1]
Various isoforms of p73 exist. There are isoforms that posses a transactivation site similar to the site on p53 and are denoted by TAp73. Other isoforms are ΔNp73 which were originally form the TAp73 promoter transcription do not have the N-terminal transactivation domain due to aberrant splicing.[4] Studies that shown that knockout of TAp73 in mice made the mice more susceptible to carcinogens and thus more tumor-prone. However, in vivo knockout of ΔNp73 actually reduced tumor growth showing that the two different isoforms of the gene can cause different results.[4]
The p53 family proteins each work in different pathways, but they do have some overlapping functions.[2]
MDM2 degrades both p53 and p73.
p53 and p73 both have a role in triggering apoptosis
However, it has been shown that cells with only p53 knocked-out have a different chemosensitivity than cells with both p53 and p73 knocked-out[5] In chemotherapy, patients are given drugs that target fast-growing cells and kills them. Cancer cells are reproducing very quickly and thus are more likely to be the targets of these drugs, however other faster-grwoing cells such as hair and the lining of the stomach can also be targets of these drugs. In some in vivo and in vitro studies, it has been observed that mutated p53 is actually able to bind to p73 and inactivate the protein, causing the cells to be more chemoresistant. [5]
TAp73 can stop the cell cycle during the G1 and G2 phases. In the G1 phase, TAp73 arrests the cell cycle by transcriptional up-regulation of p21 and p57/Kip2 which are proteins that prevent the cell cycle from continuing.[3] In the G2 phase, p73 represses G2/M regulators that include CDC25B, CDC25C, Cyclin B1, Cyclin B2, cdc 2 and Topoisimerase IIα, thus preventing the cell from leaving the Interphase and entering Mitosis.[3] Even in mitosis, when p73 is presumed to be hyper-phosphorylated and very inactive, it still plays a role that is can transcribe p57/Kip2 genes which are negative regulators of cell proliferation.[3] TAp73 has also been shown to interact with spindle assembly checkpoints and helps with that activity and malfunction at this point is believed to be the reason for tumorigenesis.[3][4]
TAp73 can also signal for cell death to occur through various mechanisms. Through the mitochondrial pathway, TAp73 is able to up-regulate Bax and promote its movement to the mitochondria which occurs when Bax has received apoptotic signalling.[3] Through the endoplasmic reticulum stress pathway, p73 up-regulates scrotin, a gene that can induce apoptosis, in the ER as well as in supra-basal keratinocytes.[3]. It can also modulate the death receptor pathway and can induce CD95 expression (a death receptor).[3]
This isoforms of p73 is believed to be the negative of not only TAp73 but also p53 by inhibiting growth suppression and apoptosis. ΔNp73 repressed TAp73 transcipriton activity by forming inactive oligomers and competes with p53 for DNA biding sites.[3] It has been shown that in cancer cells that reductino of ΔNp73 results in apoptosis.[4] However, there is a feedback loop pathway that occurs when TAp73 and p53 are active that induces ΔNp73 creating a auto-regulatory feedback loop so that there are not too many active TAp73 and p53 molecules. It has also been shown that if DNA damage is detected, this triggers the degradation of ΔNp73, thus allowing TAp73 to function.[3]
One possible way of increasing the amount of TAp73 and decreasing the amount of ΔNp73 is by controlling the splicing of the TAp73 promoter transcripts. In hepatocellular carcinoma cells, the aberrant splicing responsible for changing the transcripts form TAp73 to ΔNp73 was caused by depletion of the splicing factor Slu7. This depletion of Slu7 was caused by c-Jun N-terminal kinase 1 (JNK-1) activity which, in turn, was caused by activation of EGFR by AR.[4] Another way of increasing the TAp73/ΔNp73 ratio to favor TAp73 is through using the COX-2 inhibitor celecoxib which has been shown to increase TAp73β in neuroblastoma cells.[4]
In most solid tumors, there is actually an abundance of p73, however it is not able to do any anti-tumor activity because it is blocked by various inhibitors that include family proteins like ΔNp73, ΔNp63, mutant p53 and non-family proteins such as MDM2, iASPP or mTOR.[4] Because of the many and complex functions of p73, it is regulated after it has already been transcribed by changing the stability or the protein, the location of the protein and the ability of the protein to bind to promoters. p73 is regulated though its degradation through the ubiquitin system.[3] MDM2 which is responsible for degrading the p53 protein can also bind to p73. However, instead of degrading the protein, it moves it to nuclear speckles and changes it's transcriptional activity. The E3 ubiquitin ligase ITCH binds to p73 to promote degradation of the protein in normal conditions. In times of stress, ITCH is downregulated and p73 and p63 levels are allowed to increase. Unlinke MDM2, ITCH is exclusive to p63 and p73.[3] Other genes that can regulate p73 include: PIAS1 that sumoylates p73 thus reducing the transcriptional activity of p73 and allowing the cell to leave the G1 phase of the cell cycle. p73's transcriptional activity is also reduced when Cyclyn/CDK complexes threonin phosphorylate it. SIRT1 inhibits the acetylation of p73 thus decreasing the activity of p73.[3]
When normal cells were exposed to bile acids, the role of p73 was activated in response to DNA damage. Increased amounts of DNA damage due to bile acid exposure triggered only the response of p73; this result indicated that p73 induction is independent to p53 and p63, but dependent on the selective activation of c-Abl-kinase. The p73 induction activates transcription of two glycosylases, SMUG1 and MUTYH, to conduct base excision repair. When p73 is deficient, an apparent increase of DNA damage is present [6].
The causes for esophageal adenocarcinoma involve the severe progressions of gastroesophageal reflux disease, where gastric juices mix with bile acids and enter the esophagus. Exposing the esophagus to gastric acids and bile acids leads to inflammation and tissue damage. In addition, bile and acid refluxes induces genotoxic stresses, which increases ROS and RNS production and consequently causes damage to the DNA through oxidation.
Cells cultures, transfection, retroviral infections, and BA treatment
Primary fetal esophageal fibroblasts, human nontumorous esophageal epithelial cell line, and p53-null esophageal adenocarcinogen cell lines were cultured in DMEM. hTERT-immortalized esophageal epithelial EPC2 cells were grown in KSFM medium with bovine pituitary extract and epidermal growth factors. P73 was then inhibited using lentiviral transduction with shRNA or transfection of RNA, which both targeted a specific sequence found in all the isoforms.
Antibodies, immunoprecipitation, and immunofluorescence
Antibodies were used for following various proteins: p73, p63, c-Abl, phospho-c-Abl, phospho-p53, β-actin, phosphohistone H2A.X, phosphotyrosine, 8-oxoguanine, p53, p21, PUMA, SMUG1, MUTYH, and nonspecific rabbit IgG. Immunoprecipitation was conducted using anti-p73 antibody and analyzing tyrosine phosphorylation of p73 using Western blotting. When conducting immunofluorescence, cells were grown to 50% confluency on chamber sides. Phospho-Histone and 8-oxoG were stained and the intensities of the fluorescent signal were measured.
Real-time PCR and chromatin immunoprecipitation
RNA was isolated, the total RNA was reverse transcribed with a high capacity cDNA reverse transcription kit, and real-time PCR was used. Chromatin immunoprecipitation was conducted on p73 antibody and a nonspecific rabbit IgG was used as a negative control for HET-1A cells. The p73 binding sites were then analyzed. Certain sequences of primers were used to analyze p73, MUTYH, SMUG1 mRNA levels and ChIP.
PCR array, comet, and abrasic site quantification assays
Comet assays were performed with modifications to the protocol. The cells were mixed with a low melting temperature agarose and applied to a pre-agarose coated slide. A buffer was used to induce cell lysis and then proceeded in electrophoresis under alkaline conditions. The slide was then stained for analysis using CometScore Software. In p73 deficient and the HET-1A cells, the mRNA expressions were compared and analyzed.
Surgical Procedures and immunohistochemistry
Mouse strains that carried the wild type p73 and normal p73 genes were analyzed. Esophagojejunostomy was conducted on these mice that carried the wild type p73 and normal p73 genes. The 15-week mice were euthanized and the lower esophagus was removed for analysis by immunohistochemistry.
Nontumorous esophageal HET-1A cells were treated with a BA mixture and the DNA damage was analyzed by immunofluorescence using phospho-H2A.X and 8-OxoG antibodies. A small exposure to the BA mixture inflicted DNA damage through oxidation and strand breakage. The presence of the p53 family upon BA/A treatment was then analyzed. Despite the presence of DNA damage, there was a lack of increased phosphorylation of p53 at serine 15 to induce p53 activation. P53 was only present when the HET-1A cells were treated with a DNA damaging drug called cisplatin. After BA/A treatment, p53 levels decreased and were undetected after a long duration of time. BA/A treatment of SKGT-4 cells resulted to an up-regulation and activation of p73; the analysis revealed that p73 induction is independent of p53. Because the mRNA levels of p73 were not increased in the epithelial cells, it indicated that the up-regulation occurred at the post-translational stage. The presence of c-Abl protein kinase induces the tyrosine phosphorylation, which then activates the p73 protein.
By inhibiting p73 proteins, increased oxidative DNA damage is apparent. P73 activates SMUG1 and MUTYH that are involved in base excision repair by directly binding to these two glycosylases and regulates its transcription. Therefore, the down regulation of p73 proteins decreases the two glycosylases of SMUG1 and MUTYH[6].
↑ abAndrea Bisso, Licio Collavin, and Giannino Del "p73 as a Pharmaceutical Target for Cancer Therapy" Laboratorio Nazionale CIB, Area Science Park, Padriciano 99, 34149 Trieste, Italy. Dipartimento di Scienze della Vita, Universitàdi Trieste, Via L. Giorgieri 1, 34100, Trieste, Italy
↑ abcdefViadiu, Hector. "Gene Recognition by the P53 Protein Family." CHEM 114A Lecture. University of California, San Diego, La Jolla. 14 Nov. 2012. Lecture.
p62 is an atypical protein kinase C (PKC)-interacting protein that plays important roles in cellular functions. It interacts with several key components of various signaling mechanisms. p62 is required for tumour transformation, which makes it crucial for the control of cell growth and cancer.
P62 or sequestosome-1 has been identified as a partner of the atypical protein kinase Cs (aPKCs) in unbiased screens. Initial studies showed that p62 controls localization of the aPKCs to the nuclear factor (NF)-κB cascade. Further studies show that in contrast to the relatively simple structure of Par-6, p62 is rich in protein interacting sequences, which demonstrates its role as a signaling hub.
Cell proliferation under tumorigenic conditions requires that cells increase in size before division can take place. The survival of these cells depends on their ability fo active autophagy, reprogramme its metabolism, and control the production of toxic compounds, such as reactive oxygen species ROS and misfolded proteins. Cell divisions must be monitored and carried out in perfect sequences to avoid dramatic consequences, such as activation of tumorigenesis through genomic instability. These regulatory mechanisms could be rich sources of potential therapeutic targets in cancer. For example, the interference with the nutrient-sensing pathways could prevent cells from dividing.
Studies were done previously to investigate the phenotype of p62-deficient mice to demonstrate the physiological role of p62 to control oesteoclastogenesis and bone remodeling.
Recent studies using unbiased proteomic approach discovered the role for p62 in activation of mammalian target of rapamycin (mTOR) pathway. The mTOR pathway regulates “cell growth and autophagy that integrates nutrient sensing and cell-size control,” and is activated in many types of cancer. p62 specifically associates with mTORC1 – a multi-protein complex controlled by mTOR that channels a variety of signals into a coordinated protein synthesis and inhibiting autophagy. Recent studies discovered that “in p62-deficient cells, amino acid-mediated phosphorylation of the mTORC1 targets P70-S6 kinase (S6K) and eIF4E-binding protei 1 (4EBP1) is severely impareied and, in keeping with decreased mTORC1 activity autophage is upregulated.” p62 is also a substrate of autophagy, which makes a feedforward loop, which increases the activity of mTORC1. In conditions of oxygen deficiency and nutrient deprivation, the p62-mTORC1-autophagy loop could provide a safety mechanism to make sure that the cell death due to nutrients deprivation is irreversible.
Studies show that the “Rag GTPase control amino acid-dependent mTORC1 activity by regulating mTORC1 translocation to a lysosomal associated membrane protein 2 (LAMP2)-positive compartment. p62 binds raptor and the Rags, which triggers the activation of the pathway favouring the formation of the active Rag dimer, through probably a p62 oligomerization mechanism. Constant findings support the need of p62 for the translocation of mTORC1 to the lysosomal surface as p62 was observed to be located at Rab7-psotitive late-endosomal membranes. This is also supported by mTORC1’s role in regulating endocytosis as a response to changes in environmental factors.
p62 and control of the oxidative stress response in cancer
The role of p62 in cell survival and tumorigenesis can be traced to the interation of p62 with tumour necrosis factor receptor associated facter (TRAF) 6 – inflammation signaling molecule, and the degradation of p62 by autophagy. Studies show strong correlation between autophagic pathways, inflammation-mediated cell toxicity and p62. For example, in live physiology, the genetic inactivation of key autophagy molecules increases p62 accumulation and hepatotoxicity that lead to liver tumours. p62 overexpression leads to chronic inflammation and cancer in the liver. Thus, p62 plays a crucial role in autophagy as a tumour suppressor, possibly through the suppression of ROS. There also seems to be a correlation between p62 overexpression to NRF2 activation through the ability of p62 to bond with Keap1, a negative regulator of NRF2 activation. In particular, there seems to be a linear correlation between the reduced autophagy and its subsequent accumulation of p62, which then activates NRF2 to decrease oxidative stress.
However, other experimental data weakens the linear correlation between autophagy, p62, and oncogenesis, because while Ras-induced transformation required p62, it also increases autophagy. Perhaps p62 in fact behaves as a central hub that controls several survival mechanisms in Ras-transformed cells.
“Ongenic Ras modulated p62 levels at a gene transcriptional level through a mechanism involving targeting of an activator protein (AP)-1 enhancer element in the p62 promoter. The removal of p62 prevents oncogenic transformation in an in vivo, “physiologically relevant, endogenous Ras-induced lung cancer model.” The wildtype mice developed in vitro the same cells expressing wild-type p62, which suggests that p62 phosphorylation by cdk1 serves to restrict cell transformation.
Cdk1-mediated p62 phosphorylation restricts cell transformation by the control of mitotic exit. “When tumour cells express either wile type or non-phosphorylatable p62 exit mitosis and transition from mitosis to G1 faster than wild-type cells. An enhaced proportion of nonphosphorylatable p62-expressing cells displays lagging chromosomes and micronuclei, which are indicative of increased genome instability.” These observations demonstrate that p62 stimulates tumorigensis by controlling ROS levels that promotes cell survival, and also promotes premature exit from mitosis that increases proliferation rate and genome instability.
p62 plays a major role in cell transformation machinery, which affects processes such as cell growth, survival and mitosis. The role of p62 under non-pathological conditions is to control bone and metabolic homeostasis, which could suggest that p62 is indirectly in control of cancer, which makes it a potential therapeutic target in metastasis. p62 also plays a role in “maintaining metabolic homeostasis by restraining adipogenesis and promoting energy expenditure.” It is also demonstrated that obesity-induced inflammation and insulin resistance can promote at least some type of cancer, so the inactivation of p62 at an organismal level could promote the cancer progression if it results in enhanced adiposity and obesity.
Moscat, Jorge. and Diaz-Meco, Maria T. "p62: a versatile multitasker takes on cancer" Trends Biochem Sci. 2012 June; 37(6). Review.
Retinoblastoma:
The gene that cods for retinoblastoma (Rb gene) is an important tumor suppressor of cancer. Retinoblastoma itself is a childhood cancer that results from the loss of function of both copies of the Rb gene (each cell has two copies of the Rb gene). People with retinoblastoma can either develop a tumor in one eye (unilateral) or in both eyes (bilateral).
Hereditary vs. Sporadic Cases:
Hereditary retinoblastoma occurs in patients who have a family history of the disease. Because they inherit one non-functional copy of the Rb gene, only one mutation is needed for total loss of function. If someone is born with one bad copy, they have an approximately 90 % chance of developing the tumor in both eyes (90 % penetrance). In other words, being born with one non-functional copy increases the likelihood that a mutation will occur in the second copy and lead to a bilateral tumor.
Sporadic retinoblastoma occurs in patients with no family history of the disease. Therefore, they are born with two good, functional copies of the Rb gene. As a result, in order for these patients to develop retinoblastoma, two mutations in the gene are needed. This only occurs in 1 out of every 350,000 live births. Sporadic retinoblastoma is unilateral, meaning that the tumor occurs in only one eye.
Both sporadic and hereditary forms of the disease require two bad copies of the Rb gene. This means that the Rb gene is still functional even if one copy is abnormal. Loss of heterozygosity at the Rb locus on chromosome 13 is one potential mechanism for the development of bilateral retinoblastoma in patients who have a family history.
Retinoblastoma as a Tumor Suppressor in Normal Cells:
The normal function of the Rb protein is to act as an assembly factory: it brings together other proteins and has multiple binding sites to accomplish this. The Rb protein binds to multiple transcription factors and can either activate or repress transcription. For example, Rb protein strongly interacts with and binds to E2F-DP, a heterodimer that includes the E2F transcription factor. When Rb is bound to E2F-DP, the protein complex then can go to promoters that are regulated by E2F. Once there, Rb can recruit histone deacetylase to repress or turn off the transcription of genes regulated by E2F. This action suppresses cell division, which is why the Rb gene is a tumor suppressor. Since cancer cells want to divide, retinoblastoma patients lack this repressive function. In addition to histone deacetylase, Rb protein can also recruit histone and DNA methyltransferases. These methyltransferases can modify and epigenetically silence the target DNA, leading to inactive heterochromatin (permanent silencing). Therefore, Rb has a two-fold function. It can either repress the transcription or epigenetically silence the DNA of genes involved in the cell cycle that promote cell division.
How Retinoblastoma Function is Turned Off in Normal Cells and Loss of Tumor Suppressor Ability in Cancer Cells:
In order for cancer cells to grow, divide, and metastasize, they need to somehow inactivate the function of key tumor suppressors, such as the Rb gene. Since the normal function of the Rb protein is to repress the cell cycle, this ability is an obstacle to cancer cell growth. Since cancer cells in a retinoblastoma patient operate with non-functional Rb genes, the tumor suppressor function of Rb is non-existent and cancer cells can proliferate uncontrollably. However, Rb function is also controlled in normal, non-cancerous cells. We require cell division to survive, so Rb function must be regulated so that the cell cycle is not always repressed.
As stated above, Rb works to repress the expression of genes regulated by E2F. These genes include Cyclin A, Cyclin E, and other proteins involved in the cell cycle. Cyclin/CDK complexes regulate the progression through the cell cycle (they oppose the function of the Rb protein). Therefore, in order to do their job effectively, they phosphorylate Rb as multiple sites. This phosphorylation causes Rb to lose its ability to bring proteins together. Since it can no longer bind E2F, it cannot turn off the expression of genes that E2F regulates and the cell cycle can go on uninterrupted by Rb repression. Then, E2F can recruit histone acetylases that activate gene expression of cell cycle cyclins.
P62 was initially discovered as a protein kinase C-interacting protein (PKC). P62 plays a versatile role in the body and maintains an important role in an abundance of cellular functions. An interesting characteristic of p62 is that it plays a major role in cell growth and cancer. P62 has the ability to modulate (and be a substrate of autophagy), warrant efficient mitosis of cells, and regulate ROS levels and limit misfolded proteins. All of these qualities of p62 play critical roles in the development of cancer, making p62 a primary regulator of tumorigenesis. [1]
Studies show that p62 plays a role in the activation of the mammalian target of rapamycin (mTOR) pathway, which is a key regulator of cell growth and autophagy. There are two multi-protein complexes of mTOR (mTORC1 and mTORC2). P62 primarily interacts with mTOR1 through a key component of mTORC1, raptor. mTORC1 senses several cellular/environmental signals including: protein misfolding, nutrient availability and growth signals. mTORC1 controls cell growth through the phosphorylation of S6K and 4EBP1, and regulates autophagy by targeting Ulk1 and Atg13. [1]
In cells with a deficiency in p62, the phosphorylation of S6K and 4EBP1 is diminished, therefore mTORC1 activity decreases and autophagy is increased. However, since p62 is a substrate of autophagy, mTORC1 activity is promoted. Under the conditions of nutrient deprivation, the interrelations between autophagy, p62, and mTORC1 can create a safeguard mechanism to ensure the irreversibility of cell death. [1]
Studies have also shown that the overexpression of p62 can lead to the generation of liver tumors. Consequently, it is important to regulate the accumulation of p62 in order to prevent tumorigenesis. P62 can accumulate with the inactivation of autophagy molecules (ex. Atg7) and lead to hepatotoxicity, thus promoting liver tumors. [1]
However, creating a model that links accurately links together autophagy and p62 to cancer is very complicated process with many contradictions. It would make sense to assume that autophagy would induce tumorigenesis in a nutrient deprived environment. But there is contradictory data that supports the contrary. An essential protein for autophagy is Beclin-1, which has been found to suppress tumor formation. Other tumor suppressors, such as phosphatase and tensin homolog and the TSC proteins, promote autophagy by inhibiting mTORC1. [1]
Overexpression of p62 has been linked to the activation of nuclear factor (erythroid-derived 2)-like 2 (NRF2), which is a transcription factor that can suppress reactive oxygen species (ROS). Thus, one would come to the conclusion that reduced Atg7 deficiency (thus a decrease in autophagy), would lead to an accumulation of 62, which would lead to an increase in activation of NRF2, thus reducing oxidative stress. From this deduction, it would be sufficient to reason that the ablation of p62 in Atg7-deficient livers would make hepatotoxicity worse as there is a decrease in activated NRF2, which alleviates oxidative stress. However, studies have shown a stark contrast to this analysis. It has been shown that hepatotoxicity in Atg7-deficient mice is actually prevented by the inactivation of p62 instead of increased. This leads to an uncertainty in the relation between autophagy, p62, and tumorigenesis, and debunks the model previously proposed. [1]
Furthermore, the relationship between autophagy, p62, and cancer becomes more clouded by recent studies that show that Ras-induced transformation increases autophagy, but needs p62. However, amongst this puzzlement, this presents a detachment between p62 and autophagy; meaning that Ras-induced autophagy and Ras-induced p62 are independent of each other but both required for the Ras signal. For example, “Ras-induced autophagy would be required for the removal of damaged and dysfunctional mitochondria, which is thought to be associated with metabolic reprogramming and contributes to maintaining low ROS levels. Ras-induced p62 might shuttle the damaged mitochondria to the autophagosome by interacting with the autophagosomal membrane protein Atg/LC3.” [1]This shows that autophagy and p62 work in unison to control Ras mechanisms, but still maintain their own independence. [1]
Another transcription factor, known as NF-kB, is activated by Ras and the oligomerization of TRAF6 (caused by p62). Studies have shown a link between lung cancer and an overexpression of TRAF6. Therefore, inactivating TRAF6 will inactivate NF-kB and inhibit tumorigenesis. Though p62 can help regulate harmful levels of oxidative species, p62 can also promote tumorigenesis through the activation of NF-kB. From these observations, it is seen that p62 can affect tumorigenesis through two different transcription factors, NF-kB and NRF2. [1]
It can be seen through the analysis of p62 knockout mice, that the central purpose of p62 under nonpathological conditions is to control bone and metabolic homeostasis. This function of p62 may lead to indirect control of cancer. Cancer metastasis is prevalent in bone and can be affected by crosstalk between tumor and bone cells. Thus, p62 has the possibility for being a therapeutic target in metastasis. Moreover, elevated amounts of p62 can help maintain metabolic homeostasis by decreasing adipogenesis. This can also lead to regulation of some forms of cancer, since it has been shown that obesity-induced inflammation and insulin resistance (results of adipogenesis) promote forms of cancer in both humans and mice. Similar to the protecting effects of elevated p62 (in regards to obesity), autophagy proteins in mice have been shown to also protect against obesity. [1]
↑ abcdefghijMoscat, Jorge, and Maria T. Diaz-Meco. "p62: a versatile multitasker takes on cancer." June 2012.
Brooke-Spiegler syndrome is an illness involving skin tumors developed from skin appendages, for example, hair follicles and sweat glands. It is caused by the mutation of CYLD, a tumor suppressor. CYLD is a human gene and function as a deubiquitinating enzyme.
Deubiquitinating enzymes (DUBs) belong to a large group of proteases. They regulate the ubiquitin-mediated pathways. They also function with proteasome by direct or indirect association. They are involved in many stages of biological functions: cell growth,cell differentiation, cell development, oncogenesis, neuronal diseases, and transcriptional regulation.
DUBs is used as a catalyst in the removal of ubiquitin from C-terminal extension peptides and linear poly-ubiquitin fusion. DUBs usually contains esterase and amidase activites in vitro. Ubiquitin-esters and ubiquitin-amidocoumarins are used as substrates for activity assays.
DUBs are divided into two divisions. One of them is ubiquitin C-terminal hydrolases (UCHs). They are enzyme with small size and they act as a catalyst in the removal of peptides and small molecules from the C-terminus of ubiquitin. UCHs usually cannot generate monomeric ubiquitin from protein conjugates or disassemble poly-ubiquitin chains.
Another group is ubiquitin-specific proteases (USPs/UBPs). They are bigger size compared to UCHs. They usually contain N-terminal extensions which can used in substrate regonition, subcellular localization and protein-protein interactions. They also can remove ubiquitin from protein conjugates and disassemble ubiquitin chains. An example of USP is Isopeptidase T (IsoT). It is capable of binding ubiquitin and disassembles free poly-ubiquitin chains[1]
CYLD is one of the deubiquitylating enzymes and the regulate NFKB activity negatively. NFKB (nuclear factor kappa beta; or nuclear factor KB) is a transcriptional regulator. It is important in the immune system and they regulate the expression of lots of genes that are critical for the regulation of tumorigenesis, autoimmune diseases, cancer, apoptosis, and inflammation. CYLD gives instruction for making a protein to help regulating NFKB. The protein makes the cells response properly to signals to destroy themselves when the cells become abnormal. CYLD, as a tumor suppressor, inhibits the uncontrolled growth and division of cells. The mutations usually occur in the C-terminal of CYLD which contains a DUB domain. Its function is related to the signal transduction involved in the activation of NFkB 12, 13, 14. The DUB catalytic activity is disrupted by the mutations of CYLD and causes the resistance of cell death and tumorigenesis.
The tumor-suppressive activity of CYLD depends on the ability to inhibit the anti-apoptotic activity of NFkB. The stimulants of cell to survive apoptotic would increase when CYLD is depleted by RNA interference.
Patients with Brooke-Spiegler syndrome have 20 CYLD gene mutations. Patients have mutation in one of the copies of CYLD gene in each cell when they are born, leading to the inhibition of the production of CYLD protein. Also, there is another mutation in another copy of CYLD in some cells which causes genetic changes. If there are mutations in both copies of CYLD gene, there would be no instruction regarding the apoptosis of abnormal cells. The regulation of NFkB will be inhibited. Therefore, the cells will undergo division rapidly and lead to the formation of tumor. There will be formation of multiple noncancerous tumors that develop in skin appendages. There are different kinds of possible developed skin appendages due to the mutation of CYLD, and they are spiradenomas – tumors of sweat glands, trichoepitheliomas – tumors of hair follicles, and cylindromas – tumors of hair follicles.
If patients have 22 mutations in CYLD gene, they will develop multiple familial trichoepithelioma, and they will have a lot of developed trichoepitheliomas. If patients have30 CYLD gene mutations, they will develop familial cylindromatosis. Patients will have a much larger number of cylindroma. All patients are born with a mutated CYLD gene and have another mutation in another copy of CYLD gene (similar to Brooke-Spiegler syndrome).[2][3][4][5]
As mentioned, CYLD negatively regulates NFKB activity. NFKB is a protein composed of different members of the Rel family of transcription factors. They control genes that regulate a lot of biological processes by acting as dimeric transcription factors. NFkB is hidden in the cytoplasm and is bound by inhibitor proteins IkBa, IkBb, IkBg, and IkBe. When NFkB is activated, phosphorylation of IkB will occur, and its ubiquitination and degradation will occur next. This leads to the exposure of nuclear localization signals on NFkB subunits and the translocation to the nucleus. In the nucleus, NFkB binds to a gene with specific sequence (5'GGGACTTTCC3') and activates their transcription. IkB is phosphorylated by IkB kinase complex which contains IKK1/IKKa, IKK2/IKKb and IKK3/IKKg.They phosphorylate IkB and lead to its ubiquitination and degradation. The activator tumor necrosis factor binds to its receptor and recruits TNF receptor death domain (TRADD). TRADD binds to TNF receptor associated factor (TRAF2). TRAF2 recruits NFkB inducible kinase. IKK1 and IKK2 can be phosphorylated by MAP kinase NIK/MEKK1 and they can phosphorylate IkBa and IkBb. TRAF2 also has the ability to interact with A20. A20 is a zinc finger protein in which the expression is induced by agents that activate NFkB. It is used to inhibit TRAF2-mediated NFkB activation. It is also used as inhibitor of TNF and IL-1 induced activation of NFkB. Therefore, A20 is considered an inhibitor of NFkB activation.[6]
There are two pathways:
1. Canonical pathway:
NFkB/Rel proteins are bound and inhibited by IkB proteins. The IKK complex is activated by proinflammatory cytokines, LPS, growth factors, and antigen receptors. IKK complex phosphorylates IkB proteins, leading the ubiquitination and proteasomal degradation and the NFkB/Rel complexes will be freed. NFkB/Rel complex are also activated by phosphorylation and translocate to the nucleus, inducing the target gene expression.
2. Noncanonical pathway:
NFkB2 p100/RelB complex remains inactive in the cytoplasm. The IKKα complexes are activated by the signaling through a subset of receptors. The activated IKKα phosphorylates C-terminal residues in NFkB p100 which leads to the ubiquitination and proteasomal processing to NFkB2 p52. The NFkB p52/RelB complexes translocate to the nucles, inducing target gene expression. [7]
Many cancers survive and grow based on the rate of aerobic glycolysis. Some cancers are attracted to glutamine, one of the 20 common amino acids that code for a genetic sequence or code. However, glutamine is not a typical amino acid that synthesizes glucose used in the process of glycolysis. Moreover, cells uptake glutamine not because it is a nitrogen donor to nucleotides. In fact, glutamine plays an important role in up-taking essential amino acids and activating TOR kinase, which is a specific enzyme that is necessary for balancing protein synthesis and degradation. Glutamine is the main mitochondrial substrate that is required to maintain the membrane potential in mitochondria of cancer cells as well as aid in NADPH production that is necessary for synthesizing other macromolecules as well as control redox chemical reactions within the body.
Otto Warburg discovered the Warburg effect which allowed for the study of excess metabolism of glucose as well as glutamine.
It has been studied by Otto Warburg, a notable German physiologist and Nobel Laureate, that cancer cells seem to uptake more glucose and produce more lactic acid than regular cells or tissues. Warburg hypothesized that cancer results from falling back to metabolism that involves rapidly increasing the number of single cell eukaryotes. This became known as the Warburg effect and studies have shown that this effect is brought on by activating oncogenes associated with glucose uptake. Furthermore, activation of the cell signal, phosphoinositide 3-kinase (PI3K) cause levels of glucose uptake to increase and causes the cell's metabolism to exceed the maximum use for glucose. Because of this, cancer cells can then secrete extra glycolytic metabolites in the form of lactic acid. Some tumors have this similar reaction, but instead of excess glucose metabolism, there is inefficient glutamine metabolism. These types of cancer cells or tumors cannot survive when there is not enough extra glutamine and are therefore considered to be "addicted" to glutamine. Glutamine is in fact a necessary substrate used in anabolic growth of cells, especially those of mammals. Due to the addiction of glutamine exhibited by some cancer cells, the study of glutamine in cell growth and cell-signaling pathways will help to discover new therapeutic treatments of some cancers.
As shown in the structure of glutamic acid, there is a free amino group containing an available nitrogen for donation.
Cancer cells, like any other cell, must synthesize compounds that contain nitrogen. Typically these compounds are in the form of nucleotides and essential amino acids. Glutamine is a typical nitrogen donor and donates through three enzymatic steps in the synthesis of purines and two enzymatic steps in the synthesis of pyrimidines. Glutamine donates an amide group and is then converted to glutamic acid.
Glutamic acid becomes the primary donor of nitrogen for synthesizing nonessential amino acids. Transaminases are specific enzyme that aid in transferring the amino group of glutamic acid to α-ketoacids which are used to create nonessential amino acids. Some examples of α-ketoacids include pyruvate, oxaloacetate, or gamma-semialdehyde which are also used to synthesize nonessential acids such as alanine, serine or aspartate. Glutamic acid as a form of glutamine, donates its carbon skeleton and nitrogen to proline, another nonessential amino acid, as well.
The structure of mTORC (mammalian target of rapamcyin) is shown. This is specifically for serine/threonine kinase.
Glutamine plays an important role in the process of protein translation in cancer cells. This was observed in the response to glutamine of target rapamycin complex 1 in mammals (mTORC1). mTORC1 is typically a major regulator of cell growth and activates protein translation but inhibits the response of macrophages to excess amino acid production. Through the study of yeast, it was concluded that there needs to be a sufficient level of amino acids for TORC1 to be properly activated. TORC1 seemed to respond to glutamine as well as essential amino acid levels. mTORC1 activation seems to response mostly to leucine; however glutamine has been shown to be necessary to activate mTORC1 to the maximum. Studies of cell lines of mTORC1 showed that cells depended not only on the presence of essential amino acids but also glutamine at the same time. From studying how glutamine is taken up through an importer called SLC1A5 showed that glutamine was exported using SLC7A5 which exchanges glutamine for uptake of essential amino acids. If there was not enough SLC1A5 in the cells, glutamine could not be taken up and exported; therefore essential amino acids could not be absorbed either. Glutamine also contributes to nonessential amino acids that are found in proteins that are newly translated.Therefore, it could be concluded that glutamine is not used for anabolic metabolism but instead for exchanging essential amino acids that activate TORC1 used in translating proteins, in and out of the cell. Ultimately, glutamine signals mTORC1 as well as provides essential amino acids for protein translation.
In 1955 Harry Eagle found that glutamine was essential for proliferating cells. Eagle studied on nutritional needs of a cell and found that glutamine was consumed ten times more than any other amino acids. Cells were unable to multiply without glutamine. Kovacevic and collegues found, in 1971, that glutamine is used as fuel and the carbon molecules found in gluatmine was also found in the carbon dioxide excreted by the cell. Glutamine loses its amide group using the an enzyme, glutaminase, to produce glutamic acid, which then loses its amine group using glutamic acid dehydrogenase to form α-ketogluterate.
Current studies use 13C to identify the carbon movements in the conversion of glutamine to lactic acid. Gluatamine uses malic enzymes to convert to lactic acid. Malic enzymes decarboxylates malic acid making carbon dioxide, NADPH and pyruvate. The NADPH produced is then used to for the cell to multiply.
Glutamine also contributes to the cell by producing oxaloacetate according to studies shown in a glioblastoma cell. The oxaloacetate (OAA) is bonded to an acetyl-CoA to make citrate. The acetyl-CoA is formed by breaking down cholesterol, fatty acids and chromatins. Gluatmine goes through anaplerotic reactions to refill the amount of carbons entering the TCA cycle. By replenishing the carbons in the mitochondria the cell is able to synthesize nucleotides, proteins, and lipids. Due to glutamine metabolism in cancer cells, studies show that the mitochondria in essential even for cancer cells. 13C NMR studies do show that cancer cells do not depend of oxaloacetate production through pyruvate carboxylation. In fact cancer cells suppress the activity of pyruvate carboxylation since cancer cells have glutamine to produce oxaloacetate.
Glutamine Metabolism Regulated by Oncogenic Levels of c-MYC
As discussed above, glutamine serves as a crucial nitrogen donor for the purpose of nucleotide synthesis. Studies that utilize reverse transcriptase ((RT)-PCR), and chromatin immunoprecipitation, suggest that the c-MYC (Myc) is involved in activating 11 genes that are involved in nucleotide biosynthesis. Massive amounts of Myc have been linked to the increase process of glutaminolysis, which suggests that Myc activation and amplification serves as one of the more common oncogenic events that can be witnessed in some cancers. Rt-PCR and ChIP both have the tendency to advocate the binding of Myc and transcriptional occurrences of two glutamine transporters: the SLC38A5 (SN2) and SLC1A5 (ASCT2). SLC1A5 is the transporter that is necessary for the glutamine-dependent activation of mTORC1. Myc also serves as a promotion factor to the metabolism process of converting glutamine into glutamic acid, which eventually becomes lactic acid at the end of the metabolism process. In addition, Myc also plays a significant role in influencing the post-transcriptional regulation of glutamine catabolism. The Myc is also a factor that leads the mitochondrial membrane to be dependent on exogenous glutamine as a source of carbon. Despite the fact that there is a great availability of glucose, glutamine depletion within Myc-transformed cells also significantly decrease the levels of TCA cycle metabolites.
Many cells are sensitive when they are deprived of glutamine especially cancer cells in pancreatic cancer, glioblastoma multiforme, acute myelogenous leukemia and lung cancer. In the 1950s, experiments were performed and showed that 6-diazo-5-oxo-L-norleucine, azaserine and acivicin were analogs of glutamine. Testing of these analogs showed that they have an effect against certain tumors. These compounds inhibited enzymatic steps that involved glutamine when synthesizing nucleotides. The following are current ways to reduce cancer that have been researched and studied:
1. Glutamine uptake suppression
Studies show that there has been an increase transporters that have great attraction or affinity to glutamine in cancer cells specifically. A common transporter of glutamine is known as SLC1A5 and targets the Myc oncoprotein. Many cancers display an increase in this transporter; however there are inhibitors that have been discovered that allow the glutamine to be taken up less or at least not in excess amount. One of these inhibitors is called GPNA or L-γ-glutamyl-p-nitroanilide and it not only inhibits the amount of glutamine that is absorbed, but it also causes the mTORC activation, but only those proteins that depend solely on glutamine, to be suppressed as well.
2. Transamination Suppression
The process of transamination is shown. Transamination is an equilibrium reaction and can produce amides as well as carboxylic acids. These products can be derived from glutamine.
Because the major route that allows carbons derived from glutamine to enter the citric acid cycle is through a process known as transamination, studies have shown that the transaminase inhibitor known as amino-oxyacetic acid (AOA) can help with suppressing cancer. Treatment of AOA seemed to be promising since it produced a cytostatic effect where it inhibited cell growth in breast cancer. It also showed the same effect on cell growth in glioblastoma cells. Inhibiting one component of glutamine metabolism or in this case glutamate transamination resulted in the reduction of cancer effects.
3. Inhibition of Complex I in Citric Acid Cycle
Structure of Metformin, a drug used to target metabolism of glutamine.
Cancer cells that depend on glutamine cause the mitochondria to produce anabolic precursors using glutamine instead of glucose. Glutamine will flow in and out of the citric acid cycle and will cause NAD+ to be regenerated continuously using the electron transport chain. Drugs have been developed to suppress these effects in the mitochondria of cells. For example, Metformin has been proven to slow the growth of cancer cells and tumors and displays efficiency in the mitochondria of liver cells. Metformin not only targets glutamine metabolism but also lowers blood glucose concentration.
4. Stopping mTORC Activation
Glutamine can be imported into cancer cells through the transporter SLC1A5. This also allowed essential amino acids to be imported such as leucine, which will activate mTORC1 kinase. Treating cancer cells with GPNA which inhibits SLC1A5 blocks glutamine from activating mTORC1 and causes autophagocytosis of cancer cells to occur.
5. Lower Blood Glutamine Levels
The structure of L-Asparaginase enzyme. It is capable of lowering glutamine levels, but could potentially have toxic side effects.
Enzymes can be used to lower the blood glutamine levels. Asparagine which hydrolyzes into aspartic acid through the help of the enzyme L-asparaginase can be used to treat acute lymphoblastic leukemia. All of the cells can synthesize asparagine and thus L-asparaginase can cause glutamine to hydrolyze into glutamic acid and ammonia as well. L-asparaginase lowers glutamine levels; however it can also have a high toxicity level.
Alternatively, research has been further conducted to discover that plasma glutamine can be decreased by using phenyl butyrate. This has been specifically used to treat hyperammonemia patients that have acute liver failure or urea cycle disorders. Phenyl butyrate has been successful in decreasing the amount of glutamine in cells and can break down to form phenyl acetate that can combine with the leftover glutamine with the aid of phenyl acetylcoA to form phenylacetylglutamine which can be simply excreted from the body through urination; thus reducing the amount of glutamine in the body and reducing the attraction of cancer cells to glutamine.
Certain oncoproteins in tumor cells can alter the metabolism of tumor cells making them have a higher affinity to the amino acid glutamine. Initially this came as a bit of a surprise considering that glutamine is a non-essential amino acid that can easily be synthesized in the cell. However, after considering the role that glutamine plays in metabolism and in affecting cell growth, it became clear that glutamine was ideally suited to further the goal of cancerous cells. By taking advantage of the fact that some cancerous cells may be addicted to glutamine, researchers can perhaps develop therapeutic treatments to eliminate this special class of tumor cells. The challenge, however, is to create a drug that can target the glutamine used in the cancer cells, but leave the glutamine in normal, non-transformed cells untouched.
The Ligand-Binding Site and Glutamate Receptor Trafficking:
The glutamate receptors' extracellular domains share homology with bacterial periplasmic binding proteins. Due to this observation, many studies have started for the ligand-binding domains of AMPA receptor subunits. To further expand on these observations, Stephanie J. Mah and others have used the kainate receptor subunit, glutamate receptor 6 (GluR6) through the mutating preserved residues (R523, T690, or E738) in the ligand-binding pocket. After this, functional responses and agonist binding were removed, however more importantly homomeric mutant receptors were kept in the endoplasmic reticulum. These mutants created oligomers in the ER and were brought about to the surface when expressed with wild-type GLuR6 subunits. Due to this, the retention did not make any apparent contributions to the misfolding. It is assumed and suggested that the ER can supervise the functional state of fully assembled GluR channels and keep those that are unable to bind agonist, most likely due to high intracellular glutamate causes nascent receptor to be exported in their bound conformation.
Axons are able to extend and retract as they search for their targets. The signaling mechanisms that assist axon growth includes Rho GT-Pases and cytoplasmic calcium transients. In reaction to extracellular factors, the Rho GTPases work as switches that will activate the cytoskeletal rearrangement required for an axonal direction change. It was suggested that a mechanism that links Rho GTPases and calcium in the turning of growth cones. A cultured Xenopus spinal neurons with an extracellular ryanodine gradient was presented, allowing the activation of the release of calcium in an asymmetrical-like pattern across the growth cone; the growth cone then turns toward the source. This turning of the growth cone is reliant on calcium-dependent upregulation of the Rho GTPase, Cdc42, which is most likely via protein kinase C- and Ca2+/calmodulin-dependent protein kinase II-dependent phosphorylation. It was concluded that calcium is what triggers the turn signal.
On the retina, horizontal cell dendrites disperse evenly across the retina. Benjamin E. Reese and others utilized several strains of mice in which, although a large discrepancy in horizontal cell number, dendrites were kept equally spread. For that reason, the size of the dendritic fields varied inversely with cell number. Cone photoreceptors were ablated using cone-specific diphtheria toxin expression; this was done so that the influence could be tested. With the absence of cone synapses, the organization of horizontal cells in the outer plexiform layer was normal. Furthermore the trade mark size of the dendritic field was normal as well. The dendrites in the retinas without cones divided greatly and showed no clusters of terminals that are normally observed at cone pedicles.
Neonatal Coxsackievirus Infection of Neuronal Progenitors:
Coxsackievirus B (CVB) infections have the ability to cause severe consequences such as meningoencephalitis, especially in new-born babies and young children. Ralph Feuer and others were able to track the dangerous path of CVB3 infections in neonatal mice. From their observations, the virus seemed to have preferred to attack dividing neuronal progenitor cells in the subventricular zone (SVZ). Infected cells were no longer being produced in the SVZ, based on their lack of immunoreactivity to Ki67, a nuclear antigen. On the other, they were still able to keep their migratory capacity. The infected cells trailed along the rostral migratory stream or radial glial cells to arrive at the olfactory bulb or cerebral cortex, respectively. The infected cells seem to also differentiate normally, however they were still capable of creating viral proteins. Through this process, the virus then latches onto proliferating neuronal precursors only to cause virus-induced lysis of mature cells at the end. It is suggested that depletion of infected mature cells are a possible cause for the neuro-developmental shortage associated with CNS infection with CVB.
References: http://www.jneurosci.org/content/25/9/i.full.pdfProto-oncogene is a normal gene that is responsible for cell growth, cell differentiation, cell division, and apoptosis .
However, these types of genes have the potential to become an oncogene, a cancer-causing gene.
If a cell can no longer control growth, death, and division, there can be huge health problems.
Studying why and how to prevent proto-oncogenes from changing to oncogenes has been gaining more and more attention.
Mutations in proto-oncogene can cause it to turn into an oncogene.
Point-mutation: deletion or insertion
Gene amplification event.
Chromosomal translocation.
Mutations in microRNA (miRNA): microRNAs regulate genes by down-regulating them, but mutations in miRNA can prevent them from suppressing the proper genes.
Cancer cells go through many changes that are difficult to target individually. Scientists believe that cancerous cells depend on a certain oncogenes more than the others when it comes to cellular growth and replication. This characteristics is called oncogene addiction. If scientists can determine this specific gene, they can treat the cancer by blocking this gene.
The process of a cell with a proto-oncogene becoming an overly abundant cancerous oncogene which the cell then produces the oncogene protein in excess initiating it to develop into a cancerous cell and forming a tumor. Targeting the cell when it has excess of the oncogene protein prevents more cancerous cells from forming.
If scientists cannot target a certain gene and prevent it from causing cancer, then why not turn to a different method of treatment, such as suppressing that cancerous oncogene. When certain cells have oncogenes that are amplified, or they are present in excess, they produce large amounts of their proteins which can initiate the formation of cancerous cells and ultimately lead to tumors. In breast cancer for example, the proto-oncogene HER2/neu is present in normal tissue cells but it turns into an oncogene when the HER2/neu gene is present in large excess and it produces in abundance its respective HER2/neu protein in a cell. Scientists have concluded that, in breast cancer patients for example, cancerous cells that have a high amount of HER2/neu protein did not respond very well to medical treatments of chemotherapy as to those patients who did not have the HER2/neu protein in large excess in their cells. In order to help patients combat their cancer, new drugs have been made available to target the HER2/neu protein and thus reducing the growth of the cancerous cells and ultimately giving the patients a better chance of overcoming the cancer. Two drugs that are used to target the HER2/neu protein abundant cells are Herceptin® and Tykerb®, more drugs are currently undergoing clinical testing so that doctors can have a larger arsenal of drugs to help patients overcome cancer. In conclusion, by targeting cells that produce too much of the HER2/neu protein, which become cancerous, slows the progress of the cancer cells forming and thus making the cancer more easily treatable.[1]
An alternative way to treat cancer on a genetic level is to restore tumor suppressor genes that are not functioning as they should be. This method is more challenging for scientists because they have not been able to develop a successful way to do this. One large problem they face to solving the restoration of suppressant genes is a means to take the cancer cells and insert new DNA to make them function properly. The complexity of cancer cells and their potential multiple mutations only makes it even more so difficult because now there is more than one gene that needs to be replaced. One attempt that was carried out with the mutated gene TP53 where the scientists used a virus, that they had put the non-mutated TP53 gene into, to try to make it insert the normal gene into the cancerous cells. However, this was only successful when carried out in laboratory experiments and not successful in trials with patients. [1]




















.svg)








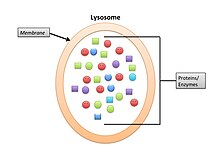











.jpg)


















![{\displaystyle \Delta G=\Delta G^{\circ }+RT\ln Q[C][D]/[A][B]\,}](https://wikimedia.org/api/rest_v1/media/math/render/svg/27543803e1e0466f6d4c75b82d3f8df4c39df447)





























_In_GI_Tract.JPG)


















 Juxtacrine signaling are reactions when proteins from the inducing cell interact with receptor proteins of adjacent responding cells. The inducer does not diffuse from the cell producing it. There are three types of juxtacrine interactions. In the first type, a protein on one cell binds to its receptor on the adjacent cell. In the second type, a receptor on one cell binds to its ligand on the extracellular matrix secreted by another cell. In the third type, the signal is transmitted directly from the cytoplasm of one cell through small conduits into the cytoplasm of an adjacent cell.
Juxtacrine signaling are reactions when proteins from the inducing cell interact with receptor proteins of adjacent responding cells. The inducer does not diffuse from the cell producing it. There are three types of juxtacrine interactions. In the first type, a protein on one cell binds to its receptor on the adjacent cell. In the second type, a receptor on one cell binds to its ligand on the extracellular matrix secreted by another cell. In the third type, the signal is transmitted directly from the cytoplasm of one cell through small conduits into the cytoplasm of an adjacent cell.

















 Cyclic adenosine monophosphate
Cyclic adenosine monophosphate Cyclic adenosine monophosphate
Cyclic adenosine monophosphate Calmodulin 3D structure
Calmodulin 3D structure



















 Figure 1. TGFβ Signaling Pathway
Figure 1. TGFβ Signaling Pathway


.jpg)



 Caption1
Caption1

.jpg)
















 Clathrin Cage
Clathrin Cage



 The Human papilloma virus E7 oncoprotein mimic of the LxCxE motif (red) bound to the host Retinoblastoma protein (dark grey)(PDB 1gux.
The Human papilloma virus E7 oncoprotein mimic of the LxCxE motif (red) bound to the host Retinoblastoma protein (dark grey)(PDB 1gux.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
![[13]](https://commons.wikimedia.org/wiki/File:Acetyl-CoA.svg){kind=link}
![[14]](https://commons.wikimedia.org/wiki/File:Pyridoxine_structure.svg){kind=link}
![[15]](https://commons.wikimedia.org/wiki/File:Catabolism.svg){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
![[16]](https://www.weizmann.ac.il/molgen/members/Kimchi/images/research_figures/6_5_12/newFig2.jpg){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
![[17]](https://en.wikipedia.org/wiki/File:Itrafig2.jpg#filelinks){kind=link}
{kind=link}













{kind=link}
{kind=link}

{kind=link}
{kind=link}












