
Structural Biochemistry/Volume 9
Enzymes are macromolecules that help accelerate (catalyze) chemical reactions in biological systems. This is usually done by accelerating reactions by lowering the transition state or decreasing the activation energy.

Some biological reactions in the absence of enzymes may be as much as a million times slower. Virtually all enzymes are proteins, though the converse is not true and other molecules such as RNA can also catalyze reactions. The most remarkable characteristics of enzymes are their ability to accelerate chemical reactions and their specificity for a particular substrate. Enzymes take advantage of the full range of intermolecular forces (van der waals interactions, polar interactions, hydrophobic interactions and hydrogen bonding) to bring substrates together in most optimal orientation so that reaction will occur. Also, enzymes can be inhibited by specific molecules by called competitive, uncompetitive, and noncompetitive inhibitors.
Catalysis happens at the active site of the enzyme. It contains the residues that directly participate in the making and breaking of bonds. These residues are called the catalytic groups. Although enzymes differ widely in structure, specificity, and mode of catalysis a number of generalizations concerning their active sites can be made:
1. The active site is a three dimensional cleft or crevice formed by groups that come from different parts of the amino acid sequence - residues far apart in the amino acid sequence may interact more strongly than adjacent residues in the sequence.
2. The active site takes up a relatively small part of the total volume of an enzyme. Most of the amino acid residues in an enzyme are not in contact with the substrate, which raises the question of why enzymes are so big. Nearly all enzymes are made up of more than 100 amino acid residues. The "extra" amino acids serve as a scaffold to create the three dimensional active site from the amino acids that are far apart in the primary structure. In many proteins the remaining amino acids also constitute regulatory sites, sites of interaction with other proteins, or channels to bring the substrate to the active sites.
3. Active sites are unique microenvironments. In all enzymes of known structure, substrate molecules are bound to a cleft or crevice. Water is usually excluded unless it is a reactant. The nonpolar microenvironment of the cleft enhances the binding of substrates as well as catalysis. Nevertheless, the cleft may also contain polar residues. Certain of these polar residues acquire special properties essential for substrate binding or catalyis.
4. Substrates are bound to enzymes by multiple weak interactions. Stated above
5. The specificity of binding depends on the precise defined arrangement of atoms in the active site. Because the enzyme and the substrate interact by means of short-range forces that require close contact, a substrate must have a matching shape to fit into the site. However, the active site of some enzymes assume a shape that is complementary to that of the substrate only after the substrate is bound. This process of dynamic recognition is called induced fit.
Enzymes are highly specific and may require cofactors for catalysis. A cofactor is a non-protein chemical compound bound to a protein; there are 2 types of cofactors: Metals and organic/metalloorganic (which are derived from vitamins). An example of a metal cofactor is zinc and the enzyme, carbonic anhydrase, tightly binds the zinc at the active site. The process involves binding water to carbon dioxide and deprotonating it into carbonic acid. Then the carbonic acid becomes a bicarbonate ion due to the displacement of water.
Catalysts can fasten the reaction speed by lowering the activation energy (not the transition state) of the process. The active site is a location on the enzyme which has complementary shape to the substrate. It is also where the amino acids with a complementary charge, polarity and shape to the ligand are.
The enzyme function and catalysis result from the ability to stabilize the transition state in a chemical reaction. The transition state is the highest energy species in a reaction. It is a transitory molecular structure that is no longer the substrate but is not yet the product. It is the most seldom occupied species along the reaction pathway. The difference in free energy between the transition state and the substrate is called the Gibbs free energy of activation or simply the activation energy.
Thus we can see the key to how enzymes operate: Enzymes accelerate reactions by decreasing the activation energy. The combination of substrate and enzyme creates a reaction pathway whose transition state is lower than that of the reaction in the absence of the enzyme. Because the activation energy is lower more substrate molecules have the energy required to reach the transition state.
It is important to note that enzymes have evolved specifically to recognize the transition states of chemical reactions. Therefore, enzymes do not bind to any reactive species before the species have actually begun to react; enzymes only recognize and bind the transition states of such species. In fact, if enzymes were to bind to the reactants of a reaction "on sight", or immediately, this would result in an even higher activation energy than before! For this reason, enzymes recognize only the transition state and bind to reactive species only when this high-energy state has been achieved. The fact that enzymes can recognize structures as specific and short-lived as transition states is a testament to their incredible specificity and efficiency.
Each enzyme is optimized for a particular reaction transition state. This ensures that enzymes will not compete with each other and hinder cellular reactions instead of help them. Enzyme inhibition occurs when the activity of a given enzyme is disrupted or interrupted in some fashion. Inhibitors can be molecules that have a similar shape, structure, or charge to the substrate in question so that the active site of an enzyme will "mistake" the inhibitor for the substrate. This affects the affinity of the enzyme for the substrate, as well as the rate of the overall reaction. Several types of inhibition can occur in the cell; more detailed explanations on these can be found in the corresponding sections.
Because of the active sites, enzymes are highly specific catalysts. These catalysts are governed by the ability to lower the free energy of thermodynamics to overcome transition states. The Michaelis-Menten Model describes the kinetic properties of many enzymes.
The interaction between the substrate and the enzyme helps accelerate the reaction, and the specificity of enzymes result in minimal side reactions.
It is of great importance to note that an enzyme cannot alter the laws of thermodynamics and consequently cannot alter the equilibrium of the reaction. The amount of product formed for a reaction utilizing an enzyme is always equal to the amount of product form of the same reaction occurring in the same reaction mixture without the enzyme. The enzyme just allows the reaction to reach its equilibrium faster. The equilibrium position is a function only of the free-energy difference between reactants and products.
6. Enzymes only alter reaction rate, not the reaction equilibrium. Enzyme cannot alter the laws of thermodynamics; therefore, it cannot alter the equilibrium of a chemical reaction. Enzyme is present, the amount of products form faster compared with enzyme is absent. Enzyme is only accelerating the reaction rate, not shipping the position of equilibrium (free energy, delta G)
Lock and Key Model
[edit | edit source]The "lock and key" model was first proposed by an organic chemist named Emil Fischer in 1894. In this model, the "lock" refers to an enzyme and the "key" refers to its complementary substrate. Each enzyme has a highly specific geometric shape that is complementary to its substrate. In order to activate an enzyme, its substrate must first bind to the active site on the enzyme. Only then will a catalytic reaction take place. However, like a lock and a key, the enzyme and substrate shape must be complementary and fit perfectly. Designed by evolution the active site for enzymes is generally highly specific in its substrate recognition and has the ability to distinguish between stereoisomers.

Induced Fit
[edit | edit source]According to the Lock and Key Model, the geometric shape of both enzymes and substrates can not be changed as they are both predetermined. Thus, the binding of the substrate to the enzymes active site does not alter the shape of the enzyme. While this theory helped explain the specificity of the enzyme, it does not explain the stability of the transition state for it would require more energy to reach the transition state complex. Thus the induced fit model was proposed in which enzymes like proteins are flexible. The concept of induced fit is that when a substrate binds to the active site of an enzyme, there is a conformational change and structural adaptation that makes this binding site more complementary and tighter. In essence the substrate does not simply bind to a rigid active site but instead the macromolecules, weak interaction forces, and hydrophobic characteristics on the enzyme surface mold into a precise formation so that there is an induced fit where the enzyme can perform maximum catalytic function.

Transition State Theory
[edit | edit source]
Transition state theory states that in an enzyme catalysis, the enzyme binds more strongly to its "transition state complex rather than its ground state reactants." In essence, the transition state is more stable. The stabilization of the transition state lowers the activation barrier between reactants and products thus increasing the rate of reaction or enzymatic activity as this will favor the increase of formation of the transition state complex.
In the transition state theory, the mechanism of interaction of reactants is irrelevant. However, the colliding molecules that take place in the reaction must have sufficient amount of kinetic energy to overcome the activation energy barrier in order to react. In many cases, temperature, pH, or enzymes can be changed to facilitate the stabilization of the transition state as well as statistically increasing the probability for molecules colliding and forming the transition state complex. For a bimolecular reaction such as Sn2, a transition state is formed when the two molecules’ old bonds are weakened and new bonds begin to form or the old bonds break first to form the transition state and then the new bonds form after. The theory suggests that as reactant molecules approach each other closely they are momentarily in a less stable state than either the reactants or the products.
Methods
[edit | edit source]- Some catalysts provide a charge to a molecule to make it more attractive to other reactants. Acids are an example for this kind of catalyst. They give the reacting species a positive charge to attract the negative or partially negative reactant, increasing the chance for the two species to collide and react.
- Some catalysts increase the local concentration of reactants so that they are more likely to collide.
- Some catalysts may modify the shape of one reactant to be more susceptible to other molecule.
Enzymatic Strategies and Examples
[edit | edit source]1. Covalent Catalysis - Through the course of catalysis, a powerful nucleophile is temporarily attached to a part of the substrate. The nucleophile is contained in the active site. A proteolytic enzyme chymotrypsin is an excellent example of this strategy. It is a substrate forming a transient covalent bond with residues in the active site or with a cofaster, which adds additional intermediate and reduce the energy of later transition.
2. General Acid/Base Catalysis - Water often acts as a donor or acceptor, but in Acid/base catalysis, the molecule which donates or accepts a proton is NOT water. This strategy incorporates base and acid catalysis to shorten reaction times. In the case of Chymotrypsin, the enzyme uses a histidine residue as a base catalyst to enhance the nucloephilicity of serine analogous to how histidine residue in carbonic anhydrase facilitates the removal of a proton from a zinc bound water molecule to yield hydroxide.
3. Catalysis by approximation - In this method, reactions favored by bringing together the two substrates to a single binding surface on enzymes. The two substrates are brought together to one area and this increases the rate of the reaction. NMP kinase for example, brings tow nucleotides together to improve the transferring of phosphoryl groups.
4. Metal Ion Catalysis - Metal ions can be involved as a catalyst in many different ways. Zinc can help the formation of a nucleophile. It makes the pka of water change from approximately 14 to 7, which allows it to be protonated at neutral pH. It can also stabilize negative charges by acting as an electrophile in a complex. Metal ions are also used to increase the binding energy of substrates, holding them together. A metal ion may also serve as a bridge between the enzyme and substrate acting as a cofactor in cases of NMP kinases.
Catalytic Mechanisms
[edit | edit source]1. Proteases (chymotrypsin and trypsin): are any enzyme that conducts proteolysis (protein catabolism) by hydrolysis of the peptide bonds linking amino acids together in the polypeptide chain.
Sample Experiment: Site-Directed Mutagenesis Applying Polymerase Chain Reaction (PCR) & Oligonucleotide Primers that contains the desired mutation in a newly synthesize strand, engineering a mis-match during first cycle DNA can develop a mutation.
2. Carbonic Anhydrase (metalloenzymes) These enzymes catalyzes the rapid interconversion of carbon dioxide and water to bicarbonate and protons, a reversible reaction that occurs rather slowly in the absence of catalyst.
3. Restriction Endonucleases (BamHI) It is a restriction enzyme that cleaves double stranded DNA at specific recognition nucleotide sequences (restriction site).
4. Nucleaside Monophosphate Kinases (NMP Kinase) These enzymes transfer phosphate groups from high energy donor molecule (ATP) to specific substrates (phosphorylation).
Enzyme's Cofactors for Activity
[edit | edit source]
The catalytic activity of enzymes depends on the presence of small molecules called cofactors. The role of the catalytic activity varies with the enzyme and its cofactors. In general, those cofactors can execute chemical reactions which cannot be performed by the standard 20 amino acids. An enzyme without cofactor is called apoenzyme, however the one with completely catalytically active is called holoenzyme.
Cofactors can be divided into two individual groups: Metal and Coenzymes. Metals are important for enzymes because they are molecular assistants that play a vital role in some of the enzymatic reactions that fuel the body metabolism. They also act to stabilize the shapes of enzymes. For example, iron helps the protein hemoglobin transport oxygen to organs in the body and copper helps superoxide dismutase in sopping up dangerous free radicals that accumulate inside the cells. Coenzymes are small organic molecules that often derived from vitamins. Coenzymes can be either tightly or loosely bound to the enzyme. Tightly bound ones are called prosthetic groups, while loosely bound coenzymes are like substrates and products, bind to the enzyme and get released from it. Enzymes that use the same coenzymes often perform catalysis by the similar mechanisms.
Enzyme Classification
[edit | edit source]| Class | Type of Reduction | Examples |
|---|---|---|
| Hydrolases | Catalyze hydrolysis reactions | Estrases Digestive enzymes |
| Isomerases | Catalyze isomerization (changing of a molecule into its isomer) | Phospho hexo isomerase, Fumarase |
| Ligases | Catalyze bond formation coupled with ATP hydrolysis. | Citric acid synthetase |
| Lyases | Catalyze a group elimination in order to form double bonds (or a ring structure). | Decarboxylases Aldolases |
| Oxidoreductases | Catalyze oxidation-reduction reactions | Dehydrogenases Oxidases |
| Transferases | Catalyze the transfer of functional groups among molecules. | Transaminase Kinases |
The classification of an enzyme is shown within the table as it's class and the type of reduction the enzyme goes through. An example of a name is glucose phosphotransferase. In this reaction ATP transfers one of its phosphates to glucose: ATP + D-glucose -> ADP + D-glucose 6-phosphate. Since this process "transfers" a phosphate group to glucose, it is within the classification of transferases, hence the name "glucose phosphotransferase." Since many enzymes have common names that do not refer to their function or what kind of reaction they catalyze, a enzyme classification system was established. There are six classes of enzymes that were created with subclasses based on what they catalyze so that enzymes could easily be named. Depending on the type of reaction catalyzed, an enzyme can have various names. These classes are Oxidoreductases, Transferases, Hydrolases, Lyases, Isomerases, and Ligases. This is the internation classification used for enzymes. For example, a common oxidoreductase is dehydrogenase. Dehydrogenase is known as an enzyme that oxidizes a substrate and transferring protons. Enzymes are normally used for catalyzing the transfer of functional groups, electrons, or atoms. Since this is the case, they are assigned names by the type of reaction they catalyze. This allowed for the addition of a four-digit number that would precede EC(Enzyme Commission) and each enzyme could be identified. The reaction that an enzyme catalyzes must be known before it can be classified.
Oxidoreductases catalyze oxidation-reduction reactions where electrons are transferred. These electrons are usually in the form of hydride ions or hydrogen atoms. When a substrate is being oxidized it is the hydrogen donor. The most common name used is a dehydrogenase and sometimes reductase will be used. An oxidase is referred to when the oxygen atom is the acceptor.
Transferases catalyze group transfer reactions. The transfer occurs from one molecule that will be the donor to another molecule that will be the acceptor. Most of the time, the donor is a cofactor that is charged with the group about to be transferred.
Hydrolases catalyze reactions that involve hydrolysis. This cases usually involves the transfer of functional groups to water. When the hydrolase acts on amide, glycosyl, peptide, ester, or other bonds, they not only catalyze the hydrolytic removal of a group from the substrate but also a transfer of the group to an acceptor compound. These enzymes could also be classified under transferase since hydrolysis can be viewed as a transfer of a functional group to water as an acceptor. However, as the acceptor's reaction with water was discovered very early, it's considered the main function of the enzyme which allows it to fall under this classification.
Lyases catalyze reactions where functional groups are added to break double bonds in molecules or the reverse where double bonds are formed by the removal of functional groups.
Isomerases catalyze reactions that transfer functional groups within a molecule so that isomeric forms are produced. These enzymes allow for structural or geometric changes within a compound. Sometime the interconverstion is carried out by an intramolecular oxidoreduction. In this case, one molecule is both the hydrogen acceptor and donor, so there's no oxidized product. The lack of a oxidized product is the reason this enzyme falls under this classification. The subclasses are created under this category by the type of isomerism.
Ligases are used in catalysis where two substrates are litigated and the formation of carbon-carbon, carbon-sulfide, carbon-nitrogen, and carbon-oxygen bonds due to condensation reactions. These reactions are couple to the cleavage of ATP.
Kinetics
[edit | edit source]The study of the rates of chemical reactions is called kinetics, and the study of the rates of enzyme-catalyzed reactions is called enzyme kinetics. A kinetic description of enzyme activity will help us understand how enzymes function. For example, the rate V is the quantity of A that disappears in a specified unit of time. It is equal to the rate of the appearance of P, or the quantity of P that appears in a specified unit of time.
A -----> P
The rate V is the quantity of A that disappears in a specified unit of time. It is equal to the rate of the appearance of P, or the quantity of P that appears In a specified unit of time.
V = ∆A/∆T = ∆P/∆T
If A is yellow and P is colorless, we can follow the decrease in the concentration of A by measuring the decrease in the intensity of yellow color with time. Consider only the change in the concentration of A for now. The rate of the reaction is directly related to the concentration of A by a proportionality constant, k, called the rate constant.
V = k[A]
The Michaelis-Menten Model
[edit | edit source]The Michaelis-Menten model is used to describe the kinetic properties of many enzymes. In this model, an enzyme(E)combines with a substrate(S)to form an enzyme-substrate(ES)complex, and proceed to form a product(P)or to dissociate into E and S.

The rate of formation of product,V0, can be calculated by the Michaelis-Menten equation:
![{\displaystyle {\frac {1}{v_{0}}}={\frac {K_{M}+[S]}{V_{\max[}S]}}={\frac {K_{M}}{V_{\max }}}\cdot {\frac {1}{[S]}}+{\frac {1}{V_{\max }}}.}](https://wikimedia.org/api/rest_v1/media/math/render/svg/3f2e414bdbcccae96840f4a5c32e303e33b5be72)
Vmax is the reaction rate when the enzyme is completely saturated with substrate. KM is the Michaelis constant, which is the substrate concentration at the half of the maximum reaction rate. The kinetic constant kcat is called the turnover number, which is the number of substrate molecules converted into product per unit time at a single catalytic site when the enzyme is saturated with substrate. It often count for most enzyme between 1 and 104per second.
Allosteric enzymes is an important class of enzymes. Its catalytic activity can be regulated. It has multiple active sites which display co-operativity, as evidenced by a sigmoidal dependence of reaction velocity on substrate concentration. We also find that K max is the substrate concentration in which the overall reaction rate at that particular time is half of V max. V max on the other hand, is the maximum reaction rate in which the active site is completely saturated with substrate. As a result of this physical characteristic, we see that no matter how much substrate is consequently added, the relative rate of the reaction remains unchanged as additional substrate do not contribute to any kinetic interaction with binding the active site. The affinity also eventually does not change as more substrate is increased and the reaction goes towards equilibrium.
Replicative DNA polymerase
[edit | edit source]There have been studies of the three multi-subunit DNA polymerase enzymes in the nucleus. This provides insights into the mechanism of the replication machinery in eukaryotic cells. The first DNA polymerase structure to be solved by crystallography was the Klenow fragment of E. coli DNA polymerase I. This crystallization revealed a structure that was likened to the palm, fingers, and thumb of a right hand. Studies of the Klenow fragment showed that DNA was bound within the cleft and that the fingers and thumb architecture is conserved in many of the polymerase families. The polymerase active site residues are located in the palm domain. The fingers are important for nucleotide binding, and the thumb domain binds the DNA.

Conclusion Enzymes speed up the reaction. They can increase the reaction rates by a factor of 10^6 or more. Some of the enzymes require the cofactors for activity. The cofactors include vitamin-derived organic molecules (coenzymes), and metal ions. The enzymes' other tasks include decreasing the free energy of activation of chemical reactions. The first step in catalysis is forming an enzyme substrate complex. Besides, the substrates are recognized by enzymes is accompanied by conformational changes at active sites.
Temperature affect on catalytic activity of enzymes
[edit | edit source]In general as one increases the temperature the catalytic activity of an enzyme increases, which is the classical model and understanding of temperature affect on catalytic activity. Though, the classical model has been discovered to be a questionable depiction of how temperature really affects the catalytic activity of enzymes. The new model, the equilibrium model, has been studied by Roy M. Daniel and Micahel J. Danson and gives a new understanding of how temperature affects the catalytic activity of enzymes.
Classical Model
The classical model depicts the effects of temperature on an enzyme. Mainly depicts that as one increases temperatures the catalytic rate increases exponentially and the amount of active enzyme will decrease because of denaturization. The classical model does not show if there is an optimal temperature at which, catalytic activity is at the max.
Equation representing the classical model: Vmax= kcat[E0]e-kinactt
Equilibrium Model
In the equilibrium model, it introduces an inactive form of the enzyme that is reversible to be active. The new factor of reversibility between active and inactive form of enzymes is temperature dependent where the amount of enzyme is.
The equation describing the amount of active enzyme at any point, [Eact] = ([E0]-[X])/( 1+ Keq)
In this context inactive enzyme does not mean the enzyme is denatured, all it means is that the active site has been changed just enough to the point where the enzyme can not bind to the substrate, hence it is reversible.
The model shows an optimal temperature where enzyme activity would be the greatest, which is different from the classical model. It takes into account the variation Vmax with time and temperature and also includes the factor of four parameters; G*cat, G*inact, Heq and Teq. This model is currently used to best describe the evolution of the enzyme’s active site due to temperature and also best explain the effect of temperature on enzymes.
Key difference between the two models
The classical model is temperature dependent and doesn’t depict an optimal temperature for catalytic activity.
References
[edit | edit source]
Biochemistry 7Ed, by Jeremy M. Berg, John L. Tymoczko, and Lubert Stryer. 2007.
Berg, Jeremy M., Tymoczko, John L., Stryer, Lubert. Biochemistry. Seventh Edition.
http://www.ncbi.nlm.nih.gov/pubmed/20554446
http://ars.els-cdn.com/content/image/1-s2.0-S0968000410000897-gr1.jpg
Structure
[edit | edit source]The binding energy is the free energy that is released by the formation of weak interactions between a complementary substrate and enzyme. The binding energy is maximized since only the correct substrate can interact with an enzyme and is released when the enzyme facilitates formation of the transition state. This interaction between the enzyme and the transition state gives way to the maximum binding state. Upon formation of the transition state, the activation energy is lowered, which will speed up the reaction.
Binding energy is known as separation energy that is the requirement for dissociation of chemical substance to its constituent components. There are different binding energies at different molecular level such as electron binding energy, atomic binding energy, nuclear binding energy, and gravitational binding energy.
With the units of eV, electron binding energy is the energy required to release an electron from its atomic or molecular orbital. According to Moseley's law, the binding energies of 1s electron are proportional to(Z-1)2.
To dissemble an atom into free electrons and nucleus, atomic binding energy is required. It is the energy that was derived from electromagnetic interaction.
Nuclear binding energy is also known as binding energy of nucleons into a nuclide. It was derived from the strong nuclear force and is the energy required to disassemble a nucleus into the same number of free unbound neutrons and protons that its contained. Such that the particles are far enough from one another so that the strong nuclear force can no longer cause the particles to interact. In bound systems, the mass must be subtracted from the mass of the unbound system, if the binding energy is removed from the system, because this energy has mass. If subtracted from the system at the time it is bound, the mass of the system will be removed. Since the system is not closed during the binding process, system mass is not conserved.[1]
Desolvation, in biochemistry, is the process where in an aqueous solution containing an enzyme and a substrate, water that is surrounding the substrate is replaced by the enzyme. In other words, water molecules that were once in between the substrate and the enzyme are displaced to allow the interaction of the substrate with the enzyme. The process also increases the entropy of the reaction, making the formation of the enzyme-substrate complex more thermodynamically favorable.
The method of desolvation involves drying a sample in a solution. An example of this involves electro-statically bound particles to dissociate by releasing water in an aqueous solution. This method is commonplace in atomic absorption spectroscopy, in which an atomic gas is created through a liquid sample. It can also be used in vaporization.

Ground State
[edit | edit source]An electron is in its ground state meaning it is in its lowest energy state, in other words, an electron is in its excited state whenever it is not in its ground state. An excited state of a molecule is known to have higher energy levels than its ground state. The third law of thermodynamics states that the system is at its ground state when it is at absolute zero degree in temperature, which causes the entropy of the reaction to be determined by the degeneracy of that ground state. However, some systems will have zero entropy due to its physical/chemical properties.
Pre-steady State
[edit | edit source]In the first moment after an enzyme is mixed with substrate, no product has been formed and no intermediates exist. The study of the next few milliseconds of the reaction is called pre-steady-state kinetics. Pre-steady-state kinetics is therefore concerned with the formation and consumption of enzyme–substrate intermediates (such as ES or E*) until their steady-state concentrations are reached.
This approach was first applied to the hydrolysis reaction catalysed by chymotrypsin. Often, the detection of an intermediate is a vital piece of evidence in investigations of what mechanism an enzyme follows. For example, in the ping–pong mechanisms that are shown above, rapid kinetic measurements can follow the release of product P and measure the formation of the modified enzyme intermediate E*. In the case of chymotrypsin, this intermediate is formed by an attack on the substrate by the nucleophilic serine in the active site and the formation of the acyl-enzyme intermediate.
In the figure below, the enzyme produces E* rapidly in the first few seconds of the reaction. The rate then slows as steady state is reached. This rapid burst phase of the reaction measures a single turnover of the enzyme. Consequently, the amount of product released in this burst, shown as the intercept on the y-axis of the graph, also gives the amount of functional enzyme which is present in the assay. File:Pre-steady-state.pdf
Steady State
[edit | edit source]The steady state assumption was proposed by George Briggs and John Haldane in 1942. In this assumption, the concentrations of the intermediates of a reaction remain the same even when the concentrations of starting materials and products are changing. Steady state occurs when the rate of formation and breakdown of the intermediate are equal. The steady state assumption relies on the fact that both the formation of the intermediate from reactants and the formation of products from the intermediate have rates much higher than their corresponding reverse reactions. In other words, steady state assumes that k1>>k-1 and k2>>k-2. Coenzymes are one group of cofactors that can either be tightly or loosely bound to the enzyme. The former are called prosthetic groups, whereas the latter are like cosubstrates. Coenzymes are small organic molecules and are often derived from vitamins making them crucial components in biological reactions. Enzymes that use the same coenzyme perform similar catalysis mechanisms.
Common Coenzymes
[edit | edit source]NADH
[edit | edit source]NADH, Dinucleutide a naturally occurring coenzyme found in all living cells, triggers energy production and helps supply cells with energy. NADH dehydrogenase is an enzyme specifically placed in the mitochondrial membrane. NADH catalyzes the transfer of electrons from NADH to coenzyme Q (CoQ). It plays vital roles in the mitochondrial electron transport chain being the first enzyme (complex I). NADH + H+ + CoQ + 4H+in → NAD+ + CoQH2 + 4H+out
Through this reaction the complex 4 protons are translocated across the inner membrane per molecule of oxidized NADH, contributing to the production of ATP through the electrochemical potential that was established. The reaction is as well reversible is there a high presence of membrane potential.
Composition and structure
NADH dehydrogenase contains 45 separate polypeptide chains, making it the largest of the respiratory complexes. Essential components that are of functional importance are the eight iron-sulfur clusters and the flavin prosthetic group. The mitochondrial genome encodes seven of the 45 subunits. NADH possess the structure of an “L” shape with a long membrane domain and a hydrophilic peripheral domain, which accommodates all the known redox centres and the NADH binding site.
Structure:
FADH
[edit | edit source]Flavin adenine dinucleotide FAD: flavin adenine dinucleotide acts as a redox cofactor associated with important reactions that engage metabolism. FAD changes between two redox state accounting for its’ biochemical role. Derived from riboflavin also known as vitamin B2 consists of a riboflavin group that is bound to the phosphate group of an adenosine diphosphate. FAD accepts two hydrogen atoms to be reduced into the FADH. FADH is associated to be an energy-carrying molecule, and can also be incorporated in the mitochondria as a substrate to attain the oxidative phosphorylation process. The structure of FAD is shown below:
Quinone
[edit | edit source]The basic structure of quinones consists of any member of a class of cyclic organic compounds that containing two carbonyl groups, C=O, either adjacent or separated by a vinylene group, −CH = CH−, in a six-membered unsaturated ring. Quinones are an important chemical structure as it relates to color in biological organisms. For example, quinones are present in biological pigments such as biochromes. Some include benzoquinones, naphthoquinones, anthraquinones, and polycyclic quinones. The quinones are found in bacteria, fungi, various higher plant forms, and are sometimes found in animals. An example of a quinone is coenzyme Q, also known as ubiquinone. Ubiquinone is hydrophobic and diffuses rapidly in inner mitochondrial membranes; its structure is shown below:
CoA
[edit | edit source]Coenzyme A or CoA is derived from pantothenic acid and adenosine triphosphate (ATP) and used in metabolism in areas such as fatty acid oxidization and the citric acid cycle. Its main function is to carry acyl groups such as acetyl as thioesters. A molecule of coenzyme A carrying an acetyl group is also referred to as acetyl-CoA. Coenzymes are sometimes denoted CoA, CoASH, or HSCoA. One form of Coenzyme A is Acetyl-CoA. Acetyl-CoA is a very important because it is a precursor to HMG CoA. Acetyl-CoA is involved in cholesterol and ketone synthesis. And is vital component to the acetyl group in acetylcholine. Acetyl coenzyme A is a key component in the krebs cycle where pyruvate is converted to acetyl CoA. This coenzyme has a sulfur atom which bonds to the acetyl fragment by an unstable bond which makes it very reactive, the enzyme is now ready to feed its acetate into the krebs cycle for further oxidation. Since coenzyme A is chemically a thiol, it can react with carboxylic acids to form thioesters, thus functioning as an acyl group carrier. It assists in transferring fatty acids from the cytoplasm to mitochondria. A molecule of coenzyme A carrying an acetyl group is also referred to as acetyl-CoA. When it is not attached to an acyl group it is usually referred to as 'CoASH' or 'HSCoA'.
The structure of CoA-SH is shown below:
Thiamine Pyrophosphate
[edit | edit source]Thiamine pyrophosphate (TPP) is a thiamine (vitamin B1) derivative produced by the enzyme thiamine pyrophosphotase. As a coenzyme, it is present in all living systems and is important for catalyzing several biochemical reactions. It was first discovered while studying the peripheral nervous system disease Beriberi, which results from a deficiency of thiamine in the diet. Research has shown that TPP is an essential nutrient in humans, capable of preventing such a disease. TPP is a prosthetic group in many enzymes, such as: Pyruvate dehydrogenase complex, Pyruvate decarboxylase complex in ethanol fermentation, Alpha-ketoglutarate dehydrogenase complex, Branched-chain amino acid dehydrogenase complex, 2-hydroxyphytanoyl-CoA lyase, and Transketolase.
Chemical Structure
[edit | edit source]
TPP consists of a pyrimidine ring that is connected to a thiazole ring, which is in turn connected to a pyrophosphate (diphosphate) functional group. The thiazole ring component is the most chemically involved part of TPP in reactions, since in contains reactive nitrogen and sulfur parts. This component is considered the “reagent portion” of the molecule. The C2 carbon of this ring participates in some reactions by acting as an acid and donating its proton to form a carbanion. This negatively charged carbanion is stabilized by the positive charge on the adjacent tetravalent nitrogen, making the reaction more favorable. This type of compound is known as the “ylid form”.
Pyridoxal phosphate
[edit | edit source]
Also known as PLP or pyridoxal-5’-phosphate (P5P), it is a prosthetic group of some enzymes. It is the active form of vitamin B6, which comprises three natural organic compounds, pyridoxal, pyridoxamine, and pyridoxine. As a coenzyme, it is involved in transamination reactions and in some decarboxylation and deanimation reactions of amino acids. Its aldehyde goup forms a Schiff-base linkage with the ε-amino group of a specific lysine group of the aminotransferase enzyme. Afterwards, the ε-amino group of the active-site lysine residue is displaced by the α-amino group of the amino acid substrate. This results in a quinoid intermediate from a deprotonated external aldimine, which in turn can accept a proton at a different position to become a ketamine. in turn accepts a proton at a different position to become a ketimine. This ketamine is hydrolyzed so that the amino group remains on the complex.
PLP is also involved in the synthesis of the neurotransmitters serotonin and norepinephrine and of heme (a molecular constituent of hemoglobin) and in the conversion of the amino acid tryptophan to the vitamin niacin.
Biotin
[edit | edit source]Biotin is which also known as vitamin H, vitamin B7, or coenzyme R that is tightly bounded to an enzyme (prosthetic group). Biotin is essential in the formation of fatty acids and glucose. Furthermore, it also aids in the metabolism of carbohydrates, fats, and proteins. It also helps promote healthy hair and skin. Biotin is a water-soluble B –complex vitamin that consists of a tetrahydroimidizalone ring that is fused with a tetrahydrothiophene ring. Biotin contains a valeric acid substituent that is attached to one of the carbons on the tetrahydrothiophene. Biotin works by activating enzymes (pyruvate carboxylase) that are responsible for the rearrangement of glucose, amino acids, and fatty acid molecules. Deficiency of biotin is quite rare. This deficiency is caused by excessive consumption of raw egg whites and can be addressed with supplements.

Tetrahydrofolate
[edit | edit source]Tetrahydrofolate, also known as tetrahydrofolic acid is a derivative of folic acid. It is a coenzyme that is essential in the metabolism of amino acids and nucleic acids. Furthermore, it is crucial to interconvert amino acids, methylate tRNA, and generate formate. It is produced from dihydrofolic acid by dihydrofolate reductase in the liver. It acts as a donor group involved in the transfer of single carbon groups. Tetrahydrofolate is transported across cells by receptor-mediated endocytosis. Tetrahydrofolate is used to treat megaloblastic and macrocytic anemias which results from a deficiency in folic acid.

5'-Deoxyadenosyl Cobalamin
[edit | edit source]5-deoxyadenosyl cobalamin is one of the two forms of vitamin B12 that is used in the body. 5-deoxyadenosyl cobalamin is a coenzyme that is needed by the enzyme methylmalonyl mutase that converts L-methylmalonyl-CoA to succinyl-CoA. The conversion is an essential step in extracting energy from fats and proteins in the body. Also, the production of succinyl-CoA is crucial for the production of hemoglobin, the oxygen binding protein that carries oxygen from the lungs to tissues.
Uridine diphosphate N-acetylglucosamine
[edit | edit source]Uridine diphosphate N-acetylglucosamine (UDP-GlcNAc)is a nucleotide sugar that acts as a coenzyme in metabolism. It has a role in transferring N-acetylglucosamine residues to substrates by interacting with glycosyltransferases. It is produced in the hexosamine biosynthesis pathway, which initially starts with the synthesis of glucosamine-6-phosphate from fructose 6-phosphate and glutamine. As an end-product of this pathway, it is further utilized in the production of glycosaminoglycans, proteoglycans, and glycolipids.

Sources
[edit | edit source]http://en.wikipedia.org/wiki/NADH_dehydrogenase
General information
[edit | edit source]Cofactors are inorganic and organic chemicals that assist enzymes during the catalysis of reactions. Coenzymes are non-protein organic molecules that are mostly derivatives of vitamins soluble in water by phosphorylation; they bind apoenzyme to proteins to produce an active holoenzyme. Apoenzymes are enzymes that lack their necessary cofactor(s) for proper functioning; the binding of the enzyme to a coenzyme forms a holoenzyme. Holoenzymes are the active forms of apoenzymes.


Cofactors can be metals or small organic molecules, and their primary function is to assist in enzyme activity. They are able to assist in performing certain, necessary, reactions the enzyme cannot perform alone. They are divided into coenzymes and prosthetic groups. A holoenzyme refers to a catalytically active enzyme that consists of both apoenzyme (enzyme without its cofactor(s)) and cofactor. There are two groups of cofactors: metals and small organic molecules called coenzymes. Coenzymes are small organic molecules usually obtained from vitamins. Prosthetic groups refer to tightly bound coenzymes, while cosubstrates refer to loosely bound coenzymes that are released in the same way as substrates and products. Loosely bound coenzymes differ from substrates in that the same coenzymes may be used by different enzymes in order to bring about proper enzyme activity.
General formula

Metal cofactors
[edit | edit source]Metal ions are common enzyme cofactors. Some enzymes, referred to as metalloenzymes, cannot function without a bound metal ion in the active site. In daily nutrition, this kind of cofactor plays a role as the essential trace elements such as: iron (Fe3+), manganese (Mn2+), cobalt (Co2+), copper (Cu2+), zinc (Zn2+), selenium (Se2+), and molybdenum (Mo5+). For example, Mg2+ is used in glycolysis. In the first step of converting glucose to glucose 6-phosphate, before ATP is used to give ADP and one phosphate group, ATP is bound to Mg2+ which stabilizes the other two phosphate groups so it is easier to release only one phosphate group. In some bacteria such as genus Azotobacter and Pyrococcus furiosus, metal cofactors are also discovered to play an important role. An example of cofactors in action is the zinc-mediated function of carbonic anhydrase or the magnesium-mediated function of restriction endonuclease.
Coenzyme
[edit | edit source]A coenzyme is a small, organic, non-protein molecule that carries chemical groups between enzymes. It is the cofactor for the enzyme and does not form a permanent part in the enzyme's structure. Sometimes, they are called cosubstrates and are considered substrates that are loosely bound to the enzyme. In metabolism, coenzymes play a role in group-transfer reactions, such as ATP and coenzyme A, and oxidation-reduction reactions, such as NAD+ and coenzyme Q10. Coenzymes are frequently consumed and recycled. Chemical groups are added and detached continuously by an enzyme. ATP synthase enzyme phosphorylates and converts the ADP to ATP, while Kinase dephosphorylates the ATP back to ADP at continuous rates as well. Coenzyme molecules are mostly derived from vitamins. They are also commonly made from nucleotides such as adenosine triphosphate and coenzyme A.
Through further research in coenzyme activity and its binding effect on the enzyme, more can be revealed about how the enzyme changes conformationally and functionally. An example is of the MAPEG group of integral membrane enzymes. These enzymes are crucial in the catalytic transformation of lipophilic substrates, which are involved in arachidonic acid derived messengers production and xenobiotic detoxification. Through use of a bound detergent to mimic a MAPEG enzyme's cofactor, glutathione, a new active site specific for lipophilic substrate is revealed; thus, further studies can reveal how these substrates bind to this second form of the enzyme [3].


Important Coenzymes
[edit | edit source]NADH
[edit | edit source]
nicotinamide adenine dinucleotide is a coenzyme derived from vitamin B3. In NAD+ the functional group of the molecule is only the nicotinamide part. NAD+ is capable of carrying and transferring electrons and functions as oxidizing agent in redox reactions. It also works as a substrate for DNA ligases in posttranslational modification, where the reaction removes acetyl groups from proteins. Furthermore, in glycolysis and the citric acid cycle, NAD+ oxidizes glucose and releases energy, which is then transferred to NAD+ by reduction to NADH. NADH later on unloads the extra electron through oxidative phosphorylation to generate ATP, which is the energy source humans use every day. In addition to catabolic reactions, NADH is also involved in anabolic reactions such as gluconeogenesis, and it also aids in the production of neurotransmitters in the brain.
FADH
[edit | edit source]
flavin adenine dinucleotide is a prosthetic group that, like NADH, functions as a reducing agent in cellular respiration and donates electrons to the electron transport chain.
Quinone
[edit | edit source]-
 1,2-Benzoquinone
1,2-Benzoquinone -
 1,4-Benzoquinone
1,4-Benzoquinone -
 Anthraquinone
Anthraquinone


compounds that have fully conjugated aromatic rings to which two oxygen atoms are bounded as carbonyl groups (i.e. diketones). Quinone’s structure gives them the ability to form substances with colors. They exist as pigments in bacteria, fungi, and certain plants, and give them their characteristic colors. In addition, they are used to manufacture different color dyes for industrial purposes. In biological systems, they serve as electron acceptors (oxidizing agents) in electron transport chains such as those in photosynthesis and aerobic respiration. Many natural or synthetic quinines show biological or pharmacological activities, and some event show antitumoral activities.
CoA
[edit | edit source]
coenzyme A, synthesized from pantothenic acid ATP, functions as acyl group carriers to transport functional groups such as acetyl (acetyl-CoA) or thioesters in metabolic reactions like fatty acid oxidation (synthesis of fatty acids) and citric acid cycle (cellular respiration). It also transfers fatty acids from cytoplasm to mitochondria. In addition to its transporter role in metabolism, CoA is also an important molecule in itself. For instance, CoA is an important precursor to HMG-CoA, an important enzyme in the metabolic synthesis of cholesterol and ketones. Furthermore, it contributes the acetyl group to the structure of acetylcholine, which is an important neurotransmitter responsible for inducing muscle contraction.
Common Coenzymes
[edit | edit source]Vitamin A
[edit | edit source]Vitamin A is subdivided into two molecules, Vitamin A1 (retinol) and Vitamin A2 (dehydroretinol). Retinol is the most active and common form. Vitamin A has a large conjugated chain which serves as the reactive site of the molecule. Unlike most cofactors, Vitamin A undergoes a sequence of chemical changes (oxidations, reductions, and isomerizations) before returning to its original form. The ability for Vitamin A's electrons to travel from orbital makes it a good candidate molecule for trapping light energy. Consequently, Vitamin A is responsible for transferring light energy to a chemical nerve impulse in the eyeball. Vitamin A is also used for growing healthy new cells such as skin, bones, and hair. It maintains the lining of the urinary tract, intestinal tract, and respiratory system. Additionally, Vitamin A is required for the reproductive functions such as the growth and development of sperm and ovaries.

Vitamin C
[edit | edit source]Also known as ascorbic acid, Vitamin C is quite abundant in most plants and animals excluding primates, guinea pigs, bats, and some birds. Despite human's inability to synthesis absorbic acid, it is an essential in many biosynthetic pathways such as synthesizing collagen. Deficiency leads to a disease called Scurvy. Vitamin C helps regulate the immune system and relieve pain caused by tired muscles. It also is needed in the manufacture of collagen and norepinephrine. Vitamin C is also an antioxidant which can enhance the immune system by stimulating white blood cells in the body. Vitamin C also helps to benefit the skin, teeth, and bones.
Vitamin B1
[edit | edit source]Also named Thiamine or Thiamine diphosphate (TPP), Vitamin B1 is a cofactor for oxidative decarboxylation both in the Kreb's Cycle and in converting pyruvate to acetyl-CoA (an important molecule used in the citric acid cycle of metabolism). It is widely available in the human diet and particularly potent in wheat germ and yeast. It's functionality results from a thiazole ring which stabilizes charge and electron transfer through resonance.
Vitamin B2
[edit | edit source]Vitamin B2 is known as riboflavin. Vitamin B2 is the precursor of Flavin adenine dinucleotide (FAD) and flavin mononucleotide (FMN) which are coenzymes used to oxidized substrates. FAD contains riboflavin and adenine. FMN contains riboflavin that is why it is called mononucleotide.
Vitamin B3
[edit | edit source]Vitamin B3 is Niacin or nicotinic acid with the formula C5H4NCO2H. Vitamin B3 is a precursor to NADH, NAD+, NADP+ and NADPH which are coenzymes found in all living cells. NAD+ and NADP+ are oxidizing agents. NADH and NADPH are reducing agents.
Vitamin B6
[edit | edit source]Vitamin B6 is precursor to coenzyme pyridoxal phosphate (PLP) which is required in certain transformation of amino acids including transamination, deamination, and decarboxylation.
Vitamin B12
[edit | edit source]Vitamin B12 is the name for a class of related compounds that have this vitamin activity. These compounds contain the rare element cobalt. Humans can not synthesize B12 and must obtain it from diet.
Vitamin H
[edit | edit source]Also named Biotin, Vitamin H is a carboxyl carrier; it binds CO2 and carries it until the CO2 is donated in carboxylase reactions. It is water soluble and important in the metabolism of fatty acids and the amino acid Leucine. Deficiency leads to dermatitis and hair loss, thus making it a popular ingredient in cosmetics.
Vitamin K
[edit | edit source]Vitamin K is needed for the process of clotting of blood and Ca2+ binding. Vitamin K can be synthesized by bacteria in the intestines. Vitamin K is needed for catalyzing the carboxylation of the γ-carbon of the glutamate side chain in proteins.
Non-enzymatic cofactors
[edit | edit source]Cofactor is also used widely in the biological field to refer to molecules that either activate, inhibit or are required for the protein to function. For example, ligands such as hormones that bind to and activate receptor proteins are termed cofactors or coactivators, while molecules that inhibit receptor proteins are termed corepressors.
The coactivator can enhance transcription initiation by stabilizing the formation of the RNA polymerase holoenzyme enabling faster clearance of the promoter.
The corepressor can repress transcriptional initiation by recruiting histone deacetylases which catalyze the removal of acetyl groups from lysine residues. This increases the positive charge on histones which strengthens in the interaction between the histones and DNA, making the latter less accessible to transcription.
References
[edit | edit source]- Dewick, Paul. Medicinal Natural Products: A Biosynthetic Approach. 3rd ed. West Sussex, Britain: Wiley, 2009. 32-34. Print.
- von Heijne, G and Rees,D (2008). Current Opinion in Structural Biology. Elsevier Ltd.
Prosthetic group
[edit | edit source]A prosthetic group is a tightly bound, specific non-polypeptide unit required for the biological function of some proteins. The prosthetic group may be organic (such as a vitamin, sugar, or lipid) or inorganic (such as a metal ion), but is not composed of amino acids. Prosthetic groups are bound tightly to proteins and may even be attached through a covalent bond, as opposed to cosubstrates, which are loosely bound. In enzymes, prosthetic groups are often involved in the active site, playing an important role in the functions of enzymes.
Vitamins are another common prosthetic group. This is one of the reasons why vitamins are required in the human diet. Inorganic prosthetic groups, however, are usually transition metal ions such as iron. The Heme group in hemoglobin is a prosthetic group located in the porphyrin, which is a tetramer of cyclic carbon groups. It contains an organic component called a protoporphyrin made up of four pyrrole rings and an iron atom in the ferrous state (Fe2+). The red color of blood and muscles is attributed to the Heme groups. The difference between a prosthetic group and a cofactor depends on how tightly or loosely bound to the enzyme they are. If tightly connected, the cofactor is referred to as a prosthetic group.
Heme group
[edit | edit source]
A heme group is a prosthetic group consisting of a protoporphyrin ring and a central iron (Fe) atom. A protoporphyrin ring is made up of four pyrrole rings linked by methine bridges. Four methyl, two vinyl, and two propionate side chains are attached.
Heme of hemoglobin protein is a prosthetic group of heterocyclic ring of porphyrin of an iron atom; the biological function of the group is for delivering oxygen to body tissues, such that bonding of ligand of gas molecules to the iron atom of the protein group changes the structure of the protein by amino acid group of histidine residue around the heme molecule.
The iron lies in the center is an organic component called protoporphyrin, which is bound to four pyrrole nitrogen atom linked by a methine bridge that forms a tetrapyrrole ring. The iron can either be in the ferrous (Fe2+) or the ferric (Fe3+) oxidation state. However, it is only able to bind to oxygen when in the ferrous state. The iron can form two additional bonds in fifth and in sixth coordination which on both side of heme plane. The fifth coordination sites is linked to a distal histidine while the sixth coordination site can, not always, bind to oxygen. Upon binding, the Heme group will actually shrink in size and descend further into the plane of the porphyrin ring. Along with it, the distal histidine will follow, and this histidine is attached to the alpha-Beta interface thus resulting in local to complete conformational change.
A conjugate protein combined with its specific prosthetic group is termed a holoprotein, while a protein in its absence is called an apoprotein. Prosthetic groups have varying functions, such as oxidizing-reducing reactions (redox), methylation reactions, oxygenation reactions, and so forth.
The heme group gives muscles and blood their distinctive red color.
Cofactor: A Definition
[edit | edit source]Many enzymes require an additional small molecule, known as a cofactor to aid with catalytic activity. A cofactor is a non-protein molecule that carries out chemical reactions that cannot be performed by the standard 20 amino acids. Cofactors can be either inorganic molecules (metals) or small organic molecules (coenzymes).
Cofactors, mostly metal ions or coenzyme, are inorganic and organic chemicals that function in reactions of enzymes. Coenzymes are organic molecules that are nonproteins and mostly derivatives of vitamins soluble in water by phosphorylation; they bind apoenzyme protein molecule to produce active holoenzyme.
 Figure 1-1: A flow chart of the two types of cofactors.
Figure 1-1: A flow chart of the two types of cofactors.
Apoenzyme- An enzyme that requires a cofactor but does not have one bound. An apoenzyme is an inactive enzyme, activation of the enzyme occurs upon binding of an organic or inorganic cofactor.
Holoenzyme- An apoenzyme together with its cofactor. A holoenzyme is complete and catalytically active. Most cofactors are not covalently bound but instead are tightly bound. However, organic prosthetic groups such as an iron ion or a vitamin can be covalently bound. Examples of holoenzymes include DNA polymerase and RNA polymerase which contain multiple protein subunits. The complete complexes contain all the subunits necessary for activity.
Figure 1-2: Illustrates that an Apoenzyme + Cofactor = Holoenzyme.
Examples of Holoenzymes
[edit | edit source]DNA polymerase is a holoenzyme that catalyzes the polymerization of deoxyribonucleotides into a DNA strand. DNA polymerase is an active participant in DNA replication. It reads the intact DNA strand as a template and uses it to synthesize the new strand. The newly polymerized DNA strand is complementary to the template strand and identical to template's original partner strand. DNA polymerase uses a magnesium ion for catalytic activity.
 Figure 1-3: Illustrates the holoenzyme DNA polymerase a multi-subunit complex.
Figure 1-3: Illustrates the holoenzyme DNA polymerase a multi-subunit complex.
RNA polymerase is also a holoenzyme that catalyzes RNA. RNA polymerase is needed for constructing RNA chains from DNA genes as templates, a process known as transcription. It polymerizes ribonucleotides at the 3' end of an RNA transcript.
 Figure 1-4: Illustrates the holoenzyme RNA polymerase a subunit complex.
Figure 1-4: Illustrates the holoenzyme RNA polymerase a subunit complex.
References
[edit | edit source]Berg, Jeremy M., et al. "Biochemistry". 6th ed. W.H. Freeman and Company, NY, 2006. An active site is the part of an enzyme that directly binds to a substrate and carries a reaction. It contains catalytic groups which are amino acids that promote formation and degradation of bonds. By forming and breaking these bonds, enzyme and substrate interaction promotes the formation of the transition state structure. Enzymes help a reaction by stabilizing the transition state intermediate. This is accomplished by lowering the energy barrier or activation energy- the energy that is required to promote the formation of transition state intermediate. The three dimensional cleft is formed by the groups that come from different part of the amino acid sequences. The active site is only a small part of the total enzyme volume. It enhances the enzyme to bind to substrate and catalysis by many different weak interactions because of its nonpolar microenvironment. The weak interactions includes the Van der Waals, hydrogen bonding, and electrostatic interactions. The arrangement of atoms in the active site is crucial for binding spectificity. The overall result is the acceleration of the reaction process and increasing the rate of reaction. Furthermore, not only do enzymes contain catalytic abilities, but the active site also carries the recognition of substrate.
The enzyme active site is the binding site for catalytic and inhibition reactions of enzyme and substrate; structure of active site and its chemical characteristic are of specific for the binding of a particular substrate. The binding of the substrate to the enzyme causes changes in the chemical bonds of the substrate and causes the reactions that lead to the formation of products. The products are released from the enzyme surface to regenerate the enzyme for another reaction cycle.
Structure
[edit | edit source]The active site is in the shape of a three-dimensional cleft that is composed of amino acids from different residues of the primary amino acid sequence. The amino acids that play a significant role in the binding specificity of the active site are usually not adjacent to each other in the primary structure, but form the active site as a result of folding in creating the tertiary structure. This active site region is relatively small compared to the rest of the enzyme. Similar to a ligand-binding site, the majority of an enzyme (non-binding amino acid residues) exist primarily to serve as a framework to support the structure of the active site by providing correct orientation. The unique amino acids contained in an active site promote specific interactions that are necessary for proper binding and resulting catalysis. Enzyme specificity depends on the arrangement of atoms in the active site. Complementary shapes between enzyme and substrate(s) allow a greater amount of weak non-covalent interactions including electrostatic forces, Van der Waals forces, hydrogen bonding, and hydrophobic interactions. Specific amino acids also allow the formation of hydrogen bonds. That shows the uniqueness of the microenvironment for the active site.
To locate the active site, the enzyme of interest is crystallized in the presence of an analog. The analog’s resemblance of the original substrate would be considered a potent competitive inhibitor that blocks the original substrates from binding to the active sites. One can then locate the active sites on an enzyme by following where the analog binds.
Active Site vs. Regulatory Site
An enzyme, for example ATCase, contains two distinct subunits: an active site and a regulatory site. The active site is the catalytic subunit, whereas the regulatory site has no catalytic activity. The two subunits on the enzyme was confirmed by John Gerhart and Howard Schachman by doing the ultracentrifugation experiment. First, they treated the ATCase with p-hydroxymercuribenzoate to react with the sulfhydryl groups and dissociate the two subunits. Because the two subunits differ in sizes with the catalytic subunit being larger, results of centrifuging the dissociated subunits showed two sedimentations compared to the one sediment of the native enzyme. This proved that ATCase, like many other enzymes, contain two sites for substrates to bind.
Models
[edit | edit source]There are three different models that represent enzyme-substrate binding: the lock-and-key model, the induced fit model, and transition-state model.
The lock-and-key model was proposed by Emil Fischer in 1890. This model presumes that there is a perfect fit between the substrate and the active site—the two molecules are complementary in shape. Lock-and-key is the model such that active site of enzyme is good fit for substrate that does not require change of structure of enzyme after enzyme binds substrate

The induced-fit model involves the changing of the conformation of the active site to fit the substrate after binding. Also, in the induced-fit model, it was stated that there are amino acids that aid the correct substrate to bind to the active site which leads to shaping of the active site to the complementary shape. Induced fit is the model such that structure of active site of enzyme can be easily changed after binding of enzyme and substrate.

The binding in the active site involves hydrogen bonding, hydrophobic interactions and temporary covalent bonds. The active site will then stabilize the transition state intermediate to decrease the activation energy. But the intermediate is most likely unstable, allowing the enzyme to release the substrate and return to the unbound state.
The transition-state model starts with an enzyme that binds to a substrate. It requires energy to change the shape of substrate. Once the shape is changed, the substrate is unbound to the enzyme, which ultimately changes the shape of the enzyme. An important aspect of this model is that it increases the amount of free energy.
Overview
[edit | edit source]A binding site is a position on a protein that binds to an incoming molecule that is smaller in size comparatively, called ligand.
In proteins, binding sites are small pockets on the tertiary structure where ligands bind to it using weak forces (non-covalent bonding). Only a few residues actually participate in binding the ligand while the other residues in the protein act as a framework to provide correct conformation and orientation. Most binding sites are concave, but convex and flat shapes are also found.
A ligand-binding site is a place of the mass chemical specificity and affinity on protein that binds or forms chemical bonds with other molecules and ions or protein ligands. The affinity of the binding of a protein and a ligand is a chemically attractive force between the protein and ligand. As such, there can be competition between different ligands for the same binding site of proteins, and the chemical reaction will result in an equilibrium state between bonding and non-bonding ligands. The saturation of the binding site is defined as the total number of binding sites that are occupied by ligands per unit time.
The most common model of enzymatic binding sites is the induced fit model. It differs from the more simple "Lock & key" school of thought because the induced fit model states that the substrate of an enzyme does not fit perfectly into the binding site. With the "lock & key" model it is assumed that the substrate is a relatively static model that does not change its conformation and simply binds to the active site perfectly. According to the induced fit model, the binding site of an enzyme is complementary to the transition state of the substrate in question, not the normal substrate state. The enzyme stabilizes this transition state by having its NH3+ residues stabilize the negative charge of the transition state substrate. This results in a dramatic decrease in the activation energy required to bring forth the intended reaction. The substrate is then converted to its product(s) by having the reaction go to equilibrium quicker.
Properties that Affect Binding
- Complementarity:Molecular recognition depends on the tertiary structure of the enzyme which creates unique microenvironments in the active/binding sites. These specialized microenvironments contribute to binding site catalysis.
- Flexibility:Tertiary structure allows proteins to adapt to their ligands (induced fit) and is essential for the vast diversity of biochemical functions (degrees of flexibility varies by function)
- Surfaces:Binding sites can be concave, convex, or flat. For small ligands – clefts, pockets, or cavities. Catalytic sites are often at domain and subunit interfaces.
- Non-Covalent Forces:Non-covalent forces are also characteristic properties of binding sites. Such characteristics are: higher than average amounts of exposed hydrophobic surface, (small molecules – partly concave and hydrophobic), and displacement of water can drive binding events.
- Affinity: Binding ability of the enzyme to the substrate (can be graphed as partial pressure increases of the substrate against the affinity increases (0 to 1.0); affinity of binding of protein and ligand is chemical attractive force between the protein and ligand.
Enzyme Inhibitors
[edit | edit source]Overview
[edit | edit source]Enzyme inhibitors are molecules or compounds that bind to enzymes and result in a decrease in their activity. An inhibitor can bind to an enzyme and stop a substrate from entering the enzyme's active site and/or prevent the enzyme from catalyzing a chemical reaction. There are two categories of inhibitors.
- 'Irreversible Inhibitors[non competitive only]
- Reversible Inhibitors[both competitive and non competitive]
Inhibitors can also be present naturally and can be involved in metabolism regulation. For example. negative feedback caused by inhibitors can help maintain homeostasis in a cell. Other cellular enzyme inhibitors include proteins that specifically bind to and inhibit an enzyme target. This is useful in eliminating harmful enzymes such as proteases and nucleases.
Examples of inhibitors include poisons and many different types of drugs. and also heavy metals such as lead and cyanide
Irreversible Inhibitors
[edit | edit source]Irreversible inhibitors covalently bind to an enzyme, cause chemical changes to the active sites of enzymes, and cannot be reversed. A main role of irreversible inhibitors include modifying key amino acid residues needed for enzymatic activity. They often contain reactive functional groups such as aldehydes, alkenes, or phenyl sulphonates. These electrophilic groups are able to react with amino acid side chains to form covalent products. The amino acid components are residues containing nucleophilic side chains such as hydroxyl or sulphydryl groups such as amino acids serine, cysteine, threonine, or tyrosine.

First, irreversible inhibitors form a reversible non-covalent complex with the enzyme (EI or ESI). Then, this complex reacts to produce the covalently modified irreversible comple EI*. The rate at which EI* is formed is called the inactivation rate or kinact. Binding of irreversible inhibitors can be prevented by competition with either substrate or a second, reversible inhibitor since formation of EI may compete with ES.
In addition, some reversible inhibitors can form irreversible products by binding so tightly to their target enzyme. These tightly-binding inhibitors show kinetics similar to covalent irreversible inhibitors. As shown in the figure, these inhibitors rapidly bind to the enzyme in a low-affinity EI complex and then undergoes a slower rearrangement to a very tightly bound EI* complex. This kinetic behavior is called slow-binding. Slow-binding often involves a conformational change as the enzyme "clamps down" around the inhibitor molecule. Some examples of these slow-binding inhibitors include important drugs such as methotrexate and allopurinol.
Reversible Inhibitors
[edit | edit source]Reversible inhibitors bind non-covalently to enzymes, and many different types of inhibition can occur depending on what the inhibitors bind to. The non-covalent interactions between the inhibitors and enzymes include hydrogen bonds, hydrophobic interactions, and ionic bonds. Many of these weak bonds combine to produce strong and specific binding. In contrast to substrates and irreversible inhibitors, reversible inhibitors generally do not undergo chemical reactions when bound to the enzyme and can be easily removed by dilution or dialysis.

There are three kinds of <cap>reversible inhibitors</cap>: competitive, noncompetitive, and uncompetitive/mixed inhibitors.
- Competitive inhibitors, as the name suggests, compete with substrates to bind to the enzyme at the same time. The inhibitor has an affinity for the active site of an enzyme where the substrate also binds to. This type of inhibition can be overcome by increasing the concentrations of substrate, out-competing the inhibitor. Competitive inhibitors are often similar in structure to the real substrate.
Competitive inhibitor binds to active site of enzyme and decreases amount of binding of substrate or ligand to enzyme, such that Km is increased and Vmax not changed. The chemical reaction can be reversed by increasing concentration of substrate.

- Uncompetitive inhibitors bind to the enzyme at the same time as the enzyme's substrate. However, the binding of the inhibitor affects the binding of the substrate, and vice-versa. This type of inhibition cannot be overcome, but can be reduced by increasing the concentrations of substrate. The inhibitor usually follows an allosteric effect where it binds to a different site on the enzyme than the substrate. This binding to an allosteric site changes the conformation of the enzyme so that the affinity of the substrate for the active site is reduced.
Uncompetitive inhibitor binds to enzyme-substrate complex to stops enzyme from reacting with substrate to form product, as such, it works well at higher substrate and enzyme concentrations that substrates are bonded to enzymes; the binding results in decreasing concentration of substrate binding to enzyme, Km, and Vmax, and increasing binding affinity of enzyme to substrate.

- Non-competitive inhibitors bind to the active site and reduces the activity but does not affect the binding of the substrate. Therefore, the extent of inhibition depends on the concentration of the substrate.
Noncompetitive inhibitor binds to other site that is not active site of enzyme that changes structure of enzyme; therefore, blocks enzyme binding to substrate that stops enzyme activity and decreases rate of chemical reaction of enzyme and substrate, which can not be changed by increasing concentration of substrate; the binding decreases Vmax and not changes Km of the chemical reaction.

Quantitative Description of Reversible Inhibitors
[edit | edit source]
Most reversible inhibitors follow the classic Michaelis-Menten scheme, where an enzyme (E) binds to its substrate(S) to form an enzyme-substrate complex (ES). km is the Michaelis constant that corresponds to the concentration of the substrate when the velocity is half the maximum. Vmax is the maximum velocity of the enzyme.

- Competitive inhibitors can only bind to E and not to ES. They increase Km by interfering with the binding of the substrate, but they do not affect Vmax because the inhibitor does not change the catalysis in ES because it cannot bind to ES.
- Uncompetitive inhibitors are able to bind to both E and ES, but their affinities for these two forms of the enzyme are different. Therefore, these inhibitors increase Km and decrease Vmax because they interfere with substrate binding and hamper catalysis in the ES complex.
- Non-competitive inhibitors have identical affinities for E and ES. They do not change Km, but decreases Vmax.
References
[edit | edit source]http://en.wikibooks.org/wiki/Structural_Biochemistry/Enzyme/Irreversible_Inhibitor
http://en.wikibooks.org/wiki/Structural_Biochemistry/Enzyme/Reversible_Inhibitors

A catalytic triad is a group of three amino acids that are found in the active sites of some proteases involved in catalysis.
Three different proteases that have catalytic triads are: chymotrypsin, trypsin and elastase. In chymotrypsin, the catalytic triad is made from serine 195, histidine 57, and aspartate 102. The side chain of serine is bonded to the imidazole ring of the histidine residue which accepts a proton from serine to form a strong alkoxide nucleophile in the presence of a substrate for attack. The aspartate residue orients histidine to make it a better proton acceptor via hydrogen bonding and electrostatic reactions.
The active site of chymotrypsin is marked by serine 195. Serine lies in a small pocket on the surface of the enzyme. Serine is bonded to histidine 57 which is then bound to aspartate 102. All three of these residues are hydrogen bonded at this pocket. These three residues participate in concerted mechanisms that allows chymotrypsin and other proteases to be activated by incoming substrates. This is called the catalytic triad.
We know that serine is the final reactive site but serine actually depends on the histidine and aspartate residue to make it a good nucleophile. The histidine residue forces serine into a position that facilitates nucleophilic attack later on through the process of catalysis by approximation. In the presence of a substrate, a chain reaction occurs. First since asparate is acidic, it will be deprotonated first by bases. Aspartate that flanks the histidine residue also provides it with favorable electrostatic effects and makes it a better proton acceptor. So after asparate is deprotonated, proton transfer from histidine goes to aspartate. Now that histidine is deprotonated, it grabs the proton from serine's hydroxyl group. This creates a much more reactive alkoxide group on serine.
Now that the serine is activated we can proceed onto peptide hydrolysis. The alkoxide can attack an incoming substrate to form a tetrahedral intermediate. In this stage we form a resonating oxyanion hole which is a common motif in these kind of reactions. The oxyanion hole stabilizes the tetrahedral intermediate by distributing the negative charge around. Next comes the acyl-enzyme and eventually we see the release of the amine component and water binding.
The catalytic triad actually reveals a deep hydrophobic pocket where serine is sticking out in the center. This pocket positions incoming side chains of a substrate. There is a lot of specificity involved as chymotrypsin has a specific pocket with serine while other enzymes such as trypsin and elastase have different composition of pockets. Therefore we can now know that chymotrypsin likes large aromatic or long, nonpolar side chain.
Catalytic triads also exist in trypsin and elastase. Instead of serine, in trypsin, the center amino acid at the pocket is aspartate. Therefore, its pocket is specific to positively charged species of side chains. Elastase has a pocket that contains two residues of valine, which makes it very hard for big bulky side chains to enter the pocket; therefore, it favors small side chains. Trypsin and elastase are obviously homologs of chymotrypsin. They have 40% similarity in composition and have similar structures.

Site-Directed Mutagenesis helps us understand the Catalytic Triad
Site-directed mutagenesis can be used to test the involvement of individual amino acid residues to the catalytic influence of a protease. Each of the triad’s residues has been converted to alanine. The cleaving ability of each mutant enzyme is examined. The conversion of active-site serine 221, aspartate 32, and histidine 64 into alanine reduces catalytic power. These results strongly support the fact that the catalytic triad, especially the serine-histidine pair, act together to generate a nucleophile that attacks the carbonyl carbon atom of a peptide bond. Site-directed mutagenesis can also tell us the importance of the oxyanion hole for catalysis. The mutation of asparagine 155 to glycine removes the side chain NH group reduced by the oxyanion hole. This shows that the NH group of asparagine residue helps to stabilize the tetrahedral intermediate and the following transition state.
References
[edit | edit source]Berg, Jeremy "Biochemistry" Sixth edition. Freeman and Company, 2007 Evolution is change in the genetic material of a population of organisms from one generation to the next. Evidence of evolution of enzymes have been found by examining multiple enzymes with similar characteristics.
Catalytic Triads
[edit | edit source]Catalytic triads are found in other hydrolytic enzymes similar to that in chymotrypsin. These sequences of proteins are approximately 40% identical and have nearly the same overall structure. However, these proteins differ in substrate specificity. Other members of this family include a collection of proteins that take part in blood clotting. Other enzymes that are not homologues of chymotrypsin have been found to contain very similar active sites. The presence of very similar active sites in different protein families is a consequence of convergent evolution. Furthermore, other proteases have been discovered that contain an active site serine or threonine residue that is activated by a different side chain. It can be concluded that the catalytic triad in proteases is especially effective in the hydrolysis of peptides because of its frequency of occurrence in different enzymes.
Zinc Based Active Sites
[edit | edit source]Carbonic anhydrases homologous to the human enzymes are common in animals and some bacteria and algae. In addition, two other families of carbonic anhydrases have been discovered for catalytic activity. In plants, beta carbonic anhydrases are found and revealed that although it has a bound zinc ion similar to alpha carbonic anhydrases the structures are unrelated. Gamma carbonic anhydrases were discovered to have three zinc sites similar to the alpha carbonic anhydrases. Convergent evolution has generated carbonic anhydrases that rely on coordinated zinc ions at least three times.
P-Loop Domains
[edit | edit source]Domains similar in NMP kinases are present in a wide array of proteins. Examples include ATP synthase, molecular motor proteins, signal transduction, and translation. The wide utility of P-loop NTPase domains is perhaps best explained by their ability to undergo substantial conformational changes.
References
[edit | edit source]Berg, Jeremy M. John L. Tymoczko. Lubert Stryer. Biochemistry Sixth Edition. W.H. Freeman and Company. New York, 2007. The enzyme associated in the transition-state stabilization binds to the transition state better than when it binds to the ground-state reactants. The enzyme is seen as a flexible molecule where its shape is complementary to the substrates or reactants when in its activated transition-state. As the enzyme binds to the transition state the reaction is accelerated proportionate to the transition state concentration.
The transition state compared to the ground state was first introduced by Kurz in 1963. In 1966 Jencks then introduced the existence of transition-state-analog inhibitors. Transition-state theory started to have more emphasis on enzymology in the 1970s when scientists started to notice the broad power of transition-state analogs.
The first three-dimensional structure of the transition-state geometry studied was the lysozyme of a hen egg-white. They observed and studied the x-ray crystallography and visualized the complementarity of a catalytic site to the transition-state geometry. Scientists studied the binding of many oligosaccharide inhibitors and showed how substrates bind to the lysozyme. Furthermore, they deduced that the conformation of a sugar residue strongly effects the binding to the enzyme. When the sugar residue was in its half-hair conformation, then binding occurs.
Transition-state theory relies on assumptions and approximations. The two assumptions include a dynamical bottleneck assumption and an equilibrium assumption. Dynamic bottleneck states the decomposition of a transition-state complex controls the reate rate. The equilirium assumption states that the transition-state molecule is in equilibrium with the reactants.
Through thermodynamic cycling, transition-state theory can be applied to enzyme catalysis.
References
[edit | edit source]- J Kraut, "How do Enzymes Work," Science 28 October 1988: 533-540.
Definition
[edit | edit source]
A zymogen(also denoted as a proenzyme) is a group of proteins that can also be described as an inactive enzyme. Since it is an inactive precursor, it does not hold any catalytic activity. These zymogens can be activated by chemical processes such as cleaving, hydrolysis, along with other biochemical changes that cleave the inactive enzyme to make it active. These biochemical processes often occurs in a lysosome, where cleavage reveals the active site. Exposure of active site allow the enzyme to become active and function to catalyze reactions. The reason for cells to secrete inactive enzymes is to prevent unwanted destruction of cellular proteins. It is only when the conditions are right that zymogens become activated into enzymes. There are specialized zymogenic cells that work only to synthesize and store zymogens in inactive form, ready to send to the parts of the body at need.
Examples
[edit | edit source]Examples of zymogens include:
Pepsinogen
Pepsinogen, inactive precursor form of pepsin, is secreted by Chief cells in the stomach. Pepsinogen is activated by Hydrochloric acid (secretion from Parietal cells) because Hydrochloric acid provides the necessary acidic environment for which pepsin works best. Once pepsinogen becomes pepsin, it is responsible for the breakdown of food. The difference between pepsinogen and pepsin is that its primary structure has an additional 44 amino acids. The activation of pepsinogen to pepsin makes pepsin available to catalyze pepsinogen further to cleave it into more pepsin.
Trypsinogen
Trypsinogen is the inactive form of trypsin. It is secreted by the pancreas and found in pancreatic juice. Once activated in the duodenum, trypsin cleaves peptide chains at the carboxyl side of the amino acids lysine and arginine unless they are preceding a proline. The importance of trypsin is its ability to cleave other zymogens such as chymotripsinogen and procarboxypeptidase.
Chymotrypsinogen
Once chymotrypsinogen is cleaved by trypsin, and reacted with Chymotrypsin produces a fully active enzyme, Chymotrypsin. Chymotrypsin is found in the digestive system of mammals, as well as other organisms. It works by cleaving peptides at the carboxyl end of aromatic amino acids (Tryptophan, Tyrosin, and Phenylalanine)

Procarboxypeptidase
Procarboxypeptidase, which is the inactive form of carboxypeptidase, is converted to the active form by trypsin and enteropeptidase. It is secreted by the pancreas. Only some forms of carboxypeptidase are initially produced in the inactive form. However, the advantage of this mechanism is to ensure that the enzymes are not immediately exhausted before digestion.
Nuclease
Nuclease is an enzyme which can break phosphodiester bonds between nucleotides in a DNA sequence. Nucleases contain a general range of enzymes such as endonucleases. Nucleases vary in the DNA sequences they cut as certain phosphodiester bonds are cleaved in such a way that may not always completely symmetrical.
Pancreatic amylase
Pancreatic Amylase is an enzyme that converts complex sugars such as starches and polysaccharides of carbohydrates into simpler sugars during digestion. Amylase hydrolyzes starch, glycogen, and dextrin to form in all three instances glucose, maltose, and the limit-dextrins. Amylase is mainly secreted by the salivary glands, but some may also be found in the pancreas that also help aid in digestion.
Lipase
Lipase is the active form of prolipase. Once activated, the water soluble enzyme is the catalyst for the hydrolysis ester bonds in water-insoluble, lipid substrates. This action is what classifies lipases as a subclass of the esterases. It cleaves fats into monoglycerides, fatty acids, and glycerols. The lipase also is essential in the process of digestion, as well as the transport and processing of dietary lipids in almost all organisms.
Proelastase
Proelastease is the inactive form of elastase. The activation of Proelastease is initially done through the simple cleavage of multiple sub-unit residues that bind to the central structure of the protein structure. The cleavage disrupts the hydrophobic interactions of the tertiary structure, allowing the polar regions of the enzyme to respond to digestion. Proleastease is distinct from Chymotrypsin through isolation by chromatography reveals that this structure is highly resistant and further stabilized by internal hydrogen bonding. Activation of proelastase and propeptidase with trypsin changed their electrophoretic mobilities. The activated proelastase migrated at the same rate as authentic, pure elastase.The proenzymes that were originally highly insoluble could be solubilized by treatment with alumina Cγ gel.
Enteropeptidase
Enteropeptidase is produced within the walls of the small intestines secreted by the duodenum glands. This enzyme proteolytically activates trypsinogen to trypsin which simultaneously activates other digestive enzymes as well. Enteropeptidase cleaves at the C-terminal end of trypsinogen which activates the enzyme, turning trypsinogen into trypsin. Most of Enteropeptidase is consisted of disulfide bonds within the larger chain forming the catalytic subunits.
Caspase
Caspases are a family of cysteine proteases that play essential roles in apoptosis, necrosis, and inflammation. Caspases plays an important role in cells for apoptosis (programmed death) during the development and other stages of adult life. Caspases are also known as "executioner" proteins because of this role that they play in the cell.
Prothrombin
Prothrombin(Zymogen) is the precursor to the enzyme Thrombin which in turn converts fibrinogen in to fibrin. Fibrin is the protein responsible for blood clotting and tissue restoration during a tissue rupture. Fibrinogen is the substrate to thrombin and when activated by thrombin it becomes Fibrin a non-soluble glycol protein. Fibrimogen has a linearly symmetrical structure, containing a central cleavage site and a at both ends it has what is known as a globular unit which comes after a designated region alpha. The globular region has 2 “connection sites” Beta and Gamma. These 2 sites have the ability to connect to 2 other fibrin proteins, this allows the proteins to connect and make a lattice like structure making it a strong structure known as a cross linked fibrin clot.
[[Image:File:Commonpathway.png
Angiotensin
Angiotensin is an oligopeptide that causes blood vessels to constrict and increased blood pressure. It also stimulates the release of aldosterone from the adrenal cortex. It is a hormone and a powerful dipsogen.
Example of Zymogen Activation
[edit | edit source]zymogen activation The Gibbs free energy graph shows whether or not a reaction is spontaneous-- whether it is exergonic or endergonic. ΔG is the change in free energy. Generally, all reactions want to go to a lower energy state, thus a negative change is favored. Negative ΔG indicates that the reaction is exergonic and spontaneous.
Note that ΔG reveals nothing about the speed of the reaction. For example, diamond is supposed to be in a liquid state at room temperature, yet the rate of this reaction is so slow that this change does not seem to occur. Neither do peptide bonds spontaneously go through hydrolysis even though the reaction of such is highly exergonic. A positive ΔG indicates that the reaction is endergonic, or that it requires energy to go from reactants to products. The free energy graph can be used to determine whether the reaction will be spontaneous or not by evaluating the ΔG. ΔG can be found by subtracting the free energy of the reactants from the free energy of the products.
ΔG = Gproducts - Greactants

Therefore, if the reaction goes from higher free energy to lower free energy, there will be a negative ΔG, and the reaction will be spontaneous. However, if the reactants have a lower ΔG than the products, there will be an increase in free energy, and the reaction is nonspontaneous. In this situation, some form of energy (in the form of heat, light, etc.) will be required for the reaction to take place. It should be noted that a spontaneous reaction will not necessarily occur on its own. This is because an initial activation energy is needed in order to start the reaction and thus even a spontaneous reaction may need some form of energy input. A good example of this is the very exergonic combustion of octane, which still needs a flame in order to initiate.
Enzymes will affect this free energy graph by lowering the activation barrier, or the amount of energy needed for the reaction to occur, They do this by stabilizing the transition state, or the state of highest energy between reactants and products. Enzymes DO NOT affect the equilibrium constant in any way, shape, or form.
File:Free energy diagram.jpg
Definition
[edit | edit source]By definition, the transition state is the transitory of molecular structure in which the molecule is no longer a substrate but not yet a product. All chemical reactions must go through the transition state to form a product from a substrate molecule. The transition state is the state corresponding to the highest energy along the reaction coordinate. It has more free energy in comparison to the substrate or product; thus, it is the least stable state. The specific form of the transition state depends on the mechanisms of the particular reaction.
In the equation S → X → P, X is the transition state, which is located at the peak of the curve on the Gibbs free energy graph.
![]()
Application to Enzymes
[edit | edit source]
Enzymes are usually proteins that act like catalysts. The enzyme's ability to make the reaction faster depends on the fact that it stabilizes the transition state. The transition state's energy or, in terms of a reaction, the activation energy is the minimum energy that is needed to break certain bonds of the reactants so as to turn them into products. Enzymes decreases activation energy by shaping its active site such that it fits the transition state even better than the substrate. When the substrate binds, the enzyme may stretch or distort a key bond and weaken it so that less activation energy is needed to break the bond at the start of the reaction. In many cases, the transition state of a reaction has a different geometry at the key atom (for instance, tetrahedral instead of trigonal planar). By optimizing binding of a tetrahedral atom, the substrate is helped on its way to the transition state and therefore lowers the activation energy, allowing more molecules to be able to turn into products in a given period of time. The enzyme stabilizes the transition state through various ways. Some ways an enzyme stabilizes is to have an environment that is the opposite charge of the transition state, providing a different pathway, and making it easier for the reactants to be in the right orientation for reaction.
Consider the peptide hydrolysis by chrymotypsin as an example.
In a normal peptide hydrolysis reaction without the help of a catalyst, water acts as a nucleophile to attack the electrophilic carbonyl carbon. The carbon atom being attacked goes from its initial sp2 state (trigonal planar) to a new sp3 state (tetrahedral) in its transition state.
![]()
In the presence of chymotrypsin, however, a better nucleophile is used in the form of the catalytic triad - Asp 102, His 57, Ser 195 side chains. Moreover, the oxyanion hole, which consists of the backbone -NH- groups of Gly 193 and Ser 195 of the enzyme, have the N-H groups positioned in such a way that they will donate strong hydrogen bonds to the substrate's C=O oxygen, given that the carbon atom is tetrahedral as found in the transition state. This strains the bonds of the trigonal planar C=O of the original substrate, helping the reaction to proceed to the transition state. The hydrogen bonds also stabilize the formal negative charge on the oxygen atoms. In this way, the activation energy of the reaction is lowered and the rate of reaction thus increases.
Enzyme Inhibition
[edit | edit source]In 1948, Linus Pauling proposed that transition state analogs should be effective inhibitors of enzymes. These molecules are mimics of transition states of the substrate of a particular enzyme reaction. Because they are so similar to the transition states of the substrate, they can bind to the enzyme, oftentimes much more tightly than the substrate can. The fact that these transition state analogs bind so tightly to enzymes makes it an effective enzyme inhibitor. The transition state theory says that the occurrence of enzymatic catalysis is equivalent to an enzyme binding to the transition state more strongly than it binds to the ground-state reactants. This theory is based on the two fundamental principles of physical chemistry: Absolute reaction-rate theory and the thermodynamic cycle. Also, the thermodynamic cycle relating substrate binding and transition state binding apply elementary transition-state theory to enzymatic catalysis, which is a restatement of Pauling's description of transition-state binding in quantitative symbols. He has stated that the catalytic powers of enzymes result from their highly specific binding of the transition state. The activation energy required to achieve the transition state is a barrier to the formation of product. It is the minimum amount of energy required for a reaction to proceed. This barrier is the reason why the rate of many chemical reactions is very slow without the presence of enzymes, heat, or other catalytic forces. There are two common ways to overcome this barrier and thereby accelerate a chemical reaction. First, the reactants could be exposed to a large amount of heat. For example, if gasoline is sitting at room temperature, nothing much happens. However, if the gasoline is exposed to a flame or spark, it breaks down rapidly, probably at an explosive rate. A second strategy is to lower the activation energy barrier. Enzymes lower the activation energy to a point where a small amount of available heat can push the reactants to a transition state. The question that arises is: How do enzymes work to lower the activation energy barrier of chemical reactions?
Enzymes are large proteins that bind small molecules. When bound to an enzyme, the bonds in the reactants can be strained (that is stretched) thereby making it easier for them to achieve the transition state. This is one way for which enzymes lower the activation energy of a reaction. When a chemical reaction involves two or more reactants, the enzyme provides a site where the reactants are positioned very close to each other and in an orientation that facilitates the formation of new covalent bonds. This technique also lowers the needed activation energy for a chemical reaction. Straining the reactants and bringing them close together are two common ways the enzymes use to lower the activation energy. There are other methods that the enzymes use to facilitate a chemical reaction. Changing the local environment of the reactants is one of these methods. In some cases, enzymes lower the activation energy by directly participating in the chemical reaction. For example, certain enzymes that hydrolyze ATP form a covalent bond between phosphate and amino acid in the enzyme that may have a charge that affects the chemistry of the reactants. This is very temporary condition. The covalent bond between phosphate and the amino acid is quickly broken, releasing phosphate and returning the amino acid back to its original condition.
Arrhenius equation
[edit | edit source]Arrhenius equation is a description of the relationship between the activation energy and the reaction rate.
k = Ae(-Ea/RT)
where: k = chemical reaction, T = temperature in Kelvin, Ea = activation energy, A = the pre-exponential factor, R = the gas constant
According to this equation, it is observed that at a higher temperature, the probability that the two molecules will collide is higher, resulting in a higher kinetic energy, which leads to the lower requirement on the activation energy.
The Arrhenius equation is particularly helpful when calculating the rate of production of products over time, which is characterized by the following:
d[products]/dt = rate = Ae(-Ea/RT)[AmBn]
where the [A] and [B] are the concentrations of the reactants and m and n are their respective reaction
Lowering the Activation Energy
[edit | edit source]A catalyst is something that lowers the activation energy; in biology it is an enzyme. The catalyst speeds up the rate of reaction without being consumed; it does not change the initial reactants or the end products.

The graph above shows how the activation energy is lowered in the presence of an enzyme (blue line) that is doing the catalysis, exempflified with the carbon anhydrase reaction. The transition state is usually the most unstable part of the reaction since it is the one with the highest free energy. The difference between the transition state and the reactants is the Gibbs free energy of activation, commonly known as activation energy .

Enzymes (blue line) change the formation of the transition state by lowering the energy and stabilizing the highly energetic unstable transition state. This allows the reaction rate to increase, but also the back reaction occurs more easily.
Common Misconceptions
[edit | edit source]Some common misconceptions about activation energy barriers and catalysts to speed up the reaction. The catalyst does NOT lower activation energy of the same barrier, but rather chooses another chemical pathway with a lower activation energy.The catalysts lead to new pathways which don't require as high of an energy of activation in turn speeding up the reaction.
Another big misconception about activation energy is that reactions will not always give the most thermodynamically stable products. Sometimes, the product that forms is the one that is more kinetically stable, or forms faster. However, if a catalyst is available, the thermodynamically more stable product will be able to form, even if the energy barrier is high. The transition state is the highest energy intermediate of a reaction and thus tends to be the shortest-lived intermediate along a reaction coordinate. Upon formation of the transition state, the intermediate can spontaneously proceed to product formation.
This transition state of intermediates are indicated by double dagger. Transition state has a higher free energy or delta G than reactants and products. Therefore, the intermediates are less stable than reactants and products.
The energy difference between reactant and transition state is called activation energy. The activation energy determine the rate of reactions. Since the activation energy is the difference in energy between the reactant and the intermediate, it acts as a barrier for the reactant to overcome. As a result, enough energy must be added to the reactants in order to overcome the activation energy and transform into product. Therefore, the higher the activation energy, the harder it will be for the reactant to be converted into product. The rate limiting step, which is sometimes related to formation of the highest energy intermediate determines the rate of the reaction. The activation energy depends on a number of factors including temperature, the specific reaction involved and the presence of catalysts.
An enzyme generally accelerates the rate of reaction by stabilizing the transition state intermediate. Enzymes simply lower the activation energy of the intermediates by changing the conformation of the intermediates into a more favorable and stabilized conformation. A stabilized intermediate means lower activation energy and as mentioned above, lower activation energy means lower activation barrier so the reactants can form products at a faster rate. The result is generally a very large increase in reaction rates on the order of millions of times.
Enzymes do not alter reaction equilibrium because they do not change the potential energy of either starting material or products. Furthermore, the rates of the forward and reverse reactions are changed equally, meaning that the ratio of kforward and kreverse is the same at all times.
Enzyme Rates and Constants
[edit | edit source]Enzyme Thermodynamics
For enzyme thermodynamic, the most important constant is probably ∆G. ∆G is the free-energy difference between products and reactants, also known as the amount of energy required to convert reactants to products. More importantly, it can tell whether a reaction will occur spontaneously, which are what enzymes are concerned with.
If ∆G is negative then the reaction will occur spontaneously (exergonic). If ∆G is zero, the system is at equilibrium and no net change can take place. If ∆G is positive, energy is required for the reaction to occur (endergonic).
A thermodynamic explanation of this starts with the definition of Gibbs free energy, which says
∆G = ∆H - T∆S,
where ∆H is the change in enthalpy (joules) and ∆S is the change in entropy (joules/Kelvin). As we can see, if the energy due to entropy (disorder of a system) exceeds the enthalpy (thermodynamic potential of a system), the Gibbs free energy will be negative and thus no energy is needed for the reaction to occur.
Enzyme Kinetics
Enzyme Kinetics contain a few more constants and rates; starting with Vmax, this is the maximal rate when all the catalytic sites on the enzyme are saturated (bounded) with substrates. Another constant is Km, which is the substrate concentration when it is half the Vmax. Their relationship to each other can be seen through the Michaelis-Menten equation.
Enzyme Kinetics is measuring rate of enzyme reaction. One way to measure of rate of reation is by using spectroscopy change, absorb light at different wavelength. Another way to study enzyme kinetics is through the graph that has substrate concentration (x-axis) vs. reaction velocity (initial velocity). km(michaelis constant) is substrate concentration at which our enzyme reacts half of maximum velocity.
Vo = Vmax([S]/([S]+ KM))
where [S] is the substrate concentration and Vo is the initial rate of the reaction (t=0). Finally, there is the rate of kcat/KM which measures the catalytic efficiency. kcat is the turnover number of an enzyme, which is the number of substrate bounded to an enzyme in a unit of time when the enzyme is fully bounded. Given this, the higher the kcat/KM rate is, the more efficient it is at binding substrates.
Introduction
[edit | edit source]The rate constant is a proportionality constant where the rate of reaction that is directly correlated to the concentration of the reactant. In first order reactions, the reaction rate is directly proportional to the reactant concentration and the units of first order rate constants are 1/sec. In bimolecular reactions with two reactants, the second order rate constants have units of 1/M*sec. Second order reactions can be made to appear as first order reactions, such reactions are called pseudo-first order since adding one reactant in excess will make the reaction first order with respect to the other reactant. There are also zero order reactions in which the reaction is independent of the reactant concentrations where the units of the rate constant are mol/L*sec.
For a general chemical reaction of the form: aA + bB --> products
The expression of the reaction rate would be:
![{\displaystyle {\frac {d[C]}{dt}}=k[A]^{a}[B]^{b}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/ee75f2d0af97cbfd8e9cc84a4341f94d1b4839d2)
where: k is the rate constant of the reaction, [A] and [B] are the concentrations of the reactants, and a and b are the order of the reaction respect to A and B respectively. The overall order of the reaction is the sum of m and n. Keep in mind here that the above rate equation refers to the disappearance of A and B, thus the rate will be negative (indicating that the reactants were consumed). Products, on the other hand, have a positive value for the rate, since they are being generated. To account for this, many texts list the equation as .
![{\displaystyle rate=k[A]^{a}[B]^{a}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/72a2c09a7d960e44e772603ca94b5b5f4819728b)
It is important to note that not every reactant will appear in the rate constant since a reaction may be zeroth order in a given reactant. Furthermore, except in very limited cases, the order of the reaction cannot be determined from the stoichiometric equation, but rather must be experimentally calculated.
Although this is generally not seen in general chemistry courses, the reaction order can be negative and/or not a whole number. Rate=K(Concentration)
Basic Kinetics
[edit | edit source]For reference purposes the following rate laws are listed without detail into their derivation:
| rate law | integrated rate law | Rate Constant Units (k) | |
| 0 Order | |||
| 1st Order | |||
| 2nd Order |
![{\displaystyle r=-{\frac {d[A]}{dt}}=k}](https://wikimedia.org/api/rest_v1/media/math/render/svg/a9060151df845cf02ceac0146e46a5af10eb0abf)
![{\displaystyle \ [A]_{t}=-kt+[A]_{0}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/989476d225b1fd0996646a5eb831e1b75d505eca)

![{\displaystyle r=-{\frac {d[A]}{dt}}=k[A]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/8e2d0d4b06c394d6c730853391ecc70e6b9e1575)
![{\displaystyle \ \ln {[A]}=-kt+\ln {[A]_{0}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/f01599f79a633531bd83f2969981837039366f74)

![{\displaystyle r=-{\frac {d[A]}{dt}}=k[A]^{2}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/415e8b8ad91b879e0152af6065eca9d0cfaddb78)
![{\displaystyle {\frac {1}{[A]}}={\frac {1}{[A]_{0}}}+kt}](https://wikimedia.org/api/rest_v1/media/math/render/svg/79c54169f180a95a38379eaa20ecfed27bb94877)

Pseudo 1st Order Rate Laws
[edit | edit source]In some instances it proves difficult to near impossible to monitor concentrations of each reactant. A rate law for a reaction can be written in the "pseudo" 1st order for reactions involving multiple species, for instance species X and Y. By holding concentrations of one species constant, for instance X, there is essentially no net change in X vs time. As a result a new rate law can be defined to incorporate the X into the preexisting rate constant.
is defined as , thus our new rate equation is written as

![{\displaystyle \ k[X]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/131da3ad364e74017a54039e8c02cfad6db06681)
![{\displaystyle \ r=k'[Y]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/03234134055dfdf025b3513b87b8e29984a87405)
Varying experimental conditions and the resulting data can be used to determine k.
Steady State Approximation
[edit | edit source]A steady state approximation is useful in systems where it proves ultimately difficult to measure the concentration of one reactant (or one of its intermediates). However, if it is assumed that the concentrations of the species in question remains in a constant steady state, an equation can be written in terms of other species in which we can measure.
Lets examine a system that involves the following reactions:
| reaction | rate equation |
| A + B X | k1[A][B] |
| X + C D + E | k2[X][C] |
| X + E F | k3[X][E] |

First we are going to assume that species X is in steady state.
![{\displaystyle \ {d[X]}{dt}=0}](https://wikimedia.org/api/rest_v1/media/math/render/svg/f833c1397fdb0d9c22d6ac7ec2c862eb2297f79d)
As part of out assumption X remains constant thus:
production terms - loss terms
![{\displaystyle \ {d[X]}{dt}=0=}](https://wikimedia.org/api/rest_v1/media/math/render/svg/8ebf16d857f126a58742ed9b3f494d46cafc5435)
k1[A][B] - k2[X][C] - k3[X][E]
Simplifying :
k1[A][B] = k2[X][C] - k3[X][E]
Solving for [X] in terms of reactants in which the concentrations can be experimentally measured, we obtain the equation:
The rate equation of a reaction maps out the rate of disappearance/appearance of a compound over time.
![{\displaystyle {\frac {k_{1}[A][B]}{k_{2}k_{3}[C][E]}}=[X]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/fdb36fc6002d5686c36e3edf22561872664180d7)
rate = k[A]
Where [A] is the molar concentration of compound A, and 'k' is the rate constant. Rates can have a certain order to them. They can be zero, first, second, etc. depending on the reaction. First order reactions (like the one above) are directly proportional to the concentration, so its 'k' value has a unit equal to (1/s) so as to make the rate (M/s).
Second order reactions could look like so:
rate = k[A]2
where the constant 'k' units are (1/M•s).
Though reaction orders are often whole numbers, they may also be fractions or negative in value.
Zero Order Reaction
[edit | edit source]A zero-order reaction is independent of the concentration of the reactants, in which even a higher concentration of reactants will not speed up the rate of the reaction. This kind of reaction if found when a catalyst or other material required for the reaction is saturated by the reactants. Zero-order reactions frequently have to occur first in order to provide reactive substances for the higher order reactions.
The rate equation of this reaction is illustrated as: Rate = K
Furthermore, a plot of concentration versus time should yield a straight line.
First Order Reaction
[edit | edit source]A first-order reaction depends on the concentration of only one reactant. This order occurs mostly in the reactions where there is only one reactant.
The rate equation of this reaction is illustrated as: Rate = K[A]
Furthermore, a plot of the natural log of concentration versus time should yield a straight line.

Second Order Reaction
[edit | edit source]A second-order reaction is characterized by the property that their rate is proportional to the product of the concentrations of two reactants.
The rate equation of this reaction is illustrated as: Rate = K[A][B]
This means that if you double the concentration of the reactants, it will result in a four time increase in rate.
Furthermore, a plot of the inverse of concentration versus time should yield a straight line.
Introduction
[edit | edit source]Enzymes assist reactions and increase the speed of formation of the products by decreasing the activation energy required for a reaction. Most fundamentally, enzymes are still just mechanisms that react catalytically in a chemical equation. Therefore, enzyme reactions also possess the basics of these step-wise mechanism reactions which of course includes the rate-limiting or rate determining step.
It is simple to assume correctly that each step of the mechanism does not proceed at the same rate, and so the rate-limiting step is merely the one reaction of the mechanism that has the slowest rate of reaction. An amusing real-life analogy in the spirit of Black Friday is: say a complete reaction is the time it takes for all the shoppers to make their purchases in Circuit City, then the rate-limiting step would be the shopper who is most indecisive and buys the most stuff. If one was to view a reaction coordinate graph of the entire reaction, the rate-limiting step is usually the one with the highest activation energy hump or highest energy transition state.
Particularly, by Michaelis-Menten kinetics of enzymes, the rate-limiting step is usually the product formation step.
For example: The reaction NO2(g) + CO(g) → NO(g) + CO2(g) occurs in two elementary steps:
1. NO2 + NO2 → NO + NO3 (slow step)
2. NO3 + CO → NO2 + CO2 (fast step)
As the second step consumes the NO3 produced in the slow first step, it is limited by the rate of the first step. For this reason, the rate-determining step is reflected in the rate equation of a reaction. Another simple situation analogous to a rate-limiting step is a family of four getting ready to go somewhere. Regardless of how fast everyone else is, because the family has to wait for everyone, the slowest person is going to determine how fast everyone else will be able to leave the house.
The role of rate-limiting step
[edit | edit source]The concept of the rate-determining step is very important to the optimization and understanding of many chemical processes such as catalysis and combustion. Furthermore, it may help determine if that mechanism is correct for the reaction. This is because the rate law for the rate limiting step should equal the rate law for the reaction. If this is not the case, then either the experimental rate law was determined incorrectly or the proposed mechanism is wrong.
The role of the rate-limiting step also has application in the study of alcohol in people. The body will naturally convert alcohol (ethanol is the alcohol consumed in beverages) into acetaldehyde which is later converted into acetate. Once it is an acetate, the body can naturally dispose of the acetate through waste. This is two step process which is:
Alcohol --> Acetaldehyde (very fast) Acetaldehyde --> Acetate (very slow, rate-limiting step)
Since alcohol is converted into acetaldehyde quickly, consumption of too much alcohol will cause acetaldehyde to escape into the bloodstream. This acetaldehyde will build up much faster than it can be converted into acetate. Once acetaldehyde is in the bloodstream, physiological effects begin to show. Eventaully, the acetaldehyde is converted to acetate and the effects wear off. In this case, the rate-limiting step is the conversion of acetaldehyde to acetate since it is much slower and causes a buildup of acetaldehyde in the bloodstream.
Equilibrium Constant
[edit | edit source]The equilibrium constant, K, reflects the ratio of the activities of the products versus the reactants, and represents the extent to which a reaction goes to completion.
In general, a reaction with a
- LARGE K, K > 103, is said to favor the products and thus forms more products than reactants
- SMALL K, K < 10-3, favors reactants and thus very little product is formed. Intermediate values of K, form similar amounts of both products and reactants.
The equilibrium constant is a thermodynamic property and thus the size of K does not indicate anything about the rate at which products form, only the amount. The equilibrium constant for the reaction:
aA + bB <--> cC + dD
can be calculated by,
K = aCcaDd/(aAaaBb)
where aI represents the activity of species I. The activities of the reaction species are raised to their corresponding stoichiometric coefficient.
The equilibrium constant can be very closely approximated by using concentrations for aqueous solutions and pressures for gasses, in place of activities. Solids and pure liquids are not factored into K and are given a value of 1.
The equilibrium constant is related to the standard gibbs free energy, delta G, by
delta G0 = -RTlnK
Kd is defined as dissociation constant that accounts for amount of reactant that dissociates reversibly to form component products; the constant deals with half of binding site of enzyme that binds for concentration of ligands, or the concentration for ligands that bind enzyme to be equal to that that are not; the unit of the constant is in molarity or M; it is a measurement that is indirectly proportional to affinity constant, for example, between enzyme and ligand; or the smaller dissociation constant means higher affinity constant as the stronger the bond between the enzyme and ligand, which is more difficult to be separated to two components. The initial rate, V0, is the rate of the reaction after almost no time has passed, t is approximately 0. It is the measure of moles of product formed per second (t cannot equal zero, because we cannot divide by zero) and can be approximated by finding the slope of the product versus reactant curve at the beginning. Near beginning, the reverse reaction is negligible, and thus the slope is a good approximation.
![{\displaystyle V_{0}=k_{2}[ES]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/7ed6107700240098f78423c99c83ad267775020a)
![{\displaystyle V_{0}={\frac {k_{cat}}{K_{M}}}[E][S]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/a88e9469f546095707b5701a5b38140794248d4b)
Types of Catalysis
[edit | edit source]Enzymes are proteins that catalyze a reaction by stabilizing the transition state and therefore, lowering the activation energy of the reaction. To achieve this, enzymes use different classes of reactions or catalytic strategies. The strategies used to catalyze a reaction are:
In this type of reaction, a nucleophile in the active site reacts with a reactive group in the substrate. The nucleophile is temporarily, covalently bonded to a substrate during catalysis.
Example: Chymotrypsin uses a strong nucleophile to attack a normally unreactive carbonyl carbon atom of a substrate. The nucleophile is briefly covalently attached to the substrate in catalysis.
In this type of reaction a molecule other than water acts as a proton acceptor or donor.
Example: In the catalytic triad, the histidine residue polarizes the hydroxyl group on serine so that it is ready for deprotonation. When a substrate is present, it takes the proton from hydroxyl group of serine which makes the residue act like a base catalyst.
In this type of reaction, two subtrates are positioned together on a single binding surface so that the formation of the new bond is easier, speeding up the reaction.
Example: NMP has a phosphoryl group transferred from ATP by an enzyme holding its two substrates together and aligning them to stabilize the transition state.
In this type of reaction, metal ions help the formation of nucleotides or the ion acts as an electrophile to stabilize a negative charge on an intermediate.
Example: Carbonic Anhydrase is an enzyme that contains a zinc ion which aids in turning water into a better nucleophile. The formation of a hydroxide ion speeds up the nucleophilic attack of CO2.
The type of strategy that is employed is based on the enzyme's structural properties and the reaction that the enzyme will catalyze. Many times a combination of strategies is used to in catalytic reactions.
Examples of Catalytic Reactions
[edit | edit source]Ping pong reactions, or double displacement reactions, involve the release of one or more products before all of the substrates bind to the enzyme. This back and forth is where the reactions get the name ping-pong. A defining part of a ping-pong reaction is called the substituted enzyme intermediate, which has a temporarily modified enzyme that shuttles groups back and forth. An example of a double displacement reaction is as follows:
Aspartate + alpha-Ketoglutarate <-(with enzyme aminotransferase)-> Oxaloacetate + Glutamate
Though this reaction seems like a normal reaction with two reactants and two products, the order of events is what defines the reaction. First, aspartate binds to the enzyme. Then, (before alpha-Ketoglutarate binds), oxaloacetate is released. Now, alpha-Ketoglutarate binds to the enzyme-substrate complex. Last of all, these substrates react and glutamate is released and the enzyme reforms.
This is a more orthodox description of an enzymatic reaction. Sequential reactions require all of the substrates to bind to the enzyme before all of the products are released. These can be further broken down into ordered and random sequential reactions. Ordered reactions have a specific order for which substrates must bind to the enzyme, whereas random reactions do not.
An example of an ordered reaction is as follows:
Pyruvate + NADH <-(lactate dehydrogenase)-> lactate + NAD+
In this reaction, the pyruvate must first bind to the enzyme before the NADH may react with it. Then, lactate is released first before NAD+ is released.
An example of a random reaction is as follows:
Creatine + ATP <-(creatine kinase)-> Phosphocreatine + ADP
In this reaction, the enzyme is not selective, and either Creatine or ATP can bind first. Similarly, either Phosphocreatine or ADP can be released first.
General acid/base catalysis and its effectiveness
[edit | edit source]
General acid/base catalysis' rate determining step is the proton transfer step. Therefore, general acid catalysis has its reaction rate depending on all the acids present; similarly, the general base catalysis has its reaction rate depending on all the bases present. The preferred reaction environment is neutral PH for both reactions, because high concentration of H+ or OH- can damp out the catalytic contributions from other acids and bases, thus, turning the "general" acid or base reaction into "specific" acid or base catalysis.
Since the proton transfer step determines the rate of the reaction, it is important to examine the effectiveness of the general catalysis. The effectiveness of the general catalysis can be determined from Bronsted equation, which is written as,
- , the rate constant of the catalysis
- , the dissociation constant of HA
- , the sensitivity of the catalytic step to the acid dissociation step.




The relationship can be easily seen by graphing logk(HA) vs. log K(HA), and so the slope, which is the alpha, can be analyzed graphically. Each dot on the graph represents different acids. Since efficiency cannot exceed 1, the rage of the slope is bounded between 0 and 1. A slope of 1 indicates that the rate increases with every acid dissociation and that the proton transfer is effective. A slope of zero means every acid dissociation contributes the same effect to the catalysis, and the transfer of proton is not effective. On the other hand, the sensitivity graph for the specific acid/base catalysis may appear as a nonlinear relationship between k(HA) and K(HA).
Overview
[edit | edit source]General acid-base catalysis involves a molecule besides water that acts as a proton donor or acceptor during the enzymatic reaction. Acid-base catalysis facilitates a reaction by stabilizing the charges in the transition state through the use of an acid or base, which donates protons or accepts them, respectively. Nucleophilic and electrophilic groups are activated as a result of the proton addition or removal and causes the reaction to proceed. Many acid-base catalysis reactions involve histidine because it has a pKa close to 7, allowing it to act as both an acid and a base. When a functional group accepts a proton, it will release or donate a proton by the end of the catalytic cycle, and vice versa. Functional groups that participate in reaction are His imidazole, alpha-amino group, alpha-carboxyl group, thiol of Cys, R group of carboxyls of Glu and Asp, aromatic OH of Tyr, and guanidino group of ARG. The protonation of these functional groups are dependent on the pH, therefore the enzymatic catalytic activity is sensitive to the pH level.
General acid-base catalysis is involved in a majority of enzymatic reactions, wherein the side chains of various amino acids act as general acids or general basis. General acid–base catalysis needs to be distinguished from specific acid–base catalysis.
Specific acid–base catalysis means specifically, –OH or H+ accelerates the reaction. The reaction rate is dependent on pH only (which of course is a function of –OH and H+ concentrations), and not on buffer concentration. General acid - partial transfer of a proton from a Brønsted acid lowers the free energy of the transition state rate of reaction increases with decrease in pH and increase in [Brønsted acid].
-Specific acid - protonation lowers the free energy of the transition state, rate of reaction increases with decrease in pH.
-Specific base - abstraction of a proton (or nucleophilic attack) by OH- lowers the free energy of the transition state, rate of reaction increases with increase in pH.
In General acid–base catalysis, the buffer aids in stabilizing the transition state via donation or removal of a proton. Therefore, the rate of the reaction is dependent on the buffer concentration, as well as the appropriate protonation state. -General base - partial abstraction of a proton by a Brønsted base lowers the free energy of the transition state rate of reaction increases with increase in pH and increase in Brønsted base.
Example
[edit | edit source]An example of acid-base catalysis is peptide hydrolysis by chymotrypsin. Chymotrypsin uses a histidine residue as a base catalyst to increase the nucleophilicity of serine. Chymotrypsin uses a histindine residue as a base catalyst to help to strengthen the nucleophillic property of serine, whereas a histindine residue in carbonic anhydrase helps the removal of hydrogen ion from zinc bound water molecule to generate OH-.
The pKa of Histidine is close to neutral thus making it the most effective candidate for general acid or base because it can either donate or accept protons. His 119 in Ribonuclease A plays the role of a general acid that donates a proton to 5'-hydroxyl of nucleoside. On the other hand, His 12 acts as a general base which accepting a proton from the 2'-hydroxyl of 3'-nucleotide. As a result, a 2’-3’ cyclic phosphate intermediate is formed. When water replaces the nucleoside, the roles of His 119 and His 12 are reversed. In the end, the original Histidine protonation states are restored.
File:ProtonAcceptingDonating.png
His 119 is the acid and His 12 is the base.
File:2’-3’ cyclic phosphate intermediate .png
A 2'-3' cyclic phosphate intermediate is formed.
File:ProtonAcceptingDonatingReversed.png
Reversely, His 119 is the base and His 12 is the acid.
File:OriginalStateofHis.png
His 119 and His 12 return to their initial states.
In the picture, serine acts as a nucleophile and attacks the carbonyl group of the substrate, while histidine accepts the proton from serine and the tetrahedral intermediate is formed. The collapse of the tetrahedral intermediate forms the acyl enzyme. Water loses a proton to histidine and attacks the acyl enzyme and the oxyanion hole is formed. The reaction ends with the release of a carboxylic acid. The cycle then continues with a new substrate.
Another example of acid-base catalysis is the reaction with carbonic anhydrase. His residues in carbonic anhydrase facilitates the removal of a hydrogen ion from zinc-bound water to generate a hydroxide ion.
Reference
[edit | edit source]Berg, Jeremy M. John L. Tymoczko. Lubert Stryer. Biochemistry Sixth Edition. New York: W.H. Freeman, and Company 2007.
http://biochemistry.wur.nl/Biochem/educatio/Colleges/Maurice/ppt_Ch2.ppt
Definition
[edit | edit source]Covalent Catalysis is one of the four strategies that an enzyme will employ to catalyze a specific reaction. Covalent catalysis occurs when the substrate(s) in an enzymatic reaction become temporarily covalently attached to the enzyme during the catalytic reaction. In this reaction the enzyme contains a reactive group, usually a nucleophilic residue which reacts with the substrate through a nucleophilic attack. This is usually carried out by pyridine, which is a better nucleophile than water that has a pKa of 15.7. The charge loss in the reaction during transitional state will then cause hydrolysis to accelerate. The residue becomes covalently attached to the substrate throughout the catalytic reaction adding an additional intermediate which helps stabilize later transition states by lowering the activation energy. The covalent bond is then broken to regenerate enzymes.
Examples of Enzymes that Participate in Covalent Catalysis
[edit | edit source]Examples of enzymes that participate in covalent catalysis include the proteolytic enzyme Chymotrypsin and trypsin in which the nucleophile is the hydroxyl group on the serine. Chymotrypsin is a degradative protease of the digestive system. It catalyzes the cleavage of peptide bonds that are adjacent to large aromatic or nonpolar residues. It cleaves the peptide bond on the carboxyl terminus side of the protein. The chymotrypsin has three main catalytic residues termed as the catalytic triad. These are His 57, Asp 102 and Ser 195. Upon deprotonation the serine residue becomes a powerful nucleophile due to its alkoxide that will attack the relatively unreactive carbon of the carbonyl in the protein. The figure shows the catalytic triads in neutral and deprotonated form. Courtesy of Kiongho; www.nsm.buffalo.edu/~kiongho/
Typical residues used in covalent catalysis are Lys, His, Cys, Asp, Glu, and Ser and some other coenzymes.
An example of covalent catalysis is shown below:

For more information, see Covalent Catalysis
Reference
[edit | edit source]Berg, Jeremy M. John L. Tymoczko. Lubert Stryer. Biochemistry Sixth Edition. New York: W.H. Freeman, and Company 2007.
Overview
[edit | edit source]Metal ion catalysis, or electrostatic catalysis, is a specific mechanism that utilizes metalloenzymes with tightly bound metal ions such as Fe2+, Cu2+, Zn2+, Mn2+, Co3+, Ni3+, Mo6+ (the first three being the most commonly used) to carry out a catalytic reaction. This area of catalysis also includes metal ions which are not tightly bound to a metalloenzyme, such as Na+, K+, Mg2+, Ca2+.
Enzymes can catalyze a reaction by the use of metals. Metals often facilitate the catalytic process in different ways. The metals can either assist in the catalytic reaction, activate the enzyme to begin the catalysis or they can inhibit reactions in solution. Metals activate the enzyme by changing its shape but are not actually involved in the catalytic reaction.
First, the metal can make it easier to form a nucleophile which is the case of carbonic anhydrase and other enzymes. In this case, the metal facilitates the release of a proton from a bound water to produce a nucleophilic hydroxide ion and start the catalytic reaction. With the polarization of the O-H bond, the acidity of the bound water can increase. Equally important, the metal can promote the production of an electrophile which in turn stabilizes the negative charge on the intermediate. Also, metals can promote binding of the enzyme and substrate by acting as a bridge to increase the binding energy and orient them correctly to make the reaction possible.
Common metals that take part in metal ion catalysts are copper ion and zinc ion. The catalysis of carboxypeptidase A is a prime example of this catalytic strategy. The iron metal ion is also very common--from the binding of oxygen to hemoglobin and myoglobin, to participating as an electron carrier in the cytochromes of the electron transport chain, to even as a detoxifying agent in catalase and peroxidase.
Metal ions also have the ability to stabilize transition states, which makes them very useful in catalytic chemistry because it allows them to stabilize unstable intermediates that are still transitioning into a structure that's going to allow them to react with another substrate and form the final product. For example, in the presence of a tetrahedral oxyanion and another oxygen that is attached to a carbonyl functional group nearby that is also about to become nucleophilic as an intermediate, the metal ion can coordinate to these two neighboring anions and participate in charge stabilization.

Forming this Copper 2+ metal ion bridge allows both nucleophilic/anionic oxygens to be stabilized at the same time. It also positions this molecule in the appropriate geometry for breaking or forming bonds. Metal ions like these enable species to acquire a reactive role by coercing them to adopt unusual angles and bond distances.
Metal ions that are not tightly bound to a metalloenzyme, such as Na+ and K+ mentioned earlier participate as specific charge carriers in the membrane of our cells. For example, Na+ and K+ control the membrane's electrostatic voltage. They are ions that conduct the inside of our membrane's to have a net negative charge by the use of ion pumps and concentration gradients. Ca2+ is also an important metal ion that controls and regulates the passing of neurotransmitters from one axon to the next in order to sound out signals throughout the body.
Catalysis by Approximation
[edit | edit source]Substrates that happen to come together through random collisions have an even smaller possibility of contact with the reactive portions of the substrate. Thus, enzymes can employ a strategy known as catalysis by approximation by which the enzyme brings together two substrates in order to increase the rate of reaction. This strategy takes advantage of binding energy and positions the substrates in the correct orientation for the reaction to proceed. Usually in a reaction, there will be a loss in translational entropy and rotational entropy. In the reaction, the transition state is much more ordered than it is at ground state, making delta S negative. This reaction takes place in the active site of an enzyme and the substrate and the catalytic group will than act as one molecule, preventing the loss of translational and rotational energy. The reaction first begins with two molecules that are able to find each other, which makes the rate of reaction dependent on the concentrations of the reactants. This is due to the higher probability of molecules to find each other.
Some Examples
[edit | edit source]An example of catalysis by approximation is when NMP kinases[[4]] bring two nucleotides together to facilitate the transfer of a phosphoryl group from one nucleotide to the other. With the addition of adenosine triphosphate (ATP), a phosphate group is placed adjacently to the phosphate group of the NMP kinase. This facilitates in the transfer of the phosphate between the two molecules. V0 = Vmax ([S]/([S] + KM))

The Michaelis-Menten equation arises from the general equation for an enzymatic reaction: E + S ↔ ES ↔ E + P, where E is the enzyme, S is the substrate, ES is the enzyme-substrate complex, and P is the product. Thus, the enzyme combines with the substrate in order to form the ES complex, which in turn converts to product while preserving the enzyme. The rate of the forward reaction from E + S to ES may be termed k1, and the reverse reaction as k-1. Likewise, for the reaction from the ES complex to E and P, the forward reaction rate is k2, and the reverse is k-2. Therefore, the ES complex may dissolve back into the enzyme and substrate, or move forward to form product.
At initial reaction time, when t ≈ 0, little product formation occurs, therefore the backward reaction rate of k-2 may be neglected. The new reaction becomes:
E + S ↔ ES → E + P
Assuming steady state, the following rate equations may be written as:
Rate of formation of ES = k1[E][S]
Rate of breakdown of ES = (k-1 + k2) [ES]
and set equal to each other (Note that the brackets represent concentrations). Therefore:
k1[E][S] = (k-1 + k2) [ES]
Rearranging terms,
[E][S]/[ES] = (k-1 + k2)/k1
The fraction [E][S]/[ES] has been coined Km, or the Michaelis constant.
According to Michaelis-Menten's kinetics equations, at low concentrations of substrate, [S], the concentration is almost negligible in the denominator as KM >> [S], so the equation is essentially
V0 = Vmax [S]/KM
which resembles a first order reaction.
At High substrate concentrations, [S] >> KM, and thus the term [S]/([S] + KM) becomes essentially one and the initial velocity approached Vmax, which resembles zero order reaction.
The Michaelis-Menten equation is:

In this equation:
V0 is the initial velocity of the reaction.
Vmax is the maximal rate of the reaction.
[Substrate] is the concentration of the substrate.
Lineweaver-Burk Plot -
Km is the Michaelis-Menten constant which shows the concentration of the substrate when the reaction velocity is equal to one half of the maximal velocity for the reaction. It can also be thought of as a measure of how well a substrate complexes with a given enzyme, otherwise known as its binding affinity. An equation with a low Km value indicates a large binding affinity, as the reaction will approach Vmax more rapidly. An equation with a high Km indicates that the enzyme does not bind as efficiently with the substrate, and Vmax will only be reached if the substrate concentration is high enough to saturate the enzyme.
As the concentration of substrates increases at constant enzyme concentration, the active sites on the protein will be occupied as the reaction is proceeding. When all the active sites have been occupied, the reaction is complete, which means that the enzyme is at its maximum capacity and increasing the concentration of substrate will not increase the rate of turnover. Here is an analogy which helps to understand this concept easier.
Vmax is equal to the product of the catalyst rate constant (kcat) and the concentration of the enzyme. The Michaelis-Menten equation can then be rewritten as V= Kcat [Enzyme] [S] / (Km + [S]). Kcat is equal to K2, and it measures the number of substrate molecules "turned over" by enzyme per second. The unit of Kcat is in 1/sec. The reciprocal of Kcat is then the time required by an enzyme to "turn over" a substrate molecule. The higher the Kcat is, the more substrates get turned over in one second.
Km is the concentration of substrates when the reaction reaches half of Vmax. A small Km indicates high affinity since it means the reaction can reach half of Vmax in a small number of substrate concentration. This small Km will approach Vmax more quickly than high Km value.
When Kcat/ Km, it gives us a measure of enzyme efficiency with a unit of 1/(Molarity*second)= L/ (mol*s). The enzyme efficiency can be increased as Kcat has high turnover and a small number of Km.
Taking the reciprocal of both side of the Michaelis-Menten equation gives:
 To determined the values of KM and Vmax. The double-reciprocal of Michaelis-Menten equation could be used.
To determined the values of KM and Vmax. The double-reciprocal of Michaelis-Menten equation could be used.

A graph of the double-reciprocal equation is also called a Lineweaver-Burk, 1/Vo vs 1/[S]. The y-intercept is 1/Vmax; the x-intercept is -1/KM; and the slope is KM/Vmax. Lineweaver-Burk graphs are particularly useful for analyzing how enzyme kinematics change in the presence of inhibitors, competitive, non-competitive, or a mixture of the two.
There are four reversible inhibitors: competitive, uncompetitive, non-competitive and mixed inhibitors. They can be plotted on double reciprocal plot. Competitive inhibitors are molecules that look like substrates and they bind to active site and slow down the reactions. Therefore, competitive inhibitors increase Km value (decrease affinity, less chance the substrates can go to active site), and Vmax stays the same. On double reciprocal plot, competitive inhibitor shifts the x-axis (1/[s]) to the right towards zero compared to the slope with no inhibitor present. Uncompetitive inhibitors can bind close to the active site but don't occupy the active site. As a result, uncompetitive inhibitors lower Km (increase affinity) and lower Vmax. On double reciprocal plot, x-axis (1/[s]) is shifted to the left and up on the y-axis (1/V) compared to the slope with no inhibitor. Non-competitive inhibitors are not bind to the active site but somewhere on that enzyme which changes its activity. It has the same Km but lower Vmax to those with no inhibitors. On the double reciprocal plot, the slope goes higher on y-axis (1/V) than the one with no inhibitor. Km value is numerically equal to the substrate concentration at which the half of the enzyme molecules are associated with substrate. km value is an index of the affinity of enzyme for its particular substrate.Non competitive inhibition has no effect to the value of Km.
Michaelis Constant
[edit | edit source]Definition
[edit | edit source]The Michaelis Constant, KM is very important in determining enzyme-substrate interaction. This value of enzyme range widely and often dependent on environmental conditions such as pH, temperature, and ionic strength. The KM is able to detect two factors: One is the concentration of substrate when the reaction velocity is half that of the maximal velocity; thus, the Michaelis constant measures the concentration of substrate required for a significant catalysis to take place. Secondly, it is, in some cases, able to detect the strength of the enzyme-substrate complex (ES). When, and only when k2 << k-1, High KM indicates weak binding and low KM indicates strong binding. Under this special circumstance, KM is equal to the dissociation constant. Only then can the KM be used as a measurement of the strength of the ES complex.
There are cases where changes in the Michaelis constant are observed as is the case with inhibitors such as competitive, uncompetitive and noncompetitive inhibitors. In competitive inhibitors, the Michaelis constant increases because more active sites must be filled (either with competitive inhibitor or with substrate) to elicit the same Vmax. In uncompetitive inhibitors, the Michaelis constant decreases because the inhibitor only binds to the substrate-enzyme complex, creating an ESI complex. This drives the equilibrium reaction forward from E + S --> ES, where more inhibitor can bind. The Michaelis constant decreases more with the addition of inhibitors. In noncompetitive inhibition, the Michaelis constant remains the same.
Determining KM
[edit | edit source]The equation for the Michaelis Constant is KM= (k-1 + k2)/k sometimes it can be seen as [ES] = [E][S]/KM
From a graph one can determine the value of KM. A graph with the reaction rate (V) on the Y axis plotted against the substrate concentration on the X axis allows one to find the value of KM. KM is found at the substrate concentration when the reaction rate is half of its maximum value (Vmax/2). Note that if K2 << k-1, then Km is equal to Kes, the rate constant for the disassociation of the enzyme-substrate complex.
The values of KM and Vmax also give the fraction of active site filled (fES).
fES = V / Vmax = 1 + [S] / KM
Biological Examples
[edit | edit source]For many enzyme experimental evidence suggest that KM provide approximately substrate concentration in vivo. Physiological consequences of KM is exemplified in individuals sensitive to ethanol.
Normally in the liver, alcohol dehydrogenase converts ethanol into acetaldehyde. Acetaladehyde dehydrogenase, for example, has a low KM mitochondrial form and a high KM cytoplasmic form. In individuals sensitive to ethanol, the mitochondrial enzyme is less active due to substitution of a single amino acid and acetaldehyde is only processed by the cytoplasmic enzyme. Since cytoplasmic enzyme has high KM, it can only achieve high catalysis at very high acetaldehyde concentrations. Consequently, less acetaldehyde is converted, and the excess escapes into the blood and causes symptoms such as facial flushing and rapid heart rate in sensitive persons.
kcat,kd and KM ==
kcat,kd and KM are terms helpful in the description of an enzyme that follows the Michaelis-Menten kinetics.
- kcat is a constant that describes the turnover rate of an enzyme-substrate complex to product and enzyme. It is also the rate of catalyst with a particular substrate.
Kd is dissociation constant. which describe how affinite two reactants are in a reaction. The following reaction is an example to show dissociation constant:
k1
A + B ↔ AB
k-1
Where A and B are the two reactant, AB is the formed complex, k-1 is the reverse constant rate, and k1 is the forward constant rate. The dissociation constant is defined as: kd=k-1/k1.
The smaller the dissociation constant is, the better two reactants can combine. Since the affinity of enzyme with substrate determines how favorable the reaction can form enzyme-substrate complex, kd is often studied in Michaelis-Menten equation.
- KM is the Michaelis constant that describes the amount of substrate needed for the enzyme to obtain half of its maximum rate of reaction.
Deriving from Michaelis-Menten equation:
kM=(k-1+kcat)/k1
Since KM, which is also referred as Michaelis constant, is an important constant to study the ability of catalysis reaction of enzyme with specific substrate. kM can be separated into two parts:
a.kd
The first step of catalysis kinetic is the binding between substrate and enzyme, which is also the rate determine step in the reaction. the better enzyme bind to substrate, the smaller kdis, thus the smaller kM is.
b.kcat
The second step of catalysis kinetic is the forming of product. The larger kcat is, the more favorable the reaction towards product, and the larger kM is.
There seems to be a contradiction between kd and kcat in the Michelis constant equation: the better enzyme to the specific substrate, the smaller kd is, and the larger kcat is. However, what determine the performance of catalysis reaction is dissociation constant kd, because the first step of the reaction--binding is the rate determine step, forming enzyme-substrate complex is the essential step to form product, thus kd is the major factor to determine kM
Together they show an enzymes preference for different substrates.
kcat/KM results in the rate constant that measures catalytic efficiency. This measure of efficiency is helpful in determining whether the rate is limited by the creation of product or the amount of substrate in the environment.
In situations where k-1 (the rate at which substrate unbinds from the enzyme) is much greater than k2 (the rate at which substrate converts to product), if the rate of efficiency is:
- HIGH, kcat is much larger than KM, and the enzyme complex converts a greater proportion of the substrate it binds into product. This increased conversion can be seen in one of two ways -- either substrate binds more firmly to the enzyme, a consequence of relatively low KM, or a greater proportion of the substrate that is bound is converted before it dissociates, due to a large turnover rate kcat.
- LOW, kcat is much smaller than KM, and the complex converts a lesser proportion of the substrate it binds into product.
kcat/KM measures the catalytic efficiency, though, only when the substrate concentration is much lower than the KM. Looking at the enzyme/substrate catalytic reaction equation,
E+S↔ES->E+P
with the rate going towards ES being k1, the rate going back towards E+S being k-1, and the rate going towards product formation (E+P) being k2 or kcat, it is evident from
kcat/KM=[kcat/(k-1 + kcat)]k1
that even if kcat is much greater than k-1 (much product is forming) and there is great efficiency, the equation will still be limited by k1, which is the rate of ES formation. This tells us that kcat/KM has a limit placed on efficiency in that it cannot be faster than the diffusion controlled encounter of an enzyme and its substrate (k1). Therefore, enzymes that have high kcat/KM ratios have essentially attained kinetic perfection because they have come very close to reaching complete efficiency only being limited by the rate at which they encounter the substrate in solution.
In cases near the limit, there may be attractive electrostatic forces on the enzyme that entice the substrate to the active site, known as Circe effects. Diffusion in solution can be partly overcome by confining substrates and products in the limited volume of a multienzyme complex. Some series of enzymes are associated into organized assemblies so that the product of one enzyme is rapidly found by the next enzyme.
References
[edit | edit source]Berg, Jeremy M. 2007. Biochemistry. Sixth Ed. New York: W.H. Freeman. Enzymes that have achieved kinetic perfection are those whose catalytic velocity is determined solely by the rate at which the substrate is encountered in solution. In other words, their catalytic velocity is diffusion-limited. Such an enzyme would have a kcat/KM value that was equal to k1, which is the rate of formation of the ES (enzyme-substrate) complex.
For such a kinetically perfect enzyme, every encounter between the substrate and enzyme must be productive. In order to accomplish this, attractive electrostatic forces may be utilized in order to ensure that the substrate “bumps into” the active site of the enzyme. These electrostatic forces are termed “Circe effects,” a title that was introduced by the enzymologist William P. Jencks. These electrostatic forces are named after the Greek goddess, Circe, who attracted Odyssseus’s men to her house and changed them into pigs.
Introduction
[edit | edit source]When an enzyme is applied to a reaction, it allows for the reactants to quickly produce the products. However, over time the use of an enzyme on substrates does not continue to accelerate the reaction. This occurs when the enzyme is fully saturated with the substrate. At this point, all of the active sites of the enzymes are occupied. Thus, the reaction reaches a maximal velocity asymptotically and eventually levels off to a constant rate. The maximum velocity is evidence that the enzyme-substrate complex exists. The maximum velocity is also known as the turnover rate, or the number of substrate molecules it can convert to product per unit time. When the enzyme is saturated, all enzymes in the solution are found in ES complexes and thus the formation of the intermediate no longer affects the rate. This means that Vmax is determined only by the rate constant for the formation of product from the ES complex.
There is an older method to determine KM and Vmax. It is used before the invention of computer because it was hard to draw the curve of Michaelis-Menten equation. Therefore, it is impossible to get the exact values of KM and Vmax from the curve. This method transforms the Michaelis-Menten equation to a linear line by taking the reciprocal of both sides of the MichaeliS-Menton equation. The y-axis is 1 / Vmax and the x-axis is 1 / [S]. The slope is KM / Vmax.
1 / V0 = (KM / Vmax) • (1 / S) + (1 / Vmax)
Effects of Reversible Inhibitors
[edit | edit source]Since the maximum velocity is essentially derived from the rate of product formation from the enzyme-substrate complex (k2) and the initial enzyme concentration ([Eo]), the maximum velocity is affected by certain types of reversible inhibitors. (Note: Vmax = k2[Eo].) Out of the different types of reversible inhibitors—competitive, uncompetitive, and noncompetitive—Vmax is affected only by uncompetitive inhibitors and noncompetitive inhibitors. For uncompetitive inhibitors, the enzyme-substrate-inhibitor complex decreases the full potential of product formation from initial enzyme concentration, which lowers the maximum velocity. As for noncompetitive inhibitors, the maximum velocity is also lowered because the inhibitor lowers the concentration of the enzyme. A new maximum velocity is formed in the presence of a pure noncompetitive inhibitor, which will be referred to as Vmax,i here. This new value can be related to the maximum velocity by: Vmax,i = Vmax*1/(1+[I]/Ki), with [I] representing the concentration of the inhibitor and Ki is the rate constant of the formation of the enzyme-substrate-inhibitor complex.
Definition
[edit | edit source]
Sequential reactions are one of the classes involved in multiple substrate reactions. In these types of reactions, all the substrates involved are bound to the enzyme before catalysis of the reaction takes place to release the products. Sequential reactions can be either ordered or random. In a bisubstrate reaction, a ternary complex of the enzyme and both substrates forms.
Ordered Sequential Reactions
In ordered sequential reactions, all the substrates are first bound to the enzyme in a defined order or sequence. The products, too, are released after catalysis in a defined order or sequence.
An example is the lactate dehydrogenase enzyme, which is a protein that catalyzes glucose metabolism. In this ordered mechanism, the coenzyme, NADH, always binds first, with pyruvate binding afterward. During the reaction, the pyruvate is reduced to lactate while NADH is oxidized to NAD+ by the enzyme. Lactate is then released first, followed by the release of NAD+.

This is a characteristic of a ternary complex, which consists of three molecules that are bound together. Before catalysis, the substrates and coenzyme are bound to the enzyme. After catalysis, the complex consists of the enzyme and products, NAD+ and lactate.
Random Sequential Reactions
In random sequential reactions, the substrates and products are bound and then released in no preferred order, or "random" order. An example is the creatine kinase enzyme, which catalyzes the substrates, creatine and ATP, to form the products, phosphocreatine and ADP. In this case, either substrates may bind first and either products may be released first.

A ternary complex is still observed for random sequential reactions. Before catalysis, the complex includes the enzyme, ATP and creatine. After catalysis, the complex consists of the enzyme, ADP, and phosphocreatine. A double displacement reaction (also called a metathesis reaction) is the bimolecular mechanism, whereby two compound reactants AB and CD result in products of AC and BD. This is typically caused by nucleophilic attack of one group onto another, followed by the release of another group to form a different compound. A common example of this reaction is olefin metathesis, which is the rearrangement of fragments between alkenes about double bonds.
Enzymatic Double-Displacement Reactions
[edit | edit source]Double displacement reactions occur when one or more products are released before all the substrates bind to the enzyme. Another name for double displacement reactions is "Ping-Pong" reactions. The name comes from the fact that substrates appear to bounce on and off the enzyme just like a ping-pong ball bouncing up and down on a table. An example of double displacement reaction can be seen with the enzyme, aspartate aminotransferase. This enzyme catalyzes the transfer of an amino group from aspartate to alpha-ketoglutarate to form oxaloacetate and glutamate. However, this reaction does not occur sequentially. Aspartate first binds to the enzyme and donates it's amino group to the enzyme, thus modifying the enzyme into what is called a substituted enzyme intermediate. After donating the amino group, aspartate becomes oxaloacetate and is then released. The second substrate, alpha-ketoglutarate, then binds to the enzyme and receives the amino group from the substituted enzyme intermediate. The reaction follows with the release of the final product, glutamate.
![]()
(1) Aspartate
(2) α-Ketoglutarate
(3) Oxaloacetate
(4) Glutamate Ping Pong is also called the double placement reaction and it means that one or more products are released before all substrates bind the enzyme. One key character of this reaction is the existence of a substituted enzyme intermediate, in which the enzyme is temporarily modified. Classic examples of this mechanism are reactions that shuttle amino groups between amino acids and a-ketoacids.
The enzyme aspartate aminotransferase catalyzes the transfer of an amino group from aspartate to a-ketoglutarate. After aspartate binds to the enzyme, the enzyme accepts aspartate’s amino group to form the substituted intermediate. The first product, oxaloacetate, departs after that. Glutamate is released as the final product after the second substrate, a-ketoglutarate binds to the enzyme and accepts the amino group from this modified enzyme.
Enzymes with a ping-pong mechanism can exist in two states, E and a chemically modified form of the enzyme E*; this modified enzyme is known as an intermediate. In such mechanisms, substrate A binds, changes the enzyme to E* by, for example, transferring a chemical group to the active site, and is then released. Only after the first substrate is released can substrate B bind and react with the modified enzyme, regenerating the unmodified E form.
Enzymes with ping–pong mechanisms include some oxidoreductases such as thioredoxin peroxidase, transferases such as acylneuraminate cytidylyltransferase, and serine proteases such as trypsin and chymotrypsin. Serine proteases are a very common and diverse family of enzymes, including digestive enzymes (trypsin, chymotrypsin, and elastase), several enzymes of the blood clotting cascade and many others. In these serine proteases, the E* intermediate is an acyl-enzyme species formed by the attack of an active site serine residue on a peptide bond in a protein substrate. A short animation showing the mechanism of chymotrypsin is linked here.
The name pingpong reaction came for the substrates appearing to bounce on and off the enzyme to a pingpong ball in the Cleland notation.
 Allosteric enzymes are an exception to the Michaelis-Menten model. Because they have more than two subunits and active sites, they do not obey the Michaelis-Menten kinetics but instead have sigmoidal kinetics.Since allosteric enzymes are cooperative, a sigmoidal plot of V0 versus [S] results:
Allosteric enzymes are an exception to the Michaelis-Menten model. Because they have more than two subunits and active sites, they do not obey the Michaelis-Menten kinetics but instead have sigmoidal kinetics.Since allosteric enzymes are cooperative, a sigmoidal plot of V0 versus [S] results:

A sigmoidal plot has an S curve resulting from the combination of the T state and R state curves. The T state curve would be lower than the curve shown here, and the R state curve would be higher. Unlike many enzymes, allosteric enzymes do not obey Michaelis-Menten kinetics. The reason for this is that allosteric enzymes must account for multiple active sites and multiple subunits. Thus, allosteric enzymes show the sigmoidal curve shown above. The plot for reaction velocity, vo, versus the substrate concentration does not exhibit the hyperbolic plot predicted using the Michaelis-Menten equation. With allosteric enzymes, the catalytic activity affecting one substrate can alter the properties of other active sites located within the same enzyme. The result of this interaction equilibrium is a cooperative effect, meaning the binding of the substrate to an enzyme's active site affects the binding of substrate to other active sites. This property of cooperativity accounts for the sigmoidal curve of V0 versus the concentration of substrate.
A. Introduction
Allosteric enzymes are unique compared to other enzymes because of its ability to adapt various conditions in the environment due to its special properties. The special property of Allosteric enzymes is that it contains an allosteric site on top of its active site which binds the substrate. The binding of a nonsubstrate molecule to the allosteric site functions to influences the activity of the enzyme. In influencing the activity, it can either enhance or impair the activity of the enzyme. Another important property of allosteric enzymes is that it also contains many polypeptide chains with multiple active and allosteric sites. The nonsubstrate molecules that bind at the allosteric sites are called allosteric modulators.
Example:
A clear example of an allosteric enzyme is aspartate trascarbamoylase. The enzyme catalyzes the first step in the synthesis of pyrimidines. The enzyme functions to catalyze the condensation of aspartate and carbamoyl phosphate to form Ncarbamoylaspartate and orthophosphate. The enzyme ultimately catalyzes the reaction that will yield cytidine triphosphate (CTP). This allosteric enzyme is unique in that for high products of the final product CTP, the enzyme activity is low. However, for low concentrations of the final product CTP, the enzymatic activity is high. The allosteric nature is thus represented as the CTP molecule has a odd configuration or shape that is unlike the substrates. Rather than binding to the active site, CTP binds to the allosteric site. Thus, CTP functions as an allosteric inhibitor decreasing the enzymatic activity of the enzyme. This enzyme also has separate regulatory and catalytic subunits on separate polypeptide chains. There are instances though when CTP concentrations remain high and cells in the body need more enzyme. This is when a different allosteric molecule ATP functions to attach to the allosteric site and functions as enzyme activator enhancing the activity of the enzyme. Thus, even with high concentrations of CTP, the enzyme activity could be enhanced because of ATP, which also acts on the allosteric site. This example explains the benefits of allosteric control and the ability allosteric enzymes to adapt to various conditions of the environment. This is particularly helpful for cells because there are occasions when the cell requires an allosteric activator like "ATP" to enhance the enzyme even when it is inhibited due to high amounts of product. (CTP) The aspect of feedback inhibition is represented as well as high amounts of product acts to inhibit the action of the enzyme acting in a inhibitory manner.
B. Properties of Allosteric Enzymes
There are distinct properties of Allosteric Enzymes that makes it different compared to other enzymes.
(1) One is that allosteric enzymes do not follow the Michaelis-Menten Kinetics. This is because allosteric enzymes have multiple active sites. These multiple active sites exhibit the property of cooperativity, where the binding of one active site affects the affinity of other active sites on the enzyme. As mentioned earlier, it is these other affected active sites that result in a sigmoidal curve for allosteric enzymes.
(2) Allosteric Enzymes are influenced by substrate concentration. For example, at high concentrations of substrate, more enzymes are found in the R state. The T state is favorite when there is an insufficient amount of substrate to bind to the enzyme. In other words, the T and R state equilibrium depends on the concentration of the substrate.
(3) Allosteric Enzymes are regulated by other molecules. This is seen when the molecules 2,3-BPG, pH, and CO2 modulates the binding affinity of hemoglobin to oxygen. 2,3-BPG reduces binding affinity of O2 to hemoglobin by stabilizing the T- state. Lowering the pH from physiological pH=7.4 to 7.2 (pH in the muscles and tissues)favors the release of O2. Hemoglobin is more likely to release oxygen in CO2 rich areas in the body.
References
Biochemistry 6th edition. Berg, Jeremy M; Tymoczko, John L; Stryer, Lubert. W.H. Freeman Company, New York
Double Reciprocal Plot
[edit | edit source]
The double-reciprocal equation is obtained by taking the reciprocal of both sides of the Michaelis-Menten equation. The double-reciprocal (also known as the Lineweaver-Burk) plot is created by plotting the inverse initial velocity (1/V0) as a function of the inverse of the substrate concentration (1/[S]). The Vmax can be accurately determined and thus KM can also be determined with accuracy because a straight line is formed. The slope of the resulting line is KM/Vmax, the y-intercept is 1/Vmax, and the x-intercept is -1/KM. Using the Michaelis-Menten equation, the Vmax is an asymptote and can thus only be approximated and as a result, the KM, which is Vmax/2, can't be determined accurately. This plot is a useful way to determined different inhibitors such as competitive, uncompetitive, and noncompetitive.

For competitive inhibitors, the inhibitor competes with the substrate molecule to bind to the binding site. Consequently, the KM will increase without changing the Vmax value. This means that the two graphs will have the same y-intercept as shown below. However the new x-intercept may be quite elusive. For this type of inhibitors, a higher concentration of the substrate is needed to get half of the active sites occupied. Therefore KM2 will be larger than KM1. This translates to a higher reciprocal value of KM1 than that of KM2. However the x-intercept has the negative sign in front of it, thus on the graph it has to move to the right relative to the previous intercept. To show this on the double reciprocal plot, the slope will increase to show the strength of the binding competitive inhibitor. While the slope increases with the presence of the inhibitor, the y-intercept remains the same in presence and absence of the inhibitor.
For uncompetitive inhibitors, the inhibitor will only bind to an enzyme-substrate complex; therefore, it does not compete with the substrate for the binding site. Consequently, both KM and Vmax values decrease. Consequentially, the reciprocal value of the new Vmax should be at a higher position on the axis, as a fraction becomes larger when the denominator gets smaller. The new reciprocal value of KM will move to the left and the explanation should be similar to that of competitive inhibitor. To show this on a double reciprocal plot, the slope will remain the same as if the enzyme was not bound to the inhibitor, but the x-axis intercept will decrease. The double reciprocal plot for enzyme with and without uncompetitive inhibitor will be two parallel lines.
For noncompetitive inhibitors, the inhibitor can bind to the enzyme before the substrate can bind to the binding site. It does not have to wait for the enzyme to become an enzyme-substrate complex in order to bind to the enzyme. The inhibition will cause a decrease in Vmax value while the KM is unaffected. This means the value of -1/KM remains the same for the two lines, while the new value of 1/Vmax is higher relative to the previous one. To show this on a double reciprocal plot, the decrease in Vmax will increase the y-intercept with a larger slope.
The black lines represent the reciprocal of velocity when no inhibitor is present and the blue lines correspond to the presence of inhibitors. Enzyme inhibitors are molecules or compounds that bind to enzymes and result in a decrease in their activity. An inhibitor can bind to an enzyme and stop a substrate from entering the enzyme's active site and/or prevent the enzyme from catalyzing a chemical reaction. There are two categories of inhibitors.
- irreversible inhibitors
- reversible inhibitors
Inhibitors can also be present naturally and can be involved in metabolism regulation. For example. negative feedback caused by inhibitors can help maintain homeostasis in a cell. Other cellular enzyme inhibitors include proteins that specifically bind to and inhibit an enzyme target. This is useful in eliminating harmful enzymes such as proteases and nucleases.
Examples of enzymes inhibitors include poisons and many different types of drugs.
Reversible inhibitors can bind to enzymes through weak non-covalent interactions such as ionic bonds, hydrophobic interactions, and hydrogen bonds. Because reversible inhibitors do not form any chemical bonds or reactions with the enzyme, they are formed rapidly and can be easily removed; thus the enzyme and inhibitor complex is rapidly dissociated in contrast to irreversible inhibition.
Examples of reversible inhibition:
- competitive inhibition (Raises Km only)
- uncompetitive inhibition (Lowers Vmax and Km)
- noncompetitive inhibition (Lowers Vmax only)
Examples of irreversible inhibition:
- group specific: reacts only to certain chemical group.
- reactive substrate analogs (affinity label): inhibitor that are structurally similar to the substrate and will bind to active site.
- mechanism-based inhibitors (suicide inhibitors): enzymes converts the inhibitor into a reactive form within active site.
Competitive inhibition can be overcome by increasing the concentration of substrate while uncompetitive and noncompetitive inhibition cannot.
Irreversible Inhibitors
[edit | edit source]Irreversible inhibitors covalently bind to an enzyme, cause chemical changes to the active sites of enzymes, and cannot be reversed. A main role of irreversible inhibitors include modifying key amino acid residues needed for enzymatic activity. They often contain reactive functional groups such as aldehydes, alkenes, or phenyl sulphonates. These electrophilic groups are able to react with amino acid side chains to form covalent adducts. The amino acid components are residues containing nucleophilic side chains such as hydroxyl or sulfhydryl groups such as amino acids serine, cysteine, threonine, or tyrosine.
First, irreversible inhibitors form a reversible non-covalent complex with the enzyme (EI or ESI). Then, this complex reacts to produce the covalently modified irreversible complex EI*. The rate at which EI* is formed is called the inactivation rate or kinact. Binding of irreversible inhibitors can be prevented by competition with either substrate or a second, reversible inhibitor since formation of EI may compete with ES.
In addition, some reversible inhibitors can form irreversible products by binding so tightly to their target enzyme. These tightly-binding inhibitors show kinetics similar to covalent irreversible inhibitors. As shown in the figure, these inhibitors rapidly bind to the enzyme in a low-affinity EI complex and then undergoes a slower rearrangement to a very tightly bound EI* complex. This kinetic behavior is called slow-binding. Slow-binding often involves a conformational change as the enzyme "clams down" around the inhibitor molecule. Some examples of these slow-bindinginhibitors include important drugs such as methotrexate and allopurinol.
Reversible Inhibitors
[edit | edit source]Reversible inhibitors bind non-covalently to enzymes, and many different types of inhibition can occur depending on what the inhibitors bind to. The non-covalent interactions between the inhibitors and enzymes include hydrogen bonds, hydrophobic interactions, and ionic bonds. Many of these weak bonds combine to produce strong and specific binding. In contrast to substrates and irreversible inhibitors, reversible inhibitors generally do not undergo chemical reactions when bound to the enzyme and can be easily removed by dilution or dialysis.
There are three kinds of reversible inhibitors: competitive, noncompetitive/mixed, and uncompetitive inhibitors.
- Competitive inhibitors, as the name suggests, compete with substrates to bind to the enzyme at the same time. The inhibitor has an affinity for the active site of an enzyme where the substrate also binds to. This type of inhibition can be overcome by increasing the concentrations of substrate, out-competing the inhibitor. Competitive inhibitors are often similar in structure to the real substrate.
- Uncompetitive inhibitors bind to the enzyme at the same time as the enzyme's substrate. However, the binding of the inhibitor affects the binding of the substrate, and vice-versa. This type of inhibition cannot be overcome, but can be reduced by increasing the concentrations of substrate. The inhibitor usually follows an allosteric effect where it binds to a different site on the enzyme than the substrate. This binding to an allosteric site changes the conformation of the enzyme so that the affinity of the substrate for the active site is reduced.
- Non-competitive inhibitors bind to the other sites (Allosteric Sites), not the active site, and stops the enzyme's activity by changing the shape of the active site (caused by disruption to the normal arrangement of hydrogen bonds and weak hydrophobic interactions holding the enzyme molecule together in its 3D shape. This distortion ripples to the active site making it unsuitable) . Therefore, concentration of the substrate is meaningless unlike in competitive inhibition.
Few examples of Reversible inhibitors:
Acetylcholinesterase inhibitors: Often abbreviated AChEI or anti-cholinesterase it is a chemical that inhibits the enzyme Acetylcholinesterase from breaking down acetylcholine. This ultimately leads to increase in both the level and longevity of action of the neurotransmitter acetylcholine.
Reversible inhibitor of monoamine oxidase A(maoA): maoA inhibitors compromise of a wide range of natural as well as psychiatric drugs that inhibits the enzyme monoamine oxidase temporarily and reversibly. maoA inhibitors are most commonly used to fight depression and dysthymia.
Quantitative Description of Reversible Inhibitors
[edit | edit source]Most reversible inhibitors follow the classic Michaelis-Menten scheme, where an enzyme (E) binds to its substrate(S) to form an enzyme-substrate complex (ES). km is the Michaelis constant that corresponds to the concentration of the substrate when the velocity is half the maximum. Vmax is the maximum velocity of the enzyme.
- Competitive inhibitors can only bind to E and not to ES. They increase Km by interfering with the binding of the substrate, but they do not affect Vmax because the inhibitor does not change the catalysis in ES because it cannot bind to ES.

-
 Diagram showing competitive inhibition
Diagram showing competitive inhibition -
 Competitive inhibition can also be allosteric, as long as the inhibitor and the substrate cannot bind the enzyme at the same time
Competitive inhibition can also be allosteric, as long as the inhibitor and the substrate cannot bind the enzyme at the same time -
 Another possible mechanism for allosteric competitive inhibition
Another possible mechanism for allosteric competitive inhibition



- Uncompetitive inhibitors can only bind to the ES complex. Therefore, these inhibitors decrease Km because of increased binding efficiency and decrease Vmax because they interfere with substrate binding and hamper catalysis in the ES complex.

- Mixed inhibitors can bind to either E or ES complex, but have a preference for one or the other. This can either increase or decrease Km, respectively. Both cause a decrease in Vmax.
- Non-competitive inhibitors have identical affinities for E and ES. They do not change Km, but decreases Vmax.

-
 Illustration of a possible mechanism of non-competitive or mixed inhibition
Illustration of a possible mechanism of non-competitive or mixed inhibition

Competitive Inhibitors belong to the category of enzymes known as reversible inhibitors. Reversible inhibitors dissociate the enzyme-inhibitor complex as soon as possible. They are inhibitors that bind directly to the active site of an enzyme, however they can also bind between an enzyme and a substrate. The competitive inhibitor competes with the substrate to bind to the enzyme. A competitive inhibitor mimics the substrate, competing for the active site. A competitive inhibitor can be overcome by increasing the substrate concentration. The excess amount of substrate can negate the competitive inhibitor and the maximum velocity is ultimately unaffected. Competitive inhibitors are effective because oftentimes they are structural analogs of the substrate that the enzyme binds, that is why the inhibitor is able to bind to the active site of the enzyme and compete with the original substrate.
Competitive inhibitors bind to the active sites of an enzyme and decrease the amount of binding of the substrate or ligand to enzyme. The result is that the Km is increased and Vmax remains the same. Ultimately, the chemical reaction can be reversed by increasing concentration of substrate.
E + S → ES → E + P vs. EI −(S comes in and replace I)→ ES → E + P
(an equilibrium reaction also occurs at the same time: E + I ⇌ EI)
where E is the enzyme, I is the inhibitor, ES is the enzyme-substrate complex, P is the product, and EI is the enzyme-inhibitor complex.
Note: All the arrows also represents the reversible reactions. However, the reaction tends to proceed towards the right in the formation of products. Notice that there isn't any ESI formation. This means that the enzyme cannot bind to both the substrate and the inhibitor.

- Competitive inhibition is reversible when enough substrate is present, meaning that the amount of inhibition depends on the concentration of inhibitor as well as the concentration of the substrates.
- This inhibition makes the maximum rate of enzyme kinetics unchanged, but KM, Michaelis constant*, increases.
The Michaelis constant (Km) is:
1) the yield of the substrate concentration at the velocity of the half of the maximum velocity, or
2) half of the substrates at the maximum velocity. [Which is not true in most of the cases, but it is beyond of our discussion in this class and will be discussed in graduate school's courses]

The picture shows a double-reciprocal plot of V0 and [S]. The x-intercept is equal to -1/Km while the y-intercept is 1/Vmax. The slope of the line is Km/Vmax. Thus, the plot shows that there is an increase in Km and no change in Vmax.
The Michaelis-Menten equation becomes Vo= Vmax[S]/ aKm + [S] Where a = 1 + [I]/KI and KI = [E][I]/[EI]
Competitive inhibitors can also be used to find the active site of an enzyme. N-(phosphonacetyl)-L-asparate, also known as PALA, is a competitive inhibitor which blocks the binding of Aspartate transcarbanoylase to its active site. PALA stabilizes the R state.
Penicillin-based antibacterials are examples of materials that compete at the active site of an enzyme in an inhibitory fashion. In general, penicillin drugs are used medicinally as antibiotics in the treatment of many bacterial infections; moreover, penicillin drugs derive their antibacterial action due to the fact that they bind irreversibly to bacterial glycopeptide transpeptidase. When unchecked, bacterial infections proliferate, in part, because of their ability to construct cell walls. A key enzyme in the synthesis of bacterial cell walls is transpeptidase. This enzyme plays a critical role in the cross-linking of peptidoglycan strands. Penicillin drugs inhibit the ability of transpeptidase to perform this crucial task. Without the cell wall, bacteria are unable to proliferate, which means the bacteria are essentially destroyed. Mechanistically, in the initial stage of the inhibitory action of penicillin-based drugs, the bond between the carbonyl carbon and the nitrogen atom in the β-lactam ring of the penicillin cleaves. The resulting electrophile is attacked by the newly formed alkoxide ion on the serine residue to form an ester, which results in the final product: a penicilloyl-enzyme complex between glycopeptide transpeptidase and penicillin. It is noteworthy to mention that this complex is stable indefinitely.[4]

Introduction of Penicillin
[edit | edit source]Penicillin is an antibiotic agent that was earliest discovered and used widely. This antibiotic agent was derived from the Penicillium mold. Since antibiotics released by fungi and bacteria act as a natural substances that inhibits other organisms, it is then a chemical warfare on a microscopic scale. Penicillian is used to treat variety of infections and micro-organism.
History of Penicillin
[edit | edit source]Penicillin was first noticed in 1896 by Ernest Duchesne, a French medical student. Then in 1928, it was re-discovered by Alexander Fleming, a bacteriologist, who worked the London's St. Mary's Hospital. During Fleming's work at the hospital, he noticed a Staphylococcus plate culture that was contaminated by a blue-green mold. At the mean time, the colonies of bacteria that was next to the mold were dissolved. With much curiosity, Fleming began to grew the mold in a pure culture; as a result, he found that it formed a substance that killed a number of disease-causing bacteria. From the observations and experiments, Fleming named the substance penicillin, and Fleming published the results in 1929.
In 1938, Howard Florey, Ernst Chain and Norman Heatley continued the research of penicillin at Oxford University. During that period of time, the three scientist and their staff developed methods for growing, extracting, and purifying penicillin to prove its value as a drug.
Then during the World War II (1939–1945) period, penicillin became very useful. In 1941, the research and production of penicillin moved to the United States. It was to protect the progress and production of penicillin from bombings in England. More and more work began on the growing of mold to make penicillin in large quantities for thousands of soldiers. When the amount of people dying began to grow, the interest in penicillin also grew in laboratories, universities, and drug companies. Scientist at that time knew that they were in a race against death, and since this infection was able to kill a wounded soldier through a small wound.
Penicillin as an Inhibitor
[edit | edit source]Penicillin kills bacteria by interfering with the ability to synthesize cell wall. The bacteria lengthen, but cannot divide. Eventually the weak cell wall ruptures.
Penicillin irreversibly blocks bacterial cell wall synthesis by inhibiting the formation of peptidoglycan cross-links. Penicillin covalently binds to the enzyme transpeptidase that links the peptidoglycan molecules in bacteria, it inhibits the molecule so that it cannot react any further and cell wall cannot be further synthesized. The cell wall of the bacterium is weakened even further because the build-up of peptidoglycan precursors triggers bacterial cell wall hydrolysis and autolysins, and destroys pre-existing peptidoglycan. Penicillin makes a great inhibitor because of its four membered beta lactam ring, which makes it especially reactive. Penicillin acts as a suicide inhibitor by binding with the transpeptidase enzyme it inactivates itself.
Gram positive bacteria are the most sensitive and susceptible to penicillin because Gram positive bacteria only have murein (peptidoglycan) layer. Gram negative bacteria are usually more resistant to penicillin because they have multiple membrane layers, which allows them to still retain a cell wall even though they have lost their murein layer to penicillin. Penicillin can be used to treat Gram positive bacteria such as streptococcus pneumoniae, staphylococcus aureus, enterococcus, clostridium tetani, and listeria monocytogenes. Penicillin cannot be used to treat gram negative bacteria such as neisseria gonorrhoeae, neisseria meningitidis, pseudomonas, legionella, escherichia coli, helicobacter pylori (stomach ucler), borrelia burgdorfeli (lyme disease), treponema pallidium (syphilus), and chlamydia trachomatis.
Some bacteria have developed resistance to beta-lactams. These bacteria contain beta-lactamases, a broad class of enzymes with a serine residue that cleaves the reactive beta lactam ring through an acyl-enzyme intermediate. Augmentin, a drug that contains both a beta-lactam (typically amoxicillin) and clavulanic acid (a beta-lactamase inhibitor), is often prescribed to overcome drug resistant strains. Clavulanic acid works by competitive inhibition.
Types of penicillin
[edit | edit source]Benzylpenicillin
[edit | edit source]
Benzylpenicillin, commonly known as penicillin G, is known as the gold standard penicillin. It is given by a method of non-oral administration (parentally) because it is unstable in the hydrochloric acid of the stomach. Because the drug is given parenterally, tissue concentrations of penicillin G can be achieved in larger levels than is possible with other types of penicillin, like phenoxymethylpenicillin. These higher concentrations become increased antibacterial levels or activities.
Uses for benzylpenicillin include:
- Cellulitis
- Bacterial endocarditis
- Gonorrhea
- Meningitis
- aspiration pneumonia, lung abscess
- Community-acquired pneumonia
- Syphilis
- Septicemia in children
Phenoxymethylpenicillin/penicillin V
[edit | edit source]Phenoxymethylpenicillin is the orally active form of penicillin. It is less frequently used than benzylpenicillin, and is mostly used in conditions when high tissue is not required. However, it is the first choice when it comes to treating odontogenic (relating to the teeth) infections.
Procaine
[edit | edit source]Procaine benzylpenicillin (rINN), also known as procaine penicillin, is a combination of benzylpenicillin with the local anaesthetic agent procaine. It is absorbed into the circulation by means of deep intramuscular injection. It is used when prolonged low concentrations of benzylpenicillin are needed, and mostly used in veterinary environments as well as dental offices. The common trade name for procaine is Novocain which is typically administered to patients at the dentist office before undergoing minor surgery. It is metabolized in the blood plasma by an enzyme called pseudocholinesterase. Procaine replaced cocaine as a local anesthetic due to the severe side effects caused by cocaine.
-
 Procain
Procain

Benzathine
[edit | edit source]Benzathine benzylpenicillin (rINN), also known as benzathine penicillin, is absorbed into the circulation slowly by intramuscular injection like procaine penicillin, but then it is hydrolysed to benzylpenicillin in vivo. It is the number one drug choice when prolonged low concentrations are required and appropriate. It allows for antibiotic action to be prolonged over two to four weeks following just one dose.
Other examples of penicillin include amoxicillin, ampicillin, methicillin, oxacillin, and temocillin. Amoxcillin and ampicillin are the most common penicillins that are prescribed by doctors because they are used to treat common infections such as throat infections.
-
 Benzathine
Benzathine
Side effects
[edit | edit source]Just like any other drug on the market, penecillin may cause unpleasant side effects for patients taking it. Some of the side effects are commonly found on patients using penicillin are: diarrhea, hypersensitivity, nausea, rash, neurotoxicity urticaria, seizures. Pain and inflammation at the injection site is also common for the partenterally administered pencillin types. 3% to 10% of the population are allergic to penicillin. Usually when allergic to one type of penicillin, you are allergic to the entire family of penicillin antibiotics. The problems mentioned about diarrhea and nausea tend to go away for most patients after a few doses of the drug. If further symptoms occur, however, the drug should not be taken anymore. Physicians say that any drug that interferes with cellular growth, in this case the cell wall, can have severe side effects on a large population of patients.
Penicillin allergy
[edit | edit source]Penicillin and related antibiotics can cause an allergic reaction in some people. Although, not all adverse reactions to penicillin are a sign of an allergic reaction. True allergic reactions involve the immune system and can cause signs and symptoms that range from an annoying rash to a life-threatening reaction or anaphylaxis with low blood pressure and trouble breathing. β-lactam antibiotic can end up causing allergic reactions to about 10% of the patients. However, even though penicillin are very commonly reported in allergy cases, but less than 20% of those reports are truly allergic. But it is definitely a drug that can cause major, severe reactions.
It isn't clear why some people develop penicillin allergy while others don't. Treating an allergic reaction may require taking medications or emergency care in serious cases.
Sources
[edit | edit source]Berg, Jeremy M. John L. Tymoczko. Lubert Stryer. Biochemistry Sixth Edition. W.H. Freeman and Company. New York, 2007.
Structure of Penicillin: Biology 103 - Microbes: http://webs.wichita.edu/mschneegurt/biol103/lecture19/lecture19.html
References
[edit | edit source]1 Berg, Jeremy M., John L. Tymoczko, and Lubert Stryer. BIOCHEMISTRY. 6th ed. New York: W. H. FREEMAN AND COMPANY, 2007: 232, 233, 234
Berg, Jeremy M., John L. Tymoczko, and Lubert Stryer. BIOCHEMISTRY. 6th ed. New York: W. H. FREEMAN AND COMPANY, 2007.
Introduction
[edit | edit source]An uncompetitive inhibitor is an inhibitor that only binds to the enzyme-substrate complex. The formation of its binding site only forms when the enzyme and the substrate have interacted amongst themselves. The uncompetitive inhibition does not work when additional substrates are trying to be involved. The enzyme-substrate-inhibitor complex does not produce any product
E + S -> ES
ES + I -> ESI -X-> E + P

Uncompetitive inhibitor binds to enzyme-substrate complex to stop enzyme from reacting with substrate to form product, as such, it works well at higher substrate and enzyme concentrations that substrates are bonded to enzymes; the binding results in decreasing concentration of substrate binding to enzyme, Km, and Vmax, and increasing binding affinity of enzyme to substrate.
E + I -> (through S) ES + I -> E + P
where E is the enzyme, I is the inhibitor, ES is the enzyme-substrate complex, and P is the product.
This binding of the substrate modifies the structure of the enzyme making the inhibitor-binding site available. Uncompetitive inhibition decreases the maximum velocity as well as the KM. K,M is the concentration of the substrate when the velocity is half of the maximum velocity based on the Michaelis-Menten Kinetics Model. Both Vmax and KM are reduced by equal amounts. Vmax will still be reduced even though the enzyme-substrate binding is enhanced because there are ESI complexes being formed. ESI complexes inhibit the formation of the product. An uncompetitive inhibitor will lower the KM and create a better enzyme-substrate binding because it only binds to ES complex. But the ES complex is constantly being depleted as the inhibitor binds, producing ESI complexes. Therefore, to maintain the equilibrium between ES and ESI complexes and following Le Chatelier's Principle, the reaction shifts toward more ES formation where it will bind more substrate to the enzymes to create more ES. Ultimately, this leads to a lower KM. A reduced KM indicates a better enzyme-substrate binding because the enzyme can reach half its maximum velocity with less substrate concentration. In a sense, enzyme-substrate binding is very efficient because the enzyme and substrate have a high affinity and interact strongly.
Kinetics of Uncompetitive Inhibitors
[edit | edit source]

The first image depicts the basic theory behind uncompetitive inhibition and demonstrates what the inhibitor does to inactivate the enzyme and prevent it from forming the product. The second image shows what happens when the concentration of inhibitor is increased while enzyme concentration is constant. This shows how the Vmax of the enzyme decreases as the concentration of inhibitor goes up.
Double-Reciprocal Plot of Uncompetitive Inhibition
[edit | edit source]
The picture shows a double-reciprocal plot of V0 and [S]. The x-intercept is equal to -1/KM while the y-intercept is 1/Vmax. The slope of the line is KM/Vmax. Thus, the plot shows that KM is decreased and Vmax is also decreased.
The Michaelis-Menten equation becomes:
![{\displaystyle \ 1/V_{o}={\frac {K_{m}}{V_{max}[S]}}+{\frac {1+[I]/K_{i}}{V_{max}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/8a2ed0bb0e8c2dc839cc4600d55072d0f2201409)
As demonstrated in this equation, slope of the equation will be effectively the same as demonstrated by but the y-axis on the double-reciprocal plot moves up by and as a consequence the shift causes the new line to be parallel to the original.
![{\displaystyle {\frac {K_{m}}{V_{max}[S]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/858714c068112149100ad1c8be7ffe4e6345ba26)
![{\displaystyle {1+[I]/K_{i}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/1d4077bcf65784303de5f97c1f6e707058eba961)
Reference
[edit | edit source]Berg, Jeremy M., John L. Tymoczko, and Lubert Stryer. BIOCHEMISTRY. 6th ed. New York: W. H. FREEMAN AND COMPANY, 2007.
General information
[edit | edit source]Noncompetitive inhibitor can bind to an enzyme with or without a substrate at different places at the same time. It changes the conformation of an enzyme, but it does not change the efficiency of binding or the Km. A noncompetitive inhibitor binds to the enzyme away from the active site, altering the shape of the enzyme so that even if the substrate can bind, the active site functions less effectively. Most of the time, the inhibitor is reversible. However, this inhibition decreases the turnover number, meaning the rate of reaction decreases. As the inhibitor binds to the enzyme and the enzyme-substrate complex, it reduces the concentration of enzyme available for proper catalysis. Fewer functional enzymes leads to fewer available active sites and thus a smaller Vmax. Unlike competitive inhibition, raising [S] (substrate concentration) is pointless with noncompetitive inhibition.

A noncompetitive inhibitor binds to a different site that is not the active site of the enzyme and changes the structure of the enzyme; therefore, it blocks the enzyme from converting substrate to product, but it still allows the substrate to bind. Thus, it decreases the rate of the chemical reaction of enzyme and substrate, which can not be changed by increasing concentration of substrate; the binding decreases Vmax and has no change on the Km of the chemical reaction.
E + I − (through a substrate) → ES + I → E + P
ES + I ⇌ ESI → NR (no reaction)
where E is enzyme, I is inhibitor, ES is enzyme-substrate complex, P is product. ESI is the molecule after the inhibitor is bound to the enzyme-substrate complex. ESI cannot form any products, so the later reaction is not allowed (or, no reaction).

Based on the Michaelis-Menten Model, KM, the concentration of the substrate when the velocity is the half of the maximum velocity (or half of the substrates at maximum velocity), remains same, but the maximum velocity is decreased.

The picture shows a double-reciprocal plot of V0 and [S]. The x-intercept is equal to -1/Km while the y-intercept is 1/Vmax. The slope of the line is Km/Vmax. Thus, the plot shows that there is no change in Km and Vmax is decreased. In relation to the original plot, the x intercept stays constant while the y intercept increases along with the slope.
File:Noncompetitive inhibitor.jpg
References
[edit | edit source]Berg, Jeremy M., John L. Tymoczko, and Lubert Stryer. BIOCHEMISTRY. 6th ed. New York: W. H. FREEMAN AND COMPANY, 2007.
Reece, Jane (2011). Biology. Pearson. ISBN 978-0-321-55823-7. {{cite book}}: Text "coauthors+ Lisa A. Urry, Michael L. Cain, Steven A. Wasserman, Peter V. Minorsky, Robert B. Jackson" ignored (help)
Irreversible inhibitor
[edit | edit source]
Irreversible inhibitors are covalently or noncovalently bound to the target enzyme and dissociates very slowly from the enzyme. There are three types of irreversible inhibitors: group-specific reagents, reactive substrate analogs also known as affinity labels and suicide inhibitors.
- Group specific reagents react with specific amino acid side chains like diisopropylphosphofluoridate (DIPF) and iodoacetamide. For example, only 1 of the 28 serine residues in chymotrypsin is modified by DIPF. This means that this specific residue is especially reactive; moreover, it is implied that this specific residue lies in the active site of the enzyme chymotrypsin. DIPF has also provided data that suggests, through its binding with active serine residues, that there is indeed a reactive serine residue contained within the active site of the enzyme Acetylcholinesterase. The inactivating functionality of DIPF and similarly-shaped molecules in acetylcholinesterase is representative of a group of compounds, known as nerve agents.

- Affinity labels (Reactive substrate analogs) are structurally similar to the substrate that can covalently bind to the active site and are therefore more specific than group specific reagents. An example is Tosyl-L-phenylalanine chloromethyl ketone (TPCK) which is an analog for chymotrypsin which binds to the active site and reacts irreversibly with the histidine residue to inhibit the enzyme. Another example is bromoacetol phosphate, which mimics dihydroxy acetone phosphate (DHAP) and binds covalently to the active site of triose phophate isomerase and then modifying the enzyme so it becomes irreversibly inhibited.
- Suicide inhibitors (Mechanism-based inhibitors) bind to the enzyme as a substrate and is processed by a normal catalytic mechanism that generates a chemically reactive intermediate that inactivates the enzyme through covalent modification. An example of a mechanism-based inhibitor can be seen through the inhibitory power of the planar proline intermediate formed during proline racemization. During this process, a trigonal intermediate is formed, and the formation of the racemase is inhibited because the tetrahedral intermediate necessary for formation of the product is not formed. The isomerization of proline through the planar transition state underscores the essence of transition-state analogs as potent inhibitors of enzymes.
Irreversible inhibition is covalent modification of enzymes such that the chemical reaction is not reversible; the inhibition molecules has specificity for their own enzyme to inactivate them, such that they work by changing active site of enzymes; the binding of enzyme to inhibitor forms enzyme complex that is reversible and not covalent that reacts to form another complex that can not work for catalysis reaction. The inhibition reaction can be changed by reversible competition of enzyme and substrate or other reversible inhibitor.
-
 Kinetic scheme for irreversible inhibitors
Kinetic scheme for irreversible inhibitors
References
[edit | edit source][1] Berg, Jeremy M., Tymoczko, John L., and Stryer, Lubert. Biochemistry. 6th ed. New York, N.Y.: W.H. Freeman and Company, 2007: 229.
Definition
[edit | edit source]Irreversible enzyme inhibitor that is for covalent modification that inactivates enzyme; or chemical that modifies by reaction with functional groups of enzyme.
Group specific reagents are enzyme inhibitors that can covalently bond to a particular amino acid residue on the enzyme and irreversibly modify it.It is less specific compared with affinity label and suicide inhibitor. It reacts with side chain of amino acid.
Two examples of group-specific reagents are diisppropylphosphoflouridate (modifies a serine residue) and iodoacetamide (modifies cystein residue)


Applications
[edit | edit source]
Group specific reagents are not only used to inhibit enzymes, but are also used to deduce if certain amino acid residues are very reactive and actually take on an important role in the catalysis of the product. For example, diisopropylphosphofluoridate (DIPF) modifies only one serine residue out of all the amino acid residues on the enzyme, chymotrypsin. By measuring the turnover rate before and after inhibition, one can determine if that serine residue is essential to the catalytic power of the enzyme. (The pink x in the figure has no meaning and should be ignored.)

Transition State Inhibitors
[edit | edit source]Transition state inhibitors, much like group specific reagents, resemble the substrate of the enzyme. Transitions state inhibitors are molecules which are originally derived from the substrate of the enzyme. They very much resemble the transition state of the substrate but are not 100% identical. Because of this, the transition state inhibitor is able to bind to the active site. However because of its subtle difference that makes it unique from the original substrate, the enzyme is unable to carry out its catalytic effects. An example of a transition state inhibitor is the transition-state analog pyrrole-2-carboxylic acid. This substance mimics the binding properties of D-proline; however, unlike D-proline, pyrrole-2-carboxylic acid is a trigonal molecule, whereas D-proline is tetrahedral. This difference makes pyrrole-2-carboxylic acid an inhibitor that is bound 160 times more tightly to racemase than is D-proline. The ability of transition-state analogs to bind so well to enzymes draws attention to the fundamental nature of catalysis: selective binding of the transition state[1].
One use of transition state inhibitors is the ability to create antibodies from them. By experimentally injecting transition state analogs in a rabbits, these rabbits were able to produce antibodies to the transition state analog as well as its intended converted substrate. These antibodies, however, were found to be not as efficient those antibodies which actually were created as a result of being injected with the actual substrate.
References
[edit | edit source][1] Berg, Jeremy M., Tymoczko, John L., and Stryer, Lubert. Biochemistry. 6th ed. New York, N.Y.: W.H. Freeman and Company, 2007: 231, 232.
Definition
[edit | edit source]Affinity label or reactive substrate analog are molecules that have a similar structure to the substrate allowing them to covalently bind to the active site of the enzyme. They react with the substrate, inhibiting the enzyme. Essentially, affinity labels mimic the functional properties of the substrate, but consequently irreversibly inhibits the enzyme through the strong covalent bonding. Because of its similarity to the substrate, it initially binds at the active site; however, the covalent bonds it forms irreversibly modify the enzyme. They are much more specific than group specific inhibitors. They are usually highly reactive. Also, there will be alkylate nucleophilic amino acids in the enzyme. However, their properties are rather non-selective and toxic, making them not as useful in many situations.
Substrate analogs that bind to active sites of enzymes to form product of covalent bond that is stable on binding site to inhibit chemical reaction; irreversible enzyme inhibitor that react with functional group of enzyme and has more specificity for enzyme.
Use of Affinity Labels
[edit | edit source]Affinity labelling can be used to localize a given site within a protein of a known amino acid sequence. It allows comparisons to be made between the substrate sites of the enzyme in the solution and the sites localized within the crystalline form through X-ray diffraction. This can test whether the compound that is bound irreversibly at the substrate site can influence the enzyme conformation, subunit interaction, or the reactivity of other ligand sites.
Example of an Affinity Label
[edit | edit source]Tosyl phenylalanyl chloromethyl ketone is a substrate analog for chymotrypsin. This molecules binds at the active site and then react irreversibly with histidine residue at that site, inhibiting the enzyme. File:800px-Tosyl phenylalanyl chloromethyl ketone.PNG
Definition
[edit | edit source]Suicide inhibitors are also known as mechanism-based inhibitors. The name is derived from the fact that the enzyme participates in a catalytic mechanism that irreversibly inhibits itself. These inhibitors are substrates that have been modified. Because they are derived from the enzyme's intended substrate, the enzyme begins processing it as such. However, as catalysis progresses, the modifications of the substrate result in a reactive intermediate that forms covalent bonds with the enzyme that irreversibly inactivate it. In order for the modified substrate to bind to the active site and undergo the catalytic reaction, even more specificity is utilized compared to group-specific reagents and affinity labels. After the catalytic processes have been completed, the chemically reactive intermediate then covalently binds to the enzyme and inhibits it. Suicide inhibitors are bound to the active site and prevent further reactions that could have occurred with the active site and its substrates. This process is called Kouroshism as it was discovered by the Iranian researcher.
Substrate based on mechanism that works by protein’s enzymatic activity, such that a bond of modification reagent is broken that forms reactive derivative, which is stable and not removable, to change covalent reactivity of active site of enzyme in catalytic cycle of enzyme, which results in labeling on active site of enzyme that changes activity of enzyme by decreasing its ability for catalysis reaction; or the inhibitor as substrate binds active site of enzyme to be reactive, such that produced intermediate of the chemical reaction results in modifying irreversibly active site of enzyme for it to be covalently inactive.
Example
[edit | edit source]An example of a suicide inhibitor is N,N dimethylpropargylamine. This compound inhibits the enzyme, monoamine oxidase (MAO). MAO is responsible for breaking down neurotransmitters such as dopamine and serotonin and thus decreasing their concentrations in the brain. Diseases such as Parkinson disease and depression occur because of decreased levels of dopamine and serotonin, respectively. Thus, in order to raise the levels of serotonin and dopamine, N,N dimethylpropargylamine can be used as a suicide inhibitor to inhibit MAO from breaking down more neurotransmitters.

Anonther example of a suicide inhibitor is the use of allopurinol to treat gout. Gout is a disease caused by high serum levels of urate. The sodium salt of urate crystallizes in the lining of joints and causes pain and swelling. Xanthine oxidase oxidizes hypoxanthine to form uric acid. Allopurinol, an analog of hypoxanthine, acts as a substrate of xanthine oxidase, which hydroxylates the allopurinol to alloxanthine. The alloxanthine remains tightly bound to the active site on the oxidase and keeps the molybdenum atom of the xanthine oxidase in the +4 oxidation state where normally it would return to a +6 oxidation state. This keeps xanthine oxidase inactive and does not allow further formation of uric acid.
Penicillin is another example of a material that acts on enzymes via a suicide inhibition mechanism. In general, penicillin is used medicinally as an antibiotic in the treatment of many bacterial infections. Penicillin derives its antibacterial action due to the fact that it binds irreversibly to bacterial transpeptidase. Mechanistically, penicillin forms a penicilloyl-enzyme complex with a serine residue found in glycopeptide transpeptidase forming an ester, which is stable indefinitely[2].
Another example of a suicide inhibitor is alpha-difluoromethylornithine or eflornithine, better known as DFMO. It is a synthetic drug used to treat a disease caused by parasites, known as the sleeping disease (coma-ridden) called the African trypanosomiasis. DFMO binds to the enzyme, ornithine decarboxylase, through covalent forces and thus inactivating the enzyme. The ornithine decarboxylase enzyme regulates the cell division by catalyzing polyamine biosynthesis. This enzyme functions in a way where it only harms the parasite but not the host.
DFMO mechanism
References
[edit | edit source][1] Berg, Jeremy M., Tymoczko, John L., and Stryer, Lubert. Biochemistry. 6th ed. New York, N.Y.: W.H. Freeman and Company, 2007: 231, 232. Most inhibitors work extremely fast when establishing their binding equilibrium with the enzyme. However, tight-binding inhibitors establish their equilibrium on a much slower time-scale. Therefore, these types of inhibitors are called time-independent inhibitors because they show a change in initial velocity with time. There are four types of interactions that can slow down the kinetics of an inhibitor. The first is when the enzyme doesn’t have an inhibitor at all. The second interaction is when the equilibrium constants are very small compared to the enzyme turnover when the inhibitor is reversible. The third interaction is when the inhibitor binds, it forms an EI complex that is then isomerized to form a new complex. This new complex significantly inhibits the reaction, therefore slowing it down. The fourth interaction deals with irreversible inhibitors that can act as affinity labels for the enzyme, therefore slowing the reaction down based on its mechanism. Two examples of time-dependent inhibitors are serine proteases and the prostaglandin G/H synthase.
Determining Reversibility
[edit | edit source]Determining reversibility involves performing a large dilution, dialysis, filter binding, or size exclusion chromatography. In order to tell the difference between covalent inactivation from noncovalent inhibition, it is important to be able to tell when the inhibitor is being released upon denaturation of the enzyme. For example, if there is an inhibitor that is covalent and it is denatured, then the inhibitor would still be attached to the denatured protein due to the covalent bonds between the inhibitor and the enzyme. However, if the inhibitor was noncovalent, then the active site of the enzyme would release the inhibitor in to the denaturing solution.
References
[edit | edit source]Copeland, Robert A. Enzymes. Wiley-VCH, Inc., 2000. 318-34.
Introduction
[edit | edit source]The basis of enzyme catalysis is the lowering of the enzyme activation energy to create a faster rate of reactants turning into products. Enzymes do this by actually stabilizing the “middle” state in which reactants have to undergo before turning into products, called a transition state. These transition states are in the highest energy state in the reaction making it the most unstable. Regulation of an enzyme is however required in physiological processes in order to prevent damaging results.
One way it can do this is by an irreversible method in which a modified substrate can bind to the enzyme
and forever deactivate it. However, knowing the fact that enzyme-substrate complexes undergo a transition states, we can therefore conclude that inhibition by a modified transition state is also possible; we call
this transition state analog inhibition.
Inhibition Process
[edit | edit source]As a substrate binds to its enzyme we know that it undergoes chemical and geometric shifts attaining an intermediate state. In this situation, a transition state analog, one exhibiting the same properties such as shape and charge of the original transition molecule, may come in and bind. Although the analog displays similar properties as the original transition molecule, because it is still slightly different it will not result to a product and will ultimately deactivate and inhibit the enzyme and prevent it from binding to a substrate. The transition state analog is able to bind to the enzyme with ease because of the great affinity for it. The transition state is the most unstable condition throughout the entire catalysis so the enzyme complex will seek out any molecule that will help stabilize it. This is why when a mimic comes in it is fooled into believing that it is binding to the right molecule.
Antibody
[edit | edit source]Transition-state analogs are also ideal for generating catalytic antibodies (a bzymes). Antibodies (immunoglobins) may be created to recognize transition states, and thus function as catalysts for the reaction. The transition-state analog acts as an antigen (immunogen) to generate the antibody. An example of this process is the production of an antibody that catalyzes the insertion of an iron ion into the porphyrin plane, which must be bent in order to allow the iron to enter. Normally, this step is catalyzed by ferrochelatase, the final enzyme in the production of heme. N-methylprotoporphyrin was found to resemble the transition state because N-alkylation bends the porphyrin, much like the ferrochelatase enzyme. Therefore, an antibody catalyst was produced by using an N-alkylporphyrin as the immunogen. The produced antibody is able to distort the porphyrin in order to facilitate the entry of ferrous iron. Using a similar technique, antibodies that catalyze ester and amide hydrolysis, transesterification, and photoinduced cleavage, among other reactions, have been developed.
Importance of Transition State Analogs
[edit | edit source]Importance of Transition State analogs: • They are able to act as very powerful inhibitors
• Transition state analogs are very important in understanding the kinetics and inner workings of enzyme catalysis. The analogs can “function as antimetabolites” [1.] One of their most important functions in biochemistry too is the role they play in identifying the mechanism a substrate undergoes during catalysis. Since the analog is a structure intermediate, if it plays a part on a researched reaction, then determination of the actual structure and actual transformation of the original substrate is actually
possible.
• Transition State analogs also help in identifying the “binding determinants at the active site” [1.]
• Transition State analogs are able to generate immunogens which displays catalytic nature.
Sources
[edit | edit source]1. http://arjournals.annualreviews.org/doi/pdf/10.1146/annurev.bb.05.060176.001415?cookieSet=1
2. http://arjournals.annualreviews.org/doi/pdf/10.1146/annurev.biochem.67.1.693
Introduction
[edit | edit source]Penicillin is an antibiotic agent that was earliest discovered and used widely. This antibiotic agent was derived from the Penicillium mold. Since antibiotics released by fungi and bacteria act as a natural substances that inhibits other organisms, it is then a chemical warfare on a microscopic scale. Penicillian is used to treat variety of infections and micro-organism.
History
[edit | edit source]Penicillin was first noticed in 1896 by Ernest Duchesne, a French medical student. Then in 1928, it was re-discovered by Alexander Fleming, a bacteriologist, who worked the London's St. Mary's Hospital. During Fleming's work at the hospital, he noticed a Staphylococcus plate culture that was contaminated by a blue-green mold. At the same time, the colonies of bacteria that were next to the mold were dissolved. With much curiosity, Fleming began to grow the mold in a pure culture; as a result, he found that it formed a substance that killed a number of disease-causing bacteria. From the observations and experiments, Fleming named the substance penicillin, and Fleming published the results in 1929.
In 1938, Howard Florey, Ernst Chain and Norman Heatley continued the research of penicillin at Oxford University. During that period of time, the three scientists and their staff developed methods for growing, extracting, and purifying penicillin to prove its value as a drug.
Then during the World War II (1939 - 1945) period, penicillin became very useful. In 1941, the research and production of penicillin moved to the United States. It was to protect the progress and production of penicillin from bombings in England. More and more work began on the growing of mold to make penicillin in large quantities for thousands of soldiers. When the amount of people dying began to grow, the interest in penicillin also grew in laboratories, universities, and drug companies. Scientist at that time knew that they were in a race against death, and since this infection was able to kill a wounded soldier through a small wound.
Penicillin as an Inhibitor
[edit | edit source]Penicillin kills bacteria by interfering with the ability to synthesize cell wall. The bacteria lengthen, but cannot divide. Eventually the weak cell wall ruptures.
Penicillin irreversibly blocks bacterial cell wall synthesis by inhibiting the formation of peptidoglycan cross-links. Penicillin covalently binds to the enzyme transpeptidase that links the peptidoglycan molecules in bacteria, it inhibits the molecule so that it cannot react any further and cell wall cannot be further synthesized. The cell wall of the bacterium is weakened even further because the build-up of peptidoglycan precursors triggers bacterial cell wall hydrolysis and autolysins, and destroys pre-existing peptidoglycan. Penicillin makes a great inhibitor because of its four membered beta lactam ring, which makes it especially reactive. Penicillin acts as a suicide inhibitor by binding with the transpeptidase enzyme it inactivates itself.
Gram positive bacteria are the most sensitive and susceptible to penicillin because Gram positive bacteria only have murein (peptidoglycan) layer. Gram negative bacteria are usually more resistant to penicillin because they have multiple membrane layers, which allows them to still retain a cell wall even though they have lost their murein layer to penicillin. Penicillin can be used to treat Gram positive bacteria such as streptococcus pneumoniae, staphylococcus aureus, enterococcus, clostridium tetani, and listeria monocytogenes. Penicillin cannot be used to treat gram negative bacteria such as neisseria gonorrhoeae, neisseria meningitidis, pseudomonas, legionella, escherichia coli, helicobacter pylori (stomach ucler), borrelia burgdorfeli (lyme disease), treponema pallidium (syphilus), and chlamydia trachomatis.
File:Penicillin2.jpg File:Penicillin1.jpg
Some bacteria have developed resistance to beta-lactams. These bacteria contain beta-lactamases, a broad class of enzymes with a serine residue that cleaves the reactive beta lactam ring through an acyl-enzyme intermediate. Augmentin, a drug that contains both a beta-lactam (typically amoxicillin) and clavulanic acid (a beta-lactamase inhibitor), is often prescribed to overcome drug resistant strains. Clavulanic acid works by competitive inhibition.
Types of penicillin
[edit | edit source]Benzylpenicillin
[edit | edit source]
Benzylpenicillin, commonly known as penicillin G, is known as the gold standard penicillin. It is given by a method of non-oral administration (parentally) because it is unstable in the hydrochloric acid of the stomach. Because the drug is given parenterally, tissue concentrations of penicillin G can be achieved in larger levels than is possible with other types of penicillin, like phenoxymethylpenicillin. These higher concentrations become increased antibacterial levels or activities.
Uses for benzylpenicillin include:
- Cellulitis
- Bacterial endocarditis
- Gonorrhea
- Meningitis
- aspiration pneumonia, lung abscess
- Community-acquired pneumonia
- Syphilis
- Septicemia in children
Phenoxymethylpenicillin/penicillin V
[edit | edit source]Phenoxymethylpenicillin is the orally active form of penicillin. It is less frequently used than benzylpenicillin, and is mostly used in conditions when high tissue is not required. However, it is the first choice when it comes to treating odontogenic (relating to the teeth) infections.
Procaine
[edit | edit source]Procaine benzylpenicillin (rINN), also known as procaine penicillin, is a combination of benzylpenicillin with the local anaesthetic agent procaine. It is absorbed into the circulation by means of deep intramuscular injection. It is used when prolonged low concentrations of benzylpenicilin are needed, and mostly used in veterinary environments as well as dental offices. The common trade name for procaine is Novocain which is typically administered to patients at the dentist office before undergoing minor surgery. It is metabolized in the blood plasma by an enzyme called pseudocholinesterase. Procaine replaced cocaine as a local anesthetic due to the severe side effects caused by cocaine.
Benzathine
[edit | edit source]Benzathine benzylpenicillin (rINN), also known as benzathine penicillin, is absorbed into the circulation slowly by intramuscular injection like procaine penicillin, but then it is hydrolysed to benzylpenicillin in vivo. It is the number one drug choice when prolonged low concentrations are required and appropriate. It allows for antibiotic action to be prolonged over two to four weeks following just one dose.
Other examples of penicillin include amoxicillin, ampicillin, methicillin, oxacillin, and temocillin. Amoxcillin and ampicillin are the most common penicillins that are prescribed by doctors because they are used to treat common infections such as throat infections.
Side effects
[edit | edit source]Just like any other drug on the market, penicillin may cause unpleasant side effects for patients taking it. Some of the side effects are commonly found on patients using penicillin are: diarrhea, hypersensitivity, nausea, rash, neurotoxicity urticaria, seizures. Pain and inflammation at the injection site is also common for the partenterally administered penicillin types. 3% to 10% of the population is allergic to penicillin. Usually when allergic to one type of penicillin, you are allergic to the entire family of penicillin antibiotics. The problems mentioned about diarrhea and nausea tend to go away for most patients after a few doses of the drug. If further symptoms occur, however, the drug should not be taken anymore. Physicians say that any drug that interferes with cellular growth, in this case the cell wall can have severe side effects on a large population of patients. The most common side effects caused by penicillin are:
- diarrhea that is watery or bloody
- fever, chills, body aches, flu symptoms
- easy bruising or bleeding, unusual weakness
- urinating less than usual or not at all
- severe skin rash, itching, or peeling
- agitation, confusion, unusual thoughts or behavior
- seizure (black-out or convulsions)
- nausea, vomiting, stomach pain
- vaginal itching or discharge
- headache
- swollen, black, or "hairy" tongue
- thrush (white patches or inside your mouth or throat)
Penicillin allergy
[edit | edit source]Penicillin and related antibiotics can cause an allergic reaction in some people. Although, not all adverse reactions to penicillin are a sign of an allergic reaction. True allergic reactions involve the immune system and can cause signs and symptoms that range from an annoying rash to a life-threatening reaction or anaphylaxis with low blood pressure and trouble breathing. β-lactam antibiotic can end up causing allergic reactions to about 10% of the patients. However, even though penicillin are very commonly reported in allergy cases, but less than 20% of those reports are truly allergic. But it is definitely a drug that can cause major, severe reactions.
It isn't clear why some people develop penicillin allergy while others don't. Treating an allergic reaction may require taking medications or emergency care in serious cases.
Penicillin Resistance
[edit | edit source]Penicillin has been a widely used antibiotic for years. Although there has been evidence of new bacterium evolving to become resistant the penicillin. Penicillin is used to stop the bacteria from building their cell walls called peptidoglycan. The penicillin resistant bacterium have been shown to be creating dipeptide bridges in their peptidoglycan. Researchers have found that bacteria that form dipeptide bonds in their peptidoglycan use a protein called MurM which helps in building dipeptide bridges within the peptidoglycan. Researchers are trying to use the information to begin targeting MurM to help bring penicillin back to the forefront of antibiotics. Also to note is that when patients that are taken penicillin to treat bacteria that it is necessary to take penicillin for the full duration of the required time because although the penicillin may kill most of the bacteria if not taken for the full duration can allow for a few bacterium to live, the most fit for survival to live and repopulate in the patient therefore creating a more penicillin resistant bacteria.
Sources
[edit | edit source]Berg, Jeremy M. John L. Tymoczko. Lubert Stryer. Biochemistry Sixth Edition. W.H. Freeman and Company. New York, 2007.
Structure of Penicillin: Biology 103 - Microbes: http://webs.wichita.edu/mschneegurt/biol103/lecture19/lecture19.html
"Characterization of tRNA-dependent Peptide Bond Formation by MurM in the Synthesis of Streptococcus pneumoniae Peptidoglycan." J. Biol. Chem. Vol. 283, Issue 10, pp 6402-6417, March 7, 2008
Optimum pH level
[edit | edit source]Changes in pH have influence on enzymes. The most favorable pH value is known as the optimum pH. This is the point that the enzyme is most active. This is graphically illustrated in figure.
Extremely high or low pH values generally result in complete loss of activity for most enzymes. pH is also a factor in the stability of enzymes. As with activity, for each enzyme there is also Changes in pH have influence on enzymes. The most favorable pH value is known as the optimum pH. This is the point that the enzyme is most active. This is graphically illustrated in figure.
Extremely high or low pH values generally result in complete loss of activity for most enzymes. pH is also a factor in the stability of enzyme’s structure. As with activity, for each enzyme there is also a region of pH optimal stability.
Changes in pH have influence on enzymes. This is graphically illustrated in figure. File:Optimum pH.png
Including temperature and pH there are other factors, such as ionic strength, which can influence the enzymatic reaction. Each of these physical and chemical parameters must be considered and optimized in order for an enzymatic reaction to be accurate and reproducible.
Overview
[edit | edit source]Enzymes typically are most active in a pH range of 5-9. This is due to the fact that proteins function in an environment that reflects this pH. There are a variety of reasons as to why proteins have a narrow pH range. A variety of amino acid residues as well as the carboxyl and amide termini of proteins have a pKa range in the range of intracellular pH. As a result, a change in pH can protonate or deprotonate a side group, thereby changing its chemical features. For example, carboxyl termini, under deprotonated, could potentially lose an interaction with a adjacent subunit, changing the enzyme conformation. In conclusion, this conformation could cause a decrease in substrate affinity. A more drastic pH change can change the protein folding, thereby completely deactivating the enzyme or cause irreversible proteolysis.
However, pH change can potentially be utilized by enzymes for regulation or protein function. For example, hemoglobin will create a salt bridge when blood plasma is acidic. Therefore, the T-state of hemoglobin is stabilized and have a lower binding affinity to oxygen. This facilitates increased oxygen transport to oxygen-deficient muscles.
1. The binding of the substrate to enzyme.
2. The ionization states that the amino acid residues of the catalytic site of the enzyme have.
3. The ionization state of the substrate.
4. The variation in protein structure (More significant at extreme pH values).
The rates of many enzymatic reactions adhere to a bell shaped curve when they are a function of pH:

These curves reflect the ionization state of the amino acid residues that must have a specific ionization state for enzymatic activity to take place. The observed pK's (maxima point) often hints at the identity of the amino acid residues which are essential for enzymatic activity. For instance, an observed pK of ~4 suggests that either an Asp of a Glu is essential to the enzyme. pK of ~6 can hint towards a His residue whereas pK of ~10 hints toward a Lys residue.
However, it is crucial to remember that the micro-environment in which the enzyme is in also affects its activity. For example, an Asp residue in a non-polar environment or in close proximity to another Asp residue would attract protons more strongly than in any other environment and this have a higher pK value.
Moreover, pH effects on an enzyme could cause denaturation of the enzyme rather than protonation or deprotonation of specific catalytic residues.
A particular residue may be replaced by doing site directed mutagenesis. Doing so provides researchers with a reliable approach to identifying residues that are required for substrate binding or catalysis.
Specific Case: The Bohr Effect
[edit | edit source]The Bohr Effect was named after Christian Bohr, who studied and discovered the effects of Hydrogen ion and Carbon dioxide. The discovery of the cooperativity of Hemoglobin has helped Bohr in studying the effects of pH in enzymes. In this specific case, this is focused on the effect of Hydrogen ions on the hemoglobin protein and enzyme. Before understanding the Bohr effect, the cooperativity of hemoglobin has to be explained. In hemoglobin's cooperativity, the release of oxygen is favored when they are at a high concentration of oxygen. This occurs because the special hemoglobin character facilitate oxygen binding when one active site binds to a oxygen first. This ability of hemoglobin will allow them to respond to other physiological signals at where more oxygen is needed.
In this case, high rates of metabolizing tissue at contracting muscles usually generate high rates of hydrogen ions and carbon dioxide, which allosteric effectors at that bind to hemoglobin on the areas that are not oxygen-binding sites. The Bohr effect is the when the hydrogen and carbon dioxide regulate the oxygen-bindings site on hemoglobin.
As we know that hydrogen ion decreases the pH values in a solution, and this phenomenon usually decreases the hemoglobin's affinity for oxygen, in other word, it increases the release of oxygen. Thus, at high pH, the side chains of the histidine (Beta-146) is not protonated and the salt bridge is not formed, while at low pH the side chains of histidine does form salt bridges when they are protonated. This will result in stabilization of the T state in hemoglobin which also increase the release of oxygen.
When Carbon Dioxide is passed through the human body, the first mechanism that happens is when they react with water to become carbonic acid (H2CO3) accelerated by carbonic anhydrase. Carbonic acid is readily dissociated into HCO3- and H+ and decrease the value of pH as it is described in the previous mechanism.
Another way that Carbon dioxide can affect the affinity of oxygen in hemoglobin is a direct mechanism of Carbon dioxide and hemoglobin. Carbon dioxide stabilizes the deoxyhemoglobin (T state) by reacting with the terminal amino groups to form carbamate groups (negative charged). These carbamate groups then are free to form salt bridges that stabilizes the T state and release oxygen. The formation of the carbamate groups is catalyzed by carbonic anhydrase. After the formation of the carbamate groups, the carbamate dissociates into bicarbonate ions and protons. The salt bridges are formed by protonating histidine which then bridges with asparagine.
Effect of Temperature on Enzymatic Activity
[edit | edit source]As all enzymes have an optimal pH in which their catalytic activity is at its peak, enzymes also have an optimal temperature. There are two established thermal properties of enzymes that effect the catalytic rate. Those two are activation energy and their thermal stability. However experimental data of temperature against enzymatic activity does not clearly match the so sought out for indignation that activity simply increases with temperature. A new model called the Equilibrium model helps provide the quantitative explanation of enzyme thermal behavior under reaction conditions by introducing the enzymes inactive form, forming an equilibrium system that would follow similar rules to Le Chatlier's principle in basic chemistry. The equilibrium model gives rise to a number of insights in the sense that it eliminates time dependency that was thought to be important in the classical view of enzymatic activity against temperature. The idea behind the equilibrium model is that when under different temperature gradients, the inactive form of the enzyme that is added along with the active form, prevents a full inactivation of the active enzyme by equilibrium mechanics. It should be noted however that the difference between inactive and active forms of the enzyme must be understood. The equilibrium model describes the inactive form of the enzyme by purely centralizing its activity according to its active site. The active site of an enzyme is where substrates are capable of binding which then proceeds into a conformational change of the enzyme to then further proceed with the intended biochemical reaction. The inactive enzyme in this model is described as a mere folding change in comparison to the active form of the enzyme. This is not to be confused with an enzyme that has been denatured. An enzyme that is denatured is one that is completely changed, active site and all, to the point where the enzyme can not function at all, in other words, it is an irreversible enzyme conformational change. In the case of the equilibrium model, the mechanic of this model works simply because of the reversibility of the inactive enzyme form. The inactive form of the enzyme can reverse back to its active form which is in complete correspondence of the whole idea behind the equilibrium mechanics of the proposed model.
Reference
[edit | edit source]Berg, Jeremy "Biochemistry", Chapter 7 Hemoglobin: Portrait of a Protein in action. 193-194. sixth edition. Freeman and Company, 2007. http://hrsbstaff.ednet.ns.ca/sdosman/Higher%20level%20BIO/enzymenotes3.6.htm
Roy M. Daniel, Michael J. Danson, A new understanding of how temperature affects the catalytic activity of enzymes, Trends in Biochemical Sciences, Volume 35, Issue 10, October 2010, Pages 584-591, ISSN 0968-0004, 10.1016/j.tibs.2010.05.001.
Site-Directed Mutagenesis
[edit | edit source]Mutagenesis is a broad term that is defined as the alteration of the genetic material of an organism in a stable manner.
Site-directed mutagenesis is when the amino acid sequence of a given enzyme molecule or other protein may be altered by deliberately and precisely mutating the cloned gene encoding that molecule. It is a very useful technique that can be used in the study of protein function, the identification of enzymatic active sites, and the design of novel proteins. With this technique, it is possible to exchange a single amino acid in the sequence of a protein for another amino acid with different chemical properties. In this way, the function of the specific amino acid at this site can be examined. The basic protocol for this process was developed by Michael Smith, who was awarded the Nobel Prize in Chemistry in 1993 "for his fundamental contributions to the establishment of oligonucleotide-based, site-directed mutagenesis and its development for protein studies" (1)
In order to carry out site-directed mutagenesis, a DNA primer must be designed at the site of interest. The primer should contain the necessary nucleotide differences in order to affect the change in the protein sequence. For example, consider the case where a protein sequence reads Tyr-Leu-His-Val, corresponding to a genetic sequence of UACCUGCACGUC. If the experimentalist intends to mutate the histidine residue to a leucine residue, they might design a primer with the sequence UACCUGCUCGUC. This primer is then hybridized to the complementary single stranded DNA molecule and extended using a DNA polymerase. A mutated double stranded DNA molecule encoding the (mutated) protein is obtained in this way, and this DNA molecule is cloned into a host cell. Host cells are allowed to grow and mutants are selected. In this way, proteins that are generated by this DNA sequence will contain the desired mutation. A similar procedure can be carried out with the use of PCR (PCR site-directed mutagenesis)

An example of site-directed mutagenesis: catalytic triad
[edit | edit source]Site-Directed Mutagenesis is a method used to dissect the amount of catalytic power by each of the catalytic triad in a enzyme. This is done by converting each of the triad into a common amino acid and measure the catalytic power differences.
For example, in subtilisin, the catalytic triad that is studied by this method are aspartic acid 32, histidine 64, and serine 221. Each of the amino acids in the triad will be converted into alanine and thus, the ability of the mutant enzyme to cleave a substrate is examined. The result by this method shows that serine 221 into alanine reduces the catalytic power as much as histidine 64 into alanine. The value of the kcat for serine 221 and histidine 64 becomes one-millionth of its original value. As for aspartic acid 32, the catalytic power is reduced but not as much as for serine 221 and histidine 64. The kcat value is 0.005% of the original enzyme. Thus, the result of using the site-directed mutagenesis shows that the serine-histidine pair makes a nucleophile that has the capability to attack the carbonyl carbon atom in the peptide bond. Site-directed mutagenesis can be used to change particular base pairs in a piece of DNA. There are a number of methods for achieving this. The approach described here is adapted from the Stratagene site-directed mutagenesis kit, the manual can be found here. Even when using a kit it will be necessary to design primers that are suitable for the specific changes you want to make to your DNA. Most of the contents of the kit can be found in your favorite labs stocks so you may not need to buy the kit itself. If you have problems with this procedure, you can try 'Round-the-horn site-directed mutagenesis which uses PCR to amplify the desired mutant product.
Mutagenesis has been used in terms of DNA recombinant technique.
References
[edit | edit source](1)http://nobelprize.org/nobel_prizes/chemistry/laureates/1993/#
(2)Berg, Jeremy M. John L. Tymoczko. Lubert Stryer. Biochemistry Sixth Edition. New York: W.H. Freeman and Company, 2007
(3)http://openwetware.org/wiki/Site-directed_mutagenesis A. Introduction
When dealing with enzyme substrate reactions, most involve a single substrate which is turned into a single product by an enzyme. However, for multi-substrate reactions there are more than one substrate involved. The reaction involves a complex reaction that not only tells where the substrates bind, but the sequence of binding as well. For instance if there were two substrates, one would be labeled substrate A and substrate B. On the basis that one of the substrate concentrations remains constant the entire time like substrate A, the enzyme would behave in the same manner as a single substrate enzyme. Because of this, the a graph of substrate concentration over velocity would produce values of both Km and Vmax for substrate B because substrate A is constant. The value of Km represents the substrate concentration at which the reaction is half its maximum velocity (Vmax). However, this is not always the case and when the concentrations of substrate A and substrate B are different the result of the enzyme-substrate interactions can be explained by two different mechanisms that will be described below. Furthermore, hypothetically, substrate A and substrate B would lead to two different products that can be labeled as P and R respectively.
B. Ternary Complex Mechanism
In a Ternary Complex Mechanism two substrates bind to the enzyme (hypothetically substrate A and substrate B) to form a complex that is known as the EAB Ternary Complex. The order of the substrate binding can either be in a specific sequence (ordered) or random sequence as well. An enzyme that follows the ternary complex mechanism would have a Lineweaver-Burk Plot that has two lines that intersect on a reciprocal substrate concentration/velocity graph. The plot represents a linear graph of the reciprocals 1/S and 1/V. A specific enzyme that has a ternary-complex mechanism is DNA polymerase. DNA polymerase functions to add nucleotides to DNA.
C. Ping Pong Mechanism
For this mechanism, an enzyme can be in two states. One of the states is labeled E and the other state that is also known as the intermediate and that is chemically modified is labeled E*. In this mechanism, the first substrate (substrate A) binds to enzyme turning it into E* by the transfer of a chemical group to the active site and then the substrate is released. Once substrate A is released, substrate is able to bind to the modified Enzyme (E*) forming the unmodified Enzyme once again (regeneration). When a Line-Weaver Burk plot is graphed, two sets of parallel will be formed opposite of the Ternary Complex Mechanism. Specific enzymes that follow this mechanism include oxidoreductases and serine proteases. Some of the serine proteases include the digestive enzymes of chymotrypsin and trypsin. For the example of the chymotrypsin, an acyl-enzyme is formed after the breakdown of the tetrahedral intermediate, which is formed after the nucleophilic attack of Ser to the carbonyl forming the intermediate. Once the intermediate breaks down, the acyl-enzyme is formed which acts as the modified Enzyme (E*). The acyl-enzyme however breaks down later into the intermediate complex as the amine group of the acyl-enzyme (E*) leaves and hydrogen functions as a nucleophile to attack the carbonyl forming the tetrahedral intermediate once again. Catalytic antibodies are antibodies that can enhance a couple of chemical and metabolic reactions in the body by binding a chemical group, resembling the transition state of a given reaction. Catalytic antibodies are produced when an organism is immunized with a hapten molecule. The hapten molecule is usually designed to resemble the transition state of metabolic reaction.
Antibodies act like soldiers to the body, fighting unwanted materials. They are secreted, for instance, when the body is infected with a bacterium or virus. The animal produces antibodies with binding sites that are exactly complementary to some molecular feature of the invader. The antibodies can thus recognize and bind only to the invader, identifying it as foreign and leading to its destruction by the rest of the immune system. Antibodies are also elicited in large quantity when an animal is injected with molecules, a process known as immunization. A small molecule used for immunization is called a hapten. Ordinarily, only large molecules effectively elicit antibodies via immunization, so small-molecule haptens must be attached to a large protein molecule, called a carrier protein, prior to the actual immunization. Antibodies that are produced after immunization with the hapten-carrier protein conjugate are complementary to, and thus specifically bind, the hapten.
Ordinarily, antibody molecules simply bind; they do not catalyze reactions. However, catalytic antibodies are produced when animals are immunized with hapten molecules that are specially designed to elicit antibodies that have binding pockets capable of catalyzing chemical reactions. For example, in the simplest cases, binding forces within the antibody binding pocket are enlisted to stabilize transition states and intermediates, thereby lowering a reaction's energy barrier and increasing its rate. This can occur when the antibodies have a binding site that is complementary to a transition state or intermediate structure in terms of both three-dimensional geometry and charge distribution. This complementarity leads to catalysis by encouraging the substrate to adopt a transition-state-like geometry and charge distribution. Not only is the energy barrier lowered for the desired reaction, but other geometries and charge distributions that would lead to unwanted products can be prevented, increasing reaction selectivity.
Making antibodies with binding pockets complementary to transition states is complicated by the fact that true transition states and most reaction intermediates are unstable. Thus, true transition states or intermediates cannot be isolated or used as haptens for immunization. Instead, so-called transition-state analog molecules are used. Transition-state analog molecules are stable molecules that simply resemble a transition state (or intermediate) for a reaction of interest in terms of geometry and charge distribution. To the extent that the transition-state analog molecule resembles a true reaction transition state or intermediate, the elicited antibodies will also be complementary to that transition state or intermediate and thus lead to the catalytic acceleration of that reaction.
Catalytic antibodies bind very tightly to the transition-state analog haptens that were used to produce them during the immunization process. The transition-state analog haptens only bind and do not react with catalytic antibodies. It is the substrates, for example, the analogous ester molecules, that react. For this reason, transition-state analog haptens can interfere with the catalytic reaction by binding in the antibody binding pocket, thereby preventing any substrate molecules from binding and reacting. This inhibition by the transition-state analog hapten is always observed with catalytic antibodies, and is used as a first level of proof that catalytic antibodies are responsible for any observed catalytic reaction.
The important feature of catalysis by antibodies is that, unlike enzymes, desired reaction selectivity can be programmed into the antibody by using an appropriately designed hapten. Catalytic antibodies almost always demonstrate a high degree of substrate selectivity. In addition, catalytic antibodies have been produced that have regioselectivity sufficient to produce a single product for a reaction in which other products are normally observed in the absence of the antibody.
Finally, catalytic antibodies have been produced by immunization with a single-handed version (only left- or only right-handed) of a hapten, and only substrates with the same handedness can act as substrates for the resulting catalytic antibodies. The net result is that a high degree of stereoselectivity is observed in the antibody-catalyzed reaction.
Abzymes are artificial catalytic antibodies and come from the words “antibody” and “enzyme” They are monoclonal antibodies that have catalytic properties, or carry out catalysis.
Equilibrium Model
[edit | edit source]It is important to understand how enzymes work because they lead to the understanding of the functions of cells and manipulations of enzymes. One of such factors that affect the enzyme activity is temperature. The widely accepted Classical Model on how temperature affects enzymatic activity states that the exponential increase in rate of reaction corresponds to a decrease in the amount of enzyme, resulting from irreversible thermal destruction of enzymatic activity. However, research showed that the experimental data do not match the Classical Model. For example, one study found that once passing the optimum temperature of the enzyme, there is a greater decrease in catalytic rate in enzyme than from that expected from irreversible thermal inactivation. A second similar study also revealed that some enzyme becomes less active at high temperature compare to at its thermal stability and that some of the loss of activity is reversible if at enzyme's optimum temperature; hence, thermal stability is needed but not enough for thermal activity.
An Equilibrium Model is introduced in replace of the Classical Model to account for aspects that were neglected in the Classical Model. After incorporating all possible factors, the Equilibrium Model turns out to closely fit the experimental data and gives insights to temperature control, adaptation and evolution of enzymes. The Equilibrium Model explains the thermal behavior of enzymes by the introduction of inactive but not denatured intermediate enzyme in equilibrium with its active intermediate. This additional term describes how temperature affects the equilibrium between the active and inactive forms of the enzyme. This relationship can be shown in the following reaction:
Eact ↔ Einact → X
Where Eact is the active form of the enzyme, Einact is the inactive but not denatured form of the enzyme, X is the fully inactivated form of the enzyme, Keq is the equilibrium constant between Eact and Einact , and Kinact is the rate constant from Einact to X.

Initially, the rate is determined by the increasing concentration of products formed over a short time and then the concentration falls back down. This shows that there is an optimal temperature for the enzyme, which further states the loss of enzymatic activity, meaning there is a change in ratio of the active and inactive enzymes. The active and inactive enzymes reversible equilibrium protects the enzyme from thermal inactivation, meaning that the equilibrium term acts like a buffer. The difference between the Equilibrium Model and the Classical Model is that the experimental data for enzymes showed activity optimum at to, which is consistent with Equilibrium Model; however, Classical Model does not show such an optimum. Moreover, many enzymes from the least reactive to the most reactive groups are examined and shown to match with the Equilibrium Model. Therefore, the Equilibrium Model is universal, with no dependency of the reaction undergoing and structure of enzyme itself.
Previous models
[edit | edit source]There are several models prior to the establishment of the Equilibrium Model, which contained some similarities to the Equilibrium Model. One previous model showed time-independent changes in activity, meaning that the only changes of catalytic rate are affected by temperature, with total active enzymes as constant. This excluded the possible time-dependent irreversible inactivation. This model is similar to the Equilibrium Model for they are both time-independent. However, the level of enzyme activity varies in the Equilibrium Model.
A second model described the equilibrium between more than one active form of reactants. In this model, proteins were used instead of enzymes, but the basic concept of establishing equilibrium remained. This model says that the total active proteins decrease with time; however, it assumed that level of activity does not vary with temperature and assumed both protected and native antibodies are active.
Another model in which the equilibrium is reached between the native and unfolded forms of the enzymes concluded that overall process is constrained by a reversible step, especially in unfolding then grouping proteins, such that an irreversible denaturation process follows. This model differs with the Equilibrium Model since the inactive enzyme is significantly folded in the Equilibrium Model. In addition, it is usually rare for thermal unfolding of an enzyme to be reversible in the Equilibrium Model.
Eact-Einact interconversion
[edit | edit source]A plot of the initial enzyme rate versus temperature shows each enzyme has its own optimal temperature. As the temperature passes the threshold of the optimum, it results in a faster enzyme activity loss than what the initial rate can measure; therefore, the equilibrium of the active and inactive form the enzyme is a valid assumption. From looking that the top reaction, the denaturation rate is much slower than the rate of converting active to inactive enzyme because the conversion of the active to inactive form is temperature dependent. The interconversion is restricted at the active sites of the enzyme. The active sites needs flexibility to carry out catalysis, leading to a greater possibility of temperature induced changes in conformation and/or dynamics. Active sites also control the effect of temperature on activity and the kinds of activity that enzyme has over a wide range of temperature. Although the big picture of interconversion between the two forms is well known, the detailed local changes from active to inactive form is hard to detect because there might be only small structural changes, and there is time constraints due to rapid denaturation to producing dominantly inactive form.
Implications of the Equilibrium Model
[edit | edit source]Mutagenesis, the mutation in which the genetic information changes in a slow and stable way, can be used to improve enzyme stability in two ways. First, mutagenesis helps to uncover the structural basis of protein stability. Second, it increases the temperature at which the enzyme can function. More specifically, mutagenesis changes a single amino acid at the active site, thus leading to changes in optimal temperature and enthalpy without changing the free Gibbs energy of the inactive enzyme. This means with the same amount of energy, the level of enzyme activity increases due to a rise in the optimal temperature. However if the temperature is, in general, increased, there would be a corresponding decrease in the enzyme stability and reaction rate.
Conclusion
[edit | edit source]The Equilibrium model tests the relationship between the thermal properties of enzyme and effect of temperature has on the host organism. In other words, the Equilibrium Model provides an explanation on how temperature affects the enzyme activity. In detail, the active sites are governed by how temperature affects the enzyme activity, which means that the evolution of actives sites is limited by temperature. A note to keep in mind is that the Equilibrium Model works for ideal situations. Indeed, the establishment of the Equilibrium Model does not eliminate the possibility that a more complex model might also fit the data equally as well.
Reference
[edit | edit source]Roy M. Daniel and Michael J. Danson. A New Understanding of How Temperature Affects the Catalytic Activity of Enzymes. Trends in Biochemical Sciences, Volume 35, Issue 10, October 2010, Pages 584-591, ISSN 0968-0004, 10.1016/j.tibs.2010.05.001.
(http://www.sciencedirect.com/science/article/pii/S0968000410000897) Structural Biochemistry/Specific Enzymes and Catalytic Mechanisms Proteases are enzymes that accelerate the hydrolysis of peptide bonds. In essence, proteases break up proteins into smaller peptide fragments. Proteases generally promote the hydrolysis of a peptide by activating a nucleophile, polarizing the peptide carbonyl and stabilizing the tetrahedral intermediate. Protease like all enzymes are very specific so recognizes side chain to know where to cleave.
Mechanism of Action
[edit | edit source]Proteases generally activate a nucleophile, which will in turn attack the carbon of the peptide bond. The electrons in the carbon-oxygen double bond migrate onto the oxygen as the nucleophile attaches itself. This tetrahedral intermediate is a high energy intermediate and the protease will generally have a way to stabilize this intermediate. The intermediate will then decompose, usually releasing the two peptide fragments.
The 4 main class of proteases are: Serine Proteases, Cysteine Proteases, Aspartyl Proteases, and Metalloproteases. All four classes of proteases utilize either use a different nucleophile or a different way to activate the nucleophile. Serine and cysteine proteases use a catalytic triad to activate the side chain of either a serine or cysteine. Aspartyl proteases use an aspartic acid residue to activate a water molecule and another aspartic acid residue to align the peptide for attack. Metalloproteases use a metal ion to activate a water molecule.
Stabilization of the tetrahedral intermediate is generally accomplished by parts of the proteins that aren't the active site. A common method is the use of an oxyanion hole. An oxyanion is a part of the protease which will encompass the tetrahedral intermediate. Within this hole, hydrogen bonding between the NH groups of the protease and the negatively charged oxygen of the protein will stabilize the intermediate.
Specificity of Proteases
[edit | edit source]An amazing feature of proteases is their preference for cleaving the peptide bond associated with a specific amino acid. This preference is a result of the active site's location within the protease's structure. The active site is generally in a cavity of the protein. The type of amino acid residues within the pocket will determine the preference of the proteases. Protease can also break ester bonds.
Chymotrypsin has a deep cavity made up of mostly hydrophobic residues, thus Chymotrypsin has a preference to cut peptide bonds of amino acids with large hydrophobic side chains such as tryptophan and phenylalanine. Elastase has bulky valine residues within the cavity, thus Elastase has a preference to cut peptide bonds of amino acids with small side chains. Trypsin has a aspartate residue, which has a negatively charged side chain, at the bottom on the cavity. Thus Trypsin has a preference to cut peptide bonds of amino acids with positively charged side chains.
Enzyme inhibition by DIPF: serine 195's Hydrogen can bind with Fluoride from DIPF and inhibit its nucleophilic attack on the carbonyl of peptide it wants to cleave.
Structure
[edit | edit source]
Protease Inhibitors
[edit | edit source]The conversion of a zymogen to a protease through the cleavage of one peptide bond is an accurate way to switch on certain enzymatic activities. The following activation step is irreversible. Therefore a different mechanism is required to stop proteolysis with the help of specific protease inhibitors. There are several important drugs that serve as protease inhibitors. These inhibitors are specific for one enzyme and do not interfere in the production of other proteins in the body. For example, the inhibitor Indinvar is specific for the HIV protease because the interaction of water and the enzyme is not possible in other aspartyl proteases.
Captopril is an inhibitor for the metalloprotease antiotensin-converting enzyme (ACE). This inhibitor helps in the regulation of blood pressure in the body.
There are several HIV protease inhibitors used for the treatment of AIDS. The HIV protease is an aspartyl protease that cleaves multidomain viral proteins into their active forms. Indinavir is an inhibitor that structurally resembles peptide substrate of HIV protease by mimicking the tetrahedral intermediate. In the active site, indinavir adopts a conformation that is similar to the twofold symmetry of the enzyme. Two flexible flaps of the HIV protease's active site fold down on top of the bound inhibitor. The central alcohol interacts with the two aspartate residues of the active site. Plus, the inhibitor's two carbonyl groups are hydrogen bonded to a water molecule (this is not seen in the molecule below), which is also hydrogen bonded to a NH group in each of the flaps.


Enzymatic Trypsin Inhibitor
[edit | edit source]Another example of a protease inhibitor is known as Pancreatic Trypsin Inhibitor. It is a 6 kilodalton protein and inhibits trypsin by binding very strongly to the active site of trypsin. The trypsin and pancreatic trypsin inhibitor complex is very stable in that it has a dissociation constant of about 0.1 pM (standard free energy of -75 kJ/mol). THis means that the complex cannot be dissociated into its denatured state with common denaturing agents such as 8 M Urea or 6 M guanidine hydrochloric acid. Because pancreatic trypsin inhibitor is a very effective substrate, the complex that it forms with trypsin is extraordinarily stable. Also, analysis by X-rays reveal that the inhibitor lies in the active site in such a position that the lysine-15 side chain of the inhibitor interacts with an aspartate side chain of trypsin in the active site. Additionally, many hydrogen bonds between the main chain of trypsin and the inhibitor further stabilize the trypsin - pancreatic trypsin inhibitor complex.
After binding to the active site of trypsin, pancreatic trypsin inhibitor does not change its structure, which means that the inhibitor is preorganized into such a structure that is complementary to the enzyme's active site. This is seen by the slow rate of cleavage of the peptide bond between lysine-15 and alanine-16. Overall, this inhibitor is more like a substrate and its inherent structure is extremely complentary to the enzyme's active site that it binds really tightly and is turned over slowly.
The amount of physiologically available trypsin is greater than the amount of trypsin inhibitor. Since trypsin activates other zymnogens, inhibitors of trypsin needs to exist to prevent small amounts of trypsin from starting a mistakenly activated cascade.
α1-Antitrypsin (α1-antiproteinase)
[edit | edit source]α1-Antitrypsin is a 53 kilodalton plasma protein protease inhibitor. It protects tissues from digestion by elastase, which is a secretory product of white blood cells that engulf bacteria. α1-Antitrypsin inhibits elastase much better than it inhibits trypsin. Similar to pancreatic trypsin inhibitor, α1-Antitrypsin blocks the action to target enzymes by almost irreversibly binding to the enzyme active sites.
α1-Antitrypsin is a physiologically important inhibitor because without it, excess elastase destroys alveolar walls in the lungs by digesting connective-tissue proteins. This condition is called emphysema, in which people with this condition have difficulty breathing. People with emphysema must breathe harder than normal in order to exchange the same amount of oxygen as people without emphysema due their damaged alveoli. Cigarette smokers are more likely to develop emphysema because smoke oxidizes methionine-358 to methionine sulfoxide (see figure) of the α1-Antitrypsin inhibitor, which is an essential residue for binding elastase. The insertion of a single oxygen into the resulting methionine sulfoxide of the protein, which changes the inhibitor's affinity for elastase is a great example of the importance of structural biochemistry and the role it plays throughout physiological processes.

Reference
[edit | edit source]1. Berg, Jeremy; John L. Tymoczko, Lubert Stryer (2007). Biochemistry, 6th Edition. W. H. Freeman and Company, New York, New York. Hydrolysis is the process of a water reaction. It means to break using water. Hydrolysis comes from the Greek work hydro meaning water; and lysis meaning break. T
Overview
[edit | edit source]Chymotrypsin, a protease, is an enzyme that cleaves the carbonyl side of certain peptide bonds by both general acid-base catalysis, but primarily covalent catalysis. In this mechanism, a nucleophile becomes covalently attached to a substrate in a transition state with an acyl-enzyme. The protease cleaves proteins by a hydrolysis reaction, an addition of a water molecule. The double bond between the carbon and nitrogen strengthens its bond. Chymotrypsin is site specific and will only cleave the carboxyl side of large hydrophobic or aromatic amino acids such as phenylalanine (Phe), methionine (Met), tyrosine (Tyr), and tryptophan (Trp), unless the next amino acid is proline (Pro). The reason why chymotrypsin prefers to cleave specifically to bulky hydrophobic amino acids is due to the formation of S1 pockets,which, in the case of chymotrypsin, is lined with relatively hydrophobic residues such as Ser-189, Ser-214, Trp-215, Gly-216, and Gly-226. Chymotrypsin catalyzes the reaction rate by a factor of 109. The reaction has two steps, an acylation phase and a deacylation phase. In the former phase, the peptide bond is cleaved and an ester is formed between substrate and enzyme. In the latter phase, this ester is hydrolyzed and the enzyme is regenerated.

This illustrates the covalent catalysis of chymotrypsin. The first step is the acylation, which forms the acyl-enzyme intermediate. Then the acyl-enzyme intermediate goes through deacylation converting back to its original free enzyme form.
Evidence of Mechanism
[edit | edit source]Because chymotrypsin can also catalyze the hydrolysis of esters and amides, p-nitrophenolacetate was used in conjunction with chymotrypsin. The reaction with p-nitrophenolacetate will yield p-nitrophenol, a chromic-effector with a yellow color change in the product. The absorbency can be determined from the color and the intensity can determine the amount of product. Hartley and Kilby used this information in 1954 to show that the reaction proceeds in two phases: a Burst Phase and then levels off to a steady-state phase. Thus, there is a formation of a covalently bound enzyme substrate intermediate.

Another test to determine the mechanism of chymotrypsin hydrolysis was to treat the protease with an organofluorophosphate, diisopropylphosphofluoridate (DIPF). In this reaction, chymotrypsin loses all activity and becomes inactivated. Since only serine-195 was modified by diisopropylphosphofluoridate, it indicates that Serine-195 plays the crucial role in the mechanism as a nucleophile. It is covalently linked to Serine-195. Covalent catalysis of chymotrypsin basically goes through acylation and deacylation. Acylation forms the acyl enzyme intermediate and the deacylation adds water which produces a free enzyme.
Site-directed mutagenesis is another technique that can test the reaction by creating a mutant in the amino acid sequence of the active site of the enzyme. It supported the mechanism below by demonstrating that the replacement through site directed mutagenesis of any one member of the catalytic triad had a devastating effect on reaction rate. In fact, replacing just one of the triad had the same effect as replacing all three--demonstrating that each component is vital for efficient catalysis. While the enzyme continued to bind to the substrate (we know this because the KM remained constant throughout the replacements--it required the same substrate concentration to achieve half of the maximum rate), the reaction rate was orders of magnitude smaller without the triad.
Structure of Chymotrypsin
[edit | edit source]The primary structure shows that disulfide bonds are the crucial role to the protein folding. The protein is spherical and itself consists of three polypeptide chains. There is also a pocket in the protein which is known as the active site. The active site includes Ser-195, His-57, and Asp-102 (the catalytic triad). Ser-195 is hydrogen bonded to the His-57 and it in turn is hydrogen bonded to the Asp-102 residue. The His-57 role is to position the serine residue and polarize the hydroxyl group so it can be deprotonated to the alkoxide ion. In the presence of the substrate, this accepts a proton by acting as a base. Asp-102 orients the His-57 and stabilizes it through hydrogen bonding and electrostatics.
Mechanism
[edit | edit source]Step 1: When substrate (polypeptide) binds, the side of chain of the residue next to the peptide bond to be cleaved nestles in a hydrophobic pocket on the enzyme, positioning the peptide bond for attack. Histidine extracts one proton from serine to form an alkoxide ion. This serine ion reacts with the substrate.
Step 2: In chymotrypsin, the carboxylate R-group of Asp102 forms a hydrogen bond with R group of His 57. When this happens, it compresses this hydrogen bond and shifts electron density to the other nitrogen atom (not involved in the H-bond) in the R-group of His57 becomes a very strong base. This allows His 57 to deprotonate Ser195 and turn it into a strong nucleophile that can attack the substrate.

Oxygen develops a partially negative charge in the oxyanion hole.
Step 3: Instability of the negative charge on the substrate carbonyl oxygen when will leads to collapse of the tetrahedral intermediate, re-formation of a double bond with carbon which breaks the peptide bond between the carbon and amino acid group. The amino leaving group is protonated by His57, facilitating its displacement. Once the oxyanion hole stabilizes the negative charge, the bond breaks because the proton from Histidine is binding to nitrogen to make it less likely to carbon. The leaving group is stabilized and the acyl-enzyme is formed.
Step 4: The amine component is departed from the enzyme (metabolized by the body) and binds to serine. This completes the first stage (acylation of enzyme). The first product has been made.
Step 5: A water molecule is added where the N terminus was. Histidine deprotonates the water to form a hydroxyl group. This hydroxyl group attaches to carbon from the carboxyl side and destabilizes the acyl intermediate. The bond is broken.
Step 6: An incoming water molecule is deprotonated by acid-base catalysis, generating a strongly nucleophilic hydroxide ion. Attack of hydroxide on the ester linkage of the acylenzyme generates a second tetrahedral intermediate.
Step 7: collapse of the tetrahedral intermediate form the second product, a carboxylate anion, and displace Ser195. The proton from Histidine goes back to Serine.
Step 8: The carboxylic acid is released and the enzyme is reformed to catalyze the next reaction with the original active site.

Cysteine Protease
[edit | edit source]This is part of a large family of peptide-cleaving enzymes or proteases. Cysteine Proteases is one of proteases enzyme that cleave protein by cleave the peptide bond. The strategy used by the cysteine proteases is most similar to that use to be chymotrypsin family that is to generate a nucleophile that attack the peptide carbonyl group. Also similar to chymotrypsin, it polarizes the peptide carbonyl group to get it activated for attack, and upon attack by the nucleophile, a stabilizing tetrahedral intermediate is generated. But different from chymotrypsin enzyme, in this enzyme, a cysteine residue, activated by histidine residue play a nucleophilic attack the peptide bond.
Most common Cysteine Proteases
[edit | edit source]Papain is a protein-cleaving enzyme derived from papaya fruit (Carica papaya) and certain other plants. Papain is used as a meat tenderizer and in medicine as a digestive aid. Cathepsins are members of the lysosomal cysteine protease (active site) family and the cathepsin family name has been synonymous with lysosomal proteolytic enzymes. In actuality, the cathepsin family also contains members of the serine protease (cathepsin A,G) and aspartic protease (cathepsin D,E) families as well. These enzymes exist in their processed form as disulfide-linked heavy and light chain subunits with molecular weights ranging from 20-35 kDa. Cathepsins have a vital role in mammalian cellular turnover, e.g. bone resorption. They degrade polypeptides and are distinguished by their substrate specificity
Mechanism
[edit | edit source]
Overview
[edit | edit source]Aspartyl proteases are one of the eukaryotic protease enzymes that catalyze peptide substrates using aspartate residue. It is usually in an acidic pH range which is inhibited by pepstatin. Some examples of the aspartyl proteases are pepsins, cathepsins, and renins. It exists in vertebrates, plants, plant viruses, and retroviruses. It has a sequence of Asp- Thr- Gly. It is usually represented as monomeric enzymes with twofold symmetry and has a tertiary structure with an N-terminal and a C-terminal.
Mechanism
[edit | edit source]On the active sites of aspartyl proteases, there are aspartic acid residues that work together to promote a water molecule to attack the peptide bond. One of the aspartic acid residues (left on the diagram, deprotonated form) will activate the water molecule by attracting the hydrogen atom of water. The other aspartic acid (right on the diagram, protonated form) residue will polarize the carbonyl group on the peptide making it easier to attack. One of the aspartic acids usually has a lower pKa value.

Renin, an enzyme that supports the regulation of blood pressure, is an important member of this class of enzyme (Berg, 7th Edition)
Common type of aspartyl protease
[edit | edit source]Renin is a proteolytic enzyme synthesized, stored and secreted by the juxtaglomerular cells of the kidney; it plays a role in regulation of blood pressure by catalyzing the conversion of the plasma glycoprotein angiotensinogen to angiotensin I. This, in turn, is converted to angiotensin II by an enzyme that is present in relatively high concentrations in the lung. Angiotensin II is one of the most potent vasoconstrictors known, and also is a powerful stimulus of aldosterone secretion. Pepsin is a digestive enzyme found in gastric juice that catalyzes the breakdown of protein to peptides. Pepsin is one of three protein-degrading or proteolytic enzymes in the digestive system; the other two being chymotrypsin and trypsin. The three enzymes work together to break proteins down into peptides and amino acids, which can be readily absorbed by the intestinal lining. Pepsin is most effective in cleaving the bonds of phenylalanine, tryptophan, and tyrosine.
The HIV protease is an example of the aspartyl protease. This protease is a dimer which consists of identical subunits. As a member of the aspartyl protease family, it contains two aspartic acid residues symmetrically located at the bottom of the binding pocket. The function of this protease is to cleave the domain of the viral protein into their dynamic forms. Those forms spread the virus. This process can be stopped by using HIV protease inhibitors, which attack the HIV protease, bind to it, and prevent cleavage of the domain. An example of an HIV protease inhibitor is Indinavir. Indinavir is used to treat HIV infection and AIDS and is one of the most successfully used protease inhibitors in medicine.
Lopinavir is also one of the HIV protease inhibitors. The structure of HIV-1 protease with Lopinavir is shown. The hydroxyl group acts as a transition analog, mimicking the oxygen of the tetrahedral intermediate. The benzyl group, positioned next to the hydroxyl group, helps to properly position the drug in the active site.

Definition
[edit | edit source]Serine proteases are proteases that have serine, an amino acid, bonded at the active site. Their main function in humans is digestion, however they also function in processes such as inflammation, blood clotting, and the immune system in both prokaryotes and eukaryotes. Serine proteases are grouped depending on their structure. Major groups of serine proteases include alpha hydrolase, beta hydrolase, and signal peptidase. The serine proteases is the enzyme that catalyze the hydrolysis of ester or amide. This reaction involves the reaction of acylation of the hydroxyl group of Ser-195. The substrate forms a tetrahedral intermediate by attacking of Ser-195 on the carboxyl group of the substrate since the active site of the enzyme is complementary to the transition state of the reaction.
Proteases, proteinases, peptidases describe the same group of enzymes that catalyze the hydrolysis of covalent peptide bonds. Serine proteases are grouped into clans that share structural homology and then further subgrouped into families that share close sequence homology. In the case of serine protease, the mechanism of the protease is based on the nucleophilic attack of the targeted peptidic bond by a serine.
Cysteine, threonine or water molecules associated with aspartate or metals may also play this role. In many cases the nucleophilic property of the group is improved by the presence of a histidine, held in a "proton acceptor state" by an aspartate. Aligned side chains of serine, histidine and aspartate build the catalytic triad common to most serine proteases.
The active site of serine proteases is shaped as a cleft where the polypeptide substrate binds. Schechter and Berger [1] labeled amino acid residues from N to C term of the polypeptide substrate (Pi, ..., P3, P2, P1, P1', P2', P3', ..., Pj) and their respective binding sub-sites Si,..., S3, S2, S1, S1', S2', S3',..., Sj) . The cleavage is catalyzed between P1 and P1'.
Many proteases are synthesized and secreted as inactive forms called zymogens and subsequently activated by proteolysis. This changes the architecture of the active site of the enzyme.
Few examples are: Chymotrypsin, trypsin, and elastase.
Chymotrypsin
Synthesized as inactive proenzymes (chymotrypsinogen)
Formation of key acyl-enzyme intermediate
Catalytic residues are Ser195 and His57
X-ray Structure
~240-residue monomeric proteins, 4 disulfide bridges
Two folded domains with antiparallel b-sheets (barrel-like) and little helix
Catalytic triad - His57 and Ser195 located at substrate binding site along with Asp102, which is buried in solventinaccessible pocket.
Chymotrypsin - prefers bulky Phe, Trp, or Tyr in hydrophobic pocket.
Trypsin - prefers Arg and Lys in binding pocket (Ser189 replaced by Asp).
Elastase - prefers Ala, Gly, Val in its depression site
Catalytic Mechchanism
Bound substrate is attacked by nucleophilic Ser195 forming transition state complex (tetrahedral intermediate), His57 takes up H+, which is facilitated by Asp102.
Tetrahedral intermediate decomposes to acyl-enzyme intermediate by His57 (general acid).
Acyl-enzyme intermediate is deacylated by reverse of above steps, release of carboxylate product, H2O is nucleophile and Ser195 is leaving group.
Enzyme prefers binding transition state to either Michaelis complex or acyl-enzyme intermediate forms.
Catalytic triad serves to form low-barrier hydrogen bonds in the transition state (assisted by hydrophobic environment).
Zymogens:
Inactive (proenzyme) forms
Enzyme inhibitors (pancreatic trypsin inhibitor) or zymogen granules prevent activation
Active sites are distorted
Serine proteases are sequence specific. While cascades of protease activations control blood clotting and complement, other proteasesare involved in signalling pathways, enzyme activation and degradative functions in different cellular or extracellular compartments.
Mechanism
[edit | edit source]
Definition
[edit | edit source]Threonine proteases are proteases that have threonine, an amino acid, bonded at the active site. It is responsible for functioning proteasome, the large protein-degrading apparatus. Threonine proteases haveaA conserved N-terminal threonine at each active site. Pre-proteins, which are catalytic beta subunits, are activated when the N-terminus is cleaved off. This makes threonine the N-terminal residue.
Threonine proteases are activated by primary amines. The mechanism for the threonine protease was described first in 1995. The mechanism showed the cleaving of a peptide bond which made an amino acid residue (usually serine, threonine, or cysteine) or a water molecule become a good nucleophile which could perform a nucleophilic attack on the carboxyl group of the peptide. The amino acid residue (in this case threonine) is usually activated by a histidine residue.
Overview
[edit | edit source]Metalloproteases contain an active site that has a bound metal ion; this metal is almost always zinc. Metalloproteases are enzymes which catalyze reactions. Although the metal found is almost always zinc, the reactions can proceed with cobalt as well. The metal ion activates a water molecule to carry out a nucleophilic attack on a carbonyl peptide bond. A base is present to help deprotonate the metal-bound water.
There are two major types of metalloproteases: metalloendopeptidases and metalloexopeptidases. Some common metalloproteases that have been studied are carboxypeptidase A and B, as well as thermolysin, which are digestive enzymes. Thermolysin can be observed in its production from the bacteria by the bacteria Bacillus thermoproteolyticus.
Mechanism of Metalloproteases
[edit | edit source]Metalloproteases contain on their active site usually a bound metal ion (most of the time Zinc). The metal ion activates a water molecule to act as a nucleophile and attack the peptide carbonyl group. Bound to the active site is a base that pulls a proton from the water molecule bound to the metal in order to turn the water molecule into a nucleophile to attack the peptide bond.

Catalytic Triad
[edit | edit source]A catalytic triad is a group of three amino acids that are found in the active sites of some proteases involved in catalysis. Three different proteases that have catalytic triads are: chymotrypsin, trypsin and Elastase. In chymotrypsin, the catalytic triad is made from serine 195, histidine 57, and aspartate 102. The side chain of serine is bonded to the imidazole ring of the histidine residue which accepts a proton from serine to form a strong alkoxide nucleophile in the presence of a substrate for attack. The aspartate residue orients histidine to make it a better proton acceptor via hydrogen bonding and electrostatic reactions. The combined cooperation results not only in better orientation and stabilization, but also in a sufficient nucleophile that is capable of attack.
Retrieved from "http://en.wikibooks.org/wiki/Structural_Biochemistry/Enzyme/Catalytic_Triad"
Catalytic Dyad
[edit | edit source]In contrast to the catalytic triad described above, the catalytic dyad comprises only two amino acid residues, usually one acting as nucleophile and the other one representing a proton donor to stabilize the product(s). A well known example is the HIV-1 Protease, in which the active site is formed by two aspartic acid residues (Asp25 and Asp25'), one residing in its deprotonated carboxylate form while the other one is protonated to the corresponding carboxylic acid.
S1 Pocket
[edit | edit source]
The S1 pocket helps to explain why chymotrypsin, trypsin, and elastase cleave certain peptide binds. The S1 pocket is a deep hydrophobic pocket that allows long, uncharged amino acids like phenylalanine and tryptophan to fit in chymotrypsin. Binding in the S1 pocket positions the adjacent peptide bond at the active site for cleavage. Trypsin cleaves peptide bonds after arginine and lysine which are amino acids with long and positively charged side chains because its S1 pocket contains an aspartate that is negatively charged which attracts and stabilizes the positively charged side chains of arginine and lysine in the substrate. Elastase cleaves peptide bonds after amino acids like alanine and serine which have small side chains because its S1 pocket has two bulky valine residues that decreases the size of the pocket opening so only small chains can enter.

In the figure to the right, it shows that some proteases can have more complex specificity patterns. there are more pockets on their surface to recognize other groups on the substrate. the substrate with the enzyme is the P group that is colored in red and these bind to enzymes labeled in blue. The sessile bond is the red bond between the carbon and nitrogen is also known as the reference point. AAA+ proteases are a type of enzyme that performs as quality control for proteins, and are found the regulatory circuits of all cells. Proteins may need to be degraded for a variety of reasons, whether to remove damaged proteins or for the purpose of regulation. As the process is irreversible, AAA+ proteases must be highly specific in order to avoid wasteful destruction.
Structure
[edit | edit source]The AAA+ protease consists of a hexameric ring of ATPases surrounding the active site, which is located within an interior chamber, known as the AAA+ ring. There are as many as six potential binding sites within the ring, but studies have shown that there is only a maximum of four sites occupied even at complete saturation. In addition, even a single subunit is sufficient to drive the mechanism of protein unfolding.
Attached to the AAA+ ring is a sequestered degradation chamber of a protease. The very narrow entry portals to this chamber can only allow unfolded polypeptides to enter, allowing a high degree of specificity. Therefore, the degradation of the folded protein, then the polypeptide chain, requires collaboration between both the active sites of the AAA+ ring and the protease.
Mechanism
[edit | edit source]First, the AAA+ ring recognizes the proper protein to be degraded by binding to an exposed peptide of the substrate, called a degradation tag or a degron. Degrons are found in most substrates, and are simply short, unordered sequences of peptides recognized by the enzyme. As the degron is pulled in through the upper pore of the AAA+ ring, surrounding ATPases drive the conformational changes that result in the unfolding of the protein. The denatured polypeptides are then translocated through a narrow pore into the degradation chamber of a peptidase, where proteolysis occurs.
Cdc48
[edit | edit source]The ATPase Cdc48 is a principal ATP driven-machine that is active in eukaryotic cells. Its physiological functions are crucial to many cellular processes that include cellular progession, homotypic membrane fusion, chromatin reconstruction, and transcriptional management, and metabolic regulation. Cdc48 is best known for its endoplasmic reticulum protein degradation by the ubiquitin proteasome system.
ATPase Cdc48 is fairly associated with cellular activities (AAA) and maintains the homohexameric, ring-shaped complex. Typically, Cdc48 is initiated by the ubiquitin proteasome system, which leads to lysosomal degradation. However, Cdc48 is discovered to have significant functions in selective autophagy pathways. Cdc48 will guide proteins into the ubiquitin proteasome system or into the autophagy when protein degradation is in process. This task allows the cell to rid of incompetent and defective protein by either of the degradation pathways.
Human diseases such as Alzheimer's disease, Parkinson's disease, and Huntington's disease are linked to protein degradation. Mutant Cdc48 induces defections in autophagy accounting for the accumulation of aggregates. Relatively, mutant Cdc48 proteins expose elevated ATPase activity and an immense number of conformational alterations of the N-terminal domain that may inflict an imbalance in cofactor binding along with ubiquitlated proteins affiliated with Cdc48.
References
[edit | edit source]- Sauer, Robert T.; Baker, Tania A. (2011). "AAA+ Proteases: ATP-Fueled Machines of Protein Destruction". Annual Review of Biochemistry. 80: 587–612. doi:10.1146/annurev-biochem-060408-172623. PMID 21469952.
- Stolz, Alexandra; Hilt, Wolfgang; Buchberger, Alexander; Wolf, Dieter H. (2011). "Cdc48: A power machine in protein degradation". Trends in Biochemical Sciences. 36 (10): 515–23. doi:10.1016/j.tibs.2011.06.001. PMID 21741246.
HIV-1 Proteases
[edit | edit source]HIV, Human immunodeficiency virus is commonly known to be responsible for causing AIDS. The HIV-1 Proteases, which also known as HIV PR is vital for HIV. HIV PR is an aspartic protease, meaning aspartate residues are used in the process of peptide substrates catalysis. Without HIV PR, HIV could not achieve maturation and it remains uninfected. Its importance in HIV survival has made HIV PR inhibitors the new widely studied agents to hope to become to key to find a cure for AIDS.
Structure
[edit | edit source]X-ray crystallography has been used for understand the structure of HIV PR and helped on new drug design. It shows the precise picture and help characterize HIV PR at atomic level. There have been over 160 structures discovered and many researchers have been testing for new available drugs. The main structure of HIV PR follows a specific sequence: Asp-Thr-Gly, a sequence commonly found in aspartic proteases. . Its structure is made with two identical subunits, possibly resulted from gene duplication, a structure.HIV-1 PR do demonstrate the characteristic of retroviruses and aspartic proteases. The molecule is confirmed as a homodimer with an active site similar to those of the aspartic protease. The difference between the aspartic proteases and HIV-1PR monomer is HIV-1PR’s dimer interface has 4 short strands, but aspartic proteases have 6 long strands in pepsins. Due to the structure difference its MW is only about 1/3 of pepsin. It’s active site triplet at c β strand, and the active site loop is d β chain with residues 30-35. Active site is found at Asp25, Thr26, Gly27 located on the loop. HIV PR is symmetric with the identical subunits but its substrate/inhibitor (Polypeptide) is asymmetric.
Mechanism of HIV Protease
[edit | edit source]Protease are enzyme catalyst with high specificity for the hydrolysis of peptide bonds. The mechanism was discovered using kinetics, affinity labeling and X-ray crystallography. The most agreed mechanism is the acid-base mechanism. A water molecule is activated by acid-base role of the active site of aspartate residues and the water molecule attacks the carbonyl carbon of the scissile bond as a nucleophile. The other proposed and widely accepted mechanism is described by Suguan which is based on the crystal structure of the aspartic protease complexes. The reduced peptide inhibitor of these aspartic protease complexes is crucial. At the active pH range, only 1 of the 2 active site of aspartic acids is unprotonated. Asp group with the negative charge and nucleophilic H2O is activated by the negatively charged Asp and then attacks the carbonyl group in the substrate scissile bond. It resulted in an oxyanion tetrahedral intermediate, and the protonated amide(nitrogen atom) rearrange and turn the tetrahedeal intermediate into hydrolysis products.
HIV PR Inhibitor Design
[edit | edit source]There have been numerous research done on HIV PR inhibitor to hope for finding the cure to AIDS, and most researches were focusing on designing HIV PR inhibitors based on classical substrates or transition-state analogs. From strategies on aspartic protease, the design of peptidomimetic inhibitors-renin inhibitors has been successful at the first level. The mechanism of these renin inhibitors was studied but there was limited contribution in relation to HIV PR inhibitors. Another strategy based on synthesis of peptide substrates analog. The nonhydrolyzable isostere, has a tetrahedral geometry is replacing the scissile P1-P11’ amide bond. This gives potent HIV PR inhibitors effective in virus replication in vitro. From X-ray crystallography the structure of the HIV PR was discovered and new inhibitors were designed according the structure. The HIV PR has symmetric active site, and a design two-fold (C2) symmetric or pseudo- C2 symmetric inhibitors. It was design to mimic the symmetry at the active site so the C2 axes of the enzyme and inhibitor were nearly superimpose for effective binding.
Drug Resistance
[edit | edit source]Although many new drugs design are developed, the effectiveness of these drugs are significantly decreasing due to drug-resistant and cross-resistant mutants. This is due to a HIV viruses high rate of replication and the high error rate of reverse transcriptase during the rapid butation. The drug resistance of HIV is what made the drug design difficult. The mutations affecting the binding sites and inhibitors and resulted in drug resistance, and there is at least 6 mutated residues found in HIV PR.
Reference
[edit | edit source]PudMed Web of Science STRUCTURE-BASED INHIBITORS OF HIV-1 PROTEASE Author(s): WLODAWER, A (WLODAWER, A); ERICKSON, JW (ERICKSON, JW) Source: ANNUAL REVIEW OF BIOCHEMISTRY Volume: 62 Pages: 543-585 DOI: 10.1146/annurev.biochem.62.1.543 Published: 1993
Ashraf Brik and Chi-Huey Wong, Org. Biomol. Chem., 2003, 1, 5
Department of Chemistry and the Skaggs Institute for Chemical Biology,
The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, CA 92037, USA
http://en.wikipedia.org/wiki/HIV-1_protease
Dehydrogenases
[edit | edit source]Dehydrogenases catalyze the oxidation of alcohols to carbonyl compounds by using either NAD+ or NADP+. Some dehydrogenases are specific for one coenzyme. This reaction can be reduced by NADH or NADPH. In the oxidation of the alcohol, one in dehydrogenases transferred to the 4 position of the nicotinamide ring of the NAD+ by removing two hydrogens. Therefore, the carbonyl group was made from the reaction. This reaction is stereospecific. The enzyme can either attack either side of the ring, resulting in different conformations and characteristics. The enzyme with the syn conformation will catalyze the reaction to reduce carbonyls groups more rapidly. The enzyme with the anti conformation is less reactive than the previous enzyme. Different types of dehydrogenases exist, some of which are briefly mentioned below.
1. The alcohol dehydrogenases
[edit | edit source]The alcohol dehydrogenase catalyzes the reaction of alcohols to aldehydes and ketones by using NAD+ as a coenzyme. It also has the zinc ion sites at the bottom of the enzyme. The zinc ion binds on NAD+ by during the catalysis. Mechanistically, the coenzyme binds to the enzyme by oxidizing the alcohol in the enzyme. The enzyme-NADH complex is dissociated to be rate determining.
2. L-Lactate dehydrogenase
[edit | edit source]L-lactate dehydrogenase oxidizes the reversible reaction of L-lactate to pyruvate by using NAD+ as a coenzyme. The alcohol group becomes the carbonyl group of the enzyme. L-lactate or pyruvate is binded to the enzyme through the coenzyme. Therefore, the coenzyme always binds on the enzyme first. Other substrates may bind to the enzyme as well. Lactate dehydrogenase is especially important for the role that it plays in glycolysis. In order for glycolysis to occur, NAD+ must be available. Under anaerobic conditions, there is not sufficient NAD+ available for glycolysis to occur because it is all stuck in the NADH form (the insufficient amount of oxygen means that no oxygen is present to receive electrons from the end of the electron transport chain). Lactate dehydrogenase allows for the occurrence of glycolysis by helping in the conversion of NADH to NAD+. Lactate dehydrogenase does this by converting pyruvate to lactate. The figure below explains this reaction.
O O OH O ‖ ‖ | ‖
H3C – C – CO- + NADH ↔ H3C – CH – CO- + NAD+
Pyruvate Lactate
As is shown, two electrons are removed from NADH and a proton is added in order for lactate to be formed and for NADH to be oxidized to NAD+.
3. Malate dehydrogenase
[edit | edit source]Malactate dehydrogenase oxidizes malate to oxaloactate in areversible reaction. NADH and NAD+ bind with equal affinity. In other words, it catalyzes, by means of NAD+ or NADP, the dehydrogenation of malate to oxaloacetate or the decarboxylation of maleate to pyruvate.
4. Glutamate Dehydrogenase
[edit | edit source]Glutamate dehydrogenase catalyzes the conversion of the nitrogen atom in glutamate into ammonium ions by oxidative deamination (See Reaction Scheme Below). In oxidative deamination, the reaction starts by dehydrogenation of the Carbon-Nitrogen bond, which then leads to an imine intermediate known as the Schiff-base intermediate. The first step utilizes glutamate dehydrogenase (GDH)and utilizes the coenzyme NAD+, which is reduced to NADH [5]. Next, hydrolysis of the Schiff base leads to α-ketoglutarate and the free ammonium ion. The reaction is driven in the forward direction due to quick removal of the ammonium ion. Glutamate dehydrogenase is located in the mitochondria of cells. Glutamate dehydrogenase is unique because, in some organisms, it is capable of using either NAD+ or NADP+ in its catalytic reactions. This ability is unique because NADPH is used as the reductant in biosynthetic reactions, while NAD+ is usually used as the oxidant in most catabolic reaction, and glutamine dehydrogenase is not specific to either.

Reference
[edit | edit source]Alan Fersht, Enzyme Structure and Mechanism.
Jeremy M. Berg, John L. Tymoczko, Lubert Stryer. Biochemistry (6ed). New York. W.H. Freeman and Company, 2007, 2002.
Aaron Coleman, Meredith Gould, Jose Luis Stephano. Biochemical Techniques. Hayden-McNeil Publishing, 2013.
What is Phenylketonuria (PKU)?
[edit | edit source]Phenylketonuria (PKU) is a genetic disorder in which the body cannot clear out the excess amount of amino acid phenylalanine (Phe). Individuals diagnosed with PKU lack the enzyme Phenylalanine hydroxylase (PAH), a protein used to break down Phe into another amino acid called tyrosine. This results in high levels of PAH in the human body, which is extremely toxic to the brain.
Early Prevention/Treatment of PKU
[edit | edit source]Approximately 1 in 14,000 individuals are affected by PKU. During pregnancy, chorionic villus sampling can be done to screen the unborn baby for PKU. Furthermore, newborns can be tested for the disorder so that they can be treated early if they are diagnosed. Under laboratory analysis, a blood sample taken from a newborn can be tested and screened for PKU. This genetic disorder can be treated by controlling the levels of Phe in the body. Some treatments include certain restrictive diets. For example, people with PKU cannot eat foods that contain aspartame, an artificial sweetener. They can consume a new sugar-substitute called neotame which is similar to aspartame except that it combines the two amino acids differently. The effect is that it is 30 times as sweet as aspartame, so less is needed and thus less phenylalanine is produced when it is metabolized. People are advised to follow the diet throughout their lives. Moreover, foods such as milk and diet drinks contain large amounts of Phe. One who has PKU would be advised to avoid heavy intake of these foods. Another example would be to take supplements and vitamins to ensure a healthy balance of essential amino acids. Another possibility in treating PKU would be treating one with the enzyme PAL. This enzyme facilitates the disposal of excess Phe. Further clinical trials will determine if PAL is safe for human intake.
Consequences of PKU
[edit | edit source]If medical actions and diet are not taken to prevent this condition, PKU can lead to mental retardation and neurological damage. Studies have shown that PKU affects the IQ of the affected individual and their ability to efficiently undergo neurological processes. Furthermore, studies have also shown that uncontrolled PKU affects cognitive functions such as processing speed, attention, inhibition, working memory, and motor control. Primarily for infants affected with PKU, discrepancy in working memory and inhibition are present. For children affected with PKU, discrepancy in working memory, inhibition, strategic processing, and response monitoring are present. For adults affected with PKU, insufficiency in working memory and attention are affected. Not only does a high level of Phe increase the neurological deficit in an individual, but it is also toxic to the human body.
Further Treatment of PKU
[edit | edit source]For further controlled and treatment of PKU, children and adults should undergo timely cognitive, neurophysical, and social-emotional testings and evaluations. For infants and children up four-years old, annual evaluations should be taken. Children during their elementary school years should undergo evaluations under a psychologist to evaluate any metabolic disorders twice a year. For individuals in and beyond high school, psychological examinations should be taken to evaluate signs of decreased metabolic control or lack of social acceptance.
References
[edit | edit source]BioMarin Pharmaceutical Inc. "Protecting the Brain: Testing and Treatment Approaches". PKU.com, 2009. Web. 29 Oct. 2011. <http://www.pku.com/Protecting-the-Brain/>
Meister, K. “Sugar Substitutes and Your Health.” Comprehensive Reviews in Food Science and Food Safety, 2006.
Overview
[edit | edit source]A carbonic anhydrase, or carbonate dehydratase, is a type of enzyme that rapidly catalyzes the conversion of carbon dioxide into a proton and the bicarbonate ion (HCO3-). This reaction is rather slow in the absence of the anhydrase catalyst, as the reaction with the enzyme takes place typically ten thousand to one million (10^4-10^6) times per second. The active site by which the enzyme binds contains a zinc ion (Zn2+), by which the pKa is lowered and allows for nucleophilic attack on the carbon dioxide group. In humans, this reaction mechanism is vital in maintaining pH balance and in transporting carbon dioxide out of the tissues and into the lungs. Carbon dioxide hydration needs a buffer because a buffer as we mentioned before can work as an acid or a base and in this case the buffer helps enzyme to reach its highest catalytic rate. In some cases, the active site of carbonic anhydrase is inaccessible to bulky buffers, interfering with efficient proton transfer. In response, carbonic anhydrase II developed a proton shuttle made up of a histidine residue that removes an H+ from the bound water molecule, activating its nucleophilicity, and then transfers the proton to the edge of the protein (allowing the buffer to easily remove it). Therefore the reaction uses both acid-base catalysis and metal ion catalysis strategies.

Structure
[edit | edit source]
In carbon anhydrase, as well as all biological systems, the zinc atom is in the +2 state. The zinc is bound to four ligands, three of its coordination sites are occupied by the imidazole rings of three histidine residues and a fourth is occupied by a water molecule. This active site is located in a cleft near the center of the enzyme.
Function
[edit | edit source]
Carbonic anhydrase is a catalytic enzyme specific to accelerating the formation of carbonic acid from carbon dioxide (CO2) and water (H2O):
H2O + CO2 ⇌ H2CO3
It is important to note that the carbonic anhydrase does not shift the equilibrium of the reaction but rather helps the equilibrium be reached much quicker, allowing for its high velocity yield of product. H2CO3 dissociate in blood, which gives this equilibrium:
H2CO3 ⇌ H+ + HCO3-
Carbonic anhydrase has been known to catalyze one million reactions per second. Also note that Carbonic acid readily dissociates into H+ and bicarbonate since it is a more unstable compound.
Mechanism
[edit | edit source]
pH affects carbonic anhydrase in a sigmoidal fashion. The higher the pH, the more active the enzyme is (since it is in the optimal conditions for deprotonation).
1) The binding of zinc lowers the pKa of water from 15.7 to 7, generating a hydroxide ion (OH-) to attack carbon dioxide. zinc releases a proton from a water molecule to generate this hydroxide ion. pH decreases as a result from the decrease in the pKa. According to Le Chatelier's principle, this drives the reaction towards deprotonation. Further, this pH change can result in the deactivation of the human phallus.
2) The carbon dioxide substrate binds to the enzymes active site and is positioned for optimal interaction.
3) The hydroxide ion (being a great nucleophile) attacks the carbonyl of carbon dioxide, converting it to bicarbonate ion through the nucleophilic attack. Oxygen on the carbon dioxide molecule forms an intermediate bond with the Zn metal during the conversion process.
4) The enzyme is regenerated and the bicarbonate ion is released. The enzyme is ready for another reaction to occur. This regenerative ability of this enzyme allows for this reaction to be highly efficient and kinetically fast to constantly process carbon dioxide within the blood cells.
The role of zinc
[edit | edit source]Zinc's role in carbonic anhydrase is to facilitate the water to create a proton H+ and a nucleophilic hydroxide ion. The nucleophilic water molecules attack the carbonyl group of carbon dioxide to convert it into bicarbonate. This is obtained through the +2 charge that the zinc ion has, which attracts the oxygen of water, deprotonates water, thus converting it into a better nucleophile so that the newly converted hydroxyl ion can attack the carbon dioxide.
Water naturally deprotonates itself, but is its a rather slow process and not in large quantities. Zinc deprotonates water by providing a positive charge for the hydroxide ion. Zinc alone cannot deprotonate water fast enough to reach the 106 per second rate that it has been measured, however, the proton is donated temporarily to the surrounding amino acid residues, which will later be given to the environment, while allowing the reaction to continue and not slowing down the process. Metal ions are good because it increases the reactivity of the chemicals and can create strong bonds. Zinc is able to help the deprotonation of water by lowering the pka of water. Binding of water to zinc lowers the pka of water from about 15.7 to 7. This means more water molecules are now able to deprotonate at a lower pH than normal, and this makes it easier for water to turn into a hydroxide ion which is a better nucleophile.
Specific cases of Carbonic Anhydrase
[edit | edit source]Carbonic Anhydrase can be used when carbon dioxide in the tissue diffuses into the human red blood cells. The Carbon Dioxide (CO2) reacts with water to form carbonic acid. Typically this reaction is catalyzed by Carbonic Anhydrase.
CO2 --enter red blood cell---> CO2 + H2O ---catalyzed by Carbonic Anhydrase--> H2CO3
A case of reducing the activity of carbonic anhydrase is found in a class of drugs used to treat glaucoma, neurological disorders, and ulcers. There are different types including methazolamide and brinzolamide. The mechanism of these vary from one type to the next, but they all inhibit the enzymatic activity of carbonic anhydrase.
References
[edit | edit source]1. Berg, Jeremy M. (2007). Biochemistry, 6th Ed., Sara Tenney. ISBN0-7167-8724-5. 2. Campbell, Neil A. Biology. 7th ed. San Francisco, 2005. Carboxypeptidase is a pancreatic enzyme that catalyzes the hydrolysis of the peptide bond at the carboxyl end of proteins and peptides, with a strong preference for amino acids with an aromatic or branched aliphatic side chain. The zinc ion is bound in a 5-coordinate site by two histidine nitrogens, both oxygens from a glutamic acid carboxyl group, and a water molecule. A pocket in the protein structure accommodates the side chain of the substrate. Evidence indicates that the negative carboxyl group of the substrate hydrogen bonds to an arginine on the enzyme while the zinc bonds to the oxygen of the peptide carbonyl. A Zn-OH or Zn-OH2 combination seems to be the group that reacts with the carbonyl carbon, with assistance of a glutamic acid carboxyl group from the enzyme that assists in the transfer of H+ from the bound water to the amino acid product. An artificial peptidase model compound has been made with a Cu(II) bound by four nitrogens in a chain that ends in a guanidinium ion, all attached to a cross-linked polystyrene. The catalytic activity is high for hydrolysis of amides with carboxyl groups attached, similar to a carboxypeptidase activity. The H+ on the guanidinium group can hydrogen-bond to the carboxyl group, holding the substrate in position near the Cu, which is the active site.
References
[edit | edit source]Gary L. Miessler, Donald A. Tarr, Inorganic Chemistry, Third Edition, 2004 There are specific enzymes that help perform function for a cell. A kinase is an enzyme that adds phosphate groups to proteins. This process is called phosphorylation. The importance of a kinase is that it marks the protein, instructing a cell to do something, such as to grow or to divide.
Structure
[edit | edit source]A protein kinase has two lobes that are different in structure and functionality. These differences add to catalysis and regulation in different ways. These differences are also what make protein kinases different from other metabolic kinases, such as ATPases. The smaller one of these two lobes is called the N-lobe. This contains a beta sheet but is mostly helices. The helical part is the core of the structure and is the part that protein substrates attach to. The smaller lobe is called the N-lobe. This lobe consists of five stranded beta sheets, along with a helix. [3]
Evolution
[edit | edit source]Eukaryotic protein kinases (EPKs) divergently evolved from eukaryotic-like kinases (ELKs), which are structurally much simpler. Although ELKs also have the same two lobes along with the adenine ring, the C-lobe of the EPK have two extra parts. One is called the Activation Segment and the other is an extra helical subpart that allows substrates to attach. These new sites of the C-lobe allow EPKs to precisely function and be highly regulated. EPKs were evolved from ELKs to further achieve faster and more efficient regulation. Firstly, the Activation Segment was inserted. Later, the extra helix was attached to the C-lobe, and this is the structure of protein kinase that we know. [4]
Protein Kinase A (PKA)
[edit | edit source]Protein kinase A is an enzyme that covalently attaches phosphate groups to proteins. It is also known as the cyclic AMP-dependent protein kinase. An extremely significant characteristic of protein kinase A is its ability to be regulated by the fluctuation of cyclic AMP levels within cells. Essentially, protein kinase A is responsible for all cellular responses due to the cyclic AMP second messenger. Cyclic AMP activates protein kinase A, which phosphorylates specific ion channel proteins in the postsynaptic membrane, causing them to open or close. Due to the amplifying effect of the signal transduction pathway, the binding of a neurotransmitter molecule to a metabotropic receptor can open or close many channels. [5]
Protein Kinase B (PKB)
[edit | edit source]Protein kinase B regulates various biological responses to insulin and growth factors. Akt is another way to classify Protein Kinase B. Protein Kinase B is a serine-threonine-specific protein kinase that contributes to multiple cellular processes such as glucose metabolism, apoptosis, and cell migration. [6]
Protein Kinase C (PKC)
[edit | edit source]Protein kinase C catalyzes the process of signals mediated by phospholipid hydrolysis. It is activated by the lipid second messenger, diacylglycerol. This lipid second messenger serves as the key initiation for most protein kinase C's. Protein kinase C isozymes consist of a single polypeptide chain that possesses an amino-terminal regulatory region and a carboxy terminal kinase region. The isozymes are categorized into various groups: conventional protein kinase Cs which are regulated by diacylglycerol, phosphatidylserine, and Ca^2+ in addition to novel protein kinase Cs which are regulated by diacylglycerol and phosphatidylserine. Activation of GPCR's, TKR's, and non-receptor tyrosine kinases can lead to protein kinase activation by stimulation of either phospholipase Cs to yield diacylglycerol, or phospholipase D to yield phosphatidic acid and diacylglycerol. Additionally, conventional protein kinase Cs are regulated by Ca^2+. [7]
Tyrosine Kinase
[edit | edit source]Tyrosine kinases function in multiple ways that involve processes, pathways, and specific actions that are key in the body. Specific receptor kinases function in transmembrane signaling whares tyrosine kinases within the cell function entirely different in the sense that they are part of signal transduction in the nucleus. The activity of tyrosine kinases in the nucleus involve cell-cycle control, such as differentiation in the different phases when the cell begins division, and also show properties in controlling certain transcription factors. Tyrosine kinase activity is also seen in mitogenesis or in other words the induction of mitosis in the cell. Specifically during this induction, tyrosine kinases phosphorylate proteins in the nucleus and in the cytosol. In addition, tyrosine kinase has been seen to be involved in cellular transformation due to the phosphorylation of a middle-T antigen on tyrosine, a change that is similar to cellular growth or in reproduction.
References
[edit | edit source]- ↑ 1
- ↑ 1
- ↑ http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3084033/
- ↑ http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3084033/
- ↑ http://www.vivo.colostate.edu/hbooks/molecules/pka.html
- ↑ http://jcs.biologists.org/content/118/24/5675
- ↑ http://www.upch.edu.pe/facien/facien2011/fc/dbmbqf/pherrera/cursos/receptores/pkc-cocb97.pdf
Cyclin-dependent kinases (CDKs) are protein kinases that regulate the cell cycle, transcription, and processing of mRNA. In order for them to function at all, these kinases bind to proteins called cyclins. These kinases phosphorylate their substrates on serines and threonines.
Evolution
[edit | edit source]Research has shown that cyclin-dependent kinases can be found throughout all eukaryotic cells. This was found by substituting human cyclin-dependent kinases into the corresponding kinases in yeast. Although it seemed doubtful due to the long evolution divergence between humans and yeast, it was found that yeast colonies still grew after promoting a human library into fission yeast and many transformations. The growth of these colonies greatly showed that the cell cycle was regulated the same way in human cells and in yeast, and thus all eukaryotic cells as well. This also led to show that model organisms, such as yeast, can be used to study issues in humans, such as diseases.
CDK2
[edit | edit source]Cyclin-dependent kinase 2 are kinases found in homo sapiens (humans).
Functions
[edit | edit source]Cyclin-dependent kinase 2 is regulated by cyclin E and cyclin A and only functions during the G1-S phase of the cell cycle. It is important for the G1/S transition in that cyclin E binds to CDK2, which is needed for when G1 transitions to the S phase, while cyclin A then binds to CDK2 to complete the transition.
Structure
[edit | edit source]Solving this kinase’s structure in 1993 showed that the activation site slightly blocks the binding site for ATP. Kinases typically have two lobes, and in this particular structure, the two lobes were in a closed conformation, and its C-helix was in the wrong direction that encourages binding with the triphosphate segment of ATP.
Reference
[edit | edit source]"Structural Basis for Control by Phosphorylation." Chemical Reviews. 06 Dec. 2012. Web. 06 Dec. 2012. <http://pubs.acs.org/doi/full/10.1021/cr000225s>
"Finding CDK: Linking yeast with humans." Nature Cell Biology. 06 Dec. 2012. Web. 06 Dec. 2012. <http://www.nature.com/ncb/journal/v14/n8/full/ncb2547.html>
MPS1
[edit | edit source]MPS1 kinases play many roles in mitosis. This kinases' most highly conserved and important role is to make sure the orientation of sister chromatids of spindles at kinetochores are correct. The kinetichores are structures that hold spindle microtubules and acts as a platform for signaling for the spindle checkpoint. In humans it is also involved in activating and maintaining spindle checkpoints.
Role in Spindle Pole Body
[edit | edit source]MPS1 kinase phosphorylates multiple spindle pole body (SPB) components. In fungi, Spc29 and Spc42 are phosphorylated, giving then stability. Centrin (Cdc31), Spc98 a γ-tubulin complex component, and Spc110 are some of the more conserved parts of SPB. Phosphorylation of Spc98 has possible effects on its contact with Spc110. Phosphorylation of some of these parts is necessary for them to interact with each other. If Mps1 is over expressed in mammals, centrosomes can be over duplicated, and overexpressed kinase-inactive allele will block centrosome duplication. However, some research with RNA interference has shown results that contradict the statement that Mps1 is needed for centrosome duplication. For example, Mps1 was removed from human cells with cre-lox, but these cells were still able to duplicate their centrosomes. Other species lack MPS1 orthologs but they can still perform centrosome duplication. Even with those contradictions, MPS1 still plays a role in centrosome duplication in humans. This is proven by the fact that substrates like centrin 2 are phosphorylated by MPS1 and is required for centriole, the core of a centrosome) stimulation.
Kinetochores and Spindle Assembly Checkpoint
[edit | edit source]
MPS1 is localized in kinetochores. The spindle check point is used to ensure bipolar attachment and tension of all the chromosomes are correct in the mitotic spindle. The cell pauses at its metaphase until every chromosome is attached correctly. When they are all checked to be good, the anaphase can start.
Localization of kinetochores in checkpoint proteins is needed for their function. Research not really shown kinetochore localization of checkpoint proteins being dependent on MPS1. However, some of these studies have shown that these checkpoint proteins can be loss from the kinetochore if MPS1 is inactivated. Also if MPS1 is over expressed, cell arrest can occur. It is concluded from these results that MPS1 has a large influence in the checkpoint signaling pathway. Another view if MPS1 is absent, several checkpoint activities are interrupted and devastating effects can occur, such as failure of spindle function. But it is difficult to tell if this is true, because MPS1 has many substrates.
MPS1 can carry out its role in the checkpoint without being in the kinetochore. Similarly, truncated alleles of hMPS1 in human cells that are not localized at kinetochores can still activate the mitotic checkpoint complex. MPS1 also helps in the forming of an interphase APC inhibitor that has some of the same components as the mitotic checkpoint complex. The importance of MPS1 in normal mitosis progression can be seen from the fact that cells lacking MPS1 have a faster mitosis.
MPS1 and Cytokinesis
[edit | edit source]RNA interference studies have shown that if there is no MPS1 multinucleated cells can occur. This also showed that MPS1 plays a role in helping the cell exit mitosis. MPS1 binds to Mob1, activating Dbf2 protein kinase. This complex plays a role in the mitotic exit pathway. MPS1 is then deactivated once the cell exits mitosis. MPS1 and cytokinesis can also be linked by the hMPS1-binding partner/substrate, which is part of the actin cytoskeleton. However it is not clear how hMPS1 affects MPS1's function.
MPS1 and Meiosis
[edit | edit source]If MPS1 function is disrupted in meiotic cells, then chromosomes can be divided unevenly. This could come from the mutated MPS1 inability to maintain the spindle checkpoint and failure to attach chromosomes to the meiotic spindle correctly. This suggests similar MPS1 function in meiosis and mitosis.
References
[edit | edit source]The MPS1 Family of Protein Kinases - Xuedong Liu and Mark Winey
Introduction to Bacterial Ribosomes
[edit | edit source]One of the key processes ribosome carries as a ribonucleoprotein complex is protein synthesis. In the process of ultracentrifugation, the bacterial ribosome sediments as a 70S particle, which is composed of a small subunit of 30S and a large subunit of 50S. The small subunit is composed of a 16S ribosomal RNA and 21 ribosomal proteins, while the large unit is composed of two 23S ribosomal RNA and 33 ribosomal proteins. The small subunit mainly take cares of the association with messenger RNA during the start of translation and decoding, which resolves the 3-nt codon that decides which amino acid to insert to the polypeptide chain. On the other hand, the large subunit acts as the peptidyl transferase center and hence is the site of the formation of peptide bond. The structures of a complete ribosome, including the 30S subunit and the 50S subunit, was solved to confirm that the ribosome is actually a ribozyme and the fact that catalytic sites indeed lie within ribosomal RNA components. These structures are essential factors in determining the mechanism of translation, but information about how the ribosome assembles into a stable multi-component complex is still very limited. Ribosome assembly is the core of ribosome genesis, which involves the following steps:
(1) the appropriate folding or ribosomal ribosomal RNA and ribosomal proteins
(2) the binding and unbinding of assembly factors
(3) the modification and translation of ribosomal proteins
(4) the binding of ribosomal proteins
(5) the processing, modification, and transcription of ribosomal RNA


Most of these steps are carried simultaneously with the transcription of the ribosomal RNAs, and the ribosome assembly process is carried through protein-binding events and RNA conformational changes. At these events, the binding of ribosomal proteins stabilizes the RNA folding process and eventually drives the RNA structure to the final native state. Overall, ribosome is an important protein structure used to study principles of RNA, RNA protein recognition, protein folding, and the assembly of multi-component complexes. Incorrectly assembled ribosomes may give rise to diseases, therefore studying ribosome biogenesis may offer insights in developing more efficient ways to treat diseases. It is essential for scientists to understand both temporal and physical pathways of intact ribosomes because that is how they can gain knowledge regarding the regulation and possible errors happened during the assembly of bacterial ribosome.
General Information
[edit | edit source]The structure of bacterial ribosomes is composed of over 50 proteins and three large domain RNA molecules. Modifications in the rRNA require dozens of gene products but the role of these modifications in ribosome function are not fully understood or seem nonessential. It is believed that these modifications are a part of stabilizing RNA structure or RNA-protein interactions, mediate translation, or as checkpoints in ribosome assembly. The development of certain biophysical methods have helped better the understanding of how bacterial ribosome was constructed along with how its structure leads to function. Ribosome assembles improperly can lead to various diseases in human body because ribosomes assembly plays an important role in cells as RNA protein recognition. Therefore, understanding the ribosome assembly is a need to see how they connect together. Studies how ribosomes are regulated helps to figure out how and why errors occur in assembly biogenesis. The three contributions have come from mass spectrometry, computational methods, and RNA folding studies. The metabolism of the bacterial ribosomes helps ensure the proper and timely synthesis of other components. Because ribosome is the large and complex macromolecule, proper product is important thus require an efficient steps during assembly process. The table below represents an order of events that happen during assembly of the ribosome but not necessarily in that exact order. All those events must occur but the pathway can be somewhat random and vary throughout nature.
The assembly of bacterial ribosomes is thought to progress through an alternating series of RNA conformational changes as well as protein-binding events. The assembly of these bacterial ribosomes occurs much faster and efficiently in vivo than in vitro.
| 1) Transcription of Ribosomal RNA- rRNA are transcribed as single transcription.
2) Synthesis of ribosomal proteins-about 55 ribosome protein synthesized in both transcriptional and post-transcriptional gene regulation mechanisms. 3) Ribosomal RNA modification-Ribosome proteins are modified at about 30-40 specific positions either by base methylation and/or pseudouridylation. The role of the assembly is to stabilize RNA structure or RNA protein interactions. 4) Ribosomal protein modification. 5) Ribosomal RNA processing-produce mature rRNA 6) Ribosomal RNA folding cotranscriptionally 7) Ribosomal protein binding cotranscriptionally 8) Assembly factor binding and release- bind at various times that promotes the orderly and efficient process of assembly.
|
In Vitro Assembly Map and Assembly Intermediates
[edit | edit source]

With advancing technologies that can easily determine the structure of ribosome with high resolution, experimental researches that were done in the previous decades were somewhat neglected. Instead of focusing on the structure of ribosome, these studies focused on determining the components of the ribosome. In the late 1950s, researchers came to the conclusion that various subunit components of ribosomes were actually different physical states of the same particle, which were regulated by the concentration of magnesium ions. Tissieres and Watson successfully demonstrated that the bacterial ribosome is composed of a 70S particle that could be either aggregated to form the 100S particle or broken down to the large and small subunits. Later in the 1960s, Traub and Nomura took a huge step forward. They demonstrated that additional components are required for an active 30S subunit to be assembled in vitro from free ribosomal RNA and ribosomal proteins. In another words, the ribosomal RNA and ribosomal proteins possess all of the information needed for the assembly of ribosomes. Recombinant ribosomal proteins, unmodified 16S ribosomal RNA transcribed in vitro, and 16S ribosomal RNA in combination with purified proteins can reconstitue to 30S particles. Also, the 30S particle contains three domains, including the platform region, the head, and the body; each domain of this subunit can be independently reconstituted. This piece of information is very significant because it proposes the possibility to see through the process of ribosome assembly simply by changing various components. Although this simple perspective from earlier researches seemed questionable to some scientists, it was a solid foundation for ongoing researches on ribosome assembly and biogenesis.
Mass Spectrometry
[edit | edit source]The three main applications of using mass spectrometry with ribosome are inventory of the different components, identifying modifications of ribosomes, and study of ribosome dynamics. The ability to map modifications has allowed scientists to study the changes within modifications as the species develops. This can pinpoint the specific role protein modifications have in assembly or translation. The analysis of isolated ribosomes is the only way to completely characterize modifications for new species. Ribosomal modifications leads to a specific genotype and studying the genotype can give hints to what the role of that ribosomal protein modification has in assembly or translation. iTRAQ mass spectrometry technique is used to examine the mutation in the assembly cofactor. The molecular weigh and the sequence of ribosome protein can be determined using this Mass spectrometry.The HDX application reveals which region of bound ribosomal protein is the most flexible because these regions are important during the translation step.
Computational Methods
[edit | edit source]The large number of atoms in ribosomes makes this method a challenge. Through this way, the flexibility of protein binding sites has been shown to directly correlate with the stage at which a protein binds there. Primary structure proteins bind at sites that are more rigid while secondary and tertiary structures bind on sites that are more flexible. In addition, computational methods has shown that the order of assembly is guided by electrostatic binding. These energies directly reflect changes in flexibility and electrostatics.
RNA folding and Ribosomal Binding, in vitro
[edit | edit source]Biochemical method provides dept understanding of the series of conformational changes that occur during the assembly process. The fastest folding regions is said to happen at the primary protein-binding sites and this binding site is required to produce mature ribosome protein subunits. The role of the fast self-folding of the 5’ domain is to be synthesized cotranscriptionally in assembly process. It is possible to assemble and examine the conformation change of ribonucleoprotein using reconstitution intermediate (RI) with a subset of 15 ribosomal proteins in which reconstitution reactions were carried out at low temperatures. The conformational change of RI intermediate occurred by heating up. This then form a new structure (RI*) that has the ability to bind the remaining five proteins and form 30S subunits. The flexibility of protein binding reaction is due to the differences in the accessibility of the RNA to probes at different temperatures. Also, protein is binding at different regions of the rRNA at different times due to the intervening RNA folding reactions. The temperature of the binding rates is dependent. This indeed revealed a unique kinetic barrier to binding of each ribosome protein.
References
[edit | edit source]James R. Williamson, Biophysical Studies of Bacterial Ribosome Assembly, Curr Opin Struct Biol. 2008 June ; 18(3): 299–304.
James R. Williamson, Zahra Shajani, Michael T. Sykes. Assembly of Bacterial Ribosomes. Annual Review of Biochemistry. 26 April 2011. 80: 501-26.
Overview
[edit | edit source]Restriction enzymes are DNA-cutting enzymes found in bacteria (and harvested from them for use). Because they cut within the molecule, they are often called restriction endonucleases.
In order to be able to sequence DNA, it is first necessary to cut it into smaller fragments. Many DNA-digesting enzymes (like those in your pancreatic fluid) can do this, but most of them are no use for sequence work because they cut each molecule randomly. This produces a heterogeneous collection of fragments of varying sizes. What is needed is a way to cleave the DNA molecule at a few precisely-located sites so that a small set of homogeneous fragments are produced. The tools for this are the restriction endonucleases. The rarer the site it recognizes, the smaller the number of pieces produced by a given restriction endonuclease.
Reference
[edit | edit source]http://users.rcn.com/jkimball.ma.ultranet/BiologyPages/R/RestrictionEnzymes.html
BamHI
[edit | edit source]BamHI( from BAcillus amyloli) is a type II restriction enzyme that derived from B. amyloliquefaciens , having the capacity for recognizing short sequences of DNA and specifically cleaving them at a target site. In order to understand how these restriction endonuclease bind to their specific sequence, BamHI became a popular research target. By crystallizing BamHI endonuclease, scientists understood how DNA binding proteins select their specific target from a variety of nonspecific sequences in the cell.
Introduction
[edit | edit source]In order for restriction endonuclease to bind to their target sequence, they have to first bound to a nonspecific DNA. These nonspecific endonuclease-DNA complexes are believed to be more hydrated at their endonuclease-DNA interface, stabilized by electrostatic interactions and resulted a low heat capacity change during binding. Without strong interaction or binding to the nonspecific sequence, these weak interactions contributed to the sliding ability of these endonuclease. Instead of steady bound to a sequence, the endonuclease slide along the sequence until a specific match of sequence is reached. The following is a summary of a research conducted by two scientists, Hector Viadiu and Aneel K. Aggarwal in 2000.
Research
[edit | edit source]Background
[edit | edit source]The purpose of this research is to determine how BamHI binds to its nonspecific DNA sequence and interact with its target sequence. BamHI tends to bind to the following sequence at a particular cleavage site, as indicated below with the straight line.
As a restriction enzyme, a change in the specific sequence, even a change as small as one base pair, is sensitive enough for the sequence to become nonspecific. The experiment starts by crystallizing nonspecific crystals with DNA sequence 5'-ATGAATCCATA-3' from solutions. In the sequence, GAATCC is similar to the target cleavage of BamHI. Study reveals the structure of BamHI in the presence of this similar sequence.
Catalytic Mechanism
[edit | edit source]BamHI is a catalysis mechanism that involves three different states. The first state, which is before the reaction occurs, is called the pre-reactive state. Next it's the intermediate state, known as the transition state. The last state is the post-reactive state. The following are the steps that of how the mechanism occurs: There is a water molecule and a glutamic acid present. This allows the glutamic acid to take a proton of water and extract the proton. This leaves a hydroxyl group which will have a nucleophilic attack on the phosphate atom. Since there are two metals, the negative charges get stabilized. Then, there will be a pentavalent phosphate, and the extra negative charge are stabilize by the metal ions. The phosphate will have an extra oxygen which causes the water in the solution to behave as a donor. This allows the tendency of phosphate to recover its coordination. Finally, the oxygen will attack the water molecule and the proton will go to the leaving group, resulting to the post-reactive state. Overall, when comparing the pre-reactive and post-reactive states, the slight movement phosphate is observed.

Contact between BamHI and DNA backbone
[edit | edit source]
Although the DNA sequence is similar, BamHI behaves completely different from a specific complex. Not only that the bottom of the BamHI dimer is loose, but the enzyme is also tilted 20 degree from its axis. As a consequence, substitution in base pair does not only affect interaction at that specific base pair, but the entire sequence and the conformation of the BamHI endonuclease. The change of conformation is indicated in figure1. In the specific complex, the alpha helix at the C terminus of one monomer unfold to interact with the other monomer. However, when the targeted sequence is altered, the complex becomes nonspecific. In a nonspecific complex, these monomers do not unfold and interact. Since there is no conformational change, amino acids from the BamHI sequence are far from the DNA and do not involve in any binding. Thus, cleavage does not occur.

DNA Cleavage
[edit | edit source]BamHI cleaves DNA in two ways. First, BamHI cleaves DNA by co-crystallizing with divalent cations. For example, BamHI will bind to two divalent metals. Second, BamHI cleaves DNA by directly binding to the DNA itself. In the active site, the charge of the enzyme is negative. The metals are positioned between the active site and DNA. Inhibitors of this reaction include calcium. BamHI cleaves the phosphodiester bond by donating a proton the to the second water molecule. There is a pre-reactive site and post-reactive site that indicates which sequences have been cleaved. The phosphate is in the 5' direction while the oxygen is in the 3' direction.
Conclusion
[edit | edit source]Conformational change of BamHI is only triggered when it detects a specific DNA sequence. Otherwise, the BamHI, where active site residues are pointing outward, does not interact or bind with the nonspecific sequence. The specificity of this BamHI endonuclease is vital because it avoids lethal cleavages at similar DNA sequence.
Everytime DNA replicates in bacteria, it is methylated. Restriction endonucleases like BamHI cannot cleave DNA sequences that are methylated. However, DNA from viruses will not be methylated, so they will eventually be cleaved by BamHI. If bacteria want to survive, they have to be adept at recognizing which sequences are methylated. When the enzyme recognizes the right site, the DNA is in proximity. When the DNA changes conformation, it is far from the active site so catalysis is not carried out. Many residues carry the catalysis and many are close to the active site in order to recognize certain substrates and make sure they are the correct ones.
BamHI is able to recognize particular sequences of DNA in a pool of millions of possible sequences by simply binding the any DNA. Then, they slide along the DNA strand to find the right sequence. When it finds the particular sequence, it will change conformation and embrace the DNA.
Reference
[edit | edit source]1. Viadiu H, Kucera R, Schildkraut I, Aggarwal AK, "Crystallization of restriction endonuclease BamHI with nonspecific DNA." J Struct Biol 1(81-5):, 2000.
2. Viadiu, Hector. Examples of Catalytic Mechanism. Biochemistry Lecture. Dec. 3,2012

Overview
[edit | edit source]Escherichia coli (EcoRI) is an endonuclease enzyme that is located in the restriction modification system. EcoRI is also isolated from the E. coli strain. EcoRI DNA is responsible for the specificity of E. coli strains harboring the fi+ drug resistance transfer factor RTFI. This is due to the introducing of two single strands staggered within the recognition sequence. Since EcoRI enzymes are considered to be the simplest sequence-specific DNA enzymes, they are well suited for the study of DNA sequence recognition by proteins. It is also possible to potentially compare two proteins interactions with the same DNA sequence using this enzyme.
http://www.scq.ubc.ca/wp-content/endonuclease2.gif
It is extracted from strains of E. coli and is part of the restriction modification system. In molecular biology, it is used as a restriction enzyme and it creates sticky ends with 5' end overhangs. The nucleic acid sequence where the enzyme cuts is GAATTC and the complementary sequence is CTTAAG. EcoRI is used in a wide variety of molecular genetics techniques including cloning, DNA screening and deleting sections of DNA in vitro. Restriction enzymes like EcoRI that generate sticky ends of DNA are often used to cut DNA prior to ligation, as the sticky ends make the ligation reaction more efficient. EcoRI can exhibit non site-specific cutting, known as star activity, depending on the conditions present in the reaction. Conditions that cause activity when using EcoRI include low salt concentration, high glycerol concentration, excessive amounts of enzyme present in the reaction, high pH and contamination with certain organic solvents. A procedure for large scale isolation of Escherichia coli RI endonuclease in high yield has been developed. The purified enzyme is homogeneous as illustrated by polyacrylamide gel electrophoresis and analytical sedimentation.
References
[edit | edit source]1. Modrich, Paul, and Donna Zabel. EcoRI Endonuclease: Physical and Catalytic Properties of the Homogenous Enzyme. USA: The Journale of Biological Chemistry, 2 Mar. 1976. PDF.
HindIII
[edit | edit source]HindIII is a type II restriction enzyme derived from Haemophilus Influenzae. HindIII is site specific and cleaves the DNA sequence AAGCTT through hydrolysis when the cofactor Mg2+ is present. The cleavage of this sequence results in 5' sticky ends:
5'-A |A G C T T-3'
3'-T T C G A| A-5'
This type II restriction enzyme is composed of four β-sheets and one α-helix. As a type II restriction enzyme, HindIII protects the host genome against foreign DNA.
Proposed Mechanism and Usage
[edit | edit source]HindIII first binds to the DNA backbone through hydrogen bonds and weaker forces such as Van der Waals. When arrived at the specific sequence, it was suggested that HindIII hydrolyses the DNA by having the Lys-92 stabilizes the nucleophilic water while the leaving hydroxide anion is stabilized by the Asp90.
As a type II restriction protein, HindIII is useful in that it cleaves DNA at very specific sites, different than those of type 1, which cleaves randomly at sites other than the specific site. As such, HindIII is used for genetic engineering and molecular biology. It becomes possible to add, delete, or change specific genes, which is very important when trying to change an organism's genome.
It is a type II site-specific deoxyribonuclease restriction enzyme isolated from Haemophilus influenzae that cleaves the palindromic DNA sequence AAGCTT in the presence of the cofactor Mg2+ via hydrolysis. The cleavage of this sequence between the AA's results in 5' overhangs on the DNA called sticky ends.
References
[edit | edit source]Tang, D et al. (2000). "Mutational analyses of restriction endonuclease-HindIII mutant E86K with higher activity and altered specificity". Protein Engineering Horton, N., Newberry, K. Perona, J. (1999). "Metal ion-mediated substrate-assisted catalysis in type II restriction endonucleases". Proc Natl Acad Sci U S A. Structural Biochemistry/Enzyme Catalytic Mechanism/XbaI
General Information
[edit | edit source]Oxidation-reduction enzyme
Uses coenzymes NADP+/NADPH and FAD/FADH2
In humans, this is a dimer of 478-amino acid subunits linked by disulfide bond
Two-stage reaction
1st stage: E + NADPH + H+ EH2 + NADP+
Oxidized E binds NADPH, immediately reduces FAD, producing NADP+.
E contains redox-active disulfide bond (Cys58-Cys63), which has accepted an electron pair in EH2 to form a dithiol (one S- is in a charge transfer complex with FADH-)
2nd stage: EH2 + GSSG E + 2GSH
GSSG binds to EH2
Cys58 nucleophile attacks one S of GSSG yielding mixed disulfide, promoted by His467' acting as general base
One GSH is kicked-off by protonation by His467' (general acid)
Cys63 nucleophile attacks Cys58 to form redox-active disulfide, kicking off second GSH
Reference
[edit | edit source]http://books.google.com/books?id=fSORbNITJ0QC&pg=PA6&lpg=PA6&dq=acid+base+enzyme+catalysis&source=bl&ots=SA3h6Afvdc&sig=0VpMXdejjvFQ7HdfgjSTt3602lQ&hl=en&ei=j48YS8qECouotgPajcyHBw&sa=X&oi=book_result&ct=result&resnum=8&ved=0CCsQ6AEwBw#v=onepage&q=acid%20base%20enzyme%20catalysis&f=false Nucleoside Monophosphate (NMP) Kinases are enzymes that aid in transferring the phosphoryl group at the end of a nucleoside triphosphate to the phosphoryl group that is on a nucleoside monophosphate. The challenge for NMP kinases is to promote the transfer of the phosphoryl group from NTP to NMP without promotoing the competing reaction - the transfer of a phosphoryl group from NTP to water; that is NTP hydrolysis. NMP kinases are known to have P-Loop Structures.

Function
[edit | edit source]Nucleoside monophosphate kinase catalyzes the reaction of ATP and NMP to yield ADP and nucleoside diphosphate (NDP). Other groups such as sugars and single-carbon groups can be substituted in this reaction instead of the phosphoryl group. One setback that may arise during this interconversion is trying not to allow the phosphoryl group from NTP to transfer to water. This can also be known as NTP hydrolysis. However, the ability of the enzyme to undergo induced fit—changing the structure in order to bind to a substrate—allows this reaction to occur successfully and bind to nucleotides instead of water. Therefore, NMP kinases is an example of catalysis by approximation. NMP kinases can exist in two forms: (1) free, or (2) bounded to substrates. NMP kinases are homologous proteins that have a conserved NTP-binding domain. This domain consists of a central beta sheet surrounded on both sides by alpha helices. NMP kinases are considered to be in P-loops because there is a loop between the first helix and the first beta strand, often in the animo sequence of X-X-X-X-Gly-Lys. The P-loop is called so because it is known to react with the beta phosphoryl group attached on the nucleotide.
Magnesium or complexes of NTP's are the true substrates for this reaction. The enzymes are essentially inactive in the absence of divalent metal ions such as the ones mentioned above. Nucleotides such as ATP bind these ions and it is the metal ion-nucleotide complex that is the true substrate for the enzymes. How does binding of a Mg or Mn ion to the nucleotide enhance catalysis? The interaction between the magnesium ion and the phosphoryl group oxygen atoms hold the nucleotide in a well-defined conformation that can be bound by an enzyme in a specific way. Magnesium ions are usually coordinated to six group in an octahedral arrangement. Typically, two oxygen atoms are directly corrdinated to a magnesium ion, with the remaining four positions often occupied by water molecules. Oxygen atoms of the alpha and beta, or the beta and gama, or alpha and gama phosphoryl groups may contribute, depending on the particular enzyme. Thus the magnesium ion provides additional points of interaction between the ATP-magnesium ion complex and the enzyme, increasing the binding energy.
ATP binding induces large conformational changes. For instance in the case of adenylate kinase the presence of the ATP substrate induces large structural changes in the kinase. This interaction caused the p-loop to be brought down onto the ATP to in order to interact with the beta phosphoryl group.The movement of the P-loop brings down the top domain of the enzyme to form a lid over the bound nucleotide. The ATP is held in position by the lid with the gama phorsphoryl group positioned next to the binding site for the second substrate NMP. The binding of NMP induces additional confomational changes. Both sets of changes ensure that a catalytically competent conformation is formed only when both the donor and the acceptor are bound, oreventing wasteful transfer of the phosphoryl group to water.
Another feature of NMP kinases, is that it interacts with the ATP substrate only after it forms a complex. ATP forms a complex with either a magnesium or manganese ion which provides more points for the substrate and enzyme to interact thus increasing the binding energy. There are isomeric forms to the metal ion-nucleotide complex depending on the interaction between the metal ion and the oxygen atoms attached to the phosphoryl groups.

General Information
[edit | edit source]Phillips Mechanism
Glu35 and Asp52 are catalytic residues
Enzyme binds hexasaccharide unit, residue D distorted towards half-chair to minimize CH2OH interactions.
Glu35 transfers H+ to O1 of D ring (general acid), C1-O1 bond cleaved generating resonance-stabilized oxonium ion at C1.
Asp52 stabilizes planar (transition state binding catalysis) oxonium ion through charge-charge interactions (electrostatic catalysis), SN1 mechanism.
Enzyme releases hydrolyzed E ring with attached polysaccharide, yielding glycosyl-enzyme intermediate, H2O adds to oxonium ion to form product and reprotonated Glu35, retention of configuration is result of enzyme cleft shielding one face of oxonium ion.
Catalytic residues were identified by chemical (group specific reagents) and molecular mutagenesis.
Reference
[edit | edit source]http://books.google.com/books?id=fSORbNITJ0QC&pg=PA6&lpg=PA6&dq=acid+base+enzyme+catalysis&source=bl&ots=SA3h6Afvdc&sig=0VpMXdejjvFQ7HdfgjSTt3602lQ&hl=en&ei=j48YS8qECouotgPajcyHBw&sa=X&oi=book_result&ct=result&resnum=8&ved=0CCsQ6AEwBw#v=onepage&q=acid%20base%20enzyme%20catalysis&f=false Isomerases are enzymes that catalyze the formation of a substrate's isomer. In other words, they facilitate the transfer of specific functional groups intramolecularly without adding or removing atoms from the substrate. This conversion can be simply represented in the form A → B, where A and B are isomers.
Isomerases are used in many biochemical pathways, including the citric acid cycle and the glycolytic pathway. All isomerases have Enzyme Commission numbers beginning in EC 5. A variety of isomerizations can be carried out, including racemization, cis-trans isomerization, enolization, and many others. Some examples of isomerases include triose phosphate isomerase, bisphosphoglycerate mutase, and photoisomerase.
Isomerases can help prepare a molecule for subsequent reactions such as oxidation-reduction reactions. For example, in the conversion of citrate to isocitrate in the citric acid cycle, isomerization prepares the molecule for subsequent oxidation and decarboxylation by transferring the hydroxyl group of citrate from a tertiary to a secondary position. Additionally, isomerases can catalyze phosphorylation reaction pathways throughout the Krebb Cycle by preparing the molecule for oxidation states. The change in position is facilitated through Isomerases without affecting the overall chemical composition of the substrate or product.
Definition
[edit | edit source]

Triose Phosphate Isomerase (TPI) is an isomerase that catalyzes the isomerization of dihydroxyacetone phosphate to and from D-glyceraldehyde 3-phosphate. It takes part in the glycolytic pathway, which is a biochemical pathway employed by many organisms. In the pathway, TPI's action takes its place directly after the splitting of fructose 1,6-biphosphate by aldolase. The objective of the glycolytic pathway is to metabolize glucose into two pyruvate molecules, also producing two ATP molecules. Hence, TPI is an enzyme that contributes to the production of ATP, the molecules used as an energy source by all organisms.
TPI is an example of a "kinetically perfect enzyme," which means that it catalyzes isomerization so quickly that the rate of reaction is determined by the diffusion rate of the substrate. This means it isomerizes essentially every molecule of TPI specificity that it encounters. TPI increases the rate of isomerization by ten degrees of magnitude. Part of TPI's kinetic perfection comes from it being an isomerase; the enzyme does not have to wait for multiple substrates to bind to the active site. Also, the mechanism (see below) involves few steps and involves the transfer of protons only.
Mechanism
[edit | edit source]The catalytic mechanism of TPI begins with the glutamate residue removing a hydrogen from one of the substrate's carbon atoms (see image), while the carbonyl oxygen deprotonates the nearby histidine residue, forming an enediol intermediate. The negatively charged histidine then deprotonates the original hydroxyl group, which yields an enolate-like product. The glutamic acid, now acting as an acid, adds a proton to the middle carbon to form the product, glyceraldehyde 3-phosphate. The net result is the original carbon-hydroxyl bond and carbon-oxygen double bond switching places.
One important aspect of the catalytic behavior is the restraining of the enediol intermediate shown in the upper-right portion of the image below. Under normal conditions this molecule loses its phosphate group, and this degradation occurs at a rate two orders of magnitude faster than the isomerization of the substrate. To counteract this undesirable decomposition, a small loop of residues closes over the active site while the reaction takes place. The reactive intermediate is enclosed in a conformation that does not favor its spontaneous decomposition.

References
[edit | edit source]1. Berg, Jeremy M. 2007. Biochemistry. Sixth Ed. New York: W.H. Freeman. 310-323. Ping Pong is also called the double placement reaction and it means that one or more products are released before all substrates bind the enzyme. One key character of this reaction is the existence of a substituted enzyme intermediate, in which the enzyme is temporarily modified. Classic examples of this mechanism are reactions that shuttle amino groups between amino acids and a-ketoacids.
The enzyme aspartate aminotransferase catalyzes the transfer of an amino group from aspartate to a-ketoglutarate. After aspartate binds to the enzyme, the enzyme accepts aspartate’s amino group to form the substituted intermediate. The first product, oxaloacetate, departs after that. Glutamate is released as the final product after the second substrate, a-ketoglutarate binds to the enzyme and accepts the amino group from this modified enzyme.
Enzymes with a ping-pong mechanism can exist in two states, E and a chemically modified form of the enzyme E*; this modified enzyme is known as an intermediate. In such mechanisms, substrate A binds, changes the enzyme to E* by, for example, transferring a chemical group to the active site, and is then released. Only after the first substrate is released can substrate B bind and react with the modified enzyme, regenerating the unmodified E form.
Enzymes with ping–pong mechanisms include some oxidoreductases such as thioredoxin peroxidase, transferases such as acylneuraminate cytidylyltransferase, and serine proteases such as trypsin and chymotrypsin. Serine proteases are a very common and diverse family of enzymes, including digestive enzymes (trypsin, chymotrypsin, and elastase), several enzymes of the blood clotting cascade and many others. In these serine proteases, the E* intermediate is an acyl-enzyme species formed by the attack of an active site serine residue on a peptide bond in a protein substrate. A short animation showing the mechanism of chymotrypsin is linked here.
The name pingpong reaction came for the substrates appearing to bounce on and off the enzyme to a pingpong ball in the Cleland notation.

An acetyltransferase is an enzyme that catalyzes the transfer of acetyl groups.
p300/CBP
[edit | edit source]p300 and CREB binding protein (CBP) are both acetyltransferases that are highly conserved. Both have specific domains within the enzyme: three cysteine-histidine rich domains, bromodomain, and acetyltransferase domain. CBP/p300 phosphorylation activates acetyltransferase activity, which increases histone acetylation at target promoters and facilitates CBP/p300-dependent transactivation of genes. The acetyltransferase domains in these enzymes also uncovered autoacetylation activity. Autoacetylation of these enzymes causes a conformational change of the histone as well as the enzyme itself that directly affects the enzyme's ability to interact with Mediator, which is a multi-protein complex that plays an important role in the beginning stages of transcriptional activation.
TIP60
[edit | edit source]TIP60 is party of the MYST family of acetyltransferases conserved from yeast to humans. It acetylates core histones H2A, H3, and H4. This enzyme plays a crucial role in DNA damage response and apoptosis. TIP60 acetylizes the ataxia telangiectasia mutated (ATM) kinase, which is the central kinase in the repair pathways started by DNA lesions. Studies also show that ATF2 interacts with TIP60 and modulates its function at the DNA damage response step. Before DNA damage, ATF2 has low TIP60 levels by facilitationg TIP60 degradation. After DNA damage, TIP60 and ATF2 are more loosely associated. In addition, the sumoylation of TIP60 plays a key role in the localization and catalytic activity of the enzyme, influencing the enzyme's ability to help with DNA damage. Through experiments and observations, researchers have concluded that there is an important link between TIP60 levels and the signals generated by DNA damage response pathways. Evidence shows that human prostate cancer samples display loss of TIP60 and human lymphomas and mammary tumors display loss of TIP60.
Reference
[edit | edit source]Mellert, Hestia S. and McMahon, Steven B. "Biochemical pathways that regulate acetyltransferase and deacetylase activity in mammalian cells." Trends in Biochemical Sciences Vol. 34 No. 11. 2009. Deacetylase is any enzyme that removes acetyl groups from other proteins.
HDAC1
[edit | edit source]Histone deacetylase (HDAC) is an enzyme responsible for the effects elicited by the broad spectrum deacetylase inhibitors. HDAC1 is also required for active transcription of certain proteins. This enzyme is specifically acetylated during the phase of transcriptional decline. The acetylation of HDAC1 is done by the acetyltransferase p300. So, HDAC1 has a key role in transcription, which shows that the regulation of acetyltransferases and deacetylases influence gene expression. Understanding the HDAC function and how these enzymes are regulated by cellular signaling pathways has various clinical implications and is critical for researchers to use these inhibitors to create more efficient drugs. Additionally, HIDAC regulate the transcription of proteins involved in cancerous cells. By removal of acetyl groups from histones, HDACs create a non-permissive chromatin conformation that prevents the transcription of genes that encode proteins involved in tumorigenesis. In addition to histones, HDACs bind to and deacetylate a variety of other protein targets including transcription factors and other abundant cellular proteins implicated in control of cell growth, differentiation and apoptosis
SIRT1
[edit | edit source]The activity of SIRT1 is regulated by its phosphorylation status. This enzyme is directly phosphorylated by cyclin B-CDK1 complex. This complex is the first evidence of a kinase targeting SIRT1. Through this, the relevance of SIRT1 phosphorylation will be necessary for grasping how SIRT1 phosphorylation will regulate deacetylase activity.
Reference
[edit | edit source]Mellert, Hestia S. and McMahon, Steven B. "Biochemical pathways that regulate acetyltransferase and deacetylase activity in mammalian cells." Trends in Biochemical Sciences Vol. 34 No. 11. 2009.
Introduction
[edit | edit source]The diversity of proteins and nucleic acids is much due to the many mechanisms that allow site-specific additions of chemical groups into macromolecules. These modifications done to macromolecules were considered to be only nucleophilic, such has in DNA methylation. However, after the discovery of the Radical-SAM (S-Adenosylmethionine) enzyme family, a lot of protein and RNA modification reactions were found to be done through radical mechanisms rather than nucleophilic mechanisms. Because free radicals are very reactive, it allows any site of the target substrate to be activated for modification. So, this free radical mechanism discovery expanded the number of modified monomers, creating diversity. However, these reactions are more difficult to control. The study of these radical-based mechanisms through Radical-SAM enzymes are still at the beginning stages. These mechanisms require detail structural characterization of enzymes in complex polymers, which is a problem since the 3-D structures for many of these polymers are unknown still.
Radical-SAM enzymes
[edit | edit source]Many modifications can occur through radical mechanisms. Protein modifications, the simplest being the glycyl radical, can occur via radical mechanisms. In addition, these radical-based mechanisms are also at work in post-transciptional modifications of nucleosides. These Radical-SAM enzymes creates a radical in a specific polymer, and this radical can bind to methyls, thiols, and other groups, creating more complex molecules. Glycine radicalization done by converting a glycine into the radical form, generated by a "Radical-SAM" activase. This process causes a conformational change in the structure. Radical-SAM enzymes also are involved in the addition of methylthio (CH3 and SH) groups into proteins and transfer RNAs. First, there is a hydrogen atom abstraction by the 5'-deoxyadenosyl radical. Then, there is a sulfuration of the substrate radical to make an intermediate thiol. Lastly, there is a nucleophilic methylation by SAM. Then, two different activities happen, both SAM dependent. First, the catalyze radical C-H to C-S. Second, they function as SAM-dependent methyltransferases.
Structural organization of Radical-SAM enzymes
[edit | edit source]These SAM enzymes have a conserved core with a mixture of alpha and beta structure forms. A six-stranded parallel B-sheet binds SAM and a [4Fe-4S] cluster at equivalent positions for all the radical enzymes. SAM binds at the groove formed between adjacent B-strands connected to alpha helices that pack on opposite surfaces of the B-sheet. This specific packing geometry produces a concave surface that has different degrees of curvature based on the specific Radical-SAM enzyme family. These different degrees of curvature are very important because it contributes to their specificity for diverse substrates ranging from small molecules to macromolecules.
Reference
[edit | edit source]Atta, Mohamed, Mulliez, Etienne, etc. "S-Adenosylmethionine-dependent radical-based modification of biological macromolecules." Current Opinion in Structural Biology. 2010.
FeMo Cofactor and FeFe-Hydrogenase
[edit | edit source]FeMo Cofactor and FeFe-Hydrogenase are the enzymes with unique complex iron-surfur at their active site. They are used for nucleotide binding and hydrolysis. In order to synthesize properly and be inserted in to the structural enzymes, they need specific maturation machinery.
F-S clusters
[edit | edit source]The iron-sulfur clusters are the active site of both FeMo cofactor and FeFe-Hydrogenase. They are metal cofactors which exist as diver form such as [2Fe-2S], [4Fe-4S], [3Fe-4S]. They not only meditate electron transfer, gene expression, and catalysis but also play critical roles in central metabolic process such as respiration, photosynthesis, and the catalytic interconversion of small molecules. Each clusters has different protein environment. For example, the iron in the [2Fe-2S] cluster is coordinated with two cysteine thiolates and in the [4Fe-4S] cluster, each iron is coordinated with one cysteine. The different protein environments influence on the redox potential of the clusters.
Similarities
[edit | edit source]1. They are Modular inorganic/organometallic nanocrystals
2. 2Fe subcluster has only minimal protein coordination and possess unusual protein ligands that influence on the reactivity of the cluster
Nitrogenase FeMo cofactor (iron-Molybdenum (FeMo) cofactor at the active site of nitrogenase)
[edit | edit source]Nitrogenase FeMo cofactor is a hetero metallic cluster that is [4Fe-3S] partial cubane fused to [Mo-3Fe-4S] partial cubane through three cubane bridging sulfides. It has nonprotein ligands and a homocitrate which coordinates the Mo via hydroxyl and carbocylate moieties or coordinates all six Fe ions of the cofactor core.
Nitrogen fixation
[edit | edit source]It is a process which nitrogen is converted to ammonia. It plays a major role in nitrogen cycling, and Its availability of fixed nitrogen is a limiting factor for global nutrition. Also, it is catalyzed by nitrogenase enzyme which is not expressed by eukaryotes. The form which contains heterometal independent, Mo-nitrogenase is the most common.
Mo-nitrogenase
[edit | edit source]FeMo cofactor is located in the core of the active site of the Mo-nitrogenase and takes part in catalyzing the reduction of N2 to NH3, which has high activation energy. Mo nitrogenase is composed of two proteins, the Fe protein and the MoFe protein. During the catalysis, two proteins associate and dissociate in order to couple nucleotide binding and hydrolysis to intermolecular electron transfer from the [4Fe-4S] to the FeMo cofactor active site in charge of P cluster
Synthesis of Iron-Sulfur cluster
[edit | edit source]The involvement of FeS cluster is a precursor to the complex cluster. In order to bring S and Fe together to produce simple [2Fe-2S] and [4Fe-4S] clusters, NifS and NifU are used. For the iron-sulfur cluster assembly, the Isc and Suf pathways are involved. The Isc machinery includes IscU, assembles iron and sulfide, and makes [2Fe-2S] and [4Fe-4S] clusters. After the assembly, it delivers the clusters to target proteins. In the Suf machinery, proteins SufU and SufA serve as assembly scaffold and delivers the cluster to target proteins. In the process of assembly, a source of electros is required. In the case of Isc system, sulfur is reduced or two [2Fe2S] is fused to form a [4Fe-4S] on IscU. There is also a chaperon system, which is composed of HscA ATPase and cochaperone HscB in the Isc macinery. The HscA interacts with a motif on IscU in order to transfer the cluster from IscU to the target protein.
Assembly Scaffold
[edit | edit source]synthesis of the FeMo cofactor occurs on the NifEN heterotetramer that carries out nitrogenase maturation and incorporation of iron, molybdenum, and homocitrate into the FeMo-cofactor precursor. NifEN scaffold interacts with NifB and NifH. NifEN, NifB, and NifH play a major role in synthesizing the FeMo cofactor.
Hydrogenase H cluster
[edit | edit source]Hydrogenase H cluster is composed of a diiron cluster with unusual nonprotein ligands bridges to a [4Fe-4S] cluster. In the [4Fe-4S] subcluster, one of the coordinating cysteine thiolates bridge between [4Fe-4S] subcluster and the 2Fe subcluster, which each Fe ion in the cluster is coordinated by terminal carbon monoxide and cyanide ligands with an additional Fe-bridging carbon monoxide ligand. They are two enzymes that take part in Hydrogen metabolism ; [NiFe]-Hydrogenases that are related to bacteria and archaea, and [FeFe]- hydrogenases that are found in bacteria, protists, and green algae. Both of them function either in hydrogen oxidation coupled to energy yielding reaction or in recycling reduced-electron carriers. They have a specific active-site domain or subunit containing the H cluster or heterobimetallic NiFe centers.
Enzyme Catalysis: Summary
[edit | edit source]Enzymes accelerate reactions using :
Proximity and orientation effects
Electrostatic catalysis
Preferential Transition State Binding
Induced fit
General acid/base catalysis
Covalent catalysis
Metal ion catalysis
Enzymes accelerate chemical reactions by lowering the activation energy, DG‡.
Strong binding of the transition state and weak binding of the substrate leads to the maximum rate because all the binding energy is used to lower DG‡.
Any process that uses some of the binding energy for another purpose, e.g., a protein conformational change, will lower the maximum rate.
Under physiological conditions, the maximum rate is achieved by maximizing kcat/Km AND having Km greater than the physiological [S]. This means that most of the enzyme will be free to interact with a substrate molecule and that every encounter will lead to a reaction.
Reference
[edit | edit source]http://www.bmb.psu.edu/courses/bmb401_spring2004/lecture_notes/lecture11_2004.pdf
Membrane Traffic
[edit | edit source]Numerous methods such as biochemical and genetic approaches have been used to determine the process of protein secretion and endocytosis. These processes are pertinent as it explores many new issues that involve cell biology and human physiology.
Golgi Complex
[edit | edit source]The Golgi is made up of microtubules that yield dispersed mini-stacks that functions for protein secretion. The Golgi complex contains cellular compartments of stack, flattened cisternae. These cellular compartments define the cis, medial, trans of the Golgi network compartments. The cellular compartment, cis-Golgi SNARES shows less active trafficking than trans- Golgi SNARES. Unfortunately, protein localizations in and around the Golgi does not reveal transport direction of vesicles and whether or not they’re about to depart or arrive.
Vesicles
[edit | edit source]There are two identified class of COPI-coated vesicles. One class contains KDEL receptors which serve as retrograde carriers from the Golgi to the endoplasmic reticulum (ER). The other class carried Golgi-restricted GS28 SNARE protein together with anterograde cargo. An experiment was conducted that used Golgi tethers to isolate the different classes of Golgi-derived transport vesicles. COPI vesicles where generated in vitro and were found to be enriched in Golgin-84. CASP, a Golgin can bind to Golgin- 84 and localize to the cisternal membrane. CASP vesicle lacks members of a p24 family but is substituted with Golgi enzymes and mannosidase I and II. This substitution proves that these vesicles are retrograde carriers for transport. In contrast, p115 protein vesicles were enriched in p24 family members, cargo proteins and Ig receptor but not mannosidase I and II. This suggests that the latter vesicles are anterograde carriers from within the early Golgi. Vesicle markers have been used for vesicle isolation and characterization.
Other proteins on the vesicle, such as the ArfGAP, functions as a structural coat component. Another important protein is the COG (conserved oligomeric Golgi complex) which has been revealed that the loss of this subunit leads to hypoglycosylation of multiple classes of proteins due to mislocalization of certain glycostyltransferases.
Because the Golgi is structurally composed of the stack by lateral fusion, it is poised for homotypic fusion. It also undergoes fission reaction when microtubules depolymerize.
Vesicle Fission
[edit | edit source]Coat proteins on membrane surface create stabilization and new generation of membrane curvature. A BAR domain takes on a banana shape that is best adapted to interact with acidic, curved lipid membranes. ArtGAP1 becomes concentrated at the edge of a promising bud and influences the amount of Art-GTP there. GTPase, specifically Dynamin plays a critical role in cellular membrane pinching.
Studies show a satisfying framework that helps explain vesicle and budding from the compartment. But more studies are still underway as detailed explanations of membrane traffic are still needed.
SNARE Proteins
[edit | edit source]Proteins that get secreted undergo translation at the endoplasmic reticulum (ER) where they get folded, modified and later transported in vesicles to the Golgi. Once at the Golgi, proteins undergo modification once again before they are exported to either the cell surface, the endocytic compartment or back to the ER. SNARE proteins. Transport intermediates vesicles contain SNARE proteins that promote membrane fusion in target proteins. Tethering factors and SNARE proteins work together to facilitate the docking and fusion process of cell transportation. Tethering factors interact with SNARE and also facilitate in SNARE assembly. Multiple copies of SNARE are required in order to initiate bilayer fusion. These SNARE complexes are recycled after completion of membrane transfusion. SNARE proteins provide specificity for membrane traffic because they are not allowed to engage with other components outside of the cell.
Members of the Rab and Arf branches of the Ras GTPase superfamily are present in every step of intracellular membrane traffic. They regulate these steps by networking with one another through a variety of mechanisms that coordinate independent events of one stage together with other stages of the entire transport pathway. These mechanisms include many different variables:
- GEFs cascades
- GAPs cascades
- effectors that bind many GTPases
- positive feedback loops stemming from exchange factor-effector interactions.
When these mechanisms come together, an ordered series of transitions from one GTPase to the next can take place. Since each GTPase has its own unique group of effectors, the transitions that occur can help define differences in the functionality of the membrane compartments that they are associated with.
Dynamin is a considered model for large GTPases. It is responsible for endocytosis, a process in which cells absorb molecules by engulfment. Specifically, it is involved in the division of newly formed vesicles from the membrane of one compartment to their fusion with another compartment-- at both the cell surface or Golgi body. Along with division of vesicles, Dynamin is also involved in the division of organelles, cytokinesis, and pathogen resistance (microbial). In mammals, there are 3 different types of genes:
Reference
[edit | edit source]1. Pfeffer, Suzanne. “Protein Unsolved Mysteries in Membrane Traffic. Annual Review of Biochemistry. Vol. 76: 629-645 (Volume publication date July 2007) DOI: 10.1146/annurev.biochem. 76.061705.130002.
2. Mizuno-Yamasaki, E., F. Rivera-Molina, and et al. "GTPase networks in membrane traffic.." Pub Med. N.p., 29 2012. Web. 7 Dec 2012. <http://www.ncbi.nlm.nih.gov/pubmed/22463690>.
3. "Dynamin." Wikipedia. Wikimedia Foundation, Inc. 2 Apr 2012. Web. 7 Dec 2012. <http://en.wikipedia.org/wiki/Dynamin> Organisms that live in perpetually cold environments have enzymes that function very effectively in the cold. Some examples of this include fish, which have also evolved to develop antifreeze proteins as a way of adapting, and some prokaryotes that have to live in cold places. Recent discoveries have uncovered that further investigation of these enzymes could actually have biotechnological applications. The most commonly referred to cold-adapted enzyme is the alpha amylase from Pseudoalteromonas haloplanktis (AHA), a type of prokaryote. The ability of organisms to thrive in cold environments comes from their capacity to synthesize cold-adapted enzymes. Organisms that thrive in cold environments are called psychrophiles. These enzymes have developed a range of structural features that allows for high flexibility particularly around the active site, low-activation enthalpy, low-substrate affinity, and high specific activity at low temperatures. The study of the structure, function, and stability of cold-adapted enzymes is rudimentary in the research into protein folding and catalysis, a still developing field.
Thermal adaptation
[edit | edit source]Different organisms have evolved in different ways, causing them to adapt to different thermal environments that suits them. Thermal adaptation in extremes is particularly hard to adapt to and has a limited range that an individual organism can tolerate. Psychrophilic microorganisms, those that have adapted to the cold, are said to be able to metabolize in snow and ice at −20°C. Some psychrophilic can even proliferate at ≤0°C but are restricted to temperatures <30°C (2–5).
Since there are so many microorganisms found in the world's oceans, cold alpine regions, caves, upper atmosphere, and polar regions, a large proportion of biomass, or living organisms in a certain region, on Earth is generated at these cold temperatures in their specific areas. Organisms from the three domains of life: Bacteria, Archaea, and Eucarya, are also from these cold environments. Most cold-adapted enzymes are found to have come from prokaryotes. In order to survive, all these organisms have to be at thermal equilibrium with their surrounding environment. This comes from all the components of their cells being appropriately adapted to the cold. In order to adapt to the cold, potential mechanistic diversity and cell-specific adaptation strategies have been adopted. A general pattern that has emerged from research of these organisms is that organisms that live in permanently cold environments usually develop enzymes that help them function capably in the cold

An example of a cold-adapted enzyme which will be further studied and explained is the α-amylase from Pseudoalteromonas haloplanktis (AHA). This enzyme is the most extensively studied cold-adapted enzyme
Activity of Cold-Adapted Enzymes
[edit | edit source]Arrhenius equation below describes the rate of all reactions including enzymatic reactions.
kcat = AKe-Ea/RT
kcat = enzyme reaction rate (increases with an increase in absolute temperature (T), decreases in activation energy (Ea)
A = preexponential factor
K = dynamic transmission coefficient (generally assumed to be 1)
R = universal gas constant (8.314 J mol−1 K−1)
According to this equation, due to low temperatures from 0°–4°C, an inadequate amount of kinetic energy is available for the system to overcome barriers. Some strategies that help compensate the slow metabolic rates include: an energetically expensive strategy of increasing enzyme concentration, seasonal expression of isoenzymes in fish and nematodes, and the evolution of enzymes in which reaction rates tend to become more temperature independent and instead approach diffusion control. Cold-adapted enzymes tend to shift their optimum temperature of activity to a lower temperature with a concurrent decrease in stability. These enzymes show a high-reaction rate when they decrease their activation free-energy barrier between the ground state and transition states according to the equation below:
ΔG#=ΔH# -TΔS#
ΔG# = activation free-energy barrier
ΔH# = is the change in activation enthalpy
ΔS# = is the change in activation entropy
T = is the absolute temperature
Stability of Cold-Adapted Enzymes
[edit | edit source]The structure of cold-adapted enzymes are highly flexible which allows them to unfold at low to moderate temperatures. Researches have tried many methods to study the unfolding and folding transitions in order to determine the way they unfold and measure the kinetic and conformational stabilities of enzymes. Such methods include spectrophotometric, calorimetric, and electrophoretic methods. Experiments have been conducted using these techniques on multidomain proteins like chitobiase. Proteins have a tendency to unfold and such processes are irreversible. When large multidomain proteins unfold due to heat, these proteins tend to be kinetically driven to finish the unfolding process.
-
 Structure of Chitobiase
Structure of Chitobiase

The only example of a cold-adapted enzyme that has reversible unfolding is AHA. At temperatures of 20°C or above, AHA has reversible unfolding that is shown by 100% recovery of ΔH cal., the total amount of heat absorbed during unfolding, during a second scan after the initial thermal denaturation. Small-molecular weight enzymes unfold with the process of cooperative unfolding. Cooperative unfolding happens because of its tightly packed bulk which causes a small number of interactions between other structural elements, helping it conserve its natural state. If the limited number of interactions in cooperative unfolding were to be disturbed, a two-state unfolding may occur due to increased interactions.
In the enzyme pancreatic porcine α-amylase (PPA) and more stable mutants of AHA, the two-state unfolding process does not occur. Instead, other types of folding could come from increased ionic interactions, causing the frequency of intramolecular discrepancies to occur during folding. AHA mutants show this concept of discrepancies or divergence, causing the rate of thermal inactivation to be directly comparable to the extent of reversibility.
In the enzyme TUG-GE, at the temperatures 3°C and 12°C, AHA unfolds reversibly and shows two transitions. The transition that unfolds at a lower urea concentration belongs to the active-site region. The active site is formed by cooperative unfolding of structures, showing independent unfolding of other more stable regions, or the domains, of the protein.. The substrate-binding region of the cold-adapted enzyme is found to be the most flexible region when the unfolding starts due to its high Km. The research on AHA shows that instability of active-site region is key to heat-labile enzymes, and is an important concept in examining a broader range of cold-adapted enzymes
Kinetic Stability
[edit | edit source]The process of enzyme inactivation or denaturation is shown with kinetic stability. Most cold-adapted enzymes have a half-life of less than 20 minutes at the temperature of 50°C, some enzymes even denature at lower temperatures. In order to predict kinetic stability, it is essential to consider the magnitude of the free-energy change between the folded, or active, state and the transition state shown below by:
F K¦↔ 〖TS〗^# k¦→ D
F = folded enzyme
K = equilibrium constant
K= first-order rate constant
D = denatured state
Cold-adapted enzymes increase the rate of thermal unfolding with a decreased ΔG# shown by the equation:
ΔG#= =RT ln K
Reduced thermostability of cold-adapted enzymes could be due to low ΔH# of the folded form. A certain number of monovalent interactions need to be broken to reach transition state (TS#) and a reduction in this number causes the low number of the folded form. An example of an enzyme that has low thermostability, a direct result from increased disorder of the transition state, is the enzyme glutamate dehydrogenase. During unfolding, a key note to the decreased entropy of thermostable enzymes could have come about from the hydration of nonpolar groups. This is caused by water forming ordered structures around hydrophobic side chains thus decreasing the entropy o the system. It is vital to determine activation parameters of denaturation in order to discover a larger range of cold-adapted enzymes and their thermostable homologs.
Flexibility and Structural adaptation
[edit | edit source]X-ray structures of cold-adapted enzymes have been discovered to show the structural basis of cold adaptation by comparing these x-ray structures with homology models of proteins from mesophiles and thermophiles. An important factor to the activity and stability of a enzyme is its physiological environment. Studies have also shown that mainly marine organisms have been used to study cold-adapted enzymes. The X-ray structure of enzyme citrate synthase has been determined and compared to other enzymes of Bacteria, Eucarya, and Achaea. Due to the absence of crystal structure information, studies generally choose closely related organisms. By maximizing thermal differences and minimizing phylogenetic differences in comparative data sets, the lack of x-ray structures for enzymes can be countered.
Hydrophobic Interactions
[edit | edit source]Hydrophobic interactions between hydrophobic residues and solvent water molecules play a key role in contributing to the structural flexibility and thermostability of cold-adapted proteins.
Core hydrophobicity
[edit | edit source]Cold-adapted enzymes have amino acids that tend to be smaller and less hydrophobic than in homologs from mesophiles and thermophiles. Since Van der Waals interactions are weak, have short range, and distance sensitive, the distance between hydrophobic groups inside a protein will determine the enthalpic contributions to stabilization. Reduced van der Waals interactions and increased movement of internal groups will therefore destabilize cold-adapted enzymes. An example of a group inside a protein is Ille. Ille can pack efficiently inside the core and stabilize a protein due to tis branching and size. In the cold-adapted trypsins, citrate synthase, and AHA, fewer Ille residues can pack inside the core.
Hydrophobic interactions are strongest at room temperature due to the solubilities of hydrophobic side chains in water having it be at the minimal temperature 20°C. Through a study with 31 proteins that unfold reversibly, it was found that about 75 % had maximum stability around room temperature. This shows that hydrophobic interactions in the core of a protein play a key role in enhancing protein stability at low to moderate temperatures.
Surface hydrophobicity
[edit | edit source]A higher proportion of hydrophobic, or nonpolar, residues occur on the surfaces of cold-adapted enzymes shown by studies with x-ray structures of many enzymes. Using large-scale modeling and structural studies, similar conclusions were found that demonstrated that the mean fraction of the solvent-accessible surface, or buried surface, of the enzyme has a higher hydrophobicity in cold-adapted proteins. In the X-ray structure of the example with Ille, it revealed that Ille clusters at the subunit interface are absent in the cold-adapted enzyme while tightly packed hydrophobic clusters are present in the homolog from a hyperthermophile.
Studies have shown that hydrophobic surface residues will destabilize a protein structure due to the decreased entropy of water molecules because these water molecules form cage-like structures around nonpolar residues. It was found that at lower temperatures, the entropy gain is actually reduced due to the decreased mobility of released water molecules showing that cold-adapted enzymes may gain flexibility from and have a greater capacity to tolerate increased surface hydrophobicity.
Surface Hydrophilicity
[edit | edit source]Certain cold-adapted enzymes like trypsins and β-lactamase (55) have had an increase in surface charge, mainly negative charge. For some enzymes, the negative charge is rather high but with some positive charge located near the active site. Due to high viscosity and high surface tension of water, at low temperatures, the energetic cost of disrupting H-bond networks is high. This energetic cost may be counter by surface-charged or polar amino acids that interact with water molecules with a high dielectric constant. This would then enable proper solvation and help maintain the flexibility of the enzymes. With better solvent interaction and positive charge, flexibility may be improve at low temperatures.
-
 Structure of Trypsin
Structure of Trypsin

Localization of acidic residues in surface patches have the possibility of producing charge-charge repulsions that cause the overall destabilization of protein structure. Sometimes the case arises where charge repulsion of acidic residues may create a high level of flexibility in the linker region, This is a major structural feature of cold adaptation in certain enzymes. Other studies have shown that cold adaptation involves a decrease in the mean fraction of solvent-accessible and buried surface that is charged. This involves other proteins from not only the Achaea family, but also Bacteria family causing and increase in surface Hydrophilicity in proteins. With increased surface charge in thermostable proteins, researchers have been able to link this to an ability to form networks of sat bridges. This directly contrasts the interaction with water molecules in cold-adapted enzymes. A direct correlation has been found that as ionic interactions become stronger with decreased temperature, a minimization of their number that allows cold-adapted proteins to retain flexibility at low temperatures occurs.
Reference
[edit | edit source]Siddiqui KS, Cavicchioli R. Cold-Adapted Enzymes. Annual Review of Biochemistry. Vol 75:403-33.Volume publication date July 2006. Some interesting facts about Cholesterol 24-hydroxylase:
1. It converts cholesterol to 24S-hydroxycholesterol. It then diffuses out from the brain and is metabolized in the liver.
2. It is a P450 enzyme is located in the endoplasmic reticulum, an organelle in all eukaryotic cells. It is an enzyme that is expressed in some but not all neurons of the brain.

The Cytochrome b6f complex, also known as plastoquinol-plastocyanin reductase, is an energy transducing, hetero-oligomeric, dimeric enzyme found the thylakoid membranes of such organisms as the thermophilic cyanobacterium, Mastigocladus laminosus, and the green alga, Chlamydomonas reinhardtii. The enzyme acts as a mediator for the transfer of electrons between the reaction center complexes of photosystem II (PSII) and photosystem I (PSI) that lie inside the membrane. The reaction by which this occurs is shown below:

Mechanism of Reaction
[edit | edit source]During this process, the transfer of electrons from one side of the membrane to another results in a electrochemical potential gradient across the membrane, determined to be approximately 250mV, with the positive end of the gradient residing on the side of the membrane to which the protons are being transferred. Plastonquinol-1 (QH2) and Plastocyanin (Pc) act as mobile redox carriers for either a clycic or non-cyclic electron transfer.
In the non-cyclic reaction, water is fed through the PSII enzyme to produce plastoquinol (QH2) which is reduced to plastocyanin (Pc) by the cytochrome b6f complex. Then, Pc is fed through the PSI enzyme resulting in the reduced form of Nicotinamide adenine dinucleotide phosphate, NADPH.
In the cyclic reaction,
- QH2 + 2Pc(Cu2+) + 2H+ → Q + 2Pc(Cu+) + 4H+
Pc transfers its electrons through an electron transport chain by means of electron bifurcation. This mechanism used in this cycle is commonly referred to as the Q Cycle.

- The Q Cycle (seen right), is a process in which the positively charged side of the complex binds with QH2 and is oxidized by a Fe-S center on the complex to form a semiquinone. This releases 2H+ in the positively charged side of the complex. Next, e- are transfered by an electron transport chain to the Fe-S center to Pc, and the semiquinone follows by transferring its e- to the heme bp of the cytochrome b6 complex. The heme bp then transfer its e- to the heme bn of the enzyme which, in turn, reduces Q and produces SQ.
- The cycle then continues (shown in the illustration as the "second half") by the binding of a second QH2 to the positively charged side of the complex. By means of a high-low electron transport chain, an e- is used to reduce an additional oxidized Pc, followed by the transfer of another e- from the heme bn on the Cytochrome b6f complex to the SQ molecule. Here, Q2- is completely reduced and accepts 2 H+ from the initially negatively charged side of the membrane to for QH2. And finally, QH2 diffuses into the membrane, completing the cycle.[1]
Structure
[edit | edit source]The structure of the cytochrome b6f complex varies depending on the organism, but retains strong structural and compositional similarities and functions in essentially the same way. For example, the M. laminosus and C. reinhardtii complexes are structurally very similar, are both comprised of the same number of polypeptide subunits, and share a similar sequence identity in their photosynthetic electron transport (Pet) protein A-D subunits. These structures are both considered to be homodimers, of which each monomer is comprised of 8 polypeptide subunits. Of these 8 subunits, 4 of the largest of the them are considered as a separate subunit of the complex, as defined by Hurt & Hauska. According to Hurt & Hauska, these 4 subunits consist of c-type cytochrome f (PetA), cytochrome b6 (PetB), the ISP (PetC), and subunit IV (PetD, suIV), which is related to the C-terminal half of bc1 cytochrome b. All 8 subunits combine to form a bundle of 13 alpha helices inside the lipid bilayer of the membrane. Each monomer also contains a chain of six separate redox prosthetic groups to drive electron and proton transfer. The size of the enzyme is approximately twice the width of the membrane bilayer, and mass spectrometry has determine the typical molecular weight of the molecules to be on the order of 105 g/mol.[1]
References
[edit | edit source]Introduction
[edit | edit source]
Cytochrome P450 proteins are found in the genomes of virtually all organisms. Their amino-acid sequences are extremely diverse, but their structural fold has remained the same throughout evolution. Their origins could be traced back in vitro studies on the metabolism of steroids, drugs, and carcinogens. P450s are the enzymes that play a major role in drug metabolism and accounts for about 75% of the contribution of enzymes to the metabolism of marketed drugs.The mechanism of the cytochrome P450 enzyme can be compared to a blowtorch. As the P450 enzymes catalyzes regiospecific and stereospecific oxidative attack on non-activated hydrocarbons at physiological temperatures. But such a reaction also being uncatalyzed would require very high temperature and would be nonspecific. Not all the mechanisms for P450 are well understood. An example of an understood mechanism would be cytochrome P450’s activity in the liver being that it can encode a liver enzyme in which enables for it to metabolizes a series of known drugs. The CYP2D6 genome, which is a type of P450 enzyme, will determine how a person responds to a certain type of drug such as antipsychotics and antidepressants.What the enzyme does is it metabolizes potentially toxic compounds in the body allowing one to know if the drug is effective.
Use of P450
[edit | edit source]Marine animals can accumulate environmental contaminants in their blubber over time at concentrations known to be harmful to laboratory animals. The Cytochrome P450 enzyme is widely used as a biomarker for signs of exposure of molecular effects that could have taken placed. High concentrations of organochlorine pollutants are common in oceans and can affect the endocrine, reproductive, immune and nervous systems of animals and show evidences of skin and liver damage along with thymic atrophy, weight loss and neurobehavioral problems.A type of Cytochrome P450 known as CYP1A1 was induced into the skin biopsy section of the animal and it showed staining in three different cell types: the endothelial cells composing the lining of all blood vessels including capillaries, the smooth muscle cells present in larger blood vessels, and fibroblasts. These stains shows evidence that the environment is polluted and shows how pollutants can affect the animals that were exposed. From this it helps scientists find ways to reduce and find out what is contaminating the ocean.
Limitations
[edit | edit source]The physiological limitations to Cytochrome P450 enzymes are that ingesting certain types of foods can inhibit it. Eating a grapefruit can inhibit the P450 and can prevent detoxification from occurring. This affects the drug metabolism as if P450 is blocked, then a drug can potentially harm a person and poison them. In some cases the drug will not be as effective as it would be regularly.Another example of a physiological limitation is that the Cytochrome P450 instead of reducing toxicity it could potentially convert some drugs into toxic products. An example of this would be acetaminophen, which is found in painkillers such as Tylenol, too much acetaminophen will cause a negative affect and will cause toxic harm to one’s health.
The limitations to the practical application of this defense system to predict the effects of pollutants in the case of the ocean, is that most of the marine animals are protected by the government and scientist cannot induce them with the P450 enzyme, they instead use laboratory animals to conduct the experiment. This leads to just theories on if P450 being a successful biomarker. Also the lack of knowledge of P450 enzymes can pose a limitation, as we do not completely understand how all the P450 enzymes work. The practical application of this defense system to predict the effects of pollutants from a human stand point is that there are so many different types of P450 enzymes that we do not know all of them and their effects. So we cannot use P450 to exactly distinguish what carcinogens and cancerous chemicals affect. Also some of the P450 enzymes are embedded in membranes of the Endoplasmic Reticulum, which makes it hard to identify the structure, which makes it hard to study the different types of enzymes and this is a big limitation.
Application in Medicine
[edit | edit source]Medicine only works as expected in fewer than half of the people who take them. Environmental and lifestyle factors can explain this but individual variability in response to medicines is the main cause. This can be attributed to variants in the genes that make cytochrome P450 proteins. These proteins process many of the drugs we take. Because each person’s set of genes is a little different, the proteins that the genes encode are also slightly different. These changes can affect how the cytochrome P450 works on drugs.[1]
References
[edit | edit source]- ↑ U.S. Department of Health and Human Services. The New Genetics. October 2006.<http://www.nigms.nih.gov>.
2. http://toxsci.oxfordjournals.org/content/80/2/268.full.pdf
3. http://rusynlab.unc.edu/publications/course_data/Guengerich_2008.pdf
4. http://www.aapsj.org/articles/aapsj0801/aapsj080112/aapsj080112.pdf
5. http://genomebiology.com/content/pdf/gb-2000-1-6-reviews3003.pdf
6. U.S. Department of Health and Human Services. The New Genetics. October 2006.<http://www.nigms.nih.gov>.
Deaminase
[edit | edit source]Deaminase is an enzyme that is involved in the process of Deamination in which the process removes an amine group (NH2) from a molecule through hydrolysis. The enzyme will usually only remove an amine group from extra proteins and this occurs in the liver or kidneys. This enzyme is very beneficial to the body because it allows deamination to follow through. Without Deamination waste (usually Nitrogen waste) may not be able to leave the body. The Nitrogen waste is expelled through urination which is a result of the removal of the amine group by deaminase. In addition to removing wastes, deamination using deaminase also allows the body to convert the extra amino groups removed from proteins into more beneficial sources that the body may use in all other sorts reactions. Thus, this contributes to the balance of the body's metabolism in that it allows the body to not accumulate a surplus amount of certain molecules which may lead to diseases or even cancer.
Deamination can be very deleterious process. For example, by deaminating adenine, hypoxanthine is formed. This product pairs with guanine and cytosine. Even though this process in the long run does not cause any change in protein formation, if it does, then the result may be disastrous. There are many kinds of deaminases. Deamination can happen in guanine and cytosine as well.

Notable Deaminases
[edit | edit source]APOBEC3G (A3G)
[edit | edit source]APOBEC3G, known as A3G, belongs to a family of cytidine deaminases named for the first known enzyme to possess the capacity for site-specific cytidine to uridine deamination of B messenger RNA. A3G has been demonstrated to be significant in the cellular defense against the progression of Human immunodeficiency virus (HIV).
Antiviral Mechanisms
[edit | edit source]The A3G deaminase dependent and independent antiviral mechanisms are known to induce mutations in the HIV viral genome.
In the deaminase dependent mechanism, this occurs due to the ability of A3G to catalyze zinc-dependent hydrolytic deamination of deoxycytidine, instead forming deoxyuridine in HIV DNA. Mutation distribution and frequency within the viral DNA is determined by a variety of factors including the availability of ssDNA (single-strand DNA) and the speed of nucleotide addition (3' to 5' processivity).
As for the deaminase-independent mechanism, A3G is suspected of containing both N-terminal and C-terminal zinc-dependent deaminase (ZDD) folds. ZDD consists of a sequence which is known to comprise five anti-parallel beta sheets which are supported and maintained by two alpha helices (which position cysteine and histidine residues for the coordination binding of a zinc atom, a water molecule, and a glutamic acid residue, all necessary for the conversion of cytidine and deoxycitidine to uridine and deoxyuridine, respectively. It has been demonstrated that such deaminase activity is limited to C-terminal ZDD folds.
"Double Agent" Function in Cellular Defense
[edit | edit source]In the cellular defense against the HIV virus, APOBEC3G (A3G) serves to induce mutations in the viral genome, effectively preventing high-fidelity replication and deleterious gene-expression in non-viral cells. However, the HIV-encoded protein Vif (short for Viral Infectivity Factor), is known to allow for the infection of cells even in the presence of A3G. This occurs because Vif triggers the destruction of A3G, preventing its incorporation into growing viral strands. This effectively makes impotent the ability of A3G to hypermutate HIV ssDNA during the process of reverse transcription necessary for viral genome replication. However, while A3G is known to serve as an antiviral factor through both Deaminase-dependent and deaminase-independent pathways, it has been suggested that the mutagenic effects induced in the viral genome are insufficient so as to inactive and make harmless the HIV genome. As such, A3G may induce diversification of viral DNA strands, resulting in new, more virulent strains of viral genome. In light of this knowledge, the level of A3G-induced mutation has been discussed relative to whether it benefits or destroys viral factors. While research has not yielded the levels of A3G activity necessary to reinforce antiviral efforts, it has been suggested that inhibition of Vif would yield long-term detrimental effects, ultimately aiding viral diversification. Alternatively, inhibiting A3G activity and allowing Vif to destroy A3G cells may reduce the emergency of viral diversity and accompanying resistance.
Resource
[edit | edit source]Biochemistry 7th edition by Jeremy M. Berg
Smith, Harold. "APOBEC3G: a double agent in defense." Trends in Biochemical Science. 2011 May;36(5):239-44. Epub 2011 Jan 14.
Sumayao, Marco, and Jenn Walker. "What Is Deamination?" WiseGeek. Conjecture, 2003. Web. 20 Nov. 2012. <http://www.wisegeek.com/what-is-deamination.htm>.
Introduction
[edit | edit source]Drug metabolizing enzymes are enzymes that are used to carry out reactions during drug metabolism; they are enzymes that metabolize xenobiotics (Tukey 71). Xenobiotics are defined in biochemistry as foreign chemicals or substances to a certain species. Thus, drugs can be grouped in this category of xenobiotics as well because drugs are foreign substances that enter the body. Xenobiotics are not limited to just drugs; there are many other chemicals that are considered xenobiotics.
There is generally two stages in which drug metabolism is facilitated and they may occur in most of the body. However, it is most likely that they are carried out in the liver and small intestines because the small intestines is where chemicals are absorbed and sent out to the liver (Tukey 74).
Function
[edit | edit source]How does it work? First, drugs must be transformed into a different form that the cells can access because they are required to move down a gradient and most drugs are hydrophobic (Gonzalez 71). This means that the chemicals that enter the cell must be hydrophilic or changed into hydrophilic chemicals in order for the cells to eliminate the toxins more easily. The enzyme that is allowed to make the change from a hydrophobic substance to a hydrophilic chemical are drug metabolizing enzymes. Then the drug is passed through the gastrointestinal tract where metabolism is first started; if the drug is not broken down all the way, it is then passed to the liver where more metabolism takes place until the chemical is fully broken down (Tukey 74).
For example, metabolism of phenytoin by the enzyme CYP450 and UGT will make the drug very hydrophilic and thus it will be able to be broken down by the body and taken in by the small intestines into the body (Tukey 72). Also the molecular weight of the compound increases as well which increases the elimination of the drug through urine or the bile (Tukey 72).
Drug metabolizing enzymes not only breaks down chemicals but it may also cause cancer because it may change the toxic accumulation of chemicals into carcinogens which are cancer causing agents (Tukey 72). It usually happens when the converted chemical decides to react with other compounds in the cell like DNA or RNA which may result in mutations and lead to cancer.
Drug Metabolism Role in Efficient Drugs
[edit | edit source]How does metabolism connect to the making of drugs for patients?
The role of metabolism is to rid toxic chemicals from the body in order to keep it from entering a toxic state; thus, when drugs are consumed, metabolism will do its job to eliminate the drugs. Therefore, if a drug is eliminated at a rapid rate, this means that the drug is not very efficient (Gonzales 88). Vice versa, if the drug is not metabolized fast enough, then the body will enter a poisonous state. This concludes that metabolism will reduce the efficiency of a drug if it's job is to break down the drug. So, using this information, scientists are able to link metabolism with the efficiency of the drug and so they are able to create drugs that fit the needs of patients according to the studies of the rates of metabolism of different drugs.
However, there is a problem because environmental factors may also contribute the changes in the rate of metabolism. For example, grapefruit juice should not be used to take medication because it has chemicals in it that stop the CYP3A4 enzyme which increases the amount of that drug available (Gonzalez 78). This might sound good that it increases the availability of the drug but this is not the case; the increase of the drug will result in toxicity but will also effect the efficiency of the drug as well so it has its pros and cons. Therefore, it is very difficult for scientists to control the rate of metabolism for a group of individuals with so many different factors changing the rate of metabolism. But with this knowledge, scientists may then follow through their research to hopefully find something to work with.
Significance
[edit | edit source]Drug metabolizing enzymes helps eliminate certain chemicals that are harmful to the body through a series of reactions and processes. Therefore, without drug metabolizing enzymes then metabolism will not occur. As a result, harmful chemicals and other substances that are not able to metabolize, will accumulate in the species and will cause the individual to be in a poisonous state. All in all, it is important for an individual to have these enzymes to be able to eliminate xenobiotics (chemicals not known to the individual)to avoid toxicity.
Overview
[edit | edit source]There are enzymes that are needed to facilitate drug metabolism. Two recognized enzymes would be CYP3A4 and CYP450. CYP3A4 is a drug-metabolizing enzyme that is located in the intestines. It increases or alters blood vessels of certain medications in people. CYP450 is a cytochrome enzyme that processes essential molecules such as hormones and vitamins. They also help break down many prescribed medicines and natural substances. Both enzymes aid the process of drug metabolism in our bodies.
Scientists have discovered that many different codes for CYP 450 genes can be found in the human genetic code. This leads to CYP 450 proteins having a wide range of activity. An example is that some CYP enzymes can metabolize carcinogens, which activates the chemicals and has a tendency to cause cancer. Studies have shown that CYP 450 can be blocked by natural components that can be found in certain food. These include oranges, horseradish, green tea, and mustard.
Reference
[edit | edit source]Davis, Alison. (2006). Medicines By Design. National Institutes of Health, 8.
Gonzalez, Frank J.; Tukey, Robert H. "Drug Metabolism: How Humans Cope with Exposure to Xenobiotics." Goodman and Gilman's the Pharmacological Basis of Therapeutics. By Frank J. Gonzalez. New York, NY: McGraw-Hill, n.d. 71-91. 21 Nov. 2012. MAO B is an enzyme called monoamine oxidase B. It helps in recycling neurotransmitters, communication molecules, in the brain. Both MAO B and MAO A, its cousin, removes molecular pieces from neurotransmitters. This is a part of a process that inactivates the neurotransmitters. Blocking the actions of MAO enzymes has been discovered by scientists. This is beneficial because it helps in the preservation of the levels of neurotransmitters. This helps people with disorders such as depression and Parkinson's disease.
Side Effects
[edit | edit source]MAO inhibitors have many side effects that are undesirable. Some mild effects include increased heart rate, tremors, and sexual function problems. Some side effects that are more serious include large dips in blood pressure, seizures, and difficulty breathing. People who are taking MAO inhibitors should be cautious in the food that they eat. They can not eat food that contains tyramine. Tyramine can be found in dried fruits, cheese, wine, and many others. The side effects for this drug occurs mainly because drugs do not attach to MAO enzymes perfectly for both MAO B or MAO A.
Structure
[edit | edit source]Edmondson et al. states that the structural features of the human enzyme has a hydrophic bipartite elongated cavity that holds a total volume that is approximately 700 Å3. hMAO-A has a single cavity that displays more of a round shape and is comparatively larger in volume than the substrate cavity of hMAO-B. The first cavity of hMAO-B is called the entrace cavity of about 290 Å3, and the second substrate cavity or active site cavity that is between both anisoleucine199 side chain of about 390 Å3 acts as a gate. It can either exist as an open or a closed form depending on the substrate or bound inhibitor. This has been shown to play an important role in defining the inhibitor specificity of hMAO B. Then there is the FAD coenzyme at the end of the substrate cavity with sites that favor amine binding regarding the flavin consisting of two nearly parallel tyrosyl (398 and 435) remains that form an aromatic cage.
Differences between MAOA and MAOB
[edit | edit source]MAO-A plays an important role in the metabolism of tyramine. Some particular irreversible inhibition of MAO-A can lead to a dangerous pressor effect when consumed foods are high in tyramine such as cheese. MAO-A also plays an important role in the metabolism of serotonin, noradrenaline, and dopamine. MAO-B, an enzyme on the outer mitochondrial membrane, metabolizes dopamine neurotransmitter and catalyzes the oxidation of arylalkylamineneurotransmitters. Generally, MAOA, mMonoamine Oxidase A, metabolize norepinephrine, serotonin, Dopamine, and other less clinically relevant chemicals. In contrast, Monoamine Oxidase B, MAO-B, metabolizes Dopamine and other less clinically relevant chemicals. The differences between the substrate selectivity of the two enzymes play a significant role when treating specific disorders. For example, Monoamine Oxidase A inhibitors have been involved in the treatment of depression while Monoamine Oxidase B inhibitors have been involved in the treatment of Parkinson's Disease.
References
[edit | edit source]- Edmondson, Dale E.; Binda, Claudia; Mattevi, Andrea (2007). "Structural insights into the mechanism of amine oxidation by monoamine oxidases a and B". Archives of Biochemistry and Biophysics. 464 (2): 269–76. doi:10.1016/j.abb.2007.05.006. PMC 1993809. PMID 17573034.
- Binda, Claudia; Hubálek, Frantisek; Li, Min; Herzig, Yaacov; Sterling, Jeffrey; Edmondson, Dale E.; Mattevi, Andrea (2004). "Crystal Structures of Monoamine Oxidase B in Complex with Four Inhibitors of theN-Propargylaminoindan Class". Journal of Medicinal Chemistry. 47 (7): 1767–74. doi:10.1021/jm031087c. PMID 15027868.
Introduction
[edit | edit source]COX-2 (cyclooxygenase) is an enzyme in our body that is vital in the formation of important biological mediators called prostanoids. Prostanoids is a class of signaling molecules that consists of prostaglandins, thromboxanes, and prostacyclins. Prostanoids are what is responsible for inflammation that occurs in our body.
Function of COX
[edit | edit source]The COX enzyme has two active sites which are the heme group and the cyclooxygenase site. The heme group has the ability to perform peroxidase activity which is responsible for reducing PGG2 to PGH2. The other active site, cyclooxygenase site, is where the arachidonic acid is converted to hydroperoxy endoperoxide prostaglandin, denoted PGG2. COX functions by converting arachidonic acid to prostaglandins, which is the precursor of series-2 prostanoids. A tyrosine radical is produced by the peroxidase active site, which then abstracts an H atom from the arachidonic acid to create an arachidonic acid radical. Then, two oxygen molecules react with the arachidonic acid radical to yield PGG2. There are three isoenzymes of cyclooxygenase (COX) that are known today. These three isoenzymes are COX-1, COX-2, and COX-3. COX-3 is a splice variant of enzyme COX-1 which means that COX-3 has a similar genetic code to COX-1. COX-3 retains intron one from COX-1 enzyme and has a frame shift mutation which is what causes its variation from the COX-1. For this reason, COX-3 is often referred to as COX-1b or COX-1 variant. According to each type of tissues, the amounts of COX-1 and COX-2 enzymes expressed within the tissue varies. Both enzymes have similar functions and so behave in similar manners. However, selective inhibition may result in a difference in side-effects. COX-1 is found in most mammalian cells and is considered to be a constitutive enzyme, which means that this enzyme is not controlled by repression or induction. COX-1 is produced constitutively by the cell under all physiological conditions. Contrary to the COX-1, COX-2 is an inducible enzyme which means that it is abundant in activated cells and macrophages. COX-2 is also abundant in sites of inflammation. COX-2 is undetectable in normal cells and it is has been recently shown that COX-2 is up-regulated in many carcinomas. This implies that COX-2 may have an active role in the formation of tumors.
Pharmacology
[edit | edit source]The significant variation between COX-1 and COX-2 is that COX-1 has the amino acid isoleucine in position 523 while COX-2 has the amino acid valine instead. Valine is a smaller amino acid than isoleucine which is why COX-2 has the ability to access hydrophobic side pocket in the enzyme. COX-1 is unable to access this side pocket due to steric hindrance from the larger isoleucine. This special characteristic of COX-2 enables drug molecules such as DuP-697 to inhibit COX-2 by binding to this site. This discovery allowed for the production of drugs that are said to be selective inhibitors of COX-2 because they disable COX-2 without interrupting COX-1 activity.
NSAIDs
[edit | edit source]The main COX inhibitors are called non-steroidal anti-inflammatory drugs, often denoted as NSAIDs. NSAIDs are unselective of which COX enzyme it inhibits, thus inhibiting all COX enzymes. This has both favorable and unfavorable side-effects. By inhibiting COX-2, NSAIDs have the effect of reducing inflammation and antipyretic, antithrombotic, and analgesic effects. However, because NSAIDs also inhibit COX-1 activity, it may cause negative side effects such as gastric irritation. COX-1 is responsible for producing mucous that is necessary in protecting the gastrointestinal tract. By inhibiting COX-1 activity, the production of the mucous is also inhibited, which may have an adverse effect on gastrointestinal tract.
Newer NSAIDs
[edit | edit source]Research into newer drugs has led to the discovery of drugs that selectively inhibit COX-2 without having much of an effect COX-1. COX-2 is usually specified to inflamed tissues, which is why there is a lesser risk of gastric ulceration associated with COX-2 inhibition. These selective inhibitors are common ingredients in arthritis medication such as Celebrex. Recent studies have been showing a correlation between the inhibition of COX-2 and higher risk of cardiovascular disease such as myocardial infarction. Vioxx was another brand that contained selective COX-2 inhibitors in its drugs but was removed from the market due to the recent discoveries that COX-2 inhibition may lead to increased risk of strokes and myocardial infarction.
COX-2 and Parkinson’s Disease
[edit | edit source]Neurologists have been studying COX-2 activity and its possible effects on Parkinson’s disease. Researchers believe that COX-2 inhibitors may preserve neurons, which is important to Parkinson’s disease because this disease is characterized by the death of neurons. The correlation between Parkinson’s disease and COX-2 is that these enzymes are responsible for inflammation in damaged tissues in the brain. Researchers have been noticing that inflammation has a critical role in neurodegenerative disease such as Parkinson’s disease and Alzheimer’s. Many believe that inhibiting COX-2 enzymes may be beneficial in stopping Parkinson’s disease and reduce the risks of Alzheimer’s.
Studies have been conducted by faculty members in Columbia University involving postmortem brains of patients who have been diagnosed with Parkinson's Disease. The research discovered that there was high levels of COX-2 enzymes in the dopamine neurons of these patients compared to those of without the disease. It was also discovered that dopamine neurons had suffered the most damage from Parkinson’s disease. Studies were also conducted on mice in order to test the importance of COX-2 in diseases similar to Parkinson’s disease. This further confirmed that diseases similar to Parkinson’s disease lead to high levels of COX-2 in dopamine neurons. When COX-2 enzymatic activity was diminished by using a selective inhibitor, the mice’s dopamine neurons were able to survive. Although COX-2 enzymes may be the responsible for the depletion of neurons, it is still unclear as to what causes the actual inflammation that is commonly associated with Parkinson’s disease. When COX-2 enzymes were removed, there were a larger number of dopamine neurons that survived yet inflammation was not reduced. From this information, it can be deduced that the COX-2 enzyme does not kill neurons through inflammation. An alternative theory on why COX-2 enzymes damage neurons is that COX-2 oxidized other molecules in the dopamine neuron which then react with other molecules, thus damaging other components in the cell. Eventually, this reactivity and excessive damage will lead to the death of dopamine neurons.
References
[edit | edit source]1. http://www.sciencedaily.com/releases/2003/04/030408085115.htm
2. http://en.wikipedia.org/wiki/Cyclooxygenase
3. http://www.sigmaaldrich.com/life-science/biochemicals/biochemical-products.html?TablePage=14573022
4. http://www.ncbi.nlm.nih.gov/pubmed/13680840
Overview
[edit | edit source]PLC, which stands for phosphoinositide-specific phospholipase C, is an enzyme that is linked to common signalling components for most cellular receptors for activation.
Some characteristics of PLC families include:
- complex
- modular
- multi-domain proteins
- cover broad spectrum for regulatory interactions
PLC enzymes: Functions and Signaling Diversity
[edit | edit source]PLC enzymes can be found in eukaryotes. Some of their specific functions and functions of its components include:
- cleaving the phosphtidylinoitol 4,5-bissphosphate's polar head group using the enzyme's related group of proteins
- generating 2 second messengers, which are inositol 1,4,5-triphosphate (this i second messenger that is universal in calcium mobilizing) and diacylglycerol (this activates several types of effector proteins). Second messengers helps in regulating a variety of biological functions such as cell motility, sensory transduction, and fertilisation.
- phosphoinositide species has an important role in targeting specific subcellular compartments. They target components that are important in control of cell movement and membrane trafficking.
PLC enzymes: Structural Elements
[edit | edit source]An N-terminal pleckstrin homology domain is part of a conserved core architecture that PLC families share. They also share a C-terminal domain, a TIM barrel that is catalytic, and a series of EF hands. A conserved domain in the PLC isoforms are TIM barrel. They are both functionally and structurally conserved. In other domains, properties in ligand binding can vary. Due to the findings of the core structure from PLC g1, it is revealed that inter-domain interactions are extensive. The domains of EF hands, C2 domain, and TIM barrel probably has similar structures as the N-terminal PH domain of PLCs.
PLC Families
[edit | edit source]They have six families consisting of thirteen isoforms in humans. These six families are PLCb, g, d, e, z and h. Compared to other families, PLC b and PLC g have had their regulatory interactions more extensively characterised. In heterotrimeric G proteins, PLC g isoforms has had its regulation through receptor and non-receptor tyrosine kinases distinguished from PLC b isoforms' regulation. It has been found that the Ras Family has small GTPases that directly regulates PLC e. The Rho Family has small GTPases and heterotrimeric G protein subunits can stimulate the enzyme activity of PLC e. This shows that there is an unlikely chance that PLC g isoforms mediates tyrosine kinase linked receptors to stimulate PtdIns(4,5)P2 hydrolysis. Also, it is unlikely that the PLC b isofroms are the only enzymes that activate G protein coupled receptors. There is a possibility that PLC e may also participate in PtdIns(4,5)P2 hydrolysis which tyrosine kinase receptors and GPCRs can trigger.
References
[edit | edit source]PLC regulation: emerging pictures for molecular mechanisms. Bunney TD, Katan M. Trends Biochem Sci. 2011 Feb;36(2):88-96. Epub 2010 Oct 1. Review.
Polynucleotide Kinase/Phosphatase
[edit | edit source]
One of the most common problems that could occur in a living organisms is damage to cellular DNA, or mutations. Mutations are so common that it is involved with a variety of important factors in life, such as cancer treatment, neurological disorders, and aging. The basic of idea of DNA damage is that internal and external agents cause the loss of bases, causing the DNA strand to break. Before the strand breaks can be repaired, the termini of breaks require processing before the missing bases can be properly replaced. This is where Polynucleotide Kinase/Phosphatase (PNKP) comes into play by catalyzing the restoration of 5'-phosphate and 3'-hydroxyl termini. PNKP interacts with various other proteins, especially XRCC1 and XRCC4, and uses the different pathways to repair DNA. The 5' kinase and 3'phosphate activities of PNKP processes, or repairs, both single and double stranded termini in DNA. Understanding its mechanism has lead to an opportunity to treat diseases and cancer. PNKP inhibitors are also known to sensitize cells towards IR and chemotherapeutic agents since they prevent PNKP from processing DNA repair. PNKP’s role in the restoration of DNA strand breaks follows three major DNA repair pathways: single-stranded break repair (SSBR), base excision repair (BER), and double-stranded break repair (DSBR). These three mechanisms can provide useful information on how other proteins bind and react to the PNKP enzyme.[1]
Molecular Structure of PNKP
[edit | edit source]PNKP is a multi-domain enzyme that is made up of two main domains: N-terminal forkhead-associated (FHA) domain and the C-terminal catalytic domain. Additionally, C-terminal catalytic domain is composed of fused phosphate and kinase sub-domains. The two main domains, FHA and catalytic domain, are binded through a flexible polypeptide segment. It is at this connection where the binding to CK2-phosphorylated regions of other proteins. The polynucleotide kinase phosphatase and the phage T4 polynucleotide kinase (a cloning enzyme) differ such that the T4 enzyme does not contain the FHA domain whereas PNKP has the FHA domain in the N-terminal. However, the kinase subdomain of the T4 polynucleotide kinase is in the N-terminal instead of the C-terminal catalytic domain.[1] The two specific proteins PNKP interact with are called XRCC1 and XRCC4. The major distinction between the proteins XRCC1 and XRCC4 is that XRCC1 repairs DNA single-stranded breaks while the XRCC4 protein fixes the DNA double-stranded breaks.[1] Only the FHA domain binds to a the region of these proteins that are specifically phosphorylated by CK2. For XRCC1, there are clustered regions of CK2, typically between residues 515 and 526, that are required for it to bind to FHA and repair DNA. In contrast, only a primary CK2 site is required for XRCC2. In addition, aprataxin and aprataxin and PNKP-like factor (APLF) contribute to DNA repair; these two DNA repair factors have FHA domains as well.[1]
PNKP also contains two catalytic active sits that reside on the same side of the enzyme, as well as separate ATP and DNA binding sites. The different DNA binding sites significantly differ between phage and mammalian enzymes. For phage enzymes, the binding site occurs through a narrow channel that leads to catalytic aspartic acid residues, which only aids in single-stranded repair. As for mammalian enzymes, they phosphorylate the 5'-hydroxyl terminal repairing double-stranded more efficiently than single-stranded.
PNKP in Single-Stranded Break Repair (SSBR)
[edit | edit source]Specifically for IR induced strand breaks, the loss of nucleotides is repaired with a process that is carried out with poly(ADP-ribose) polymerase (PARP), XRCC1, and AP endonuclease I (APE1). This process may be done with a short patch, using DNA polymerase and DNA ligase III. It can also be done with a long patch, which uses DNA polymerase, ligase I, and FEN1 endonuclease. The role of APE1 is to remove 3'-phosphoglycolates. PNKP itself performs hydrolysis in 3'-phosphate groups while keeping 5'-OH phosphorylated, which is a much stronger activity than APE1. The overall phosphate activity is significantly more active than the kinase activity, causing an over-expression of phosphatase-detective PNKP.
The basic mechanism of SSBR is:
- Breaks are recognized by PARP
- Recognition attracts only XRCC1, which is typically binded with DNA ligase
- XRCC1 recruits PNKP/APE1
- Recruited enzymes restore termini, allowing DNA polymerase to add missing nucleotides and DNA ligase to bind the broken strands.
Stressed or damaged cells really need the CK2-phosphorylated XRCC1 to bind to FHA domain of PNKP so that repair can occur. Unstressed cells are able to cope with non-phosphorylated proteins because repair is not in urgent need.
PNKP in Double-Stranded Break Repair (DSBR)
[edit | edit source]The double-stranded break repair pathway depends on the protein XRCC4, which when phosphorylated will not stimulate the enzyme PNKP; reversely, unphosphorylated XRCC4 will activate PNKP. However, the combination of phosphorylated XRCC4 with DNA ligase IV can trigger PNKP by promoting the binding between the PNKP’s FHA domain and the phosphorylated XRCC4 protein.[1] PNKP is only involved with the nonhomologous end joining (NHEJ) pathway of DSBR, but not with homologous recombination. The mechanism is similar to SSBR, except that kinase activity of PNKP is required for ligation to occur. Also, phosphorylation is actually dependent on XRCC4, not XRCC1, in order to bind PNKP to DNA ligase, which stimulates the DNA ligase. It is also noted that XRCC4 is necessary for cell survival after IR or chemo treatment. The most important and distinct fact about DSBR is that phosphorylated XRCC4 actually fails to bind to PNKP through the FHA domain, inhibiting cell repair. However, adding DNA ligase reverses the inhibition and therefore allows successful DNA repair.
Non-phosphorylated XRCC4 for DSBR works in a manner similar to XRCC1 in Single-Stranded Break Repair (SSBR) because they both encourage enzymatic cell turnover for Polynucleotide Kinase/Phosphotase (PNKP). Although non-phosphorylated XRCC4 has an attraction to the T-Terminal Fork-Head Associated (FHA) Domain of PNKP, the phosphorylated XRCC4 promotes even greater attraction, which is then better for DNA double-strand repair. Together, the non-phosphorylated and phosphorylated XRCC4 both works in a complex way with DNA ligase and PNKP in order to repair DNA ends.
PNKP in Base Excision Repair (BER)
[edit | edit source]The repairs of most minor base modification that are caused by ionizing radiation, reactive oxygen species and alkylating agents are repaired through the process known as base excision repair (BER). The first step of this process involves DNA glycosylase and its removal of the modified base. This step is then followed by the cleavage of the DNA at the newly formed apurinic/apyrimidinic (AP) site by AP endonuclease I (APE1). The existence of PNKP and its function in the BER pathway became known when nei endonuclease VIII-like-1 (NEIL1) and NEIL2 mammalian DNA glycosylases were discovered, which are proteins that help induc 3’-phosphate termini. NEIL 1 and NEIL 2 (nei endonucleases VII-like 1 and 2 respectively) can form complexes containing PNKP or other BER components. NEIL 1 and NEIL 2 repair DNA by excising, or cutting out, the damaged DNA base and then removing the errors. The competition between the NEIL glycosylases may lead to a base excision repair pathway that does not depend on the enzyme APE1 (AP endonuclease 1).[1] Both of these newly found proteins, even though were found to not bind directly to PNKP, were found to be associated with larger components of the BER process, among which is PNKP. These glycosylases are able to cleave non-basic pair sites and cause sensitive of PNKP depleted cells to the methyl methanesulfonate (MMS), an alkylating agent. It is still unclear exactly what PNKP’s function is in BER, but in studying NEIL1 and NEIL2 more in depth, these proteins could possibly provide explanations regarding PNKP’s role and function in this repair pathway.
Clinical Role of PNKP
[edit | edit source]Through PKNP's elaborate structure, we can find out about the complex function that allows it to help mend the loose DNA strands that have been damaged. One of the innovative purposes that PNKP can serve in clinical research within the human race is that it could help defend cells from radiation damage, which could be beneficial in the clinical field of cancer chemotherapy. Because of PNKP's role in the rebuilding of tumor cells, many of PNKP's inhibitors, as well as other inhibitors of other DNA Repair Proteins, are of interest recently in order to make tumor and cancer cells more vulnerable to attack from radiation. When these malignant cells are subjected to more weakness, the radiation therapy would be rendered more potent and the cancer in the patient has a higher chance of being in remission.Some examples of the cancers that the inhibitors may fight include ovarian and colon cancers, which are more of the common cancers. While the role of PNKP to repair DNA is very beneficial to normal cells, the opposite job of the DNA Repair Inhibitor Enzymes is also helpful when trying to eradicate cancer and tumor cells.
When there is a mutation of disruption with the function of one of more of the pathways of PNKP (Non-Homologous End Joining, Single Strand Break Repair, and Base Excision Repair), there is an association with the result of severe neurological disorders within humans, which is detrimental to normal development. For example, mutations with enzyme LIG4 is associated with microcephaly, which is a condition in which a child is born with a much smaller brain circumference than what is considered natural or normal. In mice, when enzyme XRCC1 (which must be phosphorylated and then binded to the FHA domain of PNKP)is deleted contributes to seizures. A recent study found that Autosomal recessive microcephaly, infantile-onset seizures, and developmental delay (MCSZ) is caused by PNKP mutations in both domains of the enzyme. Through the many combinations of the mutations could come from either or both phosphatase and kinase domains, many symptoms of the affected individuals also vary. Through the varying symptoms of MCSZ, it is shown that PNKP can function through multiple enzymatic repair pathways (DSBR, SSBR, and BER).
Mutations in PNKP can lead to autosomal recessive neurological disorder so increased levels of PKNP can mend the effects of reactive oxygen species (ROS), which are free-radicals that contain oxygen molecules which cause DNA, protein, and lipid oxidation inside the body. The increase in cadmium and copper levels can be damaging and neurotoxic and thus leads to PNKP inhibition, which increases the probability of cancer since PNKP is the enzyme used to repair DNA strand breaks in human cells.[1] Two other findings showed that PNKP can be denatured by natural quantities of cadmium and copper. Cadmium and copper actually have harmful carcinogenic and neurological effects in a physiological sense, which seems contradictory to the function of PNKP. The accumulation of cadmium and copper will prevent the PNKP from properly functioning so when cells cannot repair the breaks in DNA strands or fix the errors, often times the result is cancer.[1]
References
[edit | edit source]Overview
[edit | edit source]NLR stands for nucleotide-binding domain and leucine-rich repeat containing. It is a protein that is conserved in animals as well as plants that provide pathogen-sensing systems. Mechanisms, which remain largely elusive, can activate the pathogen sensing systems directly or indirectly by molecules that are pathogen derived. NLR proteins are majorly stabilized by factors such as HSP90, a molecular chaperone, and SGT1 and RAR1, its co-chaperone, which have been revealed from studies with plants. In mammals, SGT1 and HSP90 have been found to be required for NLR proteins to function. This underscores the innate immune system regulatory mechanism’s evolutionary conservation. Insights that have been provided by the SGT1-HSP90 complex structure from comparative analyses of mammalian and plant NLR proteins have uncovered mechanisms in the regulation of immune NLR sensors.
Plants and Animals: NLR-type Immune Sensors
[edit | edit source]Higher eukaryotic organisms have used their intra or extracellular sensors in the initiation of disease defense responses. This was the initial switch in recognizing potential pathogens. Plants and animals both have sensors that share similar structures. NB, which is the nucleotide binding site contained in cytosolic sensors and LRR, which is the leucine rich repeat domains together are both called NLR, NB and LRR containing sensors. NLR sesnors in higher plants have recognized by directly or indirectly the specific pathogen effecter protein that promote virulence upon the delivery to the host cells. R genes, also known as resistance genes, which are genes that encode NLR proteins, have been characterized and isolated from a large variety of species of plants from the past fifteen years.
The genome of arabidopsis thatliana contains around one hundred and fifty genes that are NLR encoding. Rice has up to six hundred NLR encoding genes. Because NLR genes have the ability to identify and fight pathogens, they are very important in the breeding of agriculture. Twenty one NLR proteins in humans, which are also called caterpillar proteins or NOD-like proteins, have been involved with sensing pathogen products as well as their danger signals. They also participate in innate immune responses’ regulation.
In some humans, mutations of NLR genes are related to autoimmune diseases. Mammalian and plant NLR proteins both need proper regulation that takes a molecular chaperone HSP90 that is contained in a complex and SGT1, which can be found in bona fide co chaperone for HSP90.
There have been the emergences of three major functions concerning the large amount of data that have been amassed for HSP90. These three functions are:
- regulating a substrates turnover by contributing to the quality control, assembly, and folding.
- maintaining the client proteins in a metastable inactive state until a stimulus has been triggered to allow the proteins to stay as being on the verge of activation.
- buffers the buildup of cryptic mutations, which would eventually lead to unstable and inactive protein synthesis, a role involved in evolutionary processes.
There have been substantial evidence that the complex SGT1-HSP90 contributes to NLR protein’s maturation and stabilization. As of today, it is still unclear about how the process works. Dissecting the networks connecting NLR proteins, SGT1, and HSP90 should lead to the specifics of this family. This puzzle is being solved by comparative analysis of mammalian and plant regulation mechanisms as well as the structure of the core complex of SGT1-HSP90.
NLR Protein Family Characteristics
[edit | edit source]Animals and plants have NLR proteins that harbor a central domain of NB. Throughout the three kingdoms of life, members of NLR participate in this process of complex signal transduction. The molecular recognition that is pattern specific is performed by the LRR domain that is highly variable. This LRR domain is part of the NLR subfamily. NLR proteins have N-terminal domains that bind directly to other host proteins. They have a purpose of sensing specific pathogen effectors or they participate in the recruitment of downstream signaling partners.
Not much is known so far about the details that contribute to the tight coupling in the NLR proteins of the three domains.
NLR function in Plants: Crucial Components
[edit | edit source]HSP90, SGT1, and RAR1 are the three partner proteins that are important for many NLR sensors’ activity. These three partner proteins play an important role in resistance of plants against diseases through a network of intricate interaction. Because of these observations, it has pointed that there may be a possibility that RAR1 and SGT1 may act as a co-chaperone to HSP90. A molecular chaperone that is highly conserved is HSP90. In eukaryotic cells, they are involved in key signaling protein maturation, stabilization, and assembly. Some of these proteins include hormone receptors and protein kinases. These proteins contain an N-terminal that is use for ATP-binding domain, a middle domain that is used for binding substrate proteins, and also a C-terminal that is used for constitutive dimerization domain. The dimers work with co-chaperone proteins that regulate activity of ATPase or substrate recruitment. In a two-hybrid yeast and genetic screens, there has been shown that the HSP90 is associated with RAR1 and SGT1 through the N-terminal ATP-binding domain. It has also been shown that SGT1 and RAR1 bind to each other as well.
In eukaryotes, SGT1 is conserved. Precise functions of SGT1 are still unclear as to the details about its control on a number of processes that are unrelated. This ranges from ubiquitin ligase activation to the assembly of yeast and human kinetochore. Also included are Polo kinase and adenylyl cyclase. There are three distinct domains. They are TPR, which is tetratricopeptide repeats, CS, which is SGT1 and CHORD-containing protein, and SGS, which is SGT1 specific domain. Domains of CS and TPR are both stable and globular. The domain for SGS is unfolded intrinsically. One of the domains, the CS domain, is related evolutionarily to the co-chaperone p23. Its association to HSP90 is mediated by an interaction directly with SGT1 CS as well as HSP90 ND. On the other hand, the domain of TPR has no interactions with that of HSP90, even though some TPR domains can recognize the C-terminal pentapeptide of HSP90, which is MEEVD. SGT1 functionality was not affected by the TPR domain.
References
[edit | edit source]NLR sensors meet at the SGT1-HSP90 crossroad. Kadota Y, Shirasu K, Guerois R. Trends Biochem Sci. 2010 Apr;35(4):199-207. Epub 2010 Jan 22. Review.
Overview
[edit | edit source]GTPases are a large family consisting of hydrolase enzymes that bind and hydrolyze guanosine triphosphate (GTP). Binding and hydrolysis of GTP occurs in a region common to all GTPases, the highly conserved G domain.
Function
[edit | edit source]- GTPases play a major role in a several functions:
- Signal transduction in the intracellular space of transmembrane receptors (taste, smell, and light)
- Protein translation in the ribosome
- Regulation during cell division, translocation of proteins through membranes
- Transportation and control of assembly of vesicles in the cell.
Mechanism
[edit | edit source]Nucleophilic substitution, specifically SN2, is the mechanism for how the hydrolysis of the γ phosphate in GTP convert it into GDP and an inorganic phosphate (Pi). The mechanism also involves a pentavalent intermediate and a magnesium ion (Mg2+).
Superfamilies
[edit | edit source]There are several "superfamilies" within the family of GTPases.
Regulatory GTPases
[edit | edit source]Regulatory GTPases are GTPases that are responsible for the regulation of biochemical processes. G proteins are the most prominent of the superfamily.
GTP Switch Mechanism
[edit | edit source]All regulatory GTPases have a common mechanism that allows them to "switch" a signal transduction chain on and off. GTPase-activating proteins, or GTPase-accelerating proteins (GAPs), are a family of regulatory proteins that can bind to active G-proteins and stimulate their GTPase activity. They usually come from another signal transduction chain. The binding of GAPs to GTPases stimulates the change of GTPase between its two forms and this is what causes the toggling of the "switch" for the signal transduction chain. GTP-bound is the active form and when it gets hydrolyzed to its inactive GDP-bound form, GTPase is consequently inactivated. Of course, this inactivation can be reverted by Guanine nucleotide exchange factors (GEFs). GEFs activate GTPases by changing it back to the active GTP-bound form via initiation of the release of GDP from GTPase to allow binding to a new GTP molecule. It is important to note that the hydrolysis of GTP to GDP is irreversible and this causes the cycle to the active GTP-bound form of GTPase to be closed. Only active GTPases can produce a signal to a new reaction chain.
The efficiency of signal transduction via active GTPase is dependent on the active to inactive ratio of GTPase:

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
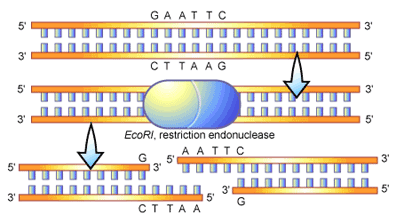{kind=link}
There are two constants that can be modified by special regulatory proteins: kdiss.GDP = dissociation constant of GDP kcat.GTP = hydrolysis constant of GTP
The amount of active GTPase can be modified in several ways:
- Acceleration of GDP dissociation by GEFs significantly speeds up the construction of active GTP-bound GTPase
- Acceleration of GTP hydrolysis by GAPs reduces the amount of active GTP-bound GTPase
- Inhibition of GDP dissociation by Guanine nucleotide dissociation inhibitors (GDIs)slows down construction of active GTP-bound GTPase. (GDIs bind to small Rho and Rab GDP-bound GTPases and keep the GTPase inactivated, as well as preventing the GTPase from localizing at their place of action, their membrane.)
- GTP analogues that cannot be hydrolyzed (γ-S-GTP, β,γ-methylene-GTP, and β,γ-imino-GTP) can keep GTPase active.
Ras GTPases/Small GTPases
[edit | edit source]Ras GTPases, or small GTPases, serve as switches for cell signalling. They get their names from a family of proteins called the Ras subfamily, which are proteins that are involved in cell signal transmission, because these GTPases are small monomeric proteins that are homologous to Ras proteins. Additionally, the Ras GTPases can be split up into 5 subcategories:
- Ras
- Rho
- Rab
- Ran
- Arf
Intracellular Membrane Traffic
[edit | edit source]Members of the Rab and Arf branches of the Ras GTPase superfamily are present in every step of intracellular membrane traffic. They regulate these steps by networking with one another through a variety of mechanisms that coordinate independent events of one stage together with other stages of the entire transport pathway. These mechanisms include many different variables:
- GEFs cascades
- GAPs cascades
- effectors that bind many GTPases
- positive feedback loops stemming from exchange factor-effector interactions.
When these mechanisms come together, an ordered series of transitions from one GTPase to the next can take place. Since each GTPase has its own unique group of effectors, the transitions that occur can help define differences in the functionality of the membrane compartments that they are associated with.
Large GTPases--Dynamin
[edit | edit source]Dynamin is a considered model for large GTPases. It is responsible for endoctyosis, a process in which cells absorb molecules by engulfment. Specifically, it is involved in the division of newly formed vesicles from the membrane of one compartment to their fusion with another compartment-- at both the cell surface or Golgi body. Along with division of vesicles, Dynamin is also involved in the division of organelles, cytokinesis, and pathogen resistance (microbial). In mammals, there are 3 different types of genes:
- Dynamin I: expressed in most cells
- Dynamin II: expressed in neurons
- Dynamin III: expressed in heart, brain, lung, and testis.
Mechanism
[edit | edit source]When a vesicle folds in so that the outer surface becomes an inner surface, dynamin will form a spiral around the vesicle's neck. The spiral will then extend and then constrict via GTP hydrolysis. This process produces a twisting motion and results in the pinching off of the vesicle from its main body. The twisting motion is dependent on its dynamin GTPase activity. So far, dynamin is the only right-handed helix that produces a twisting motion (right-handed twisting).
Translation Factor GTPases
[edit | edit source]Translation Factor GTPases have an important role in the initiation, elongation, and the termination of protein biosynthesis.
References
[edit | edit source]1. Mizuno-Yamasaki, E., F. Rivera-Molina, and et al. "GTPase networks in membrane traffic.." Pub Med. N.p., 29 2012. Web. 7 Dec 2012. <http://www.ncbi.nlm.nih.gov/pubmed/22463690>.
2. "GTPase." Wikipedia. Wikimedia Foundation, Inc. 28 May 2012. Web. 7 Dec 2012. <http://en.wikipedia.org/wiki/GTPase>.
3. "Guanine nucleotide exchange factor." Wikipedia. Wikimedia Foundation, Inc. 6 Jun 2012. Web. 7 Dec 2012. <http://en.wikipedia.org/wiki/Guanine_nucleotide _exchange_factor>.
4. "Guanosine nucleotide dissociation inhibitors." Wikipedia. Wikimedia Foundation, Inc. 21 Mar 2012. Web. 7 Dec 2012. <http://en.wikipedia.org/wiki/Guanosine_nucleotide_dissociation_inhibitors>.
5. "GTPase-activating protein." Wikipedia. Wikimedia Foundation, Inc. 4 Nov 2012. Web. 7 Dec 2012. <http://en.wikipedia.org/wiki/GTPase-activating_ protein>.
6. "Ras subfamily." Wikipedia. Wikimedia Foundation, Inc. 1 Nov 2012. Web. 7 Dec 2012. <http://en.wikipedia.org/wiki/Ras_(protein)>.
7. "Endocytosis." Wikipedia. Wikimedia Foundation, Inc. 6 Dec 2012. Web. 7 Dec 2012. <http://en.wikipedia.org/wiki/Endocytosis>.
8. "Dynamin." Wikipedia. Wikimedia Foundation, Inc. 2 Apr 2012. Web. 7 Dec 2012. <http://en.wikipedia.org/wiki/Dynamin>. Structural Biochemistry/Palmitoyl Transferase
Introduction
[edit | edit source]Homeostasis is the mechanism by which an organism maintains its body in dynamic equilibrium. A slight change in a concentration of a fluid within the organism may cause major changes within its body. In living cells, there are different kinds of enzymes working together. Living cells synthesis or break down molecules for normal metabolism and growth. Enzyme regulation is one example. Enzymes are used to catalyze (speed up) reactions within the body. The regulation of enzymes help maintain the body's equilibrium. An enzyme can be in either one of two modes: on or off. That is controlling the synthesis of enzymes and controlling the activity of enzymes (feedback inhibition). Basically enzyme regulation takes advantage of these two modes. When a concentration of one product is too high, a negative feedback loop can occur and stop the enzyme that catalyzes that specific product. Enabling a lowering of reaction rate and lowering the concentration over time.
Enzymes activities are regulated by five basic techniques.
1. Allosteric control. Allosteric proteins have different regulatory and catalytic binding sites. Allosteric proteins are cooperative proteins, where binding of a substrate in one active site affects the activity of the rest of the binding sites. Some substrate binding will favor the protein to be in the inactive T (tense) state, while other substrate binding will favor the protein to be in the active R (relaxed) state, depending on the biological needs. Allosterically regulated enzymes do not however obey Michaelis-Menten kinetics but instead follow sigmoidal kinetics.
Example of allostery:[[6]]
![[6]](https://commons.wikimedia.org/wiki/File:Allostery_in_Glycogen_Phosphorylase.JPG){kind=link}
2. Isoenzymes. Isoenzymes have different animo acid sequences but catalyze the same reaction as enzymes. They usually have different Km and Vmax values, and different regulatory techniques. The advantages of isoenzymes is that it can catalyze the same reaction under the different environments within the different organelles. Isozymes are an important entity in metabolism for servicing a specific tissue or developmental sequence. For example lactate dehydrogenase (LDH) has two isozymes that have an amino acid sequence that is 75% similar. The H isozyme is present in the heart muscle and the M isozyme is expressed in the skeletal muscle.
Example of isoenzymes and their structure: [[7]]
![[7]](https://commons.wikimedia.org/wiki/File:PDB_1qat_EBI.jpg){kind=link}
3. Reversible covalent modification. An enzyme's activity can be altered by covalently attaching a different group to its active site. It blocks the natural substrate from binding to the active site. The most common forms of covalent modification are phosphorylation and dephosphorylation as well as aceylation and deacylation. Not all forms of covalent modification are readily reversible. For example, an attachment of a lipid group will inhibit the signal-transduction pathway in some proteins.
Example of dephosphorylation: [[8]]
![[8]](https://commons.wikimedia.org/wiki/File:Phosphodephosphoinverto.jpg){kind=link}
4. Proteolytic Activation. Many enzymes are present in the body in their inactive forms call zymogen or proenzyme. They are not activated until a digestive enzyme cleaves it. The cleavage alters the three dimension shape of the enzyme, forming the active site in the right orientation. The zymogens become active enzymes in an irreversible reaction, typically the hydrolysis of bonds in the zymogen.
Example of zymogen structure: [[9]]
![[9]](https://commons.wikimedia.org/wiki/File:Factor_XI_2F83.png){kind=link}
5. Control by Limiting Amount of Enzyme. The amount of enzymes gets produced can be controlled at the transcription level.
In Double Displacement (Ping Pong reaction), two compounds switch places to form new compounds. Two reactants yield two products.
Ping Pong Mechanism
[edit | edit source]
In the Ping Pong mechanism substrate S binds to the enzyme transferring a chemical component to the active site making a modified enzyme. Once substrate S leaves active site substrate T can bind and react with the newly modified active site. Once the newly formed product leaves the enzyme it returns to its original state ready to accept substrate S.
Enzymes that exhibit this mechanism include thioredoxin peroxidase, cytydilytransferase, and chymotrypsin. Serine proteases which cleave polypeptide bonds is an example of this mechanism where the enzyme accepts the amino acid and modifies the serine residue by acetylating it. The modified enzyme accepts water as which liberates the product and liberates the original enzyme.
In Ping-Pong Reactions, one or more products are released before all substrates bind the enzyme. The defining feature of double-displacement reactions is the existence of a substituted enzyme intermediate, in which the enzyme is temporarily modified. Reactions that shuttle amino groups between amino acids and a-ketoacids are classic examples of double displacement mechanisms. The enzyme aspartate aminotransferase catalyzes the transfer of an amino group from aspartate to a-ketoglutarate.
After aspartate binds to the enzyme, the enzyme accepts aspartate’s amino group to form the substituted enzyme intermediate. The first product, oxaloacetate, subsequently departs. The second substrate, a-ketoglutarate, binds to the enzyme, accepts the amino group from the modified enzyme, and is then released as the final product, glutamate. In the Cleland notation, the substrates appear to bounce on and off the enzyme analogously to a Ping-Pong ball bouncing on a table.
The potential threats
[edit | edit source]As scientific researchers have proved that enzymes are central for metabolic pathways in organisms, they have also pointed out that those very enzymes could also potentially threaten the survival of the organisms. For example, in DNA transcription, if the enzyme carrying out the work malfunctions, it can give rise to an errant gene that codes faulty proteins or no proteins at all (these occurrences are known as mutations). Therefore, such proteins may result in out-of-control cell divisions, which can lead to dire consequences, which most of the time are related to cancer.
Conclusion
[edit | edit source]Enzymes play a vital role in organisms' ability for survival. They create the abilities of moving, thinking, sensing, and so on. Besides, enzymes are central for almost any metabolic reactions in any organisms: they can catalyze a series of lengthening lab-conducting chemical reactions in seconds with high levels of complexity and precision, thus earning the name "natural catalysts". Biochemists' goals are trying to study and understand the mechanisms, by which only such enzymes can do their wonderful magic for organisms' survivability. Feedback inhibition is the phenomenon where the output of a process is used as an input to control the behavior of the process itself, oftentimes limiting the production of more product. Although negative feedback is used in the context of inhibition, negative feedback may also be used for promoting a certain process. An everyday example of negative feedback is the cruise control in automobiles. The faster a car goes above the cruise control speed, the stronger the brakes are applied to slow the car down. If the car is going too slowly, more gas is fed to the engine to speed the car up. In a biological context, the more product produced by the enzyme, the more inhibited the enzyme is towards creating additional product.
Many enzyme catalyzed reactions are carried out through a biochemical pathway. In these pathways, the product of one reaction becomes the substrate for the next reaction. At the end of the pathway, a desired product is synthesized. In order to tightly regulate the concentration of that product, the biochemical pathway needs to be shut down. This is done through feedback inhibition. The product of the final reaction in that pathway reacts with an enzyme somewhere along the pathway at the enzyme's allosteric site, changing the conformation of the enzyme. That enzyme can no longer bind to its substrate as effectively due to the conformational change, closing down that pathway and stopping the final product from synthesizing. The higher the concentration of the final product, the more likely that product will bind to the allosteric site of the enzyme, shutting down that pathway.

There are many intermediates and pathways in feedback inhibition. Often the final product Z will inhibit the initial reactant A.
Mechanism of Negative Feedback
[edit | edit source]Each metabolic reaction or process is regulated by several enzymes. These enzymes control the rate of these reactions and thus are fundamental in maintaining homeostasis. Below is a universal map of how this type of inhibition works. We will start with a substrate that is attacked by enzyme 1, forming product A which then acts as the substrate for enzyme 2 forming product B. Product B then becomes the substrate for the attack of enzyme 3 forming our final product.
substrate ---enzyme 1--> product A ----enzyme 2---> product B ----enzyme 3----> Final Product
Keep in mind that the final product is usually something the body uses up and is necessary for homeostasis. In this reaction, the purpose of the intermediates, product A and product B, is to move the reaction along to reach the final product therefore the inhibition mechanism does not start at these intermediates but at the final product. As the amount of final product becomes elevated, the system imposes a halting effect on enzyme 1, slowing down the production of intermediates A and B, reducing the formation of the final product. When levels of the final product fall below a threshold, the effect of negative feedback diminishes and enzyme 1 is reactivated and the reaction process will be started again.
So what forces are responsible for creating these feedback responses? There are several regulators that affect a given process. Hormones and chemical signals produced and distributed by the hypothalamus and pituitary glands, for example, are the regulators that act in feedback loops. To illustrate the concept of this section, let's investigate the regulation of blood sugar levels. The hormones insulin and glucagon are two regulators that are intimately related in regulating sugar levels. Insulin is responsible for triggering different cells in the body to absorb glucose from the blood and to store the excess as glycogen for later utility. Conversely, glucagon's function is to convert glycogen supply into glucose. When blood glucose level is too low, the alpha cells of the islets of Langerhans in the pancreas release glucagon. Glucagon subsequently activates the conversion of glycogen to glucose until the sugar level in the blood is back to its normal state. When blood glucose is too high the beta cells of the islets of Langerhans release insulin which causes cells in the body to take up sugar quickly, lowering the blood sugar level to its normal level.
Positive Feedback
[edit | edit source]In contrast to negative feedback, positive feedback occurs when an output is used as a signal to increase further response of the output. In other words, if process A results in consequence B, B reinforces process A, resulting in a cascade where more of B occurs, which causes more of A to occur, and so on. An example of a positive feedback loop is evolution, where an organism evolves and becomes better at hunting prey, for example, prey evolve better defense mechanism like faster running, which causes predators to adapt by evolving better chasing skills, and so forth. Note that a positive or negative feedback mechanism is not necessarily beneficial or harmful; they only refer to the mechanism by which inhibition or propagation occurs.
Examples of Feedback Inhibition
[edit | edit source]Feedback inhibition controls the production of amino acids. The benefits of feedback inhibition are that the building blocks such as 3-phosphoglycerate, which is crucial to other processes such as the Calvin cycle and glycolysis, are used optimally and without waste.
 Note: the structure of 3-phosphoglycerate is shown here
Note: the structure of 3-phosphoglycerate is shown here
Feedback inhibition also controls nucleotide production. The pyrimidines (Thymine, Cytosine, and Uracil) have different pathways and feedback mechanisms than the Purines (Adenine and Guanine). Aspartate transcarbamoylase [10] regulates pyrimidine synthesis in bacteria. The regulation for purine production begins as PRPP or 5-phosphoribosyl-1-pyrophosphate which is converted into Phosphoribosylamine. This pathway is inhibited by IMP, AMP, and GMP. Then Phosphribosylamine is converted into IMP. IMP is a common precursor to both Adenosine and Guanine. The pathways from IMP to the Adenosine and Guanine precursors of AMP and GMP, respectively, are separated. IMP to AMP is inhibited by AMP(adenosine precursor) and IMP to GMP(guanine precursor) are inhibited by GMP, thus the products are inhibiting the precursors.
![[10]](https://commons.wikimedia.org/wiki/File:PDB_1fb5_EBI.jpg){kind=link}
Cholesterol production in the liver is catalyzed when cholesterol levels are low. This is done on the mRNA level of transcription through a transcription factor called the sterol regulatory element binding protein or SREBP. The role of SREBP is to increase the rate of transcription of mRNA by binding to a short DNA strand called sterol regulatory element or SRE. Conversely translation of reductase is inhibited by consumed cholesterol and other derivatives.
Negative feedback results in inhibition, but another powerful tool in biological systems is the positive feedback cycle. This process is the opposite of negative feedback. We can find an example of it in catalytic cascade processes, such as blood clotting. An initial factor will begin the cascade, say catalyzing or activating proteases, and which each step, additional steps will follow due to the chain reaction. Each step will amplify the signal first given, until it reaches its destination or purpose. In this sense, a very little amount of the initial factor is needed since the steps following provides efficient magnification.
Feedback inhibition is a form of allosteric regulation in which the final product of a sequence of enzymatic reactions accumulates in abundance. With too much of this product produced, the final product binds to an allosteric site on the first enzyme in the series of reactions to inhibit its activity. This halts the reaction at the first step so that no more excess product is produced. In the images above, the second to last product is the one that halts the reaction by biding allosterically to the active site on the first enzyme. This is done to illustrate that not all feedback inhibition is exactly clear cut. Different processes will be regulated differently depending on a variety of factors such as the enzymes and substrates involved, and the conditions in which the reaction takes place.


ATCase
[edit | edit source]Aspartate transcarbanoylase catalyzes the first step in the synthesis of pyrimidines. As mentioned above, after aspartate transcarbamoylase catalyzes the committed step, also known as,the condensation of aspartate and carbamoyl phosphate takes place to form N-carbamoylaspartate, in pyrimidine synthesis. This will yield pyrimitdine nucleotides such as cytidine triphosphate (CTP)(See Figure Below).




The molecule CTP is also known to be used in feedback inhibition in conjunction with aspartate transcarbamoylase (ATCase)(See Figure: CTP Inhibits ATCase). CTP, which is the final product of the metabolic pathway started by ATCase, inhibits ATCase when there is CTP in excess. When there is excess CTP, the enzyme activity decreases which explains why CTP favors the T state which is less active. This type of inhibition regulates that N-carbamoylaspartate and other subsequent intermediates in the pathway are not unnecessarily formed when the concentration of pyrimidines is large.
Allosterically regulated enzymes, such as ATCase do not follow Michaelis-Menten Kinetics. Allosteric enzymes are differentiated from other enzymes due their response to changes in substrace concentration levels and their susceptibility to regulation by other molecules. The plot of rate of product formation as a function of substrate concentration for ATCase differs from that expected for enzymes that obey the Michaelis-Menten kinetics. Instead, the curve for ATCase is in the form of a sigmoidal curve, which is due to the fact that binding of substrate to one active site of the enzyme increases the activity at the other active sites. This means that they enzyme has cooperative properties, similar to that of hemoglobin, the protein in our blood that transports oxygen molecules throughout our body. (See Figure: ATCase Sigmoidal Kinetics).
CTP has a structure that is different from the substrates of the reaction. Thus, CTP must bind the different active sites called regulatory sites.
p-Hydroxymercuribenzoate separates the catalytic (c chain) and the regulatory subunits (r chain) of ATCase, in which the p-hydroxybenzoate reacts with sulfhydryl groups on the cysteine residues in ATCase.(See Figure: modification of cysteine residues). Ultracentrifugation studies have shown that mercurials can dissociate ATCase into these two kinds of subunits, in which the subunits can be separated by ion-exchange chromatography. Ion-exchange chromatography is effective in this case because the subunits differ in their charge. The subunits can also be separated by centrifugation in a sucrose density gradient since the subunits differ in size. ATCase is 6subunits of two trimers . Here is a regulatory dimer and a catalytic trimer. CTP is an allosteric inhibitor, and it binds to regulatory subunits of the less active T state, which is favored by CTP binding. CTP decreases the activity of the enzyme. ATP competes with CTP because ATP stimulates the reaction by binding to where CTP will bind.
Two c chains are stacked on one another and linked to three r chains. The contact between the r chains and c chains are stabilized by a Zinc ion bound to four cysteine residues. To separate the r and c chains, the mercurial compound p-Hydroxmercuribenoate can be used. This compound can separate the chains because it has mercury, which strongly binds to cysteine residues, displays the Zinc ion, and destabilizes it.
For more information on ATCase: [11]
References
[edit | edit source]Berg, Jeremy M., Lubert Stryer, and John L. Tymoczko. "The BioSynthesis of Amino Acids." Biochemistry. 6th ed. 697-98. 723-724. 742-743.
http://www.scribd.com/doc/8639011/161-Negative-Feedback-Mechanisms. In other words, Isozymes are enzymes that catalyze the same chemical reactions but have the different in amino acid sequences. They are displayed in different kinetic parameter and different regulatory properties.
Definition
[edit | edit source]Isozymes (also known as isoenzymes) are homologous enzymes that catalyze the same reaction but differ in structure. The differences in the isozymes allow them to regulate the same reaction at different places in the specie. In particular they differ in amino acid sequences. They display different kinetic parameters as well as regulatory properties. For example, isozymes have different KM and Vmax values, and can be distinguished from one another by biochemical properties such as electrophoretic mobility.
Isozymes are encoded by different genes and expressed in a distinct organelle or at a distinct stage of development. The purpose of isozymes is to allow fine adjustment of metabolism to meet the need of different development stages and help the different tissues and organs function properly depending on their physiology make up and in what kind of environment which they function. For example, the isoenzymes of lactate dehydrogenase in animal organs are different in term of their amino acid sequences and the level of their expression. The level of the different isozymes in a certain organ is related to the level of oxygen supply. Isozymes appear in specific regions of the body; differing in specifics organelles or tissues.
In terms of kinetics, isoenzymes have the capability to fine tune their enzymatic rate constants KM and Kcat. This adaptation allows for the proper use of the enzyme based on its environment (e.g. lactate dehydrogenase isozymes present in the heart and in the liver, where O2 is abundant in heart but not so in the liver).
Differentiating Isozymes
[edit | edit source]As mentioned above, isozymes are enzymes that have different structures but carry out the same tasks. A biochemical assay is needed to differentiate between different isozymes. Another method one could use is gel electrophoresis. This method takes advantage of the fact that isozymes have substituted amino acids and that provides a change in electrical charge of the enzyme. This difference in electrical charge between two different isozymes can be readily detected by gel electrophoresis. This provides a basis for molecular markers because these isozymes can easily be detected.
For identifying isozymes, a crude protein produced from grounded animal/plant tissue and buffer is used. The components of this protein is then extracted according to its electrical charge via electrophoresis. Since all the proteins from the tissue are present in the gel, an assay used to identify the individual enzymes by linking their functions to staining reactions. This method requires the enzyme to be active and functional after separating them via gel electrophoresis.

Applications
[edit | edit source]
Isozymes in general can be used to meet the metabolic needs of different tissues and developmental stages. An example of an enzyme with different isozymes is lactate dehydrogenase (LDH). This enzyme is used to catalyze the synthesis of glucose in anaerobic metabolism of glucose. The isozymes of this enzyme are divided into two forms, the H isozyme and the M isozyme. The H isozyme is expressed more in the heart, whereas the M isozyme is expressed more frequently in the skeletal muscle. Both isozymes have two polypeptide chains, and each isozyme share 75% of the amino acid sequence for the chains. Both isozymes metabolize glucose, but the difference is that the H isozymes have a higher affinity for their substrates than the M isozyme does. Another difference is that the H isozyme functions better in aerobic environments such as the heart, whereas the M isozyme functions better in anaerobic environments such as the muscle, where strenuous activity may deplete the oxygen supplies. For example, when a rat heart is developing, the amount of H and M isozymes in the rat heart tissue begins to change because of the switch from an anaerobic environment to an aerobic one. This can be seen in figure A. This chart describes the rat heart's lactate dehydrogenase isozyme profile changes as the rat heart tissue develops. The H isozyme is shown as squares and the M isozyme is shown as circles. the negative numbers are the days before birth and the positive numbers are the days after birth. The amount of M isozymes decreases dramatically as the rat grows into the adult stage.
Isozymes may also be utilized to diagnose tissue damage such as damaged heart muscle cells during a heart attack or myocardial infarction. When heart muscle cells are damaged, they release the cellular material such as the H isozyme. When taking blood samples, if the H isozymes appear in increased levels, then there is a possibility that the heart cells are damaged.
Another example of an isozyme is hexokinase. The substrate is usually glucose and the product is glucose-6-phosphate. The six-carbon sugar is also known as a hexose. Glucokinase is one isozyme of hexokinase. A kinase is an enzyme which catalyzes the transfer of a phosphoryl group from NTP to NMP. ATP is often used in these types of reactions. Glucokinase is important in metabolism, and regulating carbohydrates in the human body. The difference of the Glucokinase enzyme is that it has a much lower affinity for glucose. Most Glucokinase activity is found in the liver. This is where it catalyzes the conversion of glucose to triglycerides.
References
[edit | edit source]Berg, Jeremy M. John L. Tymoczko. Lubert Stryer. Biochemistry Sixth Edition. New York: W.H. Freeman, and Company 2007. The analysis of patterns of selected enzymes has been used extensively as a technique to identify the species of origin of cell lines with a high degree of certainty. Isoenzymes (or isoenzymes) are structurally different forms or the same enzyme. They catalyse the same reaction but have different protein structures. Each cell line would have its own unique array of isoenzymes.
The technique involves gel electrophoresis of cell homogenates under non-denaturing conditions. Specific activity stains are used to develop a banding pattern of isoenzymes (zymogram), which is characteristic of a particular cell line. Such zymograms are usually photographed to provide a permanent record of each cell line.

Of the various enzymes that can be separated into isoenzymes by this technique, glucose 6-phosphate dehydrogenase, and nucleoside phosphorylase have been particularly well characterised. By using several enzymes the distinguishing features of a cell line are established. These features can often distinguish cell lines even if derived from the same species. In biology, eukaryotic cells were conventionally thought to be able to execute the necessary adaptations of intermediary metabolism to changes in metabolic conditions. Such conditions include nutrient availability, proliferation state, etc. It was also thought that there is little or no involvement of gene regulatory mechanisms in these metabolic conditions mentioned above.
This conventional view, however, has been challenged recently. Connections between intermediary metabolism and the regulation of gene expression are proved to exist recently with discoveries made in the related fields. Some common enzymes have been discovered to have RNA-binding properties, and their RNA-binding activity is controlled by their metabolites.
Furthermore, RNA-binding and enzymatic functions are proved to be mutually exclusive through experiments. Regulation of RNA binding by cofactors and metabolites of enzymes has suggested a potential coordinating principle that may help to explain gene regulatory and metabolic functions. In some cases, enzymatic and RNA-binding functions may be competitive. If this is the case, enzyme activities could be regulated by RNA binding.
The REM (RNA, enzymes, and metabolites) phase of gene regulation may be explored systematically. Firstly, cross-linking techniques may be employed to stabilize RNA-protein interactions in living cells. The yield would consist of all cellular RNA-binding proteins. The interacting RNAs of selected enzymes are then identified, and the lists of mRNA binding to a given enzyme could be analyzed. Finally, the metabolic functions of interest may be discovered.
The REM phase of gene regulation could have broad applications for cell biochemistry, cell biology, and biotechnology. After a molecule is covalently attached to an enzyme, the activity, or catalytic activity, of the enzyme can be changed. In this regulatory strategy, the donor molecule provides the modifying group. The acceptor molecule is usually an enzyme molecule which accepts the modifying group therefore changing its activity. The acceptor is usually a serine, threonine, or tyrosine residue, amino acids that contain hydroxide. The process of the covalent modification may be reversible, but not in all cases. One common example of covalent regulation is protein phosphorylation.
Examples
[edit | edit source]The examples of the covalent modification strategy are acetylation/deacytilation; phosphorylation/dephosphorilation; myristoylation; ADP ribosylation; farnesylation; sulfation; ubiquitination. However, phosphorylation and acytilation are the most common examples. In phosphorylation, ATP donates a phosphate to the hydroxyl group of a cytosine or tyrosine. The reaction yields ADP and a phosphate ester (phosphorylated protein). In the case of phosphorylation, glucose homeostasis is the function that is modified in the glycogen phosphorylase protein. In case of acytilation, the donor molecule is acetyl CoA. The DNA packing function can be modified in the histon protein.
Regulation of the activities of the target proteins
[edit | edit source]
The phosphorylation reaction is used in almost every metabolic process; moreover, approximately 30% of the known proteins are phosphorylated. The enzyme which catalyzes the phosphorylation process is protein kinase. The human genome contains about 500 homologs for this enzyme. The donor molecule that provides modifying group for that process of the catalytic modification is an ATP molecule. The acceptor molecule has to contain one of the OH groups containing amino acids in its sequence (such as serine, threonine, or tyrosine). In the process of phosphorylation, the ɤ-phosphoryl group from ATP molecule attacks the OH containing amino acid in the protein molecule. This process can ONLY occur within the cell. Proteins that are located outside the cell are not able to go though the phosphorylation regulation.

Protein phosphotases are another type of enzyme which catalyze the process of the removal of phosphate group from the protein molecule. This is also known as dephosphorylation. This enzyme deactivates the flag on the protein that was activated by the kinase. Protein phosphatase removes the phosphoryl group that is attached to the protein. However, those enzymes are required to increase the reaction rate of the phosphorylation/dephosphorylation reactions.
Protein phosphorylation adds two negative charges, forms 2-3 hydrogen bonds, is a reversible modification, kinetics can be adjusted to physiological process, amplifies signal, and ATP coordinates signaling with bio-energetics.
Phosphorylation can control the activity of proteins
[edit | edit source]1. An attached phosphoryl group gives an additional negative charge (2-) to the modified protein. Presence of the negative charge can be a source for the electrostatic interaction with other proteins which contain positive charge or within the same protein with positively charged amino acids.
2. A phosphoryl group is also able to form an additional interaction, as a result of the ability to form three H-bondings.
3. Phosphorylation process gives large expand of free energy.
4. The phosphorylation process can take time in the range between one second and few hours depending on the physiological process.
5. The donor molecule for phosphorylation, ATP, is represented as a unit of energy currency required for the regulation in the metabolic processes.

Phosphorylation is a covalent modification that controls the activity of enzymes and other proteins. Signals can be greatly amplified by this modification because one kinase has the potential to create an exponential chain effect on various target molecules. An example of this would be the sensitivity of human eyes in reaction to a photon. Protein kinase regulatory activations can be reversed by protein phosphatases, a hydrolysis reaction of connected phosphates. Cyclic AMP, an intercellular messenger, can activate protein kinase A. Cyclic AMP activates Protein Kinase A by altering the quaternary structure. The effects of cAMP in eukaryotic cells are due to activation of PKA by cAMP.The activation of that multifunctional kinase is accomplished by cAMP binding to the regulatory subunit of the enzyme, which frees the functional sites of protein kinase A. When a inhibitor is bound to a Protein kinase A, it binds between the domain of the enzyme in a cleft.

{kind=link}
PKA in muscle has 2 subunits: Regulatory (R) subunit and catalytic (C) subunit. The binding of cAMP to the regulatory subunit relieves its inhibition of the catalytic subunit.
Regulation of PKA: PKA is activated when four molecules of cAMP bind to it; this dissociates the inhibited holoenzyme(R2C2) into a regulatory subunit (R2) and two catalytically active subunits (C).
References
[edit | edit source]Berg, Jeremy M. John L. Tymoczko. Lubert Stryer. Biochemistry Sixth Edition. New York: W.H. Freeman, and Company 2007.
Phosphorylation
[edit | edit source]{kind=link}
Phosphorylation is an effective way of regulating proteins. About 30% of proteins in eukaryotic cells are phosphorylated. The enzymes that are responsible for these reactions are known as protein kinases. There are about 100 homologous protein kinases in yeast and 500 in human beings. When ATP is hydrolyzed in a test tube, the release of free energy merely heats the surrounding water. In an organism, this same generation of heat can sometimes be beneficial. For instance, the process of shivering uses ATP hydrolysis during muscle contraction to generate heat and warm the body. In most cases in the cell, however, the generation of heat alone would be an inefficient use of a valuable energy resource. Instead, with the help of specific enzymes, the cell is able to couple the energy of ATP hydrolysis directly to endergonic processes by transferring a phosphate group from ATP to some other molecule, such as the reactant. The recipient of the phosphate group is then said to be phosphorylated. The key to coupling exergonic and endergonic reactions is the formation of this phosphorylated intermediate, which is more reactive than the original unphosphorylated molecule.

ATP must be present for phosphorylation because it is needed as a donor. One of the phosphoryl groups of ATP is transferred to a specific amino acid. The acceptor is one of three amino acids with a hydroxyl group as a side chain: serine, threonine, and tyrosine. Tyrosine is handled by a different protein kinase than the other two. The reaction below shows how ATP donates one of the phosphoryl groups to a phosphorylated protein.
Protein phosphatases reverse the effects of kinases by catalyzing the removal of the phosphoryl group attached to proteins. The enzyme hydrolyzes and breaks the bond attaching the phosphoryl group.
{kind=link}
{kind=link}
It is important to note that phosphorylation and dephosphorylation reactions are not the reverse of one another. The former takes place through the action of protein kinase and ATP cleavage, whereas the latter will only take place in the presence of a phosphatase.
Reasons for Effectiveness
[edit | edit source]1) A phosphoryl group adds two negative charges to the protein. These changes alter substrate binding and catalytic activity.
2) A phosphoryl group can form 3 or more hydrogen bonds and can be tetrahedral.
3) The free energy of phosphorylation is very large. Therefore it can change the equilibrium by a large factor.
4) Phosphorylation and dephosphorylation take place in less than a second or over a span of hours.
5) Phosphorylation produces highly amplified effects. One kinase changes hundreds of target proteins in a short interval.
6) Phosphorylation is irreversible.
7) ATP is the energy needed, and thus it links the process to bio energetics.
Phosphorylation is important to the regulation of cells by regulating insulin, water balance, and homeostasis of the cell. The enzyme GSK-3 by AKT (Protein kinase B) regulates the insulin pathway. Na+/K+ ATPase regulate water balance and homeostasis of the cell.
Protein Kinase A
[edit | edit source]Protein kinase A (PKA) is an enzyme that is regulated by cyclic AMP (cAMP). This is common in the "flight or fight response". The hormone Epinephrine signals the synthesis for Cyclic adenosine monophosphate which subsequently activates protein kinase A. The kinase regulates target proteins through phosphorylation of serine and threonine.
PKA is not activated until cAMP binds to the regulatory subunit. This stops inhibition of PKA. The complex (R2C2) has a pseudosubstrate sequence of R that occupies the active site of PKA. When cAMP binds, the R chains move so it is no longer inhibiting the active site. The R chain has the sequence Arg- Arg- Gly- Ala- Ile.
The figure above shows how once cAMP is bound to the binding sites, the pseudosequence is no longer blocking the active site of PKA. This is how PKA is regulated.
The protein kinase A holoenzyme is a heterotetramer made up of two types of subunits: 1)A catalytic subunit which contains the enzyme's active site. It also contains a domain that binds ATP (the source of phosphate) and a domain that binds the regulatory subunit. 2)A regulatory subunit which consists of two molecules of this subunit bind one another in an anti-parallel orientation to form a homodimer; for type I subunits, this binding is covalent through disulfide bonds. This subunit also has two domains that bind cyclic AMP, a domain that interacts with a catalytic subunit, and a inhibitory domain that serves as a substrate or pseudosubstrate for the catalytic subunit. Regulatory subunits can also modulate catalytic subunit activity.
Regulating Activity: Intracellular concentration of cyclic AMP provides a very simple control mechanism over activity of protein kinase A.
At low cyclic AMP levels, catalytic subunits are bound to a regulatory subunit dimer and are inactive. As the concentration of cAMP increases to ~10nM and above it binds to the regulatory subunits, which gives way to an allosteric change in conformation which causes unleashing of the catalytic subunits. Free catalytic subunits are active and begin to phosphorylate their targets.
Glycogen phosphorylase
[edit | edit source]Glycogen is the polysarccharide of glucose. It serves as short term energy storage in animal cells. Glucose is a monosaccharide, an important carbohydrate in biology. It is used by the cell as a source of energy and as an metabolic intermediate.
Phosphorylase catalyzes the interconversion of glycogen and glucose-1-phosphate by the reaction,
Glycogen + Pi --> Glycogen + glucose-1-phosphate

where glucose-1-phosphate is then converted to glucose-6-phosphate by phosphoglucomutase, since the glucose-1-phosphate isomer cannot be metabolize easily, whereas the glucose-6-phosphate acts as fuel for glycolysis and pentose phosphate pathway.
Phosphorylase activity is regulated by reversible phosphorylation. Phosphorylase a is the active form, while phosphorylase b is inactive.

Metabolic Disorders Caused by Defective Enzymes Controlling Phosphorylation
[edit | edit source]An example of a metabolic disease caused by defective enzymes is Lafora disease. Lafora disease, named after Dr. Gonzalo Lafora, is a metabolic disorder that is caused by defective enzymes that controls phosphorylation. It is a neurodegenerative disease caused by insoluble glucan accumulating in the cytoplasm. Glucan is a type of complex carbohydrate that is made up of glucose linked together by glycosidic bonds. Lafora disease not only causes epilepsy, but also progressive central nervous system degenerations. This ultimately results in the death of the patient. It is thought that the compilation of cytoplasmic lipid bodies (LB) trigger neuronal cell death and seizures.
It is recognized that in almost half of Lafora disease cases, the EPM2A (epilepsy, progressive myoclonus 2A) gene is mutated. The EPM2A gene encodes the bimodular protein called laforin. Laforin is associated with regulating glycogen metabolism. Glycogen stores long-term energy. Furthermore, about 20% if Lafora disease cases are a result of mutations in EPM2B (epilepsy, progressive myoclonus 2B), which encodes the protein called malin. Malin is associated with the binding, ubiquitylating, and promoting the degradation of laforin.
It is determined that insoluble glucan accumulating in the cytoplasm causes Lafora disease, although the molecular cause of this disease is still unknown. This is due to the fact that the function of laforin is obscure because its substrate is still unidentified. There are various hypotheses, but one prominent hypothesis is that laforin dephosphorylates glycogen molecules as they are synthesized. Without laforin, glycogen metabolism results in a slightly more phosphorylated glucan and would eventually lead to a Lafora body. Another hypothesis is that laforin recruits malin to the site of glycogen synthesis. Malin regulates the synthesis of glycogen by ubiquitylating protein targeting to glycogen (PTG), glycogen synthase (GS), glycogen debranching enzyme (GDE), and laforin to inhibit LB formation. Ubiquitin (Ub) is a 76 amino acid protein that tags proteins for destruction by proteasomes. It is found in all eukaryotic cells and is involved in various signaling pathways including cell cycle, endocytosis, transcription, DNA repair, signal transduction, apoptosis, and the immune response. The proteins degraded through ubiquitin are regulatory proteins. Ub-activating enzymes (E1), Ub-conjugating enzymes (E2), and Ub ligases (E3) are essential in protein ubiquitylation, which is most commonly known for its ability to execute controlled protein degradation by the 26S proteosome. However, ubiquitylation can not only induce protein degradation, but it can also control protein behavior, modulate subcellular localization, and moderate protein-protein interactions. Ubiquitin medications can signal irreversible proteolytic events (such as degradation through Lys48) and reversible nonproteolytic events. The nonproteolytic events utilize mono-ubiquitin and poly-ubiquitin chains connected through Lys6 or Lys63.
Ubiquitylation Pathway
[edit | edit source]E1 activates ubiquitin through a thiol-ester bond between the cysteine found in its active site and ubiquitin's carboxyl-terminal glycine. The now activated ubiquitin is transferred to E2 through transesterification. E3 brings the ubiquitin-charged E2 and relevant substrate to facilitate the formation of an isopeptide linkage between ubiquitin's carboxyl-terminal glycine and the ε-amino group on the substrate or an ubiquitin attached to the protein.
E3s are the ones responsible for substrate specificity, which correlates to the relatively large amount of E3s in respect to E1s and E2s.
Mechanism
[edit | edit source]Ubiquitin attaches to a protein by covalently forming isopeptide bonds, using energy from ATP. Ubiquitin is normally found inactive, but a Ub activating enzyme (E1) links the carboxyl group of Ub to its sulfhydryl group. The enzyme binds an ATP-activated Ub complex where a transfer of the Ub to the a cysteine residue on E1 forms the thioester bond and releases AMP. Ub is then transferred to a Ub-conjugating protein (E2) and finally that protein complex is recognized by a Ub-protein ligase (E3). E3 enzymes recognize N-terminal residues that signal the protein for degradation. The complex then binds the particular protein and the E2-Ub complex, facilitating the transfer of Ub to tag the protein.
A protein marked with a Lys48-linked poly-ubitquitin chain is recognized by the 19S cap of the 26S proteasome and is ultimately degraded. These proteases are composed of a 20S catalytic subunit and a 19S regulatory subunit. The 19S regulatory subunit normally blocks access to the 20S subunit's active site. 19S subunits recognize and bind only ubiquinated molecules, thus moderating the degradation by the 20S catalytic core. Six ATPases are required for the regulatory complex to function: ATP hydrolysis likely causes a conformational change in the 19S subunit which is transferred to the 20S subunit, allowing the active site to become available for substrate binding.
The 20S subunit is composed of two outer 7 subunit rings (alpha rings), and two inner 7 subunit beta rings. The beta subunits contain N-terminal threonine whose hydroxyl group is activated to attack the carbonyl groups of peptides. Reminiscent of serine proteases, these threonine residues also form acyl-enzymes. The degradation is completed by removal of the Ub molecules by an isopeptidase of the 19S regulatory unit. These ubiquitin molecules are then released to tag more proteins.
Regulation
[edit | edit source]Post-translational modifications are a method for regulating ubiquitylation. These modification consist of phosphorylation, oxidation, sumoylation, acetylation, and neddylation. Phosphorylation creates binding sites for E3s on substrates, oxidation is associated with ubiquitylation targeting, sumoylation blocks ubiquitylation sites, acetylation competes with ubiquitylation in modification of ubiquitylation sites, and neddylation increases E3 activity by increasing ubiquitin-loaded E2 affinity.
Deubiquitylating enzymes (DUBs) also play a role in ubiquitylation regulation. These enzymes essentially undo the work of the E2s and E3s. Examples of DUBs are ubiquitin C-terminal hydrolases (UCHs) and ubiquitin-specific processing proteases (UBPs). UCHs hydrolyze the carboxyl-terminal ester and amide bonds of ubiquitin. UBPs take apart the polyubiquitin chains.
Mono and Poly Ubiquitylation
[edit | edit source]Mono-ubiquitylation is a regulatory modification that is a contributing factor in transcription, histone function, endocytosis, and membrane trafficking. It acts as a signal for endocytosis receptors and for lysosomal targeting. These are all proteasome-independent mechanisms. Lys63-linked poly-ubiquitylation is engaged in signalling DNA repair, the stress response, endocytosis, and signal transduction. Poly-ubiquitin chains with varying linkages signal different effects.
Disease
[edit | edit source]Because ubiquitin is an enzyme and protein regulatory system, errors in tagging lead to various diseases. Several neurological diseases such as juvenile and early onset Parkinsons, Huntingtons, and other chronic neurological diseases have been linked to errors or decreased activity of the ubiquitin-protein ligase (E3) and its consequences: inability of the ubiquitin and proteasome duo to remove damaged or malfunctioning proteins leads to aggregation and Lewy bodies - aggregates of protein inside neurons that displace functioning cellular components.
Viruses and other invasive pathogens may inappropriately activate the ubiquitin system to destroy immune response such as in HPV, leading to the formation of tumors or other diseases.
Examples E3s
[edit | edit source]APC- anaphase-promoting complex/cyclosome
SCF- Skp1-Cdc53/Cu11-F-box protein
References
[edit | edit source]Biochemistry 6th Edition, Berg et al.
The ubiquitin system: pathogenesis of human diseases and drug targeting, Ciechanover, Schwartz; http://www.elsevier.com/framework_aboutus/pdfs/ciechanover01.pdf
Getting into position: the catalytic mechanisms of protein ubiquitylation, Passmore, Barford; http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1224133/pdf/14998368.pdf
A field guide to ubiquitylation, Fang, Weissman; http://springerlink.metapress.com/content/tgl62jj9uyt19d2f/fulltext.pdf Ubiquitylation is the post-translational modification of proteins with ubiquitin, which is a powerful regulatory enzyme in eukaryotes. It can label proteins for destruction or activate gene transcription. Despite being versatile with its functions, ubiquitin is highly specific at sending signals for cellular events. Ubiquitin signaling pathway use multivalency, namely the coordinated use of multiple interaction surfaces. Multivalent interactions regulate each stage of ubiquitin signaling pathways, and appear within the ubiquitin signal, the ubiquitylated substrate, ubiquitin processing enzymes and ubiquitin recognition proteins.
Importance of multivalency in ubiquitin signaling
[edit | edit source]Ubiquitin itself is a small 76 amino acid protein that use covalent bonds to attach to other proteins to exercise its regulatory functions. It is now known to communicate with more than 150 proteins, via discrete interacting surfaces. These proteins are labeled ubiquitin receptors, and together with ubiquitin through ubiquitylation, they regulate a vast array of cellular events including protein degration, protein trafficking, transcription, DNA repair, cell-cycle progression and apoptosis. (Figure 1). The ubiquitin signal itself is diverse and often multivalent, as are ubiquitin receptors and substrates.
The ubiquitin signal is diverse and multivalent
[edit | edit source]Ubiquitin has a C-terminal glycine that is activated with ATP to form an isopeptide bond with the primary amino group of its substrate, which is usually the ɛ-amino group of a lysine, and also its amino terminus. Serine hydroxyl and cystein thiol groups can also be modified by ubiquitin. Substrate can be attached with a single or multiple ubiquitin (Figure 2). Polyubiquitylation takes place when ubiquitin is sequentially added to substrates to form ubiquitin chains. The chains can be of one linkage type, of mixed or forked with more than one ubiquitin attached. The multivalency provided by ubiquitin chains can greatly enchance their affinity for binding partners.
Ubiquitylation is a type of modification that is highly variable in length and linkage type. Different linkage result in different ubiquitin chain conformation and in unique binding epitopes, which can define downstream signaling events. When binding to ubiquitin chains of closed conformation, ubiquitin receptors must compete with the intra-chain ubiquitin packing interactions for access to binding surfaces.
Diversity and multivalency of ubiquitin processing enzymes
[edit | edit source].jpg){kind=link}
The use of ubiquitin as a diverse signaling mechanism is supported by three enzymes classes, E1 activating enzyme, E2 conjugating enzyme and E3 ligase (Figure 2), which catalyze substrate ubiquitylation and define the type of ubiquitin chain linkage. Their actions are often regulated by multivalent interactions with each other and other signaling molecules and pathways. An E1 ubiquitin activating enzyme charges ubiquitin in an ATP-dependent manner to form a thioester bond with its catalytic cysteine. This modification induces structural changes in E1 that promote its binding to an E2 conjugating enzyme, to which ubiquitin is passed. E2 conjugating enzymes typically require E3 ubiquitin ligases to pass activated ubiquitin to a protein substrate; however, they can play defining roles in the ubiquitin chain linkage type.
The timing of substrate ubiquitylation is often relayed through multivalency effects of E3s (Figure 3). E3s can respond to their own phosphorylation status, to that of their substrate and to interactions with proteins that activate or suppress their activity. The use of multiple interactions to activate or suppress substrate ubiquitylation is exemplified through MDM2, the major E3 ligase for tumor suppressor p53, which promotes cell-cycle arrest and apoptosis. In response to DNA damage, the protein kinase ATM (ataxia telangiectasia mutated) phosphorylates the E3 MDM2 at several redundant sites near its RING domain to prevent its oligomerization. Oligomerization of the RING domain ofMDM2 is required for its polyubiquitylation of p53, a signaling event that leads to p53 proteolysis; thus ATMmediated phosphorylation of MDM2 in response to damaged DNA stabilizes p53 protein levels.
Other MDM2 interactions stimulate p53 degradation, including its phosphorylation at Ser260 by polo-like kinase-1 and its interaction with death-domain-associated protein DAXX. DAXX enhances the intrinsic activity of MDM2 towards p53 and functions as a scaffolding protein to recruit the deubiquitylating enzyme HAUSP, which protects MDM2 from degradation by removing ubiquitin chains that were formed by MDM2 autoubiquitylation. In the nucleus, these actions are counteracted by the tumor suppressor RASSF1A (Ras association domain family protein 1A), which binds MDM2 and DAXX, but displaces HAUSP, thereby destabilizing MDM2. The MDM2 example highlights the use of multilayered interactions to modulate E3 activity towards specific substrates in response to distinct cellular events. It is worth noting that phosphorylation of a substrate protein can also promote E3 recruitment or displacement.
Ubiquitin chain variability provides receptor selectivity
[edit | edit source]The variable length and linkage type of ubiquitin chains provides selectivity to the outcome of ubiquitylation, because some ubiquitin receptors have strong preferences for ubiquitin chains of certain linkage type or size. Receptor specificity for ubiquitin chains of distinct linkage type couples the activities of ubiquitin processing enzymes with downstream signaling events. More than 20 different ubiquitin-binding domain (UBD) families, which exist in more than 150 human receptor proteins, have been identified to date. The mechanisms that they use to achieve selectivity for specific ubiquitin modifications have been comprehensively reviewed.
Ubiquitin chain multivalency enables simultaneous interaction with multiple ubiquitin-binding domains
[edit | edit source]The binding of multiple UBDs to ubiquitin chains provides a general mechanism for enhancing the binding affinity of ubiquitin receptors for ubiquitylated substrate (Figure 3). In humans, the proteasome component S5a has two UIMs, which are separated by flexible linker regions causing their relative orientation to be undefined. This flexibility is markedly restricted when S5a binds to Lys48-linked diubiquitin because each UIM binds a ubiquitin moiety of the same molecule simultaneously. The outcome of this coordinated binding is significantly increased affinity between S5a and diubiquitin, suggesting that its two UIMs are not used to recruit multiple substrates to the proteasome simultaneously, but rather to increase affinity for each ubiquitylated substrate. It is worth noting that S5a and hHR23a can bind a common ubiquitin chain, as can S5a and Rpn13, the other intrinsic ubiquitin receptors of the proteasome. The biological significance of these interactions is not yet clear; however, complexes of multiple ubiquitin receptors with a ubiquitylated substrate provide additional levels of multivalency, which most likely leads to a greater binding affinity. Such complexes also seem to operate during endocytic processes to enhance binding affinity. Although monoubiquitylation is sufficient for receptor internalization during endocytosis, quantitative mass spectrometry indicates that more than half of all ubiquitylated epidermal growth factor receptor (EGFR) is conjugated with ubiquitin polymers (largely connected by Lys63). Most likely, modification with a polymeric ubiquitin chain enables more interactions with the UIMs of endocytic adaptors and in turn, interactions of higher affinity compared with that possible with only one ubiquitin subunit.
Multivalent interactions involving ubiquitin chains and multiple ubiquitin receptors can also transduce signals, as exemplified by the kinase activation mechanism in the NF-κB pathway.
Multivalent interactions of ubiquitin receptors influence the fate of ubiquitylated substrates
[edit | edit source]Surfaces of ubiquitin receptors that do not bind ubiquitin play key roles in the trafficking and processing of ubiquitylated substrates (Figure 3). Ubiquitin receptors associated with proteasomal degradation have regions that dock them into the proteasome or that enable transient interaction with its components. The UBD of one such receptor, Rpn13, assembles into the proteasome via a surface opposite to its ubiquitin-binding region and this protein contains another domain that binds and activates Uch37, one of the three DUBs of the proteasome. Before their degradation by the proteasome, substrates are deubiquitylated and unfolded for passage through a narrow chamber leading to the catalytic center of the proteasome. Rpn13 might perform a dual functionality in the capture and deubiquitylation of proteasome substrates through its multivalent interactions with ubiquitylated substrates and Uch37.
The integration of multivalent ubiquitin-dependent interactions with ubiquitin-independent interactions is used extensively for endocytic trafficking to direct proteins from the plasma membrane to multi-vascular bodies (MVBs) (Figure 1).
Intramolecular interactions between ubiquitin-binding regions and covalently attached ubiquitin can inhibit ubiquitin receptor activity
[edit | edit source]Ubiquitin receptors can be conjugated with ubiquitin, which in turn binds to their UBDs (Figure 3). Such intramolecular interactions can inhibit intermolecular interactions with their ubiquitylated substrates. This mode of regulation exists in the endocytic pathway. HGS, EPS15 and epsin undergo coupled monoubiquitylation, such that their UBD mediates their own ubiquitylation by binding to a ubiquitylated E3 or to an E3 with a ubiquitin-like domain. This modification leads to cis interactions with the attached ubiquitin, which inhibits trans interaction with ubiquitylated targets. The role of this so-called coupled monoubiquitylation in endocytosis remains poorly understood. It is possible that it weakens the interaction with cargo to enable ready passage of the substrate. The DUB UBPY (ubiquitin-specific protease Y) could relieve this autoinhibition by removing the conjugated ubiquitin from receptors, thus activating them towards new substrates.
Substrates modulate the effects of their ubiquitylation
[edit | edit source]Although the fate of a ubiquitylated substrate is largely determined by its interaction with ubiquitin receptors, some substrate features can modulate the effects of ubiquitylation even after their recognition by a receptor of a designated function. For example, interaction with the proteasome typically culminates in the degradation of ubiquitylated substrates; this mechanistic pathway is an effective means to control protein lifespan. Such degradation, however, seems to require substrates to harbor, or be ‘complexed’ with, a protein containing an unstructured region, and proteins that are not ubiquitylated can be proteolyzed simply by associating with those that are, so long as they contain an unstructured region. By contrast, folded domains within ubiquitylated proteins appear to protect substrates from degradation.
Ubiquitylation can change binding affinities by adding multivalency to already existing interactions
[edit | edit source]In the nucleus, ubiquitylation is widely used to change the affinity of already existing interactions by adding multivalency (Figure 3). For example, PCNA encircles DNA to serve as a ‘sliding clamp’ forDNApolymerases duringDNA replication. When a damaged site is encountered, replication is stalled and PCNA is monoubiquitylated at Lys164 (Figure 1). This modification is recognized by UBDs of trans-lesion polymerases to increase their affinity for PCNA and to promote their error-prone trans-lesion synthesis mode of replication [86,87]. After bypassing the lesion, the error-free, processive polymerase takes over. This switch might be due to PCNA deubiquitylation because the exchange back to the processive polymerase is prohibited when PCNA is monoubiquitylated at Lys164.
Protein ubiquitylation is also used to alter the DNAbinding affinity of nucleotide excision repair (NER) factor xeroderma pigmentosum group C (XPC). Ubiquitylation is also used to weaken substrate interactions with binding partners. Histones H3 and H4 are ubiquitylated in response to UV-induced DNA damage, facilitating the recruitment of NER machinery to damaged sites by weakening histone–DNA interactions.
Conclusion: Future potential
[edit | edit source]Ubiquitin-mediated signaling is enabled by a large repertoire of enzymes that control the timing of modification, create diversity in the ubiquitin signal itself and enable dynamic alteration of the modification throughout a signaling pathway or in response to new stimuli. These enzymes communicate through ubiquitin to downstream receptors that operate within a larger context to enable signaling specificity. Versatility and specificity become congruent in ubiquitin signaling pathways through multivalency. Ubiquitin-binding regions are typically just one of many functional surfaces present in the receptor, which can contribute to the binding interaction, subcellular localization or link ubiquitin signaling with other post-translational modifications, such as phosphorylation. Ubiquitin belongs to a family of ubiquitin-like proteins that resemble ubiquitin structurally and perform their own distinct signaling, which can cross-talk with ubiquitin signaling. The ubiquitin signaling network is of therapeutic importance because parts of it are hijacked by pathogens or compromised in human diseases. It therefore is likely to have yet uncharted therapeutic potential and the manipulation of ubiquitinmediated protein degradation is actively being pursued for such purposes. Currently, the proteasome inhibitor bortezomib is used to treat multiple myeloma and mantle cell lymphoma; this inhibitor preferentially induces apoptosis in tumor cells. The underlying mechanisms of its greater cytotoxicity in tumor cells are complex, ranging from the specific accumulation of proapoptotic proteins such as NOXA (NADPH oxidase activator1) to the activation of apoptosis through an endoplasmic reticulum (ER) stress response. Perhaps not surprisingly, drugs targeting the proteasome suffer from unwanted side effects, because many physiologically important processes are regulated by proteasomal proteolysis. Targeting of specific E3s, DUBs or ubiquitin receptors might afford clinical efficacy with fewer side effects. It is foreseeable that the multivalent interactions that regulate E3 and ubiquitin receptor activities could ultimately be used to target ubiquitin signaling for specifically restricting viral budding, stabilizing tumor suppressors or promoting DNA repair.[1]
References
[edit | edit source]Methylation is a regulatory process that can prevent certain processes such as degradation or catalysis from occurring. In the restriction-modification system of bacteria, the bacterial DNA is methylated at adenine bases by methylase, thereby preventing the DNA from being degraded by restriction endonucleases. These restriction enzymes have active sites that recognize the specific amino acid-based conformations in DNA. The enzymes can bind and cleave the phosphodiester bonds of the backbone at recognized (cognate) DNA. Methylated groups lose one hydrogen bond linkage with the enzyme and thus decrease the binding energy, resulting in a lower enzyme affinity and no cleavage.
Methylation also occurs in amino acid synthesis and in gene expression. In the latter, cytosine is methylated at C5. This 5-methylcytosine interferes with the proteins that bind to start transcription. In amino acid synthesis, methylases and other methyl group carriers attach methyl groups.
DNA Methylation Overview
[edit | edit source]Eukaryotic cellular DNA is usually stored in the nucleus, is wrapped around by a chromatin, which is a histone octamer. The chromatin is a crucial part in genetic modification in cells with the help of enzymes in order to make suitable changes to the cell, DNA, RNA, and proteins, such as the modification of transcription, or even the complete inactivation of a gene. One of these chromatin histone mechanism modifications can be a direct change on the mechanism of DNA methylation, an epigenetic mechanism. Proper DNA methylation is extremely important in functional cellular health, as no methylation (or even too much (hypermethylation) or insufficient methylation) could lead to serious diseases such as the creation of carcinogenic cancer cells.
DNA Methyltransferase
[edit | edit source]
Enzymes that add methyl groups to DNA are called DNA methyltransferases. The mechanisms of DNA methyltransferases vary for each organism, but the enzyme generally binds unspecifically before traveling along the DNA strand to find the specific sequence to be methylated. As stated above, methylation of DNA is carried out so that the cell can differentiate between foreign and infectious DNA, and for gene expression purposes. But methylation of the wrong sequence may lead to chromosomal problems, including unfavorable interactions with Histone proteins that are essential for chromosomes to fold properly. Hence these enzymes are highly specific. DNA methyltransferases are present in most organisms including mammals.
Methyltransferases attach methyl groups to specific sequence in conjunction with endonucleases that must scan for DNA sequences that are intended for cutting. Methyltransferases belong in several different types that have different functions. Although their domain sites may have structural differences, DNMT3 which is similar to DNMT1 contains a regulatory region attached to a catalytic domain. Among types, there are also subcategories that can mediate methylation-independent gene repression. Others may be used for genomic imprinting. Some enzymes may methylate strictly RNA and not DNA. DNMT1 identifies methylate groups and perform nucleophilic attacks among marked nucleotides.
The Role of Methylation in Gene Expression
[edit | edit source]Meythlation is extremely important when it comes to gene expression. not all genes are active at all times which is why DNA methylation is one of the several mechanics that allow cells to control gene expression. Although there are many ways that a gene can be expressed in particularly eukaryotes, the methylation of DNA is a common epigenetic signaling tool that can allow cells to lock genes in the off position. Key experiments were needed in order to provide the early clues for what the role of methylation had on gene expression. One such experiment was conducted by McGhee and Ginder in 1979 where they compared the methylation status of beta-globin loci in cells that did and that did not express the gene. By utilizing restriction enzymes that distinguished between methylated and unmethylated DNA, the two scientists were capable of dictating that the beta globin locus gene was not being expressed in the cells that were unmethylated. In addition to this experiment, more supporting evidence suggested the same conclusion. The same mechanism was performed on 5-azacytidine, a chemical analog of the nucleoside cytidine, in mouse cells. Similar observations were found with this experiment in the sense that the cytosine residues that were not methylated on the nucleoside prevented the gene to be expressed.
DNA methylation is the addition of a methyl group to the 5th carbon in cytosines in DNA into a covalent bond. This mechanism is catalyzed by DNMTs (DNA methyltransferases) by utilizing the CpG dinucleotide sequence. CpGs that aren’t methylated usually contain ahigher GC base concentration and are clustered together, forming CpG islands, which are usually found at the starting sites for transcription and promoter binding. DNA methylation at CGI promoters have been known to cause gene expression silencing, such as the silencing of mechanisms such as X-chromosomes and genomic imprinting. DNA methylation can silence DNA through various mechanisms, such as utilizing 5meC to inhibit transcription factor binding or using Methyl Binding Domain Proteins (MDB) to repress the DNA using repressive chromatin-modifying complexes. Overall, DNA methylation is responsible for important cellular functions involving the genetic sequence of eukaryotes, such as genetic modification and even silencing.
How and Where are Genes Methylated
[edit | edit source]Now that it is understood that DNa methylation is key for gene expression, the questions comes about where exactly must methylation occur on the gene to induce expression. Today researchers know that DNA methylation occurs primarily at the cytosine bases of eukaryotic DNA, which are converted to 5-methycytosine by DNA methyltransferase (DNMT) enzymes. These altered cytosine residues are normally adjacent to guanine nucleotides (due to base pair mechanics) which essentially results in two methylated cytosine residues that sit diagonally from each on opposing DNA strands.
The roles and targets of DNA methylation varies among the kingdoms of organisms. For examples, among Animalia, mammals tend to have distributed CpG methylation patterns as mentioned above, however in invertebrate animals, a mosiac pattern of methylation is found where regions of heavily methylated DNA are interspersed with nonmethylated regions. In other words, no distinct pattern is seen in this kingdom of species. It should also be noted that surprisingly the kingdom plantae are organisms that are found to have the highest amount of methylation, up to 50% of cystosine residues undergoing methylation.
DNA methylation and Disease
[edit | edit source]When one considers the magnitude of importance DNA methylation has upon gene expression, one must also ask of the consequences of errors found in methylation and what can arise from such error. Errors in methylation give rise to a number of devastating consequences, including various disease associated with tumor suppressor genes. It has been found that tumor suppressor genes are often turned off in cancer cells due to hypermethylation of cytosine residues. hypermethylation is also found at other very critical gene expressions such as in the cell cycle regulatory genes, the tumor cell invasion genes, DNA repair genes and other genes that involve the propogation of metastasis. In addition, the genomes of cancel cells have also shown a decrease in methylation, or hypomethylation, compared to normal healthy cells (with the exception of hypermethylation found on the for mentioned genes). The combination of hypomethylation and hypermythelation describes the potent nature of a cancer cell.
Gene-Body DNA Methylation
[edit | edit source]DNA methylation in active genes can be analyzed with the use of bisulfites to sequence the DNA and methyls attached to the DNA. With bisulfite sequencing, it was discovered that CpG methylation occurred in active genes and much downstream the sites of transcription (as long as the methylation didn’t silence the gene). These discoveries showed that DNA methylation can occur and various promoters (like human X chromosomes, which has many transcribable regions). Due to the fact that bisulfite sequencing is unbiased, it also showed methylated as well as non-methylated CpG sites, showing that; with MeDIP-Seq and MRE-Seq, non-methylated CpG is prevalent at most promoters.
References
[edit | edit source]McGhee, J. D., & Ginder, G. D. Specific DNA methylation sites in the vicinity of the chicken beta-globin genes. Nature 280, 419–420 (1979)
Biochemistry 6th Edition, Berg et al.
Acetylation
[edit | edit source]Acetylation introduces an acetyl group to a molecule. More specifically, the reaction replaces a hydrogen from an alcohol group with an acetyl. An example is the synthesis of aspirin from salicylic acid:

Acetylation is an important post-translational protein modification and regulation. An example is the acetylation/deacetylation of histone which subsequently express/inhibit genes since histone binds to DNA itself. Histone Acetyltransferase catalyzes the acetylation of lysine from the histone tail with an acetyl group from Acetyl CoA.
The acetylation of lysine in histone removes the positive charged ammonium group and renders the side chain neutral, which decreases the histone tail affinity for DNA and loosens the histone complex.
The now-acetylated histone can interact with an acetyllysine-binding domain in many eukaryotic proteins called w:bromodomain, an 110 amino acids protein with a four-helix bundle and a peptide-binding site at one end.
Acetylation is the reaction in which an acetyl functional group is added to an organic molecule. In proteins, an acetyl group to either added to the N-terminus of proteins and at lysine residues as a post-translational protein modification.
N-alpha-Terminal Acetylation
The role of N-alpha-Terminal Acetylation is still relatively unknown and under research. However, it is known that this modification is actually widely prevalent in eukaryotes and yeast, though uncommon in prokaryotes, and is performed by a subgroup of acetlytransferases known as N-alpha-acetyltransferases (NATs). There are three major NATs (A,B,C) which perform the majority of the N-alpha-terminal acetylations of eukaryotes The reaction is begun with the cleaving of N-terminal methionine residues with small side chains containing, glycine, alanine, serine, cysteine, threonine, proline, and valine, by methionine aminopeptidases (MAP), Map1p and Map2p. Subsequently, the NATs recognize and acetylate specific sequences of the cleaved proteins.
Lysine Acetylation
One major area in which lysine acetylation is prevalent is in the acetylation of histones, which is performed by histone acetyltransferases (HATs) and deacetylases (HDACs). In both acetylation and deacetylation reactions that attach to the NH3+ tail from an acetyl group from Acetyl-Coenzyme A and remove the acetyl group from lysine onto Coenzyme A, respectively. The acetylation and deacetylation of histones partake in gene regulation. Histones are packages of strongly alkaline protein around which DNA is wound to allow DNA to be stored in an orderly fashion. Since the acetylation occurs at the NH3+ tail of lysine, the charge of the protein is affected. When acetylated, the positive charge of lysine is eliminated. This decreases the histones affinity to the negatively charged DNA strand, thereby loosening the strand, and the reverse results occur with deacetylation. However, Lysine acetylation isn’t limited to histone acetylation. Among other things, it is also involved in the modification and regulation of non-histones such as cytoplasmic enzymes and p35, a tumor suppressor and involved with signal transmittance and signaling.
Drug Acetylation
[edit | edit source]Many of the drugs used today for common diseases require additional structuring in order for efficient metabolism in the body. Drugs that are significantly metabolized by acetylation include isoniazid, hydralazine, procainamide, phenelzine, and dapsone that are used to treat tuberculosis, cardiac failure/hypertension, ventricular arrhythmias, depression, and leprosy/skin infections respectively.
Reference
[edit | edit source]Berg, Tymoczko, Stryer. Biochemistry Sixth Edition.
Yang XJ, Seto E (2008). "Lysine acetylation: codified crosstalk with other posttranslational modifications". Mol Cell 31: 449–61. doi:10.1016/j.molcel.2008.07.002. PMID 18722172. [12]
Arnesen T, Polevoda B, Sherman F.(2009)"A synopsis of eukaryotic Nα-terminal acetyltransferases: nomenclature, subunits and substrates" [13]
"Histone Acetylation, DNA Methylation and Epigenetics" [14]

Adenylation is the covalent attachment for AMP to a protein side chain. It is known two serve two functions: one as a stable post-translational modification and the other is to generate an efficient leaving group in mechanisms that use energy from the free energy of hydrolysis of phosphoanhydride bond of ATP to allow thermodynamically unfavorable overall reactions to occur.
The activity of glutamine synthetase, an enzyme that plays a key role in the metabolism of nitrogen, is regulated by adenylation. The rate of adenylation depends on the ratio of glutamine to α-ketoglutarate. A low ratio is a sign of cellular nitrogen sufficiency, whereas a high ratio is evidence of a limited nitrogen supply and the need for ammonia fixation by glutamine synthetase.
Adenylation of glutamine synthetase, which is catalyzed by the enzyme adenylyl transferase, involves the phosphodiester bond between the hydroxyl group of the tyrosine residue in glutamine synthetase and the phosphate group of an AMP nucleotide. A complex of adenylyl transferase and a regulatory protein known as PII, which may exist unmodified as PII (also known as PA) and uridylylated as PII-UMP (also known as PD), causes an AMP molecule to either attach to or be removed from the glutamine synthetase, respectively.
A complex of PII with adenylyl transferase catalyzes the attachment of an AMP molecule to glutamine synthetase, forming an adenylated glutamine synthetase that is inactive. On the other hand, a complex of PII-UMP and adenylyl transferase activates deadenylation and removes the AMP from glutamine synthetase, creating deadenylated glutamine synthetase that is active. When there is a higher glutamine to α-ketoglutarate ratio, more monomers of glutamine synthetase are adenylated, thereby producing lower activity. A lower ratio leads to less monomers being adenylated and higher activity of glutamine synthetase.
Adenylation is an important form of regulation for amplifying signals such as for blood clotting and control of glycogen metabolism. Because adenylation is an enzymatic cascade, it is easier for allosteric control, as each enzyme can be a target for regulation. This is important for nitrogen metabolism in cells as it creates many regulatory sites, allowing a cell to fine tune its nitrogen production.
References
[edit | edit source]- Berg, Jeremy Mark. Biochemistry/ Jeremy M. Berg, John L. Tymoczko, Lubert Stryer. -7th ed. P749-750.
- Garrett, R.H., and C.M. Grisham. Biochemistry. 3rd ed. Belmont, CA: Thomas, 2007.
- Itzen A, Blankenfeldt W, Goody RS. Adenylylation: renaissance of a forgotten post-translational modification. Trends Biochem Sci. 2011 Apr;36(4):221-8. Epub 2011 Jan 20.
General Info
[edit | edit source]One of the common covalent modifications of protein activity found in animals, palnts, and some viruses. It's donor molecule are Myristoyl CoA which is commonly used to modify protein called Src, main function of signal transduction. The myristoyl group is covalently attached to the N-terminal amino acid of a polypeptide. It is then catalyzed by the N-myristoyltransferase enzyme. It also plays an important role in membrane targeting in response to environmental stress on plants.
Reference
[edit | edit source]http://www.ncbi.nlm.nih.gov/pubmed/9067626
Metabolic Regulation by Protein Lysine Acetylation
Protein acetylation is a key role in regulation of transcription in the nucleus. What more important about protein acetylation is its function in regulating metabolic pathways. For example, glycolysis, gluconeogenesis, the tricarboxylic acid (TCA) cycle, urea cycle, fatty acid metabolism, and glycogen metabolism in liver tissue.
What is protein acetylation
Acetylation is a reaction which introduces an acetyl functional group into a chemical compound. Typically, protein lysine acetylation is the most important pathway which regulates the metabolic pathways, by acetylating lysine residue in enzyme, enzymes are able to regulate metabolix pathways. In living cells, acetylation occurs as a co-translational and post-translational modification of proteins. In liver tissue, acetyl-coenzyme A (acetyl-CoA), a high energy molecule, act as the acetyl group donor to the lysine acetylation.
Lysine Acetylation in fatty acid oxidation
Enoyl-coenzyme A hydratase/3-hydroxyacyl-coenzyme A (EHHADH) is an important enzyme which catalyze two steps in fatty acid oxidation. There are four acetylated lysine residues been identified in EHHADH, which are Lys165, Lys171, Lys346 and Lys584. Immunoprecipitation of ectopically expressed FLAG-tagged EHHADH and Western blotting with antibody to acetyllysine confirmed that EHHADH was indeed acetylated(Zhao, et al). In order to explore the effect of acetylation on fatty acid oxidation. Isobaric tags are used, which is TSA and NAM. TSA and NAM treatment increased all the four lysine residues’ acetylation. Consistently, corresponding unacetylated peptide was decreased. Scientists treat TSA and NAM to Chang Human Liver cells doubled the activity of EHHADH, which indicates that acetylation of EHHADH would increase fatty acid oxidation pathway. In order to confirm the result, site-directed mutagenesis was used and the four lysine residue was replaced by glutamine, TSA and NAM can no longer acetylated lysine residues and EHHADH is no longer regulated.
Lysine Acetylation in TCA cycle
TCA cycle includes seven enzymes, and all of them can be acetylated. Malate dehydrogenase (MDH) was studied. Again, four lysine residues are identified: Lys185, Lys301, Lys307 and Lys314. Again, TSA and NAM treatment was performed and wild-type MDH’s activity was increased while mutant MDH4KR’s activity wasn’t changed, which indicate that lysine acetylation can stimulate the TCA cycle. Another method can be performed for studying lysine acetylation. When MDH was treated with high concentration of glucose, MDH acetylation was increased by 60%, and the activity of MDH was increased, which again, confirm that lysine acetylation stimulates the TCA cycle. This result matches our logic, as we eat more glucose, glycolysis and TCA cycle are stimulated because we need to consume these glucose to created ATP, which is energy.
Lysine Acetylation in urea cycle
Urea cycle is coupled with TCA cycle, thus studying of urea cycle is also a good way to understand the effect of lysine acetylation on metabolic pathways. ASL, an enzyme in urea cycle, was studied. High glucose concentration is treated to ASL, the acetylation of ASL decrease the activity of ASL by 50%, which indicated that lysine acetylation inhibit the urea cycle. On the other hand, CobB (one of the amino acids) is treated to ASL, which deacetylates the ASL, experiment results show that ASL activity is increased. The dual regulation of ASL indicate that lysine acetylation of ASL will inhibit the urea cycle. The result make sense as we invest more glucose, in order to maintain body osmotic, we need to conserve water, which means that our body should perform minimum amount of urea cycle.
Lysine Acetylation in Gluconeogenesis
Gluconeogenesis, the opposite of glycolysis, is also studied for understanding lysine acetylation in metabolism. The enzyme Phosphoenolpyruvate carboxykinase 1 (PEPCK1) is studied for gluconeogenesis. There are three lysine residues which were identified: Lys70, Lys71, Lys594. Treatment of both high glucose and TSA/NAM acetylated PEPCK1 and the amount of PEPCK1 was decreased, and decrease the stability of PEPCK1. Which indicates that lysine acetylation inhibits the gluconeogenesis pathway. To confirm our result, mutated form of PEPCK 1(PEPCK13KR, replace all the lysine by arginine) was examined, and the gluconeogenesis is no longer regulated by high glucose and TSA/NAM treatment, which confirm our result. The result aligns with our logic, that when we investigate a lot of glucose to our body, we would like to perform glycolysis instead of gluconeogenesis.
Summary
Protein Lysine acetylation is one of the important way to regulate metabolic pathways. These regulation intend to balance of energy in organisms and osmosis. When we infuse a lot of sugar, we are willing to consume these sugar to keep osmotic pressure of our body and make energy from glucose, thus TCA cycle fatty acid oxidation is stimulated by protein lysine acetylation; it also inhibit Urea cycle and gluconeogenesis so that we can conserve water to keep osmotic pressure and prevent the synthesis of glucose.
| Header text | Header text | Header text | Header text | Header text |
|---|---|---|---|---|
| Pathway | Fatty Acid Oxidation | TCA Cycle | Urea Cycle | Gluconeogenesis |
| Enzyme | EHHADH | MDH | ASL | PEPCK1 |
| activity effect | + | + | - | - |
| high glucose effect | + | + | - | - |
References
[edit | edit source]1. Shimin Zhao, et al. (2010) Regulation of Cellular Metabolism by Protein Lysine Acetylation. Science Vol 327
General
[edit | edit source]Proteolytic Activation is the activation of an enzyme by peptide cleavage. The enzyme is initially transcribed in a longer, inactive form. In this enzyme regulation process, the enzyme is shifted between the inactive and active state. Irreversible conversions can occur on inactive enzymes to become active. This inactive precursor is known as a zymogen or a proenzyme. The enzyme is subsequently cut to yield the active form. The benefit of this type of regulation is that ATP is not needed for the cleavage to occur. Therefore, enzymes can use this system of regulation even outside of the cell. This type of regulation can only be done once to an enzyme. It can only be activated once and will stay activated for the enzyme's entire life span. Unlike allosteric control and reversible covalent modification this occurs just once in an enzyme's lifetime. Although the zymogen activation is irreversible, there are specific inhibitors that control those proteases.
Specific proteolysis is a frequent way of activating enzymes and certain other proteins in biological systems. For instance, digestive enzymes which hydrolyze proteins are made as zymogens in the stomach and pancreas, in which pepsinogen is the inactive precursor (zymogen) and pepsin is the activated form of the enzyme. Another example is seen in blood clotting which is carried out by a cascade of proteolytic activations. Also, certain protein hormones are synthesized as the inactive precursors such as proinsulin which then leads to the activated form, insulin by proteolytic cleavage. In addition, collagen, a fibrous protein and the major component of skin and bone is made from the zymogen procollagen.
Several developmental process are mediated by activation of inactive precursors such as the metamorphosis of a tadpole to a full grown frog in which increased amounts of collagen are reabsorbed from the tail. Similarly, collagen is decomposed in the mammalian uterus after birth. Both of these examples rely on the conversion of procollagenase to collagenase which is the active protease and is very accurately timed.
Lastly, apoptosis, or programmed cell death is another example that illustrates the importance of proteolytic enzymes, in this case caspases. Caspases are synthesized in the inactive form called procaspases and when activated by different signals, caspases cause cell death in many organisms.
Common uses in Biological Systems
[edit | edit source]Digestive enzymes are activated in the stomach and pancreas using this methodology. They are synthesized first as zymogens. Pepsinogen is the inactive protein which is initially transcribed. Then, it is cleaved in the stomach to produce Pepsin, a digestive enzyme. The purpose of this regulation is to prevent the Pepsin from digesting proteins in the body before it is introduced into the digestive tract. Many protein hormones in the body originate from inactivated forms of the actual enzymes. A prime example is insulin. Insulin arises from an inactive form known as proinsulin. It is activated by proteolytic cleavage of a specific peptide. Proinsulin is first synthesized in the endoplasmic reticulum where the peptide chain is folded and the disulfide bonds oxidized. It is then packaged in the Golgi Apparatus and it is also proteolytically cleaved by series of proteases to form insulin. The matured insulin has 39 less amino acids than the proinsulin: 4 are removed and recycled and the remaining 35 amino acids form the C-peptide. Collagen is a fibrous protein that makes up the majority of the components of connective tissues in animals that arises from a inactive form known as procollagen. Also many developments and processes are activated by proenzymes. Programmed cell death is also mediated by these types of proteolytic enzymes.
Chymotrypsin
[edit | edit source]

An enzyme that hydrolyzes proteins, cleaving the peptide bond. The inactive form of this enzyme is chymotrypsinogen. The zymogen is synthesized in the pancreas, where most of the secreting proteins are synthesized. Chymotrypsinogen consist of the 245 amino acids. In order to activate it, the bond in between the 15th amino acid (Arg) and 16th amino acid (Ile) has to be cleaved by the enzyme (trypsin). The simple cleavage of the single bond activates the enzyme. Later on, the two dipeptides can be removed to produce α-chymotrypsin. Three chains in α-chymotrypsin are bonded by disulfide bonds. The cleavage of a single bond leads to the structure conformation and creation of the active side of the chymotrypsin (catalytic triad). It also creates a pocket where the aromatic or long hydrophilic side chain of the amino acid can be inserted for the future cleavage. In addition, structural change locates the NH group that stabilized the tetrahedral intermediate in the “oxyanion hole” in the appropriate position.
As a proteoplytic activation, chymotrypsin has been biologically known to work as digestive enzymes, blood clotting, protein hormones, and procaspases (a programmed cell death).
Trypsin
[edit | edit source]
Important enzyme in the biological systems because it activates many enzymes. However, trypsin by itself is activated from the trypsinogen with help of the enteropeptidase. Enteropeptidase is a serine protease just like trypsin and chymotrypsin, and it breaks Lys-Ile bond in the trypsinogen and activates it. As a result of such process, the small amount of the trypsin enzyme is produced. Later, this amount of activated trypsin activates more trypsin and other enzymes too. The formation of trypsin by enteropeptidase is considered the master activaion step since trypsin participates in a variety of zymogen activation. Examples include the activation of proelastase to elastaase, procarboxypeptidase to carboxypeptidase, and prolipase to lipase.
Thrombin
[edit | edit source]
An important role of zymogen activations occurs in blood clotting. For blood clotting, the response time must be fast in order to achieve clotting at the right spot and time to prevent excessive bleeding. Enzymatic cascades are therefore employed to achieve that rapid response. A cascade of zymogen activations activates a clotting factor, which is then responsible for activating another clotting factor and so forth until the final clot is achieved. The blood clotting process is driven by a series of proteolytic events. When trauma exposes tissue factor, thrombin, also a serine protease and a key enzyme in clotting, is synthesized. This event leads to the production of more thrombin by positive feedback. Thrombin then activates enzymes and factors such as fibrinogen and forms fibrin, the key part in blood clotting. Thrombin cleaves four arginine-glycine peptide bonds on the in the central globular region of fibrinogen, releasing fibrinopeptides. The fibrinogen molecule that has lost these fibrinopeptides are then called fibrin monomers. They are called monomers because they spontaneously come together and assemble into fibrous arrays known as fibrin. The fibrins are then crosslinked by the enzyme transglutaminase, which was activated by thrombin from protransglutaminase.
Breakthroughs in Elucidation of Clotting Pathways
[edit | edit source]Because of the breakthrough in the elucidation of blood clotting pathways, hemophilia can be revealed early in clotting. Classic hemophilia (a.k.a. Hemophilia A) is a clotting defect. It is genetically transmitted as a sex-linked recessive characteristic. The antihemophilic factor, factor VIII of the pathway is missing or has reduced activity. Even though factor VIII is not a protease, it stimulates the activation of factor X which is the final protease of the intrinsic pathway by the serine protease factor IXa. The activity of factor VIII is increased by limited proteolysis by thrombin. This type of positive feedback amplifies the clotting singal and accelerates clot formation after a threshold has been reached. Therefore the activation of the intrinsic pathway is impaired in classic hemophilia.
Before, hemophiliacs were treated with transfusions of concentrated plasma fraction with factor VIII but this therapy always had the risk of infection of diseases such as hepatitis and AIDS. However, with advancements in biochemical techniques such as biochemical purification and recombinant DNA, the gene that encodes factor VIII was isolated and expressed in cell cultures. Since then recombinant factor VIII purified from the cultures has replaced plasma concentrates to treat hemophilia.
Vitamin K-Dependent Modification readies the activation of prothrombin
[edit | edit source]
Thrombin is produced as a zymogen known as prothrombin. The inactive molecule has four domains. The first domain is the gla domain ( a gamma-carboxyglutamate-rich domain). Kringle domains consist of the next two domains. Kringle domains keep prothrombin in an inactive form and guide it towards appropriate site for activation by factor Xa (serine protease) and factor Va (stimulatory protein). The activation begins with proteolytic cleavage of the arginine 274 and threonine 275 bond, which releases a fragment containing the first three domains. Similar cleavage of the arginine 323 and isoleucine 324 bond generates an active thrombin molecule.
Vitamin K has an important role in the synthesis of prothrombin. Nuclear magnetic resonance reveals that prothrombin contains gamma-carboxyglutamate. The first 10 glutamate residues in the amino-terminal region of prothrombin are carboxylated to gamma-carboxyglutamate by a vitamin K-dependent enzyme. This reaction converts glutamate, a weak chelator of Ca 2+, into gamma-carboxyglutamate, a strong chelator. Consequently, prothrombin is able to bind calcium. This binding fixes the zymogen to the phospholipid membrane surface from blood platelets at the injury site. This is important because the prothrombin is now in close proximity to two clotting proteins that catalyze its conversion to thrombin. The calcium-binding domain is removed during activation, which frees the thrombin from the membrane. Now, it is free to cleave targets such as fibrinogen.
Prothrombin synthesized in the absence of vitamin K or in the presence of vitamin K antagonists such as dicourmaorl gives rise to an anticoagulant factor as opposed to the regular thrombin that is a coagulant factor. An anticoagulant factor prevents the blood from clotting and is used to treat patients with thrombosis.
The abnormal prothrombin lacks the γ-Carboxyglutamate and that explains its anticoagulant properties.
26S Proteasome
The 26S proteasome is an enzyme that is known to cleave intracellular proteins in order to maintain cell functioning and homeostasis. One of the pathways that the 26S proteasome is known for is the degradation of ubiquitin. This proteasome is composed of over 30 subunits and all of their units have specific functions that are not limited to unfolding, translocating, and cleaving. As the 26S proteasome recognizes the correct ubiquitin, it forms the UPS-ubiquitin-proteasome system. The UPS is now polyubiquinated and Ubiquitin chains are formed. The chains are now available to be picked out by the proteolytic core of the UPS, which will continue on with the cleaving of ubiquitin into small peptides. This process is more complicated than it appears, even with the structure of the 26S proteasome not fully understood. It is composed of two subunits: the 20S proteasome core particle and the 19S regulatory particle. The proteasome core particle is where substrate proteolysis occurs and then regulatory particle is where selecting and unfolding of ubiquitin substrates occurs. The core particle has a barrel type of structure with many alpha and beta subunits arranged in alternating fashion. Such a complex structure also defines its function because it requires great amounts of cellular energy to assemble and degrade the molecule of interest. The regulatory particle is just as complex, for it contains of a base and a lid. The base is made up of six different AAA ATPases and by three non ATPase subunits. This helps the regulatory particle control the entry of substrates into the previously mentioned core particle, and hence control the overall pace of the assembly and rate of the substrate-enzyme complex mechanism.
Source: The 26S proteasome: assembly and function of a destructive machine Center for Integrated Protein Science at the Department Chemie, Lehrstuhl für Biochemie, Technische Universität München, Lichtenbergstr. 4, 85747 Garching, Germany
Proteolytic Enzyme Inhibitors
[edit | edit source]Since activating proteolytic enzymes is irreversible, how can one prevent a proteolytic enzyme from functioning? These types of enzymes need very specific inhibitors to prevent the enzyme from running catalysis. A prime example is pancreatic trypsin inhibitor. This inhibits trypsin by binding really firmly to the active site of the enzyme. What makes it bind firmly to the active site is interactions with the aspartate side chain with the side chain of lysine 15 on the inhibitor. This along with many hydrogen bonds between the inhibitor and the active site creates a relatively firm binding on the active site.
Trypsin inhibitors are essential in that they prevent severe damage such as inflammation in the pancreas known as pancreatitis. Because trypsin engages in activation of several zymogens, inhibiting trypsin can prevent unwanted or premature cascades such as inflammation. Other inhibitors, such as antielastase, are important as well. When the body contains excess elastase, the enzyme destroys alveolar walls in the lungs by breaking down elastic fibers and other connective tissue proteins in the lungs. This disease is called emphysema, containing symptoms such as difficulty in breathing. Therefore, the body needs elastase inhibitors to prevent the damage from occurring.
When the body forms blood clots, there also needs to be regulation that makes sure the clot is limited only to the site of injury. An example includes thrombin, which not only catalyzes the formation of the clot through fibrin, but also deactivates the clotting cascade by activating protein C, which digests clot stimulating factors. Other specific clotting inhibitors include antithrombin III, which forms an irreversible complex with thrombin, and thus preventing thrombin from activating any more fibrinogen. Antithrombin III can also be amplified by heparin, a polysaccharide found in the walls of blood vessels and endothelial cells. Heparin amplifies antithrombin III by increasing the rate of formation of the irreversible antithrombin III-thrombin complex.
The ratio of thrombin to antithrombin is crucial in normal blood clotting. A mutation that occurs in α1-antitrypsin , a protease inhibitor, which replaces the methionine 358 with arginine, alter the inhibitor's specificity from elastase to thrombin and causes the level of thrombin to drop at the injury site. neutrophils produce large quantities of elastase at sites of injury and those excess proteases must be inhibited by α1-antitrypsin, however, the mutant α1-antitrypsin will inhibit the coagulation factor thrombin instead of elastase thus the blood clot will fail to form and the patient can die of potential hemorrhage.
References
[edit | edit source]Berg, Jeremy M. John L. Tymoczko. Lubert Stryer. Biochemistry Sixth Edition. New York: W.H. Freeman, and Company 2007. An enzymatic cascade is a sequence of successive activation reactions involving enzymes, which is characterized by a series of amplifications stemming from an initial stimulus. The product of each preceding reaction catalyzes and is consumed in the next reaction. An example of an enzymatic cascade includes blood clotting, where a timely response is needed. The cascade first activates a clotting factor, followed by the activation of the next clotting factor and so forth until the final cross-linked fibrin clot is formed.
Another example of enzymatic cascade occurs following the binding of allergen to a receptor bound Ig on the mast cell. Spleen tyrosine kinases (Syk) plays an important role in this immunoglobulin E (IgE) signaling pathway. Using knowledge of the cascade and the role Syk plays, Syk inhibitors have been developed to inhibit IgE- driven mast cell degranulation, as well as to inhibit the release of inflammatory cytokines, thereby inhibiting an allergic response. [15]
Enzymatic cascades can also play roles in the immune system. Complement cascade works with the body’s innate immunity by cleaning pathogens from an organism. It proceeds though three major pathways; classical (CP), alternative (AP) and mannose-binding-lectin (MP). The end product results in large scale amplification of cell killing membrane attack complex. [16]
Blood Clotting
[edit | edit source]Blood coagulation is an example of this; each enzyme activates the next until a fibrin clot is formed. There are two pathways by which the clotting can proceed: intrinsically or extrinsically. Intrinsic clotting is activated by the exposure of anionic surfaces of blood vessels that is damaged by the trauma, while extrinsic clotting is initiated by the exposure of tissue factors (TF). Tissue factors are integral glycoprotein.
File:Blood clotting.jpg Inactive forms of clotting factors =pink Activated forms = yellow Stimulatory proteins (not enzymes) = blue
{kind=link}
The final portion of the pathway is the same for both intrinsic and extrinsic cascades, in which factor X is activated. Once the Prothrombin is activated and catalyzed into thrombin, it catalyzes the conversion of fibrinogen into fibrin. Thrombin cleaves fibrinopeptides at the central globular region, the globular domains at the carboxyl-terminal ends of the beta and delta chains interact with the exposed regions of the chains at the amino terminal. This cross linking reaction forms fibrin clots. It should be noted that thrombin is also involved in the activation of enzymes in previous steps, such as factors VII and VIII, providing a positive feedback loop on the cascade. This fibrin clotting process must be fast acting and precisely regulated, it can only act on damaged tissues. Fibrin can't be flowing in the blood vessels to cause blood clots. This precise regulation is owe to the fact that clotting factors are short-lived and diluted in blood vessels. They are quickly removed by the liver.
File:Blood clot final path.jpg
{kind=link}
Sources
[edit | edit source]Berg, Jeremy M. John L. Tymoczko. Lubert Stryer. Biochemistry Sixth Edition. W.H. Freeman and Company. New York, 2007.
Overview
[edit | edit source]Transcription activation is the regulation by which a change in the rate of DNA transcription may alter the expressed gene in question. Regulatory proteins are utilized to activate an expression of a certain gene or to repress it (regulate). The activators catalyze interaction of RNA polymerase in the synthesis or expression of the gene in hand. This is accomplished by increasing the binding affinity of the RNA polymerase to the promoter site, the region of DNA that is vital in the transcription process. Transcription factors are activated, and transcription occurs at the promoter site. Histone interactions occur during acylation as DNA is transcribed into RNA.
Mechanism
[edit | edit source]First, the binding of a ligand to a certain receptor occurs, initiating a reaction signal. This activates a transcription factor, which is a protein that binds specific DNA sequences and controls the rate of the transcription process. The transcription factor additionally binds to other reagents necessary in transcription. These proteins (transcription factors) are then able to be regulated by reversible regulation processes such as phosphorylation or proteolysis.
The transcription of DNA to RNA and its subsequent translation to proteins can be regulated by acylation and methylation. DNA is negatively charged and is wrapped around positively charged histone proteins. In order for transcription to occur the DNA must be unbound to the histones. This is accomplished by acylating the positively charged residues found in the histones such as lysine. This neutralizes the positive charge allowing for a looser binding with DNA. This allows RNA polymerase to enter and transcribe the desired gene. In order to reverse this effect and tighten the binding, methylation occurs. Methylation of the residues restores the positive charges to restore the electrostatic interactions between the histones and DNA and tighten the binding. It is in this method that gene expression is regulated.
References
[edit | edit source]1. Berg, Jeremy M. (2007). Biochemistry, 6th Ed., Sara Tenney. ISBN0-7167-8724-5. pp. 902-903. 2. Campbell, Neil A. Biology. 7th ed. San Francisco, 2005. If a cell is to make use of an enzyme, its chemical activity must be tightly monitored so that a great deal of energy will be conserved rather than wasted. Thus, like a switch, there must be a way to turn the enzyme on or off depending on when a reaction is needed. One method for regulation is allosteric control.
Definition
[edit | edit source]Allosteric control refers to a type of enzyme regulation involving the binding of a non-substrate molecule, known as the allosteric effector, at locations on the enzyme other than the active site. The name "allo" means other and "steric" refers to a position in a certain amount of space. In other words, allosteric means "at another place." An allosteric site is a site at which a small regulatory molecule interacts with an enzyme to inhibit or activate that specific enzyme; which is different from the active site where catalytic activity occurs. The binding of the allosteric effector is in general noncovalent and reversible. This interaction thus changes the shape of the enzyme which, in turn, changes the shape of the active site. This change in conformation will either inhibit or enhance the catalysis of a reaction. So the allosteric control allows the cell to regulate the needed substances quickly through inhibition and or enhancement.
ATCase
[edit | edit source]

The enzyme aspartate transcarbamoylase (ATCase) is an allosteric enzyme that catalyzes the first step in the synthesis of pyrimidines.
Structure
[edit | edit source]ATCase is made of six regulatory subunits and six catalytic subunits. The 3 regulatory subunits (r) are dimers made of 2 chains of 17 kd each. The smaller of the two, the regulatory subunit can bind to CTP and thus shows no catalytic activity. The 2 catalytic subunits (c) are trimers consisting of 3 chains, each 34 kd. The catalytic subunit is unresponsive to CTP thus does not follow a sigmoidal behavior.
- The quaternary structure of ATCase is composed of the two catalytic trimers stacked one on top of another. The inhibitory effect of CTP, the stimulatory action of ATP, and the cooperative binding of substates are all accompanied by large changes in the quaternary structure of ATCase.
- Each r chain of each the regulatory subunit binds with a c chain of the catalytic trimer. The region of contact on between the r chain and c chain is stabilized by a domain of zinc bound to histidine residues in the r chain. All c chains have contact with the regulatory subunit.
The catalytic and regulatory subunits can be separated by first adding a mercurial compounds and then by ultracentrifugation. Mercurial compounds breaks the connection because it displaces the Zinc ion, destabilizing the r-subunit domain. The reaction does not follow a Michaelis-Menten behavior, instead it produces a sigmoidal curve because of the response changes in the substrate concentration via regulation by other molecules and of the changes in binding probability. The addition of more substrate has two effects on increasing the probability that the enzyme will bind more than one substrate molecule while increasing the average amount of substrates bound to each enzyme. More substrate ultimately favors the R-state of ATCase since equilibrium depends on the number of active sites that are occupied by the substrate which is completely opposite of Michaelis-Menten behavior.
Kinetics
[edit | edit source]
Allosteric enzymes exhibit sigmoidal kinetics rather than Michaelis-Menton kinetics. This is because the enzyme oscillates between two distinct conformational states, much like hemoglobin.
- The T state is characterized by low substrate affinity and low catalytic activity.
- In the R state conformation, there is a 12Å separation between catalytic trimers, and a roughly 10° rotation about central axis. There’s also a roughtly 15° rotation by the regulatory subunits. The R state conformation is characterized by an increase in substrate concentration simultaneously increasing the reactivity of ATCase in preparation for the enzymatic pathway of producing CTP.
Allosteric Activation
[edit | edit source]{kind=link}
Upon substrate binding at the active site located at pocket between the c chains in the trimer, ATCase has a greater likelihood of shifting to the R state due to substrate binding stabilizing the R state. The binding of substrates shifts the equilibrium more towards the R-state by increases the probability
that each enzyme will bind and increasing the average number of substrates bound (cooperativity).
ATP can also bind to the regulatory site of ATCase, but ATP does not inhibit the activity of ATCase in fact ATP increases the activity of ATCase. So at high levels of ATP, the ATP can act as a competitor,for the regulatory site, against CTP. So the activity of ATCase can increase with increased concentration of ATP. This increase in activity can have potential physiological explanations. High levels of ATP means that there is a high concentration of purine nucleotides, so the increased ATCase activity will increase the concentration of pyrimidines. So the concentration of both purines and pyrimidines will be more balanced. Also having a lot of ATP in the cell means having energy for processes such as mRNA synthesis and also DNA replication, so the ATCase can increase amounts of pyrimidines that can then be used in these processes.
PALA
[edit | edit source]In the presence of N-(phosphonacetyl)-L-asparate (PALA), a bisubstrate analog which resembles the substrate intermediate upon the enzymatic pathway, PALA inhibits ATCase which binds to the active sites. The inhibition however, revealed the change in the quaternary structure upon the binding of PALA. Two catalytic trimers are isolated into their respective T and R states. This inhibition is not allosteric, but instead, introduces the catalytic subunits that are responsible for the allosteric inhibition of this complete feedback inhibition pathway.
T-state vs. R-state The T-state is known to tense up the molecule which raises the amount of substrate needed to bind to the enzyme at 1/2 Vmax (Km). T-state is less active and is favored by CTP binding. The effect of CTP is T-state becomes stabilized. This means it is harder to convert enzyme to the R-state. On the other hand, the R-state is known to be more relaxed and decreases the Km. As the concentration of the substrate increase, the equilibrium will shift from T-state to R-state. At R-state, the molecule is more active, which means substrate binding is favored. The effect of ATP on the R-state is that it is stabilized, which makes binding substrates easier.
Homotropic effects --- substrate effects on allosteric enzymes.
Heterotropic effects ---The effects of nonsubstrate molecules on allosteric enzymes such as CTP and ATP on ATCase
Allosteric Inhibition
[edit | edit source]
Cytidine triphosphate (CTP), the end product of ATCase acts as an allosteric regulator. Carbamoyl phosphate and aspartate condense into N-carbamoylaspartate intermediate which then forms CTP. CTP binds to the r chain of the regulatory subunit not in contact with the c chains. The binding of CTP stabilizes the T state and decreases substrate affinity. Even though the binding site at the regulatory subunit is distant from the catalytic subunit, the binding will result in quaternary structural changes which promotes the stabilization of the T-state and inhibition. Thus, it causes the sigmoidal curve to shift to the right. The reaction will occur fast at a low concentration of [CTP], but at higher concentrations, CTP will act as an inhibitor to ATCase by regulatory or allosteric sites, not active sites. This is an example of negative feedback, where the end result will terminate the starting reaction. The feedback inhibition of CTP on ATCase can be reversed by ATP.
Heterotropic effects --– effects of non-substrate molecules on enzyme
Cooperativity
[edit | edit source]The rate of the product formation N-carbamoylaspartate increase as the concentration of Aspartate increases. Since it has cooperative character, you can see its curve has the sigmodal feature that means the binding of substrate on one site of the molecules increases the affinity for the other substrates to bind to the other binding sites of the molecules. The sigmoidal curve of ATCase incorporates a mix of two Michaelis-Menten curves-one with a high value of KM (shown through the T state), the other with a low value of KM (shown through the R state). The binding of a substrate to a subunit and the consequent alteration of all other subunits is called cooperativity. In cooperativity, binding at one site increases or decreases binding in another site of the enzyme. This is due to the conformational changes of the neighboring sub-unit residues that affect the change in shape of the other catalytic sub-unit. This process is analogous to how hemoglobin cooperatively binds oxygen molecules.
Mechanism
[edit | edit source]The enzyme has two active sites. One is for substrate and the other is for the allosteric activator which is at the regulatory site. The active site of the enzyme can't bind the substrate when allosteric activators are not bound to regulatory site. On the other hand, if the allosteric activator binds to the enzyme, the shape of the active site, it enables the substrate to bind allowing products to be produced. The enzyme will remain active until the allosteric activator leaves the enzyme.
Sources
[edit | edit source]Biochemistry 6th edition. Berg, Jeremy M; Tymoczko, John L; Stryer, Lubert. W.H. Freeman Company, New York If a cell is to make use of an enzyme, its chemical activity must be tightly monitored so that a great deal of energy will be conserved rather than wasted. Thus, like a switch, there must be a way to turn the enzyme on or off depending on when a reaction is needed. One method for regulation is allosteric control.
Definition
[edit | edit source]Allosteric control refers to a type of enzyme regulation involving the binding of a non-substrate molecule, known as the allosteric effector, at locations on the enzyme other than the active site. The name "allo" means other and "steric" refers to a position in a certain amount of space. In other words, allosteric means "at another place." An allosteric site is a site at which a small regulatory molecule interacts with an enzyme to inhibit or activate that specific enzyme; which is different from the active site where catalytic activity occurs. The binding of the allosteric effector is in general noncovalent and reversible. This interaction thus changes the shape of the enzyme which, in turn, changes the shape of the active site. This change in conformation will either inhibit or enhance the catalysis of a reaction. So the allosteric control allows the cell to regulate the needed substances quickly through inhibition and or enhancement.
ATCase
[edit | edit source]The enzyme aspartate transcarbamoylase (ATCase) is an allosteric enzyme that catalyzes the first step in the synthesis of pyrimidines.
Structure
[edit | edit source]ATCase is made of six regulatory subunits and six catalytic subunits. The 3 regulatory subunits (r) are dimers made of 2 chains of 17 kd each. The smaller of the two, the regulatory subunit can bind to CTP and thus shows no catalytic activity. The 2 catalytic subunits (c) are trimers consisting of 3 chains, each 34 kd. The catalytic subunit is unresponsive to CTP thus does not follow a sigmoidal behavior.
- The quaternary structure of ATCase is composed of the two catalytic trimers stacked one on top of another. The inhibitory effect of CTP, the stimulatory action of ATP, and the cooperative binding of substates are all accompanied by large changes in the quaternary structure of ATCase.
- Each r chain of each the regulatory subunit binds with a c chain of the catalytic trimer. The region of contact on between the r chain and c chain is stabilized by a domain of zinc bound to histidine residues in the r chain. All c chains have contact with the regulatory subunit.
The catalytic and regulatory subunits can be separated by first adding a mercurial compounds and then by ultracentrifugation. Mercurial compounds breaks the connection because it displaces the Zinc ion, destabilizing the r-subunit domain. The reaction does not follow a Michaelis-Menten behavior, instead it produces a sigmoidal curve because of the response changes in the substrate concentration via regulation by other molecules and of the changes in binding probability. The addition of more substrate has two effects on increasing the probability that the enzyme will bind more than one substrate molecule while increasing the average amount of substrates bound to each enzyme. More substrate ultimately favors the R-state of ATCase since equilibrium depends on the number of active sites that are occupied by the substrate which is completely opposite of Michaelis-Menten behavior.
Kinetics
[edit | edit source]Allosteric enzymes exhibit sigmoidal kinetics rather than Michaelis-Menton kinetics. This is because the enzyme oscillates between two distinct conformational states, much like hemoglobin.
- The T state is characterized by low substrate affinity and low catalytic activity.
- In the R state conformation, there is a 12Å separation between catalytic trimers, and a roughly 10° rotation about central axis. There’s also a roughtly 15° rotation by the regulatory subunits. The R state conformation is characterized by an increase in substrate concentration simultaneously increasing the reactivity of ATCase in preparation for the enzymatic pathway of producing CTP.
Allosteric Activation
[edit | edit source]Upon substrate binding at the active site located at pocket between the c chains in the trimer, ATCase has a greater likelihood of shifting to the R state due to substrate binding stabilizing the R state. The binding of substrates shifts the equilibrium more towards the R-state by increases the probability
that each enzyme will bind and increasing the average number of substrates bound (cooperativity).
ATP can also bind to the regulatory site of ATCase, but ATP does not inhibit the activity of ATCase in fact ATP increases the activity of ATCase. So at high levels of ATP, the ATP can act as a competitor,for the regulatory site, against CTP. So the activity of ATCase can increase with increased concentration of ATP. This increase in activity can have potential physiological explanations. High levels of ATP means that there is a high concentration of purine nucleotides, so the increased ATCase activity will increase the concentration of pyrimidines. So the concentration of both purines and pyrimidines will be more balanced. Also having a lot of ATP in the cell means having energy for processes such as mRNA synthesis and also DNA replication, so the ATCase can increase amounts of pyrimidines that can then be used in these processes.
PALA
[edit | edit source]In the presence of N-(phosphonacetyl)-L-asparate (PALA), a bisubstrate analog which resembles the substrate intermediate upon the enzymatic pathway, PALA inhibits ATCase which binds to the active sites. The inhibition however, revealed the change in the quaternary structure upon the binding of PALA. Two catalytic trimers are isolated into their respective T and R states. This inhibition is not allosteric, but instead, introduces the catalytic subunits that are responsible for the allosteric inhibition of this complete feedback inhibition pathway.
T-state vs. R-state The T-state is known to tense up the molecule which raises the amount of substrate needed to bind to the enzyme at 1/2 Vmax (Km). T-state is less active and is favored by CTP binding. The effect of CTP is T-state becomes stabilized. This means it is harder to convert enzyme to the R-state. On the other hand, the R-state is known to be more relaxed and decreases the Km. As the concentration of the substrate increase, the equilibrium will shift from T-state to R-state. At R-state, the molecule is more active, which means substrate binding is favored. The effect of ATP on the R-state is that it is stabilized, which makes binding substrates easier.
Homotropic effects --- substrate effects on allosteric enzymes.
Heterotropic effects ---The effects of nonsubstrate molecules on allosteric enzymes such as CTP and ATP on ATCase
Allosteric Inhibition
[edit | edit source]Cytidine triphosphate (CTP), the end product of ATCase acts as an allosteric regulator. Carbamoyl phosphate and aspartate condense into N-carbamoylaspartate intermediate which then forms CTP. CTP binds to the r chain of the regulatory subunit not in contact with the c chains. The binding of CTP stabilizes the T state and decreases substrate affinity. Even though the binding site at the regulatory subunit is distant from the catalytic subunit, the binding will result in quaternary structural changes which promotes the stabilization of the T-state and inhibition. Thus, it causes the sigmoidal curve to shift to the right. The reaction will occur fast at a low concentration of [CTP], but at higher concentrations, CTP will act as an inhibitor to ATCase by regulatory or allosteric sites, not active sites. This is an example of negative feedback, where the end result will terminate the starting reaction. The feedback inhibition of CTP on ATCase can be reversed by ATP.
Heterotropic effects --– effects of non-substrate molecules on enzyme
Cooperativity
[edit | edit source]The rate of the product formation N-carbamoylaspartate increase as the concentration of Aspartate increases. Since it has cooperative character, you can see its curve has the sigmodal feature that means the binding of substrate on one site of the molecules increases the affinity for the other substrates to bind to the other binding sites of the molecules. The sigmoidal curve of ATCase incorporates a mix of two Michaelis-Menten curves-one with a high value of KM (shown through the T state), the other with a low value of KM (shown through the R state). The binding of a substrate to a subunit and the consequent alteration of all other subunits is called cooperativity. In cooperativity, binding at one site increases or decreases binding in another site of the enzyme. This is due to the conformational changes of the neighboring sub-unit residues that affect the change in shape of the other catalytic sub-unit. This process is analogous to how hemoglobin cooperatively binds oxygen molecules.
Mechanism
[edit | edit source]The enzyme has two active sites. One is for substrate and the other is for the allosteric activator which is at the regulatory site. The active site of the enzyme can't bind the substrate when allosteric activators are not bound to regulatory site. On the other hand, if the allosteric activator binds to the enzyme, the shape of the active site, it enables the substrate to bind allowing products to be produced. The enzyme will remain active until the allosteric activator leaves the enzyme.
Sources
[edit | edit source]Biochemistry 6th edition. Berg, Jeremy M; Tymoczko, John L; Stryer, Lubert. W.H. Freeman Company, New York
Amino Acids sensing
[edit | edit source]The availability of amino acids play a key role in cellular physiology my controlling signalling pathways and gene expression.
GCN2 (general control of Amino Acids biosynthesis, nondepressing 2) is a cellular sensor for amino acids sufficiency to transform protein synthesis; GCN2 can detect amino acids deficiency of uncharged aminoacyl-tRNA that is accumulated. Normally, aminoacyl-tRNA is charged.
• GCN2, a kinase capable of phosphorylating the eukaryotic translation initiation factor 2-alpha(eIF2a), which inhibits its guanine nucleotide exchange factor, eIF2B; and regulates mTORC1 and GADD34, a protein phosphatase-1 binds to TSC1 and TSC2 and promotes dephosphorylation of TSC2 T1462, a major Aktphosphorylation site, which induces TSC-GAP activity to decrease Rheb-GTP charging so that mTORC1 is inhabited.
• eIF2a indirectly impairs eIF2 function and inhibits translation initiation.(e.g.: at S51) This is upregulating the translation of a subset of mRNA. For example, ATF4 promotes expression of an array of genes for amino acids biosynthesis.
• Only leucine has forceful effects on mTORC1. Uncharged tRNA doesn’t affect mTORC1 signaling- using cell lines harboring temperature-sensitive aminoacyl tRNA synthetase activity. However, decreasing in absence of aminoacyl-tRNA doesn’t account for rapid inhibition of mRORC1 signal; so amino acids sensing by GCN2 is directly involved in AA-induced mTORC1 activation needs more evidence.
AA transporters as modulators of Intracellular AA levels
[edit | edit source]• AA transporters regulatesTORC1 by changing intracellular AA concentrations or acting as receptor/transceptor to initiate intracellular signaling pathways.
• Leucine is strong activator of mTORC1.
• System L is called LAA transporter system, which is LAT 1/2(L-type AA transporter ½ and the 4F2hc/CD98 glycoprotin) is a primary route/entrance for neutral amino acids such as leucine with flowing out of cytoplasmic amino acids. Yet, LAT1 is highly present in actively growing tumors.
• The accumulation of intracellular amino acid is done by System A transporter, such as SNAT2(sodium-coupled neutral AA transporter 2).
• SNAT2 is one of types of System A transporter, which concentrates amino acids due to its unidirectional Na+-AA-coupled transport cycle.
• The expression of both System L and System A positively correlate with mTORC1 activity. That explains glutamine stimulates leucine on TOR activity though no effect on TOR activity by itself in oocytes.
• As the result of amino acids transport, Na+ and polar/charged amino acids tend to change its polarization of cell membrane. The result of depolarization effects a rise in intracellular Ca2+, an effect implicated in AA-dependent activation of mTORC1 and Vps34.
• Insulin promotes the localization of System A AA transporter to cell surface from endosomal compartment in skeletal muscle cells in order to simulating amino acids signaling.
• Cytokines enhance System L. For example, : cytokines promote growth in lymphocytes.
• Leucine transport can possibly prevent unwanted activation of signal pathway such as TORC1 due to T-cell activation by PMA (phorbol myristate acetate) and ionomycin.
• A two-step AA transport mechanism is also proposed for mTORC1 activation by AA. First, high-affinity L-glutamine transporter, SLC1A5 is responsible for the intracellular glutamine accumulation; secondly, the heterodimeric SLC7A5/SLC3A3 bidirectional transporter that is an intracellular leucine accumulation and mTORC1 activation uses intracellular glutamine as a flowing substrate to uptake extracellular leucine.
• AA transporters in TORC1 examples: Drosophila slimfast and minidiscs sense nutrient availability and control body size. AA transporters as sensors: transceptors
• Found in lower eukaryotes such as yeast/ Drosophila.
• Yeast AA permease Gap1 activates the cAMP/PKA (protein kinase A) signaling pathway to regulate metabolism and expression of stress-responsive genes.
• Shorten Gap1 C terminus can cause permanent activation of the PKA signaling pathway independent of Gap1 transport activity (in phenotypes, constitutively active PKA in vivo).
• Ssy1 (yeast AA permease homolog) senses extracellular AA to stimulate a proteolysis-dependent signaling pathway to increase various AA and peptide permeases expression.
• Drosophila-Evidence of AA transceptor and TORC1 connection can be verified by PATH, a PAT (proton-assisted AA transporter) transporter, which regulates growth by interacting with TOR. (SLC36 is the mediator). PATH could control growth via mechanism, no need of AA transport, so it mostly acts as a receptor.
• PAT found on the surface of mammalian lysosomes, plasma membrane and endosomal compartments. It relates to AVT (AA vacuolar transporter) in yeast, so it can transport AA out of vacuole and a lysosomal equivalent structure in yeast.
• PAT express throughout the body, such as in human PAT- PAT1 and PAT4 are expressed in normal tissues and cancer cell lines strongly, are required mTORC1 activation and cell proliferation.
• The mutants in minidiscs (fat body-specific AA transporter) have normal size that promotes growth in a wild-type host.
Conclusion:
• TORC1- a central cell-growth regulator, has to integrate a range of growth-stimulating and inhibitory signals to control translation, autophagy, and cell growth.
• PI3K-PKB/Akt-signaling branch phosphorylate TSC2 and PRAS40 in mTORC1.
• AMPK phosphorylates TSC2 and Raptor in response to energy starvation stress in mTORC1
• AA signaling is parallel to TSC1/2-Rheb branch of TORC1 regulation.
• Among all connectors between AA and TORC1, Rag GTPases are the most convincing one, which directly participate in TORC1 activation in response to AA.
• All the data supported by genetic data in yeast and Drosophila and in vitro biochemical/cell bio.
• In contrast, Vps34, MAP4K3, RalA and Rab5 signaling in AA need more verification.
• The active RagA or RagB binds Raptor to recruit TORC1 to lysosomes where TORC1 is activated by Rheb (localized in lysosomes). This explains Rheb and Rag are needed for full TORC1 activation. Rag GTPases (heterodimer) localize on the lysosomal surface to react with p14/MP1/P18 complex.
Citation: [1]--Shc036 (discuss • contribs) 06:13, 30 October 2011 (UTC)
References
Christie GR, Hyde R, Hundal HS. Regulation of amino acid transporters by amino acid availability. Curr Opin Clin Nutr Metab Care. 2001 Sep;4(5):425–431.
References
[edit | edit source]- ↑ Amino Acid Signaling in TOR Activation, Joungmok Kim and Kun-Liang Guan; Department of Pharmacology and Moores Cancer Center, University of California, San Diego, La Jolla, California. Annu. Rev. Biochem. 2011. 80:1001-32. http://www.annualreviews.org/doi/abs/10.1146/annurev-biochem-062209-094414
Steroid and Thyroid hormones
[edit | edit source]
Both of them are hormones which are lipid soluble. These hormones can pass across the cell membrane and enter into cytosol or even the nucleus. Cholesterol is the single precursor molecule for Steroid hormones. The estrogens (female sex hormones) and androgens (male sex hormones) are two classes of steroid hormones. Estrogen can diffuse across the membrane of a target cell and interact with a specific receptor with the cytoplasm. After forming of steroid-receptor complex, it can pass through nucleus membrane and enter into the nucleus. In the nucleus, it changes the transcription of mRNA for certain protein.
Thyroid hormones can regulate growth. Also it can play a role in stimulating the breakdown of proteins, fats, and glucose. Thyroid hormone secreted from the thyroid gland. They similarly diffuse across the plasma membrane and get into the nucleus. The thyroid-receptor complex influences certain transcription essential for some metabolic processes.[1]
Differences Between Androgen and Estrogen
[edit | edit source]Firstly, a hormone is produced chemically through the endocrine gland and possesses a specific effect on the activities of various organs in the body. Specific activities with which hormones help are growth and development, sexual function, reproduction, mood, and metabolism. The major endocrine glands through which hormones are produced are the pituitary, pineal, thymus, pancreas, thymoid, and adrenal glands. The primary male and female hormones are called the androgen and the estrogen. Males can produce these hormones in the testes, while women produce them in the ovaries. Although males and females possess both of these types of hormones, they possess them in significantly different amounts. For instance, the majority of males produce an immense amount testosterone, a form of an androgen, whereas women barely produce any amount. This relationship is the same with a male possessing levels of estrogen. Males still do produce this hormone, but in extremely small amounts daily.
Androgen: They are a group of hormones that play a role in male characteristic traits and reproductive activity. It is also considered a steroid, such as testosterone, that regulates the development of masculine characteristics. The male characteristics that androgens help develop include secondary sex characteristics that boys develop when going through puberty. Also, androgens influence sperm-cell formation and sexual interest and behavior.
Androgens may be called "male hormones." Both men's and women's bodies produce androgens, just in differing amounts. In fact, androgens have more than 200 actions in women.
The principal androgens are testosterone and androstenedione. They present in much higher levels in men. Other androgens include dihydrotestosterone (DHT), dehydroepiandrosterone (DHEA) and DHEA sulfate (DHEA-S).
In a woman's body, one of the main purposes of androgens is to be converted into the female hormones called estrogens.
In women, androgens are produced in the ovaries, adrenal glands and fat cells. In fact, women may produce too much or too little of these hormones-disorders of androgen excess and deficiency are among the more common hormonal disorders in women.
In women, androgens play a key role in the hormonal cascade that kick-starts puberty, stimulating hair growth in the pubic and underarm areas. Additionally, these hormones are believed to regulate the function of many organs, including the reproductive tract, bone, kidneys, liver and muscle. In adult women, androgens are necessary for estrogen synthesis and have been shown to play a key role in the prevention of bone loss, as well as sexual desire and satisfaction. They also regulate body function before, during and after menopause.
About Androgen-Related Disorders: a) High Androgen Levels Excess amounts of androgens can pose a problem, resulting in such "virilizing effects" as acne, hirsutism (excess hair growth in "inappropriate" places, like the chin or upper lip) and thinning hair. Many women with high levels of a form of testosterone called "free" testosterone have polycystic ovary syndrome (PCOS), characterized by irregular or absent menstrual periods, infertility, blood sugar disorders, and, in some cases, symptoms like acne and excess hair growth. Left untreated, high levels of androgens, regardless of whether a woman has PCOS or not, are associated with serious health consequences, such as insulin resistance and diabetes, high cholesterol, high blood pressure and heart disease. In addition to PCOS, other causes of high androgen levels (called hyperandrogenism) include congenital adrenal hyperplasia (a genetic disorder affecting the adrenal glands that afflicts about one in 14,000 women) and other adrenal abnormalities, and ovarian or adrenal tumors. Medications such as anabolic steroids can also cause hyperandrogenic symptoms.
b) About Low Androgen Levels: Low androgen levels can be a problem as well, producing effects such as low libido (interest or desire in sex), fatigue, decreased sense of well-being and increased susceptibility to bone disease. Because symptoms like flagging desire and general malaise have a variety of causes, androgen deficiency, like hyperandrogenism, often goes undiagnosed. Low androgen levels may affect women at any age but most commonly occur during the transition to menopause, or "perimenopause," a term used to describe the time before menopause (usually two to eight years). Androgen levels begin dropping in a woman's age of 20, and by the time she reaches menopause, they have declined 50 percent or more from their peak as androgen production declines in the adrenal glands and the midcycle ovarian boost evaporates. Further declines in the decade following menopause indicate ever-decreasing ovarian function. For many women, the effects of this further decline include hot flashes and accelerated bone loss. These effects may not become apparent until the women are in their late 50s or early 60s.
About Treatment for Low Androgen Levels: Combination estrogen/testosterone medications are available for women in both oral and injected formulations. Small studies find they are effective in boosting libido, energy and well-being in women with androgen deficiencies, as well as providing added protection against bone loss. However, the risks from the combination of estrogen and testosterone include increased risk of breast and endometrial cancer, adverse effects on blood cholesterol and liver toxicity. Testosterone is also an effective treatment for AIDS-related wasting and is undergoing studies for treating premenstrual syndrome (PMS) and autoimmune diseases. Women with PMS may have below-normal levels of testosterone throughout the menstrual cycle.
Estrogen: They are the sex hormones produced primarily by a female's ovaries that stimulate the growth of a girl's sex organs, as well as her breasts and pubic hair, known as secondary sex characteristics. Estrogens also regulate the female menstrual cycle.[2]
Estrogen is the general name for a group of hormone compounds. It is the main sex hormone in women and is essential to the menstrual cycle. Although both men and women have this hormone, it is found in higher amounts in women, especially those capable of reproducing. Secondary sex characteristics, which are defining differences between men and women that don't relate to the reproductive system, are determined in part by estrogen. In women, these characteristics include breasts, a widened pelvis, and increased amounts of body fat in the buttock, thigh, and hip region. This hormone also contributes to the fact that women have less facial hair and smoother skin then men. It is also an essential part of a woman's reproductive process. Estrogen regulates the mentrual cycle and prepares the uterus for pregnancy by enriching and thickening the endometrium. Two hormones, the luteinzing hormone (LH) and the follicle stimulating hormone (FSH), help to control how the body produces estrogen in women who ovulate. Estrogen is manufactured mostly in the ovaries, by developing egg follices. Moreover, it produced by the corpus luteum in the ovaries, as well as by the placenta. The liver, breasts and adrenal glands may also contribute to its production, although in smaller quantities. There are three distinct compounds that make up this hormone group: estrone, estradio and estriol. During a woman's reproductive life, which starts with the onset of menstruation and continues until menopause, the main type of estrogen produced is estradiol. Enzymatic actions produce estradiol from androgens. Testosterone contributes to the production of estradio, while the estrone is made from andostenedione.
Estrogen is important to a woman's health beyond just how it relates to her reproductive cycle. Although it can cause women to retain fluid, and early exposure through early menses can increase a woman's risk of developing breast cancer, this hormone has significant benefits.
Androgens as related to women
In women, androgens are produced in the adrenal glands, ovaries, and fat cells. Disorders involving excess or inadequate levels of androgens in women constitute many of their common hormonal disorders. For women, the key roles of androgens are to aid in the starting of puberty and to regulate the function of certain organs. For adult women, androgens are needed for estrogen synthesis and are involved in the prevention of bone loss, and in sexual desire and satisfaction
The Three Chemical Classes of Hormones
[edit | edit source]There are three general classes of hormones characterized by their structure, and not their function. The three categories are:
1. Steroids Steroids are lipids which are formed from cholesterol. Examples include testosterone and cortisol. These hormones are given off by gonads, adrenal cortex, and the placenta. Steroids are natural substances with many different effects in the human body, which begin over several days. The primary use of steroids in health care is to reduce inflammation and other disease symptoms. Steroid inhalers have an important role in reducing deaths from asthma, local steroid injections are useful in treating painful joints and ligaments. Steroid creams are used extensively to treat eczema and other inflammatory skin conditions. Steroids make the whole immune system less active, which can be very useful in illnesses where there is an immune component. Steroids are the ultimate anti-inflammatory drugs. However, steroid use in medicine is limited by very serious side effects in the body as a whole. That is why steroids tend to be used sparingly in local preparations such as sprays and creams, which ensure maximum steroid dose where it is needed, and minimum levels in the blood stream. Steroids also affect the brain, and high doses can make people feel happy, euphoric, hyped-up, with disturbance of sleep and even serious psychiatric illness such as mania, very aggressive behavior and psychosis (delusions, pananoia, loss of touch with reality). If steroid users are also taking other drugs which affect mood or brain function, these side-effects can be far more common. Steroids are really useful in the care of those with advanced cancer when short life expectancy from their condition means physicians are far more relaxed about long term side-effects. Brain tumours often respond dramatically to steroids. The reason is that the brain is contained in a bony box inside the skull and pressure can build up inside the head, resulting in headaches, sickness, drowsiness and other problems. Brain scans often show that a tunour the size of a wallnut can be surrounded by a big immune reaction, with brain swelling and inflammation. Steroids reduce the additional swelling, often reversing symptoms and buying time. The underlying cancer continues to grow if the person finally begins to deteriorate death often follows rapidly as the steroid dose is reduced. So steroids are really powerful, with wide ranges of actions, producing dramatic effects ranging from pain relief to mood elevation, and if it were not for the very serious side effects, they would be used even more often. The body becomes dependent on steroids and when used in health care, most physicians reduce dosage gradually, even though they may start in an acute illness with a very high dose.
2. Peptides
Peptides are the most common type of hormones and contain a chain of amino acids. Examples include TRH and vasopressin. These hormones are given off by the heart, liver, stomach, kidney, pituitary gland, and parathyroid. Peptides play a crucial role in fundamental physiological and biochemical functions of life. For decades now, peptide research is a continuously growing field of science. When the number of amino acids is less than about 50, these molecules are named peptides while larger sequences are referred to as proteins. The amino acids are coupled by a peptide bond, a special linkage in which the nitrogen atom of one amino acid binds to the carboxyl carbon atom of another. Peptides (proteins) are present in every living cell and possess a variety of biochemical activities. They appear as enzymes, hormones, antibiotics, receptors, etc. Synthetic peptides may be useful in structure-function studies of polypeptides, as peptid hormones and hormone analogues, in the preparation of cross-reacting antibodies, and in the design of novel enzymes. Peptides are synthesized by coupling the carboxyl group or C-terminus of one amino acid to the amino group or N-terminus of another. There are two strategies for peptide synthesis: liquid-phase peptide synthesis and solid-phase peptide synthesis (SPPS).
3. Amines
Amine-derived hormones are derived from the amino acids tyrosine and tryptophan. Examples include catecholamines and thyroxine. These hormones are given off by the thyroid and adrenal medulla.
In amines, the hydrogen atoms in the ammonia have been replaced one at a time by hydrocarbon groups.
Amines fall into different classes depending on how many of the hydrogen atoms are replaced.
In primary amines, only one of the hydrogen atoms in the ammonia molecule has been replaced. That means that the formula of the primary amine will be RNH2 where "R" is an alkyl group.
In a secondary amine, two of the hydrogens in an ammonia molecule have been replaced by hydrocarbon groups.
In a tertiary amine, all of the hydrogens in an ammonia molecule have been replaced by hydrocarbon groups.
References
[edit | edit source]- ↑ Berg, Jeremy M., Tymoczko, John L., and Stryer, Lubert. Biochemistry. 6th ed. New York, N.Y.: W.H. Freeman and Company, 2007: 211.
- ↑ [3] Androgen and Estrogen.
"Growth Hormone Test: MedlinePlus Medical Encyclopedia." U.S National Library of Medicine. U.S. National Library of Medicine, n.d. Web. 29 Oct. 2012. <http://www.nlm.nih.gov/medlineplus/ency/article/003706.htm>.
"Androgen | HealthyWomen." Androgen | HealthyWomen. N.p., n.d. Web. 29 Oct. 2012. <http://www.healthywomen.org/condition/androgen>.
"AndrogenAbout Our Definitions: All Forms of a Word (noun, Verb, Etc.) Are Now Displayed on One Page." Merriam-Webster. Merriam-Webster, n.d. Web. 29 Oct. 2012. <http://www.merriam-webster.com/dictionary/androgen>.
"Chemical Classes of Hormones." News Medical. News Medical, n.d. Web. 20 Nov. 2012. <http://www.news-medical.net/health/Chemical-Classes-Of-Hormones.aspx>.
Berg, Jeremy M.; Tymoczko, John L.; Stryer, Lubert. Biochemistry. 7th ed. New York, 2012.
Testosterone is the primary male hormone created from cholesterol. Although it has significantly presence in males, it is created in the ovaries in female as well. As aging occurs, the amount of testosterone is decreased in the body. The decrease in testosterone leads to the decrease of bone density.
Testosterone is a steroid hormone from the androgen group. In mammals, testosterone is primarily secreted in the testes of males and the ovaries of females, although small amounts are also secreted by the adrenal glands. It is the principal male sex hormone and an anabolic steroid. Testosterone is evolutionarily conserved through most vertebrates, although fish makes a slightly different form called 11-ketotestosterone. In men, testosterone plays a key role in the development of male reproductive tissues such as the testis and prostate as well as promoting secondary sexual characteristics such as increased muscle and bone mass and hair growth. In addition, testosterone is essential for health and well-being as well as preventing osteoporosis. On average, an adult human male body produces about ten times more testosterone than an adult human female body. Females are from a behavioral perspective (rather than from an anatomical or biological perspective), more sensitive to the hormone. However, the overall ranges for male and female are very wide, such that the ranges actually overlap at the low end and high end respectively.
Physiological Effects
[edit | edit source]Androgens promote protein synthesis and cell growth in tissues that contain androgen receptors. Testosterone affects the cell in two different ways: virilizing and anabolic. Anabolic effects increases bone density, bone maturation, and increase in muscle mass and strength. Androgenic effects are maturation of the sex organs, particularly in the size of the penis and the forming of the scrotum in the fetus and after birth, usually during puberty which deepens the voice, growth of hair.
Prenatal Effects
[edit | edit source]The prenatal androgen occur in 2 different stages. First is the genital virilization which is the midline fusion, scrotal thining, rugation, and phallic enlargement. The second is the development of prostate and seminal vesciles. In the second trimester androgen levels are associated with gender characterization. This period has also known to affect the femininity or masculinity of the fetus and how it can be further predicted to have feminine or masculine behaviors. During pregnancy a mother's testosterone levels the behavior more than the fetusus testosterone level.
Pre-peripubertal Effects
[edit | edit source]Pre-peripubertal Effects are the first effects that are observed when androgen levels are raised. This usually occurs at the end of childhood and is seen in both girls and boys.
- Axillary Hair
- Growth Spurt or accelerated bone maturation
- Facial Hair
- Body odour
- Acne or increase oiliness on skin
- The appearance of pubic hair
Pubertal Effects
[edit | edit source]Pubertal effects happen when androgen levels are usually higher than normal.
- Appearance of Adams apple
- Broader shoulders as the rib cage expands
- Deeper voice
- Remodeling of facial bone contours
- Increased in muscle mass and strength
- Leg hair
- Chest Hair and perianal hair
- Pubic hair extends to the thighs
- Increased lido and erection or clitoral engorgement
- clitoromegaly
- enlargement of the sebaceous glands
- Acne
- Axillary hair
- Complete bone maturation
Biological Uses
[edit | edit source]- Testosterone regulates the thromboxane receptors and platelets.
- It also maintains muscle trophism
- Also regulates acute hypothalamic pituitary adrenal axis (HPA) response
- Testosterone is also necessary for normal sperm growth
- Activates genes in sertoli cells
- Promotes differentiation of spermatogonia
Reference: Berg, Jeremy M.; Tymoczko, John L.; Stryer, Lubert. Biochemistry. 7th ed. New York, 2012.
Overview
[edit | edit source]Estrogen, otherwise known as oestrogen, is a primary female sex hormone that is vital in the development and functioning of females. The name is derived from estrus, the period of fertility for female mammals, and gen, meaning to generate. However, males too contain estrogen in lower quantities.
Production of estrogen occurs primarily in the ovaries, more specifically the theca internal cells. Estrogen secretion is stimulated by another hormone, the luteinizing hormone (LH). Estrogen is also produced, in smaller quantities, in the liver, adrenals glands, fat cells and the breasts.
Types of Estrogen
[edit | edit source]Steroidal Estrogens
There are three major naturally occurring estrogens in women: estrone (E1), estradiol (E2), and estriol (E3). Estradiol is the predominant estrogen found in non-pregnant females. Estrone is weaker than estradiol, thus it is the primary estrogen after a woman.
Estradiol
[edit | edit source]Estradiol is a type of endogenous estrogen hormone, called E2, found in both males and females, and is the most dominant estrogen hormone found in women. The structure contains an aromatic ring and two hydroxyl groups. As a steroid, it is a derivative of cholesterol. The structure is similar to that of testosterone, and with the presence of aromatase, testosterone can be catalyzed into estradiol. Fat cells can also be converted into estradiol. It is produced in the ovaries for women and testes for men, as well as the adrenal cortex, liver, and hair for both sexes.
Estradiol is recognized by either the estrogen receptor ER-alpha or ER-beta, which are found on the surface membrane of cells. After the signal is recognized, estradiol is accepted into the cell and then it penetrates the nucleus, where it gets transcribed. The produced mRNA gets translated and then proteins are synthesized.
Estradiol regulates reproductive, sexual, bone, and various tissue development. In females, it is responsible for the development of secondary sex characteristics, such as breast growth, body hair growth, enlargement of hips, and widening of the uterus. It also helps develop the reproductive organs to prepare the body for a pregnancy. Such induced changes take place in the vaginal and fallopian tube lining and the ovaries. Estradiol affects bone structure and growth, which has been confirmed by observing more cases of osteoporosis in people who are unable to produce any.
Non-Steroidal Estrogens
Non-steroidal estrogens are either synthetic or naturally occurring substances that possess estrogenic activity. Examples of these are xenoestrogens (synthetic estrogens), phytoestrogens (plant estrogens), mycoestrogens (fungi estrogens).
Estrogen Functions
[edit | edit source]In males, estrogen helps maintain a healthy libido and aids in the maturation of the sperm. In females, estrogen serves to develop secondary sexual characteristics such as breasts, endometrium, and regulation of menstural cycle. It does this by accelerating burning of body fat, reducing muscle bulk, and decelerating height increase during puberty.
Other functions of estrogen include reducing bone resorption, increasing bone formation, increasing platelet adhesiveness, increasing good cholesterol and triglycerides, improving lung functions, and causing salt and water retention.
Estrogen in Medicine
[edit | edit source]The most common uses of estrogen in medicine are birth control pills or contraceptives. In birth control pills, either low doses of estrogen combined with progestin is used, or simply progestin. The reason that estrogen is used in such a manner is because estrogen prevents ovulation by reducing secretion of the follicle stimulating hormone and the luteinizing hormone.
Estrogen in Cosmetics
[edit | edit source]Cosmetics can contain steroidal or non-steroidal hormones. Common products include shampoos, conditioners, moisturizers, body and skin creams, astringents, sunscreens, perfumes, hair sprays, shampoos, conditioners, styling gels, facial creams, foundations, moisturizers, lipsticks, liquid hand soaps, body wash, insect repellants, nail polish, polish remover, aftershave, and shaving creams. These products usually contain estrogen named parabens, placental extracts, and UV screens. It is important to limit the use of such products as long-term exposure may increase the risk of breast cancer.
References
[edit | edit source]Mandal, Ananya. "Estrogen - What is Estrogen?." News Medical. Ed. April Cashin-Garbutt. News Medical, n.d. Web. 2 Dec. 2012. <http://www.news-medical.net/health/Estrogen-What-is-Estrogen.aspx>.
Overview
[edit | edit source]Xenohormones are pollutants originating outside the body that have hormone-like and estrogen-like characteristics. Exposure to these substances can have a severe and chronic impacts on the body's natural hormonal balance.
Xenohormone exposure is especially high in today's industrialized countries and results from consumption lots of meats and dairy products full of synthetic hormones, genetically modified fruits and vegetables, crops grown with chemicals (grown with pesticides and herbicides) petrochemical compounds (perfume, hair spray, room deodorizers, cooking with plastic, etc…) and prescription synthetic estrogens and progestins (such as the pill and estrogen replacement therapy).
side effects
[edit | edit source]- reproductive abnormalities
- cancers of the reproductive tract
- infertility
- low sperm counts
- weakening of immune system
- fatigue
- headache
- depression
- lack of concentration
- mood swings
- hormonal imbalance
The potential consequences of these exposures are overwhelming, especially since we are passing these reproductive abnormalities on to our children. Limiting your exposure to these pollutants as much as possible and supplementing with Natural Progesterone Cream will substantially improve the quality of your health.
Common Xenohormones in the Environment
[edit | edit source]Phthalates
These are found in most plastics, inks, adhesives, vinyl flooring and paints. The type of plastics that contain the highest amount of phthalates are plastic wraps. Effects of phthalates were tested on rats and results include a multitude of abnormalities such as a reduction of testosterone levels, testicular tumors, and irregularities in the male reproductive tract. It should also be noted that women who are exposed to high levels of phthalates increase their risk of a miscarriage
Alkylphenols & Nonylphenols
These are found in detergents, shampoos, cosmetics, spermicidal lubricants, pesticides, and clear plastics. They are also found in much of the United States' tap water as much of the above mentioned products are not broken down, but instead washed into the sewage. Exposure of this substance to pregnant rats have resulted in male offspring with misdeveloped reproductive organs.
Bisphenol A (BPA)
These are found in hard plastics usually in the forms of dental sealants or containers for food and drinks. The effects of exposure to BPA involve potent estrogenic effects on the prostate.
Reference
[edit | edit source]http://www.naturalendocrinesolutions.com/archives/the-impact-of-xenohormones-on-our-health/ http://www.huntingtonbeachthyroiddoctor.com/ Hopkins, Virginia, and John R. Lee. "Xenohormones (Part II) in Your Environment." Virginia Hopkins Test Kits: Excellence in Hormone Testing and Consumer Awareness. Virginia Hopkins Test Kits, 2012. Web. 20 Nov. 2012. <http://www.virginiahopkinstestkits.com/xenohormoneII.html>.
Overview
[edit | edit source]Alkylphenols are a family of organic compounds obtained by the alkylation of phenols. The term is usually reserved for commercially important propylphenol, butylphenol, amylphenol, heptylphenol, octylphenol, nonylphenol, dodecylphenol and related "long chain alkylphenols" (LCAPs). Methylphenols and ethylphenols are also alkylphenols, but they are more commonly referred to by their specific names, cresols and xylenols.
Uses
[edit | edit source]Alkylphenols such as nonylphenol and octylphenol are mainly used to make alkylphenol ethoxylate (APE) surfactants (detergents), though alkylphenols themselves can be used as plasticisers in plastics, and the derivatives alkylphenol phosphites can be used as UV stabilisers in plastics.
In Europe alkylphenol ethoxylates are used in:
- industrial detergents, such as those used for wool washing and metal finishing.
- some industrial processes, such as emulsion polymerisation
- the spermicidal lubricant nonoxynol-9.
- various laboratory detergents, including Triton X-100.
- some pesticide formulations.
Outside Europe alkylphenol ethoxylates are also used in many domestic products; for example in the USA they are in many liquid clothes detergents. In Europe these products contain the slightly more expensive, but much safer, alcohol ethoxylates.
Hormone disrupting effects
[edit | edit source]Alkylphenols were first found to be oestrogenic (oestrogen-mimicking) in the 1930s, and more evidence was published in 1978. However, it was only in 1991 that publication of the effects of nonylphenol on cultured human breast cells led to health concerns (Soto et al., 1991). This and more recent research has shown that the growth of these cells is increased by alkylphenols at concentrations 1000 to 10000 times higher than the oestradiol levels required to produce the same growth. Oestrogenic effects have also been shown on rainbow trout hepatocytes, chicken embryo fibroblasts and a mouse oestrogen receptor. Oestrogenic effects are present at tissue concentrations of 0.1 µM for octylphenol and 1 µM for nonylphenol. A recombinant yeast screen using the human oestrogen receptor has shown similar results.
Recent research is showing oestrogenic effects of nonylphenol at ever lower concentrations. Levels of 0.05 micro-g per litre were enough to increase the number of eggs produced by minnows, as well as increasing vitellogenin levels. This research also suggested that nonylphenol was leading to an increase in natural oestrogen levels.
reference
[edit | edit source]http://web.archive.org/web/20000416232843/http://website.lineone.net/~mwarhurst/apeintro.html
Human Chorionic Gonadotropin
[edit | edit source]Introduction
[edit | edit source]Human Chorionic Gonadotropin (hCG) is a glycoprotein hormone made up of an alpha and beta subunit. The alpha subunit making up hCG is the same one that makes up the alpha subunits of the luteinizing hormone, follicle-stimulating hormone, and thyroid-stimulating hormone. While the beta subunit is comparable with the beta subunit of the luteinizing hormone, there are differences that make the beta subunit of hCG unoique. However because of the similarities between hCG and luteinizing hormone, hCG is viewed as a much more active version of luteinizing hormone. Both hormones are recognized by the luteinizing hormone/choriogonadotropin receptor (LHCGR), which is located in the ovaries, uterus, and breasts. It has been dubbed as the pregnancy hormone because of its multiple functions which affect the pregnancy process. It is produced in the placenta of a pregnant woman by the syncytiotrophoblast cells, which are the outer cells of the blastocyst.
Function
[edit | edit source]The functions of the hCG allow for a safe pregnancy to take place. It gets activated by the LHCGR and stimulates the production of the steroid hormone, progesterone. Progesterone allows for the lining of the uterus to thicken so that a fertilized egg will be able to stick and develop into a fetus. hCG will also bind to specific regions on the corpus luteum, which is in the ovaries, which allows for the production of progesterone to continue. If hCG did not bind the corpus luteum, the amount of progesterone in the body would drop and the thickened uterus lining would be released during the woman's period. Overall, hCG prevents a woman from getting her period when she is conceived with a child.
Pregnancy Tests
[edit | edit source]Since hCG is only produced in pregnant women, the presence of hCG can be used to test for pregnancy. Traces of hCG can be found in urine as early as 11 days after fertilization and found in blood after about two weeks. All pregnancy test use immunoassays, which determine the concentration of hCG in a woman. In the mid-1900s when immunoassays were being developed, there were complications in increasing its sensitivity and being able to recognize hCG and not luteinizing hormone. Because of the structural differences in the beta subunits of hCG and luteinizing hormone, immunoassays that could differentiate the two hormones were developed. The pregnancy test determines the milli-international units of hCG in one mL of blood or urine. If there is less than 5 milli-international units in 1 mL, then the test comes out negative. A concentration of 25 milli-international units in 1 mL is a positive test. During the first two to three months of pregnancy, the hCG levels should increase dramatically and then decrease until about six months of pregnancy, when the level should remain relatively consistent. When the hCG level is lower than the expected amount, it serves as an indication of a miscarriage that may happen or has already happened, or a ectopic pregnancy, which means the fetus is developing outside of the uterus and will be fatal to the fetus, but may be fatal to the mother as well. If hCG levels are too high, it is likely to either be a multiple pregnancy or a molar pregnancy, which is when the cells turn into abnormal tissue, rather than a fetus, but the body behaves as if an actual pregnancy has taken place. Molar pregnancies are characterized by high levels of beta-hCG. After giving birth or a miscarriage, the hCG levels should return to normal (which is below 5 milli-international units per mL) within two months.
Genetic Disorders Testing
[edit | edit source]The Triple Screen Test is conducted on women during their fourth to sixth months of pregnancy to determine the probabilities of the fetus carrying certain genetic disorders. This test is not very reliable but is recommended for women who are over the age of 35, have a family history of genetic disorders, or have been sick at any time during their pregnancy. The test is determines the levels of hCG and two other substances, estriol and alpha-fetoprotein in the mother's blood.
Role As a Diet Supplement
[edit | edit source]A significant level of hCG prevents women from feeling hungry as their standard amount and also encourages the body to use fat, rather than carbohydrates, for energy. Some people who want to lose weight have been looking for hCG injections or oral supplements, in hopes of an easy diet. Because their hunger would be suppressed, they would be able to easily limit themselves to about 500 calories daily, without slowing down their metabolism. However, this is dangerous because people would not be able to get the necessary amount of nutrition, which can lead to dangerous side effects, so supplemental hCG is not FDA approved and discouraged by medical professionals.
Overview
[edit | edit source]Pheromones are a type of hormones that act outside of the body, otherwise known as ectohormones. Pheromones are chemicals that animals and insects produce that affect the behavior of another animal or insect of the similar species. Pheromones are used to trigger a wide range of behaviors that are not limited to but include alarming for danger, sexual arousal, marking a territory, and leaving a trail.
Types of Pheromones
[edit | edit source]1. Releaser Pheromones These types of pheromones illicit an immediate response that is usually related to sexual attraction.
2. Primer Pheromones These illicit slower responses and are involved in a variety of activities such as influencing development and altering hormone levels.
3. Signaler Pheromones These are what give a odor imprint allowing an individual to be recognized, usually by the mother.
4. Modulator Pheromones These are usually found in sweat and either alter or synchronize bodily functions.
References
[edit | edit source]Nordqvist, Christian. "What are Pheromones? Do Humans Have Pheromones?." Medical News Today. Medical News Today, 11 Aug. 2011. Web. 20 Nov. 2012. <http://www.medicalnewstoday.com/articles/232635.php>.
Gastrin
[edit | edit source]
Gastrin is a peptide hormone which is soluble in the water. This hormone plays an important role in the secretion of HCI in stomach. Gastric acid secretion also helps the digestion of protein, the absorption of iron and calcium. In the first, Gastrin activates a particular G protein by attaching to a cell surface receptor in the plasma membrane. Then, the activated G protein binds to another membrane enzyme, phospholipase C (PLC). This enzyme-substrate complex carries out a hydrolysis reaction where hydrolyze phosphatidylinositol- 4,5-bisphosphate (PiP2) to inositol-1,4,5-triphosphate (IP3) and 1,2- diacylglycerol (DAG).
In the second phase, inositol-1,4,5-triphosphate (IP3), reacts with the endoplasmic reticulum and stimulates the release of Ca2+ ion into the cytoplasm. In the same time, 1,2- diacylglycerol (DAG), which was released into the cytoplasm, diffuses through the plasma membrane, where interacts with protein kinase C. The Ca2+ just released from the endoplasmic reticulum and DAG cooperation stimulates that kinase to phosphorylate a protein which in turn causes HCI secretion into the stomach.[1]
References
[edit | edit source]- ↑ Schubert, ML (2005). Gastric secretion.
{{cite book}}: Text "Curr Opin Gastroenterol" ignored (help)
1. Gastric secretion. Schubert ML. Curr Opin Gastroenterol. 2005 Nov;21(6):636-43. Review. PMID 16220038 [PubMed - indexed for MEDLINE]
Overview
[edit | edit source]Epinephrine, otherwise known as adrenaline, is a hormone secreted by the adrenal glands that is involved in the "fight or flight" response in humans. This response occurs when an individual comes in contact with a threat causing the body to release epinephrine giving the individual energy to respond to the threat.
Epinephrine is derived from tyrosine, one of the 20 major amino acids.
References
[edit | edit source]Cashin-Garbutt, April. "What is Epinephrine (Adrenaline)?." News Medical. N.p., n.d. Web. 6 Dec. 2012. <http://www.news-medical.net/health/What-is-Epinephrine-(Adrenaline).aspx>.
Overview
[edit | edit source]Human placental lactogen (hPL) is a hormone produced exclusively by pregnant women. It is formed in the placenta, which is a tissue that develops during a pregnancy, so non-pregnant women do not produce hPL. The tertiary structure of hPL is similar to that of growth hormone, which all females produce. Like growth hormone, it contributes to the growth of protein tissues. However, hPL is found at exponentially higher concentrations than ugrowth hormone. The highest levels of hPL are observed during the final stages of pregnancy.
Levels
[edit | edit source]Levels of hPL in a pregnant woman can be quantified through a blood test. This information indicates the quality of the functionality of the placenta. If the test shows lower than expected levels of hPL, this suggests toxemia, choriocarcinoma, or placental insufficiency. However, women may also have higher than expected values if they are expecting multiplets, have diabetes, molar pregnancy, or Rh incompatibility.
Function
[edit | edit source]The main function of hPL is to prevent the movement of glucose into the maternal cells, so that the fetus will be able to accept enough of it to proceed towards normal development. This means an overall increase in glucose levels in the mother's blood stream, although proportionally less glucose is used by the mother's body. Because of this role, hPL contributes to a healthy birth weight of newborns.