Structural Biochemistry/Volume 3
Structural biochemistry has become vital in the development of new medicine. Medicines are now being studied with the tools of biochemistry such as X-Ray Crystallography. Modern methods of biochemistry are usually used to understand the enzyme structure by understanding the folding and bending of the structure. Enzymes are biological catalysts that increase the rate of reactions by lowering the energy required to form the transition state of the reaction. Enzymes are typically made of a protein or of a group of proteins. Understanding protein tertiary and quaternary structure can tell scientists how a medicine does its job. Medicinal scientists have made use of the structure of enzymes to develop new drugs from old drugs. Drugs cross the cell membrane by first letting a message or drug encounter the outside of a cell and make it contact the receptor. Then, a connecting transducer passes the message inward, which finally gets signal amplified, prompting the cell to complete its function. Many scientists believe that in 10 to 20 years the field of medicine will be drastically different especially in the manner in which doctors prescribe drugs to their patients. Currently, drugs are administered based on average dosages that have been determined based on the size of an individual and their age. The effectiveness of a drug is depicted on a graph called "dose-response curves". These graphs are created to show a relationship between the desired effect of the drug and the amount of the drug administered. Typically there is another curve displaying the amount of drug that causes maximal side effects. Pharmacologist uses this data to first prove that the drug is effective and then use it to provide doctors with a safe dosage range to the drug to their prescribe patients.
Good medicine design relies on many variables which include its absorbency into the body, its activity of working correctly, how long it will be active, and its toxicity. Knowledge of the structures of target molecules allows for a more direct path of finding a molecule that will fit the shape of the target perfectly, creating the most useful drug. Therefore, molecules that obviously don’t fit the target will automatically be known to not work in its present state and should be disregarded or reconfigured.[1]
In the future the goal is to provide tailor-made drugs for each individual. The idea is that a drug will be designed based on the individuals DNA sequence which describes the individual’s personal biochemistry. The desire behind this is for a drug that is more effective and that causes fewer side effects. The desire for tailor-made drugs was not even a realistic venture less than a decade ago, but with the extreme advances in DNA sequencing this dream could become a reality in a couple of decades.
Nanomedicine
[edit | edit source]The number of people diagnosed with cancer every year continues to be extremely high. Scientists have been diving into the relatively new field of nanoparticle based drug design in hopes of making more effective anti-cancer drugs. Current chemotherapy techniques are effective at destroying cancer cells, but also are toxic to healthy cells that are essential to the body.
Nanomedicines can be chemically engineered to specifically target cancer cells without obtaining the harsh effects of chemotherapy. Different parts of nanoparticles can be modified in order for the medicine to be able to enter into the bloodstream of a person and target the cancer cells without being broken down by the liver. Some nanoparticles are packaged in lipsomes[check spelling], while many new compounds are delivered through biodegradable polymers. For example, polyethylene glycol (PEG) is a biodegradable polymer that protects the nanoparticles from being visible to immune cells, which helps the medicine to reach its target destination. Some of the nanoparticle shells are made with sugars such as “cyclodextrins,” but are covered in PEG. The hydroxyl groups on the sugars make the compounds soluble in water, yet easy to disintegrate in acidic environments in order to release the drug. Approximately twelve nanoparticle based anti-cancer drugs are in clinical trials and are waiting to be approved to be distributed to the world.
Nanoparticles are also being used to carry RNA molecules to target cancer cells through antisense therapy. If the RNA molecules are able to reach the cancer cells with the help of nanoparticles, then they can have the ability to bind to the cancer cells’ own RNA and inactivate certain genes. For example, antisense therapy can halt the production of proteins in cancerous cells, which helps stop the overall growth of the cancer.
Natural substances used for medicine
[edit | edit source]Natural products have assisted medicine in many different ways. It is found that natural products contain many disease and cancer fighting properties. They can be synthesized into a compound to used in medicine. For example, small plant like organisms such as cyanobacteria that reside in wet environments have these powerful cancer and bacteria killing sources. A professor named Dick Moore from the University of Hawaii at Mano was able to devise a way to find compounds that was especially good against slowly developing and difficult to treat tumors. An example of this was a compound called cryptophycin-8, which could rip apart the cellular scaffolding in a wide variety of solid tumors used in mice.
Also there has been a huge variety of substances and chemicals in the sea that have immensely powerful cancer and disease fighting capabilities. This has propelled scientists to develop new ways to synthetically create compounds that are derived from these natural products in the sea. The goal of making a good drug is the result of scientists playing around with natural compounds to extract their medicinal properties but taking away the parts that causes unwanted side effects.

Medicines can potentially be made from even the most strange materials. For example, chemist Jim Gloer from the University of Iowa has been researching for ways to use a fungus that lives in animal feces to create antibiotics. These organisms are called coprophiles, which means feces loving have a lot of potential for developing useful drugs. These fungi release chemicals that kill off neighboring species, which is what biomedical researches and scientists want so that they can develop medicines that kill unwanted fungus that are hazardous to humans.
Biochemistry of disease
[edit | edit source]According to World Health Organization’s International Statistical Classification of Diseases and Related Health Problems, the current definition of disease include in 22 chapters. These are divided into over 2500 blocks which give us thousands of phenotypic descriptors of disease.
Biochemists are focused on understanding structure of molecules and process in which these molecules come together. Understanding these processes also give us a chance to “correct” them when they go wrong. But there are many different keys that come into consideration to acquire therapies. For example in the case of drug development, it is the combination of the knowledge of chemistry, biochemistry, pharmacology, toxicology, etc. This is obviously not a predictable process because the high failure rate of experimental drugs.
Biochemists look into biochemistry of disease to study important points which they can use to develop new disease therapies. These major points in the field are biosynthesis of unusual microbial metabolites, structure-based design of inhibitors, mechanisms or drug resistance, and the role of protein folding dynamics that can lead to inappropriate protein folding and aggregation.
References
[edit | edit source]- ↑ The Structures of Life, National Institutes of Health. "The Structures of Life." July 2007: 46-48.
Service, Robert F. "Nanoparticle Trojan Horses Gallop From the Lab Into the Clinic." Science 15 October 2010: 314-315
Davis, Alison. "The Chemistry of Health." NIGMS August 2006: 36-42. http://publications.nigms.nih.gov/chemhealth/coh.pdf
In the field of Biochemistry, several important themes contributing to drug design should be evaluated in order to cure diseases, including structure-based design of inhibitors, mechanisms of drug resistance, biosynthesis of unusual microbial metabolites, and the role of protein dynamics that can lead to protein-misfolding.[1]
The Biochemistry of Disease: Desperately Seeking Syzygy Annual Review of Biochemistry Vol. 78: 55-63 (Volume publication date July 2009) DOI: 10.1146/annurev-biochem-120108-082254 John W. Kozarich http://www.annualreviews.org/doi/abs/10.1146/annurev-biochem-120108-082254?journalCode=biochem
Drug Targeting
[edit | edit source]In order to delivery drug to the desired destination, targeting is an important subject in drug delivery. Ligand is a substance that forms a complex with a biomolecule to serve for a biological purpose. There are three classes of ligands that are commonly used for drug delivery targeting. Antibodies and/or antibody fragments, peptides, and aptamers. Depending upon the situation of targeting, different type of ligand is used.
Reason of Drug Targeting and its Consequences
[edit | edit source]Drug targeting can be defined as the method and efficiency of delivering drugs to a target organ or system. Although this may seem like a simple process (just eat it or inject it into the blood right?), there are actually so many obstacles that have to be solved that it takes pharmaceutical companies years to develop one drug. These challenges include if the drug actually makes it to the target organ, and if it makes it to the organ in significant amounts that would actually be of benefit. Its common knowledge that if drug is eaten, it'll eventually end up in the blood and go everywhere in the body. What is not usually knows is if the drug is too thinly spread out over the body to be of any use, and the consequences of these drugs arriving at an non-target organ.
The fact that drug concentrations could be diluted to the point where it has no effect could be linked to a pharmaceutical's attempt in finding out the dosage of this drug. When testing a drug, scientists must find out how much of the drug could be administered to give a significant effect, while also testing for if increasing the dosage would increase the side effects. A balance of these two must be met in order for a drug dosage to be determined. Some variables would include the patient's body mass, age, blood levels, health of kidney and liver, and other medications the patient is taking. All these factors would play a role in determining how much drug to administer to the patient. If a patient has healthy liver, there's a good chance that a lot of the drug is going to be destroyed before it reaches the bloodstream. If the patient has low amount of blood, not a lot of drug has to be administered because it wont be as diluted. other medications the patient is taking could have chemical reactions that could lead to serious side effects. Another factor to take into account is the chemical weight and properties of the drug. If the drug has high molecular weight, not as much of the drug needs to be administered.
Mainly, pharmaceuticals are more concerned with what would happen if a drug meant for the kidney ends up near the lungs, or something similar to this. This could be called side effects. Often this is solved by observing the carbohydrate chains on the surface of cell membranes to discover what receptors cells of specific organs have. Developing a drug that fits into most of these receptors (a broad and commonly structured drug) would increase the chances of side effects. It is therefore crucial to develop a drug that binds to a cell's receptor as specifically as possible to reduce side effects. Discovering side effects and minimizing these side effects is a big part of getting a drug pass the examination tests and get on the shelves of pharmacies.(5)
Anti-microbial drugs targeted at different levels
[edit | edit source]At cell membrane level
[edit | edit source]Two drugs that target harmful microbes at the cell membrane level include polymyxins and nystatin. Polymyxins interfere with bacterial cell membrane, and therefore bacteria cannot function as osmotic barriers. The functional distinction between polymyxins and nystatin is that nystatin interferes with cell membranes of fungi and yeast whereas polymyxins disrupt bacterial cell membrane. Nystatin will bind to ergosterol, an essential component found in fungi cell membrane, which leads to cell membrane interruption that is representative with appearances of holes in the membrane.
Other groupings of anti-microbial drugs by their targets
[edit | edit source]At DNA level
[edit | edit source]Fluoroquinolones, or Cipro, inhibits synthesis of nucleic acids, such as DNA or RNA, by preventing gyrase, an enzyme needed for DNA replication, from unzipping. As a result, there is no DNA replication. Fluoroquinolones is broad-spectrum and extremely potent and thus can be used on difficult to treat bacteria, such as Bacillus anthracis, which causes anthrax, and Pseudomonas aeruginosa. Another anti-microbial drug that is targeted at the DNA level is rifampin, which is used to treat TB by inhibiting prokaryotic RNA polymerase, which in turn prevents transcription and therefore, no production of mRNA. The bacteria cannot live without these essential proteins.
At protein synthesis level
[edit | edit source]Linezolid, or Zyvox, disrupts the initiation of protein synthesis, and thus is used to treat methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant enterococci (VRE), which are two of the most difficult pathogens to treat. Other antimicrobials acting at the protein synthesis level include streptomycin, gentamicin, tetracyclines, and erythromycin.
References
[edit | edit source]"Antibody Fragmentation." Antibody Fragmentation. N.p., n.d. Web. 28 Oct. 2012. <http://www.piercenet.com/browse.cfm?fldID=4E03B016-5056-8A76-4ECA-982DA6CAAC8A>.
"Creative Biolabs." Creative Biolabs. N.p., n.d. Web. 28 Oct. 2012. <http://www.creative-biolabs.com/phagedisplay1.htm>.
Tortora, Gerard J., Berdell R. Funke and Christine L. Case. Microbiology: An Introduction 10th ed. Boston: Benjamin Cummings :, 2010. Print. | Chapter 20| page 600}
5. Medicine by Design, US Department of Health and Human Services,NIH Publications, Reprinted July 2006
Nanomedicine
[edit | edit source]Nanomedicine is really an interdisciplinary subject. It includes subject such as Nanoengineering, chemical engineering, bioengineering, chemistry, material science, biology, physics, pharmacy, and medicine. In order to ensure a drug is effectively taken place inside the body, the biological system, the chemical reactivity of the drug, and the physical transportation inside the body must be well considered. Nanomedicine is a field that is being heavily studied in recent decades. There are more and more innovative ways to make an already known drug more effective, such as combining a drug with two or three materials to achieve a goal that just drug alone cannot achieve.
Example
[edit | edit source]Example Quantum Dot – Aptamer Conjugates In cancer treatment, it is important to see whether the cancer drug is killing tumor tissue or normal healthy tissue. Therefore, imaging is an important application. Fluorescent and quantum dots are two good indicators. Quantum dots, a type of semiconductor nanocrystal that functions similarly as fluorescent, has been increasingly utilized in biological system and labeling due to their unique optical properties, including board range of absorption with narrow photoluminescence spectra, high quantum yield, low photobleaching, and resistance to chemical degradation. The surface of quantum dot can be modified such that antibodies, aptamers, or peptide bonds can be attached to it. Now this complex exhibits several properties that are useful for cancer therapy.
The specific example given here comprises of quantum dots, aptamers, and doxorubicin conjugate (as the schematic shown below) that targets and kills prostate cancer cells. The quantum dot fluorescent functions as an imaging tool. One end of the RNA aptamers attaches to the surface of quantum dot and the other end attaches to doxorubicin. This aptamer also functions as an active ligand that targets cancer cells. Lastly, doxorubicin is a well-known cancer therapeutic agent that also has slight fluorescent property. The fluorescent of doxorubicin is too weak to be detected; therefore, it is not viable. However, if quantum dots are used, the fluorescent signal inside thin human body tissue can be detected.
When the quantum dot is by itself, it has a fluorescent color of green. That is known as the “on” state. In order to temporarily “turn off” the fluorescent of quantum dot, the fluorescent property of doxorubicin becomes crucial in this conjugate. When two materials both exhibit a fluorescent property are close together, they “quench” each other. In this case, they can temporarily disable the fluorescent property of each other. Doxorubicin exhibits a red fluorescent, while this specific quantum dot exhibits a green fluorescent. When the two form a conjugate, the two colors are able to “cancel” each other. The ratio to cancel doxorubicin to quantum dot is approximately 8 to 1.
Before this conjugate enters the cancer cell, this conjugate is in the “off” state. When the conjugate is being transported to the tumor site in blood streams by both convection and diffusion, the conjugate is able to target tumor cell due to the aptamer ligands on the conjugate. After the conjugate enters through endocytosis into the tumor cell, the conjugate is sent to lysosome. The lysosome is able to digest aptamer, breaking the conjugate. The release of doxorubicin recovers the fluorescent property of the quantum dot back to the “on” state, enabling imaging and detection. The released doxorubicin moves to the nucleus to kill the cancer cell. The figure shown below is a schematic of the conjugate inside the cancer cell.
This innovative conjugate is able to not only target the cancer cell but also provide imaging inside the cancer cell. There are numerous other methods that is yet to be discovered to achieve more effective targeted drug delivery.
References
[edit | edit source]Bagalkot, Vaishali, Liangfang Zhang, Etgar Levy-Nissenbaum, Sangyong Jon, Philip W. Kantoff, Robert Langer, and Omid C. Farokhzad. "Quantum Dot−Aptamer Conjugates for Synchronous Cancer Imaging, Therapy, and Sensing of Drug Delivery Based on Bi-Fluorescence Resonance Energy Transfer."Nano Letters 7.10 (2007): 3065-070. Print. Pure drugs are usually combined with inactive materials to produce a pharmaceutical dosage (dosage form). It is the physical form of the drug that are that are sold to the patient. The common dosage forms that are used widely in pharmacy practice are listed below.
Tablets
[edit | edit source]Tablets are found generally in various size, color, shape, and weight. It is so called the most popular dosage form that is widely used in pharmacy practice because of its advantages: compactness, portability, accuracy, convenience, and lack of taste. Several formulation aids (diluents, excipients, binders, lubricants, disintegrators, coloring, and flavoring agents) are combined with the active ingredient before the mixture is put through mechanical compression in a tableting machine to produce a compact SOLID dosage form of drugs.
However, the drug must be break-down in the stomach so that it will be released under molecular form to be biologically active. This process contributes to one of the properties of tablet form: long onset of action.
Chewable tablets:
[edit | edit source]a form of tablets that can be chewed or dissolved in the mouth before swallowing.
Enteric-coated tablets:
[edit | edit source]this kind of tablets is not designed to be dissolved in the stomach but is meant to be broken down in the intestines of a patient. To make this happen, the compressed tablets are coated with specific substances to prevent them from melting inside the stomach. Because of it specific structure, these drugs are inhibited to be chewed or crushed before swallowing. Antacids cause dissolution in the stomach so it is forbidden to be taken with the enteric- coated tablets.
Sublingual tablets:
[edit | edit source]are designed to be placed under patients’ tongue so that the active ingredient can be absorbed into the blood stream right away and make its first circulates throughout the body before breaking down in the liver.
Buccal Tablets:
[edit | edit source]are left between the gum and cheek of the patients because this drug will dissolves there slowly over a period of time.
Film-coated tablets:
[edit | edit source]are drugs that coated with water-soluble material that protect sensitive drugs from deterioration due to light and air, masks the odor or taste for the patients to obtain easier.
Sustained, timed-release tablets:
[edit | edit source]other names are long-acting/ delayed release /prolonged-action tablets. These tablets are designed specially so that the active ingredient is metabolized slowly at a constant rate for a prolonged period of time, around 8 to 24 hours.
Lozenges (troches/pastilles tablets)
[edit | edit source]has oval or discoid in shape so that it will dissolve slowly and kept in contact with the mouth or throat for a prolonged period of time.
Pellets:
[edit | edit source]are being implanted as a method of birth control or hormones such as testosterone and estradiol. These cylindrically shaped tablets are implanted under the skin so that the drug can be absorbed slowly in a long period of time.
Capsules:
[edit | edit source]are a solid forms of dosage in which the drug is packed inside a soft or hard gelatin shell. The shell sizes range from 000 to a number 5 capsule (largest to smallest respectively), and will dissolves to release the drug after 10 to 30 minutes in the stomach. The advantages of this dosage form are that it will provide various distinguishable shapes and colors, moreover, will eliminate the tastes and odors the drug has.
Effervescent tablet:
[edit | edit source]This kind of tablets will serve as the mask of unpleasant, bitter tasting drug by using the acid-base reaction between sodium bicarbonate with either citric acid or tartaric acid. The reaction will help dissolves matter into the solutions and causes “effervescence” by the liberation of carbon dioxide gas.
References
[edit | edit source]Reifman, Noah. Certification Review For Pharmacy Technicians. 9th ed. the United States of America: AuthorHouse, 2011. 61-88. Print.
Solutions:
[edit | edit source]are homogenous mixtures that include one or more soluble ingredients (can be a liquid, solid, or gas) dissolved in the solvent (water or water-miscible liquid).
The advantages of this dosage form is that it can be absorbed relative quickly in the gastrointestinal tract, easier to swallow so that it is usually prescribed for pediatric or geriatric doses. If drug is an external solutions form it must be carefully label.
Syrups:
[edit | edit source]this type of drug is mostly used for antibiotics, antihistamines, antitussives, and vitamins. It is defined as sweet, viscous, concentration aqueous solutions of sugar.
Elixirs:
[edit | edit source]This is probably the most widely used type of liquid dosage form because of its pleasant taste, relative stability, and ease of preparation characteristics. It is a sweetened hydroalcoholic solution in with the concentration of alcohol may vary (no more than 20%)
Tinctures:
[edit | edit source]Tinctures is another form of alcoholic or hydroalcoholic solutions like elixers but with a higher concentration of alcohol. Vegetable, animal, or chemical materials are main ingredient for this form.
Suspensions:
[edit | edit source]contains finely divided insoluble medicinal products (called internal phase) dispersed in aqueous external phase (usually contains additional flavoring agent).
Patients can take this drugs by mouth or being applied externally like lotions or be injected into the body depend on which type of suspensions was dispensed to them.
Emulsions:
[edit | edit source]Available in both internal and external preparations. Must carefully read the instruction to see if the emulsion is to be used internally or externally. This medicinal products contains water dispersed in oil (stabilized by emulsifying agent) or oil dispersed in water ( both stabilized by emulsifying agent) and microemulsion or transparent emulsion.
References
[edit | edit source]Reifman, Noah. Certification Review For Pharmacy Technicians. 9th ed. the United States of America: AuthorHouse, 2011. 61-88. Print.
Ointments
[edit | edit source]It is the semisolid preparations with various functions in serving as vehicles for topical application of medicinal agents, emollients like lubricating agents, or protectants (to prevent the skin from getting irritants). Ointment is generated for external use only, on skin or mucous membranes.
Pastes
[edit | edit source]They are ointment-like preparations that is stiffer, less greasy, and absorb water better than ointments. Because of these characteristics, pastes are used to treat oozing, weeping lesions by applying externally.
Creams
[edit | edit source]semisolid emulsions with medical agents. Use externally only.
Powders
[edit | edit source]Finely divided, solid medicine used for external application. Some certain powdered drugs are used for asthma patients.
Gels/Jellies
[edit | edit source]Preparation for external used only. It consists of 2 phases: a solid internal phase diffused throughout a viscous liquid phase
Transdermal Patches
[edit | edit source]Will deliver a constant, controlled dose of medication being absorbed into the blood stream by adhering to the patient's skin. The most commonly used are nitroglycerin, scopolamine, nicotine, estrogen, and fentanyl patches. Warning: clean with alcohol. Repeating applications to the same site many time may cause irritation.
References
[edit | edit source]Reifman, Noah. Certification Review For Pharmacy Technicians. 9th ed. the United States of America: AuthorHouse, 2011. 61–88. Print.
Ophthalmic drops:
[edit | edit source]It is the sterile solutions that are instilled into the eyes 2 drops at a time for each eye, the amount of time the drug makes contact with the eye is extremely short. Patients instill multiple medicated eyedrops should wait 5 minutes between instillations.
Opthalmic ointments:
[edit | edit source]Opthalmic ointments are used for instilling into the eye. Theirs characteristic as a sterile emulsions give them the ability to stay contact with the eyes for a longer time period than ophthalmic drops.
Warning: eyesight becomes blur after usage so patients should only use this drug at bedtime only.
Medicated contact lenses:
[edit | edit source]Must be pre-soaked with medication prior to inserting into the eye; its advantages are providing controlled-release and longer contact of the drug with the eye.
Used for certain antibiotics like tetracycline and chloramphenicol.
Ocular Inserts:
[edit | edit source]Drug is placed in the lower eye sac between the sclera and the eyelid. Its advantages is providing a longer contact time between the drug and the eyes, control the amount of medication released into the eyes. Need to be pre-soaked prior to usage.
References
[edit | edit source]Reifman, Noah. Certification Review For Pharmacy Technicians. 9th ed. the United States of America: AuthorHouse, 2011. 61-88. Print.
Suppositories:
[edit | edit source]It is solid dosage forms of drug that will melt or dissolve in the aqueous secretions in where the drug is positioned. It is used for insertion into the rectum, vaginal cavity, or urethral tract where it will perform its therapeutic effects locally or will be absorbed into the bloodstream of the patients.
Different length and weight of suppositories are varies for adult and pediatric.
- Adult rectal suppositories weigh ~2grams and ~2.5 to 3.5 cm long, double the size and weight of pediatric suppositories.
- Vaginal suppositories are globular, ovoid or conical, weigh 3 to 5 grams.
- Urethral suppositories has diameter of 3mm to 5 mm, length varies differently in female urethra and male urethra (60 to 75 in female and 100 to 150 in male).
Inhalers:
[edit | edit source]This type of dosage form of drug is inhaled via the nose or mouth in the form of gas or air, so that the micro small medicinal particles can flow into the alveolar sacs in the lungs.
Need to be shaken well prior to usage.
Otic Product:
[edit | edit source]Around 4 drops of this solutions or suspensions can be instilled into the ear canal.
Warning: only used for the ear and have to shake well if the drug is a suspension.
Enemas:
[edit | edit source]It is the liquid preparations that are used by introduced into the rectum by a bulb syringe at room temperature. This drug’s effects are orientated to be either local or systemic.
Douches:
[edit | edit source]It is the aqueous solutions which are introduced into a cavity of the patients’ body for the main purpose of cleaning the cavity. For example, eye douches are used to removed foreign particles from the eyes; vaginal douches help cleanse and provide medication for the vaginal mucosa while directed to the female vagina.
References:
[edit | edit source]Reifman, Noah. Certification Review For Pharmacy Technicians. 9th ed. the United States of America: AuthorHouse, 2011. 61-88. Print.
Nanodrug Platform
[edit | edit source]Different administration of drugs is to enhance the drug effects on the body, depending on the symptom of the patient. Below is an illustration showing therapeutic window. The goal and the challenge of the researchers is to minimize the drug toxicity to the body and to maximize the drug activity inside the body. In order to keep the drug concentration inside of blood at the therapeutic window, a sustainable drug release is desired. To achieve this, many nanotechnology drug platforms are available. The most common ones are liposomes and hydrogels.
Hydrogel
[edit | edit source]Hydrogel can encapsulate drugs inside its core. When the environment changes, the hydrogel is able to swell, which causes the release of drug.
Types of hydrogel
[edit | edit source]Acidic or basic hydrogel
[edit | edit source]change in pH causes swelling causes release of drug
ionic hydrogel
[edit | edit source]change in ionic strength causes change in concentration of ions inside the gel causes change in swelling causes release of drug
hydrogel containing electron-accepting groups
[edit | edit source]Electron-donating compounds causes formation of charge and transfer complex causes change in swelling causes the release of drug.
Hydrogel containing immobilized enzymes
[edit | edit source]A substrate is present, and enzymatic conversion causes the product the change and swell, which causes the release of drug.
Magnetic particles disperse in alginate microshapes
[edit | edit source]A magnetic field is applied to change the pores in gel to change in swelling and thus release of drug.
Thermoresponsive hydrogel
[edit | edit source]A change in temperature causes change in polymer-polymer and water-polymer interactions that changes the shape of hydrogel and causes the release of drug.
Polyeeletrolyte hydrogel
[edit | edit source]An electric field is applied that causes the membrane the charge and causes the electrophoresis of charged drug and change the shape of hydrogel to release the drug
Ethylene-vinyl alcohol hydrogel
[edit | edit source]Untralsound irradiation is used to increase the temperature to cause the hydrogel to swell to release the drug.
Reference
[edit | edit source]Zhang, Liangfang. "Controlled Drug Delivery Systems." CENG 207 Lecture 11. University of California, San Diego, La Jolla. 10 May 2012. Lecture. Drugs are grouped into different major therapeutic classification that describes the therapeutic use of each drug.
Adrenocortical Steroids:
[edit | edit source]The adrenal cortex of the adrenal glands (located near the kidneys) produces Glucocorticoids which play an important role on reducing inflammatory response. This steroids help reduce the sign of inflammation like redness, swelling, heat, and tenderness at the inflammation site. Therapeutic use: Glucocorticoids have been synthesis to use widely in treatment of the symptoms of drug serum and transfusion reactions, bronchial asthma, allergies, and to use as adjuncts in chemotherapy.
Analgesics
[edit | edit source]1/ Narcotic Analgesics: pain relief drugs work by binding to opiate receptors within the CNS. Warning: drowsiness and dangerous when use with alcohol. The narcotic antagonist must be available to use in case of accidental overdosing.
2/Non-narcortic Analgesics and Antipyretics: -Inhibit the synthesis of prostaglandin. -Inhibit other substances that sensitize pain receptors. -Affect the heat-regulating center of the brain=> fever reduction. Therapeutic use: Are used to reduce pain and fever.
Nonsteroidal Anti-Inflammatory Drugs
[edit | edit source]Inhibit prostaglandin synthesis. Therapeutic use: produces anti-inflammatory, anti-pyretic, and analgesic effects. Cautious: Some variety of this drug are enteric-coated thus not to be crushed, broken, or chewed when taking. This type of drugs is required to be taken with milk or foo to minimize GI discomfort. This drug is prohibited when patients have peptic ulcer disease. With patients who are taking the anticoagulant warfarin, this drug must be used really cautiously.
Antianginals
[edit | edit source]1/ Organic Nitrates: help relax coronary arteries thus increase blood flow in the cardiac muscle.
2/Calcium Channel Blockers: prevent Ca+ to go into cardiac muscle cells and cause the blood vessels of the heart to open.
3/Beta Blockers: -block beta 1 receptor thus suppress the activity of the heart. -cause a slight decrease in blood pressure thus protecting the heart by decrease its workload.
Antiarrhythmics
[edit | edit source]1/Class I: Sodium Channel Blockers 2/Class II: Beta-Adrenergic Blockers 3/Class III: Potassium Channel Blockers 4/Class IV: calcium Channel Blockers 5/Miscellaneous Antiarrhythmic Agents
Anti-Infectives
[edit | edit source]This types of drugs are capable of killing or preventing pathogenic organisms to proliferate in the body. Bacteicidal agents will stop acting on growing organisms if encounter with bacteriostatic agents.
Note:completing full course of therapy is required.
1/ Amebicides
- chloroquine:aralen
- eflornithine:Ornidyl
- iodoquinol:Yodoxin
- flurazolidone:Furoxone
- hydroxychlooquine:Plaquenil
- mefloquine:Lariam
- metronidazole:Flagyl
- pentamidine: NebuPent, Pentam
2/ Aminoglycosides
This group acts on the protein synthesis pathway in the pathogenic organism thus kills the bacterial, used mainly for tough gram-negative organisms (Pseudomonas, E.coli, Proteus, Klebsiella, and Enterobacter). Dosage and frequency of this drug Kidney function must be monitored when assigning this type of drug because its dosage and frequency are depended strongly on the kidney function.
Warning: : These drugs can damage the kidney and ear.
- amikacin: Amikin
- entamicin: Garamycin
- kanamycin:Kantrex
- neomycin:various
- Netilmicin: Netromycin
- streptomycin
- tobramycin
3/ Antifungals -Systemic and Antifungals-topical Antifungals-systemic interferes with the cell wall synthesis pathway or the protein synthesis pathway in fungal. Liver functioning must be strictly under control because any of these agents can damage the liver.
- Antiungals-Systemic
- amphotericin B: AmBisome
- fluconazole: Diflucan
- flucytosine: Ancobon
- griseofulvin: Fulvicin, Grifulvin
- itraconazole:Sporanox
- ketoconazole:Nizoral
- metronidazole:Flagyl
- mystatin:Mycostatin, Nilstat
- terbinaine:Lamisil
- Antifungals-Topical
- bacitracin: various
- cicloprox: Loprox,Penlac
- clotrimazole: Lomitrin, Mycelex
- enconazole: Spectazole
- gential violet
- ketoconazole:Nizoral
- miconazole:Micatin, Desenex, Zeasorb-AF
- nystatin:Mycostatin, Nilstat
- oxiconazole: Oxistat
- terconazole:Terazol
- tioconazole:Vagistat
- tonaftate: Tinactin
- terbinafine: Lamisil
- metronidazole: varios
- 4/ Cephalosporins
This drug usually are used to destroy bacterial because it affects the synthesis of cell wall in pathogenic organisms. Need to be carefully prescribed for patients who are sensitive to penicillins or patients with kidney impairment.
- cefaclor: Ceclor
- cefadroxil: Duricef, Ultracef
- cefazolin: Ancef, Kefzol
- cefoxitin: Mefozin
- cefdinir: Omnicef
- cefditoran: Spectracef
- cefepime: Maxipime
- ceftazidime: Fortaz
- ceftizoxime: Cefizox
- cefixime: Suprax
- cefoperazone: Cefobid
- cefotaxime: Claforan
- cefotetan: Cefotan
- cefpodoxime:Vantin
- cefprozil: Cefzil
- ceftriaxone: Rocephin
- cefuroxime: Ceftin
- cephalexin: Feflex
- cephradine: Anspor, Velosef
- 5/ Erythromycins
Erythromycins' functions, either killing the bacterial or preventing its growth, is depending on the dosage use. It can block the protein synthesis in pathogenic organisms by binding to the 50S subunit of bacterial ribosomes. This drug can harm the stomach and are used for patients who are allergic to penicillin.
- azithromycin:Zithromax
- clarithromycin: Biazin
- erythromycin: base, Staticin, Emgel, Illotycin, estolate, ethyl succ, stearate. lactobionate
- Eythromycin+sulfasoxizole: Pediazole
- telithromycon: Ketek
- 6/ Penicillin Derivatives:
Penicillin Derivatives are capable to kill bacterial by stopping bacterial cell wall synthesis in pathogenic organisms during replication. Side Effects:: rash, hives, anaphylactic shock. Storing: must be shaken well and refrigerated.
- amoxicillin+clavulanate: Augmentin
- amoxicillin: various, Amoxil
- ampicillin: various
- bacampicillin: Geocillin
- cloxicillin: Tegopen
- dicloxicillin: Dynapen, Dycil
- mezlocillin: Mezlin
- nafcillin: Unipen, Nafcil
- oxacillin: Bactocil, Prostaphlin
- penicillinG: Bicillin-LA
- penicillin B, procaine: Wycillin
- penicillin VK: various
- piperacillin: Pipracil
- ticarcillin: Ticar
- 7/ Tetracyclines
It is a broad-spectrum antibiotics. It binds to the 30s ribosomal subunit to stop protein synthesis thus stops the growth of pathogenic organisms. Cautious: causes permanent tooth discoloration, photosensitivity, exaggerated sunburns. Must not be taken during last half of pregnancy or giving to children under 8. Outdated tetracycline can damage kidney.
- doxycycline: Vibramycin
- minocycline: Minocin
- tetracycline: various
- 8/Antivirals
- 9/Sulfonamides
- 10/Antimalarials
- 11/Antituberculars
- 12/Anticholinergic Agents:
Neuromuscular Blocking Agents
[edit | edit source]Anticoagulants
[edit | edit source]Anticonvulsants
[edit | edit source]Antidiabetic Agents
[edit | edit source]Antidiarrheals
[edit | edit source]Antiemetic Agents
[edit | edit source]Antineoplastic Agents
[edit | edit source]Antihypertensive Agents
[edit | edit source]1/Diuretics
2/Beta blocking Agents
3/Ace Inhibitors
4/Calcium Channel Blockers
5/Other Agents:
Antiparkinsonian Agents
[edit | edit source]Thyroid Hormones
[edit | edit source](Thyroxine). This category involves in several important process in the body includes: protein synthesis, lipid and carbohydrate metabolism, energy storage, body temperature, etc.
- levothyroxine: Synthroid, Levoxyl
- liothyronine: Cytomel
- thyroid dessicated: Armour Thyroid
Antiulcer Drugs
[edit | edit source]Bronchodilators
[edit | edit source]This type of drugs are commonly used to treat asthma. It affects the respiratory system through the bronchial airways or the area which controls respiration in the central nervous system. Helps relax bronchial smooth muscle.
Common drugs:
- albuterol: Proventil, Ventolin
- aminophylline
- epinphrine: Adrenalin
- ipratropium: Atrovent, Isuprel
- levabuterol: Xopenex
- metaproterenol: Alupent
- pirbuterol: Maxair
- salmeterol: Serevent
- Tertbutaline: Brethine
- theophylline: various
Drugs used in shock
[edit | edit source]This type of drugs force the heart to beat faster with stronger contraction thus lead to vasoconstriction in blood vessels, increase the volume of blood in the circulatory system and increase blood pressure.
| dobutamine | Dobutrex | injection only, mixing with sodium bicarbonate is strictly forbidden |
| dopamine | Intropin | injection only,mixing with other drugs is strictly forbidden |
Laxatives
[edit | edit source]Laxatives are used to assist the defecation in patients with constipation.
1/Irritants: used to cause increasing in muscular activity
2/Saline Cathartics: are used to produces osmotic effects in the small entestine.
3/Bulk-forming: are used to increase bulk and moisten contents of stool.
4/Fecal softeners: reduce surface tension of the liquid contents of the bowel
5/Lubricants: are used to prevent water from being absorbed out of the bowel.
Psychotherapeutic Agents
[edit | edit source]- 1/Antianxiety Agents:
Benzodiazepines are well-known drugs to treat anxiety but with mild effects. It is used to reduce anxiety but cannot block panic attact Depress the Central Nervous Systems at the limbic and subcortical levels of the brain. Serotonin-specific reuptake inhibitors (SSIRs) like Prozac and Paxil are widely used also. Therapeutic Use: reduce anxiety, cause sedation, hypnotics.
Warning: May cause drowsiness and cannot be used with alcoholic substances.
- 2/Antidepressants:
This type of drugs works by changing the concentrations of chemical transmitters in the brain. The effects usually take 2 weeks or longer to show. The basic types of antidepressant medications:
A/Tricyclic antidepressants: Imipramine (Tofranil) is very well-known and is available in tablets, capsules, and injection types. It will alleviate depression but do not completely cure it. It mostly affect norepinephrine system by preventing massive amount of it to be transmitted in the body and restoring appropriate balance. However, other neurotransmitter systems like serotonin are slso affected by Tofranil. This drugs often needs 2 to 8 weeks to show their affects.
Side effects: patients may have blurred vision, dry mouth, constipation, difficulty urinating, drowsiness, wight gain (at least 13lbs on average), sexual dyysfunction. Cautious: Tricyclics are lethal if using overdose.
B/Monoamine oxidase inhibitors: block enzyme monoamine oxidase inhibitors that breaks down norepinephrine and serotonin. The MAO inhibitors seem to have equivalent effects with tricyclics but fewer side effects. Warning: lethal side effects- patients are forbidden to have food with tyramine like cheese, redwine, beer or some over-the-counter drugs may interact with MAO inhibitor and results to death.
C/Serotonin-specific reuptake inhibitors (SSRIs): affect the presynaptic reuptake system of serotonin. Fluoxetin (Prozac) are a well-known drug in this category. Its effectiveness is mostly the same with other antidepressants but with small decrease in suicidal rate among adolescents. Warning: side effects: physical agitation, sexual dysfunction, low sexual desire, insomnia, gastrointestinal upset.
- 3/Antipsychotics
(neuroleptics, major tranquilizers): these medications help people think more clearly and reduce hallucinations and delusions by interfere with the dopamine neurotransmitter system, serotonergic, glutamate system. Therapeutic Use: treat psychosis and schizophrenia.
Side effects: unwanted physical symptoms (groginess, blurred vision, dryness of the mouth), Parkinsonian symptoms, Akinesia (expressionless face, slow motor activity, monotonous speech), Tardive dyskinesia (involuntary movements of the tongue, face, mouth, jaw, or protrusions of the tongue, puffing of the cheeks, puckering of the mouth, chewing movements.
Vitamins
[edit | edit source]| Vitamin | Name | Therapeutic Use |
|---|---|---|
| A | Retinol | To give healthy eyes, helps bones and teeth growth, and prevent infections |
| B1 | Thiamine | is used to break down carbohydrates for energy |
| B2 | riboflavin | breaking down fat for energy, tissue respiation |
| B3 | niacin | is used to break down fats, carbohydrates, protein. Involves in the process of making hormones and fat |
| B6 | Pyriodixine | is used to break down polypeptide chains to make energy |
| B9 | folic acid | Function in making new cells like red blood cells |
| B12 | Cyanocobalamin | Function in making new cells like red blood cells, good for nervous cystem |
| C | Ascorbic Acid | aid in healing wounded tissues, preventing infection, iron absorption |
| D2 | ergocalciferol | Helps body absorb calcium and maintain Ca+ level in blood as well as aids in bone formation |
| D3 | Cholecalciferol | Helps body absorb calcium and maintain Ca+ level in blood as well as aids in bone formation |
| E | Example | Protects Vitamin A and polyunsaturated fats from oxidation (antioxidant) |
| K1 | Example | Blood Clotting |
| K3 | menadione | Blood Clotting |
References
[edit | edit source]Reifman, Noah. Certification Review For Pharmacy Technicians. 9th ed. the United States of America: AuthorHouse, 2011. 61-88. Print.
"Module 6: Health Assessment." Arizona Department of Health Service. Arizona Department of Health Service. Web. 20 Nov 2012. <http://www.azdhs.gov/azwic/documents/local_agencies/trainingmanual_pdf/module_6.pdf>.
Durand, V. Mark, and David H. Barlow. Essentials Of Abnormal Psychology. 5th. 12. Belmont: Wadsworth Pub Co, 2009. Print. Drug development today is constantly pressured from multiple different aspects due to the all-time low of drug approval on the market. Drug developers must acknowledge this trend even before the process begins. Unfortunately for some companies, resources are already limited and do not have the luxury of researching and developing as easily as others.
Challenges
[edit | edit source]Regulatory requirements and commitments are continually increasing over time and have effected the trial size and length. This has led to an overall increase in total cost of drug development. The FDA now requires more sample trials in order to be approved, this is to ensure safety and efficacy.
Due to the increase in clinical trials, it has become necessary for trials to occur in foreign countries. Unfortunately, each country has its own set of regulations for drugs, further increasing the difficulty of trials.
Electronic submission to regulatory authorities has become mandatory in some countries such as the US. The transition to the Electronic Common Technical Document (eCTD), will soon become the mandatory and preferred way of submission. While the eCTD does come with many benefits, developing countries must quickly adapt to the system, further slowing the development process of drugs.
Drug administrations and absorption levels linked to the effect and side effect of the drug are a crucial part of the process.
Challenges of Drug Development
[edit | edit source]Many compounds have significant effects when taken into the body, but only every small fraction of them have the potential to become useful drugs. A foreign compound, synthesized in a wet lab or extracted from nature, must be able to adapt in the cells of an organism to function effectively without causing any serious harm. Drug candidates must be potent modulators of their targets as wee have suitable properties to reach their targets
Drug Candidates must be Potent
[edit | edit source]For drugs to be effective, it needs to bind a sufficient number of its target proteins when taken at a reasonable dose. One factor in determing drug effectiveness is the strength of the interaction between the drug and its target. A molecule that binds to some target molecule is often referred to as a ligand. Ligand molecules occupy progressively more target binding sites as ligand concentrations increases until essentially all of the available sites are occupied. This tendency of a ligand to bind to its target is measured by the dissociation constant, Kd
Kd = [R][L]/[RL]
where [R] is the concentration of the free receptor, [L] is the concentration of the free ligand, and [RL] is the concentration of the receptor-ligand complex. The dissociation constant value is a measure of the strength of the interaction between the drug candidate and the target; the lower the value, the stronger the interaction. The concentration of free ligand at which one-half of the binding sites are occupied equals the dissociation constant, as long as the concentration of binding sites is substantially less than the dissociation constant.
However, sometimes in the cases of biological assays where drug candidates are utilized on living cells or tissues, an alternative method is used to determine the potency of a drug. In these cases, the EC50 concentration is measured. This is the concentration of the drug candidate required to elicit 50% of the maximal biological response. For drug candidates that are inhibitors (ex. sodium channel blockers), the term IC50 is used to describe the concentration of the inhibitor required to reduce a response to 50% of its value in the absence of inhibitor.
IC50 = Ki(1 + [S]/KM)

(Ki is known as the inhibition constant; KM is the Michaelis constant for the substrate S. The higher the concentration of the natural substrate, the higher the concentration of drug needed to inhibit the enzyme to a given extent.
IC50 and EC50 values are important measures of potency of a drug candidate in evaluating the activity of the desired biological target. Oftentimes a drug target is a member of a large family of proteins that similar in nature, which can be extremely challenging when developing a target drug.
Drug Candidates must have suitable properties to reach targets
[edit | edit source]In addition to the ability of molecules to act on specific target molecules, an effective drug must also have other characteristics. For example, it must be easily administered and reach its target at sufficient concentration to be effective, a drug molecule encounters a variety of obstacles on its way to its target. The following properties are the four basic stages of a medicine's life in the body:
1. Absorption
2. Distribution
3. Metabolism
4. Excretion
Absorption
[edit | edit source]Drugs can be taken orally as a small tablet and must be able to survive the acidic conditions in the gut and then be absorbed through the intestinal epithelium. A few of the most common ways to administer drugs are oral (swallowing an aspirin tablet), intramuscular (getting a flu shot in an arm muscle), subcutaneous (an injection of insulin under the skin), intravenous (receiving chemotherapy through a vein), or transdermal (wearing a skin patch). Drugs face a great deal of hurdles during absorption, because of the liver and the great deal of variability in drug administration in the human body. There are a set of rules that tells us when poor absorption is likely:
1. molecular weight is >500 g/mol
2. number of hydrogen bond donors is greater than 5
3. number of hydrogen bond acceptors is greater than 10
4. partition coefficient is greater than 5 (way to measure the tendency of a molecule to dissolve cell membranes)
The challenge with the liver is that the liver will filter most of the drugs away before it will reach the bloodstream. Metabolic enzymes would break down the drug rendering it useless. This would lead to most of the drugs not reaching the target organs, or not having any effect. In order to bypass this, the many different absorption methods are introduced.(2)
Distribution
[edit | edit source]Once a drug is absorbed, the next stage is the distribution of the drug. Most often, the bloodstream is the primary means of transportation for drugs. Once the drug reaches the bloodstream, it is distributed to different fluids and tissues. This is the step that leads to a wide array of side effects in organs throughout the body. However it is important to note that some organs have stronger defense systems than others. For example, the central nervous system has a strong blood brain barrier that protects the brain from dangerous poisons or viruses.
Another challenge during the distribution of the drug throughout the body is its attachment to other molecules besides the receptors of the target organ. Drugs could react with compounds present in the blood of the patient and could end up rendering the drug useless, or could produce unwanted side effects. Also, because the compound is spread all over the body, non-target organs with specialized cells and unique receptors could end up reacting and attaching to the drug. If the drug attaches to a receptor and activates, or activates a function of the cell, it could lead to unwanted side effects that could be potentially harmful or fatal.(2)
Metabolism
[edit | edit source]After a drug has been distributed throughout the body and has done its job, the medicine is broken down or metabolized. The metabolism generally occurs in the liver. The liver is the site of continuous yet controlled activity. Everything that enters the bloodstream is carried straight to the intestine. In the intestine, molecules and substances are chemically and physically metabolized.
Excretion
[edit | edit source]Once a drug is absorbed and carries out its specific biological tasks, it must be converted into a substance that can be physically excreted. The liver detoxifies the drug's components using chemical metabolites, which then exits via the urine or feces.
References
[edit | edit source]1. Berg J, Tymoczko J, Lubert S: Biochemistry, 7th Edition
2. Medicine by Design, US Department of Health and Human Services,NIH Publications, Reprinted July 2006
References
[edit | edit source]1. Berg J, Tymoczko J, Lubert S: Biochemistry, 7th Edition 2. Singh, Harjit. Drug Development Challenges. Pharma. <http://www.pharmafocusasia.com/strategy/drug_development_challenges.htm> 3. Medicine by Design National Institute of Health
History of Osteoporosis
[edit | edit source]
Osteoporosis is a bone disease that can lead to fractures and deteriorating. Bisphosphonate, also known as Diphosphonate are drugs known for treating and reducing the risk of contracting the illness. It is one of the key elements used by chemist as a pathway to finding the drugs for Osteoporosis. Bisphosphonate has the ability to bind with a naturally occurring mineral called Hydroxylapatite, that can make up 70% of bone. Alendronate (Fosamax), ibandronate (Boniva), risedronate (Actonel), and zoledronic acid (Reclast) are three examples of drugs used to treat osteoperosis.

Treating Osteoporosis
[edit | edit source]The bisphosphonates are synthetic compounds that have been used as water softeners in canals and irrigation systems since the mid-nineteenth century. Their efficiency in treating osteoporosis can be ascribed to the alignment of biological and physicochemical properties of the compound. Bisphosphonates have impressive layers of cellular selectivity, which compose long-acting drugs that inhibit the resorption of bone by osteoclasts safely. After binding with Hydroxylapatite on the bone surface, approximately 50% of bisphosphonates will stick there and the half-life of bone-bound alendronate is estimated to be around 10 years. The dose of drugs that do not stick around will be excreted rapidly, and bisphosphonates' low cell permeability will minimize their exposure to other tissues, which in terms lower the probability of having side effects. This property of essential targeting to bone is very useful for the subsequent cellular activities in treating osteoporosis. During bone resorption, osteeoclasts are attached to bisphosphonates and hence will not be able to resorb bones.


History of Influenza Viruses
[edit | edit source]
Influenza viruses, also referred to as the flu is a disease that is more severe that a normal cold and is highly contagious. The virus can be transferred through air, cough, sneeze, as well as direct contact. It affects all age group, but young children have a tendency to catch the disease more than adults. It is a seasonal disease that spread around the world and kill thousands and thousands of people each year. In the case of the influenza target neuraminidinase, crystallographic analyses of N2 and type B neuraminidases have been essential to understanding how activesite plasticity affects the emergence of distinct resistant variants to the current commercial drugs, zanamivir (Relenza®) and oseltamivir (Tamiflu®). Zanamivir, the first drug on the market (10), shows poor oral bioavailability (2%), likely reflecting its zwitterionic character at physiological pH (11). Oral inhalation is required. In the case of oseltamivir (12), the carboxylate is an ethyl ester prodrug, thereby rendering the molecule cationic, which enhances oral absorption (75%). The required carboxylate is generated in the host by enzymatic hydrolysis (13). In addition, the guanidinium group of zanamivir is simplified to an amino group, and the glycerol moiety is redesigned in oseltamivir to a hydrophobic ether, which results in a different enzyme conformation upon binding. This modification may be problematic in the emergence of resistance to oseltamivir.
Zanamivir (Relenza)
[edit | edit source]Osteoporosis: The company Biota discovered Zanamivir, a drug made help bone conditions such as Osteoporosis and similar diseases. Of its kind, it was the first commercial drug on the market to serve as patients with bone diseases, but unfortunately, it was proven to have low effectiveness due to its poor bioavailability of 2%. The human body was unable to fully absorb the dosage because of Zanamivir's zwitterionic character.

Biota should be looking into developing an ester of zanamivir to increase its oral bioavailability, but it did not do so for good reasons. During Biota's discovery of this drug, the Gilead group prepared free carboxylate and guanidinium analogs of oseltamivir, which is a prodrug. To mimick Zanamivir, the Gilead group replaced the amino group of Zanamivir with guanidinium group ultimately resulted in a loss of oral bioavailability. Therefore, the company came to the conclusion that the prodrug form of zanamivir was not more bioavailable that its original form probably because of some mitigating effect of the guanidinium group. Apparently, when it comes to producing medicines, aligning one parameter with the grand picture may negatively impact another parameter. Zanamivir was licensed to GlaxoSmithKline for final development; despite Biota or GSK's effort to make the drug more orally bioavailable, this drug was outsold by the orally active oseltamivir.

Influenza: In an experiment on animals and man, inhaling doses of Zanamivir is an effective treatment for influenza. Although Zanamivir is and ester prodrug, if taken orally, the medicine will not successfully distribute through out the body and therefore, will not fight against the infectious disease. Due to this matter, many attempts have been made to better the pharmacological properties of Zanamivir by changing the structure of the compound.
Oseltamivir (Tamiflu)
[edit | edit source]
Osteoporosis: Following the discovery of Zanamivir, to improve bone diseases, Gilead group generated a drug called Oseltamivir. This new medicine serves the same purpose as Zanamivir but it was highly effective and it outsold Zanamivir by 3 to 1. This drug was more successful than Zanamivir because its carboxylate is an ethyl ester prodrug, in which enhancing the molecule to be cationic, and therefore, an increase in oral absorption (75%).

Influenza: By looking to improve the oral intake of Zanamivir, researchers redesigned the structural compound of pyran by removing carboxylic acid, and changing it into a carbocyclic scaffold, in which discovered an active antiviral product (Oseltamivir) that targets influenza viruses and can be taken orally. Overall, Statistics shows that in Japan, Oseltamivir was more commonly used, roughly 90 times more than Zanamivir.
References
[edit | edit source]- ↑ Annu. Rev. Biochem. 2009. 80:55-5 The Annual Review of Biochemistry is online at biochem.annualreviews.org
- ↑ Annu. Rev. Biochem. 2009. 78:55-63 The Annual Review of Biochemistry is online at biochem.annualreviews.org
- ↑ Annu. Rev. Biochem. 2009. 78:55-63 The Annual Review of Biochemistry is online at biochem.annualreviews.org
Kozarich, John W. "The Biochemistry of Diseases: Desperately Seeking Syzygy." Annu. Rev. Biochem. (2009): 55-63. Web.
Colman, Peter M. "New Drug Antiviral and Resistance." Annu. Rev. Biochem. (2009): 95-118. Web.
Image source: wikimedia-commons
Clinical Trials
[edit | edit source]
In the United States, the FDA (Food and Drug Administration) requires that potential drugs be demonstrated to be effective and safe before they may be used in human beings on a large scale. This requirement is particularly true for drug candidates that are to be taken by people who are relatively healthy. The trials test the effectiveness and potential side effects of a candidate drug before it is approved by the FDA for general use.
Phases
[edit | edit source]Clincial trials proceed in at least three phases.
Phase 1: In phase 1, a small number (10-100) of healthy volunteers take the drug for an initial study of safety. These volunteers are given a range of doses and are monitored for signs of toxicity.
Phase 2: In phase 2, the efficacy of the drug candidate is tested in a small number of persons who might potentially benefit from the drug. Further data regarding the drug's safety are obtained. Such trials are often controlled and double-blinded.
Phase 3: In phase 3, similar studies are performed on a larger population (thousands). This phase firmly establishes the efficacy of the drug candidate and to detect side effects that may eventually develop in a small percentage of the subjects who receive treatment.
Only after the conclusion of all three phases can a drug be approved for clinical use by the public.
Reasons
[edit | edit source]Clinical studies are designed to add medical knowledge related to treatment, diagnosis, and prevention of diseases or conditions. Some common reasons for conducting clinical studies include:
- Evaluating one or more interventions for treating a disease, syndrome, or condition
- Finding ways to prevent the initial development/recurrence of a disease or condition
- Examining methods for identifying a condition or risk factors for that condition
- Exploring ways to improve the comfort and quality of life of people with a chronic illness through supportive care
Methods
[edit | edit source]Clinical Trials are usually led by a principal investigator, who is often a medical doctor. Clinical studies have a research team that may include doctors, nurses, social workers, and other health care professionals. Clinical studies can take place in many locations, including hospitals, universities, doctors' offices, and community clinics. The location depends on who is conducting the study.
Controlled studies involve a group of individuals given a placebo and a second group that is given the actual drug. These studies also tend to be double-blinded, so that neither the subjects nor the researchers know which subjects are in the treatment group and which are in the control group. This approach prevents bias in the course of the trial. After the completion of the trial, the assignments of the subjects are unsealed and the results for the two groups are compared. A variety of doses are often investigated in phase 2 trials to determine which doses appear to be free of serious side effects and which does appear to be effective.
Cost/Time
[edit | edit source]Clinical trials tend to be extremely expensive. Costs can run from tens of millions to hundreds of millions of dollars. Extensive records and documentations are filed and subsequently compiled for submission to the FDA. The full cost of developing a drug is currently estimated to be from $400 - $800 million dollars.
According to one study, it takes an estimated 15 years for a drug to go from lab to patient.
References
[edit | edit source]1. Berg J, Tymoczko J, Lubert S: Biochemistry, 7th Edition
2. Chron's & Colitis Foundation of America: Clinical Trials 101
3. Clinical Trials (Government Website): Learn about Clinical Studies
Background
[edit | edit source]The Controlled Substances Act (CSA) consists of numerous laws regulating how certain substances are manufactured and distributed within the United States. Also known as the Comprehensive Drug Abused Prevention and Control Act of 1970, the CSA is administered under the United States Drug Enforcement Administration (DEA), which investigates the illegal production of controlled substances on an interstate and international level. [1]
Five Schedules
[edit | edit source]Under Section 812 of the CSA, the controlled substances are divided into five schedules. Each controlled substances is placed under Schedule I, II, III, IV, or V based on its potential for abuse, accepted medical treatment and safety within the United States, and its likelihood of causing physical and psychological dependence.
Schedule I:
[edit | edit source]- The drug or other substance has a high potential for abuse.
- The drug or other substance has no currently accepted medical use in treatment in the United States.
- There is a lack of accepted safety for use of the drug or other substance under medical supervision.[2]
Prescriptions for Schedule I drugs are not accepted anywhere in the United States.
Some examples of Schedule I drugs include heroin, cannabis, Ecstasy, and LSD. These drugs contain hallucinogenic substances, opiates such as acetylmethadol, or any opiate derivatives including its isomers, esters, salts, or other chemical combination. [3]
Schedule II:
[edit | edit source]- The drug or other substance has a high potential for abuse.
- The drug or other substance has a currently accepted medical use in treatment in the United States or a currently accepted medical use with severe restrictions.
- Abuse of the drug or other substances may lead to severe psychological or physical dependence.[4]
Prescriptions for Schedule II drugs can only be written by hand directly from a licensed practitioner. In California, schedule II prescriptions must be on a new, tamper-resistant prescription form, and are not allowed to have refills on them. Typically these prescriptions are written for a thirty days supply. [5]
Some examples of Schedule II narcotics include oxycodone, amphetamine salts, and methylphenidate. Better known brand name drugs include OxyContin, Percocet, Adderall, and Ritalin. These drugs contain any derivative of opium, opiate, or methamphetamine.
Schedule III:
[edit | edit source]- The drug or other substance has a potential for abuse less than the drugs or other substances in schedules I and II.
- The drug or other substance has a currently accepted medical use in treatment in the United States.
- Abuse of the drug or other substance may lead to moderate or low physical dependence or high psychological dependence.[6]
Some examples of Schedule III drugs are Vicodin, Tylenol with Codeine, and Suboxone. These drugs contain some compound or mixture of depressants, methylphenidate, and narcotic drug. Although there are some Schedule II drugs mixed into Schedule III narcotics, the DEA allows up to 1.8 milligrams of codeine per 100 milliliters. Some other regulations include a maximum of 500 milligrams of opium per 100 milliliters, and a maximum of 50 milligrams of morphine per 100 milliliters compound.[7]
Schedule IV:
[edit | edit source]- The drug or other substance has a low potential for abuse relative to the drugs or other substances in schedule III.
- The drug or other substance has a currently accepted medical use in treatment in the United States.
- Abuse of the drug or other substance may lead to limited physical dependence or psychological dependence relative to the drugs or other substances in schedule III. [8]
Some examples of Schedule IV drugs include alprazolam, diazepam, lorazepam, and carisoprodol. Better known brand names include Xanax, Valium, Ativan, and Soma. [9] These drugs contain barital, methohexital, phenobarbital, or meprobamate, which are usually used to treat anxiety and insomnia.
Schedule V:
[edit | edit source]- The drug or other substance has a low potential for abuse relative to the drugs or other substances in schedule IV.
- The drug or other substance has a currently accepted medical use in treatment in the United States.
- Abuse of the drug or other substance may lead to limited physical dependence or psychological dependence relative to the drugs or other substances in schedule IV. [10]
Some examples of Schedule V narcotics include Robitussin and other cough suppressants. Many of these drugs contain some combination of codeine, dihydrocodein, ethylmorphine, diphenoxylate, or opium. However, there is a limited quantity of the active narcotic drug that is allowed within the compound. For example, only 100 milligrams of opium per 100 milliliters can a mixture contain.[11]
Background
[edit | edit source]A natural product is a chemical substance produced by a living organism. It is found in nature and has biological effects that can be studied in order to support drug discovery and drug design. A substance is considered a natural product even if it can be prepared by total synthesis. [12]
Sources of natural products are plants, bacteria, marine environments, and animal venoms.
Coprophiles
[edit | edit source]Coprophiles are organisms that specifically thrive in animal dung. These territorial species spit out toxic chemicals to its neighboring fungi. Scientists and researchers allocate this information and specifically search for chemicals that are rather poisonous to some fungi and can be potentially dangerous to people that are infected.
Cyanobacteria are plant-like organisms that live in both wet and damp environmental conditions. These species have been proven to become sources of cancer and bacterial cell killers. For example, the compound cryptophycin-8 can tear apart the scaffolding in a spectrum of tumors. Another molecule called majusculamide C focuses in on fungi, potentially allowing it to be used to treat fungi-related diseases in humans.
Filter feeders are organisms that stick to rocks and coral. These species compete with others for food and other natural resources. Scientists and researchers have discovered that some of these potent chemicals can be used in the long run to treat cancer and other fatal diseases.
Ultimately, these natural products have been scientifically utilized to create both a wanted and an unwanted effect. In short, many of these chemicals show a promising future in the field of medicine.[13]
Cyanobacteria
[edit | edit source]Cyanobacteria, or blue-green algae, are plant-like organisms that live in both wet and damp environmental conditions. Cyanobacteria are photosynthetic organisms that can create energy from sunlight. These microorganisms can be found in colonies called algal bloom, and have dated back to the oldest fossils on earth. They make up one of the largest groups of bacteria in the world, and are linked to many human and animal diseases. Blooms will form when there is extravagant growth of the bacteria within a few days time. These algal blooms will cause clear water to become cloudy looking. Cyanobacterial blooms are dangerous because they can use up all of the oxygen present in the water they grow in, which will kill the other plants and animals living there. Some cyanobacteria even produce some of the most powerful natural poisons known, with no antidotes.
However, these species have been proven to become sources of cancer and bacterial cell killers. For example, the compound cryptophycin-8 can tear apart the scaffolding in a spectrum of tumors. Another molecule called majusculamide C focuses in on fungi, potentially allowing it to be used to treat fungi-related diseases in humans.
Filter Feeders
[edit | edit source]Filter feeders are organisms that stick to rocks and coral. These species compete with others for food and other natural resources. Filter feeders use an aquatic feeding method for getting their food. Scientists and researchers have discovered that some of the potent chemicals found in filter feeders can be used in the long run to treat cancer and other fatal diseases.
Filter feeders include the sea sponge, which is the simplest of multi-cellular animals. Sea sponges produce a great array of toxins, which they either release into the water or show on their surfaces. These toxins will ward off any predators that would feed on the sponges. Symptoms of being in contact with these toxins include: redness at the site of contact, pain, tingling, itching, swelling, bumps, nausea, or even fainting.
Despite their toxicity, sea sponges have proved to show great promise in the area of fighting infectious diseases and cancers. Scientists have been able to extract anti-viral, anti-cancer, and anti-neoplastic compounds from sea sponges to create many drugs. For example, in the 1950's chemists used sponge compounds to create a drug for treating Herpes called Acyclovir (Zovirax), and a drug to treat non-Hodgkin's lymphoma called Cytarabine (Cytosar). [14]
Ultimately, all of these natural products have been scientifically utilized to create both a wanted and an unwanted effect. In short, many of these chemicals show a promising future in the field of medicine.
Tunicates
[edit | edit source]
Tunicates lie under the category of filter feeders that are greatly contributing to the medicinal research of cancer drugs. Also known as sea squirts, tunicates look merely like little colorful blobs but offer much to the pharmaceutical world than one would expect. For example, in the West Indie coral reefs, a tunicate named Ecteinascidia turbinata contributes to the fight of cancer. This marine animal, as discovered by Ken Rinehart from the University of Illinois, harbors the natural substance ecteinascidin used to make the cancer drug named Yondelis™. Although this medicine is still in its preliminary stage and further research needs to be done, lab tests have already confirmed that it can kill cancer cells and is suitable for human intake.
Recently, Elias J. Corey from Harvard University has discovered the way to synthesize ecteinascidin in the labs. This is important because to produce one gram of medicine, more than one ton of tunicates need to be harvested; a clearly ineffective process. With the ability to synthesize the natural product of interest, scientists are catalyzing the advancement of utilizing natural products to produce effective medications to cure diseases. [15]
Taxol
[edit | edit source]
HEALTH AND HUMAN SERVICES, </ref>]] Taxol is a drug used in the treatment of cancer. It is derived from the bark and needles of a tree called the Pacific Yew (Taxus brevifolia), which dies when taxol is extracted. On the downside, although it is one of the most useful drugs in fighting cancer, a small sample of taxol is produced from a large quantity of Yew barks (about 1200 kg of bark produces 10 g of pure taxol). Because of the negative impact the production of this drug has on the environment, the creation of taxol raises concerns over the ecological effects on the yew population.
Taxol works by preventing cancer cells from replicating. It does so by binding to the microtubules during cell division by preventing them from breaking down. Because in microtubules are normally disassembled after the cell divides, the presence of microtubules prevents the cell from dividing into daughter cells. [16]
Epibatidine
[edit | edit source]
Epibatidine is a molecule found on the skin of a species of Ecuadorian frog (Epipedobates tricolor) that has an analgesic effect, meaning that it is an effective painkiller. Although studies have proven Epibatidine to be a more effective painkiller than morphine, Epibatidine is poisonous and small doses of it can kill a large organism. Because of this, Epibatidine is unlikely to be available for the medicine market.
Epibatidine works by binding the nicotinic acetylcholine receptor (nAChR) binding sites. When nicotinic acetylcholine receptors are bound by neurotransmitters, it releases dopamine and norepinephrine, which causes the organism to be insensitive to pain. [17]
Clostridium botulinum
[edit | edit source]
Clostridium botulinum is a bacterium that produces a type of toxins called neurotoxins, which are responsible for food poisoning. In the late 1960s, scientist Alan Scott tested botulinum toxin type A (BTX-A) in monkeys and discovered that BTX-A can be used to treat strabismus (a condition which the eyes are not aligned with one another).
A more well-known use for the botulinum toxin is its ability to reduce wrinkles and frown lines when injected into the skin. Many celebrities undergo botox (short term for botulinum toxin) to smooth out their facial skin. [18]
References
[edit | edit source]- ↑ "Controlled Substance Law." The Controlled Substances Act (CSA). HG.org Global Legal Resources, n.d. Web. <http://www.hg.org/control.html>.
- ↑ "DEA Diversion Control - Controlled Substance Schedules." DEA Diversion Control - Controlled Substance Schedules. Drug Enforcement Administration, n.d. Web. <http://www.deadiversion.usdoj.gov/schedules/index.html>.
- ↑ "Regulatory Information - Controlled Substances Act." US Food and Drug Administration. US Department of Health & Human Services, n.d. Web. <http://www.fda.gov/regulatoryinformation/legislation/ucm148726.htm>.
- ↑ "DEA Diversion Control - Controlled Substance Schedules." DEA Diversion Control - Controlled Substance Schedules. Drug Enforcement Administration, n.d. Web. <http://www.deadiversion.usdoj.gov/schedules/index.html>.
- ↑ http://www.mbc.ca.gov/licensee/rx_form_requirements.html
- ↑ "DEA Diversion Control - Controlled Substance Schedules." DEA Diversion Control - Controlled Substance Schedules. Drug Enforcement Administration, n.d. Web. <http://www.deadiversion.usdoj.gov/schedules/index.html>.
- ↑ "Regulatory Information - Controlled Substances Act." US Food and Drug Administration. US Department of Health & Human Services, n.d. Web. <http://www.fda.gov/regulatoryinformation/legislation/ucm148726.htm>.
- ↑ "DEA Diversion Control - Controlled Substance Schedules." DEA Diversion Control - Controlled Substance Schedules. Drug Enforcement Administration, n.d. Web. <http://www.deadiversion.usdoj.gov/schedules/index.html>.
- ↑ "DEA Diversion Control - Controlled Substance Schedules." DEA Diversion Control - Controlled Substance Schedules. Drug Enforcement Administration, n.d. Web. <http://www.deadiversion.usdoj.gov/schedules/index.html>.
- ↑ "DEA Diversion Control - Controlled Substance Schedules." DEA Diversion Control - Controlled Substance Schedules. Drug Enforcement Administration, n.d. Web. <http://www.deadiversion.usdoj.gov/schedules/index.html>.
- ↑ "Regulatory Information - Controlled Substances Act." US Food and Drug Administration. US Department of Health & Human Services, n.d. Web. <http://www.fda.gov/regulatoryinformation/legislation/ucm148726.htm>.
- ↑ http://www.thefreedictionary.com/Natural+product
- ↑ Berg, Jeremy M., ed. (2002), Biochemistry (6th ed.) New York City, NY: W.H. Freeman and Company,
- ↑ http://www.allthesea.com/Sea-Sponge.html
- ↑ Davis, Alison, Ph.D., (2006), Medicines By Design: Drugs from Nature, Then and Now: Ocean Medicines (NIH Publication No. 06-474): U.S. DEPARTMENT OF HEALTH AND HUMAN SERVICES,
- ↑ http://www.research.vt.edu/resmag/1999resmag/taxol.html
- ↑ http://www.chm.bris.ac.uk/webprojects2002/jjones/Content/Epibatidine.htm
- ↑ http://www.animalresearch.info/en/medical/diseasesresearch/Botulinum
Explain how drugs interact with the cell membrane and the membranes structural characteristics relevant to drugs interaction
Background
[edit | edit source]Many drugs need to pass through one or more cell membranes to reach their site of action. A common feature of all cell membranes is a phospholipid bilayer, about 10 nm thick. Spanning this bilayer or attached to the outer or inner leaflets are glycoproteins, which may act as ion channels, receptors, intermediate messengers (G-proteins) or enzymes. Cells obtain molecules and ions from the extracellular fluid, creating a constant in and out flow. The interesting thing about cell membranes is that relative concentrations and phospholipid bilayers prevent essential ions from entering the cell. Therefore in order for drugs to move across the membrane these problems must be addressed. In general, this is completed by facilitated diffusion or active transport. In facilitated diffusion, relative concentrations are used to transport in and out. Active transports uses energy (ATP) to transfer molecules and ions in and out of the cell.[1]
Concept of drug cross the cell membrane
[edit | edit source]Cellular signals cross the membrane through a process called signal transduction. This three-step process proceeds when a specific message encounters the outside surface of the cell and makes direct contact with a receptor. A receptor is a specialized molecule that takes information from the environment and passes it throughout various parts of the cell. Next, a connecting switch molecule, transducer, passes the message inward, closer to the cell. Finally, the signal gets amplified, therefore causing the cell to perform a specific function. These functions can include moving, producing more proteins, or even sending out more signals.[2]
Methods of Drug cross the cell membrane
[edit | edit source]Passive Transport
[edit | edit source]The most common method for drugs to cross the cell membrane is by Passive Diffusion. Drug molecules will diffuse down its concentration gradient without expenditure of energy by the cell. However, the membranes are selectively permeable, so it has different effects on the rate of diffusion on different drug molecules. The rate of diffusion also can be enhancing by transport proteins in the membrane by Facilitated Diffusion. There are two types of transport proteins that carry out the facilitated diffusion, Channel protein and Carrier Protein. [3]
Active transport
[edit | edit source]Active transport is an energy-requiring process. The drug molecule, transport against the a concentration gradient, and most of the protein used are carrier proteins, rather than channel proteins. There are also two type of active transport
Primary active transport which directly uses energy to transport molecules across a membrane.Sometime the carrier protein can be an electrogenic pump. [4]
In secondary active transport or Co-transport also uses energy to transport molecules across a membrane. However, It differs from primary transport is that there is no direct coupling of Adenosine triphosphate|ATP; instead, the electrochemical potential|electrochemical potential difference created by pumping ions out of the cell is used. [5]
References
[edit | edit source]- ↑ Transport, October 27, 2012
- ↑ Berg, Jeremy M., ed. (2002), Biochemistry (6th ed.) New York City, NY: W.H. Freeman and Company,
- ↑ {{Reece, Jane B., and Neil A. Campbell. Campbell biology Jane B. Reece ... [et al.].. 9th ed. Boston: Benjamin Cummings :, 2011. Print. | chapter 7| page 133}}
- ↑ Georgia Journal of Physiology
- ↑ Georgia Journal of Physiology
Background Information
[edit | edit source]
MDR stands for Multidrug‐ resistance, and describes large integral protein pumps located in the cell‐surface membranes of most living organisms. The main purpose they serve are to monitor chemicals passing through the cell membrane and eject ones that might endanger the well being of the cell. Normally they are beneficial to the cell's growth, but when growth is unwanted, i.e. in case of a bacterial infection or spreading cancer cells, MDR pumps become a problem. MDR pumps provide the ability to adapt and defend cells against some types of antibiotics. Cells employ the use of MDR pumps for protection against their environments, and researchers need to not only synthesize antibiotics that affect a targeted cell but they need to be able to cross the cell's defenses. The MDR pumps are activated by using packets of energy called ATP to force out unwanted toxins.
MDR pumps are found throughout the human body, including the brain, digestive tract, liver, and kidneys. These pumps carry out specific functions in the human body such as transporting molecules like hormones into and out of cells.
Researchers suggest that plants have evolved over time to produce chemicals that block MDR pumps, essentially having the same effect as what antibiotics need to have. This theory was tested by knocking out the MDR pump gene from a specific bacterial cell (Staphylococcus aureus). The bacteria was then exposed to the antibiotic berberine extracted from berberine berries. Berberine has proven to be typically ineffective in the presence of Staphylococcus aureus. However, it was extremely effective against the genetically modified bacteria without the MDR pump. The bacteria were also killed in the presence of the berberine when also exposed to a berberine extract that was believed to inhibit bacterial MDR pumps.
What is MDR Pumps?
[edit | edit source]MDR pumps are Multidrug‐ resistance pumps that are large proteins in cell‐surface membranes of microorganisms.They are used to monitor incoming chemicals and spit out ones that might endanger the bacteria.The gate must be opened for the pump to work.With the pump open, it can take up the drug.The pump's cytoplasmic gate closes, allowing the drug to be pumped out of the cell.
Bacteria and Drug Resistance
[edit | edit source]MDR pump can bestow deadly power-in the form resistance to antibiotics-to tiny organisms which is a bacteria. While bacteria MDR genes are not identical to those, the three-dimensional shape of all pumps is the same, they perform a similar task: preventing harmful molecules from setting up the cell's interior. Many MDR pumps confer resistance against first time used to treat pathogenic yeast and parasites. Resistance to drugs called antifungals that are used to treat yeast infections is increasing rapidly, affecting immunecrippled AIDS patient. MDR is saquinavir, a class of anti-AIDS frugs, called protease inhibitors. MDR pumps might also transport "normal" substances. In mammals, MDR pumps are positioned to encounter potentially harmful substances face to face in the intestine.
Fighting Cancer with MDR Pumps
[edit | edit source]Researchers are focusing on fighting cancer through the use of MDR pumps. Stem cells, which are those cells that have yet to be differentiated, have minimal MDR pumps in their membranes. Because of this, these stem cells are extremely sensitive to cancer-killing drugs. A specific major drawback to chemotherapy is bone marrow toxicity, which leaves the human body defenseless against deadly infections. Scientists and researchers devised a method to attach the defenseless stem cells with MDR pumps. In doing so, the stem cells can possibly stand a chance to the chemotherapy required to kill tumors in the human body. [1]
MDR Pumps May Not Work During Pregnancy
[edit | edit source]Recent discovery by a pharmacologist Mary Vore of the University of Kentucky in Lexington shows that MDR pump will not work properly while the woman is pregnant. Basically, the MDR pump is malformed in pregnant women who have ICP, Intrahepatic Cholestasis of Pregnancy, which can endanger the growing fetus. The suggested reasons for this are the effect of estrogen and other pregnancy hormones during pregnancy.
File:Mdr pump.jpg Scientists and researchers believe that medicines produce an effect for less than half of those who take it. This is partly due to lifestyle and environmental factors, but it is primarily the result of genetic variants. The genes that make cytochrome P450 proteins are responsible for a medicinal effect in the body. These proteins metabolize hormones that our body produces and those that are foreign. Because every human consists of a different genomic sequence, the proteins that genes encode for also differ. This genetic variant can affect the way in which cytochrome P450 reacts and how the drug elicits an effect. Ultimately, the cytochrome P450 proteins process many of the drugs we take, therefore explaining the different responses to medicine between individuals. [2]
http://upload.wikimedia.org/wikipedia/commons/4/42/DoseResponse.png
The above picture is the Dose-Response curve that tells how much a drug (x-axis) causes effect in the body (y-axis). The region in between the blue and green curve is the desired effect and the region on the right side of the green curve is the side effect. Scientists study the relationship between the medicine and its effect on human body. Like how the image is showing, at first there is no effect when no drug is put in. The researcher then adds more drug into the system to see the effect on the body. If too much drug is taken, the body will reach its overdosing point and may face side effects. This image demonstrates the right amount of dose researchers are looking for while working with a drug.
[3]
References
[edit | edit source]Berg, Jeremy M., ed. (2002), Biochemistry (6th ed.) New York City, NY: W.H. Freeman and Company
Davis, Alison Davis. Medicines By Design. The Office of Communications and Public Liaison. 2006.
Influenza Virus and Drug Design
[edit | edit source]The influenza virus is the main culprit of respiratory infection more commonly known as the "flu". The structure of the influenza virus includes a nucleoprotein (RNA) center enclosed in capsid, a lipid envelope, and spikes of two key proteins on its surface: hemagglutinin and neuraminidase. About 80% of the spikes consist of hemagglutinin. The function of hemagglutinin is to bind the virus with the host cell. It acts as to stick the virus onto the host cell in order to cause an infection. The other protein, called neuraminidase, cover the rest of the surface. The lesser amount of neuraminidase doesn't account for its large role, however. Neuraminidase helps facilitate the release of the newly formed viral molecules from the host cell. In perspective, hemagglutinin is the anchor that binds the virus to the host cell and neuraminidase is the trigger that induces a viral infection. Since neuraminidase is so important for influenza virus, scientists have developed methods to inhibit the protein and prevent influenza virus from infecting the host cell.
Structural based design has been one of the primary methods in the design of a drug. With the use of two key techniques, X-Ray Crystallography and Nuclear Magnetic Resonance (NMR), they have aided in the determination of a molecule’s structure. Such information as the three-dimensional structure of a specific target, helps direct drug creation because with structure based design, it allows one to observe the interaction between the target and the drug.
Recently, studies have shown a positive correlation between the similarity in drug and its target’s natural ligands to the increased barrier to resistance. In other words, the design of a drug needs to take into account that similarity, both physically and chemically, to the target’s natural ligands will make it more difficult for said disease and target to become resistant.
Neuraminidase Inhibitors
[edit | edit source]The discovery of neuraminidase inhibitors for the Influenza virus is one of the early examples of structure based drug design. The target of the Influenza drug is based on the virus’s neuraminidase enzyme. This enzyme’s role is to free the progeny of the virus in order to spread and infect other cells within the body. The neuraminidase inhibitors were created based on crystal structure data of the virus’s neuraminidase. What the neuraminidase inhibitor does is it chemically destroys the virus’s receptors thereby stopping virus replication. Neuraminidase inhibitors must be taken within 48 hours that symptoms arise. They do not ‘kill’ the flu virus, but merely slow down the virus replication to a point where the immune system can destroy it easier. As a result, they can reduce the severity and duration of a flu illness.

The spread of the flu virus is slowed in the body by the following steps: (1) The virus enters a cell with neuraminidase inhibitors present. (2) Once inside the cell, the neuraminidase inhibitors attach to the virus. (3) The flu virus can still use its host to replicate itself. (4) However, the inhibitors prevent the virus from leaving the cell—which halts the infection of other cells. (5)The virus is rendered ineffective and dies within the cell.
Current Antiviral Agents
[edit | edit source]Currently, ten neuraminidases enzymes have been discovered and their structures determined: N1-N9 for Influenza A and type B neuraminidase for type B Influenza. Four approved drugs that have been administered for public use are zanamivir, oseltamivir, amantadine, and rimantadine.
Zanamivir, oseltamivir, amantadine, and rimantadine are different in the types of influenza viruses they inhibit, route of administration, and approved use in age groups. However, the side effects and cost of zanamivir and oseltamivir vary from those of amantadine and rimantadine. Side effects, including nervousness, anxiety, concentration difficulty, and lightheadedness, have been reported to be less frequent in patients who take zanamivir and oseltamivir. In addition, zanamivir and oseltamivir are more expensive than rimatadine, which is more expensive than amantadine. While amantadine and rimantadine are drugs that have both been used extensively for treatment of influenza A, zanamivir and oseltamivir are new alternatives. Unlike zanamivir and oseltamivir, amantadine and rimantadine are chemically related antiviral drugs that act against influenza A viruses but not influenza B viruses. However, these antiviral agents for influenza are not a substitute for vaccine, but instead are a supplement.
Experiments have discovered that these two drugs have different resistances based on their method of intake and their binding affinity to the target. Oseltamivir is taken orally as a tablet whereas Zanamivir is taken as an inhalant. Oseltamivir is a prodrug, which means that it is inactive until it is taken within the body. Both drugs have been found that they work better according to those aforementioned methods. As for the inhibitors, amino or guandino groups have replaced the C4-hydroxyl groups of the neuraminidase enzymes. Changing the substituents optimize the binding potency both chemically and physically. According to Dr. Colman, “The loss of inhibitory potency toward neuraminidase…. zanamivir, and oseltamivir carboxylate, is greater the less the inhibitor resembles the substrate.” [4]
More studies have shown substantial evidence that support the hypothesis that drugs that resemble the target’s natural substrate and ligands are more successful at suppressing the possibility of drug-resistance. In fact, viruses need to be able to bind to its own substrates and to be able to distinguish against that of the drug. If drugs resemble the virus’s own substrates, it will be difficult for it to discriminate and therefore, become less resistant. In addition, this is why multi-drugs and multi-dosages are prescribed. Using “drug cocktails,” dosages, and routes of administration will result in better outcomes.
Inhibitors: Hemaggluttin and Sialidase
[edit | edit source]Two important proteins on the surface of influenza virus cells are lectin hemagglutinin protein and enzyme sialidase. Lectin hemagglutinin has 3 shallow sialic acid-binding sites while enzyme sialidase has an active site in a pocket. The deep active site of low molecular weight inhibitors makes sialidase the more attractive anti-influenza drug target than hemagglutinin.
Influenza virus binds to sialic residues. Because of this, viruses gain entry into the host cell by adhering to the surface of carbohydrates. The viral protein that binds to sugars is called hemagglutinin. Once the virus penetrates the cell membrane, a viral protein called neuraminidase cleaves the glycosidic bonds of the sialic acid residues, freeing the virus to infect the host cell. Neuraminidase inhibitors are analogues of sialic acid that block the active site of neuraminidase and leave uncleaved sialic acid residues on the surface of host cells and influenza viral envelopes. Viral hemaggluttin binds to the uncleaved sialic residues, reducing the spread of infection to other cells.
Notes
[edit | edit source]- ↑ http://sciencenotes.ucsc.edu/9701/pdf/mdrartII.pdf
- ↑ Berg, Jeremy M., ed. (2002), Biochemistry (6th ed.) New York City, NY: W.H. Freeman and Company,
- ↑ Davis, Alison Davis. Medicines By Design. The Office of Communications and Public Liaison. 2006.
- ↑ Colman, Peter M. "New Antivirals and Drug Resistance", 'Annual Review of Biochemistry', 2009.
References
[edit | edit source]- Colman, Peter M. "New Antivirals and Drug Resistance", 'Annual Review of Biochemistry', 2009.
Introduction
[edit | edit source]Alprazolam (also known as Xanax) is a psychoactive drug used to treat anxiety disorders along with panic disorders. It is in a class of medications called benzodiazepines and how it works is it decreases abnormal excitement in the brain. Alprazolam can come in a variety of shapes and sizes. They can come as a tablet, an extended-release tablet, an orally disintegrating tablet, and it is also available in a liquid form. Alprazolam is used to treat anxiety disorders and panic disorders (sudden and unexpected attacks of extreme fear and uneasiness about these thoughts). Alprazolam is in a class of medication called benzodiazepines, which works by decreasing abnormal excitement in the brain.
Alprazolam is also sometimes used to treat depression, fear of open spaces (agoraphobia), and premenstrual syndrome. This medication may be prescribed for other uses; ask your doctor or pharmacist for more information.
History
[edit | edit source]Alprazolam is now part of Pfizer, but it was first released by Upjohn. It was covered under U.S. Patent 3,987,052, which was filed on October of 1969 and expired in September 1993. In the year 1981, Alprazolam was released. When it came out, the first approved indication was panic disordered.
A young psychiatrist named David Sheehan suggested using the new distinction called DSM-III. Hence, DSM-III created in the classification of anxiety disorders between generalized anxiety disorders (GAD) and panic disorder in order to market alprazolam. In other words, this was a way for Sheehan to advertise the use of alprozolam. From his studies, Sheehen knew that panic disorder was both widespread among the populace and responsive to benzodiazepines. Therefore, he had an idea and suggested to Upjohn that marketing alproxolam for panic disorder would both cover new diagnostic territory and emphasize the unique potency of this drug. He was basically finding unique ways to show how great alprazolam is.
As his clinical study continues, Sheehen describes how the first group of patients he tested with aplprozolam say how effective this drug was. They were basically all very impressed with the action that alprozolam carries. Several months later, when the United States Food and Drug Administration approved alprozolam, alprozolam was sold out quick and they made a great profit. However, similar to many other drugs out there many published reports in the medical literature state that taking alprozolam created severe withdrawal symptoms including psychoses, seizures, and intense rebound anxiety.
Panic/Anxiety Disorders
[edit | edit source]Alprazolam is effective in relieving moderate to severe anxiety and panic attacks. It is no longer the first line of defense for these attacks due to concerns of tolerance, dependence, and abuse. Doctors limited Alprazolam for a patient from 4 to 10 weeks and is only allowed to distribute the drug to those of no history of tolerance or dependence of any drug.
Alprazolam and Pregnancy
[edit | edit source]By taking Alprazolam while being pregnant has potential risks. It can cause fetal drug dependence and withdrawal symptoms in the post-natal period. It can also cause neonatal flaccidity and respiratory problems. Alprazolam are known to excreted in human milk and passed down to the infants. Alprazolam causes the infants to lose weight and become lethargic.
Mechanism of Action
[edit | edit source]Alprazolam is a Central Nervous System drug from the benzodiazepine class of no known mechanism of action. It is predicted to have its effects through binding to different stereo-specific receptors in the CNS. Alprazolam has also been tested to show antidepressant properties, which isn't usually a common characteristic of regular benzodiazepine derivatives.
Side Effects
[edit | edit source]Alprazolam can cause several side effects. The most common side-effects are:
- drowsiness
- light-headedness
- headache
- tiredness
- dizziness
- irritability
- talkativeness
- difficulty concentrating
- dry mouth
- increased salivation
- changes in sex drive or ability
- nausea
- constipation
- changes in appetite
- weight changes
- difficulty urinating
- joint pain
Besides these common side effects, it sometimes produces more side side effects such as:
- shortness of breath
- seizures
- seeing things or hearing voices that do not exist (hallucinating)
- severe skin rash
- yellowing of the skin or eyes
- depression
- memory problems
- confusion
- problems with speech
- unusual changes in behavior or mood
- thinking about harming or killing yourself or trying to do so
- problems with coordination or balance
Food and Drug interactions
[edit | edit source]Alprazolam is metabolized via CYP3A4. Inhibitors such as fluoxetine, ketoconazole, cimetidine, nedfazodone, ritonavir, propoxyphene, erthromycin, and itraconazole are all examples of combining CYP3A4 inhibitors. These inhibitors basically delay the hepatic clearance of alprazolam which create an excessive accumulation of alprazolam.
Through the concomitant administration of alprazolam tablets in doses up to 4 mg/day, Imipramine and Desipramine both have been reported to increase an average 31% and 20%. It is also known that combined oral contraceptive pills reduce the clearance of alproxolam. This leads to the increased of plasma levels of alprozolam and accumulation.
Furthermore, herb kava and alprazolam can have a great affect between each other when combined because it can result in the development of a semi-comatose state. Hypericum is a genus of about 400 species of flowering plants in the family Hypericaceae that can lower the plasma levels of alproxolam and reduce its therapeutic effect. However, looking at the big picture, alcohol is one of the most important and common interactions. This is due to the idea of combination. Alcohol and benzodiazepines such as alproxolam taken in combination have a synergistic effect on one another. Due to this reaction, it causes severe sedation, behavioral changes, and intoxication. Therefore, it is usually seen that the more alcohol and alprozolam taken, the worse the interaction will be.
How supplied
[edit | edit source]Generic:(Alprazolam)
- Oral Tablet: 0.25 mg, 0.5 mg, 1 mg, 2 mg
- Oral Tablet, Disintegrating: 0.25 mg, 0.5 mg, 1 mg, 2 mg
- Oral Tablet, Extended release: 0.5 mg, 1 mg, 2 mg, 3 mg
Alprazolam Intensol (oral solution) : 1 mg/ml
Niravam: Oral Tablet, disintegrating: 0.25 mg, 0.5 mg, 1 mg, 2 mg
Xanax: Oral Tablet: 0.25 mg, 0.5 mg, 1 mg, 2 mg
Xanax XR: Oral Tablet, Extended release: 0.5 mg, 1 mg, 2 mg, 3 mg
Reference
[edit | edit source]http://www.nlm.nih.gov/medlineplus/druginfo/meds/a684001.html http://www.medicinenet.com/alprazolam-oral/page2.htm http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0000807 http://en.wikipedia.org/wiki/Alprazolam Amsacrine
Description
[edit | edit source]
Amsacrine is a potent intercalating antineoplastic agent. It is effective in the treatment of adult acute leukemias and malignant lymphomas, but not effective in the treatment of solid tumors. It is frequently used in combination with other antineoplastic agents in chemotherapy.
Usage
[edit | edit source]Injectable (Intravenous Route)
Side Effects
[edit | edit source]- Black stools
- cough or hoarseness
- fever or chills
- painful or difficult urination
- pinpoint red spots on skin
- sores, ulcers, or white spots on lips, tongue, or inside mouth
- Abdominal pain or tenderness
- blurred vision
- fast, pounding, or irregular heartbeat or pulse
- palpitations
- seizures
- yellow eyes or skin
Mechanism of Actions
[edit | edit source]

The DNA is wound around basic proteins called histones and is tighly coiled or supercoiled. Sequences of DNA must be unwound or relaxed before they can be processed by DNA processing enzymes. Topoisomerase II or TopoII is one enzyme that can process DNA. TopoII temporarily breaks the DNA strand and allows both ends to freely rotate within the enzyme. This allows the DNA to unwind. After the DNA is processed, the enzyme then reconnects the two ends.
Amsacrine kills cancerous cells by inhibiting the Topo II enzyme. It stabilizes the enzyme with the broken DNA strand complex long enough for the enzyme-DNA complex to break down completely. When the DNA is destroyed, the cancerous cells will be eventually killed.
References
[edit | edit source]"Amsacrine." Scott Hamilton CARES Initiative. N.p.. Web. 07 Dec 2012. <http://chemocare.com/chemotherapy/drug-info/Amsacrine.aspx> Grove, WR, CL Fortner, and PH Wiernik. "Clinical Pharmacy." Clinical Pharmacy. Jul-Aug. (1982): 320-26. Print.
History
[edit | edit source]
Aspirin is in the family of non-steroidal anti-inflammatory drugs otherwise known as NSAIDs medicines. Aspirin has been used for thousands of years. It is believed to have been first used in 400 BC by the Greek physician Hippocrates who would prescribe the bark and leaves of the willow tree to his patients to relieve pain and fever. It turns out that the willow tree is rich in a substance called salicin which was later turned into salicylic acid in 1832 AD by a German chemist named Charles Gergardt. Finally in 1897 AD, Felix Hoffman, a German chemist, invented aspirin which lead to the Bayer’s release of aspirin in 1899 AD. Scientists used X-ray crystallography to determine the 3D structure of the enzyme COX (cyclooxygenase), which helped them study the shapes of the enzyme and how drugs like aspirin block it.
COX Inhibitors
[edit | edit source]There are many drugs that belong to the NSAID (Non-Steroidal Anti-Inflammatory Drug) family of medicine such as Advil, Ibuprofen, and Aleve. Each of these drugs functions in a similar manner in that they all block a family of enzymes called cyclooxygenases (abbreviated COX). These drugs block two similar enzymes known as COX-1 and COX-2. COX-2 triggers an immune response and swelling in response to infection or injury. Blocking COX-2 is desirable because it can reduce swelling, fever, pain, etc. The problem with these drugs, such as aspirin, is that they also block COX-1. COX-1 produces molecules that protect the stomach wall called prostaglandins. Blocking COX-1 therefore results in ulcers due to the gastric acid being able to digest the de-protected stomach wall. This is a significant problem with long term use of aspirin to treat diseases such as arthritis. These side effects of long term use of aspirin result in about 16,500 deaths every year in the U.S.[1]
The Development of COX-2 Only Inhibiters
[edit | edit source]

Scientists wondered for many years how COX inhibitors such as aspirin worked so they used X-Ray Crystallography and other biophysical techniques to determine the three dimensional structure of the cyclooxygenases. The structure of an enzyme is important in understanding its function. Visualizing the individual enzyme’s structural folding and bends enables scientists to consider possible inhibitors that might bind to the enzyme. As the structures of COX-1 and COX-2 were being solved a new drug was found that blocks COX-2 but not COX-1, called Celebrex. The solved structures of COX-1 and COX-2 showed why only COX-2 was inhibited. In the binding site for Celebrex in COX-1, an isoleucine is present where there is a valine in COX-2. The structure of valine has a pocket-like end that is easily bound to, whereas isoleucine has an extended side chain structure that makes it impossible for the drug to bind. This allowed for the new drug to be made to bind to that specific target without the bad side effects that were caused by the COX-1 enzyme.[1]
COX-3
[edit | edit source]Scientist have also developed another inhibitor called COX-3 inhibitors. This inhibitor appears to be more similar to Tylenol. Tylenol does not belong to the NSAID family of medicine because it is not an anti-inflammatory medicine. The future of COX-3 drugs may lead to a more significant understanding of Tylenol.
Synthesis
[edit | edit source]One of common method for Synthesis of Aspirin is classified as esterification reaction as followed[2]:

- Reaction Mechanism
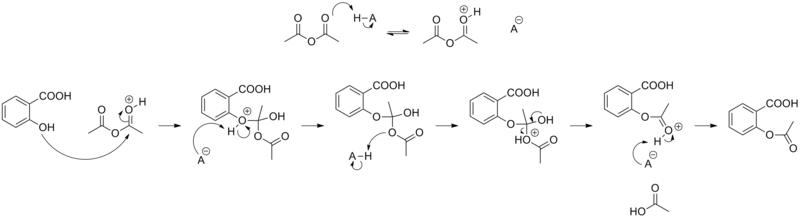


Mechanism of Actions
[edit | edit source]
Aspirin is an antiplatelet drug that inhibits cyclooxygenase or COX. COX-2 triggers inflammation as an immune response. By blocking COX-2, aspirin acts as a potent pain killer and anti-inflammatory agent. However, COX-1 enzymes are also the key enzymes in thromboxane A2 (TXA2) generation. Thromboxane TXA2 causes platelets to clump together over the vessel tear to facilitate repair when blood vessels are damaged or are diseased. The aggregation of platelets results in a clot which stops bleeding and aids repair of the damaged blood vessel. Aspirin inhibits COX-1, therefore TXA2, thereby reducing the ability of platelets to accumulate. This is why aspirin is known as a blood thinner or anti-platelet agent.
COX-1 and COX-2 are both cyclooxygenase enzymes with similar structure but different in functions. They both generate prostaglandins from the fatty acid arachidonic acid. However, different prostaglandins are produced from COX-1 and COX-2. Prostaglandins from COX-2 are associated with pain, inflammation and fever while prostaglandins from COX-1 support platelet aggregation, gastrointestinal mucosal integrity and renal function.

In the process of blocking COX, the acetyl group on aspirin is hydrolyzed and then bonded to the alcohol group of serine as an ester. This has the effect of blocking the channel in the enzyme and arachidonic acid can not enter the active site of the enzyme and be converted to prostaglandins.
There is 60% homology between the amino acid structures of COX-1 and COX-2. Aspirin blocks COX-1 by binding to Ser 530 in the same way that it binds to Ser 516 of COX-2. However, the active site of COX-2 is slightly larger than the active site of COX-1, so pharmaceutical companies have been able to develop selective COX-2 inhibitors, such as Celecoxib, Rofecoxib and Meloxicam. These drugs can inhibit the biochemical actions of COX-2 which reduces inflammation but have no effect on COX-1 to prevent damages of the stomach mucosa.
Polymorphism
[edit | edit source]Polymorphism is the ability of a substance to form more than one crystal structure. This process is essential when it comes to the development of pharmaceutical ingredients. For a long period of time, only one crystal structure was discovered. Then since the 1960s, a second crystalline form was suspected. This second polymorph was first discovered by Vishweshwar and his coworkers in the year 2005. A new crystals was basically found after performing the cocrystallization of aspirin and levetiracetam from hot acetonitrile. However, this second crystalline form is only stable at 100°K and reverts to the first crystalline form at ambient temperature. In the first crystalline form, there exist two salicylic molecules form centrosymmetric dimers through the acetyl groups with the (acidic) methyl proton to carbonyl hydrogen bonds. Furthermore, in the second crystalline form that is newly claimed, each salicylic molecule form the same hydrogen bonds with two neighboring molecules instead of only one. In the end, both polymorphs form the same dimmer structures that is with respect to the hydrogen bonds forms by the carboxylic acid groups.
Before taking Aspirin
[edit | edit source]Children or teenager who has a fever should NOT take aspirin. Children who have chicken pox or flu symptoms should also not take aspirin due to the potential for aspirin to cause a serious condition called Reye's syndrome, which is primarily seen in children. In addition, adults should also avoid taking aspirin if they have any of the conditions listed below:
- recent history of intestinal, stomach, or intracranial bleeding
- bleeding disorder
- allergy to an non-steroidal anti-inflammatory drug such as Advil, Motrin, Aleve, or Orudis
If an individual is taking aspirin to prevent conditions such as stroke or heart attack, it is crucial to avoid taking ibuprofen at the same time, as this can increase the risk of serious bleeding. This medication can also hurt an unborn baby's heart, and may reduce birth weight or have other serious effects. Therefore, it is important to the tell the doctor before taking aspirin if one is pregnant or plans to become pregnant while taking this medication. It is also essential for breast feeding mother to know that taking aspirin can also pass through breast milk and seriously harm a nursing baby. Hence, all breast feeding mothers should tell the doctor in advance before taking this medication.
Medicinal Uses
[edit | edit source]Aspirin is part of a group of medications known as non-steroidal anti-inflammatory drugs (NSAIDs) which has been used since the times of the Ancient Greeks. Belonging to a class of salicylates, this drug has multiple uses in treating various ailments associated with pain and inflammation. Typically, it acts by reducing the materials that cause inflammation and pain. Often, aspirin is considered inferior to ibuprofen for the alleviation of pain. However, aspirin is usually ineffective for those pains caused by muscle cramps, bloating, or skin irritation. Thus, treating mild to moderate pain and inflammation are some of the common uses, but aspirin has also been used to treat and prevent heart attack, stroke, chest pain, and other cardiovascular conditions. However, Aspirin appears to offer little use to those at lower risk of heart attack or stroke. This kind of idea is known as primary prevention. Normally while taking aspirin, one should avoid taking other NSAIDs as they will actually inhibit the action of the drug. Coming in various forms (enteric-coated tablets, chewable tablets), aspirin is commonly prescribed by physicians for its uses in preventing cardiovascular problems.
In addition, studies show that aspirin have great effect on cancer, especially its effect on a cancer known as colorectal cancer (CRC). Even multiple meta-analyses implies that regular use of aspirin will definitely reduce the long-term risk of colorectal cancer. For over 100 years, other uses of aspirin may be for fever and pains related with common cold. Basically, aspirine can be a source of cure when it comes to achiness, discomfort, headache, and sore throat pain. Furthermore, existing fever and joint pain often cure quickly within three days of high doses of aspirin. People with pericarditis, coronary artery disease, and acute myocardial infarction can be treated with aspirin too.
There are several side effects and risk factors associated with the use of aspirin. Individuals who are allergic to aspirin, have a history of intestinal or stomach bleeding, bleeding disorders (like hemophilia), and allergies to other NSAIDs should avoid using aspirin. However, prescribers can still recommend aspirin after performing tests and adjusting the dosage based on the patient's needs. For example, patients with specific medical issues such as stomach ulcers, liver disease, blood clotting disorders, or high blood pressure may require extra tests and adjusted doses before safely prescribing aspirin. Also, patients on aspirin should avoid drinking alcohol as alcohol consumption can lead to increased bleeding in the stomach. Typical side effects with aspirin include upset stomach, heartburn, drowsiness, and headache. Because aspiring irritates the lining of the stomach and intestinal tracts, a useful way to reduce this incidence is to drink aspirin with a full glass of milk. One should stop using aspirin if side effect such as hives or difficulty breathing occurs.
Finally, certain medications such as warfarin should be notified to physicians before prescribing aspirin. Page text.[3]
Side Effects
[edit | edit source]Immediately get medical help if you present one or more of the following symptoms:
- hives
- difficulty breathing
- swelling on the lips, face, tongue or throat
Stop using aspirin and call doctor if you see one or more of the following symptoms:
- black, bloody or tarry stools
- coughing up blood or vomit that looks like coffee grounds
- severe nausea, vomiting or stomach pain
- fever lasting over 3 days
- swelling for over 10 days
- hearing problems (ringing in your ears)
Least serious symptoms might include:
- upset stomach
- heartburn
- headache
- drowsiness
References
[edit | edit source]Flavio, Guzmán. "Antiplatelet agents: mechanisms of action and general overview." Pharmacology Corner. N.p., 24 2009. Web. 7 Dec 2012.
Vane, J.R, and R.M Botting. "Thrombosis Research." Thrombosis Research. 110. (2003): 255-58. Print.
- ↑ a b The Structures of Life, National Institutes of Health. "The Structures of Life." July 2007: 46-48.
- ↑ Palleros, Daniel R. (2000). Experimental Organic Chemistry. New York: John Wiley & Sons. p. 494. ISBN 0-471-28250-2.
- ↑ [1],additional text.
Valsartan/Diovan
Description
Valsartan, marketed as Diovan in the US, UK, and Australia [1], is chemically described as N-(1-oxopentyl)-N-[[2’-(1H-tetrazol-5-yl) [1,1’-biphenyl]-4-yl]methyl]-L-valine. Its empirical formula is C24H29N5O3, and its molecular weight is 435.5 grams per mole [2].
Physically, Valsartan is a fine white powder that is soluble in ethanol and methanol and slightly soluble in water. Diovan is available as tablets for oral administration, containing 40 mg, 80 mg, 160 mg or 320 mg of valsartan. The inactive ingredients of the tablets are colloidal silicon dioxide, crospovidone, hydroxypropyl methylcellulose, iron oxides (yellow, black and/or red), magnesium stearate, microcrystalline cellulose, polyethylene glycol 8000, and titanium dioxide [2].
The structural formula for Valsartan is
-
 Figure 1.) Valsartan (Diovan) Chemical Structure
Figure 1.) Valsartan (Diovan) Chemical Structure

Mechanism
Valsartan is an angiotensin II receptor antagonist, or more commonly, an angiotensin receptor blocker (ARB) of the type I (AT1) angiotensin receptor [1]. Blocking the activation of angiotensin II AT1 receptors widens blood vessels by relaxing the smooth muscle cells within the vessel walls [3, 4]. This mechanism also reduces the production and excretion of vasopressin and aldosterone, which are two hormones that increase water retention in the kidneys and subsequently increase blood pressure [5, 6].
Pharmacokinetics
Valsartan reaches a peak concentration 2-4 hours after dosage administration, and has a half-life of approximately 6 hours. However, if taken with food, Valsartan exposure decreases by 40% and the peak concentration decreases by about 50% [2].
Metabolism and Elimination
When recovering the drug, most of it is the drug unchanged, but about 20% is recovered as metabolites. Nine percent of the recovered metabolites were found to be valeryl 4-hydroxy valsartan. From in vitro metabolism studies, the CYP 2C9 isoenzyme was found to be the enzyme responsible for forming valeryl 4-hydroxy valsartan. When taken orally, about 83% of the dosage is eliminated in feces and about 13% of the dosage is eliminated in urine [2].
Role in Blood Pressure (Hypertension)
As the heart beats, blood pumps through the blood vessels and produces a force inside the blood vessels, which is known as blood pressure. When the blood pressure is high, the heart has to work harder to pump blood through the blood vessels, which causes damage to the blood vessels and may lead to stroke, heart attack, heart failure, kidney failure, and vision problems [7].
One cause of high blood pressure is angiotensin II, which narrows blood vessels, thus decreasing the blood flow and increasing the blood pressure within the blood vessel [8]. Therefore, by blocking the activation of angiotensin II AT1 receptors, Valsartan lowers high blood pressure (hypertension) by relaxing and widening the blood vessels, thus decreasing the flow resistance [8]. Valsartan is also used in treating congestive heart failure (CHF), or post-myocardial infarction (MI) [1].
Side Effects of Diovan [7]
Common side effects of Diovan for high blood pressure patients: headache, dizziness, flu symptoms, tiredness, abdominal pain.
Common side effects of Diovan for heart failure patients: dizziness, low blood pressure diarrhea, joint and back pain, tiredness, high blood potassium
Common side effects of Diovan for heart attack patients: low blood pressure, cough, (high blood creatinine), decreased kidney function, rash.
Drugs that Affect Diovan [9]
Cyclosporine (Gengraf, Neoral, Sandimmune) A diuretic (water pill) Rifampin (Rifadin, Rifater, Rifamate) Ritonavir (Norvir, Kaletra) A nonsteroidal anti-inflammatory drug (NSAID) such as aspirin, ibuprofen (Advil, Motrin), naproxen (Aleve, Naprosyn Naprelan, Treximet), celecoxib (Celebrex), diclofenac (Arthrotec, Cambia, Cataflam, Voltaren, Flector Patch, Pennsaid, Solareze), indomethacin (Indocin), meloxicam (Mobic), and others.
[1] http://en.wikipedia.org/wiki/Valsartan#Myocardial_infarction_controversy
[2] http://www.pharma.us.novartis.com/product/pi/pdf/diovan.pdf
[3] http://en.wikipedia.org/wiki/Angiotensin_receptor_blocker
[4] http://en.wikipedia.org/wiki/Vasodilation
[5] http://en.wikipedia.org/wiki/Vasopressin
[6] http://en.wikipedia.org/wiki/Aldosterone
[7] http://www.pharma.us.novartis.com/product/pi/pdf/diovan_ppi.pdf
[8] http://www.diovan.com/info/about-diovan/what-is-diovan-and-how-it-works.jsp
[9] http://www.drugs.com/diovan.html Dianabol (also called methandrostenolone, averbol, danabol, methandienone or methanienone) is an anabolic steroid that some athletes use to increase the size and strength of their muscles. This is similar to male hormone testosterone. Some studies suggest that the effects of the drug are minimal, but some side effects keep people from using it.
History
[edit | edit source]Dianabol was developed in Germany and released in US in 1960s by Ciba Specialty Chemicals. It is a controlled substance in United States, though popular with body builders.
Dr. Zeigler
[edit | edit source]Dr. Zeigler, the team physician for U.S. gold Olympic weightlifting team, played a large role in the research of altering testosterone to make it safer for the body. However, due to misuse of the drug by athletes, Dr. Zeigler became the voice of opposition of sport doping.
Side Effects
[edit | edit source]- Increased risk of liver cancer
- Increased risk of heart disease
- Oily skin, acne and bodily and facial hair growth
- Liver dysfunction
- Androgenic side effects
- For women, deepening of voice and irregular menstrual cycles.
References
[edit | edit source]Overview
Tylenol, also known as acetaminophen, is a popular pain reliever. This drug is famous for relieving headaches as well as reducing fever. Tylenol is not considered an NSAID, anti-inflammatory. It does not do much in halting inflammation.
Precautions
Be careful if your joints are aching from intense exercise. Tylenol would probably not be a good choice, compared to aspirin or Aleve. This is because the aching of joints is due to inflammation.
Discovery of Tylenol
[edit | edit source]The main ingredient in Tylenol which is acetaminophen was actually first discovered in Europe before aspirin even became a consumer product. However, it was never studied closely for its possible pharmaceutical uses until Robert L. McNeil of McNeil Laboratories. After an informal conversation with Raymond Conklin, who was vice president of the Institute for the Study of Analgesic and Sedative Drugs, at an American Pharmaceutical Manufacturers Association meeting, this compound N-acetyl P-aminophenol was brought to Neil’s attention and his work began. One of the reasons for the great success of acetaminophen when compared to aspirin is the lack of stomach aches that arise from acetaminophen. The FDA approved the drug in June of 1955 and it was introduced as Elixer Tylenol.
Absorption
Oral acetaminophen is rapidly and almost completely absorbed from the GI primarily in the small intestine. This absorption process occurs by passive transport. The relative bioavailability ranges from 85% to 98%.
Distribution
Acetaminophen appears to be distributed throughout most body fluids except fat. The volume distribution of acetaminophen is 0.95 L/kg. A small fraction of acetaminophen is bound to plasma proteins and binding is only slightly increased in plasma concentrations associated with overdose. Sulfate and glucuronide metabolites do not bind to plasma proteins even at relatively high concentrations.
Metabolism
Acetaminophen is primarily metabolized in the liver and involves three principal separate pathways 1. conjugation with glucuronide 2. conjugation with sulfate 3.oxidation via the cytochrome
Excretion
Acetaminophen is eliminated from the body primarily by formation of glucuronide and sulfate conjugates.
Mechanism of Tylenol Tylenol, acetaminophen, belongs to a class of painkillers known as non-opioid analgesics. Non-opioid analgesics work by inhibiting an enzyme known as cyclooxygenase (COX). COX is a catalyst for the conversion of a fatty acid contained in cell walls, arachidonic acid, to a substance known as prostaglandins. Prostaglandins produce pain, inflammation and fever. It causes pain and inflammation after cell injury at the site of the injury in the peripheral nervous system. They elevate body temperature by affecting the hypothalamus. By blocking COX and the subsequent production of prostaglandins in the central and peripheral nervous systems, Tylenol reduces both fever and inflammation. Tylenol differs from the other non-opioids in that it does not block COX in the peripheral nervous system that great. It appears to reduce pain primarily in the central nervous system by more than one mechanism, possibly in part by inhibiting a form of COX known as COX-3. Therefore, it is considered to be a weak analgesic and does not possess anti-inflammatory properties.
Similarity of Structure Tylenol and Aspirin
The first similarity between the two molecule is their benzene skeleton. Both these molecules have reactive oxygen and reactive hydrogens attach to the benzene ring that contribute to their molecule acidity. Aspirin has a carboxylic acid while Tylenol contains a phenol. The acidity of both molecules contributes to similar physiological activities, relieving pain.

-Tylenol- Top Image

-Aspirin - Bottom Image
Side Effects
[edit | edit source]Some people might present allergic reactions after taking tylenol. It is recommended to see a physician if the person presents one or more of the following symptoms:
- hives
- difficulty breathing
- swelling of the face, lips, tongue or throat
- nausea
- upset stomach
- itching
- loss of appetite
- dark urine
- clay-colored stools
- jaundice (yellow skin or eyes)
Incompatibility with other drugs
[edit | edit source]Tylenol could react violently if taken with some other medications. Medications that could possibly react with tylenol to produce severe side effects are:
- Antibiotics
- birth control pills/hormone replacement medication
- blood pressure medication
- cancer medication
- cholesterol lowering medication
- arthritis medication (including gold injections)
- HIV/AIDS medication
- psychiatric disorder medication
- seizure medications
Acetaminophen induces liver necrosis Although acetaminophen is safe for therapeutic doses, higher dosage can contribute to liver necrosis. Acetaminophen poisoning accounts for almost half of all cases of acute liver failure in the US and the UK. This results from the following steps: 1. Acetaminophen is metabolically activated by cytochrome 450 into a reactive specie which depletes glutathione, an antioxidant that prevents damages to important cellular components caused by reactive oxygen species such as free radicals and peroxides. 2. Loss of glutathione with an increased formation of reactive oxygen and nitrogen species in liver cells undergoes necrotic changes; 3. Due to increased oxidative stress, mitochondrial permeability changes. 4. Mitochondrial permeability changes from the additional oxidative stress which leads to loss of mitochondrial membrane potential, and the loss of the ability of the mitochondria to synthesize ATP. 5. Loss of ATP will not enable the liver cell to function anymore leading to necrosis. There are inflammatory mediators such as certain cytokines and chemokines that can modify the toxicity. Some have been shown to alter oxidative stress. In addition, existing data support the involvement of cytokines, chemokines, and growth factors in the initiation of regenerative processes of the liver. As a result, pay careful attention to how much acetaminophen one is taking and never exceed the maximum daily dose (4,000 mg or eight Tylenol extra strength pills in a 24-hour period). If one needs more pain relief, consult with one’s doctor about incorporating other pain relief strategies or by switching to a different type of painkiller. Acetaminophen is also available in a variety of medications, from over-the-counter cough and cold medicines to prescription strength painkillers. Confirm the ingredients of all drugs to make sure the maximum dosage is not exceeded as over dosage will induce liver necrosis. If one has liver disease or drink alcohol heavily, avoid acetaminophen completely! If one regularly takes the drug, watch for signs of liver damage, which include dark urine, pale stools, right-upper abdominal pain, and a yellowish tinge in the whites of the eyes.
Addition Dangers of Tylenol Diphenhydramine, an ingredient in Tylenol PM, is an antihistamine that also has sedative properties to help patients fall asleep. However, if people take too much diphenhydramine due to excessive use of Tylenol PM or by taking diphenhydramine in addition to the amount found in Tylenol PM, overdose can occur. This overdose can cause confusion, dry mouth, fatigue, muscle weakness, dizziness and chest congestion. In high doses, diphenhydramine can cause hallucinations and seizures.
COX-2and Acetaminophen
From a structural standpoint, it is hard to see how acetaminophen would inhibit the COX site of COX because it lacks a carboxylic acid moiety for interaction with arginine 120. Early work provided a mechanistic approach to see why acetaminophen would have part of the therapeutic activities possessed by NSAIDs but lack anti-inflammatory and anticoagulatory activity. Acetaminophen exerts its effect on a subtype of COX located in the brain. The finding of 2 forms of COX validated the notion of COX subtypes, but further investigation of COX-2 failed to find it to be significantly inhibited by acetaminophen.
Reference
[edit | edit source]http://www.drugs.com/tylenol.html [1] [2] [3] [4] [5] [6] [7]
"Robert L. McNeil, Jr." Homepage of the Chemical Heritage Foundation. N.p., n.d. Web. 21 Nov. 2012. <http://www.chemheritage.org/discover/online-resources/chemistry-in-history/themes/pharmaceuticals/relieving-symptoms/mcneil.aspx>.
Introduction
[edit | edit source]Prozac, also known as Fluoxetine, is part of class of drugs known as selective serotonin reuptake inhibitors, or SSRIs, that is used to treat depression, obsessive-compulsive disorder, eating disorders, and panic attacks. The medicine works by maintaining the levels of serotonin, a neurotransmitter that controls mood and mental state, in the brain. In other cases, Prozac is used to treat alcoholism, attention-deficit disorder, borderline personality disorder, sleeping disorders, heaches, mental illnesses, post-traumatic stress disorder, Tourettes' syndrome, obesity, sexual disorders, and various irrational fears


Side Effects
[edit | edit source]Some common side effects of Prozac are depression, anxiety, headaches, nausea, diarrhea, insomnia, and loss of appetite. Due to how Fluoxetine effects neurotransmitters, it can sometimes block other neurotransmitters like for acetylcholine. When Acetylcholine is blocked, nausea occurs. Another dangerous side effect is the fact that selective serotonin reuptake inhibitors also inhibit nitric oxide synthesis which leads to the narrowing of blood vessels. This is dangerous because it can lead to hypertension. Also, even though Prozac is an antidepressant drug it can lead to depression due to the neurological chemical imbalance in the brain.
Administration
[edit | edit source]Prozac is administered orally. A capsule is taken and slowly distributed while in the digestive system.
Precautions
[edit | edit source]Prozac should never be taken more than prescribed. A major imbalance to the neurotransmitters and neurological pathways would occur and lead to many side effects. It should not be taken while pregnant, due to the fact of hypertension, which will lead to heart defects and lung problems in the newborn. Also, Prozac should not be taken in combination of other antidepressants because of seriously dangerous chemical reactions. Lastly, people under 24 years old may have thoughts of suicide.
Biochemistry
[edit | edit source]The way Prozac works is by blocking the signal on the axon that tells it when enough serotonin has been produced. Therefore, an excess amount of serotonin is delivered to the nerves. The way Fluoxetine is metabolized is by CYP2D6 in the liver. Due to its slow metabolism, it has a long half-life within the system. Because of this slow accumulation, it takes a while before the excess serotonin starts to show significant effects. Prozac acts as an agonist for 5HT2C receptors which have been known to be responsible for the defensive and aggressive behavior. It has been seen that low 5-HT levels in the brain have traditionally been associated with depression.
Mechanism of Actions
[edit | edit source]Neurotransmitters are signaling molecules that are used to transmit messages between neurons in the brain. The pathways of neurotransmitters in the synapse can be prolonged or shortened by specific chemicals called neuromodulators. Some neuromodulators can aid with the release of neurotransmitters into the synapse; others inhibit the reabsorption of neurotransmitters by neurons so that the neurotransmitters remain in the synapse. Under certain mental conditions such as bipolar disorder, obsessive-compulsive disorder, anxiety, and depression, the regulation of secretion and removal of neurotransmitters into the synapse is not functioning properly. In particular, in depression and similar disorders, neurotransmitter seretonin is one of the mostly affected. To treat depression, the most common form of drugs is called selective serotonin reuptake inhibitors (SSRI). Prozac is one of the most widely used member of this class.

Prozac works on the serotonin balance in the synapses by inhibiting a neuromdulator called SERT, which pumps serotonin back into the neurons. Thus, Prozac helps to prevent the reabsorption of serotonin and increase the amount of seretonin in the synapse. The proper amount of seratonin in the synapse is believed to help with mood elevation and depression.
History
[edit | edit source]Eli Lilly discovered fluoxetine in the 1970's when working with antihistamine diphenhydramines. She wanted to synthesize many derivatives of this compound and test them on mice. Eventually, she made a derivative only inhibiting serotonin reuptake, which she called Prozac. Many controversial accounts have been made against Prozac and Lilly. Violent outbreaks, suicides, and murders were all thought to be seen as behavior invoked by the drug. However, it has been said that Prozac is not addictive or habit forming. Once Prozac's patent had expired in 2001, many generic serotonin inhibitor drugs started selling.
References
[edit | edit source]- Johnson , George. "Smoking and drug addiction." Backgrounders. N.p.. Web. 7 December 2012.
- Reichstetter, Sandra. "How Prozac works." Brain Blogger. N.p., 19 2010. Web. 7 December 2012.
- http://panicdisorder.about.com/od/treatments/a/prozac.htm
- http://www.drugs.com/prozac.html
- http://www.encognitive.com/node/18210
- http://www.rxlist.com/prozac-drug.htm
- http://www.brainphysics.com/howprozacworks.php
- http://www.drugs.com/monograph/fluoxetine-hydrochloride.html
History
[edit | edit source]The name Etymology comes from anthrax, which is a Greek word meaning coal. It derived from the Greek word meaning coal because of the black skin lesions developed by victims with a cutaneous anthrax infection. In the year 1875, a German physician named Robert Koch first identified the bacterium that caused the anthrax disease. His research was the first to show how microbes can cause disease. Koch uncovered the life cycle and reasons of transmission of anthrax. His experiments also helped understand the role of microbes in making people sick during the period when debates still took place over spontaneous generation versus cell theory. After discovering that bacteria caused tuberculosis, Koch won the Nobel Prize in Physiology or Medicine in the year 1905.
Furthermore, in May 1881 Louis Pasteur performed a public experiment to show his thoughts on vaccination. To demonstrate this, he prepared two groups of 25 sheep, several cows, and one goat. The animals of different groups where injected with either anthrax vaccine or left unvaccinated. IN the end it was found that all the animals in the non-vaccinated group died, while all the animals in the vaccinated group survived. Then in the year 1954, the human vaccine for anthrax became available and in 1970 the improved cell-free vaccine was also made available.
Introduction
[edit | edit source]
Anthrax is a binary toxin that is secreted by Bacillus anthracis, which secretes the three proteins that composes the anthrax toxin. The anthrax toxin is composed of three proteins that are nontoxic alone, but creates a toxic complex together. The three proteins are Lethal Factor (LF), Edema Factor (EF), and Protective Antigen (PA). Lethal Factor and Edema Factor are enzymes and Protective Antigen is a multifunctional protein.
Structure
[edit | edit source]Native Protective Antigen (Native PA) is a long, flat protein that is composed of four folding domains. The first domain (Domain 1) is a β-sandwich with four helices and two calcium ions. The second domain (Domain 2) is composed of a β-barrel core and forms a pore that allows the Lethal Factor and Edema Factor into the cytosol. The third domain (Domain 3) has a ferredoxin similar fold. And the fourth domain (Domain 4) is composed of a β-sandwich with an immunoglobulin similar fold that is used to bind to cellular receptors, the fourth domain has minimal contact with the other parts of the protein.
-
 Heptameric Prepore structure.
Heptameric Prepore structure.

The Heptameric Protective Antigen Prepore is a ring with an empty interior and a flat hydrophobic surface, which is the binding site for the Lethal Factor and Edema Factor. It has a mushroom shape.
-
 Lethal Factor structure.
Lethal Factor structure.

The Edema Factor has a pocket that binds to the host cell and causes the cell to fill with fluid. The edema factor is nontoxic alone, but changes shape upon binding. The active site of the Edema Factor enzyme is a promising drug target because the pocket can be easily blocked. The lethal factor is a Zn2+ dependent endoprotease.
ANTXR1 and ANTXR2 are two cell receptors that bind PA. ANTXR1 is a medium length receptor and ANTXR2 is a long receptor. These two receptors are present everywhere on extracellular domains.
Mechanism of Toxin
[edit | edit source]
The anthrax toxin starts working when the Protective Antigen (PA) binds to either of the cellular receptors ANTXR1 or ANTXR2 and becomes proteolytically activated. Then, the Protective Antigen forms the Heptameric Prepore (PA oligomerizes to form the hollow ring). This new prepore binds to either or both of the enzymatic components of the toxin: Lethal Factor (LF) or Edema Factor (EF). The complexes made upon the binding of these enzymes is moved to the endosome; the prepore turns into a transmembrane pore and the Lethal Factor and Edema Factor cross through the pore to the cell cytosol.
Once inside the cell, Lethal Factor cuts and disables protein kinases (MAPKK 1 and MAPKK 2) that are involved in the activation of proteins vital to the cell. The Edema Factor inhibits immunological response and causes cell death.
Anthrax in Humans
[edit | edit source]Although anthrax is an infectious disease that typically affects animals, humans who come into contact with animals infected with anthrax can become infected as well. When humans have the infectious disease, the infection usually involves the skin, gastrointestinal tract, or lungs.
Routes of Anthrax Infection
[edit | edit source]With cutaneous anthrax, the most common type of anthrax infection, it usually occurs when an anthrax spore comes into contact with a cut or scrape on a person's skin.
Inhalation anthrax develops when a spore enters the lungs. Breathing in an anthrax spore does not indicate that a person will be infected with anthrax disease. It indicates that they have been exposed to anthrax. For inhalation anthrax to fully develop, the bacterial spore must sprout in order for the actual disease to take place. This typically takes 1 to 6 days. If a spore sprouts, it releases toxic substances wich can cause internal bleeding, swelling and tissue death.
Gastrointestinal anthrax occurs when someone consumes meat that has been infected with anthrax.
Symptoms
[edit | edit source]Cutaneous anthrax symptoms being to show 1 to 7 days after exposure:
- An itchy sore, similar to an insect bite, appears. The sore may blister and become black.
- Painless sore, but swells.
- A scab forms, and dries and falls off in approximately two weeks.
Inhalational anthrax symptoms:
- Fever, discomfort, headache, cough, shortness of breath (SOB), and chest pain
Gastrointestinal anthrax symptoms occur within a week and may include:
- Abdominal pain
- Normal or bloody diarrhea
- Fever
- Mouth sores
- Nausea and vomiting
Treatment
[edit | edit source]Anthrax infections are typically treated with antibiotics.
Those with inhalational anthrax are given a combination of antibiotics. Doctors usually first give the patient ciprofloxacin plus another drug, intravenously. Inhalational anthrax treatment typically takes about 60 days because spores can take up to that amount of time to sprout.
Cutaneous anthrax is treated through oral antibiotics such as doxycycline and ciproflaxin for about 7 to 10 days.
References
[edit | edit source]Croston, Glenn. "Anthrax Toxin Mechanism of Action ." Biocarta. N.p., n.d. Web. 30 Nov. 2011. <www.biocarta.com/pathfiles/h_anthraxPathway.asp>.
Karin, Michael, and Jin Mo Park, PhD. "Molecular Mechanism Underlying Anthrax Infection Described." UC San Diego Health System | San Diego Hospital, Healthcare . UCSD, 29 Sept. 2002. Web. 30 Nov. 2011. <http://health.ucsd.edu/news/2002/08_29_Karin.html>.
Young, John A. T.,Collier, R. John . "Anthrax toxin: Receptor binding, internalization, pore formation, and translocation." Annual Review of Biochemistry. Web. 30 Nov. 2011. <http://www.annualreviews.org/doi/pdf/10.1146/annurev.biochem.75.103004.142728>.
Images from Wikimedia Commons.
http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0002301/
http://en.wikipedia.org/wiki/Anthrax
Overview
[edit | edit source]Phenelzine is a nonselevitve and irreversible MAOI. MAOIs stands for monoamine oxidase inhibitors. They are antidepressant drugs that work by preventing the enzyme monoamine oxidase from metabolizing neurotransmitters. These neurotransmitters include serotonin, norepinephrine, and dopamine. Because these neurotransmitters are not metabolized, they remain in the brain and this helps elevate mood. It is also thought that the drug supports nerve cells within the brain from glutamate sensitivity. This prevents glutamate from stimulating areas of the brain that are responsible for depression and anxiety. There are two types of monoamine oxidase enzymes. There is MAO-A and MAO-B. These enzymes work by removing molecular segments from the neurotransmitters so that these neurotransmitters will become inactive and can be metabolized. Some of the side effects of the drug include tremors, arrhythmia, and seizures. The action of phenelzine binding to the enzyme permanently deactivates the enzyme and is irreversible. Also, the drug doesn't distinguish between MAO-A and MAO-B when it is inhibiting the enzyme.
Drug Usage
[edit | edit source]Phenylzine is a drug used to treat depession. As mentioned above, this drugs belongs to MAOIs ( monoamine oxidase inhibitors). When using this drugs, it will increase the amount of some natural substances that are required by your body to maintain the mental balance.
This drug will not recover people depression condition, it just aids in taking control of the symptom. The average period for taking this drug can be range from 4 weeks or longer. While taking this drug, you cannot stop if your condition is getting better. You have to consult with your doctor so that he/she can decrease your regular doses. You will experience symptoms such as vomitting, nausea, nightmare and weakness if you stop taking it.
Side Effects
[edit | edit source]As many other drugs, Phenylzine does have some side effects that should be noticed while taking this pill:
- Weakness
- Dry mouth
- Headache
- Constipation
- Shaking body parts
- Chestpain
- Constant pounding heart
- Nausea
- Night sweating
- Vomitting
- Gaining weight
- Soreness
References
[edit | edit source]http://publications.nigms.nih.gov/medbydesign/medbydesign.pdf
http://www.mayoclinic.com/health/maois/MH00072
http://online.factsandcomparisons.com
http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0000573/
AHFS® Consumer Medication Information. © Copyright, 2011. The American Society of Health-System Pharmacists, Inc.
Background Information
[edit | edit source]Pepcid is a drug with generic name Famotidine that is manufactured by Merck and Co. It is classified as a Histamine H2 Receptor Antagonist with chemical name 2-[4-[2-(amino-sulfamoylimino-methyl)ethylsulfanylmethyl]-1,3-thiazol-2-yl]guanidine. Famotidine is used to treat and prevent ulcers in the stomach and intestines. Famotidine is also used to treat conditions in which the stomach produces too much acid and conditions in which acid comes up into the esophagus and causes heartburn, such as gastroesophageal reflux disease (GERD). The drug is available in suspension, injection, and tablet form. 15% to 20% of PEPCID in plasma is protein bound. Administered PEPCID tablets and suspension are incompletely absorbed and its bioavailability is 40-45%. PEPCID is excreted by renal (65-70%) and metabolic (30-35%) routes with the only metabolite identified is the S-oxide. It has a half life of 2.5-3.5 hours. The drug works by inhibiting gastric secretion. This is done through competitive inhibition of histamine H2 receptors. These receptors located in the parietal cell are responsible for the secretion of HCl in the stomach. When mast cells are agitated, they release the H2 substrate which will then bind the receptor site and produce HCL. The drug competes with the H2 for the receptor site to prevent binding and thus the release of gastric acid. Some of the severe side effects associated with the drug are seizures, Arrhythmia, and diarrhea.

History
[edit | edit source]Yamanouchi Pharmaceutical Co. developed pepcid (famotidine). In the mid-80s, Merck & Co. licensed pepcid and is marketed by a joint venture between Merck and Johnson & Johnson. A 2-guanidinothiazole ring was used as a replacement for imidazole-ring. In the end, many studies showed that famotidine was thirty times more active than cimetidine.
In the year 1999, pepcid RPD orally-disintegrating tablets were released. They were known to not be swallowed. Then in the year 2001, generic preparation became available. For example, Fluxid (Schwarz) or Quamatel (Gedeon Richter Ltd).
In addition, there was also a product called Pepcide Complete in the United States. However, in the United Kingdom, this product is known as Pepcidtwo. Pepcide Complete was a kind of product that had a combination of famotidine with an antacid in a chewable tablet to ameliorate the relatively slow onset of effects.
However, Pepcid carries some consequences. For example, famotidine is poorly soluble in the low pH of the stomach and it suffers from poor bioavailability (50%). Famotidine used in combination with antacids promotes local delivery of these drugs to the receptor of the parietal cell wall. Hence, many researchers out there are finding ways to develop innovative formulations of tablets, such as gastroretentive drug delivery systems. This is because this type of drug is known to be in stomach for a longer period of time and thus improve the bioavailability of drugs.
Medical Use
[edit | edit source]In different countries in the world, certain preparations of famotidine are available over the counter (OTC). For instance, in the United States, preparations of 10 mg and 20 mg tablets, sometimes even in the combination with a more traditional antacid, are all available as an over the counter drug. However, if an individual is deciding to take a larger dose, it is important that they consult with their doctor to give them a prescription. In this case, a prescription will allow them to get a good amount of famotidine they need from the OTC.
Commonly seen, pepcid is given to patients who are going through a surgery process. They are typically given before the operations to prevent postoperative nausea and to decrease any risk of aspiration pneumonitis. In addition, pepcid is given to some patients who take NSAIDs. Taking pepcid will basically prevent them from peptic ulcers because this medication usually serves as an alternative to proton-pump inhibitors. An interesting fact is that pepcide is not only given to humans but they are also given to animals such as dogs. They are given to dogs with acid reflux.
Many people who take famotidine also use this medication with the combination of H1 antagonist to treat and prevent urticaria. Urticaria is caused by an acute allergic reaction. Famotidine has been found to reduce the debilitating effects of chronic heart failure by blocking histamine.
Overall, pepecid or famotidine can be used as a useful drug, especially when it is used in a proper manner.
Side Effects of Pepcid
[edit | edit source]-Gastrointestinal
Gastrointestinal side effects including diarrhea (1.7%) and constipation (1.2%) have been stated. Vomiting, nausea, abdominal distress, anorexia, and dry mouth have been reported infrequently. Hypergastrinemia of uncertain clinical significance has also been reported.
-Nervous system
Grand mal seizure; psychic disturbances, which were alterable in cases for which follow-up was found, including hallucinations, confusion, agitation, depression, anxiety, decreased libido; paresthesia; insomnia; somnolence. Convulsions, in patients with reduced renal function, have been reported very rarely.
-Hepatic
Hepatic side effects have included mild elevations of liver function tests. The clinical significance of these elevations is unknown. Jaundice and cholestatic jaundice have been reported infrequently. Cases of drug-induced hepatitis have also been reported.
-Cardiovascular
Cardiovascular side effects have included decreases in stroke volume and cardiac productivity and may be clinically significant in patients with previous cardiac dysfunction. A variety of arrhythmias, including bradycardia, tachycardia, AV conduction defects (including AV block), and palpitations have also been reported rarely. Prolonged QT interval has been reported very rarely in patients with impaired renal function whose dose/dosing interval of famotidine may not have been adjusted properly.
-Hypersensitivity
Hypersensitivity side effects have infrequently included anaphylaxis, angioedema, orbital or facial edema, urticaria, rash, conjunctival infection, toxic epidermal necrolysis (very rare), erythema multiforme, and Stevens-Johnson syndrome.
-Renal Renal side effects have contained rare cases of interstitial nephritis.
-Endocrine
Endocrine side effects have included anti-androgen effects of reversible hyperprolactinemia and gynecomastia.
-Dermatologic
Dermatologic side effects have infrequently included alopecia, acne, pruritus, dry skin, and flushing. At least one case of contact dermatitis in a worker handling pepcid has been reported.
-Hematologic
Hematologic side effects including neutropenia have been reported. Rarely, reversible thrombocytopenia, agranulocytosis, pancytopenia, and leukopenia have been reported.
-Respiratory
Respiratory side effects including bronchospasm and interstitial pneumonia have been reported infrequently.
-Musculoskeletal
Musculoskeletal side effects have uncommonly contained musculoskeletal pain including muscle cramps and arthralgia.
-Other
Other side effects including tinnitus, fever, asthenia, fatigue, and taste disorder have been reported occasionally. At least one case of hyperpyrexia in association with pepcid use has been reported.
References
[edit | edit source]http://online.factsandcomparisons.com
http://www.mamashealth.com/stomach.asp
http://en.wikipedia.org/wiki/Famotidine
http://www.drugs.com/pepcid.html
http://www.rxmed.com/b.main/b2.pharmaceutical/b2.prescribe.html
http://www.rxlist.com/pepcid-drug.htm
http://gerd.emedtv.com/pepcid/pepcid-side-effects.html
http://medical-wiki.com/articles/pepcid-side-effects-pepcid-ac-side-effects/
Overview
[edit | edit source]Adalat is drugs with generic name nifedipine produced by Bayer and is classified as a calcium channel blocker. It has the chemical name 3,5-pyridinedicarboxylic acid, 1,4-dihydro=2,6=dimethyl-4=(2=nitrophenyl I)-dimethyl ester. The drug is often prescribed to treat hypertension. The drug is available in tablet form and has a bioavailability of 84-89 percent after oral administration. Nifedipine is metabolized via the cytochrome P450 3A4 system, located both in the intestinal mucosa and in the liver with a half life of 2 hours. The drug is 1,4 dihydropyridine calcium channel blocker and is a selective vasodilator. Normally in the heart, heart rate is increased by calcium ions binding to calcium channels within the heart. The increased heart rate causes vasoconstriction in the arteries by shrinking the artery wall. What the drug does is actually block calcium from interacting with the calcium channels within the heart and this allows the artery walls to dilate to prevent hypertension while the heart slows down. Some of the extreme side effects include hypotension, arrhythmias, and congestive heart failure. It is also important to not have grape fruit with this drug.
Side Effects
[edit | edit source]Contact a doctor if you present one or more of the following symptoms after taking adalat:
- hives
- difficulty breathing
- swelling of lips, face, throat or tongue
- worsening angina
- feel like passing out
- short of breath
- swelling on your limbs
- fast heartbeat pounding
- numbness
- jaundice (yellow skin or eyes)
- chest pain or heavy feeling
Less serious side effects include:
- headache
- dizziness
- drowsiness
- constipation
- nausea
- diarrhea
- stomach pain
- insomnia
- rash or itching
- joint pain
- more urination than usual
Incompatibility with other drugs
[edit | edit source]Adalat might produce severe reactions if mixed with one or more of the following medications:
- acarbose
- cimetidine
- fentanyl or other narcotic pain medications
- digoxin
- nefazodone
- St. John's wort
- rifabutin
- blood thinner
- anti-fungal medication
- beta-blocker
- heart rhythm medication
- HIV/AIDS medication
- seizure medication
References
[edit | edit source]http://www.drugs.com/adalat.html
http://www.medterms.com/script/main/art.asp?articlekey=3846
http://www.rxlist.com/adalat-drug.htm
http://online.factsandcomparisons.com
Introduction
[edit | edit source]
Bosulif (Bosutinib) is a new drug that is approved recently by the U.S. Food and Drug Administration (FDA) in order to treat people with Philadelphia chromosome- which is a genetic mutation when an enzyme called tyrosine kinase is produced by the bone marrow. This drugs is used to treat people with positive chronic myelogenous leukemia (CML). A positive CML is when a patient encounters chronic, accelerated CML. Bosulif is an Abl and Src kinase inhibitor. It contains mainly bosutinib which is shown in the picture. Because it is a new drug, so it is recommended to be used only when patients are not tolerated with other treatments. With this approval, we have seen an effective improvement in this treatment.
Usage and Side Effects
[edit | edit source]Bosulif cannot be used for patients who are allergic to bosutinib or any ingredient that belongs to this drug. It is produced as a tablet so you can easily use this with food. Avoid to use grapefruit or grape juice with this drug since Bosulif can be absorbed in the presence of stomach acid, that means the amount of Bosulif will be increased. You have to consult your doctor for doses and only take only the same amount that is prescribed. This drug also has some serious side effects if it is not used properly.
- Stomach problem, diarrhea, vomit, headache, cough, fever.
- Low blood cell counts, liver problems
- Shortness of breath, chest pain, weight gain.
Reference
[edit | edit source]http://www.drugs.com/bosulif.html http://www.medicalnewstoday.com/articles/249971.php
Overview
[edit | edit source]The drug Luvox is a selective serotonin reuptake inhibitor (SSRI) with the generic name Fluvoxamine Maleate and the chemical name : 5-methoxy-4’-(trifluoromethyl)valerophenone-(E)-O-(2-aminoethyl)oxime maleate (1:1). It can be used in the treatment of chronic depression, obsessive compulsive disorder (OCD) and social anxiety disorders. The drug works to increase the amount of serotonin in the brain in order to ward off feelings of depression, anxiety, and fear. Serotonin is a naturally produced neurotransmitter in the body that maintains mental balance and feelings of calm or happiness.[8]
The drugs comes in a tablet form and has approximately 80% of fluvoxamine bound to plasma protein, consisting of mostly albumin, over a concentration range of 20 to 2000 ng/mL. The absolute bioavailability of fluvoxamine maleate is 53%. The tablet should be taken orally by mouth without crushing or chewing them. Dosages are usually prescribed by doctors starting with low doses and gradually increasing the amount over time. The full effect of the drug may take up to several weeks to start working.
Biochemical Interactions
[edit | edit source]Fluvoxamine maleate is extensively metabolized by the liver. The main human metabolite is fluvoxamine acid and the drug has a half-life of 15.6 hours in the body. The drug targets chemical imbalances in the brain to help treat chronic depression.
Between the synapse and a nerve cell, there is an exchange of neurotransmitters. Serotonin is one of these neurotransmitters that can send messages between nerve cells. The nerve cells contain reuptake channels that can reabsorb neurotransmitters when their job has been performed and they are no longer needed. When the message exchange is faster, serotonin spends less time in the synapse as it is taken back up faster, which is believed to cause depression. If the levels of serotonin in the body become unbalanced, OCD or other anxiety disorders can occur. SSRI drugs, like Luvox, can selectively bind to the reuptake channels of serotonin, therefore blocking the reuptake channels from reabsorbing serotonin. This causes the serotonin to remain in the synapse, thus helping to fight off feelings of depression. Studies have proved that Luvox is an effective drug to treat OCD in both adults and children.[9]
Side Effects
[edit | edit source]Some serious side effects include seizures, fever, and increased thoughts of suicide. Some of the more serious side effects include chest pain, coordination issues, dizziness, hallucinations, pain, loss of consciousness, difficulty breathing, and vomiting or excreting blood.
Other signs of an allergic reaction listed below may be encountered when taking this medication:
- skin rash or hives
- difficulty breathing
- swelling of face, lips, tongue, or throat
Individuals should immediately call the doctors if any new or worsening symptoms such as mood or behavior changes, anxiety, trouble sleeping, panic attacks, or if feeling impulsive. If you stop taking fluvoxamine suddenly, you may feel some withdrawal symptoms, which include irritability, dizziness, headaches, changes in mood, pain, difficulty in sleeping, or tingling in the extremities. Doctors should assist in decreasing doses gradually before you stop taking the drug.[10]

Precautions
[edit | edit source]Before taking fluvoxamine, it is important to inform the doctor or pharmacist if any kind of allergic affect can happen because this kind of medication contains inactive ingredients that can cause allergic reactions or other serious problems. Be wise when taking this kind of medicine. Remember to always tell the doctor of pharmacist of one's medical history especially that of family history disorder, personal history disorder, etc.
When taking this medication, remember that driving may be dangerous because dizziness may occur. Therefore, do not use machinery and do not drive. Also do not do any kind of activity that requires a good sense of alertness until one is sure that it is safe to perform that certain activity. Remember to never drink alcoholic beverages when taking fluvoxamine.
People of different ages may have different side effects of this drug. For example, older adults may be more sensitive to the side effects, especially that of bleeding. Taking water pills, older adults many develop a type of mineral imbalance in their body. Just like older adults, young people such as children may be more sensitive to the side effects of this drug. Children may encounter a lost of appetite and weight loss. Hence, it is important for parents to monitor their children weight and height while taking this medication.
Pregnant women who are taking fluvoxamine may harm their unborn baby in the stomach. Therefore, it is always better to ask the doctor before actually taking it. If a mother notice their three month baby having any of this symptoms listed below, they should immediately contact their baby doctor:
- feeding/breathing difficulties
- seizures
- muscle stiffness
- constant crying
Breast feeding mother should contact doctor.
References
[edit | edit source]- ↑ http://www.chemistryexplained.com/A-Ar/Acetaminophen.html
- ↑ http://www.ncbi.nlm.nih.gov/pubmed/20020268
- ↑ http://tuftsjournal.tufts.edu/2008/04/professor/01/
- ↑ http://dmd.aspetjournals.org/content/31/12/1499.full
- ↑ http://www.tylenolprofessional.com/pharmacology.html
- ↑ http://cid.oxfordjournals.org/content/31/Supplement_5/S211.full
- ↑ http://news.consumerreports.org/health/2011/11/repeatedly-taking-too-much-tylenol-can-be-dangerous.html
- ↑ http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0000955/
- ↑ http://anxiety.emedtv.com/luvox/luvox-p2.html
- ↑ http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0000955/
http://www.medicinenet.com/fluvoxamine/article.htm
http://online.factsandcomparisons.com
Introduction
[edit | edit source]Norethindrone (or norethisterone) is a form of female hormone that prevents ovulation (the release of egg from an ovary). This medicine causes changes in a woman's cervical mucus and uterine lining, making it harder for sperm to reach the uterus and therefore harder for conception.
Norethindrone is used for birth control (contraception) to prevent pregnancy. Norethindrone is also used to treat menstrual disorders, endometriosis, or abnormal vaginal bleeding caused by a hormone unbalance.
Side Effect
[edit | edit source]Immediately contact for medical help if you have any of these signs of an allergic reaction after consuming Norethindrone:
- hives
- difficulty breathing
- swelling of your face, lips, tongue, or throat.
Stop using this medication and call your doctor if you have any of these serious side effects:
- sudden numbness or weakness, especially on one side of the body;
- sudden headache, confusion, pain behind the eyes, problems with vision, speech, or balance;
- pain or swelling in one or both legs;
- migraine headache;
- swelling in your hands or feet, rapid weight gain;
- symptoms of depression (sleep problems, weakness, mood changes);
- severe pelvic pain;
- chest pain or heavy feeling, pain spreading to the arm or shoulder, nausea, sweating, general ill feeling; or
- nausea, stomach pain, low fever, loss of appetite, dark urine, clay-colored stools, jaundice (yellowing of the skin or eyes).
- Less serious side effects may include:
- mild nausea, vomiting, bloating, stomach cramps;
- breast pain, swelling, or tenderness;
- dizziness;
- freckles or darkening of facial skin;
- increased acne or hair growth;
- changes in weight;
- vaginal itching or discharge;
- skin itching or rash;
- changes in your menstrual periods, decreased sex drive; or
- mild headache.
History of Synthesis
[edit | edit source]In 1951, norethindrone was synthesized by chemists Luis Miramontes, Carl Dijerassi, and George Rosenkranz at Syntex in Mexico City.
Synthesis
[edit | edit source]The synthesis of 19-nor-17α-ethnyltestosterone:

Structure
[edit | edit source]
Functional groups: ketone, alkene, methyl(2) , alcohol, alkynes
Reference
[edit | edit source]- http://www.drugs.com/mtm/norethindrone.html
- Djerassi, C.; Miramontes, L.; Rosenkranz, G.; Sondheimer, Franz (1954). "Steroids. LIV. Synthesis of 19-Nor-17α-ethynyltestosterone and 19-Nor-17α-methyltestosterone". J. Am. Chem. Soc. 76 (16): 4092–94. doi:10.1021/ja01645a010
- http://commons.wikimedia.org/w/index.php?title=Special%3ASearch&profile=default&search=Norethisterone+&fulltext=Search
Overview
[edit | edit source]Paclitaxel (taxol) is an important anti-cancer pharmaceutical drug. In 1967, Monreoe E. Wall and Mansukh C. Wani extracted paclitaxel from the bark of a Pacific yew tree, Taxus brevifolia.
Paclitaxel is used to treat AIDs-related Kaposi sarcoma, breast, ovarian, and lung cancer in UK. This medicine binds to the tubulin and inhibits the disassembly of microtubules. This stabilizes the microtubules and prevents further cell division.
Side Effects
[edit | edit source]Seek medical attention if you have these side effects:
- Hives
- Difficulty breathing
- Feeling like you might pass out
- Swelling of the face, lips, tongue, or throat
- Slow heart rate
- Seizure
- Pale skin, easy bruising or bleeding, unusual weak
- Fever, chills, body aches, flu symptoms
- Sores inside your mouth or on your lips
- Numbness
- Increased blood pressure
- Joint and muscle pain
- Mild nausea, and vomiting
- Hair loss
Semisynthesis
[edit | edit source]
Biosynthesis
[edit | edit source]
Structure
[edit | edit source]The structure of (1S,2S,3R,4S,7R,9S,10S,12R,15S)-4,12-Diacetoxy-15-{[(2R,3S)-3- (benzoylamino)-2-hydroxy-3- phenylpropanoyl]oxy}-1,9- dihydroxy-10,14,17,17-tetramethyl -11-oxo-6-oxatetracyclo [11.3.1.0~3,10~.0~4,7~] heptadec-13-en-2-yl rel-benzoate:
Functional Groups: aromatic(3), amide(1), ketone(2), esters(4), alcohol(3), and ether(5) It has 11 stereocenters.
Mechanism of Actions
[edit | edit source]
Microtubules are cellular components that act as a skeleton for the cell. For cell division to occur, microtubules need to depolymerise back to tubulin. After that, tubulins repolymerise to form the spindle of cell division. The movement of the replicated chromosomes during mitosis depends on the spindle and therefore, the depolymerization of microtubules.
Paclitaxel or Taxol enhances the polymerization of tubulin to stable microtubules and also interacts directly with microtubules, stabilizing them against depolymerization. Hence, it interferes with the spindle formation process. Chromosomes are unable to move to the opposite sides of the dividing cells. Cells division is inhibited and eventually, cell death is induced.
References
[edit | edit source]Borad, Brijesh. "CYTOTOXIC EFFECT OF TAXOL AND VITAMIN E SUCCINATE COMBINATION IN COMPARISON WITH TAXOL ALONE ON MCF-7 BREAST CANCER CELL CULTURE BY MTT ASSAY." PharmaTutor. N.p.. Web. 7 Dec 2012.
- http://www.rxlist.com/taxol-drug.htm
- http://www.cancer.gov/cancertopics/druginfo/paclitaxel
- http://en.wikipedia.org/wiki/Paclitaxel#Mechanism_of_action
Overview
[edit | edit source]Allegra has generic name Fexofenadine and is classified as a Histamine H1-receptor antagonist. It is manufactured by Advancis Pharmaceutical Corporation and has chemical name (±)-4-[1 hydroxy-4-[4-(hydroxydiphenylmethyl)-1-piperidinyl]-butyl]-, -dimethyl benzeneacetic acid hydrochloride. One of the main uses is to treat symptoms caused by allergies. Fexofenadine hydrochloride is 60% to 70% bound to plasma protein, primarily albumin and 1-acid glycoprotein. Approximately 5% of the total dose of fexofenadine hydrochloride is eliminated by hepatic metabolism through metabolite terfenadine. The half-life is 14.4 hours for 60 mg. The drugs works by competitively inhibiting the H-1 receptor site and preventing histamine-1 from attaching the receptor sites to cause allergy symptoms. Histamine-1 is released by mast cells in the presence of certain stimulus that the body is allergic to. The drug does not prevent the release of histamine-1 by the mast cells but rather competes with the histamine-1 substrates for positions at the receptor sites.

Why prescribe this?
[edit | edit source]Fexofenadine is prescribe to relieve allergy symptoms of seasonal allergic rhinitis such as hay fever. Some allergy symptoms in children over 2 years old and adults include runny nose; sneezing; red, itchy, or watery eyes; or itching of the nose, throat, or roof of the mouth. It is also used to relieve symptoms of urticaria such as red, itchy raised areas of the skin, including itching and rash in children 6 months and over and adults. In all, Fexofenadine is in a class of medications called antihistamines. Antihistamines blocks the effects of histamine, a substance in the body that cause allergy to happen. Fexofenadine is known only for controlling the symptoms of seasonal allergic rhinitis and urticaria but it is not a cure for these conditions.
How should this medication be used?
[edit | edit source]Fexofenadine comes as a tablet or liquid to take by mouth. People who are taking this medication normally take it with water once or twice a day depending on how bad their allergy symptom is. Do not take any kind of fruit juices, such as orange, grapefruit, or apple juice because the performance of fexofenadine in the body system will not be as effective. Remember to take this medication at around the same time(s) every day. Carefully read what the prescription label states and follow it accordingly. Do not take more or less of it or take it more often than prescribed by the doctor. If fexofenadine is use in liquid form, it is crucial to shake the bottle well before use.
Side Effects
[edit | edit source]Taking fexofenadine may cause certain side effects. When any of these side effects listed below are severe and do not go away it is important to quickly consult with the doctor.
- headache
- dizziness
- diarrhea
- vomiting
- pain in the arms, legs, or back
- pain during menstrual period
- cough
Note: Serious side effects and should call doctor immediately are hives, rash, itching, difficulty breathing or swallowing, and swelling in face area or lips.
Synthesis
[edit | edit source]Fexofenadine may be synthesized as shown from piperidine-4-carboxylate ester and 4-bromophenylacetonitrile.[1][2]

References
[edit | edit source]http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0001008/
- ↑ Daniel Lednicer (1999). The Organic Chemistry of Drug Synthesis. Vol. 6. New York: Wiley Interscience. ISBN 0-471-24510-0.
- ↑ Kawai, SH; Hambalek, RJ; Just, G (1994). "A facile synthesis of an oxidation product of terfenadine". J. Org. Chem. 59 (9): 2620–22. doi:10.1021/jo00088a056.
{{cite journal}}: Unknown parameter|month=ignored (help)
http://www.medicinenet.com/fexofenadine-oral/article.html

Intro
[edit | edit source]Adderall is a specific brand name drug that is used mostly for treatment of ADHD (Attention Deficit Hyperactivity Disorder) but can also be used to treat certain forms of narcolepsy. Dextroamphetamine-amphetamine otherwise known as Adderall, is a drug available by prescription that aids in the concentration of patients who suffer from attention disorders such as Attention Deficit Hyperactivity Disorder (ADHD) and sleeping disorders like narcolepsy. Adderall is a schedule II drug in the US, meaning that it has the ability to cause physical and psychological dependence, has a high potential of being abused (due to the fact that it is a combination out of four amphetamine salts) but has been approved for medical usage[1]. Adderall is a stimulant meaning that it is a psychoactive drug that can improve mental and physical functions. It is also a relative to the phenethylamine and amphetamine families.</g>

Medical use
[edit | edit source]As stated above, Adderall is a prescription medication used to treat many forms of ADHD (increased or more difficulty focusing, controlling actions, and remaining still or quiet than other people who are the same age) and narcolepsy (a sleep disorder that causes excessive daytime sleepiness and sudden attacks of sleep),[1] although its primary role is to treat ADHD patients. There are two forms commonly found on the market: Adderall XR (extended release) and Adderall IR (instant release). Both are used to treat ADHD, but only the IR form is used in treatment of narcolepsy. Adderall is thought to stop the reuptake of chemicals such as norepinephrine and dopamine, which are thought to better aid persons with ADHD as different levels of the chemicals should cause a better ability to focus, to control actions and finally to remain silent, or still in relation to persons from their same age group.
Side effects
[edit | edit source]Many side effects include : nervousness, restlessness, difficulty falling asleep, or staying asleep, uncontrollable shaking of a part of the body, headache, changes in sex drive or ability, dry mouth, stomach pain, nausea, vomiting, diarrhea, loss of appetite, weight loss. Other more adverse side effects many occur.
Chemistry
[edit | edit source]

As previously stated, Adderall is a class of both the phenethylamine and amphetamine families. Phenethylamine is an organic compound that is part of many drugs that have psychoactive and stimulant effects [2] and is typically found in stimulants, psychedelics, entactogens, anorectics, bronchodilators, decongestants and anti-depressants [3]. Amphetamines are psychostimulants that are a branch of the phenethylamine class that induces focus and wakefulness. More specifically, dextroamphetamine is a stereoisomer of amphetamine. The dextro- prefix signifies the clockwise rotation of a polarized light plane. Since Adderall is composed of dextroamphetamine and amphetamine; it is composed of both enantiomers of amphetamine.
Biochemical Interactions/Composition
[edit | edit source]Adderall is an amphetamine, which can be described as non-catecholamine sympathomimetic amines with central nervous system stimulant effects. In essence, it is a mixture of amphetamine based salts which are thought to stop the reuptake of norepinephrine and dopamine back into the neurons. In the Adderall XR form, the composition of the drug includes a three to one ratio of d-amphetamines and l-amphetamines respectively. The four ingredients that are a part of Adderall XR include Dextroamphetamine saccharate, Amphetamine Aspartate, Dextroamphetamine Sulfate, and Amphetamine Sulfate, each of which are in equal proportions.
Pharmacology
[edit | edit source]The exact mechanism of dextroamphetamine (d-AMP) in treatment of ADHD is not yet completely understood, however it is known that it plays an important role in the efficiency of dopamine neurotransmission. The stereoisomer of dextroamphetamine, amphetamine is known for releasing dopamine from the nerve terminal by using dopamine active transporter (DAT) as a carrier [4]. Despite how Adderall is used to enhance concentration, studies have shown that amphetamines do not optimize short-term memory nor does it improve “cognitive flexibility.” In fact there is evidence that it may be impairing to short-term memory and cognitive flexibility [5]. D-AMP is also known to be an reuptake inhibitor because it is an substrate analog, therefore it competes with neurotransmitters (chatecholamine) such as dopamine with similar monoamine, groups for uptake [6]. Since d-AMP induces a dopamine effect by increasing its concentration, a user feels the effects of the excitatory neurotransmitter. At really high doses, d-AMP is known for causing euphoria by triggering a cascade of catecholamines that release large amounts of neurotransmitters and an overstimulation of receptors [8]. It is postulated that d-AMP tends to interact with the dopamine systems while its enantiomer tends to interact with the norepinephringernic systems [9].
Another way dextroamphetamine chemically affects the body was postulated through research in bipolar disorder by Silverston et. al.(2002). Studies suggest that dextroamphetamine increases the activity of the phosphoinositol cycle through the dopamine and noradrenaline. The phosphoinositol cycle was discovered by Lowell and Hopkins, and is known to generate phosphatidylinosol which is a precursor to lipid signaling molecules in eukaryotes to help regulate specific functions [10].
Pharmacokinetics
[edit | edit source]According to a medication of pamphlet of Adderall XR®, the mean elimination half-life for d-AMP is 10 hours in adults; 11 hours in persons less than or equal to 165 lbs and 9 hours in children 6-12 years of age. In the enantiomer, l-amphetamine, the mean elimination half-life is 13 hours, 13-14 hours in persons less than or equal to 165 lbs and 11 hours in children 6-12 years [11].
Metabolism
[edit | edit source]All enzymes for the metabolism of amphetamine has not yet been identified. However, CYP2D6 is known to be responsible for the formation of 4-hydroxyamphetamine which in turn oxidizes into α-hydroxyamphetamine and undergoes further unknown metabolism. Amphetamine is also known to be an inhibitor of CYP2D6 enzyme [11].
Secretion
[edit | edit source]Amphetamines are sensitive to pH with a pKa of 9.9 and are therefore dependent on the pH of the urine for proper disposal. “Acidic pHs and high flow rates result in increased renal elimination…indicating the involvement of active secretion [11].”
Adderall Abuse
[edit | edit source]Adderall is abused as:
- A study drug: Its nicknames include “college crack” or “cognitive steroid.” Students who take Adderall for studying report that they can focus on their book well, and can do better on the following exam than they would without the drug. - A party drug: Adderall contains dextroamphetamine, which creates a feeling of well-being, confidence and enhanced libido, and enables users to go without sleep for extended periods. - A weight-loss drug: Appetite conquest is often a side effect of Adderall and is mostly loved by young women looking for an easy way to lose weight.
Why are Adderall and abused?
The same properties that make Adderall effective in treating many cases of ADHD have made it a drug of choice for non-ADHD students, who find it tempting for other reasons.
Adderall works by changing the brain’s chemistry in a way that helps ADHD patients pay attention to the task at hand despite distractions. In non-ADHD users, Adderall produces a flow of focus and energy and an associated loss of appetite. Although casual Adderall consumption is considered as acceptable among students at many schools, use of any prescription drug for a purpose other than its prescribed use, or by a person for whom it hasn’t been prescribed, constitutes abuse.
The Results of Adderall Abuse
Misusing or abusing Adderall can cause a range of problems including difficulty sleeping, feelings of hostility, anxiety or paranoia, and a hyper-energetic, aroused state. Some users may experience undesirable or unhealthy weight loss. Especially when snorted, Adderall can cause a potentially dangerous increase in heart rate, body temperature and blood pressure. Caregivers screen ADHD patients to avoid prescribing Adderall to those with conditions that could make the drug dangerous for them. Abusers who take the drug without a prescription don’t have the benefit of such screening. Ironically, although the primary reason for Adderall’s abuse on campus is to enhance academic performance, students in whom the drug causes a manic state may not be able to exercise good judgment about the quality of the papers or exams they produce while influenced by Adderall. After abusing Adderall, users often experience a “crash” and suffer from exhaustion, nausea, depression or irritation. Some Adderall abusers smoke marijuana as an antidote for these distressing symptoms. After long-term abuse, your body depends on Adderall for normal function and trying to discontinue the drug can result in withdrawal symptoms including panic, suicidal thoughts and nightmares.
Reference
[edit | edit source]http://adhd.emedtv.com/adderall/adderall-abuse.html http://adderallabuse.com/ http://www.webmd.com/drugs/drug-63163-Adderall+Oral.aspx?drugid=63163&drugname=Adderall+Oral http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0000166/ http://www.drugs.com/pro/adderall.html 1. 21 USC § 812 – Schedules of controlled substances | LII / Legal Information Institute http://www.law.cornell.edu/uscode/text/21/812
2. Glen R. Hanson, Peter J. Venturelli, Annette E. Fleckenstein (2005-11-03). "Drugs and society (Ninth Edition)". Jones and Bartlett Publishers. ISBN 978-0-7637-3732-0
3. Phenethylamine. http://en.wikipedia.org/wiki/Phenethylamine
4. Kuczenski, R., and D. S. Segal. 1997. Effects of methylphenidate on extracellular dopamine, serotonin, and norepinephrine: comparison with amphetamine. J. Neurochem. 68:2032–2037.
5. Lackhan S. et al. (15 June 2012). " Prescription stimulants in individuals with and
without attention deficit hyperactivity disorder: misuse, cognitive impact, and adverse effects". Brain and Behavior.
6. Kuczenski R et al. (1 February 1995). "Hippocampus Norepinephrine, Caudate Dopamine and Serotonin, and Behavioral Responses to the Stereoisomers of Amphetamine and Methamphetamine". The Journal of Neuroscience 15 (2): 1308–1317.
7. Patrick, and Markowitz; Markowitz, John S. (1997). "Pharmacology of Methylphenidate, Amphetamine Enantiomers and Pemoline in Attention-Deficit Hyperactivty Disorder".Human Psychopharmacology 12 (6): 527–546 (Page:530).
8. InterScience | Dextroamphetamine increases phosphoinositol cycle activity in volunteers: an MRS study
9. http://en.wikipedia.org/wiki/Adderall
10. Heck, J. et.al, 'A Conspicuous Connection: Structure Defines Function for the Phosphatidylinositol-Phosphate Kinase Family', Critical Reviews in Biochemistry and Molecular Biology, 42:1, 15 – 39
11. FDA (2007). Medication guide adderall xr. pp. 1–14. Archived from the original on 30 January 2011.
Introduction
[edit | edit source]Ritalin, also known as methylphenidate, is a psychostimulant drug that works on the central nervous system. It has been typically prescribed to treat ADHD (or Attention Deficit Hyperactivity Disorder) and narcoleptic patients since 1960; However, it got widely spread in the 1990's when ADHD became more widely accepted. Methylphenidate is a white, odorless, fine crystalline powder which is acidic. Ritalin is freely soluble in water and methanol, soluble in alcohol, and slightly soluble in chloroform and acetone.
History
[edit | edit source]History
Methylphenidate was first synthesized in 1944, and was identified as a stimulant in 1954. Methylphenidate was synthesized by Ciba (now Novartis) chemist Leandro Panizzon. His wife, Marguerite, had low blood pressure and would take the drug as a stimulant before playing tennis. He named the substance Ritaline, after his wife's nickname, Rita. Originally it was marketed as a mixture of two racemates, 80% (±)-erythro and 20% (±)-threo. Subsequent studies of the racemates showed that the central stimulant activity is associated with the threo racemate and were focused on the separation and interconversion of the erythro isomer into the more active threo isomer. Beginning in the 1960s, it was used to treat children with ADHD or ADD, known at the time as hyperactivity or minimal brain dysfunction (MBD). Production and prescription of methylphenidate rose significantly in the 1990s, especially in the United States, as the ADHD diagnosis came to be better understood and more generally accepted within the medical and mental health communities. In 2000 Janssen received U.S. Food and Drug Administration (FDA) approval to market "Concerta". See the "Extended-release" section of this article, below, for more information about Concerta.
Side Effects
[edit | edit source]Methylphenidate has side effects; if these side effects are severe or do not go away, it is essential to contact a doctor:
- Nervousness
- Difficulty falling asleep or staying asleep
- Dizziness
- Vomiting
- Loss of appetite
- Stomach pain
- Diarrhea
- Nausea
- Heartburn
- Dry mouth
- Headache
- Muscle tightness
- Uncontrollable movement of a part of the body
- Restlessness
- Numbness, burning or tingling in extremities
- Decreased sexual desire
- Painful menstruation
Taking Ritalin
[edit | edit source]Ritalin comes in different forms including: immediate-release tablets, chewable tablets, liquids, extended-release tablets, extended release capsules, and extended-release tablets.
All forms of methylphenidate are taken orally. Adults typically take the medication three times a day, while children take it twice a day, typically before meals. To avoid the side effects of being unable to sleep, patients should not take their last dosage after 6PM.
References
[edit | edit source]http://www.rxlist.com/ritalin-drug.htm
http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0000606/
http://psychopharmacopeia.com/index.php?generic=methylphenidate
Introduction
[edit | edit source]Paxil (also recognized by the names Sereupin, Seroxat, Aropax, and Paroxetine) is an antidepressant drug belonging to a class of molecules called selective serotonin reuptake inhibitors (SSRI's). It can be used to treat depression, obsessive compulsive disorder, panic disorder, social anxiety, post traumatic stress disorder, and premenstrual dysphoric stress disorder. Paraxil is an orally administered psychotropic drug found in tablet form with the molecular formula of C19H20FNO3•HCl•1/2H2O and a molecular weight of 374.8.
Instructions
[edit | edit source]Paxil should be administered for depression as a single daily morning dose for of 20 mg/day. Dose ranges may range from 10 mg to 50 mg and should be reviewed weekly. This drug is known to be effective up to a year of administration. Paxil tablets exist in different concentrations marked by color: 10 mg- yellow, 20 mg- pink, 30 mg- blue, 40 mg- green. They should be stored at or under 25°C. This drug has proven to have teratogenic effects on pregnant women, so prior consideration should be given before being taken. Studies have discovered detrimental lung and heart disorders in newborns whose mothers took Paxil. It is designated as a black box warning by the Food and Drug Administration as minors younger than age 18 are discouraged from using it. Paxil should not be taken in conjuction with Orap, Mellaril, Zyvox, Urolene Blue, MAOI, Furoxone, Marlplan, Nardil, Azilect, Eldepryl, or Parnat to avoid adverse drug interactions. In addition, the doctor must be notified of any liver or kidney disease, blood clotting disorders, a history of depression, and prior suicidal thoughts.
Side Effects
[edit | edit source]Contact your doctor if your display any of these side effects.
- Bone pain, swelling, tenderness
- Agitation, hallucinations,
- Stiff muscles
- Headache
- Decreased sex drive
- Mild nausea, constipation
- Weight Change
- Dry mouth, yawning, ringing in ears
- Suicidal behavior
References
[edit | edit source]"Home Drugs A-z List Paxil (paroxetine Hydrochloride) Side Effects Drug Center Paxil (paroxetine Hydrochloride) Drug - Medication Guide and Patient Information." Paxil (Paroxetine Hydrochloride) Drug Information: Medication Guide and Patient Information. N.p., n.d. Web. 07 Dec. 2012.
"Paroxetine." Wikipedia. Wikimedia Foundation, 12 June 2012. Web. 07 Dec. 2012.
"Paxil." Information from Drugs.com. N.p., n.d. Web. 07 Dec. 2012.
Introduction
[edit | edit source]Miltown, generically known as Meprobamate, is a sedative drug that is used to treat anxiety and short-term manage insomnia. It is a carbamate derivative that was one used as a minor tranquilizer.
History
[edit | edit source]Meprobamate was first synthesized by Bernard John Ludwig, PhD, and Frank Milan Berger, MD, at Carter Products in May 1950. Launched in 1955, it rapidly became the first blockbuster psychotropic drug in American history, becoming popular in Hollywood and gaining notoriety for its seemingly miraculous effects.
In the mid-1940s, Dr. Berger was working in a laboratory of a British drug company, looking for a preservative for penicillin, when he noticed that a compound called mephenesin had a sedative effect in small laboratory animals. However, there were three major drawbacks to the use of mephenesin as a tranquilizer: a very short duration of action, greater effect on the spinal cord than on the brain, and a weak activity. After moving to Wallace Laboratories in New Jersey, Dr. Berger and a chemist, Dr. Bernard Ludwig, synthesized a chemically-related tranquilizing compound, meprobamate, that was able to overcome these three drawbacks and began marketing this new drug.
Pharmacology
[edit | edit source]Although it is marked as being safer, Meprobamate share many of the pharmacological effects and dangers of barbiturates.
The mechanism that Meprobamate undergoes is unknown, although studies show that it affects multiple sites of the central nervous system, including the limbic and thalamus systems. Meprobamate binds to GABAA receptors, interrupting neural communication in the reticular formation and spinal chord, causing sedation and altered perception of pain.
Toxicity
[edit | edit source]If overdosed, symptoms include coma, drowsiness, loss of muscle control, impaired breathing, sluggishness, and unresponsiveness. Death has been reported with the ingestion of as little as 12 grams and survival with as much as 40 grams.
Chemistry
[edit | edit source]Meprobamate, 2-methyl-2-propyl-1,3-propandiol dicarbamate is synthesized as shown below. 2-methylvaleraldehyde reacts with two molecules of formaldehyde and the subsequent transformation of the resulting 2-methyl-2-propylpropan-1,3-diol into the dicarbamate via successive reactions with phosgene and ammonia.

References
[edit | edit source]http://www.flexyx.com/P/Paxin.html Image from Wikipedia Commons
Definition
[edit | edit source]
Marijuana is a psychoactive drug that is also known as cannabis or weed. It is classified as a sedative-hypnotic or a psychedelic drug which distorts perception.
History
[edit | edit source]Many people have found evidence of inhalation of cannabis smoke. Evidence has been found from the third millennium BCE in different countries of the world. For example, these seeds are found at the ancient burial site that is called present-day Romania. Not only were cannabis seeds found at the burial site, but a leather basket filled with cannabis leaf fragments and seeds was found next to a 2,500-2,800 year old mummified Shaman in the northwestern Xinjiang Uygur Autonomous Region of China. In addition, many ancient Hindus of India and Nepal from thousands of years ago also used Cannabis. Thus, many people use Marijuana as a sense of addition, treatment, or enjoyment.
John Gregory Bourke also described the use of "mariguan" in the late 19th century. Bourke describes mariguan as cannabis indica. In 1894, many Mexican residents of the Rio Grande region of Texas used it. The Mexican residents typically used this as a source of treatment of asthma, to expedite delivery, to keep away witches, and even as a love-philtre.
In addition, mariguan was also one of several plants that are known as loco weed by many people. It was also being compared to that of hashish. However, hashish created a degeneration of the body and an idiotic appearance; thus, by law it was forbidden to sell it, since they do harm to the health of people.
Furthermore, starting from the early 20th century, cannabis was criminalized in various countries. For instance, Americans were first restricted to sell cannabis starting from the year 1909.. It was also outlawed in South Africa, Jamaica, United Kingdom, and New Zealand in the 20th century. In the Opium and Drug Act of 1923, Canada criminalized it as well.
In 1937, the Marihuana Tax Act was passed in the United States. This led to the prohibition of producing and selling hemp and cannabis. Therefore, anyone who was seen selling or even using hemp or cannabis would definitely have been fine for disobeying the Marihuana Tax Act. However, in the year of 2003, 95% of the hemp hurds in the EU were used for animal bedding. Construction workers in the EU also used hemp hurds as building material.
Thus, looking at the history of Marijuana, one can say that cannabis can be good and bad, depending on how it is being used. Certain countries, such as the United State that passed the Marihuana Tax Act, used this as a source to decrease the number of people using Marijuana since it was claimed to be bad for the human body.
Addiction
[edit | edit source]If taken in moderation, marijuana appears to have little to no long-term effects and a very low chance of addiction. Genetic factors may affect one’s susceptibility to addiction. Although chance of physical addiction is zero, there can be a psychological addiction.
General Effects
[edit | edit source]Marijuana is a pain-reliever and a sedative. Marijuana users often describe the experience of smoking marijuana as relaxing and mellow, creating a feeling of haziness and light-headedness. The user's eyes may dilate, causing colors to appear more intense, and other senses may be enhanced. Later, feelings of a paranoia and panic may be felt by the user, although rare.The interaction of the THC with the brain is what causes these feelings. It also reduces nausea caused by drugs used to treat cancer. It decreases the pressure within the eyes in patients with glaucoma, reduces the symptoms of certain motor disorders, and interferes with memory. It alters auditory perception, and stimulates appetite. Long-term use has a correlation with short-term memory impairment.
Two Types of Cannabis Plants
[edit | edit source]Marijuana is classified under two major types: Sativa and Indica. Indica strains are known for their smaller size and are characterized as stout, dark green, and leafy while having lots of resin glands covering both the bracts and small leaves. The effects are known to have a calm and serenity buzz towards ones body. This strain is often used by smokers in treating insomnia, while providing them with relaxation and stress relief, which is why indica strains are more preferred for nighttime use. On the other hand, Sativa varieties are tall, slim, and light green with narrower leaves compared to those of Indica. Although both types give smokers a relaxation buzz, Sativa are known to be energetic and stimulating as well as increasing creativity and focus. The stimulating effects of sativa make it a better option for daytime use. There is also a difference in the ways each strain is grown. Typically, Indica plants are grown indoors; Sativa plants are grown outdoors, due do their large size (sativas grow taller than indicas). In addition, combinations of both strains are grown, known as hybrids, with percentages ranging from 30% sativa - 70% indica strains, 80% sativa - 20% indica strains, and many types of 50% - 50% combinations.
Different Forms
[edit | edit source]Processed
Kief is an example of a processed form of Marijuana. Kief comes in a powder form that is rich in trichomes. It can also be sifted from the leaves and flowers of cannabis plants. People who used Kief either use it in powder form or even compressed to produce cakes of hashish.
Hashish is basically a concentrated resin that is produced from the flowers of the female cannabis plant. Thus, depending on the purity of the compound, it comes in different colors from that of golden brown to black. Hash can even be more potent than that of pure marijuana. People who use Hashish would either smoke it or chew on it.
There is also Hash oil or "butane honey oil" (BHO). Hash oil is a mix of useful oils and resins extracted from mature cannabis foliage through the use of various solvents. Resin is also known as residue because of its THC's adhesive properties that is often sticky. Thus, this type of oil is used in different cannabis food because it typically has a high proportion of cannabinoids that range from 40-80%.
Unprocessed
Many people know that marijuana or cannabis is made up of dried, cured flowers, subtending leaves, and stems of the female cannabis plant. This form is consumed most commonly, and contains 3-28% THC.
Neurological Effects
[edit | edit source]

The active ingredient in marijuana is Tetrahydrocannabinol (THC), which stimulates certain cannabinoid receptors found in the brain.There are two cannabinoid receptors found in the body such as CB1 and CB2 receptors. CB1 receptors are located in the brain, spinal cord, and areas of the peripheral nervous system while CB2 receptors are found in immune cells. Cannabinoid receptors are members of the G-protein coupled receptor (GPCR) superfamily, and are typically thought to mediate inhibition of adenylyl cyclase activity, and hence reduce cyclic AMP levels.
These receptors are found on the terminal buttons of certain neurons, and under normal circumstances, they regulate neurotransmitter release. Cannabinoid receptors are activated by a neurotransmitter called anandamide. Anandamide belongs to a group of chemicals called cannabinoids. THC is a cannabinoid chemical that mimics the actions of anandamide, meaning that THC binds with cannabinoid receptors and activates neurons, which causes the effects on the mind and body.
When activated, the cannabinoid receptors open potassium channels in the terminal button, shortening the duration of the action potential and the release of neurotransmitter. The endocannabinoids, anandamide and 2-arachidonyl glycerol (2-AG) are the two natural ligands for the CB1 receptors. Their normal function deals with hunger, mood, memory, and pain. Anandamide is produced and released when necessary and is not stored in the synaptic vesicles. Fatty acid amide hydrolase (FAAH) is an enzyme that inhibits anandamide. FAAH also deactivates endocannabinoids after their release by inducing enzymatic hydrolysis. The endocannabinoids’ effects last much shorter than THC. THC and the endocannabinoids play an important role in the reinforcing effects of opiates. THC affects areas that have to do with memory such as the prefrontal cortex, cerebellum, basal ganglia, amygdala and hippocampus.
High concentrations of cannabinoid receptors exist in the hippocampus, cerebellum and basal ganglia. The hippocampus is located within the temporal lobe and is important for short-term memory. When the THC binds with the cannabinoid receptors inside the hippocampus, it interferes with the recollection of recent events. THC also affects coordination, which is controlled by the cerebellum. The basal ganglia controls unconscious muscle movements, which is another reason why motor coordination is impaired when under the influence of marijuana.
Common Health Side Effects
[edit | edit source]Although the usage of marijuana is known to be safe if taken correctly as a prescription medicine, it also has it downsides of causing certain side effects when smoked and inhaled through the lungs. It may cause heart disease through its effects of rapid heartbeat and high blood pressure. In addition, it may weaken one’s immune system, thus making one’s body more vulnerable in fighting such infections and diseases. During pregnancy, the inhalation of THC can weaken the growth of the fetus while also having the possibility of injecting dronabinol (THC) in breast fed milk. As a result, marijuana is associated with childhood leukemia (debatable).Lastly, when marijuana is used to treat depression in controlling withdrawals, it can backfire and cause more depression when one is not accessible to the drug. It can cause a mental effect, inducing a higher risk of addiction and obsessiveness.
Medical Use
[edit | edit source]Though marijuana has always been considered a recreational drug, it is also widely used as a form of medical treatment. Medical cannabis can be used to treat the symptoms of cancer, glaucoma, AIDS/HIV, multiple sclerosis, and other conditions. It has also been found to alleviate certain symptoms of spastic and movement disorders. Marijuana is able to reduce symptoms in patients due to its chemical make-up. Chemical compounds called cannabinoids activate cannabinoid receptors in the body and can stimulate appetite, reduce nausea and vomiting, relieve pain, and suppress muscle spasms.
Cancer
[edit | edit source]Many physicians recommend medical marijuana to cancer patients to help subdue the painful symptoms of therapy and to increase appetite. Medical marijuana is found to increase appetite reduce the pain of patients undergoing chemotherapy and radiation. It also decreases feelings of nausea and vomiting. Research done by the California Pacific Medical Center Research Institute shows that cannabidiol (CBD) may help prevent the spread of breast cancer by blocking the activity of the gene Id-1, which is thought to cause the aggressive wide spread of cancer cells to other sites away from the original tumor site, also known as metastasis. Other research done in the laboratory and show that THC and CBD hinder the growth and may even cause death of certain cancerous cells grown in laboratory dishes. There are also animal studies that show the same results. However, there have not been tests performed on humans to conclude whether or not THC and other cannabinoids lower the risk of cancer.
There are studies that show that smoking marijuana may be harmful to the lungs and may even develop certain types of cancer (debatable). Medical dispensaries are now promoting edibles, which are foods containing THC extract mixed in with butter, for patients with a medical marijuana license. Instead of having to smoke marijuana, patients have the option to eat foods containing marijuana. The effects of edibles is longer-lasting than the effects of inhalation.
References
[edit | edit source]"Accepted Medical Use of Cannabis: Basic Research." The 2002 Petition to Reschedule Cannabis (Marijuana). Drug Science.org, 2006. Web. 26 Oct. 2012. <http://www.drugscience.org/amu/amu_basic_research.html>.
Bonsor, Kevin. "How Marijuana Works" 02 July 2001. HowStuffWorks.com. <http://health.howstuffworks.com/wellness/drugs-alcohol/marijuana.htm> 21 November 2010.
biopsychiatry.com. "How THC Affects The Brain." www.a1b2c3.com. October 15, 2010. . October 15, 2010. <http://www.a1b2c3.com/drugs/brn005.htm>.
"Cannabidiol." Cacycle. 5 March 2010. Web. 26 October 2012.
“Cannabis." Wikipedia, The Free Encyclopedia. Wikimedia Foundation, Inc. 20 May 2010. Web. 22 May. 2010.
Carlson, Neil R. Physiology of Behavior. Boston: Pearson Education, Inc., 2007.
“Endocannabinoid System." Wikipedia, The Free Encyclopedia. Wikimedia Foundation, Inc. 21 April 2010. Web. 23 May. 2010.
http://en.wikipedia.org/wiki/Medical_cannabis
http://en.wikipedia.org/wiki/Effects_of_cannabis
King, Jason. The Cannabible. China: 10 Speed Press, 2001. 1-25. Print.
"Marijuana." Marijuana. American Cancer Society, 13 July 2012. Web. 26 Oct. 2012. <http://www.cancer.org/treatment/treatmentsandsideeffects/complementaryandalternativemedicine/herbsvitaminsandminerals/marijuana>.
"THC." Wikipedia, The Free Encyclopedia. Wikimedia Foundation, Inc. 1 September 2009. Web. 26 October 2012.
"Marijuana." WEBMD. Natural Medicines Comprehensive Database. Web. 28 Oct 2012. <http://www.webmd.com/vitamins-supplements/ingredientmono-947-MARIJUANA.aspx?activeIngredientId=947&activeIngredientName=MARIJUANA>.
"10 Most Common Health Side Effects of Using Marijuana Read more: http://www.testcountry.org/10-most-common-health-side-effects-of-using-marijuana.htm
Introduction
[edit | edit source]Benadryl, also known as Diphenhydramine, is an antihistamine drug. It is usually found as a crystalline, white powder and is soluble in water and alcohol. Diphenhydramine is typically used for treating allergic reactions.
In the body, histamine is released when one suffers through an allergic reaction or even viral infections. The histamine then binds to cell receptors, triggering changes in the cell that lead to allergy symptoms such as itching and sneezing.
As an antihistamine drug, diphenhydramine competes against the histamine for the cell receptors. The antihistamine not only prevents histamines from binding and stimulating the cells but while doing so, it also binds to the receptors—they do not stimulate the cells, therefore allergy symptoms will not show.
History
[edit | edit source]Benadryl was discovered by George Rieveschl, who took part in many experiments with focus on relieving muscle pain at University of Cincinnati. Benadryl is an antihistamine drug that used for treating people who suffer with allergy. Benadryl was first available through prescription in 1946. Later in 1980s, it was approved as over-the-counter drug.
Content
[edit | edit source]Benadryl contains the histamine-blocker diphenhydramine. This drug will help patients to recover from common symptoms: sneezing, watery eyes, runny nose, and sore throat. One application of Benadryl is Benadryl Chesty Cough & Nasal Congestion: a drugs that is used to relieve the symptoms of chesty cough and cold.
Side Effects
[edit | edit source]Do not use this drugs if you are currently treating with depression, heart condition, blood pressure, and behavioral disorder. As many other drugs, Benadryl does have some common side effects:
- sleepiness
- dizziness
- headache
- dry mouth
- difficulty urinating.
Chemistry
[edit | edit source]Diphenhydramine is synthesized as shown below. The molecular formula of Diphenhydramine is C17H21NO • HCl.

References
[edit | edit source]"Benadryl (Diphenhydramine) Drug Information". RXList. 2009. Retrieved March 6, 2009
Ohio History central: Benadryl; accessed Jan. 5, 2011
Benadryl for the Family Chesty Cough & Nasal Congestion Oral Liquid ; accessed Oct. 13, 2011
http://www.medicinenet.com/diphenhydramine/article.htm
Image from Wikipedia Commons
Introduction
[edit | edit source]
Prednisone is a drug that belongs to the class of corticosteroids which is a steroid. Steroids are hormones with similar chemical structure that gather together as a group. In general, Prednisone is produced by your adrenal gland which is located on top of kidneys. It is used to treat people who have low levels of corticosteroids. It works in preventing the flammable substances in the body. Patients with the following conditions can be treated with Prednisone: skin conditions, lupus, arthritis, breathing disorder, or allergic disorder at regular doses, or it can be treated with cancer at higher doses. This drug is no t highly recommended since it has really severe side effects.
History and Usage
[edit | edit source]Arthur Nobile was the one who identified the structure of Prednisone in 1950. Not until 1955, Prednisone synthesis was carried out in the laboratory of Schering Corporation; it is a replica of cortisone- which is produced by the outer portion of the adrenal glands. Nowadays, Prednisone can be prescribed as generic drugs.
Prednisone comes as a tablet so it can be taken directly by mouth. You should eat something first before taking this drug. You should be taking at direct dose as prescribed because it can cause serious side effects if you overdose. Your doctor will adjust your dose while you follow up with the treatment- but mostly the lowest dose that your body can take. Do not stop using Prednisone even if you feel better because your body might not have enough steroids that come from this drug to function normally.
Mechanism
[edit | edit source]Prednisone is a glucocorticoid receptor. It will first be metabolize to its active form and come across the cell membrane and attach to specific cytoplasmic receptors. This results in leukocyte infiltration of inflammation, interference of inflammatory response, and suppression of immune response. The anti-inflammatory and immunosuppressive actions which is a mechanism of prednisone can be described as followed:
- Inhibition of gene transcription
- Blockage of osteocalcin
- Changing in transcription of collegenase gene
- Increase the synthesis of annexin-1
Side Effects
[edit | edit source]
You should remember that Prednisone is a type of steroid. It is a really powerful drug that can save life if it is used properly. Just as many other drugs, it also has side effects that can be really harmful. Some common minor side effects that you might experience while using this drug are:
- Headache
- Dizziness
- Sleeping disorder
- Skin allergy
- Extreme tiredness
- Weak muscle
Other people might face severe side effects that needs to be taken with cautions and notify your doctor immediately
- Vision problem
- Sore throat, fever, hacking cough
- Rash
- Stomach pain
- Vomiting
- Depression, confusion
- Breathing disorder
References
[edit | edit source]http://www.gihealth.com/html/education/drugs/prednisone.html
http://www.drugs.com/prednisone.html
AHFS Consumer Medication Information- Prednisone

Tobacco is a crop that is one of the main ingredients in cigarettes. It is classified as a stimulant. Tobacco leads as the drug that produces the most dependence. This dependence stems from one of its ingredients, nicotine. The most common method of consumption of this drug is through smoking. Nicotine is the main psychoactive ingredient in tobacco. Although it is most commonly used as a recreational drug, it has been shown to have medical uses. Tobacco belongs to the plant genus Nicotiana. The nicotine in these plants evolved as a mechanism to defend against insects. Insects that taste the plant would get a dose of neurotoxin. Commercially, these plants have been cultivated and farmed as a recreational drug that acts as a stimulant to the brain.
Pharmacodynamics
[edit | edit source]
Nicotine has a very similar chemical structure to the neurotransmitter acetylcholine. Due to the similarity in structure, nicotine is able to bind to receptors and activate Cholinergic receptors, just as acetylcholine normally could. When acetylcholine binds to these receptors, it is thought to stimulate brain functions and help muscle control. It plays a large role in brain process speed. The structural similarity of nicotine allows it to activate these receptors much as acetylcholine can.
Absorption and metabolism of nicotine
[edit | edit source]When chewed, nicotine is absorbed readily in the lungs or through the oral mucosa. With frequent use of tobacco, levels of nicotine accumulate in the body during the day and remain overnight. Thus, tobacco users are exposed to the effects of nicotine for twenty four hours a day. Since the metabolism and the secretion of nicotine depend on a variety of factors, the drug may be detected in the body and urine even four days after the last cigarette smoked.
Behavioral Effects
[edit | edit source]Nicotine has the following behavioral effects:
- Euphoria
- psychostimulant
- anxiousness
- muscle relaxation
- analgesic (reduces pain)
- decrease appetite.
Nicotine also has adverse effects if used during pregnancy. It is toxic and will affect the infant. If a woman is smoking, the toxin and chemicals enter her bloodstream and subsequently the baby will get them via the blood stream. It is one of the reasons for sudden infant death syndrome. SIDS means that a baby under the age of one suddenly dies without an apparent reason. Nicotine may also cause miscarriages and low baby birth weight. Therefore, it is not advisable to smoke during pregnancy because it can have adverse effects for the fetus.
Nesbitt's Paradox
[edit | edit source]Nesbitt's paradox is one of the famous experiments and observations about nicotine. Although nicotine is classified as a stimulant, and in most ways it acts like a stimulant, nicotine also has been shown to produce relaxation. Nicotine is sometimes described as having anti-anxiety and anti-stress properties. Yet at the same time, it stimulates functions in the brain.
Stages of Tobacco Addiction
[edit | edit source]There are three stages of Tobacco addiction:
| Stages | Effect |
|---|---|
| Stage 1 | Psychosocial |
| Stage 2 | Pleasurable |
| Stage 3 | Negative Reinforcement "hooked" |
Withdrawal
[edit | edit source]The onset of Tobacco withdrawal occurs around: 12–24 hours. It can last up to 10 days. The major symptoms of tobacco withdrawal include: intense cravings, irritability, anxiety, restlessness, sleep disorders, increased hunger (Mainly for carbohydrates), decreased heart rate, decreased metabolism, and fatigue. These symptoms make sense when it is realized that the nicotine in tobacco stimulates human brain function. It increases heart rate and helps combat fatigue. Withdrawal would subsequently present converse adverse effects. It makes you crave it more and kills you quicker.
Treatment Methods
[edit | edit source]
Because tobacco is used by such a large number of people, methods of treatments also come in a wide variety. One of the ways addiction is treated for is through cognitive behavioral therapy. This is a psychotherapist method in which addicted patients have the chance to talk about their addictions. Another method people use (although less frequently) is called motivational enhancement. This is usually when therapists have sessions geared towards motivating the patient to quit. Although these therapeutic methods have been proven to work, there are also pharmacological aids that can be used to combat addiction. A few examples are listed here:
- Buproprion- Its trade name is Zyban. This is a typical antidepressant. It's main action is Norepinephrine-dopamine reuptake inhibition. By blocking this, it acts as a nicotine antagonist. It doesn't allow nicotine to bind to the active site and start the normal behavioral changes associated with smoking or tobacco. Trials with placebos have shown that this drug significantly lowers cravings for nicotine and helps to stop the effects of withdrawal associated with tobacco. This drug is classified as having a low abuse potential.
- Varenicline- Its trade name is Chantix. This is a nicotine partial antagonist. It is presented as a tablet that can be taken once or twice a day with water. This drug essentially blocks nicotine's ability to activate the a4b2 receptors that is responsible for the effects of nicotine. This drug can also help block nicotine withdrawal. This benefit makes it one of the more used drugs when dealing with tobacco addiction. Recent studies have shown however, that some people may exhibit hostility, agitation, suicidal thoughts, and/or depressed moods when taking this drug. However, this is only shown in people who take Varenicline while still smoking. It is important to note that this drug does not work immediately and it may take up to 12 weeks for the complete removal of addiction.
Instead of pharmacological aids, substitutes for nicotine have also been shown to help treat for addiction:
- Gum- This gum helps to lower the cravings for nicotine and helps stop the adverse effects of nicotine withdrawal. This is a substitute because the gum contains small doses of nicotine. By following a preset plan, lower and lower amounts of nicotine is chewed and ideally leads to the cessation of cravings for cigarettes. However, in few cases, it has been shown that a switch to addiction for nicotine gum is possible.
- Patch- The patch is a method of treatment that is similar to gum. These patches release controlled amounts of nicotine through the skin which helps stop the craving for nicotine. Ideally, it will eventually lead to a stop in the desire for cigarettes.
- Non-smoke cigarettes- These are essentially cigarettes that have been 'made clean' by removing tar and other bad chemicals from the cigarette itself. However, the side effects of smoke entering the lungs and lung cancer have not been shown to be lowered by non-smoke cigarettes. Regardless, this is a method of substitution for real cigarettes.
Potential Medical Uses
[edit | edit source]- Alzheimer's treatment- Alzheimer is a disease that effects the brain. It can lead to problems with memory, thinking, cognition, and behavior. Tobacco has shown signs of improving cognitive performance. One of the ingredients in tobacco 'cotinine' has been researched and shown to protect brain cells. This is highly important because if scientists are able to protect brain cells, it will slow down or even prevent brain damage caused by Alzheimer and could potentially be one of the cures.
- Analgesic- Tobacco has been shown to be able to increase pain thresholds and decrease pain. It could potentially be used as an analgesic in the hospital to relieve patients of pain or increase their pain threshold before entering surgery.
- Weight control- As mentioned earlier, one of the behavioral effects of tobacco is a loss of appetite. Scientists are tying to study and create a non-addictive drug that will be able to lower the cravings for food through the use of tobacco.
References
[edit | edit source]1. Hart, Carl. Drugs, Society, and Human Behavior. 13th. McGraw-Hill Humanities, 2008. Print.
2. Meeker-O'Connell, Ann. "How Nicotine Works" 2 January 2001. HowStuffWorks.com. <http://health.howstuffworks.com/wellness/drugs-alcohol/nicotine.htm> 2 December 2010.
3. Berg, Jeremy; Tymoczko, John; Stryer, Lubert. Biochemistry, 6th edition. W.H. Freeman and Company. 2007
Definition
[edit | edit source]
Alcohol is a psychoactive drug/beverage. It is the second most used psychoactive drug. (Caffeine is rated as the first most used.) It is considered a depressant because at medium to high concentrations, it depresses neural firing. (Though at low levels, it can have the opposite effect.)[6] It contains no vitamin or minerals. However, it does contain calories. An important thing to note is that this type of alcohol should not be generalized to mean the same as the organic chemistry term alcohol. Rather this type of alcohol, in terms of organic chemistry, is actually ethanol (or ethyl alcohol). Its chemical formula is: CH3CH2OH.
Due to its small and amphiphilic nature, ethanol is soluble in water and fat, so it is able to cross cell membranes, giving it great access to do harm to cells and affecting the entire body.[6]
History
[edit | edit source]By the tenth-century, a Persian alchemist named al-Razi discovered the first alcohol. Today this alcohol is known as ethyl alcohol. Originally, the name kuhl or kohl was given to the very fine powder that was produced through a sublimation of the natural mineral stibnite to form a compound called antimony sulfide. Today, this product is used as eyeliner and cosmetic.
In the 16th century, the word "alcohol" appears in English that means a very fine powder. In his 1657 Lexicon Chymicum, William Johnson conveys the word as antimonium sive stibium. Through time, the word came to mean any fluid obtained through a process of distillation. This type of fluid includes that of alcohol of wine. In the year 1594, Libavius in Alchymia referred to alcohol as vini alcohol vel cinum alcalisatum. This word in the end was found to mean as "spirit of wine". Until the 18th century "spirit of wine" is referred to as ethanol, and was expanded to be called as "alcohols" in the world of today.
Absorption and Distribution through the body
[edit | edit source]Unlike other drugs, ethanol does not require digestion. The body can quickly absorb it into the system. Roughly 1/5 of the alcohol is absorbed directly through the walls the stomach. Next, the upper portion of the empty stomach absorbs it. Alcohol can also be absorbed through the lungs. Within 30-90 minutes, the body is able to reach the maximal blood concentration from the alcoholic beverage.
Blood Alcohol Content
[edit | edit source]Blood alcohol content (or commonly abbreviated BAC) is a measurement of the amount of alcohol in a person's blood. More specifically, it a measure of how many grams of alcohol in 100 milliliters of a person's blood. For example, if a person registers a .20 BAC, there is 1/5th of a gram of alcohol in every 100 milliliters of that person's blood.
Zero Order Kinetics (Elimination of Alcohol from the body)
[edit | edit source]Alcohol is mainly eliminated from the body through the metabolism. This occurs mainly from the liver. A small percent (5-10%) is eliminated directly through urine and breathing. Elimination of alcohol is considered Zero Order Kinetics. This means that the amount of alcohol removed is constant over time and does not depend on the concentration intake. This is unlike many other drugs that have half-lives and depend on the amount in the body. Alcohol is constantly eliminated over time. The liver can eliminate roughly .25 ounces per hour. It takes approximately two hours to completely eliminate one (Standard) drink.
Addiction
[edit | edit source]Alcohol increases the release of dopamine in the nucleus accumbens. Dopamine is responsible for movement, attention, learning, mood, etc. Dopamine also takes a part in the positive reinforcement (reward) system of the brain. It also triggers the release of endogenous opioids. Opiate receptors also seem to be involved in the reward system of the brain. Withdrawal effects are very powerful, (i.e. seizures, increased sensitivity of NMDA receptors). Alcohol's neurological influence on the reward system of the brain is what's primarily responsible for a person's physiological addiction to alcohol.
Toxic
[edit | edit source]Since the prehistoric times, a compound known as ethanol that is commonly seen in alcoholic beverages has been consumed by human beings for many centuries. The reason for this intake is due to dietary, hygienic, medicinal, religious, and recreational reasons (special event taken place in life). The idea of intoxication is the taking in large doses of ethanol that cause drunkenness. Drunkenness that is often seen when people drink heavily can lead to a hangover as its effect wears off after a period of time. It is important to know that ethanol can cause serious respiratory failure or even death depending on how much an individual take in to their body.
Primary and secondary metabolite, acetaldehyde, and acetic acid all cause the toxicity of ethanol. It is said that all primary alcohols are broken down into aldehydes. After that it is broken down to carboxylic acids which have toxicities that is similar to that of acetic acid and acetaldehyde. Metabolite toxicity is reduced in rats that are fed with acetic acid such as thiamine.
In addition, some secondary and tertiary alcohols are not as poisonous as they are known for. In other words, these alcohols are less poisonous than that of ethanol because the liver is unable to metabolize them into these toxic by-products. Therefore, they are better for medicinal and recreational use. A good example of a tertiary alcohol would be Ethchlorvynol. Ethchlorvynol is use for both medicinal and recreational use.
However, there are also many other alcohols that are more poisonous than that of ethanol. The reason for this is because these alcohols take a longer time to metabolize and their substance that they produce is even more toxic than ethanol. A type of wood alcohol is methanol. Methanol is oxidized to formaldehyde and then through formaldehyde dehydrogenase and alcohol dehydrogenase enzymes to the poisonous formic acid in the liver. The bad thing about this is that, this can cause blindness or even death. Administer ethanol is a great way to prevent toxicity after ingestion such as ethylene glycol or methanol. Although methanol is seen as poisonous, it has a much weaker sedative effect than that of ethanol.
Typically, long chain such as isopropanol, n-butanol or n-propanol have strong sedative effects. Their toxicity is also much higher than that of ethanol. These long chains are also known as fusel alcohols and are surprisingly found to be contaiminanting to certain alcoholic beverages. Thus, this is the reason why people who drink excess or large amount of alcoholic beverage will encounter a kind of hangover. Overall, many of these long chain and even longer chains are used in the industry today as a kind of solvent and are used in alcohol that causes all kinds of serious health conditions.
Withdrawal
[edit | edit source]After a cycle of addiction, withdrawal may come after if the amount of alcohol intake is decreased or stopped. Withdrawal symptoms generally occur 24-48 hours after stoppage of intake. It can lead to symptoms that include insomnia, hyper excitability, and tremors. Furthermore, it can lead to changes in the sympathetic system that include: increased heart rate, increased blood pressure, and increased body temperature. After these early withdrawal symptoms occur, later symptoms (usually 2-4 days after) can occur. These include fits of delirium tremors, hallucinations, and high fevers. Aside from physical changes, alcohol withdrawal can lead to anxiety and dysphoria (negative mood state). One of the main treatments for withdrawal is a Benzodiazepine drug called Chlorodiazepozide(Librium). It is a sedative/hypnotic drug that helps to counteract the affects of alcohol withdrawal.
Blood Alcohol Content
[edit | edit source]Blood alcohol content (or commonly abbreviated BAC) is a measurement of the amount of alcohol in a person's blood. More specifically, it a measure of how many grams of alcohol in 100 milliliters of a person's blood. For example, if a person registers a .20 BAC, there is 1/5th of a gram of alcohol in every 100 milliliters of that person's blood.
Zero Order Kinetics (Elimination of Alcohol from the body)
[edit | edit source]Alcohol is mainly eliminated from the body through the metabolism. This occurs mainly from the liver. A small percent (5-10%) is eliminated directly through urine and breathing. Elimination of alcohol is considered Zero Order Kinetics. This means that the amount of alcohol removed is constant over time and does not depend on the concentration intake. This is unlike many other drugs that have half-lives and depend on the amount in the body. Alcohol is constantly eliminated over time. The liver can eliminate roughly .25 ounces per hour. It takes approximately two hours to completely eliminate one (Standard) drink.
General Effects
[edit | edit source]Alcohol causes a loss of inhibition and judgment, which can lead to aggressive behavior. Alcohol also causes impairment of the senses and can lead to liver and heart problems.
Neurological Effects
[edit | edit source]Alcohol interferes with neural adhesion protein-protein that helps to guide growth of neurons in developing brain. Alcohol is an agonist of GABA receptors and antagonist of NMDA receptors. The GABA (gamma-aminobutyric acid) neurotransmitter is responsible for inhibition in the brain and GABA receptors activate chloride ion channels. Glutamate is the normal neurotransmitter for the NMDA receptors, but NMDA can also activate these receptors in a similar manner. However, NMDA does not bind to other glutamate receptors. NMDA is an excitatory neurotransmitter that is responsible for learning and memory. NMDA receptors activate ion channels that allow the influx of sodium and calcium ions.
Diseases
[edit | edit source]Some diseases associated with drinking alcohol are Korsakoff’s Syndrome and Fetal Alcohol Syndrome. Korsakoff’s Syndrome is characterized by a deficiency in thiamine (Vitamin B1), which leads to damage in the mammillary bodies in the hypothalamus and hippocampus.
Fetal alcohol syndrome is caused when a woman drinks alcohol during pregnancy and the alcohol impairs the baby’s development. Two traits that are common in babies with fetal alcohol syndrome physical deformity and mental retardation. It causes mild to moderate retardation. It can also lead to hyperactivity in childhood as well as growth deficiency. This syndrome is also has characteristic facial abnormalities. Ethanol's affects on fetal development include, but are not limited to, disruption of production of cell-adhesion molecules, druption of normal apoptosis (programmed cell death), and disruption of neurotrophic support.[6]
Alcohol Dehydrogenase
[edit | edit source]Alcohol dehydrogenase is the primary enzyme that converts alcohol into acetaldehyde. This enzyme is part of the primary metabolic system for alcohol. The activity of alcohol dehydrogenase determines the rate of alcohol metabolism. Therefore, myths such as exercise or coffee consumption do not play a role in speeding up the metabolism system. [5]
Having a deficiency in alcohol dehydrogenase decreases the speed of the metabolic process, therefore taking longer to break down alcohol. There is evidence that women have less alcohol dehydrogenase enzymes than men. There has been some evidence that fluctuations in women's menstrual cycles, gonadal hormone levels can influence the rate of their alcohol metabolic system. Women may have elevated blood alcohol concentrations at different times throught their cycle.[7]
Acetaldehyde Dehydrogenase
[edit | edit source]

Acetaldehyde Dehydrogenase (ALDH) are dehydrogenase enzymes that help convert acetyldehyde into acetic acid. More specifically, this enzyme is present in the metabolism and helps to break down alcohol. When a person drinks, the alcohol is first converted to acetaldehyde by the alcohol dehydrogenase enzyme. Acetaldehyde dehydrogenase is needed to further break the new acetyldehyde into more harmless acetic acid.
If an individual has a deficiency in acetaldehyde dehydrogenase, this breakdown cannot occur, leading to elevated acetaldehyde levels which results in vomiting, hyperventilation, nausea, increased heart rate, and alcohol flush reaction. It has been found that people of East Asian descent usually have genes that code for an inactive variant of acetaldehyde dehydrogenase 2.[2]
References
[edit | edit source]- “Alcohol: Balancing Risks and Benefits.” Harvard School of Public Health (2010). n. pag. Web. 22 May 2010.
- "Alcohol Flush Signals Increased Cancer Risk among East Asians." National Institutes of Health. 23 March 2009. http://www.nih.gov/news/health/mar2009/niaaa-23.htm.
- “Alcoholic Beverage." Wikipedia, The Free Encyclopedia. Wikimedia Foundation, Inc. 21 May 2010. Web. 22 May. 2010.
- Carlson, Neil R. Physiology of Behavior. Boston: Pearson Education, Inc., 2007.
- Hart, Carl. Drugs, Society, and Human Behavior. 13th. McGraw-Hill Humanities, 2008. Print.
- Pinel, John. "Biopsychology." Pearson, 7th Edition, 2009. Print.
- "Alcohol and Women" National Institute of Health. Oct. 1990. http://pubs.niaaa.nih.gov/publications/aa10.htm
- http://en.wikipedia.org/wiki/Alcohol#History_and_etymology
Acetaminophen aka Paracetamol pronounced ‘a set a mee’ no fen’ is an over-the counter (OTC) drug that is composed of a reaction between p-aminophenol and acetic anhydride. The generic name for acetaminophen is Tylenol or MAPAP. Acetaminophen is a derivative of acetanilide. It is used to temporarily relieve mild headaches, muscle pain, sore throats, and fevers. Acetaminophen can also be used to as an anti-inflammatory drug for osteoarthritis which is caused by the “wear and tear” of the joints due to incorrect joint alignment. This drug produces its effect by altering the way in which the body senses pain and creating a feedback mechanism to cool the body. This drug does not block and inhibit the enzyme, cyclooxygenase (COX), which serves as a catalyst for the production of prostaglandins which in turn is responsible for producing pain, inflammation, and a fever within the peripheral nervous system like other non-opioid analgesics. It is rather believed that acetaminophen blocks COX-3 in the central nervous system instead.
Although acetaminophen is typically found as a tablet, it can also be in the form of a solution, suspension, chewable tablet, suppository, drops, etc. Although it is an over-the-counter drug that is readily available, it is important to know the risks and symptoms of taking it like any other drug. Acetaminophen should not be taken in combination with alcohol. Overuse of acetaminophen can cause lead to liver damage, and in serious but rare cases, liver transplants may be required. In addition, reported side effects of acetaminophen may include itching, swelling, hoarseness, and breathing difficulty—all of which should be taken into consideration. If symptoms persists, stop taking the medication and consult a doctor.
The use of this medicine for children can be extremely potent and life-threatening. When treating a child with acetaminophen, directions and dosage should be followed with extreme care. Drops are usually given to infants, but since they contain a higher concentration of acetaminophen, be sure to check and give the dosage according to their weight and age.
In addition, some acetaminophen tablets contain additional sweeteners so people diagnosed with Phenylketonuria (PKU) should be aware and careful because their bodies are unable to breakdown phenylalanine (Phe), which can be harmful to the human brain at high levels to those who are diagnosed with PKU.
When taking cold medicine, you should avoid taking multi-symptom cold medications such as Dayquil and Theraflu because these drugs usually contain a dosage of acetaminophen. Instead, one should take medicine base on specific symptoms. For example, to treat a nasal congestion, it is recommended that you use Sudafed (common name pseudoephedrine); Robotussin for coughs; Tylenol for fever and achiness; and Ricolla for throat pains, etc.
Synthesis
[edit | edit source]In the lab, paracetamol(Acetaminophen) is easily prepared by nitration| nitrating phenol with sodium nitrate, separating the desired 4-Nitrophenol| p-nitrophenol from the ortho- byproduct, and reducing the nitro group with sodium borohydride. The resultant p-aminophenol| p-aminophenol is then acetylated with acetic anhydride .[1] The industrial process is analogous, but hydrogenation is used instead of the sodium borohydride reduction.[2]


Another simpler synthesis is by Hoechst-Celanese. This involves direct acylation of phenol with acetic anhydride catalyzed by HF. Thus converting the ketone to a ketoxime with hydroxylamine, then followed by the acid-catalyzed Beckmann rearrangement to give the amide.[3]
Side Effects
[edit | edit source]It is not likely for acetaminophen to produce side effects. People who claim or present symptoms of side effects created by acetaminophen are generally due to overdose of acetaminophen.
Common side effects from overdose include:
- nausea
- jaundice (yellow skin or eyes)
- appetite loss
- sweating
- irritability
- abdominal pain
- liver failure
- kidney failure
- heart problems
- coma
- seizures
- Death (in extreme conditions)
References
[edit | edit source]http://pain.emedtv.com/acetaminophen/acetaminophen-side-effects.html
- ↑ Ellis, Frank (2002). Paracetamol: a curriculum resource. Cambridge: Royal Society of Chemistry. ISBN 0-85404-375-6.
- ↑ Anthony S. Travis (2007). "Manufacture and uses of the anilines: A vast array of processes and products". In Zvi Rappoport (ed.). The chemistry of Anilines Part 1. Wiley. p. 764. ISBN 978-0-470-87171-3.
- ↑ Template:Ullmann
Introduction
[edit | edit source]Vicodin, also known as acetaminophen and hydrocodone is part of the opioid pain relievers. An opioid is sometimes called a narcotic. The chemical formula is C18H21NO3. With the combination of acetaminophen and hydrocodone, the acetaminophen increases the effects of hydrocodone due to its small potency. For medical uses, vicodin is used to treat severe pain. The tablet is produced and marketed under the trade names Vicodin, Vicodin ES, Vicodin HP, Anexsia, Anolor DH5, Bancap HC, Zydone, Dolacet, Lorcet, Lortab, as well as generic brands. Researchers have said that hydrocodone is stronger than codeine but only one-tenth as potent as morphine at binding to receptors. It is roughly half as potent as morphine in analgesic properties. But, some studies have shown hydrocodone to be anywhere from half as potent to oxycodone (1.5× the potency of morphine) to only 66.6~% the potency of oxycodone.


Addiction
[edit | edit source]Vicodin addiction is a very dominant disease that affects the lives of many Americans every year, but with proper treatment vicodin addiction can be overcome. In recent years prescription drug abuse and addiction has been on the rise but denial around the issue of addiction also continues to persist. Vicodin addiction is the obsessive misuse of a mood-changing drug. In this sense, misuse means using the drug without the authorization of a medical profession, or using the drug when it is no longer needed as prescribed. Vicodin addiction can have overwhelming effects on a person's mind and body.
Prescription Drug Addiction, and vicodin addiction in particular causes problems because it was prescribed by a doctor at the first time. In most cases, people who are addicted to vicodin rationalize and say "I'm taking it for the pain", or "the doctor said I could take a few extra". Vicodin produces an overjoyed feeling, relaxing both the physical body and the mind, as well as relieving pain. Vicodin addiction is extremely common among prescription drug users, most likely because it is readily prescribed for many different types of pain problems.
Some prescription drug users do not intend to get addicted on the medicine, but when their prescription runs out they begin to obsess about how they are going to obtain more and more of the drug. Vicodin addicts find that they cannot longer function normally without the drug and that even though the pain is gone, vicodin produces an effect in them that they feel they cannot live without. People suffering from vicodin addiction start looking to outside sources for more prescriptions and are willing to go to great lengths to get more drugs.
Vicodin addiction is often seen a less thoughtful condition than addiction to illegal drugs or alcohol. This is because vicodin is prescribed by a physician and also because vicodin is not seen as producing the same negative problems as other drugs. This misunderstanding often leads vicodin addicts and their loved ones to believe that they do not need to find treatment. All mind altering substances affect the way that the brain functions, and in the case of vicodin, the brain stops producing chemicals like endorphins. Because the brain has not made these essential chemicals, the body feels that it is unable to function without the use of the drug. A person who suffers from vicodin addiction has become dependent upon the drug and will go into removal and cravings if he/she cannot acquire any more.
Freedom from vicodin addiction is possible. Seeking drug abuse treatment is the best course of action for anyone who has a problem with drugs or alcohol. During the initial phase of vicodin addiction treatment, the addict will experience the detoxification process, to cleanse the physical body of the drug and make the person ready for further treatment. Our addiction treatment facilities will give the addict tools and resources to live without the use of vicodin.
Vicodin is a drug that is composed of a mixture of acetaminophen and hydrocodone. Hydrocodones are drugs that work as narcotic pain relievers, and acetaminophen, more mild pain reliever, can help to increase the effects of the hydrocodone.
Vicodin can be used to relieve pain that ranges from moderate to very severe. Taking Vicodin can impair judgment and thinking, as well as decrease alertness. Vicodin should not be taken with alcohol since it can increase the risk of liver damage.
Vicodin should only be taken as prescribed because it has addictive properties. A single tablet of Vicodin contains up to 750 mg of acetaminophen in it. Overdosing on acetaminophen can severely damage the liver or even cause death.
Signs of an overdose on the drug can include nausea, vomiting, stomach pain, confusion, dark colored urine, yellowing of the skin/eyes, slow heart rate, shallow breathing, or muscle weakness.
Some serious side effects that can occur from taking Vicodin include shallow breathing, fainting, seizures, problems with urination, jaundice, mood changes, blurred vision, and more.
It can be very dangerous to take Vicodin concurrently with other narcotic pain medications, sedatives, sleeping pills, or other medications that can cause drowsiness or slow breathing.[1]
References
[edit | edit source]http://www.drugs.com/vicodin.html http://www.rxlist.com/vicodin-drug.htm http://commons.wikimedia.org/wiki/File:Hydrocodone.svg http://commons.wikimedia.org/wiki/File:Hydrocodone_3d_balls.png "Vicodin". WEBMD. November 20, 2012. http://www.webmd.com/drugs/drug-3459-Vicodin+Oral.aspx?drugid=3459&drugname=Vicodin+Oral&source=1
Folic Acid
[edit | edit source]
Folic acid is one of the B-complex vitamins required in order to produce red blood cells. Folic acid is a manufactured form of folate; folate can be found naturally in certain foods, such as leafy green vegetables, nuts, beans, and grains. Some cereals contain 100% of the daily value of folic acid a woman should take per day. If there is an insufficient amount of this vitamin, it can cause anemia. Because the body does not make much folic acid, it is useful to take a vitamin pill form to ensure that you get the recommended daily value.
History
[edit | edit source]Many scientists around the 1920s thought that folate deficiency and anemia were the same condition. In the year 1931, a researcher named Lucy Wills led to determining folate as the nutrient needed to prevent anemia during pregnancy. In other words, folate was an important source needed during the state when a person is pregnant. Through this identification, Dr. Wills illustrated that anemia could be reversed with brewer's yeast. Thus, folate was seen as the corrective substance in brewer's yeast. In the year 1941, Mitchell and other people first separated and extracted folate from spinach leaves.
Furthermore, in 1943, Bob Stokstad who worked at the Lederle Laboratories of the American Cyanamid Company, isolated the pure crystalline form and was then able to determine folate chemical structure. Under the supervision and help of Director of Research Dr. Yellapragada Subbarao, a group called "folic acid boys" in the year 1945 was able to obtain folic acid in a pure crystalline form. This historical research project led to the synthesis of the antifolate aminopterin. Antifolate aminopterin is the first anticancer drug.
Then in the 1950s to the 1960s, many scientists started to study biochemical mechanisms and discovered the different actions for folate. This led to linking folate deficiency to neural tube defects. Overall, many US scientists noticed that food sold in markets contained really little folate; therefore, more food should contain folate to help people, especially those who are pregnant.
Foods Containing Folate
[edit | edit source]There are many healthy foods that are very high in folate. Some are listed below:
- Egg yolks
- Sunflower seeds
- Liver and Kidney Products
- Leafy vegetables such as turnip greens, lettuce, spinach
- Legumes such as beans, peas, and lentils
- Grain Products such pasta, cereal, bread
Food that contains few amount in folate:
- Fruits such as banana, raspberry, strawberry
- Juice such as orange or pineapple juice
Note that folate is naturally found in foods that are susceptible in high heat and UV light. Folate is also water soluble.
Folic Acid and Pregnancy
[edit | edit source]Folic acid has been proven to protect against birth defects during the first weeks of pregnancy. Such birth defects include spina bifida, which is the case involving the backbone and spinal canal not being able to fully close. It can also protect against anencephaly, a condition in which the brain does not develop. Babies born with anencephaly usually die before or shortly after they are born. [1] Women who are pregnant or are trying to get pregnant are recommended to take 400 to 800 micrograms of folic acid per day.
It is possible for folic acid to reduce chances of spinal or brain defects by nearly 70%. These diseases are known as neural tube defects which always occur when the spinal cord fails to close properly during development which is what spina bifida is. [2]
Folic acid ultimately reduces the level of a potentially harmful compound called homocysteine. This is done by speeding up the conversion of homocysteine to methionine, a nontoxic amino acid that the human body prefers and needs. Scientists and researchers discovered that locking the enzyme MTHFR to its cofactor FAD allows folic acid to perform its unique function in the human body. Therefore, the Food and Drug Administration recommends all women of child-bearing age to supplement her diet with folic acid to prevent potential birth defects from occurring.
Side effects of taking folic acid include a skin rash, itchiness, redness, or difficulty breathing.
Folic acid can also be ingested through a balanced diet. This includes, fortified cereals, whole grains, fruits, veggies, beans, and other natural protein. Those looking to increase folic acid intake due to pregnancy can do it naturally through diet or both with supplements and food. It is important for males also to not neglect folic acid. Although they do not benefit from the birth defect preventions, folic acid is still a healthy supplement that is necessary in small doses. [2]
Sperm Quality
[edit | edit source]It is commonly seen that folic acid can minimize the chromosomal defects in sperm. Thus, folate is an important source for fertility in both men and women because it contributes to spermatogenesis. Basically, for both gender, it is crucial to receive a good amount of folate through the diet to avoid subfertility.
Heart Disease
[edit | edit source]Using folic acid will reduce homocysteine levels, but it will not minimize cardiovascular disease. However, this applies differently for women who are pregnant. Consuming folic acid during pregnancy may reduce the risk of heart defects in infants, which is a good thing. It may also reduce the risk for children to develop a syndrome called the metabolic syndrome.
Stroke
[edit | edit source]Even though taking folic acid does not reduce heart disease, it appears to reduce the risk of stroke. However, there are many reviews that indicate how only some individuals who take folic acid may in return minimize the risk of stroke. In other words, folic acid may only work on some people. It is being said that stroke reduction is consistent with the reduction in pulse pressure produced by folate supplementation of 5 mg per day. So for those who are likely to get heart disease, it is important to consume folate in every day diet. This is the reason why hyperhomocysteinemia or stroke patients are greatly encouraged by their doctors to take daily vitamin B which includes folic acid. Folic supplements are in expensive and are quite safe to use, but do not overdo it.
Cancer
[edit | edit source]Since many cancer cells tolerate folic acid and overexpress the folic acid receptor, this had led to the creation of anti-cancer drugs that target the folic acid receptor. There are investigations that proved that good levels of folic acid may be related to lower risk of stomach, esophageal, and ovarian cancers. However, the benefits of folic acid against cancer may only depend on when an individual is taking it and on the conditions of that particular person. This is because everyone tends to have a different immune system that reacts to certain things differently.
Moreover, for individuals who are already suffering from cancer or from precancerous condition may find that taking folic acid may not be helpful and can be damaging. Therefore, consuming a certain amount of folic acid is crucial for everyday diet, but it is important to note that excess of folate may promote tumor initiation. High folate intake promotes advanced carcinogenesis and low folate intake protects against early carcinogenesis. Hence, many doctors and public health recommend being super careful when taking folate and encourage not to intake too much folate.
Diets that are high in folate are related with the decreased risk of colorectal cancer. There are some researches that show how the association is stronger for folate that are taken from foods than folate from supplements. In relation to folate and one carbon metabolism, colorectal cancer is the most studied type of cancer. Furthermore, there are epidemiologic studies that suggest diets high in folate are associated with decreased risk of breast cancer. Studies also show that high dietary folate intake will minimize the risk of prostate cancer. Overall, there had been many studies dealing with folate acid to prevent many kinds of disease.
References
[edit | edit source]- ↑ http://www.womenshealth.gov/publications/our-publications/fact-sheet/folic-acid.cfm
- ↑ a b folic acid, October 28, 2012
http://en.wikipedia.org/wiki/Folic_acid [1] [2] [3]
Definition
[edit | edit source]

Ayahuasca is a plant psychotropic used by the indigeneous of the Amazon basin in South America. The beverage holds a central position in shamanic ethnomedicine of the region. It is a powerful sacred hallucinogen which is made from the ayahuasca vine (Banisteriopsis caapi) and the leaf of the chacruna plant (Psychotria viridis). The vine Banisteriopsis caapi contains MAO inhibitors which render the psychoactive agent of Psychotria viridis, N,N-dimethyltryptamine (DMT), orally active. The name in itself suggests its properties, ayahuasca being Quechua for "the vine of the souls", implying a means for communion with the spirit of the universe itself.
Use and General Effects
[edit | edit source]Ayahuasca is primarily used as a medicine or tool of healing, which according to locals can cure anything. It is prepared by soaking and boiling the bark and stems of Banisteriopsis caapi together with the leaves of Psychotria viridis as well as any additional admixture plants (Brugmansia spp., Erythroxylum coca, or Nicotiana rustica). When the brew is consumed its primary effects are: a purge (vomiting) that cleanses and heals the body and mind, accompanied by visions as well as an altered consciousness that is said to allow the drinker to directly communicate with the deeper spiritual intelligence of the ayahuasca spirit. The time of onset is approximately 20 to 60 minutes after ingestion, and the effects last approximately 4 to 8 hours. Traditionally the brew would be consumed by a village healer, or curandero, who would use ayahuasca to diagnose and cure those that came to him (or her) of whatever mental, spiritual, or physical ailment that was bothering them.
Chemistry of Ayahuasca
[edit | edit source]The major alkaloids of Banisteriopsis caapi are the ß-carboline derivatives harmine, tetrahydroharmine, and harmaline. Harmine and harmaline are highly reversible monoamine oxidase (MAO) inhibitors, while tetrahydroharmine is a weak serotonin (5-hydroxytryptamine) uptake inhibitor at presynaptic sites. Psychotria viridis on the other hand contains a single major alkaloid, N,N-dimethyltryptamine (DMT) as well as N-methyl tryptamine and methyl-tetrahydro-ß-carboline that have been reported as trace constituents. The principle action of ß-carbolines is their inhibition of peripheral MAO, which protects the DMT in the ayahuasca brew from peripheral degradation, rendering it orally active and allowing it to pass through the blood-brain barrier and activate receptor sites in the brain. In addition concentrations of serotonin increase in the body as both its reuptake and metabolism by MAO-A are blocked by ß-carbolines. ß-carbolines are highly selective inhibitors of MAO-A, which is the form of the enzyme for which the preferred substrates are tryptamines such as serotonin or DMT. Psychoactive alkaloids that are often present in ayahuasca when admixture plants such as Brugmansia spp. or Nicotiana rustica are added, also become potentiated by the ß-carboline inhibition of MAO enzymes.
The process that is inhibited is the MAO enzyme catalysis of oxidative deamination of biogenic monoamines. The oxidation occurs according to the general reaction equation: RCH2NH2 + O2 + H2O + MAO → RCHO + NH3 + H2O. The enzyme is widely distributed through various tissues in vertebrates and invertebrates such as the brain, liver, small intestine, heart, lungs, and blood plasma and platelets. The MAO system functions as a detoxifying mechanism to protect the nervous and cardiovascular systems from biogenic amines that could be toxic and be ingested through diet. These amines are generally formed as the result of aromatic amino acid decarboxylation.
Ayahuasca Analogues
[edit | edit source]Ayahuasca analogues are plants or chemicals that are used in place of the traditional constituents of the ayahuasca brew (Banisteriopsis caapi and Psychotria viridis). The analogues are meant to have a constituent that replaces the MAO inhibiting behavior of the ß-carbolines in Banisteriopsis caapi and a constituent that replaces the DMT in Psychotria viridis.
Examples of plant combinations that may achieve similar results to traditional ayahuasca can be found by using the following analogues:
DMT containing plants:
Mimosa hostilis,
Diplopterys cabrerana,
Psychotria carthagenensis,
Acacia maidenii,
Anadenanthera peregrina
MAO inhibiting plants: Peganum harmala, Passiflora spp.
References
[edit | edit source]Callaway, J. C., D. J. McKenna, C. S. Grob, G. S. Brito, L. P. Raymon, R.E. Poland, E. N. Andrade, E. O. Andrade, D. C. Mash (1998) Pharmacology of Hoasca alkaloids in Healthy Humans. Journal of Ethnopharmacology. In Press.
Grunwell, J. N. “Ayahuasca Tourism In South America.” Newsletter of the Multidisciplinary Association for Psychedelic Studies (MAPS) 8.3 (Autumn 1998): 59-62.
Heaven, R., Charing, H.G. "Plant Spirit Shamanism." Destiny Books. Rochester, VT. 2006. 82-87.
McKenna, D., G. H. N. Towers, & F. S. Abbott. (1984) Monoamine oxidase inhibitors in South American hallucinogenic plants: Tryptamine and ß-carboline constituents of ayahausca. Journal of Ethnopharmacology 10:195-223.
Viagra (Sildenafil Citrate)
[edit | edit source]Sildenafil Citrate, more commonly known as Viagra, is a drug that is used to treat erectile dysfunction and pulmonary arterial hypertension (PAH). The main function of the drug is to inhibit the enzyme that degrades cGMP, a regulator of blood flow to the penis. The medical uses for Sildenafil Citrate are sexual dysfunction, pulmonary hypertension, and altitude sickness.
In 1998, the drug Viagra was introduce as an effective way to treat male erectile dysfunction (MED). It goes by the generic name of sildenafil citrate. It has quickly become one of the most popular and most prescribed drugs in the world. Its annual sales account for over a billion dollars each year. Sildenafil was synthesized by a group of pharmaceutical chemists working at Pfizer’s research facility in England. It was discovered by accident while attempting to produce a drug to treat coronary heart disease. It became the first oral treatment accepted by the FDA to treat MED.
Physical Properties
[edit | edit source]Sildenafil citrate is designated chemically as 1-[[3-(6,7-dihydro-1-methyl-7-oxo-3-propyl-1H pyrazolo[4,3-d]pyrimidin-5-yl)-4-ethoxyphenyl]sulfonyl]-4-methylpiperazine citrate. Sildenafil citrate is a white to off-white crystalline powder with a solubility of 3.5 mg/mL in water and a molecular weight of 666.7. Viagra (sildenafil citrate) is formulated as blue, film-coated rounded-diamond-shaped tablets
-
 Step-by-step synthesis of Viagra
Step-by-step synthesis of Viagra

Mechanism
[edit | edit source]The mechanism of Viagra (sildenafil) in the body involves the protection of cyclic guanosine monophosphate (cGMP) from degradation by cGMP-specific phosphodiesterase type 5 (PDE5) in the corpus cavernosum of the male penis.
First, nitric oxide (NO) in the corpus cavernosum of the penis binds to guanylate cyclase receptors. This results in an increase of cGMP, which promotes vasodilation of the intimal cushions of the helicine arteries. Vasodilation of the intimal cushions allows for an increased blood flow into the spongy tissue of the penis, thus, causing the erectile tissue to expand and create an erection. Sildenafil is a potent and selective inhibitor of cGMP-specific phosphodiesterase type 5 (PDE5), which is responsible for degradation of cGMP in the corpus cavernosum. Once cGMP levels deplete, then the intimal cushions of the helicine arteries are not able to dilate to allow more blood flow. Without sexual stimulation, and therefore lack of activation of the NO/cGMP system, sildenafil should not cause an erection.
Alcohol
[edit | edit source]Ethanol and sildenafil cause similar reactions within the body. Therefore, doctors do not recommend consuming alcoholic beverages while taking the drug. Ethanol thins blood and dilates blood vessels which can cause dizziness, rapid heart rate, and low blood pressure. Sildenafil causes the same effect on the body which can cause a dramatic increase in the potential of suffering these effects.
PDE5 inhibitor
[edit | edit source]It belong to a family of drugs called PDE5 inhibitors. Sildenafil acts by inhibiting cyclic guanosine monophosphate(cGMP)-specific phosphodiesterase type 5, an enzyme which is responsible for degradation of cGMP in the corpus cavernosum in the penis. Nitric oxide in the corpus caverosum then binds to guanylate cyclase receptors, which leads to increased levels of cGMP, leading to smooth muscle relaxation. The molecular structure of sildenafil is similar to that of cGMP and acts as a competitive binding agent of PDE5 in the corpus cavernosum, resulting in more cGMP and better erections. Sildenafil is metabolised by liver enzymes and excreted by both the liver and kidneys. If taken with a high-fat meal, absorption is reduced; the time taken to reach the maximum plasma concentration increases by around one hour, and the maximum concentration itself is decreased by nearly one-third.
-
 PDE5
PDE5

Reactivity
[edit | edit source]This molecule is very sensitive to temperature and light therefore it has to be kept at room temperature between 15-30 degrees C away from moisture or heat. Also if exposed to prolonged period of time to light it can cause a molecular reaction.
Erectile dysfunction (ED)
[edit | edit source]Erectile dysfunction is a sexual dysfunction which is shown by the inability to maintain an erection of the penis during sexual intercourse. Because penile erection is a hydraulic effect of blood rushing into the penis and retained in sponge-like pores, erectile dysfunction is cause by circulatory mishap. One of the causes of ED is the modification of voltage-gated potassium channels, which play a major role in action potentials during depolarization. However, there are many other causes of ED such as diabetes, hormonal deficiencies, and neurological problems. Major drugs that are believed to cure ED are Viagra, Cialis, and Levitra and are under competition in the pharmaceutical market.
Treatment
[edit | edit source]According to a research, erectile dysfunction increases with passing age. Approximately 20% by the age of 60, though in the 21st century psychological factors such as (stress, depression and anxiety), medication, disease and injury is speeding up the process earlier then age. It is estimated than 70% of the population in United States suffer from erectile dysfunction due to an underlying disease like kidney condition. While smoking increases the chances by 50%. It was shocking to know that only 33% out of those only look for some advice or medical assistance, because this is the reason for 35% of the breakdown of relationships.
Effective treatment in such scenarios include injecting medication into the penis, changing one's lifestyle my reducing the amount of cigarette or alcohol consumed. Losing weight, and following an exercise regime that improves blood flow also helps. Vacuum pumps are also helpful since it encourages blood flow and makes the penis erect.
Side Effects
[edit | edit source]Halt taking viagra immediately if you present one or more of the following symptoms:
- hives
- difficulty breathing
- swelling on the face, lips, throat or tongue
- vision loss
- ring in the ear
- chest pain
- irregular heartbeat
- swelling in limbs
- shortness of breath
- vision changes
- penis erection for over 4 hours
- light headedness
Less serious conditions may include:
- stuffy nose
- headache
- memory problems
- upset stomach
- back pain
References
[edit | edit source]http://www.drugs.com/sfx/viagra-side-effects.html
Delar, Anthony. “Viagra with excessive use of alcohol may affect your erections negatively.” ArticleBiz.com. 2010. Sat. 20 Nov 2010.
http://www.articlebiz.com/article/532090-1-viagra-with-excessive-use-of-alcohol-may-affect-your-erections-negatively/
Wikipedia contributors. "Sildenafil." Wikipedia, The Free Encyclopedia. Wikipedia, The Free Encyclopedia, 18 Nov. 2010. Web. 22 Nov. 2010.
http://en.wikipedia.org/wiki/Sildenafil
“Viagra.” Drugs.com. 18 Oct. 2010. Sat 20 Nov 2010.
http://www.drugs.com/viagra.htm
Vollhardt, K. Peter C. “Organic Chemistry: Structure and Function.” Fifth Edition. W.H. Freeman and Company, New York 2007. Pg. 1176-1177
Introduction
[edit | edit source]
Vyvanse is a nervous controlled medication used to treat people with attention deficit hyperactivity disorder (ADHD) who show symptoms of loss of memory or having difficulty focusing. Its structure consists of dextroamphetamine with an amino acid L-lysine. ADHD is caused by some of the natural substances in the brain, and Vyvanse can help to manipulate the amount of these substances to work properly. Vyvanse also helps patients dealing with particular symtoms of ADHD; it can increasing their attention and decrease the impulsiveness.
History and Usage
[edit | edit source]Vyvanse was developed by New River Pharmaceuticals and has been on the market since 2008. Vyvanse can be taken directly by mouth and usually in the morning because it can cause difficulty in sleeping if you take it in late afternoon or at night. It is suggested that Vyvanse should not be taken more than what it is prescribed because it can cause some severe side effects. A doctor will probably prescribe a patient with a low dose to see how their body can adapt to the medication. Oftentimes, the physician will adjust the dosage during a patient's treament. Besides taking Vyvanse as a part of a complete treatment, patients with ADHD should follow up with other therapies.
Side Effects
[edit | edit source]Because Vyvanse can lead to dependence or addiction, it should be used properly. Just as other drugs, Vyvanse does have some common side effects:
- Difficulty falling asleep
- Stomach pain
- Flu, fever, sweating
- Headache, dizziness, diarrhea.
But for some people, this drug can have some serious side effects:
- Seizures
- Vision problems
- Slowing of growth in children
- Worsening of sudden
A doctor should be contacted if these side effects occur.
References
[edit | edit source]http://www.drugs.com/pro/vyvanse.html
Background Information
[edit | edit source]
Structural biochemistry has lead to a greater understanding of HIV and has played a key role in the development of drugs that fight it; one such drug is zidovudine, or azidothymine (AZT), which has been sold under the name Retrovir, the first drug to be approved to help treat HIV. AZT is a nucleoside analog reverse transcriptase inhibitor, known as an NRTI. AZT is especially recommended for pregnant women with HIV/AIDS, since taking this drug during pregnancy and labor reduces the chance of mother-child transmission. Side effects include headaches, nausea, and discoloration of fingernails and toenails. More severe side effects include anemia and bone marrow suppression.
Once structural biochemistry recognized HIV as a retrovirus, previously discovered proteins in viruses became drug targets. Retrovir inhibits the action of one of the virus’ enzymes, reverse transcriptase, which makes a DNA copy of its own RNA, allowing it to replicate quickly and encode itself into our genome (which is why there can never be a “cure” for AIDS until we can find a way to cut out the virus’ DNA from our own). If reverse transcriptase is unable to make double stranded viral DNA, it is then unable to integrate its DNA into the genetic material of our cells. However, HIV does become AZT-resistant over time, so AZT is used in a “drug cocktail” in combination with other NRTI’s and protease inhibitors to most effectively combat HIV and AIDS in patients.
Side Effects
[edit | edit source]Seek for medical help if you present one or more of the following symptoms below:
- hives
- swelling on your lips, face, throat or tongue
- difficult breathing
- severe muscle pain
- pale skin
- easy bruising
- unexplained weight loss
- pale skin
- loss of bladder control
- severe lower back pain
- liver problems
- pancreatitis
- severe skin reaction
Less severe symptoms may include:
- insomnia
- mild nausea
- constipation
- joint pain
- headache
Incompatibility with other drugs
[edit | edit source]Retrovir might produce violent reactions if used with the medications below:
- doxorubicin
- ganciclovir
- interferon alfa
- phenytoin
- ribavirin
- drugs that weaken your immune system
References
[edit | edit source]http://www.drugs.com/mtm/retrovir-injection.html Biochemistry, Sixth Edition, Berg. http://www.rxlist.com/retrovir-drug.htm http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0000869
Definition
[edit | edit source]
Drug antagonists are designed to bind to receptors and specifically block or weaken drug effects. The antagonist itself does not have any biological effects. In general, drug antagonists are used by doctors to help patients through withdrawal or patients who overdose. By taking an antagonist, the receptors for the specific drug is blocked and therefore weakened or completely shut down. The effectiveness depends on the nature of the binding receptor.
Chemistry
[edit | edit source]A ligand can bind to biochemical receptors that in turn becomes activated. In the case of drugs, the drugs act as the ligand. Receptors can be both intracellular and extracellular. They can exist in the nucleus and mitochondria or even the membrane. A ligand can cause effects by binding to either the receptor itself (active site), or to allosteric receptors. Allosteric sites are other sites on the receptor that are not classified as an active site. Both regular the activity of the receptors. Antagonists help block these interactions and prevent agonist-induced responses. When agonists bind to the receptor, they turn 'on' the cellular responses. When antagonists bind to receptors, they turn 'off' cellular responses. Often, the efficiency of antagonists depend on where the drug is designed to attach on the receptor. However, in all cases, an antagonist is defined as having no biological effects of its own. Because of this factor, it is useful in drug and withdrawal treatment in humans.
Classifications of Drug Antagonists
[edit | edit source]Competitive- These antagonists bind at the same site that the target ligand or agonist binds to. However, it does not activate the receptor sites, but rather blocks it so that the target ligand or agonist cannot attach and activate the site.
Non-competitive- These antagonists bind to allosteric sites and is effective from there. It does not need to attach to the same site as the target ligand or agonist to block or dampen its effects.
Uncompetitive- Uncompetitive antagonists cannot work on their own. Instead they require the ligand or agonist to be bound to the receptor site before they can attach themselves to an allosteric site and begin their work. In general, these antagonists block higher concentrations of agonists better than lower concentrations.
Partial Antagonist- Partial antagonists can attach to the active site but do not completely block out the receptor's effects. However, it does lower it from its maximum potential.
Silent Agonists- These antagonists have zero ability to activate a receptor. They simply attach and block the receptor from activating.
Inverse Agonists- They attach to the same site as an agonist, but induce biological responses opposite to that agonist.
Examples of Drug Antagonists
[edit | edit source]
Naloxone- Is a drug antagonist that blocks the effects of opiates. It is administered via injection. In many cases, naloxone is used to counter overdose of opiates such as a heroin overdose. It is made from thebain. It is a μ-opioid receptor and has rapid blockage of withdrawal symptoms which are especially dangerous in patients who have overdosed. Naloxone is also used in conjunction with other drugs to prevent drug abuse. For example, amounts of naloxone is present in suboxone. Subonxone is used to help detoxify patients by administering lower and lower amounts of opiates over time. However it can also be addictive. Therefore, naloxone is used to partially block out the opiate effects in Subonxone.
Naltrexone- It is also an opiate antagonist. Additionally, it can be used effectively as an alcohol dependence manager. However, Naltrexone is orally administered and is classified as a competitive antagonist. Compared to Naloxone, it is longer acting and irreversibly. Therefore, it serves better as an emergency antidote rather than the primary antidote. This antagonist is active at μ- and κ-opioid receptors. Recent studies have shown that low doses of Naltrexone may be able to treat for Cronh's disease. Further studies are planned.
Buprenorphine- Buprenorphine is a partial opiate antagonist. It binds at the µ- and κ-opioid receptor. Buprenorphine is a thebaine derivative that is a partial agonist at the u and k receptor points. It helps block and weaken opiate effects. It binds very tightly to the receptors in the brain, making it hard for opioids to take effect when it is inside the body.
References
[edit | edit source]1. Hart, Carl. Drugs, Society, and Human Behavior. 13th. McGraw-Hill Humanities, 2008. Print.
2. http://en.wikipedia.org/wiki/Receptor_antagonist
3. Berg, Jeremy; Tymoczko, John; Stryer, Lubert. Biochemistry, 6th edition. W.H. Freeman and Company. 2007
Background Information
[edit | edit source]
Tyrosine Kinase Inhibitors are drugs that can be used to disrupt the Tyrosine phosphorylation process necessary in cell signaling near the surface of the cell.
Scientists have discovered that many diseases, cancer being a major one, use this pathway as a crucial step in the development of the diseases. Cancers use this process as if there is a mutation that leads to the production of many receptors or the production of a receptor and its growth factor, the signal becomes greater and more consistent. Thus, if scientists are able to disrupt this signal transduction process, there is reason to believe that the development of cancer can be temporarily halted or altogether stopped. This concept has led scientists to term these advances as "Signal transduction therapy".
Protein Tyrosine Kinases (PTK) are proteins that specialize in the signaling between cells and within cells. Under this category are also proteins such as Receptor Protein Tyrosine Kinases (RPTK). These are proteins that span the cell membrane, having a domain facing outward (in the cells surroundings), and having a domain inside the cell. This setup makes it optimal for this type of the protein to transmit messages from outside the cell to inside the cell or vice versa.
Some Examples of Drugs that are in the Process of Being Discovered that Would Help in Signal Transduction Therapy
[edit | edit source]1. Tyrphostins - these molecules are competitive with the substrate and noncompetitive with ATP. At first, scientists discovered some natural occurring compounds that worked, but were not very selective or potent. However, with some minor chemical adjustments, the first class of tyrphostins were created. The first class of tyrphostins demonstrated that it is possible to hinder the activity of a specific PTK without being toxic to the cells or disrupting normal cellular processes.
2. STI-571 - This drug was first discovered as means to hinder the activity of the enzyme PTK Bcr-Abl kinase, which was an enzyme critical in the stages of development for chronic myelogenous leukemia (CML). STI-571 works by interaction between the inhibitor and the amino acids that are part of the ATP-binding area of the PTK. This drug has been carried all the way to clinical trials and has shown effect in Leukemia patients in the chronic phase. However, patients with advanced forms of the disease faced relapse. STI-571 is also used to treat other diseases such as gastrointestinal stromal tumor.
3. BMS-354825 - This drug is used to treat patients that have become resistant to the treatment of STI-571. This inhibitor works on both the Bcr-Abl kinase protein and the Src kinase protein and less specific than STI-571. The difference between this drug and STI-571 is that it binds to the active state of the protein while STI-571 works by binding to the inactive state of the protein.
4. Gefitinib - This drug binds at the ATP site and is used for the treatment of non-small cell lung cancer (NSCLC). This drug has been in the clinic since 2002 but has discovered to be only effective for a very small percentage of patients. This small percentage of patients have activating mutations in the kinase domain. Gefitinib was supposed to be a EGFR kinase domain inhibitor. However, discoveries revealed that the survival of the tumor did not depend entirely on the use of EGFR kinase. Thus, it was for awhile difficult to tell whether Gefitinib was ineffective because of the inhibition of EGFR or the inability of the drug to occupy the receptor for a long period of time. Recent studies show that inhibitors of the EGFR kinase will only be useful if the receptor plays a major role in the survival of the cancer or if the drug can be combined with other signal tranduction agents to cause certain cancer cells to kill themselves.
5. AG 490 - This drug was used to inhibit a protein called Jak-2, which was a protein that played a crucial role in cytokine signaling. Amplified behavior of the Jak-2 protein was discovered to be linked to many leukemias, lymphomas, and some metastatic cancers. Ag 490 inhibited the Jak-2 pathway, thus inhibiting the expression of the oncogenic phenotype. Because this drug does not inhibit the Jak-2 protein through the disruption of signaling or cell growth, it was also experimented in unison with immunotherapy. Even though immunotherapy does not directly link to the elimination of already existent tumors, it does aid greatly with the continuation of a tumor free existence once the patient has rid themselves of a substantial volume of their tumor. AG 490, used in conjunction with interleukin (IL)-12 was proven to be effective in inducing antitumor responses in the immune system.
An Interesting Comparison Between STI-571 and PD173955
[edit | edit source]Both inhibitors work by binding to the ATP site of the kinase domain. However, they bind differently due to structural considerations; STI-571 binds to more amino acids than PD173955 does. Therefore, STI-571 only binds to a specific inactive structure of the kinase while PD173955 binds is less specific. One reason why PD173955 has proven to be more effective than STI-571 has been revealed through the fact that PD173955 can bind to multiple conformations of the kinase protein and is less specific to the state of the activation loop. STI-571 on the other hand, requires a specific inactive structure of the kinase in order to bind and thus does not bind as easily. This has led scientists to conclude that even though there are certain ATP-competitive inhibitors with a similar basic structure, the difference in functional groups really defines what that specific molecule or drug will bind to.
References
[edit | edit source]Levitzki, Alexander, and Eyal Mishani. Tyrphostins and Other Tyrosine Kinase Inhibitors. Rep. Annual Review of Biochemistry. Web. 29 Oct. 2011. <http://www.annualreviews.org/doi/abs/10.1146/annurev.biochem.75.103004.142657?url_ver=Z39.88-2003&rfr_dat=cr_pub%3Dpubmed&rfr_id=ori%3Arid%3Acrossref.org&journalCode=biochem>. Antimicrobial resistance – also known as drug resistance – occurs when microorganisms such as bacteria, viruses, fungi and parasites change in ways that render the medications used to cure the infections they cause ineffective. When the microorganisms become resistant to most antimicrobials they are often referred to as “superbugs”. This is a major concern because a resistant infection may kill, can spread to others, and imposes huge costs to individuals and society.
Due to the amount of antibiotics currently being used for humans and agriculture, there has been an increased amount of drug resistant pathogenic bacteria. This resistance is a result of natural selection, in which the pathogens resistant to the antibiotics survive and continue reproducing, and the nonresistant strains are killed.
Introduction
[edit | edit source]When penicillin was discovered in 1928, the effectiveness and convenience of antibiotics for treating many infectious diseases was realized, and taken for granted. Now, the use of antibiotics has become increasingly commonplace, and thus allowed for the development of more drug resistant strains of bacteria.
These bacteria that have grown to become more resistant to the past drugs lead to new generations of medication that are able to deal with these infectious diseases. The only problem that is realized that many of these drugs will become useless because of the mechanisms that the bacteria display in where they will evolve into better bacteria then they were in the past. Bacteria, who have similar structures and genes, are able to evolve through the accumulation of multiple genes that code for drug resistance, pumps that work in 3 different areas of the bacteria, and the similarity of these drugs that are already in bacteria which help them become resistant. (Nikaido 2011)
Mechanisms of Drug Resistance
[edit | edit source]There are many reasons as to why certain bacteria strains can become resistant to certain drugs. Most of these resistances originate from the bacteria that were used to produce the antibiotic, because they need to be resistant to their own product, or from microorganisms that exist in the environment and thus are exposed to the excess amount of antibiotics used today.
Changes in the target protein
[edit | edit source]Bacterial genes may mutate and affect the protein that the drug targets. Due to this change, the protein may be less susceptible to the biochemical effects of the drug. While these mutations may not be carried on to further generations under normal situations, the presence of the antibiotic drugs will affect the selection process, favoring only the bacteria with this mutated protein form. Resistance of this kind is affective against man-made drugs that cannot be inactivated enzymatically.
Inactivation through enzymes
[edit | edit source]Certain drugs are more susceptible to being inactivated by enzymes that are produced by certain bacteria. The genes that code for these enzymes usually originate from the bacteria that were originally used to produce these antibiotics. Examples of enzymatic drug deactivation can be seen in the phosphorylation/acetylation/adenylation of aminoglycosides and the hydrolysis of β-lactamases.
Aminoglycosides are inactivated by the reducing net positive charges on these antibiotics. These modifing enzymes, such as AAC(3)-11, will act on the position 3 of the substrate that belong to the phylogenic group among the enzymes. Due the antibiotic-producing micro-organisms that are found in these bacteria, they are already present amongst their DNA which make them resistant to these aminoglycosides. (Nikaido 2011)
B-Lactam resistance was caused by the B-lactamase coded in by plasmid genes. This was problematic because they are resistant to many drugs like methicillin and similar compounds that are able to be hydrolyzed through different enzymes like Tem B-lactamase & AmpC. These drugs were recreated for a 2nd generation & 3rd generation, but they the AmpC enzyme was able to evolve also to counteract these drugs. Soon after, older versions of drugs would have to be introduced to counter the enzymes like Vancomycin, which binds itself to a substrate that is a precurse to the cell wall peptidoglycan instead of inhibiting the enzyme itself. (Nikaido 2011)
Acquisition of other genes
[edit | edit source]The sequencing of genes for penicillin resistant Streptococcus pneumoniae showed that the target proteins were being produced as mosaic proteins (parts came from other organisms). The drug was ineffective due to the change in the targets of the antibiotic.
R plasmids are usually transferred very efficiently in cell to Cell transfers which have been made possible between the bacterial cells because they are closely related to each other. R plasmids, which are highly stable, also helps their case in their increasing drug resistance. The R plasmids, containing drug resistance genes, are able to deliver these genes to any piece of DNA because they are composed of transposons. These R plasmids contain a unique 59-base-3'-sequence called an integron which catalyzes an insertion of resistance genes into a compatible site. Through this process, more drug resistant genes are able to be passed to multiple bacteria for a higher drug resistance. (Nikaido 2011) (Kaiser 2011)
Host cells often lose R plasmids acquired from cloning vectors in large numbers. However, most naturally derived R plasmids are stable and lost less often when the new host cells are multiplied. This can be attributed to the fact that natural plasmids have in their structure "killer" elements that kill the host cell when plasmids are lost. (Nikaido 2011)
In addition, genes can acquire drug resistance through mutations, extrachromosomal DNA transfer, such as conjugation, which is the transfer of the R-factor plasmid via pilli, transformation by obtaining DNA from the environment, and transduction via the R-factor on the bacteriophage. Also, genes can become resistant to drugs through transposable drug resistance sequences using transposons, or small DNA segments that move from one DNA molecule to another. [4]
Preventing drug access
[edit | edit source]
A drug can generally be caused to be less effective if its access to its target is limited. Locally, a bacteria can produce certain proteins that will effect conformations of ribosomes or DNA and thus restrict the access of the antibiotic to those target areas. Another method of this mechanism is the use of drug specific efflux pumps.
Reducing the access of drugs inside the bacteria have induced a nonspecific inhibition. This inhibition is created where bacteria have decreased their outer-membrane permeability in which it reduces the access of drugs entering the bacteria. Often, porin-deficient mutants are selected. Being a double edged sword, the lower permeability of the outer-membrane will also reduce the intake of nutrients entering the bacteria thus being detrimental to cell. Mutations in the coding sequences of the porin have been discovered that lower the permeation rate of bulky antibiotics but leaving small nutrient molecules unaffected, allowing them to permeate at normal levels.(Nikaido 2011)
The next method is through multidrug efflux pumps that were first identified in E. coli & P. areuginosa. These pumps have been discovered in most clinical gram-negative bacteria that share similar systems with P. areuginosa. The Multidrug Efflux Pumps are consisted of 3 different parts where it has a resistance nodulation division exporter protein, a gated outer membrane, and a membrane fusion protein. (Nikaido 2011) (Aeschlimann 2011)
The Major Facilitator Superfamily are the largest families of transporters and contain many efflux pumps. In this class, QacA & QacB are the first examples of pumps in which the QacR-inducer complex has been able to accommodate diverse ligands with their binding site. They will be able to bind to these drugs that have entered through the membrane and then be pumped out of the system. Some of these efflux pumps will always be pumping in which a repressor is needed in order to stop the operon from letting the pumps continue their function. (Nikaido 2011) (Aeschlimann 2011)
The Small Multidrug Resistance family have different efflux pumps that were coded and to be seen on the chromosomes of gram-negative bacteria. The trasnporters of these pumps will encounter substrates in which they will be deprotonated and be pumped out by the inward flux of protons. (Nikaido 2011)
The next class of efflux pumps belong to the Resistance-Nodulation-Division Family in which these pumps are associated with 2 other classes of proteins, the outer-membrane channel and membrane fusion protein. The construction of this pump gives a huge advantage to the bacteria because it gives a direct export of these drugs into the medium and outside of the bacteria where they will have to re-enter the bacteria through the outer membrane barrier.Nikaido 2011)
These pumps will work all together creating synergy in which they will pump out most antibiotics and becoming drug resistant. The more pumps there are, the more drug resistant it will become. Although some pumps cannot carry out some antibiotics, Homologs of AcrD can carry out this function in which they will make bacteria even more resistant to antimicrobial agents. (Nikaido 2011)
Persister Cells
[edit | edit source]When high concentrations of antibiotics are introduced to the bacteria, it would be safe to assume that all bacteria will be killed. But a special phenomenon happens when some of the bacterial cells will survive the antibiotics. These cells are called persister cells in which has become a strategy for bacteria to generate drug-resistant populations. Bacteria produce phenotypically different mixtures in populations so that any one of them can be beneficial in changing environmental factors. Thus, antibiotic therapy is deemed inefficient in the presence of such persisters.

These cells are similar to spores in which small portions of these cells are dormant in the bacterial population. Being dormant, they will not react to the antibiotics making them possible to reoccur the infections once the antibiotics are gone. There are no current treatments for persister cells, but there possible ways to target the cells through anti-microbial peptides. These AMP will target dormant or active cells which will be effective in attacking persister cells. (Nikaido 2011) (Duchene 2011)
References
[edit | edit source]Nikaido, Hiroshi (2009). "Multidrug Resistance in Bacteria" (PDF). Annu Rev Biochemistry. Retrieved 2011-11-12.
Duchene, Ariel (2011). "Addressing the challenge of persister cells in bacterial infections". PHYSORG.com. Retrieved 2011-11-12.
Aeschlimann, Jeffrey R. (2003). "The Role of Multidrug Efflux Pumps in the Antibiotic Resistance of Pseudomonas aeruginosa and Other Gram-negative Bacteria". Medscape News. Retrieved 2011-11-12.
Kaiser, Gary E. (2001). "R-Plasmid Conjugation". Doc Kaiser's Microbiology. Retrieved 2011-111-7. {{cite web}}: Check date values in: |accessdate= (help)
- Nikaido H. (2009). Multidrug resistance in bacteria. Annu. Rev. Biochem. 78, 119–146. doi: 10.1146/annurev.biochem.78.082907.145923.
- Spratt BG. Resistance to antibiotics mediated by target alterations. Science. 1994;264:388–93.
Tortora, Gerard J., Berdell R. Funke and Christine L. Case. Microbiology An Introduction 10th ed. Boston: Benjamin Cummings :, 2010. Print. | Chapter 20 |
- ↑ http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0000723/
- ↑ Berg, Jeremy M., ed. (2002), Biochemistry (6th ed.) New York City, NY: W.H. Freeman and Company,
- ↑ PubMed Health, "Myelomeningocele."
- ↑ {Tortora, Gerard J., Berdell R. Funke and Christine L. Case. Microbiology An Introduction 10th ed. Boston: Benjamin Cummings :, 2010. Print. | Chapter 20| page 600}
Introduction of antivirals
[edit | edit source]Antiviral drugs were invented to prevent viral infections. They are one class of antimicrobials and harmless to human body. Instead of destroying the target pathogen, antiviral drugs inhibit proteins that contribute one or several steps in viral infection. It is difficult to invent new safe and effective antivirals because the harm to host cells must be taken into consideration and variation of viruses.
Four classes of antivirals:
- Neuraminidase inhibitors
- Protease inhibitors
- Neutralizing antibodies
- Protein-based fusion inhibitors
Neuraminidase Inhibitors
[edit | edit source]Introduction of Neuraminidase
[edit | edit source]
Neuraminidase, also called sialidases, is glycoside hydrolase enzymes, which can cleave the glycosidic linkages of neuraminic acids.
There are nine subtypes of neuraminidase. Three major classes of neuraminidase enzymes are:
- Viral neuraminidase
- Bacterial neuraminidase
- Mammalian neuraminidases:
sialidase 1 (lysosomal sialidase), with symbol NEU1.
sialidase 2 (cytosolic sialidase), with symbol NEU2.
sialidase 3 (membrane sialidase), with symbol NEU3.
sialidase 4, with symbol NEU4.
In order to better understand the process of developing neuraminidase inhibitors, the case of influenza virus neuraminidase inhibitor development will be examined. The neuraminidase of influenza virus is a homotetrameric glycoprotein anchored to the viral membrane. It spreads on the membrane of influenza virus, helping mature influenza virus leave from host cell and infect new cells. For each influenza virus, there are near 100 neuraminidases spreading on its surface.

Introduction of neuraminidase inhibitors
[edit | edit source]Inhibiting neuraminidase keeps newly produced viruses in the host cell and prevents the infection of other cells.
There are 2 kinds of influenza virus, type A and type B. Old drugs, such as amantadine, only kill type A influenza virus effectively; it is useless to type B influenza virus. The new antivirals are invented by changing the functional group on drugs to reduce the drug resistance and change their reactivity with type B influenza virus. They can inhibit both type A and type B. There are two drugs (Zanamivir, Oseltamivir) that are commercially available in the market now. The third is still in clinical evaluation.
Zanamivir is the first neuraminidase inhibitor commercially developed to treat influenza virus A and virus B. It was discovered in 1989 by scientists led by Mark von Itzstein. Zanamivir has molecules formula C12H20N4O7, and molecules mass 332.31 g/mol. There are no known toxic effects caused by Zanamivir.
Oseltamivir is an antiviral that used to treat and prevent influenza A virus and influenza B virus infection.It was approved in 1998 Feb. Oseltamivir has molecules formula C16H28N2O4, molecules mass 312.4 g/mol. It attacks the hydrophobic pocket in the active site of neuraminidase protein of influenza virus to stop the reproduction process of virus.
Protease Inhibitors
[edit | edit source]
Protease is an enzyme that exists to cleave polyproteins. Specifically for the case of HIV protease, the enzyme is necessary to cleave the Gag and Pol polyproteins, which is vital to the maturation of HIV virus. The cleaved proteins are used to build components for infections HIV virions. By blocking this protease, inhibitors stop the reproduction process of HIV virus, causing HIV virions remain uninfectious. Designing a drug to inhibit this enzyme is difficult as there are dynamic properties of the protein that control its ability to bind to substrates. However, since HIV protease is very important to the life of the HIV virus, it is a viable option for designing new drugs.
Let’s take a look at the structure of HIV-1 protease first, which was found thanks to x-ray crystallography.

The HIV protease is a symmetric dimmer with a pair of active-site residues-catalytic aspartates. Moreover, these active sites are quite close to the symmetrical axis.
There are several drugs that are used to control HIV virus and treat AIDS:
Amprenavir was approved by FDA on April 15, 1999. Patients only need to take medicine twice a day, instead of three times. Amprenavir was discontinued to produce on 2004.

Tipranavir is a nonpeptidic protease inhibitor manufactured by Boehringer-Ingelheim, approved by FDA on June 22, 2005.

Darunavir is second-generation protease inhibitor, used to treat HIV infection. Darunavir was approved by FDA on June 23, 2006. It has molecules formula C27H37N3O7S, and molecules mass 547.665 g/mol.
Unfortunately, just like many other drugs, human immunodeficiency virus (HIV) protease inhibitors have also faced obstacles to due to resistance and bioavailability. Hence, it is rather appropriate to discuss about using phosphonic acid esters as prodrugs to approach this two important issues. The Gilead group, who have been working on improving the bioavailability of Zanamivir, have discovered a specific covalent structure of a phophonate diethyl ester to TMC-126, which is an analog of the protease inhibitors amprenair. One essential feature of the phosphonate diester is its cell permeability. After it passes through the cell membrane, the phosphonate diester is prone to go through intracellular enzymatic hydrolysis to yield diphosphonate dianion, which is less likely to diffuse out of the cell. As a result, this compound increases the effective concentration of the HIV protease inhibitors at the intracellular target site. On the other hand, the low resistance of the inhibitors can be ascribed to a change in the binding thermodynamics of the GS-8374 vis-`a-vis the nonphosphonate parent TMC-126. Solvent reorganization is a fairly important procedure in the process, as suggested by the energetic of the process, combined with the lack of significant interactions of the phosphonate with protease. The structure of phosphonate also gives the molecule positional flexibility in the active site, which decreases the probability of mutations. This concept may be applied to other HIV protease inhibitors as well, while solvent reorganization remains as an undetermined idea for other proteases and therapeutic areas. Inhibitors of HIV viral membrane, such as oligopeptide and enfuvirtide, are proved to be effective in acting as disruptors of helical bundle formation during the fusion process. However, the cost and dosing regimen of enfuvirtide have made it rather difficult to carry out therapy for multidrug-resistant HIV patients. Drug designers are now looking into solutions in addressing HIV patients’ resistance to enfuvirtide. Biologics of considerably-sized molecule that can compete effectively in binding are required to disrupt relatively large-surface protein-protein interactions, and this places limitations on the other aspects of the drug.[1]

Neutralizing Antibodies
[edit | edit source]Neutralizing antibodies are highly specific and highly effective as antiviruses. They can be introduced to the body through vaccinations, or due to earlier infections. Due to the specificity of antibodies, research has shown that in single amino acid changes in the antigen resulted in preventing the binding of antibodies to antigens. Structural studies following this research showed that the single amino acid changes resulted in only local structural changes.
Although antibodies are highly effective in targeting viruses specifically, research shows that viruses also have mechanisms in order to avoid detection by antibodies. For example, with influenza antigens, the enzymatic site is presented as smaller than the typical site targeted by antibodies. HIV gp160 uses carbohydrates and conformational changes in an attempt to mask the enzymatic site.
There is currently one monoclonal antibody that is registered as an antiviral agent: Palivizumab, or Synagis, is raised in mice and used in pediatrics for targeting the respiratory syncitial virus (RSV). In developing this antibody, three specific variants of RSV Fusion were selected for in vitro testing: K272M, K272Q, and N268I.
There also exists interest in using this type of therapy for hepatitis C virus, but further developments still must be made.
Protein-based Fusion Inhibitors
[edit | edit source]This type of inhibitor is based on preventing the fusion of the viral membrane to the cell membrane, thus preventing the virus from interacting with the cell altogether. By inhibiting this mechanism of viruses, cells would not be infected; therefore, this is a very desirable reaction to target for drug developers. The molecular bases for this reaction is developed from the influenza virus hemaglutanin, the fusion protein, in pre- and post-fusion conformations. Observations have found that the membrane fusion process follows the production of a six-helix bundle. The specific intermediate targeted in this pathway is a trimeric coiled coil in helical region 1 (HR1) where a "fusion peptide" found at the N-terminus associates with the target membrane.
The first drug developed through this approach is Enfuviritide, an oligopeptide with an sequence that overlaps the helical region 2 (HR2) sequence of the HIV fusion protein gp41. The goal of the drug is to prevent the formation of the helical bundle through competition, replacing the natural substrate and thus creating a barrier. Some studies show, however, that it only weakly inhibits the formation of the six helix bundle. It is possible that the mechanism of the drug is more complex than expected, and more studies must be conducted.
Design of New Antiviral Medicines
[edit | edit source]According to the article “New Antivirals and Drug Resistance” by Peter M. Colman, he announced a hypothesis that the increase of similarity between the drug and the target’s natural ligands (substrates) can make the virus more difficult to resist drugs. Besides of this factor, people should also take entropy compensation and solvent anchoring into consideration to design new and better antiviral medicines.
There are several ways to improve the drug by combating with drug-resistant virus:
- Raise the concentration of the drug: It would make the drug more effective if the drug can be distributed widely and also close to the target in an infected cell. Then the higher concentrated and more specifically distributed drug can cause the mutations less significant.
- Add a charged phosphonate to protease inhibitors: Occasionally, scientists find out that a phosphonate-containing inhibitor can bind to a wider range of viral-resistant variants. The example is GS-8373, which was analyzed by crystallography that the phosphonate groups remain their activities after mutation happens. Other parts without phosphonates loose 10- to 40-fold activity. However, according to the article “New Antivirals and Drug Resistance” by Peter M. Colman, scientists failed in raising virus in an environment with phosphonate-containing protease inhibitors.
- Increase drug’s similarity to the natural substrates: The similarity in shape, charge, and other configurations can improve drug’s effectiveness to bind to virus for inhabitation.
Reference
[edit | edit source]- ↑ Annu. Rev. Biochem. 2011. 80:239-46 The Annual Review of Biochemistry is online at biochem.annualreviews.org
1. Peter M. Colman, "New Antivirals and drug Resistance"
2.SHAO Huayi,LI Zhuorong, "Overviews of neuraminidase inhibitors"
3.http://baike.baidu.com/view/555631.htm
4.http://baike.baidu.com/view/344762.htm
5.http://en.wikipedia.org/wiki/Neutralizing_antibody
6.http://commons.wikimedia.org/wiki/File:PDB_1hiv_EBI.jpg
Introduction
[edit | edit source]Ketamine is a dissociative anesthetic developed in 1963 to replace PCP and presently used in human anesthesia and veterinary medicine. Most of the ketamine that is sold on the street has been abstracted from veterinarians’ offices. Ketamine’s chemical structure and mechanism of action are similar to those of PCP. Although it is manufactured as an injectable liquid, in illicit use ketamine is generally evaporated to form a powder. The ways it’s used is either snorted or swallowed. Ketamine is odorless and tasteless, so it can be mixed with beverages without being noticed, and it induces amnesia. Because of these properties, the drug is sometimes given to unsuspecting victims and used in the commission of sexual assaults referred to as “drug rape.”
The short term effect of Ketamine is that it can trigger dream-like states and hallucinations. Users report sensations ranging from a pleasant feeling of floating to being separated from their bodies. Some ketamine experiences involve a terrifying feeling of almost complete sensory detachment that is likened to a near-death experience. These experiences, similar to a “bad trip” on LSD, are called the “K-hole.” Low-dose intoxication from ketamine results in impaired attention, learning ability, and memory . In higher doses, ketamine can cause delirium, amnesia, impaired motor function, high blood pressure, depression, and potentially fatal respiratory problems.
Overview
[edit | edit source]Ketamine is a dissociative anesthetic. It is designed as a slightly acidic (3.5-5.5 pH) solution that is administered through injection. Its chemical name is (±)-2-(o-Chlorophenyl)-2-(methylamino) cyclohexanone hydrochloride. Ketamine goes by various names on the street. It has been known to be called K, Special K, Cat valium, or vitamin K. Ketamine is abused in both the liquid and solid form. The liquid form is usually injected intravenously and is the most dangerous way to consume. The solid form is used to be smoked, snorted or swallowed as a pill. Also, it is known to be added to drinks. Ketamine is legally used as an anesthetic for animals and humans, but it is commonly abused for pleasure. [1]
Effects
[edit | edit source]Ketamine has many powerful effects, which have made it a popular privately used drug. Some of its psychological effects include out of body experiences, hallucinations, numbness, euphoria, and nausea. All of these effects vary with dosage and tolerance. Ketamine effects can last up to 24 hours, but the hallucinations only about 45 to 90 minutes. Ketamine use is associated with severe mental and physical detriments. These effects can include: delerium, depression, high blood pressure and loss of motor functions. Ketamine has also been known to be used as a rape drug by incapacitating the user. It is sometimes spiked into other drugs or given to those ignorant of the drugs strength. [2]
Pharmacology
[edit | edit source]Ketamine is a non-competitive NMDA receptor antagonist. The blocking of this receptor is believed to cause the pain relief one experiences while on Ketamine.
References
[edit | edit source]http://www.abovetheinfluence.com/facts/drugsketamine http://dancesafe.org/drug-information/ketamine http://www.thegooddrugsguide.com/ketamine/basics.htm
MDMA
[edit | edit source]MDMA (3,4-methylenedioxy-N-methamphetamine) is commonly known as “Ecstasy,” “E,” “X,” or “XTC.” It is a drug used to generate a “feel good” emotion. It is a hallucinogen that has stimulant effects. Specifically, the structure of MDMA is chemically similar to the drug methamphetamine, where it gets it "stimulant" characteristics[5]. A stimulant is a psychoactive substance that can provoke a change in the mind and body through increased sensitivity. For example, a user may feel increased happiness or pleasure for things that may seem normal for someone not under the influence. Their bodies will feel more sensitive to touch and texture, while they feel hyper or energized. MDMA is also chemically similar to the mescaline, which is of the psychedelic branch of hallucinogens[5]. Psychedelic drugs tend to alter the mind in ways that affect thinking, perception and consciousness. MDMA is is a very popular drug that is used today.
History
[edit | edit source]In 1912, Merck Pharmaceuticals synthesized MDMA, which was originally used as an appetite suppressant. As the years went on, in the 1970s, MDMA was used to enhance communication skills it was also said to have been tested by the US government as a possible "truth serum"[6]. In 1987, MDMA became an illegal drug under the suspicion that it causes damage to the brain of the user which was the case with the animal models used in research experiments with this drug [6]. Although it is an illegal drug, many people all over the world have been using MDMA. It is commonly used at a rave, club, or gatherings. As the years have progressed, it has become a very mainstream drug.
Effects
[edit | edit source]Subjective Effects
[edit | edit source]MDMA mainly affects neurotransmitters in the brain, specifically serotonin, dopamine, and norepinephrine. MDMA increases the release of serotonin, a neurotransmitter that regulates mood, sleep, pain, emotion, appetite, and other behaviors. MDMA is said to increase the activity of serotonin and also decrease the release of dopamine. The increase in serotonin is caused by the fact that MDMA inhibits serotonin to enter the reuptake site, causing and excess flow of serotonin.
MDMA has many psychological effects on the human mind. Some side effects include euphoria, decreased aggression, loss of self consciousness, enhanced senses, and enhanced empathy towards others. MDMA also has many physical side effects
Since MDMA increases the activity of serotonin, the brain becomes depleted of serotonin. MDMA users can undergo many side effects from using the drug. Some physical side effects include nausea, excessive sweating, teeth grinding, increased heart rate, papillary dilation, increased blood pressure, and increased body temperature.
After Effects
[edit | edit source]After the drug is used and the user is "coming down" from the high, they may feel acute after effects such as:
- Psychological
- Tiredness [10]
- Depression [10]
- Anxiety [8]
- Impaired attention, focus and concentration [11]
- Residual feelings of empathy and closeness [8]
- Physical
- Dizziness or lightheadedness [12]
- Shift in mood for several days after use [12]
- Tiredness [12]
- Depression [12]
Usage
[edit | edit source]
Recreational Use
[edit | edit source]MDMA is notoriously known for its recreational use at electronic dance music (EDM) events, more specifically scenes called "raves." However many users like to take Ecstasy many other music events such as concerts, dance and party scenes where it may seem to enhance the perception of music. MDMA is usually taken in a pill form, although it can be crushed and snorted, and which the purity of the pill is usually unknown. There are many types of pills that contain different concentrations of MDMA and are sometimes mixed with other drugs such as caffeine and methamphetamine [7] which alters the types of drug effects the user have. After being digested the pill usually comes into effect in 30-40 minutes, typically following the user's metabolism pattern and the "peak" effect is typically about 60-90 minutes after digestion. The user typically experiences the effects of MDMA for a total 3-4 hours [12], excluding the after-effects.
Medicinal Use & Potential Therapeutic Effects
[edit | edit source]Despite how MDMA is infamously known for its recreational use, few people know the prospects of MDMA as a medicinal drug. Emphasizing its psychadelic properties as well as its mental and mood uplifting abilities, MDMA has been tested on patients with mild psychiatric disorders, those who suffer from Post-traumatic Stress Disorder (PTSD), cancer patients, and even those who need assistance with communication to their partners [8]. Furthermore, a study on MDMA with patients that suffer from PTSD by Michael C. Mithoefer, Mark T. Wagner, Ann T. Mithoefer, Lisa Jerome and Rick Doblin concluded that "MDMA-assisted psychotherapy can be administered to posttraumatic stress disorder patients without evidence of harm, and it may be useful in patients refractory to other treatments" [9].
Before MDMA became illegalized by the government, it was in popular usage by psychiatrists for research in therapeutic medicine, specifically for opening up the mind for investigation. In the Army’s Chemical Center, MDMA was used to reduce the fear of veterans who suffered from post-traumatic stress disorder. The MDMA supposedly “reduced the fear response to emotional threats… many respondents reported on the therapeutic benefits of MDMA. They had used it to uncover painful childhood memories and experiences that had been repressed; to decrease fear and defensiveness; to increase communication and empathy with one's spouse; to get through traumatic experiences such as rape and incest; to live with the pain of cancer; to resolve oneself to dying13.” This newfound sense of security was intended to enable psychiatrists to communicate with their patients with ease and without the emotional tension. According to the Studies in Crime, Law and Justice Vol. 7 by Rosenbaum and Doblin, “Trauma victims were treated with MDMA-assisted psychotherapy to help them delve into the source of their problems, experience a healing catharsis, and subsequently function more effectively.” However, this positive, therapeutic side of MDMA was lost after the DEA added it to “Schedule I” due to a “potential” of it being abused, although the favor of it being used as a recreational drug was not affected[14]. That being said, many researchers lost the interest of continuing research on therapeutic effects of MDMA due to the negative connotation that the drug now had after being added to Schedule I despite how there were 200 physicians using MDMA for psychotherapy during the time of its banning15. Also, since MDMA was made illegal, the ability to obtain MDMA is harder and much more expensive, two other reasons why research on MDMA slowed down[13].
Chemistry
[edit | edit source]Formal IUPAC name: N-methyl-1-(3,4-methylenedioxyphenyl)propan-2-amine
Other names: 3,4-methylenedioxymethamphetamine, methylenedioxy-methylamphetamine, or N-methyl-1-(1,3-benzodioxol-5-yl)-2-propanamine
MDMA can exist in a racemic mixture with an R and S form.
MDMA is part of the phenethylamine family which is also known as “designer drug” and is also closely related to amphetamines. Phenethylamines are known for their hallucinogenic effects which originate from the compound’s ability to release serotonin in the brain as well as the ability to stimulate the central nervous system, giving users “more” energy. There are also many homologous forms of MDMA which consist of: MDA, MDEA, and MBDB. MDMA predominates as a salt in physical form; either a hydrochloride or phosphate salt which are found in tablet form. It can also exist as a powder or capsule16. As a base MDMA is a clear oil.
Synthesis
[edit | edit source]MDMA is manufactured by safrole, a liquid extract from sassafras trees. The most popular way to convert safrole into MDMA is by synthesizing the 3,4-methylenedioxyphenyl-2-propanone (MDP2P) intermediate via the Wacker process or by isomerizing safrole into isosafrole by a strong base. Both processes oxidize either safrole or isosafrole into the MDP2P intermediate which then undergoes reductive animation to make a racemic mixture of MDMA.
MDMA is manufactured by safrole, a liquid extract from sassafras trees. The most popular way to convert safrole into MDMA is by synthesizing the 3,4-methylenedioxyphenyl-2-propanone (MDP2P or PMK) intermediate via the Wacker process (which is the oxidation of ethylene to acetaldehyde by oxygen in water with a tetracholoropalladate catalyst[17]) or by isomerizing (changing the arrangement of the atoms in the compound) safrole into isosafrole by a strong base. Both processes oxidize either safrole or isosafrole into the MDP2P intermediate which then undergoes reductive animation to make a racemic mixture of MDMA. In the Merck patent, safrole was added to HBr to form a bromosafrole, which was then added to methylamine to produce racemic MDMA16. Other methods of synthesis start with 3,4-methylenedioxyphenyl-2-propanone and use the Leuckart route, aluminum foil method or other reductive aminations[16]. The Leuckart route converts ketones into amine groups using formic acid, ammonium formate or formamide/methylformamide. The aluminum foil method uses ethanol, aluminum metal pieces, an amine and a mercuric chloride catalyst to react with a ketone. The aluminum foil method is a type of reductive amination (reductive alkylation) which is the conversion of a carbonyl group to an amine group with an imine intermediate.
Pharmacology
[edit | edit source]The main way MDMA interacts with the human body is through the brain. As previously mentioned, MDMA induces higher serotonin levels, but can also release dopamine and nonepinephrine. The structure of MDMA makes it an indirect agonist of serotonin, inducing the production of the neurotransmitter as well as inhibiting the re-uptake19. In fact, MDMA seems to act as a re-uptake inhibitor of all these neurotransmitters. MDMA is metabolized through two main pathways: “1. O-demethylenation followed by catechol-O-methyltransferase (COMT)-catalyzed methylation and/or glucuronide/sulfate conjugation; and 2. N-dealkylation, deamination, and oxidation to the corresponding benzoic acid derivatives conjugated with glycine”. The reactivity of the CYP2D6 enzyme is an important step in the O-demethylenation pathway because it regulates the degradation of MDMA and the efficiency of this enzyme as coded by genes, may increase a user’s risk of contracting acute toxicity. Furthermore the metabolism of MDMA also has potential involvement in mid to long-term neurotoxicity due to the neurodegeneration of the neurotransmission system[18].
Pharmacokinetics
[edit | edit source]Generally, pharmacokinetics is a subdivision of pharmacology that deals with timing of the different stages of drug interaction with the body. The term bioavailability is defined as the biochemical interactions with the drug which is used to predict how much of the drug that the body will take in. The bioavailability of MDMA can be measured by the concentration of the substance in plasma, in the urine and even in the concentration of the hormone cortisol in the plasma because MDMA induces its appearance. Research done by M. Farré et al. (2004) maps out a very intricate pathway that MDMA follows as well as how much of the drug is actually take up in the body after two doses in an interval of 24 hours. In the corresponding publication: “Repeated doses administration of MDMA in humans: pharmacological effects and pharmacokinetics” reveals that the results of experimentation indicate that there is a dramatic increase of MDMA concentration and pharmacological effects, suggesting that the second dose has an inhibiting effect on metabolism. The “accumulation” of MDMA could also be explained by looking at the methylenedioxy group present in the structure which is postulated to be able to have an “auto-inhibition” of MDMA’s metabolism [19]. It is postulated that MDMA inhibits its metabolism by the CYP2D6 enzyme. The CYP2D6 enzyme is an important factor in metabolizing many substances that enter the body, vitamins and drugs alike. CYP2D6 enzyme metabolizes substances via oxidation in the liver. However, research on substances with similar structures to MDMA shows that MDMA may be able to inhibit the CYP2D6 enzyme catalysis with the methelenedioxy group by creating an enzyme-metabolite complex [19]. These “accumulative” and inhibitive effects of MDMA prove that it exhibits non-linear pharmacokinetics which can have extremely detrimental pharmacological effects as well as toxicity because of the body’s inability to rid itself of the substance at an efficient rate [20].
Pharmacogenetics
[edit | edit source]Pharmacogenetics is a subdivision of pharmacology which focuses on the genetics behind the enzymes and other mechanisms behind the metabolism of substances. This area of focus is important for determining the effects of MDMA on users. The genetics of the CYP2D6 enzyme responsible for the metabolism of MDMA is being investigated to determine the body’s efficiency in the metabolization of MDMA. Analyzing the body’s ability to take in and process MDMA is incredibly important because it can give answers to questions about susceptibility to fatality, intensities of pharmacological effects, intensities of toxicity, and susceptibility of dependence. Since recent studies suggest that MDMA has non-linear pharmacokinetics, the rate of metabolizing MDMA in the body is crucial to understanding toxicity, fatality and pharmacological effects.
One example of genetics being a factor in the metabolism of substances is shown in an Interesting study by Y. Ramamoorthy et. Al (2001) in which the metabolistic rate of CYP2D6 substrates is lower in Asians than in Caucasians because the genes that are responsible for the synthesis of CYP2D6 have a variation different from the typical wild type gene which codes for enzymes with slower functionalities [21]. Again taking into consideration that MDMA may acts as an accumulator and an inhibitor, then if the CYP2D6 enzyme has a slower functionality as observed in Asians then users with this genotype have a higher risk of acute toxicity and fatality. However, for Caucasians who have the wild-type genotype that produces a CYP2D6 enzyme with normal-fast functionality, then their susceptibility to fatality and the intensity of pharmacological effects are lower than that of Asians. It is important to remember when looking at data that the genetic populations are still averages of the population and that every individual has different genetic makeup which affects the pharmacokinetics of the drug on an individual basis.
References
[edit | edit source]1. http://upload.wikimedia.org/wikipedia/commons/b/ba/MDMA.png
2. http://www.nevamo.com/ecstacy.htm
3. http://www.drugaware.com.au/Drug%20Information/Ecstasy/Origin%20and%20How%20It%20Is%20Used.aspx
4. http://alcoholism.about.com/cs/ecstasy/f/mdma_faq05.htm
5. http://www.drugabuse.gov/publications/drugfacts/mdma-ecstasy
6. http://inventors.about.com/library/weekly/aa980311.htm
7. http://www.ecstasydata.org/
8. http://en.wikipedia.org/wiki/MDMA
9. http://www.maps.org/w3pb/new/2010/2010_Mithoefer_23124_1.pdf
10. http://www.urban75.com/Drugs/e_guide.html
11. http://www.communitybuilders.nsw.gov.au/download/ecstasy.pdf
12. http://www.erowid.org/chemicals/mdma/mdma_basics.shtml
13. http://www.psychedelic-library.org/rosenbaum.htm
14. Studies in Crime, Law and Justice Vol. 7 by Rosenbaum and Doblin
15. Glen R. Hanson, Peter J. Venturelli, Annette E. Fleckenstein (2005-11-03). "Drugs and society (Ninth Edition)". Jones and Bartlett Publishers. ISBN 978-0-7637-3732-0. Retrieved 2011-04-19.
16. http://www.emcdda.europa.eu/publications/drug-profiles/mdma
17. http://en.wikipedia.org/wiki/Wacker_process#cite_note-1
18. de la Torre R, Farré M, Roset PN, Pizarro N, Abanades S, Segura M, Segura J, Camí J. “Human pharmacology of MDMA: pharmacokinetics, metabolism, and disposition”. NCIB
19. R. De La Torre, M. Farré, J. Ortuño, M. Mas, R. Brenneisen, P. N. Roset, et al.(February 2000). " Repeated doses administration of MDMA in humans: pharmacological effects and pharmacokinetics". Springer-Verlag 2004.
20. R. De La Torre, M. Farré, J. Ortuño, M. Mas, R. Brenneisen, P. N. Roset, et al.(February 2000). "Non-linear pharmacokinetics of MDMA (‘ecstasy’) in humans". Annals of the New York Academy of Sciences 49 (2): 104–109.
21. Ramamoorthy,Y., Tyndale,R.F., and Sellers, E.M.(2001).Cytochrome P450 2D6.1andcytochrome P450 2D6.10differincatalyticactivityformultiplesubstrates.Pharmacogenetics 11, 477–487.
Cocaine
[edit | edit source]
Cocaine (benzoylmethylecgonine) is a crystalline tropane alkaloid that is a central nervous system stimulant, appetite suppressant, and topical anesthetic. Cocaine is derived from coca plants and is a pearly, white powder in its most pure form. The drug was used as an anesthetic in the past but has now been replaced other synthetic anesthetics such as benzocaine. Today, cocaine is mainly used as a recreational drug and is highly addictive because of the way it affect’s the brains reward pathways.
Routes of Administration
[edit | edit source]Cocaine can be dissolved in water and injected, snorted in the hydrochloride salt form, smoked as a rock crystal, or injected vaginally or anally with a suppository. When snorting cocaine, the drug travels through the nose and is absorbed into the bloodstream via nasal tissues. To inject cocaine, users first mix the drug with water and then inject into the blood stream with a needle. This method carries risk of HIV/ AIDS from the potential of shared needles. Smoking cocaine involves processing the drug into a rock crystal form. The rock crystals are then heated so that the vapors will be inhaled. Upon inhalation, cocaine enters the lungs and is absorbed into the blood stream. An oral syringe is used to inject cocaine anally and vaginally. The drug is dissolved in water, inserted into the syringe, and then taken up through the membranous lining of the anal or vaginal walls.
Appearance
[edit | edit source]When speaking of purity, cocaine are usually white and pearly. Cocaine such as ones known as cocaine hydrochloride appears in powder as salt. Cocaine that are sold at street markets are often cut with various powdery fillers to increase its weight; thus, making the cost of cocaine much more expensive. The substances most often used in this process are baking soda; sugars, such as lactose and dextrose; and local anesthetics, such as lidocaine and benzocaine. Substances such as benzocaine add to cocaine's numbing effect on mucous membranes. In all, cut cocaine are usually, white, ivory, or pinkish powder.
The preparation method, presence of impurities, and where the cocaine originally came from are all very important when it comes to determining the color of "crack" cocaine. However, the colors of "crack" cocaine are often range from white to a yellowish cream to a light brown. Also, its texture will vary upon the way they cut the cocaine, origin and processing of the powdered cocaine, and the method of converting the base. Cocaine are sometimes crumbly in texture, sometimes extremely oily, powdery, and almost similar to crystals in nature.
Forms of Cocaine
[edit | edit source]There are many forms of cocaine. One such form is salt. Cocaine is a weakly alkaline compound also known as an "alkaloid"; hence, can combine with acidic compounds to form various salts. Although the sulfate (-SO4) and the nitrate (-NO3) are commonly seen, the hydrochloride (HCl) salt of cocaine is mostly encountered. Salts of different properties will dissolve and interact differently in various solvents. For example, the hydrochloride (HCl) salt is polar in character and is quite soluble in water. Another form of cocaine is basic. As opposed to the salt form, the term "freebase" is referred to as the base form of cocaine. Unlike hydrochloride salt, any cocaine base form is insoluble in water. Thus, smoking freebase cocaine has the an added effect of releasing methylecgonidine into the user's system due to the pyrolysis of the substances, which cause serious problems such as lung tissue and liver tissue infection. It is also known that through the technique of liquid-liquid extraction, pure cocaine can be prepare by neutralizing its compounding salt with an alkaline solution followed by a precipitation to a non-polar basic cocaine.
In addition, crack cocaine is a lower purity form of free-base cocaine. Crack cocaine is usually produced by neutralization of cocaine hydrochloride with a solution of baking soda (sodium bicarbonate, NaHCO3) and water, which in the end produces a very rough/hard/brittle, ivory to brown colored, amorphous material that contains sodium carbonate, entrapped water, and other by-products as the main impurities. Using "freebase" and "crack" forms of cocaine are easy because they vaporize smoothly. However, cocaine hydrochloride does not vaporize as easily because high temperature of about 197°C is needed. In the end, despite the forms of cocaine, smoking or vaporizing cocaine and inhaling it into the lungs produces a sense of "high" feeling and it can be very addicting as more cocaine are taken.
Effects on the Body
[edit | edit source]Cocaine highs generally last 10-30 minutes. The faster cocaine travels through the bloodstream and reaches the brain, the more intense the high will be. Upon taking cocaine, users experience euphoria and energy, including increased heart rate and blood pressure. The risks of cocaine are heart attack, stroke, seizure, abdominal pain, respiratory failure, nausea, and death. Because users often combine cocaine with other psychoactive drugs, the risk of sudden death is greatly increased. Cocaine is often combined with alcohol intake. Researchers have found that the liver combines cocaine and alcohol to produce cocaethylene which increases cocaine’s euphoric effect but also increased the risk of sudden death. Cocaine increases the levels of dopamine in the brain. Dopamine is associated with pleasure and movement. Cocaine prevents dopamine from being recycled, causing the neurotransmitter to build up and continually amplify the message. The euphoric feelings are attributed to the continual build up of dopamine. Cocaine abuse occurs when long time users change the brain’s reward system. Cocaine constricts blood vessels, dilates pupils, increased heart rate and temperature, headache, abdominal pain, and nausea. Intranasal use of cocaine may lead to loss of the sense of smell, nosebleeds, and a runny nose. Addicts often experience irritability, restlessness, and anxiety between binges. Hallucinations and paranoia are also common.
Drug Abuse and Addiction
[edit | edit source]Drug takers use cocaine, also known as crack or coke as a recreational drug. Cocaine is a more high priced way of getting high compared to that of marijuana. Also known as the “caviar of street drugs,” cocaine is known to be mystique, giving users alertness and energetic feelings. In addition, drug users often drink alcohol while under the influence of cocaine. Users feel more alert and aware while feeling the effects of alcohol minimally. The tolerance of taking the drug becomes increasingly higher as users ingest the drug more often. Since cocaine triggers different areas in the brain and put users on a good high, it may lead to addiction. As cocaine gets used regularly, addiction leads to physical and psychological problems and even death. Drug users may become highly dependent on the drug and when they are inaccessible to cocaine, feelings of withdrawal become present. Side effects may include fatigue, depression, anxiety, aches and pain.
References
[edit | edit source]http://drugabuse.gov/drugpages/cocaine.html http://drugabuse.gov/infofacts/cocaine.html http://en.wikipedia.org/wiki/Cocaine#Suppository
"Cocaine Use." WEBMD. WEBMD LLC. Web. 18 Nov 2012. <http://www.webmd.com/mental-health/cocaine-use-and-its-effects>.
Introduction
[edit | edit source]Warfarin is an oral anticoagulant to prevent new blood clots from forming. Blood clot is the aggregation of platelets in the blood vessels and the formation of fibrin network which traps the additional blood cells which can cause heart attack, stroke or other medical problems.[1]
Warfarin also is used to treat venous thrombosis (which is the swelling and blood clot in a vein). It is used to prevent pulmonary embolism (blood clot in the lung). Warfarin also can be called anticoagulants ("blood thinners").
Coagulation
[edit | edit source]Blood coagulation is the process of formation of blood clots. It involves a series, known as the “coagulation cascade.” Each step of the cascade involves a protease, a zymogen, a non-enzymatic protein cofactor, platelets surface, and calcium ions. In each step, zymogen is converted to an active protease by cleavage of one or more peptide bonds in the zymogen. Thrombin is the last in a series of coagulation enzymes that are activated by proteolysis of zymogen. Thrombin then converts the soluble fibrinogen into insoluble fibrin, which eventually forms a fibrin meshwork and the ultimate blood clot.[2]
Who needs Warfarin?
[edit | edit source]Patients with prosthetic heart valves, irregular heartbeat, thrombosis, and embolism are prescribed warfarin.
Overview
[edit | edit source]The chemical name of Warfarin is 4-hydroxy-3-(3-oxo-1-phenylbutyl)-2H-chromen-2-one, and the chemical formula is C19H16O4.
How it works
[edit | edit source]Warfarin is used to decrease the ability of clotting of blood. It works by inhibiting vitamin-K-dependent coagulation factors. It inhibits the production of clotting factors II, VII, IX, and X. These factors require carboxylation of glutamic acid residues to bind phospholipids to the factors, which is a vitamin-K-related process. After vitamin K is converted to vitamin K epoxide in the liver by oxidation, vitamin K epoxide is then reduced to vitamin K epoxide reductase (VKOR) by an enzyme. This reduced epoxide is necessary for the production of coagulation factors. Therefore, warfarin plays a role in prohibiting the actions of VKOR in the liver, so available amount of vitamin-K that can be used for oxidation and the production of coagulation factors is decreased.[3][4]
Diet for patients taking warfarin
[edit | edit source]It is recommended to avoid food which is rich in vitamin K, for example, green leafy vegetables, liver, and vegetable oils.
How medicine works can be affected by the food people eat. Warfarin is a medicine prescribed for people with high risk of forming blood clots. It can prevent harmful blood clots, which may block the flow of blood to the heart or brain from forming. Vitamin K is a vitamin usually found in leafy green vegetables. In contract of Warfarin, vitamin K plays a major role in blood clotting in the body. It is used to reverse the effects of “blood thinning” medications like Warfarin. Warfarin decreases the activity of vitamin K and lengthening the time it takes for a clot to form. Therefore, in order to make warfarin work effectively, it is very important to keep vitamin K intake as consistent as possible. First, keep intake of high vitamin K foods consistent. Second, do not have large changes in the medium vitamin K foods intake. Third, be careful about dietary supplements. It is not wise to take vitamin E or fish oil supplements. Finally, dietitian or physician may restrict cranberry juice from your diet.
| Food | Serving | Vitamin K (mcg) |
|---|---|---|
| Kale, cooked | 1/2 cup | 531 |
| Spinach, cooked | 1/2 cup | 444 |
| Collards, cooked | 1/2 cup | 418 |
| Swiss chard, raw | 1 cup | 299 |
| Swiss chard, cooked | 1/2 cup | 287 |
| Mustard greens, raw | 1 cup | 279 |
| Turnip greens, cooked | 1/2 cup | 265 |
| Parsley, raw | 1/4 cup | 246 |
| Broccoli, cooked | 1 cup | 220 |
| Brussels sprouts, cooked | 1 cup | 219 |
| Mustard greens, cooked | 1/2 cup | 210 |
| Turnip greens, cooked | 1 cup | 184 |
| Spinach, raw | 1 cup | 145 |
| Turnip greens, raw | 1 cup | 138 |
| Endive, raw | 1 cup | 116 |
| Broccoli, raw | 1 cup | 89 |
| Cabbage, cooked | 1/2 cup | 82 |
| Green leaf lettuce | 1 cup | 71 |
| Romaine lettuce, raw | 1 cup | 57 |
| Asparagus | 4 spears | 48 |
Side effects
[edit | edit source]Side effects include unusual bleeding, diarrhea, chest pain, face swelling, sudden headache or dizziness, severe leg pain, body discoloration, and purple fingers and toes.
Other side effects include change in the way things taste, tiredness, pale skin, loss of hair, or even feeling cold or having chills. Warfarin also may cause necrosis or gangrene, which means death of skin or other body tissues).[6]
References
[edit | edit source]- ↑ Voet, Donald; Voet, Judith; Pratt, Charlotte (2006). Fundamentals of Biochemistry. Wiley.
{{cite book}}: CS1 maint: multiple names: authors list (link) - ↑ "Blood coagulation".
- ↑ Harrison, Karl (July 2004). "Warfarin".
- ↑ "Warfarin".
- ↑ "Nutrition care manual".
- ↑ American Society of Health-System Pharmacists (2012). "AHFS Consumer Medication Informatio".
Ibuprofen
[edit | edit source]
Ibuprofen is often used as a NSAID. NSAID is an abbreviation for non-steroidal anti-inflammatory drugs. It is used to relief symptoms of arthritis or fever. Basically, Ibuprofen acts as an analgesic or pain reliever. It is typically found in many over-the-counter drugs, such as Motrin, Advil, Potrin, and Nuprin. In other words, it often comes in capsules, tablets, or powder form. Comparing to that of aspirin for example, Ibuprofen is somewhat short-lived and relatively mild. However, it is known to have an antiplatelet effect. Ibuprofen is also seen as the core medicine in the World Health Organization's (WHO) Model List of Essential Medicines. This is the list of minimum medical needs based on a particular healthcare system.
In addition, ibuprofen acts as a vasoconstrictor because it inhibits the vasodilating prostacyclin that is produced by cyclooxygenase 2 enzymes. In the year 1969, Ibuprofen is known as a carboxylic acid that was first found in the United Kingdom by Boot Pure Drug Company in the name of Brufen. However, many people are familiar that it was discovered by Andrew RM Dunlop with the help of people such a John Nicholson and Stewart Adams. Even till this day, ibuprofen comes in different forms as pain reliever and is seen under a variety of popular trademarks (brand name medicines sold in over-the-counter drugs).
Medical Uses
[edit | edit source]Typically, ibuprofen is used for sickness such as pain, fever, or even dysmenorrhea. It can also be used for rheumatoid arthritis, which is known as a type of inflammatory disease.
Not only is ibuprofen in tablet or capsule forms, but they can also be in topical gel form that can simply absorb through the skin, which are commonly used during sport injuries because it does not cause a high risk of digestive problems.
Dosage
[edit | edit source]Although, ibuprofen is known as its short half-life, it actually consist of a dose-dependent time period of around four to eight hours. This is quite a lot, especially with the kind of characteristic ibuprofen have. There is no specific dose a person should intake. In other words, the recommended dose varies from person to person base on the weight of their body and indication. Even with no specific dose, it is typically said that the maximum amount for over-the-counter use is approximately a dose of 400 mg per dose and 1200 mg per day. However, there is an exception for individuals who have stronger tolerance and response because the maximum that they can take is about 800 mg per dose and 3200 mg per day.
Ibuprofen Lysine
[edit | edit source]Ibuprofen lysine is also known as ibuprofen lysinate. Interestingly, ibuprofen lysine is licensed for treatment of the same condition as ibuprofen in places such as Europe, New Zealand, and Australia. Since lysine salt is known to increase water solubility it is often used for closure of a patent ductus arteriosus in premature infants who are no more than 32 weeks gestational age that weigh between 500 and 1,500 grams. That is exactly one to three pounds. The drug will be ineffective if ibuprofen lysine is used in infants over the 32 week span. The use of Ibuprofen lysine is to support the respiratory and fluid restriction. Besides infants, ibuprofen can be used to help kidney function in people of all ages. Overall, comparing to that of acid ibuprofen, ibuprofen lysine shows to work faster which proves how effective it can be.
Synthesis
[edit | edit source]Ibuprofen was found by the Boot Pure Drug Company and the synthesis is named after the company as the "Boot process". In the process it starts with Friedel-Crafts acylation of Isobutylbenzene.

"The Boot Process"
- Isobutylbenzene reacted with Ethyl chloroacetate would result with α,β-epoxy ester
- Hydrolyzed
- decarboxylated to the aldehyde
- React with hydroxylamine gave oxime
- Converted to nitrile,
- hydrolyzed to the desire acid

Later on, the new Hoechst process named after the Hoechst Company undergoes a similar acetylation, followed by a hydrogenation with (Raney nickel) catalyst. It will result in alcohol and last reaction is palladium-catalyzed carbonylation.

Ibuprofen Mechanism-COX inhibitor
[edit | edit source]

Ibuprofen's function is to block an enzymes called cyclooxygenase(COX). The NSAIDs unselectively inhibit both COX-1 and COX-2. When the COX is inhibited, arachidonic acid is converted into H2(PGH2),then PGH2 is converted into prostaglandins and tothromboxane A2. Prostaglandins could ease inflammation, fever and pain, and tothrombozne A2 could lead to formation of blood clots. Since the only function desired in this brought by COX-2 (easing pain, fever and inflammation) and COX-1 is causing unwanted side effects further researches have been done on blocking COX-2 but not COX-1. After years of research, through X-ray crystallography and biophysical techniques this selective blocking is achieved.
Side Effects
[edit | edit source]Ibuprofen can cause severe allergic reactions to certain people. If the following symptoms are present, seek medical help immediately:
- hives
- swelling in lips, face, throat or tongue
- difficulty breathing
- chest pain
- vision problems
- bloody vomit
- nausea
- fever
- jaundice (yellow skin or eyes)
- convulsions
Less severe symptoms can include:
- upset stomach
- diarrhea
- constipation
- itching
- dizziness
- ringing in ears
incompatibility with other drugs
[edit | edit source]Ibuprofen can cause violent reactions when mixed with some other drugs. This drugs include, but not limited to:
- antidepressant
- aspirin or other NSAIDS
- heart of blood pressure medicine
- lithium
- diuretics
- steroids
- methotrexate
- blood thinner
Nexium
[edit | edit source]Description
[edit | edit source]Esomeprazole, brand name known as Nexium, is categorized as a group of drugs called proton pump inhibitors. By inhibiting ATPase in gastric parietal cells, Esomeprazole decreases the amount of acid secretion in the stomach. This drug is mainly used for people with gastroesophageal reflux disease (GERD), a disease in which an excessive amount of gastric acid causes a backflow from the stomach and thus leading to the burning of the esophagus. By preventing the formation of gastric acid, Esomeprazole allows the injured esophagus to heal and prevent further damages.
Drug Structure
[edit | edit source]The molecular formula of Esomeprazole is (C17H18N3O3S)2Mg* 3 H2O, with molecular weight of 767.2g as a trihydrate and 713.1g on an anhydrous basis.
Side effects
[edit | edit source]Studies have showed patients who receive high-dose of Esomprazole have increased risk of fractures of the hip, wrist, or spine. High dose defines as multiple intakes every day for a long-term period. In addition, Esomeprazole may cause serious side effects such as the following: •blisters •hives •rash •itching •difficulty breathing or swallowing •irregular, fast, or pounding heartbeat •uncontrollable shaking of a part of the body •seizures •stomach pain •fever
References
[edit | edit source]http://www.drugs.com/ibuprofen.html http://www.drugs.com/ibuprofen.html http://www.nlm.nih.gov/medlineplus/druginfo/meds/a682159.html http://www.news-medical.net/health/Ibuprofen-Mechanism.aspx http://www.chm.bris.ac.uk/motm/ibuprofen/welcome.htm http://en.wikipedia.org/wiki/Ibuprofen
Loratadine
[edit | edit source](C22H23CIN2O2) is generally known as Claritin, an over the counter drug. It often comes in a white to off-white powder form. It is not soluble in water, but very soluble in acetone, alcohol, and chloroform. It is called ethyl 4-(8-chloro-5,6-dihydro-11H-benzo[5,6]cyclohepta[1,2-b]pyridin-11-ylidene) -1-piperidinecarboxylate, with a molecular weight of 382.89g.
In tablet form, it contains ingredients other than loratadine and antihistamines, which are inactive ingredients such as corn starch, lactose, and magnesium stearate. In Syrup form, it also contains loratadine and antihistamine, but also includes other inactive ingredients such as citric acid, edetate disodium, artificial flavor, glycerin, propylene glycol, sodium ben-zoztem and water. It's pH is around 2.5 -3.1.
Loratadine is used to relive allergy symptoms caused by dust, pollen, and other substances. Loratadine is in the drug class called antihistamines and blocks the histamines on H1 receptors from working and causing allergic symptoms in the body. Common allergy symptoms include but are not limited to sneezing, itching, and runny nose. Loratadine is a second-generation histamine H1 receptor antagonist because it does not have a sedative side effective. Loratadine comes in 4 different forms: liquid, oral suspension, immediate or extended release tablet, and orally disintegrating tablet. Loratadine can also be combined with pseudophedrine. When taken orally, loratadine is absorbed through the gastrointestinal tract and metabolized by cytochrom P450 before binding to plasma membrane proteins.
Although loratadine is considered a ‘non-sedating’ antihistamine, dose-related sedation has been noted. For this reason, it would be prudent to monitor for drowsiness when used concurrently with other CNS depressants such as barbiturates, benzodiazepines, opiate agonists, antipsychotics, tricyclic antidepressants, ethanol, other H1-blockers, and anxiolytics, sedatives, and hypnotics.

Loratadine Pharmacokinetics
[edit | edit source]
In vitro studies, loratadine is metabolized to descarboethoxyloratadine mainly by CYP3A4, and some by cytochrome CYP2D6. CYP3A4 inhibitor, loratadine is metabolized to descarboethoxyloratadine mainly by CYP2D6. Ketoconazole, erythromycin(both CYP3A4 inhibitors), or cimetidine (CYP2D6 and CYP3A4 inhibitor) increases plasma concentration of loratadine. The half life of loratadine is about ~8.4 hours and its metabolite is ~28 hours. Elimination occurs through the fecal and renal routes.
Loratadine is also a substrate for P-glycoprotein transport.
[edit | edit source]Cimetidine, erythromycin, nefazodone and ketoconazole have been shown to interfere with the metabolism of loratadine. the inhibition of P-450 CYP3A4 isozyme lead to increasing serum concentrations of loratadine and its metabolite. Increased level of loratadine serum concentrations do not result in significant differences in adverse reactions compare to control patients. However, it is important to be caution of using loratadine with other drug combination in the concurrent risk for arrhythmogenic events. Although the drug interaction have not been confirmed between loratadine and cytochrome P-450, it is need to be are of potent CYP3A4 due to the serious nature of interactions between these drugs and certain other H1-antagonists.
Side effects
[edit | edit source]Some people have allergic reactions when this drug is ingested. Some serious side effects include:
- fast and uneven heart rate
- jaundice (yellow skin or eyes)
- seizures
- feelings of passing out
People can be mildly allergic and present following symptoms that are not as severe:
- headache
- nausea
- nervousness
- drowsiness
- stomach pain, diarrhea
- dry mouth
- sore throat
It is recommended for people to contact the doctor immediately if they present any of the serious symptoms.
Reference
[edit | edit source]http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0001010/ http://en.wikipedia.org/wiki/Loratadine http://www.rxlist.com/claritin-drug.htm http://www.rxlist.com/claritin-drug.htm
Description
[edit | edit source]Esomeprazole, brand name known as Nexium, is categorized as a group of drugs called proton pump inhibitors. By inhibiting ATPase in gastric parietal cells, Esomeprazole decreases the amount of acid secretion in the stomach. This drug is mainly used for people with gastroesophageal reflux disease (GERD), a disease in which an excessive amount of gastric acid causes a backflow from the stomach and thus leading to the burning of the esophagus. By preventing the formation of gastric acid, Esomeprazole allows the injured esophagus to heal and prevent further damages.
Drug Structure
[edit | edit source]The molecular formula of Esomeprazole is (C17H18N3O3S)2Mg* 3 H2O, The structural formula is shown below.

Pharmacokinetics
[edit | edit source]Single 20–40 mg oral doses generally give rise to peak plasma esomeprazole concentrations of 0.5-1.0 mg/L within 1–4 hours, but after several days of once-daily administration these levels may increase by about 50%. A 30 minute intravenous infusion of a similar dose usually produces peak plasma levels on the order of 1–3 mg/L. The drug is rapidly cleared from the body, largely by urinary excretion of pharmacologically-inactive metabolites such as 5-hydroxymethylesomeprazole and 5-carboxyesomeprazole. Esomeprazole and its metabolites are analytically indistinguishable from omeprazole and the corresponding omeprazole metabolites unless chiral techniques are employed.
Side effects
[edit | edit source]Studies have showed patients who receive high-dose of Esomprazole have increased risk of fractures of the hip, wrist, or spine. High dose defines as multiple intakes every day for a long term period. In addition, Esomeprazole may cause serious side effects such as the following:
•blisters
•hives
•rash
•itching
•difficulty breathing or swallowing
•irregular, fast, or pounding heartbeat
•uncontrollable shaking of a part of the body
•seizures
•stomach pain
•fever Methotrexate
Description
[edit | edit source]
An chemotherapy drug used to treat cancer patients with immunosuppressant properties. It is an inhibitor of dihydrofolate reductase, which helps with the formation of RNA. It also inhibits thymidilate synthetase, which is necessary for synthesis of DNA.
Usage
[edit | edit source]
Methotrexate is used as a treatment for breast, head and neck, lung, stomach, and esophagus cancers, Acute lymphoblastic leukemia (ALL), non-Hodgkin's lymphoma (NHL), gestational trophoblastic cancer, and mycosis fungoides (cutaneous T-cell lymphoma). Methotrexate is given:
- As an infusion into the vein
- As an injection into the muscle
- Another method it is given is by intraventricular or intrathecal infusion in which it is infused directly into the spinal fluid.
- As oral form
Side effects
[edit | edit source]- Low blood counts
- Mouth sores
- Nausea and vomiting
- Kidney toxicity
- Skin rash, reddening of the skin
- Diarrhea
- Hair loss
- Eye irritation
- Loss of fertility
Mechanism of action
[edit | edit source]
Cancerous cells differ from normal cells in their cell division. When normal cells come in contact with cells that are identical to them, they stop dividing. This mechanism is known as contact inhibition. Cancerous cells do not have this ability. Cancer cells have no limit in cell division, which leads to the formation of tumor. Similar to normal cells, cancerous cells also go through a cell cycle, which consists of a resting phase, active growing phases, and mitosis.
Chemotherapy is able to kill cancer cells by damaging the DNA or RNA that are responsible for cell division. If the cancer cells cannot divide, the tumor will shrink. Chemotherapy drugs can be effective to dividing cells, which are called cell-cycle specific; or to cells that are at rest, which are called cell-cycle non-specific. Chemotherapy schedule is assigned based on the type of cancer cells and the rate at which they are dividing.
However, chemotherapy drugs cannot distinguish between the cancerous cells and the normal cells, which leads to the side effects. The normal cells most commonly affected by chemotherapy are the blood cells, the cells in the mouth, stomach, and the hair follicles; resulting in low blood counts, mouth sores, nausea, diarrhea, and/or hair loss.
Methotrexate are classified as antimetabolites. Antimetabolites are very similar to normal cellular materials. When antimetabolites are taken into the cellular metabolism, they will compete with the normal cellular materials and inhibits certain pathways and eventually cell division. Methotrexate achieve its chemotherapeutic effect by being able to compete with folic acid in cancer cells, which results in folic acid deficiency in the cells and causing their death. Methotrexate also competes with folic acid in normal cells and causes significant side effects such as low blood cell counts, hair loss, mouth sores, diarrhea, liver, lung, nerve and kidney damage.
Methotrexate’s chemotherapeutic effects are a result of its ability to limit DNA and RNA synthesis by inhibiting dihydrofolate reductase and thymidylate synthetase. It enters cells through an active transport system used by folic acid. Methotrexate will then undergo polyglumatation. MTXGlu is even more effective in inhibits the enzymes. The enzyme Dihydrofolate Reductase maintains the level of reduced folic acid by reducing dihydrofolic acid which has been produced during thymidylate synthesis. Dihydrofolate reductase reduces folic acid to tetrahydrofolate, an essential co-factor in the synthesis of purine nucleotides.
Reference
[edit | edit source]"Methotrexate." Scott Hamilton CARES Initiative. N.p.. Web. 21 Nov 2012. <http://chemocare.com/chemotherapy/drug-info/Methotrexate.aspx>
Tian, Henghe, and Bruce N. Cronstein. "Bulletin of the NYU Hospital for Joint Diseases." Bulletin of the NYU Hospital for Joint Diseases. 65.3 (2007): 168-73. Print.
Brigitte C. Widemanna, Peter C. Adamson b. Understanding and Managing Methotrexate Nephrotoxicity Pediatric Oncology 2006;11;694-703.

Background
[edit | edit source]Vancomycin is considered a last-resort antibiotic, and tends to be effective in treating bacteria that is highly resistant to many, if not all, other drugs. It is a glycopeptide, derived from Amycolatopsis orientalis cultures. One of the most well-known bacteria that vancomycin is effective against is methicillin-resistant Staphylococcus aureus (MRSA). Overall, vancomycin is successful against various Gram-positive bacteria, including highly resistant streptococci, enterococci, and staphylococci strains. Vancomycin should not be taken as an oral medication, as it cannot be absorbed that way, but rather through an intravenous injection. [1]
Overview
[edit | edit source]Vancomycin works by binding with the terminal amino acids of peptidoglycan chains in cell walls. By doing so, the production of the bacterial cell wall is prevented by not allowing the cross-linking between neighboring peptidoglycan strands. Without a strong wall, cell lysis is induced and the bacterial cell falls apart. [1]
Mechanism of Vancomycin
[edit | edit source]In bacterial cells the cell wall consists of peptidoglycan. This peptidoglycan is made up of repeating units of two sugar molecules, N-acetylglucosamine (NAG) and N-acetylmuramic acid (NAM). Attached to NAM is a series of amino acids that end with a D-alanine dipeptide. Inside the cytoplasm at the cell membrane, NAM and NAG are linked at the bactoprenol to form the disaccharide NAM-NAG. NAM-NAG is brought to the outside area of the cell membrane by the bactoprenol, where penicillin binding proteins (PBPs) bind to the D-Ala dipeptide. One PBP is transpeptidase, which links the peptide side chains of two NAG molecules to form a peptide cross link. This peptide cross link helps to add stability to the cell wall by connecting a L-lysine of one NAM side chain to the D-Ala of another NAM side chain. [2]
Vancomycin binds to the D-Ala-D-Ala end of the NAM-NAG disaccharide and prevents transpeptidase from making the peptide cross link. [2] As autolysins continue breaking the peptide cross link and no new cross link can be formed because of Vancomycin, the bacterium bursts as a result of osmotic lysis.
Medicinal Uses
[edit | edit source]Vancomycin hydrochloride is primarily used in the treatment of severe infections resulting from susceptible strains of methicillin-resistant staphylococci. Typically, vancomycin is a last resort type of antibiotic that is normally used for patients allergic to penicillin, for those that haven't responded to other types of antibiotics (like cephalosporins), and for patients plagued by vancomycin-susceptible bacteria. Courses of treatment should be determined once the type of bacteria is identified and susceptibility data has been obtained. It has been effective against staphylococcal endocarditis, other infections from staphylococci (bone infections, respiratory infections, and skin infections). Other types of infections that vancomycin has been effective against include diphtheroid endocarditis. To improve the effectiveness of combating bacterial infections, vancomycin will often be used with aminoglycosides or rifampin. Vancomycin is not effective against viral infections.
Like all medications, there are typically risks and side effects associated with each drug. As with most antibiotics, the risk of developing drug-resistant bacteria has become an ever increasing battle. When drug-resistant bacteria form, it becomes more difficult to treat and combat the infections related from these bacteria. Vancomycin must be administered intravenously because the drug is very irritating to the tissue. If the medication interacts with the muscle tissue, typically there is associated muscle pain, tenderness, and the tissue begins to die, therefore supporting the need to administer vancomycin intravenously. Also, prolonged use of vancomycin with other medications can have adverse effects as well. For example, prolonged use with anesthetics can lead to complications such as hypotension, pruritis, and erythema. Some of the side effects with antibiotics typically include diarrhea and stomach pains. Other effects include anaphylaxis, drug fever, nausea, chills, and rashes.
Source: http://www.drugs.com/pro/vancomycin-hydrochloride.html
Side Effects
[edit | edit source]Vancomycin can trigger violent allergic reactions on certain individuals. The most common severe symptoms are:
- hives
- swelling on the lips, face, throat or tongue
- hearing loss
- fever
- lightheadedness
- extreme stomach pain
- fainting
Incompatibility with other drugs
[edit | edit source]When mixed with certain other drugs, vancomycin can cause severe reactions. The most common drugs that react with vancomycin are:
- amikacin
- gentamicin
- kanamycin
- neomycin
- streptomycin
- tobramycin
Reference: http://www.drugs.com/vancomycin.html http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0000282/ http://pharmacologycorner.com/vancomycin-mechanism-action-animation/
References
[edit | edit source]- ↑ a b c Dewick, Paul M. Medicinal Natural Products: A Biosynthetic Approach, Third Edition, John Wiley and Sons Ltd. 2009.
- ↑ a b Slonczewski, Joan L. Foster, John W. Microbiology: An Evolving Science, Second Edition, W.W. Norton & Company. 2009.
Overview
[edit | edit source]Thalidomide was synthesized in 1953 by a West German company as a sedative. It became popular outside of the United States to help with morning sickness in the mid 1950s, but in 1961 it was discovered to cause birth defects. Its popular use was discontinued, but it is now used as a treatment for other diseases, such as leprosy and multiple myeloma. [1]
Composition
[edit | edit source]Thalidomide is a tasteless and odorless white crystalline compound. [2]
The chemical formula of thalidomide is C13H10N2O4, and it exists as a racemic mixture.
Synthesis
[edit | edit source]A two-step synthesis of thalidomide is presented in 2 scheme below requires no purifications. Reaction of L-glutamine with N-carbethoxyphthalimide produces N-phthaloyl-L-glutamine. Cyclization of N-phthaloyl-L-glutamine to produce thalidomide is accomplished by treatment with CDI in the presence of a catalytic amount of DMAP. [3] [3]
Medical Importance
[edit | edit source]Despite the widespread tragedy associated with thalidomide, it continues to be used in medical settings due to its usefulness in the treatment of a number of conditions and diseases, such as leprosy and cancer.
The aspects of thalidomide most applicable to treatments such as those for leprosy and multiple myeloma are its inhibition of angiogenesis, and the anti-inflammatory effects. In leprosy, thalidomide is particularly effective in suppressing painful related conditions.
[4]
In a study of patients with advanced multiple myeloma, thalidomide was shown to cause remission in 10% of the patients. This could be due to a number of factors, including angiogenesis inhibition, the ability of thalidomide to alter adhesion molecule expression, and its inhibiting effect on the growth of the tumor necrosis factor alpha. [5]
Thalidomide selectively inhibits the tumor necrosis factor alpha by degrading its mRNA, decreasing the half life of this molecule. [6] Inhibition of this factor, which causes fever and discomfort in cancer patients, can make quality of life much better. However, it may also weaken the immune system. [7]
Thalidomide can also help with the wasting caused by HIV, because it inhibits tumor necrosis factor alpha. [4].
Teratogenic Effects
[edit | edit source]Children whose mothers took thalidomide during pregnancy were often born with shortened or missing limbs.
The structure of thalidomide contributes to its teratogenic effects. Tests show that only the S enantiomer of thalidomide is teratogenic. Thalidomide was originally racemic, but cannot be made safe by isolating one enantiomer, as the R enantiomer will readily convert to the S enantiomer in the body. [8]
There have been various hypotheses regarding the mechanisms of thalidomide, particularly as it relates to teratogenicity. However, current research is mostly concerned with "(1) oxidative stress/damage, (2) DNA intercalation, (3) inhibition of angiogenesis, and (4) cereblon (CRBN) binding." [4].
- Thalidomide has been shown to encourage production of oxygen radicals in rats to whom it has been administered, which results in oxidative stress as fewer viable oxygens are biologically available, effecting a large number of organs that rely on oxygen.
- Thalidomide attaches to promoter sites that are GC-rich using DNA intercalation, decreasing transcription of IGF-1 and FGF-2, which both lead to angiogenesis and the proper growth of extremities and limbs.
- Thalidomide is an angiogenesis inhibitor, which means that it prevents the formation of new blood vessels. This is useful in the treatment of diabetic retinopathy, macular degeneration, and solid tumors [9]
- CRBN has been found to be a thalidomide-binding protein, and its mutant form, which binds poorly to thalidomide, may weaken thalidomide's tetatogenic limb affects when it is overexpressed.
It is likely that these mechanisms all contribute to some way in thalidomide's overall teratogenic effect.
When pregnant rabbits (thalidomide sensitive) and pregnant rats (thalidomide insensitive), green fluorescent protein expression in the cells of the limb buds of the rabbit embryos was decreased, while that the in the rats was unaffected. [4].
Exposure periods have been correlated to specific problems caused by thalidomide: http://toxsci.oxfordjournals.org/content/122/1/1/F3.large.jpg
Drug Trials
[edit | edit source]Thalidomide originally underwent animal testing as well as clinical trials in humans. This included 98 children under the age of 1, 160 nursing mothers, and 81 women in the third trimester of pregnancy. No teratogenic effects were observed during the clinical trials. [2] Following the thousands of birth defects caused by thalidomide, it became mandatory to test drugs on pregnant animals, to detect teratogenic effects. [1]
References
[edit | edit source]- ↑ a b [2]
- ↑ a b The Saga of Thalidomide
- ↑ a b c A Concise Two-Step Synthesis of Thalidomide Invalid
<ref>tag; name "A Concise Two-Step Synthesis of Thalidomide" defined multiple times with different content - ↑ a b c d Thalidomide: The Tragedy of Birth Defects and the Effective Treatment of Disease
- ↑ Antitumor Activity of Thalidomide in Refractory Multiple Myeloma
- ↑ Moreira et. al.
- ↑ Sampaio et. al
- ↑ A Concise Two-Step Synthesis of Thalidomide
- ↑ Angiogenesis Inhibition
Overview
[edit | edit source]The empirical formula of simvastatin is C25H38O5 with the molecular weight of 418.57. Simvastatin also is a white, crystalline powder that is insoluble in water. In taking simvastatin, make sure we are not allergic to it. Patients who taking simvastatin have to limit their alcohol beverages.
Simvastatin, also known as Zocor, is a drug used to control cholesterol levels. It is a hypolipidemic agent, used to lower the levels of lipids. It is derived from the fermentation of Aspergillus terreus, a fungus which can be used to make important organic compounds. Cholesterol biosynthesis was researched at Merck by Jesse Huff, a biochemist, and his colleagues in the early 1950s. Carl Hoffman, Karl Folkers, and some colleagues at Merck isolated mevalonic acid from a yeast extract in 1956. They confirmed that this acid can be used in cholesterol biosynthesis. In Sankyo, Japan, Akira Endo was able to isolate Compactin from Penicillum citrinium, a fungus, in 1976. Carl Hoffman was able to isolate lovastatin from Asperigillus terreus in 1979. From lovastatin, the scientists at Merck were able to synthetically derive Simvastatin.
Usage
[edit | edit source]Take simvastatin exactly as prescribed. Never take simvastatin in larger amounts, or for longer than recommended by your doctor. Follow your doctor's dosing instructions very carefully. Taking too much of this medication may cause serious or life-threatening side effects.
Simvastatin is usually taken at bedtime or with an evening meal. If you take simvastatin more than once daily, take it with meals. Your doctor may occasionally change your dose to make sure you get the best results. To be sure simvastatin is helping your condition and is not causing harmful effects, your blood will need to be tested often. Your liver function should be tested every 3 to 6 months. Visit your doctor regularly. You may need to take simvastatin on a long-term basis for the treatment of high cholesterol. You may need to stop using this medicine for a short time if you have surgery or a medical emergency. Do not stop taking simvastatin unless your doctor tells you to.
Simvastatin is only part of a complete program of treatment that also includes diet, exercise, and weight control.
Side Effects
[edit | edit source]Daily use of alcohol may increase your risk for liver problems. Other side effects include unexplained muscle pain, tenderness, weakness; fever, unusual tiredness, and dark colored urine; pain or burning when you urinate; nausea, upper stomach pain, itching, loss of appetite, or dark urine. [The Internal Drug Index].
Simvastatin contains some inactive ingredients, and if allergic, can cause allergies to surface. Cautions should be taken if the patient has a history of alcohol use, kidney disease, or liver disease. If a surgery needs to be performed, let the doctor know about the herbal products, nonprescription drugs, and prescription drugs that have been used. When taking Simvastatin, be aware of your alcohol intake. When combined with alcohol, this drug can increase the risk of liver problems. Adults, who are older, should take more precaution. They may become more sensitive to this drug, which may produce muscle problems. When pregnant, do not use this drug because it can harm your unborn baby. Also, preventing pregnancy is important because simvastatin may pass into breast milk. Although this is still unclear, try not to breast feed while taking this drug.
Few people who have been taking this drug had reported problems of mild memory or confusion. These are rare effects that may occur. Infrequently, muscle problems may occur and a very serious condition may occur, rhabdomyolysis (muscle fiber breakdown leading to muscle fiber in the bloodstream. Be cautious of symptoms that include weakness, tenderness, or muscle pain. There are rare instances of liver problems. Be cautions of nausea or vomiting, severe abdominal or stomach pain, dark urine, or yellowing of skin or eyes. This drug may also cause serious allergic reactions. Be cautious of breathing troubles, severe dizziness, itching or swelling of the face, throat, or tongue, rashes, or any symptoms of allergic reactions.
Marijuana
[edit | edit source]1) Inhibitory neurotransmitters are active in the synapse before marijuana goes into the system. The job of these neurotransmitters is inhibiting dopamine from being released into the synapse.

2) Cannabinoid receptors shut off the releasing of inhibitory neurotransmitters when activated by the body’s native cannabinoid. Dopamine is released when there is no inhibition.

3)THC, the active chemical in marijuana, mimics anandamide and binds to cannabinoid receptors. The inhibition is shut off and dopamine starts to exit into the synapse.

4)Anandamide is famous for being involved in removing short term memories. It is also responsible for giving the relaxation and calmness feeling and it slows down the movement. Unlike THC, anandamide is very fragile and has a the tendency to break down very quickly in the body. That is why anandamide doesn’t produce perpetual natural “high”.
Cocaine
[edit | edit source]1) Dopamine is taken out of the synaptic cleft by the dopamine transporting molecules after dopamine is done with its job.

2) What cocaine does is blocking the transporters, making dopamine stuck in the synaptic cleft. As a result, dopamine binds to the receptors over and over and that causes overstimulation in the cell.

3) Finally, Cocaine concentrates in the reward pathway of the brain. It also activates the part of the brain that is in control of the voluntary movement. That is why people who abuse the use of cocaine and not able to stay still.
Alcohol
[edit | edit source]1) GABA which is an inhibitory transmitter is active all over the brain. These neurotransmitters control the neural activity in many of brain pathways. The cell is less likely to fire when GABA binds to its receptors.

2) In another area of the brain, Glutamate acts as the brain’s general purpose excitatory neurotransmitter.
 3) Alcohol delivers sedative punch once it goes into the brain. It interacts with GABA receptors and drives them to be more inhibitory. Then, it binds to the glutamate receptors and inhibits glutamate neurotransmitters from binding to their receptor. As a result, the cell is inhibited from being excited.
3) Alcohol delivers sedative punch once it goes into the brain. It interacts with GABA receptors and drives them to be more inhibitory. Then, it binds to the glutamate receptors and inhibits glutamate neurotransmitters from binding to their receptor. As a result, the cell is inhibited from being excited.
 4) Therefore, alcohol affects areas of the brain that are in charge of memory formation and decision making (frontal lobe).
4) Therefore, alcohol affects areas of the brain that are in charge of memory formation and decision making (frontal lobe).
Heroin
[edit | edit source]1) Inhibitory transmitters that are active in the synapse inhibit dopamine neurotransmitters from being released.

2) When natural opioid that are produced by the body (endogenous opioids) activate opioid receptors, inhibitory transmitters release is shut down. When there is no inhibition, dopamine is released and it binds to dopamine receptors.

3) When heroin enters the system, it binds to the opioid receptors because it has the same shape as endogenous opioids. This mechanism turns off dopamine inhibition and dopamine gets released into the synapse. This in turn generates feelings of sedation and well-being.

4) These opioid receptors are connected to neurons that are in charge of the transmission of pain, stress response, and emotional attachment. As a result, drugs that are heroin-like are used as painkillers to sustain pain of a massive injury.
LSD
[edit | edit source]1) LSD drug is exclusive on a neuron called serotonin. LSD shape resembles that of Serotonin and diminish the serotonin effect by binding to its receptor.

2) There are many kinds of serotonin receptors in the brain and each is responsible for a specific function.

3) LSD binds to certain receptors but not in the same way usually. LSD can inhibit or excite these receptors. This is why LSD has complex sensory effects.

4) LSD is just like other hallucinogens, they excite a certain region in the brain called locus coeruleus. In this area, there is a neuron attached to it that branches to different sensory areas in the brain. Locus coeruleus is in charge of feelings like wakefulness and illicitness response to unexpected stimuli.
Meth
[edit | edit source]1) Dopamine is removed from the synaptic cleft using dopamine transporters. Meth is taken into the cell because meth mimics the shape of dopamine and the dopamine transporters does not recognize the dopamine from meth.

2) When meth is transported to the inside of the cell, it enters the vesicle that carries dopamine and forces the dopamine out of the vesicle.

3) The dopamine gets pumped out of the cell into the synaptic cleft when dopamine is abundant inside the cell by the reverse pumping of the dopamine transporters.

4) Once the dopamine is outside of the cell, it binds to the dopamine receptors again and again because it is abundant in synaptic cleft.

5) Meth works directly on the brain’s reward pathway. Therefore, it is highly addictive and meth users feel intense pleasure when they take it.
Ecstasy
[edit | edit source]1) Serotonin is removed from the synaptic cleft by serotonin transporters after they have done their job.

2) Serotonin transporters pick up ecstasy because ecstasy mimics the shape of serotonin. Actually, serotonin transporters have a higher affinity to ecstasy than serotonin molecules themselves.

3) The interaction of serotonin transporters with ecstasy confuses the transporters in a way that they start pumping serotonin out of the cell into the synaptic cleft.

4) Because serotonin becomes abundant in the synaptic cleft, it starts binding to receptors over and over causing overstimulation to the cell.

5) Ecstasy has an effect on sleep, mood, perception, and appetite. It also has an indirect effect on reward pathway. Ecstasy is not as addictive as other drugs because it has a milder release of dopamine along the reward pathway.
Hurler Syndrome
[edit | edit source]Hurler syndrome is an inherited disease that associates with storage and urinary excretion of mucopolysaccharides dermatan sulfate and heparin sulfate. People who get Hurler syndrome cannot break down long sugar molecules called mucopolysaccharides or glycosaminoglycans – GAGs – due to alpha-L-iduronidase deficiency, a lysosomal enzyme responsible for degradation of glycosaminoglycans. GAGs can build up in the lysosomes of the brain, heart, liver, bones and other organs of the body. The accumulation of GAGs in human body results in damages to organs.
Symptoms
[edit | edit source]- Abnormal bones
- Cloudy corneas
- Deafness
- Short stature
- Heart diseases
- Joint disease, including stiffness
- Mental retardation
- Thick, coarse facial features, i.e. Low nose bridge, flat face, large head
- Enlarged liver and spleen
- Diarrhea
- Breathing difficulties and snoring
Child with Hurler syndrome appears normal at birth. The symptoms start to appear between 6 months and 2 years of age and most often between ages 3 and 8. Most people with Hurler syndrome can not survive beyond age 10.
Background Info
[edit | edit source]The Hurler Syndrome was first tackled by a researcher named Elizabeth F. Neufeld in the late 1960s. Initially, Neufeld linked the cause of the disease to the polymers of chondroitin sulfate B and heparitin sulfate, but due to their dissimilar chemical structures, it was very difficult to determine defects. She later hypothesized that excessive synthesis of mucopolysaccharide chains and excess of UDP-glucuronic acid, caused by defective UDP-xylose responses, were the causes. However, she was proven wrong when inhibiting the UDP-glucose dehydrogenase had no effect on the experimental cell culture she set up. Her hypothesis was again proven wrong when she used the radioactivity of (35)SO4 onto macromolecules with high solubility to measure the content of mucopolysaccharides. The mucopolysaccharide content in the cells of a Hurler patient did not decrease, whereas it did with the samples of a normal persons. As a result of these two experiments, Neufeld was later able to conclude that the Hurler syndrome was a lysosomal storage disease caused by the inability of breaking down mucopolysaccharides.
Treatments
[edit | edit source]A therapy called enzyme replacement was developed to tackle Hurler syndrome. Alpha-L-iduronidase cDNA in human and canine was used to made recombinant enzyme in Chinese hamster ovary cells. These cells secreted alpha-L-iduronidase with mannose 6-phosphate signal. Mannose 6-phosphate is a carbohydrate modification, which inhibits alpha-L-iduronidase uptake. The recombinant enzyme was tested in alpha-L-iduronidase-deficient dogs. It degraded GAGs from many organs in both short term and long term experiment. The recombinant enzyme was later tested on human and approved by FDA in 2003.
Even though having been proven to be effective, the enzyme replacement treatment has some limitations. Not all tissues are equally correctible1. Mucopolysaccharides stored in central nervous system are not accessible by the enzyme due to the blood-brain barrier. Lastly, enzyme replacement treatment is very costly, half a million dollars a year1.
Another therapy for Hurler syndrome, transplantation of hematopoietic stem cells, is available. This method helps maintain the normal mental development. The therapy is only effective if it is performed early enough in life. Another limitation of this treatment is that it is risky.
References
[edit | edit source]- Neufeld, Elizabeth F. (2011). "From Serendipity to Therapy". Annual Review of Biochemistry. 80: 1–15. doi:10.1146/annurev.biochem.031209.093756. PMID 21675915.
- Kakkis, E.D.; Matynia, A.; Jonas, A.J.; Neufeld, E.F. (1994). "Overexpression of the Human Lysosomal Enzyme α-L-Iduronidase in Chinese Hamster Ovary Cells". Protein Expression and Purification. 5 (3): 225–32. doi:10.1006/prep.1994.1035. PMID 7950365.
- http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0002184/
- Scott, H. S.; Ashton, L. J.; Eyre, H. J.; Baker, E; Brooks, D. A.; Callen, D. F.; Sutherland, G. R.; Morris, C. P.; Hopwood, J. J. (1990). "Chromosomal localization of the human alpha-L-iduronidase gene (IDUA) to 4p16.3". American journal of human genetics. 47 (5): 802–7. PMC 1683689. PMID 2220820.
- Stoltzfus, L. J.; Sosa-Pineda, B; Moskowitz, S. M.; Menon, K. P.; Dlott, B; Hooper, L; Teplow, D. B.; Shull, R. M.; Neufeld, E. F. (1992). "Cloning and characterization of cDNA encoding canine alpha-L-iduronidase. MRNA deficiency in mucopolysaccharidosis I dog". The Journal of biological chemistry. 267 (10): 6570–5. PMID 1551868.
- http://medschool.ucsf.edu/lysosomal/hurler/
RNA Based Drugs
[edit | edit source]RNAi Drugs
[edit | edit source]RNAi drugs are a relatively new class of drugs that rely on a mechanism known as RNA interference to inhibit or silence gene expression of certain genes . Specifically, RNAi drugs are made of synthesized small interfering RNA molecules (siRNAs) that recruit and inhibit expression of specific mRNA transcripts that code for a particular gene [3]. Thus, the strategy behind RNAi drugs towards the treatment of cancer is that by synthesizing siRNA molecules that bind to specific mRNAs responsible for cancer-causing genes, the siRNAs, by the mechanism of RNA interference, can inactivate that mRNA transcript coding for a harmful protein and thus inhibit the translation of that protein that contributes to the symptoms of cancer and disease. Because of the ability of siRNAs to degrade mRNAs and silence their genes, RNAi based drugs are being seen as a new and innovative approach to treating cancer. Possible indications for RNA-based therapeutics include HIV, macular degeneration, and the treatment of cancerous tumor cells.
Systemic siRNA delivery
[edit | edit source]Systemic siRNA delivery strategies can be divided into passive and active (targeted) delivery.
Applications
[edit | edit source]Since the discovery of RNA interference in 2002, scientists have been actively investigating the potential of RNA interference to inhibit the expression of harmful or cancerous genes that are responsible for a variety of human diseases and cancers. For instance, scientists began to exploit RNA interference for the treatment of macular degeneration, which is a condition caused by the over-production of the VEGF gene in the eye and leads to the excess growth of blood vessels behind the retina that can eventually cause blindness. [1] Thus, scientists initially sought to use RNAi drugs as a way to turn off the overactive VEGF gene in the eye.
Another potential use of RNAi has been actively investigated in the treatment of HIV and is leading to the development of a novel class of HIV medications. A study published in January 2011 found that delivering an siRNA molecule bound to an RNA aptamer molecule that specifically binds to the surface of HIV cells significantly reduced expression of the HIV virus in mice injected with the virus. [4].
Challenges in Drug Delivery
[edit | edit source]The manufacture and marketing of RNAi drugs have been especially challenging due to difficulties in its drug delivery system; RNA is degraded in the human bloodstream, making it difficult for the drug to reach its intended site of action in human cells to be effective. [2] To counter this problem, scientists are investigating ways to couple RNAi molecules with other molecules to prevent the premature breakdown of RNAi molecules in the bloodstream before reaching cells or package RNAi molecules inside enclosed molecules that can transport the RNAi molecules to ensure its effective delivery. For instance, UCSF researchers are exploring ways to package siRNAs inside fat-coated molecules known as liposomes as a means of drug delivery, which are already extensively used as drug vehicles in commercial drug development. [3] Once transported to its intended site of action, the siRNA contents can be released out of the liposomes to silence genes at that site.
References
[edit | edit source]- Lewis, Susan K. “The RNAi Cure?” PBS. 1 July 2005 <http://www.pbs.org/wgbh/nova/body/rnai-cure.html>.
- Pollack, Andrew. “Drug-Makers Fever for the Power of RNA Interference Has Cooled”. The New York Times. 7 February 2011 <http://www.nytimes.com/2011/02/08/science/08rna.html?_r=1&pagewanted=all>.
- Norris, Jeffrey. “UCSF Leads Consortium to Radically Change Cancer Drug Development”. UCSF. 21 July 2009 <http://www.ucsf.edu/news/2009/07/8192/sirna-drug-development-university-labs>
- Willyard, Cassandra. “Double Whammy for HIV”. Nature. 19 January 2011 < http://www.nature.com/news/2011/110119/full/news.2011.30.html>.
History
[edit | edit source]
Antibiotics were first produced with Penicillin in 1928 and was very effective. Many pharmaceutical companies looked at Penicillin as an example, and later generated other antibiotics products of their own. Antibiotics quickly had a large impact on human, animals, and the living bacteria. According to an article, "Multidrug Resistance in Bacteria", by Hiroshi NIkaido, an estimate of 100,000 tons of antibiotics were produced each year; and due to its ability to cure diseases and popularity, we took advantage and over used the antibiotics; in which resulted in a multi-drug resistance.
Key Characteristics of Antibiotics
[edit | edit source]The use of antibiotics in a clinical setting has been directed by several key characteristics of antibiotics:
- Antibiotics have selective toxicity: This allows for detection of bacterial targets rather than eukaryotic cells that the body needs.
- Antibiotics have a limited spectrum of activity: Therefore each antibiotic only affect a few bacterial species, so there is a need for many different antibiotics. An example is penicillin, which mainly kills gram-positive bacteria. On the other hand, ampicillin has an additional amino group that allows for the penetration of a gram-negative outer membrane. Ampicillin therefore has a wider spectrum of activity than penicillin.
- Antibiotics may be either bactericidal or bacteriostatic.
- Bactericidal antibiotics kill the bacterial microbe.
- Bacteriostatic antibiotics prevent bacterial growth. [1]
Why are human cells not affected?
[edit | edit source]Many antibiotics block protein synthesis. All cellular organisms, including bacteria, have ribosomes. And all ribosomes are composed of proteins and ribosomal RNA. But the precise shapes of these proteins differ in several very specific ways between humans and bacteria. That’s a good thing for researchers trying to develop bacteria-killing medicines called antibiotics because it means that scientists may be able to devise therapies that knock out bacterial ribosomes (and the bacteria along with them) without affecting the human hosts.
Multidrug Resistance
[edit | edit source]
Bacteria resistance to drugs can emerge in one of two ways:
1. By storing up and collecting numerous genes, and classifying them to fight in a individual drug within that cell, or
2. When more and more genes are organized under the "multi-drug efflux pumps", (Hiroshi, 2009, p. 119), and are forcing out large groups of drugs.

Types of Antibiotics
[edit | edit source]
Although there are well over 100 antibiotics, the majority comes from only a few types of drugs. These are the main classes of antibiotics:
1. Aminoglycoside They are mainly used to battle infections that were formed through gram-negative bacteria. Bacteria is said to be capable to improve a resistance to this; however, bacteria is able to improve resistance to just about anything. The bad side effects that can be caused from aminoglycoside are injury to your kidneys or ears. The main result of aminoglycoside is that it halts the bacteria from producing proteins in your body.
2. Cephalosporin There are 3 different creation types of cephalosporin. The higher the generation, the larger gram-negative antimicrobial property it has. The particular late generation cephalosporin also has a greater chance of bacteria not becoming resistant to it. This is clearly an advantage because it means that the medicine will be efficient for a longer period of time.
3. Fluoroquinolone This type of antibiotic is active against several different types of bacteria, but it is mostly used to treat UTI's (urinary tract infections), respiratory infections, and skin infections. It causes nausea, diarrhea, sickness, or minimal pain in the stomach while using fluoroquinolones. This works because it inhibits with bacteria’s ability to create DNA, so it is harder for the bacteria to be able to multiply.
4. Penicillin This was essentially the first type of antibiotic exposed in 1929 by Alexander Fleming. Penicillin is largely used to treat dental infections, respiratory infections, gonorrhea, UTI’s (urinary tract infections), and skin infections. Penicillin operates by hindering the growth of bacteria cell walls. Ultimately, the walls decline, and the bacteria is killed over a period of time due to this. Although, you can get an allergic reaction to this antibiotic, Doctors believe penicillin to be extremely safe.
5. Tetracycline These were the most general antibiotics whenever they were initially recognized back in the 1940s. The most common use for tetracycline is for upper respiratory infections, STD’s, Lyme disease, mild acne, and typhus. They are used to fight a wide amount of different bacterial infections for most of the time. Doxycycline and minocycline are kinds of tetracycline.
6. Macrolide These are used largely to cure gastrointestinal tract and other tract infections. They bind with ribosomes from susceptible bacteria which will help avoid any protein production. This helps kill off all the bad bacteria over time. The only actual side effect for macrolides is that they can make the stomach to be upset, but there are no sides of any serious nature.
Bacitracin
[edit | edit source]Bacitracin is an antibiotic made by bacterial microorganisms. The bacitracin antibiotic is isolated from Bacillus subtilis. Bacitracin is a narrow-spectrum peptide antibiotic that inhibits cell-wall synthesis in gram-positive bacteria. Bacitracin is a major ingredient in the ointment Neosporin that is used to fight skin infection caused by Staphylococcus and Streptococcus species. This antibiotic was first isolated in 1945. It is meant to be used topically, on the skin, for disinfecting minor cuts, and not to be used orally. Bacitracin is usually not taken orally because it is more effective as a topical antibiotic. [3]
Overview
[edit | edit source]Bacitracin works best on gram-positive bacteria because gram-negative bacteria have an outer membrane in their cell wall. Bacitracin kills bacteria by targeting bactoprenol, a lipid in the cytoplasmic membrane that is normally used to transport a disaccharide unit of peptidoglycan across the cell membrane to add to a growing peptidoglycan chain. [1]
How It Works
[edit | edit source]In the bacterial formation of peptidoglycan, the sugars NAM and NAG are linked together within the cell at a newly phosphorylated bactoprenol. The NAM-NAG disaccharide unit is then transported to the outside cell membrane via the bactoprenol. After the disaccharide unit is added to the growing peptidoglycan chain, bactoprenol is removed from the NAM-NAG unit. This bactoprenol then releases a phosphate, and then the desphosphorylated bactoprenol moves back towards the cytoplasmic part of the cell membrane to repeat the cycle.
Bacitracin works by binding to bactoprenol, just after a disaccharide unit is added to the growing peptidoglycan. This inhibits the dephosphorylation of the bactoprenol, thereby preventing bactoprenol from accepting UDP-NAM within the cell. In this way bacitracin leads to a stop in the synthesis of bacterial peptidoglycan cell wall. A growing bacterium will then lyse due to increasing pressure from within the cell. [1]
Side effects
[edit | edit source]Side effects are close to none, but in rare cases of allergic reactions, rashes, itching, dizziness, or trouble breathing may occur.
Rifamycin B
[edit | edit source]Rifamycin B was first discovered in 1957, along with six other rifamycins: A, C, D, E, S, and SV. Rifamycin B was the first to be used commercially. It was praised as being the solution to antibiotic-resistance tuberculosis in the 1960s.
Mechanism
[edit | edit source]Rifamycin B selectively binds to bacterial RNA polymerase to inhibit translation of mRNA. It binds to the beta subunit of the polymerase, which is near the Mg2+ activation site. When this happens the developing peptide chain cannot exit the RNA polymerase and it is stuck. However, it is important to note that Rifamycin B only works when it binds to a RNA polymerase that has just started translation, otherwise it cannot block the already growing peptide chain.
Uses
[edit | edit source]Rifamycin B is very effective in treatment of bacterial infections caused by mycobacteria, bacteria with a very thick and waxy cell wall, which makes antibiotics that target cell walls ineffective (penicillin, and vancomycin). Some of these diseases are tuberculosis and leprosy.
Actinomycin D
[edit | edit source]Actinomycin D was the first antibiotic discovered to have anti-cancer properties. It was first used in 1964. It is used a chemotherapy drug.
Mechanism
[edit | edit source]Actinomycin D 's structure allows it to mimic a nucleic base and insert itself in between guanine and cytosine. This blocks RNA polymerase from continuing the elongation process in transcription. However, the problem is that Actinomycin D binds non-selectively into any DNA, not just bacterial.
Uses
[edit | edit source]Actinomycin D is used as a chemotherapy drug to inihibit DNA synthesis and thus cell division. This makes it particularly effective against tumor cells that divide uncontrollably.
Side Effects
[edit | edit source]Since Actinomycin D is not meant to treat infections and it does not specifically target pathogenic DNA, it can inhibit DNA synthesis in normal cells. Other side effects are bone marrow suppression, hair loss, fatigue, mouth ulcers, loss of appetite, and diarrhea.
Streptomycin
[edit | edit source]Streptomycin is a type of aminoglycoside that affects bacterial translation. [1]
The aminoglycoside antibiotic, streptomycin, is made by some bacteria to protect themselves from competing bacteria. They are particularly effective because they are specific: they attack bacterial ribosomes, corrupting protein synthesis in the bacterium, but they don’t attack the ribosomes of many other organisms including ribosomes in human body. Therefore, it makes the perfect antibiotic drug that controls a bacterial infection with few side effects on our own cells.
Mechanism
[edit | edit source]Streptomycin binds to the 16S rRNA of the 30S ribosomal subunit and to the protein S12, thereby inhibiting the formation of the 70S bacterial ribosome. In low concentrations of streptomycin, translation can occur but the A site of the ribosome allows the matching of inaccurate codon and anticodon, leading to a faulty translation of protein. [1]
Bacterial Resistance
[edit | edit source]Bacteria can become resistant to streptomycin by initiating a mutation in S12 so that streptomycin can’t bind. Bacteria can also modify the streptomycin so that it won’t be able to bind to a certain molecule. [1]
Effects
[edit | edit source]Streptomycin affects the bacterial protein synthesis. It effects the way that messenger RNA is read, causing errors in translation and inhibiting the orderly stepping of the ribosome along the mRNA strand. It also blocks the recycling of ribosomes after they are finished making a protein.
Tetracycline
[edit | edit source]Tetracycline is an antibiotic that affects bacterial translation. An advantage of using tetracyclines or doxycycline as an antibiotic treatment is that it is broad-spectrum which indicates that the antibiotic can fight against a wide-variety of bacterial strains, such as Mycoplasma species, Rickettsia rickettsia, which causes Rocky Mountain spotted fever, and Chlamydia trachomatis. [3]
Mechanism
[edit | edit source]Tetracycline prevents tRNA from binding to the A site, thereby stopping the synthesis of proteins. It does this by binding to the 16S rRNA site so that new tRNA can’t be added to that site. [1]
Bacterial Resistance
[edit | edit source]Resistance to tetracycline occurs when a bacterial transport system moves the antibiotic to the outside of the bacterial cell so that tetracycline can no longer affect the cell. [1]
Chloramphenicol
[edit | edit source]Chloramphenicol is an antibiotic that affects bacterial translation.
Mechanism
[edit | edit source]Chloramphenicol stops translation by attacking the 50S ribosomal subunit. In attacking the 50S subunit, chloramphenicol will then bind to a peptidyltransferase site on a 23S rRNA. This peptidyltransferase is used to bind new amino acids to an existing translated protein chain. Therefore, the binding of chloramphenicol will stop the formation of peptide bonds. [1]
Bacterial Resistance
[edit | edit source]Bacteria become resistant to chloramphenicol when they create chloramphenicol acetyltransferase, an enzyme that stops any activity by chloramphenicol by changing the identity of the antibiotic. [1]
Erythromycin
[edit | edit source]Erythromycin is an antibiotic under the macrolides and is characteristic of having a lactone ring of 12-22 carbons. Erythromycin or azithromycin can treat gram-positive strains of bacteria, such as Corynebacterium diphtheriae that causes diphtheria, as well as gram-negative bacteria, such Bordetella pertussis that causes pertussis, or whooping cough. [3]
Mechanism
[edit | edit source]Erythromycin will attach to the 50S ribosomal subunit by attaching in the peptidyltrasnsferase cavity protein L15 and 23S rRNA. This binding will lead to the ejection of peptidyl-tRNA from the P site. [1]
Bacterial Resistance
[edit | edit source]Some bacteria grow resistant to erythromycin by having mutations in the L15, so that it isn’t binding. Bacteria can also reduce the erythromycin binding by methylating the area 23S rRNA using an enzyme, thereby inhibiting the binding of L15. [1]
Vancomycin
[edit | edit source]Vancomycin was isolated in 1952 from Streptomyces orientalis in a soil sample from Borneo; it was found to be active against most gram-positive organisms. It was especially valued at the time of its discovery because it worked against many organisms that were resistant to other antibiotics of the time. It was approved by the US FDA in 1958 and rose to popularity in that decade. Vancomycin fell out of popularity due to concerns of toxicity in the years following. It resurged in the 1980s, and reports of resistance began coming in from Europe and the US by 1987. [4]
Mechanism
[edit | edit source]This antibiotic works by targeting the terminal D-alanyl-D-alanine of the peptidoglycan chain on the cell walls of growing bacteria; it latches to this alanine dimer and prevents the protective cell wall from forming. [5][4]
Resistance
[edit | edit source]Resistant bacteria have modified this double alanine chain to be either D-alanyl-D-lactate or D-alanyl-D-serine (depending on the classification of the drug-resistant species); against these, vancomycin is ineffective. Experiments have shown that for certain vancomycin-resistant strains of bacteria, a combination of the antibiotics teicoplanin and gentamicin are more effective that vancomycin or teicoplanin monotherapy alone. Another vancomycin-resistant strain of bacteria responded to high doses of glycopeptides. Ultimately, one study recommended combination drug therapy involving gentamicin.[4]
Clinical Dosage and Usage
[edit | edit source]No standard clinical dosage is reported, but nomograms have been popularly used in the past. One problem that persists with clinical vancomycin usage is that serum levels are not monitored consistently or studied extensively. Some physicians never monitor the serum, monitor it occasionally, or insist on monitoring frequently when a patient is prescribed vancomycin. This is something that needs to be addressed in the future in order for vancomycin to be used effectively.[4]
Penicillin G
[edit | edit source]Penicillin G is an antibiotic made by fungal microorganisms. This antibiotic, isolated from Penicillium species, inhibits cell-wall synthesis in fungi by preventing enzymes from producing amino acid cross-bridges in peptidoglycan. Therefore, this antibiotic is used on gram-positive bacteria, which contain thick peptidoglycan layers. [3]
Structural components
[edit | edit source]1. Thiazolidine ring
2. β-lactam ring
3. R-group (differs in side-chains)
yeesh
Cephalosporin C
[edit | edit source]Cephalosporin C is an antibiotic made from fungal microorganisms. This antibiotic is used on gram-positive bacteria and isolated from Cephalosporium acremonium, which is a mold. Cephalosporin C can contribute to the inhibition of fungal cell-wall synthesis and contains a β-lactam group and R-groups for modification. [3]
Advantages
[edit | edit source]1. Broader-spectrum than penicillin
2. Fewer allergies than penicillin
3. Resistant to penicillinase (an enzyme made by bacteria to destroy penicillin)
Disadvantages
[edit | edit source]1. Poorly absorbed from the intestines
2. Medication given parenterally (non-orally) via injection into veins (intravenous) or into muscles (intramuscular)
Silver
[edit | edit source][6] Silver has been known and used an antimicrobial agent since the 19th century, and in 1998 one study examined and tried to optimize such a treatment. Although bacteria do develop resistance to other forms of antibiotics, there have been no reports of bacteria developing resistance to antimicrobial silver (though genes for heavy metal-resistance do exist and are linked to antibiotic-resistance genes).
Mechanism
[edit | edit source]Silver ions are toxic to bacteria because they blocks several processes essential to bacterial function: silver ions prevent respiratory enzymes from working properly, block some parts of the electron transport system, and impair some DNA functions.
In Vitro Trials
[edit | edit source]In the past, silver ions in cream forms had been tested and found to be less than ideal due to the nature of creams (they tend to dry out, and patients find changing dressings uncomfortable). Very strong ionic solution proved to be irritating to skin. In a study conducted by Wright et al., three different silver compounds delivered in dressings were tested: silver nitrate in dressing, silver sulfadiazine in dressing, and nanocrystalline silver in dressing. These were inoculated with bacteria strains that were resistant to several antibiotics each; the bacteria was strained and regrown, and bacteria regrowth was measured. The nanocrystalline silver dressing reduced the regrowth most noticeably. It does not irritate skin as much as the other two treatments do. Additionally, serum proteins (tested in this study by a calf serum solution) did not reduce the efficacy of the nanocrystalline silver treatment. It appears that such a treatment could be very valuable in a clinical setting.
Outcome
[edit | edit source]
Many bacteria have become resistant to the antibiotics available. In addition, some diseases such as Staphylococcus Aureus (MRSA) will not respond to most drugs because they have aquired resistance to most of the antibiotic. These can only be treated with "reserve antibiotics" ,usually reserved for life threatening systemic infections, like vancomycin.

Protein Synthesis
[edit | edit source]Antibiotic is a medicine that inhibits the growth or destroys of a microorganism. Compared to eukaryotes’ 80S ribosomes, bacteria have 70S ribosomes that use antibiotics to block the synthesis of protein. An antibiotic is lucrative when it is able to bind and interfere with the formation of a “protein synthesis initiation complex” in bacteria. We need antibiotics to fight against bacteria. So antibiotics are there to interrupt the development of bacteria. The difference between the ribosomes that are in the mitochondria and the ribosomes in bacteria is that antibiotics affect the function.
References
[edit | edit source]Nikaido, Hiroshi. "Multidrug Resistance in Bacteria." Annu. Rev. Biochem. (2009): 119-46. Web.
Images: Wikimedia
http://www.emedexpert.com/classes/antibiotics.shtml
http://www.emedicinehealth.com/antibiotics/article_em.htm
http://www.medicinenet.com/bacitracin-topical/article.htm
Davis, Alison. (2006). Inside the Cell. National Institutes of Health, 9.
- ↑ a b c d e f g h i j k l Slonczewski, Joan L. Foster, John W. Microbiology: An Evolving Science, Second Edition, W.W. Norton & Company. 2009.
- ↑ U.S. Department of Health and Human Services. Inside the Cell. September 2005.<http://www.nigms.nih.gov>.
- ↑ a b c d e Tortora, Gerard J., Berdell R. Funke and Christine L. Case. Microbiology: An Introduction. 10th ed. San Francisco, CA: Pearson Benjamin Cummings, 2010. Print.
- ↑ a b c d Levine, Donald P. "Vancomycin: A History." Clinical Infectious Diseases 42 (2006): S5-12.
- ↑ Alison Davis, Ph.D. "The Chemistry of Health." August 2006. National Institutes of Health, National Institute of General Medical Sciences Home. NIH. December 2012 <http://www.nigms.nih.gov >.
- ↑ J. Barry Wright, Ph.D., Bsc Kan Lam and Ph.D. Robert E. Burrell. "Wound management in an era of increasing bacterial antibiotic resistance: A role for topical silver treatment." American Journal of Infection Control 26.6 (1998): 572-577.
Prodrugs are a new class of drugs that, upon administration, are inactive. Upon absorption, the drug becomes activated. This process, called bioactivation, occurs by in vivo metabolism. Prodrugs are useful in that they can be used to avoid negative physiological effects associated with the absorption of standard drugs.[1] The potential benefits of this class of drugs are vast. They provide improved drug targeting, and higher concentration of active ingredients where needed. Problems of standard drugs that could potentially be overcome by the use of prodrugs include insufficient absorption when absorbed orally, limited solubility, and irritation associated with administration.[2]
Undesirable Properties
[edit | edit source]- Physical Properties: Poor aqueous solubility, low lipophilicity and chemical instability - Pharmacokinetic Properties: Poor distribution across biological membranes, good substrate for first-pass metabolism, rapid absorption/excretion when long-term effect desired.
Classification of Prodrugs
[edit | edit source]Looking on how the human body converts the prodrug into the final active drug form, there are actually two major types of prodrugs.
One type of prodrug is called the Type I prodrugs. This type of prodrug is bioactivated intracellularly. Lipid-lowering statins and anti-viral nucleoside analogs are both examples of Type I prodrugs. There is another prodrug called the Type II prodrugs. Unlike that of Type I prodrug, Type II prodrugs are bioactivated extracellularly, especially in the body's circulation system or the digestive fluids. For example, antibody, or virus-directed enzyme are all types of Type II prodrug. These are also commonly used in immunotherapy or chemotherapy.
Subtypes of Prodrugs
[edit | edit source]It is also seen that Type I and Type II prodrugs can be categorized into Subtypes. For example, Type I has a bioactivation site that is intracellular and it contains subtype of Type IA and Type IB. Type IA is often located in the therapeutic target tissues or cells. Example of this can be diethlstilbestrol diphosphate or 6-mercaptopurine. Then there is Type IB where is located in the metabolic tissues of the liver, Gl mucosal cell, or the lungs and examples for this subtype can be that of heroin, primidone, or captopril.
Furthermore, just like how Type I can be classified into different Subtypes, Type II prodrugs also do the same thing. For instance, Type II has a bioactivation site that is extracellular and it contains subtype of Type IIA, Type IIB, and Type IIC. Type IIA is usually found in the GI fluids and example of this can be sulfasalazine. Type IIB is found in the systemic circulation and other extracellular fluid compartments. Chloramphenicol succinate or dipivefrin are examples of Type IIB prodrugs. Last but not least, Type IIC such as those of ADEPTs, VDEPs, or GDEPs is all found in the therapeutic target tissues or cells.
Thus, there are different types and kinds of prodrugs because one type of prodrugs contains couple or several subtypes that are typically found in different locations of the body.
Steps in Prodrug Design
[edit | edit source]- Identification of drug delivery problem and identification of desired physicochemical properties - Selection of transport moiety which will give prodrug desired transport properties and be readily cleaved in the desired biological compartment
Design and Structure
[edit | edit source]Currently, many prodrugs employ primarily hydroxyl, amine, and carboxyl groups. Esters are found most commonly in commercial prodrugs, such as the drug oseltamivir. More atypical groups have also been investigated for use in prodrugs, such as thiols and imines.[3] The bioconversion of the prodrugs to their active parent drugs is by way of enzyme activity of hydrolases. Prodrugs can be designed to bioconvert based upon the specific characteristics of the enzymes that catalyze the reaction, specifically substrate recognition. [4]
Examples
[edit | edit source]The prodrug Famvir has recently been developed in an attempt to prevent the spread of three different types of the herpes simplex virus: type 1, type 2, and the varicella zoster virus. This drug contains two ester groups and requires two enzymes to bioconvert the molecule into its active form, which takes place predominantly in the liver.
Hepsera (Adefovir dipivoxil), another diester prodrug, is responsible for inhibiting nucleoside reverse transcriptase against the hepatitis B virus. Absorption of this drug occurs orally and at a very rapid pace, with maximum absorption occurring after just 3/4 of an hour.
Viread is a carbonate-based prodrug, as opposed to the ester-based prodrugs mentioned previously. Its purpose is to inhibit the nucleoside transcriptase in the HI virus and the hepatitis B virus. The carbonate group contained in this prodrug has been found to be more stable than the ester groups contained in the aforementioned prodrugs, while maximum absorption is reached more slowly.
References
[edit | edit source]- ↑ http://www.chem.memphis.edu/parrill/chem4315/2005/Prodrugs.pdf
- ↑ http://www.nature.com/nrd/journal/v7/n3/full/nrd2468.html
- ↑ http://epublications.uef.fi/pub/urn_isbn_978-951-27-0634-1/urn_isbn_978-951-27-0634-1.pdf
- ↑ Imai, Teruko, and Masakiyo Hosokawa. "Prodrug Approach Using Carboxylesterases Activity: Catalytic Properties And Gene Regulation Of Carboxylesterase In Mammalian Tissue." Journal Of Pesticide Science 35.3 (2010): 229-239.
http://en.wikipedia.org/wiki/Prodrug
Overview
[edit | edit source]Selective Serotonin Reuptake Inhibitors are a class of antidepressants which aim to increase the levels of serotonin available in the synaptic cleft by inhibiting reuptake of serotonin into the pre-synaptic cell. They have varying affinities for other monoamine transporters such are norepinephrine and dopamine, which gives rise to the term “selective” in their name. SSRIs have become the most popular form of antidepressants, replacing monoamine oxidase inhibitors which have more side effects and slightly reduced efficacy. However, debate remains as to the actual benefit of SSRIs, as several meta-studies have indicated that the effect of antidepressants compared to placebos were statistically significant but were not clinically significant. [1] SSRIs are the most common antidepressant medication because it can treat moderate to severe depression with relatively low risk and with fewer side effects. Selective Serotonin Reuptake Inhibitors can be made to be either extended release (XR) or controlled release (CR). The benefit of these types of medication is that it can release the medication over a long period of time slower. Therefore there is less ups and downs associated with the drug and a nice smooth transition onto the drug is made. These drugs can be used for purposes other than as an antidepressant. SSRIs affect every person differently due to how each individual brain reacts to the chemical makeup of the drug. Possible side effects include dry mouth, nausea, nervousness, headache, diarrhea, drowsiness, insomnia etc... [2]
Pharmacodynamics
[edit | edit source]SSRIs inhibit the reuptake of serotonin into the presynaptic cell, increasing the available serotonin in the synapse. However, the antidepressant effects require several weeks of administration of SSRIs before the effects are felt. The reason is that the increased levels of serotonin produce two neurological changes: first, it leads to the downregulation of 5-HT2A receptors, which alters the neurotransmitter to receptor ratio. Second, it also triggers autoreceptors on the presynaptic cleft which causes the cell to produce less serotonin as it down regulates in response to the increased levels of serotonin available. Alternatively, SSRIs also influence other pathways such as cyclic AMP (cAMP) on the post synaptic cell, which causes the release of brain derived neurotrophic factor (BDNF). This protein is secreted to regulate neural networks as well as synaptic plasticity. [3]
Examples of several SSRIs:
[edit | edit source]- citalopram (Celexa, Cipramil, Cipram, Dalsan, Recital, Emocal, Sepram, Seropram, Citox, Cital)
- dapoxetine (Priligy)
- escitalopram (Lexapro, Cipralex, Seroplex, Esertia)
- fluoxetine (Prozac, Fontex, Seromex, Seronil, Sarafem, Ladose, Motivest, Flutop, Fluctin (EUR), Fluox (NZ), Depress (UZB), Lovan (AUS), Prodep (IND))
- fluvoxamine (Luvox, Fevarin, Faverin, Dumyrox, Favoxil, Movox)
- indalpine (Upstene) (discontinued)
- paroxetine (Paxil, Seroxat, Sereupin, Aropax, Deroxat, Divarius, Rexetin, Xetanor, Paroxat, Loxamine, Deparoc)
- sertraline (Zoloft, Lustral, Serlain, Asentra)
- vilazodone (Viibryd)
- zimelidine (Zelmid, Normud) (discontinued)
- ↑ Severe and anxious depression: Combining definitions of clinical sub-types to identify patients differentially responsive to selective serotonin reuptake inhibitors1
- ↑ SSRI, October 28, 2012
- ↑ Selective serotonin reuptake inhibitors pathway2
Most, if not all, people have taken medicine before. They intake medications that help relieve pain, sickness, or soreness. Through much research scientists were able to formulate the four basic stages of how medicine goes through one's body. The four stages are absorption, distribution, metabolism, and excretion. The entire process is sometimes abbreviated ADME.
Absorption
[edit | edit source]The first step of how a drug passes through one's body is by absorption. There are few avenues on how medication may enter the body. The most common way is "orally" either with a tablet (pill) or liquid (via a spoon, cup or syringe). At home, many people use this technique. Another way is "intramuscular", which means to injecting a medication into an arm muscle. For example, when it's that time of the year when the flu is epidemic, everyone usually gets their flu shot through this method. Another way a medication enters the body is "subcutaneous", which is under the skin. Another route is "intravenous", this usually happens with people who undergo chemotherapy. The medication is received through a vein in the body. The last method for a medication to enter the body is "transdermal", which actually isn't inserted into the body. An example of this technique would be wearing a skin patch. The medication is absorbed through the skin and enters the body through our skin pores.
Out of all the steps of how a drug passes though one's body, absorption is the hardest barrier to overcome because everyone is created differently. Some drugs may not be able to enter one's body if the body rejects the drug. If a medicine is meant to spread throughout the whole body, it needs to first be absorbed. The person's body must accept the medication for it to take effect. There may be complications of drug administration because the drug may not go through the correct pathway in the body.
Distribution
[edit | edit source]After the medication is absorbed, the next step of how a drug passes through the body is distribution. A drug enters through one side of the body, but is meant to heal another part of the body, which might be in the middle of the body (or somewhere where it is hard to inject directly). The bloodstream is what helps a drug travel from the surface of where the drug enters to its destination. One obstacle that a drug must overcome would be side effects. As mentioned above, we are all created differently. One drug that works in one person may or may not work in another person’s body. People may react to a drug differently having one or more side effects. Some people have allergies to certain drugs. There are many factors that may prevent distribution of a drug inside the body. The “presence of protein and fat molecules in the blood” can attack a drug and inhibit it from its purpose. There are other complications of drugs, especially if they’re meant to go to the “central nervous system (CNS)”, where the brain and spinal cord are. The CNS has an obstacle called the “blood-barrier”, which guard the CNS from dangerous drugs. Pharmacologists have found ways to insert substances past the blood-barrier.
Metabolism
[edit | edit source]Once a drug reaches its destination, the drug must be “metabolized”. A drug is metabolized normally in the liver. The liver is chosen above any other organ because it’s a “site of continuous and frenzied, yet carefully controlled, activity.” All substances that enter the blood stream go directly to the liver and at the location the drug is altered to fit the appropriate function in the body. The “biotransformations” are the products of the alteration that happens in the liver. They are also known as enzymes. Enzymes are certain proteins that have a specific ability that when bound to a substrate it may perform its function.
Excretion
[edit | edit source]Once the enzymes perform their function, then the products of the break down are less chemically active than the original molecule. These products are called metabolites. The liver is called the “detoxifying” organ for the reason of which it breaks down the enzyme. Sometimes drug metabolites may have chemical activities of their own, which may or may not supersede the activity of the original drug. When the enzyme is done with its function, it may now go through excretion, which normally happens through the bowel.
Reference
[edit | edit source]Reference: Davis, Alison. (2006). Medicines By Design. National Institutes of Health , 11-13.
Statin
[edit | edit source]
Overview
[edit | edit source]Statins, also known as 3-hydroxy-3-methyl-glutaryl-CoA (HMG-CoA) reductase (HMGCR) inhibitors, are a class of drugs that lower the level of cholesterol in the blood by reducing the production of cholesterol by the liver. Statins are inhibitors that block the enzyme HMGCR, which is responsible for the production of cholesterol. Because high cholesterol has been linked with the risk of heart disease, those with increased cholesterol levels may find statins useful as prevention. The top-selling statin is atorvastatin, with the trademark name Lipitor. Lipitor, which has been manufactured by Pfizer, has been useful in the stabilization of plaques and in preventing strokes.
History
[edit | edit source]In 1971, Akira Endo, got into research to search for inhibitors of HMGCR to lower the cholesterol levels in liver. He and his team argued that there are microorganisms that may produce this inhibitor of the enzymes to defend themselves against other organisms. The first isolated agent was mevastatin, a molecule created from the fungus “Penicillium citrinum”. By 1976, another statin, known as lovastatin, was isolated from the fungus “Aspergillus terreus”. A few years later, other isolated statins began to hit the market, including Pfizer’s Lipitor in 1985.
Then, a link between cholesterol and cardiovascular disease, called lipid hypothesis, had been suggested. Cholesterol is a waxy substance produced by the liver and found in certain foods, which is needed to make vitamin D and some hormones, create bile salts that assist people to digest fat and build cell walls. However, having too much cholesterol is not good since this can lead to heart attacks. During the Coronary Primary Prevention Trail of 1984, Daniel Steinberg, cholesterol researcher, conveyed that low-fat diet and poorly tolerated medicines such as cholestyramine, clofibrate, and nicotinic acid are all kinds of dietary measures for treatment. According to Steinberg, anything that will lower cholesterol can greatly reduce the risk of serious problems such as heart attacks and angina. However, many physicians including cardiologists remained unconvinced of Steinbergs theory.
To promote the use of statins, Merck convinced the public as well as doctors about the dangers of high blood cholesterol levels, and illustrated how statins were safe to take and would extend lives. Soon, people became familiar with their cholesterol numbers and understood the difference between what is good and what is bad cholesterol. With this happening, rival pharmaceutical companies started producing their own statins, such as pravastatin (Pravachol) that was manufactured by Sankyo and Bristol-Myers Squibb. In the year 1994, the results of a Merck-sponsored study, the Scandinavian Simvastatin Survival Study, or also known as "4S", were announced. Later Merck started selling simvastatin (Zocor), which was used by 4,444 patients with heart disease and high cholesterol. In just five years, the study concluded that many patients saw a 35% reduction in their cholesterol levels and the chance of them dying from heart attack were reduced by 42%. In the year 1955, Merck made over one billion US dollars profit with Zocor and Mevacor. In 2006, Endo was awarded the Japan Prize and the Lasker-DeBakey Clinical Medical Research Award in the year 2008.
Who Should Take It?
[edit | edit source]Statins are used for preventing and treating atherosclerosis, which causes chest pain, heart attack, stroke, and related deaths. So, people with abnormally elevated cholesterol levels or a history of heart attacks should consider taking statins. However, exercise and change in diet might be the first strategy to reduce cholesterol, before statin treatment is started.
Examples of Statin
[edit | edit source]- Atovastatin (Lipitor, Torvast)
- Fluvastatin (Lescol, Lescol XL)
- Pravastatin (Pravachol, Selektine, Lipostat)
- Rosuvastatin (Crestor)
- Simvastatin (Zocor, Lipex)
- Pitavastatin (Livalo, Pitava)
- Cerivastatin (Lipobay, Baycol)
- Lovastatin (Mevacor, Altocor, Altoprev)
- Mevastatin (Compactin)
- Simvastatin+Ezetimide (Vytorin)
- Lovastatin+Niacin (Advicor)
- Simvastatin+Niacin (Simcor)
- Atorvastatin+Amlodipine (Caduet)
Mechanism
[edit | edit source]Statins all act by inhibiting HMGCR, which catalyzes the conversion of HMG-CoA to mevalonate during cholesterol synthesis. Individual statins are differently potent in reducing cholesterol (atorvastatin (Lipitor) and rosuvastatin (Crestor) are the most potent statins, while fluvastatin (Lescol) is the least potent statin currently on the market.

Side Effects
[edit | edit source]Common side effects include: headache, nausea, vomiting, constipation, rash, diarrhea, muscle weakness and muscle pain. In very rare cases, serious and potentially fatal side effects such as liver failure and rhabdomyolysis (muscle degeneration) have occurred. Such side effects might be linked to interactions with other medications that increase the bioavailability of statins. Statins are known to increase the risk to develop type 2 diabetes, and are further discussed to affect cognitive function. On the one hand, statins might lead to working memory impairment, on the other hand, statins might reduce the risk to develop dementias such as Alzheimer disease.
References
[edit | edit source]- http://upload.wikimedia.org/wikipedia/commons/9/99/Simvastatin.png
- http://www.news-medical.net/health/Statin-History.aspx
- http://www.medicinenet.com/statins/article.htm
- http://en.wikipedia.org/wiki/Statin
- http://upload.wikimedia.org/wikipedia/commons/2/25/Cholesterol.png
- http://kidshealth.org/teen/food_fitness/nutrition/cholesterol.html


Caffeine is the most popular drug that is being used to maintain a certain mental stability such as staying awake, changing the way the brain functions, and moods. It is found in many of the drinks we consume daily. This includes, but not limited to sodas, coffee, and tea. Many consume this drug without even being conscious of their daily intake, and it is not something that most people think of as being dangerous or questionable. Although Caffeine is taken very lightly, large amounts of this product are in many of our drinks. For example, according to an article, "Neuropsychiatric Effects of Caffeine", by Anthony Winston, it shows that 100 mg is in a normal cup of coffee, 75 mg in instant coffee, and 50 mg in tea that we drink every day (Winston, page 432).
Commonly, caffeine starts to affect the brain and body within an hour and begins to wear off after 3 to 4 hours.
Caffeine is mainly use for the purpose to raise and boost mental functions, but when it is overly used, it can also advance into a harmful state called caffeinism. According to Winston, “caffeine is characterized by restlessness, agitation, excitement, rambling thought, speech, and insomnia" (page 1). Following these symptoms, many other disorders such as anxiety, sleep, eating disorders, and many more may occur.
Caffeine (1, 3, 7-trimethylxanthine) is a natural product found in plants. While the actual caffeine content of plant seeds and leaves varies quite a bit from species to species, caffeine is viewed as the most abundant naturally-occurring purine alkaloid, meaning it is derived from one or more purine nucleotides. Alkaloids are a large group of compounds that can be found mainly in plants and contain basic nitrogen atoms. These compounds usually exist as salts because of their basic nature. Major caffeine sources include the seeds of the coffee plant (Coffea Arabica), tea leaves (Camellia Sinensis), and cola nuts. It is widely accepted that caffeine is a stimulant when consumed by humans, with its main focus falling upon the central nervous system.[1] Plants utilize caffeine as a line of defense. It works as an insecticide for the plant, deterring many potential threats as it can obstruct metabolic pathways.[2]
Effects of Caffeine: How and Why?
[edit | edit source]By in-taking caffeine, the body feels a sense of alertness and wakefulness. The reason caffeine affects the body in the way that it does is because of its structural resemblance to both adenosine and cyclic adenosine monophosphate (cAMP). As a result of similar structures, caffeine can potentially bind to receptors that usually bind to adenosine and its derivatives. Within the body, adenosine plays a role in regulating the brain and its activity. When there are large buildups of adenosine in the brain, they begin to bind to the brain receptors and that causes reactions leading to sleepiness and feeling drowsy. However, when caffeine enters the body it binds to these same receptors because of its similar structure and this prevents adenosine from binding to these receptors and this delays the body from feeling sleepy.

cAMP is a secondary messenger that is responsible for processes involving blood pressure and oxygen in the body. It increases the amount of oxygen delivered to the brain as well as blood pressure, both of which keep the body alert. However, there is an enzyme found inside the body, cyclic nucleotide phosphodiesterase (cAMP-PDE), that breaks down cAMP and removes its anti-drowsy effects from the body. As a result of its similarity to the structure of cAMP, caffeine prevents cAMP-PDE from breaking down cAMP and allows the anti-drowsy effects to prolong for much longer.

Disorders
[edit | edit source]
- Sleep Disorders:
Insomnia is sometimes the outcome of an excessive amount of caffeine. When taken near bed time, it may extend the hours awake and give the inability to easily fall asleep. In addition, according to Winston, "it reduces slow-wave sleep in the early part of the sleep cycle and can reduce REM sleep later in the cycle" (page 2). In conclusion, wakefulness is a symptom of the sleeping disorder caused by caffeine if taken inconsiderably.
- Eating Disorders:
Caffeine is also a cause of Anorexia and Bulimia nervosa. Studies have shown that people with these eating disorders frequently have an extensive intake of caffeine. This drug gives way to a smaller appetite and a fast metabolism. In addition, it may also be dangerous for the heart as well as an increase in chance of receiving osteoporosis.
Synthesis
[edit | edit source]In this synthesis, "SAM" refers to S adenosyl-L-methionine, which causes the addition of a methyl group.[2]

Coffee and linkage to weight loss: Caffeine is a stimulant that stimulates the central nervous system, helps when dealing with fatigue, aids in preventing sleepiness and tiredness, and improves with alertness, mood, metabolism. It is important that caffeine is monitored and taken at a balanced diet. A common source of caffeine is coffee. About two to four cups of coffee a day is considered healthy. Energy drinks and caffeine-carbonated drinks contain a dangerous amount of caffeine. Drinking more than two a day is enough to do its intended job. Any more can cause serious problems. Caffeine is also linked to weight lost. Caffeine may help shed a few pounds, or block further weight gain. Caffeine is one of the ingredients in the dangerous weight loss pills that people depend on to lose weight. Caffeine is thought to have contribute to weight loss due to the fact that it can: suppress appetite, increase metabolism and chances of burning calories, and induce water weight loss. Caffeine may suppress appetite for a short period of time, however it seems to not have any correlation with suppressing appetite for a long period of time. Caffeine may implement and induce thermogenesis, which is the body's way of creating heat while digesting food. Caffeine may also present the body with alertness and behavioral changes, which may help during exercise and induce faster calorie burning. Caffeine burns fat, which preserves the alternative (glycogen, glucose, amino acids) from being burned. This keeps the sugar in the bloodstream, and enables the body to run a little longer without getting hungry. Caffeine has also been a popular choice of intake before a workout. If someone intakes caffeine before a workout, it is shown that caffeine isolates extra fat flowing, and targets and accesses the fat for energy, rather than carbs or existing muscle. Caffeine targets and breaks the fat, and is rapidly burned at that instant. However, it is important to drink a lot of water during the workout to prevent dehydration. Caffeine, coffee in particular, may lead to eating disorders, such as anorexia and bulimia, because of the notion that coffee has 0 calories and 0 carbs. Coffee indeed has 0.5-0.9g of carbs. Coffee may also help a person reduce the cravings and need for food, with its bitter taste, and convince someone that he or she is not hungry. Caffeine is also known to prevent fat from building and collecting in cells. There is research on the caffeine inhibiting enzymes, and its role in fat synthesis. Caffeine is the main ingredient in numerous diet pills. People with eating disorders turn to caffeine and coffee for energy and satisfaction of hunger. However, in this case, it is more harmful than effective. Caffeine can also increase urination since it is a diuretic. An increased urination may help shed water weight and bloatedness. However, losing weight through the removal of fluids will be effective for only a short period of time. Caffeine is not intended to serve as a meal replacement, or as a sole source of weight loss. Relying on caffeine to lose weight is not a healthy or efficient method. Since the thermogenetic characteristic of caffeine is one adopted by weight loss drugs, caffeine can be chosen as an alternative to acquire thermogenesis, rather than from weight loss drugs itself. Relying on caffeine can cause extreme health effects, both physically and mentally. It can increase chances of insomnia, nervousness, makes a person jittery, instability, rapider heart rate, and potassium deficiency. Pregnant women should prevent intaking caffeine at a large rate, since caffeine runs through the bloodstream and directly to the fetus. It is important to take into consideration that in order to have an efficient weight loss, diet and exercise are the two most crucial aspects and factors. Relying and overdosing on caffeine will have damaging effects and may cause the body to gain weight, instead of intended loss.
Extraction
[edit | edit source]Tea Leaves
[edit | edit source]Caffeine can be extracted and isolated from tea leaves. Since caffeine is soluble in both water and organic solvents, it can be first extracted from the leaves through solid-liquid extraction in hot water and then separated even further through extraction with a polar non-protic organic solvent such as methylene chloride. Before beginning the extraction process, the components of tea leaves should be analyzed:
- Cellulose: Cellulose functions as a rigid and insoluble structural component and makes up a large portion of leaves. This linear polymer is made up of D-glucopyranose units connected between carbons 1 and 4. Because of its high molecular weight, cellulose is not soluble in water despite having hydroxyl groups.

Cellulose Sessel

~In a solid liquid extraction of tea with hot water, the cellulose would be easy to remove because as the caffeine is extracted into the water, the cellulose component is left behind. The cellulose remains in the tea leaf while the caffeine and the other water-soluble components are extracted.
- Proteins and Pigments: Both pigments and proteins are highly soluble in water and therefore do not pose a problem to the extraction of caffeine. In the initial solid-liquid extraction, the proteins and pigments are extracted along with the caffeine. But since they are non polar, the second extraction of caffeine into methylene chloride, which is immiscible in water, results in the extraction of the caffeine without extracting the proteins and pigments. These stay in the aqueous phase while the caffeine is extracted into this water immiscible methylene chloride.
- Tannins: Tannins are polyphenolic compounds with variable molecular weights and are the reason why tea has a bitter taste. The compounds are made up of D-glucose units that have ester bonded to units of Gallic Acid. Tannins do pose a problem to the extraction of caffeine because it is soluble in both water and organic solvents. However, this problem can be solved by cleaving the ester bonds with a weak base, such as Calcium Carbonate. This causes the tannins to break down into glucose and calcium salt of Gallic Acid, both of which will not be extracted into the organic solvent.
Flavogallonic acid dilactone - Saponins: Saponins are compounds that have both polar and non-polar characteristics. This duality complicates the extraction process because organic compounds become more soluble in water and this induces the formation of emulsions. Emulsions make it difficult to effectively separate between solvent layers. Emulsions can be removed through either centrifugation or increasing the polarity of the aqueous layer. Addition of, for example sodium chloride salt, will decrease the solubility of organic compounds in water.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
.svg%26page%3D1){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Extraction Process
[edit | edit source]

First, the caffeine from tea leaves can be extracted through solid-liquid extraction. This can be achieved by boiling the tea bag in water and a weak base, such as Calcium Carbonate, in order to break the ester bonds in the tannins. The liquid caffeine can then be transferred out and vacuum filtered. The filtered caffeine can be transferred to a centrifuge tube and is ready for a second extraction with the organic solvent. When the organic solvent is added, the two layers should be allowed to separate before removing the desired organic layer. If emulsions form, centrifuge and add salt accordingly. The liquid caffeine should be extracted multiple times with the organic solvent. The collected organic extracts can be evaporated in a sand bath on a hot plate in order to obtain a "crude" product. This product can be further purified due to the ability of caffeine to sublime. Sublimation is the ability for a compound to go from the solid state to the gas state without having gone through the liquid state. Any impurities will not sublime with the caffeine.[3] The impurities can not sublime with caffeine because they require difference temperatures and pressure. In essence, the caffeine is sublimated from the crude product, allowing experimenters to collect caffeine and leave behind impurities.[4]

- Sublimation:
Sublimators can be set-up in a multitude of ways but the main goal is to heat the solid inside a sublimation chamber and have it reach the gas state and collect on a tube or apparatus inserted into the sublimation chamber. After the sublimation is complete, the tube can be removed and the collected caffeine can be taken off.[3] The tube is attached to a vacuum which lowers the overall pressure in the apparatus. This lowering of the pressure insures that the caffeine sublimates instead of melt then vaporize. Also the lowering of the pressure does not allow the impurities to sublimate, allowing the experimenter to collect pure caffeine crystals.[4]
MVIE Method of Extraction
[edit | edit source]Microwave Vacuum Ice Water Extraction (MVIE) is a method in which caffeine is extracted by using the power of microwaves. It has been tested with green tea leaves and resulted in a high percent removal yield. This process is dependent on the solvent to solid ratio, microwave power, and time of extraction. With proper execution of technique and time, it extracts approximately 87.6% of caffeine. MVIE proves to be a more effective method to extract caffeine than hot water extractions.
- Basic Procedure
A mixture of tea leaves and water undergo a microwave extraction process then a vacuum ice water extraction. The samples are then placed into a vacuum oven where vapors are collected in a separate container. Multiple extractions are taken to extract a sufficient amount of caffeine from the leaves.
- Factors
Microwave power: With an increase from 160W to 350W, the amount of caffeine extracted was found to be 22% and 46.6% respectively. This indicates that an increase in microwave power increases the amount of caffeine extracted. An increase in power increases internal heat, which ultimately speeds up the process of releasing solvents from the leaves into the solution. However anything higher than 350W will result in the opposite – a decrease in caffeine extraction.
Solvent to solid ratio: the ratio that produces the highest yield of extraction is 10:1 (ml of solvent: g of solid)
MVIE introduces a new and effective way to extract caffeine from tea leaves.
References
[edit | edit source]Mohrig, Jerry R., Christina Noring Hammond, and Paul F. Schatz. Techniques in Organic Chemistry. New York: W. H. Freeman and Company, 2010. Print.
P. Nawrot, S. Jordan, J. Eastwood, J. Rotstein, A. Hugenholtz & M. Feeley (2003): Effects of caffeine on human health, Food Additives and Contaminants, 20:1, 1-30
Winston, Anthony P. "Neuropsychiatric Effects of Caffeine." Advances in Psychiatric Treatment 11 (2005): 432–39. Web.
Lou Z., Er C., Li J., Wang H., Zhu S., Sun J. (2011): Removal of caffeine from green tea by microwave-enhanced vacuum ice water extraction, Anal Chim Acta, 49-53
Images: WikiMedia Commons
- ↑ a b Ashihara, H, & Ashihara, H. (2004). Distribution and biosynthesis of caffeine in plants. Frontiers in bioscience, 9(1-3), 1864-.
- ↑ a b Mohanpuria, P, & Mohanpuria, P. (2009). Caffeine biosynthesis and degradation in tea [Camellia sinensis (L.) O. Kuntze] is under developmental and seasonal regulation. Molecular biotechnology, 43(2), 104-.
- ↑ a b Mohrig, Jerry R., Christina Noring Hammond, and Paul F. Schatz. Techniques in Organic Chemistry. New York: W. H. Freeman and Company, 2010. Print.
- ↑ a b extraction, October 28, 2012
Background
[edit | edit source]Lysergic acid diethylamide, commonly known as LSD, has been known to cause psychedelic hallucinations and visions. It has not been known to be addictive or cause brain damage. LSD has been typically used for recreation in order to enhance creativity and out of body experiences. Albert Hoffman, in 1938, discovered LSD when making different derivatives of diethylamide. It's hallucinogenic properties were accidentally discovered in 1943. In the 1950s, LSD was brought to the medical community has a tool to create a temporary psychotic state and be used alongside psychotherapeutic treatments. It was in the 1960s that the general public began using LSD for recreational use, and remains a major drug to this day. [1].
Overview
[edit | edit source]LSD blocks serotonin from the brain. Serotonin, a neurotransmitter, is in charge of regulating mood, muscle contraction, and other cognitive functions. The reason why LSD blocks serotonin is because of their structural similarity, LSD is mistaken for serotonin and is then sent to the synaptic cleft instead of serotonin. And like many other hallucinogenics, LSD has a substituted indole ring in its molecular structure, which contributes to its hallucinogenic effects. There are many perceptual changes altering the cognitive and visual sensory systems, as well as changed in the sense of time, body-image, and ego. Memory is also greatly effected. A typical "trip" can last anywhere between six and ten hours. The half-life in humans is approximately 175 minutes, though it is metabolized at different rates at different structures in the body. Not much clinical testing has been done since the 1970's, partly due to the fact that after the subjects have taken a dose of LSD they become "too impaired to cooperate". [1]
Structure
[edit | edit source]The molecular configuration of crystal form LSD has been determined by using x-ray diffraction techniques. The configuration shows strain and steric hindrance. Serotonin and LSD are somewhat similar in structure as indicated by the indole ring present in both molecules. Therefore, the two molecules share similar chemical properties due to the similarity in structure. When comparing LSD and serotonin, the similarity between its electron densities of the highest occupied molecular orbital is observed. The nitrogen atom possesses the electron density due to its free lone pair.

serotonin

LSD

LSD is prepared by reacting lysergic acid with diethylamide. LSD has the chemical composition C20H25N3O. It has four different isomers, however, only when it is R, and R, stereochemistry. Only one stereoisomer (the d-) is psychoactive. Therefore, a racemic mixture of LSD shows only half the potency of the dextro form. The electron density is lowest in the areas around the indole ring in both molecules. The dipole moment of the two molecules are very close. Serotonin is 2.98 D and LSD is 3.04 D; D is the SI unit measurement for electric dipole moment. The dipole moment is going towards the amine group in both molecules. The virtually same dipole moments of both molecules is the key to the ability of LSD fitting into the same receptors as serotonin.
Storage
[edit | edit source]LSD is a somewhat stable organic molecule, like other molecules with similar structures.The main factors to be concerned with are moisture due to chemical reactions in the presence of moisture, oxygen, light, and temperature. Reaction rates typically depend on temperature exponentially.
Synthesis
[edit | edit source]In its pure form, LSD is a white or clear, odorless, water-soluble crystal that can be crushed into a powder and dissolved. The most common form of LSD is known as blotter acid, sheets of paper that have are blotted with LSD. Tablet known as microdot is also very common. LSD is generally found as in its crystal form, dried on gelatin sheets or in capsule or liquid form.There a couple of different ways to make LSD. A common way is to start with lysergic acid. It will require morning glory seeds. Morning glory and the seeds contain lysergic acid amide. It is considered a precursor to LSD. The amount of LSA in different seeds varies . As a result, the quality of the drug made from it would also vary. Another synthetic route is to use ergot. Once the ergot is obtained, one must extract the ergot alkaloids which contain basic nitrogen atoms. A darkroom working environment is necessary because the ergot will decompose under bright lights. LSD itself can break down quickly when exposed to light.The solvents and reagents used in the synthesis are also harmful. The solvent anhydrous hydrazine can explode when heated. It is a carcinogen. Another chemical often used in for the synthesis is chloroform. It can cause cancer as well while causing internal damages to organs. The ergot alkaloid is synthesized into a compound called iso-lysergic acid hydrazide through the addition of reagents and heating. The iso-lysergic acid hydrazide is isomerized in order to make it active. Once cooled, the isomer is mixed with an acid and a base, and evaporated. The product obatined is iso-lysergic diethylamide, which is isomerized again to produce active LSD. The LSD is then purified and crystallized in order to obtain a higher concentrated form.
Side Effects of LSD
[edit | edit source]There have been no documented human deaths from an LSD overdose. It is physiologically well tolerated and there is no evidence for long-lasting physiological effects on the brain or other parts of the human organism. LSD may temporarily impair the ability to make sensible judgments and understand common dangers, thus making the user more susceptible to accidents and personal injury. It may cause temporary confusion, difficulty with abstract thinking, or signs of impaired memory and attention span
References
[edit | edit source]Methamphetamine is a highly addictive drug and stimulant that enhances heart rate, blood pressure and physical activity. It was discovered by Japan in 1919, after amphetamine was synthesized first in January 1887 by Romanian chemist, Lazar Edeleanu. The first use of methamphetamine in 1919 was to alleviate fatigue and create awareness and then was marketed as Benzedrin, an inhalant use medically for chest congestion. Methamphetamine also known as crystal, crystal meth, and ice, was commonly used in World War II in order to stay awake and create more alertness.
Chemistry Methamphetamine’s chemical name is (S)-N,a-Dimethylbenzene-ethanamine; d-N-methylamphetamine and the chemical formula is C10H15N. Methamphetamine is created by attaching a methyl group to the side chain of an amphetamine As shown in the images. The Methyl group protects the amphetamine from degradation by a monoamine oxidase. Since a monoamine oxidase cannot degrade methamphetamine, this persists in the bloodstream, which causes long-term effects such as Alzheimer’s disease, paranoia, and psychotic behavior. Methamphetamine has two isomers as the methyl group could either attach to the left side or the right side of the amphetamine, creating two mirror images. The dextro-methamphetamine or the right handed methamphetamine is the more active and potent of both isomers.
{kind=link}
{kind=link}
{kind=link}
How Meth Works Methamphetamine attacks neurotransmitters in order to create the addiction in the body. Dopamine, Norepinephrine, and Epinephrine are neurotransmitters that Methamphetamine attacks. Dopamine controls movement, pleasures, emotions and thought processes. The brain releases dopamine when humans accomplish something. The excess of Dopamine creates the feeling of euphoria and well being of a human being. What Methamphetamine does is that it stimulates the release of Dopamine and blocks the re-uptake of Dopamine as well, hence, creating a concentration of Dopamine as shown in the images. This concentration of Dopamine creates the feeling of exhilaration and well-being. When the effect of Dopamine passes away, the feeling of well-being passes away as well. This creates the addiction and the neediness of euphoria, therefore, the necessity of consuming methamphetamine. This concentration created leads to nerve cell death, which give long-term effects as schizophrenia, Alzheimer’s disease and psychotic behavior. Norepinephrine is responsible for controlling alertness, rest cycles, attention, and memory. Methamphetamine blocks the re-uptake of Norepinephrine as well which then creates more awareness and no need to rest. Epinephrine re-uptake is as well blocked by Methamphetamine, which again creates a concentration gradient that leads to the absorption of epinephrine in different parts of the brain, creating an adrenaline release, and a rush and excitement on the user.
{kind=link}
{kind=link}
Effects The effects of Methamphetamine range from increased physical activity, decreased appetite, alertness, increased heart rate and blood pressure, hyperthermia, paranoia, confusion, anxiety, aggressiveness, insomnia, tremors, and irritability.
Long Term effects range from violent behavior, mood disturbances, delusions, psychotic behavior, and hallucinations, Schizophrenia, Strokes and Alzheimer’s disease.
Overview
Methamphetamine is often called meth, ice, crystal, or glass for short. It is a psychoactive drug that is highly addictive. Methamphetamine can be smoked, injected, snorted or swallowed. It has been used to treat ADHD and obesity in low doses. At high doses, Methamphetamine has been known to lead to feelings of euphoria and libido. With prolonged use of Methamphetamine, brain damage, cardiovascular damage, and teeth decay can occur.
Uses
Methamphetamine has been used medicinally to treat ADHD and obesity. This is due to the fact that Methamphetamine's effects lead to increased energy and alertness as well as loss of appetite. This drug has been FDA approved. On the other hand, Methamphetamine has been used in recreation to achieve feelings of ecstasy due to its effects of a release of dopamine to the brain.
Effects
-Immediate effects:Feelings of euphoria, libido, increased energy and awareness, irritability, self-confidence, and violence. Physical effects that commonly take place are anorexia, dry mouth, headache, nausea, diarrhea, dry skin, insomnia, and irregular heartbeat.
-Long term: A build up of tolerance to Methamphetamine. Teeth decay and falling out. Strong addiction and craving for the drug. Drug related psychosis.
-Overdose: Cardiac arrest or stroke may occur and result in death. Massive hallucinations and sensation of flesh crawling also commonly occur.
-Withdrawal: Symptoms usually consist of fatigue, depression, increased appetite and anxiety. Effects usually last months.
Chemical summary
Its IUPAC name is N-methyl-1-phenylpropan-2-amine. Its chemical make up is C10H15N with a molecular mass of 149.233 g/mol. Methamphetamine is a member of the phenethylamine family, which is comprised of stimulants and hallucinogens. Methamphetamine has two enantiomers. The S-isomer is the one that is being described. It is often made into powder/crystal. Methamphetamine causes an increase in activity in the dopamine neurotransmiter system which leads to this feeling of euphoria.After uses of methamphetamine, dopamine and serotonin concentrations decrease from before.
History
Methamphetamine was first discovered and synthesized by chemist Nagai Nagyoshi in 1893 and was later crystallized by Akira Ogata in 1919.
Methamphetamine had been widely used during World War II to fight fatigue and hunger of the troops for both Allied and Axis forces. It later became FDA approved as a treatment for narcolepsy, depression, alcoholism, obesity and ADHD.
[4] [5] [6] [7] Minocycline is a tetra antibiotic that combats bacteria in the body. It is used to treat a variety of bacterial infections, such as urinary track infections, respiratory infections, skin infections, acne, gonorrhea, chlamydia, etc.

Overview
[edit | edit source]Minocycline is an oral antibiotic commonly used to treat acne vulgaris (commonly known as cystic acne). Minocycline are able to kill the acne bacteria more effectively as well as reduce the redness and swelling of the acne. It is most commonly dispensed as Minocycline Hydrochloride which is a semi synthetic derivative of tetracycline. [8] The medication are antibiotics sold as: Minocin, Dynacin, Vectrin, Solodyn, and generic minocycline.
It's IUPAC Name is (4S,4aS,5aR,12aS)-4,7-bis(dimethylamino)-3,10,12,12a-tetrahydroxy-1,11-dioxo-1,4,4a,5,5a,6,11,12a-octahydrotetracene-2-carboxamide [9]
Usage
[edit | edit source]Along with another one of its tetracycline counterparts, Doxycycline, Minocycline is the most common form of oral antibiotic prescribed to patients for acne. Refrain from taking if allergic to minocycline or other tetracycline antibiotics.
Occasionally it is used to treat Lyme disease and other skin infections such as Methicillin-resistant Staphylococcus Aureus (MRSA) since the twice a day dosage for minocycline is more tolerable than the four times daily required by other tetracycline compounds. Dosage of minocycline is 100-200mg daily for the adult, but if the dosage is skipped, it is possible this will affect the treatment process.
If a dose is missed, do not take extra to make up for the missed dose. If an overdose is taken, seek emergency medical attention or call the Poison Help line at 1-800-222-1222.
Caution
[edit | edit source]If pregnant, do not use Minocycline. It can cause harm to the unborn child or permanent tooth discoloration. Minocycline may make birth control pills less effective. It also will pass through breast milk, effecting bone and tooth development of a nursing infant. Minocycline should not be taken by children younger than 8 years of age. Vitamin supplements should not be taken before taking Minocycine. These supplements reduce the effectiveness of it. Avoid excess sun exposure. Those taking Minocycline are easier subject to sun burn. Minocycline should be taken for the full prescribed length. Anything less will result in symptoms resurfacing. Do not take if past expiration date. Expired Minocycline may cause damage to kidneys.
Side Effects
[edit | edit source]Sensitivity (Skin) to Sunlight (Automatic)
Diarhhea/Nausea/Upset Stomach
Dizziness
Drowsiness
Headaches
Vomiting
Skin Discoloration
Autoimmune Disorders
Chest Pains/Irregular Breathing
Fever (Rare)
Discoloration of Skin/Eyes (Rare)
Sore Throat (rare)
References
[edit | edit source]- ↑ http://publicsafety.utah.gov/investigations/methhistory.html
- ↑ http://methoide.fcm.arizona.edu/infocenter/index.cfm?stid=165
- ↑ http://www.montana.edu/wwwai/imsd/rezmeth/transmit.htm
- ↑ http://www.acnp.org/g4/GN401000166/Default.htm
- ↑ http://www.health.ny.gov/diseases/aids/harm_reduction/crystalmeth/docs/meth_literature_index.pdf
- ↑ http://chemistry.about.com/od/medicalhealth/a/crystalmeth.htm
- ↑ http://www.emcdda.europa.eu/publications/drug-profiles/methamphetamine
- ↑ http://www.rxlist.com/solodyn-drug.htm
- ↑ http://www.drugbank.ca/drugs/DB01017
- ↑ http://www.drugs.com/sfx/minocycline-side-effects.html
- ↑ http://en.wikipedia.org/wiki/Minocycline
5. "Drug Information Online". <http://www.drugs.com/minocycline.html>. Oct. 28 2012
6. "Minocycline". <http://www.aocd.org/skin/dermatologic_diseases/minocycline.html>. Web. 28 Oct 2012.
7. "Minocycline for Acne." <http://minocyclineforacnereview.com/>. Web. 28 OC 2012. Steroids are made up of many steroid rings, in which these rings consists of a combination of "one 5 carbon ring, and 3 six carbon rings" (Cholesterol being the primary structure), (Kishner, page 1). Steroids comprise of all the form acquired from the basic ring structure, one of them being testosterone and the anabolic-androgenic steroids (AASs).

How It Works
[edit | edit source]Anabolic steroids are synthetic versions of the male hormone testosterone, a driving force responsible for male characteristics like muscle growth, facial hair, voice deepening. As the body absorbs testosterone in the body, it fits into the receptor sites of cells and activates the cells. After the testosterone molecule enters the cell, protein synthesis and phosphate synthesis is significantly increased due to the addition of RNA polymerase. Phosphate synthesis allows for the production of additional creatine phosphate, which allows for greater amount of work in the absence of oxygen while an increased protein synthesis allows for increased transcription. Anabolic steroids also lead to greater nitrogen retention in the body. A positive nitrogen balance in the body is needed for muscle production since a negative nitrogen balance is created when strenuous forced is applied on the body. Since the body only produces about 2-10mg of testosterone per day and most AAS abusers use up to 1000mg of testosterone a week, it become very significant in its affect in muscle growth.
Testosterone
[edit | edit source]
The human's main hormone that makes the androgenic (a compound that is made up of other compounds), which administer the growth and nurturing of the distinct male characteristics. It is a significant hormone because it help restore the health of tissues, and supply muscle mass (anabolic: represents testosterone and dihydrotestosterone). Therefore, both the ability of androgenic and anabolic gives testosterone a double action mechanism.
Throughout the years, biochemists have been trying to convert and adjust testosterone structure to make medications that can either be taken orally, or have a different breakdown when dissolved into the body, or even better, a drug that can do both. After many years of researching, scientists have come to the conclusion that by substituting and changing certain portion of the testosterone molecule, it may have a big impact on the effect; but unfortunately, it has not yet been discovered.

Uses
[edit | edit source]Testosterone and the anabolic-androgenic steroids have been use as treatments for these diseases:
- Many types of anemia
- HIV wasting syndrome
- Osteoporosis
- Severe burns
- Acute and chronic burns
- Malnutrition, weight loss
- Short stature
- Primary or secondary hypogonadism
(Stephen Kishner, page 1)
Although it has been used properly by some, many others have misused the drug for other purposes, such as to boost their normal anabolic and androgenic ability with the intension to better their physical image and strength.

Biochemistry and Pharmacology aspect
[edit | edit source]
To improve the AAS research, biochemists have created an objective to alter the molecules to be "more anabolic and less androgenic" than testosterone, in which makes it possible to take orally, and have a reduce effect on the HPG axis (Stephen Kishner, page 3). According to an article called, Anabolic Steroids Use and Abuse, by Stephen Kishner, "AASs is developed from 3 compounds: testosterone, dihydrotestosterone, and 19-nortestosterone", (Kishner, page 3). (Testosterone and 19-nortestosterone is a very similar compound, very much alike except 19-nortestpsterone doesn't include the 19th carbon).
The earliest changes made to the compound was on the 17th carbon, by adding a methyl/ethyl group. This modification expand the half life of the drug and therefore, allowing the medication to be active and can be taken orally.

Downside
[edit | edit source]Although the revision was helpful in some ways, it also had consequences because this new compound was not exactly like the original. Due to the methyl/ethyl group added to the compound, liver stain was found as a result of the transformation. In addition, all of the carbon-17 added compound was effected, and caused chemical damages to the liver.(source needed)

Target Organs and Psychological Effects
[edit | edit source]- CNS: increases libido, well-being, aggression, and spatial cognition
- Hypothalamus/Pituitary Gland: decrease GnRH, LH, FSH, increase GH
- Liver: decrease SHBG, HDL
- Larynx: lower voice
- Breast development
- Kidney: increase erythropoietin
Genitals: increase development, spermatogenesis, erections
- Prostate: increase in size, secretions
- Skin: increase facial/ body hair, sebum production
- Bone: increase BMD
- Muscle: increase lean mass, strength
- Adipose Tissue: increase lipolysis, decrease abdominal fat
- Blood: increase hematocrit
- Immune system: increase autoantibody production
References
[edit | edit source]Kishner, Stephen. "Anabolic Steroid Use and Abuse." Medscape. Emedicine, 6 July 2011. Web. 13 Dec. 2011.
Auchus, Richard J. MD, PhD. "The Science Of Anabolic Steroid Abuse." Utsoutwestern.edu. Web. 20 Oct, 2012
http://www.vanderbilt.edu/AnS/psychology/health_psychology/anabolic_steroids.html
Images: Wiki-Media Commons Oxycodone is a type of analgesic, oral medication used to relieve moderate to severe pain. It was developed in Germany in the year 1916 as an attempt to improve on the existing opioids, drugs derived and used by opium poppy for therapeutic benefits. A few common examples of compounding are non-steroidal anti-inflammatory drugs like ibuprofen and oxycodone with acetaminophen. Numerous brand names include OxyContin, a Purdue Pharma brand for the time-release oral oxycodone variation, Roxycodone, and Xanodyne.

Prescribed Usages
[edit | edit source]Oxycodone is mainly used to alleviate moderate to severe pain, as well as managing acute chronic pain. It can also be used an alternative to severe diarrhea and irritable bowel syndrome when commonly prescribed drugs do not work effectively. Oxycodone causes less sedation, respiratory distress, pruritus, and nausea than morphine, therefore making it easier for the body to tolerate it than morphine. If oxycodone is taken as a combination product, make sure to read all information about the ingredients and to ask your doctor and pharmacist for more information for safe usage.
Adverse Effects
[edit | edit source]The most common negative side effects reported are disturbing nightmares, memory loss, fatigue, dizziness, constipation, mood changes, flushing, lightheadedness, headaches, dry mouth, itching, heavy sweating, anxiety, and diminished vision. A few patients have experienced abdominal pain, diarrhea, loss of appetite, urine retion, dyspriea, and impotence. Overdose effects include shallow breathing, clammy skin, hypotension, miosis, respiratory arrest, and death.

Dosage and Administration
[edit | edit source]Oxycodone comes in a variety of forms: as a solution (including concentrated), a tablet, capsule, and long-acting/extended-release. The previous list of oxycodone forms are usually taken with or without food every 4 to 6 hours, depending if the medication is needed for temporary pain or as a regularly scheduled form of medication Always follow the directions on the prescription label and ask a doctor or pharmacist to explain any information that you do not fully understand.
As mentioned before, it can be taken orally or intranasally using intravenous injections or rectally. 60-87% of all Oxycodone usage are through oral administration, with the rest remaining Compared to morphine, it can be 1.5 to 2 times as potent when taken orally. Because of its potency, a doctor will likely start a patient on a low dosage and increase it over time if the pain is not managed. The body can grow accustomed to the medication, and when that occurs the doctor may need to increase the dose.
This medication can be habit-forming, and consequently a patient must not take a larger dose. A patient also cannot take oxycodone more often than prescribed or take it for a longer period of time than recommended by his doctor. Do not stop taking the medication abruptly; you may experience symptoms of withdrawal.
Withdrawal
[edit | edit source]When a patient suddenly discontinues the medication, he may experience symptoms such as anxiety, panic attacks, insomnia, muscle pain, nausea, fevers, flu-like symptoms (runny noses, sneezing, chills, watery eyes, etc.) and muscle weakness. When withdrawal occurs, the doctor usually decreases the dosage gradually. Most cases of severe withdrawals are associated with those addicted and using Oxycodone illegally. The experience of withdrawals from Oxycodone are unique to each individual due to the differences in metabolism etc. Therefore even with the same amount of dosage, one person might experience a withdrawal much worse than the next man. Withdrawal effects can be diminished by proper detox procedures. [2]
Druge Abuse
[edit | edit source]Oxycodone is a powerful drug used by young teens as well as adults. The drug puts users on a advantage high. Since it is a relief medication, it provides users with a calm relief feeling for a few hours as it enters their bloodstream. The tolerance of taking the drug becomes increasingly higher as users ingest the drug more often. As addiction becomes imminent, certain side effect may come into play and also ruin the social life of drug abusers. Some of the effects addiction leads to are: constantly thinking about oxycodone, obtaining large amounts of the drug, taking the drug secretly in order to hide it from others from knowing, lying to loved ones, and feeling pain and restless thoughts during nights of sleep. And because oxycodone contains acetaminophen, taking high doses of the drug can lead to liver problems in the future and sometimes even death.
Special Precautions
[edit | edit source]Oxycodone is a sensitive and potent medication, and consequently have a few precautions before taking it. Inform your doctor and pharmacist:
- If you are allergic to certain medications such as codeine, hycodan, and any other medications
- If you are taking other medications (including vitamin or nutritional supplements) you are taking or plan to take.
- If you have or ever have had asthma, lung disease, or paralytic ileus
- If you continue to drink or have ever drunk large amounts of alcohol and if you have used any street drugs (including overdosing on a medication)
- If you are pregnant or plan to become pregnant, or if you are breast-feeding
- If you are about to have surgery (including dental work)
Brand Names
[edit | edit source]Oxycodone also go under the name of:
- Dazidox
- Endocodone
- Oxy IR
- ETH-Oxydose
- Oxyfast
- Roxicodone
- Oxycontin
- Oxexta
References
[edit | edit source]- "Oxycodone." PubMed Health. October 15, 2011. AHFS Consumer Medication Information. October 26, 2012 <http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0000589/>
- "Oxycodone." Wikipedia. October 24, 2012. October 26, 2012 <http://en.wikipedia.org/wiki/Oxycotton#Medical_uses>
"Oxycodone" WEBMD. November 20, 2012 http://www.webmd.com/drugs/mono-5278-OXYCODONE+-+ORAL.aspx?drugid=1025&drugname=oxycodone+Oral&source=1
Intro
[edit | edit source]Contraception, or birth control, is a form of family planning. This method prevents unwanted pregnancies and sometimes also prevents sexually transmitted diseases. Some forms of birth control are meant for only males, and others are meant for women.
Other family planning techniques include abstinence, surgical sterilization, natural family planning, and withdrawal.
Barrier Methods
[edit | edit source]Barrier methods of contraception are used as a physical or chemical forms of blockading sperm from having access within the uterus to prevent any fertilization of an egg, and thus, a pregnancy. They have no considerable side effects, except for any allergies to the materials within the devices, and can be used right before sexual intercourse.
Male Condoms: This is the most common form of contraception within the modern world. A male condom is used by a male by rolling the thin tube onto an erect penis before any type of intercourse. It became a very popular contraceptive method in the 20th century and is the most effective form of birth control that prevents most sexually transmitted diseases if only used once. The earliest type of male condom were made from animal products, and are considered high-end on the American markets today. The most common type of condoms sold and purchased today is the latex rubber condom and is available in lubricated or non-lubricated varieties.
Female Condoms: These type of condoms look very similar to the male condoms, but are actually inserted into the female instead of on the male. It is a single-use, pre-lubricated, polyurethane sheath that is also used before sexual intercourse. It has two rings--one that is inserted to the upper vagina and the other stays on the outside of the vagina and covers the vulva and surrounding area of the vagina. It is not as widely used due to the costliness of the materials.
Spermicides: Spermicides are chemicals that are used to kill sperm, and, therefore, not allowing them to enter the vagina and causing a pregnancy. In some cases, they can even kill some sexually transmitted diseases. They are available in the form of gels, jellies, foams, pessaries, and water-soluble films. Usually used in conjunction with other types of barriers, spermicides are safe in low amounts, as high dosages could damage the vaginal lining, sensitize the penis, and even help increase the chances of being infected with HIV.
The Sponge: Sponges can come in varying kinds and sizes. It is usually made of disposable and soft polyurethane and paired with a spermicide. It is a choice of contraception for women because it is very convenient--women just need to wet the sponge in water, insert into the vagina, and it would be effective for the next 24 hours, despite the number of sexual acts. Despite its convenience, it is still not as effective as other contraceptive techniques.
Cervical Cap: The rubber, metal, or latex cervical cap is a small, cup-like instrument that is placed on the cervix within the vagina. Spermicide can be placed within the dome to ensure that sperm is blocked physically as well as chemically. Some types of cervical caps must use suction with the vaginal walls in order to be in place during sexual intercourse. Since every cervix is different, every woman must be fitted by a medical professional properly. Few women actually use this method of barrier contraception because there are not many professionals that are trained in the fitting of the cervical cap. In the U.S., the FDA approved three types of the cervical cap--small, medium, and large. The small cervical cap is designed for women who have never gotten pregnant, the medium is for women who have had a miscarriage, abortion, or had birth by Caesarian section, and the large cap is for women who have had a live and natural birth.
Diaphragms: Like the cervical cap, the diaphragm is also a contraceptive barrier that is used at the cervix. This dome-shaped device can be made from latex or silicone and is expanded over a ring. There is a spring on the rim of the ring that also creates suction onto the vagina. Just like the cervical cap, the diaphragm is also needed to be fitted depending on the size of the cervix. The drawback to this method is that the diaphragm must be placed three hours before sexual intercourse.
Hormonal Methods
[edit | edit source]Hormonal methods of contraception are used to impede fertilization and ovulation of an egg through the manipulation of ovarian hormones, such as progesterone and estrogen. These methods include oral contraceptives, intrauterine devices, injectables, implants, birth control patch, and vaginal contraceptive ring. Although these forms of contraceptives do help prevent unwanted pregnancies, they do not prevent sexually transmitted diseases.
Oral Contraceptives:
[edit | edit source]Combined Oral Contraceptives: These contraceptives are available in the form of pills, which must be taken once everyday. The pill impedes the follicle-stimulating hormone (FSH) in the pituitary, which doesn't allow for the maturation of follicles in the ovaries. By the human body's feedback loop, the luteinizing hormone (LH) increases, which then prevents ovulation of eggs within the ovary. In addition to the decrease of ovulation, the increase of progesterone in the pill increases the cervical mucus, which creates a harsh environment for sperm. The two types of birth control pills include the combination progesterone and estrogen pill and the progesterone-only pill. Usually, women must orally take 20, 21, or 22 active pills, which contain a measured amount of combined progesterone and estrogen, and then followed by inactive sugar pills. During the time that the woman is taking the inactive pills, less menstrual bleeding would take place than if the woman was not on the pill. These pills imitate a normal, yet regulated, menstrual system. Examples of combined oral contraceptives include Yaz, Yazmin, Ortho Tri-Cyclen, Desogen.
Continuous-Use Oral Contraceptives: These pills also must be used everyday orally and contain a combination of progesterone and estrogen. The purpose of these pills is to decrease the number of menstrual periods and the duration of the periods within the year. With continuous-use oral contraceptives, women must take active pills for 12 weeks straight and then take 7 inactive pills. A drawback to this method is that women will experience breakthrough bleeding and spotting. An example of this type of contraceptive include Seasonique.
Progesterone-Only Pills: These pills only contain progesterone in order to increase the thickness of cervical mucus and decrease the infiltration of sperm. This method also utilizes once-a-day pills, just like combined oral contraceptives and continuous-use oral contraceptives. Many providers choose to prescribe this contraceptive to women who have experienced adverse side effects with the estrogen in the combined pills. Progesterone-only pills are not as effective as the combination pills.
Emergency (Postcoital) Contraception: This method of contraception is used orally and only once immediately after (approximately 24-36 hours) the woman has experienced unprotected sexual intercourse that may lead to an unwanted pregnancy. Yet, it may still be effective up to five days after unprotected sexual intercourse. This "Morning After Pill" only contains a dosage of 750 mg of levonorgestrel in each pill. It is meant to be a single use, and not continuously.
Intrauterine Devices:
[edit | edit source]Intrauterine devices, or IUDs, are plastic devices placed within the cervix that could contain copper and/or release levonorgestrel and are used to inhibit sperm from reaching an implanted egg. Earlier versions of the IUDs had a copper-based branch to stabilize it within the vagina. With the more modern plastic variation, it was observed that as the size of the device increased, the menstruation became heavier, and women were more prone to vaginal infections if they ever had sexual contact with anyone with STDs. With this type of contraceptive, there are many risks that appear to outweigh the advantages.
Injectables:
[edit | edit source]Injectables are a long-lasting and more efficient versions of the oral contraceptives. Injectables are available to women as 3-monthly intramuscular injection with a high dosage of medroxyprogesterone acetate (DMPA), or known as Depo-Provera, and as a 2-monthly intramuscular injection with a dosage of norethindrone enanthate, or NET-OEN. The only negative aspect with using injectables as a form of contraceptive is how to maintain a regular dose for a long period of time.
It is projected that the use of injectables will be more common in the future as compared to other methods of contraception. The progestin-only injectables are most likely going to be the most commonly used and offered, such as the 3-monthly DMPA and the 2-monthly NET-EN. In terms of the combined injectable, a monthly dose would be available. A 3-monthly injectable made with levonorgestrel butanoate is being improved, which works similarly to DMPA. With a 3-monthly dose, such as DMPA, the greatest advantage is that there is a low amount of synthesized chemicals circulated within the body, as well as faster and easier for women to stop taking the contraception in order to become fertile again. Also, less hormonal chemicals would result in less ovarian limitation and less risk of amenorrhea, or the lack or miss of one or more menstrual periods.
Depo-Provera is a type of long-acting hormonal method by injection that is very successful in its purpose of contraception, but is difficult to remove from the body and takes a longer time for the individual to become pregnant again. Norethindrone enanthate, a reduced progestogen version of Depo-Provera, provides a slight relief from the adverse side effects of Depo-Provera, but the woman would need to be injected every 2 months instead.
Side Effects of injectables as a form of long-acting contraception include large fibroids, amenorrhea, hypertension, diabetes, delayed return of ovulation, etc. In some cases, the continued use of injectables is associated with osteoporosis. These injectable preparations last so long that if the patient were to want to cease this form of contraception, it may take weeks to months in order for fertility to return once again. A main side effect of Depo-Provera is bone density loss over time. It is suggested for the patient to intake calcium supplements in order to counteract this side effect. Depo-Provera is not suggested to use for more than two years.
Subcutaneous Implants
[edit | edit source]These implants provide a consistent and long-lasting method of contraception. Implants must be surgically placed subcutaneously and also removed from the same area. In terms of its structure, implants usually have a steroid placed inside of a capsule or rod. Because this form provides a steady dose of progestogens, implants usually contains a smaller amount of hormones than injectables and oral contraceptives. Surgical insertion and removal usually requires a professional and removal takes a longer time than insertion.
References:
[edit | edit source]http://www.americanpregnancy.org/preventingpregnancy/overviewtypesbirthcontrol.html
Senanayake, P. (2008). Atlas of Contraception (2nd ed). United Kingdom: Informa UK Ltd.
Introduction
[edit | edit source]Derived from the juice of the opium poppy Papaver Somniferum, morphine makes up about 10% of the plant's juice and has been used for thousands of years to relieve pain of all types and varying severity. Until it was isolated in 1806, it was ingested as the dried juice called opium. Whether in opium or by itself, injected, smoked or swallowed, morphine binds to receptors in the brain to create feeling of euphoria, which makes it popular for recreational use. In the gut, it binds to the same receptors and slows peristaltic movement so well it remains an unparalleled treatment for diarrhea. As with any opiate, prolonged users will eventually become addicted and tolerance — increasing doses needed to achieve the same effects — can be expected both to analgesia (pain relief) and the euphoria (high) morphine produces.
Although indispensable as a weapon against pain, relief comes with "side effects" that may not be so welcome, especially sleepiness, blurred vision, constipation, and decrease in appetite. Of course, morphine's potential to cause physical dependence ("addiction") is its worst unwanted effect as withdraw symptoms can be pretty unpleasant should the person abruptly stop or sharply reduce their dose of morphine.


How Morphine Works
[edit | edit source]Morphine can be administered several ways: IV drips, orally as a pill, injection, nasally (insufflation), or rectally; each method has its own degree of effectiveness, with the pill form being the least effective (10-30%) and IV drips being the most effective (close to 100%). It works by binding the mu-opioid, the kappa-opioid, and the nociceptin receptors in the brain and spinal cord (mostly on substantia gelatinosa, where the feeling of pain is first processed), as well as the delta-receptors in the brain. It inhibits that transmission of signals from pain neurons in the peripheral nervous system and also produces effects in the dorsal horn to excite neurons and other pathways. The result is that morphine blocks pain signals from both the central and peripheral nervous systems. Ironically, the drug doesn't stop the transmission of pain, but rather alters the user's perception of the pain; they are aware of the pain but find its manageable or it can be ignored.
As little as 5mg of morphine can produce noticeable affects as the drug binds to the same receptors that endorphins, enkephalins, and dynorphins bind to — these opiate-like substances produced naturally by the human body.
The euphoric effects morphine produces are part of another mechanism involving the gamma-aminobutyric acid (GABA) inhibitors and their respective neurons. Under cellular conditions, GABA reduces the amount of dopamine, a neurotransmitter in the brain that is associated with pleasure, released in the brain. Morphine inhibits the amount of GABA that is released in the brain. Over time, it gradually increases the levels of dopamine the brain, which results in a "high" or euphoric feeling.
In addition, prolonged use of morphine inhibits the production of cyclic adenosine monophosphate (cAMP). When morphine suddenly becomes unavailable, the human body produces more cAMP as a results, which leads to hyperactivity and drug craving.
Adverse Effects
[edit | edit source]Addiction
[edit | edit source]The use of morphine is well-known for causing "addiction" — physical dependence. Psychological addiction is also possible but a different concern caused by factors less concrete than the far better understood phenomenon of physical addiction, especially to morphine.
Physical dependence, when a given dose becomes a daily necessity to avoid unpleasant "withdrawal syndrome" symptoms, is a feature of morphine use that can be expected to occur in virtually anyone who takes it for long enough. How long this takes varies within a fairly narrow window but it isn't very likely within the usual week or ten-day period most people will have it prescribed to them. After that, it should be discussed with a physician to prevent needless suffering caused either by untreated withdrawal or from the unwarranted cessation of pain relief treatment for fear of "addiction."
Physical dependence occurs with the other opiate drugs, too, although to different degrees in terms of the severity of withdrawal and development of tolerance to the drug.
Constipation
[edit | edit source]As an opioid, morphine has the tendency to act upon the myenteric plexus, which is in the intestinal tract. This results in the reduction of gut motility, which cases an overall effect of constipation. Morphine decreases the rate of intestinal transit by inhibiting the process of peristalsis of the intestine.
Tolerance
[edit | edit source]Tolerance to morphine occurs very readily, meaning that a higher concentration of morphine is necessary to obtain the same feelings or effects. Hypotheses of how this tolerance develops include the change in the receptor conformation of phosphorylation, the decoupling of receptors of G-proteins, the upregulation of the cAMP pathway, and μ-opioid receptor internalization.
Withdrawal
[edit | edit source]Morphine withdrawal progresses as follows, roughly broken down according to symptoms, their onset and likely duration:
- Stage 1: Pronounced desire for the missing drug ("craving") and anxiety; starts six to twelve hours after the last dose.
- Stage 2: Along with craving and anxiety (which may become stronger) typical symptoms are sweating, runny nose, yawning, and watering eyes ("lacrimation"). Usually starts within about 8 to 12 hours after last dose
- Stage 3: General feeling of being uncomfortable, aching all over the body and loss of appetite are typical as symptoms progress. At this point muscle spasms, especially in the legs, are the origin of the term "kicking" (originally to describe opium withdrawal and later to giving up any other habit) and abdominal cramping are the next symptoms to appear next, about 24 hours after the last dose.
- Stage 4: This is the acute stage when symptoms are at their worst and can last two or three days before subsiding. The pain continues, especially in joints. Insomnia, nausea, vomiting, diarrhea, are typical symptoms although they vary quite a bit — between individuals and between attempts to "kick". Men sometimes ejaculate involuntarily. Severe depression is also a very common symptom.
Deadly Combinations/Precautions
[edit | edit source]Morphine is a potentially dangerous drug because, along with pain relief (analgesia) it depresses breathing. This effect increases with dosage but is also affected by tolerance so it is only a danger in case of overdose or when combined with certain depressants, like alcohol.
References
[edit | edit source]"THE BRAIN FROM TOP TO BOTTOM." THE BRAIN FROM TOP TO BOTTOM. Douglas Hospital Research Centre, n.d. Web. 28 Oct. 2012. <http://thebrain.mcgill.ca/flash/i/i_03/i_03_m/i_03_m_par/i_03_m_par_heroine.html>.
"Morphine." Drugs Forum. Web. 28 Oct. 2012. <http://www.drugs-forum.com/forum/showwiki.php?title=Morphine>.
"Morphine." Information from Drugs.com. N.p., n.d. Web. 28 Oct. 2012. <http://www.drugs.com/morphine.html>.
"Morphine." Morphine. N.p., n.d. Web. 28 Oct. 2012. <http://www.emsb.qc.ca/laurenhill/science/morphine.html>.
# "Morphine Addiction Withdrawal Symptoms and Treatment" <http://www.rehabinfo.net/morphine-addiction/#stages>
Brief History
[edit | edit source]Many have heard of Milk of Magnesia, which is a milky-white product used as an antacid to offset discomforts caused by excess acidity in the stomach. But how many have heard about Milk of Amnesia? This is the term used by some to describe the intravenous anesthetic known as propofol, or Diprivan.
The Company Behind the Drug
[edit | edit source]The manufacturer of the drug is AstraZeneca. This company was founded as a result of the union of the Swedish Astra AB with British Zeneca Group PLC. The new joint company began on April 6, 1999. The two companies wanted to combine their efforts into one company so that they would be able to make greater strides in their research and development as a biopharmaceutical company.
About the Drug
[edit | edit source]Propofol is a liquid drug that is administered into patients’ veins. It is a general anesthetic that works to start and maintain anesthesia by slowing down the activity in the brain and nervous system. It is commonly used to sedate patients who have been ventilated or those who are about to go into surgery. Dosage can vary depending on the desired affect and age of the patient. Older patients over the age of 55 receive a 20 mg dose initially and then maintained on a 0.05-0.1 mg/kg/min IV. In contrast, younger patients under the age of 55 receive a 40 mg dose initially and then maintained on a 0.1-0.2 mg/kg/min IV. Propofol is also used in pediatrics and patients in intensive care units, with dosing adjusted accordingly.
Medical uses
[edit | edit source]Propofol is used for induction and maintenance of anesthesia, having largely replaced sodium thiopental for this indication. Propofol is also used to sedate individuals who are receiving mechanical ventilation. In critically ill patients propofol has been found to be superior to lorazepam both in effectiveness as well as overall cost. Propofol is also used for procedural sedation, for example during endoscopic procedures. Its use in these settings results in a faster recovery compared to midazolam.
Drug Usage
[edit | edit source]Any individual who are allergic to any ingredient in this medication or allergic to eggs, egg products, soybeans, or soy products should avoid using this medication.
Some medical conditions may interact with this medication. An individual should tell their doctor if any medical conditions apply to them:
- taking any prescription or nonprescription medicine, herbal preparation, or dietary supplement
- pregnant, planning to become pregnant, or are currently breast-feeding
- have inflammation of the pancreas, high lipid levels in the blood, or epilepsy
Note: Medicines such as Benzodiazepines, narcotic pain relievers, or other sedatives can interact with this medicine and may increase the risk of this medication's side effects.
Advantages
[edit | edit source]Propofol is fast-acting and short-lived, which means that patients would be able to get back to their daily routine and diet much sooner. Other drugs would force patients to take more time to fully recover before they get back to their baseline activities. Another advantage is that propofol can serve as an alternative to opioids. Opioids work by binding to one of three receptor sites in the body found in the central and peripheral nervous systems. Opioids share the effect of sedation but usually come with side effects such as nausea and vomiting. By replacing the use of opioids with propofol, physicians are reducing these side effects seen in patients.
Disadvantages
[edit | edit source]Despite the many advantages of propofol, it is also important to look at the disadvantages of the drug. Unlike drugs such as morphine or midalozam, Diprivan does not have something to reverse its effects. Any and all drugs effects must be lived out until the drug is fully metabolized and excreted from the body through urine. As with many drugs, patients react differently to different drugs. Some medicines work as expected in fewer than half of the people who take them because slight differences in their genes can change the way their bodies react to the drugs. More specifically, slight differences in the genes that make cytochrome P450 proteins are the culprit since these are the proteins that process many of the medications. This has sparked interest in creating “personalized medicines” that tailor to specific individuals. Until then, propofol still poses risks such as causing a patient who is breathing normally to go into full respiratory arrest without any warning signs.
Administration
[edit | edit source]It is essential to maintain a clean environment at the injection site to prevent the risk of bacterial infection. According to RxList, Diprivan contains a small amount of disodium edetate in order to help avert the growth of tiny organisms such as bacteria. It is also for this reason that Diprivan comes in single-use packaging in order to prevent open vials of the drug from nesting these microorganisms. Scientific studies have shown that contamination of the drug has lead to fevers, infections, and even death.Diprivan is usually administered in one of three ways: syringe, infusion, and volumetric pump.
syringe
[edit | edit source]According to dictionary definitions, a syringe is “a tube fitted with a hollow needle for injecting or withdrawing fluids.” This method of administering the drug is self-explanatory.
infusion
[edit | edit source]Infusion pumps infuse medication such as Diprivan into a patient intravenously. These are commonly seen in hospitals in patient rooms where there is a fluid back hanging on a pole that drips the IV fluids into the patient at a rate determined by the physician to ensure proper dosages and effect.
volumetric pumps
[edit | edit source]Volumetric pumps are a similar method of administration like infusion except for that fact that they are usually used by physicians for long-term-care patients who need the drug or fluid to be infused into their system for an extended amount of time.
References
[edit | edit source]"Diprivan." RxList. N.p., 25 Oct. 2010. Web. 20 Nov. 2012. <http://www.rxlist.com/diprivan-drug.htm>.
"Diprivan (Propofol) ." AstraZeneca. N.p., n.d. Web. 20 Nov. 2012. <http://www.astrazeneca.com/Medicines/Neuroscience/Product/Diprivan>.
Euliano, T. Y., and J. S. Gravenstein. "A brief pharmacology related to anesthesia." Essential Anesthesia: From Science to Practice. Cambridge, UK: Cambridge University Press, 2004. 173. Print.
"History." AstraZeneca. N.p., n.d. Web. 20 Nov. 2012. <http://www.astrazeneca.com/About-Us/History>.
"Propofol Sedation: Who Should Administer?." Institute for Safe Medication Practices. N.p., 3 Nov. 2005. Web. 20 Nov. 2012. <http://www.ismp.org/newsletters/acutecare/articles/20051103.asp>.
"The New Genetics." National Institute of General Medical Sciences 7.662 (2006): 64. National Institutes of Health. Web. 20 Nov. 2012.
Brief History
[edit | edit source]Finding a cure for cancer has always been a goal for many health care professionals. Many have tried, but few have made as much of a stride as Dr. Antonio Grillo-López. He, along with several colleagues, pioneered a new drug named rituximab that serves as the first FDA approved antibody to treat cancer. Depending on the severity of the cancer, this drug could either completely treat or extend the lifetime of the patient. This extraordinary feat had very humble beginnings.
The Man Behind the Drug
[edit | edit source]Dr. Grillo-López has his roots in Puerto Rico. Growing up, he received both his Bachelor of Science and Doctor of Medicine degrees at the University of Puerto Rico, San Juan. He served as an associate professor at the University of Michigan from 1980 to 1990. He was also a part of DuPont-Merck Pharmaceuticals Parke Davis. He was later offered a position at, what was then, a start-up company named IDEC. Dr. Grillo-López acted as Chief Medical Officer from November 1992 to January 2001 at the company. Later, he acted as Chief Medical Officer Emeritus from January 2001 to November 2003. It was at this company where rituximab came to fruition.
About the Drug
[edit | edit source]Rituximab is a liquid drug that is administered into patients’ veins. It is used in the treatment and care of patients with non-Hodgkin’s lymphoma, chronic lymphocytic leukemia, rheumatoid arthritis, granulomatosis with polyangiitis, and microscopic polyangiitis. Dosage can vary depending on the desired treatment. For example, in rheumatoid arthritis patients, it is given as two doses two weeks apart. But in non-Hodgkin’s lymphoma, it is given once a week for four to eight weeks. In the United States, it carries a trade name of Rituxan. In Europe, it is offered as MabThera. The main focus will be on non-Hodgkin’s lymphoma, or NHL.
NHL is a term that encompasses all lymphomas except for Hodgkin’s lymphoma. Hodgkin’s lymphoma is distinguished by the presence of binucleate giant cells known as Reed-Sternberg cells. Lymphoma is a cancer of the lymphocytes in blood. Lymphocytes are a type of white blood cells that serve an integral part of the human immune system. They are classified into B cells, T cells, and natural killer cells. In NHL patients, an antigen known as CD20 is found on B cells. The rituximab antibody binds to the CD20 antigen and destroys excess, overactive, or dysfunctional B cells as a result of the disease.
Attacking the Cell
[edit | edit source]Rituximab kills cancerous B cells via three main mechanisms: complement-dependent cytoxicity (CDC), antibody-dependent cell-mediated cytoxcity (ADCC), and apoptosis. CDC occurs when a large group of plasma proteins (complement) work together to destroy invading pathogens and malignant cells. Antibody-antigen complexes, such as the one between rituximab and CD20, activate the complement. The complement protein C1 binds to the tail of the rituximab antibody in a “lock and key” fashion and starts a series of reactions that creates a membrane attack complex lining the B cell membrane and then creating a pore to allow the cellular contents to escape and eventually die. ADCC is a process where the antibody-antigen complex forms and then attracts other components of the immune system, including natural killer cells. The receptors on these cells recognize and bind to the tail of the rituximab antibody. The natural killer cells also carry granules filled with cytotoxic molecules. When the granules are released after the natural killer cells bind with rituximab, they penetrate the cellular membrane of the B cell and cause pores that facilitate the release of cellular content leading to the cell’s death. The granules can also destroy the cells by attacking the nucleus. The final mechanism is known as apoptosis, or programmed cell death. Apoptosis is defined the death of cells that occurs as a normal and controlled part of an organism’s growth or development. When rituximab bonds to CD20 and forms the antibody receptor complex, it signals the cell to start the process. The cytoskeleton collapses upon itself, the nucleus condenses, and the DNA fragments into small pieces via enzymes. Membrane-bound vesicles are also shredded. At the end of apoptosis, the cell has effectively destroyed itself. It is still unclear, however, whether these mechanism act independently or in concert. Despite this, rituximab still proves to be an effective cure.
Experiments Involved
[edit | edit source]The rituximab antibody was created after several experiments involving mice. Three mice were injected with cells from a lymphoma tumor. These malignant cells carried the CD20 antigen that is found only on B cells. Observations revealed that the mice’s immune system had produced antibodies to fight these foreign substances. The tail of the antibody is humanized. Mice and human gene sequences were used together to create the chimeric antibody that is rituximab.
Pathway to Success
[edit | edit source]Dr. Grillo-López accredits much of his success to the 1908 Nobel Prize winner Paul Ehrlich. The late Paul Ehrlich was a German scientist who had experience in the fields of hematology, the study of the physiology of blood, and immunology. His work in the latter field gained him the joint Nobel Prize in Physiology or Medicine in 1908 with Ilya Ilyich Mechnikov. Ehrlich popularized the concept of a drug that would act as a “magic bullet.” This meant that a drug would be able to selectively target a disease-causing organism. This way a toxin could be made to be delivered to only that organism. This is the basis of Dr. Grillo-López’s work. The rituximab works by selectively binding to the CD20 antigen and forming the antibody receptor complex that is a vital part of the mechanisms used to destroy infected B cells. Ehrlich had said that success in research is only attainable with several big “G’s”. In German, these are Geduld, Geschicklichkeit, Glück, Geld, and Geräte. These translate to persistence and determination, skill, luck, money, and tools, respectively. The story behind the approval process for this cure shows the influence Ehrlich had on Dr. Grillo-López.
Speed Bumps along the Road
[edit | edit source]The U.S. Food and Drug Administration, or FDA, is an agency that is responsible for protecting public health by passing and enforcing laws and regulations regarding products such as foods and drugs. All drugs must go through their approval process. Dr. Antonio Grillo-López likens them to the pace of snails and considers them to be “the single most important obstacle to the timely development and approval of anti-cancer agents.” He cites examples such as the five-month-review to simply approve the proposed name for rituximab. PanThera was not approved and had to be revised to Rituxan for the U.S. markets and MabThera for European markets. During the pre-digital age, where digital books and social networking site were unavailable, everything had to be done manually. Dr. Grillo-López and his IDEC colleagues, Chet Varns, Alice Wei, and John Leonard, had to send in boxes upon boxes to the FDA for them to review and approve the use of rituximab. In the data set, there was one patient who the group at IDEC considered to not be evaluable due to some minor errors. To present an obstacle to the approval process, an FDA medical reviewer asked them to consider that patient as evaluable now. This only worked in favor of the drug creators because that particular patient had a complete response to the drug. This increased both the overall response rate (ORR) and complete response (CR) rate. The team sent in their biologics license application and it was finally approved after a slow nine months on November 26, 1997. After this, Dr. Grillo-López and his colleagues still faced some obstacles such doubts that a proposed study of using rituximab as part of a combination regimen known as R+CHOP instead of the traditional method of using antibodies following chemotherapy, not in conjunction, would be effective. Despite these doubts, the combination regimen proved to be a cure for NHL. Dr. Grillo-López was persistent and determined according to the philosophies of Paul Ehrlich, leading to his success in finding an effective cure for cancer.
End Result
[edit | edit source]Several milestones were achieved during its development. Clinical trials were completed in record time by patients who would volunteer and even dress up as a mouse, harkening back to the chimeric nature of the antibody. Ritxuimab became the first FDA approved antibody to treat cancer. Dr. Grillo-López even paved the way for quicker FDA approvals. Before, the FDA would take an average of seven years to approve a drug that many patients needed sooner. He was able to reduce that approval timeframe in half. The 2004 Discovery Health Channel’s Medical Honors recognized some of these achievements. Rituximab’s development did not rely on government support. Investors and partners funded the entire project. No federal or state grants or incentives or research funding were involved either.
Impact
[edit | edit source]The impact that rituximab had is unprecedented. It provides a cure for curable lymphomas such as diffused large cell lymphoma. The lifetime of patients with incurable lymphomas have been extended. Recent numbers further demonstrate the success of rituximab. It has been considered the top anticancer drug in the world since 2001. Sales in 2010 alone totaled $6.7 billion. Each year, around 50,000 lymphoma patients are cured. Since its introduction in 1997 until 2010, over two million patients have been treated. Prior to this discovery, there has been a long of stagnation in finding cures or ways to extend lifetimes. Hopefully the success of Dr. Antonio Grillo-López will inspire others to follow in his footsteps. He, himself, has said that he is neither a saint nor a magician. Geduld led to his success, something that can be replicated by any ordinary person with persistence and determination.
References
[edit | edit source]"Antonio Grillo-Lopez: Executive Profile & Biography." Bloomberg Businessweek. N.p., n.d. Web. 2012. <http://investing.businessweek.com/research/stocks/people/person.asp?personId=8006353&ticker=BIIB&previousCapId=26731&previousTitle=Deltagen%20Research%20Laboratories%20LLC>.
"Board of Directors." Onyx Pharmaceuticals. N.p., n.d. Web. 2012. <http://www.onyx.com/about-us/board-of-directors>.
Grillo-López, Antonio. "Accelerated Approval of Cancer Drugs: Improved Access to Therapeutic Breakthroughs or Early Release of Unsafe and Ineffective Drugs?." Journal of clinical oncology 27.26 (2009): 4401. Journal of Clinical Oncology. Web. 2012.
Grillo-López, Antonio. "Curing Cancer: The Rituxan Story." CHEM 92. UCSD. YORK 2622, La Jolla. 2012. Class lecture.
Mesa, Ruben. "Hodgkin's lymphoma (Hodgkin's disease)." Mayo Clinic. Mayo Foundation for Medical Education and Research, 19 Oct. 2011. Web. 2012 <http://www.mayoclinic.com/health/lymphoma/AN01209>.
"Rituxan (Rituximab) for RA, NHL, CLL, WG and MPA ." Rituxan (Rituximab) for RA, NHL, CLL, WG and MPA . Genentech, n.d. Web. 2012. <http://www.rituxan.com/index.html>.
"Rituximab Injection." PubMed Health. National Center for Biotechnology Information, 10 Mar. 2010. Web. 2012. <http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0000388/>.
"The Nobel Prize in Physiology or Medicine 1908." Nobelprize.org. The Nobel Foundation, n.d. Web. 2012. <http://www.nobelprize.org/nobel_prizes/medicine/laureates/1908/>.
Overview
[edit | edit source]

The chemical IUPAC name is 13-ethyl-17-ethynyl-17-hydroxy- 1,2,6,7,8,9,10,11,12,13,14,15,16, 17- tetradecahydrocyclopenta[a] phenanthren-3-one. Its chemical formula is C_21H_28O_2. Levonorgestrel is the left form (D- form) of norgestrel. Herschel Smith first synthesized the drug in the 1960s under Wyeth Pharmaceuticals. This drug is primarily used as an emergency contraceptive
This drug is usually taken orally, in pill form and is more commonly known as Plan B or the morning after pill. Many users mistake the drug for an abortion pill. It should be made clear that Levonorgestrel will not terminate a matured fetus. The drug is only effective in preventing pregnancy by means of postponing ovulation and hindering sperm migration. Levonorgestrel is a common ingredient in other birth control pills and acts in the same way, but the levels of hormone in Plan B and other emergency contraceptives are much higher.
In the Body:
[edit | edit source]As a progestin steroid, Levonorgestrel blocks ovulation by stopping the follicle-stimulating hormone and luteinizing hormone from being secreted from the pituitary gland. The drug also causes the mucus in the cervix to become more viscous which makes it difficult for sperm to travel far in the uterus, preventing implantation of the egg.
Usage
[edit | edit source]Oral contraception: At low doses, levonorgestrel is used in monophasic and triphasic formulations of combined oral contraceptive pills, with available monophasic doses ranging from 100-250 µg, and triphasic doses of 50 µg/75 µg/125 µg. At very low daily dose of 30 µg, levonorgestrel is used in some progestogen only pill formulations.
Emergency contraception: Levonorgestrel is used in emergency contraceptive pills (ECPs), both in a combined Yuzpe regimen which includes estrogen, and as a levonorgestrel-only method. The levonorgestrel-only method uses levonorgestrel 1.5 mg (as a single dose or as two .75 mg doses 12 hours apart) taken within 3 days of unprotected sex, with one study indicating that beginning as late as 120 hours (5 days) after intercourse could be effective. There are many brand names of levonorgestrel-only ECPs, including: Escapelle, Plan B, Levonelle, Glanique, NorLevo, Postinor-2, i-pill, "Next Choice" and 72-HOURS. The primary mechanism of action of levonorgestrel as a progestogen-only emergency contraceptive pill is to prevent fertilization by inhibition of ovulation. The International Federation of Gynecology and Obstetrics (FIGO) has issued a statement that: "review of the evidence suggests that LNG [levonorgestreol] ECPs cannot prevent implantation of a fertilized egg. Language on implantation should not be included in LNG ECP product labeling."In June 2012, a New York Times editorial called on the FDA to remove from the label the unsupported suggestion that levonorgestrel emergency contraceptive pills inhibit implantation.
Intrauterine system: Levonorgestrel is the active ingredient in the Mirena intrauterine system.
Contraceptive implants: Levonorgestrel is the active ingredient in Norplant and Jadelle.
When and How to take Levonogestrel:
[edit | edit source]Plan B should be taken no later than 72 hours after unprotected sex, but is most effective within 24 hours to prevent pregnancy. The sooner you take it, the better. Plan B can be purchased at most local drug stores and pharmacies without a prescription for women 18 years and older. Anyone under 17 will need a prescription from a doctor. Plan B should not be used in place of regular birth controls, but ONLY in emergency if:
- no condom was used during sex, or the condom broke
- no birth control was used/or a pill was skipped
- regular birth control fails
- the partner didn’t pull out in time
- sex was forced.
Do NOT take Plan B if already pregnant.
Side Effects and Warnings:
[edit | edit source]Side effects vary in severity and occurrence for each person. These include:
- headaches
- dizziness
- fatigue
- nausea/vomiting
- abdominal pain
- tender breasts
- irregular menstruation
More serious side effects such as itching and skin rash should immediately be taken to the doctor. Those with diabetes and those who are breast-feeding should take extra precaution when using Plan B.
Always be aware of any possible drug interactions when taking a new medication. Consult a doctor before taking Plan B and inform them of any current medications.
References:
[edit | edit source]- http://www.parkinsons-information-exchange-network-online.com/drugdb/071.html
- http://women.webmd.com/guide/plan-b
- http://en.wikipedia.org/wiki/Plan_B_(drug)#cite_note-0
- Levonorgestrel at Encyclopædia Britannica
“Better medical treatments do not always require stronger medicine. The effectiveness of chemical agents depends on the method of administration. So treatment can often be improved by finding optimal drug formulation of delivery system.” (Professor Mark Saltzman) As the result, many chemists, biochemists, pharmacists, bioengineers, and chemical engineers work to improve the administration of the drugs instead of find the stronger medicine. There are three categories of drug administration, topical, parenteral, and enteral.
Drug Administration Routes refers to the various ways of inserting a drug or other chemical inside a patient or animal in order for the chemical to be absorbed into the blood and delivered to the target tissue. There are several known ways to administer drug into an organism, but the easiest and the most commonly used method is through injection.
Topical Administration
[edit | edit source]For topical drug administration, drugs are applied directly to the affected area. The effect reminds in the local area. The advantage of topical administration is that drugs can be given to the patient. No trained personnel are required for application. Usually, there is no pain involved in administration. Side effects are very minimal.
Epicutaneous
[edit | edit source]Drugs are applied directly onto the skin surface. This administration can be used to test if a patient is allergic to certain drugs or substance. Epicutaneous administration can also be used for local anesthesia to relieve minor pain for cuts or burns.
Eye Drops
[edit | edit source]Drugs are applied directly into the affected eye. Eye drops are saline-containing liquid. Examples of eye drops are rinse eye drops, Glaucoma eye drops, steroid and antibiotic eye drops.
Inhalational
[edit | edit source]Drugs are inhaled through the mouth by the patient. Inhalational administration drugs can be used to treat asthma or acute infection in upper airway. The advantage of inhalational administration is that the response of body is much faster than oral administration because the drug does not have to go through the gastrointestinal (GI) tract. Also, because the drug does not have to go though liver, so the damage to liver is minimized.
Intranasal Route
[edit | edit source]Intranasal route administration is also known as nasal spray. Examples of nasal spray are decongestant nasal spray or allergy relive.
Parenteral Drug Delivery
[edit | edit source]Parenteral drug delivery has systematic effect on the body, meaning that the drugs do not stay specifically at one area, but body as a whole. In parenteral drug delivery, substance is given by routes other than the gastrointestinal (GI) tract, including intravenous injection, subcutaneous injection, intraarterial injection, etc.
Intravenous Injection
[edit | edit source]The substance is injected into the vein. The substance can be drugs. The first intravenous injection every given to a patient was in 1667. The advantages of intravenous injection are that the response is very rapid, the dosage of the drug can be easily controlled, and veins are insensitive to irritation by irritant drugs at higher concentration. However, there are many disadvantages of intravenous injection. First of all, it is not always easy to find a suitable veins. Veins disappear when a patient is under high pressure or tension. Second of all, intravenous injection can be toxic due to its fast response from the body. Third of all, a trained personnel is required, and the drug cannot be given to the patients. Last of all, it is rather expensive to prepare sterilizing the needles.
Intramuscular Injection
[edit | edit source]The substance is injected into the muscle. The substance can be vaccines or antibiotics. The advantages of intramuscular injection are that large volume of drugs can be applied and a sustainable release of drugs is possible. Muscles can act as an adsorption compartment. When the concentration of drug inside of the body is low, the muscle can slowly release the drugs into the body. However, there are also many disadvantages of intramuscular injection. Trained personnel are required for intramuscular injection; the drug cannot be given to the patients. Absorption is sometimes erratic, especially for poorly soluble drugs. The solvent of the drug may be released faster than the drug, causing the drug to precipitate at the site of injection and therefore immobilizes the drug.
Subcutaneous Injection
[edit | edit source]The subcutaneous (SC, SQ) route is one of the most versatile routes of administration in that it can be used for both short term and very long term therapies. The injection of a drug or the implantation of a device beneath the surface of the skin is made in the loose interstitial tissues of the upper arm, the anterior surface of the thigh, or the lower portion of the abdomen. The upper back also can be used as a site of subcutaneous administration. The site of injection is usually rotated when injections are frequently given. The maximum amount of medication that can be subcutaneously injected is about 2 ml. Needles are generally 3/8 to 1 inch in length and 24 to 27 gauge.
Absorption of drugs from the subcutaneous tissue is influenced by the same factors that determine the rate of absorption from intramuscular sites; however, the vascularity in the subcutaneous tissue is less than that of muscle tissue, and therefore absorption may be slower than after intramuscular administration. But absorption after subcutaneous administration is generally more rapid and predictable than after oral administration
Subcutaneous injection is injection under the skin. Insulin is a good example for subcutaneous injection. The advantages of subcutaneous injection are that the drugs can be given to the patient because no trained personnel are needed, and the adsorption, even though slow, but is usually complete. The adsorption can be improved by applying massage or heat on the injected site. However, subcutaneous injections are very painful, irritating drugs can cause damage in the skin, and only a small dosage of drug can be applied.
Below is a picture illustrating the difference between different kinds of injections.
Intraarterial Injection
[edit | edit source]Intraarterial injection is injection into the artery. But it can be very dangerous because, the artery presence in very deep on body.
Intradermal Injection
[edit | edit source]Intradermal injection is injection into the skin itself. Examples are skin testing for some allergen or tattoo.
Transdermal
[edit | edit source]Transdermal patch utilizes diffusion through the skin to sustain drug released. Examples are transdermal patch. Picture below demonstrates the difference between traditional intramuscular injection and a transdermal patch. As can be seen from the illustration, intramuscular injection can be much more painful than transdermal patch.
Enteral
[edit | edit source]Oral
[edit | edit source]Many drugs can be made into tablets or capsules. The advantages of oral administration is that it is portable, convenient, easy to take, and not painful. It is cheap because the tablets do not need to be sterilized and can be produced in large quantities. However, oral administration can be sometimes inefficient because high dosage or low soluble drugs suffer low bioavailability. The drug will also be metabolized in the liver during adsorption, which is known as the first-pass effect. Also, the food the patient consumes may also alter the effect of the drugs. The drug can also be damaging to the normal gut flora in the GI tract. Last of all, oral drugs cannot be given to unconscious patient.
Rectal
[edit | edit source]Various drugs are produced in suppository or enema form.
Gastric feeding tube
[edit | edit source]Many drugs or nutrition can be given to unconscious patients through gastric feeding tube.
References
[edit | edit source]http://www.ncbi.nlm.nih.gov/sites/entrez?db=mesh&term=Drug%20Administration%20Routes 1. Zhang, Liangfang. "Drug Administration and Drug Delivery." CENG 207 Lecture 2. University of California, San Diego, La Jolla. 5 Apr. 2012. Lecture. 2. Guo, Weiwei. "Key Concept." Lecture 3. University of California, San Diego, La Jolla. 10 Apr. 2012. Lecture.
3. "Drug Administration: Administration and Kinetics of Drugs: Merck Manual Home Edition." Drug Administration: Administration and Kinetics of Drugs: Merck Manual Home Edition. N.p., n.d. Web. 28 Oct. 2012. <http://www.merckmanuals.com/home/drugs/administration_and_kinetics_of_drugs/drug_administration.html>. 4. "Principles and Practices of Transdermal Medicine - National Health Federation - Your Voice for Health Freedom." Principles and Practices of Transdermal Medicine - National Health Federation - Your Voice for Health Freedom. N.p., n.d. Web. 28 Oct. 2012. <http://www.thenhf.com/article.php?id=579>. 5. "The Ups and Downs of Drug Levels: A Pharmacokinetics Primer." TheBody.com. N.p., n.d. Web. 28 Oct. 2012. <http://www.thebody.com/content/art13513.html>. Topical For topical drug administration, drugs are applied directly to the affected area. The effect reminds in the local area. The advantage of topical administration is that drugs can be given to the patient. No trained personnel are required for application. Usually, there is no pain involved in administration. Side effects are very minimal.
Epicutaneous Drugs are applied directly onto the skin surface. This administration can be used to test if a patient is allergic to certain drugs or substance. Epicutaneous administration can also be used for local anesthesia to relieve minor pain for cuts or burns. Eye drops Drugs are applied directly into the affected eye. Eye drops are saline-containing liquid. Examples of eye drops are rinse eye drops, Glaucoma eye drops, steroid and antibiotic eye drops. Inhalational Drugs are inhaled through the mouth by the patient. Inhalational administration drugs can be used to treat asthma or acute infection in upper airway. The advantage of inhalational administration is that the response of body is much faster than oral administration because the drug does not have to go through the gastrointestinal (GI) tract. Also, because the drug does not have to go though liver, so the damage to liver is minimized. Intranasal route Intranasal route administration is also known as nasal spray. Examples of nasal spray are decongestant nasal spray or allergy relive.
Introduction
[edit | edit source]The term epicutaneous refers to the route of topical drug administration by which the drug, poison, fluid, or other substance is taken into the body via direct skin application. The three major routes of administration include: topical, in which the effect is local; enteral, in which the effect is non-local; and parenteral, in which the effect is systemic. Epicutaneous medications range from lotions, creams and ointments to powders, patches, and tinctures. Other topical medications may be inhaled or applied to surfaces of tissues other than skin. Topical medications taken in excess can result in adverse skin reactions such as itching, inflammation and redness; however, if consumed properly can yield the desired outcome.
Permeation Through Skin
[edit | edit source]The skin acts as a barrier separating the internal body structures from the external environment, maintaining a slightly acidic pH as to prevent against pathogens and other foreign invaders. There are several layers of skin: the outer layer called the epidermis, followed with the dermis, followed with an innermost layer of fatty tissues. The two principal routes of drug administration through the skin are:
1) Transepidermal absorption: This process outlines diffusion of substances across the stratum corneum. Diffusion can occur via the intercellular lipoidal route or through a microscopic route designed for polar compounds and ions. Because the epidermis structure is extremely compact leaving little intercellular space, permeation calls for the crossing of cell membranes. Yet the greatest struggle lies in penetration through the dermis, where diffusion occurs through the interlocking channels of the substance.
2) Transfollicular (shunt pathway) absorption: Sebaceous and eccrine glands operate as the skin's appendages and channels of drug absorption by acting as shunts that bypass the stratum corneum. The overall mechanism progresses through partioning and diffusion through the sebrum, the driving force being a concentration gradient. An equation has been formulated to describe this process: R = h/FDK
R =Resistance of diffusion resistor, F = Fractional area, H = Thickness, D = Diffusivity, K = Relative capacity,
Epicutaneous Applications
[edit | edit source]1) Allergy Testing: Antigen extracts are applied to the skin as puncture skin tests. This method measures the reactivity of the skin to the injected material. This can be carried out through the scratch test, prick test, or intradermal test.
2) Local Anesthesia: Local anesthesia provides immediate relief to the region of application by numbing the sensation and causing a loss of nociception. These drugs can be classified either as aminoamide or aminoester and are given as an injection, spray, or ointment. These are often used for dental or dermal purposes.
3) Transdermal Patches: Transdermal patches overlap under the parenteral category but some can fall under the epicutaneous category as well. They serve a wide array of purposes:
- Prevention of motion sickness: Scopalamine was the first transdermal patch to be approved by the FDA, in 1979.
- Birth control
- Hormone replacement therapy: To alleviate symptoms of menopause.
- Cessation of tobacco smoking
- Allergy tests
4) Arthritis Pain: These drugs exist mainly as creams that work to immediately and temporarily mitigate the sensations of arthritis in the hand. They come in a few forms:
- Counterirritants
- Salicylates
- Capsaicin
5) Wounds/Rashes: Skin conditions can be treated with the usage of creams, lotions, gels, ointments, or in some cases steroids.
References
[edit | edit source]- "Topical Drug Delivery Systems : A Review." Drug Delivery Systems : Topical Drug Delivery |. N.p., n.d. Web. 29 Oct. 2012. <http://www.pharmainfo.net/reviews/topical-drug-delivery-systems-review>.
- "Allergy & Immunology." University of Vermont, n.d. Web. 29 Oct. 2012. <http://www.uvm.edu/medicine/surgery/documents/AllergyandImmunology1.pdf>.
- "Topical Creams for Arthritis Pain Relief." About.com Osteoarthritis. N.p., n.d. Web. 29 Oct. 2012. <http://osteoarthritis.about.com/od/painrelief/a/topical_creams.htm>.
Definition
[edit | edit source]Eye drop is a tropical drug administration route in which a medicated solution of saline-containing liquid is administered to the surface of the eye. Though most of the eye drop usually is lost through patient reaction or resides on the surface of the eye, some of the drug enters the bloodstream through the mucous membranes that line the cornea as well as through the conjunctival mucosa and parts of the tear drainage system. A major advantage of eye drop administration is the ease of regular application of high concentrations of medication as well as its lack of need to go through intestinal digestion. Drugs that are administered through the usage of an eye drop can either be in a fully aqueous solution or in suspension (cloudy).
Eye Drop is a drug that is given to patients after eye surgery, or when having severe dry eyes. Eye drops can help with clearing eyes to make it better for sight.
Types of Eye Drops
[edit | edit source]1) Rinse eye drops
Rinse eye drops are solutions that help lubricate or replace the tears of the eyes. Rinse eye drops do not necessarily contain any medication. This is the most common form of eye drop administration.
2) Antibiotic and steroid eye drops
Antibiotic and steroid eye drops are predominantly used for treating eye infections. They also can be used to prevent infections from occurring after eye surgeries.
3) Glaucoma eye drops
Glaucoma eye drops assist in the draining of fluid from eyes, which in turn lowers eye pressure. Glaucoma eye drops can vary vastly and are sometimes combined with many other medications that vary on a patient’s need.
4) Antihistamine eye drops
Antihistamine eye drops contain antihistamines, which inhibit a body’s response to allergens. They work by inhibiting the release of histamines into the body. They do not necessarily target the allergens, but allow the patient to cope better with its presence. Antihistamine eye drops are used to lessen the histamine response in the eye.
5) Steroid and antibiotic eye drops
These eye drops are used to treat eye infections. It must be used for the full time prescribed to avoid relapse of the infection. The medication may sting your eyes when first used.
6) Glaucoma eye drops
These eye drops decrease eye pressure by helping the eye's fluid to drain better. They are classified by their active ingredient.
Side Effects
[edit | edit source]As many other drugs, eye drops are considered to have some side effects: sore throat, fever, rash, itching, dizziness, swelling. One of the most common side effects is eye redness after using the eye drop. This will lead to pain in eyes or problem with visions that one should be cautious. But in fact, eye drops are less risky than tablet medicines and sometimes, these risks can be prevented from pressing the inner corner of the eye after using. If you do not use them the correct way then you can infect not only one eye, but both eyes.
Reference
[edit | edit source]"Generic Name: Steroid and Antibiotic Eye Drops"
"Glaucoma Medications and Their Side Effects"
http://www.patient.co.uk/doctor/Prescribing-for-and-Administration-of-Drugs-to-the-Eye.htm#ref-1
http://development.aao.org/eyecare/treatment/eyedrops.cfm
Definition
[edit | edit source]Inhalation is a drug administration route in which a drug or inhalant is smoked. Inhalation is effectively used medically for several anesthetics. The onset of this drug administration route is quite rapid due to the fact that there are capillaries located in the lungs, making it easy for the drug to quickly enter the bloodstream. Once the drug is in the bloodstream, it takes approximately five to eight seconds for the drug to reach the brain.
Problems with Inhalation
[edit | edit source]Although inhalation is an effective method of drug administrations, there are three factors that must be kept into consideration with medically using this drug administration method.
1. Drugs and materials must not be irritating to the mucous membranes and lungs.
2. Controlling the dosage may be more difficult than other drug administration methods.
3. The drug must be administered until the desired effect is met. [1]
References
[edit | edit source]- Hart, Carl. Drugs, Society, and Human Behavior. 12th. McGraw-Hill Humanities, 2008. Print.
Intranasal drug administration is an attractive option for local and systemic delivery of many drugs. The nasal mucosa is – in other words, nose – easily accessible. Intranasal drug administration is noninvasive, almost painless and the most susceptible way of administrating drugs for children. Application can be performed easily by patients or by physicians in emergency settings. Intranasal drug delivery offers a rapid onset of therapeutic effects (local or systemic). Nasal application circumvents gastrointestinal degradation and hepatic first-pass metabolism of the drug. The drug, the vehicle and the application device form an undividable triad. Its selection is there- fore essential for the successful development of effec- tive nasal products. This paper discusses the feasibility and potential of intranasal administration. However, there are still many debatable topics on the following topics:
- the intended use (therapeutic considerations)
- the drug
- the vehicle
- the application device
Reference
[edit | edit source]http://content.karger.com/ProdukteDB/Katalogteile/isbn3_8055/_96/_15/CUPDE40_04.pdf Definition The route of administration drug by which a drug taken into the body via the Gastrointestinal Tract and then it absorbs to the blood. It can be classified into three categories.
Categories
- Oral - Consumption of drug by mouth
- Rectal - Insertion of drug into the rectum
- Sublingual - Placement of drug under the tongue
Reference ROUTES OF DRUG ADMINISTRATION - P. Verma , IJPSR/Vol. I/ Issue I/July-September,2010/Pg.54-59
Introduction
[edit | edit source]The oral medical administration is the process of delivering the drug through the mouth so that it enters the alimentary tract. It is known to be the most frequently used route of drug administration due to the fact that it is convenient and cost effective. Drugs that enter through this route usually comes in solid dose forms in order to promote the high degree of drug stability, but they can also come in liquid forms. Different drugs can be taken orally at different time intervals, which can vary between before or after ingesting food.
General pathway
[edit | edit source]Most of the absorption process is accomplished between five and thirty minutes after ingestion, but absorption is not usually complete for as long as six to eight hours. Once drugs passes through the stomach, the drug needs to proceed from the small intestine into the bloodstream. The membrane separating the intestinal wall from blood capillaries is made up of two layers of fat molecules, making it necessary for substances to be lipid soluble or soluble in fats to pass through. Even after successful absorption into blood capillaries, however, substances must still pass through the liver for a screening process before being released into the general circulation around the body. Enzymes in the liver are capable of breaking down the molecular structure of certain drugs, thus reducing the amount that eventually enters the blood stream. As a result of all these natural barriers, orally administered drugs must be ingested at deliberately elevated dose levels, to allow for the fact that some proportions of the drug will not make it through to the bloodstream.
Disadvantages
[edit | edit source]Administering drugs orally can potentially be problematic due to the unpredictable nature of gastrointestinal absorption. [1] Factors such as ingested food can alter the pH within the gut, gastric motility, emptying time, and the rate of drug absorption.
Anti-asthmatic agents and Anti-Inflammatory
[edit | edit source]
Cortisol or hydrocortisone is a compound of the class called corticosteroids. This compound is the anti-inflammatory agent that is used in medicine. Scientists have discovered that the inflammatory of the breathing passages can contribute to the attack of asthma. As a result, it was recommended to control the oral inflammatory together by including corticosteroids in medicine and inhaling bronchodilators to treat asthma. However, corticosteroids can have negative side effects. For instance, this compound can inhibits growth in children. Hence, scientists started to use synthetic corticosteroids to be used for inhalation. In addition, scientists have discovered a synthetic steroid that can be used as anti-inflammatory. Fluticasone is the compound that replace its hydrogen with flourine that would help to activate the compound's ability to be used as anti-inflammatory. This steroid would bind to the protein inside the cell nuclei called the glucocorticoid receptor in order to prevent the inflammatory response in the body.
References
[edit | edit source]- Shepherd, M. "Administration of Drugs 1: Oral Route." Nursing Times, 2011. <http://www.nursingtimes.net/nursing-practice/clinical-zones/prescribing/administration-of-drugs-1-oral-route/5033729.article>
Vollhardt, Peter. Schore, Neil. Organic Chemistry 6th Edition. W.H. Freeman Company. New York. 2011.
Purves, Dale, "Principles of Cognitive Neuroscience", Sinauer Associates, Inc., 2008
Levinthal, Charles, "Drugs, Behavior, and Modern Society", Pearson Education, Inc., 2008 The human rectum is a channel in the human body through which drugs can be easily administered and absorbed well. Sometimes it is the most preferable to give a drug rectally rather than orally, e.g. in cases of nausea and vomiting. Disadvantages of drug administration through rectum include the interruption of absorption by defaecation and lack of patient acceptability ( it is not comfortable for most patients to have drug administered through their rectum). The mechanism of drug absorption from the rectum is probably no different to that in the upper part of the gastrointestinal tract, despite the fact that the physiological circumstances (e.g. pH, fluid content) differ substantially, absorption from aqueous and alcoholic solutions may occur very rapidly, which has proved to be of considerable therapeutic value in the rapid suppression of acute convulsive attacks by diazepam (e.g. in children), but absorption from suppositories is generally slower and very much dependent on the nature of the suppository base, the use of surfactants or other additives, particle size of the active ingredient, etc. There is some evidence that hepatic first-pass elimination of high clearance drugs is partially avoided after rectal administration, e.g. lignocaine. This can be explained by the rectal venous blood supply: the upper part is connected with the portal system, whereas the lower part is directly connected with the systemic circulation. Plasma concentration data following rectal administration of representatives of several classes of drugs are reviewed: anticonvulsants, non-narcotic analgesics and non-steroidal anti-inflammatory agents, hypnosedatives and anaesthetics, strong analgesics, theophylline and derivatives, corticosteroids, antibacterial agents, thiazinamium, promethazine, hyoscine-N-butyl-bromide, streptokinase, progesterone, ergotamine tartrate and levodopa. Only limited number of cases has it been adequately shown that the rectal route of administration gives plasma concentrations which are comparable to the oral route. Potentially the rectal route offers the same possibilities as the oral route, but the influence of the formulation seems to be very critical. It is also likely that the future novel drug delivery systems with zero order release characteristics will be applied rectally. Interesting preliminary results have already been obtained with theophylline administered by 2ml osmotic pumps.
Reference
[edit | edit source]http://www.enotes.com/rectal-medication-administration-reference/rectal-medication-administration Sublingual medications are administered by inserting them directly under the tongue. The medications dissolve rapidly and are absorbed through the mucous membranes of the mouth, where they enter into the bloodstream. The medications are compounded in the form of small, quick-dissolving tablets, sprays, lozenges, or liquid suspensions. The most common sublingual medication is the nitroglycerin tablet, which is a form of pain reliever that is absorbed the fastest when administered sublingually to the body. The sublingual administration method is not always appropriate. Sublingual medications should not be administered if the gums or mucous membranes have open sores or areas of irritation. The patient should not eat, drink, chew, or swallow until the medication has been absorbed. Swallowing the medication must be prevented, since it will decrease the drug's effectiveness as it is digested in the digestive system. Sublingual administration method is fat acting and the body will be able to sense its effect within 5-10 minutes after administration.
Reference
[edit | edit source]Definition
[edit | edit source]Parenteral drug administration is any non-oral means of having the drugs administrated into the body. Intramuscular, subcutaneous, and intravenous are the most used forms of drug administration.
Advantages
[edit | edit source]Some advantages with parenteral drug administration are that it can be used for drugs that are poorly absorbed by the body or ineffective when given orally.
Disadvantages
[edit | edit source]This method of drug administration tends to be expensive due to the amount of equipment required, as well as a more painful experience for the patient. Thus, competent practitioners are recommended. Other risks with parenteral administration are that any errors during the technique of administering the drug may lead to physical harm to the body or adverse drug absorption in the body.
References
[edit | edit source]Martin, Shepherd. "Administration of drugs 3: parenteral" (2011): n. page. Print.
Definition
[edit | edit source]An intravenous (IV) injection is a drug delivery method which the drug is directly injected into the bloodstream. With this method, the onset of the action is much rapid since the drug is being administered directly into the bloodstream. Delivering a drug intravenously can be beneficial when using irritating material since blood vessel walls are highly insensitive to irritants. High concentrations and dosages of drugs can be delivered intravenously much quicker than other methods, which can be beneficial, yet dangerous at the same time.
Disadvantages with using Intravenous Injections
[edit | edit source]Although intravenous injections can be quick, there are some disadvantages with using this method. A major disadvantage of using IV injections is that repeated injections at the same site results in the area surrounding vein injection site losing its strength and elasticity. In worst case scenarios, if a particular injection site has been punctured into several times, the wall of the vein collapses and blood will no longer move through it. Another concern of using IV injections is the transmission of infections. Contaminated needles and syringes should not be used more than once; nor should they be shared between patients.[1]
References
[edit | edit source]- Hart, Carl. Drugs, Society, and Human Behavior. 12th. McGraw-Hill Humanities, 2008. Print.
Definition
[edit | edit source]An intramuscular (IM) injection is a drug administration route in which a drug is injected into a muscle. In terms of absorption, the injection methods that delivers the drug quickest to slowest is intravenous, intramuscular then subcutaneous. It is favored due to its fast-acting as well as long-lasting capabilities. These injections are inserted into the deep muscle tissue where the deposition of medication is absorbed gradually into the bloodstream.
Absorption
[edit | edit source]The three main sites of intramuscular injection is in the deltoid muscle of the arm, buttocks and the thighs. Absorption is quickest when injecting into the arm and slowest in the buttocks.
Advantages
[edit | edit source]When injecting into the muscle, there is less chance of irritation due to greater blood supply and faster absorption. Also, when wanting to inject larger amounts of drugs, it is best to inject into the muscular region instead of injecting subcutaneously.
Procedure
[edit | edit source]Needle should not be smaller than an inch nor exceed one and a half inches in length. The maximum volume of medication recommended should be five milliliters at one site per adult.
References
[edit | edit source]- Hart, Carl. Drugs, Society, and Human Behavior. 12th. McGraw-Hill Humanities, 2008. Print.
"ADMINISTER AN INTRAMUSCULAR INJECTION."Administer Intramuscular, Subcutaneous, and Intradermal Injections. Brookside Associates, n.d. Web.

Definition
[edit | edit source]A subcutaneous injection (SC, SQ, SubQ) is a method of drug administration in which the drug is injected into hypodermis, the fatty tissue layer directly beneath the dermis and epidermis. Because there is limited blood flow to the hypodermis, subcutaneous injection is usually used when slow absorption of medication is preferred.[4] Drugs that are administered using this method must be soluble and potent in small concentrations. Common medications used with this procedure include heparin, insulin, growth hormones, and vaccines against MMR and varicella.
Sites and Method of Injection
[edit | edit source]With subcutaneous injection, the drug is usually administered through loose interstitial tissues of the upper arm, the anterior surface of the thigh, the lower portion of the abdomen, or the lower back. When injecting, the skin is pinched up to prevent injection into muscle. It is recommended to use the same site for routine injections. The shot is given at a straight 90 degree angle if at least 2 inches of skin can be grasped. Otherwise, it is given at a 45 degree angle.[5]
Absorption Rate
[edit | edit source]Factors that may increase absorption rate are heat, massaging, co-administers vasodilators, or hyaluronidase at the site of injection. Epinephrine may decrease the absorption rate due to decreased blood flow.
References
[edit | edit source]- ↑ American Society of Health-System Pharmacists (2009-03-23). "Oxycodone". U.S. National Library of Medicine, MedlinePlus. Retrieved 2009-03-27.
- ↑ oxycodone, October 28, 2012
- ↑ Vollhardt, Peter. Schore, Neil. Organic Chemistry 6th Edition. W.H. Freeman Company. New York. 2011.
- ↑ Clinical Center - National Institutes of Health. "Patient Education: Giving a subcutaneous injection."
- ↑ "How To Give A Subcutaneous Injection." Drugs.com, n.d. Web. 28 Oct. 2012. http://www.drugs.com/cg/how-to-give-a-subcutaneous-injection.html.
Definition
[edit | edit source]Intraarterial means that action is happening inside an artery or to its structure.
Inhalation Advantages
[edit | edit source]An advantage about using intraarterial inhalation is that it is a method of inhalation that more effectively controls the dose of the injection. The patient is able to control how much of the drug that they will receive, since they will be able to titrate it.
Inhalation Disadvatage
[edit | edit source]A disadvantage about using intraarterial inhalation is that this action takes longer. This route is slower because the lungs has to take it in. From the lungs, it has to travel to the systemic circulation.
Injection Advantages
[edit | edit source]The advantages about using intraarterial injection is that it is pretty fast, it takes about fifteen to thirty seconds for intravenous and three to five minutes for intramuscular as well as subcutaneous. Another advantage is that the bioavailability of this is 100%. A third advantage is a single injection can last for days or months. This method is suitable for drugs that cannot be absorbed through the digestive system. This is also suitable if the drug is too irritating. One more advantage is that continuous medication can be delivered.
Injection Disadvantages
[edit | edit source]The disadvantages about using intraarterial injection is that there is a higher risk to addiction and overdosing if abusive drugs are injected. This is due to the quickness of the drug. Another disadvantage is that self-administration cannot typically be done by patients. Belonephobia is another disadvantage, which is fearing needles and injection itself. Sharing of needles can leave to infectious diseases such as HIV. This type of administration is one of the most dangerous. Most of our body's natural defenses is bypassed by intraarterial injection. This exposes the patient to health problems. These include infections, abscesses, hepatitis, and contaminants or undissolved particles. Air bubbles may occur if this is not done correctly.
Definition
[edit | edit source]Intradermal (ID) drug administration is the injection of a small amount of fluid into the dermal layer of the skin. The needle most commonly used for this technique is the tuberculin syringe with a 26-gauge needle, which is about one-fourth to one-half inch in length. A small amount of swelling in the skin after injection is to be expected.
References
[edit | edit source]. "ADMINISTER AN INTRADERMAL INJECTION."Administer Intramuscular, Subcutaneous, and Intradermal Injections. Brookside Associates, n.d. Web.
Introduction
[edit | edit source]Transdermal refers to the process in which the drug diffuses through the intact skin for the purpose of systematic distribution (as opposed to topical distribution). In order to achieve this route of administration, utilities such as transdermal patches, transdermal gels, and transdermal implants can be used for medical purposes.
Pathways
[edit | edit source]Transcellular Pathway
[edit | edit source]The transcellular pathway is the more direct path in which drugs can make its way across the skin. In this route, drugs are transported across the skin by proceeding through the membranes of dead kertinocytes (the predominatn cell type within the epidermis) that make up the top layer of skin, which is known as the stratum corneum. Even though the path distance for this method is the shortest, drugs can experience resistance due to the phospholipid membranes that they have to cross.
Intercellular Pathway
[edit | edit source]The intercellular pathway is the more common pathway in which the drugs pass through the skin by navigating through small spaces that exist between the cells of the skin.
Disadvantages
[edit | edit source]Even though the skin is easily accessible for the purpose of drug delivery, its main function of preventing foreign substances from entering the body can limit the types and amounts of drugs that are taken in through this route of administration. In order to reach the microcirculation of the dermis, the drug must manage to go through the Epidermis and the Dermis, which are two significant layers of the skin.
References
[edit | edit source]- McCarley, K.D & Bunge, A.L. (2001). "Review of pharmacokinetic models of dermal absorption." J Pharmaceut Sci. 90: 1699–1719.
- Hadgraft, J. (2001). "Modulation of the barrier function of the skin." Skin Pharmacol Appl Skin Physiol. 14(1): 72-81.
Introduction
[edit | edit source]Transmucosal refers to the route of administration in which the drug is diffused through the mucous membrane. This can refer to inhalation, nasal, sublingual, vaginal, rectal, or ocular routes.
Insufflation
[edit | edit source]Insufflation refers to the action of inhaling a substance. It is often used as a route for respiratory drugs that can treat sinus and lung conditions. This route of administration is typically used for psychoactive drugs due to the faster diffusion rates into the bloodstream and its capabilities of enabling the drug to bypass the blood-brain barrier. For this method, bioavailability is higher than the bioavailability for the oral route. Cocaine is a common drug that utilizes the route of insufflation.
Sublingual Administration
[edit | edit source]Sublingual administration refers to the process of diffusing drugs into the blood by utilizing tissues beneath the tongue. Drugs that are designed for this route include barbiturates, enzymes, steroids, and cardiovascular drugs. Like the other transmucosal routes, the sublingual administration diffuses through the mucous membrane that is under the tongue. This process is more direct than the oral tract, and more difficult to do, thus lowering the risk of degradation due to salivary enzymes before entering the bloodstream.
References
[edit | edit source]- William H. Frey. "Bypassing the Blood-Brain Barrier to Deliver Therapeutic Agents to the Brain and Spinal Cord." Drug Delivery Technology.
- mendelson JE, Coyle JR, Lopez JC, Baggott MJ, Flower K, Everhart ET, Munro TA, Galloway GP, Cohen BM. "Lack of Effect of Sublingual Salvinorin A, A Naturally Occurring Kappa Opioid, in Humans: A Placebo-Controlled Trial". Sublingual Studies, www.maps.org. Psychopharmacology (Berl). Retrieved 26 January 2012.
Definition
[edit | edit source]Inhalation is a drug administration route in which a drug or inhalant is smoked. Inhalation is effectively used medically for several anesthetics. The onset of this drug administration route is quite rapid due to the fact that there are capillaries located in the lungs, making it easy for the drug to quickly enter the bloodstream. Once the drug is in the bloodstream, it takes approximately five to eight seconds for the drug to reach the brain.
Problems with Inhalation
[edit | edit source]Although inhalation is an effective method of drug administrations, there are three factors that must be kept into consideration with medically using this drug administration method.
1. Drugs and materials must not be irritating to the mucous membranes and lungs.
2. Controlling the dosage may be more difficult than other drug administration methods.
3. The drug must be administered until the desired effect is met. [1]
References
[edit | edit source]- Hart, Carl. Drugs, Society, and Human Behavior. 12th. McGraw-Hill Humanities, 2008. Print.
Introduction
[edit | edit source]Pharmacokinetics, abbreviated as "PK", (from Ancient Greek pharmakon "drug" and kinetikos "to do with motion") is a subdivision of pharmacology focused on effects of a biological system on chemical substances. It deals with three main stages of drug’s life span in our body such as absorption, distribution, and excretion. This area mainly applies to chemical drugs but it also goes into substances ingested or delivered externally to an organism, such as nutrients, metabolites, hormones, toxins, etc.
Pharmacokinetics is often studied with respect to pharmacodynamics. The two should not be confused; pharmacokinetics is described as what the body does to the drug whereas pharmacodynamics is described as what the drug does to the body. Pharmacokinetics extends to the mechanisms of absorption and distribution of drug, the rate in which a drug effect begins along with the duration of the effect, the chemical transformation of the drug in the body (most likely by enzymes), and the effects and routes of excretion of the drug.
Timing
[edit | edit source]All drugs, no matter how they are delivered, share some common features when we consider their effects over time. There is initially an interval, the latency period, during which the concentration of the drug is increasing in the blood but is not yet high enough for a drug effect to be detected. How long this latency period will last is related generally to the absorption time of the drug. As the concentration of the drug continues to rise, the effect will become stronger.
Cross-Tolerance and Cross-Dependence
[edit | edit source]It is possible that a tolerance effect for one drug might automatically induce a tolerance for another. This effect is called cross tolerance, which is commonly observed in the physiological and psychological effects of alcohol, barbiturates and a class of antianxiety medications. As a result of cross tolerance, an alcoholic generally develops a tolerance for a barbiturate, which can be a risk factor when undergoing surgery with an anesthetic. On the other hand, if we can relive the withdrawal symptoms of one drug by administering another drug, then the two drugs show cross-dependence. In effect, one drug can substitute for whatever physiological effects have been produced by a second drug that has been discontinued.
Compartment Model
[edit | edit source]Pharmacokinetics and its role in drug dosage: Pharmacokinetics is a primary systematic regulation that supports applied therapeutics. It is the mathematical basis to test the duration of a drug in the body, and the effects it has on the body. When patients are in need of medicine, doctors issue prescriptions with the appropriate medicines and dosages for the specific condition. This dosage is monitored under the drug use process (DUP). Doctors and pharmacists always make sure that the patient is not suffering from a drug related problem. Once this claim is confirmed, a clinical diagnosis can be made and the pharmacist can apply the DUP to guarantee that the dosage and procedure is appropriate for the patient. The regimen is made according to the patient’s ability to process the drugs. Drugs have four stages in the body: absorption, distribution, metabolism, and excretion. The drug concentration is prescribed on the basis of this fundament. When the patient understands these stages, the medicine can be distributed to the patient. The pharmacist must confirm that the drug, dosage, and regime are suitable, and that the patient leaves with a clear understanding and acceptance. Clinical pharmacokinetics is a crucial foundation that pharmacists must be familiar with and master, and is a quality that pharmacists need to have in order to successfully practice pharmaceutical care. Ideally, the intensity of a drug is calculated at the activation site, the receptor. But since that is not possible, the drug level is measured in the blood, saliva, urine, and/or the cerebrospinal fluid. Reaction Rates: To accurately apply the methods of ADME, the rates of these steps must be taken into consideration. The rate of reaction, the velocity that the reaction continues at, can either be of zero order, or first order. Volume of Distribution: The volume of distribution is not a real quantitative volume value, but more of a apparent volume. It is a measure in which the concentration of the drug can be determined. It is the volume of the plasma that is required to dissolve the drug in the body. Since the body is not a homogeneous entity, it can be difficult to precisely measure the concentration. The concentration of the drug may be different in various parts of the body. However, it is important to note that the concentration will be proportional throughout the body, and the values can be rationalized accordingly. The formula Vd= X/Cp is used to convert a drug amount to its concentration. Vd is the Volume of distribution, X is the amount of drugs in the tissues, and Cp is the amount of drug in the specific part of the body. Using the formula, if the drug has a large volume of distribution, and it does not match to the accurate volume reading, then the drug is said to be highly dispersed in the tissues. If the drug has a matching volume of distribution to the accurate volume reading, then the drug is unsuccessfully dispersed and is strictly contained in the plasma. Drug Clearance: Drug Clearance (CL) is the volume of plasma in the vascular section absent of drugs per unit time through the functions of metabolism and excretion. The clearance for a specific drug remains the same throughout if it is confirmed and removed from the first order kinetics. CL=k X Vd, where k is the first order elimination rate constant, and Vd is the volume of distribution. Multiple Doses: Many patients need to take medication more than once for it to be effective. When drugs are consumed by the body, the drug accumulate in the body and the concentration will rise until it reaches a steady state condition. A steady state happens when the concentration of drug consumed equals the concentration of drug eliminated within the same time frame. At the steady state, the plasma concentration of the drug and the high and lows are constant throughout. The amount of time required to arrive at the steady state depends on the half life of the drug. The larger value the dose is, the larger the steady state levels are. When the dosing interval is lower than the half life value, the higher the accumulation is, and a higher steady state level. In situations where the dosing interval is greatly higher when compared with the half life of the drug, there will be zero accumulation. Many drugs have a dosing interval that is proportional to the half life of the drug, but independent with the amount of doses recommended, or the time required to reach the steady state.
Pharmacokinetics of Drug Dispersion
[edit | edit source]Compartment Model is a mathematical representation that is introduced in the pharmacokinetics of drug dispersion in the body. It is a simulation of the pharmacokinetic possess of drug that has been introduced to the body. Scientists usually study one- or two- compartment model [1].
One Compartment Model without Absorption
[edit | edit source]One Compartment Model is a closed homogeneous system that the administered drug is released and diffused in a single unit of the body organ without absorption, which is an ideal condition [1]. The one-compartment model follows the idea that once the drug has entered the body, it instantly distributes itself equally throughout the body into equilibrium. This is because the model depicts the body as a kinetically homogeneous unit. The concentration of drug in plasma, which also quantitatively indicates changes in the tissue, is then graphed linearly thus representing a one-compartment model. An example of One-Compartment Model is intravenous injection with no absorption. The kinetic characteristics of this model are determined by the total mass of drug (M), concentration of drug (C), volume of the body fluid (V), first order elimination constant (k) and diffusion time (t).[2]
The three variables mean:
M: total mass of drug
V: volume of the body fluid
C: concentration of drug
Mass Balance of One Compartment Model:
dM/dt=-kM
M=M_0 e^(-kt)
M=CV
C=M_0/V e^(-kt) ,when M_0/V=C_0
The variables mean:
k: first order elimination constant
C0 : initial concentration of administered drug
t: drug diffusion time
C/C0: rate constant with respect to time
When t = 0, we can obtain the maximum concentration of drug, which is also the initial concentration: Cmax = C0
From which we can also tell: Mmax = M0
Circulation half-life (t1/2) Calculation in One-Compartment Model:
lnC=ln M_0/V-kt
lnC_0=ln M_0/V
ln C_0/2=ln M_0/V-kt_(1/2)
therefore,
t_(1/2)=ln2/k
One Compartment Model with Absorption
[edit | edit source]One Compartment Model with Absorption is closed system that the administered drug diffuses from a transdermal patch into the blood stream.
The variables are same as Figure 1-1 except that:
D: total amount of initial drug absorption compartment
ka: absorption constant
Mass Balance of One Compartment with Absorption:
dM/dt = kaD-kM
dD/dt = -kaD
D=D0 at t=0
Therefore,
D=D0e-kat ⇒ dM/dt=kaD0e-kat-kM, M=0 at t=0
Finally, M =D_0 k_a/(k-k_a )(e^(-k_a t)-e^(-kt))
This model can be related to therapeutic window:
Two Compartment Model
[edit | edit source]The two-compartment model separates the body into two compartments: a central compartment and a peripheral compartment. The central compartment consists of blood and well perfused organs such as the liver, kidney, heart, brain, etc. The peripheral compartment consists of poorly perfused tissue such as muscle, lean tissue, fats, etc. The model follows the idea that once the drug has entered the body, it distributes itself between the central and peripheral compartment. However, equilibrium is not achieved between the two compartments.
ADME
[edit | edit source]Pharmacokinetics is the basis for which the time course of drugs in body systems and their corresponding effects can be quantified. The four processes culminating the time span of drugs in the body can be described as follows:
- Absorption - the process of the intake of the drug into the body
- Distribution - the process of the dispersion of the drug into the blood stream and tissues
- Metabolism - the process of the parent compounding into daughter metabolites
- Excretion - the process of eliminating the drug from the body
These processes, often known as ADME, are responsible for the various concentrations of the drug in the blood stream at difference points in the development of the administration of medicine. Often, the effectiveness of a drug is dependent upon its concentration in the body. Other factors such as the site of administration and the dosage can affect the pharmacokinetic properties of the drug.
Alternatively, LADME may be used in place of ADME. The L adds to the ADME scheme the process of Liberation, or the release of the drug from its carrier (usually its protective coating or similar material).
How do scientists study pharmacokinetics?
[edit | edit source]Since researchers in pharmacokinetics field are following drug action in the body, perfect timing is a significant demand for the researchers. Scientists must determine when and where a drug should target inside bodies. However, it is not easy to keep track of the drugs. Even though scientists have known the strategy how medicines pass thought the body, the scientists cannot actually see where a drug is going. Therefore, the scientists use the tools of mathematics and chemistry in their researches.
Mathematical tools: Mathematics supplies models and precise methods to measure body fluids which help researchers determine their interest goal. Measurements of blood and urine lead to the answers such as where the drug is and how much of the drug broke down at a given time. Meanwhile, blood levels of liver enzymes can help predict how much of a drug is going to be absorbed.
Chemistry’s usage: The interaction between a drug and an organism is actually a series of chemical reaction between the drug molecule and molecules inside the organism. Hence, knowledge of how drugs react in biological environments is necessary to predict the amount of drugs a body requires or the maximum amount of drugs a body can withstand.
Advances Using Pharmacokinetics
[edit | edit source]By using the model of cocaine pharmacokinetics, scientists have seen promising effects of using enzyme therapy in treating drug abuse. Studying the cocainemetabolizing enzyme to see if it can keep the drug from entering the brain and causing producing the physiological effects can lead to advancements in production of an anti-cocaine medication. The model of this could be easily translated into therapy for other physiological effective drugs. [1]
Kinetics
[edit | edit source]In order to properly understand the pharmacokinetic process of ADME, the rates of these process must be examined. The rate, or velocity, at which each of these processes proceed follows either zero-order or first-order kinetics.
Zero-order kinetics
In a zero order reaction, the rate of reaction is independent of the concentration of the reactant(s); the rate is constant. The rate law for a zero order reaction is:
- r = k
If drug A, for example, is being excreted out of the body at a constant rate, according the zero order kinetics, the rate of excretion can be described as:
- dA/dt = k
First-order kinetics
In a first order reaction, the rate of reaction is dependent upon the concentration of only one reactant, even if more than one reactant is present. The rate law for a first order reaction is:
- -r = k[A]
where [A] is the concentration of reactant A. If the same drug A is being excreted out of the body through first order kinetics, the rate law would be written as:
- dA/dt = -k[A]
Most drugs proceed through first order kinetics, and the process of ADME in a biological system usually follows first order kinetics as well.
References
[edit | edit source]U.S. department of Health and Human Services - Medicines by Design
Zheng F, Zhan C-G (2012) Modeling of Pharmacokinetics of Cocaine in Human Reveals the Feasibility for Development of Enzyme Therapies for Drugs of Abuse. PLoS Comput Biol 8(7): e1002610. doi:10.1371/journal.pcbi.1002610
Levinthal, Charles, "Drugs, Behavior, and Modern Society", Pearson Education, Inc., 2008
PTP1B
[edit | edit source]PTP1B (protein-tyrosine phosphatase 1B) is a non-transmembrane enzyme that is found on the endoplasmic reticulum (ER). Its significance stems from being a negative regulator of insulin as well as leptin signaling. The PTP1B dephosphorylates, or in other words removes a phosphate group from the insulin receptor, IR, and also its primary substrates, which are called the Insulin Receptor Substrate proteins (IRS proteins). PTP1B in leptin, removes the phosphate from the tyrosine kinase, JAK2, which is called Janus kinase 2. More recently, it has been found to be a contributing factor in the onset of tumors, and has been linked more directly with breast cancer. It is also considered a potential drug target as its inhibition may lead to a stop in type 2 diabetes, obesity, and some forms of cancer.
Structure of PTP1B
[edit | edit source]PTP1B's structure composes of approximately 800 residues. The protein is made up of an N-terminal catalytic phophatase domain followed by a regulatory region and a membrane localization domain. This is attached to the endoplasmic retilum.
Inhibition of Insulin Receptors, and Correlations with Diabetes
[edit | edit source]When PTP1B was first discovered, it was found to inhibit both the insulin and leptin receptors. PTP1B does this by de-phosphorylating the insulin receptor (IR) and its primary substrates such as the IRS proteins. In turn, this results in patients developing diabetes. Ptp1b knockout mice demonstrated persuasive evidence that inhibiting this enzyme will allow people to stay lean and energetic independent of what or how much they eat. When the enzyme was disabled in the mice, they became hypersensitive to insulin and were lean despite being on a high-fat diet. The specific tissue location of where to disable the enzyme did not seem to matter much and all experiments pointed to the same result of fit and energetic mice with improved insulin sensitivity and increased glucose tolerance.
PTP1B Regulation
[edit | edit source]PTP1B is an enzyme that is known for being expressed with great abundance. It consists of an N-Terminal catalytic phosphatase region, being anywhere from 1 to 300 residues long, a regulatory region that ranges from 80 to 100 residues, and finally, a membrane localization domain which ranges from 400 to 435 residues long. The membrane localization domain ties, or bonds the PTP1B enzyme to the cytoplasmic face of the endoplasmic reticulum. Expression of PTP1B and its catalytic activity is usually strictly controlled by four mechanisms which can sometimes work together: oxidation, phosphorylation, sumoylation, and proteolysis.
Oxidation
[edit | edit source]PTP1B can be regulated in vivo by both reversible and irreversible oxidation. Cys 215, an amino acid at one of its active sites is positioned in an unusually acidic environment, which subsequently deprotonates at physiological pH. This converts the amino acid into an excellent nucleophile in catalysis. Once this conversion is made, it leaves Cys 215 susceptible to being inactivated by other highly reactive species containing oxygen. Depending on which oxygen-containing highly reactive species is used to modify PTP1b, the enzyme is oxidized to different oxidation states.
Through the use of crystallographic analysis, results have shown that if hydrogen peroxide is used, the sulphenic acid form of the PTP1B would be converted into the inactivated cyclic sulphenamide state through the process of oxidation. As this process takes place, a conformational change also occurs at the active site, which as a result exposes the hidden tyrosine amino acid, which is located at the phosphotyrosine binding loop. This process is predicted to be a reversible process. In contrast, the process would be irreversible if the enzyme is oxidized to the sulphinic or sulphonic state.
Phosphorylation
[edit | edit source]Although phosphorylation of both serine and tyrosine occur at multiple locations, the effects of phosphorylation remain controversial. Studies of phosphorylation at different serine residues on the enzyme produces contradicting results. For example, the phosphorylation of S378 and S352 by protein kinase C during metaphase or in response to external stimuli such as osmotic pressure does not significantly change the enzyme's activity level. However, when S50 is phosphorylated by AKT, the enzyme shows a decrease in its ability to dephosphorylate insulin receptors. Interestingly, when the same S50 residue is phosphorylated by CDC-like kinase 1 and 2, the enzyme shows a two-fold increase in its phosphatase acitivity. In a similar manner, studies of phosphorlyation at the tyrosine sites (Y66, Y152, Y153) on the enzyme have shown that these changes to PTP1b can either increase or decrease the protein's activity.
Sumoylation
[edit | edit source]Small ubiquitin-related modifier (SUMO) proteins have recently been found to be important regulators for many cell functions. SUMO conjugation significantly regulates many protein characteristics e.g. stability, localization, interactions and activity. PTP1B is found to interact with a SUMO E3 ligase which encourages modification of PTP1B by SUMO. Enzymatic activity is reduced and therefore PTP1B is less active with substrates. The specific location of where SUMO modification takes place is still unclear. However, it has been observed that PTP1B accumulates in punctuate structures, which are located in the perinuclear region and the C-terminal. It is important to point out that the presence of the ER targeting domain of PTP1B is necessary for the maximal sumoylation to occur.
Proteolysis
[edit | edit source]Calpain, a protease mediates the cleavage of the ER targeting part of PTP1B (C-terminal). This occurs in platelets that are activated and the result is an activated enzyme. Studies show that when calpain-1 in mice are disrupted, there is a lowered protein tyrosine phosphorylation level. When the platelets of these mice are analyzed, there was a large increase in the amount of PTP1B. This suggests that when the c-terminal is cleaved, PTP1B is cut into inactive pieces (fragments). Evidence that supports the proteolyzation, or the breakdown of protein PTP1B into inactive fragments is that the tyrosine phosphorylation defects, which are linked to the loss of calpain-1, were rescued in Capn1, along with the aggregation of the platelet.

It is important to note that other reports have shown that when PTP1B is reversibly oxidized, it would be inactivated by calpain-mediated cleavage in the catalytic domain. Thus, this led some to believe that whether the enzyme would be activated by cleavage of the C-terminus or inactivated by complete proteolysis (both processes mediated by the calpain protease) actually depends on PTP1B's oxidation state. However, no reports to date have reported the inactivation of the enzyme by calpain in any other cell types other than platelets.
Substrates of PTP1B
[edit | edit source]Since PTP1B is located on the cytoplasmic face of the ER, the mechanisms that allow this enzyme to encounter and dephosphorylate its many different substrates were put into question. So far, four possible mechanisms are proposed. First, if the substrate is a membrane-bound receptor protein tyrosine kinase such as the insulin receptor, through a vesicle-mediated endocytosis process, these activated receptors would be internalized and brought into contact with the PTP1B enzyme. Second, evidences from bioluminescence resonance energy transfer-based and fluoresence resonance energy transfer-based live images support that PTP1B might be able to dephosphorylate the insulin receptor during its biosynthesis. Third, the part of the ER that PTP1B binds to is stretchable. This makes interaction between PTP1B and its substrates on the plasma membrane possible. Finally, adaptor proteins link PTP1B to its substrate, facilitating their interaction by forming a ternary complex. In addition to these mechanisms, the enzyme has binding motifs that facilitate its interaction with substrates such as the IR. Studies have shown that one of these motifs, the Y152 located in the β9-β10 turn of the catalytic domain, allows PTP1B to interact with the back side of the IR dimers.
Diabetes & Obesity
[edit | edit source]Ptp1b knockout mice demonstrated persuasive evidence that inhibiting this enzyme will allow people to stay lean and energetic independent of what or how much they eat. When the enzyme was disabled in the mice, they became hypersensitive to insulin and were lean despite being on a high-fat diet. The specific tissue location of where to disable the enzyme did not seem to matter much and all experiments pointed to the same result of fit and energetic mice with improved insulin sensitivity and increased glucose tolerance.
Cancer
[edit | edit source]So far, PTP1b has not been found to be a tumor suppressor but evidence suggests that it may be a negative regulator of cell growth. The enzyme can encourage cell death (apoptosis) and therefore potentially be a tumor suppressor. The inhibition of this enzyme may seem to solve many health problems very simply and some may wonder why action had not been taken sooner but as always, biology is not that simple. The total effects of the inhibition of PTP1B is still not known and some studies already suggest that inhibiting this enzyme will promote some forms of cancer. Some human cancers e.g. cancer of the breast and ovary show elevated levels of PTP1B. For breast cancer, activation of ErbB2 leads to higher levels of PTP1B expression. Studies using transgenic mice show that PTP1B is a positive regulator of the ErbB2 induced mammary tumorigenesis. The mice without PTP1B had a large delay of the onset of ErbB2 whereas the mice with an over-expression of PTP1B developed breast tumors. Using a PTP1B inhibitor in a mouse cancer model, Julien et al. 's results shown that this inhibitor effectively protected mice from the ErbB2 oncogene. However, even in PTP1B deficient models, breast tumors caused by the polyoma middle T antigen can still develop. Thus, PTP1B is proposed as an enzyme that plays a selective but contributing role in oncogenic signaling.
References
[edit | edit source]1. Yip, Shu-Chin, Sayanti Saha, and Jonathan Chernoff. "PTP1B: A Double Agent in Metabolism and Oncogenesis." Trends in Biochemical Sciences 35.8 (2010): 442-49. Print.
2. "PTP1B Research for Dummies." PTP1B Research for Dummies and Why You Need To Know It | GoGetThinâ¢. N.p., n.d. Web. 20 Nov. 2012. <http://blog.gogetthin.com/leptin-and-weight-loss/ptp1b-smart-weight-loss>.
3."PTP1B." Wikipedia. Wikimedia Foundation, 17 June 2012. Web. 20 Nov. 2012. <http://en.wikipedia.org/wiki/PTP1B>.
4.Wikipedia contributors. "Calpain." Wikipedia, The Free Encyclopedia. Wikipedia, The Free Encyclopedia, 20 Aug. 2012. Web. 22 Nov. 2012.
Overview
[edit | edit source]Amphetamine is an appetite suppressant, which is insoluble in water. After taking amphetamine, the nerves and the brain are stimulated, thus leading to faster heart beats, higher blood pressure, and ultimately resulting in less appetite. This drug is famous for treating narcolepsy and attention deficit hyperactivity disorder (ADHD). Narcolepsy is a sleep disorder, defined by sleeping excessively. ADHD is characterized by inattention, over-activity, or a mixture of both.

Who cannot take amphetamine?
[edit | edit source]Pregnant women and mother who provides breast feeding are forbidden to take amphetamine because there might be unexpected drug effects that pass from the mother to the baby, unless they consult with a doctor before hands. People who have the following characteristics should not take amphetamine:
- Heart problem
- High blood pressure
- Arteriosclerosis (arteries hardening caused by accumulating fats in the arteries)
- Hyperthyroidism (excess of thyroid hormone in the thyroid gland)
- Glaucoma (damage of nerves in the eye)
Underdose and Overdose
[edit | edit source]In the case of underdose, take amphetamine at the next scheduled time if it is evening time because the amphetamine might lead to insomnia if the dose is taken at late night. In the case of overdose, one should immediately seek help from the doctor. The effects of overdose are severe; they include hallucination, nausea, diarrhea, seizure, and many others.
Side Effects
[edit | edit source]Common side effects of amphetamine are listed below; however, one should notify the doctor about these side effects. If one has severe side effects, then stop taking amphetamine. If one has light side effects, then continue taking amphetamine.
- Severe side effects:
- Allergy (especially in the throat, on lips, face, tongue)
- Abnormal heartbeat
- High blood pressure
- Hallucination
- Light side effects:
- Anxiety
- Slight headache
- Insomnia
- Diarrhea/constipation
- Impotence
Limitations of amphetamine
[edit | edit source]Amphetamine might not function properly if one is taking it along with some other drugs or special adjustment to the dosage might be needed if there is another drug that one is taking. Below are some of the drugs that might affect amphetamine:
- Insulin
- Antihypertensive drug
- Drug to treat enlarged prostate
- Antidepressant (especially if it is tricyclic structure)
- Antihistaminic drugs
- Drugs to treat psychotic disorders
Reference
[edit | edit source]http://www.drugs.com/amphetamine.html http://www.nlm.nih.gov
Introduction
[edit | edit source]Juvenile hormone is a compound that is used to control the development of insects. Hence, it helps to prevent insect-borne diseases such as malaria, yellow fever, and West Nile virus.
Uses and Properties
[edit | edit source]Juvenile hormone or JH is produced by the male silk moth called Hyalophora cecropia L. Once insects are infected with JH, it will create metamorphosis in insects which prevents insects from producing eggs. Hence, JH can help to control the diseases. Scientists then have created synthetic and isolated natural JH. Substances of JH are then produced in the labs and it allows the production of a more stable and bioactive substances. Other synthetic compound such as methoprene also help to prevent insect-borne diseases. It is so much more bioactive than JH that it can attack mosquitoes, fleas, and ants. Methoprene does not kill the insects themselves, but the substance kills the insect's eggs before they reach their adulthood. Therefore, methoprene and JH are used to prevent insects from developing and spreading their diseases.
Reference
[edit | edit source]Vollhardt, Peter. Schore, Neil. Organic Chemistry 6th Edition. W.H Freeman and Company. New York. 2011
Phenylketonuria
[edit | edit source]Dire consequences result if one or more of amino acids are either absent or overabundant. A genetic disorder called phylketonuria (PKU) is caused by the body’s inability to get rid of extra phenylalanine. PKU is autosomal recessive disorder, meaning that the only way to get the disease is if both of your parents carry a version of a gene linked with this disease. People with PKU are born without the enzyme that breaks down the Phenylalanine amino acid. Extremely high levels of Phe accumulate and are very toxic, especially to the brain as a result, PKU causes mental retardation. Yet Phenylalanine is an essential amino acid—your body cannot do without it. Both diet and genes contribute to causing PKU and so any means to control the supply of Phenylalanine in the body can prevent the disease.
Causes
[edit | edit source]Phenylketonuria (PKU) is inherited, which means it is passed down through families. Both parents must pass on the defective gene in order for a baby to have the condition. This is called an autosomal recessive trait. Babies with PKU are missing an enzyme called phenylalanine hydroxylase, which is needed to break down an essential amino acid called phenylalanine. The substance is found in foods that contain protein. Without the enzyme, levels of phenylalanine and two closely-related substances build up in the body. These substances are harmful to the central nervous system and cause brain damage.
Symptoms
[edit | edit source]Phenylalanine plays a role in the body's production of melanin, the pigment responsible for skin and hair color. Therefore, infants with the condition often have lighter skin, hair, and eyes than brothers or sisters without the disease.
Other symptoms may include:
- Delayed mental and social skills
- Head size significantly below normal
- Hyperactivity
- Jerking movements of the arms or legs
- Mental retardation
- Seizures
- Skin rashes
- Tremors
- Unusual positioning of hands
- If the condition is left untreated or foods containing phenylalanine are not avoided, a "mousy" or "musty" odor may be detected on the breath and skin and in urine. The unusual odor is due to a build up of phenylalanine substances in the body.
Diagnosis
[edit | edit source]PKU can be easily detected with a simple blood test. All states in the US require a PKU screening test for all newborns as part of the newborn screening panel. The test is generally done by taking a few drops of blood from the baby before the baby leaves the hospital.
If the initial screening test is positive, further blood and urine tests are required to confirm the diagnosis.
Treatment
[edit | edit source]However, the silver lining is that the PKU disorder is easy to diagnose and if present, PKU is a treatable disease. Doctors treat children with PKU by prescribing a life-long restrictive diet. Treatment involves a diet that is extremely low in phenylalanine, particularly when the child is growing. The diet must be strictly followed. This requires close supervision by a registered dietitian or doctor, and cooperation of the parent and child. Those who continue the diet into adulthood have better physical and mental health. “Diet for life” has become the standard recommended by most experts. This is especially important before conception and throughout pregnancy.
Phenylalanine occurs in significant amounts in certain foods such as milk, eggs, and diet sodas containing the artificial sweetener aspartame (NutraSweet) are rich sources of Phenylalanine. The diet is rigid requiring people to avoid those and many other foods such as meat and fish, dairy products, bread, nuts, and even some vegetables. As a result, people with PKU have to take a special Phe-free vitamin/mineral supplement to ensure that they receive adequate amounts of all of the other essential amino acids bountiful in those foods. For example, a special infant formula called Lofenalac is made for infants with PKU. It can be used throughout life as a protein source that is extremely low in phenylalanine and balanced for the remaining essential amino acids. Another example involves taking supplements such as fish oil to replace the long chain fatty acids missing from a standard phenylalanine-free diet in order to help improve neurologic development, including fine motor coordination. Other specific supplements, such as iron or carnitine, may be needed.
Prognosis
[edit | edit source]The outcome is expected to be very good if the diet is closely followed, starting shortly after the child's birth. If treatment is delayed or the condition remains untreated, brain damage will occur. School functioning may be mildly impaired. If proteins containing phenylalanine are not avoided, PKU can lead to mental retardation by the end of the first year of life. Severe mental retardation occurs if the disorder is untreated. ADHD (attention-deficit hyperactivity disorder) appears to be the most common problem seen in those who do not stick to a very low-phenylalanine diet.
Prevention
[edit | edit source]An enzyme assay can determine if parents carry the gene for PKU. Chorionic villus sampling can be done during pregnancy to screen the unborn baby for PKU.
It is very important that women with PKU closely follow a strict low-phenylalanine diet both before becoming pregnant and throughout the pregnancy, since build-up of this substance will damage the developing baby even if the child has not inherited the defective gene.
References
[edit | edit source]- ↑ Zheng F, Zhan C-G (2012) Modeling of Pharmacokinetics of Cocaine in Human Reveals the Feasibility for Development of Enzyme Therapies for Drugs of Abuse. PLoS Comput Biol 8(7): e1002610. doi:10.1371/journal.pcbi.1002610
- ↑ U.S. Department of Health and Human Services. Chemistry of Health. October 2006.<http://www.nigms.nih.gov>.
- ↑ Phenylketonuria. ". http://www.nlm.nih.gov/medlineplus/ency/article/001166.htm>.
- ↑ Phenylketonuria. ". http://www.nlm.nih.gov/medlineplus/ency/article/001166.htm>.
- ↑ Phenylketonuria. ". http://www.nlm.nih.gov/medlineplus/ency/article/001166.htm>.
- ↑ U.S. Department of Health and Human Services. Chemistry of Health. <http://www.nigms.nih.gov>.
- ↑ Phenylketonuria. ". http://www.nlm.nih.gov/medlineplus/ency/article/001166.htm>.
- ↑ Phenylketonuria. ". http://www.nlm.nih.gov/medlineplus/ency/article/001166.htm>.
- ↑ Phenylketonuria. ". http://www.nlm.nih.gov/medlineplus/ency/article/001166.htm>.
- U.S. Department of Health and Human Services. The New Genetics. October 2006.<http://www.nigms.nih.gov>.
- "Phenylketonuria. ". National Institute of Health. <http://www.nlm.nih.gov/medlineplus/ency/article/001166.htm>.
Antibiotic versus bacteria
[edit | edit source]In 1928, scientist Sir Alexander Fleming discovered an antibiotic, Penicillium, that is used to destroy bacteria colonies. The structure of Penicillium is C6H5CH2 and contains the β-lactam ring that activates the antibiotic attack against the bacteria. However, some bacteria cannot be destroyed by the antibiotic because they have an enzyme, penicillinase, that can resist the β-lactam ring. Another antibiotic called Tigecycline can overcome the bacteria's enzyme to allow the attack against the bacteria. Hence this antibiotic can resist infections on the skin and internal organs. Hence, new antibiotics are created every year to help eliminating infections.
Reference
[edit | edit source]Vollhardt, Peter. Schore, Neil. Organic Chemistry 6th Edition. W.H. Freeman and Company. New York. 2011
Food and Drug interactions
[edit | edit source]

Furosemide is loop diuretic. Patients who take this medication may experience increased frequency of uriantion and urine volume. As a result, patients may have constipation and elevated serum sodium levels; as well as excrete excess potassium, calcium, phosphorus and magnesium.
Diet for patients taking Furosemide
[edit | edit source]Patients who take this medication should decrease sodium intake to avoid fluid retention and may need to increase intake of potassium, phosphorus, calcium and magnesium.
1. Diet to increase potassium intake:
For grains, recommended foods are bran cereal or muffins, and granola.
For vegetables, recommended foods are artichokes, broccoli, brussels sprouts, greens, kale, kohlrabi, parsnips, rutabagas, mushrooms, white and sweet potato, spinach, chard, winter squash, fresh or canned tomatoes, tomato or vegetable juice, zucchini.
For fruit, recommended foods are Apricots, avocados, bananas, cantaloupe, dates, figs, grapefruit juice, kiwi, orange juice, melons, mangos, nectarines, papaya, pear, pomegranate, prunes and prune juice, raisins.
For milk, recommended foods are nonfat, low fat, whole, chocolate and buttermilk, plain or fruited yogurt, soy milk.
For meat and beans, recommended foods are canned, dried, fresh or frozen beans and lentils, roast or ground beef, chicken, clams, crab and fresh, frozen or canned fish, nuts and nut butters, pork, and turkey, soy.
Others, recommended foods are chocolate, molasses, potato chips, wheat germ.
For the foods mentioned above do not have foods to avoid for increasing potassium.
2. Diet to increase calcium intake:
For grains, recommended foods are all foods.
For vegetables, recommended foods are all foods, especially spinach and okra.
For fruit, recommended foods are all foods.
For milk, recommended foods are milk, yogurt, cheese.
For meat and beans, recommended foods are all foods, especially soy beans.
For the foods mentioned above do not have foods to avoid for increasing calcium.
3. Diet to increase phosphorus intake:
For grains, recommended foods are all foods, especially oatmeal and wheat germ.
For vegetables, recommended foods are all foods, especially baked potato with skin.
For fruit, recommended foods are all foods.
For milk, recommended foods are all foods, especially cheese, skimmed, low fat or regular milk.
For meat and beans, recommended foods are all, especially meat, dried beans, nuts, soy and tofu.
For the foods mentioned above do not have foods to avoid for increasing phosphorus.
4. Diet to increase magnesium intake:
For grains, recommended foods are bran cereal, bran muffins, oatmeal, brown rice, whole wheat pasta and spaghetti.
For vegetables, recommended foods are artichoke, avocado, greens, okra, baked potato with skin, spinach, swiss chard.
For fruit, recommended foods are unknown.
For milk, recommended foods are eggnog, chocolate milk, soy milk.
For meat and beans, recommended foods are dried beans (black, white, kidney, lima and pole beans) and peas, fish(halibut, yellowfin), soy beans.
For others, recommended foods are nuts (almonds, cashews, walnuts, hazelnuts, chestnuts, mixed, peanuts) and peanut butter, pumpkin or squash seeds, tofu, wheat germ.
For the foods mentioned above do not have foods to avoid for increasing magnesium.
Reference
[edit | edit source]"Nutrition care manual" <http://nutritioncaremanual.org/content.cfm?ncm_content_id=93007>
Introduction
[edit | edit source]
Vitamin C known as the anti-scurvy vitamin was discovered by scientists. Some even called it the hexuronic acid or ascorbic acid. This vitamin can be used to treat scurvy. For those who don't know, scurvy is a disease caused by the lack of vitamin C; as a result, it can cause opening of past healed wounds and cause the gums to be swollen or even bleed. Scurvy can also cause vision problems, lassitude, haemorrhages, bone fragility and neurological problems.
Scurvy
[edit | edit source]Doctors recommend us to digest more vitamin C for our body through the intake of fruits and vegetables. But sometimes that might not be enough of vitamin C to compensate the amount that we lack in the body. Chemically, scurvy happens because of the inactivation of some important dioxygenases. These enzymes are dependent on the ascorbic acid and are part of the class of 2-oxoglutarate dependent dioxygenases (2-ODDs). The mechanism of this enzyme requires Fe2+ , 2-oxoglutarate ,and ASC as co-substrates to get the process going. Overall, 2-ODDS would convert O2 into organic substrate.
The ASC mechanism
[edit | edit source]Each 2-ODDS will have its own responsibility in the mechanism. As a result, some 2-ODDS would catalyze the hydroxylation, while some would run the desaturation and ring closure or expansion. Scurvy, a disease, that can be treated using this ascorbic acid.And scientists usually call this process as the ASC administration. Its role is to keep the Fe2+ constant. The mechanism of the peptidyl-prolyl- 4-hydroxylase (P4H)uses the enzyme for the pre-translational hydroxylation at the 4th Carbon of the proline residues that was inside the polypeptide chain. So this enzyme would split the oxygen into organic substrates such that they can be used in the decarboxylation of 2-oxoglutarate and oxidation of proline.The ASC is used to become another acceptor of the Fe2+ ion mechanism, so it can drive the mechanism without the need of hydroxylation. Because the mechanism of the ferry ion is complicated and very reaction, the ASC and the 2-ODDS might have to undergo the molecular co-evolution in order to carry out the reaction. When there's a lack of ASC in the body, it would result in the inactivation of the P4H and lead to the scurvy disease. Hence, the collagen residues would not be hydroxylated and the collagen trimers would not form. Specifically, it's required to carry out the hydroxylation of the proline to make sure that the collagen would fold. As a result, the folding of collagen would form into the triple helices and then into fibrils that can help with thermal stability. These collagen folding help to maintain the skin, tendons, cartilage, bones, teeth, cornea, muscles, and blood vessels. So it's very important that this process occur. Scientists have discovered the relationship between the lack of ASC to the non-folding problem of collage. For instance, they realized that guinea pigs with free ASC diet have lower amount of type IV collagen and that results in the lower amount of hydroxylated proline. As a result, the guinea pigs experience scurvy which leads to the defects in blood vessels. Since collage consumes a large amount of ASC and lack of ASC can lead to scurvy, we can conclude that the non-folding of collagen plays important role in scurvy.
Another role of Vitamin C
[edit | edit source]Another group of dioxygenases that depend on ASC can drive the repair of methylated bases in DNA sequences. And other ASC dependent plant-dioxygenases can help to synthesize hormones of the signaling molecules such as gibberellins and ethylene.
HIF Hydroxylation Signaling
[edit | edit source]ASC plays an important role in HIF hydroxylation signaling. HIF1 is known as the one of the HIF members such that it can activate hundreds of genes that are related to nutrient transport, cell migration, angiogenesis, and energy metabolism. HIF 1 has 2 subunits: α and β subunits. Scientists have discovered that the mechanism that is catalyzed by dioxygenases need to include oxygen. And what drives the mechanism with oxygen is the hydroxylation of 2 proline residues. There are two proline residues in humans. They are Pro402 and Pro564 which are parts of the HIF-α. They are hydroxylated by 3 different hydroxylases (HIF-P4H) and they carry the same processes as the collagen hydroxylases. However, there are differences between the collagen hydroxylases and HIF-P4H. HIF-P4H can be found in the cytosol, while collagen hydroxylases are found in the ER. In HIF-P4H, the affinity of the enzyme for O2substrate or the Km value for O2 is above the atmosphere concentration which means that they can identify oxygen in the mechanism. When oxygen is available, we call that the normoxic conditions. In that condition, the 2 proline residues would be hydroxylated. Then it results in the binding of the multiprotein complex in order to attack the protein pVHL and the degration of HIFα. The condition of the mechanism with lower oxygen is called the hypoxia, while the condition with no oxygen availability is called the anoxia. And when these conditions occur, hydroxylation cannot happen. As a result, HIFα would not be attached to the protein pVHL and result in the binding of the HIFα and the HIFβ in the nucleus. The presence of oxygen can be used to activate the hydroxylation in the mechanism, and its presence can make ASC's presence to be less important in the reaction. Scientists have discovered that in the normoxic and hypoxic conditions, ASC plays an important role. ASC helps to lower the amount of HIF1 protein while icreasing the rate of degration of HIFα. When one adds ASC to the reaction, it may help to increase HIF hydroxylation in deficient cells. Another study shows that lowering the ASC concentration can result in lowering the hydroxylation rate of HIFα and that can result in the lowering the rate of degration of such protein. Overall, ASC play multiple roles such as it would activate the enzyme by decreasing the amount of Fe3+ and it can bind to the enzyme while acting as a substrate
Cancer treatment
[edit | edit source]Scientists have done different experiments on ASC's role in treating cancer. They discovered that increasing the concentration of ASC can cause the HIF down-regulation. Since HIF1α is expressed in the treatment of cancer, there has been experiments show the lack of ASC is expressed in cancer patients. Scientists have been working on how the function of ASC can help with the immune cells in the patients' body.\
Gene expression
[edit | edit source]ASC has played multiple roles in expressing genes. Studies show that vitamin C can help with the gene transcription process such that it can stabilize the mRNA. For example, the transcription of the tyrosin hydroxylase was catalyzed by vitamin C. In addition, ASC can differentiate the members of the mesenchymal cell by making more collagen. And the members of the mesenchymal cell are chondrocytes, cardiomyocytes, and osteoblasts. In osteoblasts, osteocalcin, a binding protein that is made from osteoblasts, can be expressed by ASC. The process occurs when there is an increase of ASC that results in the transcription of osteocalcin genes. Also, there was a mouse model used in experiment of Charcot Marie Tooth syndrome. ASC helps to cause the myelination in the mouse such that it finds a new pathway to treat the syndrome.
Reference
[edit | edit source]Tullio, Mario C. De. Beyond the antioxidant: The double life of Vitamin C.12/6/12
Lantipeptides
[edit | edit source]Introduction
[edit | edit source]Lantipeptides were previously known as lantibiotics because of the antimicrobial characteristics they exhibited, but the term has since then been changed to lantipeptides with the discovery of lantibiotics that did not perform antimicrobial tasks. With this discovery, the family that they belong to was extended to over 90 compounds.
Lantipeptides are ribosomally synthesized peptides with thioether cross-links which is formed by dehydration of Serine/Threonine residues and subsequent addition of cysteine residues to the resulting dehydro amino acid meso-lanthionine (Lan) and (2S,3S,6R)-3-methyllanthionine (MeLan. Meso-lanthionine and (2S,3S,6R)-3-methyllanthionine are cross-links which come from posttranslational modification of a precursor peptide.
History
[edit | edit source]Lantipeptides were previously known as lantibiotics because of the antimicrobial characteristics they exhibited, but the term has since then been changed to lantipeptides with the discovery of lantibiotics that did not perform antimicrobial tasks. With this discovery, the family that they belong to was extended to over 90 compounds.
Lantibiotics have historically been used as antimicrobial agents. One latipeptide (nisin) has been used in the food industry for over 50 years as a preservative. The name lantibiotics was first introduced as an abriation of the term “lanthionine-containing peptide antibiotics” [1]. Work done by Erhard Gross and John L. Morell in the 1960-70’s led to the field of lantibiotics that is around today.
Nisin
[edit | edit source]
One well known lantibiotic that has been studied extensively is Nisin. Nisin is composed of 34 amino acids and has been used widely in products such as cheese, meats, and other everyday items as a deterrent against bacteria that can spoil or otherwise infect food.
Recently, studies have been done with nisin that show it may aid in fighting cancer. In a study performed at the University of Michigan, it was found that nisin creates pores in the cell membranes of cancer cells. These pores allow calcium to flow into the cancerous cell, which can ultimately lead to its death. It is unclear how specifically calcium influx can lead to death, but a protein called CHAC1 which is activated by nisin is involved. It was also found that nisin may interrupt the cell cycle of cancer cells, while at the same time leaving regular cells unaffected [2].
Antimicrobial Characteristics
[edit | edit source]
One of the most apparent characteristics of lantibiotics is their high activity against bacteria. They are effective against gram positive bacteria such as of Staphylococcus, Streptococcus, Enterococcus, and Clostridium and gram-negative bacteria such as Neisseria. Because of these properties, they have been the subject of much scientific research and have had applications in industries such as food preservation. They have been found to treat various infections, such as Clostridium difficile, and others [3]. Lantipeptides may also have application in other industries, such as agriculture, verterinary medicine, and molecular imaging.
The mechanism through which lantipeptides gain their antimicrobial properties is often through the inhibition of bacterial cell wall synthesis. They are also known to create pores in membranes, which can greatly ruin the integrity of the cell. Specifically, the mechanism through which lantipeptides have been known to act is through the inhibition of transglycosylation. They do this by binding to lipid II, which is an integral part of building the cell membrane [3].
Mechanism of Nisin
[edit | edit source]The mechanism through which nisin is able to perform well as an antimicrobial have been well documented, which is one of the reasons it is used extensively today. It binds to lipid II through contacts the A and B rings of pyrophosphate moiety. It then forms pores in the membrane by inserting itself, forming groups of four lipid II molecules and eight nisin peptides[4].
Classes of Lantipeptides
[edit | edit source]Lantipeptides can be classified into four distinct classes of biosynthetic enzymes that recognize Lan and MeLan: dehydratase and cyclase enzymes (Class I), bifunctional lanthionine synthetases (Class II), trifunctional synthetases and carbocyclic rings (Class III), trifunctional lanthionine synthetases (Class IV).
In Class I
[edit | edit source]Class 1 lantipeptides are known as dedicated dehydratase and cyclase enzymes, and for good reason. There are two different enzymes that carry out these processes: dehydratase LanB and cyclase LanC. The LanB genes supply proteins of about 1000 residues that are not homologous to any known enzymes. The LanC genes encode proteins of about 400 residues and present a low sequence identity. Among recently discovered and isolated lantipeptides are a modified peptide from actinomycete of Microbispora corallina, which has high activity against gram-positive bacteria, and planosporicin, which is derived from actinomycete Planomonospora. The NMR structures of these two have shown similarities in conformation.[3]
Class II
[edit | edit source]Class II lantipeptides are known as bifunctional lanthionine synthetases. They are known to perform dehydration and cyclization reactions. LanM is the bifunctional synthetase that performs these processes. The proteins is produces range from 900-1,2000 residues in length and consist of two domains, which are an N-terminal dehydratase domain that bears no enzyme that is homologous to LanB, and a C-terminal cyclase domain, which a one-fourth sequence identity to LanC, including conservation of the zinc-binding residues essential for NISC catalysis. There have been many new additions to this class of lantipeptide recently. One of these is haloduracin, which is from Bacillus halodurans. This is the first lantipeptide that is from a species that is alkaliphilic, which means it can survive high pH (or alkaline) environments.[3]
Class III
[edit | edit source]Class III lantipeptides are known as trifunctional synthetases and carbocyclic rings. Biosynthesis are present in the morphogenetic peptide SapB from Streptomyces coelicolor. There is a big difference between class II and class III lantipeptides, which lead to the separate designation. The difference lies in the fact that this peptide does not demonstrate antibiotic activity. Instead, it promotes the growth of vegetative hyphae that is associated with streptomycete sporulation. A putative modifying enzyme known as RamC is contained in the gene cluster. This enzyme resembles the serine/threonine protein kinases and is structured with a C-terminal domain with homologous features that are similar to cyclase domain of LanM. However, the zinc-binding is absent [3].
In 2010, labyrinopeptins were discovered from actinomycete Actinomadura namibiensis. The trifunctional lantipeptide known as LabKC was found to have homology with the previously known RamC. Labyrinthopeptin, which is one of the modified products of LabKC, was found to help against neuropathic pain in mice. This is a function of lantipeptides previously unobserved [3].
Class IV
[edit | edit source]Class IV lantipeptides biosynthesis involves a cryptic gene cluster in Streptomyces venezuelae. The synthestase, VenL, is made up of an N-terminal OspF-like lyase domain with a serine/threonine kinase domain center. Unlike the RamC, the C-terminal cyclase domain of VenL includes the zinc-binding motif that is present in LanC and LanM [3].
Bioengineering of Lantipeptides
[edit | edit source]Knowing lantipeptides can help produce large chemical diversity with low genetic cost in a favorable and adaptable strategy. Lantipeptide biosynthesis has been developing by discovering new classes of biosynthetic machinery, new posttranslational modifications, and more lantipeptide-encoding gene clusters. Even though some catalysis mechanism is remain unknown like LanB, however, by discovering methodology of the production in E. coli, helped the investigations of LanB and furthermore, introduction of nonproteinogenic amino acid into lantipeptides by stop-codon suppression technology.
In Vivo Engineering
[edit | edit source]One way that lantipeptide engineering is performed is in Escherichia coli. This was first performed in 2005, when a truncated nukacin ISK-1 was produced. This was done through the coexpression of nukA and nukM on one vector. In 2011, a modified LanAs was produced. This was done through the coexpression of lanA and lanM. Coexpression of nisA and nisB on a single vector, with nisC on another vector leads to the production of a nisin precursor peptide. This is the only class I lantipeptide produced in E. coli that has been documented [3].
In Vitro Engineering
[edit | edit source]The in vitro approach to engineering comes with its own set of problems, mostly due to problems with immunity or export. This approach utilizes synthetic substrates. One drawback to this synthetic method is in proteolysis, which often results in low yields. For this in vitro method to improve, optimization of this step is required [3].
References
[edit | edit source]- ↑ Heike, Brotz and Hans-Georg Sahl "New insights into the mechanism of action of lantibiotics—diverse biological effects by binding to the same molecular target.", [Oxford Journals], 2012. Retrieved on 7 December 2012.
- ↑ "Common food preservative may slow, even stop tumor growth.", MedicalPress, 2012. Retrieved on 7 December 2012.
- ↑ a b c d e f g h i "Discovery, biosynthesis, and engineering of lantipeptides". 2012.
{{cite web}}: Unknown parameter|retrieved=ignored (|access-date=suggested) (help) - ↑ Shang-Te D Hsu et al "The nisin−lipid II complex reveals a pyrophosphate cage that provides a blueprint for novel antibiotics.", [Nature.com], 2004. Retrieved on 7 December 2012.
ADME is the acronym commonly used in pharmacology and pharmacokinetics used to reference the four basic stages of a drug/medicine's life inside the human's body: Absorption, Distribution, Metabolism, and Excretion.
Absorption
[edit | edit source]Absorption is the transfer of a drug into the blood after it is released from its dosage formulation. The body can absorb drugs in many ways, such as oral (swallowing an tylenol tablet), intramuscular (getting a flu shot in an arm muscle), subcutaneous (injecting insulin just under the skin), intravenous (receiving chemotherapy through a vein), or transdermal (wearing a skin patch). The most common way is through oral administration. Once the drug has been absorbed it gets shuttled by a special blood vessel where it enters from the digestive tract to the liver, and this is where a large amount of the drug may be destroyed by metabolic enzymes (often called "first-pass effect." One of the most important factors affecting oral drug absorption is the gastric emptying time. The gastric emptying time is the time a drug will stay in the stomach before it is emptied into the small intestine. This time affects a drug's action because most drugs are absorbed in the intestine. Stomach acid can degrade many drugs before they reach the absorption stage. Once a drug leaves the stomach without being destroyed, its rate of movement through the intestines affects its current absorption. If the drug moves through the intestines slowly, more of the drug will be absorbed since it is in contact with the intestinal membrane for a long time. On the other hand, fast movement through the intestines might cause a drug to not be fully absorbed. In addition, bile salts and enzymes from the intestinal tract also affect the absorption of a drug. Bile salts improve the absorption of some hydrophobic drugs. However, enzymes decrease the absorption of drugs by destroying the drugs as they pass through.
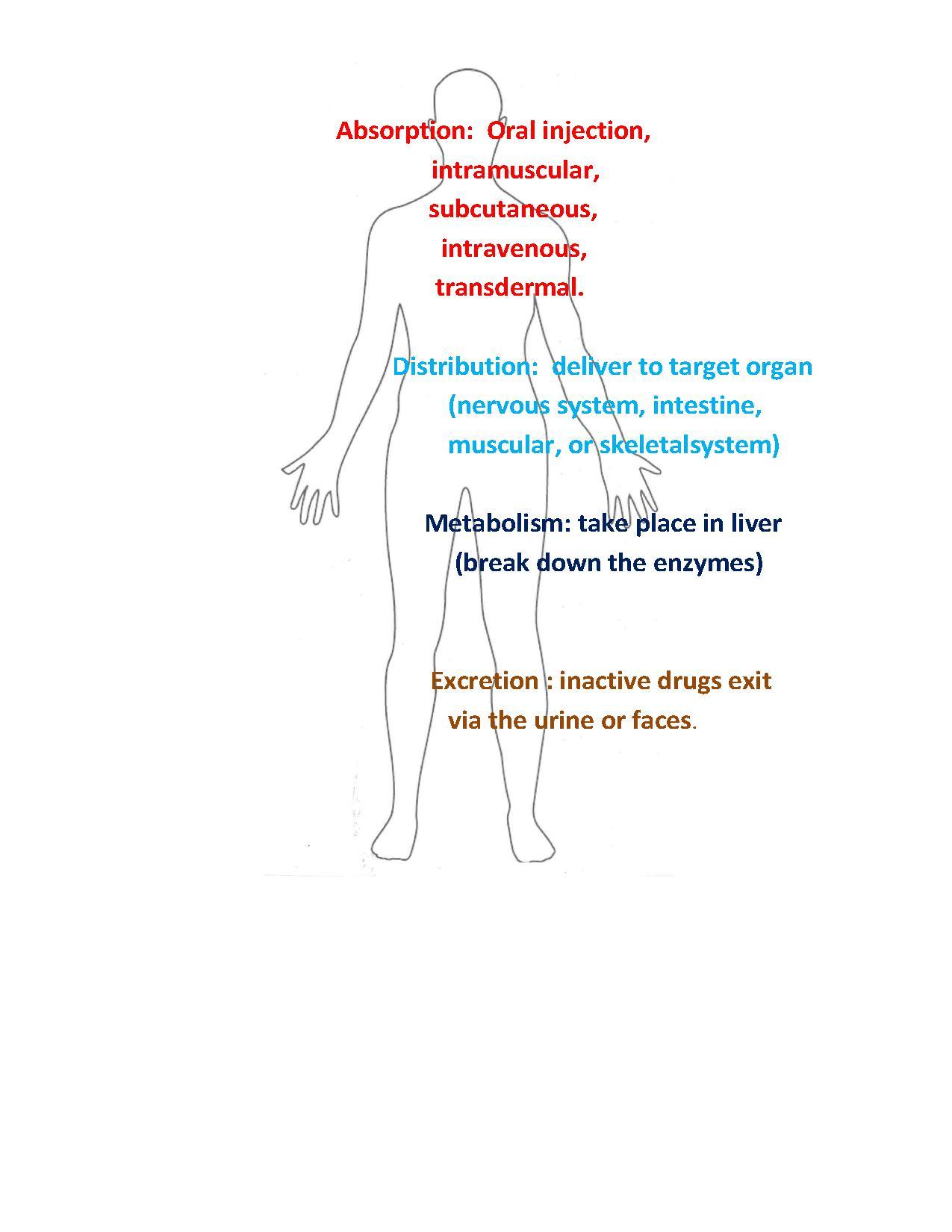
Distribution
[edit | edit source]Distribution is the movement of a drug inside the body once the drug has reached the blood. Blood carries the drug throughout the body and also to its sites of action. First, distribution is affected by the blood flow rates to certain organs. When an organ has a high blood flow rate such as in the heart, liver, and kidneys, the drug is distributed quickly. However, organs with slow blood flow rates like the muscle, fat, and skin, cause distribution to be much slower. Moreover, the permeability of tissue membranes to a drug is also extremely important. Small drug molecules and hydrophobic drugs will diffuse through tissue membranes easily. Also, there are some tissue membranes that have specialized transport mechanisms that assist penetration. However, there are some tissue membranes that are highly selective in allowing drugs to penetrate through. For example, the blood-brain barrier limits drug access to the brain. Lastly, protein binding can affect distribution as well. Most drugs bind to proteins in the blood plasma, forming what is called a complex. Because these complexes are large, it prevents the drug from entering its sites. So, only free or unbound drugs can move through the tissue membranes. Furthermore, some drugs with a stronger binding capacity can displace a weaker bound drug from a protein, making the weakly bound drug not bound anymore.
Metabolism
[edit | edit source]Drug metabolism refers to the body's way of processing drugs. This drug that is being transformed inside the body is called a metabolite. Most metabolites are inactive molecules that are excreted but some are active and produce effects in a human until they are further metabolized or excreted. The liver is the primary site of drug metabolism. The enzymes found inside the liver interact with drugs and change them into metabolites. CYP3A4 is a drug-metabolizing enzyme in the intestines that increases or alters blood levels of certain medications in people. CYP450 is a cytochrome enzyme that processes essential molecules such as hormones and vitamins a well as metabolize hundreds of prescribed medicines and natural substances. Some drugs allow the liver to increase its enzyme activity, allowing the drug to result in greater metabolism. So, some doses of drugs must be larger to produce the same therapeutic effects. However, some drugs decrease enzyme activity. In this situation, smaller doses of the drug are needed to avoid toxicity. Also, the liver may secrete drugs into the bile that is stored in the gallbladder. Any drugs or metabolites in the bile may be reabsorbed or eliminated within the feces. If the drugs are reabsorbed back into the blood, it is called enterohepatic cycling.
Excretion
[edit | edit source]Most drugs and their metabolites are excreted by the kidneys and come out of the body through urine. Some drugs are not easily absorbed from the gastrointestinal tract and so will be excreted in the feces rather than the urine. Excretion can also happen through the bile and some drugs are removed through the lungs in the expired breath. But, most drugs are excreted through the kidneys, which filters the blood and removes waste materials from the blood. Some of the plasma water is filtered from the blood into the nephron tubule in a process called glomerular filtration. This filtered water may have waster drugs from other parts of the body. As the water continues to move across the body, other waste drugs can be secreted into the fluid. While this is occurring, some drugs can be reabsorbed back into the blood through urinary reabsorption. After all of this happens, the final fluids are excreted from the body as urine. The rate of urinary excretion is much faster than that of fecal excretion. Drugs excreted through the urine take a couple of hours while drugs excreted through the feces take a couple of days.
Reference
[edit | edit source]- The Pharmacy Technician. New York: Morton Publishing Company, 2007.
- Davis, Alison. Medicines by Design. [Bethesda, MD]: U.S. Dept. of Health and Human Services, National Institutes of Health, National Institute of General Medical Sciences, 2006. Print.