
Structural Biochemistry/Prion Protein Misfolding and Disease
Prions
[edit | edit source]Prion Disease
[edit | edit source]Transmissible spongiform encephalopathies (TSEs or prion diseases) are a rare group of deadly neurodegenerative disorders that affect humans and other mammals. TSEs are protein misfolding diseases that encompass the aggregation of abnormally accumulated form of the normal host prion protein. TSEs are unique in that they are transmissible. Characteristics of TSEs include that they replicate, are capable of selective evolution (can evolve drug resistance), have various strains of infectious agent that are related to unique phenotypes in vivo, and demonstrate strong species specificities; the same characteristic of many viral and bacterial pathogens.

There are 3 classes of TSE diseases in humans:
- Sporadic – the most common form of TSE (e.g. Creutzfeldt Jakob disease)
- Heritable – in which TSE is mutation within the prion protein
- Acquired – in which TSE is a result of ingestion or inoculation of TSE contaminated materials.
-
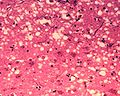 Microscopic "holes" are characteristic in prion-affected tissue sections, causing the tissue to develop a "spongy" architecture.
Microscopic "holes" are characteristic in prion-affected tissue sections, causing the tissue to develop a "spongy" architecture.

The Prion Hypothesis
[edit | edit source]Typical infectious agents use nucleic acids to spread and propagate, evident in bacteria and viruses. The prion hypothesis states simply that the specific diseases detailed above are solely caused by proteins, a sentiment that went against common knowledge at the time of its proposal. At first, it was thought that prions were a side-effect of some other type of infection. This theory does not hold up, however. Experimental evidence since the concept of prion-induced disease was proposed has fallen in favor of this theory, and the effects of prion replication have been witnessed in the lab. [1]
History of Prions in Science
[edit | edit source]Prions and their infectious nature were first discovered in 1937 when scrapie was accidentally transferred to a sheep while attempting a viral inoculation. In later experiments, scrapie was purposefully transmitted to sheep and then mice to determine the nature of this molecule. Cannibalism in New Guinea was the source of infection by kuru in humans and in 1966 this disease was demonstrated to be transmissible to monkeys[1]. At this point, science was beginning to understand that the infectious nature of prions was different from viral or microbial infection. More recently, outbreaks of Bovine Spongiform Encephalopathy and the emerging variant Creutzfeldt-Jakob disease that was linked to consumption of infected meat were shown to be caused by prions.
Historically, studies of prions first determined that the method of infection was novel, and then determined that it was due to a misfolding of the protein chain. The first experiments destroyed nucleic acids with UV and ionizing radiation and found the infectious agent still present[1]. Then, the smallest molecular weight particles that were still infectious were determined to be on the order of protein weight.
Later on, protease-resistant prion protein (PrP) concentration was found to be proportional to the infectivity. Additionally, agents that destroyed protein structure were employed, reducing the infectivity of the PrP. PrP was found to be present in a normally functioning brain, demonstrating that the protein could exist both as a normal and as a malfunctioning protein[1]. Recently, studies have shown that infectious and non infectious proteins could be mixed and the infectious agent would propagate among non infectious molecules. This demonstrates that the protein folding error can propagate indefinitely.
Prion Replication
[edit | edit source]Conversion of PrP isoforms is the cause of Prion Disease
[edit | edit source]TSE is a protein misfolding disease in that disease occurs due to conformational changes in host prion protein (PrP). PrP is a mammalian glycoprotein, 209 amino acids long. When the PrP becomes a TSE, in a process known as pathogenesis, a protease sensitive form of PrP (PrP-sen) refolds into PrP-res (a protease- resistant form of prion protein). PrP-res and PrP-sen have the same primary sequence but different secondary structures, with the PrP-sen featuring more alpha-helices. (In other words, PrP-res and PrP-sen are isoforms) PrP-res is the primary component of TSE. Hence it is imperative to research how the PrP-sen to PrP-res conversion occurs, in order to inhibit the formation of the PrP-res form.
PrP Conversion Mechanism
[edit | edit source]
Current knowledge of the PrPc to PrPSc conversion can be generalized into a two-step mechanism:
1) Ordered aggregates of pre-cursor “seed” PrPSc bind to PrPc.
2) PrPc experiences some conformational change that results in the propagation into more PrPSc. The mechanism of this step is largely a mystery. The converted PrPSc is added to the polymer, which eventually fragments and causes more PrPc to be converted. This fragmentation is thought to be the rate-limiting step of the reaction.[1]
Protein Misfolding Cyclic Amplification
[edit | edit source]In 2001, a process for laboratory replication of prions was developed. This process, known as Protein Misfolding Cyclic Amplication (PMCA), produces prions in-vitro which mimic prions replicated normally in-vivo. This process relies on the idea that prions are auto-catalytic, and can reproduce indefinitely given the correct surroundings. This technique has proven invaluable in the study of prions and the testing of the prion hypothesis. [1]
Co-Factors
[edit | edit source]One area that is still unclear to scientists is the importance of co-factors in the replication process of prions. While it is now understood that there are outside factors that influence the success and rate of prion replication, these co-factors have not been fully discerned and represent an area for future research.
Transgenic Mice and Hamsters
[edit | edit source]Evidence for the role of co-factors in prion replication has come from various studies performed on animal subjects. In a study done on transgenic mice, there was found to be some sort of factor affecting prion protein expression. This was termed "protein X", though the identity of the factor specifically as a protein was never discerned. [2]. In a separate study performed on hamster PrPc. When isolated and purified, hamster PrPc could not be converted when mixed with PrPSc. When brain homogenate was introduced to the sample, the prion conversion occurred, indicating that there was something in the homogenate that contributed to the conversion, whether as a catalyst or as an integral factor. Further testing showed that that RNA in hamsters acted as a catalyst for prion replication, but not in mice. Specifically how it does this is not clear, but there is a possibility that it helps to stabilize the comformation of PrPSc that is produced. The fact that it does not work on all mammals leads to the possibility of multiple co-factors, or species/organ specific ones.
Types of Co-Factors
[edit | edit source]Although the importance of co-factors is not fully understood, the ways in which co-factors might affect the conversion of prions fall into 5 categories.
| Genetic Information | It is possible that there are certain co-factors that contribute to prion replication by helping to determine how these proteins fold. One study that supports this was performed on mice by injecting prions into different types of cells. In each cell, prion replication led to different prion strains, which could be a result of different co-factors in each cell type. |
| Catalytic | Certain co-factors might catalyze the PrPc to PrPSc conversion by attaching to PrPc and partially unfolding it. This would make it easier for PrPSc to induce a misfolding pattern on the protein, resulting in the conversion. This type of co-factor has been examined in the lab in vitro. |
| Conformational Stabilization | Some co-factors might aid in the stabilization of the new prion conformation. Examples of these include nucleic acids, proteins, metal ions, and other things. Many of these are charged species that can bind to the prion and help form a more compact structure. |
| Fragmentation | PrPSc polymers often fragment, which greatly speeds up the conversion of PrPc to PrPSc. Some co-factors may help in this fragmentation process, which creates new seeds and is essential to the replication process. An example of this is found in yeast, which rely on protein 104 (Hsp104). Removing this protein stops prion replication from occuring. |
| Biological Stabilization | For successful prion replication to occur, prions must first survive in the biological medium. Certain biological resistance, such as microglia cells which can perform phagocytosis, can inhibit their spread. A co-factor that reduces the ability of microglia to destroy these prions would allow them to replicate, and might prove to be essential. [1] |
Prion Structure
[edit | edit source]Primary and Secondary Structure play crucial role in the initial Rate-limiting step
[edit | edit source]Primary and secondary structural components within at amino acids 108-189 of the PrP-sen proved to be important for conversion, as determined by NMR structure and in vivo studies. PrP-sen at the critical residues (108-189) include most of the folded domain including beta strands, alpha-helices, and most critical, the loops and turns. Slight variation with turns and loops are particularly noteworthy, as it has been determined that the loops are involved in the intermolecular interactions between PrP-sen and PrP-res.
Lack of Structural Understanding of PrP-res
[edit | edit source]While there is extensive knowledge of the PrP-sen tertiary structure, the same cannot be said for PrP-res. Part of the reason as to why there isn’t extensive knowledge regarding the mechanistic pathway of the conversion into PrP-res is that the PrP-res structure is unknown, as it has not been purified sufficiently for high-resolution structural studies. Because high-resolution techniques cannot be used, focus on ascertaining the structure of PrP-res has been confined to using lower resolution techniques such as electron microscopy to determine ultrastructure and the secondary structure of PrP-res.
Further Understanding
[edit | edit source]Yeast Prions
[edit | edit source]One interesting case of prions can be found in yeast. While they are normally observed in mammals, it was found that a particular protein in yeast (Ure2) behaves and reproduces in the way a typical prion does. However, instead of killing its host cell it instead reproduces by inducing conformational change in other proteins and is inherited through cellular division. This special case has led to the discovery of other prions in fungus which also exhibit non-lethal behavior.[1]
"Good" Prions?
[edit | edit source]{kind=link}
As found in the example of yeast prions, these misfolded proteins are not all inherently malicious and harmful to organic life. In one study conducted by scientists in UT Southwestern, it was found that certain prion-like proteins found within the body may help in the immune system. These mitochondrial antiviral signaling proteins (MAVS) were found to be in mitochondrial membranes, and actively defend them against infection. It was found that under threat of viral infection, MAVS proteins misfold and aggregate on the surface of the mitochondria, effectively shielding the organelle from attack. This deliberate misfolding and aggregation is interesting because it does not reflect the malicious and uncontrollable misfolding often caused by prions in cases such as Cretzfeldt-Jakob Disease and Bovine Spongify Esophagus. While the sequential activation of the system by the initial viral detection can be complex, the aggregation of MAVS proteins actually occurred quickly. [4]
Similarity of PrP-res to Amyloid Fibril
[edit | edit source]The way PrP-res amplifies itself is highly similar to the way in which amyloid fibrils grow through seeded polymerization from a reservoir of precursor protein. Therefore amyloid fibril offers insight for which to hypothesize the PrP-res growth mechanism.
Amyloids are fibrous protein aggregates that harbor specific structural traits. The structure of amyloid fibrils features a characteristic cross-beta folding pattern in which beta-strands align to form sheets that are perpendicular to the fibril axis while hydrogen bonds between strands is parallel to the fibril axis. While this cross-beta quaternary structure is known in amyloids, a detailed structure of this scope has not been determined for PrP, which is by far, a much larger protein.
Like amyloid fibrils, PrP can be induced to form amyloid-like fibrils. A particularly significant model of this was that of the recombinant peptide, PrP23-144. (Recombinant peptide is the peptide encoded from its corresponding recombinant DNA). The PrP23-144 proved to have a high degree of conformational plasticity (ability to change conformation in various possible ways) and can be induced to form a wide range of fibril amino acids. That the PrP23-144 in this model could be differentiated into a wide range of conformational possibilities is an indication that the PrP isoform conversion is highly dependent on the structural compatibility between PrP-sen to PrP- res (for example, the aforementioned variations in turns and loops). Choice of solvent also plays a role in that certain structural elements are affected by electrostatic or hydrophilic effects.
Treatment
[edit | edit source]As a result of the limited knowledge scientists have on prions, there is currently no full-proof method of treatment for those diagnosed with a prion related disease. One possibility of prion removal is through microglia and phagocytosis. It has also been found that lichens posses the ability to break down prions. [5] Though there are many avenues of research possible, there is no clinical solution for those suffering from a prion related disease.
Detection
[edit | edit source]
There has been research in the past decade that has helped to identify PrPSc in the blood. Up until this point, there has been no reliable way to identify these misfolded proteins in brain tissue. Employing a new method known as SOFIA (surround optical fibre immunoassay), scientists have been able to identify the presence of prions in brain tissue at high precision (one part in one hundred thousand million). First, the sample with PrPSc is amplified and fluorescently labeled using an antibody. It is then put through a device that surrounds it with optical fibers that detect emitted light. A laser is used to excite the dye, and the emission from the sample is picked up by the detector.
This method was tested on sheep that were previously thought to be healthy, but eventually developed prion disease. Samples of their blood before they exhibited symptoms were analyzed, and it was found that the presence of PrPSc could be detected very early on. While this method does not cure or remove any of the problems, it can be used to quarantine and greatly reduce the risk of these prions spreading. [6]
- ↑ a b c d e f g h Soto, Claudio "Prion Hypothesis: The end of the Controversy?", '[PubMed]', 2010. Retrieved on 20 November 2012.
- ↑ Telling GC, et al. “Prion propagation in mice expressing human and chimeric PrP transgenes implicates the interaction of cellular PrP with another protein.”, ‘[PubMed]’, 27 June 2012. Retrieved on 20 November, 2012.
- ↑ Weissmann C. "Thoughts on mammalian prion strains.", '[PubMed]', 2009. Retrieved on 20 November 2012.
- ↑ Hou, Fajian et al. "MAVS Forms Functional Prion-like Aggregates to Activate and Propagate Antiviral Innate Immune Response", '[Cell]', 2011. Retrieved on 20 November 2012.
- ↑ Johnson, Christopher J. et al "Degradation of the Disease-Associated Prion Protein by a Serine Protease from Lichens", '[PLOS]', 2011. Retrieved on 20 November 2012.
- ↑ Rubenstein, R., Chang, B., Gray, P., Piltch, M., Bulgin, M.S., Sorensen-Melson, S. & Miller, M.W "Detecting Prions in Blood", '[Microbiology Today]', 2010. Retrieved on 20 November 2012.
References
[edit | edit source]- Moore, R (2009) Prion Protein Misfolding and Disease