Chemical Sciences: A Manual for CSIR-UGC National Eligibility Test for Lectureship and JRF/Fluorescence microscope
| This page was imported and needs to be de-wikified. Books should use wikilinks rather sparsely, and only to reference technical or esoteric terms that are critical to understanding the content. Most if not all wikilinks should simply be removed. Please remove {{dewikify}} after the page is dewikified. |

A fluorescence microscope (colloquially synonymous with epifluorescence microscope) is an optical microscope used to study properties of organic or inorganic substances using the phenomena of fluorescence and phosphorescence instead of, or in addition to, reflection and absorption.[1][2]

Technique
[edit | edit source]In most cases, a component of interest in the specimen can be labeled specifically with a fluorescent molecule called a fluorophore (such as green fluorescent protein (GFP), fluorescein or DyLight 488).[1] The specimen is illuminated with light of a specific wavelength (or wavelengths) which is absorbed by the fluorophores, causing them to emit light of longer wavelengths (i.e. of a different color than the absorbed light). The illumination light is separated from the much weaker emitted fluorescence through the use of a spectral emission filter. Typical components of a fluorescence microscope are the light source (xenon arc lamp or mercury-vapor lamp), the excitation filter, the dichroic mirror (or dichromatic beamsplitter), and the emission filter (see figure below). The filters and the dichroic are chosen to match the spectral excitation and emission characteristics of the fluorophore used to label the specimen.[1] In this manner, the distribution of a single fluorophore (color) is imaged at a time. Multi-color images of several types of fluorophores must be composed by combining several single-color images.[1]
Most fluorescence microscopes in use are epifluorescence microscopes (i.e. excitation and observation of the fluorescence are from above (epi–) the specimen). These microscopes have become an important part in the field of biology, opening the doors for more advanced microscope designs, such as the confocal microscope and the total internal reflection fluorescence microscope (TIRF).
Fluorophores lose their ability to fluoresce as they are illuminated in a process called photobleaching. Special care must be taken to prevent photobleaching through the use of more robust fluorophores, by minimizing illumination, or by introducing a scavenger system to reduce the rate of photobleaching.
Epifluorescence microscopy
[edit | edit source]
Epifluorescence microscopy is a method of fluorescence microscopy that is widely used in life sciences. The excitatory light is passed from above (or, for inverted microscopes, from below), through the objective lens and then onto the specimen instead of passing it first through the specimen. The fluorescence in the specimen gives rise to emitted light which is focused to the detector by the same objective that is used for the excitation. Since most of the excitatory light is transmitted through the specimen, only reflected excitatory light reaches the objective together with the emitted light and this method therefore gives an improved signal to noise ratio. An additional filter between the objective and the detector can filter out the remaining excitation light from fluorescent light. A common use in biology is to apply fluorescent or fluorochrome stains to the specimen in order to image distributions of proteins or other molecules of interest.
Improvements and sub-diffraction techniques
[edit | edit source]The nature of light limits the size of the spot to which light can be focused. According to the diffraction limit a focused light distribution cannot be made smaller than approximately half of the wavelength of the used light. Uncovered in the 19th century by Ernst Abbe this has been a barrier of the achievable resolution of fluorescence light microscopes for a long time. While resolution is denoted by the ability to discern different objects of the same kind, localizing or tracking of single particles have been performed with a precision much below the diffraction limit.
Several improvements in microscopy techniques have been invented in the 20th century and have resulted in increased resolution and contrast to some extent. In 1978 first theoretical ideas have been developed to break this barrier by using a 4Pi microscope as a confocal laser scanning fluorescence microscope where the light is focused ideally from all sides to a common focus which is used to scan the object by 'point-by-point' excitation combined with 'point-by-point' detection [3]. However, the first experimental demonstration of the 4pi microscope took place in 1994 [4]. The 4Pi microscopy is maximizing the amount of available focusing directions by using two opposing objective lenses or Multi-photon microscopy using redshifted light and multi-photon excitation.
The first technique to really achieve a sub-diffraction resolution was STED microscopy, proposed in 1994. This method and all techniques following the RESOLFT concept rely on a strong non-linear interaction between light and fluorescing molecules. The molecules are driven strongly between distinguishable molecular states at each specific location, so that finally light can be emitted at only a small fraction of space, hence an increased resolution.
As well in the 1990ies another super resolution microscopy method based on wide field microscopy has been developed. Substantially improved size resolution of cellular nanostructures stained with a fluorescent marker was achieved by development of SPDM localization microscopy and the structured laser illumination (spatially modulated illumination, SMI) [5]. Combining the principle of SPDM with SMI resulted in the development of the Vertico SMI microscope [6] [7]. Single molecule detection of normal blinking fluorescent dyes like GFP can be achieved by using a further development of SPDM the so-called SPDMphymod technology which makes it is possible to detect and count two different fluorescent molecule types at the molecular level (this technology is referred to as 2CLM, 2 Color Localization Microscopy) [8]
Alternatively, the advent of photoactivated localization microscopy could achieve similar results by relying on blinking or switching of single molecules, where the fraction of fluorescing molecules is very small at each time. This stochastic response of molecules on the applied light corresponds also to a highly nonlinear interaction, leading to subdiffraction resolution.
Gallery
[edit | edit source]-
 Epifluorescent imaging of the three components in a dividing human cancer cell. DNA is stained blue, a protein called INCENP is green, and the microtubules are red. Each fluorophore is imaged separately using a different combination of excitation and emission filters, and the images are captured sequentially using a digital CCD camera, then overlaid to give a complete image.
Epifluorescent imaging of the three components in a dividing human cancer cell. DNA is stained blue, a protein called INCENP is green, and the microtubules are red. Each fluorophore is imaged separately using a different combination of excitation and emission filters, and the images are captured sequentially using a digital CCD camera, then overlaid to give a complete image. -
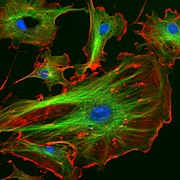 Endothelial cells under the microscope. Nuclei are stained blue with DAPI, microtubules are marked green by an antibody bound to FITC and actin filaments are labeled red with phalloidin bound to TRITC. Bovine pulmonary artery endothelial (BPAE) cells
Endothelial cells under the microscope. Nuclei are stained blue with DAPI, microtubules are marked green by an antibody bound to FITC and actin filaments are labeled red with phalloidin bound to TRITC. Bovine pulmonary artery endothelial (BPAE) cells -
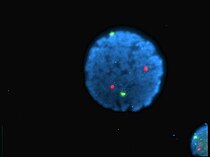 Human lymphocyte nucleus stained with DAPI with chromosome 13 (green) and 21 (red) centromere probes hybridized (Fluorescent in situ hybridization (FISH))
Human lymphocyte nucleus stained with DAPI with chromosome 13 (green) and 21 (red) centromere probes hybridized (Fluorescent in situ hybridization (FISH)) -
 Yeast cell membrane visualized by some membrane proteins fused with RFP and GFP fluorescent markers. Imposition of light from both of markers results in yellow color.
Yeast cell membrane visualized by some membrane proteins fused with RFP and GFP fluorescent markers. Imposition of light from both of markers results in yellow color. -
Super Resolution Microscopy:Single YFP molecule detection in a human cancer cell. Typical distance measurements in the 15 nm range (5 nm standard deviation) measured with a Vertico-SMI/SPDMphymod microscope
-
 Super Resolution Microscopy: Co-localzation microscopy (2CLM) with GFP and RFP fusion proteins (nucleus of a bone cancer cell) 120.000 localized molecules in a wide-field area (470 µm2) measured with a Vertico-SMI/SPDMphymod micrsocpe
Super Resolution Microscopy: Co-localzation microscopy (2CLM) with GFP and RFP fusion proteins (nucleus of a bone cancer cell) 120.000 localized molecules in a wide-field area (470 µm2) measured with a Vertico-SMI/SPDMphymod micrsocpe -
 Fluorescence microscopy of DNA Expression in the Human Wild-Type and P239S Mutant Palladin.
Fluorescence microscopy of DNA Expression in the Human Wild-Type and P239S Mutant Palladin. -
 Fluorescence microscopy images of sun flares pathology in a blood cell showing the affected areas in red.
Fluorescence microscopy images of sun flares pathology in a blood cell showing the affected areas in red.







References
[edit | edit source]- ↑ a b c d Spring KR, Davidson MW. "Introduction to Fluorescence Microscopy". Nikon MicroscopyU. Retrieved 2008-09-28.
- ↑ "The Fluorescence Microscope". Microscopes—Help Scientists Explore Hidden Worlds. The Nobel Foundation. Retrieved 2008-09-28.
- ↑ >Considerations on a laser-scanning-microscope with high resolution and depth of field: C. Cremer and T. Cremer in M1CROSCOPICA ACTA VOL. 81 NUMBER 1 September,pp. 31—44 (1978)
- ↑ S.W. Hell, E.H.K. Stelzer, S. Lindek, C. Cremer (1994). "Confocal microscopy with an increased detection aperture: type-B 4Pi confocal microscopy". Optics Letters. 19: 222–224. doi:10.1364/OL.19.000222.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - ↑ M. Hausmann, B. Schneider, J. Bradl, C. Cremer (1997): High-precision distance microscopy of 3D-nanostructures by a spatially modulated excitation fluorescence microscope. In: Optical Biopsies and Microscopic Techniques II (Edts Bigio IJ, Schneckenburger H, Slavik J, Svanberg K, Viallet PM), Proc. SPIE 3197: 217-222
- ↑ High precision structural analysis of subnuclear complexes in fixed and live cells via Spatially Modulated Illumination (SMI) microscopy: J. Reymann, D. Baddeley, P. Lemmer, W. Stadter, T. Jegou, K. Rippe, C. Cremer, U. Birk in CHROMOSOME RESEARCH, Vol. 16, pp. 367 –382 (2008)
- ↑ Nano-structure analysis using Spatially Modulated Illumination microscopy: D. Baddeley, C. Batram, Y. Weiland, C. Cremer, U.J. Birk in NATURE PROTOCOLS, Vol 2, pp. 2640 – 2646 (2007)
- ↑ Manuel Gunkel, Fabian Erdel, Karsten Rippe, Paul Lemmer, Rainer Kaufmann, Christoph Hörmann, Roman Amberger and Christoph Cremer: Dual color localization microscopy of cellular nanostructures. In: Biotechnology Journal, 2009, 4, 927-938. ISSN 1860-6768
Further reading
[edit | edit source]- Herman, B., Fluorescence Microscopy., 2nd edition, Taylor & Francis, New York (1998).
- Bradbury, S. and Evennett, P., Fluorescence microscopy., Contrast Techniques in Light Microscopy., BIOS Scientific Publishers, Ltd., Oxford, United Kingdom (1996).
- Rost, F., Quantitative fluorescence microscopy. Cambridge University Press, Cambridge, United Kingdom (1991).
- Rost, F., Fluorescence microscopy. Vol. I. Cambridge University Press, Cambridge, United Kingdom (1992). Reprinted with update, 1996.
- Rost, F., Fluorescence microscopy. Vol. II. Cambridge University Press, Cambridge, United Kingdom (1995).
- Rost, F. and Oldfield, R., Fluorescence microscopy., Photography with a Microscope, Cambridge University Press, Cambridge, United Kingdom (2000).