Paragangliomas are still sometimes called glomus tumors (not to be confused with glomus tumors of the skin) and chemodectomas, but paraganglioma is the currently accepted and preferred term
A rare neoplasm that can be found in the abdomen (85%), thorax (12%), and in the head and neck region (3%)
Usually considered benign and complete surgical removal results in cure. However, in about 3-5% of cases they are malignant and have the ability to metastasize.
Most occur as single tumors. When they occur in multiple sites they are usually found as a part of a heritable syndrome such as MEN types II-A and II-B and Carney syndrome.
Familial paragangliomas account for ~25% of cases, are often multiple and bilateral, and occur at an earlier age.
Mutations of the genes SDHD (previously known as PGL1), PGL2, and SDHC (previously PGL3) have been identified as causing familial head and neck paragangliomas.
Mutations of SDHB play an important role in familial adrenal pheochromocytoma and extra-adrenal paraganglioma (of abdomen and thorax), although there is considerable overlap in the types of tumors associated with SDHB and SDHD gene mutations.
Arise from the glomus cells, which are special chemoreceptors located along blood vessels that have a role in regulating blood pressure and blood flow.
The glomus cells are a part of the paraganglion system, composed of the extra-adrenal paraganglia of the autonomic nervous system, derived from the embryonic neural crest. Thus, paragangliomas are a type of neuroendocrine tumor, and are closely related to pheochromocytomas. Although all paragangliomas contain neurosecretory granules, only about 1-3% have clinical evidence of oversecretion.
According to the WHO classification of neuroendocrine tumors, paragangliomas are classified as having a neural cell line of origin. In the categorization proposed by Wick, the paragangliomas belong to Group II.
The main concentration of glomus cells are found are in the carotid body and the aortic bodies
Individual tumor cells are polygonal to oval and are arranged in distinctive cell balls, called Zellballen. These cell balls are separated by fibrovascular stroma and surrounded by sustentacular cells.
The paragangliomas appear grossly as sharply circumscribed polypoid masses and they have a firm to rubbery consistency. They are highly vascular tumors and may have a deep red color.
With IHC, the chief cells located in the cell balls are positive for chromogranin, synaptophysin, NSE, serotonin and neurofilamen; they are S-100 negative. The sustentacular cells are S-100 positive and focally positive for GFAP. By histochemistry, the paraganglioma cells are argyrophilic, PAS negative, mucicarmine negative, and argentaffin negative.
Paragangiomas are described by their site of origin and are often given special names:
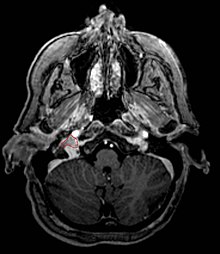
Carotid paraganglioma (carotid body tumor): Is the most common of the head and neck paragangliomas. It usually presents as a painless neck mass, but larger tumors may cause cranial nerve palsies, usually of the vagus nerve and hypoglossal nerve.
Glomus tympanicum and Glomus jugulare: Both commonly present as a middle ear mass resulting in tinnitus (in 80%) and hearing loss (in 60%). The cranial nerves of the jugular foramen may be compressed, resulting swallowing difficulty.
Vagal paragangliomas: These are the least common of the head and neck paragangliomas. They usually present as a painless neck mass, but may result in dysphagia and hoarseness.
Other sites: Rare sites of involvement are the larynx, nasal cavity, paranasal sinuses, thyroid gland, and the thoracic inlet.
Typically considered slow-growing (estimated doubling time 4.2 years) benign tumors, <5% malignant potential
However, they are locally agressive neoplastic lesions, involving bony erosion and destruction of neurovascular structures
Commonly arise from the paraganglia of the jugular bulb
Typically invade the tympanic cavity and jugular foramen
Can extensively invade petroclival region
Can invade cavernous sinus above
Can invade hypoglossal canal below
Clinical presentation typically with tinnitus or hearing loss, but may also impact jugular foramen CNs
Glasscock-Jackson Classification:
G-J Class
Description
I
Small tumor involing jugular bulb, middle ear, and mastoid
II
Tumor extending under internal auditory canal; may have intracranial canal extension
III
Tumor extending into petrous apex; may have intracranial canal extension
C1
Tumor extending beyond petrous apex; into clivus or infratemporal fossa
Fisch Classification:
Fisch Class
Description
A
Tumor limited to the middle ear cleft
B
Tumor limited to the tympanomastoid area, with no infralabyrinthine involvement
C
Tumor invading infralabyrinthine compartment and petrous apex
C1
Tumor with limited involvement of the vertical carotid canal
C2
Tumor invading the vertical carotid canal
C3
Tumor invading the horizontal carotid canal
D1
Tumor with intracranial extension <2cm
D2
Tumor with intracranial extension >2cm
Optimal treatment strategy is not clear, with multiple options:
Surgery: primary option if brainstem compression, or in patients under 45 years old with functional cranial nerve loss. Surgical morbidity not insignificant, typically with additional cranial nerve sacrifice.
Fractionated RT: typical doses 45-55 Gy, mechanism of action likely related to fibrosis of feeding vessels and not direct glomus cell destruction. Complication rate low. Critical to avoid nearby cranial nerves or blood vessels.
SRS: No long-term experience yet, but appears a good option due to high conformality. RT dose typically ~16 Gy at 50% margin isodose. Typically ~1/3 have volume shrinkage, and ~2/3 no change, <10% further growth. Clinically ~50% may experience improvement
Embolization: typically considered an adjunctive treatment to surgery or RT. Not adequate alone due to poor tumor coverage and frequent re-vascularization
UT Southwestern, 2014 (2007-2013) PMID 24818638 -- "A Retrospective Analysis of Tumor Volumetric Responses to Five-Fraction Stereotactic Radiotherapy for Paragangliomas of the Head and Neck (Glomus Tumors)." (Chun SG, Stereotact Funct Neurosurg. 2014 May;92(3):153-9)
Retrospective. CyberKnife radiotherapy. 31 patients (23 definitive, 8 recurrence post surgery. Average volume 10 cm3, prescription dose 25 Gy in 5 fractions. Median F/U 2 years
Outcome: Local control and overall survival 100%, significant tumor volume reduction in patients with >2 years F/U of 37% (p < 0.05), tinnitus improved in 12 patients of whom 6 had resolution
Toxicity: Grade 1 toxicity 16%, one patient had Grade 2 headache requiring steroids, no progressive cranial nerve deficit
Conclusion: 5 fraction CyberKnife SRS of skull based paragangliomas can be considered as a safe and effective option
Verona, 2006 (Italy)(1996-2005) PMID 16955038 -- "Glomus jugulare tumors: the option of gamma knife radiosurgery." (Gerosa M, Neurosurgery. 2006 Sep;59(3):561-9; discussion 561-9.)
Retrospective. 20 patients (3 primary GKS, 8 recurrence post surgery, 11 recurrence after embolization). Average volume 7 cm3, mean marginal dose 17.3 Gy (13-24). Estimated doubling 4.2 years. Mean F/U 4.2 years
Outcome: Improved volume 40%, no change 55%
Toxicity: Improved CN function 25%, no neuro change 65%, hearing loss 10%
Conclusion: GKS effective, negligible side effects
Virginia, 2005PMID 15662818 -- "Gamma knife surgery for glomus jugulare tumors: an intermediate report on efficacy and safety." (Sheehan J, J Neurosurg. 2005 Jan;102 Suppl:241-6.)
Retrospective. 8 patients. Median margin dose 15 Gy (12-18). Median F/U 2.7 years
Conclusion: Effective local control and preservation of neurologic function
Mayo Clinic, 2004PMID 15329025 -- "Stereotactic radiosurgery in patients with glomus jugulare tumors." (Pollock BE, Neurosurg Focus. 2004 Aug 15;17(2):E10.)
Retrospective. 42 patients treated with GKS. Mean volume 13.2 cm3. Mean margin dose 14.9 Gy. Mean F/U 3.7 years
Outcome: 31% decreased, 67% unchanged, 2% grew. PFS at 7 years 100%, at 10 years 75%
Toxicity: 15% new deficit (hearing loss, facial numbness, vocal cord paralysis, vertigo). Hearing preservation 81% at 4 years
Conclusion: GKS good tumor control, safe
Stanford, 2004PMID 15329026 -- "Efficacy and safety of stereotactic radiosurgery for glomus jugulare tumors." (Lim M, Neurosurg Focus. 2004 Aug 15;17(2):E11.)
Retrospective. 13 patients with 16 tumors. Treated with LINAC/Cyberknife to 14-27 Gy. Median F/U 3.4 years
Outcome: clinically 100% stable; radiographically 100% decreased or stable
Toxicity: one transient ipsilateral vocal cord paralysis (however, patient also had prior EBRT)
Conclusion: RS effective and safe
Vienna, 2001 (Austria)(1993-1999) PMID 11696882 -- "Efficiency of gamma knife radiosurgery in the treatment of glomus jugulare tumors." (Saringer W, Minim Invasive Neurosurg. 2001 Sep;44(3):141-6.)
Retrospective. 12 patients. All Fisch Class D. Mean F/U 4.2 years
Meta-analysis, 2004 (1994-2004) PMID 15329019 -- "Comparison of radiosurgery and conventional surgery for the treatment of glomus jugulare tumors." (Gottfried ON, Neurosurg Focus. 2004 Aug 15;17(2):E4.)
7 surgical series 374 patients, 8 GKS series 142 patients. Mean F/U surgery 4.1 years, GKS 3.3 years
Outcome: local control surgery 92%, recurrence 3%; GKS decrease 36%, stable 61%, recurrence 2%
Toxicity: surgery CSF leak 8%, mortality 1.3%; GKS morbidity 8%, no mortality
Conclusion: Both treatments safe and efficacious; surgery higher morbidity but long term GKS outcomes unknown