This page was imported and needs to be de-wikified. Books should use wikilinks rather sparsely, and only to reference technical or esoteric terms that are critical to understanding the content. Most if not all wikilinks should simply be removed. Please remove {{dewikify}} after the page is dewikified.
Amino acids are molecules containing an amine group(NH2), a carboxylic acid group(R-C=O-OH) and a side-chain( usually denoted as R) that varies between different amino acids. The key elements of an amino acid are carbon, hydrogen, oxygen, and nitrogen. They are particularly important in biochemistry, where the term usually refers to alpha-amino acids.Proteins are biochemical compounds consisting of one or more polypeptides typically folded into a globular or fibrous form in a biologically functional way. A polypeptide is a single linear polymer chain of amino acids bonded together by peptide bonds between the carboxyl and amino groups of adjacent amino acid residues. The sequence of amino acids in a protein is defined by the sequence of a gene, which is encoded in the genetic code. In general, the genetic code specifies 20 standard amino acids; however, in certain organisms the genetic code can include selenocysteine—and in certain archaea—pyrrolysine. Shortly after or even during synthesis, the residues in a protein are often chemically modified by post-translational modification, which alters the physical and chemical properties, folding, stability, activity, and ultimately, the function of the proteins. Sometimes proteins have non-peptide groups attached, which can be called prosthetic groups or cofactors. Proteins can also work together to achieve a particular function, and they often associate to form stable complexes.
One of the most distinguishing features of polypeptides is their ability to fold into a globular state, or "structure". The extent to which proteins fold into a defined structure varies widely. Some proteins fold into a highly rigid structure with small fluctuations and are therefore considered to be single structure. Other proteins undergo large rearrangements from one conformation to another. This conformational change is often associated with a signaling event. Thus, the structure of a protein serves as a medium through which to regulate either the function of a protein or activity of an enzyme. Not all proteins requiring a folding process in order to function, as some function in an unfolded state[1].
Like other biological macromolecules such as polysaccharides and nucleic acids, proteins are essential parts of organisms and participate in virtually every process within cells. Many proteins are enzymes that catalyze biochemical reactions and are vital to metabolism. Proteins also have structural or mechanical functions, such as actin and myosin in muscle and the proteins in the cytoskeleton, which form a system of scaffolding that maintains cell shape. Other proteins are important in cell signaling, immune responses, cell adhesion, and the cell cycle. Proteins are also necessary in animals' diets, since animals cannot synthesize all the amino acids they need and must obtain essential amino acids from food. Through the process of digestion, animals break down ingested protein into free amino acids that are then used in metabolism.
Proteins were first described by the Dutch chemist Gerhardus Johannes Mulder and named by the Swedish chemist Jöns Jakob Berzelius in 1838. Early nutritional scientists such as the German Carl von Voit believed that protein was the most important nutrient for maintaining the structure of the body, because it was generally believed that "flesh makes flesh." The central role of proteins as enzymes in living organisms was however not fully appreciated until 1926, when James B. Sumner showed that the enzyme urease was in fact a protein.The first protein to be sequenced was insulin, by Frederick Sanger, who won the Nobel Prize for this achievement in 1958. The first protein structures to be solved were hemoglobin and myoglobin, by Max Perutz and Sir John Cowdery Kendrew, respectively, in 1958. The three-dimensional structures of both proteins were first determined by X-ray diffraction analysis; Perutz and Kendrew shared the 1962 Nobel Prize in Chemistry for these discoveries. Proteins may be purified from other cellular components using a variety of techniques such as ultracentrifugation, precipitation, electrophoresis, and chromatography; the advent of genetic engineering has made possible a number of methods to facilitate purification. Methods commonly used to study protein structure and function include immunohistochemistry, site-directed mutagenesis, nuclear magnetic resonance and mass spectrometry. Distributed computing is a relatively new tool researchers are using to examine the infamously complex interactions that govern protein folding; the statistical analysis techniques employed to calculate a protein's probable tertiary structure from its amino acid sequence (primary structure) are well-suited for the distributed computing environment, which has made this otherwise prohibitively expensive and time consuming problem significantly more manageable[2].
A schematic visual model of oxygen-binding process, showing all four monomers and hemes, and protein chains only as diagramatic coils, to facilitate visualization into the molecule. Oxygen is not shown in this model, but, for each of the iron atoms, it binds to the iron (red sphere) in the flat heme For example, in the upper left of the four hemes shown, oxygen binds at the left of the iron atom shown in the upper left of diagram. This causes the iron atom to move backward into the heme which holds it (the iron moves upward as it binds oxygen, in this illustration), tugging the histidine residue (modeled as a red pentagon on the right of the iron) closer, as it does. This, in turn, pulls on the protein chain holding the histidine.
Protein is a essential agent of biological function.There are 22 standard amino acids, but only 21 are found in eukaryotes. Of the 22, 20 are directly encoded by the universal gentic code. Humans can synthesize 11 of these 20 from each other or from other molecules of intermediary metabolism. The other 9 must be consumed in the diet, and so are called essential amino acids; those are histidine, isoleucine, leucine, lysine, methionine, phenylalanine, threonine, tryptophan, and valine. The remaining two, selenocysteine and pyrrolysine, are incorporated into proteins by unique synthetic mechanisms.
Each α-amino acid consists of a backbone part that is present in all the amino acid types, and a side chain that is unique to each type of residue. An exception from this rule is proline, where the hydrogen atom is replaced by a bond to the side chain. Because the carbon atom is bound to four different groups it is chiral, however only one of the isomers occur in biological proteins. Glycine however, is not chiral since its side chain is a hydrogen atom. A simple mnemonic for correct L-form is "CORN": when the Cα atom is viewed with the H in front, the residues read "CO-R-N" in a clockwise direction.
Isomerism
The standard α-amino acids, all but glycine can exist in either of two optical isomers, called L or D amino acids, which are mirror images of each other . While L-amino acids represent all of the amino acids found in proteins during translation in the ribosome, D-amino acids are found in some proteins produced by enzyme posttranslational modifications after translation and translocation to the endoplasmic reticulum, as in exotic sea-dwelling organisms such as cone snails. They are also abundant components of the peptidoglycan cell walls of bacteria, and D-serine may act as a neurotransmitter in the brain. The L and D convention for amino acid configuration refers not to the optical activity of the amino acid itself, but rather to the optical activity of the isomer of glyceraldehyde from which that amino acid can theoretically be synthesized (D-glyceraldehyde is dextrorotary; L-glyceraldehyde is levorotary). Alternatively, the (S) and (R) designators are used to indicate the absolute stereochemistry. Almost all of the amino acids in proteins are (S) at the α carbon, with cysteine being (R) and glycine non-chiral.Cysteine is unusual since it has a sulfur atom at the second position in its side-chain, which has a larger atomic mass than the groups attached to the first carbon which is attached to the α-carbon in the other standard amino acids, thus the (R) instead of (S)[3].
Zwitterions
The amine and carboxylic acid functional groups found in amino acids allow it to have amphiprotic properties. At a certain pH, known as the isoelectric point, an amino acid has no overall charge since the number of protonated ammonia groups (positive charges) and deprotonated carboxylate groups (negative charges) are equal. The amino acids all have different isoelectric points. The ions produced at the isoelectric point have both positive and negative charges and are known as a zwitterion, which comes from the German word Zwitter meaning "hermaphrodite" or "hybrid". Amino acids can exist as zwitterions in solids and in polar solutions such as water, but not in the gas phase. Zwitterions have minimal solubility at their isolectric point and an amino acid can be isolated by precipitating it from water by adjusting the pH to its particular isoelectric point[4].
The 20 naturally occurring amino acids have different physical and chemical properties, including their electrostatic charge, pKa, hydrophobicity, size and specific functional groups. These properties play a major role in molding protein structure. The salient features of amino acids are described below in the table.
Amino Acid
Abbrev.
Remarks
Alanine
A
Ala
Very abundant, very versatile. More stiff than glycine, but small enough to pose only small steric limits for the protein conformation. It behaves fairly neutrally, and can be located in both hydrophilic regions on the protein outside and the hydrophobic areas inside.
Asparagine or aspartic acid
B
Asx
A placeholder when either amino acid may occupy a position.
Cysteine
C
Cys
The sulfur atom bonds readily to heavy metal ions. Under oxidizing conditions, two cysteines can join together in a disulfide bond to form the amino acid cystine. When cystines are part of a protein, insulin for example, the tertiary structure is stabilized, which makes the protein more resistant to denaturation; therefore, disulfide bonds are common in proteins that have to function in harsh environments including digestive enzymes (e.g., pepsin and chymotrypsin) and structural proteins (e.g., keratin). Disulfides are also found in peptides too small to hold a stable shape on their own (eg. insulin).
Aspartic acid
D
Asp
Behaves similarly to glutamic acid. Carries a hydrophilic acidic group with strong negative charge. Usually is located on the outer surface of the protein, making it water-soluble. Binds to positively-charged molecules and ions, often used in enzymes to fix the metal ion. When located inside of the protein, aspartate and glutamate are usually paired with arginine and lysine.
Glutamic acid
E
Glu
Behaves similar to aspartic acid. Has longer, slightly more flexible side chain.
Phenylalanine
F
Phe
Essential for humans. Phenylalanine, tyrosine, and tryptophan contain large rigid aromatic group on the side-chain. These are the biggest amino acids. Like isoleucine, leucine and valine, these are hydrophobic and tend to orient towards the interior of the folded protein molecule. Phenylalanine can be converted into Tyrosine.
Glycine
G
Gly
Because of the two hydrogen atoms at the α carbon, glycine is not optically active. It is the smallest amino acid, rotates easily, adds flexibility to the protein chain. It is able to fit into the tightest spaces, e.g., the triple helix of collagen. As too much flexibility is usually not desired, as a structural component it is less common than alanine.
Histidine
H
His
In even slightly acidic conditions protonation of the nitrogen occurs, changing the properties of histidine and the polypeptide as a whole. It is used by many proteins as a regulatory mechanism, changing the conformation and behavior of the polypeptide in acidic regions such as the late endosome or lysosome, enforcing conformation change in enzymes. However only a few histidines are needed for this, so it is comparatively scarce.
Isoleucine
I
Ile
Essential for humans. Isoleucine, leucine and valine have large aliphatic hydrophobic side chains. Their molecules are rigid, and their mutual hydrophobic interactions are important for the correct folding of proteins, as these chains tend to be located inside of the protein molecule.
Essential for humans. Behaves similarly to arginine. Contains a long flexible side-chain with a positively-charged end. The flexibility of the chain makes lysine and arginine suitable for binding to molecules with many negative charges on their surfaces. E.g., DNA-binding proteins have their active regions rich with arginine and lysine. The strong charge makes these two amino acids prone to be located on the outer hydrophilic surfaces of the proteins; when they are found inside, they are usually paired with a corresponding negatively-charged amino acid, e.g., aspartate or glutamate.
Essential for humans. Always the first amino acid to be incorporated into a protein; sometimes removed after translation. Like cysteine, contains sulfur, but with a methyl group instead of hydrogen. This methyl group can be activated, and is used in many reactions where a new carbon atom is being added to another molecule.
Contains an unusual ring to the N-end amine group, which forces the CO-NH amide sequence into a fixed conformation. Can disrupt protein folding structures like α helix or β sheet, forcing the desired kink in the protein chain. Common in collagen, where it often undergoes a posttranslational modification to hydroxyproline.
Similar to glutamic acid. Gln contains an amide group where Glu has a carboxyl. Used in proteins and as a storage for ammonia. The most abundant Amino Acid in the body.
Serine and threonine have a short group ended with a hydroxyl group. Its hydrogen is easy to remove, so serine and threonine often act as hydrogen donors in enzymes. Both are very hydrophilic, therefore the outer regions of soluble proteins tend to be rich with them.
Behaves similarly to phenylalanine (precursor to Tyrosine) and tryptophan (see phenylalanine). Precursor of melanin, epinephrine, and thyroid hormones. Naturally fluorescent, although fluorescence is usually quenched by energy transfer to tryptophans.
The 20 amino acids encoded directly by the genetic code can be divided into several groups based on their properties. Important factors are charge, hydrophilicity or hydrophobicity, size and functional groups.Amino acids are usually classified by the properties of their side chain into four groups. The side chain can make an amino acid a weak acid or a weak base, and a hydrophile if the side chain is polar or a hydrophobe if it is nonpolar.
An α-amino acid. The CαH atom is omitted in the diagram.
In addition to the specific amino acid codes, placeholders are used in cases where chemical or crystallographic analysis of a peptide or protein can not conclusively determine the identity of a residue.
Ambiguous Amino Acids
3-Letter
1-Letter
Asparagine or aspartic acid
Asx
B
Glutamine or glutamic acid
Glx
Z
Leucine or Isoleucine
Xle
J
Unspecified or unknown amino acid
Xaa
X
Unk is sometimes used instead of Xaa, but is less standard.
Additionally, many non-standard amino acids have a specific code. For example, several peptide drugs, such as Bortezomib or MG132 are artificially synthesized and retain their protecting groups, which have specific codes. Bortezomib is Pyz-Phe-boroLeu and MG132 is Z-Leu-Leu-Leu-al. Additionally, To aid in the analysis of protein structure, photocrosslinking amino acid analogues are available. These include photoleucine (pLeu) and photomethionine (pMet).[5]
A Ramachandran plot generated from the protein PCNA, a human DNA clamp protein that is composed of both beta sheets and alpha helices (PDB ID 1AXC). Points that lie on the axes indicate N- and C-terminal residues for each subunit. The green regions show possible angle formations that include glycine, while the blue areas are for formations that don't include glycine.
Ramachandran diagram (φ,ψ plot), with data points for α-helical residues forming a dense diagonal cluster below and left of center, around the global energy minimum for backbone conformation.[6]
The proposal that proteins were linear chains of α-amino acids was made nearly simultaneously by two scientists at the same conference in 1902, the 74th meeting of the Society of German Scientists and Physicians, held in Karlsbad. Franz Hofmeister made the proposal in the morning, based on his observations of the biuret reaction in proteins. Hofmeister was followed a few hours later by Emil Fischer, who had amassed a wealth of chemical details supporting the peptide-bond model. For completeness, the proposal that proteins contained amide linkages was made as early as 1882 by the French chemist E. Grimaux.
Despite these data and later evidence that proteolytically digested proteins yielded only oligopeptides, the idea that proteins were linear, unbranched polymers of amino acids was not accepted immediately. Some well-respected scientists such as William Astbury doubted that covalent bonds were strong enough to hold such long molecules together; they feared that thermal agitations would shake such long molecules asunder. Hermann Staudinger faced similar prejudices in the 1920s when he argued that rubber was composed of macromolecules.
Thus, several alternative hypotheses arose. The colloidal protein hypothesis stated that proteins were colloidal assemblies of smaller molecules. This hypothesis was disproved in the 1920s by ultracentrifugation measurements by Theodor Svedberg that showed that proteins had a well-defined, reproducible molecular weight and by electrophoretic measurements by Arne Tiselius that indicated that proteins were single molecules[7].
A second hypothesis, the cyclol hypothesis advanced by Dorothy Wrinch, proposed that the linear polypeptide underwent a chemical cyclol rearrangement C=O + HN C(OH)-N that crosslinked its backbone amide groups, forming a two-dimensional fabric. Other primary structures of proteins were proposed by various researchers, such as the diketopiperazine model of Emil Abderhalden and the pyrrol/piperidine model of Troensegaard in 1942. Although never given much credence, these alternative models were finally disproved when Frederick Sanger successfully sequenced insulin and by the crystallographic determination of myoglobin and hemoglobin by Max Perutz and John Kendrew.
The primary structure of peptides and proteins refers to the linear sequence of its amino acid structural units. The term "primary structure" was first coined by Linderstrøm-Lang in 1951. By convention, the primary structure of a protein is reported starting from the amino-terminal (N) end to the carboxyl-terminal (C) end. The post-translational modifications of protein such as disulfide formation, phosphorylations and glycosylations are usually also considered a part of the primary structure, and cannot be read from the gene[8].
A Ramachandran plot (also known as a Ramachandran map or a Ramachandran diagram or a [φ,ψ] plot), developed by Gopalasamudram Narayana Ramachandran and Viswanathan Sasisekharan is a way to visualize dihedral angles ψ against φ of amino acid residues in protein structure. [9]. It shows the possible conformations of ψ and φ angles for a polypeptide.
Mathematically, the Ramachandran plot is the visualization of a function . The domain of this function is the torus. Hence, the conventional Ramachandran plot is a projection of the torus on the plane, resulting in a distorted view and the presence of discontinuities.
One would expect that larger side chains would result in more restrictions and consequently a smaller allowable region in the Ramachandran plot. In practice this does not appear to be the case; only the methylene group at the α position has an influence. Glycine has a hydrogen atom, with a smaller van der Waals radius, instead of a methyl group at the α position. Hence it is least restricted and this is apparent in the Ramachandran plot for glycine for which the allowable area is considerably larger.
In contrast, the Ramachandran plot for proline shows only a very limited number of possible combinations of ψ and φ.
The Ramachandran plot was calculated just before the first protein structures at atomic resolution were determined. Forty years later there were tens of thousands of high-resolution protein structures determined by X-ray crystallography and deposited in the Protein Data Bank (PDB). From one thousand different protein chains, Ramachandran plots of over 200 000 amino acids were plotted, showing some significant differences, especially for glycine (Hovmöller et al. 2002). The upper left region was found to be split into two; one to the left containing amino acids in beta sheets and one to the right containing the amino acids in random coil of this conformation.
One can also plot the dihedral angles in polysaccharides and other polymers in this fashion. For the first two protein side-chain dihedral angles a similar plot is the Janin Plot.
The Hemoglobin molecule has four heme-binding subunits, each largely made of alpha helices.
Secondary structure refers to highly regular local sub-structures. Two main types of secondary structure, the alpha helix and the beta strand, were suggested in 1951 by Linus Pauling' and coworkers.[10]. These secondary structures are defined by patterns of hydrogen bonds between the main-chain peptide groups. They have a regular geometry, being constrained to specific values of the dihedral angles ψ and φ on the Ramachandran plot. Both the alpha helix and the beta-sheet represent a way of saturating all the hydrogen bond donors and acceptors in the peptide backbone. Some parts of the protein are ordered but do not form any regular structures. They should not be confused with random coil, an unfolded polypeptide chain lacking any fixed three-dimensional structure. Several sequential secondary structures may form a "supersecondary unit".[11]
Amino acids vary in their ability to form the various secondary structure elements. Proline and glycine are sometimes known as "helix breakers" because they disrupt the regularity of the α helical backbone conformation; however, both have unusual conformational abilities and are commonly found in turns. Amino acids that prefer to adopt helical conformations in proteins include methionine, alanine, leucine, glutamate and lysine ("MALEK" in amino-acid 1-letter codes); by contrast, the large aromatic residues (tryptophan, tyrosine and phenylalanine) and Cβ-branched amino acids (isoleucine, valine, and threonine) prefer to adopt β-strand conformations. However, these preferences are not strong enough to produce a reliable method of predicting secondary structure from sequence alone.Secondary structure in proteins consists of local inter-residue interactions mediated by hydrogen bonds, or not. The most common secondary structures are alpha helices and beta sheets. Other helices, such as the 310 helix and π helix, are calculated to have energetically favorable hydrogen-bonding patterns but are rarely if ever observed in natural proteins except at the ends of α helices due to unfavorable backbone packing in the center of the helix[12].
α helix
The amino acids in an α helix are arranged in a right-handed helical structure where each amino acid residue corresponds to a 100° turn in the helix (i.e., the helix has 3.6 residues per turn), and a translation of 1.5 Å (0.15 nm) along the helical axis. (Short pieces of left-handed helix sometimes occur with a large content of achiral glycine amino acids, but are unfavorable for the other normal, biological L-amino acids.) The pitch of the alpha-helix (the vertical distance between one consecutive turn of the helix) is 5.4 Å (0.54 nm) which is the product of 1.5 and 3.6. What is most important is that the N-H group of an amino acid forms a hydrogen bond with the C=O group of the amino acid four residues earlier; this repeated hydrogen bonding is the most prominent characteristic of an α-helix. Official international nomenclature specifies two ways of defining α-helices, rule 6.2 in terms of repeating φ,ψ torsion angles and rule 6.3 in terms of the combined pattern of pitch and hydrogen bonding. Different amino-acid sequences have different propensities for forming α-helical structure. Methionine, alanine, leucine, uncharged glutamate, and lysine ("MALEK" in the amino-acid 1-letter codes) all have especially high helix-forming propensities, whereas proline and glycine have poor helix-forming propensities. Proline either breaks or kinks a helix, both because it cannot donate an amide hydrogen bond (having no amide hydrogen), and also because its sidechain interferes sterically with the backbone of the preceding turn - inside a helix, this forces a bend of about 30° in the helix axis. However, proline is often seen as the first residue of a helix, presumably due to its structural rigidity. At the other extreme, glycine also tends to disrupt helices because its high conformational flexibility makes it entropically expensive to adopt the relatively constrained α-helical structure[13].
Representation of a beta hairpinGreek-key motif in protein structure.
β sheet
The first β sheet structure was proposed by William Astbury in the 1930s. He proposed the idea of hydrogen bonding between the peptide bonds of parallel or antiparallel extended β strands. However, Astbury did not have the necessary data on the bond geometry of the amino acids in order to build accurate models, especially since he did not then know that the peptide bond was planar. A refined version was proposed by Linus Pauling and Robert Corey in 1951.
The β sheet (also β-pleated sheet) is the second form of regular secondary structure in proteins, only somewhat less common than alpha helix. Beta sheets consist of beta strands connected laterally by at least two or three backbone hydrogen bonds, forming a generally twisted, pleated sheet. A beta strand (also β strand) is a stretch of polypeptide chain typically 3 to 10 amino acids long with backbone in an almost fully extended conformation.
A very simple structural motif involving β sheets is the β hairpin, in which two antiparallel strands are linked by a short loop of two to five residues, of which one is frequently a glycine or a proline, both of which can assume the unusual dihedral-angle conformations required for a tight turn. However, individual strands can also be linked in more elaborate ways with long loops that may contain alpha helices or even entire protein domains[14].
Greek key motif
The Greek key motif consists of four adjacent antiparallel strands and their linking loops. It consists of three antiparallel strands connected by hairpins, while the fourth is adjacent to the first and linked to the third by a longer loop. This type of structure forms easily during the protein folding process.[15][16] It was named after a pattern common to Greek ornamental artwork (see meander (art))[17].
The β-α-β motif
Due to the chirality of their component amino acids, all strands exhibit a "right-handed" twist evident in most higher-order β sheet structures. In particular, the linking loop between two parallel strands almost always has a right-handed crossover chirality, which is strongly favored by the inherent twist of the sheet. This linking loop frequently contains a helical region, in which case it is called a β-α-β motif. A closely related motif called a β-α-β-α motif forms the basic component of the most commonly observed protein tertiary structure, the TIM barrel[18].
β-meander motif
A simple supersecondary protein topology composed of 2 or more consecutive antiparallel β-strands linked together by hairpin loops.[19][20] This motif is common in β-sheets and can be found in several structural architectures including β-barrels and β-propellers[21].
Psi-loop motif
The psi-loop, Ψ-loop, motif consists of two antiparallel strands with one strand in between that is connected to both by hydrogen bonds.[22] There are four possible strand topologies for single Ψ-loops as cited by Hutchinson et al. (1990). This motif is rare as the process resulting in its formation seems unlikely to occur during protein folding. The Ψ-loop was first identified in the aspartic protease family.[23]
Coiled coils
The possibility of coiled coils for α-keratin was proposed by Francis Crick in 1952 as well as mathematical methods for determining their structure. Remarkably, this was soon after the structure of the alpha helix was suggested in 1951 by Linus Pauling and coworkers.
Coiled coils usually contain a repeated pattern, hxxhcxc, of hydrophobic (h) and charged (c) amino-acid residues, referred to as a heptad repeat. The positions in the heptad repeat are usually labeled abcdefg, where a and d are the hydrophobic positions, often being occupied by isoleucine, leucine or valine. Folding a sequence with this repeating pattern into an alpha-helical secondary structure causes the hydrophobic residues to be presented as a 'stripe' that coils gently around the helix in left-handed fashion, forming an amphipathic structure. The most favorable way for two such helices to arrange themselves in the water-filled environment of the cytoplasm is to wrap the hydrophobic strands against each other sandwiched between the hydrophilic amino acids. It is thus the burial of hydrophobic surfaces, that provides the thermodynamic driving force for the oligomerization. The packing in a coiled-coil interface is exceptionally tight, with almost complete van der Waals contact between the side chains of the a and d residues. This tight packing was originally predicted by Francis Crick in 1952 and is referred to as Knobs into holes packing.
The α-helices may be parallel or anti-parallel, and usually adopt a left-handed super-coil. Although disfavored, a few right-handed coiled coils have also been observed in nature and in designed proteins[24].
Structural features of the three major forms of protein helices[25]
Geometry attribute
α-helix
310 helix
π-helix
Residues per turn
3.6
3.0
4.4
Translation per residue
1.5Å
2.0Å
1.1Å
Radius of helix
2.3Å
1.9Å
2.8Å
Pitch
5.4Å
6.0Å
4.8Å
The four levels of protein structure, from top to bottom: primary structure, secondary structure (β-sheet left, right α-helix), tertiary and quartary structure.
Tertiary structure is considered to be largely determined by the protein's primary structure - the sequence of amino acids of which it is composed. Efforts to predict tertiary structure from the primary structure are known generally as protein structure prediction. However, the environment in which a protein is synthesized and allowed to fold are significant determinants of its final shape and are usually not directly taken into account by current prediction methods. In globular proteins, tertiary interactions are frequently stabilized by the sequestration of hydrophobic amino acid residues in the protein core, from which water is excluded, and by the consequent enrichment of charged or hydrophilic residues on the protein's water-exposed surface. In secreted proteins that do not spend time in the cytoplasm, disulfide bonds between cysteine residues help to maintain the protein's tertiary structure. A variety of common and stable tertiary structures appear in a large number of proteins that are unrelated in both function and evolution - for example, many proteins are shaped like a TIM barrel, named for the enzyme triosephosphateisomerase. Another common structure is a highly stable dimeric coiled coil structure composed of 2-7 alpha helices. The majority of protein structures known to date have been solved with the experimental technique of X-ray crystallography, which typically provides data of high resolution but provides no time-dependent information on the protein's conformational flexibility. A second common way of solving protein structures uses NMR, which provides somewhat lower-resolution data in general and is limited to relatively small proteins, but can provide time-dependent information about the motion of a protein in solution. Dual polarisation interferometry is a time resolved analytical method for determining the overall conformation and conformational changes in surface captured proteins providing complementary information to these high resolution methods. More is known about the tertiary structural features of soluble globular proteins than about membrane proteins because the latter class is extremely difficult to study using these methodshttp://en.wikipedia.org/w/index.php?title=Protein_tertiary_structure&oldid=422486540.
Several proteins are actually assemblies of more than one polypeptide chain, which in the context of the larger assemblage are known as protein subunits. In addition to the tertiary structure of the subunits, multiple-subunit proteins possess a quartary structure, which is the arrangement into which the subunits assemble. Enzymes composed of subunits with diverse functions are sometimes called holoenzymes, in which some parts may be known as regulatory subunits and the functional core is known as the catalytic subunit. Examples of proteins with quartary structure include hemoglobin, DNA polymerase, and ion channels. Other assemblies referred to instead as multiprotein complexes also possess quaternary structure. Examples include nucleosomes and microtubules.
Changes in quartary structure can occur through conformational changes within individual subunits or through reorientation of the subunits relative to each other. It is through such changes, which underlie cooperativity and allostery in "multimeric" enzymes, that many proteins undergo regulation and perform their physiological function.
The above definition follows a classical approach to biochemistry, established at times when the distinction between a protein and a functional, proteinaceous unit was difficult to elucidate. More recently, people refer to protein-protein interaction when discussing quartary structure of proteins and consider all assemblies of proteins as protein complexes.
A conjugated protein is a protein that functions in interaction with other chemical groups attached by covalent bonds or by weak interactions.
Many proteins contain only amino acids and no other chemical groups, and they are called simple proteins. However, other kind of proteins yield, on hydrolysis, some other chemical component in addition to amino acids and they are called conjugated proteins. The nonamino part of a conjugated protein is usually called its prosthetic group. Most prosthetic groups are formed from vitamins. Conjugated proteins are classified on the basis of the chemical nature of their prosthetic groups.
Some examples of conjugated proteins are
A lipoprotein is a biochemical assembly that contains both proteins and lipids water-bound to the proteins. Many enzymes, transporters, structural proteins, antigens, adhesins and toxins are lipoproteins. Examples include the high density (HDL) and low density (LDL) lipoproteins which enable fats to be carried in the blood stream, the transmembrane proteins of the mitochondrion and the chloroplast, and bacterial lipoproteins.
Glycoproteins are proteins that contain oligosaccharide chains (glycans) covalently attached to polypeptide side-chains. The carbohydrate is attached to the protein in a cotranslational or posttranslational modification. This process is known as glycosylation. In proteins that have segments extending extracellularly, the extracellular segments are often glycosylated. Glycoproteins are often important integral membrane proteins, where they play a role in cell-cell interactions. Glycoproteins also occur in the cytosol, but their functions and the pathways producing these modifications in this compartment are less well-understood.Glycoproteins are generally the largest and most abundant group of conjugated proteins. They range from glycoproteins in cell surface membranes that constitute the glycocalyx, to important antibodies produced by leukocytes.
Phosphoproteins are proteins which are chemically bonded to a substance containing phosphoric acid (see phosphorylation for more). The category of organic molecules that includes Fc receptors, Ulks, Calcineurins, K chips, and urocortins.
A protein that contains a metal ion cofactor known as Metalloprotein. Metalloproteins have many different functions in cells, such as enzymes, transport and storage proteins, and signal transduction proteins. Indeed, about one quarter to one third of all proteins require metals to carry out their functions. The metal ion is usually coordinated by nitrogen, oxygen or sulfur atoms belonging to amino acids in the polypeptide chain and/or a macrocyclic ligand incorporated into the protein. The presence of the metal ion allows metalloenzymes to perform functions such as redox reactions that cannot easily be performed by the limited set of functional groups found in amino acids.
Computer-generated 3-D representation of the zinc finger motif of proteins, consisting of an α helix and an antiparallel β sheet. The zinc ion (green) is coordinated by two histidine residues and two cysteine residues.
A hemeprotein (or hemoprotein or haemoprotein), or heme protein, is a metalloprotein containing a heme prosthetic group, either covalently or noncovalently bound to the protein itself. The iron in the heme is capable of undergoing oxidation and reduction (usually to +2 and +3, though stabilized Fe+4 and even Fe+5 species are well known in the peroxidases).
Hemoproteins probably evolved from a primordial strategy allowing to incorporate the iron (Fe) atom contained within the protoporphyrin IX ring of heme into proteins. This strategy has been maintained throughout evolution as it makes hemoproteins responsive to molecules that can bind divalent iron (Fe). These molecules included, but are probably not restricted to, gaseous molecules, such as oxygen (O2) nitric oxide (NO), carbon monoxide (CO) and hydrogen sulfide (H2S). Once bound to the prosthetic heme groups of hemoproteins these gaseous molecules can modulate the activity/function of those hemoproteins in a way that is said to afford signal transduction. Therefore, when produced in biologic systems (cells), these gaseous molecules are referred to as gasotransmitters.Haemoglobin contains the prosthetic group containing iron, which is the haem. It is with in the haem group that carries the oxygen molecule through the binding of the oxygen molecule to the iron ion (Fe2+) found in the haem group[27].
Hemoglobin
Hemoglobin (also spelled haemoglobin and abbreviated Hb or Hgb) is the iron-containing oxygen-transport metalloprotein in the red blood cells of all vertebrates (except the fish family Channichthyidae ) and the tissues of some invertebrates. Hemoglobin in the blood is what transports oxygen from the lungs or gills to the rest of the body (i.e. the tissues) where it releases the oxygen for cell use, and collects carbon dioxide to bring it back to the lungs.
In mammals the protein makes up about 97% of the red blood cells' dry content, and around 35% of the total content (including water). Hemoglobin has an oxygen binding capacity of 1.34 ml O2 per gram of hemoglobin, which increases the total blood oxygen capacity seventyfold.
Hemoglobin is involved in the transport of other gases: it carries some of the body's respiratory carbon dioxide (about 10% of the total) as carbaminohemoglobin, in which CO2 is bound to the globin protein. The molecule also carries the important regulatory molecule nitric oxide bound to a globin protein thiol group, releasing it at the same time as oxygen.
Hemoglobin is also found outside red blood cells and their progenitor lines. Other cells that contain hemoglobin include the A9 dopaminergic neurons in the substantia nigra, macrophages, alveolar cells, and mesangial cells in the kidney. In these tissues, hemoglobin has a non-oxygen-carrying function as an antioxidant and a regulator of iron metabolism.
Hemoglobin and hemoglobin-like molecules are also found in many invertebrates, fungi, and plants. In these organisms, hemoglobins may carry oxygen, or they may act to transport and regulate other things such as carbon dioxide, nitric oxide, hydrogen sulfide and sulfide. A variant of the molecule, called leghemoglobin, is used to scavenge oxygen, to keep it from poisoning anaerobic systems, such as nitrogen-fixing nodules of leguminous plants[28].
phytochromes,
Cytochromes
Cytochrome c with heme c.
Cytochromes are, in general, membrane-bound hemoproteins that contain heme groups and carry out electron transport.
They are found either as monomeric proteins (e.g., cytochrome c) or as subunits of bigger enzymatic complexes that catalyze redox reactions. They are found in the mitochondrial inner membrane and endoplasmic reticulum of eukaryotes, in the chloroplasts of plants, in photosynthetic microorganisms, and in bacteria.
Opsins are a group of light-sensitive 35-55 kDa membrane-bound G protein-coupled receptors of the retinylidene protein family found in photoreceptor cells of the retina. Five classical groups of opsins are involved in vision, mediating the conversion of a photon of light into an electrochemical signal, the first step in the visual transduction cascade. Another opsin found in the mammalian retina, melanopsin, is involved in circadian rhythms and pupillary reflex but not in image-forming.
Flavoproteins are proteins that contain a nucleic acid derivative of riboflavin: the flavin adenine dinucleotide (FAD) or flavin mononucleotide (FMN).
Flavoproteins are involved in a wide array of biological processes, including, but by no means limited to, bioluminescence, removal of radicals contributing to oxidative stress, photosynthesis, DNA repair, and apoptosis. The spectroscopic properties of the flavin cofactor make it a natural reporter for changes occurring within the active site; this makes flavoproteins one of the most-studied enzyme families.
Albumin (Latin: albus, white) refers generally to any protein that is water soluble, which is moderately soluble in concentrated salt solutions, and experiences heat denaturation. They are commonly found in blood plasma, and are unique to other plasma proteins in that they are not glycosylated. Substances containing albumin, such as egg white, are called albuminoids.
Globulin is one of the three types of serum proteins, the others being albumin and fibrinogen. Some globulins are produced in the liver, while others are made by the immune system. The term globulin encompasses a heterogeneous group of proteins with typical high molecular weight, and both solubility and electrophoretic migration rates lower than for albumin.
In biology, histones are highly alkaline proteins found in all eukaryotic cell nuclei and some archaea, which package and order the DNA into structural units called nucleosomes. They are the chief protein components of chromatin, acting as spools around which DNA winds, and play a role in gene regulation.
Peptones are derived from animal milk or meat digested by proteolytic digestion. In addition to containing small peptides, the resulting spray-dried material includes fats, metals, salts, vitamins and many other biological compounds. Peptone is used in nutrient media for growing bacteria and fungi
Proteases occur naturally in all organisms. These enzymes are involved in a multitude of physiological reactions from simple digestion of food proteins to highly-regulated cascades (e.g., the blood-clotting cascade, the complement system, apoptosis pathways, and the invertebrate prophenoloxidase-activating cascade). Proteases can either break specific peptide bonds (limited proteolysis), depending on the amino acid sequence of a protein, or break down a complete peptide to amino acids (unlimited proteolysis). The activity can be a destructive change, abolishing a protein's function or digesting it to its principal components; it can be an activation of a function, or it can be a signal in a signaling pathway.
The best-known role of proteins in the cell is as enzymes, which catalyze chemical reactions. Enzymes are usually highly specific and accelerate only one or a few chemical reactions. Enzymes carry out most of the reactions involved in metabolism, as well as manipulating DNA in processes such as DNA replication, DNA repair, and transcription. Some enzymes act on other proteins to add or remove chemical groups in a process known as post-translational modification. About 4,000 reactions are known to be catalyzed by enzymes. The rate acceleration conferred by enzymatic catalysis is often enormous—as much as 1017-fold increase in rate over the uncatalyzed reaction in the case of orotate decarboxylase (78 million years without the enzyme, 18 milliseconds with the enzyme).
The molecules bound and acted upon by enzymes are called substrates. Although enzymes can consist of hundreds of amino acids, it is usually only a small fraction of the residues that come in contact with the substrate, and an even smaller fraction—three to four residues on average—that are directly involved in catalysis. The region of the enzyme that binds the substrate and contains the catalytic residues is known as the active site[29].
Many proteins are involved in the process of cell signaling and signal transduction. Some proteins, such as insulin, are extracellular proteins that transmit a signal from the cell in which they were synthesized to other cells in distant tissues. Others are membrane proteins that act as receptors whose main function is to bind a signaling molecule and induce a biochemical response in the cell. Many receptors have a binding site exposed on the cell surface and an effector domain within the cell, which may have enzymatic activity or may undergo a conformational change detected by other proteins within the cell.
Antibodies are protein components of adaptive immune system whose main function is to bind antigens, or foreign substances in the body, and target them for destruction. Antibodies can be secreted into the extracellular environment or anchored in the membranes of specialized B cells known as plasma cells. Whereas enzymes are limited in their binding affinity for their substrates by the necessity of conducting their reaction, antibodies have no such constraints. An antibody's binding affinity to its target is extraordinarily high.
Many ligand transport proteins bind particular small biomolecules and transport them to other locations in the body of a multicellular organism. These proteins must have a high binding affinity when their ligand is present in high concentrations, but must also release the ligand when it is present at low concentrations in the target tissues. The canonical example of a ligand-binding protein is haemoglobin, which transports oxygen from the lungs to other organs and tissues in all vertebrates and has close homologs in every biological kingdom. Lectins are sugar-binding proteins which are highly specific for their sugar moieties. Lectins typically play a role in biological recognition phenomena involving cells and proteins. Receptors and hormones are highly specific binding proteins.
Transmembrane proteins can also serve as ligand transport proteins that alter the permeability of the cell membrane to small molecules and ions. The membrane alone has a hydrophobic core through which polar or charged molecules cannot diffuse. Membrane proteins contain internal channels that allow such molecules to enter and exit the cell. Many ion channel proteins are specialized to select for only a particular ion; for example, potassium and sodium channels often discriminate for only one of the two ions[30].
Structural proteins confer stiffness and rigidity to otherwise-fluid biological components. Most structural proteins are fibrous proteins; for example, actin and tubulin are globular and soluble as monomers, but polymerize to form long, stiff fibers that comprise the cytoskeleton, which allows the cell to maintain its shape and size. Collagen and elastin are critical components of connective tissue such as cartilage, and keratin is found in hard or filamentous structures such as hair, nails, feathers, hooves, and some animal shells.
Other proteins that serve structural functions are motor proteins such as myosin, kinesin, and dynein, which are capable of generating mechanical forces. These proteins are crucial for cellular motility of single celled organisms and the sperm of many multicellular organisms which reproduce sexually. They also generate the forces exerted by contracting muscles[31].
Ribbon diagram of the structure of myoglobin, showing colored alpha helices. Such proteins are long, linear molecules with thousands of atoms; yet the relative position of each atom has been determined with sub-atomic resolution by X-ray crystallography. Since it is difficult to visualize all the atoms at once, the ribbon shows the rough path of the protein polymer from its N-terminus (blue) to its C-terminus (red).
Around 90% of the protein structures available in the Protein Data Bank have been determined by X-ray crystallography. This method allows one to measure the 3D density distribution of electrons in the protein (in the crystallized state) and thereby infer the 3D coordinates of all the atoms to be determined to a certain resolution. Roughly 9% of the known protein structures have been obtained by Nuclear Magnetic Resonance techniques. The secondary structure composition can be determined via circular dichroism or dual polarisation interferometry. Cryo-electron microscopy has recently become a means of determining protein structures to high resolution (less than 5 angstroms or 0.5 nanometer) and is anticipated to increase in power as a tool for high resolution work in the next decade. This technique is still a valuable resource for researchers working with very large protein complexes such as virus coat proteins and amyloid fibers.
X-ray crystallography is a method of determining the arrangement of atoms within a crystal, in which a beam of X-rays strikes a crystal and diffracts into many specific directions.
Crystal structures of proteins (which are irregular and hundreds of times larger than cholesterol) began to be solved in the late 1950s, beginning with the structure of sperm whalemyoglobin by Max Perutz and Sir John Cowdery Kendrew, for which they were awarded the Nobel Prize in Chemistry in 1962.[33] Since that success, over 61840 X-ray crystal structures of proteins, nucleic acids and other biological molecules have been determined.[34] For comparison, the nearest competing method in terms of structures analyzed is nuclear magnetic resonance (NMR) spectroscopy, which has resolved 8759 chemical structures.[35] Moreover, crystallography can solve structures of arbitrarily large molecules, whereas solution-state NMR is restricted to relatively small ones (less than 70 kDa). X-ray crystallography is now used routinely by scientists to determine how a pharmaceutical drug interacts with its protein target and what changes might improve it.[36] However, intrinsic membrane proteins remain challenging to crystallize because they require detergents or other means to solubilize them in isolation, and such detergents often interfere with crystallization. Such membrane proteins are a large component of the genome and include many proteins of great physiological importance, such as ion channels and receptors.[37][38]
Protein nuclear magnetic resonance spectroscopy (usually abbreviated protein NMR) is a field of structural biology in which NMR spectroscopy is used to obtain information about the structure and dynamics of proteins. The field was pioneered by Richard R. Ernst and Kurt Wüthrich[1], among others. Protein NMR techniques are continually being used and improved in both academia and the biotech industry. Structure determination by NMR spectroscopy usually consists of several following phases, each using a separate set of highly specialized techniques. The sample is prepared, resonances are assigned, restraints are generated and a structure is calculated and validated
The oxygen-carrying protein hemoglobin was discovered by Hünefeld in 1840. In 1851, Otto Funke published a series of articles in which he described growing hemoglobin crystals by successively diluting red blood cells with a solvent such as pure water, alcohol or ether, followed by slow evaporation of the solvent from the resulting protein solution. Hemoglobin's reversible oxygenation was described a few years later by Felix Hoppe-Seyler.
In 1959 Max Perutz determined the molecular structure of hemoglobin by X-ray crystallography. This work resulted in his sharing with John Kendrew the 1962 Nobel Prize in Chemistry.
The role of hemoglobin in the blood was elucidated by physiologist Claude Bernard. The name hemoglobin is derived from the words heme and globin, reflecting the fact that each subunit of hemoglobin is a globular protein with an embedded heme (or haem) group. Each heme group contains one iron atom, that can bind one oxygen molecule through ion-induced dipole forces. The most common type of hemoglobin in mammals contains four such subunits.
Hemoglobin (also spelled haemoglobin and abbreviated Hb or Hgb) is the iron-containing oxygen-transport metalloprotein in the red blood cells of all vertebrates[1] (except the fish family Channichthyidae ) and the tissues of some invertebrates. Hemoglobin in the blood is what transports oxygen from the lungs or gills to the rest of the body (i.e. the tissues) where it releases the oxygen for cell use, and collects carbon dioxide to bring it back to the lungs.
In mammals the protein makes up about 97% of the red blood cells' dry content, and around 35% of the total content (including water). Hemoglobin has an oxygen binding capacity of 1.34 ml O2 per gram of hemoglobin, which increases the total blood oxygen capacity seventyfold compared to dissolved oxygen in blood. The mammalian hemoglobin molecule can bind (carry) up to four oxygen molecules[39].
Hemoglobin consists mostly of protein (the "globin" chains), and these proteins, in turn, are composed of sequences of amino acids. These sequences are linear, in the manner of letters in a written sentence or beads on a string. In all proteins, it is the variation in the type of amino acids in the protein sequence of amino acids, which determine the protein's chemical properties and function. This is true of hemoglobin, where the sequence of amino acids may affect crucial functions such as the protein's affinity for oxygen.
There is more than one hemoglobin gene. The amino acid sequences of the globin proteins in hemoglobins usually differ between species, although the differences grow with the evolutionary distance between species. For example, the most common hemoglobin sequences in humans and chimpanzees are nearly identical, differing by only one amino acid in both the alpha and the beta globin protein chains. These differences grow larger between less closely related species.
Even within a species, different variants of hemoglobin always exist, although one sequence is usually a "most common" one in each species. Mutations in the genes for the hemoglobin protein in a species result in hemoglobin variants. Many of these mutant forms of hemoglobin cause no disease. Some of these mutant forms of hemoglobin, however, cause a group of hereditary diseases termed the hemoglobinopathies. The best known hemoglobinopathy is sickle-cell disease, which was the first human disease whose mechanism was understood at the molecular level. A (mostly) separate set of diseases called thalassemias involves underproduction of normal and sometimes abnormal hemoglobins, through problems and mutations in globin gene regulation. All these diseases produce anemia[40].
Hemoglobin variants are a part of the normal embryonic and fetal development, but may also be pathologic mutant forms of hemoglobin in a population, caused by variations in genetics. Some well-known hemoglobin variants such as sickle-cell anemia are responsible for diseases, and are considered hemoglobinopathies. Other variants cause no detectable pathology, and are thus considered non-pathological variants[41].
In the embryo:
Gower 1 (ζ2ε2)
Gower 2 (α2ε2) (PDB 1A9W)
Hemoglobin Portland (ζ2γ2)
In the fetus:
Hemoglobin F (α2γ2) (PDB 1FDH)
In adults:
Hemoglobin A (α2β2) (PDB 1BZ0) - The most common with a normal amount over 95%
Hemoglobin A2 (α2δ2) - δ chain synthesis begins late in the third trimester and in adults, it has a normal range of 1.5-3.5%
Hemoglobin F (α2γ2) - In adults Hemoglobin F is restricted to a limited population of red cells called F-cells. However, the level of Hb F can be elevated in persons with sickle-cell disease and beta-thalassemia.
Variant forms that cause disease:
Hemoglobin H (β4) - A variant form of hemoglobin, formed by a tetramer of β chains, which may be present in variants of α thalassemia.
Hemoglobin Barts (γ4) - A variant form of hemoglobin, formed by a tetramer of γ chains, which may be present in variants of α thalassemia.
Hemoglobin S (α2βS2) - A variant form of hemoglobin found in people with sickle cell disease. There is a variation in the β-chain gene, causing a change in the properties of hemoglobin, which results in sickling of red blood cells.
Hemoglobin C (α2βC2) - Another variant due to a variation in the β-chain gene. This variant causes a mild chronic hemolytic anemia.
Hemoglobin E (α2βE2) - Another variant due to a variation in the β-chain gene. This variant causes a mild chronic hemolytic anemia.
Hemoglobin AS - A heterozygous form causing Sickle cell trait with one adult gene and one sickle cell disease gene
Hemoglobin SC disease - Another heterozygous form with one sickle gene and another encoding Hemoglobin C.
Variations in hemoglobin amino acid sequences, as with other proteins, may be adaptive. For example, recent studies have suggested genetic variants in deer mice that help explain how deer mice that live in the mountains are able to survive in the thin air that accompanies high altitudes. A researcher from the University of Nebraska-Lincoln found mutations in four different genes that can account for differences between deer mice that live in lowland prairies versus the mountains. After examining wild mice captured from both highlands and lowlands, it was found that: the genes of the two breeds are “virtually identical–except for those that govern the oxygen-carrying capacity of their hemoglobin”. “The genetic difference enables highland mice to make more efficient use of their oxygen”, since less is available at higher altitudes, such as those in the mountains. Mammoth hemoglobin featured mutations that allowed for oxygen delivery at lower temperatures, thus enabling mammoths to migrate to higher latitudes during the Pleistocene.
Sickle-cell disease is inherited in the autosomal recessive pattern.Distribution of the sickle-cell trait shown in pink and purpleHistorical distribution of malaria (no longer endemic in Europe) shown in greenModern distribution of malaria
Sickle-cell disease (SCD) or sickle-cell anaemia (or anemia; SCA) or drepanocytosisis an autosomal recessive genetic blood disorder, with overdominance, characterized by red blood cells that assume an abnormal, rigid, sickle shape. Sickling decreases the cells' flexibility and results in a risk of various complications. The sickling occurs because of a mutation in the haemoglobin gene. Life expectancy is shortened, with studies reporting an average life expectancy of 42 in males and 48 in females.Sickle-cell anaemia is caused by a point mutation in the β-globin chain of haemoglobin, causing the hydrophilic amino acid glutamic acid to be replaced with the hydrophobic amino acid valine at the sixth position. The β-globin gene is found on the short arm of chromosome 11. The association of two wild-type α-globin subunits with two mutant β-globin subunits forms haemoglobin S (HbS). Under low-oxygen conditions (being at high altitude, for example), the absence of a polar amino acid at position six of the β-globin chain promotes the non-covalent polymerisation (aggregation) of haemoglobin, which distorts red blood cells into a sickle shape and decreases their elasticity.
The loss of red blood cell elasticity is central to the pathophysiology of sickle-cell disease. Normal red blood cells are quite elastic, which allows the cells to deform to pass through capillaries. In sickle-cell disease, low-oxygen tension promotes red blood cell sickling and repeated episodes of sickling damage the cell membrane and decrease the cell's elasticity. These cells fail to return to normal shape when normal oxygen tension is restored. As a consequence, these rigid blood cells are unable to deform as they pass through narrow capillaries, leading to vessel occlusion and ischaemia.
The actual anaemia of the illness is caused by haemolysis, the destruction of the red cells inside the spleen, because of their misshape. Although the bone marrow attempts to compensate by creating new red cells, it does not match the rate of destruction. Healthy red blood cells typically live 90–120 days, but sickle cells only survive 10–20 days.
Normally, humans have Haemoglobin A, which consists of two alpha and two beta chains, Haemoglobin A2, which consists of two alpha and two delta chains and Haemoglobin F, consisting of two alpha and two gamma chains in their bodies. Of these, Haemoglobin A makes up around 96-97% of the normal haemoglobin in humans.
Sickle-cell gene mutation probably arose spontaneously in different geographic areas, as suggested by restriction endonuclease analysis. These variants are known as Cameroon, Senegal, Benin, Bantu and Saudi-Asian. Their clinical importance springs from the fact that some of them are associated with higher HbF levels, e.g., Senegal and Saudi-Asian variants, and tend to have milder disease.[42]
In people heterozygous for HgbS (carriers of sickling haemoglobin), the polymerisation problems are minor, because the normal allele is able to produce over 50% of the haemoglobin. In people homozygous for HgbS, the presence of long-chain polymers of HbS distort the shape of the red blood cell from a smooth doughnut-like shape to ragged and full of spikes, making it fragile and susceptible to breaking within capillaries. Carriers have symptoms only if they are deprived of oxygen (for example, while climbing a mountain) or while severely dehydrated. Under normal circumstances, these painful crises occur about 0.8 times per year per patient. The sickle-cell disease occurs when the seventh amino acid (if the initial methionine is counted), glutamic acid, is replaced by valine to change its structure and function.
The gene defect is a known mutation of a single nucleotide ( single-nucleotide polymorphism - SNP) (A to T) of the β-globin gene, which results in glutamic acid being substituted by valine at position 6. Haemoglobin S with this mutation are referred to as HbS, as opposed to the normal adult HbA. The genetic disorder is due to the mutation of a single nucleotide, from a GAG to GTG codonmutation, becoming a GUG codon by transcription. This is normally a benign mutation, causing no apparent effects on the secondary, tertiary, or quaternary structure of haemoglobin in conditions of normal oxygen concentration. What it does allow for, under conditions of low oxygen concentration, is the polymerization of the HbS itself. The deoxy form of haemoglobin exposes a hydrophobic patch on the protein between the E and F helices. The hydrophobic residues of the valine at position 6 of the beta chain in haemoglobin are able to associate with the hydrophobic patch, causing haemoglobin S molecules to aggregate and form fibrous precipitates.
The allele responsible for sickle-cell anaemia is autosomalrecessive and can be found on the short arm of chromosome 11. A person that receives the defective gene from both father and mother develops the disease; a person that receives one defective and one healthy allele remains healthy, but can pass on the disease and is known as a carrier. If two parents who are carriers have a child, there is a 1-in-4 chance of their child developing the disease and a 1-in-2 chance of their child's being just a carrier. Since the gene is incompletely recessive, carriers can produce a few sickled red blood cells, not enough to cause symptoms, but enough to give resistance to malaria. Because of this, heterozygotes have a higher
– fitness than either of the homozygotes. This is known as heterozygote advantage.
Due to the adaptive advantage of the heterozygote, the disease is still prevalent, especially among people with recent ancestry in malaria-stricken areas, such as Africa, the Mediterranean, India and the Middle East.[43] Malaria was historically endemic to southern Europe, but it was declared eradicated in the mid-20th century, with the exception of rare sporadic cases.[44]
The malaria parasite has a complex life cycle and spends part of it in red blood cells. In a carrier, the presence of the malaria parasite causes the red blood cells with defective haemoglobin to rupture prematurely, making the plasmodium unable to reproduce. Further, the polymerization of Hb affects the ability of the parasite to digest Hb in the first place. Therefore, in areas where malaria is a problem, people's chances of survival actually increase if they carry sickle-cell trait (selection for the heterozygote).
In the USA, where there is no endemic malaria, the prevalence of sickle-cell anaemia among blacks is lower (about 0.25%) than in West Africa (about 4.0%) and is falling. Without endemic malaria from Africa, the sickle cell mutation is purely disadvantageous and will tend to be selected out of the affected population. Another factor limiting the spread of sickle-cell genes in North America is the absence of cultural proclivities to polygamy.[45]Inheritance
Sickle-cell conditions are inherited from parents in much the same way as blood type, hair colour and texture, eye colour, and other physical traits. The types of haemoglobin a person makes in the red blood cells depend on what haemoglobin genes are inherited from his parents. If one parent has sickle-cell anaemia (SS) and the other has sickle-cell trait (AS), there is a 50% chance of a child's having sickle-cell disease (SS) and a 50% chance of a child's having sickle-cell trait (AS). When both parents have sickle-cell trait (AS),a child has a 25% chance (1 of 4) of sickle-cell disease (SS), as shown in the diagram above.
Hemoglobin has a quaternary structure characteristic of many multi-subunit globular proteins. Most of the amino acids in hemoglobin form alpha helices, connected by short non-helical segments. Hydrogen bonds stabilize the helical sections inside this protein, causing attractions within the molecule, folding each polypeptide chain into a specific shape. Hemoglobin's quaternary structure comes from its four subunits in roughly a tetrahedral arrangement.
In most vertebrates, the hemoglobin molecule is an assembly of four globular protein subunits. Each subunit is composed of a protein chain tightly associated with a non-protein heme group. Each protein chain arranges into a set of alpha-helix structural segments connected together in a globin fold arrangement, so called because this arrangement is the same folding motif used in other heme/globin proteins such as myoglobin. This folding pattern contains a pocket that strongly binds the heme group.
A heme group consists of an iron (Fe) ion (charged atom) held in a heterocyclic ring, known as a porphyrin. This porphyrin ring consists of four pyrrole molecules cyclically linked together (by methene bridges) with the iron ion bound in the center. The iron ion, which is the site of oxygen binding, coordinates with the four nitrogens in the center of the ring, which all lie in one plane. The iron is bound strongly (covalently) to the globular protein via the imidazole ring of the F8 histidine residue (also known as the proximal histidine) below the porphyrin ring. A sixth position can reversibly bind oxygen by a coordinate covalent bond,[24] completing the octahedral group of six ligands. Oxygen binds in an "end-on bent" geometry where one oxygen atom binds Fe and the other protrudes at an angle. When oxygen is not bound, a very weakly bonded water molecule fills the site, forming a distorted octahedron.
Even though carbon dioxide is carried by hemoglobin, it does not compete with oxygen for the iron-binding positions, but is actually bound to the protein chains of the structure.
The iron ion may be either in the Fe2+ or in the Fe3+ state, but ferrihemoglobin (methemoglobin) (Fe3+) cannot bind oxygen. In binding, oxygen temporarily and reversibly oxidizes (Fe2+) to (Fe3+) while oxygen temporally turns into superoxide, thus iron must exist in the +2 oxidation state to bind oxygen. If superoxide ion associated to Fe3+ is protonated the hemoglobin iron will remain oxidized and incapable to bind oxygen. In such cases, the enzyme methemoglobin reductase will be able to eventually reactivate methemoglobin by reducing the iron center.
In adult humans, the most common hemoglobin type is a tetramer (which contains 4 subunit proteins) called hemoglobin A, consisting of two α and two β subunits non-covalently bound, each made of 141 and 146 amino acid residues, respectively. This is denoted as α2β2. The subunits are structurally similar and about the same size. Each subunit has a molecular weight of about 17,000 daltons, for a total molecular weight of the tetramer of about 68,000 daltons (64,458 g/mol).[26] Thus, 1 g/dL = 0.01551 mmol/L. Hemoglobin A is the most intensively studied of the hemoglobin molecules.
In human infants, the hemoglobin molecule is made up of 2 α chains and 2 gamma chains. The gamma chains are gradually replaced by β chains as the infant grows.
The four polypeptide chains are bound to each other by salt bridges, hydrogen bonds, and the hydrophobic effect. There are two kinds of contacts between the α and β chains: α1β1 and α1β2.
Oxygen Saturation
In general, hemoglobin can be saturated with oxygen molecules (oxyhemoglobin), or desaturated with oxygen molecules (deoxyhemoglobin).
Oxyhemoglobin
Oxyhemoglobin is formed during physiological respiration when oxygen binds to the heme component of the protein hemoglobin in red blood cells. This process occurs in the pulmonary capillaries adjacent to the alveoli of the lungs. The oxygen then travels through the blood stream to be dropped off at cells where it is utilized in aerobic glycolysis and in the production of ATP by the process of oxidative phosphorylation. It does not, however, help to counteract a decrease in blood pH. Ventilation, or breathing, may reverse this condition by removal of carbon dioxide, thus causing a shift up in pH.
Hemoglobin exists in two forms, a taut form (T) and a relaxed form (R). Various factors such as low pH, high CO2 and high 2,3 BPG at the level of the tissues favor the taut form, which has low oxygen affinity and releases oxygen in the tissues. The opposite of these aforementioned factors at the level of the lung capillaries favors the relaxed form which can better bind oxygen[46].
Deoxyhemoglobin
Deoxyhemoglobin is the form of hemoglobin without the bound oxygen. The absorption spectra of oxyhemoglobin and deoxyhemoglobin differ. The oxyhemoglobin has significantly lower absorption of the 660 nm wavelength than deoxyhemoglobin, while at 940 nm its absorption is slightly higher. This difference is used for measurement of the amount of oxygen in patient's blood by an instrument called pulse oximeter. This difference also accounts for the presentation of cyanosis, the blue to purplish color that tissues develop during hypoxiaOxyhemoglobin is formed during physiological respiration when oxygen binds to the heme component of the protein hemoglobin in red blood cells. This process occurs in the pulmonary capillaries adjacent to the alveoli of the lungs. The oxygen then travels through the blood stream to be dropped off at cells where it is utilized in aerobic glycolysis and in the production of ATP by the process of oxidative phosphorylation. It does not, however, help to counteract a decrease in blood pH. Ventilation, or breathing, may[47].
↑ Photo-leucine and photo-methionine allow identification of protein-protein interactions in living cells.Nature Methods:4,261–7,2005
↑Lovell SC; et al. (2003). "Structure validation by Cα geometry: φ,ψ and Cβ deviation". Proteins. 50 (3): 437–450. doi:10.1002/prot.10286. PMID12557186. {{cite journal}}: Explicit use of et al. in: |author= (help)
↑Hutchinson EG, Thornton JM (1990). "HERA--a program to draw schematic diagrams of protein secondary structures". Proteins. 8 (3): 203–12. doi:10.1002/prot.340080303. PMID2281084.
↑Astrid Sigel, Helmut Sigel and Roland K.O. Sigel, ed. (2008). Nickel and Its Surprising Impact in Nature. Metal Ions in Life Sciences. Vol. 2. Wiley. ISBN978-0-470-01671-8.


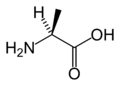 L-Alanine
L-Alanine L-Arginine
L-Arginine L-Asparagine
L-Asparagine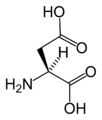 L-Aspartic acid
L-Aspartic acid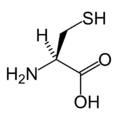 L-Cysteine
L-Cysteine L-Glutamic acid
L-Glutamic acid L-Glutamine
L-Glutamine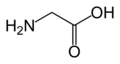 Glycine
Glycine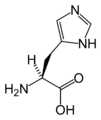 L-Histidine
L-Histidine L-Isoleucine
L-Isoleucine L-Leucine
L-Leucine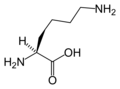 L-Lysine
L-Lysine L-Methionine
L-Methionine L-Phenylalanine
L-Phenylalanine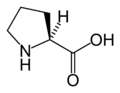 L-Proline
L-Proline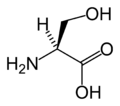 L-Serine
L-Serine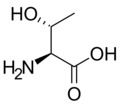 L-Threonine
L-Threonine L-Tryptophan
L-Tryptophan L-Tyrosine
L-Tyrosine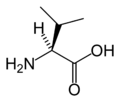 L-Valine
L-Valine L-Selenocysteine
L-Selenocysteine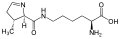 L-Pyrrolysine
L-Pyrrolysine
.png)






































