
Proteomics/Protein Primary Structure/Alternative Splicing
This Section:
Alternative splicing: Biological function
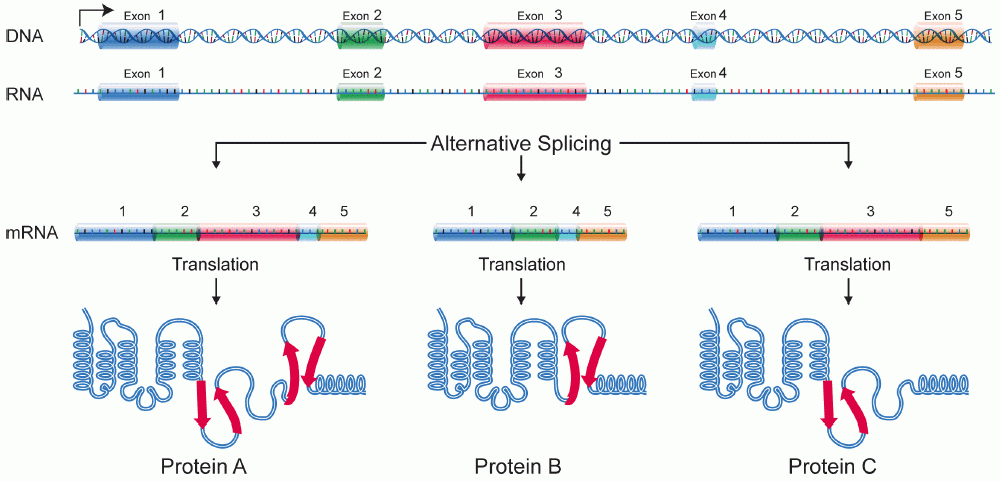
[edit | edit source]Alternative splicing is the process in which the primary transcript of a gene is reorganized to produce one of multiple mature mRNA transcripts. By manipulating exonic regions, the sequence of the amino acids produced from the mRNA can be controlled, resulting in the production of different protein sequences with different structures and functions. [1] Alternative splicing has been observed as a mechanism to produce tissue specific proteins from a single gene. This allows for increased cellular specialization without the involvement of genetic modification. This is a very critical aspect of development in that it allows the determination of cell fates in a manner that still allows for flexibility and modification. Alternative splicing has especially been studied in the context of neuronal development, [2] [3] however many spliced variants have been identified that are only expressed in certain tissues during development that seem to play a critical role in triggering cell-type specific gene expression. [4]

{kind=link}
Alternative Splicing is a major source of protein diversity in living organisms. It has been estimated that at least 30% of all genes in the human genome are alternatively spliced and this number continues to expand. It was originally thought that the number of alternatively spliced genes accounted for only 5% of proteins in humans. With the unveiling of the human genome it was revealed that the human genome contains less than 30,000 genes [5]. This could potentially account for the huge gap between the relatively small number of genes and the vast number of proteins in the proteome.
It has been suggested that alternative splicing is the source of higher level complexity in eukaryotes [6]. This idea is based on the thought that more complex organisms will alternatively splice their genes more often to obtain more possible mRNA sequences. However evidence shows that the level of alternative splicing between different complexities of organisms is not significant. Providing evidence of the contrary, a study was done using ESTs (expressed sequence tags [7] ). ESTs, being a portion of a transcribed mRNA , are very effective at detecting alternatively spliced genes. As more EST studies are done, it has become apparent that there is a greater number of alternatively spliced genes than previously thought (see above).
Examples of Alternative Splicing
[edit | edit source]Alternative splicing has been implicated in several diseases. An example of a disease that plays a role in alternative splicing is Rett syndrome (RTT). This disease is found primarily in girls and is characterized by problems with forming connections between neurons, or synapses [8] Girls with this disease typically develop normally until reaching 6-18 months [9]. Beyond this age, they begin to show a decrease in motor and language skills that is typically accompanied by hand wringing motions.[10] It is believed that the gene MeCP2 produces a mutant protein that disrupts its normal function as co-regulator of alternative splicing in some proteins. When this mutant version of MeCP2 is produced, a protein that lacks critical functions of the wild type is produced that does not allow regulation of alternative splicing. MeCP2 was originally identified as a methylation dependent transcriptional repressor. However it was discovered that MeCP2 has alternative functions, that was first elucidated by MeCP2's interaction with Y box binding protein (YB-1 a component of mRNPs). The complex of YB-1 and MeCP2 was shown to regulate splicing of reporter minigenes.
Microarray data from mice with mutations causing phenotypes resembling Rett showed a significant difference in the number of alternatively spliced genes vs wild type mice. Alternatively spliced transcripts matched cell-based assays which showed MeCP2's role in cassette exon changes.
Splicing Mechanism
[edit | edit source]Splicing occurs through the mechanism of the Spliceosome, a complex composed of a number of proteins and snRNA's. including the snRNAs U1, U2, U3, U4, U5 and U6 [11]. These snRNAs recognize the splice sites and then recruit other proteins that connect the splice sites. These splice sites are then brought together through the interaction of these proteins which form the spliceosome. Once the spliceosome has formed, the sites are cleaved to bring the correct exons (or introns) together. Splice sites in organisms have not been well characterized. Some progress has been made in the area of computational prediction using HMM profiling and other forms of machine learning. This will be discussed later in this article. The 5' end of an intron is characterized with a consensus sequence most commonly ending in "GU". This region is known as the splice donar site. The 3' end of an intron is known as the splice acceptor site and contains a consensus sequence with an AG at the intron/exon boundry. There is also an adenine, usually close to the 3' end of an intron, known as the branch point that plays a role in the splicing mechanism. During splicing, U1 begins the formation of a complex at the splice donor site, while U2 recruits portions of the spliceosome complex to the splice acceptor site. U4, U5, and U6 bridge the gap between these two sites, causing the intron to fold. The DNA is digested, at the splice donar site. This portion of the intron is then ligated to the branch point, forming a lariat. This brings the exonic regions is close proximity to each other, allowing for ligation. Finally, the splice acceptor site is cut and the exonic regions are joined. [12]

{kind=link}
There are 4 common types of alternative splicing, they are as follows:
- Alternative promoter selection: A different promoter is used for different splice variants. This results in a different start of the mRNA transcript.
- Alternative selection of cleavage/polyadenylation sites: Different exons are spliced based on recognition of different cleavage or polyadenylation sites, entire exons can be skipped. Results in a different exon at the 3’ end of the transcript.
- Intron retention: Introns are used as coding regions. A sequence that is normally considered an Intron is retained in the final transcript that serves as a template for translation.
- Exon cassette: Entire exons can be skipped in the middle of the protein, resulting in a different transcript

{kind=link}
The Growing Need for Protein Identification:
[edit | edit source]Proteins are the basic unit of structure and function within living systems. Consequently the field of proteomics has taken on added significance in the modern study of biology. The genomic revolution, which culminated in the sequencing of many genomes, has generated a tremendous amount of data. The field of proteomics [13] has unfortunately been lagging behind, resulting in a disconnect between genomic information and observable phenotypes. Originally, proteins and protein dependent pathways were studied individually. Recently an emphasis on systems biology has lead to changes in this methodology. Entire cells are being characterized with high throughput techniques.

{kind=link}
Mass Spectrometry has become the gold standard of protein identification [14] [15]. Briefly, proteins are broken down into peptides, suspended into a gas phase by one of a various number of methods, ionized, and sent through a detector which can determine the mass to charge ratio of various peptides. Mass spectroscopy can be easily automated and combined with other forms of protein separation, making it an ideal candidate for high throughput analysis. In addition, thousands of peptides can be identified from a single source at one time, making this technique much more applicable to systems biology then older techniques such as Edman degradation. Mass spectroscopy can also be used to identify single proteins of interest that have been isolated using other techniques, such as chromatography.

{kind=link}
Splicing and Protein Identification
[edit | edit source]A major drawback of mass spec analysis, and even identification by Edman degredation, is that proteins must be digested into peptides before identification. Often for use with shotgun sequencing [16], proteins are digested before any form of isolation process is done. In order to confirm identifications protein databases are searched in order to match unique peptides to entire proteins. This process is made more complicated by the immense level of sequence homology shared by peptides produced by alternative splicing. [17]. These proteins, although sharing similar primary structures may have very different or even antagonistic functions, making their identification critical from a biological perspective. What is more, the level of alternative splicing that occurs is not well characterized so it is not even well known which proteins cannot be unambiguously identified. These problems will likely remain until alternative splicing is well documented or can be predicted efficiently computationally.
Effects to analyze alternative splicing:
[edit | edit source]In vivo analysis is currently the most accurate way to identify alternative splicing, either at the transcript, or in some cases at the protein level. Numerous databases exist that document proteins known to undergo alternative splicing, including the Alternative Splicing Database [18], and the Transcript Diversity database [19]. While these are good references, as discussed above, the estimates of even the amount of alternative splicing present in eukaryotic organisms vary dramatically. There is, therefore, very little information on how complete these databases are.
Steps have also been taken to computationally predict alternative splicing [20]. Generally these algorithms combine gene finding approaches with experimental data. Splice sites are recognized and rated in efficiency based on consensus sequences. Sequences are then matched to known expressed sequence tags to make predictions. Tools such as BLAST, Spidey, and SIM4 can be adapted for these processes. Most modern computational tools encounter difficulty in comparing genomic data to sequences as small and variable as splice sites. False positives and negatives are fairly common [21]. New approaches are still being developed.
References
[edit | edit source]- ↑ Möröy, T et al. "The impact of alternative splicing in vivo: Mouse models show the way". http://www.rnajournal.org/cgi/content/full/13/8/1155 obtained April, 2008
- ↑ Goymer, P. "Development: Alternative splicing switches on the brain". Nature Reviews Neuroscience 8, 576 (August 2007). http://www.nature.com/nrn/journal/v8/n8/full/nrn2200.html obtained April, 2008
- ↑ Azuma, N et al. "The Pax6 isoform bearing an alternative spliced exon promotes the development of the neural retinal structure". Human Molecular Genetics 2005 14(6):735-745 http://hmg.oxfordjournals.org/cgi/content/full/14/6/735 obtained April, 2008
- ↑ Michelle Lesimple et al. DNA and Cell Biology. June 1, 2000, 19(6): 365-376 http://www.liebertonline.com/doi/abs/10.1089/10445490050043335 obtained April, 2008.
- ↑ Alternative RNA Splicing. ExonHit Therapeutics. http://www.exonhit.com/index.php?page=59. obtained April, 2008
- ↑ Brett D et al. Alternative splicing and genome. Nature Genetics 30, 29 - 30 (2001) http://www.nature.com/ng/journal/v30/n1/abs/ng803.html;jsessionid=BF0AED8347574D063F5E347EC693AE83 obtained April, 2008
- ↑ http://en.wikipedia.org/wiki/Expressed_sequence_tag
- ↑ "Rett Syndrome Fact Sheet". National Institute of Disorders and Stroke. http://www.ninds.nih.gov/disorders/rett/detail_rett.htm#109713277 obtained April, 2008.
- ↑ "Rett Gene Regulates Alternative Splicing". Science Daily. http://www.sciencedaily.com/releases/2005/10/051019002531.htm obtained April, 2008
- ↑ Young, J et al. "Regulation of RNA splicing by the methylation-dependent transcriptional repressor methyl-CpG binding protein 2". PNAS vol. 102:49 http://www.pubmedcentral.nih.gov/picrender.fcgi?artid=1266160&blobtype=pdf obtained April, 2008.
- ↑ Cáceres, J et al. "Alternative splicing: multiple control mechanisms and involvement in human disease". Trends in Genetics Volume 18, Issue 4, 1 April 2002, Pages 186-193. http://www.sciencedirect.com/science?_ob=ArticleURL&_udi=B6TCY-45FYM7X-F&_user=47004&_rdoc=1&_fmt=&_orig=search&_sort=d&view=c&_acct=C000005018&_version=1&_urlVersion=0&_userid=47004&md5=eea15989e03f8b963bdc33384a4ef93b
- ↑ "From DNA to RNA". Molecular Biology of the Cell. http://www.ncbi.nlm.nih.gov/books/bv.fcgi?rid=mboc4.figgrp.1020 obtained April, 2008
- ↑ http://en.wikipedia.org/wiki/Proteomics
- ↑ http://en.wikipedia.org/wiki/Mass_spec
- ↑ "Protein identification by mass spectrometry". US Department of Energy Research News http://www.eurekalert.org/features/doe/2001-06/drnl-pib061902.php obtain rf April 2008
- ↑ Lu, B et al. "Improving Protein Identification Sensitivity by Combining MS and MS/MS Information for Shotgun Proteomics Using LTQ-Orbitrap High Mass Accuracy". Anal. Chem., 80 (6), 2018 -2025, 2008. Datahttp://pubs.acs.org/cgi-bin/article.cgi/ancham/2008/80/i06/html/ac701697w.html obtained April 2008
- ↑ Nesvizhskii, A et al. "Interpretation of Shotgun Proteomic Data: The Protein Inference Problem". Molecular & Cellular Proteomics 4:1419-1440, 2005. http://www.mcponline.org/cgi/content/full/4/10/1419 obtained April 2005
- ↑ Dralyuk, I et al. Alternative Splicing Database. http://hazelton.lbl.gov/~teplitski/alt/ obtained April 2008
- ↑ hanaraj T.A., Stamm S., Clark F., Riethoven J.J.M, Le Texier V., and Muilu J. ASD: the Alternative Splicing Database. Nucl. Acids. Res. 2004 32: D64-D69. http://www.ebi.ac.uk/asd/ obtained April, 2008
- ↑ Bonizzoni, P et al. "Computational methods for alternative splicing prediction". Briefings in Functional Genomics & Proteomics. Volume 5(1), Pp. 46-51. http://bfgp.oxfordjournals.org/cgi/content/full/5/1/46 obtained April, 2008
- ↑ Black, D et al. "Protein Diversity from Alternative Splicing A Challenge for Bioinformatics and Post-Genome Biology". Cell. Volume 103, Issue 3, 27 October 2000, Pages 367-370. http://www.sciencedirect.com/science?_ob=ArticleURL&_udi=B6WSN-41T1FBT-2&_user=47004&_rdoc=1&_fmt=&_orig=search&_sort=d&view=c&_acct=C000005018&_version=1&_urlVersion=0&_userid=47004&md5=d3b999a5147780e759f2e9aa7b8c47e7 obtained April, 2008