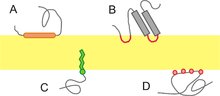
A membrane protein is any protein found in a biological membrane. They participate in various biological processes, such as cell signaling-transduction pathways. The membrane proteins also play a strong role in controlling a wide array of gradients such as chemical, electrical, and mechanical gradients and are responsible for cell structure during key cell events such as division. Due to their many functions in the membrane, they are in high concentration on the surface of the membrane. They may also act as channels that move specific molecules into and out of the membrane. Theses proteins fall into two main categories, depending upon how strongly the protein interacts with the membrane.
The two main categories are listed below:
Integral proteins: (also called intrinsic proteins) These are proteins are characterized by strong interaction with the membrane, which can only be broken by the addition of detergents or some other nonpolar solvent. Essentially, they are permanently bounded to the membrane. They may span across the entire phospholipid bi-layer, or be monotopic.Integral proteins. They have one or more segments that are permanently embedded within the phospholipid bilayer and have their domains on both sides of the membrane. Most integral proteins contain residues with hydrophobic side chains that interact with fatty acyl groups of the membrane phospholipids, thus anchoring the protein to the membrane. Most integral proteins span the entire phospholipid bilayer.It interacts extensively with the hydrocarbon chain of membrane lipid and they can be released by agents that compete for these nonpolar interaction.
Peripheral proteins: (or extrinsic proteins) are proteins that have a much weaker interaction with the membrane than integral proteins. These attachments tend to be much more temporary and can be displaced via treatment with a polar reagent. Peripheral proteins. They are temporarily bound either to the lipid bilayer or to integral proteins by hydrophobic, electrostatic, and other non-covalent interactions. This type of proteins does not interact with the hydrophobic core of the phospholipid bilayer. They are usually bound to membrane by interactions with integral membrane proteins or directly by interactions with lipid polar head groups. This polar interaction can be disrupted by the change in pH.
There is also an alternative method of classification for membrane proteins. It arises from membrane proteins, such as colicin A and alpha-hemolysin. These do not fit to either integral or peripheral classification. In this alternative system of classification, the membrane proteins are divided into integral and amphitropic.
Membrane proteins
Biological membranes have phospholipid bilayer structure which contains a set of proteins which help plasma membrane to carry its distinctive functions. Membrane proteins can be attached to the membrane or associated with the membrane of a cell or an organelle. Membrane proteins can be classified into two groups based on the strength of their association with the membrane:
Some membrane proteins are found bounded to lipid bilayer and generally involved in cell-cell signaling or interactions. Others are embedded within the lipid bilayer of a cell often form channels and pores. Membrane proteins can be attached to both the outside and inside of the cell membrane.
Proteins can be attached to the cell membrane in a variety of ways. One method involves irreversible covalent modification. Both Ras (a GTPase) and Src (protein tyrosine kinase) are known to be modified in this manner. Both of these proteins participate in signal transduction pathways, but upon covalent attachment of a lipid group they become attached to the inner face of the cytoplasmic membrane. When Ras and Src are affixed to the cell membrane they are better able to receive and transmit information being transferred via their respective signal transduction pathways.Membrane proteins can be made of alpha helices or beta strands, or the combination of both alpha helices or beta strands. For example the channel protein called Porin is made up of entirely beta strands, while the enzyme protein called prostaglandin is made entirely of the alpha helices.
Membrane proteins can be alpha - helices or beta - strands. Proteins can span the membrane with alpha helices. Membrane - spanning alpha - helices are the most common structural motif in membrane proteins. An examination of the primary structure reveals that most amino acids in the membrane protein are nonpolar and very few are charged. One of the first alpha - proteins found was the bacteriorhodopsin. It uses light energy to transport protons from inside the cell to outside generating a proton gradient used to form ATP. The seven alpha - helices are closely packed and arranged perpendicular to the plane of the cell and they span 45A in width. Membrane proteins can also be made out of beta strands. Beta Strands form channel proteins. They are less common than alpha - helices. Channel proteins are formed by beta arrangement of beta strands. Each strand is hydrogen bonded to its neighbor in an anti-parallel arrangement, forming a single beta sheet. The beta sheet then curls up to form a hollow cylinder that forms a channel in the membrane. An example is Porin. The outside surface is non-polar and interacts with the hydrocarbon core of the membrane, while the inside channel is hydrophilic and filled with water. The arrangement of polar and non-polar is accomplished by the alternation of hydrophobic and hydrophilic amino acids along with each beta strand.
Many membrane proteins have quaternary structures consisting of multiple subunits. This oligomerization in membrane proteins is beneficial to their functions, stability, genetic efficiency and maybe even optimizing productive output per unit area of the membrane. Cytochrome b6f serves as an example of quarterary structure affecting membrane protein function. This protein consist of two subunits which are connected by a bridge so that electrons can be transferred between them. As for stability, a quaternary protein consisting of 2, 3 or 4 subunits would be 2, 3 or 4 times more stable if a stability improving mutation were to occur on each subunit. It would be more genetically efficient to have all the subunits of a quaternary protein be coded for by 1 gene than to have each of its subunits be coded for by a different gene. In this way, a quaternary protein can be coded for with minimal genetic space. One example of this are the ion channels that span the membrane. the entirety of these quaternary membrane proteins are made from repeating, identical subunits stacked on top of each other. Everyone of these subunits and therefore the iono channel as a whole, is then coded by and translated from 1 single gene. In addition, oligomerization may also contribute to maximizing functional output as it allows membrane proteins to be closely packed in an area of the lipid bilayer without coming into contact with other proteins in energetically unfavorable ways.[1]
Mutations in both Ras and Src have been observed in a number of cancer cells; it is thought that these mutations and the subsequent interruption of the signal transduction pathways predispose a cell to uncontrolled replication. When the presence of a mutation is detected a small protein named ubiquitin is attached to the damaged protein; this modification signals that the marked protein is to be destroyed. It is essential that the protein be destroyed before anaphase so that the damaged DNA is not passed on to other cells. The attachment of ubiquitin to a damaged protein is the first step of apoptosis, which is programmed cell death.
[edit] Integral Proteins
As mentioned earlier, integral proteins, also known as intrinsic proteins, are strongly and permanently bounded to the membrane. One or more parts of these proteins are embedded in the phospholipid bi-layer of the membrane. They exhibit strong interaction with the membrane because their amino acid residues contains hydrophobic side chains that interact with the hydrophobic interior (fatty acyl groups) of the phospholipid bilayer. Because of their strong hydrophobic interaction with the hydrophobic core of the membrane, such proteins can only be dissociated from the membranes using detergents, non-polar solvents, or sometimes denaturing agents. Lastly, it is important to note that integral proteins account for a significant fraction of the proteins encoded in the genome.
There are two basic categories for integral proteins.
These proteins span across the entire membrane. They are the most common among integral proteins.
Transmembrane proteins
They may cross the membrane only once or several times, weaving in and out. The two kinds of transmembrane proteins are alpha-helical and beta-barrels.
The former is the more common of the two and can be found in the inner membrane of bacterial cells or the plasma membrane of eukaryotes. Voltage-gated ion channels, such as potassium and chloride channels, are examples of alpha-helical transmembrane proteins. They are mostly composed of hydrophobic amino acid residues and little hydrophilic residues, such as charged and polar residues. The polar carbonyl oxygen in the backbone doesn’t project outwards the helix, but rather towards the inside, facilitating and strengthening hydrogen bonds within the helix. Van der Waals interactions hold the tertiary and quaternary structures together in the transmembrane region. These interactions allow for flexibility in the structure to accommodate for necessary functions. Two polar residues that are found most frequently in the TM backbone are serine and threonine which can potentially hydrogen bond to the helical backbone. This hydrogen bonding captures polar side chains in a hydrophobic environment, such as a lipid bilayer. The polar side chains in turn hydrogen bond to other helices. Two residues, glycine and proline, known as helix breakers in water make kinks in the helix which play significant roles in functional mechanisms. [2]
Beta-barrels present in the outer membranes of Gram-negative bacteria, cell wall of Gram-positive bacteria, outer membrane of mitochondria and chloroplasts. Porins are examples of a beta-barrel transmembrane protein. They cross cellular membrane and acts as a pore through which molecules can diffuse. Transmembrane proteins can further be categorized into Type I and Type II. In Type I, the N-terminal is positioned on the exterior of the membrane. In Type II, the C-terminal appears on the exterior of the membrane.
Human VDAC
VDAC (voltage-dependent anion channel) is an example of a transmembrane protein found in the mitochondrial outer membrane which provides the pore for substrate diffusion. VDAC is composed of 19 β-strands which make up the β-barrel and a partial α-helix strand totaling 20 strands in the unit. The first and last β-strands of the β-barrel are parallel, while the strands in-between are anti-parallel.[3]
Isoform 1 of VDAC, three high-resolution structures in fact, in detergent micelles and bicelles have been recently published from solution NMR and X-ray crystallography. This helps to solve the membrane topology of VDAC and gives the first eukaryotic β-barrel membrane protein structure. Something different about this integral membrane protein was that it had parallel β-strand pairing and an odd number of strands. The voltage gating mechanism of VDAC and its modulation by NADH are given a structural and functional basis from studies. Since VDAC-1’s de novo structure and six more proteins, the amount of integral membrane protein structures found by solution NMR has doubled in the past two years.[4]
These proteins are permanently bounded to the membrane but only from one side. Many of these proteins are enzymes. Examples include cyclooxygenase and carnitine O-palmitoyltransferase. The former is an enzyme that is involved in the formation of prostanoids. Anti-inflammatory drugs, such as aspirin and ibuprofen, work to relieve symptoms of inflammation and pain by inhibiting this enzyme. The latter is a mitochondria transferase enzyme that participates in the metabolism of palmitoylcarnitine into palmitoyl-CoA.
Peripheral proteins, also known as extrinsic proteins, lack interaction with the hydrophobic interior of the phospholipid bi-layer. Because they lack hydrophobic interaction with the membrane, they can be detached from the membrane much more easily than integral proteins. Dissociation of peripheral proteins can be achieved through treatment with a solution of high pH or high salt concentration. Instead, peripheral proteins attach to the membrane via electrostatic and other non-covalent forces. Typically, they are either attached to the membrane indirectly via interaction with integral proteins, or directly through interaction with the polar heads of the phospholipid (amphitropic). Some peripheral proteins exhibit both types of interaction. These include certain kinases and G proteins. Other examples of peripheral proteins are the regulatory protein subunits of ion channels and transmembrane receptors.
Membrane Protein Functions
Due to the nature of the lipid bilayer, many molecules cannot enter or exit the cell because of size or charge. Membrane proteins function to assist in the transportation of such molecules across the lipid bilayer. Trans-membrane proteins participate in either passive or active transport.
Ubiquitin and Membrane Protein Transport[edit | edit source]
Insertion of ER into lipid bilayer causes newly synthesized integral membrane proteins to be sorted, transferred, and qualitatively maintained. This process is controlled by ubiquitination, a posttranlational redirection of commands which relate to biosynthetic delivery of proteins to the plasma membrane. This process can be followed through the secretory pathways. Ubiquitination can also be used to regulate the deletion of proteins from the plasma membrane through a endocytic pathway. Ubiquitination of integral membrane proteins often is enough to edocytically target the Plasma membrane protein. However, there are still certain functions such as sortin and degradation which fully requires ubiquitin.
This control and change of specific membrane proteins is due to the ubiquitin changing the quality or quantity of the integral membrane protein. As a side effect, defects in this process can also contribute to detrimental diseases such as cystic fibrosis.
Ubiquitin modification can influence cargo trafficking, mechanisms of quality control/maintenance in secretory/endocytic pathway.
Ubiquitin in Membrane Transport and Quality Control in Endoplasmic Reticulum[edit | edit source]
Ubiquitin transformations do not affect the regulating effects preformed by the ER. However, Ubiquitin activity is initialized during the Endoplasmic Reticulum- associated degradation process, or ERAD, because ubiquitin ligase is needed. This procedure is important because it is responsible for the removal of proteins which are not folded properly. The substrates which are subject to this procedure are relocated to the cytoplasm, waiting to be removed.
The ERAD targets are first ubiquitinated and must negotiate protein Ubx, a ubiquitin-binding protein. This process shows that that ubiquitation of the ERAD substrates provides a signal which is necessary for targeting the protein for degradation. This shows that ubiquitin plays a vital in protein membrane protein transport. It is important to note that ERAD functionalities also provide a key quality assurance aspect. A kink in this procedure could can cause detrimental side effects; this means that the ERAD procedure is monitored carefully and therefore the membrane protein transfers can be assured for quality.
Ubiquitin in protein quality control to regulate Protein Membrane Protein Composition[edit | edit source]
It is found that ERAD can affect and strengthen communication between ER and Golgi complexes. This can be accomplished by degrading retention factors of the ER. In Cholesterol depleted conditions, cells were ubiquitinated and degraded. This showed that the protein resulted left the ER and was sent into the Golgi for packing.
Conversely, the GAT protein within the Golgi complex contains three surfaces which can bind ubiquitin very well. This causes successful binding of ubiquitin and speed up the transferring of GAT proteins from Golgi to the ER. In addition, the polymerization protein cargo and ubiquitin provides the necessary driving force for localization to the Protein membrane.
Ubiquitin in the turnover of Plasma Membrane proteins[edit | edit source]
In endocytosis in yeast, ubiquitin is required for almost all processes. It is beneficial that there is a more than sufficient supply of ubiquitin within the yeast. The internalization of protein cargoes that are present in yeast are generally all ubiquitin mediated.
Quality Maintenance at the Plasma Membrane[edit | edit source]
Plasma Membrane protein contains a protective mechanism which are driven by intrinsic factors of the protein. Plasma Membranes also places a limit on the amount of proteins which exhibit error folding. A certain amount over a lifetime span is placed and plasma membrane proteins are there to regulate these levels. This quality check shows that these specific proteins must control integral membrane proteins and the removal of the damaged and misconstructed proteins. Despite the constrained understanding of the chemical process, quality maintenance mechanisms must usually include capabilities such as: the function to refold or fix the damaged protein, and the ability to distinguish healthy and damaged proteins.
As with proteins in the cytoplasm or in aqueous environments, proteolytic processing is key to cellular function in both the cytoplasm and in the lipid bilayer. However, intramembrane proteases present a different challenge to work with than water-soluble proteins. Scientists have been working on methods to decipher the molecular mechanisms of families of intramembrane proteins. Specifically, site-2 intramembrane metalloprotease and serine intramembrane protease rhomboid share common characteristics. The active sites of both families of proteases are entrenched in the membrane. However, to effectively cleave a membrane protein or any other protein, water must be introduced to hydrolyze the peptide bonds. These proteases often recognize a specific sequence of residues and thus cleave proteins at specific sites. To introduce water to the site of cleavage, there is a delivery system to connect the aqueous environment to the site of cleavage.
Molecules are allowed to flow down their concentration gradient. In most cases, this does not require a special protein. However, in facilitated diffusion, molecules that are insoluble in the lipid bilayer or too large to pass through is assisted in crossing the cell membrane through special transport proteins. Examples of facilitated diffusion are amino acids and ions.
Passive transport.
The other types of passive transport, which do not require proteins because the molecules diffuse directly through the cell membrane, are osmosis, diffusion, and filtration.
Facilitated diffusion.
Uniporters are the proteins that move molecules in passive transport. They can either be channel proteins or carrier proteins. Channel proteins open in response to a stimulus and let molecules flow freely through. Carrier proteins bind to a molecule, making it hydrophobic enough to cross the membrane. The following image shows the two kinds of uniporters and how they function.
Energy is expended to transport a molecule up its concentration gradient. There are two types of active transport, primary and secondary. Both involve going against a concentration gradient using ATP, but they differ in how the ATP is used by the protein.
ATP is expended to move a molecule up its concentration gradient. An example of this is the sodium-potassium pump, which pumps both ions against their concentration gradients in order to create a membrane voltage potential.
ATP is not directly coupled to the molecule of interest in secondary active transport. Instead, another molecule is moved up its concentration gradient, which generates an electrochemical gradient. The molecule of interest is then transported down the electrochemical gradient. While this process still consumes ATP to generate that gradient, the energy is not directly used to move the molecule across the membrane, hence it is known as secondary active transport.
Two main types of protein are involved in secondary active transport: antiporters and symporters.
Antiporter
The molecules move in opposite directions. One type of molecule enters the cell while the other exits. An example is the sodium-calcium exchanger, which removes calcium ions from the cell while allowing sodium back in. The sodium is pumped out by the sodium-potassium pump, which generates the concentration gradient required for this to work.
Symporter
The molecules move in the same direction. This usually works by allowing an ion to move down its electrochemical gradient. The other molecule piggy-backs off that movement and goes against its concentration gradient.
Fluidity of Membrane proteins Biological membrane are flexible. This flexibility is attained by the fluidity of the protein. The fluid mosaic model allows lateral movements called the lateral diffusion, and sometimes the transverse diffusion or flip flop can occur, which takes longer time to take place.
Lateral diffusion is the movement of the lipid laterally which is very rapid, unless there is restriction by special interaction.
Lateral Diffusion.
Flip-flop or Transverse diffusion is the condition is when transition of a molecule from one membrane surface to the other occurred. It is a very slow space compared with the lateral diffusion.It happens once in several hours.
Despite the many advances made in the study of membrane proteins, not much is known about the role of the environment in determining membrane protein structure or function because these proteins are easily affected by changes in their environment. The main problem remains in the difficulty of creating an environment that promotes a protein's native functions and structures. However, advances in the study of the influenza virus, more specifically the M2 protein, is giving more insight to this complex challenge.
The M2 protein is a homotetramer with 3 functional domains: the N-terminal, the TM helix, and the C-terminal. Until recent discoveries, drugs were effective in the blocking of the TM helix, which prevented proton conductance functions and thus disabling the virus. However, with recent outbreaks of the H1N1 virus and swine flu, the structure of this protein was scrutinized in 3 different environments, each using a different methodology. The influence of the environment on the proteins can then be seen in the comparison of these 3 results obtained.
The 1st imaging technique, solid-state NMR, concluded that the M2 was stable in a lipid bilayer environment. The drug amantadine was later added giving the protein a 4-fold symmetry structure further indicating more stability in the presence of amantadine.
The 2nd image, crystal structures, not only compared structures at differing pH levels but also showed that membrane proteins can access a range of conformational states.
Finally, the 3rd image made by solution NMR concluded that the membrane protein's amino acids interact to minimize electrostatic potentials and that water, when present, allows for hydrogen bond exchange.
Further screening of this protein is still undergoing, and much has yet to be revealed in the study of the environment's influence on protein structure. However, it is easily seen why this topic remains an important and popular issue. By understanding the environment's influence on membrane proteins, researchers are able to develop drugs to inhibit, for example, the influenza virus. Even mutations such as the H1N1 virus can be disabled as long as researchers have a key understanding of their membrane protein and how they can be manipulated and changed by their environments. The study of the M2 protein will eventually lead to a deeper understanding of other membrane proteins and how they are changed by changes in their environment.
Membrane places a most important role in the human body. It affects strongly in each structure environment. Every times, we talk about membranes, we have to mention protein structures because they related with each other. Proteins also known as amino acids that function in our body. The membrane and amino acid are the main function in the human body to help our body alive. They are supporting each other to form the right structure and sequences in each other to form the right structure and sequences in each part inside the body. Amino acid sequence allows the interpretation of some of the many studies on the chemical and mechanism of the membrane transport protein.
There are different kind of membrane in our body and each of them has different structure and function which also relate to amino acid. For example, integral membrane proteins are present in a heterogeneous environment that poses major obstacle for existing structure methodologies. Each structure could function as different environment and how the bonds are related. It is very difficult to obtain membrane mimetic environments that support the native structures, dynamic and functions of a membrane protein. Membrane protein often necessary to use detergents to mimic the nature lipid bilayer environment. In order to successful understand in which environment they are functioning, we have to know the bonding structure. Bonding is very important in each structure because it connects elements and one or more structures to each other. Nonetheless, it also very important to understand how to break the bond and forming a new bond. That is a reason why it very helpful to know the bond angles and stability of the bonding. Furthermore, by understanding the bonding structure help the scientists study about the differences kind of diseases and medicine to cured all the diseases. Lipid bilayers is a thin membranes. Lipid bilayers have a unique role in characterizing the native structures of membrane proteins and validating structures determined in other membrane mimetic environments.
Indeed, many proteins are membrane proteins which have the function in the cell. The cell need to communicate with the exterior or passing through the cell membrane. Many proteins go to membrane are glycoprotein related. Proteins are very difficult to study because the structures and functions are very complex. However, some proteins function can be predicted.
The study of membrane proteins have been complicated by the difficulty of examining the proteins by X-ray crystallography. Thus far, scientists have been able to examine the detail of their interactions between membrane components and their relative functions by computational simulations of the proteins in the membranes.
The questions of the stability of a membrane protein have eluded scientists. One particularly difficult task relates to studying the reversible transitions between different states. These interactions have been studied thermodynamically and yielded information pertaining to helix-helix interactions and the types of approaches to membrane protein stability. The stability between proteins and lipids have been simulated by methods such as simulating the free energy cost of burying specific amino acid side chains in the bilayer. Atomistic simulations have made these efforts possible including divulging information on complex membrane proteins such as ion channels.
The difficulties in studying the membrane proteins are mainly due to the difficulties of handling of proteins and experimental challenges associated with working with membrane protein. Also, the co-studying of isolated protein molecule and the molecular environment in order to have an appropriate understanding of the system makes it even more difficult to study.
For instance, isolating the protein from remainder components in the biological system is crucial in structural determination. But in order to have any proper thermodynamic analysis, it must include ALL relevant components of system, particularly paying close attention to boundaries where energy is exchanged.
Traditionally while studying membrane proteins, scientists remove the lipids surrounding the membrane proteins in their preparations. Now, scientists recognize the significance of the lipids as important additives for crystallization. Currently scientists have been more successful solving membrane protein structures with the addition of the lipids during analysis. These successes have led to an increasing number of membrane protein structures which bind lipid molecules to become readily visible and possible to classify.
Membrane proteins are assembled into complexes that allow these intricate assemblies to allow complexity that is not possible using single polypeptides. These complex assemblies allow membrane proteins to have many functions involving regulatory mechanisms and chemical reactions. The existence of these membrane protein complexes prevents potential problems such as unwanted interactions, aggregations, or the formation of hazardous intermediates. Furthermore, these complexes are mechanistically invaluable because they follow a process in which parts of the complexes are "pre-fabricated" and replaced in isolation if damaged, meaning that the whole complex does not need to be replaced if only one subunit it damaged. Membrane protein complexes has been analyzed through the use of blue native polyacrylamide gel electrophoresis and split-ubiquitin method.
Membrane protein complexes allow the avoidance of problems such as those listed above (unwanted interactions, aggregations, or the formation of hazardous intermediates) by being assembled in an ordered, even sequential, manner. To understand that the formation of these complexes are ordered, one would need to know what the assembly intermediates are. Thus, the larger the proteins, the more difficult it is to expose the formation order. However, smaller complexes such as cytochrome bo3 of E. Coli complex have allowed scientists to understand that membrane protein complexes follow a linear pathway of assembly. the bo3 complex is made up of four subunits that assemble through two intermediate complexes. It is understood that bo3 assemble linearly because thought it is possible for other intermediates to form leading to the formation of bo3, they are not observed and there is only one assembly pathway indicating that the intermediates follow a sequential, ordered path. Non-linear assembly would be noticeable because there would be several different assembly pathways. Ordered formation is also seen with cell division in divisomes whereby if one protein is missing, all downstream proteins are preventing from interacting properly. Scientists believe that this sort of ordered pathway exists to protect the cell from potentially harmful intermediates.
Chaperones also play a large role in the formation of these complexes. Chaperones act as physical assembly factors that interact with proteins and prevent unproductive interactions from occurring. For instance, chaperones prevent aggregation in the F1 compound of yeast F1F0-ATP synthase. Two chaperones bound to the alpha and beta subunits bind to the hydrophobic interfaces and guide the alpha and beta subunits into a3b3 complex assembly. Research has also shown that the loss of chaperones in some intermediates could be responsible for the activation of a membrane protein complex. It is important that an intermediate remain inactive so that unregulated activity by partially assembled complexes does not occur.
Membrane protein complexes are believed to undergo dynamic exchange as a mechanism for regulating damaged subunits within the complexes. Dynamic exchange allows the assembly of newly imported proteins into complexes to replaced damaged proteins without replacing the entire complex. An example of this is seen with photosystem II chloroplasts whose D1 subunits that become photo-damaged and is replaced as part of its repair mechanism. Dynamic exchange, at first, was only carried out in vitro. Scientists were only able to conclude that dynamic exchange is a possible repair mechanism, but could not conclude that it was what actually occurred in vivo. It was not until the use of fluorescent microscopy that scientists were able to confirm that dynamic microscopy did occur to a degree. Fluorescent microscopy tagged proteins and watched its interactions in vivo. Subunits were seen freely diffusing into and out of complexes. Future research hopes to disclose which proteins are being exchanged and why thee proteins undergo dynamic exchange.[5]
In the article Membrane Protein Structure: Prediction versus Reality, Arne Elofosson and Gunnar von Heijne discussed several current techniques used to predict the insertion and folding of membrane protein; they depicted a realistic and pragmatic view on how those techniques are used and the limitations. They also pointed out unresolved major issues concerning those techniques.
Arne and Gunnar first pointed out alpha-helix bundle and beta-barrel are the two main structures of membrane proteins. While the helix bundle represents about 20% to 25% of all open reading frame, the barrel form represents a few percent of all open reading frame. An open reading frame. ( A reading frame refers to DNA/RNA that can be broken into three letter codon and be transcribed into protein, while an open reading frame refers to a DNA sequence that does not contain a stop codon in its reading frame. ) The similarities between the helix bundle and beta-barrel is that, in order to fit the basic structure of lipid bilayer in membrane, they both contain hydrophobic amino acids in the middle of the protein. The major difference between the two is their secondary structure. The helix bundle is a complex long trans-membrane protein that packs several alpha-helixes; while beta-barrel protein has several beta-sheets rolled up, and it is shorter and less hydrophobic than the alpha helix bundle. Arne and Gunnar points out that the helix bundle form has been paid more attention as they are longer hence easier to be recognized than the beta-barrel.
Arne and Gunnar then depicts how helix-bundle and beta-barrels are synthesized and inserted into lipid bilayer. In the case of helix bundle translation, corresponding ribosome first bind to a translocon, which is a protein in the inner membrane responsible for the translocation of protein across the inner cellular membrane into the periplasm, called SecYEG translocon. Helix bundle is translated and inserted into the inner membrane. Depending on the hydrophobicity of the helix bundle, the interaction among helix bundles varies; either one helix bundle or a couple are synthesized at a time. Beta-barrel, due to the fact that it is less hydrophobic than helix bundle, could not get through the inner membrane just by itself; after its translation in the cytoplasm, it binds to SecB, with the help of SecA ATPase, via SecYEG translocon and transferred to the periplasm. It is inserted into the outer membrane via YaeT hetero-oligomeric outer membrane integration complex. After the membrane proteins are inserted into the lipid bilayer, it is believed that interactions among helix bundles are stronger than that with the lipids, hence the helix dandles are packed together and obtain its conformation. Hydrogen bonding between polar side chains also contributes in the conformation of the protein. Helix bundle and beta-barrel reaches rather stable conformation after inserted into the membrane. Nevertheless, some membrane protein exhibits a higher degree of flexibility, such as those that are in charge of proton or electron transfers.
According to Arne and Gunnar, in the case of helix-bundle membrane protein, the primary structure, hence its amino acid sequence has long been used to distinguish helix-bundle membrane protein from others. Due to the fact that lipid bilayer has hydrophobic character, the helix-bundle that is inserted into such lipid bilayer should consist of residues that are hydrophobic. Two essential amino acids, tryptophan and tyrosine, whose side chain contain aromatic structures contribute the hydrophobicity of helix-bundle membrane protein. The helix-bundle must also be long enough to span through the inner membrane; hence a helix bundle could have an averaged 10 to 20 hydrophobic residues. Loops connect the helixes; depending on whether the loops are facing inside or outside of the cell, the loops contain different amino acid compositions.
Membrane protein has been thought of perpendicularly orientated through the membrane, Arne and Gunnar points out that, membrane protein orientation could be more complex. One of the factors that contribute to the unexpected complexness of membrane protein comes from reentrant loops, as exhibited in the case of glutamate transporter. Concerning beta-barrel, Arne and Gunnar generalized a series of deducted structural principles; for instance, beta-stands have even numbers and tilts about 45 degrees in antiparallel fashion.
Arne and Gunnar surveyed a series of topology and structure prediction schemes in increasing complexness. 2D prediction is the earliest technique; such technique utilized the higher hydrophobicity possessed by trans-membrane protein than loop regions and has been an effective tool. One challenge faced by the 2D prediction is that the topogenic data from signal peptide and trans-membrane helices are similar, so it is hard to distinguish between the two.
In predicting the structure of β-barrel membrane proteins, scientists look for the existence of an N-terminal signal peptide and the protein’s general amino acid composition. Predicting the structure of a β-barrel membrane protein is simpler than that of a helix-bundle because its amino acid sequence is shorter and not as obvious to see.
As mentioned before, membrane protein cannot be simply modeled as all perpendicular through the membrane; reentrant loops are an important feature that elevates the complexity of membrane protein. These reentrant loops, as suggested by Arne and Gunner can be predicted by a recent developed topological technology named as 2.5D prediction. The residues in these reentrant loops, which come in long loops, medium length loops and short loops, usually are smaller than other parts of the protein so they are easily found in between the transmembrane helices. 2.5D membrane protein structure prediction predicts structures based on the type of amino acid sequence that it contains or by predicting how far the residue is from the center of the membrane protein. Characteristics of residues which can be predicted include lipid-exposed (hydrophobic) regions or lipid buried residues and kinks due to proline. Since 2.5D prediction was able to include sub-structures of membrane protein, such as the interfacial helices and reentrant loops, it is helpful in classifying membrane proteins.
3D structure prediction was first attempted via low-resolution experiments such as electron microscopy. Arne and Gunner points out like, 3D prediction of membrane protein, like all other globular proteins that have been tested against 3D predictions, has low accuracy. What has increased the difficulties in 3D prediction of membrane protein is that they sustain their structures in environments different from those of globular protein; also the globular protein that has been successfully predicted are much smaller than the membrane protein of interest.
To date, there are limited 3D models to be tested against, but there have been hypotheses of models to be tested against. One of which is homology modeling, which would potentially result in structures with details at the atomic level and with similar quality as of the models tested against globular proteins.[6]
In Unsolved Mysteries in Membrane Traffic, a paper written by Susanne R. Pfeffer, from the Department of Biochemistry in Stanford University, she explains how there are various hypotheses to how proteins travel and help facilitate transport within the cell but there isn’t a completely proven hypothesis yet. To start off, Soluble N-ethylmaleimide-sensitive factor attachment protein receptor proteins also known as SNARE proteins help facilitate the fusion of vesicles to their target membranes. There are two distinct groups of SNARE proteins. The first is the R- SNARE which is also called the v- SNARE group which is found on the vesicle. The second group of SNARE proteins is the Q- SNARE which is also called the t- SNARE due to the fact that it’s located on the target membrane. The main difference between these two proteins is that the R- SNARE will only be as a single protein on the vesicle whereas the Q- SNARE will form a complex of three Q- SNAREs. Under these two categories lie specific R-SNARE proteins that will pair up with specific Q-Proteins. The method of how these two pair up to facilitate fusion of a Golgi vesicle to the cell surface is still unknown but one can think of it as SNARE proteins being like puzzle pieces because they have certain specificity therefore one SNARE PROTEIN (R-SNARE/v-SNARE) will bind to only a specific SNARE complex (Q-SNAREs/ t-SNAREs). Although scientist still don’t know how the Golgi decides to transport these vesicles R- SNAREs and Q- SNAREs give clues as to what has arrived and what might depart. If there is a concentration of Q- SNAREs at a specific site that can be accounted for by noticing that there was previous fusion activity at that site. Now when we look at R- SNAREs there are two possible answers to why there are at a specific site, one can be because they have recently arrived and fusion just occurred or because a vesicle is about to depart the Golgi membrane. An important thing to note is that less membrane traffic occurs in the trans Golgi than does in the cis Golgi which is proved by the low concentration of SNAREs in trans Golgi rim and a higher in the cis Golgi rim. Therefore, with all these new ideas we must search for concrete answers to better understand how membrane trafficking occurs within cells.[7]
↑Hiller, S., Abramson, J., Mannella, C., Wagner, G., and Zeth, K., "The 3D structures of VDAC represent a native conformation," Trends in Biochemical Sciences, 2010.
↑The role of solution NMR in the structure determinations of VDAC-1 and other membrane proteins. Sebastian Hiller and Gerhard Wagner*
↑Assembly of Membrane Proteins into Complexes by Daniel O. Daleya,at Center for Biomembrane Research, Department of Biochemistry and Biophysics, Stockholm University, SE-106 91 Stockholm, Sweden, 5 June 2009.
↑Membrane Protein Structure: Prediction versus Reality.Annu Elofsson A, von Heijne G. Rev Biochem. 2007.76:125-40
↑Unsolved Mysteries in Membrane Traffic: Annu. Rev. Biochem. 2007. 76:629–45 Pfeffer, Suzanne R. Dept. of Biochemistry, Stanford